development of an electrotransformation protocol for genetic manipulation of clostridium...
TRANSCRIPT

Development of an electrotransformation
protocol for genetic manipulation of Clostridium pasteurianum
Michael E Pyne1, Murray Moo-Young1, Duane A Chung1,2* and C Perry Chou1*
Published: 9 April 2013
www.ncbi.nlm.nih.gov/pubmed/23570573

Abstract• Industrial biofuels proliferation and co-integration with fossil
fuels. Reducing the production cost,increasing Revenues.
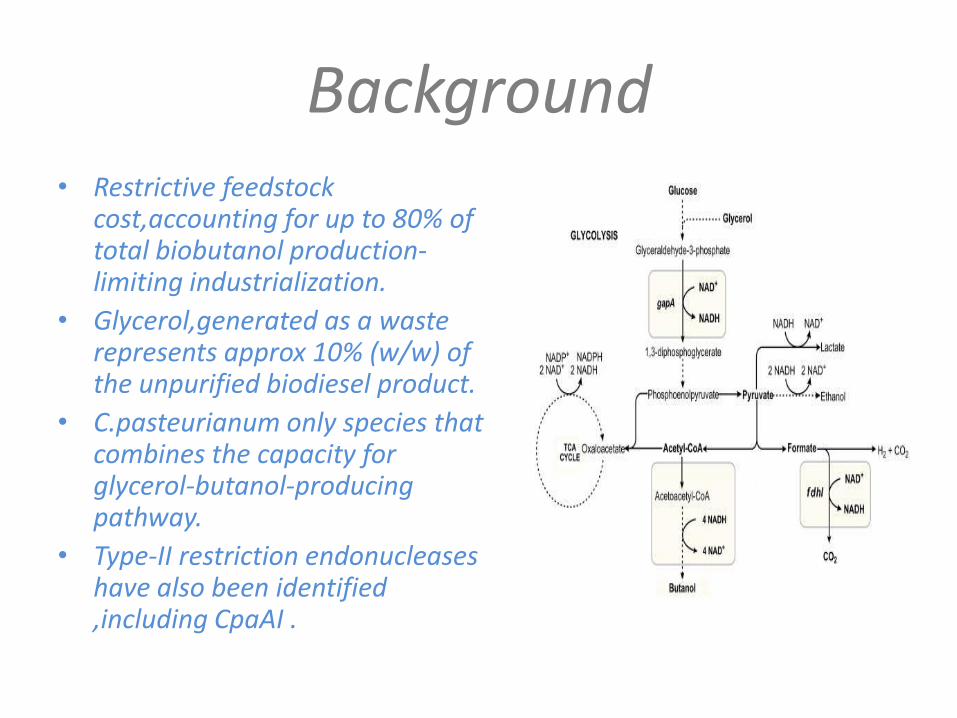
• Anaerobe Clostridium pasteurianum, the only microorganism known to convert glycerol alone directly into butanol.
• The development of an electrotransformation protocol permitting high-level DNA transfer to C. pasteurianum ATCC 6013.
• Key factors affecting-CpaAI restriction-modification system, cell-wall-weakening using glycine,ethanol-mediated membrane solubilization, field strength of the electric pulse, and sucrose osmoprotection.
Background• Restrictive feedstock
cost,accounting for up to 80% of total biobutanol production-limiting industrialization.
• Glycerol,generated as a waste represents approx 10% (w/w) of the unpurified biodiesel product.
• C.pasteurianum only species that combines the capacity for glycerol-butanol-producing pathway.
• Type-II restriction endonucleaseshave also been identified ,including CpaAI .
Protection of plasmid DNA from CpaAIrestriction
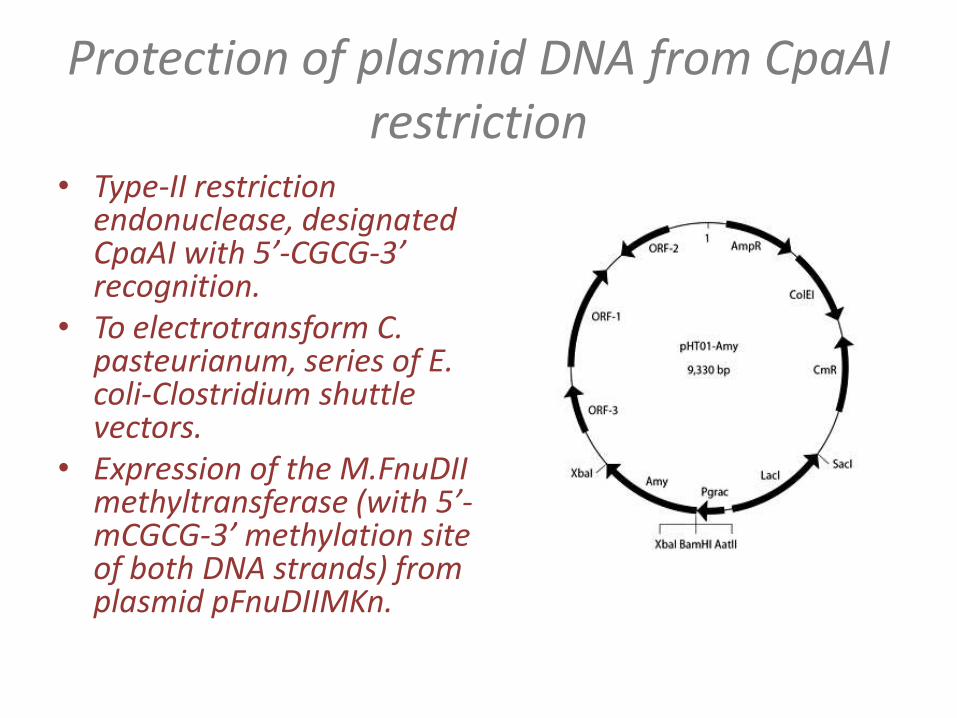
• Type-II restriction endonuclease, designated CpaAI with 5’-CGCG-3’ recognition.
• To electrotransform C. pasteurianum, series of E. coli-Clostridium shuttle vectors.
• Expression of the M.FnuDIImethyltransferase (with 5’-mCGCG-3’ methylation site of both DNA strands) from plasmid pFnuDIIMKn.

