goodpasture autoantibodies unmask cryptic epitopes … · goodpasture autoantibodies unmask cryptic...
TRANSCRIPT

1
GOODPASTURE AUTOANTIBODIES UNMASK CRYPTIC EPITOPES BYSELECTIVELY DISSOCIATING AUTOANTIGEN COMPLEXES LACKING
STRUCTURAL REINFORCEMENT: NOVEL MECHANISMS FORIMMUNE PRIVILEGE AND AUTOIMMUNE PATHOGENESIS*Dorin-Bogdan Borza, Olga Bondar, Selene Colon, Parvin Todd,
Yoshikazu Sado†, Eric G. Neilson, and Billy G. Hudson From the Division of Nephrology, Department of Medicine, Vanderbilt University School of
Medicine, Nashville, Tennessee, 37232, and †Shigei Medical Research Institute, Okayama, Japan. Running Title: Goodpasture antibodies dissociate autoantigen complexes.
Address correspondence to: Dr. Dorin-Bogdan Borza, S-3223 MCN, Division of Nephrology, Department of Medicine; Vanderbilt University School of Medicine; Nashville, Tennessee 37232. Phone: 615 322-7298; fax: 615-343-7156; e-mail: [email protected]
Rapidly progressive glomerulonephritis in Goodpasture’s disease is mediated by autoantibodies binding to the non-collagenous NC1 domain of IV) collagen in the glomerular basement membrane. Goodpasture epitopes in the native autoantigen are cryptic (sequestered) within the NC1 hexamers of the 3 4 5(IV) collagen network. The biochemical mechanism for crypticity and exposure for autoantibody binding is not known. We now report that crypticity is a feature of the quaternary structure of two distinct subsets of 3 4 5(IV) NC1 hexamers: autoantibody-reactive M-hexamers, containing only monomer subunits; and autoantibody-impenetrable D-hexamers, composed of both dimer and monomer subunits. Goodpasture antibodies only breach the quaternary structure of M-hexamers, unmasking the cryptic epitopes, whereas D-hexamers are resistant to autoantibodies under native conditions. The epitopes of D-hexamers are structurally sequestered by dimer reinforcement of the quaternary complex, which represents a new molecular solution for conferring immunologic privilege to a potential autoantigen. Dissociation of non-reinforced M-
3 4 5(IV) hexamers by Goodpasture antibodies is a novel mechanism whereby pathogenic autoantibodies gain access to cryptic B cell epitopes. These findings provide fundamental new insights into immune privilege and the molecular mechanisms underlying the pathogenesis of human autoimmune Goodpasture’s disease.
The refinement of immunological privilege has been a work in progress over a number of decades. Privilege originally implied a location anatomically protected from attack by the immune system, such as the eye, brain, or testes (1). It is now recognized that the pathophysiology of protectiveness involves multiple mechanisms, including cytokines that inhibit innate or adaptive immunity (2), systemic regulatory T cells (3), and local cytotoxic mechanisms producing apoptosis of effector T-cells induced by local FasL-Fas interactions (4). What has not been widely appreciated is that the molecular sequestration of self-antigen in complex structures can be a critical mechanism of immunologic privilege. The autoantigen of Goodpasture’s (GP1) disease is a prototypic example.
In GP disease, autoreactive B cells, with assistance from helper T cells, produce autoantibodies that bind to the glomerular basement membrane (GBM), causing rapidly progressive glomerulonephritis and renal failure (5). GP autoantibodies passively transfer anti-GBM disease to monkeys or human kidney allografts (6). These autoantibodies are directed against the 3 chain of collagen IV (7) and recognize two major conformational epitopes (EAand EB) located within the carboxyl-terminal noncollagenous (NC1) domain (8,9). In the GBM, the 3(IV) chain associates specifically with the
4(IV) and 5(IV) chains (10,11), forming a triple helical protomer. The 3 4 5(IV) protomers self-assemble into a network through end-to-end associations. At the C-terminus, two protomers are linked head-to-head by interactions of their
JBC Papers in Press. Published on May 24, 2005 as Manuscript M504050200
Copyright 2005 by The American Society for Biochemistry and Molecular Biology, Inc.
by guest on May 27, 2018
http://ww
w.jbc.org/
Dow
nloaded from

2
trimeric NC1 domains, forming a stable NC1 hexamer structure, which is the native form of the autoantigen targeted by GP antibodies. The NC1 hexamer complex can be excised from the insoluble collagen IV network by cleavage with collagenase for in vitro studies.
The GP epitopes are cryptic and structurally sequestered within the NC1 hexamer. Whereas a small amount of GP antibody can bind isolated NC1 hexamers under native conditions, maximal antibody binding requires unmasking of the cryptic epitopes. In tissue sections, this is achieved by pretreatment with acid urea (12), and in vitro,by exposing hexamers to dissociating conditions (13,14). In vivo unmasking of the cryptic GP epitopes by putative pathogenic factors, such as oxidative stress (15), is thought to be critical in the mechanism underlying the initiation of the disease.
The biochemical mechanism for the epitope crypticity remains an enigma, although some contributing factors have been described. Near the junction between the triple-helical and NC1 domains, the quaternary structure of the NC1 hexamer hides a part of the EA epitope along a ridge adjacent to the 5NC1 domain, and the EBsite is partly hidden along a ridge adjacent the
4NC1 domain (11). Within the EA site, several residues critical for GP antibody binding have been identified (16,17). A homology-based model of the 3 4 5(IV) hexamer reveals that some GP epitope residues (Val-27, Gln-57) have restricted accessibility in the quaternary structure, which may prevent GP antibody binding to hexamers by steric hindrance (11).
In the present study, we investigated the mechanism for sequestration of GP epitopes and how autoantibodies gain access to them. The results demonstrate that crypticity is a feature of the quaternary structure of two distinct subsets of
3 4 5(IV) NC1 hexamers, which differ in the degree of reinforced stabilization. GP antibodies selectively dissociated non-reinforced M-hexamers, composed of monomer subunits only. In contrast, D-hexamers, reinforced by the presence of dimer subunits, were impenetrable by GP antibodies under native conditions. Sequestration of GP epitopes by structural reinforcement of D-hexamers represents a novel molecular mechanism for conferring immune privilege to a potential autoantigen.
Materials and Methods
Materials – Recombinant (r-) human 1 and 3NC1 domains of type IV collagen were expressed in human embryonic kidney 293 cells and purified as described (8). Human GBM was purified by detergent extraction from the renal cortex of pooled normal kidneys not used for transplantation (Midwest Organ Bank, Kansas City, KS). NC1 hexamers solubilized from the GBM by digestion with collagenase (Worthington, Lakewood, NJ) were purified by ion-exchange on DE-52 and gel-filtration on S-300 columns (11). The NC1 monomer and dimer fractions of human and bovine GBM hexamers were separated by gel filtration after acid dissociation, and re-associated into NC1 hexamers in vitro, as described (10). Human GP IgG autoantibodies were affinity-purified from sera or plasmapheresis fluid of patients with biopsy-proven anti-GBM disease, sequentially using immobilized protein G and human r- 3NC1 monomers (14). Normal human IgG and the non-GP IgG fraction of GP sera were used as negative controls. Use of human tissues and sera were approved by the Institutional Review Board at Vanderbilt University. ELISA – GP sera or purified GP IgG antibodies were assayed by indirect ELISA as described (8). A specific sandwich ELISA assay for 3NC1 domains was developed using immobilized GP IgG as capture antibodies and 3NC1-specificmAbs as detection antibodies. The assay was performed in 96-well Maxisorp microtiter plastic plates coated overnight with affinity-purified human GP IgG (1 g/ml in carbonate buffer, pH 9.6) and blocked with 0.2% bovine albumin. Samples containing 3NC1 domains were diluted in incubation buffer (Tris-buffered saline, pH 7.4, with 1 mg/ml bovine albumin and 0.05% Tween-20) and allowed to bind to immobilized GP antibodies for 1 h at 37°C in duplicate wells. To unmask cryptic GP epitopes, some antigen samples were treated for 30 minutes with either acid (0.1 M glycine, pH 2.2) or 6M guanidinium chloride (14) prior to the assay. The 3NC1domains captured by immobilized GP antibodies were detected by sequential incubations with mAb EB3 (2 g/ml), alkaline phosphatase-conjugated anti-mouse IgG that had no cross-reactivity with human IgG (Rockland, Gilbertsville, PA), and
by guest on May 27, 2018
http://ww
w.jbc.org/
Dow
nloaded from

3
chromogenic substrate (p-nitrophenol phosphate). Absorbance at 405 nm was measured with a Spectramax 190 plate reader (Molecular Devices, Sunnyvale, CA). The results represent the average of duplicate determinations which differed by less than 5%. All experiments were repeated at least twice. Binding curves were obtained by non-linear regression by fitting the absorbance values (A) at various concentrations of antigen X to a single-site binding model, A=Amax*[X]/(Kd+[X]), where Amaxis the maximal binding and Kd, the apparent dissociation constant, is the concentration of antigen at half-saturation. Comparisons between binding curves after various treatments were performed by global curve fitting, followed by F-test. Statistical analyses were performed using GraphPad Prism software, version 4.05. Immunoprecipitation and Western blotting – Affinity-purified GP IgG (~10 g) was incubated with native GBM NC1 hexamers (5-10 g) in 200
l Tris-buffered saline, pH 7.4, overnight at room temperature or for several days at 4°C. Dissociated NC1 subunits, prepared by treating hexamers with 6M guanidinium chloride for 15 minutes at 60°C, were diluted at least 20-fold prior to incubation with GP IgG for ~1 hour. Analysis of NC1 hexamers by immunoprecipitation with Mab3 (Wieslab AB, Lund, Sweden) was performed as described (10). Antibody-antigen complexes were precipitated with protein G-agarose beads, solubilized in sample buffer, and analyzed by immunoblotting, along with the unbound NC1 domains in supernatant. All samples were resolved into component NC1 monomers and dimers by sodium dodecylsulfate polyacrylamide gel electrophoresis (SDS-PAGE) in replicate 4-22% gradient gels under non-reducing conditions. After transfer to Immobilon P and blocking with casein,
1- 5 NC1 domains were identified by immuno-blotting with specific mAbs, as described (10,11). Molecular modeling and graphic representation – The molecular model for the ( 3 4 5)2 NC1 hexamer is a full-atom homology model built from the ( 1 2 1)2 NC1 hexamer crystal structure, PDB code 1M3D (18), and optimized by a full-atom molecular dynamics simulation (J.P. Cartailler; unpublished data) using the NAMD software package (19). The GP IgG model is idealized from the crystal structure of an intact IgG; PDB code 1IGT (20).
