anemia hemolitica
TRANSCRIPT
UNIVERSIDAD NACIONAL EXPERIMENTAL “FRANCISCO DE MIRANDA”
ÁREA CS DE LA SALUDPROGRAMA: MEDICINA
MORFOFISIOPATOLOGIA III
Anemia Hemolítica
• Karen Malpica• Franklin
Chirino• Yaritza Villa• Jesús Arias
• Marelys Córdoba
• Mariangel Gallardo
• Joanni Duran
HEMOLISIS
La hemólisis (eritrocateresis) es el fenómeno de la desintegración de los eritrocitos (glóbulos rojos o hematíes). El eritrocito carece de núcleo y orgánulos, por lo que no puede repararse y muere cuando se «desgasta». Si la destrucción de los GR es superior a la velocidad de regeneración medular, sobreviene la anemia.
La anemia hemolítica es una afección en la cual hay un número insuficiente de glóbulos rojos en la sangre, debido a su destrucción prematura. Existen muchos tipos específicos de este tipo de anemia, los cuales se describen de manera individual.
ANEMIA HEMOLÍTICA

CLASIFICACIÓN
Según el Mecanismo de
Hemolisis.
Alteraciones Extrínsecas
(Extracorpusculares)
Mediadas por anticuerpos• Anemias Inmunohemolitica.Por traumatismos mecánicos en los eritrocitos.• Anemias hemolíticas microangiopáticas:
purpura trombótica trombocipénica, coagulación intravascular diseminada.
• Anemias hemolíticas por microtraumatismos en el corazón.
Infecciones: paludismo.Por agentes químicos: intoxicación por plomo.Por secuestro en el sistema mononuclear fagocitico: hiperesplenismo.

CLASIFICACIÓN
Según el Mecanismo de
Hemolisis.
Alteraciones Intrínsecas
(Intracorpusculares) de los eritrocitos.
HereditariasAlteraciones en las membranas de los eritrocitos.• Trastornos del citoesqueleto de la
membrana: esferocitosis, eliptocitosis.• Trastornos de la síntesis de lípidos:
aumento selectivo de lecitina en la membrana.
Déficit enzimático de los eritrocitos.Trastornos en la síntesis de la hemoglobina.• Déficit de síntesis de la globina:
síndromes talasémicos.AdquiridasDefecto de la membrana: hemoglobinuria paroxística nocturna.
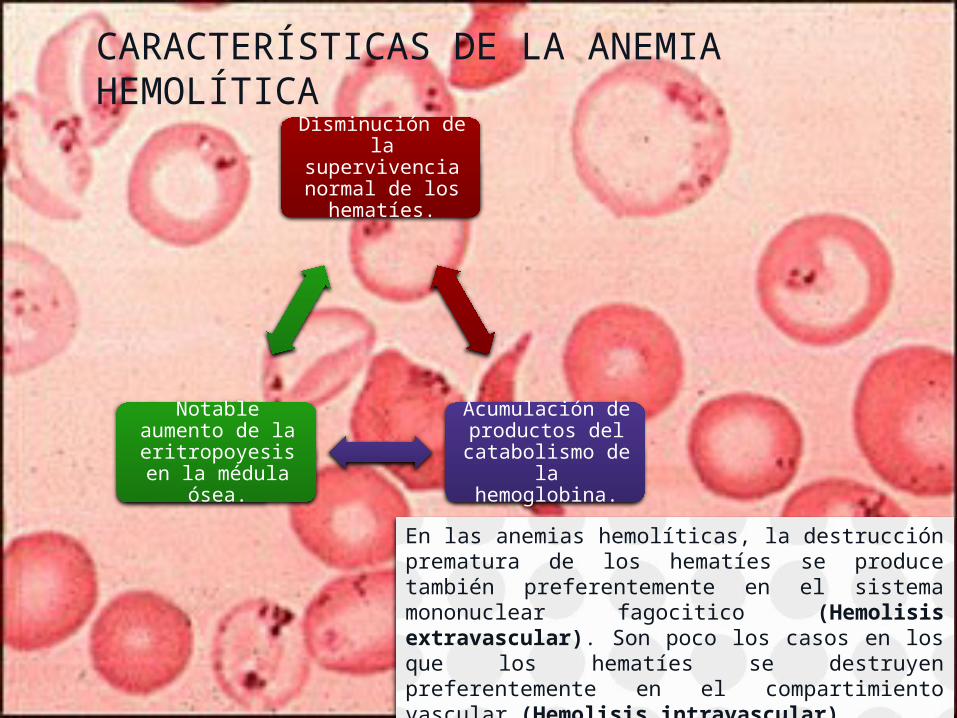
CARACTERÍSTICAS DE LA ANEMIA HEMOLÍTICA
Disminución de la supervivencia normal de los
hematíes.
Acumulación de productos del
catabolismo de la hemoglobina.
Notable aumento de la
eritropoyesis en la médula ósea.
En las anemias hemolíticas, la destrucción prematura de los hematíes se produce también preferentemente en el sistema mononuclear fagocitico (Hemolisis extravascular). Son poco los casos en los que los hematíes se destruyen preferentemente en el compartimiento vascular (Hemolisis intravascular).

