retinitis pigmentosa
TRANSCRIPT

RETINITIS PIGMENTOSA
Othman Al-Abbadi, M.D

Retinitis Pigmentosa
• Inherited diffuse retinal degenerative diseases• Most common hereditary fundus dystrophy (1/5.000)• Initially predominantly affecting the rod
photoreceptors, with later degeneration of cones• May occur as –AD –AR –XLR–Sporadic

Modes of inheritence
• XLR is the least common but most severe form, and may result in complete blindness by the third or fourth decades• AR disease can also be severe• Sporadic cases may have a more favourable
prognosis, with retention of central vision until the sixth decade or later • AD disease generally has the best prognosis

Symptoms
• Nyctalopia and dark adaptation difficulties are frequently presenting symptoms• Mid-peripheral then far-peripheral field loss• reduced central vision (late)• Cataract• Photopsia

Signs
• Triad of• bone-spicule retinal pigmentation• arteriolar attenuation• ‘waxy’ disc pallor

Progression
• Bilateral mid-peripheral intraretinal perivascular ‘bone-spicule’ pigmentary changes with arteriolar narrowing• Then a gradual increase in density of the
pigment • Anterior and posterior spread• Peripheral pigmentation may become severe,
with marked arteriolar narrowing and disc pallor

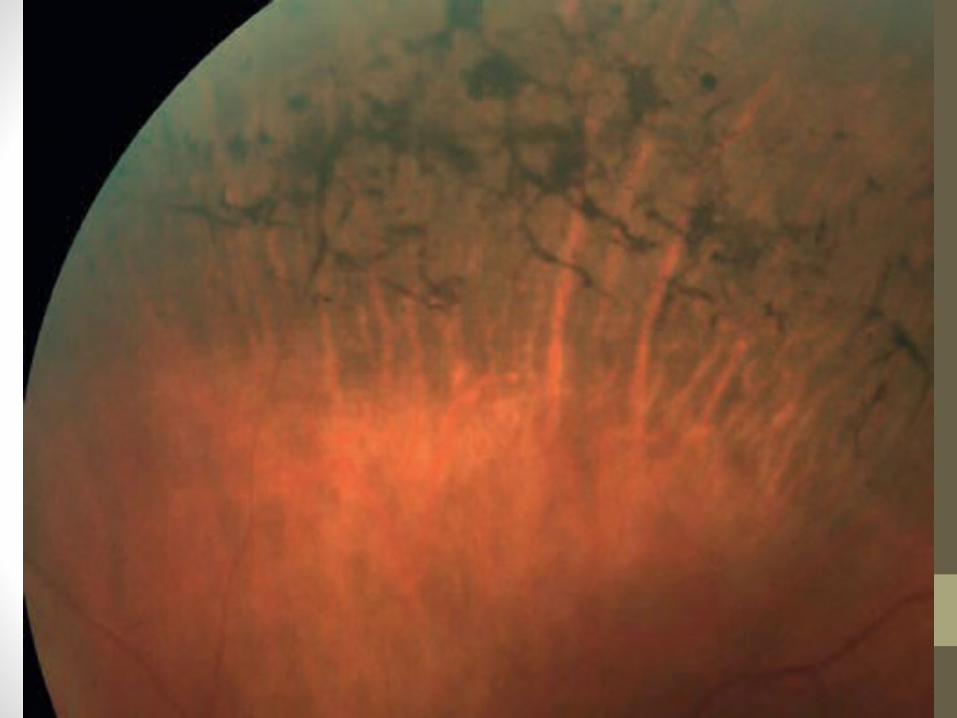


• The macula may show • Atrophy• epiretinal membrane formation• cystoid macular oedema (CMO)
• Myopia is common.• Optic disc drusen occur more frequently

Complications
• PSCC• OAG• Keratoconus • PVD• Intermediate uveitis• Exudative RD

Investigations
• ERG• EOG• Perimetry• OCT• Genetic analysis

ERG
• ElectroRetinoGram• Components• A wave photoreceptors• B wave muller cells + bipolar cells• C wave RPE

ERG
• Sensitive diagnostic test
• Early reduced scotopic rod and combined responses
•With progression photopic responses also reduce


EOG
• Measurement of standing potential between the cornea and the retina
measurement of function of the RPE and photoreceptors • Abnormal in RP• However, ERG is considered a more sensitive
test for detection of photoreceptor function and consequently EOG is not routinely done

Visual field
• Characteristically shows a ring scotoma in the mid-periphery of the visual field • Start as a group of isolated scotomas around 20
degrees from fixation, and gradually coalesce to form a partial followed by a complete ring• Useful in monitoring the progression of disease
and document the status of legal blindness

