immunotherapy for antineutrophil cytoplasmic antibody–associated vasculitis: challenging the...
TRANSCRIPT

Review
Immunotherapy for antineutrophilcytoplasmic antibody–associatedvasculitis: challenging the therapeuticstatus quo?Xavier Bosch1*, Antonio Guilabert2*, Gerard Espinosa3 and Eduard Mirapeix4
1 Department of Internal Medicine, Hospital Clınic, Institut d’Investigacions Biomediques August Pi i Sunyer (IDIBAPS), University
of Barcelona, Barcelona, Spain2 Department of Dermatology, Hospital Clınic, Institut d’Investigacions Biomediques August Pi i Sunyer (IDIBAPS), University of
Barcelona, Barcelona, Spain3 Department of Autoimmune Diseases, Hospital Clınic, Institut d’Investigacions Biomediques August Pi i Sunyer (IDIBAPS),
University of Barcelona, Barcelona, Spain4 Department of Nephrology, Hospital Clınic, Institut d’Investigacions Biomediques August Pi i Sunyer (IDIBAPS), University of
Barcelona, Barcelona, Spain
Anti-neutrophil cytoplasmic antibodies (ANCA) are IgGantibodies directed against antigenic constituents ofneutrophils contained in the azurophilic granules andmonocyte lysosomes. Systemic vasculitis with ANCA[ANCA-associated vasculitides (AAV)] is a subgroup oflife-threatening inflammatory disorders affecting small-to medium-sized vessels; immunosuppressants and glu-cocorticoids represent the current therapeutic mainstay.Although these agents have improved patients’ survival,25% present with severe adverse events, and standardtherapy does not sustain remission. Therefore, an unmetneed for safer and more effective therapies hasprompted interest in biological agents. Continuousadvances in the knowledge of AAV pathogenesis arepaving the way to new biologicals that are now awaitingtesting.
Anti-neutrophil cytoplasmic antibody–associatedvasculitis: Wegener’s granulomatosis, Churg-Strausssyndrome and microscopic polyangiitisAnti-neutrophil cytoplasmic antibodies (ANCA) are IgGantibodies directed against antigenic constituents of neu-trophils contained in the azurophilic granules andmonocytelysosomes. After indirect immunofluorescence of ethanol-fixed neutrophils, two characteristic immunostainingpatterns can be observed: cytoplasmic (C-ANCA) and peri-nuclear (P-ANCA) (Figures 1a and 1b, respectively). Mostcases of C- and P-ANCA are directed against proteinase 3(PR3) and myeloperoxidase (MPO), respectively.
Systemic vasculitides are uncommon life-threateningdisorders where vascular inflammation of an autoimmunenature causes occlusion and ischemia of affected tissues.Among these diseases, vasculitis with serum positivity forANCA [ANCA-associated vasculitis (AAV)] is a well-established subgroup of disorder that affects small- (e.g.
Corresponding author: Bosch, X. ([email protected]).* These authors contributed equally to this work.
280 1471-4906/$ – see front matter � 2008 El
capillaries) to medium-sized vessels (e.g. renal arteries).Wegener’s granulomatosis (WG), microscopic polyangiitis(MPA) and Churg-Strauss syndrome (CSS) are the threeforms of AAV [1]. The incidence of AAV in the developedworld ranges from 2.4 cases permillion (CSS) to 3.6 (MPA)to 8.5 (WG) [2]. AAV are more frequent among whites(�90% of cases), and the mean age of presentation is 55years [2]. The clinical manifestations of AAV are diverseand sometimes overlapping. Necrotizing and crescenticglomerulonephritis (NCGN) – a severe form of renal vas-cular disorder that produces rapidly progressive renalinsufficiency – and pulmonary capillaritis are commonfeatures of WG and MPA. In MPA, lung involvementusually manifests as a diffuse pulmonary hemorrhage,which commonly leads the patient to respiratory insuffi-ciency. In contrast to MPA, patients with WG and CSSexhibit granulomatous destructive inflammation of therespiratory tract, which in the case of CSS is rich ineosinophils. Early in the course of the disease, CSSpatients present asthma and peripheral blood eosinophi-lia and, subsequently, the lung and the heart may beinfiltrated by eosinophils, producing eosinophilic pneumo-nia and rapid-onset heart failure, respectively. [2] In CSS,vasculitic damage, which appears late in the disease, isusually less severe than in MPA and WG. Other organsthat can be affected in AAV include peripheral nerves,skeletal muscle, skin and joints. [3]
Between 80% and 90% of WG and MPA patients haveANCA, mainly directed against PR3 in the case of WG andagainst MPO in the case of MPA [4]. The few WG patientswho are ANCA negative mostly have a granulomatousdisease limited to the respiratory tract, although ANCAoften appear later [5]. ANCA are detected in �40% of CSSpatients, with a predominance of MPO-ANCA [6]. Inaddition to their unquestionable value as disease markers,ANCA are now believed to play a major role in the de-velopment of necrotising vasculitis in these disorders.However, thus far, there are no data showing that ANCA
sevier Ltd. All rights reserved. doi:10.1016/j.it.2008.03.001 Available online 24 April 2008

Figure 1. (a) Characteristic cytoplasmic anti-neutrophil cytoplasmic antibodies (C-
ANCA) and (b) perinuclear (P-ANCA) patterns of ANCA binding. Indirect
immunofluorescence of ethanol-fixed neutrophils. Original magnification, �1000
(a) and �2500 (b).
Review Trends in Immunology Vol.29 No.6
are the driving force of granuloma formation in WG andCSS.
An unmet need for treatment of AAVCurrently, immunosuppressants (e.g., cyclophosphamideor methotrexate) in combination with glucocorticoids arethe mainstay of AAV treatment [7]. Although dramaticallyimproving patient survival, they are far from perfect, with�25% of patients having treatment-related severe adverseevents (e.g. infections and malignancy) [8]. Furthermore,these drugs do not sustain remission efficaciously (there isa 50% relapse rate [9]), meaning that AAV become chronic,relapsing disorders, with accumulative, irreversible organdamage occurring because of repetitive episodes of diseaseactivity leading to intensification of toxic immunosuppres-sants. Approximately 10% of AAV patients [10] are refrac-tory for the standard immunosuppressant combinationand are at high risk of death [7]. Therefore,more efficaciousand safer alternative treatments are needed to improve theglobal outcome of AAV patients. This, together withgreater insight into AAV pathogenesis, has prompted in-terest in biological agents. A more ’pathogenesis-oriented’therapeutic approach could overcome the deficiencies ofcurrent therapies by ablating key immune pathways withless toxicity [11]. We will discuss the role of the biologicaltreatments most used in AAV, paying special attention totheir pathogenesis and the immunological rationale ofbiological agents.
Pathogenesis of ANCA-associated vasculitisGranuloma formation in WG
WG usually starts as a granulomatous disease of therespiratory tract, and in most patients, progresses tosystemic disease with PR3-ANCA–associated vasculitis[12]. This suggests that WG may start with an aberrantcell-mediated immune response to an exogenous orendogenous antigen in the respiratory tract, which resultsin granuloma formation, with humoral autoimmunity toPR3 occurring at a later stage [13]. However, granulomatain WG and their relationship with systemic vasculitis ispoorly understood. No ANCA animal model has yet repro-duced the typical granulomata of WG [14,15].
Multiple cell types are present in WG granulomas in-cluding PR3+ cell clusters (neutrophils and monocytes)surrounded by antigen-presenting cells and abundantTh1-type CD4+CD28- effector memory T cells (TEMs).Maturing B and plasma cells are also present, suggestingneoformation of lymphoid like tissue (Figures 2a and 2c)[16].