HEMOLISIS INTRAVASCULAR
Se produce cuando los eritrocitos normales
sufren lesiones mecánicas, fijación del
complemento o una agresión de tóxicos
exógenos.
El traumatismo de los hematíes puede
ocurrir cuando hay prótesis valvulares
mecánicas o trombos alojados en la
microcirculación.
Los hematíes viven normalmente de 90 a 120 días en la sangre
circulante.
Hemoglobinemia.
Hemoglobinuria.
Metahemalbuminemia.Ictericia.
Hemosideriuria.
Se manifiesta por:

HEMOLISIS INTRAVASCULAR
• Cuando la hemoglobina pasa al plasma, se une inmediatamente a una globulina (haptoglobina) formando un complejo que impide su excreción por la orina, ya que esos complejos son eliminados rápidamente por el sistema reticuloendotelial.
• En todos los casos de hemolisis intravascular se observa una disminución característica de la haptoglobina sérica.
• Cuando la haptoglobina se agota, parte la hemoglobina libre o no unida se oxida rápidamente, formando metahemoglobina, y ambas hemoglobina y metahemoglobina, se excretan por los riñones proporcionando un color castaño rojizo a la orina (hemoglobinuria y metahemoglobinuria).
• Las células del túbulo proximal pueden reabsorber y catabolizar gran parte de la hemoglobina filtrada, pero el resto se escapa por la orina.
• El hierro liberado por la hemoglobina puede acumularse dentro de las células tubulares y producir hemosiderosis del epitelio tubular renal.
• Al mismo tiempo, los grupos HEM derivados de los complejos se catabolizan en el sistema mononuclear fagocitico, produciendo, en ultimo termino ictericia.
• En las anemias hemolíticas, la bilirrubina sérica no está conjugada y el grado de hiperbilirrubinemia depende de la capacidad funcional del hígado, así como la velocidad de la hemolisis.
• Si el hígado es normal, raras veces hay ictericia intensa. • La elevada excreción de la bilirrubina por el hígado al tubo digestivo produce
mayor formación y excreción fecal de urobilina.

HEMOLISIS EXTRAVASCULAR
Se produce siempre que los hematíes
sufren lesiones, se convierten en
elementos “extraños” o se vuelven menos
deformables.
Para que los hematíes atraviesen con éxito los sinusoides esplénicos deben adoptar cambios morfológicos de intensidad extrema, de modo que los hematíes poco deformables son incapaces de realizar ese transito y son secuestrados en los cordones del bazo, y seguidamente, fagocitados.
En la hemolisis extravascular es
evidente que no hay Hemoglobinemia,
hemoglobinuria ni los correspondientes
cambios intravasculares.
El catabolismo de los eritrocitos en las células fagocíticas provoca anemia e
ictericia que, por los demás, son
indistinguibles de las causadas por la
hemolisis intravascular.