RESULTS
Validation of the cryptic nature of the GP epitopes—The concept of cryptic GP epitopes is based on the observation that maximal binding of GP antibodies required the dissociation of NC1 hexamers by protein denaturants (13-15). In previous studies, this was usually assayed by indirect ELISA, comparing the binding of GP antibodies to GBM hexamers absorbed to a plastic solid-phase with or without pre-treatment with guanidinium chloride. However, pitfalls in this assay potentially undermine the concept of cryptic properties. First, immobilization of proteins by adsorption to the plastic surface of ELISA plates can alter the antigen conformation (21), obscuring the features that confer autoantibody binding to native NC1 hexamers. Second, the heterogeneity of typical preparations of human GBM NC1 hexamers, containing ~15% 3 4 5 hexamers and ~84% 1 2 hexamers (11), along with the cross-reactivity of GP antibodies toward 1NC1 domains2, introduces an ambiguity with regard to the identity of the antigen binding GP antibodies.
In the present study, both pitfalls were avoided by using a capture ELISA for 3NC1 domains. In this assay, the soluble antigen (as recombinant NC1 monomers, native NC1 hexamers, or their subunits) was captured by affinity-purified GP IgG adsorbed onto a solid phase, and then GP-bound 3NC1 domains were specifically detected with mAbs reacting exclusively with 3NC1. Several anti- 3NC1 mAbs were found to detect GP-bound 3NC1domains specifically and sensitively (data not shown). For consistency, all experiments shown were performed using mAb EB3 (11) as detecting antibody. The concentration dependence for the binding of human r- 3NC1 monomers to purified GP antibodies from three different patients is shown in Fig. 1A. Half-saturation occurred at antigen concentrations ranging from 18 ± 3 to 30 ± 4 ng/ml (~0.7-1.1 nM), indicating a very high affinity of interaction with the GP antibodies. Negative controls showed the absence of non-specific interactions, demonstrating the specificity of the assay. Neither binding of r- 3NC1 monomers to non-GP human IgG, nor binding of r- 1NC1 monomers to GP antibodies were detected (Fig. 1, A and B). Thus, capture ELISA
by guest on May 27, 2018
http://ww
w.jbc.org/
Dow
nloaded from

4
provides a specific and sensitive assay for 3NC1binding to GP antibodies, while preserving the native conformation of the antigen in solution.
Using the newly developed capture ELISA, the crypticity of 3NC1 epitopes in human GBM NC1 preparations containing a mixture of 1 2and 3 4 5 hexamers could be assayed without interference from 1NC1 (Fig. 1B). The extent of
3NC1 binding to GP IgG was compared after reaction with native versus acid-treated NC1 hexamers. Similar results were obtained with four different GP antibodies. A relatively small amount of 3NC1 binding to GP IgG was consistently measured upon reaction with native NC1 hexamers (18% ± 2%, relative to r- 3NC1 positive control), which increased ~3-4 fold after acid-treatment of hexamers (to 60% ± 8%; p<0.001). This increase in antigen binding reflects the exposure of 3NC1 cryptic epitopes, likely caused by the disruption of the hexamer quaternary structure under dissociative conditions. However, it is not known whether the concomitant disruption of the secondary/tertiary structure of dissociated subunits also plays a role in unmasking the GP epitopes. This was addressed next.
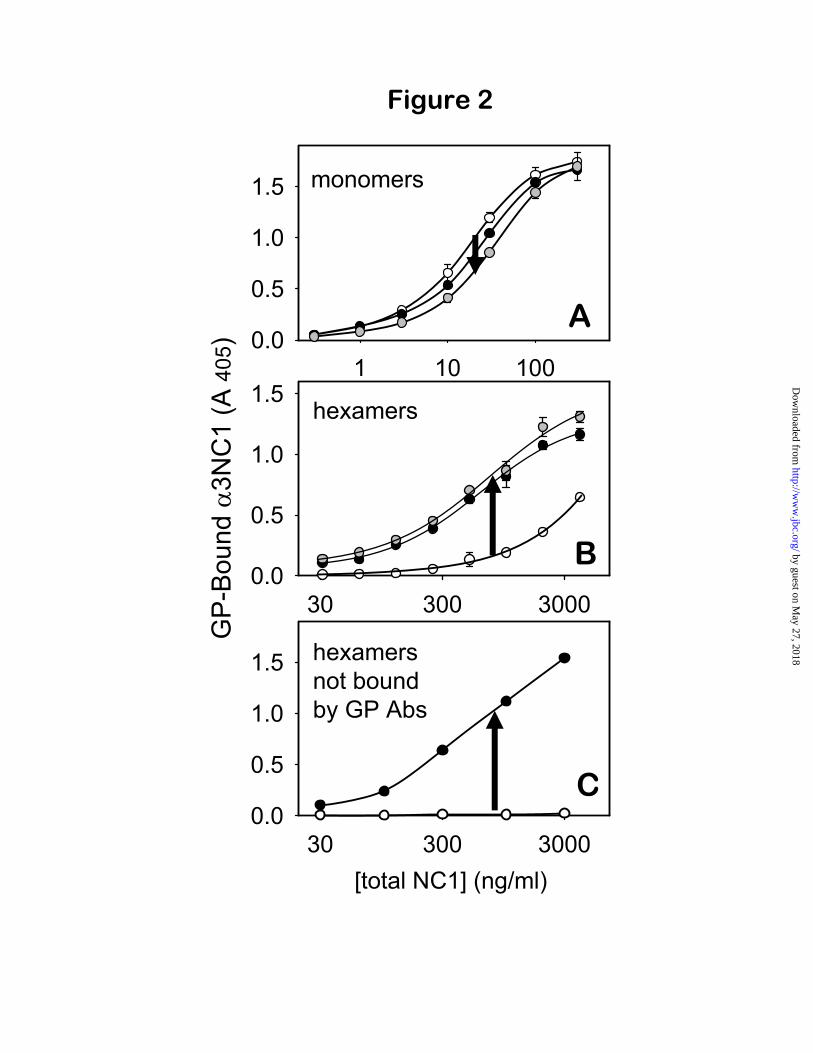
The crypticity of GP epitopes depends on the antigen quaternary structure—To determine whether dissociating conditions unmask GP epitopes by inducing changes in the antigen secondary/tertiary structure, the binding to GP IgG of human r- 3NC1 monomers (expressed in mammalian cells for native folding) was compared with and without acid-treatment (Fig. 2A). The binding of r- 3NC1 monomers to GP IgG was slightly decreased after acid treatment, shifting the binding curve toward higher concentrations of antigen, equivalent to a 25% increase in the apparent Kd (from 17.1 ± 0.9 to 21.6 ± 2 ng/ml; p=0.005). A comparable decrease in reactivity was produced by treatment of 3NC1 monomers with 3M guanidinium chloride. Thus, dissociating conditions slightly degrade the binding capacity, demonstrating that the GP epitopes are not cryptic in the 3NC1 monomers.
