farmacovigilancia periodic safety update report module vii gvp azierta
TRANSCRIPT

FarmacovigilanciaPSUR / PBRER.
Good Vigilance Practices (GVP). Módulo VII

Good Vigilance Practices (GVP)
Medidas dirigidas a facilitar la actuación de losespecialistas de farmacovigilancia.
Se encuentran divididas en módulos que hacen referencia alos diferentes (aunque conectados) procesos de lafarmacovigilancia. Todos han sido creados por expertos,teniendo en cuenta los intereses y comentarios de todoslos stakeholders.
Puedes consultar más información en la Web de la EMA

1. Cronología de implementación2. ¿Qué es un informe periódico de seguridad (PSUR)?3. Objetivos del PSUR4. Periodicidad, Fechas de referencia en la UE, y
Puntos de información cierre5. Excepciones de envío de PSURs6. Evaluación individual del PSUR (PSUSA)7. Resumen del módulo VII de las GVP8. Cómo revisar un PSUR
GVP Módulo VII: PSUR/PBRER

1. Cronología de implementación

2. ¿Qué es un informe periódico de seguridad (PSUR)?
Un Informe Periódico de Seguridad o Periodic Safety Update Report(PSUR) es un documento de farmacovigilancia que expone laevaluación del beneficio- riesgo de un medicamento que presentanlos Titulares de Autorización de Comercialización en determinadasfechas durante la Fase de Post-Autorización.
PBRER (Periodic Benefit Risk Evaluation Report) hace referencia al PSUR desde su implementación en Europa a través del modulo VII de las GVP.

3. Objetivo del PSUR (1)
I. Presenta un análisis completo de nueva información de losriesgos, y cuando sea el caso, de nuevos hallazgos sobre losbeneficios para actualizar la valoración de riesgo-beneficio delmedicamento.
II. Contiene una evaluación de toda la información acumulada,relevante y disponible sobre un medicamento
• Compara si la nueva información coincide con la yaexistente.
• Resume la nueva información que pueda tener impactosobre el perfil de seguridad.
• Recopila nueva información sobre eficacia y efectividad.• Desarrolla una nueva evaluación beneficio-riesgo (con
toda la nueva información).

4. Periodicidad (1)
Los PSUR deben ser desarrollados:• Inmediatamente tras petición expresa de Autoridades
Competentes.• De acuerdo con la frecuencia definida por la EURD list.
Excepción – En algunos casos la frecuencia y fechas pueden variarsiempre y cuando:
• sea una condición para la comercialización y serán fijadas en lalista de Fechas de Referencia de la UE (EURD List).
• lo exige la Autoridad Sanitaria• no está incluido en la EURD list por estar registrado en un único
país (purely national procedure)
El envío se debe realizar:• Hasta los 70 días en intervalos de hasta 12 meses• Hasta 90 días en intervalos de más de 12 meses.

4. Fecha de nacimiento internacional (IBD) y Fechas de Cierre (DLP) (3)
Se conoce como Fecha de Nacimiento Internacional (IBD de sussiglas en inglés: International Birth Date) a la fecha de la primeraautorización de comercialización para cada producto medicinal encualquier país del mundo.LA DLP (Data Lock Point) es la fecha de cierre de datos a incluir enel informe, marca el final del periodo del PSUR.

La Lista de Fechas de Referencia de la UE (EURD) es una lista donde serecopilan los principios activos y la combinación de principios activospara las cuales se deben enviar PSURs.
• Legalmente vinculante desde que el Módulo VII se hizoefectivo el 2 de julio de 2012.
• La periodicidad se define según una aproximación enfunción del riesgo.
La Lista de Fechas de Referencia de la UE fue desarrollada parafacilitar la armonización de las Fechas de Cierre (Data Lock Points oDLPs) y la frecuencia de envío de los PSURs para medicamentos quecontienen el mismo principio activo o la misma combinación desustancias activas sujetas a diferentes autorizaciones decomercializacion, autorizadas en más de un Estado Miembro.
4. Lista de fechas de referencia (4)

