microarray data analysis tool using java and...
TRANSCRIPT

MICROARRAY DATA ANALYSIS TOOL USING JAVA AND R
By
Vasundhara Akkineni B.Tech, University of Madras, 2003
A Thesis Submitted to the Faculty of the
Graduate School of the University of Louisville In Partial Fulfillment of the Requirements
for the Degree of
MASTER OF SCIENCE
Department of Computer Engineering and Computer Science University of Louisville
Louisville, Kentucky
May 2006

MICROARRAY DATA ANALYSIS USING JAVA AND R
By
Vasundhara Akkineni B.Tech, University of Madras, 2003
A Thesis Approved on
April 14, 2006
By the following Thesis Committee:
Dr. Eric C. Rouchka, Thesis Director
Dr. Dar-jen Chang
Dr. Thomas Knudsen
ii

DEDICATION
Dedicated to my parents
Mr. Sarat Kumar Akkineni
and
Mrs. Surya Rani Akkineni
Thanks for everything, papa and ma.
iii

ACKNOWLEDGEMENTS
First, I would like to thank my thesis director, Dr. Eric C. Rouchka for his direction,
assistance and guidance. I would also like to thank the members of my thesis committee,
Dr. Dar-jen Chang and Dr. Thomas Knudsen for their time. I thank Tim Hardin,
Elizabeth Cha, Yamini Rudraraju and Eric Stutzenberger for making the Bioinformatics
lab an enjoyable place to come to each day. Additional thanks to the other members of
the Bioinformatics Research Group. I thank all the friends I have made over the years at
the University of Louisville. I have learned a lot from each one of you. Finally, I thank
my parents with all due respect for their love, support and encouragement. Support for
this project was provided by NIH-NCRR grant # P20 RR16481 (Nigel Cooper, PI).
iv

ABSTRACT
MICROARRAY DATA ANALYSIS USING JAVA AND R
VASUNDHARA AKKINENI
APRIL 14, 2006 Microarray technology has become an essential tool in functional genomics for
monitoring the expression of many genes in parallel. Gene expression values obtained
from microarray experiments help biologists to understand the way in which a cell
responds to varying conditions (including, but not limited to development over time,
response to environmental stimuli, or disease states) by analyzing the increase or
decrease in the expression level of genes. We have developed web-based software that
provides biologists with several statistical solutions for analyzing gene expression data.
This platform independent java servlet first performs normalization of the gene
expression values in order to eliminate any systematic bias in the measured intensity
values arising from the microarray process. Several normalization methods like Total
Intensity Normalization, Median Normalization and Lowess Normalization have been
implemented. After normalization, visualization of the experimental data can be
performed using scatter plots, MA plots, RI plots and image maps of the intensity ratios.
For detection of genes which are differentially expressed the software provides fold-
change detection and t-test techniques. The tool also provides the users the ability to
create a workflow of the different analysis tools used to study the uploaded
v

data. All the statistical routines used in this software were developed in R called from
Java code. This software is a freely available tool to statistically analyze microarray
experiments.
vi

TABLE OF CONTENTS
DEDICATION……………………………………………………………………………iii ACKNOWLEDGEMENTS………………………………………………………………iv ABSTRACT……………………………………………………………………………….v LIST OF TABLES………………………………………………………………………..xi LIST OF FIGURES...…………………………………………………………….……...xii 1. INTRODUCTION ...................................................................................................... 1
1.1 Overview of molecular biology .......................................................................... 4
1.2 DNA.................................................................................................................... 4
1.3 RNA .................................................................................................................... 6
1.4 mRNA................................................................................................................. 6
1.5 Gene .................................................................................................................... 7
1.6 Central Dogma of Molecular Biology ................................................................ 7
1.7 MicroRNA (miRNA) .......................................................................................... 8
1.8 Microarrays ......................................................................................................... 9
2. MICROARRAY ANALYSIS TECHNIQUES......................................................... 13
2.1 Microarray data analysis ................................................................................... 13
2.2 Log ratios .......................................................................................................... 16
2.3 Normalization ................................................................................................... 17
2.3.1 Total intensity normalization .................................................................... 17
2.3.2 Median normalization ............................................................................... 18
2.3.3 Lowess normalization ............................................................................... 18
2.4 Scatter plot ........................................................................................................ 19
vii

2.5 MA plot............................................................................................................. 20
2.6 RI plot ............................................................................................................... 21
2.7 Difference between MA and RI plots ............................................................... 22
2.8 Identifying differentially expressed genes ........................................................ 23
2.8.1 Fold change............................................................................................... 23
2.9 Clustering.......................................................................................................... 24
2.10 Types of clustering............................................................................................ 24
2.10.1 Hierarchical clustering .............................................................................. 25
2.10.2 Dendrogram .............................................................................................. 26
2.10.3 Heat maps.................................................................................................. 27
3. LITERATURE REVIEW ......................................................................................... 29
3.1 Bioconductor..................................................................................................... 29
3.2 TM4 and MIDAS.............................................................................................. 31
3.3 BASE: BioArray Software Environment.......................................................... 32
3.4 WebArray: an online platform for microarray data analysis ............................ 32
3.5 SNOMAD (Standardization and Normalization of MicroArray Data)............. 33
4. IMPLEMENTATION SPECIFICS .......................................................................... 35
4.1 R........................................................................................................................ 35
4.1.1 Statistics and R.......................................................................................... 36
4.1.2 R and Windows™..................................................................................... 36
4.2 Rserve ............................................................................................................... 37
viii

4.2.1 Installation of Rserve ................................................................................ 38
4.3 Java ................................................................................................................... 39
4.3.1 Java language ............................................................................................ 39
4.3.2 Java platform............................................................................................. 39
4.4 Java servlets ...................................................................................................... 40
4.5 JSP (Java Server Pages) .................................................................................... 42
4.6 JDBC (Java Database Connectivity)................................................................. 42
4.7 MySQL ............................................................................................................. 43
4.8 Apache Tomcat ................................................................................................. 44
5. OBJECTIVES AND RESULTS............................................................................... 46
5.1 Uploading experiment data ............................................................................... 47
5.2 Normalization methods..................................................................................... 48
5.3 Data visualization.............................................................................................. 50
5.3.1 Scatter plot ................................................................................................ 50
5.3.2 MA plot..................................................................................................... 51
5.3.3 RI plot ....................................................................................................... 52
5.4 Creating a process pipeline ............................................................................... 53
5.5 Identifying genes of interest.............................................................................. 57
5.5.1 Fold change cut-off ................................................................................... 57
5.6 Clustering genes................................................................................................ 60
5.7 Top and bottom intensity ratios ........................................................................ 61
ix

5.8 Search genes...................................................................................................... 63
5.9 Output results as files........................................................................................ 65
6. CONCLUSIONS....................................................................................................... 67
6.1 Possible improvements ..................................................................................... 68
REFERENCES…………………………………………………………………………..70
APPENDICES…………………………………………………………………………...72
CURRICULUM VITAE…………………………………………………………………77
x

LIST OF TABLES
Table 2.1 - Gene expression matrix with raw gene expression data................................. 14
Table 2.2 - Gene expression matrix with intensity ratio values........................................ 15
Table 2.3 - Gene expression matrix with log 2 intensity ratio values ............................... 16
Table 3.1 - Bioconductor packages................................................................................... 30
Table 5.1 - Color coding scheme for differentially expressed genes using fold change .. 57
xi

LIST OF FIGURES
Figure 1.1 - An overview of the formation of proteins....................................................... 4
Figure 1.2 - DNA double helix structure ............................................................................ 5
Figure 1.3 - Formation of mRNA ....................................................................................... 6
Figure 1.4 - Central Dogma of Molecular Biology............................................................. 8
Figure 1.5 - Preparation of microarrays............................................................................ 10
Figure 2.1 - Process of obtaining a gene expression matrix ............................................. 13
Figure 2.2 - Effects of lowess normalization .................................................................... 19
Figure 2.3 - A scatter plot ................................................................................................. 20
Figure 2.4 - An MA plot ................................................................................................... 21
Figure 2.5 - An RI plot...................................................................................................... 22
Figure 2.6 - Construction of a two-dimensional dendrogram representing a hierarchical
cluster of related genes...................................................................................................... 26
Figure 2.7 - A heat map with a dendrogram and a color key............................................ 28
Figure 4.1 - R command line interface on startup ............................................................ 37
Figure 4.2 - Java servlet execution process ...................................................................... 41
Figure 4.3 - The three-tier architecture of a JDBC connection......................................... 43
Figure 5.1 - Sample data file for analysis ......................................................................... 47
Figure 5.2 - Process of uploading data files...................................................................... 48
Figure 5.3 - Normalized data file using total intensity normalization .............................. 49
Figure 5.4 - Normalized data file using median normalization ........................................ 49
xii

Figure 5.5 - Total intensity normalization scatter plots and text files .............................. 50
Figure 5.6 - matrix of scatter plots with a zoomed out portion for two specific
experiments ....................................................................................................................... 51
nn×
Figure 5.7 - matrix of MA plots with a zoomed out portion for two specific
experiments ....................................................................................................................... 52
nn×
Figure 5.8 - matrix of RI plots with a zoomed out portion for two specific
experiments ....................................................................................................................... 53
nn×
Figure 5.9 - Steps for pipelining analysis ......................................................................... 54
Figure 5.10 - Pipeline screen to define a sequence of routines to be performed on the data
........................................................................................................................................... 55
Figure 5.11 - Results screen after submitting the pipeline screen .................................... 56
Figure 5.12 - Color based image map of the gene’s intensity ratios between two
experiments ....................................................................................................................... 59
Figure 5.13 – Individual gene details................................................................................ 59
Figure 5.14 - Heat map with a dendrogram to represent expression clusters ................... 61
Figure 5.15 - User selected columns for calculating the intensity ratios and the number of
top/bottom genes needed................................................................................................... 62
Figure 5.16 - Results showing top 10 ratios and bottom 25 ratios between two
experiments ....................................................................................................................... 63
Figure 5.17 - Search screen with the list of genes from the uploaded data file ................ 64
Figure 5.18 - Output screen for a search done on gene information................................. 65
xiii

1. INTRODUCTION
Two complementary advances, one in knowledge and one in technology, are
greatly facilitating the study of gene expression and the discovery of the roles played by
specific genes in the development of disease. As a result of the Human Genome
Project[1], there has been an explosion in the amount of information available about the
DNA sequence of the human genome, including identification of a large number of genes
within these previously unknown sequences. The challenge currently faced by scientists
is to find a way to organize and catalog this vast amount of information into a usable
form. The full impact of the Human Genome Project will be realized only after the
functions of the new genes are discovered.
With this vast amount of information comes the need for tools to make sense of
the data. This led to the second advance which facilitated the identification and
classification of the DNA sequence information and the assignment of functions to these
new genes- the DNA microarray technology. With the invention of the DNA chip,
researchers have gone from looking at genes one at a time to tens of thousands at a
time[2]. In order to really understand a genome, scientists need to understand how genes
interact with each other and which genes are present under different conditions. This can
be done by measuring the amount of each mRNA present in the cell. Microarrays enable
us to measure this for thousands of genes simultaneously. With the aid of a computer, the
amount of mRNA bound to the spots on the microarray is precisely measured, generating
1

a profile of gene expression in the cell. Microarrays generate huge amounts of valuable
data and the handling and analysis of such data is becoming one of the major bottlenecks
in the utilization of the technology. The raw microarray data are images, which have to
be transformed into gene expression matrices—tables where rows represent genes,
columns represent various samples such as tissues or experimental conditions, and
numbers in each cell characterize the expression level of the particular gene in the
particular sample. These matrices have to be analyzed further, if any knowledge about the
underlying biological processes is to be extracted and this forms the basis for my thesis-
microarray data analysis.
The data analysis process constitutes the analysis of the gene expression matrix
using either supervised or unsupervised methods. Among the many statistical packages
available for data analysis, ‘R’ is a statistical package which is widely used for the
analysis of microarray data[3]. Several open source software are available which perform
data analysis using R functionality as their base. Most of these packages either require
some hands on programming experience and syntactical knowledge of the software in
order to perform the analysis of the microarray data or are platform dependent and not
universally available for all types of users.
The Bioinformatics Research Group (BRG) [http://kbrin.a-
bldg.louisville.edu/brg/], which is a joint collaboration between the Speed School of
Engineering and the School of Medicine at the University of Louisville, came up with the
initiative for developing user-friendly software that can be used by biologists who
generally lack programming knowledge. This thesis work is concentrated on developing a
web based java tool which allows users to upload their data files in the format of a gene
2

expression matrix and then performs normalization of the data, produces plots to
visualize the data, perform clustering of similar patterns of differentially expressed genes
and lets users to save their results to a text file.
It should be noted that a good understanding of these methods and the biology
behind the data is needed to choose the most appropriate for solving a particular problem.
The rest of chapter one is devoted to an overview of molecular biology, including a
discussion of DNA, RNA, genes and microarrays. Chapter two discusses microarray
analysis techniques, including an overview of log ratios, normalization, visualization
plots, differentially expressed genes using the fold change method, and clustering.
Chapter three is a literature review of existing microarray data analysis software, their
drawbacks and how the system being developed caters to the needs of the user who lacks
programming expertise. Chapter four gives a detailed description of the software used for
the development of the microarray analysis tool. Installation and implementation
specifics are also covered in a detailed manner. An overall discussion of the system being
developed in the form of its objectives and the results obtained is dealt with in chapter
five. Conclusions and further improvements to the microarray data analysis tool are
discussed in chapter six. A detailed glossary of terms is also available as part of this
thesis for the reader’s reference.
3

