ankara Ünİversİtesİ bİlİmsel araŞtirma projesİ...
TRANSCRIPT

EK-11
ANKARA ÜNİVERSİTESİ BİLİMSEL ARAŞTIRMA PROJESİ
KESİN RAPORU
Proje Başlığı
Deneysel glioma modellerinde sürekli intrakranial suberoylanilide hydroxamic asid inhibitörlerinin
infüzyonu ve yüksek grade’li glial tümörlerde mevcut gen defektlerine yönelik kemoterapi
protokollerinin geliştirilmesi
Proje Yürütücüsünün İsmi
Prof.Dr.Yücel Kanpolat
Türkiye Bilimler Akademisi, Başkan
Yardımcı Araştırmacıların İsmi
Doç.Dr.Hasan Çağlar Uğur
Ankara Üniversitesi Tıp Fakültesi Nöroşirürji Ana Bilim Dalı/
Öğretim Üyesi
Prof.Dr.Ali Savaş
Ankara Üniversitesi Tıp Fakültesi Nöroşirürji Ana Bilim Dalı/
Öğretim Üyesi

Doç.Dr.Aylin Okçu Heper
Ankara Üniversitesi Tıp Fakültesi Patoloji Anabilim Dalı/ Öğretim Üyesi
Uzm.Dr.Mevci Özdemir
Diyarbakır Ergani Devlet Hastanesi/ Beyin ve Sinir Cerrahi Kliniği
Ankara Üniversitesi Bilimsel Araştırma Projeleri Ankara - " 2010"

Projenin Türkçe ve İngilizce Adı: Deneysel glioma modellerinde sürekli intrakranial suberoylanilide
hydroxamic asid inhibitörlerinin infüzyonu ve yüksek grade’li glial tümörlerde mevcut gen defektlerine
yönelik kemoterapi protokollerinin geliştirilmesi
Continuous intracranial administration of suberoylanilide hydroxamic acid in an orthotopic glioma
model and improvement of chemotherapy protocols in terms of gentic defects in human malignant
gliomas
Özet
Gliomalar primer beyin tümörlerinin %60’ını oluştururlar (1, 2). Dünya Sağlık Örgütü (WHO)
bu tümörleri atipi, mitoz, endotelial proliferasyon ve nekroz gibi histopatolojik özelliklerine göre
evrelendirmiştir. Tümör bu özelliklerden hiçbirisini taşımıyorsa evre 1, birisini (genellikle atipi)
taşıyorsa evre 2, ikisini taşıyorsa evre 3, daha fazlasını taşıyorsa evre 4 olarak evrelenmektedir. Grade I
tümörler, tanım gereği bu histopatolojik özelliklerden bağımsız olarak değerlendiren özel glial tümör
tipleridir (pilositik astrositom gibi). Tümörde hafif sellülarite artışı ve hafif atipi bulunuyorsa evre 2,
belirgin sellülarite artışı, atipi bulguları, mitoz sayısında artış içeriyorsa evre 3, nekroz ve endotelyal
proliferasyon gösteriyor ise evre 4 olarak değerlendirilmektedir. Evre 1 ve 2, düşük evreli tümörler
olarak tanımlanmıştır. Bu tümörler yavaş büyürler ve iyi sınırlıdırlar. Grade I olarak kabul edilen
juvenil pilositik astrositoma, pleomorfik ksantoastrositoma ve subependimal dev hücreli astrositoma iyi
sınırlı astrositomlar olarak değerlendirilirler. Tümörlerin grade’i arttıkça daha infiltratif bir görünüm
sergilerler. Grade III ve IV glial tümörler ise yüksek grade’li glial tümörler olarak değerlendirilir, bu
grup içinde de anaplastik astrositoma, glioblastoma multiforme, anaplastik oligodendroglioma ve
anaplastik ependimoma yer alır (1, 3, 4, 5, 6, 7, 8).

Epidemiyolojik çalışmalar ailesel kanser hikâyesinin olmasının, SSS de tümör gelişme riskini
arttırdığını göstermektedir. SSS tümörlü hastaların ortalama %15’inde ailesel malignite hikayesi
bulunmaktadır (6, 9, 10, 11).
Hücre siklusundaki özellikle DNA sentezinin olduğu S fazında pek çok genin düzenlediği
karmaşık supresyon ve aktivasyon noktaları mevcuttur. Malignite gelişim sürecinde tümör süpresor
genlerde allel kaybı onkogenlerde ise amplifikasyon olmaktadır. Önce retinoblastoma geni ve p53
sonrasında da bir çok tümör supressör gen (p16, p15, PTEN….) bulunmuştur. Onkogenler
protoonkogenlerin mutasyonel varyasyonlarıdır ve hücre proliferasyonunu uyarırlar. Bunların
başlıcaları Ras, MDM-2, PDGF, CDK-4’tür. Tümor süpressor genler, özelikle DNA sentezi
aşamasında mutasyonel DNA ortaya çıktığı dönemde apopitozu indükler. Tümör supressor
genlerindeki fonksiyon kaybı için genin karşılıklı her iki allelinin mutasyona uğraması gerekir (10, 12,
13, 14, 15, 16, 17).
Erişkin yaş grubundaki primer beyin tümörlerinin % 40-45’ini yüksek gradeli astrositomalar
oluşturur. Bu sebeple çalışma grubunda sadece yüksek gradeli tümörler ele alınmıştır (18).
Anaplastik astrositomalar gliomların yaklaşık %30’unu oluşturur, ortalama görülme yaşı 46’dır
ve belirtilerin ortaya çıkış süresi ortalama 16 aydır. Astrositomalarda 17. kromozomun kısa kolundaki
(17p) p53 geninin (telomerden yaklaşık 15-20 santimorgan uzaklıkta 53 kiloDalton ağırlığında, 11
ekson ve 10 introndan oluşan protein sentezleyen supressor gen) inaktivasyonunun gliomun
gelişmesinde önemli bir yeri vardır. Yedinci kromozomda aberasyon ve fazlalık sıklıkla saptanır.
Ayrıca 1. kromozomda TP73 geni (p53 benzeri tümör supressör gen), 9. kromozomda MTS 1 ve 2
geni, 13. kromozomda VEGF reseptörleri ve retinablastom tümör süpressör geni yitimi söz konusudur.
Yirmiikinci kromozomun PDGFb trombosit orjinli büyüme faktörü yanı sıra NF-2 gen lokusuna göre
daha telometrik yerleşimli “c-sis” onkogeni ve 10. kromozomun supressor genlerinin kaybının da rolü
büyüktür. Onuncu kromozomun her iki kolunda sekans kayıplarıda bildirilmiştir (11, 19, 20, 21, 22).