HEMOLISIS EXTRAVASCULAR
Los niveles de
haptoglobina en
plasma están
siempre
disminuidos.
Los cambios morfológicos que aparecen son idénticos a los de
la hemolisis intravascular, excepto que la eritrofagocitosis
produce generalmente hipertrofia de las células del
sistema mononuclear fagocitico, y esto puede causar
esplenomegalia.

MORFOLOGÍA DE LA ANEMIA HEMOLÍTICA
Las anemias y descenso de la tensión de oxigeno en los tejidos estimulan
la producción de eritropoyetina.
Producen un notable aumento del numero
de normoblastos en la médula ósea.
A veces el estimulo es tan intenso que aparece
hematopoyesis extramedular.
La eritropoyesis compensadora acelerada va seguida de intensa reticulocitosis en la sangre periférica.
La gran cantidad de bilirrubina que se
excreta por el hígado favorece la formación de
cálculos pigmentarios (colelitiasis).
Si el proceso se cronifica, los hematíes o
la hemoglobina fagocitada acaban
produciendo hemosiderosis, que
habitualmente se limita al sistema mononuclear
fagocitico.

HEMOGLOBINURIA PAROXÍSTICA NOCTURNA
• A pesar de su rareza, este proceso ha fascinado a los hematólogos porque es el único ejemplo de anemia hemolítica que presenta un defecto adquirido, en lugar de hereditario, de la membrana de los hematíes.
• Las células sanguíneas les falta un gen : PIG-A este permite que el glucosil-fosfatidilinositol (GPI) ayude a que ciertas proteínas se fijan a las células.
• La ausencia de tallos GPI hace a la célula anormalmente susceptible a lisis mediada por complemento.
• Sin el PIG-A importantes proteínas no pueden fijarse a la superficie de las células y protegerlas de sustancias destructivas en la sangre.
• El trastorno de estos glóbulos rojos ocurre a nivel de las células pluripotenciales (mutación somática), por lo que es común ver afectadas a otras líneas celulares (plaquetas y granulocitos).

FISIOPATOLOGÍA
• Las células hematológicas son sensibles al
complemento porque carecen de la presencia
de sustancias que inhiben la acción del
complemento sobre la membrana del hematíe,
al faltar dicha sustancia pequeñas activaciones
del complemento aun fisiológicas pueden
ocasionar destrucción de la membrana no solo
de GR sino también de leucocitos y plaquetas:
pancitopenia.
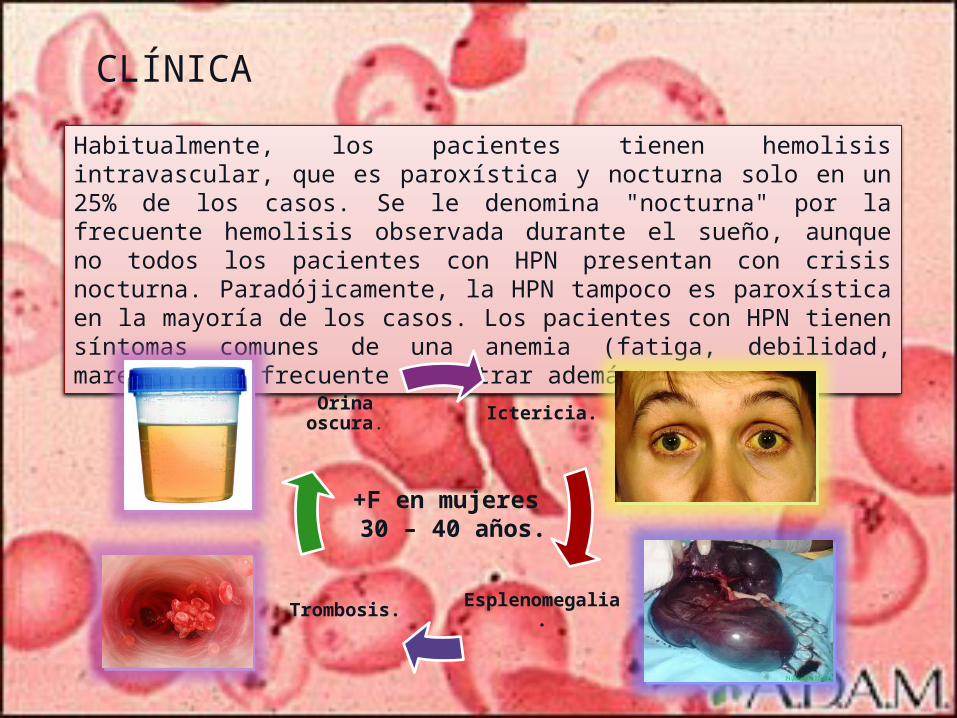
CLÍNICA
Habitualmente, los pacientes tienen hemolisis intravascular, que es paroxística y nocturna solo en un 25% de los casos. Se le denomina "nocturna" por la frecuente hemolisis observada durante el sueño, aunque no todos los pacientes con HPN presentan con crisis nocturna. Paradójicamente, la HPN tampoco es paroxística en la mayoría de los casos. Los pacientes con HPN tienen síntomas comunes de una anemia (fatiga, debilidad, mareos) y es frecuente encontrar además:
Ictericia.
Esplenomegalia.
Trombosis.
Orina oscura.
+F en mujeres 30 – 40 años.

