microarray database resource for designing...
TRANSCRIPT

MICROARRAY DATABASE RESOURCE FOR DESIGNING CUSTOM MICROARRAYS
By
Ravi Shrikanth Gundlapalli M.S. University of Louisville, 2005
A Thesis Submitted to the Faculty of the
Graduate School of the University of Louisville in Partial Fulfillment of the Requirements
for the Degree of
MASTER OF SCIENCE
Department of Computer Engineering and Computer Science University of Louisville
Louisville, Kentucky
December 2005



DEDICATION
This thesis is dedicated to my parents
Mr. Subbarayudu Gundlapalli
and
Ms.Vijaya Kumari Gundlapalli
whose love and support made this possible for me.
iii

ACKNOWLEDGEMENTS
I owe this work to many people. I would like to thank my mother who has been
my inspiration all through my education. Many thanks are needed for Dr Eric C.
Rouchka, for the instruction and guidance provided during this long process. I would also
like to thank the members of my thesis committee, Dr. Nigel G. F. Cooper, Dr. Ahmed
Desoky. Additional thanks go out to Tim Hardin, Elizabeth Cha, Vasundhara Akeneni
and other members of the Bioinformatics Research Group. The work for this thesis was
performed in the University of Louisville’s Bioinformatics Labarotary. Support for this
work as well as equipment for the Bioinformatics lab is provided through Kentucky
Biomedical Research Infrastructure Network (NIH-NCRR grant # P20 RR16481; Nigel
Cooper, PI).
iv

ABSTRACT
MICROARRAY DATABASE RESOURCE FOR DESIGNING CUSTOM
MICROARRAYS
RAVI SHRIKANTH GUNDLAPALLI
NOVEMBER 29, 2005
Global gene expression monitoring with microarrays provides a vast
amount of biological information. Computers play a central role in many aspects of
microarray analysis, including the layout design of customized arrays that can be used by
printing robots to create customized microarrays for a myriad of different systems. We
have developed an integrated software called “MIDAR” that provides optimized design
solutions for customized microarrays based on user specified inputs. These include the
genes and gene groups, the number of replicates and the type of chip design (oligo or
cDNA). Users can additionally associate experimental data to stored chip designs. This
readily provides researchers with the gene expression level, and an interactive image map
that allows the user to view the included oligo/primer detail information and list specific
data details for each spot. MIDAR incorporates the Gene Ontology (GO) database to
provide for selection of genes based on a common characteristic such as biological
function, molecular process, and cellular component. MIDAR is a fully featured
community-driven solution to the challenge of microarray design, data management and
analysis. The main contribution of this thesis work deals with the design solutions for
custom microarrays as well as the incorporation of visual analysis approaches.
v

TABLE OF CONTENTS
DEDICATION …………………………………………………………………………iii ACKNOWLEDGEMENTS…………………………………………………………….iv ABSTRACT...…………………………………………………………………………..v LIST OF FIGURES……………………………………………………………………..vii NOMENCLATURE………………………………………………………………….…viii
1. INTRODUCTION….…………………………………………………………....1
2. LITERATURE REVIEW…...…………………………………………………...5
2.1 GENES…………………………………………………………………..5
2.2 GENETIC CODE………………………………………………………..10
2.3 CENTRAL DOGMA OF MOLECULAR BIOLOGY.…….…………...11
2.4 THE HUMAN GENOME PROJECT……………..…………………….13
2.5 GENE DISCOVERY THROUGH EXPRESSED SEQUENCE TAGS...15
2.6 dbEST : A DESCRIPTIVE CATALOG OF ESTs...…………………….18
2.7 GENE EXPRESSION PROFILING……………………………………..18
2.8 HYBRIDIZATION AND GENE EXPRESSION………..……………...20
3. MICROARRAYS..………………………………………………………………21
3.1 COMMERCIAL ARRAYS...……………………………………………25
3.2 EXPERIMENTAL DESIGN…………………………………………….29
3.3 APPLICATION OF MICROARRAYS…………………………………35
3.4 AVAILABLE MICROARRAY TOOLS...……………………………...35
4. MIDAR – DESIGN ASPECTS………………………………………………….38
5. MIDAR – A SCENARIO…..……………………………………………………51
6. FUTURE – MICROARRAYS AND MIDAR…..………………………………56
REFERENCES………………………………………………………………………58
CIRRICULUM VITAE..…………………………………………………………….60
vi

LIST OF FIGURES
Figure 1.1 - Overview of Microarray Analysis….……………………………………….2
Figure 2.1 - Structure of a Gene………………………………………………..………...6
Figure 2.2 - Exons and Introns in DNA Sequence……………………………………….7
Figure 2.3 - Spiral Structure of DNA…………………………………………………….8
Figure 2.4 - mRNA Processing…………………………………………………………..9
Figure 2.5 - Central Dogma of Molecular Biology………………………………………12
Figure 2.6 - Overview of Protein Synthesis……………………………...………………16
Figure 2.7 - Overview of how ESTs are Generated……………...………………………17
Figure 3.1 - Image of a Portion of a Microarray that has been Hybridized and Scanned..22
Figure 3.2 - Combinatorial Chemistry Approaches to Microarray Manufacturing…...…24
Figure 3.3 - Distribution of Types of Microarrays…………………………….………...27
Figure 3.4 - Experimental Approach for Microarray Analysis………….……………….30
Figure 3.5 - Five Steps of Microarray Analysis……………………………….………....32
Figure 3.6 - Hybridizing the Two Samples………………………….…………………...34
Figure 3.7 - Scanning the Microarray.…………………………………………………...34
Figure 4.1 - Database Design……………….……………………………………………42
Figure 4.2 – Data Header for Uploading Gene Information……………………………..43
Figure 4.3 – Explanation of Code………………………………………………………..45
Figure 5.1 - Screenshot Depicting User Input for Chip Design…….……………………51
Figure 5.2 - Screenshot Depicting the Custom Array Design and the Primer/Oligo
Information of a Specific Spot on the Array……….…………………………………….52
Figure 5.3 - Screenshot Depicting Marked Spots Associated with a Gene of Interest.….53
Figure 5.4 - Screenshot Depicting the Activated Custom Array.………………………..54
Figure 5.5 - Screenshot Depicting a Comparative Analysis of a Single Custom Array
from Two Different Experiments..……..………………………………………………...55
vii

NOMENCLATURE
Affymetrix: A company that has revolutionized manufacturing process for
microarrays. It uses semiconductors to produce high-density arrays called
GeneChip.
Agilent: Another popular manufacturer of microarrays with area of research in
MEMS, nanotechnology and Life Sciences.
Amino acids: The building blocks of proteins.
BASE: Abreviated for BioArray Software Environment, is a free web-based
solution for analyzing huge amounts of data obtained from microarrays.
cDNA: Abbreviated for complimentary DNA, is a stable compound of mRNA and
contains only the coding regions of DNA. The sequence is a reverse
compliment of mRNA.
Codon: A single unit of genetic code that is made up of three (triplet) nucleotide
bases in a DNA or RNA molecule specifying a single amino acid.
DNA: The molecule that encodes genetic information. DNA is a double stranded
molecule made of two twisting, paired strands held together by weak
bonds between base pairs of nucleotides
EST: Abbreviated for Expressed Sequence Tags, these are the sequences
obtained from either sides of a cDNA sequence.
viii

Exon: The coding regions of DNA.
Gene: The fundamental physical and functional unit of heredity. A gene is an
ordered sequence of nucleotides located in a particular position within the
genome that encodes a specific functional product (i.e., a protein or RNA
molecule).
Genetic Code: The sequence of nucleotides, coded in triplets (codons) along with the
mRNA that determines the sequence of amino acids in protein synthesis.
A gene’s DNA sequence can be used to predict the mRNA sequence, and
the genetic code can in turn be used to predict the amino acid sequence.
Genome: All the genetic material of a particular organism, its size is generally given
as its total number of base pairs or as its total number of genes.
Genomic Era: The new era in genetic research featuring rapid acquisition and integration
of increasingly advanced genetic information resulting from the progress
and completion of Human Genome Project.
Human Genome Project: It was initiated by the government of United States for DNA
sequencing of the human genome.
Intron: The non-coding regions of DNA.
LAD: Abbreviated for Longhorn Array Database, is an open source, MIAME-
complaint version of Stanford Microarray Database (SMD).
MADAM: Abbreviated for MicroArray Data Management is a software package
available from The Institute of Genomic Research. It helps in tracking
experimental parameters and results associated with microarrays.
Microarray: An ordered array of microscopic elements on a planar substrate that allows
ix

the specific binding of genes or gene products.
mRNA: A molecule that can move from the nucleus to the cytoplasm of cells that
serves as the crucial connecting message between information contained
in the gene and protein synthesis. The structure of RNA is similar to that
of DNA. The mRNA molecule serves as a template for the specific amino
acid sequence of a protein.
Nucleotide: The basic subunits of DNA or RNA. Thousands of nucleotides are linked
to form a DNA or RNA molecule. The four nucleotides in DNA contain
the bases adenine (A), guanine (G), cytosine (C), and thymine (T). In
nature, base pairs form only between A and T and between G and C; thus
the base sequence of each single strand can be deduced from that of its
partner.
Oligonucleotide: Small single stranded segments of DNA typically 20-30 nucleotide
bases in size which are synthesized in vitro.
PCR: Abbreviated for Polymerase Chain Reaction, is the process where RNA
and DNA polymerase enzymes link together into DNA and RNA chains.
Perl: A programming language released by Larry Wall, borrows heavily from
C, sed, awk, shell scripting and others, is gaining immense popularity
among open source developers.
PHP: Another widely open-source programming language primarily for server-
side programming and developing dynamic web pages.
Photolithography: It is a process used in semiconductor device fabrication to transfer a
pattern from a photo mask to the surface of a substrate.
x

Probe: Labeled molecule in solution that reacts with a complimentary target
molecule on the substrate.
Protein: A large molecule composed of one or more chains of amino acids in a
specific order; the order is determined by the base sequence of nucleotides
in the gene that codes for the protein. Proteins are required for the
structure, function and regulation of the body’s cells, tissues and organs,
and each protein has unique functions. Examples are hormones, enzymes
and antibodies.
Ribosome: A cytoplasmic organelle that serves as the molecular machine on which
polypeptide synthesis from mRNA occurs.
Sequencing: Determination of the order of nucleotides (base sequences) in a DNA or
RNA molecule.
SMD: Abbreviated for Stanford Microarray database is a database used to
publish microarray data and for public access of microarray data.
SNP: Abbreviated for single nucleotide polymorphism is a common sequence
variant containing a one-base-pair change relative to the normal gene.
TM4: A suite of software available with The Institute of Genomic Research
developed to address all data associated with microarray design and
analysis.
Transcription: The synthesis of an mRNA copy from a sequence of DNA (a gene), the
first step in gene expression.
Translation: The process in which the genetic code carried by mRNA directs the
synthesis of proteins from amino acids.
xi

tRNA: A class of RNA that recognizes the triplet nucleotide coding sequences of mRNA
and carries the appropriate amino acid to the ribosomes, where proteins are assembled
according to the genetic code carried by mRNA.
xii

