sorption of methanol/mtbe and diffusion of methanol in 6fda–oda polyimide
TRANSCRIPT

Sorption of Methanol/MTBE and Diffusion of Methanol in6FDA–ODA Polyimide
H. DENNY KAMARUDDIN,1 WILLIAM J. KOROS2
1 Air Liquide, MEDAL, L.P., 305 Water Street, Newport, Delaware 19804-2410
2 Department of Chemical Engineering, University of Texas at Austin, Austin, Texas 78712
Received 5 February 1999; revised 22 May 2000; accepted 24 May 2000
ABSTRACT: This article discusses the diffusion and solubility behavior of methanol/methyl tert-butyl ether (MTBE) in glassy 6FDA–ODA polyimide prepared from hexaflu-oroisopropylidene 2,2-bis(phthalic anhydride) (6FDA) and oxydianiline (ODA). Thediffusion coefficients and sorption isotherm of methanol vapor in 6FDA–ODA polyimideat various pressures and film thicknesses were obtained with a McBain-type vaporsorption apparatus. Methanol/MTBE mixed-liquid sorption isotherms were obtained byhead-space chromatography and compared with a pure methanol sorption isothermobtained with a quartz spring balance. Methanol sorption isotherms obtained with thetwo methods were almost identical. Both methanol sorption isotherms obeyed thedual-mode model at a lower activity, which is typical for glassy polymer behavior. TheMTBE was readily sorbed into the polymer in the presence of methanol, but the MTBEsorption isotherm exhibited a highly nonideal behavior. The MTBE sorption levels werea strong function of the methanol sorption level. Methanol diffusion in the polymer wasanalyzed in terms of the partial immobilization model with model parameters obtainedfrom average diffusion coefficients and the dual-mode sorption parameters. Simpleaverage diffusion coefficients were obtained from sorption kinetics experiments,whereas the dual-mode sorption parameters were obtained from equilibrium methanolsorption experiments. An analysis of the mobility and solubility data for methanolindicated that methanol tends to form clusters at higher sorption levels. © 2000 JohnWiley & Sons, Inc. J Polym Sci B: Polym Phys 38: 2254–2267, 2000Keywords: polyimide; sorption; diffusion; methanol; MTBE
THEORY
Sorption in Glassy Polymers
The physical properties of an amorphous polymerare significantly different in the glassy and rub-bery states. In the rubbery state above the glass-transition temperature, polymer chain segmentsare highly mobile, with a behavior similar to thatof a highly viscous liquid. The free movements of
the polymer segments result in the formation ofshort-lived microscopic transient gaps betweenpolymer chain segments. Below the glass-transi-tion temperature, the polymer chain segmentshave much lower mobility but are still able toperform subtle vibrational motions around moreor less fixed random lattice positions. This re-stricted motion in glassy polymers allows muchmore sensitive size and shape discrimination be-tween penetrants compared with the same poly-mer in the rubbery state. The abrupt loss of seg-mental mobility causes microscopic packing de-fects to be frozen into the matrix at the glass-transition temperature. The frozen microscopic
Correspondence to: W. J. Koros (E-mail: [email protected])Journal of Polymer Science: Part B: Polymer Physics, Vol. 38, 2254–2267 (2000)© 2000 John Wiley & Sons, Inc.
2254

packing defects are sometimes referred to as un-relaxed volume and indicate a nonequilibrium na-ture for glassy materials. The unrelaxed volumeprovides a driving force for the polymer chains topack better through a protracted aging process.Because the segmental motion of the polymerchain is minimal, it can take many years for thepolymer packing to approach the equilibrium con-dition. Sorption, diffusion, and permeation prop-erties can be affected significantly by such densi-fication of the polymer matrix.1–3
Sorption isotherms in rubbery polymers followHenry’s law behavior at low penetrant activity.Pure-component sorptions in glassy polymers canbe modeled with the simple dual-mode sorptionmodel, which idealizes glassy polymers as havingtwo distinct types of sorption environments, un-relaxed volume (defects) and dense matrix.4–9
Components sorbed in the unrelaxed-volume sitesare well described by a Langmuir isotherm,whereas those occupying the dense matrix aredescribed by a Henry’s law model at a low activ-ity. The dual-mode model for single-componentsorption is shown in eq 1, and equations for thebinary mixture components are shown in eqs 2and 3:
CA 5 kDApA 1C9HAbApA
1 1 bApA(1)
CA 5 kDApA 1C9HAbApA
1 1 bApA 1 bBpB(2)
CB 5 kDBpB 1C9HBbBpB
1 1 bApA 1 bBpB(3)
kDi is the Henry’s law constant that characterizessorption in the dense region of the polymer ma-trix, pi is the partial pressure of the component, biis a measure of the affinity of the penetrant forthe Langmuir packing defect sites, and C9Hi is theLangmuir capacity constant for component i.When mixtures that exhibit nonideal gas-phasebehavior are described, fugacity or activity in-stead of partial pressure should be used.
Fickian Diffusion
The diffusion of penetrant through a dense poly-meric matrix exhibits Fickian behavior when thetimescale of the diffusion process is either muchlarger or much shorter than the relaxation time of
the polymer chains. The kinetics of a Fickiansorption and desorption process for a single com-ponent in a flat sheet of thickness, ,, can be de-scribed with either an infinite series of incompleteerror function complement terms or an infiniteseries of exponential function terms for the case ofa concentration-independent diffusion coeffi-cient.10 For a concentration-dependent diffusioncoefficient, these expressions can still be used asreasonable approximations if the average appar-ent diffusion coefficient, Dap, for the concentra-tion interval of interest is used. For strongly con-centration-dependent cases, the appropriate rela-tion between Dap and the local concentration-dependent coefficient can be quite complex.
The error function form is often referred to asthe short-time solution because only a single term(eq 4) is required for Mt/M` # 0.6. However, theexponential form is often referred to as the long-time solution because one term of the infiniteseries is required, which leads to the simple ex-pression in eq 5 for Mt/M` $ 0.6. Either formreduces to eq 6 under the special condition ofMt/M` 5 0.6.