Epidermal biüyüme faktörü olarak da bilinen ve 7. kromozomda yer alan “Erb Bl” onkogeni ile
birlikte “src”, “ros”, “n-myc”, “c-myc”, “ets”, “erb A2” ve “mel” onkogenlerinin aktivasyonları da
gösterilmiştir. Anaplastik astrositomun tipik moleküler biyolojik özelliği 17. ve 7. kromozomlardaki
anomalilere eşlik eden ARFtümör supressor geni aberasyonudur. Ayrıca retinoblastoma supressor gen
lokusu E2F transkripsiyon faktörünü regüle eder. Anaplastik astrositomalarda bunun kaybı söz
konusudur. Bütün bunlardan ayrı olarak 20. , 3. , 6. , 9. , 11. ve hatta seks kromozomlarında bile
morfolojik kayıplar bildirilmiştir (23, 24, 25, 26, 27, 28).
Glial tümörlerin tedavisi cerrahi eksizyon, post-operatif radyoterapi ve kemoterapiden oluşur.
Rekürrensinde re-operasyon, kemoterapi, interstisyel brakiterapi yada radyocerrahi uygulanabilir. Tüm
tedavi seçenekleri uygulandığında glioblastomalarda ortalama yaşam süresi ne yazık ki 1 yıldan
kısadır. Kesin tedavisi yoktur. Cerrahiye eklenen her tip ek tedavi ile yaşam süresi ortalama 2 yılı
aşmaz. Median yaşam süresi sadece cerrahi uygulananlarda 4 ay, cerrahi+radyoterapide 9,25 ay,
cerrahi+radyoterapi+kemoterapi uygulananlarda ise 10 aydır. Reoperasyon ve/veya ek tedaviler yaşam
süresini 9-12 aya kadar uzatabilir (29, 30).
Glioblastoma multiforme tüm beyin tümörlerinin % 20-25’ini oluştururlar. Kromozom anomali
çalışmaları sonucunda GBM Tip I ve Tip II olmak üzere klinik prognozları da farklı olan iki ayrı
antiteden bahsedilmektedir. Tip I GBM (sekonder GBM), daha çok genç yaş (30-40) grubunda ortaya
çıkar ve düşük grade’li glial tümörlere sekonder olarak gelişir. 17p(-) ve 10q(-) tarzındaki kromozom
anomalilerinin gelişiminin basamaklı seyrettiği ve astrositom- anaplastik astrositom- GBM şeklindeki
oluşumdan sorumlu olduğu düşünülmektedir. Tip II GBM (GBM de novo, primer GBM ), Tip I’e göre
daha ileri yaşlarda (60) ortaya çıkar ve öncesinde bir glioma öyküsü yoktur. Onuncu kromzomun uzun
kolunda delesyon ve 7. kromozomda EGFR artışı söz konusudur (31, 32, 33, 34, 35, 36).
Plazminlerle ve amplifiye edilmiş genlerle moleküler kolonileştirme yöntemleri kullanılarak
glioblastoma multiformede 12.kromozomda “gli” isimli bir onkogenin varlığı gösterilmiştir. Yine
12.kromozom kaynaklı p53’ü inaktive eden bir onkoprotein olan MDM 2’nin de glioblastoma