DIAGNOSTICO• CSC: pancitopenia • Signos de hemolisis:• Reticulocitosis• Aumento de la LDH: como
consecuencia de la destrucción de hematíes.
• Aumento de bilirrubina indirecta. Por el mismo motivo de la LDH
• Hemosideriuria • Prueba COOMBS• Uroanálisis• Prueba de hemolisis de la sacarosa:
detecta aumento de la fragilidad de los GR al hincharse en una solución iónica baja
• Anemia hemolítica:• Aumento de LDH• Aumento de bilirrubina indirecta• Disminución de haptoglobina• Elevación de reticulocitos
TRATAMIENTO
• Esteroides: Inhibir el sistema inmunitario.
• Transfusiones sanguíneas.• Acido fólico, hierro. • Anticoagulantes para
prevenir la formación de coágulos.

ANEMIA INMUNOHEMOLITICA
• Las anemias hemolíticas de este grupo se deben a mecanismos extracorpusculares. Aunque se les puede llamar anemias hemolíticas autoinmunitarias, es mejor la denominación de anemias inmunohemoliticas porque, en algunos casos, la reacción inmunitaria se desencadena al tomar un fármaco.
• Cualquiera que sea el anticuerpo, para distinguir a las anemias inmunohemoliticas de otras clases de anemias hemolíticas es necesario demostrar los anticuerpos dirigidos contra los hematíes.

ANEMIA INMUNOHEMOLITICA POR AC CALIENTES
• Éstos suelen ser anticuerpos IgG que tienen la peculiaridad de activarse a la temperatura del cuerpo.
• Es la forma mas frecuente de anemia hemolítica inmunitaria. • El proceso es idiopático y primario en el 50% de los pacientes; en
el otro 50% hay un proceso predisponente de fondo o algún fármaco que ha provocado en forma secundaria de anemia hemolítica la mayoría de los autoanticuerpos son inmunoglobulinas G (IgG); solo a veces se encuentran anticuerpos IgA.
• +F en mujeres.• En esta forma de enfermedad hemolítica, el proceso que destruye
los hematíes no es, en su mayor parte, una hemolisis intravascular, sino que depende de que los hematíes recubiertos de IgG se unan a los receptores Fc de los monocitos y de los macrófagos del bazo, y sufran cambios que los convierten en esferocitos.
• Esos cambios se originan cuando los hematíes recubiertos de IgG pierden porciones de su membrana durante los intentos de fagocitosis.
• Seguidamente los esferocitos son secuestrados y eliminados por el bazo, el principal sitio donde se destruyen los hematíes en este proceso. De ahí que esta forma de anemia se caracterice por una moderada esplenomegalia.

