anemia hemolitica-enzimopatias
TRANSCRIPT

Universidad Autónoma de Puebla.
Ciencias Químicas Q.F.B.
Teoría de Hematología I.
Trabajo: Anemia Hemolítica (enzimopatias)
Integrantes: Viridiana Martínez Ramos Tania Balderas HernándezJoel Lara OcotitlaIxchel España CarreraJose Alberto Aldape AguayoAnahi Llanes Sánchez

Anemia HemolíticaEs una afección en la cual hay un número insuficiente de glóbulos rojos en la sangre, debido a su destrucción prematura.

Los eritrocitos, además de transportar el oxígeno y el dióxido de carbono en la sangre, realizan otras importantes funciones metabólicas. Su metabolismo se reduce a las rutas glucolítica y de las pentosas fosfato, al ciclo del 2,3-bisfosfoglicerato y a unas pocas reacciones de oxidorreducción y de la ruta de rescate de nucleótidos.

Las anemias hemolíticas se caracterizan por:

El acortamiento de la supervivencia normal de los hematíes, es decir, la destrucción prematura de los hematíes

La acumulación de los productos del catabolismo de la hemoglobina

Un notable aumento de la eritropoyesis en la médula ósea, en un intento de compensar la pérdida de hematíes que se manifiesta clínicamente por aumento del índice de formación de reticulocitos a más del triple de lo normal.

Anemia hemolítica es una afección en la cual hay un número insuficiente de glóbulos rojos en la sangre, debido a su destrucción prematura.
1)No hay datos morfológicos que sugieran una alteración de la membrana eritrocítica2)No existe evidencia sugestiva de trastorno en la síntesis dela hemoglobina 3)La reacción de Coombs directa es negativa, descartando la presencia de algún fenómeno inmunológico.




ClasificaciónLas anemias hemolíticas se pueden dividir en tres maneras. La causa de la destrucción acelerada de los hematíes puede considerarse debida a: Un defecto molecular (hemoglobinopatía o enzimopatía) intrínseco al hematíeUna alteración en la estructura y función de la membrana Un factor ambiental, como los traumatismos mecánicos o la acción de un autoanticuerpo. Los procesos hemolíticos se pueden clasificar en hereditarios y adquiridos.

EnzimopatiasLas deficiencias en algunas de las enzimas que catalizan las reacciones de dichas rutas originan enzimopatías, porque, al carecer de núcleo y orgánulos celulares, no son capaces de compensar el defecto enzimático sintetizando las enzimas, ni utilizar otras rutas alternativas.

Los eritrocitos maduros no tienen núcleo, mitocondrias, ni retículo endoplásmico, por ello no pueden sintetizar proteínas, glucógeno ni lípidos y son incapaces de llevar a cabo la fosforilación oxidativa. En consecuencia, su metabolismo, muy reducido, queda limitado.

Enzimopatias de las rutas metabólicas

Las enzimopatías en los eritrocitos provocan alteraciones en las funciones de estas células, porque al carecer de la maquinaria de síntesis de proteínas y poseer un metabolismo muy sencillo, no son capaces de compensar el defecto enzimático produciendo la enzima mutada, ni de utilizar otras rutas alternativas.

Aldolasa (ALD)
La deficiencia en la aldolasa (ALD) causa una anemia moderada que se agrava por el aumento de la concentración de F1,6BP en los eritrocitos y con infecciones en el tracto respiratorio. Se han descrito tres isoenzimas codificadas por distintos genes, siendo la isoenzima A la única presente en los eritrocitos. El residuo de ácido aspártico situado en la posición 128 tiene un papel primordial en la estructura espacial y en la función catalítica de la enzima.

Triosa fosfato isomerasa (TPI)
La deficiencia en triosa fosfato isomerasa (TPI) origina un conjunto de manifestaciones clínicas, desde anemia hemolítica y anormalidades neuromusculares a complicaciones cardíacas que, en niños, conducen inevitablemente a la muerte. La enzima mutada de eritrocitos muestra poca actividad y estabilidad y provoca la acumulación de dihidroxiacetona fosfato.

Fosfoglicerato quinasa (PGK)
La fosfoglicerato quinasa (PGK) es la única enzima de la ruta glucolítica codificada por un gen localizado en el cromosoma X. Las manifestaciones clínicas más usuales de una enzimopatía de esta clase son anemia hemolítica, alteraciones neurológicas y retardo mental. Se conoce la estructura primaria de la enzima de eritrocitos humanos que consta de 417 aminoácidos.