oluşumunda rol aldığı gösterilmiştir. Glioblastomada ayrıca erbBl, sis, ros, neu, onkogenlerinin varlığı
ve 9.kromozomun kısa kolunda kayıp gösterilmiştir. Glioblastoma multiforme olgularının yaklaşık
%80’inde 10.kromozomda tam ya da kısmi kayıplar bildirilmiştir. Kısmi kayıplardaki bu defektif
bölgenin 10.kromozomun uzun kolunun distalinde prostat kanseri ve melanom için de defektif olarak
bildirilen 24-26 arası bölümdür. Glioblastoma supressor geni de 1997 yılında iki ayrı araştırıcı grubu
tarafından tanımlanmıştır. Bunlar PTEN (phosphate-tensin) ve MMAC 1 (mutated in multiple
advanced cancers-1) genleridir. Bunlaradan PTEN daha yaygın bir kullanım alanı bulmuştur. Son
çalışmalar ile bu lokusun kesin tanımı 10q23.3 olarak tescil edilmiştir (37, 38, 39, 40).
Son dönemlerde bu hastalığın ortalama yaşam süresini arttırmak için birçok yeni tedavi histon
deasetilaz (HDAC) inhibitörlerinin kullanımı gündeme gelmiştir. HDAC inhibitörleri akciğer, kolon,
mesane, meme, prostat, over, endometrium ve hematolojik sistem malignitelerinde kullanılmıştır. Buna
rağmen HDAC inhibitörlerinin gliomalar üzerine etkisi hakkında çok az yayın bulunmaktadır. HDAC
ve histon asetil transferaz (HAT) histonlardaki asetilasyon basamağını düzenlemektedir. Histon
deasetilaz ve histon asetil transferaz tarafından yapılan spesifik histonların amino gruplarının reversibl
asetilasyonu gen ekspresyonunun düzenleyici mekanizmasındaki en önemli basamaktır. HDAC
aktivitelerinin inhibisyonu asetile edilmiş nükleer histonların deasetilasyonu ile sonuçlanır. SAHA
(suberoylanilide hydroxamic acid) Bcl-2 familyasının preapopitotik üyelerinden olup apopitozisi
selektif olarak arttırır ve aynı zamanda proliferasyonuda engeller. Endotelyal hücrelerde ve kanser
hücrelerinde anjiogenezis inhibisyonu ile ilgili genlerin SAHA inhibitörlerinde varlığı gösterilmiştir
(41, 42, 43).
Delesyon veya mutasyonlarla inaktive edilmiş HAT varlığında insan tümörlerinin artışının
bağlantılı olduğu gösterilmiştir. Bu bağlamda bakıldığında SAHA ilerde klinik uygulamaları olma
potansiyeli olan bir ajan olarak görülmektedir (44, 45, 46).
Oligodendrogliomaların genetik analizi sonrasında elde edilen sonuçlar göstermiştirki 1p 19q
delesyonuna sahip tümörler kemoterapiye çok iyi yanıt vermektedir ve beklenen yaşam süresi 1p 19q

delesyonu olmayan tümörlere göre çok daha uzundur. Projenin ikinci basamağının amacı yüksek evreli
glial tümörlerin tedavisinde benzer bir ilişkiyi açığa koyabilmektir (47, 48, 49).
II. Amaç ve Kapsam
İnsan kaynaklı yüksek grade’li glial tümörlerde CGH ile genetik analiz yapılması. Bu tümör
dokularından cell line oluşturulması ve tespit edilen genetik anomaliler ile değişik kemoteropatiklere
olan yanıt arasında fark olup olmadığının araştırılması.
III. Materyal ve Yöntem
İnsan glioblastoma cell line oluşturulması:
Glioblastoma multiforme ön tanısı ile ameliyat edilmesi kararlaştırılan hastalara ameliyat öncesi
çalışmaya katılabilmeleri konusunda bilgi aktarılmıştır. Çalışmaya katılmak isteyen bireylere etik kurul
tarafından onayı alınmış bilgilendirilmiş hasta onam formları imzalatılmıştır.
Tümör parçasının tümörijenik hücre populasyonu içerebileceği ve hücre kültürü yapılabileceği patolog
tarafından saptandıktan sonra ilgili doku materyali 30 dakika içerisinde hücre kültürü laboratuarına
getirilerek primer kültür protokolü uygulanmaya başlanmıştır. Bazı hastalardan tümör dokusu yanında
ameliyat sırasında tümöre ulaşmak için çıkartılan normal doku örnekleri de alınmıştır. Bu durum
hakkında da hastalara ameliyat öncesinde bilgilendirme yapılmıştır.
Protokol:
Tümör dokusunun bir kısmı hücre kültürü amacıyla laminar flow kabin içerisinde ayrılmıştır. Ayrıca,
tümör ve normal beyin dokusuna ait parçaların bir kısmından Trizol protokolüyle RNA elde edilmiştir.
Geri kalan kısımlar ise azot muamelesini takiben -80°C’ye kaldırılmıştır.