In WG, there is an alteration of T-cell differentiation,with T cells lacking the co-stimulatory molecule CD28being detected early in localized disease with furtherexpansion as WG generalizes [17]. These cells displayfeatures of TEMs with several potent effector functions[18]. Being a major source of Th1-type cytokines [princi-pally tumor necrosis factor a (TNF-a) and interferon g
(IFN-g)], CD4+CD28-T cells are thought to be the drivingforce of granuloma formation [19,20]. These cells expressb2-integrin and the Th1-type CC-chemokine receptor 5(CCR5) [21] and also have cytotoxic potential [22]. None-theless, the cause of CD4+CD28 T-cell proliferationremains unclear and might be an antigen (possiblyPR3)-driven process and/or the result of T regulatory celldysfunction in genetically susceptible individuals. In fact,Abdulahad et al. [23] have recently observed that patientswith WG in remission have an expanded number of regu-latory T cells with a defective suppressive function, whichmay be an important factor accounting not only for thedevelopment of WG but also for its relapsing nature.
It has recently been postulated that WG granulomamight not be simply a haphazard, deleterious aggregationof immune cells but could also represent a lymphoid struc-ture ultimately responsible for PR3-ANCA production. Forinstance Voswinkel et al. [24] found B lymphocyte–richfollicle-like aggregates within the granulomatous lesions ofendonasal specimens ofWG patients in the vicinity of PR3+
cells and plasma cells (Figure 2a, c). Analysis of the immu-noglobulin gene repertoire showed autoreactive B cellswith potential affinity for PR3. Chronic secretion ofTNF-a and IFN-g in the granuloma might provide a favor-able proinflammatory environment for the neogenesis ofgerminal centre-like structures, where T and B cellscooperate to break tolerance and maintain antibody pro-duction against PR3. An interesting theory of PR3-directedautoimmunity has recently been proposed. This involvescPR3, the ‘complementary peptide’ of PR3 encoded by theantisense strand of the PR3 gene [25]. If the immunesystem is exposed to cPR3, antibodies against it canappear, which in turn generate anti-idiotype antibodiesthat cross-react with PR3. Accordingly, PR3-encoding gene
281
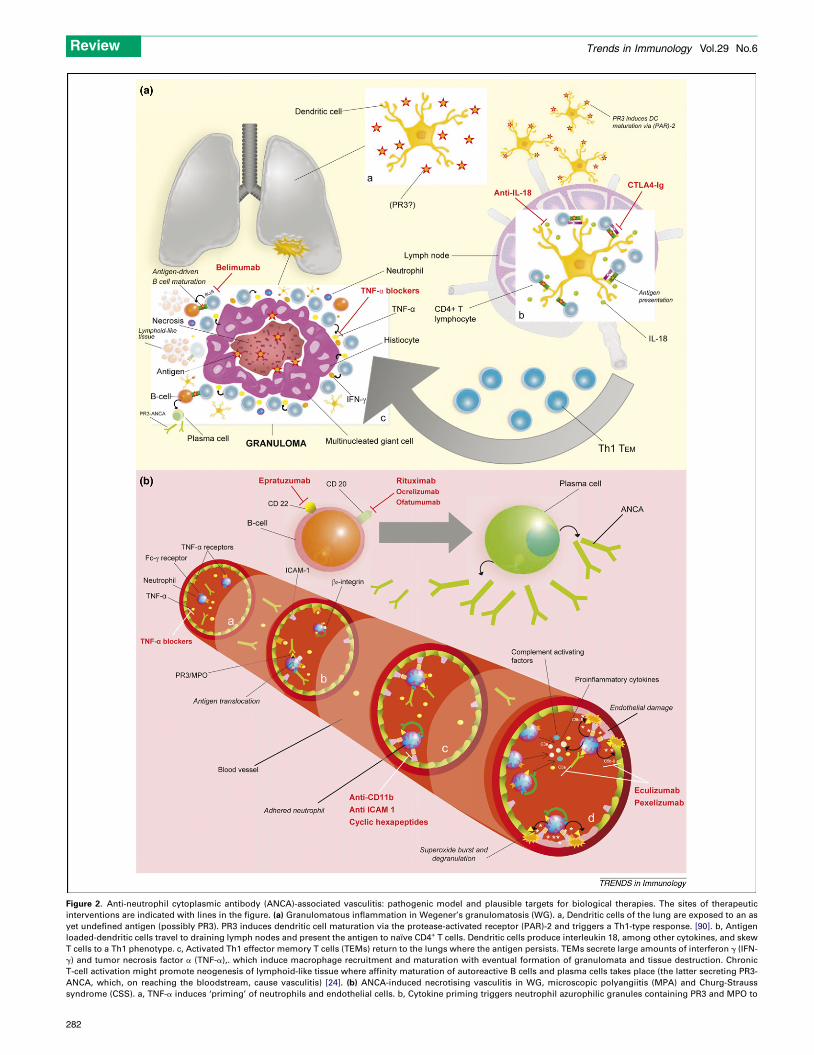
Figure 2. Anti-neutrophil cytoplasmic antibody (ANCA)-associated vasculitis: pathogenic model and plausible targets for biological therapies. The sites of therapeutic
interventions are indicated with lines in the figure. (a) Granulomatous inflammation in Wegener’s granulomatosis (WG). a, Dendritic cells of the lung are exposed to an as
yet undefined antigen (possibly PR3). PR3 induces dendritic cell maturation via the protease-activated receptor (PAR)-2 and triggers a Th1-type response. [90]. b, Antigen
loaded-dendritic cells travel to draining lymph nodes and present the antigen to naıve CD4+ T cells. Dendritic cells produce interleukin 18, among other cytokines, and skew
T cells to a Th1 phenotype. c, Activated Th1 effector memory T cells (TEMs) return to the lungs where the antigen persists. TEMs secrete large amounts of interferon g (IFN-
g) and tumor necrosis factor a (TNF-a),. which induce macrophage recruitment and maturation with eventual formation of granulomata and tissue destruction. Chronic
T-cell activation might promote neogenesis of lymphoid-like tissue where affinity maturation of autoreactive B cells and plasma cells takes place (the latter secreting PR3-
ANCA, which, on reaching the bloodstream, cause vasculitis) [24]. (b) ANCA-induced necrotising vasculitis in WG, microscopic polyangiitis (MPA) and Churg-Strauss
syndrome (CSS). a, TNF-a induces ‘priming’ of neutrophils and endothelial cells. b, Cytokine priming triggers neutrophil azurophilic granules containing PR3 and MPO to
Review Trends in Immunology Vol.29 No.6
282

Box 1. Anti-neutrophil cytoplasmic antibody–induced
neutrophil intracellular signalling
Studies on neutrophil intracellular signaling pathways that are
activated on anti-neutrophil cytoplasmic antibody (ANCA) binding
have focused on the machinery leading to the respiratory burst. To
trigger superoxide generation, a simultaneous engagement of
ANCA F(ab’)2 (to proteinase 3 or myeloperoxidase) and Fc (to Fc-g
IIa and IIIb receptors) is needed, with b2-integrin (CD18/CD11b)–
intercellular adhesion molecule-1 binding permitting Fc- g receptor
to propagate the signal [4]. Each binding activates independent but
complementary intracellular cascades. The Fc-g receptor binding
leads to the activation of tyrosine kinases (including Src and Syk, clb
and probably phospholypase C-g) [91–93]. ANCA F(ab’)2 binding
activates inhibitory G proteins, phosphatidyl inositol-3 kinase type
1-g, protein kinase B and p21ras protein activator [92]. Downstream
mediators of both pathways converge on the p21ras, which seems
to be a pivotal mediator controlling the neutrophil respiratory burst
[32,92]. With regard to other ANCA-mediated functions, recent
investigations have revealed that neutrophil cytoskeletal changes
require Fc-g IIa receptor engagement, tyrosine phosphorylation and
calcium fluxes, whereas the b2 integrin–dependent pro-adhesive
phenotype is mediated mainly by F(ab’)2 binding that induces
activation of diacylglycerol kinase and production of phosphatidic
acid. [93]
Review Trends in Immunology Vol.29 No.6
complementary sequences have been identified inmicroorganisms including Staphylococcus aureus, hencesupporting the role of infectious agents as triggering fac-tors of PR3 autoimmunity (molecular mimicry). Chronicnasal carriage of S. aureus has indeed been related to WG.