1.1 Overview of molecular biology
Every cell in an organism contains a full set of chromosomes and identical genes.
At a given point of time, only a subset of these genes is active. These genes define certain
unique properties of a cell type. The information contained in the DNA is transcribed into
messenger RNA (mRNA) molecules, which are then translated into proteins, which
perform most of the important functions of the cell. Figure 1.1 illustrates this process.
Cell Nucleus
Chromosome
Protein Gene (DNA)Gene (mRNA), single strand
Cell Nucleus
Chromosome
Protein Gene (DNA)Gene (mRNA), single strand
Figure 1.1 - An overview of the formation of proteins
1.2 DNA
Deoxyribonucleic Acid (DNA) is the basis for the building blocks encoding the
information of life. A single stranded DNA molecule, called a polynucleotide or
oligomer, is a chain of small molecules called nucleotides. There are four different
nucleotides, or bases: adenosine (A), cytosine (C), guanine (G) and thymine (T).
4
Stringing together a simple alphabet of four characters together we can get
enough information to create a complex organism. The ends of the polynucleotide are
marked either 3’ or 5’. The general convention is to label the coding strand from 5’ to 3’
(left to right). For instance, the following is a polynucleotide:
5’ G→T→A→A→A→G→T→C→C→C→G→T→T→A→G→C 3’
DNA can be either single-stranded or double stranded. When DNA is double-
stranded, the second strand is referred to as the reverse complement strand.
Complementary bases are determined by which pairs of nucleotides can form bonds
between them. In the case of DNA, A binds to T, and C binds to G. For the
polynucleotide given above, the double-stranded polynucleotide is as follows:
5’ G→T→A→A→A→G→T→C→C→C→G→T→T→A→G→C 3’
| | | | | | | | | | | | | | | |
3’ C←A←T←T←T←C←A←G←G←G←C←A←A←T←C←G 5’
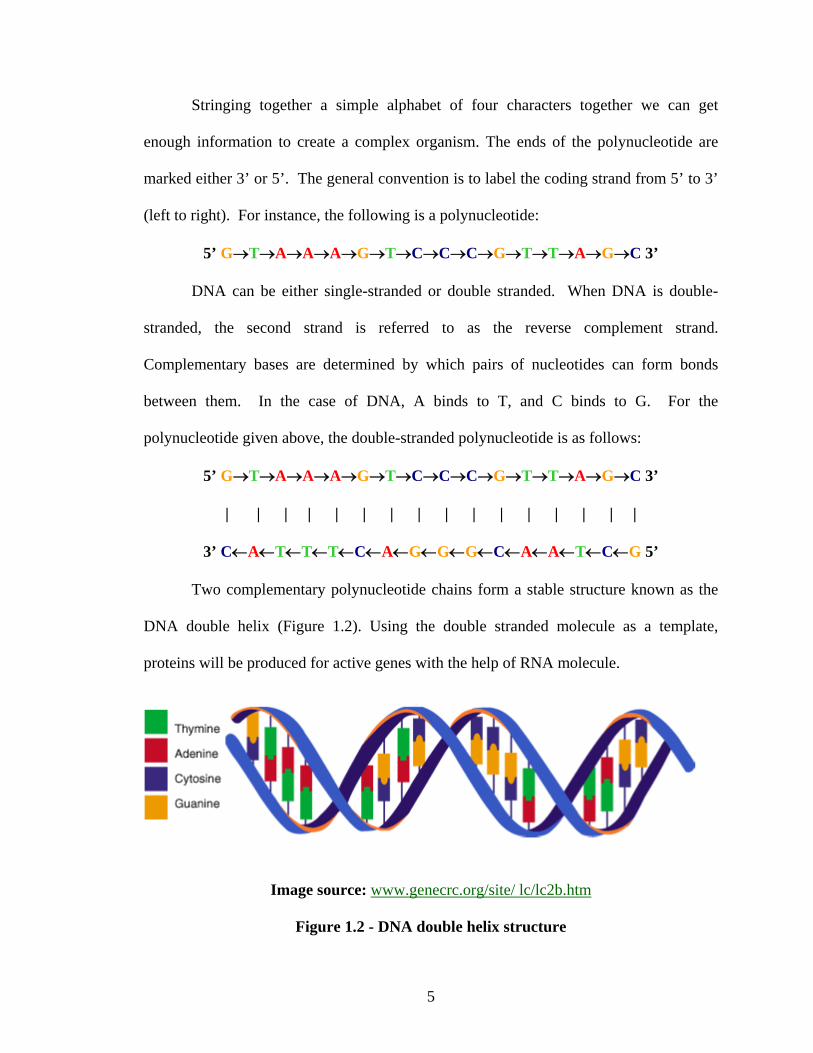
Two complementary polynucleotide chains form a stable structure known as the
DNA double helix (Figure 1.2). Using the double stranded molecule as a template,
proteins will be produced for active genes with the help of RNA molecule.
Image source: www.genecrc.org/site/ lc/lc2b.htm
Figure 1.2 - DNA double helix structure
5

1.3 RNA
Ribonucleic Acid (RNA) is similar to DNA in the fact that it is constructed from
nucleotides. However, instead of thymine (T), an alternative base uracil (U) is found in
RNA. RNA can be found as double-stranded or single-stranded, and can also be part of a
hybrid helix where one strand is an RNA strand and the other is a DNA strand. RNA is
important in the cell and contributes in a variety of ways. One of the most important
roles of RNA is in protein synthesis. Two of the major RNA molecules involved in
protein synthesis are messenger RNA (mRNA) and transfer RNA (tRNA).
1.4 mRNA
Messenger RNA (mRNA) is a linear molecule encoding genetic information
copied from DNA molecules. DNA is copied into a single stranded mRNA molecule by
the transcription process. This occurs as follows. Genes consist of coding regions called
exons and non-coding regions called introns. mRNA processing removes introns and
splices the exons together. Processed mRNA can be translated into a protein sequence.
Source: http://www.ebi.ac.uk/microarray/biology_intro.html
Figure 1.3 - Formation of mRNA
6

Therefore, in order to determine which genes are active in a cell (i.e., those that are
producing a protein product) one can measure the amount of mRNA present. This gives
an approximation of the activity of individual genes in a cell.
1.5 Gene
A gene can be described as the physical and functional unit of heredity that carries
information from one generation to the next[4]. A gene can be thought of as the DNA
sequence necessary for the synthesis of a functional protein or RNA molecule. Proteins
are important components of the body that determine how the different kinds of
molecules in the body are organized and act. Thus, proteins play a key role in the way we
look and the also in making us a unique individual. Genes are expressed as proteins, a
complex process consisting of two main steps: Each gene (DNA) is converted
(transcribed) into messenger RNA (mRNA), RNA that serves as a template for protein
synthesis. The resulting mRNA then guides the synthesis of a protein through a process
called translation. Thus isolating the mRNA helps us to find expressed genes from the
human genome.
1.6 Central Dogma of Molecular Biology
The Central Dogma of Molecular Biology states that the region of a double
stranded DNA molecule that corresponds to a gene is copied, or transcribed, to a
complementary single stranded mRNA molecule[5]. The single stranded mRNA
molecule then gets translated to a protein (Figure 1.4). If mRNA molecules can be
identified, the expression level of the corresponding genes can be determined.
7

Source: http://www.accessexcellence.org/RC/VL/GG/images/central.gif
Figure 1.4 - Central Dogma of Molecular Biology
1.7 MicroRNA (miRNA)
A miRNA is a form of single-stranded RNA which is typically 20-25 nucleotides
long, and is thought to regulate the expression of other genes[6]. miRNAs are RNA genes
which are transcribed from DNA, but are not translated into protein. The DNA sequence
that codes for a miRNA gene is longer than the miRNA. This DNA sequence includes the
miRNA sequence and an approximate reverse complement. When this DNA sequence is
transcribed into a single stranded RNA molecule, the miRNA sequence and its reverse-
complement base pair to form a double stranded hairpin loop which is a primary miRNA
structure (pri-miRNA). Pri –miRNAs are processed in the nucleus into hairpin RNAs
called Pre-miRNAs. The pre-miRNA molecule is then actively transported out of the
nucleus by a carrier protein. Thus through a mechanism that is not fully characterized, the
8

bound mRNA remains untranslated resulting in reduced expression of the corresponding
gene.
The function of miRNAs appears to be in gene regulation. miRNAs have been
reported to be critical in the development of organisms; they are differentially expressed
in tissues and are involved in viral infection processes. In the past two to three years, a
great deal of effort has gone in understanding how, when and where miRNAs are
produced and functions in cells, tissues and organisms. Several research groups have
provided evidence that miRNAs may act as key regulators of processes as diverse as
early development, cell proliferation and cell death, apoptosis and fat metabolism and cell
differentiation. There is speculation that the role of miRNAs in regulating gene
expression could be as important as that of transcription factors. The discovery of
miRNAs and their functions has added insight into how gene regulation is much more
complex than the Central Dogma of Molecular Biology previously led biologists to
believe.
1.8 Microarrays
Microarrays, developed in the lab of professor Patrick Brown at Stanford, in the
early 1990’s, took molecular biology by storm[7]. They are small slides spotted with
fixed samples of DNA, each for a different gene. When a researcher prepares a labeled
cell extract and incubates it with the slide, messengers in the sample anneal to the fixed
DNA, showing which genes in the sample are active. Microarray technology helps to
identify genes that are expressed under different conditions such as during the stages of a
cell cycle, under different environmental conditions, under diseased states at a particular
9

time, or under different tissue or cell types. A microarray is typically a glass slide, on to
which DNA, cDNA or Oligonucleotide molecules are attached at fixed locations (spots).
There may be tens of thousands of spots on an array, each containing a huge
number of identical DNA molecules of varying lengths. For gene expression studies, each
of these molecules ideally should uniquely identify a single gene in the genome.
Microarrays are used to compare gene expression levels in two different samples, for
example, a cell in a healthy state and a diseased state. A microarray employs the ability of
a given mRNA molecule to bind specifically to, or hybridize to, the complementary DNA
from which it is originated.
Source: http://www.bioteach.ubc.ca/MolecularBiology/microarray/index.htm
Figure 1.5 - Preparation of microarrays
10