ANEMIA INMUNOHEMOLITICA POR AGLUTINAS FRIAS
• Esta variedad de anemia Inmunohemolitica se debe a anticuerpos IgM caracterizados por su gran afinidad para unirse a los hematíes a temperaturas bajas (0 a 4 oC).
• Estos anticuerpos aparecen de forma aguda durante la fase de recuperación de ciertas infecciones, como la neumonía por micoplasmas y la mononucleosis infecciosa. También secundarios a neoplasias.
• Esta forma de anemia hemolítica desaparece espontáneamente y raras veces provoca manifestaciones de hemolisis.
• Las manifestaciones clínicas aparecen cuando el paciente se expone al frío, y desaparece al calentar las partes afectadas.
• La obstrucción vascular causada por los conglomerados hematíes produce palidez, cianosis de las partes expuestas al frio y fenómeno de Raynaud.

ANEMIA HEMOLÍTICA CONGÉNITA
Se reconocen tres tipos:1- Defectos de membrana del hematíe.2- Enzimopatías o trastorno del metabolismo del hematíe.3- Trastorno de la hemoglobina.
Defecto de membrana del hematíeNormal: GR presenta una capa externa de lípidos que aísla el hematíe e impide la permeabilidad excesiva al agua.
Por dentro de esta capa lipídica se encuentra un citoesqueleto de proteínas (proteína + importante: espectrina) que mantiene los lípidos en la membrana del gr y facilita la adaptación de la morfología del gr en las diferentes zonas de la microcirculación.

ESFEROCITOSIS HEREDITARIA
• Esta es la anemia hemolítica mas frecuente.• Este trastorno hereditario se caracteriza por un defecto intrínseco
de la membrana eritrocitaria, que vuelve esféricos a los hematíes, y vulnerables a su secuestro y destrucción por el bazo.
• En un 75% aproximadamente de los casos, la herencia es de tipo autosómico dominante. El resto de los pacientes tiene una forma autosómica recesiva de la enfermedad, que es mucho mas grave que la forma dominante.
• +F al norte de Europa.• Se producen glóbulos rojos en forma de esfera y se
descomponen en forma prematura (anemia hemolítica crónica).• Es un trastorno causado por un gen defectuoso. • A veces, el proceso se manifiesta en la primera infancia, pero es
frecuente que su diagnóstico no se haga hasta la vida adulta.

FISIOPATOLOGÍA• Trastorno en la proteína de membrana
(espectrina) que ocasiona una falta de fijación adecuada de lípidos a la membrana del hematíe.
• Como consecuencia de la pérdida de lípidos de membrana se produce un aumento de la permeabilidad de la membrana al sodio y al agua lo que produce hinchazón del hematíe que da lugar a la forma esférica del GR.
• Esferocitos carecen de capacidad de adaptación a la microcirculación, por lo que al llegar a los sinusoides esplénicos quedan atrapados y son destruidos.
• En las mutaciones que afectan la integridad del citoesqueleto, los hematíes bicóncavos normales pierden fragmentos de membrana. Para adaptarse a la perdida de superficie el GR adopta una forma esférica.
• Los esferocitos son menos deformables de lo normal y por eso son atrapados en los cordones del bazo y fagocitados por los macrófagos.

MORFOLOGÍA
• El rasgo mas llamativo de esta enfermedad puede que sea la forma esteroidea de los hematíes, que en las extensiones se manifiesta por unos eritrocitos anormalmente pequeños y que carecen de su habitual palidez central.
• La esferocitosis es característica, pero no patognomónica, pues también se ve en las anemias hemolíticas autoinmunitarias.
• Es característico de la EH un aumento moderado del bazo (500 a 1000 g); son pocas las anemias hemolíticas en la que la esplenomegalia es tan intensa o tan frecuente.
• Eritrofagocitosis en los cordones congestivos.

