le syndrome de turcot : à propos d’un cas
TRANSCRIPT

J Radiol 2009;90:842-4© Éditions Françaises de Radiologie, Paris, 2009
Édité par Elsevier Masson SAS. Tous droits réservés lettre neuroradiologie
Le syndrome de Turcot : à propos d’un cas
R Sanou (1), R Anxionnat (1), L Taillandier (2), MA Bigard (3), D Regent (4) et S Bracard (1)
Key words: Turcot syndrome. Glioblastoma. Colon carcinoma. Familial adenomatous polyposis. HNPCC.
Mots-clés : Syndrome de turcot. Glioblastome. Cancer colorectal. Polypose adénomateuse familiale. HNPCC.
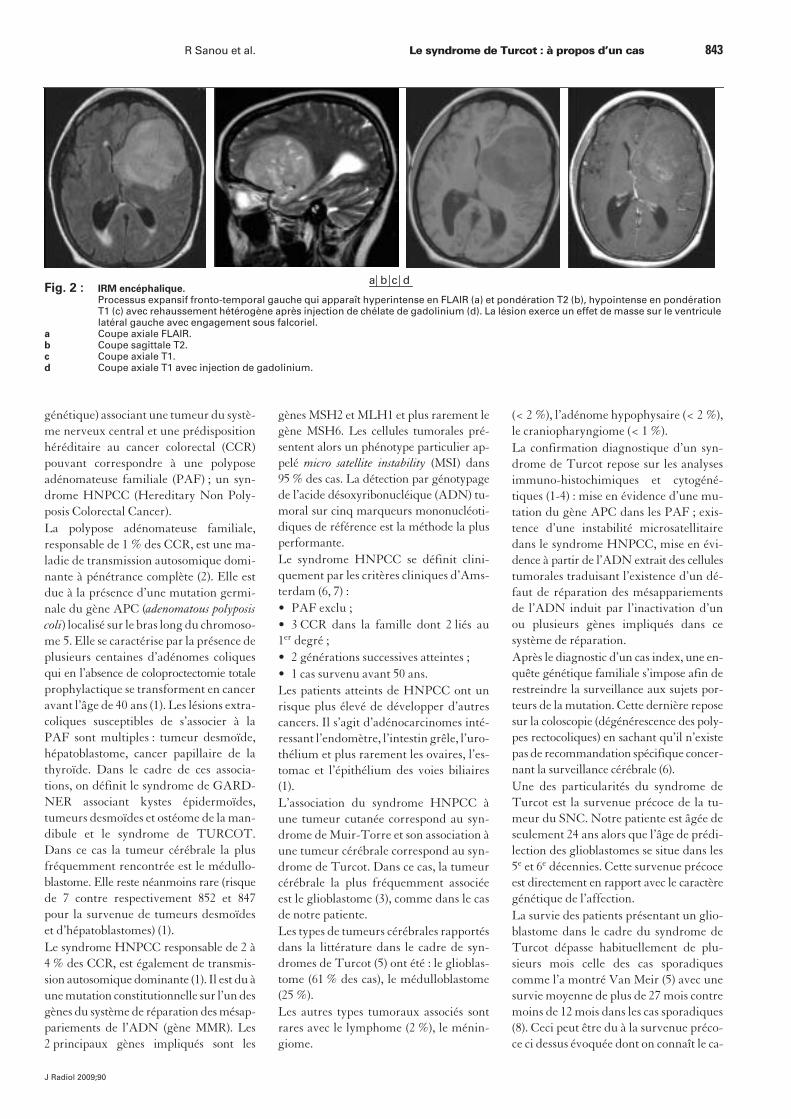
Par la suite, elle a présenté un syndromed’hypertension intracrânienne avec criseépileptique apparement généralisée inau-gurale. Devant ces symptômes, un biland’imagerie fut rapidement réalisé. En to-modensitométrie a été découverte une vo-lumineuse masse tumorale de localisationfronto-temporale gauche responsabled’un effet de masse avec engagementsous-falcoriel. Cette lésion se rehaussaitaprès injection de produit de contrasteavec un aspect hétérogène. En IRM, la lé-sion apparaissait en hypersignal T2 etflair hétérogène avec foyers de nécrosemultiples et prise de contraste après injec-tion de gadolinium (fig. 2). Un gliomemalin a été évoqué en première intention.Dans les suites immédiates la patienteétait prise en charge en neurochirurgie oùfut réalisée une exérèse partielle. L’analyseanatomopathologique a permis de confir-mer le diagnostic suspecté de glioblasto-me. La patiente a par la suite bénéficiéd’un traitement par chimio-radiothérapieconcomitantes. Peu après le début de laradiothérapie, elle a présenté de façon ful-minante un tableau d’hypertension intra-crânienne ayant entraîné son décès.