OCT
• to detect • CMO• Epiretinal membrane• vitreomaular traction

Treatment
• Many treatments have been explored without proven benefit• These include various vitamins and minerals,
vasodilators, tissue therapy with placental extract, cortisone, cervical sympathectomy, injections of a hydrolysate of yeast RNA, ultrasound, transfer factor, dimethyl sulfoxide, ozone, muscle transplants, and subretinal injections of fetal retinal cells

Atypical retinitis pigmentosa
• Syndromic• Usher syndrome• Kearns–Sayre syndrome• Bassen–Kornzweig
syndrome• Refsum disease• Bardet–Biedl syndrome
• Non-syndromic• Retinitis pigmentosa sine
pigmento• Retinitis punctata
albescens• Sector retinitis pigmentosa• Leber congenital
amaurosis• Pigmented paravenous
chorioretinal atrophy

Usher syndrome
• About 15% to 20% of affected individuals with retinitis pigmentosa have associated hearing loss • There are three major types;• Type I (75%) which features profound congenital
sensorineural deafness and severe RP with an extinguished ERG in the first decade plus unintelligible speech & vestibular ataxia• Type III (2%), with progressive hearing loss, vestibular
dysfunction and relatively late-onset pigmentary retinopathy

Kearns–Sayre syndrome
• Part of chronic progressive external ophthalmoplegia• Mitochondrial inheritance• Abnormalities include• Ptosis• diffuse disturbance of the RPE• ERGs that are usually reduced in amplitude• respiratory distress• heart block which may require a pacemaker

Bassen–Kornzweig syndrome
• Abetalipoproteinaemia• Malabsorption of fat and fat-soluble vitamin• Develops FTT in infancy• The fundus exhibits scattered white dots
followed by RP-like changes developing towards the end of the first decade; there may also be ptosis, ophthalmoplegia, strabismus and nystagmus

Refsum disease
• The patient accumulates exogenous phytanic acid• Findings include a peripheral neuropathy, ataxia, an increase
in CSF protein with a normal cell count, and retinitis pigmentosa• All have elevated serum phytanic acid• A defect exists in the conversion of phytanic acid to alpha-
hydroxy phytanic acid which results in its accumulation• Treatment consists of restricting not only animal fats and
milk products (i.e., foods that contain phytanic acid) but also green leafy vegetables containing phytol

Bardet–Biedl syndrome
• Includes RP, mental retardation, polydactylism, apple-shaped obesity, and hypogonadism
• Almost 80% have severe changes by the age of 20 years

Retinitis pigmentosa sine pigmento• Sine pimento = Without pigment
• Absence or paucity of pigment accumulation
• May subsequently appear with time
• Functional manifestations are similar to typical RP


Retinitis punctata albescens
• Albescens = whitish• Scattered whitish-yellow spots, most
numerous at the equator, usually sparing the macula, and associated with arteriolar attenuation • Nyctalopia and progressive field loss occur


Sector retinitis pigmentosa
• Sectoral RP• AD• Characterized by involvement of inferior
quadrants only• Progression is slow (many cases are
apparently stationary)• Unilateral RP can also occur


Leber congenital amaurosis
• Severe rod-cone dystrophy• The commonest genetically defined cause of
visual impairment in children• ERG is usually non-recordable even in early cases• Systemic associations include• mental handicap, deafness, epilepsy, central
nervous system and renal anomalies, skeletal malformations and endocrine dysfunction

• Presentation• Blindness at birth or early infancy• associated with roving eye movements or
nystagmus• Photoaversion• Cataract• Hypermetropia • Nestagmus

• Signs are variable but may include:• Absent or diminished pupillary light reflex• The fundi may be normal in early life apart from mild
arteriolar narrowing• Initially mild peripheral pigmentary retinopathy, salt and
pepper changes, and less frequently yellow flecks• Severe macular pigmentation• Pigmentary retinopathy, optic atrophy and severe
arteriolar narrowing in later childhood• Oculodigital syndrome: constant rubbing of the eyes
may cause orbital fat atrophy with enophthalmos, and subsequent keratoconus or keratoglobus



Pigmented paravenous chorioretinal atrophy• Pigmented paravenous chorioretinal atrophy• Usually asymptomatic and non-progressive• ERG is normal• Paravenous bone-spicule pigmentation together
with sharply outlined zones of chorioretinal atrophy that follow the course of the major retinal veins• Changes may also encircle the optic disc• The optic disc and vascular calibre are usually
normal