1. INTRODUCTION
Imagine trying to solve a jigsaw puzzle without knowing if you have all of the
pieces. This is the dilemma faced by scientists in the field of molecular medicine when
attempting to understand how human genes and their protein products interact with one
another to lead to normal biological functions, how these functions can break down in
various disease states, and how normal functions can be restored through molecular
intervention. My dilemma could partly be attributed to my background being computer
engineering and not biology. The following material is a basic overview of genomics and
is meant to define the boundaries of a puzzle whose solution is going to be my thesis
work and is within my grasp, though not in my hands.
With the completion of Human Genome project [1], and cataloguing of thousand
of genes already (estimates ranging from approximately 64,000 to 80,000 genes have
been advanced [2]), the biggest challenge to scientists now is to understand how these
genes work – what regulates their activity and how they interact with each other and the
environment. In other words, there are available huge amounts of data relevant to the
puzzle, but solving the puzzle remains a bioinformatics challenge.
The challenge of determining the function of genes was well accepted and in
came a capability to analyze gene expression level of thousand of genes simultaneously
in a single experiment quickly and efficiently. As we have seen earlier that thousand of
genes and their products in a given living organism function in a complicated and
1

orchestrated way that creates the mystery of life. However, the traditional methods in
molecular biology generally work on one gene in one experiment basis [3], which means
that throughput is very limited and the whole picture of gene function is hard to obtain.
Microarray technology monitors the whole genome on a single chip so that researchers
can have a better picture of the interactions among thousand of genes simultaneously.
Microarray technology is being considered as the best method for examining
global aspects of genomic data and gene expression profiling. The microarray data
essentially added another dimension in bioinformatics with its infinite possibilities of
expression change comparison in various conditions.
Source: - bldg6.arsusda.gov/benlab/ microarrays002.jpg
Figure 1.1 – Overview of Microarray Analysis
2

Microarray analysis (Fig. 1.1) involves DNA sequences from two different
samples – one a normal sample and the other a test sample. They are then combined
together on a slide typically of a size of microscope slide. The samples then come
together and bind to complementary sequences and express themselves. Once the genes
have been expressed they are scanned and all the information is fed to a computer for
further analysis.
Computers play a central role in many aspects of microarray analysis, including
design informatics. Microarray printing robots can be directed through software to
manufacture microarrays with myriad physical characteristics, and this design flexibility
is critical for the study of different methods, and the development of new assays. Though
this software is most pertinent to researchers who make their own arrays, those who
purchase off-the-shelf microarrays are also served well by understanding design
informatics. The location of the microarray on the printing substrate, the number of
features and genes, the number of pins and samples, and content maps are among the
topics addressed in this software.
A critical aspect of microarray production is the design considering space
optimization to produce high – density arrays for a given set of gene samples and number
of replicates to be present. The software available with robotic spotters translates user
input parameters into a set of instructions in robotic language for building arrays. These
softwares do not offer design capabilities in which spotting parameters and grid
configurations can be chosen for a given set of samples and replicates. Presently various
solutions have to be derived manually in most academic laboratories.
User-friendly software that can be used by experts and novice alike would
3

simplify and aid rapid design of microarrays. We at the University of Louisville have a
joint collaboration between the Speed School of Engineering and School of Medicine to
address various bioinformatics issues through the Bioinformatics Research Group (BRG)
[kbrin.a-bldg.louisville.edu/brg/]. BRG initiated a multifaceted project - Microarray
database resource (MIDAR) - wherein researchers and students of the group will be
investigating efficient ways to design, analyze and store biological data resulting from the
experiments conducted on microarrays.
While the MIDAR system will include the ability to design, store and analyze
microarray experiments from various custom and commercial packages, this thesis work
will concentrate on developing optimized design techniques for microarray chips
followed by their simulation with the experimental values.
4

2. LITERATURE REVIEW
The genetic blueprint is carried in the genome, an improbable assembly of DNA
bases, genes, and chromosomes. Cells pass an exact copy of the genome to other cells
during cell division, and the blueprint is inherited during reproduction. Human genomes
are structurally complex, and minor changes in the sequence can produce disease. Each
cell in the human body contains the same genomic sequence as every other cell, but gene
expression varies greatly from cell to cell. Transcription and translation convert gene
information into proteins, and DNA replication synthesizes exact copies of the genomic
sequence. DNA sequencing technology [1;4] has recently afforded the sequence of the
entire human genome, as well as sequences of dozens of other organisms, including
viruses, bacteria, yeast, worms, insects, plants and rodents. This chapter provides a
survey of genes and genomes, and offers a glimpse into exciting and fast-moving field of
genomics.
2.1 GENES
All humans are basically the same, yet we are unique, with different traits that
allow us to stand out as individuals. Some are tall, some are short, some are fair, and
some are dark. These physical similarities and differences are due to similarities and
differences in our genetic instructions. Our own set of genetic instructions, our genes
5

determine our particular traits, inherited from our parents.
Genes are instructions inside you that tell your body what to look like, and how to
work. There are genes, which tell your hair to be curly or straight, genes, which tell your
body to grow tall (or not so tall!), genes, which tell your stomach how to digest food,
genes that account for every little detail of your body! Of course, your body is also
affected by the things you do and the things that go on around you. If you dye your hair,
you will look different. If you do not eat a healthy diet, you might not grow as tall. We
call the things outside your body that can affect it ‘environmental influences’[5].
Source: www.biotec.or.th/Genome/ whatGenome.html
Figure 2.1 – Structure of a Gene
Technically genes are continuous segments of genomic DNA constructed from
four nucleotide building blocks (Fig. 2.1). Each gene encodes a specific mRNA and
protein, the latter of which imparts biological function in cell. Genes in higher eukaryotes
6

such as humans, contain exons and introns [6]. An exon is gene segment that is copied
into mRNA and maintained after mRNA processing, and an intron is a gene segment that
is copied into mRNA, but removed from the mature mRNA before protein synthesis (Fig.
2.2). Genes in lower eukaryotes, such as yeast, are essentially devoid of introns, and
bacteria do not contain any introns at all. The presence of introns in complex organism,
but not in simple systems, suggests an evolutionary role for these noncoding gene
sequences, potentially in alternative splicing.
Figure 2.2 – Exons and Introns in DNA Sequence
Genes are composed of double-stranded DNA, and gene size is measured in a unit
known as base pair, corresponding to one nucleotide of double stranded DNA. The
genetic blueprint of virtually every organism in the biosphere is stored in the
biopolymeric molecule known as deoxyribonucleic acid (DNA). DNA is composed of a
long string of nucleotides, each of which contains one of four bases (A, G, C or T), a
deoxyribose sugar, and a phosphate group [7]. Nucleotides are joined together in a
covalent manner in the cell to build linear DNA sequences. A typical human gene and
chromosome contain approximately 20,000 and 100,000,000 nucleotides [2] respectively.
Different nucleotides can be strung together to form a polynucleotide. However, the ends
of the polynucleotide are different, meaning that each polynucleotide sequence will have
directionality. The ends of the polynucleotide are marked either 3’ or 5’. The general
convention is to label the coding strand from 5’ to 3’ (left to right).
DNA can be either single-stranded or double stranded. DNA chains that bond to
7

each other through A-T and G-C interactions are known as complimentary strands. The
chemical process by which complementary strands bind into a double-stranded molecule
is known as hybridization. Complimentary strands of DNA form a spiral molecule or
double helix (Fig. 2.3), whereby the two interwoven chains coil around a center axis like
a spiral staircase. Two complementary polynucleotide chains form a stable structure
known as the DNA double helix. For the polynucleotide given above, the double-
stranded polynucleotide is as follows:
5’ G→T→A→A→A→G→T→C→C→C→G→T→T→A→G→C 3’
| | | | | | | | | | | | | | | |
3’ C←A←T←T←T←C←A←G←G←G←C←A←A←T←C←G 5’
Source: www.genecrc.org/site/ lc/lc2b.htm
Figure 2.3 - Spiral Structure of DNA
The DNA encodes the genetic blueprint of an organism, and the blueprint is
converted into protein information using ribonucleic acid (RNA) as a molecular
intermediary. Certain classes of RNA also play a role in protein synthesis. RNA is similar
to DNA, but possesses some novel structural, physical and functional properties.
Ribonucleotides are identical to deoxyribonucleotides, except that the RNA building
blocks contain uracil (U) instead of thymine (T) and ribose instead of deoxyribose as the
sugar. RNA chains are generally much shorter than DNA chains; most RNA molecules
contain 70-10,000 ribonucleotides [8]. Unlike DNA, which forms a double stranded
8

double helix, nearly all RNA molecules are single stranded. RNA is important in the cell
and contributes in a variety of ways. Two of the major RNA molecules involved in
protein synthesis are messenger RNA (mRNA) and transfer RNA (tRNA).
mRNA encodes the genetic information as copied from the DNA molecules.
Transcription is the process in which DNA is copied into an RNA molecule. The
resulting linear molecule is an mRNA transcript (Fig. 2.4). In eukaryotic cells, before the
mRNA can be translated into a protein, it needs to be modified. The nature of most
eukaryotic genes is that the genes are created in pieces, where coding regions, called
exons, are interspersed with noncoding regions, called introns. One of the steps in
processing the mRNA is to remove the intronic regions and to splice together the coding,
or exonic regions. The processed mRNA can then be transported from the nucleus and
translated into a protein sequence.
Source: http://departments.oxy.edu/biology/Stillman/bi221/111300/processing_of_hnrnas.htm
Figure 2.4 - mRNA Processing
9