Lista de Referencia en la Unión Europea
4. Lista de fechas de referencia en la UE (EURD) (5)

La Lista de Fechas de Referencia de la UE es un documento dinámicoque será revisado siempre y cuando lo considere el PRAC, el CHMP o laCMDh como respuesta a la aparición de nueva información deseguridad, nuevas sustancias autorizadas y peticiones recibidas porTitulares de Autorización de Comercialización.
Las sustancias pueden ser añadidas o eliminadas según se estime. LaEURD se actualiza mensualmente por lo que los Titulares deAutorización de Comercialización deben estar pendientes del estado dela lista.
Los PSURs deben de ser enviados inmediatamente siempre y cuando lorequieran las autoridades regulatorias.
4. Lista de fechas de referencia en la UE (EURD) (6)

4. Lista de fechas de referencia en la UE (EURD)Principales actores involucrados (7)
12
El grupo de coordinación para el reconocimiento mutuo yprocedimiento descentralizado de humana, examina cualquiercuestión relativa a la Autorización de comercialización de unproducto medicinal en dos o mas miembros estado de acuerdocon el procedimiento de reconocimiento mutuo odescentralizado.
Cuerpo ejecutivo de la UniónEuropea responsable deproporcionar un marco legal,implementar decisiones,mantener tratados y gestionar eldía a día de la Unión Europea.
Ayuda a proteger y promover la salud en Europa a través de la evaluación de medicamentos de uso humano o veterinario.
Autoridades Sanitarias de cada Estado Miembro
El comité para productos medicinales de uso humano es ungrupo de la EMA responsable de examinar temasrelacionados con los medicamentos de uso humano.
CHMP
El comité de la EMA que es responsable de asesorar ymonitorizar temas en temas de seguridad paramedicamentos de uso humano.
EMA
PRAC
Autoridades Nacionales Competentes (NCA)

5. Excepciones de envío de PSURs
Los genéricos, los medicamentos de uso bien establecido ymedicamentos tradicionales a base de plantes están exentos deenviar los PSURs, salvo:
La autorización de comercialización tiene como condición elenvío de PSURs.
EL PSUR es requerido debido a la escasa información deFarmacovigilancia o a la falta de PSURs de un principioactivo determinado después de que la autorización decomercialización ha sido garantizada (p. ej.: Cuando elproducto medicinal “referente” se deja de comercializar).
Productos donde los PSURs no son enviados de forma rutinaria seespera que los titulares de autorización de comercialización evalúende forma continua la seguridad del producto y envíen cualquiernueva información de seguridad que pueda impactar en el perfilbeneficio-riesgo o en la información del producto.