The vasculitic pole of the spectrum: pathogenic role of
ANCA
WG, MPA and CSS almost always present necrotisingvasculitis involving different organs. The strong clinicalassociation between MPO-ANCA and PR3-ANCA andAAV, together with compelling evidence from in vitroand animal models, suggests a critical role for these anti-bodies in the pathogenesis of vascular damage in AAV.
ANCA antigens are mainly located within the cyto-plasmic granules of resting neutrophils, ’sheltered’ fromcirculating ANCA. However, when neutrophils areprimed by TNF-a (and by other cytokines or microbialproducts), MPO and PR3 traffic to the external leaflet ofthe neutrophil plasma membrane, facilitating ANCAbinding [26,27] (Figure 2b, a and b). This preliminarylow-grade inflammation step may occur in vivo as a resultof infection [28]. In 1990, Falk et al. [26] demonstratedthat ANCA binding triggered the release of reactive ox-ygen species and toxic granule enzymes by TNF-a–primed neutrophils. Subsequently, ANCA-activated neu-trophils were found to be able to kill cultured endothelialcells [29,30]. In vitro studies have yielded additionalevidence of an orchestrating role for ANCA in a neutro-phil-mediated vascular injury model. Although themajority of ANCA antigens are located in the cytoplasmof unstimulated neutrophils, a small quantity is presentconstitutively in the plasma membrane. Interestingly, ithas been shown that the baseline level of PR3 surfaceexpression in resting neutrophils varies widely betweenindividuals, and there have been reported higher degreesof PR3 surface expression in WG patients compared withcontrols [31]. One can easily speculate on how a higherconstitutive surface expression of PR3 could increase thelikelihood of PR3-ANCA binding. The neutrophil intra-cellular signaling cascades activated by ANCA bindingare different from those of the normal inflammatoryresponse (Box 1).
ANCA are also believed to influence neutrophil–endo-thelial interactions. A coated endothelial cell flow modelusing intravital microscopy has shown that primed neu-trophils exposed to ANCA firmly adhere and transmigrateto TNF-a–primed endothelium instead of undergoingrolling adhesion [32]. Calderwood et al. [33] found thatneutrophil adhesion and transmigration ismediated by b2-integrins. Rather than an increase in b2-integrins on theneutrophil surface (which is actually induced by cytokinepriming), ANCA binding promotes a conformationalchange in the CD11b subunit that makes the moleculemore active and enhances ligand binding [32] (Figure 2b,
relocate to the cell membrane and allow binding by ANCA. In addition, TNF-a induces
Simultaneous interaction of ANCA with their antigen and Fcg receptor prompts severa
cytokine-primed endothelium. d, Once adhered, ANCA induce the neutrophil respirator
promote neutrophil secretion of proinflammatory cytokines and activates complement f
perpetuating the pathological process. PR3, proteinase 3; MPO, myeloperoxidase;
lymphocyte–associated antigen 4-immunoglobulin; TEM, effector memory T cells; ICAM
c). ANCA has been shown to induce changes in the neu-trophil actin cytoskeleton resulting in changes in shapeand capillary sequestration, hence favoring adhesion tosmall vessel endothelium [34].
Only once adhered to endothelium are ANCA-activatedneutrophils able to release reactive oxygen species andlytic enzymes (including MPO and PR3), damaging vesselwalls [5,27]. Furthermore, ANCA-activated neutrophilsand monocytes also release proinflammatory cytokines[interleukin 1b (IL-1b), IL-6, IL-8, TNF-a, monocyte che-moattractant protein 1 and leukotriene B4], which acti-vate and recruit more inflammatory cells includingmonocytes and T cells (see Box 2), amplifying and perpe-tuating a deleterious immune response [35] (Figures 2band 2d). Noninflammatory physiological mechanisms ofneutrophil clearance also seem to be altered in the pre-sence of ANCA-induced signals. For instance, ANCA bind-ing dysregulates normal neutrophil apoptosis by delayingthe expression of surface phosphatidylserines, which areessential for macrophage-dependent cell removal.Because of this delayed clearance, ANCA-coated neutro-phils undergo secondary necrosis with release ofadditional inflammatory mediators [36]. Moreover,ANCA-opsonized apoptotic neutrophils enhance the rateof uptake by macrophages, which results in them upregu-lating TNF-a [37].
In vivo evidence for AAV: humans and animal models
The pathogenic role of ANCA in vivo was convincinglydemonstrated in 2002, when Xiao et al. [38] passivelyadministered murine anti-MPO IgG to Rag2�/� (recombi-nase activating gene 2) mice (which lack functioning T or B
enhanced expression of adhesion molecules (e.g. ICAM-1) by endothelial cells. c,
l effector functions including firm adhesion (and not rolling) of neutrophils to the
y burst and degranulation, which ultimately causes vasculitic damage. ANCA also
actors, leading to the recruitment of more inflammatory cells, thus amplifying and
DC, dendritic cell; (PAR)-2, protease activated receptor; CTLA4-Ig, cytotoxic T
-1, intercellular adhesion molecule-1.
283

Box 2. Role of effector T cells in vasculitis
Besides their role in anti-neutrophil cytoplasmic antibody (ANCA)
production (ANCA are high-affinity class-switched antibodies), it is
now suspected that effector T cells can accompany ANCA and thus
amplify vascular damage. In fact, T cells are frequently found in
biopsies of vasculitic areas and some T cell–targeted therapies have
reversed manifestations of vasculitis [23]. Marinaki et al. [94] have
found an association between persistent CD4+ T-cell activation and
disease severity in both Wegener’s granulomatosis and microscopic
polyangiitis. In addition, a recent animal model of myeloperoxidase
(MPO)-ANCA–induced necrotizing and crescentic glomerulonephri-
tis has provided a mechanistic basis for CD4+ T cells in this disease
[95]. In light of these results, the authors speculated on the existence
of a synergistic combination of both humoral and cellular auto-
immunity to MPO. Initially, ANCA induces glomerular neutrophil
infiltration and degranulation with MPO release. Subsequently,
MPO-specific CD4+ T cells are activated, and together with macro-
phages, exacerbate pathology (see Table 1).
Review Trends in Immunology Vol.29 No.6
lymphocytes) and resulted in the development of focalnecrotising, crescentic glomerulonephritis (NCGN) with-out immune deposits (indistinguishable from humanANCA-associated glomerulonephritis), with �15% of glo-meruli suffering damage. The addition of bacterial lipopo-lysaccharide (LPS) increased the number of glomeruliaffected and augmented TNF-a levels [39]. This seemsto confirm in vitro hypotheses underlining the importanceof synergistic TNF-a priming for effective neutrophil acti-vation and how co-infection might exacerbate ANCAeffects. In this regard, the role of toll-like receptors inneutrophil priming may also deserve future investigation.Subsequent mice experiments have demonstrated anessential role for both neutrophils [40] and the alternativepathway (AP) of complement [41,42] in AAV (Table 1). Theactivation of the AP – perhaps by ANCA-activated-neu-trophil release of properdin (a positive regulator of AP,which acts by increasing the stability of AP convertases) –might create a proinflammatory loop that amplifies vas-cular damage by both recruiting additional neutrophilsand direct cytotoxicity (Figure 2b, d).
In 2005, Little et al. [43] developed a Wistar-Kyoto ratmodel in which focal NCGN and pulmonary capillaritiswere induced after immunisation with purifiedhuman MPO. The investigators also explored the effectsof MPO-ANCA on the induction of leucocyte–endo-thelium interactions using intravital microscopy ofmesenteric venules. Administration of CXCL-1 chemo-kine (a rat homolog of IL-8) in the mesenterium of bothimmunised and naıve rats (which received purified IgGfrom sera of MPO-immunised rats) led to increasedleucocyte adherence and transmigration with microvas-culature focal haemorrhage at chemokine applicationsites. This experiment (despite reinforcing the patho-genic effect of ANCA) confirmed previous in vitro flowmodels showing that ANCA (in collaboration with asynergistic proinflammatory stimulus) promote neutro-phil adhesion to endothelium in vivo. In contrast toaccumulated evidence from MPO-ANCA animal models,there is no convincing in vivo evidence of PR3-ANCApathogenicity.