Doku kültürü:
1-Doku parçaları laminar flow kabin içerisinde %10 Fetal Bovine Serum (FBS), 4mM L-glutamin,
100U/ml penisilin, 10µg/ml streptomycin içeren medyum (yüksek glikoz) içerisinde 1mm3 hacme
kadar kesilerek küçültülmüştür. Bu işlem sırasında damar parçaları ve nekrotik bölgeler çıkarılmıştır.
2-Doku parçaları içerisinde 10ml %10 FBS’li medyum içeren 50ml’lik falkona aktarılmıştır. Falkon
içerisinde pastör pipeti kullanılarak yapılan karıştırma ve pipetaj yardımıyla parçaların daha iyi
ayrılması sağlandı.
3-Parçalar bu medyum içerisinde 15 dakika bekletildi.
4-15 dakika sonunda parçalar falkon tüpünün alt kısmına çöktü ve üst kısım parçalara yaklaşılmadan
pastör pipeti yardımıyla atıldı. Tekrar 10ml’ye kadar ilgili medyum eklendi, 2. ve 3. basamak işlemleri
tekrar edildi. Bu yıkama işlemleri en az 3 kez tekrarlandı.
5- Son yıkama işlemi sonunda doku parçalarının üzerine 4 ml %40 FBS içeren (yüksek glikoz)
medyum eklendi ve karıştırıldı. Bu karışımın en fazla 1ml’si 25cm2’lik flasklara aktarıldı. Her flaskta
ortalama 20-25 doku parçası olmalıdır. Flasklar %5 CO2’li nemlendirilmiş, 37 ˚C’lik etüve kaldırıldı.
6- Hücreler 48 saat her hangi bir müdahale yapılmaksızın etüv içerisinde bekletildi. Bu aşama doku
parçalarının yüzeye tutunmalarını sağlamak içindir. Doku parça sayısının 25’in üzerinde olması doku
parçalarının homojen biçimde dağılmalarını ve tutunmalarını, medyum miktarının 1ml’nin üzerinde
olması doku parçalarının tutunmalarını engellemektedir.
7- 48 saat sonunda inverted mikroskop yardımıyla hücreler incelendi. Tututan doku parçaları var ise
flaskta yer alan medyumun üst kısmı atılır ve 2ml %40 FBS içeren medyum eklenir.
8-Bu işlem 48 saatte bir yenilendi. Doku parçalarında çıkan hücreler çoğalarak flaskın %50-60’ını
kapladığı zaman pasaj işlemi yapıldı.
9- %0,05’lik Tripsin/EDTA kullanılarak yapılan pasaj işlemi sonrasında ve hücreler %10’luk FCS
içeren medyuma konuldu ve bu aşamada standart hücre kültürü işlemleri uygulandı.

10- Birinci pasaj sonrasında hücrelerde immünositokimyasal yöntemle GFAP (Glial Fibrillary Acidic
Protein) boyaması yapıldı. Bu işlem çoğaltılan hücrelerin gerçek tümör hücresi olduğunu göstermek
amacıyla yapıldı. Bu boya yalnızca glial kökenli hücrelere özgü bir proteini işaretlemektedir.
11- İlk 2 pasaj sonrasında hücreler aynı şartlarda 75cm2’lik flasklara aktarılmış ve sonraki pasajlarda bu
flasklar kullanılmıştır. Deney başlangıcında yer alan kan, epitel ve fibroblastlar pasaj sayısı arttıkça
ortadan kaybolmuş ve glial kökenli hücreler seçilmiştir.
12- Pasajı yapılan hücrelerden bir kısmı likit nitrojen (%90 FBS / %10 DMSO) içinde dondurulmuştur.
Sitotoksisite ve Canlılık Analizleri
Hastalardan elde edilen ve immün boyamayla glial kökenli hücre olduğu saptananlarda çeşitli ilaçlar
kullanılarak MTT ile sitotoksisite ve Trypan mavisiyle de canlılık analizi yapılmıştır. Bu çalışma GBM
olduğu bilinen ticari hücre serilerinde de yapılmıştır. Hücre serilerine GBM tedavisinde en çok
kullanılan Temozolomide (Temodal, Schering-Plough) verilerek MTT ve canlılık analizi yapılmıştır.
Real-Time PCR (Gerçek Zamanlı PZR)
MTT ve Trypan mavisi boyama sonuçlarına göre hücrelere çeşitli dozlarda ilaç kombinasyonları
uygulanmıştır. Özellikle uygulamalar 48 saat üzerinden temozolomid kullanılarak yapılmıştır. 48.saatin
sonunda hücrelerden RNA elde edilmiştir. Elde edilen RNA’lar spektrofotometrik olarak ölçüldükten
sonra cDNA sentezi yapılmıştır. Elde edilen cDNA’lar kullanılarak GBM tedavisinde ilaç direncinden
sorumlu bazı genlerin ekspresyonları araştırılmıştır. Bu analizde kullanılan Real-Time cihazına ait
optimizasyon görüntüleri aşağıda gösterilmektedir.

Sonuçlar
Doku ve Hücre Kültürü:
Elde edilen primer kültürlere ait bazı şekiller aşağıda sunulmuştur.
Şekil I- Doku kültüründe flaska tutunmuş bir tümör parçasından çıkan hücrelerin görünümü-1
Şekil II- Doku kültüründe flaska tutunmuş bir tümör parçasından çıkan hücrelerin görünümü-2

Şekil III- Doku kültüründe flaska tutunmuş bir tümör parçasından çıkan hücrelerin görünümü-3
Şekil IV- Doku kültüründe flaska tutunmuş bir tümör parçasından çıkan hücrelerin görünümü-4