Chez cette jeune patiente, il existe ainsiune association rare de tumeur cérébralemaligne et cancer colique sur probableprédisposition génétique aux cancers co-lorectaux. Cette association a fait évoquerun syndrome de Turcot.Une analyse immunohistochimique de lapièce opératoire a été réalisée afin deconfirmer le lien entre les 2 lésions. Elle aconsisté en l’étude de l’expression des pro-téines MLH1, MSH2, MSH6, PM2. Elle aconclu à la perte de l’expression de la pro-téine MSH6 orientant vers l’existenced’une instabilité des microsatellites (MSI)constatée chez les sujets présentant uneprédisposition au cancer colorectal no-tamment de type HNPCC.Ainsi, notre patiente ne répond pas auxcritères cliniques de l’HNPCC (1) tout enprésentant les anomalies génétiques cor-respondantes.
DiscussionLe syndrome de Turcot (4) est un syndromehéréditaire rare (environ 160 cas décritsdont une minorité repose sur un diagnostic
Fig. 1 : Coloscopie.a, b Polypes rectaux.c Polypes plans du colon trans-
verse.
a bc
e syndrome de Turcot est un syndro-me héréditaire rare associant une tu-meur du système nerveux central et
une prédisposition héréditaire au cancercolorectal (CCR). Le diagnostic de cette af-fection repose sur des critères cliniques etendoscopiques désormais bien codifiés. Lepronostic à court ou moyen terme est som-bre en raison de l’association d’une tumeurgénéralement maligne du SNC. Mais l’in-térêt de ce diagnostic repose sur le déclen-chement de l’enquête génétique familialeet sur le suivi des sujets à risque qui sontexposés à plusieurs affections malignes.
Cas cliniqueNous présentons le cas d’une patienteâgée de 24 ans, sans antécédent familialdigestif, suivie depuis 2 ans pour polypesrécidivants du rectum avec initialementabsence de polypes coliques.Les polypes rectaux ont fait l’objet d’exé-rèses endoscopiques. La dernière colosco-pie retrouvait de nombreux polypes rec-taux, et montrait l’apparition de polypescoliques droit et transverse (fig. 1). Àl’œsogastroduodenoscopie, il existait parailleurs une polypose glandulokystiquegastrique. Les biopsies réalisées retrou-vaient à l’étude anatomopathologique unedégénérescence en carcinome in situ despolypes rectaux et au niveau du colon despolypes adénomateux avec dysplasies delégère à modérée. La patiente bénéficiaitalors d’une colectomie totale en étant consi-dérée comme cas index d’une prédispo-sition génétique aux cancers colorectaux.