CLÍNICA
Anemia
EsplenomegaliaIctericia
En un 20 a 30% de pacientes, la enfermedad es casi asintomática porque la ligera destrucción de los hematíes queda compensada por el aumento de eritropoyesis.
Esta evolución clínica más o menos estable puede estar salpicada por las crisis aplásica (desencadenadas habitualmente por una infección por parvovirus de los precursores eritroides de la medula ósea), que provocan la interrupción temporal de la eritropoyesis; esto se manifiesta por un brusco empeoramiento de la anemia por desaparición de los reticulocitos en la sangre periférica.

DIAGNOSTICO Y TTO
• Antecedentes familiares.• Signos hematológicos.• Datos de laboratorio de la esferocitosis:
expresados por un aumento de fragilidad osmótica.
• Esplenomegalia.• Reticulocitos.• Aumento de la fragilidad osmótica del
GR.• Frotis de sangre periférica: esferocitosis.• CSC: muestra anemia.• Coombs directo: negativo.• Coombs indirecto: negativo.• Bilirrubina elevada y LDH: aumentado.• Esferocitos con un aumento de CHCM:
hay pérdida de la membrana del hematíe que ocasiona disminución de la superficie del mismo sin que haya trastorno en la formación de HB por lo que la concentración de HB de cada hematíe se encuentra elevado.
• VCM: normal o disminuido.
Depende de la evolución:• Transfusiones de
sangre para normalizar las cifras de hematíes.
• Esplenectomía para controlar la enfermedad.

ESPLENOMEGALIA
• Bazo normal no supone amenaza para los gr normales
• Bazo atrapa y destruye los glóbulos rojos con defectos
mínimos
Esplenomegalia:
• Exagera las condiciones adversas a la que se ven
expuestos GR.

ESPLENOMEGALIAS
SE CLASIFICAN:
• Enfermedades infiltrativas: trastornos
mieloproliferativos. Linfomas o enfermedades por
deposito: enfermedad de gaucher.
• Enfermedades inflamatorias sistémicas causantes de
hipertrofia esplénica.
• Enfermedades que causan esplenomegalia
congestiva.

ANTICUERPOS ERITROCITARIOS
• Hemolisis inmunitaria en el adulto: puede estar inducida
por tres tipos generales de anticuerpo.
• Aloanticuerpos: adquiridos por transfusiones sanguíneas o
embarazos y dirigidos contra los hematíes transfundidos.
• Ac reactivos a la temperatura corporal y dirigidos contra los
propios hematíes del paciente.
• Ac reactivos en frio y dirigidos contra los propios GR del
paciente.

ANEMIA HEMOLÍTICA DEL RECIÉN NACIDO
• Se presenta con mayor frecuencia cuando una madre con sangre factor RH negativo concibe un bebé con un padre RH positivo.
• Esta situación puede provocar graves problemas si los glóbulos rojos del feto llegan a estar en contacto con la sangre de la madre, aunque este no es un suceso común en el transcurso de un embarazo normal, excepto durante el parto, cuando la placenta se desprende y la sangre del bebé entra en contacto con la sangre de la madre.
• La incompatibilidad Rh afecta al 5% de los matrimonios. Un 10% de las madres Rh negativo se sensibiliza después de su primer embarazo; el 30% lo hacen después del segundo embarazo, y un 50% con posterioridad al tercero. El riesgo de sensibilización post aborto es 2%, y después de un aborto provocado es de un 4 a un 5%.
La eritroblastosis fetal, también llamada enfermedad hemolítica del recién nacido (HDN) es un trastorno sanguíneo en la que una madre produce anticuerpos durante el embarazo que atacan los glóbulos rojos de su propio feto, cuando la madre y el bebé tienen tipos de sangre Rh diferentes. En este caso la madre tendría sangre Rh negativo y el feto Rh +, heredado del padre.

ANEMIA HEMOLÍTICA DEL RECIÉN NACIDO
• La razón de este problema es que el sistema inmune de la madre considera a los eritrocitos Rh positivo del bebé como "extraños", y dignos de ser atacados por el sistema inmune materno, el cual responde desarrollando anticuerpos para combatir los glóbulos rojos del bebé.
• Una vez desarrollado el ataque contra los antígenos Rh positivos el sistema inmune de la madre guarda esos anticuerpos de forma indefinida por si dichas células extrañas vuelven a aparecer en contacto con la sangre materna, y con esto se produce la "sensibilización Rh" de la madre.