A microarray contains many DNA sequences, and the expression levels of
thousands of genes can be determined in a single experiment by measuring the amount of
mRNA bound onto each spot of the array. Arranged systematically, the particular
sequences can be identified by the location of the spots on the slide.
For two channel experiments, the relative abundance of each of the gene-specific
sequences in two RNA samples (test and reference) may be estimated by fluorescently
labeling the samples, mixing them and hybridizing them to the sequences on the glass
slide. The two samples of mRNA from the cells (target) are reverse transcribed into
cDNA, and labeled using two different dyes (red Cyanine 5 and green Cyanine 3).
Usually, the reference sample is labeled Cy3 and the test sample with Cy5. The mixture
reacts with the spotted cDNA sequences (probes). This results in cDNA sequences from
the targets and the probes base-pairing with one another. After this hybridization step is
complete, the microarray is placed in a scanner, consisting of lasers with different
wavelengths, a microscope and a camera. The slide is scanned twice, first using one
colored laser and then the second. Laser light excites the fluorescent dyes, Cy3 is excited
by green laser light and Cy5 is excited by red laser light[4]. Green spots indicate that the
test substance has lower activity than the reference substance, red spots indicate that the
test substance is more abundant than the reference substance; yellow spots mean that
there is no change in the activity level between the two populations of test and reference
substance. Black represents areas where neither test nor control substance has bound to
the target DNA. The process of creating and labeling a microarray can be observed in
Figure 1.5.
11

Having an introduction to the central dogma of molecular biology, genes,
microarrays, their preparation process and uses, the next chapter introduces microarray
data analysis and the techniques used to analyze microarray data for obtaining useful
information and knowledge about the underlying biological processes.
12

2. MICROARRAY ANALYSIS TECHNIQUES
2.1 Microarray data analysis
Analysis of microarray data is performed to identify which genes are involved in
the process being studied. It involves statistical analysis by various graphical and
numerical means to select differentially expressed (DE) genes or to find groups of genes
whose expression profiles can reliably classify the different RNA sources into
meaningful groups. The analysis of gene expression data is performed by constructing the
gene expression matrix that describes spot quantitations from different hybridizations.
The process of constructing a gene expression matrix from the raw microarray data is
summarized in Figure 2.1.
Figure 2.1 - Process of obtaining a gene expression matrix
13

A gene expression matrix is a matrix, in which the first column represents the
gene names, and the subsequent columns represent the different experimental conditions
and the cell values usually represent the gene expression value for the given experiment.
Given in Table 2.1 is a gene expression matrix with sample gene expression values.
Exp 1 Exp 2 Exp 3 Exp 4 Exp 5 Exp 6
Gene 1 403 409.3 611.5 569.2 536.6 580.2
Gene 2 757.3 574.4 826.7 595.3 755.2 956
Gene 3 284.4 327.3 421.6 336.6 391.3 412.6
Gene 4 2314.2 1685.3 2264.7 2204.1 2233.1 2458.4
Gene 5 1574.5 1273 1484.6 1321.2 1474.7 1774.1
Gene 6 2333.7 1796.8 2464.5 2372.5 2095.9 2735.7
Table 2.1 - Gene expression matrix with raw gene expression data
The cell values can also be the intensity ratios of the particular experiment with a
preset experiment value. In the gene expression matrix in Table 2.2, the cell values are
the intensity ratios of all the other experiments with experiment 1, calculated from the
expression matrix of Table 2.1.
14

Exp 2/Exp 1 Exp 3/Exp 1 Exp 4/Exp 1 Exp 5/Exp 1 Exp 6/Exp 1
Gene 1 1.015633 1.51737 1.412407 1.331514 1.439702
Gene 2 0.758484 1.091641 0.786082 0.997227 1.26238
Gene 3 1.150844 1.482419 1.183544 1.375879 1.450774
Gene 4 0.728243 0.97861 0.952424 0.964955 1.062311
Gene 5 0.808511 0.942903 0.839124 0.936615 1.12677
Gene 6 0.769936 1.056048 1.016626 0.898102 1.172259
Table 2.2 - Gene expression matrix with intensity ratio values
Data analysis is based on the hypothesis that there are biologically relevant
patterns to be discovered in the data. The microarray data analysis process depends on the
analysis of the gene expression matrix using both supervised and unsupervised methods.
Most data analysis methods use raw expression values, intensity ratios or both for their
analysis routines. Data analysis methods take these huge sets of data as input and produce
both visual and numerical results for interpretation and further analysis. The most
commonly used microarray data analysis methods include log ratios of the gene intensity
data in order to spread the values across a given range, normalization to identify and
remove bias from the data, diagnostic plots of the microarray data for visualization
purposes, and methods to identify differentially expressed genes and clustering of genes
with similar behavior patterns. Each of these analysis techniques are discussed in detail in
the following sections.
15

2.2 Log ratios
A logarithmic transformation produces a continuous spectrum of values and treats
up and down regulated genes evenly across a range [8]. Equation 2.1 shows the formula
for calculating log2 ratios.
i
ii
GRX 2log= [Equation 2.1]
Where i=1, 2…..Ngenes and Ri , Gi are the measured intensity values for gene ‘i’ from two
different experimental conditions.
By using log2 values, X=0 represents equal expression, X=1 represents up
regulation by a factor 2, X=-1 down regulation by a factor 2, X=2 up regulation by factor
4, and so on. Additionally, calculating log 2 values spreads the values more evenly across
the intensity range and provides better visualization of the data and it tends to make the
variability of data more constant over the intensity range[9]. Given in Table 2.3 are the
log2 values of the intensity ratios calculated in Table 2.2.
Exp 2/Exp 1 Exp 3/Exp 1 Exp 4/Exp 1 Exp 5/Exp 1 Exp 6/Exp 1
Gene 1 0.02237883 0.60157266 0.49815582 0.413067216 0.52577046
Gene 2 -0.39880918 0.12649896 -0.34724803 -0.004006164 0.33614569
Gene 3 0.20269214 0.56795340 0.24311371 0.460353645 0.53682236
Gene 4 -0.45750812 -0.03119360 -0.07032387 -0.051465694 0.08720612
Gene 5 -0.30666134 -0.08481948 -0.25304488 -0.094472262 0.17219357
Gene 6 -0.37718928 0.07867587 0.02378897 -0.155049228 0.22929092
Table 2.3 - Gene expression matrix with log 2 intensity ratio values
16

2.3 Normalization
The goal of normalization is to identify and remove any systematic bias in the
measured fluorescence intensities, arising from variation in the microarray process rather
than from biological differences between the RNA samples or the printed probes[10;11].
Sources of bias include:
• labeling efficiencies of the dyes
• different amounts of Cy3 and Cy5 labeled mRNA
• scanning parameters
• spatial or plate effects, print tip effects, etc.
In the normalization process, a normalization factor (also referred to as scaling
factor) is calculated and is multiplied to all the values of an experiment. Either of the
experiments which are being compared can be multiplied with the normalization factor.
This process is the same as taking a constant value away from the log of the normal ratio.
2.3.1 Total intensity normalization
Total intensity normalization computes the normalization factor by summing the
measured intensities in both the experiments considered[10]. This is shown in Equation
2.2,
∑
∑
=
==array
kk
array
kk
totalN
G
NR
N
1
1 [Equation 2.2]
where Narray is the total number of genes, Gk and Rk are the measured intensity values of
the kth gene in both the experiments. The intensities are then rescaled such that Gk’ = Ntotal
17

Gk and Rk’= Rk and the normalized expression ratio for each feature are calculated
(Equation 2.3).
k
k
totalk
kk
GR
NGRT 1
''' == [Equation 2.3]
This is equivalent to
)(log)(log)'(log 222 totalkk NTT −=
2.3.2 Median normalization
In median normalization the normalization factor is found by calculating the
median of the array in question. Hence the equation becomes
akk medianTT −= )(log)'(log 22 where a is the experiment array.
The advantage of using the median normalization is that it is insensitive to outliers which
occur commonly in microarray data sets.
2.3.3 Lowess normalization
Lowess stands for Locally Weighted Linear Regression. It is also referred to as
Loess. Lowess uses a linear regression model whereas Loess uses a quadratic regression
model. The lowess normalization procedure subtracts a Lowess regression curve from the
data to normalize it[10;12].
18

Figure 2.2 - Effects of lowess normalization
A Lowess curve is first drawn on the RI Plot. The lowess curve is calculated by a
regression process which calculates the dependence of the ratio on the intensity and puts
it in a mathematical context.
The dependence, for each gene (i) is calculated by observing its distance
from the curve. On subtracting the dependence from the observed log
)( ixy
2 ratios, the
equation becomes:
))(^2(log)(log)()(log)'(log 2222 ikikk xyTxyTT −=>−= [Equation 2.4]
Figure 2.2 shows the effect of lowess regression on a set of data. The plot on the right
hand side is the RI plot itself and the plot on the left hand side is the RI plot fitted with
the lowess curve. Lowess detects the systematic deviations in the RI plot and corrects
them by carrying out a local weighted linear regression function given by Equation 2.4,
and uses this function, point by point, to correct the measured ratio values. The results of
applying such a lowess correction can be seen in the left hand side plot of Figure 2.2.
Lowess analysis is used as a normalization method that can remove intensity dependant
effects in the log2 ratio values.
2.4 Scatter plot
The scatter plot is an important graphical tool for studying the spread and linearity
of data[8]. In its simplest form, two variables are plotted along the axes, and marks are
19
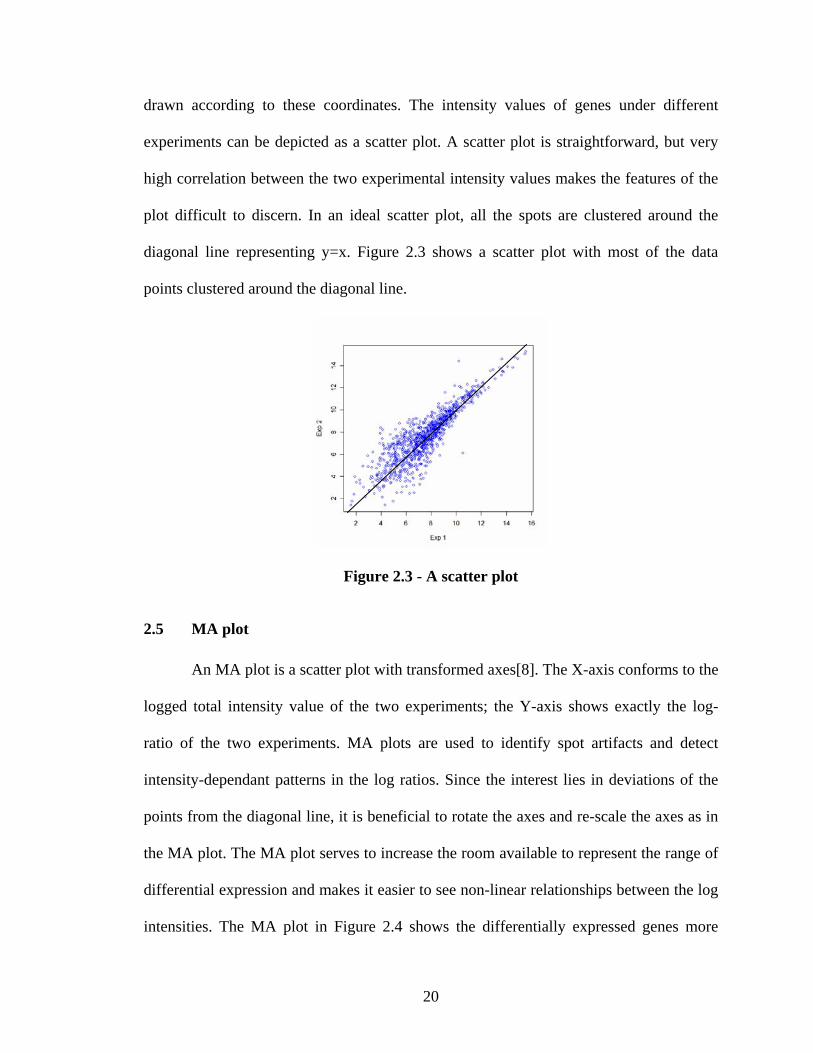
drawn according to these coordinates. The intensity values of genes under different
experiments can be depicted as a scatter plot. A scatter plot is straightforward, but very
high correlation between the two experimental intensity values makes the features of the
plot difficult to discern. In an ideal scatter plot, all the spots are clustered around the
diagonal line representing y=x. Figure 2.3 shows a scatter plot with most of the data
points clustered around the diagonal line.
Figure 2.3 - A scatter plot
2.5 MA plot
An MA plot is a scatter plot with transformed axes[8]. The X-axis conforms to the
logged total intensity value of the two experiments; the Y-axis shows exactly the log-
ratio of the two experiments. MA plots are used to identify spot artifacts and detect
intensity-dependant patterns in the log ratios. Since the interest lies in deviations of the
points from the diagonal line, it is beneficial to rotate the axes and re-scale the axes as in
the MA plot. The MA plot serves to increase the room available to represent the range of
differential expression and makes it easier to see non-linear relationships between the log
intensities. The MA plot in Figure 2.4 shows the differentially expressed genes more
20