When the long-time analysis method is used,that is, plotting, [ln(1 2 Mt/M`)] versus [p2t/,2]gives a straight line for Mt/M` . 0.6 if Fickiankinetics apply. The slope of this plot is equal tothe apparent diffusion coefficient of the permeantin the polymer, Dap. The two methods can workequally well; however, when the value of the half-time is small, long-term analysis is the experi-mentally preferred method of the two:
Mt
M`5 4FDapt
p,2 G 0.5
(4)
lnF1 2Mt
M`G 5 lnF 8
p2G 2 Fp2tDap
,2 G (5)
t1/2 50.0492,2
Dap(6)
As mentioned previously, eqs 4–6 assume con-stant diffusivity, which is referred to as the ap-parent diffusion coefficient, Dap. For rubberypolymers, in the absence of plasticization andclustering, sorption and desorption should giveidentical apparent diffusion coefficient values.Nevertheless, higher values of the diffusion coef-ficient obtained from the sorption and desorptionexperiments does not prove that plasticization ofthe polymer by the penetrant is occurring. Any
DIFFUSION AND SOLUBILITY OF METHANOL/MTBE 2255

diffusion coefficient form that increases with in-creasing local concentration leads to a similarresult, with higher apparent diffusion coefficientsdetermined from sorption/desorption experi-ments. The so-called partial immobilizationmodel satisfies this criterion.11
For single-component diffusion that can be de-scribed adequately by the dual-mode sorptionmodel, the effective local diffusion coefficient, Deff,of the solute can be described by eq 7. By defini-tion, the diffusive flux is equal to the product ofDeff and the concentration gradient, dC/dx, of thepermeant, as shown in eq 8. The concentration ineq 8, C, is the total local sorbed concentration ofthe penetrant. The so-called partial-immobiliza-tion model expresses the flux in a mathematicallyconvenient form by attributing equal mobility tothe penetrant in the Henry’s law population and acharacteristic fraction, F, of the Langmuir popu-lation.4–7,12–13 Working with this so-called mobileconcentration, CM, simplifies mathematical ma-nipulations. This mathematical formalism is ef-fectively equivalent to a more physically appeal-ing physical situation in which the entire Lang-muir population has a lower intrinsic mobilitythan that of the Henry’s law population, but bothare linked by local equilibrium. The mobile con-centration of a penetrant in single-componentpermeation is defined in eq 9 in terms of the localfugacity, ƒ, of the component. If the concentrationgradient is based on CM, the diffusive flux is equalto the product of the mobile concentration gradi-ent and the diffusion coefficient of the Henry’s lawpopulation, DD (eq 9). Equations 8 and 10 aremathematically equivalent:
Deff 5
DDS1 1FK
~1 1 aCD!2D1 1
K~1 1 aCD!2
(7)
where a 5bkD
J 5 2Deff
dCdx (8)
CM 5 CD 1 FCH 5 CDS1 1FK
1 1 aCDD
5 kDfS1 1FK
~1 1 bf!D (9)
J 5 2DD
dCM
dx (10)
The simple average value of the diffusion coeffi-cients, Deff, obtained from the sorption and de-sorption experiments (eq 11) and the dual-modesorption parameters can be used to estimate thepartial immobilization parameters, DD and F.14
The parameter F 5 DD/DH equals the ratio of thediffusion coefficient of the Henry’s law and Lang-muir populations. The simple arithmetic averageof the apparent diffusion coefficients from sorp-tion and desorption (Deff) experiments is a usefulmeasure of penetrant diffusivity over a given pen-etrant concentration range between Ci and Cf :
Deff 5Dap
s 1 Dapd
2 (11)
In this case, a reasonable estimate for Deff can bemade with eq 12:
Deff 5
ECi
Cf
Deff~c! dC
ECi
Cf
dC
5
ECDi
CDf
Deff~c!dCdCD
dCD
ECDi
CDf dCdCD
dCD
(12)
where
dCdCD
5 1 1K
~1 1 aCD!2 (13)
Substituting eqs 7 and 13 into 12 and integratingthe conditions CDi
5 0 and CDf5 CD yields
DeffS1 1K
~1 1 bff!D 5 DDF
K~1 1 bff!
1 DD (14)
Equation 14 can be used to describe the kineticsof the sorption experiment in which the pene-trant-free polymer film is exposed to a step con-centration change by exposure of the sample to anexternal fugacity, ff . Plotting Deff (1 1 K/~1 1 bff!)versus K/(1 1 bff ), gives a straight line of slopeDDF and an intercept of DD.
2256 KAMARUDDIN AND KOROS

EXPERIMENTAL METHODS
6FDA–ODA polyimide was prepared in our labo-ratory by reacting hexafluoroisopropylidene 2,2-bis(phthalic anhydride) (6FDA) and oxydianiline(ODA) according to methods employed by Husk etal.15 The imidization of the polyamic acid wasdone chemically. A schematic of the polymer re-actions and its physical properties are illustratedin Figure 1. Polymeric films were prepared by6FDA–ODA polyimide polymer solutions beingdeposited inside a stainless steel casting ringplaced on top of a glass plate. The concentrationand amount of the polymer solution deposited canbe varied to obtain the desired film thicknesses.The glass plate was leveled with a bubble levelerprior to the depositing of the polymer solution toensure uniform film thickness. After the polymersolution was deposited inside the stainless steelcasting ring, a glass funnel was immediately usedto cover the casting ring. The end of the glassfunnel was covered with a KimWipet tissue toprevent dust from contaminating the polymer so-lution inside the casting ring during the evapora-tion step. The nascent membrane was dried atroom temperature overnight and placed in a vac-
uum oven at 100 °C for 24 h. The vacuum oventemperature was then increased to 285 °C andmaintained for 3 h. The polymer film thicknesseswere measured with a micrometer and by aknown area of the films being weighed. Bothmethods provided similar results.
Figure 2. Schematic of the McBain-type quartz-spring sorption apparatus.
Figure 1. Schematic of 6FDA–ODA polyimide and its physical properties.
DIFFUSION AND SOLUBILITY OF METHANOL/MTBE 2257
The equilibrium sorption isotherm and diffu-sion coefficients of methanol in 6FDA–ODA poly-imide at 40 °C were obtained with a McBainquartz-spring balance system. This system is of-ten used for investigating vapor sorption and dif-fusion into polymeric materials.16 The schematicof the sorption system is shown in Figure 2. Thesorption apparatus is vacuum-tight, and its maincomponents are a pressure transducer, quartz-spring, vapor-source vial, jacketed sorption cham-ber, and vacuum pump. The vapor sorption appa-ratus is made of glass, with the exception of thestainless steel flexible tubing connecting theBaratront pressure transducer to the apparatus.The sorption chamber is jacketed with water,which was connected to a temperature-controlledbath. The rest of the nonjacketed parts wereheated with an electrical-tape heater to preventcondensation of vapors during the sorption/de-sorption experiment. The absolute pressure in thesorption apparatus was measured with a Bara-tront pressure transducer.
At the start of each set of experiments, thepolymer film was left overnight inside the sorp-tion chamber under vacuum at 40 °C to ensurecomplete desorption of water vapor that might bepresent in the film. When the polymer film wasready, the valve leading to the vacuum pump wasclosed, and solvent vapor was carefully intro-duced to the sorption chamber until the desiredvapor pressure was reached. A liquid nitrogentrap was attached between the sorption appara-tus and the inlet of the vacuum pump to preventback diffusion of the vacuum pump oil vapors.The sorption kinetics and thermodynamics exper-iments were carried out with various film thick-nesses ranging from 21 to 67 mm.
EXPERIMENTAL RESULTS
The head-space gas chromatography experi-mental procedures utilized to obtain methanoland methyl tert-butyl ether (MTBE) mixed-liq-uid sorption isotherms is discussed in refs. 17and 18.