In contrast to 3NC1 monomers, exposure of NC1 hexamers to dissociating conditions caused a dramatic increase in binding to GP antibodies (Fig. 2B). The overall effect was a large shift of the binding curves toward lower antigen concentrations, equivalent to an 8-fold decrease in
the apparent Kd after acid-treatment, and a 10.6-fold decrease after guanidinium treatment (p<0.0001). Overall, these findings show that the GP epitopes are cryptic only in 3 4 5 NC1 hexamers, but not in 3NC1 monomers, thus demonstrating that their crypticity is dependent on the quaternary structure of the autoantigen.
Two subsets of ( 3 4 5)2 NC1 hexamers have distinct reactivities toward GP antibodies—The small amount of binding under native conditions and the 8-10 fold enhancement of binding after dissociation suggested the existence of two distinct subsets of 3 4 5 NC1 hexamers: reactive hexamers that bind GP antibodies under native conditions, and impenetrable hexamers that do not react unless dissociated into subunits. Thishypothesis was tested by immunoprecipitating human GBM hexamers with purified GP antibodies under native conditions. The GP-bound fraction, absorbed to protein G beads (the immunoprecipitate), corresponds to the subset of reactive 3 4 5-hexamers that bind GP antibodies under native conditions (open circles in Fig. 2B). In contrast, the GP-unbound fraction (the supernatant) consisted of 3 4 5-hexamers that were impenetrable by GP antibodies under native conditions (Fig. 2C, open circles). In impenetrable hexamers, the GP epitopes were fully sequestered and inert toward GP antibodies, until the quaternary structure was disrupted by dissociating conditions (Fig. 2C, closed circles).
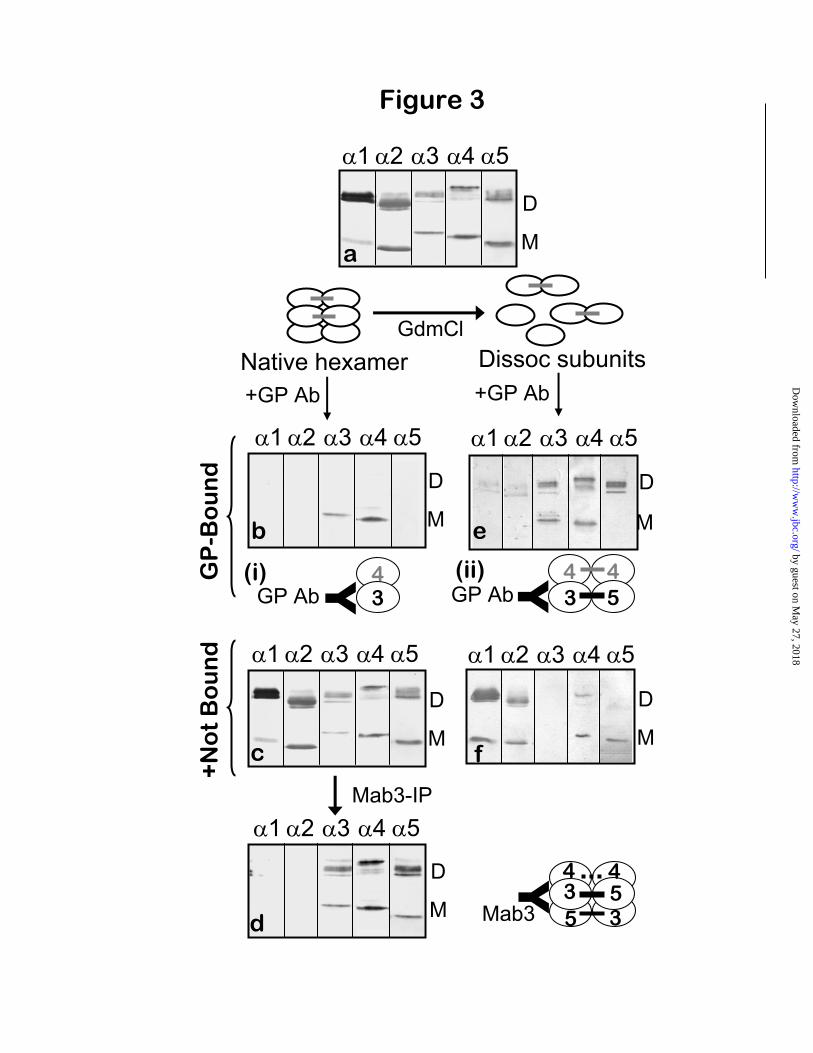
Subunit composition of GP-reactive hexamers—To explore the structural basis for the different reactivity toward GP antibodies of the two subsets of 3 4 5 NC1 hexamers, their subunit composition was determined. The GP-bound and GP-unbound fractions were analyzed by SDS-PAGE and immunoblotting with mAbs specific for 1- 5 NC1 domains. The reference sample, total GBM hexamers (Fig. 3A), contained
1 and 2 NC1 monomers and homodimers (derived from ( 1 2 1)2 NC1 hexamers), along with 3, 4 and 5 monomers, and 3/ 5 and
4/ 4 dimers (derived from ( 3 4 5)2 NC1 hexamers), as previously reported (11).
After the reaction of GBM hexamers with GP antibodies under native conditions, the GP-bound fraction was composed of 3 and 4 NC1 monomers, but devoid of 5NC1 monomers, 4-
4, and 3- 5 NC1 dimers (Fig. 3B). The same
by guest on May 27, 2018
http://ww
w.jbc.org/
Dow
nloaded from

5
results were obtained with affinity-purified GP antibodies from three other patient sera (data not shown). Co-precipitation of 4NC1 monomers was due to their non-covalent association with
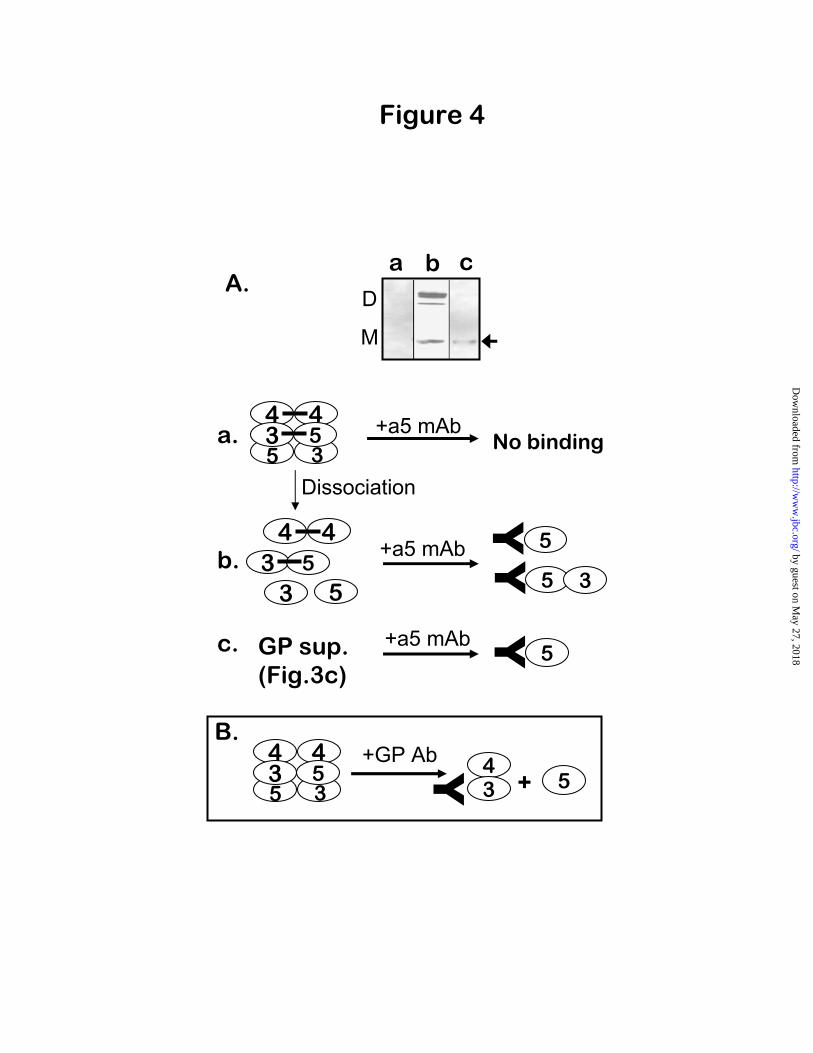
3NC1 monomers, because GP antibodies did not bind to purified 4NC1 monomers (data not shown). Since 3 and 4NC1 monomers only form hexamers together with 5NC1 monomers (10), we investigated whether the binding of GP antibodies caused the release of free 5NC1 subunits. This was verified using mAb H52 (22), which recognizes a cryptic epitope within the
5NC1 domain, and binds only to isolated 5NC1 monomer and dimer subunits (Fig. 4A, lane b) but not to intact hexamers (Fig. 4A, lane a). Importantly, immunoprecipitation with mAb H52 revealed the presence of free 5NC1 monomers (but not 5NC1 dimers) in the GP-unbound supernatant from the reaction of GBM hexamers with GP antibodies (Fig. 4A, lane c). Because mAb H52 could not react with 5NC1 from intact hexamers, the 5NC1 monomers it precipitated from the GP-supernatant could only have been produced by the dissociation of GP-reactive hexamers upon antibody binding to 3/ 4NC1 monomers (Fig. 4B). Therefore, the subset of GP-reactive hexamers was originally composed of 3,
4, and 5NC1 monomers, but devoid of dimer subunits (henceforth designated as M- 3 4 5hexamers). Thus, under native conditions, GP antibodies reacted with M- 3 4 5 hexamers with dissociation of the quaternary complex: the interactions between 3 and 5 monomer were disrupted upon antibody binding, whereas those between 3 and 4 monomers were retained.