tRNA molecules develop a well-defined three-dimensional structure, which is
critical in the creation of proteins. Attached to each tRNA molecule is an amino acid
(which will be discussed momentarily). The amino acid to be attached is determined by a
three base sequence called an anticodon sequence, which is complementary to the
sequence in the mRNA. Translation is the process in which the nucleotide base sequence
of the processed mRNA is used to order and join the amino acids into a protein with the
help of ribosomes and tRNA.
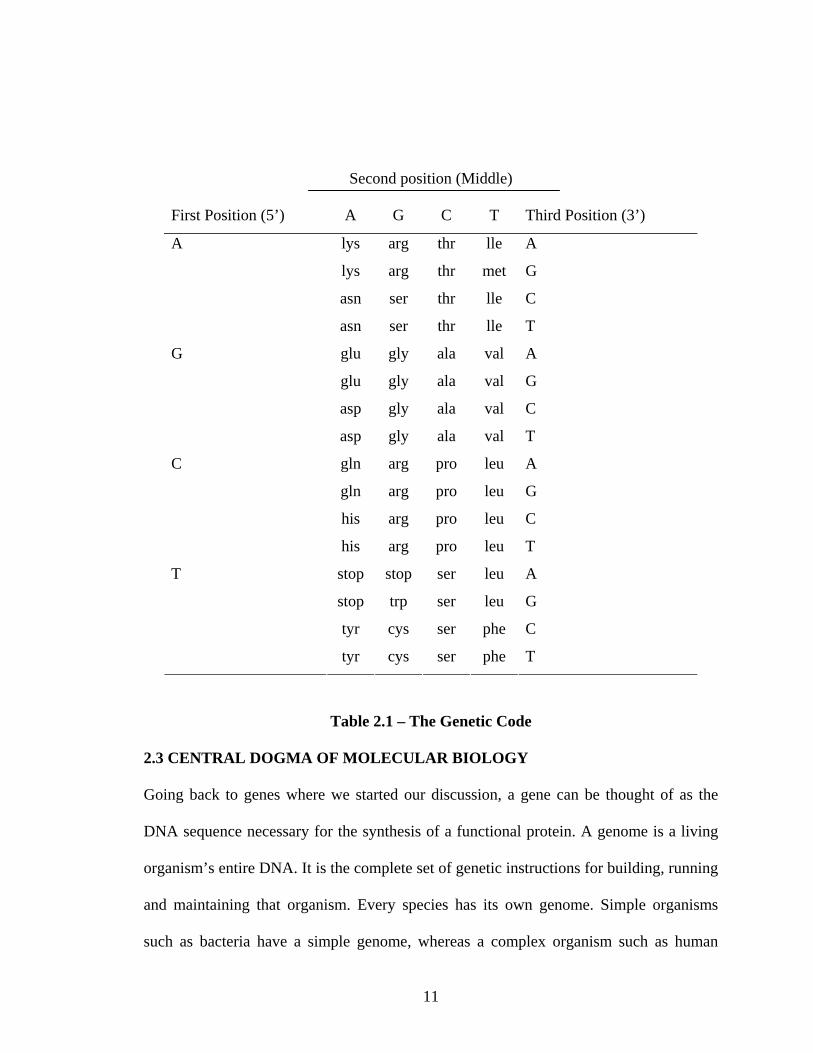
2.2 GENETIC CODE
Genetic information is stored in a fundamental unit of genetic information known
as a codon. A codon, or triplet, contains three successive nucleotides that are read by the
cellular machinery in a 5’ or 3’ manner to specify an amino acid. There are 64 possible
combinations of the four nucleotides, and all 64 codons are used in the cell. The cellular
“conversion table” between codon and amino acid is known as the genetic code (Table
2.1). Of the possible combinations, 61 codons specify the 20 amino acids; and the
remaining 3 combinations are stop codons, the genetic signals in the code that signal
termination.
Thus the flow of genetic information is from the DNA directing the synthesis of
RNA, and RNA which in turns directs the synthesis of protein. This flow of genetic
information from nucleic acids to protein has been called the Central Dogma of
Molecular Biology [9] (Fig. 2.5).
10
Second position (Middle)
First Position (5’) A G C T Third Position (3’)
lys arg thr lle A
lys arg thr met G
asn ser thr lle C
A
asn ser thr lle T
glu gly ala val A
glu gly ala val G
asp gly ala val C
G
asp gly ala val T
gln arg pro leu A
gln arg pro leu G
his arg pro leu C
C
his arg pro leu T
stop stop ser leu A
stop trp ser leu G
tyr cys ser phe C
T
tyr cys ser phe T
Table 2.1 – The Genetic Code
2.3 CENTRAL DOGMA OF MOLECULAR BIOLOGY Going back to genes where we started our discussion, a gene can be thought of as the
DNA sequence necessary for the synthesis of a functional protein. A genome is a living
organism’s entire DNA. It is the complete set of genetic instructions for building, running
and maintaining that organism. Every species has its own genome. Simple organisms
such as bacteria have a simple genome, whereas a complex organism such as human
11

species has a relatively large genome with about 30,000 genes [2]. And interestingly, in
any two humans, 99.9% of their DNA is identical [2].
DNA
↓
RNA
↓
PROTEIN Source: http://www.people.virginia.edu/~rjh9u/dnaprot.html
Figure 2.5 - Central Dogma of Molecular Biology
The entire set of genetic instructions is so large that the 0.1% variation allows for
millions of these possible differences among individuals. These tiny fractions of DNA
where variation occur, leads to the enormous diversity that makes each of us unique. Yet,
the same variation that causes the differences in our appearance also leads to differences
in our likelihood of getting any particular disease. Knowledge about the effects of DNA
variation between individuals can lead to better understanding of disease and to advances
in medicine.
The cells of our bodies are made of different kinds of molecules, such as water,
minerals, proteins, sugars fats and DNA. Of these, proteins are particularly important
because they are the fundamental components of the body that determine how all of the
molecules are organized and how they act. Thus, proteins play a key role in the way we
12

look and in the way we grow. DNA acts as a molecular code for making these proteins.
The DNA in each gene provides the instructions for making one protein, or sometimes, a
few related proteins. However, only about 1/60th of the entire genome directly codes – or
provides the instructions – for making proteins [5]. The rest of DNA in our genomes
helps direct when and where in the body each gene should be used. Taken together, all of
the DNA of the genome can be thought of as a blueprint for a human being.
In the past, doctors and scientists did not have the benefit of a human genetic
blueprint to help them better understand sickness and develop appropriate treatments. It is
much similar to the following scenario – If a house needs a repair or maintenance,
mechanics and engineers can consult the blueprint when analyzing a problem and avoid
unnecessary work, or, more importantly, avoid worsening the problem. Similarly, the
blueprint of the human body provided through the Human Genome project will help
analyze problems when something goes wrong with a person. In the future, when a doctor
is treating someone who is sick, he or she will be able to consult the patient’s genetic
blueprint in order to determine what variation of genes that patient has and prescribe a
particular treatment that he or she knows is most likely to be effective for that individual.
This will also help doctors avoid prescribing a drug that could cause a serious side effect.
2.4 THE HUMAN GENOME PROJECT
Understanding the potential of a human genome, The United States started the
Human Genome project in 1990 with the ambitious goal of sequencing the human
genome within the next fifteen years [4]. The project’s new research strategies and
experimental technologies have generated a steady stream of ever-larger and more
13

complex genomic data sets that have poured into public databases and have transformed
the study of virtually all life processes. The genomic approach of technology
development and large-scale generation of community resource data sets has introduced
an important new dimension into biological and biomedical research. Interwoven
advances in genetics, comparative genomics, high throughput biochemistry and
bioinformatics are providing biologists with a markedly improved repertoire of research
tools that will allow the functioning of organisms in health and disease to be analyzed
and comprehended at an unprecedented level of molecular data.
The project’s huge success is based on the fact that only about 2% of total bases
make up the protein coding portions of our genes; the remaining 98% is of unknown
function and often referred to as junk DNA [10]. Thus, sequencing the genome may not
be the most efficient way to generate a catalog of human genes. As Brenner put it, “If
something like 98% of the genome is junk, then the best strategy would be to find the
important 2% and sequence it first” [10].
A number of investigators have advocated large scale sequencing of the
transcription products of genes in the form of complimentary DNA (cDNA) clones, as a
prelude to sequencing of entire human genome and then came the era of high –
throughput cDNA sequencing, initiated in 1991 by a landmark study from Venter and
colleagues [11]. The basic strategy involves selecting cDNA clones at random and
performing a single automated sequencing read from one or both ends of their inserts.
They introduced the term “Expressed Sequence Tag” (EST) to refer to this new class of
sequence, which is characterized by being short (typically around 400 bases) and
relatively inaccurate (around 2% error).
14

2.5 GENE DISCOVERY THROUGH EXPRESSED SEQUENCE TAGS
EST’s provide researchers with a quick and inexpensive route for discovering
new genes, for obtaining data on gene expression and regulation, and for constructing
genome maps. The idea is to sequence bits of DNA that represent genes expressed in
certain cells, tissues, or organs from different organisms and use these tags to fish a gene
out of a portion of chromosomal DNA by matching base pairs. The challenge associated
with identifying genes from genomic sequences varies among organisms and is
dependent upon genome size as well as the presence or absence of introns, the
intervening DNA sequences interrupting the protein coding sequence of a gene. And
most of the human genome is composed of introns interspersed with a relative few DNA
coding sequences, or genes, thus making gene identification difficult among humans.
Genes are expressed as proteins, a complex process composed of two main steps.
As seen in the Central Dogma of molecular Biology each gene (DNA) must be converted,
or transcribed, into messenger RNA (mRNA), RNA that serves as a template for protein
synthesis. The resulting mRNA then guides the synthesis of a protein through a process
called translation. Interestingly, mRNAs in a cell do not contain sequences from the
regions between genes, nor from the non-coding introns that are present within many
genes. Therefore, isolating mRNA is key to finding expressed genes in the vast expanse
of the human genome (Fig. 2.6).
15

Source: http://www.ncbi.nlm.nih.gov/About/primer/est.html
Figure 2.6 - Overview of Protein Synthesis
The problem, however, is that mRNA is very unstable outside of a cell; therefore,
scientists use special enzymes to convert it to complementary DNA (cDNA) [11]. cDNA
is a much more stable compound and, importantly, because it was generated from a
mRNA in which the introns have been removed, cDNA represents only expressed DNA
sequence. Once cDNA representing an expressed gene has been isolated, scientists can
then sequence a few hundred nucleotides from either end of the molecule to create two
different kinds of EST’s. Sequencing only the beginning portion of the cDNA produces
what is called a 5' EST (Fig 1.8). A 5' EST is obtained from the portion of a transcript
that usually codes for a protein. These regions tend to be conserved across species and do
not change much within a gene family. Sequencing the ending portion of the cDNA
molecule produces what is called a 3' EST (Fig 2.7). Because these ESTs are generated
from the 3' end of a transcript, they are likely to fall within non-coding or untranslated
regions (UTRs), and therefore tend to exhibit less cross-species conservation than do
coding sequences [11].
16