The clearest evidence for the pathogenicity of ANCA inhumans is the reported case of a newborn (whose motherpresented active MPO-ANCA MPA at delivery) who devel-
284
oped pulmonary haemorrhage and glomerulonephritis48 h after birth. Cordal blood was positive for MPO-ANCAat similar levels to that of the maternal serum. Treatmentof the newborn led to vasculitis resolution and negativisa-tion of serum ANCA [44,45].
Rationale and clinical efficacy of biologicals in ANCA-associated vasculitisTNF-a blockers: infliximab and etanercept
There is compelling in vitro and in vivo evidence for acentral role of TNF-a in AAV pathogenesis. TNF-a med-iates granuloma formation (and maybe lymphoid-like tis-sue neogenesis) in WG and is necessary for endothelialactivation and neutrophil priming in ANCA-mediated vas-cular damage. TNF-a is increased in serum and vasculiticfoci in active disease, with a return to normal levels duringremission [46,47]. Furthermore, anti-TNF-a antibodieshave been shown to prevent [39] and even strongly attenu-ate [43] ANCA-induced NCGN in two animal models.These findings strongly suggest that inhibition of TNF-acould be beneficial in different stages of AAV.
Infliximab is a high-affinity chimeric monoclonal anti-TNF-a antibody that blocks cytokine receptor ligation andneutralizes biological activity [48]. It can bind to two TNF-a molecules, and up to three antibodies can bind to onetrimeric TNF-a molecule, thus blocking all the TNF-abinding sites. Infliximab can also bind monomeric TNF-a, preventing formation of the trimer [49]. Four open-labeltrials of infliximab including 51 patients with AAV havebeen performed [8,50–52]. Most patients had refractorydisease and were taking corticosteroids and at least one ortwo immunosuppressant drugs at infliximab introduction.In all but one trial [51], patients continued with immuno-suppressants while on infliximab. This, together with thevariable treatment and follow-up times and the varyingdefinitions of remission and relapse between studies, makedefinitive conclusions difficult. However, infliximabachieved a remission rate of up to 80% with only 14% ofrelapses [49]. Clinical responses were observed within 2weeks to 2months, and the reduction inmeanBirminghamVasculitis Activity Score (BVAS) – a consensus activityindex – was significant and clinically relevant in all stu-dies. In general, infliximab was well tolerated, with infec-tions occurring in six cases.
Etanercept is a fusion protein composed of two extra-cellular p75 TNF-a human receptor domains linked to theFc portion of human IgG1. It forms a one-to-one bond withthe active trimeric soluble TNF-a molecule, leaving onebinding site free [49]. In 2001, one open-label trial foundetanercept to be effective in WG [53], and a larger random-ized, placebo-controlled trial (WGET trial) [54] wasdesigned to evaluate the efficacy of etanercept in inducingand maintaining remission in 180 WG patients. Etaner-cept or placebo was added to standard therapies at eachclinical stage. The trial demonstrated no additional effectof etanercept because there were no significant differencesbetween groups in the time to or duration of remission [54].There was a significantly higher incidence of cancer inpatients treated with etanercept [55]. All etanercept-trea-ted patients who developed solid-organ tumors alsoreceived cyclophosphamide during the trial. However,
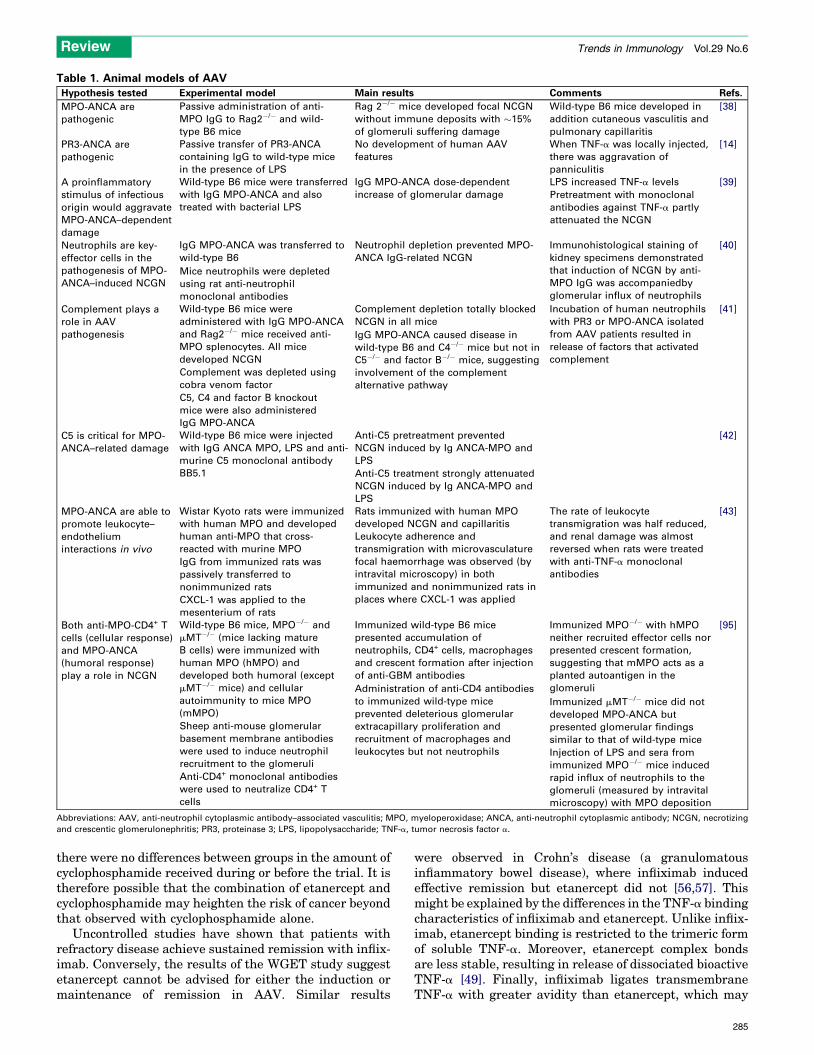
Table 1. Animal models of AAV
Hypothesis tested Experimental model Main results Comments Refs.