Subunit composition of GP-impenetrable hexamers—In contrast to the GP-reactive fraction, the GP-unbound fraction containing impenetrable hexamers comprised all subunits present in the control sample, including 3, 4 and 5NC1monomers, 3- 5 NC1 heterodimers, and 4- 4NC1 homodimers. Analysis of this fraction by immunoprecipitation with Mab3 (Fig. 3D), which recognizes a non-cryptic epitope on 3NC1(17,23), produced the same profile previously reported for total GBM hexamers (10). Therefore, the impenetrable hexamers were composed of
3NC1 monomers and dimers co-existing with 4and 5NC1 monomers and dimers. Judging from
the intensity of staining, the relative proportion of NC1 monomers to dimers was reduced in the GP-impenetrable hexamers, compared to control GBM hexamers. The GP-impenetrable hexamers were designated as D- 3 4 5 hexamers, since they were distinguished from M-hexamers by the presence of 3, 4 and 5 NC1 dimers, in addition to 3, 4 and 5 NC1 monomers.
Disruption of the quaternary hexamer structure by low pH renders all 3NC1 subunits reactive with GP antibodies—Upon exposure to low pH, the impenetrable D- 3 4 5 hexamers dissociate, allowing GP antibodies to bind to
3NC1 dimer and monomer subunits. Immuno-precipitation of acid-dissociated hexamers by GP antibodies yielded 3- 5 and 4- 4 NC1 dimers along with 3 and 4 NC1 monomers (Fig. 3E). The 3 NC1 monomers and 3- 5 NC1 dimers subunits of dissociated hexamers were completely precipitated by GP antibodies, since they were not detected in the GP-unbound supernatant (Fig. 3F). In contrast, 4NC1 monomers and homodimers were present in both the immunoprecipitate and the unbound fraction, suggesting a reversible association with 3NC1 subunits, whereas 5NC1monomers were present exclusively in the unbound fraction. The absence of 5NC1 monomers from the GP-bound fraction further confirms that the binding of GP antibodies to
3NC1 subunits disrupts the 3/ 5 interactions, as found for the M- 3 4 5 hexamers. These findings imply that within the intact D- 3 4 5hexamers, the epitopes of 3NC1 monomer and dimer subunits are totally inaccessible for binding GP antibodies, whereas after epitope unmasking by hexamer dissociation by low pH, both the
3NC1 monomer and dimer subunits can bind GP antibodies. Importantly, this indicates that 3- 5NC1 dimers are impenetrable toward GP antibodies only in the context of an NC1 hexamer complex, but not as isolated NC1 subunits. Thus, within the D- 3 4 5 hexamers, the presence of dimers rendered the GP epitopes of 3NC1subunits inaccessible for antibody binding.
Overall, these findings reveal a distinction between the quaternary structures of the two hexamer subsets. One subset, composed of monomer subunits only (M- 3 4 5 hexamers), binds GP antibodies under native conditions
by guest on May 27, 2018
http://ww
w.jbc.org/
Dow
nloaded from

6
causing dissociation of subunits. The other subset is composed of dimer and monomer subunits (D-
3 4 5 hexamers) and is impenetrable toward GP antibodies unless dissociated into subunits by low pH or protein denaturant. The findings also reveal the ability of GP antibodies to disrupt the quaternary structure of M- 3 4 5 hexamers, causing dissociation into subunits and unmasking of epitopes, but not of D- 3 4 5 hexamers, indicating that the NC1 dimer subunits ( 3- 5heterodimers and 4- 4 homodimers) prevent the antibody-induced dissociation of hexamers.
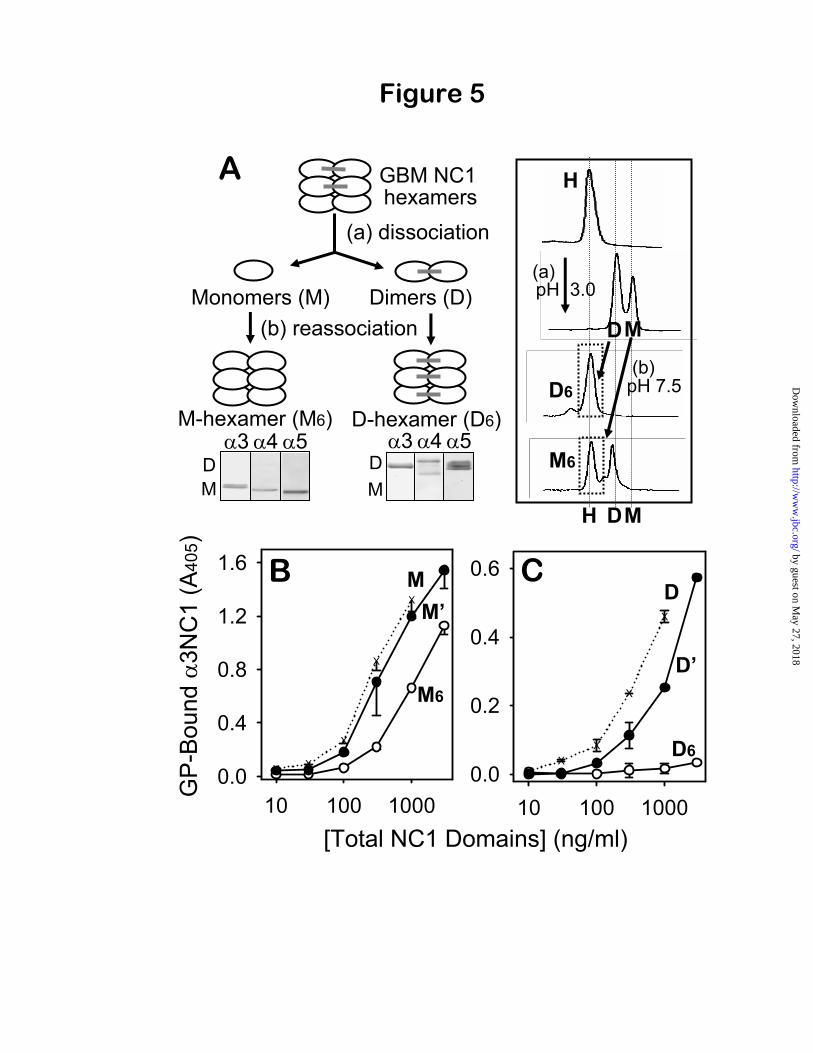
Verification of the two hexamer subsets and their distinct binding to GP antibodies—Usingreconstituted M- 3 4 5 and D- 3 4 5hexamers, the properties of the two hexamer subsets were further investigated with regard to their reactivity with GP antibodies. We have previously shown that homologous M- 1 2 and D- 1 2 hexamers can each be reconstituted from monomer and dimer subunits that were isolated from acid-dissociation of native 1 2 hexamers (24). In the present study, this strategy was used to produce in vitro reconstituted M- 3 4 5hexamers from NC1 monomer subunits and D-
3 4 5 hexamers from NC1 dimer subunits of human GBM. The composition of reconstituted hexamers was verified by Western blot (Fig. 5A), confirming that reassociated M- 3 4 5 hexamers contained 3, 4 and 5 monomer subunits only, and reassociated D- 3 4 5 hexamers contained
3, 4, and 5 dimer subunits only. The GP antibody reactivity of isolated GBM
monomers (due to 3NC1 monomers) was substantially reduced but not completely abolished upon reconstitution into M- 3 4 5 hexamers, indicating that GP epitopes become less accessible in M-hexamers. The restricted accessibility of GP epitopes within the quaternary structure was confirmed by a 3-fold increase in binding to GP IgG after acid-dissociation of reconstituted M-hexamers (Fig. 5B). In contrast, the GP antibody reactivity of isolated GBM dimers (due to 3- 5NC1 dimers) was totally abolished upon assembly into D- 3 4 5 hexamers, indicating that GP epitopes became completely inaccessible (cryptic) under native conditions within the reinforced quaternary structure (Fig. 5C). Re-dissociation of D-hexamers restored reactivity with GP
antibodies. Overall, the reconstitution studies verified that: a) 3, 4 and 5 NC1 monomers can assemble into M-hexamers, while 3- 5 and
4- 4 NC1 dimers can assemble into D-hexamers; and b) under native conditions, M-hexamers can still bind GP antibodies, but D-hexamers are impenetrable unless first dissociated by denaturing conditions. Therefore, NC1 dimers render the cryptic epitopes of D-hexamers totally inaccessible for binding GP antibodies.