ANEMIA HEMOLÍTICA EN EL RECIÉN NACIDO
• Lo más preocupante de esto es que los anticuerpos de la madre atacan y destruyen los glóbulos rojos del feto (hemólisis), y esto da por resultado que el bebé se vuelva anémico y como consecuencia el cuerpo del bebé intenta producir más glóbulos rojos de forma más rápida, para compensar la deficiencia producida por la hemólisis inmune, esto hace que sus órganos se agranden, siendo perjudicial para su desarrollo.
• Además de esto, a medida que los glóbulos rojos se destruyen, se forma una sustancia llamada bilirrubina que es difícil para los fetos deshacerse de ella. Es posible que la bilirrubina se acumule en su sangre, tejidos y fluidos corporales, trastorno que se denomina hiperbilirrubinemia, por el que la piel y los tejidos del bebé se tornan amarillentos.
Kernicterus:Forma más grave de exceso de bilirrubina y se produce por su acumulación en el cerebro. Puede provocar convulsiones, dañar el cerebro, producir sordera y la muerte.

DIAGNOSTICO
• Laboratorio• COOMBS

LABORATORIO
• Bilirrubina indirecta: Elevada.
• Haptoglobulina sérica: Baja.
• Hemoglobinuria.
• Hemosiderina en la orina.
• Aumento del urobilinógeno urinario y
fecal.
• Conteo de reticulocitos absoluto
elevado.
• Conteo de glóbulos rojos y
hemoglobina bajos.
• LDH en suero elevado.

TRATAMIENTO
Durante el embarazo.• Transfusiones intrauterinas de glóbulos rojos en la
circulación del bebé.
Después del nacimiento.• Transfusiones de sangre y/o líquidos por vía endovenosa.• Ayuda respiratoria por medio de oxígeno o respirador
mecánico y exsanguíneo.• En casos excepcionales, si la incompatibilidad es grave y el
bebé se encuentra en peligro, se puede realizar una serie de transfusiones especiales de sangre (denominadas exanguinotransfusiones) mientras el bebé está en el útero materno o después del parto.
-La Hb normal está constituida por cuatro cadena globina y cuatro núcleos de HEM.-Hematíe adulto: 96% de la Hb esta constituida por Hb A1, formada por dos cadenas alfa y dos cadenas b, un 3% por Hb a2 y 1% Hb F.
Los síndromes talasémicos son un grupo heterogéneo de trastornos mendelianos, caracterizados todos ellos por la falta de síntesis de una de las dos cadenas de globinas, alfa o beta, de la
HbA.
La talasemia beta consiste en un déficit de síntesis de la cadena beta, mientras que la
talasemia alfa se debe al déficit de síntesis de la
cadena alfa.
Las consecuencias hematológicas de la disminución de la síntesis de una cadena de globina dependen no solo de la baja concentración de hemoglobina intracorpuscular (hipocromía), sino también del exceso relativo de la otra cadena.
Por ejemplo, en la talasemia beta, sobran cadenas alfa. Como consecuencia, las cadenas alfa sobrantes tienden a agregarse y formar inclusiones dentro de los eritrocitos y sus precursores, provocando la destrucción prematura de los eritroblastos que están madurando en la medula ósea (eritropoyesis ineficaz) y la lisis de los hematíes maduros en el bazo (hemolisis).
El defecto hemolítico es provocado por el exceso de cadena de globina que se sintetiza normalmente, que al no poder unirse a la cadena de globina que se sintetiza defectuosamente, precipita en el interior del hematíe, ocasionando la lesión del mismo y una hemólisis, además de eritropoyesis ineficaz. La herencia es autosómica recesiva.
SÍNDROMES TALASÉMICOS