Methanol Vapor Sorption Isotherm
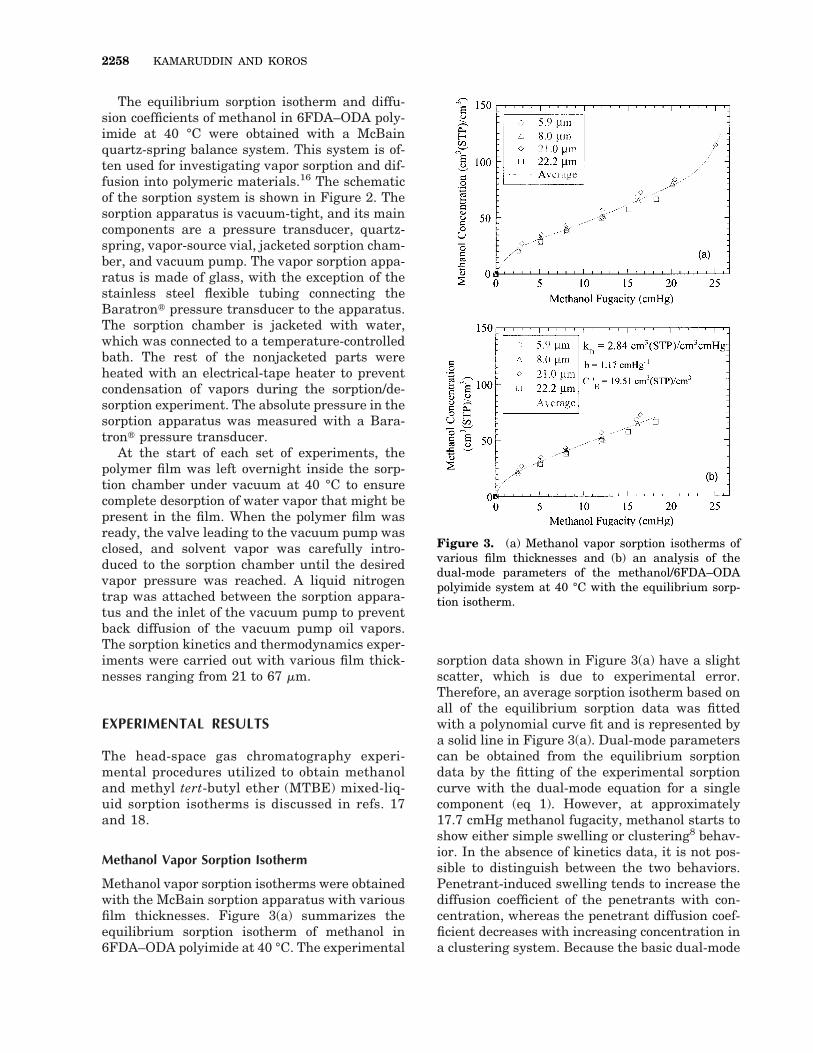
Methanol vapor sorption isotherms were obtainedwith the McBain sorption apparatus with variousfilm thicknesses. Figure 3(a) summarizes theequilibrium sorption isotherm of methanol in6FDA–ODA polyimide at 40 °C. The experimental
sorption data shown in Figure 3(a) have a slightscatter, which is due to experimental error.Therefore, an average sorption isotherm based onall of the equilibrium sorption data was fittedwith a polynomial curve fit and is represented bya solid line in Figure 3(a). Dual-mode parameterscan be obtained from the equilibrium sorptiondata by the fitting of the experimental sorptioncurve with the dual-mode equation for a singlecomponent (eq 1). However, at approximately17.7 cmHg methanol fugacity, methanol starts toshow either simple swelling or clustering8 behav-ior. In the absence of kinetics data, it is not pos-sible to distinguish between the two behaviors.Penetrant-induced swelling tends to increase thediffusion coefficient of the penetrants with con-centration, whereas the penetrant diffusion coef-ficient decreases with increasing concentration ina clustering system. Because the basic dual-mode
Figure 3. (a) Methanol vapor sorption isotherms ofvarious film thicknesses and (b) an analysis of thedual-mode parameters of the methanol/6FDA–ODApolyimide system at 40 °C with the equilibrium sorp-tion isotherm.
2258 KAMARUDDIN AND KOROS

model (eq 1) does not treat clustering or plastici-zation, the simple dual-mode parameters wereobtained with sorption data obtained only up to17.7 cmHg methanol fugacity. The solid line inFigure 3(b) represents the fitted dual-modemodel. A summary of the dual-mode and partialimmobilization parameters is presented in TableI. A discussion of the partial immobilization pa-rameters is presented later in the article.
Conditioning and Hysteresis of 6FDA–ODAPolyimide Caused by the Sorption of Methanol
Conditioning and hysteresis behavior caused bygas sorption in glassy polymers has been docu-mented by previous researchers.19–23 The totalsample volume of glassy polymers was increasedafter CO2 was sorbed at high pressure. It washypothesized that the increase in volume wascaused by disruption of chain packing in thedensely packed segments comprising the majorityof the polymer matrix (also referred to as Henry’senvironment in the dual-mode terminology).Sorption among the densely packed polymer seg-ments required dilation of the matrix to accom-modate the penetrant. Upon complete desorptionof the permeants, it is believed that some loca-tions in the dilated matrices were unable to com-pletely relax back to their original position,thereby leading to additional excess volume incomparison with the initial polymer state beforesorption. This phenomenon is referred to as con-ditioning. The permeability and solubility coeffi-cients of a conditioned polymer are higher thanthose of the fresh polymer for pressures lowerthan the conditioning pressure. The permeationproperties of a conditioned polymer changed onlyslightly over a period of 3 months, indicating thatthe sorption-induced defects were long-lived.21,23
The conditioning effects of methanol on the6FDA–ODA polyimide studied here appears sim-
ilar to CO2 conditioning of polycarbonates. Thesorption of methanol in fresh and methanol-con-ditioned 6FDA–ODA polyimide is shown in Fig-ure 4. The polymer was conditioned at a methanolfugacity of 25 cmHg. Figure 4 clearly shows thatthe sorption isotherm of the conditioned polyim-ide is higher than that of fresh polyimide. Subse-quent sorptions into the methanol-conditionedpolyimide after 48 h indicated only a slight addi-tional change in the sorption characteristics ofthe polymer. It seems reasonable that the cause ofthe methanol-conditioning effects on 6FDA–ODApolyimide was similar to what was proposed byFleming and Koros.19–21 Fleming showed that theLangmuir capacity constant, C9H, of CO2-condi-tioned polycarbonate increases as a function ofthe conditioning pressures, whereas the Henry’slaw and Langmuir affinity constants, kD and b,were constant. This seems to suggest that CO2conditioning increases the polycarbonate unre-laxed volume related to packing defects.
It was postulated that the sorption-induced de-fects resulting in the conditioning response weremore subtle than the preexisting Langmuir de-fects responsible for the concave sorption iso-therms seen in the fresh polymer samples.19–21
The size of these sorption-induced defects werebelieved to be smaller than the size of the pene-trant responsible for causing these molecular-sizedefects because of partial relaxation following re-moval of the penetrant. The hypothesis concern-ing the size of the sorption-induced defects wasbased on the swelling experiments as interpretedwithin the framework of the simple dual-modesorption model. This model assumes that swellingin glassy polymer is solely based on the sorption
Table I. Summary of the Methanol/6FDA–ODAPolyimide Dual-Mode and Partial ImmobilizationParameters at 40 °C
Parameters
kD 2.84 cm3 (STP)/(cm3cmHg)b 1.17 cmHg21
C9H 19.51 cm3 (STP)/cm3
DD 1.15 3 1029 cm2/sF 0.27
Figure 4. Conditioning effect of methanol at 25 cmHgon 6FDA–ODA polyimide.