Terapia
Se puede afirmar que no existe un tratamiento definitivo en las enzimopatías eritrocitarias. La terapia que más se ha utilizado en niños para paliar los efectos de una deficiencia, cuando los valores de hemoglobina son muy bajos, son las transfusiones de sangre.

Después de la niñez muchos pacientes se adaptan a un nivel compensado de hemólisis que no suelen requerir transfusión, a menos que existan otras complicaciones como un estado infeccioso, embarazo u otras condiciones.

El trasplante de médula ósea puede aliviar los síntomas en células hematopoyéticas, debería incluir la transferencia de la enzima desde células de la médula ósea normales a células deficientes, bien por contacto célula- célula o por liberación de la enzima en el plasma. Se demostró que trasplante de médula ósea es eficaz, al menos, en niños.

DEFICIT DE PIRUVATO QUINASAEl déficit de piruvato quinasa, se trata de un trastorno genético de herencia autosómica recesiva.También es conocida como anemia hemolítica no esferocitica

Es un trastorno en el cual los glóbulos rojos se destruyen mas rápido de lo que la medula ósea pude producirlos (hemolisis).


La PK es la enzima que cataliza una de las etapas más importantes de la glucólisis transformando el fosfoenolpiruvato (PEP) a piruvato, proceso en que se produce una molécula de ATP

FISIOPATOLOGIA
El déficit de PK en los glóbulos rojos es una enfermedad autosómica recesiva causada por mutaciones en el gen PKLR que se localiza en el cromosoma 1 (1q21).

Altera la capacidad energética del eritrocito, dificultando la formación o utilización del ATP.Como consecuencia se produce un aumento en la concentraciones intracelulares de todos los metabolitos de la ruta por encima del bloqueo, particularmente 2,3 difosfoglicerato.Cuando disminuye la capacidad energética del eritrocito éste envejece prematuramente y es eliminado de la circulación sanguínea

Conforme un glóbulo rojo deficiente de PK envejece hay una progresiva reducción de la glicólisis que va en paralelo a la gradual degradación de la enzima lo que lleva a una depleción de ATP y a hemólisis

Manifestaciones clínicas
Hemolisis crónica Ictericia Esplenomegalia Reticulocitosis

DIAGNOSTICO El diagnostico se basa en la en la
presentación clínica (anemia, cambios en el bazo como aumento de tamaño y congestión, ictericia…)
Citometria hemática Recuento de reticulocitos Niveles de bilirrubina Medición de la actividad enzimática de
la PK

DIAGNOSTICO G6PD
Nivel de bilirrubina Hemograma Determinación de actividad G6PD sobre
papel de filtro mediante tecticas fluorescentes.
Método de indofenoldiclorofenos (DPIP). Se detecta de G6PD por decoloración de marcador en un tiempo especifico

ANEMIA HEMOLÍTICA POR DEFICIENCIA DE
G6PD

los glóbulos rojos carecen de núcleo y pierden sus mitocondrias en la medida en que maduran
utilizan vías alternativas para mantener estables los niveles de ATP y de poder reductor necesarios para cumplir sus funciones vitales

La glucosa-6-fosfato deshidrogenasa (G6PD) interviene en la primera reacción de la ruta de las pentosas.
catalizando la conversión de glucosa 6-fosfato (G6P) en 6- fosfogluconato (6PG) y obteniendo NADPH a partir de la nicotinamida adenina dinucleótido fosfato (NADP).
principal fuente de obtención de NADPH en los eritrocitos humanos

Deficiencia de G6PD: Variantes
De acuerdo con su nivel de actividad, las variantes de la enzima, se agruparon en cinco clases, que son:
Clase 1: Deficiencia severa de la enzima con anemia hemolítica crónica no esferocítica (CNSHA).
Clase 2: Deficiencia enzimática severa (1- 10% actividad residual) asociado con anemia hemolítica aguda.
Clase 3: Deficiencia enzimática moderada (10%- 60%, actividad residual).
Clase 4: Actividad Normal (60%-150%). Clase 5: Actividad enzimática por encima de lo normal (>150%).