Source: http://www.ncbi.nlm.nih.gov/About/primer/est.html
Figure 2.7 - Overview of how ESTs are generated
Since ESTs represent a copy of subjective part of a genome, that is expressed,
they have proven themselves again and again as powerful tools in the hunt for genes
involved in hereditary diseases. ESTs also have a number of practical advantages in that
their sequences can be generated rapidly and inexpensively, only one sequencing
experiment is needed per each cDNA generated, and they do not have to be checked for
sequencing errors because mistakes do not prevent identification of the gene from which
the EST was derived.
ESTs employed for gene identification - To find a disease gene using this
approach, scientists first use observable biological clues to identify ESTs that may
correspond to disease gene candidates. Scientists then examine the DNA of disease
patients for mutations in one or more of these candidate genes to confirm gene
identity[12]. Using this method, scientists have already isolated genes involved in
Alzheimer's disease, colon cancer, and many other diseases. It is easy to see why ESTs
17

will pave the way to new horizons in genetic research.
Thanks to EST, there is explosion in information about DNA sequences of the
human genome. Scientists have identified large number of novel genes using ESTs.
Although important goals of any sequencing project may be to obtain a genomic
sequence and identify a complete set of genes, the ultimate goal is to gain an
understanding and when, where and how a gene is turned on, a process commonly
referred to as gene expression.
2.6 dbEST: A DESCRIPTIVE CATALOG OF ESTs
Scientists at NCBI created dbEST [13] to organize, store, and provide access to
the great mass of public EST data that has already accumulated and that continues to
grow daily. Using dbEST, a scientist can access not only data on human ESTs but
information on ESTs from over 300 other organisms as well. Whenever possible, NCBI
scientists annotate the EST record with any known information. For example, if an EST
matches a DNA sequence that codes for a known gene with a known function, that gene's
name and function are placed on the EST record. Annotating EST records allows public
scientists to use dbEST as an avenue for gene discovery. By using a database search tool,
such as NCBI’s BLAST, any interested party can conduct sequence similarity searches
against dbEST.
2.7 GENE EXPRESSION PROFILING
Chips containing hundreds, thousands, or even tens of thousands of genes
arranged in parallel are revealing the function of genes and relationships between genetic
18

and biochemical pathways that are virtually impossible to group by any other means.
Organisms express their genes at a relatively constant rate until the products encoded by
these genes are needed for a specific function. When needed, genes are activated or
repressed rapidly and in a dramatic fashion changing by 10-, 100- or even 1000-fold or
more depending on the particular gene and the strength of the regulatory cue [8]. The
expression of genes changes in response to a wide spectrum of signals, including
hormones, chemicals, nutrients, stress, changes in cell division and development, light
simulation and the like, provides a gene expression that is characteristic for a given
physiological state.
Because gene expression correlates specifically and tightly with function, it is
possible to infer the function of genes and the interaction of pathways by documenting
the expression of those genes are turned up or down in a given physiological state. Gene
regulation provides a selective evolutionary advantage by conserving cellular building
blocks (e.g.: - nucleotides, amino acids) and enzymatic machinery (e.g.: - transcription
factors, polymerase) until they are needed, and by allowing the organism to adapt to a
plethora of different environmental conditions to which it is exposed during its lifetime.
When cells are subjected to elevated temperature, for example, heat shock genes
are activated to protect cellular proteins from thermal damage [8]. Disease states, drug
treatment, different developmental stages, and many other processes can be examined by
cataloging gene expression profiles. Understanding how genes are expressed in normal
and diseased cells can help uncover novel potential targets for therapies. The logical
extension of this concept is to build comprehensive gene expression databases for each
organism that contain expression profiles for each gene across thousands of different
19

conditions, thereby allowing biological exploration to take place predominately by means
of a computer.
2.8 HYBRIDIZATION AND GENE EXPRESSION
A single stranded DNA molecule with a known sequence is labeled with a
radioactive isotope or fluorescent dye. This is used as a “probe” to detect a fragment of
DNA or mRNA, with the complimentary sequence. In order to determine if a gene is
expressed in a particular tissue, use the following “Northern blot” procedure could be
used:
1 Make a fluorescent dye probe by using a small piece of gene A
2 Isolate mRNA from all the tissues of interest
3 Bring the mRNA to a solid medium (like nylon filter)
4 Hybridize the probe the filter
5 If gene is expressed in the tissue, see a fluorescent signal on the filter
6 Thus one could detect the present of a particular DNA or RNA
But the process above and most other traditional methods in molecular biology
generally work on a “one gene in one experiment” basis, which means that the
throughput is very limited and the “whole picture” of gene function is hard to obtain.
There is a need for methods that can handle these huge sets of available data in a global
fashion, and that can analyze such large systems.
20

3. MICROARRAYS
With the completion of Human Genome project, and cataloguing of thousand of
genes already, the biggest challenge to scientists now is to understand how these genes
work – what regulates their activity and how they interact with each other and the
environment. A technology that is reshaping molecular biology, gives the scientists the
capability to analyze expression level of thousand of genes simultaneously in a single
experiment quickly and efficiently. As we have seen earlier that thousand of genes and
their products in a given living organism function in a complicated and orchestrated way
that creates the mystery of life. However, the traditional methods in molecular biology
generally work on “one gene in one experiment” basis, which means that the throughput
is very limited and the “whole picture” of gene function is hard to obtain. The microarray
technology monitors the whole genome on a single chip so that researchers can have a
better picture of the interactions among thousand of genes simultaneously.
The magic of microarray analysis is sweeping through the agricultural and
medical sciences, replacing traditional biological assays based on gels, filters, and
purification columns with small glass chips containing tens of thousands of DNA and
protein sequences. Microarrays function like biological microprocessors, enabling the
rapid and quantitative analysis of gene expression patterns, patient genotypes, drug
mechanisms, and disease onset and progression on a genomic scale.
21

A microarray is a small analytical device that allows genomic exploration with
speed and precision unprecedented in history of biology. Schena and co-workers first
developed them at Stanford University in early 1990’s [14]. Glass chips containing tens
of thousands of genes are used to examine fluorescent samples prepared by labeling
messenger RNA (mRNA) from cells, tissues and other biological sources. Molecules in
the fluorescent sample react with cognate sequences on the chip, causing each spot to
glow with intensity cognate sequences on the chip, causing each spot to glow with
intensity proportional to the activity of the expressed gene. The enormous capacity of
these miniature devices allows the analysis of entire human genome in a single
experiment. Since patterns of gene expression correlate strongly with function,
microarrays are providing unprecedented information on human disease, aging, drug and
hormone action, mental illness, diet, and many other clinical matters. Microarrays can
also be used to find alterations in gene sequences, paving the way for a new era of genetic
screening, testing and diagnostics.
Source: http://www.soe.ucsc.edu/~sugnet/microarray/microarray_FAQ.html
Figure 3.1 - Image of a Portion of a Microarray that has been Hybridized and
Scanned
A microarray is an ordered array of microscopic elements on a planar substrate
that allows specific binding of genes or gene products. Microarray is a new scientific
22

word derived from the Greek word mikro (small) and the French word arayer
(arranged)[8]. Microarrays also known as biochips, DNA chips, and gene chips contain
collection of small elements or spots arranged in rows and columns. To qualify as a
microarray, the analytical device must be (1) ordered, (2) microscopic, (3) planar, and (4)
specific. Devices that fulfill only a subset of these criteria do not afford the advantages of
microarrays, do not qualify as microarrays, and should not be considered as such.
A typical microarray would consist of a regular microscope glass slide that
contains thousands of microscopic quantities of PCR products of cDNA [14] or synthetic
oligonucleotides of genes. Each spot should represent one specific exon or one gene in
the genome. Thanks to recent development of high-speed robotic printing, once all the
nucleotide sequences are ready, mass production of microarray slides is now possible for
many different experiments. A computer then precisely measures the amount of sample
(mRNA) bund to each spot on the microarray, generating a profile of gene expression in
the cell. Microarray takes advantage of two basic technologies.
1) One is binding between single stranded DNA sequence with its complementary
sequence (Base pairing or hybridization, i.e. A-T and G-C for DNA; A-U and G-
C for RNA)
2) Another is using fluorescent probe to visualize difference in cDNA level which in
turn represents mRNA level.
Currently microarrays come in many different types but they are two main
fabrication methods.
1. Synthesize DNA probes separately, using PCR for cDNAs [15] or chemical
synthesis for oligonucleotides. Then a robot is used to spot these DNA probes
23

onto microarrays into very small grids. The substrate for microarrays can be glass,
plastic or even nylon membranes. Most labs use glass microscope slides since this
method is comparatively cheap and flexible. Some related technologies use ink-jet
like printers to spray oligonucleotide probes on the microarrays.
2. Synthesize DNA oligonucleotides [3] directly on the microarray using UV-masks
and photo-activated chemistry (Fig. 3.2). Currently the company Affymetrix is the
only commercial company using combinatorial chemistry approaches. The
technique used is as follows: deprotect sites that will have the next base (A,C,T,
or G) bound to them using UV light, then bind the next base to those sites and
repeat with a different base. To direct which sites will be deprotected, Affymetrix
uses a photolithographic mask which only lets the UV light activate certain sites.
Image source: - http://www.cse.ucsc.edu/~sugnet/microarray/microarray_FAQ.html
Figure 3.2 – Combinatorial Chemistry Approaches to Microarray
Manufacturing
Using this technology Affymetrix is able to build up very large arrays of
oligonucleotides in parallel. However due to synthesis efficiencies the longest
oligonucleotide probes that Affymetrix makes are 25 nucleotides long.
24

3.1 COMMERCIAL ARRAYS
There are various available companies manufacturing commercial microarrays.
Here are three of the popular commercial microarray platforms:
• Affymetrix (GeneChip®)
• Agilent Technologies (Agilent SurePrint)
• Amersham Biosciences (CodeLink™)
Over the past few years oligonucleotide GeneChip arrays, commercially produced
by Affymetrix, have become widely used by the scientific community to study genome
wide gene expression. More recently, Agilent and Amersham Biosciences
commercialized each a new microarray platform, also based on oligonucleotides. Both
technologies give high quality, good resolution and advanced gene information. Because
these technologies are, or will become, standards in life science research, it is important
to provide the scientific community with a privileged access to these technologies.
Affymetrix: Leveraging technologies adapted from the semiconductor industry,
the manufacture of GeneChip arrays use photolithography and solid-phase chemistry to
produce arrays containing hundreds of thousands of oligonucleotide probes packed at
extremely high densities. The probes are designed to maximize sensitivity, specificity,
and reproducibility, allowing consistent discrimination between specific and background
signals, and between closely related target sequences. Affymetrix also provides the
researcher access to array content information, including probe sequences and gene
annotations via the NetAffx™ Analysis Center. This center enables researchers to
correlate their GeneChip® array results with array design and annotation information
(www.affymetrix.com).
25