The relative increase in GP reactivity of reconstituted M- and D-hexamers after acid dissociation permitted an estimation of the relative proportion of M- and D-hexamers in the human GBM preparation. The GP reactivity of native human GBM hexamers, solely due to native M-hexamers, was ~10% relative to their isolated subunits (Fig. 2B). Dissociated M-hexamers were 3-fold more reactive than native M-hexamers (Fig. 5B), thereby accounting for ~30% of the overall GP reactivity of dissociated GBM hexamers. Thus, the balance of ~70% of total GP reactivity was due to 3NC1 subunits of impenetrable D-hexamers, rendered reactive by dissociation. Therefore, a relatively small fraction of 3(IV) collagen exists in the GBM in a form that can be targeted by GP antibodies, and a majority occurs as impenetrablecomplexes not reactive with antibodies.
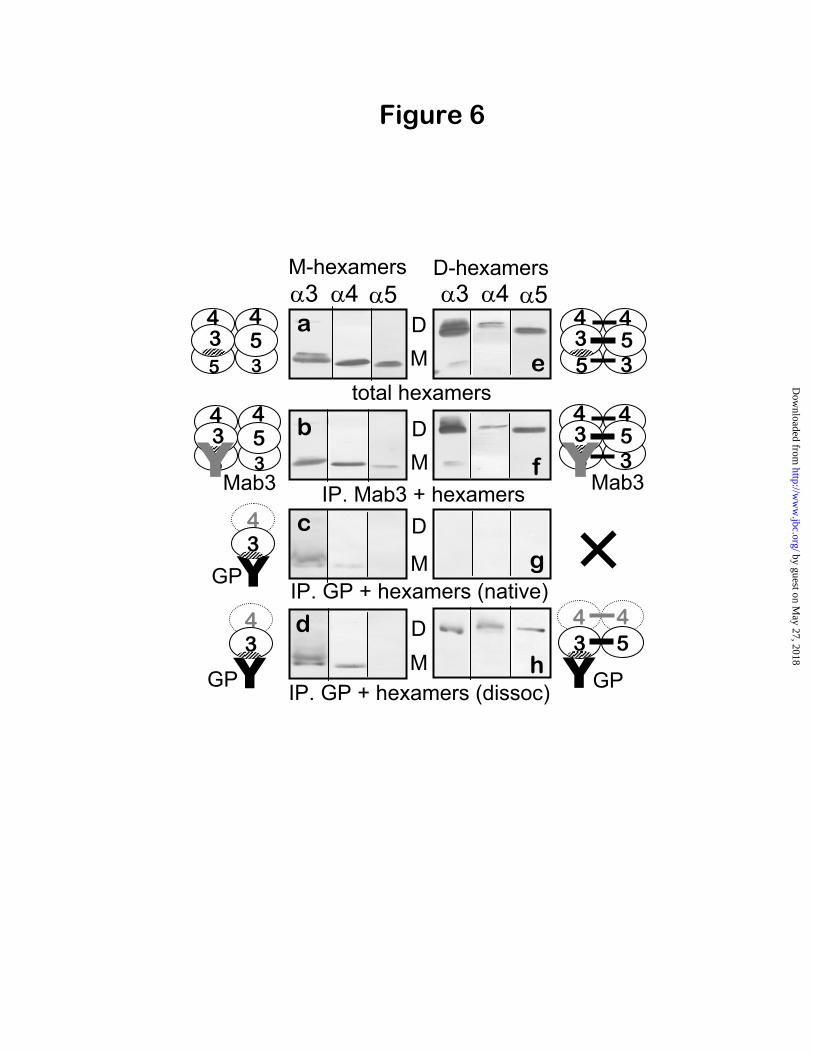
Verification of the capacity of GP antibodies to dissociate M-hexamers but not D-hexamers—The capacity of GP antibodies to dissociate M-
3 4 5 hexamers but not D- 3 4 5 hexamers was further investigated by immunoprecipitation of reconstituted M- 3 4 5 and D- 3 4 5hexamers. The reconstituted hexamers (Fig. 6) were prepared from NC1 monomers and dimers isolated from the bovine GBM because of the very limited availability of human GBM. Bovine GBM hexamers, which can be prepared in large quantities, have been well characterized and shown to be similar to human GBM hexamers with regard to subunit organization (10) and cryptic properties of the GP epitopes (13). The reconstituted M- 3 4 5 hexamers (Fig. 6, a) were analyzed with Mab3 antibody, which binds to 3NC1 subunits, but co-precipitated the 4 and
5 NC1 monomers along with 3NC1 monomers (Fig. 6, b), indicating that the 3, 4 and 5 NC1 domains occurred together in the reconstituted M-hexamers. Likewise, within the reconstituted D-
by guest on May 27, 2018
http://ww
w.jbc.org/
Dow
nloaded from

7
3 4 5 hexamers (Fig. 6, e), 3- 5 heterodimers occurred together with the 4- 4 homodimers (Fig. 6, f). Under native conditions, M-hexamers reacted with GP antibodies, which bound to the
3/ 4 NC1 monomer complex while displacing 5NC1 monomers (Fig. 6, c); this confirms the
pattern of binding observed for native M- 3 4 5hexamers from human GBM. In sharp contrast, reconstituted D-hexamers did not bind GP antibodies under native conditions (Fig. 6, g), although a complex of 3- 5 and 4- 4 NC1 dimers bound to GP IgG after dissociation (Fig. 6, h). Together, these findings verify the capacity of GP antibodies to dissociate M- 3 4 5 hexamers and unmask GP epitopes for binding by displacing
5NC1 monomers, while retaining the interaction between the 3NC1 and 4NC1 monomers. Moreover, these findings demonstrate that the impenetrably of D- 3 4 5 hexamers to GP antibodies is due to the dimers subunits, which stabilize the quaternary structure and prevent hexamer dissociation by GP antibodies.
DISCUSSION
This study identified several novel features of the biochemical mechanisms underlying epitope crypticity and how GP antibodies bind to native NC1 hexamers of collagen IV. First, a newly developed specific capture ELISA provided a strategy to validate the cryptic properties of GP epitopes and to ascertain that their crypticity is dependent on the hexamer quaternary structure. Second, the difference in binding capacity under native and dissociative conditions revealed the existence of two distinct subsets of hexamers differing in quaternary structure: a) reactive M-
3 4 5 hexamers, composed of NC1 monomer subunits only, which binds GP antibodies under native conditions, and b) impenetrable D- 3 4 5hexamers, composed of dimer and monomer subunits, which do not bind autoantibodies unless dissociated into subunits by low pH or protein denaturants. Third, the disruption of the interactions between 3 and 5NC1 monomer subunits demonstrated the capacity of GP antibodies to selectively dissociate M- 3 4 5hexamers (but not D- 3 4 5 hexamers), unmasking cryptic epitopes in the 3NC1 subunit. Finally, the impenetrably of D- 3 4 5 hexamers
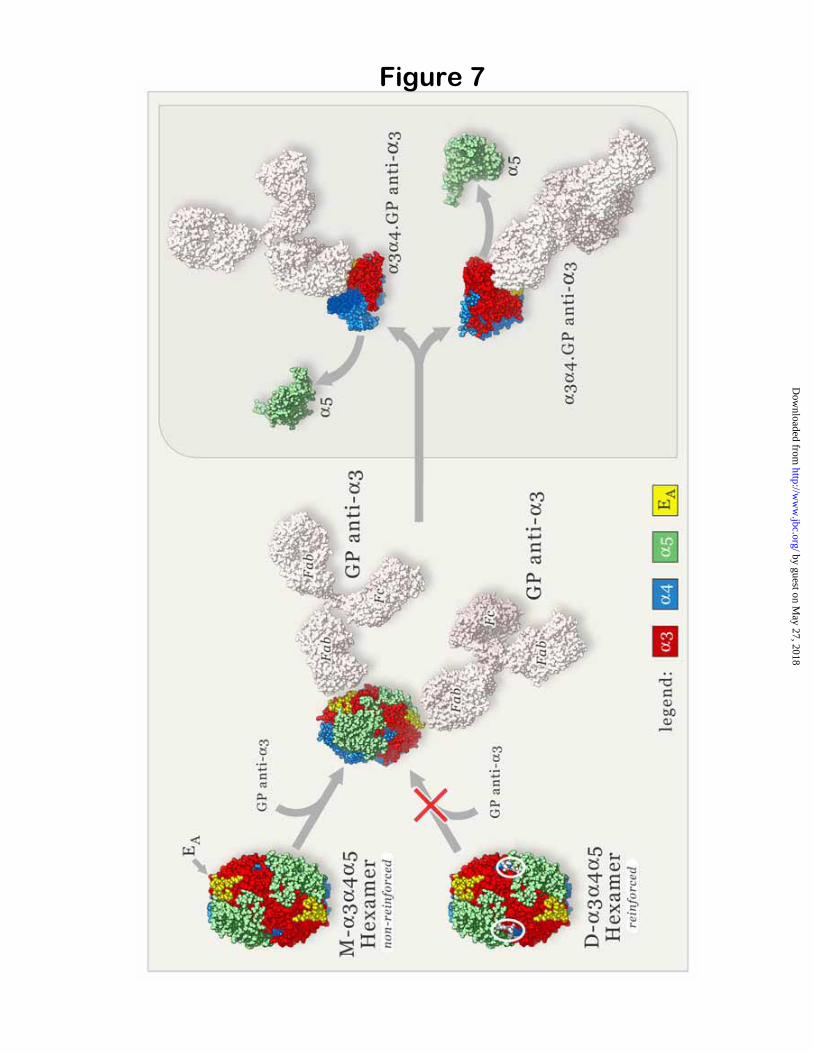
for GP antibodies under native conditions, contrasting with the reactivity of M- 3 4 5hexamers, suggests that resistance to dissociation by GP antibodies is due to NC1 dimers stabilizing the hexamer quaternary structure. Together, these finding provide the foundation for a new paradigm for the mechanism of immune privilege in the natively occurring GP autoantigen and its interaction with autoantibodies (Fig. 7).