DIFFUSION AND SOLUBILITY OF METHANOL/MTBE 2259
of the Henry’s law population, whereas the Lang-muir sorption does not contribute to polymerswelling (dilation). In this ideal limit, the volumedilation of the polymer as a function of the sorp-tion of A is shown in eq 15:
SDVV0
D 5 kDAfAS V̂A
22,400D (15)
where DV is the sorption-induced dilation and V0is the total volume of the unswollen polymer. Thedilation of the unconditioned polycarbonate as afunction of the CO2 fugacity showed a linear re-lationship, as suggested by eq 16. However, dila-tion data of CO2-conditioned polycarbonate didnot show a linear relationship as a function of theCO2 fugacity at lower fugacity. Moreover, the di-lation of the CO2-conditioned polymer is higherthan the predicted value, which suggests thatsorption-induced defects are smaller in size thanthe penetrant. Sorbing into these sorption-in-duced defects, therefore, can be envisioned to re-quire the dilation of the polymer chain, althoughto a lesser degree than that of sorption into asimple Henry’s law (dense) environment. There-fore, the physical sorption-related properties of aconditioned polymer are different from those ofthe unconditioned polymer. In a sense, in additionto the Henry’s law and Langmuir sorption sites, anew intermediate scale of sorption site is believedto be available in a conditioned polymer. This newsorption site possesses unrelaxed free volumethat is somewhat similar to the Langmuir site,but sorption into the site also requires some dila-tion of the polymer matrix, which is Henry-like.Clearly, these sharp distinctions are simplistic,but they are quite useful for considering the semi-quantitative evolution in morphology due to theconditioning of glassy materials such as thosestudied here. The increase in the polymer freevolume as a result of the sorption-induced defectsshould tend to lower the enthalpy of sorption ofthe conditioned polymer, and this expectation isindeed met, as discussed later.
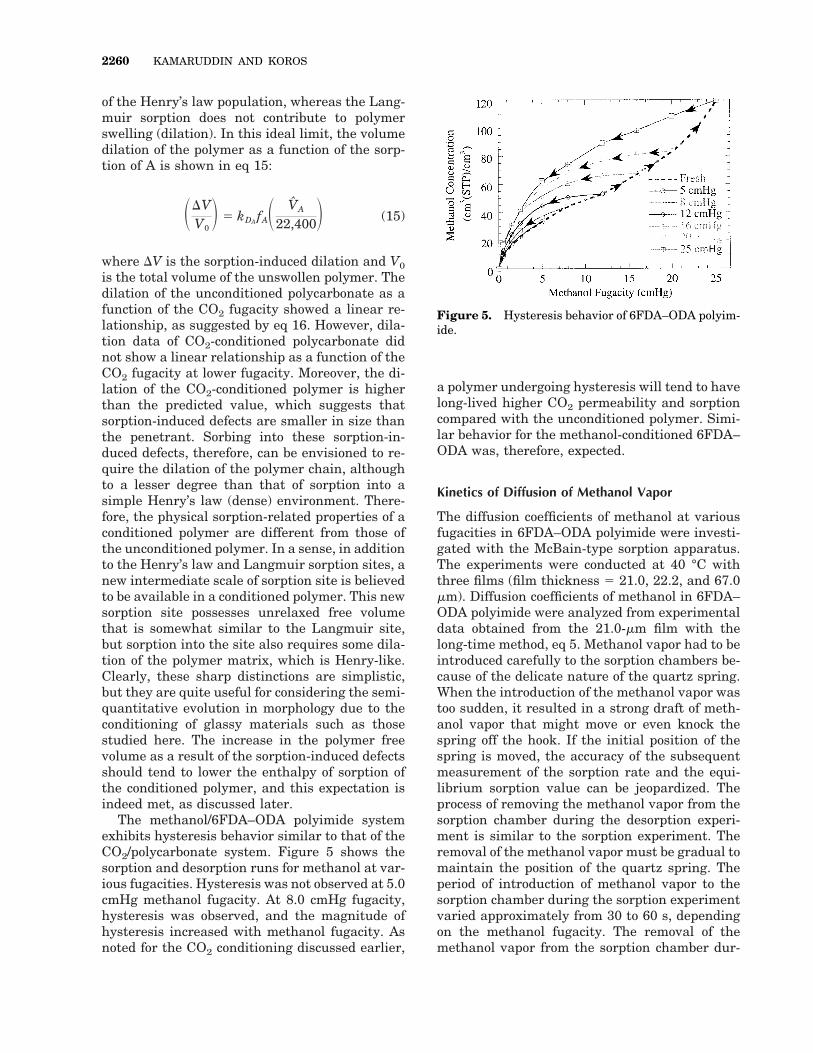
The methanol/6FDA–ODA polyimide systemexhibits hysteresis behavior similar to that of theCO2/polycarbonate system. Figure 5 shows thesorption and desorption runs for methanol at var-ious fugacities. Hysteresis was not observed at 5.0cmHg methanol fugacity. At 8.0 cmHg fugacity,hysteresis was observed, and the magnitude ofhysteresis increased with methanol fugacity. Asnoted for the CO2 conditioning discussed earlier,
a polymer undergoing hysteresis will tend to havelong-lived higher CO2 permeability and sorptioncompared with the unconditioned polymer. Simi-lar behavior for the methanol-conditioned 6FDA–ODA was, therefore, expected.
Kinetics of Diffusion of Methanol Vapor
The diffusion coefficients of methanol at variousfugacities in 6FDA–ODA polyimide were investi-gated with the McBain-type sorption apparatus.The experiments were conducted at 40 °C withthree films (film thickness 5 21.0, 22.2, and 67.0mm). Diffusion coefficients of methanol in 6FDA–ODA polyimide were analyzed from experimentaldata obtained from the 21.0-mm film with thelong-time method, eq 5. Methanol vapor had to beintroduced carefully to the sorption chambers be-cause of the delicate nature of the quartz spring.When the introduction of the methanol vapor wastoo sudden, it resulted in a strong draft of meth-anol vapor that might move or even knock thespring off the hook. If the initial position of thespring is moved, the accuracy of the subsequentmeasurement of the sorption rate and the equi-librium sorption value can be jeopardized. Theprocess of removing the methanol vapor from thesorption chamber during the desorption experi-ment is similar to the sorption experiment. Theremoval of the methanol vapor must be gradual tomaintain the position of the quartz spring. Theperiod of introduction of methanol vapor to thesorption chamber during the sorption experimentvaried approximately from 30 to 60 s, dependingon the methanol fugacity. The removal of themethanol vapor from the sorption chamber dur-
Figure 5. Hysteresis behavior of 6FDA–ODA polyim-ide.
2260 KAMARUDDIN AND KOROS

ing the desorption process took approximately 15to 30 s.