Agilent technologies print high-quality oligonucleotide microarrays using
SurePrint technology. These oligonucleotide microarrays are manufactured using
Agilent’s non-contact in situ synthesis process of printing 60-mere probes, base-by-base.
Up to 22,000 oligonucleotides per microarray currently can be synthesized. Each
microarray is uniquely bar-coded and data about the microarray and gene identification is
stored in an accompanying (GEML) microarray layout. GEML, or Gene Expression
Mark-up Language, is an XML-based open standard format that preserves expression
profile information consistently even when used under different database schemes,
allowing researchers to compare new data with existing data from other microarray
platforms (www.agilent.com).
CodeLink™ Bioarray Platform offers a high-precision microarray solution in a
high-density format. Presynthesized and functionally validated 30 mere-oligonucleotide
probes are piezo-electrically deposited onto a proprietary 3-D aqueous gel matrix.
Attachment is accomplished through covalent interaction between the amine-modified
group present on the 5' end of the oligonucleotide and the activated functional group
present in the gel matrix. The 3-D gel matrix provides an aqueous environment, allowing
for maximal interaction between probe and target. The 3-D platform achieves sensitivity
down to approximately one transcript per cell, and a minimum detectable fold change as
low as 1.3-fold with 95% confidence (www.amershambiosciences.com).
Different types of microarrays - Oligonucleotide microarrays are one of the
commonly used microarrays, finding wide use in a variety of applications, including gene
expression profiling and genotyping. Oligonucleotides are single stranded 15- to 70-
26

nucleotide molecules made by chemical synthesis, and these synthetic targets produce
high specificity and good signal strength in hybridization reactions. More than one
quarter of all microarray publications to date use oligonucleotide are the target molecules
[8]. Complimentary DNA and oligonucleotide microarrays both exploit the chemical
process of hybridization to generate microarray signals. Oligonucleotide and cDNA
microarrays fall into a broader category known as nucleic acid microarrays (Fig 3.3),
which encompasses microarrays containing any type of DNA or RNA as the target
material.
Figure 3.3 – Distribution of Types of Microarrays
Differences between different types of microarrays - The different types of
microarrays each have their own peculiarities and no one has published any sort of study
rigorously comparing the different technologies. However there are some inherent
strengths and weaknesses to each technology.
• The Affymetrix chemistry is great, but their technology is very expensive and
fairly inflexible. The basic Fluidics station and scanner are over $100,000 and
27

then each Gene Chip is around $5,000. The reason that it is inflexible is that if
you do not like their arrays and wants to make your own the cost for a new
photolithographic mask can be over a million dollars. That said, the technology is
very robust and Affymetrix's chemistry is reproducible and allows the detection of
SNPs and other small features in the DNA.
One thing to note is that Affymetrix is not currently doing co-hybridizations,
which make it very important, and challenging to normalize between the
experimental and control gene chips. That is to say that Affymetrix does not
produce ratios; each probe produces only an absolute intensity.
• Spotting DNA on glass microscope slides is relatively inexpensive and very
flexible. However the spotting process itself is inherently variable. Also most
microarrays produced in this manner use cDNAs as their probes. Using cDNAs
has a couple of technical problems.
1. You need a copy of that DNA to start with so you can use PCR to produce
your probes. This is a major point as we know the sequence of whole
genomes but we do not have unique cDNA libraries that span genomes
2. When using a cDNA as a probe you get a very long sequence to bind to
which makes it impossible to discern between genes that are more than
80% similar, and forget about detecting SNPs
It is possible to spot oligos on glass slides and save yourself a lot of PCR and
avoid the above limitations.
28

The technique that allows the spotting technology to sidestep the issue of
variability in spotting and other concerns is the use of co-hybridizations. This technique
is covered in greater detail later in this document but the main concept is to use relative
RNA expression levels instead of absolute expression levels. To accomplish this two
separate RNA samples are used: an "experimental" and a "reference". Each RNA is
labeled with a different fluorescent dye, and then the two samples are mixed and
hybridized at the same time to the microarray. When the microarray is scanned, number
of photons in the experimental dye's spectrum is compared to the number of photons in
the reference dye's spectrum. Many variations in spot size, probe concentration and other
issues are cancelled out in this manner.
3.2 EXPERIMENTAL DESIGN
Microarray analysis differs from traditional research in a number of striking ways,
one of which is the relationship between the amount of experimental time required and
the amount of data obtained. Traditional experimental approaches based on gels and
filters blots required a relatively large amount of experimental time (Fig 3.4) to obtain a
small volume of data, whereas microarray analysis affords vast quantities of data with
relatively little experiment time. Microarrays purchased commercially provide an
extreme example, allowing a single researcher to generate millions of datum points (Fig
3.4) in a few weeks. This paradigm shift and upside down relationship between
experimentation and data output places tremendous importance on sound experimental
design in microarray analysis. Properly designed experiments that include the right
experimental components and controls enable researchers to avoid the data avalanche that
can quickly bury the uninitiated.
29

Every microarray experiment should contain a positive control, a negative control,
and an experiment component. A positive control is a microarray element or substrate
that provides a readable signal, irrespective of the results obtained from the experimental
component of the assay. Readable results from the positive controls greatly improve the
capacity to evaluate the experimental data, particularly if negative results are obtained
from the experimental components
Microarray Analysis Traditional Research
DataData
Experimental time Experimental
time
Figure 3.4 – Experimental Approach for Microarray Analysis
Intense signals from the positive controls exclude trivial explanations for a failed
experiment, such as defect in hybridization, washing, scanning, or data analysis. No
formal conclusions can be drawn from a negative result in a microarray experiment
unless the positive controls produce readable signals.
A negative control is a microarray element or substrate that provides little or no
readable signal, irrespective of the results obtained from the experimental component of
the assay. Negative results add confidence to the experimental data by excluding or
reducing the possibility that a nonspecific biochemical event (e.g.: - cross-hybridization)
is producing the signals at the experimental locations. It is risky to draw conclusions
concerning the experimental components of a microarray assay if the assay does not
30

include one or more negative controls.
The experimental component of a microarray assay corresponds to the new
information that is sought in a given experiment. The experimental data contain
information regarding gene expression patterns, genotypes, and other biological
processes or pathways. Microarray assays that contain positive controls, negative
controls, and an experimental component yield reliable experimental data that can be
quantified, mined, and modeled using an increasing powerful collection of software tools.
Experimental design can be simplified by understanding the five basic steps in the
microarray analysis cycle: a biological question, sample preparation, a biochemical
reaction, detection and data analysis and modeling [8] (Fig
3.5).
31

1
Biological Question
Figure 3.5 - Five Steps of the Microarray Anal
Microarrays are used in the following manner - The basic pr
1. Isolate the RNA you are interested in and the RNA from
can come from any cells. It is important to realize tho
tissues or any heterogeneous cells may lead to results t
How do patterns of gene expression compare in root and leaf tissue?
2
Data Analysis & modeling
Quantitate data, calculate ratios, cluster
Detection
Select channels, laser settings, produce images
Biochemica
Hybridizationprocessing, bwashing
Microarray Analysis
“Lifecycle”
Sample Preparation
5
4
32
mRNA isolation, probe labeling, PCR,microarray manufacture
ysis Cycle
otocol [16] is as follows:
your control. The RNA
ugh that the RNA from
hat reflect changes in the
l Reaction
, substrate locking,
3

composition of the sample rather than in changes due to the experimental
hypothesis.
2. Label the RNA. Usually this means performing a reverse transcriptase reaction
and incorporating dye that has been linked to a DNA nucleotide. However some
protocols, i.e. Affymetrix's, call for an amplification of the RNA and labeling of
the RNA itself. For microarrays on nylon membranes usually the label is
radioactive.
Figure 3.6 – Hybridizing the Two Samples
3. Hybridize the labeled target to the microarray (Fig 3.6). This consists of placing a
solution containing the labeled target on the microarray and letting it sit for a
period of hours. This allows a given target to find its probe on the microarray and
bind to it. Usually this is carried out a specific temperature to minimize non-
specific binding of target to the probes on the microarray.
4. Remove the hybridization solution and wash the microarray. The washing can be
done at different salt and detergent concentrations to minimize non-specific
binding. In general solutions with lower salt concentrations weaken the DNA base
33

paring and are referred to as "more stringent" and vice versa for higher salt
concentrations.
5. Once the microarray has been washed it is time to scan the microarray. Scanning
is just quantitizing how much target bound to the DNA probe on the microarray.
Most microarrays use fluorescent dyes and are scanned in the following manner
(Fig 3.7):
Figure 3.7 – Scanning the Microarray
1. laser is used to excite the fluorescent dye; the photons coming from the
dye are captured using lenses to focus the light and a photo multiplier tube
(PMT) to quantitative how many photons are being captured.
2. The resulting number for that section of the microarray is translated into
one pixel of a 16-bit .tiff file. The more pixels per centimeter, the better
the resolution of the resulting .tiff image. It is important to note that .tiff
files are uncompressed and file formats like .jpeg and .gif which
compresses data should not be used for storage of results.
34