clearly than the scatter plot in Figure 2.3. If an MA plot clearly shows the dependence of
the log ratio M on overall spot intensity A, this suggests that intensity or ‘A’ dependent
normalization method may be preferable.
Figure 2.4 - An MA plot
2log1log 22 ExpExpM −= )2log1(log21
22 ExpExpA +=
2.6 RI plot
A ratio-intensity (RI) plot is also a scatter plot like the MA plot that shows the
intensity specific effects for all the genes by plotting the log ratio as a function of the
product of the intensities[9;12]. RI plots are used to determine if there is a rough
correlation between the total intensity of a spot and its ratio. The easiest way to visualize
intensity-dependent effects, and the starting point for the lowess analysis described in
section 2.3.3, is to plot the measured log2 (Exp 1/Exp 2) for each gene as a function of the
log10 (Exp 1*Exp 2) product intensities. This ‘R-I’ plot can reveal intensity-specific
artifacts in the log2 (ratio) measurements which can be eliminated using lowess
21

normalization method. Under the assumption that most genes are not differentially
expressed, most of the points in the RI plot should fall along the horizontal line. Figure
2.5 shows an RI plot where a large number of genes which are not differentially
expressed fall along the horizontal line, and a number of differentially expressed genes
are scattered away from the horizontal line.
Figure 2.5 - An RI plot
2.7 Difference between MA and RI plots
The MA plot and RI plot are used to check if the data exhibits an intensity dependent
structure. RI plots and MA plots are used in an alternative manner by scientists.
In an MA plot, plot M=log 2 (R/G) Vs A= (1/2) log 2 (R*G)
In a RI plot, plot R= log 2 (R/G) Vs I=log 10 (R*G)
where R and G are two different experiments.
The type of plots used for analysis is a source of confusion due to the fact that the RI plot
looks very similar to an MA plot. It is important to know that MA plots are similar to RI
plots but are not the same. RI plots are most commonly used to show the effect of lowess
22

normalization. MA plots are used instead of scatter plots because they serve to increase
the room available to represent the range of differential expression and makes it easier to
see non-linear relationships between the log intensities.
2.8 Identifying differentially expressed genes
One of the main goals of microarray experiments is to identify differentially
expressed (DE) genes[11]. It will be practical to identify a limited number of genes which
are the most likely candidates. This set of DE genes can be further analyzed using
clustering techniques, etc.
2.8.1 Fold change
The fold change detection is a simple approach where a fixed fold-change-cutoff
interval is used to find genes which are differentially expressed.[10; 13]
21
sampleinvalueExpressionsampleinvalueExpressionchangeFold = [Equation 2.5]
If a gene’s experimental log-ratio exceeds the upper cutoff interval boundary, then
it is marked as significant and over expressed. If a gene’s experimental log-ratio falls
below the lower cutoff interval boundary, then it is marked as significant and under
expressed. Genes with experimental log-ratios in the range of the interval are marked for
regular behavior. Important factors to be considered in fold change method are what
cutoff should be used and should the cutoff be the same for all the genes. Though it is a
very straightforward method for classifying genes, the fold change method has the
disadvantage of not considering variability. Hence, genes with large variances are more
likely to make the cutoff just because of noise. For poorly expressed genes, small changes
23

in intensity can lead to large calculated fold changes. And it is not a statistically based
method.
2.9 Clustering
Microarray experiments deal with a large amount of data, which has to be stored
and analyzed. Therefore a general idea is to reduce the dimensionality of the data. The
basic concepts in clustering are to try to identify and group together similarly expressed
genes and then try to correlate and interpret the observations at the biological end. The
basic principles in gene clustering are:
1. Organize the data into a small number of homogeneous groups.
2. Find similar expression patterns of genes. Both low and high expression level
genes can be placed in the same cluster if their expression profiles have similar
shape.
2.10 Types of clustering
Clustering can be hierarchical or flat, as well as agglomerative or divisive[10].
Agglomerative processes start out by considering each object as a separate cluster and
proceed to group the most similar objects in an iterative fashion until all the data are
included. Divisive methods start out with the complete set of data as one large group, or
cluster, and proceed by partitioning the objects starting with those that are most
dissimilar. Based on their background principle, the different types of clustering methods
available are Hierarchical agglomerative clustering[9;10], Hierarchical divisive
clustering[9;10], k-means clustering and self organizing maps (SOM’s)[9;10;13].
24

2.10.1 Hierarchical clustering
The clustering method used for analysis in this tool is hierarchical clustering. The
hierarchical clustering algorithm uses a bottom-up approach where it iteratively joins the
two closest clusters starting from a single cluster[9;10;13]. After each step, a new
distance matrix between the newly formed clusters and the other clusters is recalculated.
For a set of N genes to be clustered, and a NN × distance matrix, the hierarchical
clustering is performed as follows:
1. Assign each gene to a cluster of its own.
2. Find the closest pair of clusters and merge them into a single cluster.
3. Compute the distances between the new cluster and each of the old clusters.
4. Steps 2 and 3 are repeated until all the genes are clustered.
The distance matrix is calculated by considering the shortest distance from any
member of one cluster to any member of the other cluster. Hierarchical clustering has
become popular for the following reasons:
• Hierarchical clustering techniques are meaningful to cluster data at the experiment
level rather than at the level of individual genes. Such experiments are most often
used to identify similarities in overall gene expression patterns in the context of
different treatment regimens.
• The analysis reveals groups of similar genes that can be studied in greater depth.
• It is possible to visualize the data in a hierarchical way using interactive computer
programs.
While intuitively appealing as a method, hierarchical clustering is not an efficient
method for very large gene expression matrices as the full distance matrix of all pair-wise
25

distances has to be calculated in advance, which for n objects takes an order of n2 steps.
Hierarchical clustering is also less suitable for noisy data.
2.10.2 Dendrogram
Hierarchical clustering can be represented as a tree called a dendrogram[9;10].
Source 1: http://www.awprofessional.com/articles/article.asp?p=357695&seqNum=4&rl=
Figure 2.6 - Construction of a two-dimensional dendrogram representing a hierarchical cluster of related genes
26

By cutting the dendrogram at a particular height will give the different clusters and the
ze of the clusters. The dissimilarity of the clusters is proportional to the length of the
ertical lines projecting from each cluster. Figure 2.6 is an example of how a dendrogram
f clusters is obtained.
Each column represents a different experiment, each row a different spot on the
icroarray. The height of each link is inversely proportional to the strength of the
orrelation. Relative correlation strengths are represented by integers in the
ccompanying chart sequence. Genes 1 and 2 are most closely coregulated, followed by
enes 4 and 5. The regulation of gene 3 is more closely linked with the regulation of
enes 4 and 5 than any remaining link or combination of links. The strength of the
orrelation between the expression levels of genes 1 and 2 and the cluster containing
enes 3, 4, and 5 is the weakest (relative score of 10). (Adapted from: Jeffrey Augen,
ioinformatics and Data Mining in Support of Drug Discover," Handbook of Anticancer
rug Development. D. Budman, A. Calvert, E. Rowinsky, editors. Lippincott Williams
2.10.3 Heat maps
A heat map is a color image with a dendrogram attached to the left side and to the
top of the image[10;14]. The rows and columns plotted in the heatmap are re-ordered
based on the restrictions imposed by the dendrogram. Each row in the heatmap represents
a gene and the columns represent the different experiments to which the gene is
subjected. The colors in the heat map simply represent the values in the gene expression
matrix. One can observe from a heat map (Figure 2.7) that genes with similar gene
expression profiles (i.e. strings of similar colors) are grouped close together.
si
v
o
m
c
a
g
g
c
g
"B
D
and Wilkins. 2003)
27

Figure 2.7 - A heat map with a dendrogram and a color key
The next chapter is a literature review of existing microarray data analysis tools,
and their advantages and disadvantages in terms of ease of use, availability and
functionality. It also includes a discussion about the motivation for the developed
microarray data analysis tool.
28

3. LITERATURE REVIEW
There are several commercial and non-commercial solutions as well as a growing
body of freely available open source software for analyzing microarray data. A review of
some popular open source microarray data analysis tools is presented here including
Bioconductor, TM4, MIDAS, BASE, WebArray and SNOMAD.
3.1 Bioconductor
Bioconductor is an open source project for computational biology[15]. The main
focus is to d on analysis.
Biocon
s at least one vignette, a document that provides a textual,
sk oriented description of the package’s functionality and can be used interactively.
lthough initial efforts focused primarily on DNA microarray data analysis, many of the
ftware tools are general and can be used broadly for the analysis of genomic and
xpression data. Bioconductor has adopted object-oriented programming as its primary
rogramming paradigm.
he main features of the Bioconductor project are:
Use of R to provide a wide range of statistical and graphical methods for the
analysis of genomic data.
eliver high-quality infrastructure and end-user tools for expressi
ductor is built completely on R[3;14] and R packages. A list of the different types
of packages available is given in Table 3.1. In addition to providing genomic data
analysis tools, Bioconductor has excellent integrated, dynamic documentation. Each
Bioconductor package contain
ta
A
so
e
p
T
29

Help integrate biological literature data from PubMed and LocusLink with the
analysis of genomic data.
Allows the development of extensible, scalable and interoperable software.
Provide high qual le research.
nalysis tool with a simple user interface, which does not require
the user upload data for analysis and
dow o the
web-ba e focus of this thesis.
ity documentation and reproducib
Provide training in computational and statistical methods for the analysis of
genomic data.
Task Packages
General programming tools Biobase, graph, tkWidgets, reposTools,
rhdf5
Annotation AnnBuilder,
Table 3.1 - Bioconductor packages
Although Bioconductor has the advantage of building on the existing toolkit of
statistical applications, it is command line based which is imposing for many users. The
tkWidgets package provides some functionality for creating GUI’s, but even that requires
additional programming.
The need for an a
annotate
Graphics Geneplotter, hexbin
Preprocessing microarray data Affy, marrayClasses, marrayInput,
marrayNorm, marrayPlots, marrayTools
Differential gene expression Genefilter, multtest, ROC
any kind of programming skills, and instead lets
nl ad the results in a point-and-click fashion, is the main factor in developing
sed application which is th
30

3.2
alysis suite of tools was developed to provide the
mic of the
mic ajor applications,
Mic a
System Multiexperiment Viewer (MeV). Since the focus of this project is
in array data analysis, the discussion is confined to MIDAS.
M alysis System
MIDAS is a java application which pr users an intuitive interface to design
an sses combining one or more ring steps. MIDAS
reads “.tav” (TIGR ArrayViewer file type, w mn, tab-delimited text
fo purposes o
a single slide) files generated by TIGR Spo ia
M les include lo ormalization. It
also includes background- ate analysis and filtering,
and the
s the data in tav format. While TM4 overcomes some of the
limitati
accessed through web, instead of
TM4 and MIDAS
The TM4 microarray an
roarray community with a comprehensive set of tools to handle all aspects
roarray process[16]. The TM4 suite of tools consist of four m
ro rray Data Manager (MADAM), TIGR_Spotfinder, Microarray Data Analysis
(MIDAS), and
micro
IDAS: Microarray Data An
ovides
alysis proce normalization and filte
hich is an eight-colu
rmat developed at TIGR for the f storing the intensity values of the spots on
tfinder or retrieved from the database v
ADAM. Normalization modu wess and total intensity n
and quality- control trimming, replic
identification of differentially expressed genes using intensity dependent Z-scores
and user defined fixed fold-change cut-offs. MIDAS provides scatter plots that illustrate
the effects of each algorithm on the data. When the normalization and filtering steps are
complete, MIDAS output
ons of a command-line driven system, it has the disadvantage of requiring users to
maintain current copies of the software locally and to update the system as it evolves.
Thus the need for an analysis tool which can accept data as a simple text file,
instead of program specific formats and which can be
31

maintaining local copies of the program on a user’s computer, has been another
motiva
BASE was developed using a web-based approach which closely integrates a data
management system with a data analysis system[16]. Since expression analysis tools are
evolving rapidly, BASE has a plug-in architecture that allows new modules to be easily
added for data transformation, analysis, or visualization. BASE incorporates a data
analysis interface that allows users to define an analysis method that passes data through
multiple routines and to create transformed datasets and subsets. This allows the original
unmodified data to be analyzed in a number of ways to create multiple analyses. BASE
allows data to be visualized in a variety of ways. Unmodified and transformed datasets
can be plotted interactively as scatter plots, displayed in histograms, or viewed as tables.
Though BASE minimizes the software update problem through its web-based approach,
it has the disadvantage that it loses a good deal of the graphical functionality that local
applications can provide.
The motivation for creating a pipeline process in the application being developed
comes from the analysis method of BASE. Also, though not yet implemented, the
integration of the data analysis module with a data management system as done in BASE
is a good future improvement.
ting fact in developing this application.
3.3 BASE: BioArray Software Environment
3.4 WebArray: an online platform for microarray data analysis
WebArray offers a convenient platform for biologists to access several cutting-
edge microarray data analysis tools[17]. WebArray runs on a LAMP system (Linux +
32