Şekil V: Farklı hastalara ait dokularadan elde edilen astrsoit kökenli hücreler
Şekil VI- a) GFAP İmmün boyama öncesinde glial hücreler b) GFAP İmmün boyama sonrasında glial hücreler
a b
a b
Şekil VI- a) GFAP İmmün boyama öncesinde glial hücreler b) GFAP İmmün boyama sonrasında glial hücreler
MTT ve Canlılık Testi
Hastalara ait hücrelere ve ticari olarak alınan hücre serilerinde Temozolomide ait sitotoksisite ve
canlılık testi sonuçları birbirine yakın bulunmuştur. Temozolomid’in 250, 500 ve 1000µM’lık
konsatrasyonlarının toksik olduğu yapılan 24, 48 ve 72. saat testlerinde gösterilmiştir. Hastalar ait
hücrelerin IC50 değerleri Graphad Prizm programı ile hesaplanmıştır.
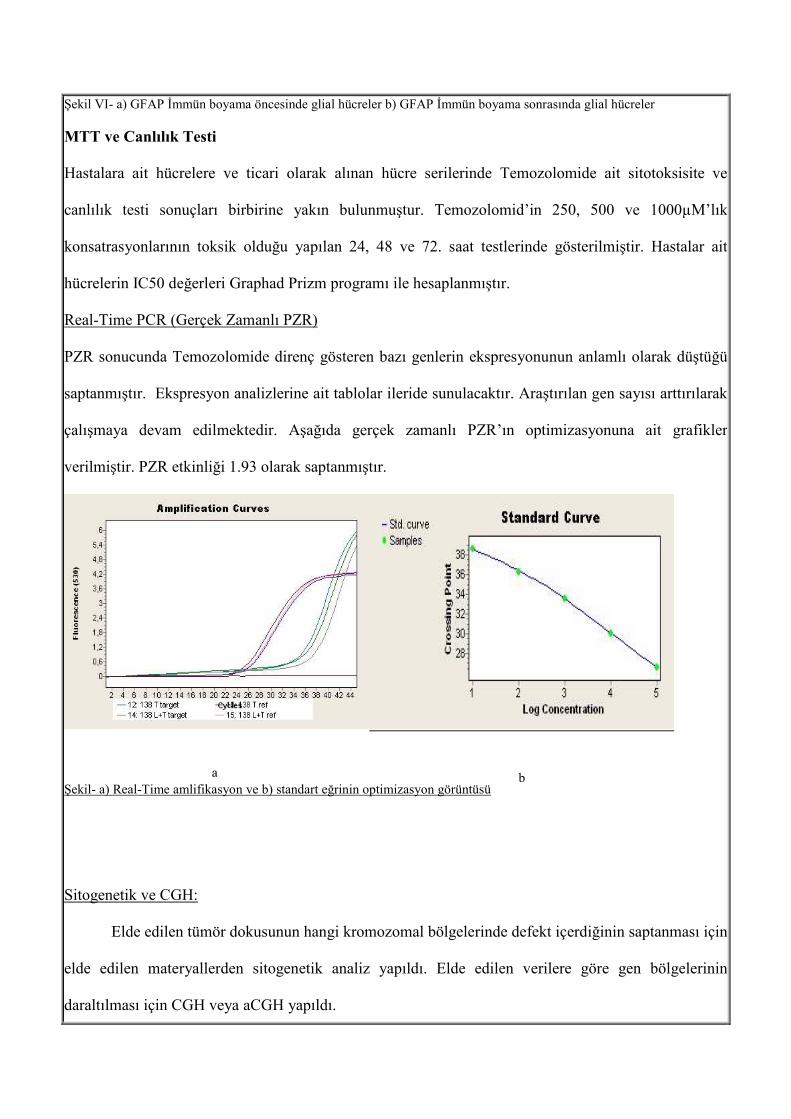
Real-Time PCR (Gerçek Zamanlı PZR)
PZR sonucunda Temozolomide direnç gösteren bazı genlerin ekspresyonunun anlamlı olarak düştüğü
saptanmıştır. Ekspresyon analizlerine ait tablolar ileride sunulacaktır. Araştırılan gen sayısı arttırılarak
çalışmaya devam edilmektedir. Aşağıda gerçek zamanlı PZR’ın optimizasyonuna ait grafikler
verilmiştir. PZR etkinliği 1.93 olarak saptanmıştır.
Şekil- a) Real-Time amlifikasyon ve b) standart eğrinin optimizasyon görüntüsü
Sitogenetik ve CGH:
Elde edilen tümör dokusunun hangi kromozomal bölgelerinde defekt içerdiğinin saptanması için
elde edilen materyallerden sitogenetik analiz yapıldı. Elde edilen verilere göre gen bölgelerinin
daraltılması için CGH veya aCGH yapıldı.
a b