3. The resulting image is analyzed by finding the spots and comparing the
differences between chips (if the hybridization contained only one fluor)
or the ratio of the two fluors for co hybridization experiments. How these
differences are normalized, compared and interpreted is beyond the scope
of this document.
3.3 APPLICATION OF MICROARRAYS
Two trends in microarray research are the diversification of the assays and the
worldwide spread of the technology. Gene expression applications account for 81% of
the scientific publications to date [8], but microarrays are being used for many other
purposes, including genotyping, tissue analysis, and protein studies. Microarray assays
for genetic and infectious diseases may improve health care by providing rapid and
affordable genotyping data for treatable and curable illnesses.
3.4 AVAILABLE MICROARRAY TOOLS
Currently there are numerous tools available for microarray analysis, but there are
very few available for the design of microarrays. What follows is a list of few of the
available tools and their properties.
Microarray DAta Management (MADAM) - MADAM [17] is software available
from The Institute of Genomic Research (TIGR). It guides users through the microarray
process from RNA procurement to data analysis, offering intelligent forms to simplify the
tracking of experimental parameters and results that are essential for the interpretation of
expression results in downstream analyses. MADAM is platform independent and has
been tested on Microsoft Windows, Linux, Unix, and Mac OS X successfully. Additional
35

tools – TIGR Spotfinder, MIDAS, MeV are made available along with MADAM, thus
making a suite of software – TM4 (S. Dudoit, R.C. Gentleman and J. Quackenbush).
MIDAS provides for normalization of data and MeV provides for analysis of normalized
data whereas Spotfinder helps in image analysis.
TM4 comes close to your requirements but fails in that it only helps in tracking
the experimental parameters rather than help provide experimental parameters, thus
failing to design the chip. TM4 does not provide for designing chips, does not allow for
selection of genes that go on the chip and does not either allow for other design
parameters. However it is capable of tracking them.
BioArray Software Environment (BASE) - BASE is a comprehensive free web
based database solution for the massive amounts if data generated by microarray analysis.
It was developed at the Department of Theoretical Physics, Lund University [18]. It has
been designed entirely using free software – Linux OS, MYSQL database, Apache web
server, Java/C++/PHP languages. It manages bio material information, raw data and
images and provides integrated and “plug –in”-able normalization, data viewing and
analysis tools. BASE can be installed on a local server, which can be accessed via any
web browser using personal logins with administered access levels. It allows for data to
be visualized using various plots, histograms and tables.
Again, BASE deals entirely with the analysis aspect and does well but does not
provide any for design aspect.
Stanford Microarray Database - SMD [19] is a research tool for hundred of
Stanford researchers and their collaborators. It provides for unrestricted access to
microarray data published by SMD users. It has the ability to store, retrieve, display and
36

analyze the complete raw data produced by various microarray platforms and image
analysis software packages. Softwares have been implemented to increase the ease with
which data from SMD can be published adhering to accepted standards and as well
increase the accessibility of published microarray data to the general public.
Longhorn Array Database (LAD – Patrick J Killion, Gavin Sherlock, Vishwanth
R Iyer) - The Longhorn Array Database (LAD) [20]is a MIAME [21] compliant
microarray database that operates on PostgreSQL and Linux. It is a fully open source
version of the Stanford Microarray Database (SMD), one of the largest microarray
databases. LAD provides a simple, free, open, reliable and proven solution for storage
and analysis of two-color microarray data. LAD stores raw and normalized data from
microarray experiments, as well as their corresponding image files. In addition, LAD
provides interfaces for data retrieval, analysis, and visualization.
SMD and LAD both provide for features that are helpful for data analysis. None
of the above databases provide for both – the design and analysis of microarrays. Having
both the aspects in one place helps researchers with effective study of microarrays.
37

4.MIDAR – DESIGN ASPECTS
Personal computers have taken up residence on the desks of virtually every
scientist in the world, and larger workstations are common equipment in most research
departments. This chapter explores the myriad different faces of MIDAR as a tool for
electronic resource and biological databases, sequence and design informatics, data
quantization, mining and modeling.
Electronic Resources - The linking of microarray scientists worldwide via the
internet is speeding technological advance like never before. The internet, which supports
the World Wide Web (WWW) and electronic mail (e-mail), allows scientists to share
microarray protocols and data, obtain commercial products, and send electronic messages
quickly and economically. A host of electronic support services, including an electronic
library of microarray citations and Pub Med, are provided in MIDAR to assist scientists
in keeping pace with the rapidly expanding scientific literature.
Biological Databases - MIDAR allows microarray scientists to access biological
databases with extensive content on genes and genomes. These databases are useful in
selecting sequences for microarray manufacture and in interpreting the results of
microarray experiments. This section describes the backend of MIDAR, various available
genomic databases and how they are being implemented.
38

Microarray scientists commonly use three types of nucleotide sequence databases.
Many laboratories, companies and universities have small in-house databases of select
content, which is available on a limited basis. These sequence databases are useful for
focused projects, but tend to be somewhat limited in terms of the amount of content.
Several large biotechnology companies, including Celera Genomics (Rockville, MD) and
Incyte Genomics (Palo Alto, CA), possess large private databases of genomic and
expressed sequence tags (EST) information, providing data on a paid basis. The third
source of sequence information for microarray experimentation is found in GenBank
(www.ncbi.nlm.nih.gov), a public sequence database maintained by NCBI. For MIDAR,
we have implemented the first and last type of nucleotide sequence databases – small in-
house databases and public sequence databases. Small in-house database is incorporated
to provide for individual laboratory purposes. Scientists in laboratories can upload those
genes in their area of study as custom gene groups and use them for chip design. After all
MIDAR is for custom chips, chips with a subset of genes (the ones they are interested for
their research). Public databases cater to the general needs of the scientists. We have
incorporated GO (Gene Ontology) database, since GO database is a standard database
recognized by most laboratories.
Genes are well organized as groups based on biological process, molecular
function or cellular component, allowing user to choose genes based on their
characteristics. This feature is very helpful in microarray design because microarrays are
used to study genes that share some kind of common characteristics. The use of GO terms
by several collaborating databases facilitates uniform queries across them. The controlled
vocabularies are structured so that you can query them at different levels: for example,
39

you can use GO to find all the gene products in the mouse genome that are involved in
signal transduction, or you can zoom in on all the receptor tyrosine kinases. This structure
also allows annotators to assign properties to gene products at different levels, depending
on how much is known about a gene product.
One of the future directions for MIDAR is to involve KEGG [22] pathways for
gene selection. KEGG pathways are becoming increasingly popular and will provide
readily with pathways that most researchers would be interested in. KEGG pathways
database is a collection of graphical diagrams (KEGG pathway maps) representing
molecular interaction networks in various cellular processes. Each reference pathway is
manually drawn and updated regularly. Organism-specific pathways are computationally
generated based on the KEGG Orthology (KO) assignment in individual genomes.
If these databases provide for gene information, we still need to store the chip
information, primer and oligonucleotide sequences and other information concerning chip
design. So we have added few tables to one of the database to provide for custom chip
information.
Database design - For every chip that is being designed we will have to store a
unique id, description, designerId, genes involved, their concentration levels, number of
replicates and the number of columns and rows. We will also have to store the x, y
coordinates of individual gene on the chip so that we could replicate later when the chip
is re opened. All this cannot be stored in one table as it would lead to large amounts of
redundant data. After applying normalization techniques, we were able to successfully
place the attributes in two different tables with no redundant data. There was a similar
problem with storing the primer/oligo information of genes. Since there could be more
40

than one primer/oligo sequence for a particular gene, storing them along with other gene
attributes would lead to redundant data. Similarly applying the normalization techniques
we were able to rightly group the attributes.
As the work progressed, there were a few more needs and new tables had to come
over. These new requirements also added of new attributes to the existing tables. Though
this would take the redesigning of the entire database, we had to go for it. Finally after all
the changes, designing and redesigning, the database design follows in the next section
(Figure 4.1).
Designing the front-end - After designing the database, it was time to design a
front-end based on these databases. Basically front-end had to cater to two requirements –
populating the databases and providing for design of custom chips.
Populating the databases - The GO database is available as an SQL dump at
http://www.godatabase.org/dev/database/. It can be downloaded anytime and the database
can be constructed. That leaves us to the small in-house database. There are two things
that needed to be uploaded – the gene attributes and then the primer/oligo information. In
order to upload the gene information, we designed forms that would help upload data, the
only requirement being that the data be available in a tab-delimited format and follows
the specified data header. Different laboratories follow varied conventions in representing
gene data, so uploading data would be near impossible if there is no common standard.
For this purpose, a header was developed which defines gene attributes and the order they
need to be present. This approach helps uploading data from varied laboratories. The data
header is explained in figure 4.2.
Users could upload primer/oligo information in the same file or use Mprime [23]
41

Concentration ConcentrationId Value Units
Gene GeneId Description GBAcc GBID GeneCode Seq molType GeneGroup GeneSubgroup GeneSubunit CuratedCitations Diseases
Primers PrimerId GeneId LeftPrimerSeq RightPrimerSeq ProductSeq LeftPrimerLen RightPrimerLen RightPrimerBeg RightPrimerEnd LeftPrimerBeg LeftPrimerEnd LeftPrimerTM RightPrimerTM LeftPrimerGC RightPrimerGC Methods
Oligos OligoId GeneId OligoSeq OligoLen OligoBeg OligoEnd OligoTM OligoGC Methods
Person PersonId LastName FirstName Address1 Address2 City State
ChipGenes ChipId GeneId Primer_OligoId ConcentrationIdXCoord YCoord
Chip ChipId ChipDescriptionChipPIID ChipDesignerId ChipType NumSpots NumCols GeneGroup NumReplicates
Figure 4.1- Database Design
to generate primer/oligo information. MPrime is an external program, which is designed
by Dr Rouchka and his team to generate primers and oligos given a gene sequence.
Primer and oligo sequences are required for microarray design. They are the ones that are
actually placed on the chip and not the whole gene. So finding primer and oligo
sequences are very much needed in MIDAR. MPrime was readily available and provided
42

for primer and oligo sequence generation. And our team developed it so incorporating it
in my project was not a hassle. More over reusing code is the order of the day and is
advised in object-oriented concepts.
Data Header
GeneCode
GBAcc
Description
Oligo Sequence/s
Left Primer Sequence
Right Primer Sequence
Figure 4.2 – Data Header for Uploading Gene Information
MPrime was developed in C/C++ and MIDAR is being developed in VB.Net.
Both the programming languages are from two different programming paradigms – one
being the object oriented and the other being event driven programming. After getting
excited about the reusability and availability, this was little dampening. Moreover
VB.Net is not one of the best languages to implement searching and sequencing tens and
thousands long strings. However after some research on the internet, I found more than
one way to incorporate C++ code into my application.
Incorporation of C++ code into VB.NET - One way of doing this is making a
DLL (Dynamic Link Library) of the C++ code, which could later be used, by making an
instance of it in VB.Net code. There were few additions I had to make in the code. They
are as follows. Generally any C++ program would contain two files - .cpp file and .h file.
43

The .cpp file contains the implementation of the program while .h contains the
declarations of the member variables and functions. In order to incorporate C++ program
into VB.net, you have to define one other file - .Def file. This would act as an interface
and would expose all the member functions to objects defined in VB.Net code.
A sample .Def file would like the following
LIBRARY [DLL NAME]
EXPORTS
[FUNCTION NAME]
One could export more than one function. By using the DLL along with these
function names one could make a call to them and use them as and where required. The
.h files needs a little addition too. We have to add _stdcall keyword to the function
declaration after the return type and before the function name. This is needed to ensure
that function uses the same way as VB for passing variables about and working with the
stack. A sample header file would look like the following
int _stdcall function1 { return 1; } The above code is compiled as a DLL and now this program is available for external use,
it can be a VB.Net or VC++ or anything else for that matter.
The above DLL can be used in VB.Net by making an API call. A sample API call
would look like the following (Figure 4.3). Once the API call has been created, the
function can be called normally as you would call any other function.
Private Declare Sub Sleep Lib "Kernel32.dll" (By Val dwMilliseconds As Long)
44