MPO-ANCA are
pathogenic
Passive administration of anti-
MPO IgG to Rag2�/� and wild-
type B6 mice
Rag 2�/� mice developed focal NCGN
without immune deposits with �15%
of glomeruli suffering damage
Wild-type B6 mice developed in
addition cutaneous vasculitis and
pulmonary capillaritis
[38]
PR3-ANCA are
pathogenic
Passive transfer of PR3-ANCA
containing IgG to wild-type mice
in the presence of LPS
No development of human AAV
features
When TNF-a was locally injected,
there was aggravation of
panniculitis
[14]
A proinflammatory
stimulus of infectious
origin would aggravate
MPO-ANCA–dependent
damage
Wild-type B6 mice were transferred
with IgG MPO-ANCA and also
treated with bacterial LPS
IgG MPO-ANCA dose-dependent
increase of glomerular damage
LPS increased TNF-a levels [39]
Pretreatment with monoclonal
antibodies against TNF-a partly
attenuated the NCGN
Neutrophils are key-
effector cells in the
pathogenesis of MPO-
ANCA–induced NCGN
IgG MPO-ANCA was transferred to
wild-type B6
Neutrophil depletion prevented MPO-
ANCA IgG-related NCGN
Immunohistological staining of
kidney specimens demonstrated
that induction of NCGN by anti-
MPO IgG was accompaniedby
glomerular influx of neutrophils
[40]
Mice neutrophils were depleted
using rat anti-neutrophil
monoclonal antibodies
Complement plays a
role in AAV
pathogenesis
Wild-type B6 mice were
administered with IgG MPO-ANCA
and Rag2�/� mice received anti-
MPO splenocytes. All mice
developed NCGN
Complement depletion totally blocked
NCGN in all mice
Incubation of human neutrophils
with PR3 or MPO-ANCA isolated
from AAV patients resulted in
release of factors that activated
complement
[41]
Complement was depleted using
cobra venom factor
IgG MPO-ANCA caused disease in
wild-type B6 and C4�/� mice but not in
C5�/� and factor B�/� mice, suggesting
involvement of the complement
alternative pathway
C5, C4 and factor B knockout
mice were also administered
IgG MPO-ANCA
C5 is critical for MPO-
ANCA–related damage
Wild-type B6 mice were injected
with IgG ANCA MPO, LPS and anti-
murine C5 monoclonal antibody
BB5.1
Anti-C5 pretreatment prevented
NCGN induced by Ig ANCA-MPO and
LPS
[42]
Anti-C5 treatment strongly attenuated
NCGN induced by Ig ANCA-MPO and
LPS
MPO-ANCA are able to
promote leukocyte–
endothelium
interactions in vivo
Wistar Kyoto rats were immunized
with human MPO and developed
human anti-MPO that cross-
reacted with murine MPO
Rats immunized with human MPO
developed NCGN and capillaritis
The rate of leukocyte
transmigration was half reduced,
and renal damage was almost
reversed when rats were treated
with anti-TNF-a monoclonal
antibodies
[43]
IgG from immunized rats was
passively transferred to
nonimmunized rats
Leukocyte adherence and
transmigration with microvasculature
focal haemorrhage was observed (by
intravital microscopy) in both
immunized and nonimmunized rats in
places where CXCL-1 was appliedCXCL-1 was applied to the
mesenterium of rats
Both anti-MPO-CD4+ T
cells (cellular response)
and MPO-ANCA
(humoral response)
play a role in NCGN
Wild-type B6 mice, MPO�/� and
mMT�/� (mice lacking mature
B cells) were immunized with
human MPO (hMPO) and
developed both humoral (except
mMT�/� mice) and cellular
autoimmunity to mice MPO
(mMPO)
Immunized wild-type B6 mice
presented accumulation of
neutrophils, CD4+ cells, macrophages
and crescent formation after injection
of anti-GBM antibodies
Immunized MPO�/� with hMPO
neither recruited effector cells nor
presented crescent formation,
suggesting that mMPO acts as a
planted autoantigen in the
glomeruli
[95]
Sheep anti-mouse glomerular
basement membrane antibodies
were used to induce neutrophil
recruitment to the glomeruli
Administration of anti-CD4 antibodies
to immunized wild-type mice
prevented deleterious glomerular
extracapillary proliferation and
recruitment of macrophages and
leukocytes but not neutrophils
Immunized mMT�/� mice did not
developed MPO-ANCA but
presented glomerular findings
similar to that of wild-type mice
Anti-CD4+ monoclonal antibodies
were used to neutralize CD4+ T
cells
Injection of LPS and sera from
immunized MPO�/� mice induced
rapid influx of neutrophils to the
glomeruli (measured by intravital
microscopy) with MPO deposition
Abbreviations: AAV, anti-neutrophil cytoplasmic antibody–associated vasculitis; MPO, myeloperoxidase; ANCA, anti-neutrophil cytoplasmic antibody; NCGN, necrotizing
and crescentic glomerulonephritis; PR3, proteinase 3; LPS, lipopolysaccharide; TNF-a, tumor necrosis factor a.
Review Trends in Immunology Vol.29 No.6
there were no differences between groups in the amount ofcyclophosphamide received during or before the trial. It istherefore possible that the combination of etanercept andcyclophosphamide may heighten the risk of cancer beyondthat observed with cyclophosphamide alone.
Uncontrolled studies have shown that patients withrefractory disease achieve sustained remission with inflix-imab. Conversely, the results of the WGET study suggestetanercept cannot be advised for either the induction ormaintenance of remission in AAV. Similar results
were observed in Crohn’s disease (a granulomatousinflammatory bowel disease), where infliximab inducedeffective remission but etanercept did not [56,57]. Thismight be explained by the differences in the TNF-a bindingcharacteristics of infliximab and etanercept. Unlike inflix-imab, etanercept binding is restricted to the trimeric formof soluble TNF-a. Moreover, etanercept complex bondsare less stable, resulting in release of dissociated bioactiveTNF-a [49]. Finally, infliximab ligates transmembraneTNF-a with greater avidity than etanercept, which may
285

Review Trends in Immunology Vol.29 No.6
induce apoptosis or lysis of membrane TNF-a–expressingimmune effector cells [58–60].
B-cell depletion with rituximab
Rituximab is a chimeric monoclonal anti-CD20 antibodythat induces both complement and antibody-dependentdestruction of all B-lineage cells with the exception ofplasma and pre-B cells, which do not express surfaceCD20. Four weekly infusions of rituximab deplete periph-eral B cells for �6–12 months. Rituximab has shown verypromising effectiveness in non-Hodgkin B cell lymphoma,rheumatoid arthritis and systemic lupus erythematosus[61]. The rationale for rituximab use in AAV rests on theassumption that elimination of immediate precursors ofCD20� plasma cells could interfere with their replacement,leading to transient pathogenic antibody removal andhealing of vasculitis. This presupposes that ANCA areproduced by short- and not long-lived plasma cells [62].In addition, rituximab might block B cell–dependent auto-immune arms unrelated to autoantibody production suchas cytokine secretion, antigen presentation and inter-actions with T cells and other antigen presenting cells,thus hypothetically producing widespread immunologiceffects [16]. In AAV patients, the number of activatedcirculating B lymphocytes correlates with disease activityand extent of involvement [63]. Rituximab has mostly beenused to treat refractory disease in small case series and afew uncontrolled studies [64–72]. Most patients were tak-ing simultaneous drugs (including initial high-dose gluco-corticoids [67,69,70] and immunosuppressants) at the timeof rituximab introduction. Rituximab addition led toclinical response in 64 of 72 patients, with 83% experien-cing complete remission by 6 months. In addition, ritux-imab favored the tapering (or withdrawal) of concomitantmedication. Of note, nonresponders exhibited mostly gran-ulomatous manifestations (e.g. retroorbital granulomas).Rituximab universally induced total depletion of circulat-ing B cells by 1 month, and ANCA titers generallydecreased, but only 50% of patients achieved negativisa-tion. Some patients experienced complete remission withno significant decrease in ANCA titers or before ANCAdrop [66,68,71]. Approximately one third of patients suf-fered a relapse in the follow-up, and retreatment wasgenerally successful [73]. Relapses were frequently pre-ceded by a rise in ANCA titers and, in all but one case, bycirculating B-cell recovery that occurred within 6–12months [64,68]. Rituximab was well tolerated, and fewserious infusion reactions or infections were reported. B-cell depletion led to a decrease in IgM levels and a slightreduction in IgG.
Although preliminary, these results raise questionsabout AAV pathogenesis. It seems that granulomatousfeatures are resistant to rituximab, which confirms thecurrent concept that B cells and ANCA have a negligiblerole in initiating or sustaining granuloma-dependentdamage [62]. However, rituximab seems to effectively healvasculitis, apparently by preventing generation of ANCA,although some patients respond with B-cell depletion butwithout ANCA clearance. Similarly, rituximab failed toreduce anti–doubled-stranded DNA and antiphospholipidantibodies in patients with systemic lupus erythematosus
286
[11]. This challenges the theory of ANCA production byshort-lived plasma cells. The lack of negativisation in onehalf the cases suggests that a significant proportion ofANCA may be produced by autoreactive long-lived plasmacells. The ANCA isotype (IgG) and the presence of somaticmutations point to long-lived plasma cells as the source ofANCA [62]. Some authors even suggest that the negativi-sation of ANCA and the good clinical response observed insome trials was merely caused by high doses of concomi-tant glucocorticoids [11,62]. Another plausible explanationis that the beneficial effect of rituximab depends more onthe inhibition of B-cell functions other than antibody pro-duction (e.g. the B-cell influence on T-cell functions)[68,74].