Apache + MySQL + Python) system. Background computations are mostly done by R
scripts. The currently implemented functions of WebArray were based on limma (Linear
Models for Microarray Analysis) and affy package from Bioconductor, the spacings
LOESS histogram (SPLOSH) method, PCA-assisted normalization method and genome
mapping method. WebArray incorporates these packages and provides a user-friendly
interface for accessing a wide range of key functions of limma and others, such as spot
quality weight, background correction, graphical plotting, normalization, linear modeling,
empirical bayes statistical analysis, false discovery rate (FDR) estimation, and
chromosomal mapping for genome comparison. Microarray analysis using WebArray can
be executed in three steps: 1) uploading and managing files; 2) selecting datasets and
methods for analysis, 3) browsing results. A good help document is also available with
detailed annotation of all the functions of WebArray. Thus WebArray is an excellent free
open source software for microarray analysis that can be used by an average biologist
after some training.
ardization and Normalization of MicroArray Data)
on to the regular transformations and visualization tools,
SNOMAD includes two non-linear transformations which correct bias and variance
which are non-uniformly distributed across the range of microarray element signal
intensities: 1) local mean normalization; and 2) local variance correction (Z-score
generation using locally calculated standard deviation).
3.5 SNOMAD (Stand
SNOMAD is an interactive, user-friendly web-application which can be accessed
freely via the internet with any standard HTML browser[18]. SNOMAD is a collection of
algorithms for the normalization and standardization of gene expression datasets derived
from diverse sources. In additi
33

The SNOMAD tool is available at -
http://pevsnerlab.kennedykrieger.org/snomad.htm. No programming expertise or
software installation is required. Users can upload their gene expression data and specify
the transformations they wish to apply on their data. Results come in the form of both a
text file containing numeric values and image files of graphs of the data corresponding to
all the transformations.
WebArray and SNOMAD are two user-friendly tools available in the market for
microarray data analysis. But they have their own disadvantage of having limited
functionality, confined to a certain set of routines that the user can perform on the data.
They do not have the scope for adding new R programs to the already existing system. In
such a case, biologists tend to use multiple tools for obtaining the required results. This
lack of extensibility formed another motivation for the development of the application
under discussion. Thus all these above discussed factors led to the development of the
current application to provide a solution to the community driven need for an easy to use,
readily available and extensible microarray data analysis tool, which uses R routines for
analysis.
34

4. IMPLEMENTATION SPECIFICS
In this chapter, the software implementation specifics for the microarray analysis
tool are discussed. A brief introduction to the R package, which forms the base for the
statistic
4.1 R
R is a powerful software environment for data manipulation, calculation and
graphical display. It is a GNU General Public License project similar to the S language.
The name is partly based on the first names of the first two authors (Robert Gentleman
and Ross Ihaka), and partly a play on the name of the Bell Labs language ‘S’[3;14].
supports a wide range of statistical techniques including descriptive statistics,
linear and nonlinear modeling, classical statistical tests, probability distributions, analysis
of variance (ANOVA), time series analysis, classification, clustering, robust regression
and maximum likelihood.
al analysis of the microarray data is given. Description of Java which has been
used to develop the user interface, and information about Rserve, which is the plug-in
used to connect to R from Java are also given. Some background about MySQL database
and the JDBC connection needed to connect to a database from Java code is also
provided. A clear understanding of the software is needed to understand the
implementation techniques discussed in this thesis.
R
35

R is extensible via user defined functions written in its own language, or through
the use of dynamically loaded modules written in other languages. It can be used with
Linux, UNIX and Microsoft Windows™.
4.1.1 Statistics and R
Most of the statistical techniques have been built into the base R environment and
many more are supplied in the form of packages. There are about 10 packages called
standard packages which are supplied with R and many more can be downloaded from
the Comprehensive R Archive Network (CRAN) website (http://cran.r-project.org).
The major difference between R and other statistical systems is that in R, the
statistical analysis is performed as a sequence of steps with the results of every step
stored in objects. In systems like SAS and SPSS copious output is obtained from a
regression or analysis whereas R will give minimal output and store the results in a fit
ct f further processing by R functions.
4.1.2 R and Windows™
The latest version of R for Windows™ can be downloaded from the CRAN
website. The version used for development of this project is R 2.1.1. A full installation of
R on Windows™ takes up to 50 MB of disk space. To install, double click on the icon for
rw2011.exe and follow the instructions. R installed in this way can be started from the
start menu or by double clicking the R shortcut. To add packages to the existing R
system, download the packages from the CRAN website and unzip them into the
R/rw2011/library folder directly.
obje or
36

Figure 4.1 - R command line interface on startup
erted into native data types.
• Persistent connections until the connection are closed.
4.2 Rserve
Rserve [19]is a TCP/IP server which allows other programs to use R facilities
from various languages without the need to initialize R or link against R library. Rserve
supports remote connection, authentication and file transfer. Typical use of Rserve is to
integrate R backend for computing statistical models, plots, etc from other applications.
The features of Rserve include:
• R initialization is not necessary.
• Most R data types are conv
37

• Offers client independence since the client is not linked to R.
• Rserve provides some basic security in the form of encrypted user/password
authentication.
• Rserve allows transferring files between the client and the server.
Rserve itself is the server which responds to requests from the clients. It listens
for incoming connections and processes incoming requests. A client framework was also
developed – JRclient. JRclient is a client suite which allows a java application to access
Rserve. It was developed in java. It provides automatic type translation for most objects
such as int, double, arrays, string or vector and classes for special R objects such as
RList, RBool, etc. The idea behind the separation of client/server side allows handling
multi-threading better when linking to R library directly.
4.2.1 Installation of Rserve
R 1.5.0 or to be able to use
AIX and Windows™. The Windows™ version
of Rserve was used for development. Although Rserve works on Windows™, it is not the
recommended since Windows™ lacks important features that make the separation of
namespaces possible. Therefore Rserve for Windows™ allows only one connection at a
time and all subsequent connections share the same namespace.
Installation process for Windows™:
1. Make sure to download the proper binary based on the version of R.
2. Copy the binary Rserve.exe to the same directory where R.dll is located. By
default it is in the R\rw2011\bin folder.
3. Run rserve.exe to start the server and to make connections to R.
higher needs to be installed on your system in order
Rserve. Rserve works on Linux, Solaris,
38

Rserve was developed by Simon Urbanek, a researcher at AT&T Research labs.
Any e
(General Public License).
4.3
This microarray data analysis tool was mostly developed using Java in order to
provide a platform independent solution. The IDE (Integrated Development
Environment) used for code development is Eclipse SDK 3.1.1. Other IDE’s that can be
used are Borland’s JBuilder or Netbeans.
ple object-oriented, distributed,
interpreted, robust, secure, architecture neutral, portable, high-performance,
multithreaded, and dynamic language[20]. A program written in java is both compiled
and interpreted. A java compiler generates an architecture independent object file
executable on any system supporting the java runtime environment. The object code
consists of bytecode instructions designed to be both easy to interpret on any machine
and easily translated into native machine code at load time. So compilation takes place
only once, interpretation occurs each time the program is executed.
rogram runs.
Som
operati e. The java platform differs from these
on interested to contribute to the project can do so since it is released under GPL
Java
4.3.1 Java language
Java as described by Sun Microsystems is, a sim
4.3.2 Java platform
A platform is the hardware or software environment in which a p
e popular platforms like Windows™, Linux, Mac OS, etc. are a combination of the
ng system and the underlying hardwar
39

platform
parts:
1. The Java Virtual Machine (JVM)
2. The Java Application Programming Interface (API)
The JVM is the interpreter and the runtime system, which lets java programs run
on any hardware-based platform where it has been already ported to. The API is a large
collection of ready-made software components that provide several capabilities. It is a
grouped up collection of libraries of related classes and interfaces. These libraries are also
4.4
Servlets[21] are java programs that run on a web server and build web pages.
Servlets provide a component-based, platform-independent method for building web-
based applications. Servlets are server- and platform- independent which leaves us free to
select any server, platform and tools for running our application.
s based on the fact that it is a software-only platform that runs on top of other
hardware-based platforms.
The java platform has two
known as packages[20].
Java servlets
40

Source: http://cs.nmu.edu/~jeffhorn/Classes/CS122/Figures/javaTranslation.gif
Figure 4.2 - Java servlet execution process
form that specifies
method=POST. To be a servlet, a class should extend HttpServlet and contain the doGet
and the doPost methods to handle the GET and POST requests respectively. Both these
methods take two arguments: an HttpServletRequest and an HttpServletResponse. The
HttpServletRequest has methods to handle all incoming information such as form data,
HTTP request headers, and the client’s hostname. The HttpServletResponse lets you
specify outgoing information such as HTTP status codes, content-type, cookies and most
importantly lets you post document content back to the client. The two important
packages that have to be imported into the servlet file are:
Servlets are class files which handle GET and POST requests. GET requests are
the usual type of browser requests for web pages in HTTP, when a user types a URL on
the address line or follows a link from a web page. Servlets also handle POST requests,
which are generated when someone submits an HTML
41

1. javax.servlet (for HttpServlet) and
2. javax.servlet.http (for HttpServletRequest and HttpServletResponse).
4.5 JSP (Java Server Pages)
Java Server Pages[20;21] is a technology that lets you mix regular, static HTML with
dynamically-generated HTML. You simply write the regular HTML in the normal
manner, using whatever web-page building tools you normally use. You can then enclose
the code for the dynamic parts in special tags which start with “<%” and end with “%>”.
A JSP is saved with a .jsp extension and it can be invoked just like any other normal web
page. Though it appears to be a normal HTML file, a JSP acts like a servlet behind the
scene
.6 JDBC (Java D
s.
4 atabase Connectivity)
JDBC[20;21] defines how a java program can communicate with a database.
JDBC API provides two packages – java.sql and javax.sql. By using JDBC API, one can
connect to any database, send queries to the database and process the results.
JDBC architecture defines the different layers to work with any database and Java.
1. JDBC API interfaces and classes which are at top most layer (to work with java)
2. A driver which is at the middle layer (maps java to database specific language)
3. A database at the bottom (to store physical data)
42