L
(1) Service de Neuroradiologie diagnostique et thérapeu-tique, Hôpital Central, CHU de Nancy, 29, avenue du Maréchal de Lattre de Tassigny, 54035 Nancy cedex. (2) Service de Neurologie, Hôpital Central, CHU de Nancy. (3) Service de Gastroentérologie, CHU de Nancy, hopitaux de Brabais, allée de Morvan, 54511 Vandœuvre-lés-Nancy Cedex. (4) Service de Radiologie, Adultes, CHU de Nancy, Hôpitaux de Brabois, Allée de Morvan, 54511 Vandœuvre-lès-Nancy cedex.Correspondance : R SanouE-mail : [email protected]
J Radiol 2009;90
R Sanou et al. Le syndrome de Turcot : à propos d’un cas 843
génétique) associant une tumeur du systè-me nerveux central et une prédispositionhéréditaire au cancer colorectal (CCR)pouvant correspondre à une polyposeadénomateuse familiale (PAF) ; un syn-drome HNPCC (Hereditary Non Poly-posis Colorectal Cancer).La polypose adénomateuse familiale,responsable de 1 % des CCR, est une ma-ladie de transmission autosomique domi-nante à pénétrance complète (2). Elle estdue à la présence d’une mutation germi-nale du gène APC (adenomatous polyposiscoli) localisé sur le bras long du chromoso-me 5. Elle se caractérise par la présence deplusieurs centaines d’adénomes coliquesqui en l’absence de coloproctectomie totaleprophylactique se transforment en canceravant l’âge de 40 ans (1). Les lésions extra-coliques susceptibles de s’associer à laPAF sont multiples : tumeur desmoïde,hépatoblastome, cancer papillaire de lathyroïde. Dans le cadre de ces associa-tions, on définit le syndrome de GARD-NER associant kystes épidermoïdes,tumeurs desmoïdes et ostéome de la man-dibule et le syndrome de TURCOT.Dans ce cas la tumeur cérébrale la plusfréquemment rencontrée est le médullo-blastome. Elle reste néanmoins rare (risquede 7 contre respectivement 852 et 847pour la survenue de tumeurs desmoïdeset d’hépatoblastomes) (1).Le syndrome HNPCC responsable de 2 à4 % des CCR, est également de transmis-sion autosomique dominante (1). Il est du àune mutation constitutionnelle sur l’un desgènes du système de réparation des mésap-pariements de l’ADN (gène MMR). Les2 principaux gènes impliqués sont les
gènes MSH2 et MLH1 et plus rarement legène MSH6. Les cellules tumorales pré-sentent alors un phénotype particulier ap-pelé micro satellite instability (MSI) dans95 % des cas. La détection par génotypagede l’acide désoxyribonucléique (ADN) tu-moral sur cinq marqueurs mononucléoti-diques de référence est la méthode la plusperformante.Le syndrome HNPCC se définit clini-quement par les critères cliniques d’Ams-terdam (6, 7) :• PAF exclu ;• 3 CCR dans la famille dont 2 liés au1er degré ;• 2 générations successives atteintes ;• 1 cas survenu avant 50 ans.Les patients atteints de HNPCC ont unrisque plus élevé de développer d’autrescancers. Il s’agit d’adénocarcinomes inté-ressant l’endomètre, l’intestin grêle, l’uro-thélium et plus rarement les ovaires, l’es-tomac et l’épithélium des voies biliaires(1).L’association du syndrome HNPCC àune tumeur cutanée correspond au syn-drome de Muir-Torre et son association àune tumeur cérébrale correspond au syn-drome de Turcot. Dans ce cas, la tumeurcérébrale la plus fréquemment associéeest le glioblastome (3), comme dans le casde notre patiente.Les types de tumeurs cérébrales rapportésdans la littérature dans le cadre de syn-dromes de Turcot (5) ont été : le glioblas-tome (61 % des cas), le médulloblastome(25 %).Les autres types tumoraux associés sontrares avec le lymphome (2 %), le ménin-giome.