IFN-a in the treatment of CSS
Proliferating eosinophils play a pivotal role in the patho-genesis of CSS by infiltrating tissues and causing damagevia release of lytic proteins. CSS patients have been trea-ted with IFN-a since the 1990s because of its capacity tosuppress overreactive Th2-immune responses and itsinhibitory effect on eosinophil effector functions [6]. IFN-a receptors are detectable on both CD4+ T cells and eosi-nophils, and their ligation inhibits eosinophil-activating T-cell cytokine production (including IL-5) and the release ofeffector molecules by eosinophils [75]. To date, the effec-tiveness of IFN-a has not been studied systematically.Short-term benefits have only been reported in smallstudies totaling eight patients [76–78]. Furthermore, lo-ng-term use has been associated with leukoencephalopa-thy [16], which, in our opinion, may limit the use of IFN-afor CSS in the future.
Conclusions and future directionsAmong available biologicals, only rituximab and infliximabhave been trialed with apparent initial success in theclinic. The high expectations generated by rituximab arebeing tested in two prospective randomized trials (RITUX-VAS and RAVE) designed to confirm the short-term safetyand efficacy in granulomatous and vasculitic disease andclarify the mechanism of action of the drug. This mightprovide more insight into the role of B cells in AAVpathogenesis [73]. Similarly, infliximab may deserve aclinical trial to elucidate whether – in contrast to etaner-cept and by virtue of its differences – it is a useful agent inany of the disease states of AAV. In the meantime, theseagents should be restricted to patients who prove refrac-tory to standard immunosuppressants or patients includedin clinical trials.
The need for better drugs, and the continuous advancesin the knowledge of the pathogenesis of AAV, is fuellingimmunotherapy research in this group of autoimmunediseases. Some novel agents are awaiting testing in AAVpilot studies. Agents containing cytotoxic T lymphocyte–associated antigen 4-immunoglobulin (CTLA4) haveachieved good results in rheumatoid arthritis [79], psor-iasis [80] and renal transplantation patients [81]. Abata-cept and belatacept are fusion proteins (combining theIgG1 Fc fragment with the extracellular domain of humanCTLA4) that bind to CD80 and CD86 (with higher avidityin the case of belatacept) and prevent these co-stimulatory

Review Trends in Immunology Vol.29 No.6
molecules from interacting with CD28 for full T-cellantigen–mediated activation and clonal expansion [79](Figure 2a, b). A diminished frequency of homozygosityfor the shortest allele of the CTLA4 microsatellite poly-morphism (AT)n (a crucial allele formaintenance of normallevels of CTLA4 expression and balance between T-cellactivation and inhibition) has been reported in WG [82],making co-stimulatory signals an attractive target fornovel therapeutics.
Agents intended to neutralise IL-18 might also have arole in AAV, because several pathogenic steps can betargeted at once. IL-18 induces Th1 responses andenhances IFN-g production (Figure 2a, b) and, morespecifically, IL-18 is upregulated in renal biopsies ofpatients with active disease. Furthermore, in vitro studieshave found that IL-18 can prime neutrophils (via themitogen-activated protein kinase p38) for ANCA-mediatedsuperoxide burst in a manner similar to TNF-a [83].
IL-5 is known to be the most relevant Th2-cytokineinvolved in eosinophil proliferation, maturation and fullactivation of eosinophils and levels of this cytokine runparallel to disease severity [84]. Similarly, activation of Tcells from CSS patients induces the production of higheramounts of IL-5 compared with healthy controls [85].Therefore, the neutralization of IL-5 with mepoluzimab(a monoclonal antibody against IL-5) could be beneficial inCSS.
With regard to B-cell therapies, ocrelizumab, ofatumu-mab (both humanised anti-CD20 antibodies) and epratu-zumab (humanised anti-CD22 antibody, a surface receptorspecifically expressed on B cells) could provide more effec-tive and long-lasting B-cell depletion without the devel-opment by the patient of neutralizing anti-chimericantibodies [73,86]. B-cell activating factor belonging tothe TNF family (BAFF) B-cell activating factor (a T-cellcytokine critically involved in B-cell maturation and sur-vival) is increased in patients with active WG [87]. There-fore, anti-BAFF antibodies (belimumab) could subtlymodulate T- and B-cell interaction in AAV without B-celldepletion (Figure 2a, c). BAFF levels are elevated withrituximab therapy, suggesting that the addition of belimu-mab could enhance B cell suppression [73].
In 2007, Huugen et al. [42] proved that monoclonalantibodies against the complement component C5 cansignificantly attenuate MPO-ANCA NCGN, probably byinhibiting neutrophil recruitment, paving the way for itsuse in AAV. Inhibition of C5 cleavage with eculizumab (ahumanised monoclonal antibody) or pexelizumab (itssingle chain version) has produced clinical benefit in par-oxysmal nocturnal haemoglobinuria and ischaemia-reper-fusion injury, respectively, with good adverse-eventprofiles [88].
ANCA-mediated leucocyte–endothelium interactionsrepresent an exciting area to explore. Neutrophil fixedadhesion via b2-integrin– intercellular adhesion mol-ecule-1 (ICAM-1) binding is a prerequisite for the neutro-phil respiratory burst. Therefore, monoclonal antibodiesagainst CD11b and/or ICAM-1 would prevent neutrophil-related primary endothelial damage. (Figure 2b, c). Sim-ilarly, peptides (cyclic hexapeptides) can be designed toselectively bind and inactivate crucial adhesion molecules
[89]. Interfering with the specific neutrophil signallingcascades induced by ANCA is another promising strategy.Blockade of the crucial downstreammediator p21ras by itsselective inhibitor farnesylthiosalicylic acid might neutral-ise noxious superoxide production and vascular necrosis.Gene therapy might be the last frontier in the treatment ofAAV. Gene transfer (using viral vectors or transfectedinflammatory cells) would allow the delivery of a steadynontoxic concentration of a desired factor (cytokine naturalinhibitors or even enzymes catalysing a therapeutic pro-duct) specifically at the disease site.
AcknowledgementsWe thank Monica Perez-Poquet and Jose Manuel Mascaro, Jr. fortechnical support in the development of manuscript figures.
References1 Jennette, J.C. et al. (1994) Nomenclature of systemic vasculitides.