Ideally, the introduction and removal of themethanol vapor to (or from) the sorption chamberoccurs as a step change; however, this is physi-cally unrealistic with the experimental proce-dures described earlier. Therefore, at the begin-ning of the sorption and desorption process, themethanol vapor fugacity in the sorption chambersomewhat varied with time. In such a case, Crankand Park10 showed that Mt/M` versus t1/2 willexhibit sigmoid sorption curves. Therefore, theanalysis of the diffusion coefficient of methanol asa function of its fugacity for the 21.0-mm film wasconducted with the long-time analysis. The sig-moid sorption curve can also indicate non-Fickianbehavior. However, plotting Mt/M` as a functionof t0.5/, should normalize the sorption curves ifFickian transport applies. Figures 6 and 7 showthe sorption and desorption curves obtained withfilms of various thicknesses at approximately 8.0and 16.5 cmHg methanol fugacities. These sorp-tion and desorption figures of different film thick-
nesses show good agreement and Fickian scalingbehavior. The slight discrepancy between the de-sorption curve of the 22.2-mm film and those ofthe 21.0- and 67.0-mm films was within experi-mental error. It was probably due to the slightdifferences between the removal rates of metha-nol vapor from the sorption chamber during thedesorption experiments.
Examples of the long-time analysis performedon the 21.0-mm film are shown in Figure 8. Thedata in these figures correspond to Mt/M` . 0.6.The diffusion coefficients of methanol obtainedfrom the long-time analysis are summarized inFigure 9 and Table II. The kinetics data clearlyindicate that the diffusion coefficient of methanolincreases as a function of fugacity up to approxi-mately 17 cmHg; then, the diffusion coefficientstarts to decrease with fugacity.
In some systems, when the penetrant concen-tration reaches a certain value, some of the pen-
Figure 6. (a) Sorption and (b) desorption of methanolwith 21.0-, 22.2-, and 67.0-mm 6FDA–ODA polyimidefilms at 8.0–8.8 cmHg methanol fugacity.
Figure 7. (a) Sorption and (b) desorption of methanolwith 21.0- and 67.0-mm 6FDA–ODA polyimide films at16.4 and 16.5 cmHg methanol fugacity.
DIFFUSION AND SOLUBILITY OF METHANOL/MTBE 2261
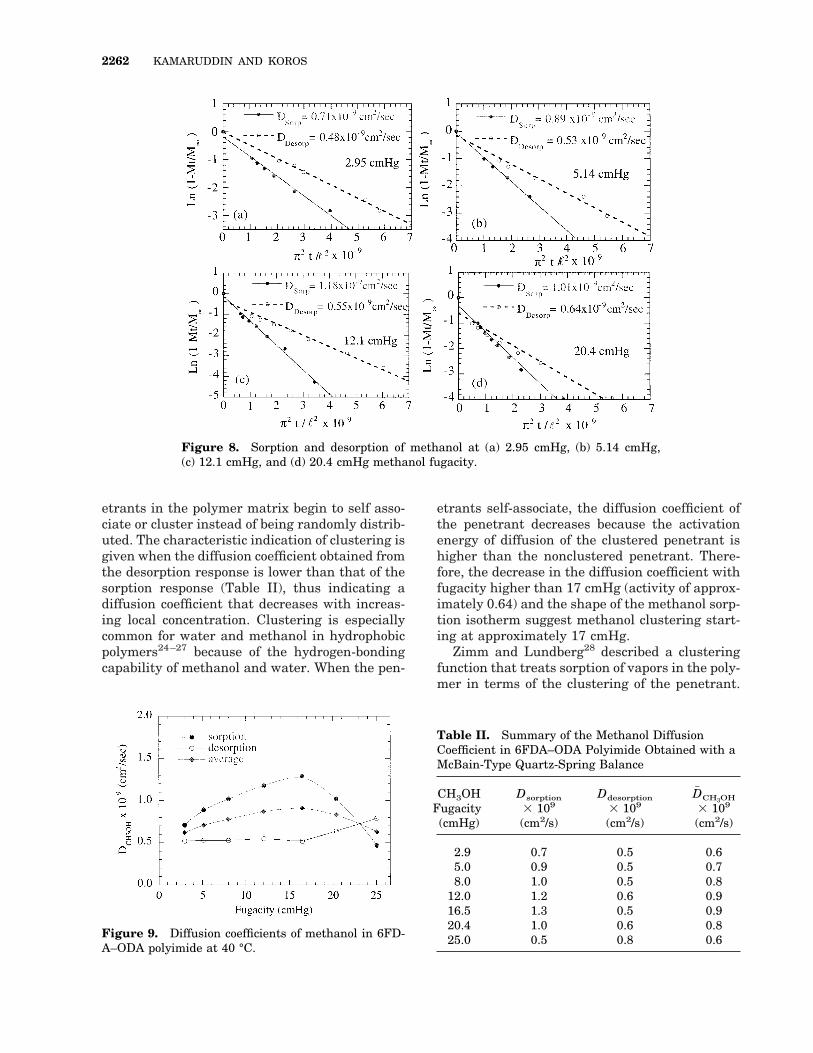
etrants in the polymer matrix begin to self asso-ciate or cluster instead of being randomly distrib-uted. The characteristic indication of clustering isgiven when the diffusion coefficient obtained fromthe desorption response is lower than that of thesorption response (Table II), thus indicating adiffusion coefficient that decreases with increas-ing local concentration. Clustering is especiallycommon for water and methanol in hydrophobicpolymers24–27 because of the hydrogen-bondingcapability of methanol and water. When the pen-
etrants self-associate, the diffusion coefficient ofthe penetrant decreases because the activationenergy of diffusion of the clustered penetrant ishigher than the nonclustered penetrant. There-fore, the decrease in the diffusion coefficient withfugacity higher than 17 cmHg (activity of approx-imately 0.64) and the shape of the methanol sorp-tion isotherm suggest methanol clustering start-ing at approximately 17 cmHg.
Zimm and Lundberg28 described a clusteringfunction that treats sorption of vapors in the poly-mer in terms of the clustering of the penetrant.
Figure 8. Sorption and desorption of methanol at (a) 2.95 cmHg, (b) 5.14 cmHg,(c) 12.1 cmHg, and (d) 20.4 cmHg methanol fugacity.
Figure 9. Diffusion coefficients of methanol in 6FD-A–ODA polyimide at 40 °C.
Table II. Summary of the Methanol DiffusionCoefficient in 6FDA–ODA Polyimide Obtained with aMcBain-Type Quartz-Spring Balance
CH3OHFugacity(cmHg)
Dsorption
3 109
(cm2/s)
Ddesorption
3 109
(cm2/s)
D# CH3OH
3 109
(cm2/s)
2.9 0.7 0.5 0.65.0 0.9 0.5 0.78.0 1.0 0.5 0.8
12.0 1.2 0.6 0.916.5 1.3 0.5 0.920.4 1.0 0.6 0.825.0 0.5 0.8 0.6
2262 KAMARUDDIN AND KOROS

They derived a clustering function, GAA/V̂A,which is defined as follows:
GAA
V̂A
5 2~1 2 fA!SaA/fA
aAD 2 1 (16)
where GAA, V̂A, and fA are the cluster integral,partial molar volume, and volume fraction of pen-etrant A, respectively. Equation 16 can be used toanalyze sorption data to measure the clusteringtendency of the penetrant. When the value ofGAA/V̂A . 21, is greater than 21, penetrant clus-tering is likely, and when GAA/V̂A , 21, is lessthan 21, the penetrant is most likely to be ran-domly distributed, which means that strong at-tractive interaction between penetrants is un-likely. The parameter GAAfA/V̂A represents theaverage number of penetrant molecules in thevicinity of a given penetrant in excess of the mean
concentration. The function (GAAfA/V̂A 1 1) rep-resents the average size of the clusters.