Source: http://www.dbmsmag.com/9610i06.html#figure1
Figure 4.3 - The three-tier architecture of a JDBC connection The three main interfaces provided by the JDBC API to work with databases are:
connection functionality.
2.
hich comes
4.7
conjunction with web technology server applications[22].
The database has been designed for speed, which would be useful in large transactions.
MySQL is currently the most widely installed database, a well respected product that is
more than capable of commercial operation. In fact, the entire Google search engine is
built upon MySQL technology[23]. MySQL offers most of the functionality one will
1. Connection interface provides database
Statement interface provides SQL query representation and execution
functionality.
3. ResultSet interface provides functionality for retrieving the data w
from the execution of a SQL query using Statement.
MySQL
MySQL is a very popular open source database server which is commonly used in
to create dynamic and powerful
43

expect from an RDBMS. It ensures that transactions comply with the ACID model
(Atomicity, Consistency, Isolation, and Durability), allows the building of indexes,
supports standard data types and allows for database replication, among other features.
One area where MySQL falls short is its lack of certain features like sub-queries,
constraints, views, cursors and objects. MySQL is fast, easy to use, is open source and if
the application is a web application then MySQL meshes in perfectly with most of the
web development languages. When using MySQL with java, the MySQL Connector/J
driver needs to be downloaded from MySQL’s website
[http://www.mysq
.8 Apache Tomcat
developm
application container that was created to run Servlets and Java Server Pages (JSP) in web
applications. Java m
eb pages,
servlets and JSP into a single directory structure. It can be thought of as a container
h a deployment directory where you can place all your web application files
for them
l.com/products/connector/j/].
4
Apache Tomcat (also codenamed Catalina) is a standalone Web server used as a
ent server on your desktop. The Tomcat server [21] is a java based web
ust be installed for Tomcat to operate.
Tomcat organizes all the parts of a web application such as static w
whic cts as the
to execute without any hassles.
The root folder is the deployment folder where all the static html files and JSPs
can be placed. The Servlets are placed in the ROOT/WEB-INF/Classes folder. Pros of
Tomcat are that it is an open source project, stays on top of the Servlet API
developments, and works extremely well. The cons are that it is not the fastest
implementation and that you are on your own for support.
44

In the next chapter, a detailed discussion on the developed microarray data
analysis tool is presented. The discussion is in the form of objectives of the analysis tool
and the results that have been achieved along with screen shots of the system for easier
understanding.
45

5. OBJECTIVES AND RESULTS
The aim of this thesis was to develop a freely available, platform independent
plication for visualization, normalization and analysis of microarray experiments and
so a tool which will guide the users through the steps of normalization and data analysis
such as identifying differentially expressed genes and to cluster those differentially
expressed genes into clusters of genes exhibiting similar behavior.
R, the statistical package which is freely available can be used to perform all
types of analysis on microarray data, but it has the disadvantage of being a command line
based package which requires the biologists to know the syntax of various commands and
also requires the users to be familiar with programming techniques and concepts. The
users have to in short, be well versed in R to perform efficient data analysis. Thus the
motivation for the tool comes from the need for a easy to use, point and click kind of
interface which is easily accessible over the internet, to which users can easily connect to,
upload their data files retailored to a particular format and get both numeric and visual
results for interpreting the data without having to worry about the intricacies of
programming.
I will now discuss about the different objectives of the tool and the way they were
implemented and also discuss the results of normalizing and analyzing the microarray
data using the software tool, which was developed during the course of this work.
ap
al
46

5.1 Uploading experiment data
The user can upload experimental data as a text file, which is more importantly in
the format of a gene expression m
the genes and the expression values of the genes for different experiments (which may be
different biological conditions, different time-points, etc).
atrix. The data text files should contain a listing of all
Figure 5.1 - Sample data file for analysis
As shown in figure 5.1, the gene names form the first column, the other columns
are the different experiments and the numerical values represent the gene expression data
for each gene under the different experiments. The data file can be uploaded through a
user friendly web page as shown below and it will be saved in the working directory of
the application on the server and will be used for all further analysis (figure 5.2).
47

Figure 5.2 - Process of uploading data files
5.2 Normalization methods
A wide choice of common normalization methods is offered to the user to remove
the systematic bias within the data. It is also possible to add newly developed
normalization techniques at a later stage.
All the previou e applied to the data
from the uploaded file. The normalization can be applied to more than one experimental
column or to all the experimental columns. In the case of multiple experimental columns,
all the columns are normalized with respect to the first experimental column using the
normalization method selected. The normalized data can be downloaded as a text file for
reference or for input to another system.
sly discussed normalization methods can b
48

Figure 5.3 - rmalization Normalized data file using total intensity no
Figure 5.4 - Normalized data file using median normalization
49

50
Figure 5.3 shows an example of total intensity normalization, which can be compared to
figure 5.4 (median normalization). The slight variation in the normalized data from the
different methods can be observed.
5.3 Data visualization
To get an idea about the condition of the data sets or the effects of different
normalization methods, different means of graphical display such as scatter plots, MA
plots and RI plots have been implemented.
5.3.1 Scatter plot
Plotting the log 2 intensity values of the one experiment condition versus the log 2
intensity values of another experiment condition is a common way to display the
distribution of the data. The normalization step gives two forms of output. One is the data
file with the normalized data and the second form is a scatter plot of the normalized data.
Figure 5.5 - Total intensity normalization scatter plots and text files

The graphical result of the normalization step is a nn× matrix of scatter plots
where n is the number of experimental columns to be normalized (figure 5.5 and 5.6).
The nn× matrix of scatter plots consists of scatte ent versus itself
and all the other experiments selected. Thus it can be observe om the results that the
scatter plot of an experiment versus itself is a straight line passing through the origin. The
matrix of the scatter plots has an image area map defined on it which lets users to
zoom in on a particular scatter plot for better viewing of the plots.
r plots of each experim
d fr
nn×
51
Figure 5.6 - matrix of scatter plots with a zoomed out portion for two specific
xperiments
MA plot
An alternative to the scatter plot is the MA plot with transformed axes to provide
intensity information. This tool produces MA plots for both normalized and raw data.
The MA plots are also produced as a
nn×
e
5.3.2
nn× matrix of individual MA plots where n is the
51
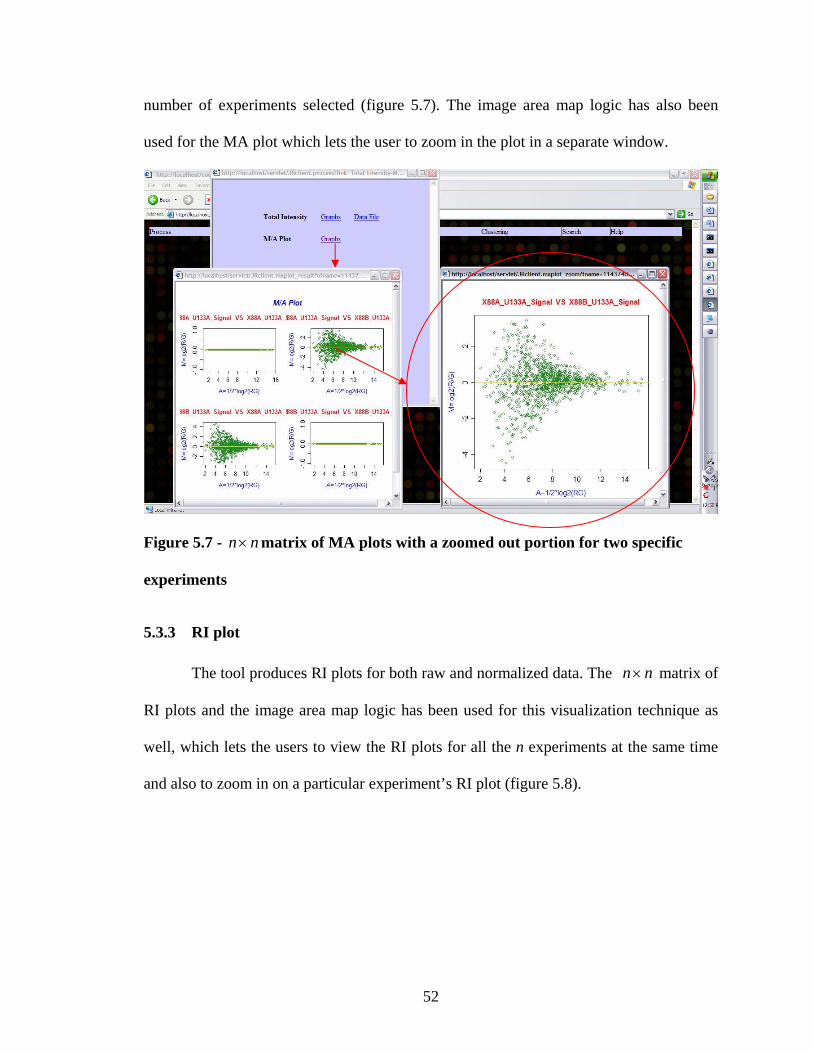
number of experiments selected (figure 5.7). The image area map logic has also been
used for the MA plot which lets the user to zoom in the plot in a separate window.
Figure 5.7 - matrix of MA plots with a z
experiments
5.3.3 RI plot
The tool produces RI plots for both raw and normalized data. The matrix of
RI plots and the image area map logic has been used for this visualization technique as
well, which lets the users to view the RI plots for all the n experiments at the same time
and also to zoom in on a particular experiment’s RI plot (figure 5.8).
nn× oomed out portion for two specific
nn×
52
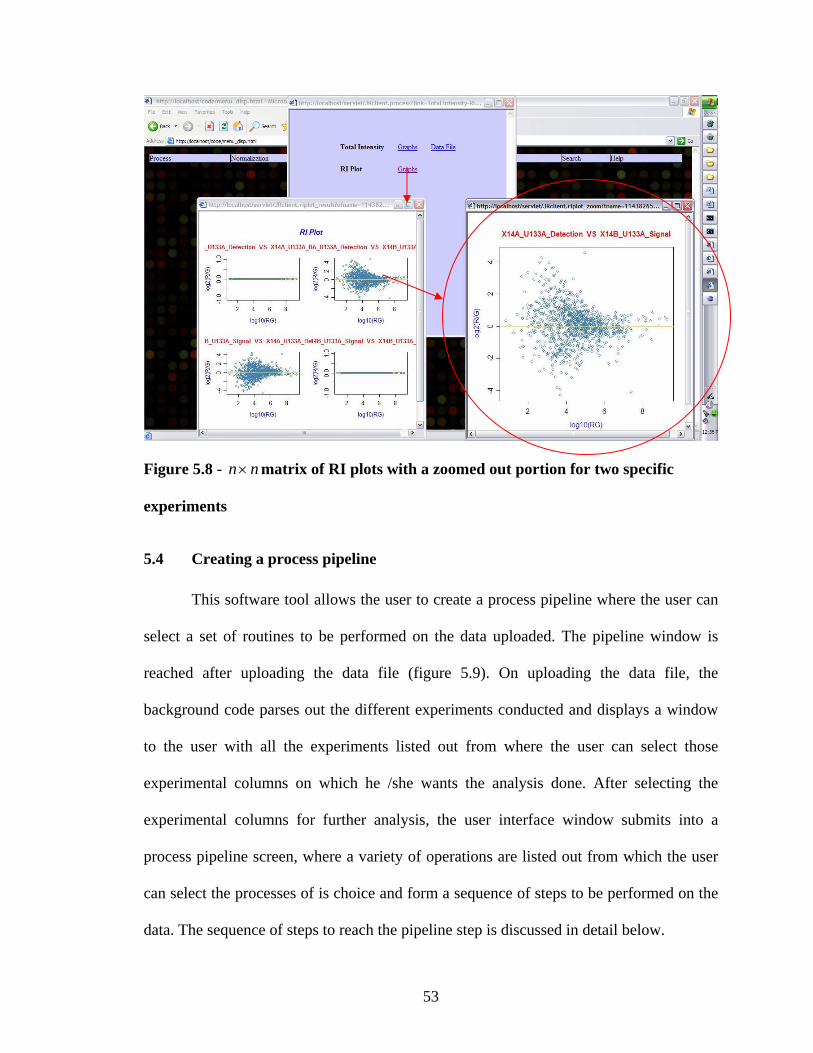
Figure 5.8 - matrix of RI plots wi
experiments
5.4 Creating a process pipeline
This software tool allows the user to create a process pipeline where the user can
a outines to be performed on the data uploaded. The pipeline window is
reached data
After selecting the
experimental columns for further analysis, the user interface window submits into a
process pipeline screen, where a variety of operations are listed out from which the user
can select the processes of is choice and form a sequence of steps to be performed on the
data. The sequence of steps to reach the pipeline step is discussed in detail below.
nn× th a zoomed out portion for two specific
select set of r
after uploading the data file (figure 5.9). On uploading the file, the
background code parses out the different experiments conducted and displays a window
to the user with all the experiments listed out from where the user can select those
experimental columns on which he /she wants the analysis done.
53