(< 2 %), l’adénome hypophysaire (< 2 %),le craniopharyngiome (< 1 %).La confirmation diagnostique d’un syn-drome de Turcot repose sur les analysesimmuno-histochimiques et cytogéné-tiques (1-4) : mise en évidence d’une mu-tation du gène APC dans les PAF ; exis-tence d’une instabilité microsatellitairedans le syndrome HNPCC, mise en évi-dence à partir de l’ADN extrait des cellulestumorales traduisant l’existence d’un dé-faut de réparation des mésappariementsde l’ADN induit par l’inactivation d’unou plusieurs gènes impliqués dans cesystème de réparation.Après le diagnostic d’un cas index, une en-quête génétique familiale s’impose afin derestreindre la surveillance aux sujets por-teurs de la mutation. Cette dernière reposesur la coloscopie (dégénérescence des poly-pes rectocoliques) en sachant qu’il n’existepas de recommandation spécifique concer-nant la surveillance cérébrale (6).Une des particularités du syndrome deTurcot est la survenue précoce de la tu-meur du SNC. Notre patiente est âgée deseulement 24 ans alors que l’âge de prédi-lection des glioblastomes se situe dans les5e et 6e décennies. Cette survenue précoceest directement en rapport avec le caractèregénétique de l’affection.La survie des patients présentant un glio-blastome dans le cadre du syndrome deTurcot dépasse habituellement de plu-sieurs mois celle des cas sporadiquescomme l’a montré Van Meir (5) avec unesurvie moyenne de plus de 27 mois contremoins de 12 mois dans les cas sporadiques(8). Ceci peut être du à la survenue préco-ce ci dessus évoquée dont on connaît le ca-
Fig. 2 : IRM encéphalique.Processus expansif fronto-temporal gauche qui apparaît hyperintense en FLAIR (a) et pondération T2 (b), hypointense en pondération T1 (c) avec rehaussement hétérogène après injection de chélate de gadolinium (d). La lésion exerce un effet de masse sur le ventricule latéral gauche avec engagement sous falcoriel.
a Coupe axiale FLAIR.b Coupe sagittale T2.c Coupe axiale T1.d Coupe axiale T1 avec injection de gadolinium.
a b c d

J Radiol 2009;90
844 Le syndrome de Turcot : à propos d’un cas R Sanou et al.
ractère favorable sur le plan pronostiquepour les gliomes malins (6).
ConclusionLe syndrome de Turcot est une situationrare qu’il faut évoquer devant l’associa-tion tumeur cérébrale et d’une tumeurdigestive. Son pronostic est sombre essen-tiellement en raison de la composanteneuro-oncologique. Au delà du diagnos-tic chez un cas index, une enquête géné-tique familiale puis un suivi des patientsporteurs de la mutation doivent systéma-tiquement être réalisés.Le rôle de l’imagerie réside ici dans le dia-gnostic et le suivi de la lésion cérébrale, leradiologue doit connaître cette affectionafin de suggérer une enquête génétique
lors de la découverte de la lésion cérébralechez un patient suspect ou porteur d’uneprédisposition au CCR.
Références1. Laurent-puig P. Prédispositions génétiques
aux cancers colorectaux. In gastro-entérolo-gie. Paris: Éditions ellipses, 2005; p490-6.
2. Hegde MR, Chong B, Blazo ME, Chin LH,Ward PA et al. A homozygous mutation inMSH6 causes turcot syndrome. Clin Can-cer Res 2005;11:4689-93.
3. Okamoto H, Mineta T, Nakahara Y, Ichi-nose M, Shiraishi T, Tabuchi K. Molecu-lar analysis of astrocytoma associated withTurcot syndrome Type 1. Neurol MedChir (Tokyo) 2004;44:124-8.
4. Cavanee WK, Burger PC, Van Meir EG.Turcot Syndrome. In Pathology and gene-
tics, Tumors of the Nervous System. Lyon :Ed Paul Kleihues, Leslie H. Sobin, 2000 ;p 238-9.
5. Van Meir EG. Turcot’s syndrome: Pheno-type of brain tumors, survival and mode ofinheritance. Int J Cancer 1998; 75: 162-4.
6. Olschwang S, Eisinger F. Prédispositionhéréditaire au cancer colorectal et inacti-vation de la fonction de réparation desmésappariements de l’ADN. EMC – Hé-pato-Gastroenterologie 2005;2:214-22.
7. Olschwang S, Bonaïti-Pellié C, Feingold J,Frébourg T et al. Identification et priseen charge du syndrome HNPCC (heredi-tary non polyposis colon cancer). Prédis-position héréditaire aux cancers du côlon,du rectum et de l’utérus. Pathol Biol2006;54:215-29.
8. Lebrun C, Olschwang S, Jeannin S, Van-denbos F, Sobol H, Frenay M. Turcot syn-drome confirmed with molecular analy-sis. Eur J Neurol 2007;14:470-2.