The characteristic feature of D-hexamers is the presence of NC1 dimer subunits resistant to dissociation by denaturing agents or SDS-PAGE. The stabilizing event associated with the formation of non-dissociable NC1 dimers is necessary to render the D-hexamers impenetrable by GP antibodies, with potential impact on both the etiology and the pathogenesis of the disease. In the absence of this structural reinforcement, the formation of the M- 3 4 5 NC1 hexamer complex brings the GP epitopes in close proximity to 4 and 5NC1 domains and limits their accessibility, but is not sufficient to avert reaction with GP antibodies. These findings establish the structural reinforcement of the quaternary structure as a novel molecular mechanism of immune privilege for potential autoantigens.
The nature of the molecular interactions underpinning the formation of NC1 dimers has been extensively studied in the homologous ( 1 2 1)2 hexamers, but the detailed mechanism remains unknown. A subset of M- 1 2 1hexamers composed of 1 and 2 NC1 monomers exists in the bovine lens capsule basement membrane, and dimer-rich D- 1 2 1 hexamers are predominant in the placenta basement membrane (24). The X-ray structures of M- and D- 1 2 1 hexamers (18,25) disproved the long-held view that NC1 dimers were generated by an inter-chain disulfide exchange. A novel thioether cross-link between opposing Met-93 and Lys-211 at the NC1 trimer-trimer interface was proposed based on the 1.9Å X-ray crystal structure of D-
1 1 2 from placenta basement membrane (25), but no cross-links were found at a higher resolution of 1.5Å (24). However, the comparative analysis of their NC1 monomer and dimer subunits by trypsin digestion followed by mass spectrometry and Edman degradation is consistent with a cross-link between Met-93 and a Lys-211 post-translationally modified to hydroxylysine3.
by guest on May 27, 2018
http://ww
w.jbc.org/
Dow
nloaded from

8
An analogous interactions (represented in Fig. 7 by white ellipses at the trimer-trimer interface of reinforced hexamers) may play a key role in the structural reinforcement of D- 3 4 5 hexamers.
Within the GBM, only the subset of M-3 4 5 NC1 hexamers lacking reinforcement of
the quaternary structure has the propensity to react with circulating GP autoantibodies under native conditions. Disruption of the quaternary structure upon autoantibody binding provides a mechanism whereby pathogenic antibodies can gain access to cryptic epitopes under native conditions. The GP antibodies unmask cryptic epitopes on the 3NC1 monomer by displacing the 5NC1 subunit, while leaving the 3NC1 monomer in complex with the
4NC1 monomer. Selective dissociation of 5 but not of 4NC1 monomers from susceptible hexamers implicates the immunodominant EAepitope (14) as both the culprit as well as the victim. Our results are consistent with exposure of the EA epitope, because it is located near the junction of the 3 and 5NC1 monomers within a hexamer structure, and is inaccessible for binding when bound to the 5NC1 monomer (11). In contrast, the EB epitope is located near the junction of 3 and 4NC1 monomers and is inaccessible when 3NC1 is bound to the 4NC1 monomer. Thus, the findings implicate the immunodominant GPA antibodies, targeting the EA epitope (8), in the dissociation event. This may provide a rationale for the observation that the prognosis for kidney survival is inversely correlated with titers of GPAbut not of GPB antibodies (9).
In conclusion, our findings provide new insights into the pathogenic mechanisms of GP disease. Although 3(IV) collagen is present in the thymus and available for induction of T cell tolerance (26), autoreactive T-cells still escape elimination and circulate (27). The crypticity of the GP epitopes in tissue basement membranes makes it unlikely, however, that autoreactive T
cells in the peripheral immune system will expand without an external precipitating event. Putative pathogenic factors include exposure to solvents (28,29), inhaled smoke (30), or endogenous reactive oxygen species (15). Decondensation of
3 4 5 hexamers by non-immune mechanisms in a host expressing immune susceptibility genes (31,32) might activate residual auto-reactive helper T cells that expand the B cell repertoire against GP epitopes encoded by the 3(IV) collagen chain (5). Our results suggest that GP antibodies could further damage the GBM by accelerating epitope exposure. Circulating GP antibodies would readily target newly synthesized
3 4 5(IV) collagen protomers in the GBM before the structural stabilization of the hexamers could occur, thus perpetuating the inflammatory response. This may explain the therapeutic effect of plasmapheresis, whereby removal of circulating autoantibodies would break this vicious circle, facilitating GBM repair.
Differences among individuals in the 3NC1 monomer-dimer ratio in kidneys and lungs may also account for the variability in lung hemorrhage associated with glomerulonephritis. By Western blot, whereas 3NC1 monomer subunits are uniformly present and relatively constant in the human GBM from different individuals, they are frequently not found in the lung alveolar basement membranes (33). The absence of 3NC1 monomers—implying an absence of GP-reactive M- 3 4 5 hexamers—in the lungs of some patients may explain the phenotype of anti-GBM disease without pulmonary hemorrhage. Finally, the well-recognized difficulty in inducing anti-GBM disease in mice may be related to the relative impenetrability of their GBM hexamers, which in turn is determined by the higher content of 3NC1 dimers in the murine GBM, compared to that found in the human kidneys (34).
REFERENCES
1. Ferguson, T. A., and Griffith, T. S. (1997) Immunol Rev 156, 167-184 2. D'Orazio, T. J., and Niederkorn, J. Y. (1998) J Immunol 160, 2089-2098 3. Sonoda, K. H., Exley, M., Snapper, S., Balk, S. P., and Stein-Streilein, J. (1999) J Exp Med 190,
1215-1226 4. Griffith, T. S., Brunner, T., Fletcher, S. M., Green, D. R., and Ferguson, T. A. (1995) Science 270,
1189-1192
by guest on May 27, 2018
http://ww
w.jbc.org/
Dow
nloaded from

9
5. Hudson, B. G., Tryggvason, K., Sundaramoorthy, M., and Neilson, E. G. (2003) N Engl J Med 348,2543-2556
6. Lerner, R. A., Glassock, R. J., and Dixon, F. J. (1967) J Exp Med 126, 989-1004 7. Saus, J., Wieslander, J., Langeveld, J. P., Quinones, S., and Hudson, B. G. (1988) J Biol Chem 263,
13374-13380 8. Netzer, K. O., Leinonen, A., Boutaud, A., Borza, D. B., Todd, P., Gunwar, S., Langeveld, J. P., and
Hudson, B. G. (1999) J Biol Chem 274, 11267-11274 9. Hellmark, T., Segelmark, M., Unger, C., Burkhardt, H., Saus, J., and Wieslander, J. (1999) Kidney
Int 55, 936-944 10. Boutaud, A., Borza, D. B., Bondar, O., Gunwar, S., Netzer, K. O., Singh, N., Ninomiya, Y., Sado,
Y., Noelken, M. E., and Hudson, B. G. (2000) J Biol Chem 275, 30716-30724 11. Borza, D. B., Bondar, O., Todd, P., Sundaramoorthy, M., Sado, Y., Ninomiya, Y., and Hudson, B. G.