Sitogenetik analiz:
Her bir flasktan 5 ml süspanse kültür alınarak 15 ml’lik konikal tüplere aktarıldı. 0.04µg/ml son
konsantrasyonda kolşisin eklenen tüpler 4 saat 37°C'de bekletildi. Örnekler 900 rpm'de 10' santrifüjde
çevrildikten sonra üst faz atıldı ve gevşetilen çökelti üzerine konulan hipotonik (0.075 M KCl) ile 5
ml'ye tamamlandı. Otuz dakika 37°C'de bekletilen tüpler yine aynı devirde 10' çevrilip ve üst faz
atıldıktan sonra çöküntü bu kez Carnoy fiksatifi (metanol: asetik asit 3:1, Merck #6008 ve #56) ile 5
ml'ye tamamlandı. Fiksatif ile yıkama işlemi 3 kez tekrarlandı. Son üst faz atıldıktan sonra çöküntü
temiz, ıslak ve soğuk lamlar üzerine yayıldı. %5 Giemsa ile boyanan preparatlarda 20 metafaz
değerlendirildi (Rooney ve Czepulkowski, 1992b).
CGH:
Test ve referans DNA’ların “Nick Translation” (NT) yöntemi ile işaretlendi. Fragman
boylarının 300-3000 bç arasında olduğundan emin olmak için, NT ürünlerinin 8’er µl’si, %1’lik agaroz
jele yüklenecek 90 V’da yaklaşık 45 dakika olacak şekilde elektroforez işlemi gerçekleştirildi.
CGH hibridizasyonu için CGH Hybridization Reagents Kit’i kullanıldı. Normal bir bireye ait lenfosit
kültüründen elde edilmiş hedef preparatlar (Vysis, 30-806010) inverted mikroskop (Olympus) altında
metafaz yoğunluğu ve kalitesi açısından değerlendirildi ve optimum hibridizasyon için uygun alanlar
belirlendi. Preparatlar dehidrate ve denatüre edildi ve 55°C’deki “hot plate”de kurutuldu. Her bir prob
karışımı 10µl olacak şekilde, preparat üzerinde daha önceden belirlenmiş alanlara uygulandı ve
üzerlerine 22x22mm lamel kapatılarak 37 °C’lik etüvde, nemli ve ışık almayan kapalı bir kutuda
yaklaşık 96 saat inkübe edildi. Lamellerin uzaklaştırılmasından sonra, preparatlar, 75°C’deki su
banyosunda bulunan yıkama solüsyonlarından geçirildi. Kurumuş prepratlar üzerinde prob uygulanan
bölgelere 10 µl DAPI II konarak lamel ile kapatıldı.
Preparatların değerlendirilmesi Olympus BX61 mikroskobunda yapıldı. Görüntü analizi için

Cytovision 3.1 yazılım programı (Applied Imaging) kullanıldı. Her bir hücre için sırasıyla DAPI,
spectrum-green ve spectrum-red filtreleri kullanılarak 3 ayrı görüntü elde edildi ve kalite kontrol
kriterlerini geçen hücreler analize dahil edildi. Her örnekte optimum 20 metafaz için bu işlem
tekrarlandı. Her hücre için karyotip işlemi bitirildikten sonra, çalışılan örneğin elde edildiği kişinin
cinsiyetine uygun standart referans aralığı seçilerek %95 güven aralığında CGH profilleri
değerlendirildi.
Sitotoksite çalışması:
Oluşturulan cell line’larda günümüzde kullanılan kemoteropatikler ve deneysel olarak deneme
aşamasında olan diğer ajanların insan glioma cell line’larındaki etkileri CCK-8 assay ile saptandı.
Sitogenetik analizlerden elde edilen veriler ile sitotoksisite çalışmalarından elde edilen istatisiksel
olarak değerlendirildi ve farklı yolakların etkilendiği durumlar için farklı kemoterapi kullanımının
klinik sonuçlarını etkileyip etkilemediği konusunda veri oluşturulmaya çalışıldı.
IV. Analiz ve Bulgular
Çalışmaya alınan hastalar tamamlanmış olup testler devam etmektedir. Bulguların daha sağlıklı
yorumlanması için testlerin neticelenmesi ile birlikte kesin sonuç bildirilecektir.
V. Sonuç ve Öneriler
Çalışmanın tamamlanması ile birlikte glioma tedavisine ışık tutacak yeni bulgulara ulaşılması
beklenmektedir. Testlerin tamamlanması ile birlikte sonuç ve öneriler ek rapor halinde
sunulacaktır.

VI. Kaynaklar
1. Baek SY, Kim SR, Bae MK, Hwang JW, Kim JS, Choi YH, et al: Trichostatin A increases the
thermosensitivity of human glioblastoma A172 cells. Neurosci Lett 396:230-234, 2006
2. Bansal K, Liang ML, Rutka JT: Molecular biology of human gliomas. Technol Cancer Res
Treat 5:185-194, 2006
3. Burzynski SR, Janicki TJ, Weaver RA, Burzynski B: Targeted therapy with antineoplastons
A10 and AS2-1 of high-grade, recurrent, and progressive brainstem glioma. Integr Cancer
Ther 5:40-47, 2006
4. Butler LM, Agus DB, Scher HI, Higgins B, Rose A, Cordon-Cardo C, et al: Suberoylanilide
hydroxamic acid, an inhibitor of histone deacetylase, suppresses the growth of prostate cancer
cells in vitro and in vivo. Cancer Res 60:5165-5170, 2000
5. Canes D, Chiang GJ, Billmeyer BR, Austin CA, Kosakowski M, Rieger-Christ KM, et al:
Histone deacetylase inhibitors upregulate plakoglobin expression in bladder carcinoma cells and
display antineoplastic activity in vitro and in vivo. Int J Cancer 113:841-848, 2005
6. Chinnaiyan P, Vallabhaneni G, Armstrong E, Huang SM, Harari PM: Modulation of radiation
response by histone deacetylase inhibition. Int J Radiat Oncol Biol Phys 62:223-229, 2005
7. Chintagumpala MM, Friedman HS, Stewart CF, Kepner J, McLendon RE, Modrich PL, et al:
Open-label simultaneous radio-chemotherapy of glioblastoma multiforme with topotecan in
adults. J Neurooncol 77:193-198, 2006
8. Cohen LA, Marks PA, Rifkind RA, Amin S, Desai D, Pittman B, et al: Suberoylanilide
hydroxamic acid (SAHA), a histone deacetylase inhibitor, suppresses the growth of carcinogen-
induced mammary tumors. Anticancer Res 22:1497-1504, 2002
9. Combs SE, Heeger S, Haselmann R, Edler L, Debus J, Schulz-Ertner D: Treatment of Primary
Glioblastoma Multiforme with Cetuximab, Radiotherapy and Temozolomide (GERT) - Phase