The clustering tendency of methanol in 6FDA–ODA polyimide at a higher methanol activity wasanalyzed with eq 16 and is summarized in Figure10(a). Figure 10(a) shows the clustering functionas a function of the methanol activity. As dis-cussed previously, when the value of GAA/V̂A isgreater than 21, clustering is likely. Figure 10(a)shows that methanol tends to cluster at a meth-anol activity of approximately 0.67 (ai 5 fi/f i
sat),which is consistent with the diffusion coefficientdata indicating the diffusion coefficient starts todecrease at a methanol activity of approximately0.64 (17 cmHg). This corresponds rather closely tothe activity at which the maximum in the diffu-sion coefficients is seen in Figure 9. Figure 10(b)shows the average number of penetrants forminga cluster as a function of the activity. At an activ-ity of 1, approximately 60% of the methanol formsclusters.
The partial immobilization parameters, DDand DH, can be obtained with eq 14. The analysisrequires average diffusion coefficient data and thedual-mode sorption parameters. The average dif-fusion coefficient data were obtained from thelong-time kinetics experiment, whereas the dual-mode parameters were obtained from the equilib-rium vapor sorption data discussed in a later partof the article. The partial immobilization theo-ry4,11,13 assumes constant Henry’s law and Lang-muir diffusion coefficients. According to the pre-vious analysis, methanol tends to form clustersstarting at approximately 17.7 cmHg. The clus-tering of methanol violates the assumption of thepartial immobilization theory, that is, constant
Figure 10. (a) Tendency of methanol clustering as afunction of its activity and (b) the average number ofmolecules clustered as a function of the methanol ac-tivity.
Figure 11. Analysis of the partial immobilization pa-rameters of methanol in 6FDA–ODA polyimide.
DIFFUSION AND SOLUBILITY OF METHANOL/MTBE 2263

penetrant diffusivity (DD and DH). Therefore, thekinetic experimental data used for the analysis ofthe partial immobilization parameters were re-stricted to less than 17.7 cmHg methanol fugacityto satisfy the assumptions of the basic partialimmobilization theory. Figure 11 shows the anal-ysis of the partial immobilization parameters viaeq 15. The intercept is the diffusion coefficient ofthe Henry’s law population, DD, and the interceptis equal to the value of (F DD). The parameter DDwas 1.09 3 1029 cm2/s, DH was 3.38 3 10210
cm2/s, and the DH/DD ratio was F 5 0.27.
Mixed-Liquid Methanol/MTBE Sorption
Nonideal Behavior of Methanol and MTBE Mixture
Methanol and MTBE form nonideal liquid mix-tures. The fugacity of component i, ƒi, in a liquidmixture can be related to its molar fraction in theliquid, xi, by the following equation:
fi 5 gixipisat (17)
where pisat and gi are the saturation vapor pres-
sure and activity coefficient of component i, re-spectively. For an ideal mixture, the activity co-efficient is equal to 1, and eq 17 becomes Raoult’slaw. The greater the deviation is from unity forthe activity coefficient, the greater the departureis from the ideal mixture behavior.
The activity coefficient of methanol and MTBEmixtures can be calculated with various methods,such as UNIQUAC, Wilson, NRTL, and UNIFAC.29
In general, all of these methods yield reasonableapproximations for the activity coefficients of thecomponents in the mixtures. A computer programto handle the UNIFAC calculation method wasutilized to account for the nonideal behavior ofmethanol/MTBE liquid mixtures. The UNIFACcomputer program was developed by Goldbergeret al.30 Pure-component vapor pressures were cal-culated with the Wagner equation described inref. 31.
The activity coefficients of methanol andMTBE as a function of the methanol concentra-tion are shown in Figure 12(a). The accuracy ofthe calculated methanol and MTBE fugacitieswith the UNIFAC program and Wagner equationwas compared with methanol/MTBE liquid–va-por equilibrium experimental data obtained fromGmehling et al.32 Figure 12(b) shows that thecalculated and experimental vapor–liquid equi-
librium data of the methanol/MTBE mixtures arein good agreement.
Methanol/MTBE Mixed-Liquid Sorption Isotherm
The methanol/MTBE mixed-liquid sorption iso-therms in 6FDA–ODA polyimide at 40 °C as afunction of the fugacity are shown in Figure 13.The mixed-liquid sorption isotherm of methanolclearly shows dual-mode behavior, which is typi-cal for a glassy polymer. The mixed-liquid sorp-tion isotherm of methanol is almost identical tothe pure methanol vapor sorption isotherm ob-tained with the McBain-type balance. Figure 13also shows a comparison of the pure methanolvapor and mixed-liquid methanol sorption iso-therms. The comparison of the pure and mixed-methanol sorption isotherms18 indicate a slightdeviation from the dual-mode model. According to
Figure 12. (a) Methanol and MTBE activity coeffi-cients as a function of the MTBE concentration and (b)a comparison of the MTBE concentration in the vaporphase calculated with UNIFAC and methanol/MTBEvapor–liquid equilibrium data.
2264 KAMARUDDIN AND KOROS

the dual-mode model, the binary component sorp-tion isotherm should be lower than the single-component sorption isotherm because the modelpredicts that competition for the Langmuir sorp-tion sites reduces the Langmuir population and,thus, the total concentration. The mixed-metha-nol and pure methanol sorption isotherms areapproximately equal, which is inconsistent withthe dual-mode model prediction. The deviation ofthe experimental data and dual-mode modelmight be caused by experimental error, a slightchange in the physical or thermodynamic naturesof the polymer that affected the dual-mode pa-rameters of methanol, or both. Because the dis-crepancies are small and within experimental er-ror, it is impossible to determine their cause.
The MTBE sorption isotherm in Figure 13shows neither a simple rubbery sorption isothermnor a glassy-type sorption isotherm. The MTBEsorption level in 6FDA–ODA polyimide at anMTBE activity of unity was too small to be de-tected even after approximately 3 months in a20-mm film. However, the sorption level of MTBEincreased markedly with the methanol sorptionlevel. Because MTBE activity decreases as meth-anol activity increases, a steady increase in theMTBE sorption level with the methanol activityclearly suggests a strong dependency of theMTBE sorption on the methanol sorption level inthe polymer. However, the MTBE sorption levelhas to eventually approach 0 as the MTBE activ-ity approaches 0 (100% methanol). Therefore, theMTBE sorption level has to reach a maximumvalue and decrease with methanol activity. Themaximum MTBE sorption level is at approxi-
mately 17–19 cmHg methanol fugacity. Sorptionisotherms similar to MTBE have been observed inother systems.33–36
Pope37 measured and compared methane sorp-tion isotherms in unconditioned, CO2-condi-tioned, and conditioned-exchange polystyrene.The conditioned-exchange system resembles thehysteretic system described earlier for CO2 expo-sure to polycarbonate. The sorption isotherm ofmethane in a conditioned-exchange polystyrenewas obtained first by the conditioning of the poly-mer with CO2. Pure methane was next introducedto purge the sorption chamber until all of the CO2in the polymer and sorption chamber was re-placed with pure methane. In essence, the condi-tioned-exchange procedure probes the property ofthe polymer matrix after it has been exposed topenetrants capable of disrupting the chain pack-ing; methane is sorbing into the polymer withoutallowing packing disruptions due to CO2 swellingto relax significantly back to their initial state.Figure 14 shows a comparison of CH4 sorptionisotherms in unconditioned, conditioned, and con-ditioned-exchange polymers as a function of theCH4 pressure. The increase in the sorption levelof the conditioned-exchange polymer is consider-ably higher than that of the conditioned polymer.This is because the increase in the polymer freeand unrelaxed volumes in the conditioned-ex-change polymer is higher than that of the condi-tioned polymer.