(2002) J Biol Chem 277, 40075-40083 12. Yoshioka, K., Michael, A. F., Velosa, J., and Fish, A. J. (1985) Am J Pathol 121, 156-165 13. Wieslander, J., Langeveld, J., Butkowski, R., Jodlowski, M., Noelken, M., and Hudson, B. G. (1985)
J Biol Chem 260, 8564-8570 14. Borza, D. B., Netzer, K. O., Leinonen, A., Todd, P., Cervera, J., Saus, J., and Hudson, B. G. (2000) J
Biol Chem 275, 6030-6037 15. Kalluri, R., Cantley, L. G., Kerjaschki, D., and Neilson, E. G. (2000) J Biol Chem 275, 20027-20032 16. Gunnarsson, A., Hellmark, T., and Wieslander, J. (2000) J Biol Chem 275, 30844-30848 17. David, M., Borza, D. B., Leinonen, A., Belmont, J. M., and Hudson, B. G. (2001) J Biol Chem 276,
6370-6377 18. Sundaramoorthy, M., Meiyappan, M., Todd, P., and Hudson, B. G. (2002) J Biol Chem 277, 31142-
31153 19. Kale, L., Skeel, R., Bhandarkar, M., Brunner, R., Gursoy, A., Krawetz, N., Phillips, J., Shinozaki, A.,
Varadarajan, K., and Schulten, K. (1999) J Comp Phys 151, 283-312 20. Harris, L. J., Larson, S. B., Hasel, K. W., and McPherson, A. (1997) Biochemistry 36, 1581-1597 21. Schwab, C., and Bosshard, H. R. (1992) J Immunol Methods 147, 125-134 22. Sado, Y., Kagawa, M., Kishiro, Y., Sugihara, K., Naito, I., Seyer, J. M., Sugimoto, M., Oohashi, T.,
and Ninomiya, Y. (1995) Histochem Cell Biol 104, 267-275 23. Johansson, C., Butkowski, R., and Wieslander, J. (1991) Connect Tissue Res 25, 229-241 24. Vanacore, R. M., Shanmugasundararaj, S., Friedman, D. B., Bondar, O., Hudson, B. G., and
Sundaramoorthy, M. (2004) J Biol Chem 279, 44723-44730 25. Than, M. E., Henrich, S., Huber, R., Ries, A., Mann, K., Kuhn, K., Timpl, R., Bourenkov, G. P.,
Bartunik, H. D., and Bode, W. (2002) Proc Natl Acad Sci U S A 99, 6607-6612 26. Wong, D., Phelps, R. G., and Turner, A. N. (2001) Kidney Int 60, 1777-1783 27. Salama, A. D., Chaudhry, A. N., Ryan, J. J., Eren, E., Levy, J. B., Pusey, C. D., Lightstone, L., and
Lechler, R. I. (2001) J Am Soc Nephrol 12, 1908-1915 28. Bombassei, G. J., and Kaplan, A. A. (1992) Am J Ind Med 21, 141-153 29. Stevenson, A., Yaqoob, M., Mason, H., Pai, P., and Bell, G. M. (1995) QJM 88, 23-28 30. Donaghy, M., and Rees, A. J. (1983) Lancet 2, 1390-1393 31. Burns, A. P., Fisher, M., Li, P., Pusey, C. D., and Rees, A. J. (1995) Qjm 88, 93-100 32. Kalluri, R., Danoff, T. M., Okada, H., and Neilson, E. G. (1997) J Clin Invest 100, 2263-2275 33. Yoshioka, K., Iseki, T., Okada, M., Morimoto, Y., Eryu, N., and Maki, S. (1988) Clin Exp Immunol
74, 419-424 34. Heidet, L., Borza, D. B., Jouin, M., Sich, M., Mattei, M. G., Sado, Y., Hudson, B. G., Hastie, N.,
Antignac, C., and Gubler, M. C. (2003) Am J Pathol 163, 1633-1644
by guest on May 27, 2018
http://ww
w.jbc.org/
Dow
nloaded from

10
FOOTNOTES
* Figure 7 was produced by J.P. Cartailler of Symmation LLC (Nashville, TN). This work was supported by grants P01 DK65123 (to D.B.B.), R01 DK46282 (to E.G.N.), and R37 DK18381 (to B.G.H.) from the National Institutes of Health, and by the Carl W. Gottschalk Research Scholar Award (to. D.B.B.) from the American Society of Nephrology.
1The abbreviations used are: GBM, glomerular basement membrane; GP, Goodpasture; NC1, the noncollagenous domain of type IV collagen
2In indirect ELISA using equal amounts of recombinant monomers, cross-reactivity of GP antibodies toward 1NC1 is 7 ± 4% of the binding to 3NC1 (ref. 8), but reaches 33 ± 11% when 1NC1 is in 10-fold excess over 3NC1 (unpublished observations)—as found in human GBM NC1 hexamers. This cross-reactivity may be an artifact of indirect ELISA, because, as shown in Fig. 3, GP antibodies do not immunoprecipitate 1NC1 domains from solution.
3R.M. Vanacore, D.B. Friedman, A.J.L. Ham, M. Sundaramoorthy, and B.G. Hudson. Manuscript submitted to J. Biol. Chem.
FIGURE LEGENDS
Fig. 1. A specific capture ELISA assay for 3NC1 binding to GP antibodies. Panel A: Binding of r-3NC1 monomers to immobilized GP antibodies, affinity-purified from three patient sera (filled circles),
or to normal human IgG (open circles), used as negative control, was measured by capture ELISA using anti- 3NC1 mAb EB3 as detecting antibody. Panel B: The crypticity of GP epitopes was assayed by capture ELISA by comparing the binding of native vs. dissociated human GBM hexamers (500 ng/well) to four different GP antibodies (cross-hatched bars). There was no binding to normal human IgG (solid bars). Recombinant 3NC1 monomers (50 ng/well) were used as positive control, and an excess of r-
1NC1 monomers (500 ng/well) as negative control.
Fig. 2. Dissociating agents unmask cryptic GP epitopes on 3 4 5NC1 hexamers but not on 3NC1monomers. Binding of r- 3NC1 monomers (A), untreated GBM hexamers (B), and GBM hexamers pre-absorbed with GP antibodies (C) to immobilized GP antibodies was measured by capture ELISA. GP-absorbed hexamers (C) were prepared by incubating GBM hexamers overnight with an excess of purified GP antibodies, under native conditions, then removing the immune complexes using immobilized protein G. Binding curves were compared for native, untreated antigens (open symbols) vs. antigens exposed to dissociating conditions (gray symbols: 6 M guanidinium hydrochloride; filled symbols: 0.1 M glycine, pH 2.2). Arrows indicate an increase or a decrease in GP reactivity induced by dissociating agents.
Fig. 3. Subunit composition of GP-bound and GP-unbound fractions of human GBM hexamers. NC1 hexamers from human GBM (a) were incubated with GP antibodies under native conditions (left) or after acid dissociation (right). The GP-bound fractions from native (b) or dissociated (e) hexamers were precipitated with protein G beads and resolved by SDS-PAGE into monomer (M) and dimer (D) subunits, the identity of which was established by Western blot. For each sample, five replicate strips were individually stained with chain-specific mAbs against the 1- 5 NC1 domains. The supernatants containing the GP-unbound fractions from native (c) and dissociated (f) hexamers were analyzed in parallel. The GP-unbound fraction from native hexamers (c) was further analyzed by immunoprecipitation with Mab3 (d), which binds to NC1 hexamers containing 3NC1 subunits.
Fig. 4. Binding of GP antibodies to reactive GBM hexamers releases free 5NC1 monomers. Panel A: Native human GBM hexamers (a), hexamer subunits produced by hexamer dissociation with 6 M guanidine hydrochloride for 15 minutes at 60°C (b), and the GP-unbound fraction (the supernatant) from the reaction of GP antibodies with native hexamers (c), were immunoprecipitated with rat mAb H52,
by guest on May 27, 2018
http://ww
w.jbc.org/
Dow
nloaded from

11
recognizing a cryptic epitope on the 5NC1 domains. The presence of 5NC1 domains in the H52-precipitated fractions was determined by Western blot using an anti- 5NC1 mouse mAb, Mab5. Panel B: Because native NC1 hexamers do not react with mAb H52, precipitation of 5NC1 monomers by H52 from the GP-unbound supernatant indicates the presence of free 5NC1 monomers, which must have been dissociated by GP antibodies binding to hexamers composed of 3, 4 and 5NC1 monomers.
Fig. 5. Binding of GP antibodies to M- and D-hexamers reconstituted from NC1 monomers and dimers. Panel A: Preparation of M- and D-hexamers is illustrated schematically (left). The NC1 monomer (M) and dimer (D) subunits of human GBM hexamers were separated by gel filtration at pH 2.5, then re-associated in vitro into M-hexamers (M6) and D-hexamers (D6). Composition of M- and D-hexamers was verified by Western blot for 3- 5 NC1 domains. Gel-filtration profiles of native, dissociated, and re-associated hexamers are shown in the inset (right). Panel B: Binding of monomers (M, dotted line), M-hexamers (M6, open symbols), and acid-dissociated M-hexamer subunits (M’, closed symbols) to GP antibodies was measured by capture ELISA. Panel C: Binding of dimers (D, dotted line), D-hexamers (D6, open symbols), and acid-dissociated D-hexamer subunits (D) to GP antibodies was measured by capture ELISA.