“Private” We know that this method can only be used in this code block. It is
irrelevant as far as the declaration goes, this is just for VB’s sake.
“Declare” This means that this is only a method header and not the entire
method.
“Sub” Again we know this means it is a subroutine and does not return a
value.
“Sleep” This is the name of the method. It does not have to be the same as the
method name in the DLL however if it differs then an “Alias” clause
must also be added to the declaration, for simplicities sake though
it’s best to just use the same name.
“Lib” This means we are going to give VB the library name (DLL file.)
‘ “Kernel32.dll” ’ The name of the DLL file that contains the method.
“(“ We know this indicates that we are going to declare some parameters
(If any) that need to be passed to this method.
“ByVal” This means we are passing the value of the variable passed to the
method, not the address of its value in memory. This is important to
get right in the function declarations since it can cause big problems
later on.
“dwMilliseconds” This is the name of the parameter, this is just for VBs sake, it does
not have to be the same as it is declared in the DLL but it makes
sense to do so, no alias needs setting if it is not the same as in the
DLL.
“As Long” This means we are setting the type and thus how much memory to
send over to the DLL when we pass a value to it, again this is very
important to get these matched up with the DLL correctly.
“)” Finally we know this means we are done with parameters and as
there is no return value since we are dealing with a subroutine
declaration.
Figure 4.3 – Explanation of Code
45

Another way of doing the same is make an executable of the C++ program and
calling the executable from VB.Net code. This would not give as much flexibility as the
above method would give. The above method would give access to all member variables
and functions and the program would be treated as “in-process” process. However for
MIDAR there is no much need of flexibility. So I have adapted to the second owing to its
simplicity and “fitting the bill” feature without much work. The second method would
require an object of System.diaognostics.process to make a call to exe. The gene
sequences would be written to a file, which would serve as input. Similarly the output
will be written to file from where one could upload the primer/oligo sequence to the
database. This completes the processes in regards to uploading gene information and
primer/oligo sequences. Now lets look into designing the custom chip.
An input form is provided for the user to make his/her choice as to the set of
genes required, the concentration levels, the number of replicates and few other
parameters required for chip design. MIDAR is a huge gene database and to extract the
needed information would require some complex queries, which retrieve data efficiently.
The GO database contains vast amounts of gene information over several tables. An
example query that list all the genes exhibiting a common biological process
select gene_product.symbol from term as rchild,term as ancestor, graph_path,association,gene_product " _
& "where graph_path.term2_id = rchild.id and " _
& "graph_path.term1_id = ancestor.id and " _
& "association.term_id = rchild.id and " _
& "association.gene_product_id = gene_product.id and " _
& "ancestor.name = '" & strFunction & "' order by symbol"
Where strFunction is some chosen term definition such as …
46

The query involves multiple joins and a self-join of the term table. Initially a self-
join of term table is made and is then joined with graph_path table to obtain parent-child
relationship of the terms. The resultant join is then joined with association table to obtain
the associated genes pertaining to a term.
As the project progressed, the list of genes had to be refined more. The above
query will list all genes pertaining to a term, all genes belonging to all genomes. What
people are really interested in is the list of genes pertaining to a genome rather than all
genomes. This would result in few more joins to the above query thus slowing it down to
a great extent. To avoid this situation what we have done is split the process into two
queries. The first query will list only terms associated with a specific genome. The
second query would list genes pertaining to the particular term. The choices we had were
one big query involving large number of joins, or two descent queries with manageable
number of joins. We have made the choice of the second so that the response time
remains constant, predictable and better compared to one big query.
A search text box has been provided for users to search for terms of specific
interest. The search is dynamic and refreshes with every character you specify and gets
you closer to your desired term search.
Layout of selected genes on the custom array - Serial and Random - In serial
arrangement all the genes along with their replicates are placed sequentially one after the
other in the order they were selected. In a random layout, the genes and their replicates
are placed randomly. That is to say a gene along with its replicates can be present
anywhere on the chip. The choice of either of the layouts can be made based on user
requirements. If the user desires that the genes can be placed anywhere irrespective of
47

their location on the chip, then they would go for the serial arrangement. It has to be
understood that gene spots located on the corners may not be expressed, as they should be
owing to its corner location. So if a gene and its replicates are all placed together one
corner and they are not expressed, there could be two possibilities – one they are not
actually expressed and the other though they are expressed they are dimmed because of
their corner location. So the choice rests with the user as to a serial layout or a random
layout.
As discussed earlier a typical custom array would contain tens of thousands of
genes and potentially upwards of hundred thousand spots on a single microarray. MIDAR
is being developed in VB.NET and the type of control to be chosen to represent a spot on
the array was a tough decision to be made. The spots needed to have a click event so that
a mouse click on a spot would allow the user to view the included oligo/primer detail
information and list specific data details for each spot. So the initial decision was to have
a button, it had a click event and it had a background property using, which the intensity
value could be represented. It had a tag property using which a mouse over event would
quickly show brief information – gene name and concentration levels. The choice seemed
to be ideal, until we built chips with large number of spots, a typical number being five
thousands. We needed chips that should provide for at least ten thousand spots. Hence the
decision was reverted and we chose graphics to do the part.
Each spot on the array was a circle, which is being drawn using the graphic pen. It
could generate ten thousand spots in no time and performed the same for even larger
number of spots. So it seemed to be a good choice. But then it does not have a click
event, it is just a drawing on the form. There is no background or tags associated with it.
48

It did provide for scalability but lacked the necessary features. In order to add this
functionality, I implemented arrays to store all the information and associated the array
index with the position of the drawing on the chip. The position was in turn tied to the
pixels of the spot. Thus I could relate a mouse click event of the form to the array by
obtaining the pixels of the clicked area and transforming into location and thus index of
the array. Initially the transformations took some time to understand but once I realized
the logic, it was easy to incorporate in my code. In all it was a good decision to choose a
graphic circle as it provided for everything though it was tedious initially. A custom array
contains tens of thousands of genes. One would want to readily refer to a particular gene
or a gene group. One would want to know where the gene is distributed along with its
replicates on the chip, what are the intensity values, what are the corresponding
oligonucleotide sequence and so on. Well we have provided for this feature. One could
specify the gene or gene group of specific interest and a color to mark these genes and
could readily see them marked on the array. Users could find all the information they
need by simply clicking on the marked ones.
Experiment data obtained from a microarray contain intensity values
corresponding to each spot on the custom array. These values vary a great deal. The
range at times could be several thousands. To represent such a wide range of intensity
values on our software all we have is 255 colors. So data needs to be normalized, but then
again the range being so varied, the normalized values obtained are not distributed
uniformly. So a sorting approach was chosen. Using this approach all the intensities are
ranked according to their values. And then these values are normalized to 255 colors.
These represent a close resemblance to a real expressed custom array.
49

Microarray analysis generally involves conducting experiments on the same chip
at different time frames and different time conditions. One needs to study all this data to
understand as to what genes are expressed under what conditions. We have currently
provided for comparison of custom arrays under different time frames. Users could
associate one set of experiment values to the chip with one color (green). And then
associate another set of experiment values with a different color (red). We could then run
a logarithmic ratio of both the experiment values and associate with the chip. The spots
which are close to either green or red would be the ones interested ones as they were
either expressed or under expressed under different conditions. The ones close to yellow
were expressed under both the conditions and may not be of much interest.
50

5.MIDAR – A SCENARIO
Lets start with designing a custom microarray. The application opens up with an
input form (Fig. 5.1), wherein a user can specify his/her required inputs. After specifying
the chip description and ownership, the user could either choose genes from the standard
GO database or the custom gene groups. The genes in GO terms are grouped in terms of
biological process, cellular component and molecular function. The user could start with
specifying the genome type he/she is interested, followed by the GO term type. The user
could then scroll down the list or search for a GO term by using the search box. It is the
same with selecting custom gene groups. The user can then specify other parameters such
as number of replicates, positive and negative controls, and number of columns and rows.
Figure 5.1 – Screenshot Depicting User Input for Chip Design
51
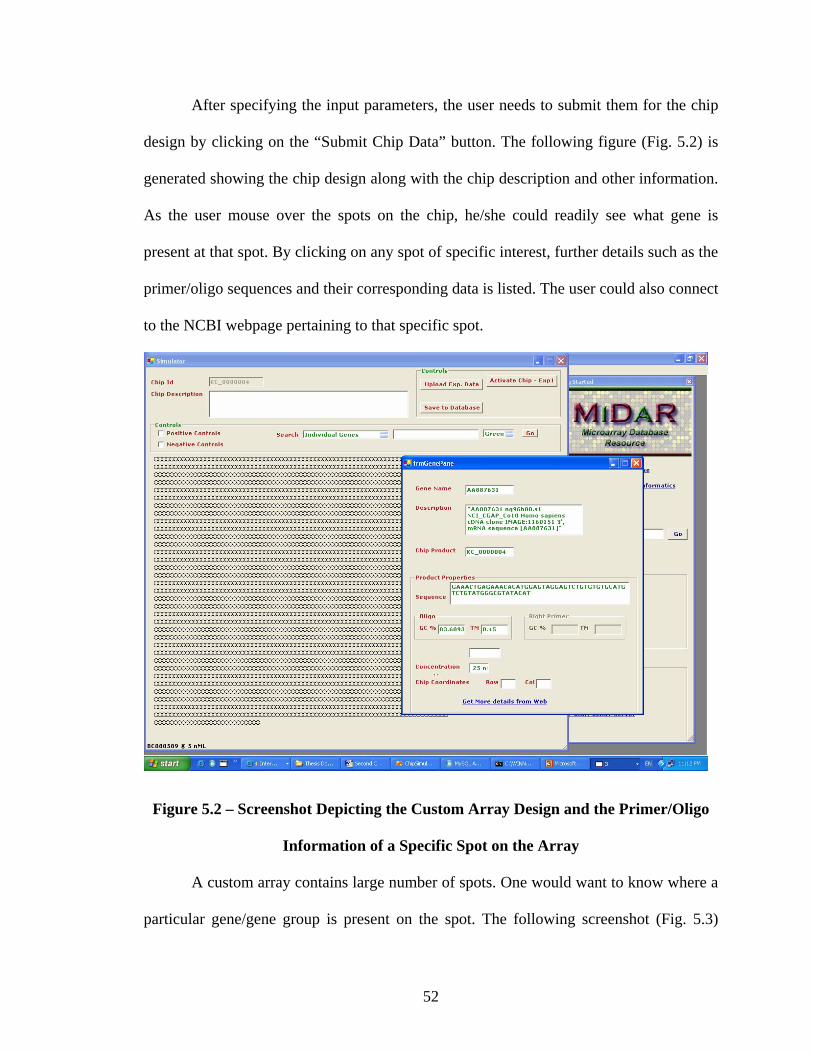
After specifying the input parameters, the user needs to submit them for the chip
design by clicking on the “Submit Chip Data” button. The following figure (Fig. 5.2) is
generated showing the chip design along with the chip description and other information.
As the user mouse over the spots on the chip, he/she could readily see what gene is
present at that spot. By clicking on any spot of specific interest, further details such as the
primer/oligo sequences and their corresponding data is listed. The user could also connect
to the NCBI webpage pertaining to that specific spot.
Figure 5.2 – Screenshot Depicting the Custom Array Design and the Primer/Oligo
Information of a Specific Spot on the Array
A custom array contains large number of spots. One would want to know where a
particular gene/gene group is present on the spot. The following screenshot (Fig. 5.3)
52