DEFICIENCIA DE GLUCOSA-6-FOSFATO DESHIDROGENASA
es un desorden hereditario ligado al cromosoma X. El defecto es causado por mutaciones en el gen G6PD
se manifiesta cuando hay alteraciones en el ambiente:ingestión de sustanciasInfecciones
Deficiencia se expresa por completo
Heterocigota.
La actividad enzimática media de la G6PD puede ser: normal moderadamente reducida deficiente

coenzima NADPHEs un donante de
electrones.Glutatión
Eritrocitosglutatión reducido
Eritrocitos maduros
protege a los eritrocitos de lesiones por agentes oxidantes :anión superóxido (O2)
peróxido de hidrógeno (H2O2)
radical hidroxilo (OH)
oxidantesAcumulación genera daño: lípidos y proteínas celulares.
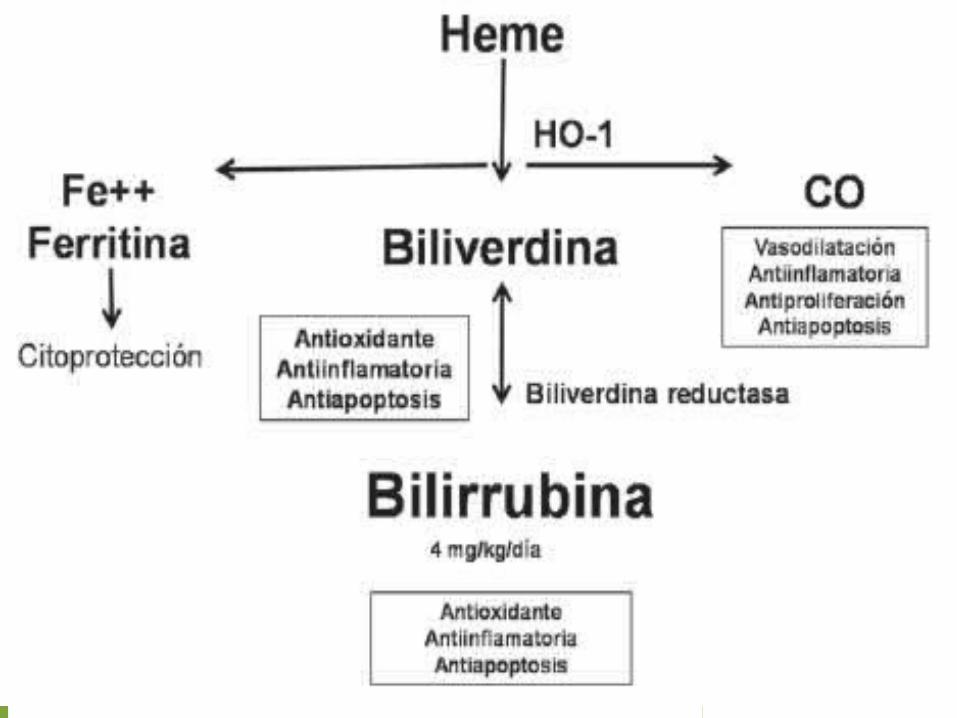
Este proceso es evitado por glutatión reducido (GSH) el cual convierte el peróxido de hidrógeno (H2 O2 ) en agua (H2O) a través de la enzima glutatión peroxidasa.
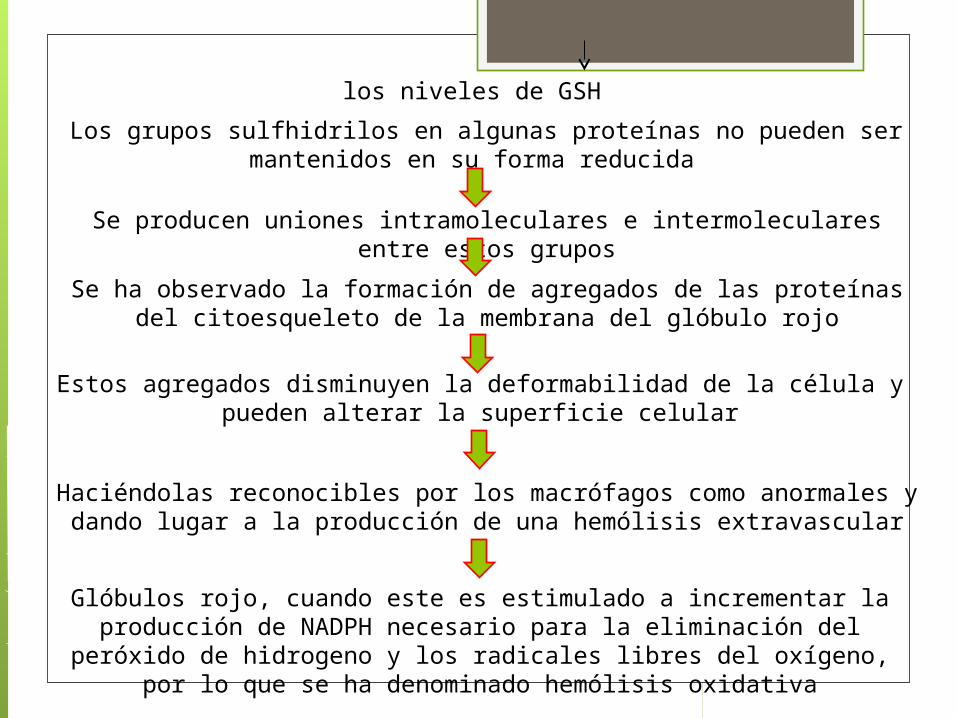
los niveles de GSH
Los grupos sulfhidrilos en algunas proteínas no pueden ser mantenidos en su forma reducida
Se producen uniones intramoleculares e intermoleculares entre estos grupos
Se ha observado la formación de agregados de las proteínas del citoesqueleto de la membrana del glóbulo rojo
Estos agregados disminuyen la deformabilidad de la célula y pueden alterar la superficie celular
Haciéndolas reconocibles por los macrófagos como anormales y dando lugar a la producción de una hemólisis extravascular
Glóbulos rojo, cuando este es estimulado a incrementar la producción de NADPH necesario para la eliminación del peróxido de hidrogeno y los radicales
libres del oxígeno, por lo que se ha denominado hemólisis oxidativa