Figure 5.9 - Steps for pipelining analysis
The columns parsed out from the uploaded file, which represent the different experimental columns.
The columns transferred
double right arrow button, will be subjected to
here by clicking on the
further analysis routines.On clicking the Submit button, the window which lets the user select a pipeline of processes to be performed on the data uploaded, will open up.
Form
54

igu
rocesses listed as list boxes, which the user can perform on the uploaded data. The first
ategory is the normalization routines from where the user can select a particular
ormalization method and apply it to the data. The second category is the various
isualization plots available to view the distribution of data. The third list box forms the
ird category of processes called “Ratios and Clustering” which consists of finding the
p and bottom intensity ratios between experiments, a color coded representation of the
differentially expressed y the clustering of the
ost differentially expressed genes into groups with the same pattern of behavior. The
igure 5.10 - Pipeline screen to define a sequence of routines to be performed on the
data
In the pipeline window as shown in the figure 5.10, there are three categories of
rocesses listed as list boxes, which the user can perform on the uploaded data. The first
ategory is the normalization routines from where the user can select a particular
ormalization method and apply it to the data. The second category is the various
isualization plots available to view the distribution of data. The third list box forms the
ird category of processes called “Ratios and Clustering” which consists of finding the
p and bottom intensity ratios between experiments, a color coded representation of the
differentially expressed y the clustering of the
ost differentially expressed genes into groups with the same pattern of behavior. The
FF re 5.10 - Pipeline screen to define a sequence of routines to be performed on the
data
In the pipeline window as shown in the figure 5.10, there are three categories of
pp
cc
nn
vv
thth
toto
genes by the fold change method and finall genes by the fold change method and finall
mm
Thca
The list box with the sequence of processes to be performed on the data.
Buttons to change the order of the processes or to delete a selected process.
e three tegories of
routines available r analyzing the
data fo
Submit to get results.
Button for transferring routines from LHS to RHS.
55

buttons with the right arrows by the side of each list box of methods is used to select that
ethod into the pipeline list box on the right hand side. The “Up”, “Down” and “Delete”
uttons on the right hand side of the pipeline window are used to change the sequence of
e processes lined up in the “Selected Order of Execution” list box. In order to move a
articular process up to the beginning of the pipeline, the user has to select the process
nd click on the “Up” button. Similarly, for moving a particular process to a later stage of
e pipeline, the user has to select the process and click on the “Down” button. The
Delete” button is used for deleting a particular process from the pipeline. On clicking
the “Submit” button the pipeline is taken into the system and the sequence of steps are
performed on the data. On completion of the pipeline, the results screen is displayed as a
parate window as shown in figure 5.11.
m
b
th
p
a
th
“
se
Figure 5.11 - Results screen after submitting the pipeline screen
Click on this link to
data file. get the normalized
Click on the links to get visual results.
56

5.5 Identifying genes of interest
One of the most important goals of microarray technology is the search for new
target genes. Methods have been provided to detect differentially expressed (DE) genes.
The techniques implemented in these methods are fold change detection and statistical t-
test.
A certain threshold value is set with which the log
5.5.1 Fold change cut-off
ous colors are assigned to represent the genes
log intensity ratio values.
2 values of the intensity ratios
of a gene between different experiments, are compared. If a gene’s log 2 intensity ratio
value exceeds the threshold value it is marked as differentially expressed (DE) in that
given experiment. In this software tool, vari
based on the gene’s 2
Cutoff Range Color coding in RGB(red, green, blue) Color image< -2.0 (153, 0, 0)
5.12 −<−≥ and (255, 0, 0) 0.15.1 −<−≥ and (255, 51, 0) 7.00.1 −<−≥ and (255, 103, 0) 5.07.0 −<−≥ and (255, 204, 51) 3.05.0 −<−≥ and (255, 204, 102) 1.03.0 −<−≥ and (255, 255, 204)
0.01.0 <−≥ and (255, 255, 230) 1.00.0 <≥ and (235, 255, 230) 3.01.0 <≥ and (204, 255, 153) 5.03.0 <≥ and (153, 255, 0) 7.05.0 <≥ and (10 , 255, 0) 3 0.17.0 <≥ and (51, 255, 0) 5.10.1 <≥ and (51, 204, 0) 0.25.1 <≥ and (0, 153, 0)
0.2≥ (0, 51, 0)
Table 5.1 - Color coding scheme for differentially expressed genes using fold change
57

The ignment of a color was done foass r easier interpretation of data. The different ranges
of the
The process of assigning colors to the different genes for a given experiment is as
follows. The intensity ratios of all the genes between the two experiments the user has
log 2 values of the intensity ratios are then calculated. The
log 2 v
provided as a tool tip (figure 5.12). On
clicking a r b tensity
ratios of the gene in other experiments and f the gene’s expressio value in all
the experiments can be vie ed (figure 5.1
log 2 intensity ratio values and the corresponding color coding assigned is given
below.
selected are calculated. The
alues are then compared with the ranges in Table 5.1. to determine a range and
then a corresponding color is assigned to the gene, thus categorizing it as either regularly
expressed, under expressed or over expressed. All the genes are plotted as matrix of
color images, where when a user does a mouse over on a particular color image, the gene
corresponding to it and the intensity value is
particular colo ox, a window opens up where the gene name, the in
a line plot o n
w 3).
58

Figure 5.12 - Color based image map of the gene’s intensity ratios between two
experiments
Figure 5.13 – Individual gene details
59

60
5.6 Clustering genes
Clustering of genes allows biologists to identify genes which exhibit similar
behavior patterns over a set of experiments. Modules which perform clustering of genes
using hierarchical clustering technique have been implemented. The outputs obtained
from the clustering module are a few groups which contain genes which behave similarly.
This software tool performs clustering in the following way. The background
code calculates the intensity ratios of the gene expression values for all the experimental
columns selected by the user. The intensity ratios are calculated with respect to the very
first experimental column. Then the background code checks for those genes whose log 2
intensity ratios across all the experiments is greater than the upper limit of 2.0. It selects
all m
into groups of gene with the same pat r. The cluster of genes are displayed
sing a heat map which uses a dendrogram to show the gene clusters and also a color key
e a color-based illustration of the gene in
In the case of huge data file
ough the heat map can accommodate all the genes and their corresponding
lusters, it appears very clumsy and illegible. In order to solve this problem, the top fifty
ected and then clustered into similar pattern
er is not looking at a large number of genes clustered together,
ut can expect a more refined clustering of the most differently behaving genes.
mploying such a clustering method, the tool tends to overlook the other gene clusters
which might be carrying be understood that the
clustering information of all the genes is present and the tool is applying the clustering
those genes and then applies hierarchical clustering technique in order to cluster the
tern of behavio
u
to provid tensity ratio values.
s, the number of genes selected for clustering may be
large and th
c
most differentially expressed genes can be sel
groups. This way, the us
b
By e
some significant information. It should

routi to all the genes anne d not just the top fifty genes. But the number of genes used for
displaying the clustered information are the ones which are most differentially expressed
in order to make the heat map (Figure 5.14) easier to view and understand.
Figure 5.14 - Heat map with a dendrogram to represent expression clusters
5.7
2
Top and bottom intensity ratios
The tool has two methods implemented for finding the top and bottom 10, 25, 50,
75 and 100 genes based on their log intensity ratios between different experiments. The
user can select the experiments for which the ratio has to be calculated for and also select
the number of top/bottom ratios needed which can be either of 10, 25, 50, 75 and 100.
Both these methods return a ordered list of genes and their intensity ratio values. The
purpose of these two methods is to provide a quick way to determine those genes which
are the most over/under expressed in a set of selected experiments.
61

or calculating the intensity ratios and the F ure 5.15 - User selected columns fig
number of top/bottom genes needed
The result screens obtained for the selections done in figure 5.15 are shown in figure
5.16. The results are displayed in the form of a table with the gene name and its
log 2 intensity ratio value.
62

Figure 5.16 - Results showing top 10 ratios and bottom 25 ratios between two
experiments
5.8
A module has bee m thousands of rows of
different
he search also returns the results of a t-test on the gene
expression profile which consists of the confidence interval for a regular gene expression
value and also the up and down regulated gene expression values based on the confidence
interval.
Search genes
n implemented for searching genes fro
gene expression information (figure 5.17). The search is done by the gene name and
returns as output, a line plot showing the behavior of the gene expression for the
experimental conditions. T
63

Figure 5.17 - Search screen with the list of genes from the uploaded data file
n intelligent one which does not require the user to scroll through
thousan
The search is a
ds of rows of gene names. Keying in the first few letters of the gene name will
highlight the gene in the displayed list. On submitting the above servlet by clicking on the
“View Graph” button, the user can view in a new window, a line plot of the gene
expression values for all the experiments in the data file, and a color coded representation
of the results of a t-test on the gene’s expression profile. The resulting screen is shown in
figure 5.18.
64
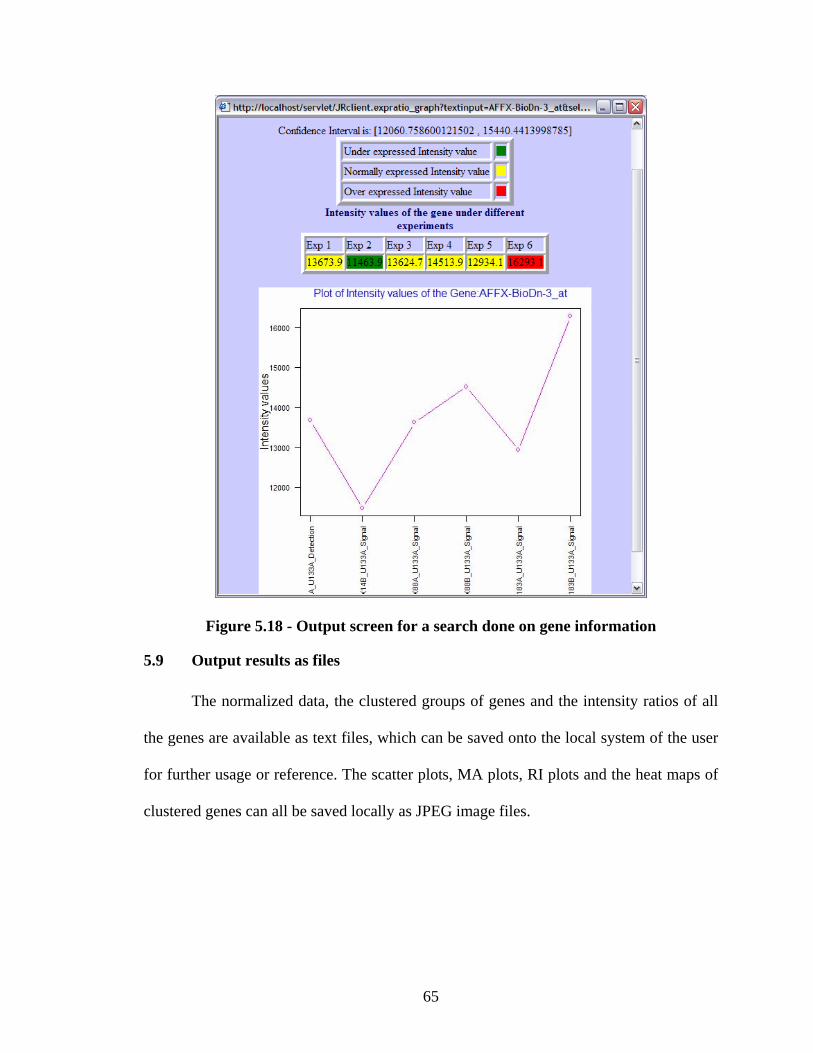
Figure 5.18 - Output screen for a search done on gene information
5.9 Output results as files
The normalized data, the clustered groups of genes and the intensity ratios of all
the genes are available as text files, which can be saved onto the local system of the user
for further usage or reference. The scatter plots, MA plots, RI plots and the heat maps of
clustered genes can all be saved locally as JPEG image files.
65