Fig. 6. Immunoprecipitation analysis of reassociated M-hexamers and D-hexamers. M-hexamers (a-d) and D-hexamers (e-h), reassociated from isolated bovine GBM NC1 monomers and dimers, were analyzed by immunoblotting with mAbs to 3, 4, and 5 NC1 domains, directly (a, e) and after immunoprecipitation with Mab3 (b, f) or GP antibodies (c, g). Acid-dissociated M- and D-hexamers were also immunoprecipitated with GP antibodies (d, h).
Fig. 7. A new paradigm for the interaction of GP autoantibodies with natively occurring ( 3 4 5)2 NC1 hexamers within the GBM. Space-filling structural models for the ( 3 4 5)2 NC1 hexamers and IgG molecules are shown at atomic scale. The subset of M- 3 4 5 hexamers lacking reinforcement reacts with GP antibodies (GP anti- 3) with concomitant disruption of the quaternary structure. One possible mechanism of reaction is illustrated. In NC1 hexamers, certain epitope residues in the EA region (yellow) of the 3 NC1 subunits (red) are masked by the proximity to 5 NC1 subunits (green), whereas other residues are accessible and may initially bind autoantibodies with lower affinity. An antibody-induced conformational change in the 3NC1 could then cause the dissociation of 5NC1 monomers, completely unmasking the EA epitope and allowing unrestricted, high-affinity binding of autoantibodies to 3NC1monomers. Alternatively, autoantibodies may competitively displace 5NC1 subunits because they have higher affinity for 3NC1, reaching the same final state. In contrast, the subset of reinforced D- 3 4 5hexamers is impenetrable to GP antibodies under native conditions. The putative site of cross-link, proposed to involve Met-93 and Lys-211 (or Hyl-211) by analogy to D- 1 2 1 hexamers, is represented by white ellipses at the NC1 trimer-trimer interface of the reinforced hexamer. This reinforcement represents a novel molecular mechanism of immune privilege for potential autoantigens.
by guest on May 27, 2018
http://ww
w.jbc.org/
Dow
nloaded from
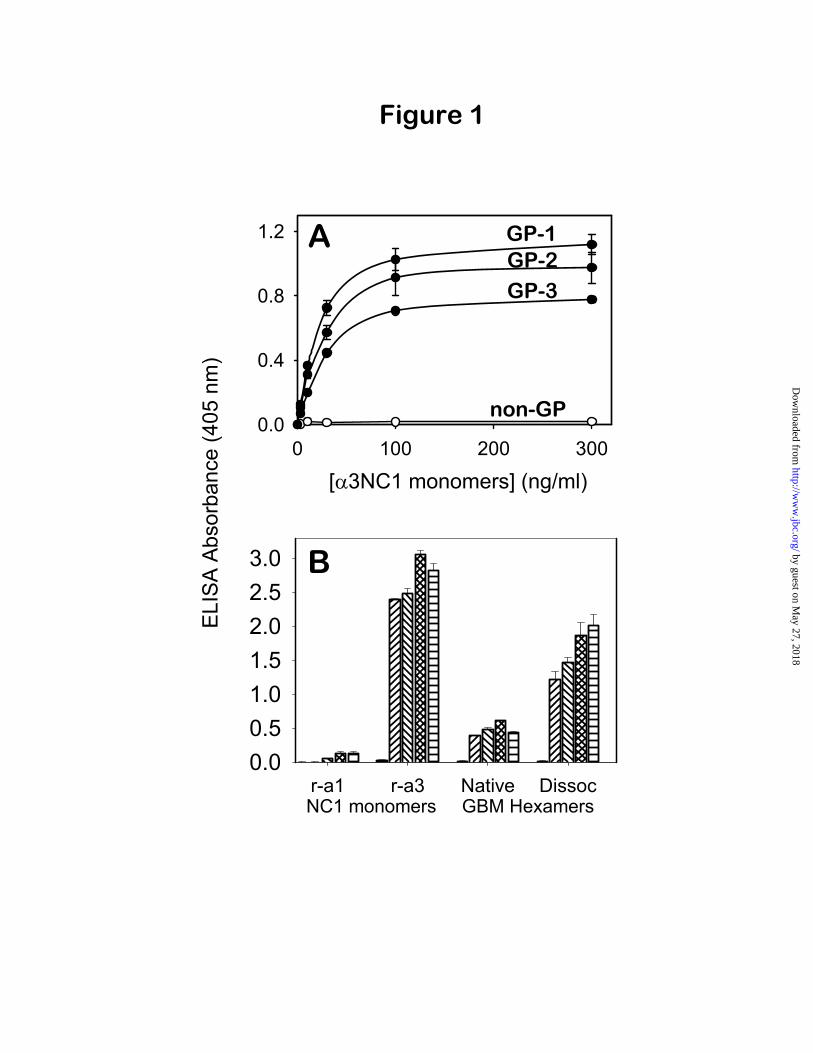
Figure 1
ELI
SA
Abs
orba
nce
(405
nm
)
r-a1 r-a3 Native Dissoc0.00.51.01.52.02.53.0
NC1 monomers GBM Hexamers
0 100 200 3000.0
0.4
0.8
1.2
[ 3NC1 monomers] (ng/ml)
B
A GP-1GP-2GP-3
non-GP
by guest on May 27, 2018
http://ww
w.jbc.org/
Dow
nloaded from
1 10 1000.0
0.5
1.0
1.5
[total NC1] (ng/ml) 30 300 3000
0.0
0.5
1.0
1.5
30 300 30000.0
0.5
1.0
1.5
GP
-Bou
nd3N
C1
(A 4
05)
hexamers
monomers
B
C
hexamersnot boundby GP Abs
A
Figure 2
by guest on May 27, 2018
http://ww
w.jbc.org/
Dow
nloaded from
1 2 3 4 5
Mab3-IP
a
b
c
d
D
M
D
M
D
M
D
M
1 2 3 4 5
D
M
D
M
1 2 3 4 5
4Y
GP Ab
GdmClNative hexamer Dissoc subunits
GP
-Bo
un
d+
No
t B
ou
nd
4 43 5
Y
GP Ab
1 2 3 4 51 2 3 4 5
1 2 3 4 5
+GP Ab +GP Ab
e
f
54
334
5Mab3
Y
(i) (ii)3
Figure 3
by guest on May 27, 2018
http://ww
w.jbc.org/
Dow
nloaded from
a b c
D
M
a.
b.
c. GP sup.(Fig.3c)
5
Y
5
Y
5
Y
3
No binding+a5 mAb
+a5 mAb
+a5 mAb
43
4535
4
33
4
55
Dissociation
43 +
+GP Ab
A.
B.43
4535
Y
5
Figure 4
by guest on May 27, 2018
http://ww
w.jbc.org/
Dow
nloaded from
10 100 10000.0
0.2
0.4
0.6
XX
X
X
X
[Total NC1 Domains] (ng/ml)10 100 1000
GP
-Bou
nd3N
C1
(A40
5)
0.0
0.4
0.8
1.2
1.6
X X
X
X
X
B C
3 4 5DM
DM
3 4 5
GBM NC1 hexamers
D-hexamer (D6)M-hexamer (M6)
Dimers (D)Monomers (M)(b) reassociation
(a) dissociation
A
pH 3.0
pH 7.5
H
DM
D6
M6
(a)
(b)
D6
MM’
M6
D
D’
H
DM
Figure 5
by guest on May 27, 2018
http://ww
w.jbc.org/
Dow
nloaded from
3 4 5 3 4 5
total hexamers
IP. Mab3 + hexamers
IP. GP + hexamers (native)
IP. GP + hexamers (dissoc)
DM
DM
D
M
DM
M-hexamers D-hexamers
3554
34
5 3
43
Y4 43 5
Y
54
334
5
54
334
53554
34
5 3
43
Y
Mab3 Mab3
GP
GP GP
YY
a
b
c
d
e
f
g
h
Figure 6
by guest on May 27, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Neilson and Billy G. HudsonDorin-Bogdan Borza, Olga Bondar, Selene Colon, Parvin Todd, Yoshikazu Sado, Eric G.
immune privilege and autoimmune pathogenesisautoantigen complexes lacking structural reinforcement: Novel mechanisms for Goodpasture autoantibodies unmask cryptic epitopes by selectively dissociating
published online May 24, 2005J. Biol. Chem.
10.1074/jbc.M504050200Access the most updated version of this article at doi:
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
by guest on May 27, 2018
http://ww
w.jbc.org/
Dow
nloaded from