It is postulated that the strong dependence ofthe MTBE sorption as a function of the methanolsorption level in the polymer is due to the condi-tioning behavior, conditioned-exchange behavior,or both, such as that seen in CO2/CH4–polysty-
Figure 13. Methanol and MTBE mixed-liquid sorp-tion isotherms and a comparison with a pure methanolsorption isotherm obtained with a McBain-type sorp-tion apparatus.
Figure 14. Methane sorption isotherms of uncondi-tioned (fresh), conditioned, and conditioned-exchangepolystyrene at 35 °C.
DIFFUSION AND SOLUBILITY OF METHANOL/MTBE 2265
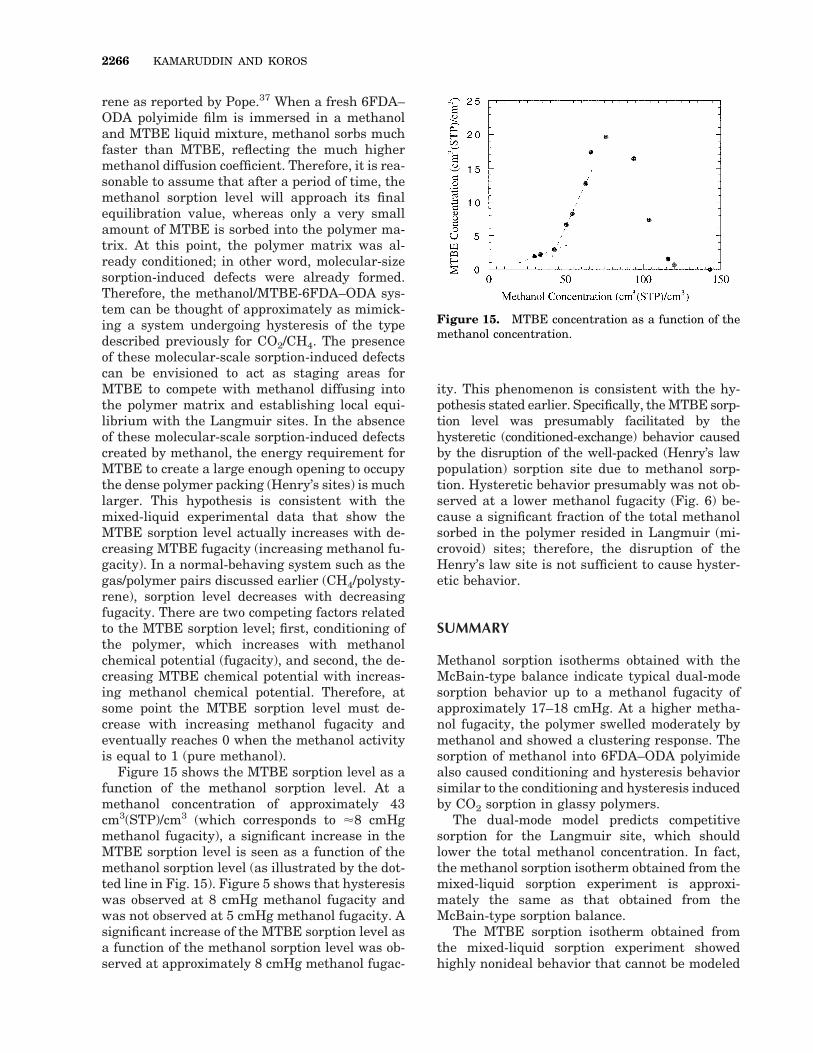
rene as reported by Pope.37 When a fresh 6FDA–ODA polyimide film is immersed in a methanoland MTBE liquid mixture, methanol sorbs muchfaster than MTBE, reflecting the much highermethanol diffusion coefficient. Therefore, it is rea-sonable to assume that after a period of time, themethanol sorption level will approach its finalequilibration value, whereas only a very smallamount of MTBE is sorbed into the polymer ma-trix. At this point, the polymer matrix was al-ready conditioned; in other word, molecular-sizesorption-induced defects were already formed.Therefore, the methanol/MTBE-6FDA–ODA sys-tem can be thought of approximately as mimick-ing a system undergoing hysteresis of the typedescribed previously for CO2/CH4. The presenceof these molecular-scale sorption-induced defectscan be envisioned to act as staging areas forMTBE to compete with methanol diffusing intothe polymer matrix and establishing local equi-librium with the Langmuir sites. In the absenceof these molecular-scale sorption-induced defectscreated by methanol, the energy requirement forMTBE to create a large enough opening to occupythe dense polymer packing (Henry’s sites) is muchlarger. This hypothesis is consistent with themixed-liquid experimental data that show theMTBE sorption level actually increases with de-creasing MTBE fugacity (increasing methanol fu-gacity). In a normal-behaving system such as thegas/polymer pairs discussed earlier (CH4/polysty-rene), sorption level decreases with decreasingfugacity. There are two competing factors relatedto the MTBE sorption level; first, conditioning ofthe polymer, which increases with methanolchemical potential (fugacity), and second, the de-creasing MTBE chemical potential with increas-ing methanol chemical potential. Therefore, atsome point the MTBE sorption level must de-crease with increasing methanol fugacity andeventually reaches 0 when the methanol activityis equal to 1 (pure methanol).
Figure 15 shows the MTBE sorption level as afunction of the methanol sorption level. At amethanol concentration of approximately 43cm3(STP)/cm3 (which corresponds to '8 cmHgmethanol fugacity), a significant increase in theMTBE sorption level is seen as a function of themethanol sorption level (as illustrated by the dot-ted line in Fig. 15). Figure 5 shows that hysteresiswas observed at 8 cmHg methanol fugacity andwas not observed at 5 cmHg methanol fugacity. Asignificant increase of the MTBE sorption level asa function of the methanol sorption level was ob-served at approximately 8 cmHg methanol fugac-
ity. This phenomenon is consistent with the hy-pothesis stated earlier. Specifically, the MTBE sorp-tion level was presumably facilitated by thehysteretic (conditioned-exchange) behavior causedby the disruption of the well-packed (Henry’s lawpopulation) sorption site due to methanol sorp-tion. Hysteretic behavior presumably was not ob-served at a lower methanol fugacity (Fig. 6) be-cause a significant fraction of the total methanolsorbed in the polymer resided in Langmuir (mi-crovoid) sites; therefore, the disruption of theHenry’s law site is not sufficient to cause hyster-etic behavior.