Proposal of an international consensus conference. Arthritis Rheum.37, 187–192
2 Seo, P. and Stone, J.H. (2004) The antineutrophil cytoplasmicantibody-associated vasculitides. Am. J. Med. 117, 39–50
3 Jennette, J.C. and Falk, R.J. (1997) Small-vessel vasculitis.N. Engl. J.Med. 337, 1512–1523
4 Bosch, X. et al. (2006) Antineutrophil cytoplasmic antibodies. Lancet368, 404–418
5 Kallenberg, C.G. et al. (2006) Mechanisms of disease: pathogenesis andtreatment of ANCA-associated vasculitides. Nat. Clin. Pract.Rheumatol. 2, 661–670
6 Pagnoux, C. et al. (2007) Churg-Strauss syndrome. Curr. Opin.Rheumatol. 19, 25–32
7 Bosch, X. et al. (2007) Treatment of antineutrophil cytoplasmicantibody associated vasculitis: a systematic review. JAMA 298, 655–669
8 Booth, A. et al. (2004) Prospective study of TNFalpha blockade withinfliximab in anti-neutrophil cytoplasmic antibody-associated systemicvasculitis. J. Am. Soc. Nephrol. 15, 717–721
9 Langford, C.A. (2003) Treatment of ANCA-associated vasculitis. N.Engl. J. Med. 349, 3–4
10 Hellmich, B. et al. (2006) Advances in the therapy of Wegener’sgranulomatosis. Curr. Opin. Rheumatol. 18, 25–32
11 Sarraf, P. et al. (2006) Wegener’s granulomatosis: is biologic therapyuseful? Curr. Rheumatol. Rep. 8, 303–311
12 Bacon, P.A. (2005) The spectrum of Wegener’s granulomatosis anddisease relapse. N. Engl. J. Med. 352, 330–332
13 Sarraf, P. and Sneller, M.C. (2005) Pathogenesis of Wegener’sgranulomatosis: current concepts. Expert Rev. Mol. Med. 7, 1–19
14 Pfister, H. et al. (2004) Antineutrophil cytoplasmic autoantibodiesagainst the murine homolog of proteinase 3 (Wegener autoantigen)are pathogenic in vivo. Blood 104, 1411–1418
15 Specks, U. (2000) Are animal models of vasculitis suitable tools? Curr.Opin. Rheumatol. 12, 11–19
16 Lamprecht, P. et al. (2007) Current state of biologicals in themanagement of systemic vasculitis.Ann. N. Y. Acad. Sci. 1110, 261–270
17 Voswinkel, J. et al. (2005) Is PR3-ANCA formation initiated inWegener’s granulomatosis lesions? Granulomas as potentiallymphoid tissue maintaining autoantibody production. Ann. N. Y.Acad. Sci. 1051, 12–19
18 Lamprecht, P. et al. (2004) CD28- T cells display features of effectormemory T cells in Wegener’s granulomatosis. Kidney Int. 65, 1113
19 Komocsi, A. et al. (2002) Peripheral blood and granulomaCD4(+)CD28(�) T cells are a major source of interferon-gamma andtumor necrosis factor-alpha in Wegener’s granulomatosis. Am. J.Pathol. 160, 1717–1724
20 Csernok, E. et al. (1999) Cytokine profiles in Wegener’sgranulomatosis: predominance of type 1 (Th1) in the granulomatousinflammation. Arthritis Rheum. 42, 742–750
21 Lamprecht, P. et al. (2003) Differences in CCR5 expression onperipheral blood CD4+CD28- T-cells and in granulomatous lesionsbetween localized and generalized Wegener’s granulomatosis. Clin.Immunol. 108, 1–7
287

Review Trends in Immunology Vol.29 No.6
22 Appay, V. et al. (2002) Characterization of CD4(+) CTLs ex vivo. J.Immunol. 168, 5954–5958
23 Abdulahad, W.H. et al. (2007) CD4-positive effector memory T cellsparticipate in disease expression in ANCA-associated vasculitis. Ann.N. Y. Acad. Sci. 1107, 22–31
24 Voswinkel, J. et al. (2006) B lymphocyte maturation in Wegener’sgranulomatosis: a comparative analysis of VH genes from endonasallesions. Ann. Rheum. Dis. 65, 859–864
25 Preston, G.A. et al. (2005) New insights that link microbes with thegeneration of antineutrophil cytoplasmic autoantibodies: the theory ofautoantigen complementarity. Curr. Opin. Nephrol. Hypertens. 14,217–222
26 Falk, R.J. et al. (1990) Anti-neutrophil cytoplasmic autoantibodiesinduce neutrophils to degranulate and produce oxygen radicals invitro. Proc. Natl. Acad. Sci. U. S. A. 87, 4115–4119
27 Jennette, J.C. et al. (2006) Pathogenesis of vascular inflammation byanti-neutrophil cytoplasmic antibodies. J. Am. Soc. Nephrol. 17, 1235–1242
28 Kallenberg, C.G. (2007) Antineutrophil cytoplasmic autoantibody-associated small-vessel vasculitis. Curr. Opin. Rheumatol. 19, 17–24
29 Savage, C.O. et al. (1992) Autoantibodies developing tomyeloperoxidase and proteinase 3 in systemic vasculitis stimulateneutrophil cytotoxicity toward cultured endothelial cells. Am. J.Pathol. 141, 335–342
30 Ewert, B.H. et al. (1992) Anti-myeloperoxidase antibodies stimulateneutrophils to damage human endothelial cells. Kidney Int. 41, 375–383
31 Schreiber, A. et al. (2003) Membrane expression of proteinase 3 isgenetically determined. J. Am. Soc. Nephrol. 14, 68–75
32 Morgan, M.D. et al. (2006) Anti-neutrophil cytoplasm-associatedglomerulonephritis. J. Am. Soc. Nephrol. 17, 1224–1234
33 Calderwood, J.W. et al. (2005) ANCA induces beta2 integrin and CXCchemokine-dependent neutrophil-endothelial cell interactions thatmimic those of highly cytokine-activated endothelium. J. Leukoc.Biol. 77, 33–43
34 Tse, W.Y. et al. (2005) ANCA-induced neutrophil F-actinpolymerization: implications for microvascular inflammation. KidneyInt. 67, 130–139
35 Day, C.J. et al. (2003) New developments in the pathogenesis of ANCA-associated vasculitis. Clin. Exp. Rheumatol. 21, S35–S48
36 Csernok, E. (2003) Anti-neutrophil cytoplasmic antibodies andpathogenesis of small vessel vasculitides. Autoimmun. Rev. 2, 158–164
37 Moosig, F. et al. (2000) Opsonization of apoptotic neutrophils by anti-neutrophil cytoplasmic antibodies (ANCA) leads to enhanced uptakeby macrophages and increased release of tumour necrosis factor-alpha(TNF-alpha). Clin. Exp. Immunol. 122, 499–503
38 Xiao, H. et al. (2002) Antineutrophil cytoplasmic autoantibodiesspecific for myeloperoxidase cause glomerulonephritis and vasculitisin mice. J. Clin. Invest. 110, 955–963
39 Huugen, D. et al. (2005) Aggravation of anti-myeloperoxidase antibody-induced glomerulonephritis by bacterial lipopolysaccharide: role oftumor necrosis factor-alpha. Am. J. Pathol. 167, 47–58
40 Xiao, H. et al. (2005) The role of neutrophils in the induction ofglomerulonephritis by anti-myeloperoxidase antibodies. Am. J.Pathol. 167, 39–45
41 Xiao, H. et al. (2007) Alternative complement pathway in thepathogenesis of disease mediated by anti-neutrophil cytoplasmicautoantibodies. Am. J. Pathol. 170, 52–64
42 Huugen, D. et al. (2007) Inhibition of complement factor C5 protectsagainst anti-myeloperoxidase antibody-mediated glomerulonephritisin mice. Kidney Int. 71, 646–654
43 Little, M.A. et al. (2005) Antineutrophil cytoplasm antibodies directedagainst myeloperoxidase augment leukocyte-microvascularinteractions in vivo. Blood 106, 2050–2058
44 Bansal, P.J. and Tobin, M.C. (2004) Neonatal microscopic polyangiitissecondary to transfer of maternal myeloperoxidase-antineutrophilcytoplasmic antibody resulting in neonatal pulmonary hemorrhageand renal involvement. Ann. Allergy Asthma Immunol. 93, 398–401
45 Schlieben, D.J. et al. (2005) Pulmonary-renal syndrome in a newbornwith placental transmission of ANCA. Am. J. Kidney Dis. 45, 758–761
46 Noronha, I.L. et al. (1993) In situ production of TNF-alpha, IL-1 betaand IL-2R in ANCA-positive glomerulonephritis. Kidney Int. 43, 682–692
288
47 Tesar, V. et al. (1998) Cytokines and adhesion molecules in renalvasculitis and lupusnephritis.Nephrol.Dial. Transplant.13, 1662–1667
48 Knight, D.M. et al. (1993) Construction and initial characterization of amouse-human chimeric anti-TNF antibody. Mol. Immunol. 30, 1443–1453
49 Mukhtyar, C. and Luqmani, R. (2005) Current state of tumour necrosisfactor {alpha} blockade inWegener’s granulomatosis.Ann. Rheum. Dis.64 (Suppl 4), iv31–iv36