6. Evaluación individual de PSUR en la UE (PSUSA) (1)
El informe PSUSA es una evaluación única de sustancias activas deproductos autorizados por el procedimiento centralizado yproductos autorizados de forma nacional. Conduce a decisioneslegalmente vinculantes: Mantenimiento, variación, suspensión, yrevocación de la autorización.
Objetivos:Armonizar y actualizar la evaluación de seguridad y beneficio-riesgode medicinas en la UE; incrementar el uso compartido de recursosentre autoridades competentes; evaluar a través de la PRAC conayuda de CHMP o CMDh en caso de acciones regulatorias.
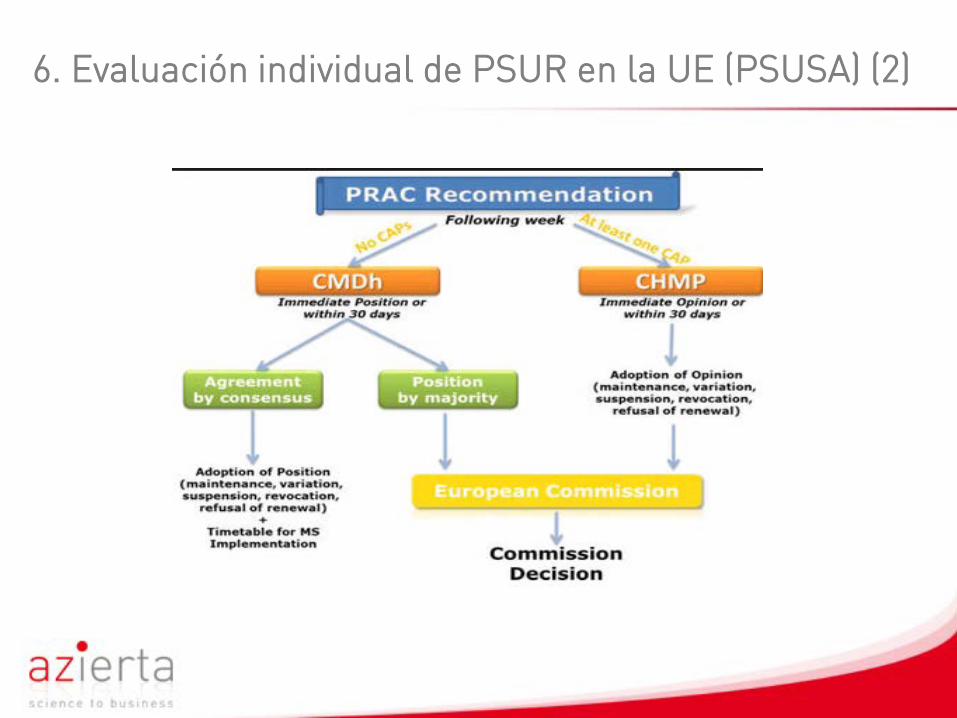
6. Evaluación individual de PSUR en la UE (PSUSA) (2)

6. Evaluación individual de PSUR en la UE (PSUSA) (3)
La EMA publica una lista de resultados para NAPs en su website,mientras los resultados para CAPs son publicados como parte delinforme de evaluación pública de cada principio activo.
Cualquier cambio en la información de un producto, como resultado deuna evaluación PSUR es implementado directamente por CAPs y através de la variación apropiada a nivel nacional por NAPs.
Periodic safety update report single assessments

7. Resumen del Módulo 7 de las GVP
El Módulo VII de las GVP incluye las directrices en cuanto a objetivo,formato y contenido de los PSUR, así como recomendaciones parasistemas de calidad y formación del personal involucrado en lagestión y desarrollo de los PSURs.
Evaluación científica del perfil beneficio-riesgo Resumen de toda la información científica/clínica incluyendo las
búsquedas bibliográficas. Información de ventas/prescripción para calcular la exposición del
paciente.
La redacción de los PSURs debe ser coherente con la terminologíaMedDRA (Medical Dictionary for Regulatory Activities).

8. Cómo revisar un PSUR/PBRER (1)
Los PSUR deben ser revisados por un experto en seguridad queconozca en profundidad el desarrollo de un PSUR, los procesosinvolucrados en la unidad de Farmacovigilancia y medicamento.
Calidad /Auditorías / Inspecciones
• Todas las instrucciones y recomendaciones recibidas por lasAutoridades Sanitarias, ya sea en los PSUR o en cualquier email,deben ser tenidas en cuenta.
• Asegurate de conocer en profundidad la terminología (CCSI, etc.)• La persona que revise el PSUR debe prestar especial atención a
señales, poblaciones especiales (mujeres embarazadas, niños,ancianos, etc.), antiguas y nuevas indicaciones, cambios en los textosdel producto, cambios en los riesgos, etc.)

8. Cómo revisar un PSUR/PBRER (2)
• Revisión de eventos adversos graves (SAE) y reacciones adversas(ADR).
¿Alguna sorpresa o señal en los SAE / ADRs del periodo delinforme?
¿Existen hallazgos en poblaciones específicas de pacientesdefinidas por enfermedad, edad, grupos vulnerables, etc.?
¿Ha habido algún cambio en la información de los riesgos obeneficios conocidos previamente?

En Azierta somos expertos en Farmacovigilancia, si quieresconocernos visita:
Azierta
O síguenos en: Azierta @Azierta