I/II Trial: Study Protocol. BMC Cancer 6:133, 2006
10. DeAngelis LM: Brain tumors. N Engl J Med 344:114-123, 2001
11. Entin-Meer M, Rephaeli A, Yang X, Nudelman A, VandenBerg SR, Haas-Kogan DA: Butyric
acid prodrugs are histone deacetylase inhibitors that show antineoplastic activity and
radiosensitizing capacity in the treatment of malignant gliomas. Mol Cancer Ther 4:1952-
1961, 2005
12. Eyupoglu IY, Hahnen E, Buslei R, Siebzehnrubl FA, Savaskan NE, Luders M, et al:
Suberoylanilide hydroxamic acid (SAHA) has potent anti-glioma properties in vitro, ex vivo
and in vivo. J Neurochem 93:992-999, 2005
13. Fandy TE, Shankar S, Ross DD, Sausville E, Srivastava RK: Interactive effects of HDAC
inhibitors and TRAIL on apoptosis are associated with changes in mitochondrial functions and
expressions of cell cycle regulatory genes in multiple myeloma. Neoplasia 7:646-657, 2005
14. Giussani C, Carrabba G, Pluderi M, Lucini V, Pannacci M, Caronzolo D, et al: Local
intracerebral delivery of endogenous inhibitors by osmotic minipumps effectively suppresses
glioma growth in vivo. Cancer Res 63:2499-2505, 2003
15. Gui CY, Ngo L, Xu WS, Richon VM, Marks PA: Histone deacetylase (HDAC) inhibitor
activation of p21WAF1 involves changes in promoter-associated proteins, including HDAC1.
Proc Natl Acad Sci U S A 101:1241-1246, 2004
16. Hockly E, Richon VM, Woodman B, Smith DL, Zhou X, Rosa E, et al: Suberoylanilide
hydroxamic acid, a histone deacetylase inhibitor, ameliorates motor deficits in a mouse model
of Huntington’s disease. Proc Natl Acad Sci USA 100:2041-2046, 2003
17. Hsi LC, Xi X, Lotan R, Shureiqi I, Lippman SM: The histone deacetylase inhibitor
suberoylanilide hydroxamic acid induces apoptosis via induction of 15-lipoxygenase-1 in

colorectal cancer cells. Cancer Res 64:8778-8781, 2004
18. Kamitani H, Taniura S, Watanabe K, Sakamoto M, Watanabe T, Eling T: Histone acetylation
may suppress human glioma cell proliferation when p21 WAF/Cip1 and gelsolin are induced.
Neuro-oncol 4:95-101, 2002
19. Kelly WK, O'Connor OA, Krug LM, Chiao JH, Heaney M, Curley T, et al: Phase I study of an
oral histone deacetylase inhibitor, suberoylanilide hydroxamic acid, in patients with advanced
cancer. J Clin Oncol 23:3923-3931, 2005
20. Kelly WK, Richon VM, O'Connor O, Curley T, MacGregor-Curtelli B, Tong W, et al: Phase I
clinical trial of histone deacetylase inhibitor: suberoylanilide hydroxamic acid administered
intravenously. Clin Cancer Res 9:3578-3588, 2003
21. Kim EH, Kim HS, Kim SU, Noh EJ, Lee JS, Choi KS: Sodium butyrate sensitizes human
glioma cells to TRAIL-mediated apoptosis through inhibition of Cdc2 and the subsequent
downregulation of survivin and XIAP. Oncogene 24:6877-6889, 2005
22. Kim MS, Blake M, Baek JH, Kohlhagen G, Pommier Y, Carrier F: Inhibition of histone
deacetylase increases cytotoxicity to anticancer drugs targeting DNA. Cancer Res 63:7291-
7300, 2003
23. Kim MS, Kwon HJ, Lee YM, Baek JH, Jang JE, Lee SW, et al: Histone deacetylases induce
angiogenesis by negative regulation of tumor suppressor genes. Nat Med 7:437-743, 2001
24. Komatsu N, Kawamata N, Takeuchi S, Yin D, Chien W, Miller CW, et al: SAHA, a HDAC
inhibitor, has profound anti-growth activity against non-small cell lung cancer cells.Oncol Rep
15:187-191, 2006.
25. Leon SP, Folkerth RD, Black PM: Microvessel density is a prognostic indicator for patients
with astroglial brain tumors. Cancer 77:362-372, 1996