CARACTERÍSTICAS CLÍNICAS Anemia hemolítica congénita no esferocítica
El recién nacido es anémico y presenta íctero.
En ocasiones la concentración de hemoglobina es normal y la hemólisis está compensada
El estrés oxidativo producido por el déficit en la producción de NADPH por la deficiencia en la actividad de G6PD y, por consiguiente, en el mantenimiento de los niveles de glutatión reducido, puede llevar a una dramática caída en los niveles de hemoglobina.
una infección aguda o la administración de una droga oxidante precipita los episodios hemolíticos

Íctero neonatal
está asociada con la deficiencia de la G6PD.
está asociada con la deficiencia de la G6PD (variante deficiente con actividad enzimática muy disminuida).
La transferencia a través de la placenta de fármacos y compuestos químicos tomados por la madre ha estado implicada, en ocasiones, como la causa del íctero neonatal.
Lo que implica una relación directa con la presencia de un estrés oxidativo, provocado por una disminución en la defensa antioxidante del eritrocito.

Favismo
Hemólisis aguda que se desarrolla en algunos individuos después de la ingestión de habas.
Se puede aseverar que todo individuo que presenta favismo es deficiente de G6PD
Una hemólisis aguda donde el conteo de eritrocitos cae por debajo de 1,0 x 10 12/L.
Los glóbulos rojos son vistos cuerpos de Heinz.
Variante B- de la enzima G6PD

El glóbulo rojo sufre un daño oxidativo producido por un agente químico y entre los que se han identificado están: pirimidina, aglicón, divicina e isouramil en combinación con el ácido ascórbico

Anemia hemolítica inducida por infecciones
particularmente importante son la hepatitis tipo viral, la neumonía y la fiebre tifoidea.
la generación de peróxido de hidrógeno por los neutrófilos polimorfo nucleares puede provocar una disminución en la cantidad de glutatión reducido
cuya función es eliminar del glóbulo rojo la acumulación de metabolitos que oxidan a los grupos sulfhídrilos formados por el estrés oxidativo
por lo que disminuye la capacidad protectora de la célula.
la activación de los neutrófilos interviene directamente en la peroxidación de los lípidos de la membrana y provoca de forma directa la destrucción de la célula.

Caso clínico

Varón, 2 años y 7 mesespeso de nacimiento: 3.015 g
talla: 49,5 cmmadre e hijo con igual grupo sanguíneo O
Antecedente
Hospitalización 8 días en el período neonatal por hiperbilirrubinemia de 25,1 mg/dL sin causa evidente.
Caso clínico:
palidez progresiva dolor abdominal fiebre y vómitos.

Exámenes
revelaron anemia
a ecotomografía abdominal mostro hepatomegalia leve y dilatación de vía biliar mínima.

El paciente había ingerido habas frescas 48 h previo al inicio de los síntomas
Sin ingesta de fármacos.
Se planteó
probable Favismo.
Se manejó con hidratación intravenosa con volúmenes altos, se transfundió con glóbulos rojos 10 cc/ kg y ácido fólico oral.

Post transfusión quedó con Hto: 25%, Hb: 8,1 gr/dL, reticulocitos: 11,6%.
A las 48 h se mantenía estable con persistencia de los reticulocitos elevados 14,2%.
Test de detección cualitativo de G6PD (Tonz y Betke) confirmó la deficiencia de la enzima.
Evolucionó en buenas condiciones generales y al mes se encontraba sin anemia ni signos de hemolisis.