Beta Talasemias
TALASEMIA MAYOR
Los genes de la talasemia beta son los más frecuentes y la talasemia mayor es la variedad más común en los países mediterráneos, partes de África y el Sureste Asiático.Gran disminución de síntesis de cadena b ocasiona un descenso de síntesis de Hb A1, con aumento de la formación de Hb A2 y Hb F.
Anemia grave produce aumento de eritropoyetina que da lugar a hiperplasia de médula ósea: malformaciones óseas en niños (pseudoquistes en manos y pies, deformidad de cráneo como cráneo en cepillo.
Aumento de la Hb F la cual tiene mayor afinidad por el oxigeno ocasiona una defectuosa oxigenación hacia los tejidos y por tanto hipoxia tisular crónica que aumenta la formación de eritropoyetina produciéndose mayor hiperplasia medular y un aumento de absorción de Fe++ con la consiguiente hemosiderosis secundaria.
SÍNDROMES TALASÉMICOS

CLÍNICALa evolución clínica de la talasemia beta mayor es breve porque los niños, salvo que sean transfundidos, sufren retraso del crecimiento y fallecen a temprana edad por las graves consecuencias de la anemia.
La afectación cardiaca debida a la progresiva sobrecarga de hierro con hemocromatosis secundaria es una causa importante de muerte incluso en los pacientes que, por lo demás podrían sobrevivir gracias a las transfusiones.
Las manifestaciones clínicas pueden producirse partiendo de las alteraciones morfológicas y hematológicas. Si sobreviven lo bastante, la cara de los pacientes se vuelve mas grande y algo deforme. Suele haber hepatoesplenomegalia.
Las transfusiones de sangre no solo mejoran la anemia, sino que también suprimen los fenómenos secundarios relacionados con la eritropoyesis excesiva.
TALASEMIA MAYOR

Diagnóstico
• Tratamiento
•Sospechar en un paciente con hemólisis congénita severa, microcitosis e hipocromía.Se confirma:
•Electroforesis de Hb: descenso de Hb A1 y aumento de HbA2 y Hb F.
•Con transfusiones y quelantes, muchos pacientes llegan al tercer decenio de vida.
•Elección: Trasplante de médula ósea ya que se trata de una enfermedad genética.Esplenectomía.
TALASEMIA MAYOR

Esta forma es mucho mas frecuente que la talasemia mayor, y afecta lógicamente a los mismos grupos étnicos.
Los individuos con rasgo talasémico permanecen casi siempre asintomáticos, y la anemia, si la tienen es leve.
En el frotis de sangre periférica suelen encontrarse algunas alteraciones de los hematíes, como hipocromía, microcitosis, punteado basófilo y dianocitico.
La medula ósea muestra una ligera hiperplasia eritroide. Un hallazgo característico en la electroforesis de la hemoglobina es el aumento de la HbA2, que puede representar el 4 a 8% de toda la hemoglobina (normal, 2.5 +/- 0.3%).
Los niveles de HbF pueden ser normales o ligeramente altos.
TALASEMIA MENOR O RASGO TALASÉMICO

Diagnóstico
El diagnostico del rasgo de talasemia beta es importante por dos motivos:
1. Para distinguirlo de la anemia hipocrómica microcítica por déficit de hierro.
2. Para el consejo genético.
La importancia de distinguir el rasgo talasémico de una anemia ferropénica reside en que esta ultima mejora administrando hierro, mientras que aquel puede empeorar.
La distinción suele lograrse determinando el hierro sérico, la capacidad total de fijación de hierro y la ferritina sérica.
TALASEMIA MENOR O RASGO TALASÉMICO

Son las formas clínicas más raras. Estos trastornos se caracterizan por la síntesis disminuida o nula de las cadenas de globina alfa.
Como normalmente hay cuatro genes de la globina alfa, la gravedad de los síndromes clínicos es muy variable, según el número de genes afectados..
La disminución de síntesis de cadena alfa produce una formación de tetrámeros de cadena gamma (Hb Barts) en los recién nacidos y tetrámeros de cadenas beta (Hbh) en adultos.
ALFA TALASEMIA
GRACIAS…