26. Lin RJ, Nagy L, Inoue S, Shao W, Miller WH Jr, Evans RM: Role of the histone deacetylase
complex in acute promyelocytic leukaemia. Nature 391:811–814, 1998
27. Marks P, Rifkind RA, Richon VM, Breslow R, Miller T, Kelly WK: Histone deacetylases and
cancer: causes and therapies. Nat Rev Cancer 1: 194–202, 2001
28. Munshi A, Kurland JF, Nishikawa T, Tanaka T, Hobbs ML, Tucker SL,et al: Histone
deacetylase inhibitors radiosensitize human melanoma cells by suppressing DNA repair
activity. Clin Cancer Res 11:4912-4922, 2005
29. Munster PN, Troso-Sandoval T, Rosen N, Rifkind R, Marks PA, Richon VM: The histone
deacetylase inhibitor suberoylanilide hydroxamic acid induces differentiation of human breast
cancer cells. Cancer Research 61: 8492–8497, 2001
30. O'Connor OA, Heaney ML, Schwartz L, Richardson S, Willim R, MacGregor-Cortelli B, Curly
T, et al: Clinical experience with intravenous and oral formulations of the novel histone
deacetylase inhibitor suberoylanilide hydroxamic acid in patients with advanced hematologic
malignancies. J Clin Oncol 24:166-173, 2006
31. Qian DZ, Kato Y, Shabbeer S, Wei Y, Verheul HM, Salumbides B, et al: Targeting tumor
angiogenesis with histone deacetylase inhibitors: the hydroxamic acid derivative LBH589. Clin
Cancer Res 12:634-642, 2006
32. Qian DZ, Wang X, Kachhap SK, Kato Y, Wei Y, Zhang L, et al: The histone deacetylase
inhibitor NVP-LAQ824 inhibits angiogenesis and has a greater antitumor effect in combination
with the vascular endothelial growth factor receptor tyrosine kinase inhibitor
PTK787/ZK222584. Cancer Res 64:6626-6634, 2004
33. Reardon DA, Rich JN, Friedman HS, Bigner DD: Recent advances in the treatment of
malignant astrocytoma. J Clin Oncol 24:1253-1265, 2006

34. Richon VM, Sandhoff TW, Rifkind RA, Marks PA: Histone deacetylase inhibitor selectively
induces p21WAF1 expression and gene-associated histone acetylation. Proc Natl Acad Sci U S
A 97:10014-10019, 2000
35. Sambucetti LC, Fischer DD, Zabludoff S, Kwon PO, Chamberlin H, Trogani N, et al: Histone
deacetylase inhibition selectively alters the activity and expression of cell cycle proteins leading
to specific chromatin acetylation and antiproliferative effects. J Biol Chem 274:34940-34947,
1999
36. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al: Radiotherapy
plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 352:987-996,
2005
37. Schmidt KF, Ziu M, Schmidt NO, Vaghasia P, Cargioli TG, Doshi S, et al: Volume
reconstruction techniques improve the correlation between histological and in vivo tumor
volume measurements in mouse models of human gliomas. J Neurooncol 68:207-215, 2004
38. Schmidt NO, Ziu M, Carrabba G, Giussani C, Bello L, Sun Y, et al: Antiangiogenic therapy by
local intracerebral microinfusion improves treatment efficiency and survival in an orthotopic
human glioblastoma model. Clin Cancer Res 10:1255-1262, 2004
39. Suzuki T, Nagano Y, Kouketsu A, Matsuura A, Maruyama S, Kurotaki M, et al: Novel
inhibitors of human histone deacetylases: design, synthesis, enzyme inhibition, and cancer cell
growth inhibition of SAHA-based non-hydroxamates. J Med Chem 48:1019-1032, 2005
40. Takai N, Kawamata N, Gui D, Said JW, Miyakawa I, Koeffler HP: Human ovarian carcinoma
cells: histone deacetylase inhibitors exhibit antiproliferative activity and potently induce
apoptosis. Cancer 101:2760-2770, 2004
41. Takai N, Desmond JC, Kumagai T, Gui D, Said JW, Whittaker S, et al: Histone deacetylase
inhibitors have a profound antigrowth activity in endometrial cancer cells. Clin Cancer Res

10:1141-1149, 2004
42. Weidner N, Semple JP,Welch WR, Folkman J: Tumor angiogenesis and metastasis-correlation
in invasive breast carcinoma. N Engl J Med 324:1-8, 1991
43. Wetzel M, Premkumar DR, Arnold B, Pollack IF: Effect of trichostatin A, a histone deacetylase
inhibitor, on glioma proliferation in vitro by inducing cell cycle arrest and apoptosis. J
Neurosurg 103(6 Suppl):549-556, 2005
44. Zhang C, Richon V, Ni X, Talpur R, Duvic M : Selective induction of apoptosis by histone
deacetylase inhibitor SAHA in cutaneous T-cell lymphoma cells: relevance to mechanism of
therapeutic action. J Invest Dermatol 125:1045-1052, 2005
45. Dai C., Holland E.C. Glioma Models Biochimica et Biophysia Acta 1551 M19-M27, 2001
46. Furnari B, Su Huang H.J., Caneve W.K : Molecular Biology of Malignant Degeneration of
Astrocytoma Pediatric Neurosurgery 24:41-49, 1996
47. Hegi M.E., Klein M.A., Rüedi D., Chene P, Hamou M.F., Aguzzi A: p53 Transdominance But
No Gain Of Function in Mouse Brain Tumor model. Cancer Research 60:3019-3024, 2000
48. Barker F.G, Chen P, Furman F, Aldape K.D, Michael S.B, Edwards And Mark A: P16 deletion
and mutation analysis in human brain tumors. Journal of Neuro-Onkoloji 31:17-23, 1997
49. Ugur HC, Sayan AE, Kanpolat Y, Ozturk M. Expression of TAP73 and ∆NP73 in malignant gliomas. Oncology Rep (B) 11(6): 13337-1341, 2004.