provides for the same. Users can type in the input box their specified interest of genes,
choose a color to mark them and submit the information. The application would then
mark all the spots on the array associated with the gene/ gene group.
Figure 5.3 – Screenshot Depicting Marked Spots Associated with a Gene of Interest
The following screenshot (Fig. 5.4) is the result after activating the custom array.
Users associate the custom array chip design with the experiment values obtained by
conducting the experiment on a real custom array of same design. The color of the spots
shows the intensity values obtained by scanning the custom array after the experiment
was done. The intensity depicts the expression levels of a gene and one could readily see
the genes which are over expressed or which are under expressed.
53

Figure 5.4 – Screenshot Depicting the Activated Custom Array
MIDAR currently provides for comparison of intensity values of a custom array
obtained from two different conditions (Fig. 5.5). This feature would readily identify the
genes of interest for this custom array. The ones which are more of green or more of red
indicate the ones which are either expressed in one sample or under expressed in another.
The other spots (more of yellow) are expressed in both the samples, thus are not area of
interest.
54

Figure 5.5 – Screenshot Depicting a Comparative Analysis of a Single Custom Array
from Two Different Experiments
55

6. FUTURE – MICROARRAYS AND MIDAR
Human beings are gene machines, responsive to a myriad of genetic and
environmental signals, and these signals are integrated by each person to produce
resultant phenotypes that manifest as mental and physical wellness, normal and aberrant
behavior, disease predisposition, longevity, and so forth. Medical practice has succeeded
in identifying and curing disease using a host of clinical procedures, but medicine is
lacking in the sense that many illnesses, therapies, and drugs are poorly understood at the
genomic level.
The use of microarray technology in a clinical setting holds the promise of
providing detailed molecular information to the physician, enabling more informed
decision-making and better health care. Chips could be used to profile patients for
predisposition to drug and alcohol abuse, eating disorders, depression and schizophrenia.
The biochemical and physiological changes that accompany physical exercise, poor diet,
substance abuse, antidepressant and antipsychotic medications, and many other
environmental and chemical stimuli that alter body chemistry and probably detectable by
microarray. As microarray analysis is used extensively in clinical research and genetic
screening, testing and diagnostics, it makes sense to consider whether future visits to the
doctor’s office might include one or more microarray tests as components of the physical
examination. Microarrays are of great potential and a general awareness needs to created
among young students. MIDAR can help as a model for a virtual in “silico” experiment.
56

We would need to be able to apply a virtual probe set in which most of the genes on the
chip would have matching probes assigned random values. However, we "spike" values
for our favorite genes of interest, say 20 genes or so. So, we need to develop the two tools
that would allow this in silico modeling. These would include
• a way of generating the virtual probe set and assigning random values for
all genes in the probe set
• a way of generating a sub list from the virtual probe set for our genes of
interest, and a way of re-assigning or "spiking" their values.
We see this particular use of the tool as a way to market it to biology classes throughout
the US and beyond.
MIDAR also needs to expand on the analysis part of the microarrays. Scientists
seeking to harness the potential of microarrays are often challenged by the prodigious
quantities of data produced. There is a need for support to provide for quantizing and
normalizing these huge datasets. Data obtained from microarrays reflect various
conditions (time course, drug dosage, tissue specificity) and different time frames. If they
need to compared and studied they need to be normalized so that they all stand on the
same plain. Various data visualization techniques need to be developed to better illustrate
the analysis of experiment data.
In all MIDAR is a community driven solution developed to aid the design and
testing of custom chips and an aid to the evaluation and analysis of commercial chips.
57

REFERENCES
[1] J. C. Venter, M. D. Adams, E. W. Myers et. al., "The sequence of the human genome," Science, vol. 291, no. 5507, pp. 1304-1351, Feb.2001.
[2] C. Fields, M. D. Adams, O. White, and J. C. Venter, "How many genes in the human genome?," Nat. Genet., vol. 7, no. 3, pp. 345-346, July1994.
[3] J. DeRisi, L. Penland, P. O. Brown, M. L. Bittner, P. S. Meltzer, M. Ray, Y. Chen, Y. A. Su, and J. M. Trent, "Use of a cDNA microarray to analyse gene expression patterns in human cancer," Nat. Genet., vol. 14, no. 4, pp. 457-460, Dec.1996.
[4] E. S. Lander, L. M. Linton, B. Birren, C. Nusbaum et. al.,"Initial sequencing and analysis of the human genome," Nature, vol. 409, no. 6822, pp. 860-921, Feb.2001.
[5] "From Blueprint to you," NCBI,2005.
[6] W. Gilbert, "Why genes in pieces?," Nature, vol. 271, no. 5645, p. 501, Feb.1978.
[7] J. D. Watson and F. H. Crick, "A structure for deoxyribose nucleic acid. 1953," Nature, vol. 421, no. 6921, pp. 397-398, Jan.2003.
[8] F. Crick, "Central dogma of molecular biology," Nature, vol. 227, no. 5258, pp. 561-563, Aug.1970.
[9] S. Brenner, "The human genome: the nature of the enterprise," Ciba Found. Symp., vol. 149, pp. 6-12, 1990.
[10] M. D. Adams, J. M. Kelley, J. D. Gocayne, M. Dubnick, M. H. Polymeropoulos, H. Xiao, C. R. Merril, A. Wu, B. Olde, R. F. Moreno, and ., "Complementary DNA sequencing: expressed sequence tags and human genome project," Science, vol. 252, no. 5013, pp. 1651-1656, June1991.
[11] M. S. Boguski, T. M. Lowe, and C. M. Tolstoshev, "dbEST--database for "expressed sequence tags"," Nat. Genet., vol. 4, no. 4, pp. 332-333, Aug.1993.
[12] E. D. Green and P. Green, "Sequence-tagged site (STS) content mapping of human chromosomes: theoretical considerations and early experiences," PCR Methods Appl., vol. 1, no. 2, pp. 77-90, Nov.1991.
58

[13] M. Schena, Microarray Analysis John Wiley & Sons, 2005.
[14] M. Schena, D. Shalon, R. W. Davis, and P. O. Brown, "Quantitative monitoring of gene expression patterns with a complementary DNA microarray," Science, vol. 270, no. 5235, pp. 467-470, Oct.1995.
[15] M. S. Boguski, C. M. Tolstoshev, and D. E. Bassett, Jr., "Gene discovery in dbEST," Science, vol. 265, no. 5181, pp. 1993-1994, Sept.1994.
[16] D. J. Lockhart, H. Dong, M. C. Byrne, M. T. Follettie, M. V. Gallo, M. S. Chee, M. Mittmann, C. Wang, M. Kobayashi, H. Horton, and E. L. Brown, "Expression monitoring by hybridization to high-density oligonucleotide arrays," Nat. Biotechnol., vol. 14, no. 13, pp. 1675-1680, Dec.1996.
[17] L. Zhang, W. Zhou, V. E. Velculescu, S. E. Kern, R. H. Hruban, S. R. Hamilton, B. Vogelstein, and K. W. Kinzler, "Gene expression profiles in normal and cancer cells," Science, vol. 276, no. 5316, pp. 1268-1272, May1997.
[18] L. H. Saal, C. Troein, J. Vallon-Christersson, S. Gruvberger, A. Borg, and C. Peterson, "BioArray Software Environment (BASE): a platform for comprehensive management and analysis of microarray data," Genome Biol., vol. 3, no. 8, p. SOFTWARE0003, July2002.
[19] C. A. Ball, I. A. Awad, J. Demeter, J. Gollub, J. M. Hebert, T. Hernandez-Boussard, H. Jin, J. C. Matese, M. Nitzberg, F. Wymore, Z. K. Zachariah, P. O. Brown, and G. Sherlock, "The Stanford Microarray Database accommodates additional microarray platforms and data formats," Nucleic Acids Res., vol. 33, no. Database issue, p. D580-D582, Jan.2005.
[20] P. J. Killion, G. Sherlock, and V. R. Iyer, "The Longhorn Array Database (LAD): an open-source, MIAME compliant implementation of the Stanford Microarray Database (SMD)," BMC. Bioinformatics., vol. 4, p. 32, Aug.2003.
[21] T. B. Knudsen and G. P. Daston, "MIAME guidelines," Reprod. Toxicol., vol. 19, no. 3, p. 263, Jan.2005.
[22] M. Kanehisa and S. Goto, "KEGG: kyoto encyclopedia of genes and genomes," Nucleic Acids Res., vol. 28, no. 1, pp. 27-30, Jan.2000.
[23] E. C. Rouchka, A. Khalyfa, and N. G. Cooper, "MPrime: efficient large scale multiple primer and oligonucleotide design for customized gene microarrays," BMC. Bioinformatics., vol. 6, p. 175, July2005.
59

CIRRICULUM VITAE
RAVI SHRIKANTH GUNDLAPALLI
Date of Birth July 20, 1981
Place of Birth Hyderabad, India
Undergraduate Study Jawaharlal Nehru Technological University B.Tech. Computer Science and Information Technology 1999-2003 Graduate Study University of Louisville M.S. Computer Engineering and Computer Science 2003-2005 Experience Intern, GE Energy - Somersworth NH
(September 2004 – August 2005) Student Assistant – University of Louisville (June 2004 – August 2004)
60