50 Booth, A.D. et al. (2002) Safety and efficacy of TNFalpha blockade inrelapsing vasculitis. Ann. Rheum. Dis. 61, 559
51 Bartolucci, P. et al. (2002) Efficacy of the anti-TNF-alpha antibodyinfliximab against refractory systemic vasculitides: an open pilot studyon 10 patients. Rheumatology (Oxf) 41, 1126–1132
52 Lamprecht, P. et al. (2002) Effectiveness of TNF-alpha blockade withinfliximab in refractory Wegener’s granulomatosis. Rheumatology(Oxf) 41, 1303–1307
53 Stone, J.H. et al. (2001) Etanercept combined with conventionaltreatment in Wegener’s granulomatosis: a six-month open-label trialto evaluate safety. Arthritis Rheum. 44, 1149–1154
54 Wegener’s Granulomatosis Etanercept Trial (WGET) Research Group.(2005) Etanercept plus standard therapy for Wegener’sgranulomatosis. N. Engl. J. Med. 352, 351–361
55 Stone, J.H. et al. (2006) Solid malignancies among patients in theWegener’s Granulomatosis Etanercept Trial. Arthritis Rheum. 54,1608–1618
56 Akobeng, A.K. and Zachos, M. (2004) Tumor necrosis factor-alphaantibody for induction of remission in Crohn’s disease. CochraneDatabase Syst. Rev. 2004, CD003574
57 Sandborn, W.J. et al. (2001) Etanercept for active Crohn’s disease: arandomized, double-blind, placebo-controlled trial. Gastroenterology121, 1088–1094
58 Scallon, B. et al. (2002) Binding and functional comparisons of twotypes of tumor necrosis factor antagonists. J. Pharmacol. Exp. Ther.301, 418–426
59 Mitoma, H. et al. (2004) Binding activities of infliximab and etanerceptto transmembrane tumor necrosis factor-alpha. Gastroenterology 126,934–935
60 Van den Brande, J.M. et al. (2003) Infliximab but not etanerceptinduces apoptosis in lamina propria T-lymphocytes from patientswith Crohn’s disease. Gastroenterology 124, 1774–1785
61 Flossmann, O. et al. (2006) Should rituximab be used to treatantineutrophil cytoplasmic antibody associated vasculitis? Ann.Rheum. Dis. 65, 841–844
62 Sneller, M.C. (2005) Rituximab and Wegener’s granulomatosis: are Bcells a target in vasculitis treatment? Arthritis Rheum. 52, 1–5
63 Popa, E.R. et al. (1999) Differential B- and T-cell activation inWegener’s granulomatosis. J. Allergy Clin. Immunol. 103, 885–894
64 Brihaye, B. et al. (2007) Adjunction of rituximab to steroids andimmunosuppressants for refractory/relapsing Wegener’sgranulomatosis: a study on 8 patients. Clin. Exp. Rheumatol. 25,S23–S27
65 Tamura, N. et al. (2007) Two cases of refractory Wegener’sgranulomatosis successfully treated with rituximab. Intern. Med. 46,409–414
66 Aries, P.M. et al. (2006) Lack of efficacy of rituximab in Wegener’sgranulomatosis with refractory granulomatous manifestations. Ann.Rheum. Dis. 65, 853–858
67 Keogh, K.A. et al. (2006) Rituximab for refractory Wegener’sgranulomatosis: report of a prospective, open-label pilot trial. Am. J.Respir. Crit. Care Med. 173, 180–187
68 Smith, K.G. et al. (2006) Long-term comparison of rituximab treatmentfor refractory systemic lupus erythematosus and vasculitis: Remission,relapse, and re-treatment. Arthritis Rheum. 54, 2970–2982
69 Stasi, R. et al. (2006) Long-term observation of patients with anti-neutrophil cytoplasmic antibody-associated vasculitis treated withrituximab. Rheumatology (Oxford) 45, 1432–1436
70 Keogh, K.A. et al. (2005) Induction of remission by B lymphocytedepletion in eleven patients with refractory antineutrophilcytoplasmic antibody-associated vasculitis. Arthritis Rheum. 52,262–268
71 Eriksson, P. (2005) Nine patients with anti-neutrophil cytoplasmicantibody-positive vasculitis successfully treated with rituximab. J.Intern. Med. 257, 540–548

Review Trends in Immunology Vol.29 No.6
72 Omdal, R. et al. (2005) Anti-CD20 therapy of treatment-resistantWegener’s granulomatosis: favourable but temporary response.Scand. J. Rheumatol. 34, 229–232
73 Golbin, J.M. and Specks, U. (2007) Part 2: Synopsis of B-lymphocytetargeted therapy of ANCA-associated vasculitis.Clin. Exp. Rheumatol.25, S74–S76
74 Eisenberg, R. and Looney, R.J. (2005) The therapeutic potentialof anti-CD20 ‘‘what do B-cells do? Clin. Immunol. 117, 207–213
75 Gross, W.L. (2002) Churg-Strauss syndrome: update on recentdevelopments. Curr. Opin. Rheumatol. 14, 11–14
76 Tatsis, E. et al. (1998) Interferon-alpha treatment of fourpatients with the Churg-Strauss syndrome. Ann. Intern. Med.129, 370–374
77 Simon, H.U. et al. (2003) Clinical and immunological effects of low-doseIFN-alpha treatment in patients with corticosteroid-resistant asthma.Allergy 58, 1250–1255
78 Termeer, C.C. et al. (2001) Low-dose interferon alfa-2b for thetreatment of Churg-Strauss syndrome with prominent skininvolvement. Arch. Dermatol. 137, 136–138
79 Genovese, M.C. et al. (2005) Abatacept for rheumatoid arthritisrefractory to tumor necrosis factor alpha inhibition. N. Engl. J. Med.353, 1114–1123
80 Abrams, J.R. et al. (2000) Blockade of T lymphocyte costimulation withcytotoxic T lymphocyte-associated antigen 4-immunoglobulin(CTLA4Ig) reverses the cellular pathology of psoriatic plaques,including the activation of keratinocytes, dendritic cells, andendothelial cells. J. Exp. Med. 192, 681–694
81 Vincenti, F. et al. (2005) Costimulation blockade with belatacept inrenal transplantation. N. Engl. J. Med. 353, 770–781
82 Zhou, Y. et al. (2004) An analysis of CTLA-4 and proinflammatorycytokine genes in Wegener’s granulomatosis. Arthritis Rheum. 50,2645–2650
83 Hewins, P. et al. (2006) IL-18 is upregulated in the kidney and primesneutrophil responsiveness in ANCA-associated vasculitis. Kidney Int.69, 605–615
84 Schonermarck, U. et al. (2000) Circulating cytokines and soluble CD23,CD26 and CD30 in ANCA-associated vasculitides. Clin. Exp.Rheumatol. 18, 457–463
85 Hellmich, B. et al. (2005) Proinflammatory cytokines andautoimmunity in Churg-Strauss syndrome. Ann. N. Y. Acad. Sci.1051, 121–131
86 Walsh, M. and Jayne, D. (2007) Rituximab in the treatment of anti-neutrophil cytoplasm antibody associated vasculitis and systemiclupus erythematosus: past, present and future.Kidney Int. 72, 676–682
87 Krumbholz, M. et al. (2005) BAFF is elevated in serum of patients withWegener’s granulomatosis. J. Autoimmun. 25, 298–302
88 Trendelenburg, M. (2007) Complement inhibition by anti-C5antibodies–from bench to bedside and back again. Swiss Med. Wkly.137, 413–417
89 Selamet, U. et al. (2007) ANCA-associated vasculitis: new optionsbeyond steroids and cytotoxic drugs. Expert Opin. Investig. Drugs16, 689–703
90 Csernok, E. et al. (2006) Wegener autoantigen induces maturation ofdendritic cells and licenses them for Th1 priming via the protease-activated receptor-2 pathway. Blood 107, 4440–4448
91 Rarok, A.A. et al. (2003) Neutrophil-activating potential ofantineutrophil cytoplasm autoantibodies. J. Leukoc. Biol. 74, 3–15
92 Williams, J.M. and Savage, C.O. (2005) Characterization of theregulation and functional consequences of p21ras activation inneutrophils by antineutrophil cytoplasm antibodies. J. Am. Soc.Nephrol. 16, 90–96
93 Williams, J.M. et al. (2007) Antineutrophil cytoplasm antibody-stimulated neutrophil adhesion depends on diacylglycerol kinase-catalyzed phosphatidic acid formation. J. Am. Soc. Nephrol. 18,1112–1120
94 Marinaki, S. et al. (2006) Persistent T-cell activation and clinicalcorrelations in patients with ANCA-associated systemic vasculitis.Nephrol. Dial. Transplant. 21, 1825–1832
95 Ruth, A.J. et al. (2006) Anti-neutrophil cytoplasmic antibodies andeffector CD4+ cells play nonredundant roles in anti-myeloperoxidasecrescentic glomerulonephritis. J. Am. Soc. Nephrol. 17, 1940–1949
289