SUMMARY
Methanol sorption isotherms obtained with theMcBain-type balance indicate typical dual-modesorption behavior up to a methanol fugacity ofapproximately 17–18 cmHg. At a higher metha-nol fugacity, the polymer swelled moderately bymethanol and showed a clustering response. Thesorption of methanol into 6FDA–ODA polyimidealso caused conditioning and hysteresis behaviorsimilar to the conditioning and hysteresis inducedby CO2 sorption in glassy polymers.
The dual-mode model predicts competitivesorption for the Langmuir site, which shouldlower the total methanol concentration. In fact,the methanol sorption isotherm obtained from themixed-liquid sorption experiment is approxi-mately the same as that obtained from theMcBain-type sorption balance.
The MTBE sorption isotherm obtained fromthe mixed-liquid sorption experiment showedhighly nonideal behavior that cannot be modeled
Figure 15. MTBE concentration as a function of themethanol concentration.
2266 KAMARUDDIN AND KOROS

with the dual-mode sorption model. The MTBEsorption level was a strong function of the meth-anol sorption level. The concentration of MTBEactually increases with decreasing MTBE fugac-ity. It is postulated that the increase in MTBEwith the methanol sorption level was due to theconditioned-exchange effect of methanol on thepolymer. Specifically, it is believed that sorption-induced packing defects are formed as a result ofthe sorption of methanol into the Henry’s lawsites. These hypothetical packing defects resultfrom the inability of the previously well-packedpolymer chain segments to completely relax backto their original packing condition upon completedesorption of methanol.
The kinetics of methanol sorption exhibitsFickian behavior. The kinetics of methanol sorp-tion can be modeled with the partial immobiliza-tion theory up to approximately 17 cmHg metha-nol fugacity. The kinetics of desorption above 17cmHg methanol fugacity suggests the formationof methanol clusters. The methanol diffusion co-efficient of the Henry’s law population, DD, was1.15 3 1029 cm2/s, whereas the diffusion coeffi-cient of the Langmuir population, DH, was 0.313 1029 cm2/s.
The authors would like to acknowledge and thank theDepartment of Energy, Exxon, and the SeparationsResearch Programs of the University of Texas at Aus-tin for funding this project.
REFERENCES AND NOTES
1. Pfromm, P. Ph.D. Thesis, University of Texas, Aus-tin, Texas, 1994.
2. Rezac, M. E.; Pfromm, P. H.; Costello, L. M.; Koros,W. J. Ind Eng Chem Res 1993, 32, 1921.
3. Rezac, M. E. Ind Eng Chem Res 1995, 34, 3170.4. Koros, W. J. Ph.D. Thesis, University of Texas,
Austin, Texas, 1977.5. Koros, W. J. J Polym Sci Polym Phys Ed 1980, 18,
981.6. Koros, W. J.; Patton, C. J.; Felder, R. M.; Fincher,
S. J. J Polym Sci Polym Phys Ed 1980, 18, 1485.7. Sanders, E. S.; Koros, W. J. J Polym Sci Part B:
Polym Phys 1986, 24, 175.8. Koros, W. J.; Hellums, M. W. In Concise Encyclo-
pedia of Polymer Science and Engineering; Kros-chwitz, J. I., Ed.; Wiley: New York, 1990; pp 1211–1219.
9. Koros, W. J.; Coleman, M. R.; Walker, D. R. B. AnnRev Mater Sci 1991, 22, 47.
10. Crank, J.; Park, G. S. The Mathematics of Diffu-sion; Academic: New York, 1968; Chap. 4.
11. Yang, D. K., Ph.D. Thesis, North Carolina StateUniversity, Raleigh, North Carolina, 1984.
12. Chan, A. H.; Koros, W. J.; Paul, D. R. J Membr Sci1978, 3, 117.
13. Koros, W. J.; Chern, R. T.; Stannett, V.; Hopfenberg,H. B. J Polym Sci Polym Phys Ed 1981, 19, 1513.
14. Koros, W. J.; Patton, C. J.; Felder, R. M.; Fincher,S. J. J Polym Sci Phys Ed 1980, 18, 1485.
15. Husk, G. R.; Cassidy, P. E.; Gebert, K. L. Macro-molecules 1988, 21, 1234.
16. Moaddeb, M.; Koros, W. J. J Appl Polym Sci 1995,57, 687.
17. Kamaruddin, H. D.; Koros, W. J. J Polym Sci PartB: Polym Phys, in press.
18. Kamaruddin, H. D. Ph.D. Thesis, University ofTexas, Austin, TX, 1997.
19. Fleming, G. K. Ph.D. Thesis, University of Texas,Austin, TX, 1988.
20. Fleming, G. K.; Koros, W. J. J Polym Sci Part B:Polym Phys 1990, 28, 1137.
21. Fleming, G. K.; Koros, W. J. Macromolecules 1990,23, 1353.
22. Jordan, S. M.; Koros, W. J.; Beasley, J. K. J MembrSci 1989, 43, 103.
23. Jordan, S. M. Ph.D. Thesis, University of Texas,Austin, TX, 1989.
24. Barrie, J. A.; Machin, D.; Nunn, A. Polymer 1975,16, 811.
25. Barrie, J. A.; Machin, D. J Macromol Sci Phys1969, 3, 645.
26. Yasuda, H.; Stannett, V. J Polym Sci 1962, 57, 907.27. Williams, J. L.; Hopfenberg, H. B.; Stannett, V. J
Macromol Sci Phys 1969, 3, 711.28. Zimm, B. H.; Lundberg, J. L. J Chem Phys 1956,
60, 425.29. Prausnitz, J. M.; Lichtenthaler, R. N.; de Azevedo,
E. G. Molecular Thermodynamics of Fluid-PhaseEquilibria, 2nd ed.; Prentice Hall: Upper SaddleRiver, NJ, 1986; Chapter 6.
30. Goldberger, E. W.; Harvey, R.; Fair, J. R. UNIFAC;Separation Research Programs, University ofTexas, Austin, TX, 1993.
31. Reid, R. C.; Prausnitz, J. M.; Poling, B. E. TheProperties of Gases and Liquids, 4th ed.; McGraw-Hill: New York, 1987; Chapter 7.
32. Gmehling, J.; Onken, U.; Arlt, W. Organic HydroxyCompounds: Alcohols. Behrens, D., Eckermann, R.,Eds. In Vapor–Liquid Equilibrium Data Collection;Dechema: Frankurt, Germany, 1982; Suppl 1, Vol.1, Part 2c.
33. Hauser, J.; Reinhardt, G. A.; Stumm, F.; Heintz, A.Fluid Phase Equilib 1989, 49, 195.
34. Hauser, J.; Reinhardt, G. A.; Stumm, F.; Heintz, A.J Membr Sci 1989, 47, 261.
35. Heintz, A.; Funke, H.; Lichtenthaler, R. N. Perva-poration Membrane Separation Processes; Huang,R. Y. M., Ed.; Elsevier: Amsterdam, 1991; Chapter 6.
36. Lee, C. H.; Hong, W. H. J Membr Sci 1997, 135, 187.37. Pope, D. S. Ph.D. Thesis, University of Texas, Aus-
tin, TX, 1991.
DIFFUSION AND SOLUBILITY OF METHANOL/MTBE 2267