The above discussion of the results and features of the microarray data analysis
tool, show that it is a tool which will be intuitive for all the users who have a basic
understanding of normalization and data analysis. It will be a very handy tool for all
kinds of users- biologists and software developers, mainly. It’s simple user interface and
easily accessible results, supported by a help manual will help the analysis of microarray
data a simpler and easier task.
In the next chapter, some of the potential improvements that can be added to the
tool are discussed as well as the conclusions reached about optimal usage of the tool.
66

6. CONCLUSIONS
In this thesis, a platform-independent and versatile software tool for normalizing
and analyzing microarray data was developed. The software meets the requirements that
were o
ion.
The web-based front end is accessible from any web browser and handles all user
interaction. The program with its current functionality can be used by biologists for data
analysis, but there are certain improvements still in progress.
he presented program handles a wide range of functions as listed below:
• Since normalization of data is an important concern, the software tool provides
different means of normalizing the data. The possibilities are total intensity
normalization, median normalization and lowess normalization.
• The effects of normalization can be observed by useful graphical plots like scatter
plots, MA plots and RI plots. All plots can be created before and after
normalization.
• The system provides the capability of creating a pipeline of processes to be
performed on the microarray data. The user can also subject the data to specific
individual analysis techniques incase he/she does not want to create a pipeline.
riginally set forth for a tool of this nature. The interface is user-friendly. The
system is available for access to multiple users simultaneously upon user authenticat
T
67

• To detect differentially expressed genes, this tool provides simple fold-change
detection and also statistical tests like t-test. The detected target genes can be
printed to a file, for further analysis.
• Clustering methods ar sis and are also useful for reducing
6.1 Possible improvements
As with most of the software tools, this tool has a lot of scope for improvement.
Since currently new methods for normalization and analysis are developed, it may be
useful to adapt the present system to theses needs.
Possible improvements would be:
• Further normalization methods
• Additional diagnostic plots (QQ-plot, volcano plots)
•
• ent
•
tool. As far as issues are concerned, most of them have been solved and some are still in
progress.
e widely used for analy
the amount of microarray data to a subset of genes, usually to those which are
most variable between different experimental samples. This has been achieved by
using hierarchical clustering method.
• The system is expandable for further development.
ANOVA
User managem
• More sophisticated methods for detection of differentially expressed genes
Compatibility with multiple types of input data files
Many suggestions for improvement have been provided by potential users of the
68

In conclusion, this microarray data analysis tool developed using Java and R is a
nity driven solution developed to help make the analysis of microarray data
and efficient. Additional functiona
commu
simpler lity will be added on with the continuing
dev p
elo ment by other members of the Bioinformatics Research Group (BRG).
69

REFERENCES
[1] International Human Genome Sequencing Consortium, "Finishing the uchromatic sequence of the human genome," Nature, vol. 431, pp. 931-945,
[2] Tom A.van de Goor, "A History of DNA Microarrays," Advanstar ommunications Inc., 2005.
[3] eter Dalgaard, Introductory Statistics with R Springer, 2002.
[4] ff Augen, "Bioinformatics and Transcription," in Bioinformatics in the Post-Genomic Era: Genome, Transcriptome, Proteome, and Information-Based
edicine Addison Wesley Professional, 2004.
[5] .Crick, "Central dogma of molecular biology," Nature, vol. 227, no. 5258
[6] . Ruvkun, "Molecular biology.Glimpses of a tiny RNA world," Science, vol. 94, no. 5543, pp. 797-799, Aug.2001.
[7] an Cray, "Gene Detective," 2001.
[8] i Pasanen, Janna Saarela, Ilana Saarikko, Teemu Toivanen, Martti Tolvanen, auno Vihinen, and Garry Wong, DNA Microarray Data Analysis Picaset Oy,
[9] Dov Stekel, Microarray Bioinformatics Cambridge University Press, 2003.
[10] Helen C.Causton, John Quackenbush, and Alvis Brazma, A beginner's guide. icroarray Gene expression data analysis Blackwell Publishing, 2003.
[11] ordon K.Amyth, Yee Hwa Yang, and Terry Speed, "Statistical issues in cDNA Microarray Data Analysis," Functional Genomics:Methods and Protocols, vol.
24, no. Methods in Molecular Biology, pp. 111-136, 2003.
[12] hn Quackenbush, "Microarray data normalization and transformation," nature genetics supplement, vol. 32, no. December 2002, pp. 496-501, 2002.
[13] Knudsen, Guide to Analysis of DNA Microarray Data, Second ed John iley & Sons, 2004.
eOct.2004.
C
P
Je
M
FAug.1970.
G2
D
TomM2003.
M
G
2
Jo
Steen W
70

[14] W.N.Venables, D.M.Smith, and R Development Core Team, An Introduction to R Network Theory Ltd, 2004.
[15] Robert C.Gentleman, Wolfgang Huber, Vincent Carey, Rafael Irizarry, and Sandrine Dudoit, Bioinformatics and Computational Biology Solutions Using R and Bioconductor (Statistics for Biology and Health Series) Springer, 2005.
[ Source rch
[ an online 306
vsner, "SNOMAD .
[19] ide R Functionality to
[20] gan, Java in a Nutshell O'Reilly & Associates, Inc., 1997.
pache Sams Publishing, 2003.
[
16] Sandrine Dudoit, Robert C.Gentleman, and John Quackenbush, "Open Software for the Analysis of Microarray Data," Biotechniques, vol. 34, no. Ma2003, p. s45-s51, 2003.
17] Xiaoqin Xia, Michael McClelland, and Yipeng Wang, "WebArray: platform for microarray data analysis," BMC Bioinformatics, vol. 6:Dec.2005.
[18] Carlo Colantouni, George Henry, Scott Zeger, and Jonathan Pe(Standardization and NOrmalization of MicroArray Data): web-accessible geneexpression data analysis," Bioinformatics, vol. 18, no. 11, pp. 1540-1541, 2002
Simon Urbanek, "Rserve -- A Fast Way to ProvApplications," 2003.
David Flana
[21] Bruce W.Perry, Java Servlet & JSP Cookbook O'Reilly Media Inc, 2004.
[22] Julie C.Meloni, PHP, MySQL and A
23] Robert McMillan, "Loosen the reins, says Google CEO,", 11 ed 2005.
71

GLOSSARY ANOVA (Analysis Of Variance) is a collection of statistical models and their
associated procedures which compare means by splitting the overall
cDNA mplementary DNA) DNA synthesized from mRNA or DNA by
Chromosomes
the form of one or more large macromolecules called
several
exually
ome, one from
CRAN (Comprehensive R Archive Network). A network of ftp and web servers
around the world that store identical, up-to-date versions of code and
documentation for R.
DNA (Deoxyribonucleic Acid) is a nucleic acid, usually in the form of a
double helix that contains the genetic instructions specifying the
biological development of all cellular forms of life, and most viruses.
Exons The coding regions of DNA.
observed variance into different parts.
(Co
reverse transcriptase often synthesized from a cellular extract.
The DNA which carries genetic information in cells is normally
packaged in
chromosomes. Most multicellular organisms have
chromosomes, which together comprise the genome. S
reproducing organisms have two copies of each chromos
each parent.
72

Fold change The ratio of gene expression between two samples in a microarray
experiment.
Gene Units of heredity in living organisms. They are encoded in the
organism’s genetic material (usually DNA or RNA), and control the
physical development and behavior of the organism.
Genome The whole hereditary information of an organism encoded in the
DNA. It includes both genes and non-coding sequences.
Human Genome Project
A project initiated by the government of United States for DNA
sequencing of the human genome.
Hybridization The process of combining complementary, single stranded nucleic
acids into a single molecule. Nucleotides will bind to their
complement under normal conditions, so two perfectly
complementary strands will bind to each other readily.
IDE (Integrated Development Environment). Environment used for
developing code for any application.
A list of coordinates relating to a specific image, created in order to
hyperlink areas of the image to various destinations (as opposed to a
age links to a
single destination).
Introns Non-coding regions of DNA.
Java API (Java Application Programming Interface). Collection of ready-made
software components that provide several capabilities.
JDBC (Java Database Connectivity). Set of communication protocols
Image map
normal image link, in which the entire area of the im
73

between a java program and a database.
(Joint PhotogJPEG raphic Experts Group) is a commonly used standard
JSP gy which mixes
JVM programs to
JVM ter and runtime system, which lets
LAMP
LocusLink rmation about
Microarray spots attached to a physical
Microarray experiment
ing gene expression in a system under controlled
time, stimulus, developmental stage, or
MIDAS TM4 suite.
method of lossy compression for photographic images. The file
extensions for this format are .jpeg or .jpg.
(Java Server Pages). A java programming technolo
static HTML with dynamically-generated HTML.
(Java Virtual Machine). The interpreter which lets java
run on any platform where it has already been installed.
(Java Virtual Machine). An interpre
java programs run on any platform it has been ported to.
(Linux + Apache + MySQL + Python). A platform used by the
microarray data analysis tool, WebArray.
Single query interface to sequence and descriptive info
genetic loci.
A collection of microscopic DNA
substrate such as glass, plastic or silicon chip forming an array.
Microarrays are hybridized with labeled samples and then scanned
and analyzed to generate data.
An experiment study
conditions to factors such as
dosage on a sample.
(Microarray Data Analysis System). Forms a part of the
A java application which provides users an intuitive interface to
74

design analysis processes combining one or more normalization and
miRNA
pression of other genes.
A
nthesis to undergo
which a gene
MySQL
zation
riation from the microarray
Oligo tide) Short sequence of nucleotides (<80 base pairs)
Protein
gical functions of all living cells and
PubMed
from MEDLINE and other life science journals
RNA
filtering steps.
(micro RNA) is a form of single-stranded RNA which is 20-25
nucleotides long and which regulates the ex
mRN (messenger RNA) is RNA that encodes and carries information from
DNA during transcription to sites of protein sy
translation in order to yield a gene product. The amount of any
particular type of mRNA in a cell reflects the extent to
has been “expressed”.
A very popular open source database server.
Normali The process used to standardize microarray data by removing the
effect of all sources of non-biological va
data, making them comparable.
(Oligonucleo
always single stranded to be used as probes or spots.
A complex, high-molecular-weight, organic compound that consists
of amino acids joined by peptide bonds. Proteins perform a wide
variety of structural and biolo
viruses.
A service of the U.S. National Library of Medicine that includes over
16 million citations
for biomedical articles back to the 1950s.
(Ribonucleic acid) A class of nucleic acids that consist of nucleotides
75

containing the bases- adenine (A), guanine (G), cytosine (C), and
uracil (U). An RNA molecule is typically single-stranded and can pair
Servlet
SQL guage). A standard computer language for
Microarray Data Analysis System
Transcription from DNA into
Translation
ce a specific protein according to the rules specified by the
tRNA
tide chain at the ribosomal
with DNA, another RNA molecule, or form secondary structure by
hybridizing to itself.
Rserve Rserve is a TCP/IP server which allows other programs to use R
facilities without the need to initialize or link against R library.
Java program that runs on a web server and which is used to build
web pages.
(Structured Query Lan
accessing and manipulating databases.
TM4 A suite of analysis tools developed to handle all aspects of microarray
process. Includes four major applications, Microarray Data Manager
(MADAM), TIGR_Spotfinder,
(MIDAS), and Multiexperiment Viewer (MeV).
The process in which transfer of genetic information
RNA takes place. It is the beginning of the process that ultimately
leads to the translation of the genetic code into a protein.
The second process of protein synthesis, in which mRNA is decoded
to produ
genetic code. Translation is preceded by transcription.
A small RNA chain (approximately 75 bp in length) that transfers a
specific amino acid to a growing polypep
site of protein synthesis during translation.
76

CURRICULUM VITAE
Date of Birth 20, 1982
ce of Birth
Undergraduate India
Graduate Study
ter Science
Experience
uisville, KY
VASUNDHARA AKKINENI
May
Pla Madras, India
Study University of Madras, Madras,
B.Tech. Information Technology
(1999-2003)
University of Louisville, Louisville, Kentucky
M.S. Computer Engineering and Compu
(2003-2006)
IT Digitization Intern, GE Energy, Atlanta, GA
(Jan, 2005 - July, 2005)
QA Analyst Intern, Yum! Brands Inc, Louisville, KY
(June, 2004 – Dec, 2004)
Student Assistant, University of Louisville, Lo
(Aug, 2003 – May, 2004)
77