High-level electrotransformation of C. pasteurianum
• Cell-wall-weakening-glycine, DL-threonine,lysozyme, and penicillin G.
• Individually,were screened for the effect of glycine and DL-threonine by supplying the additives in the presence of 0.25 M sucrose.
• Lysozyme and penicillin G were screened by addition at the wash stage.
• Only glycine and DL-threonine improved the electrotransformation efficiency.
• Glycine regimen with respect to concentration and duration of exposure.

High-level electrotransformation of C. pasteurianum
• Osmoprotection-0.27 M sucrose was adopted as the optimum sucrose concentration.
• Cell membrane solubilization-Ethanol added at 5 and 10% provided a 1.6- and 1.3-fold respective increase in electrotransformation efficiency.
• Electric pulse parameters-low voltages in the range of 1.8-2.0 kV,Pulse duration changes were not predictive of the effects on electrotransformation efficiency.

High-level electrotransformation of C. pasteurianum
• DNA quantity and outgrowth duration-number of transformants increase linearly between 0.5 and 5.0 μg of pMTL85141, the greatest efficiency occurred using 0.5 μg of plasmid DNA.
• The greatest electrotransformationefficiency was attained at 16 hours.
• 7.5 × 104 transformants μg-1 DNA, an increase of more than three orders of magnitude.

Preparation of protoplasts and assay of CpaAI activity
C. pasteurianum 100 ml culture (OD600 of 0.4-0.6)
+
25 ml of protoplast buffer (25 mM potassium phosphate,pH 7.0+6 mMMgSO4+15% lactose+200 μg/ml lysozyme)
Incubation for 45 minutes anaerobically at 37°C
25 ml of protoplasts (centrifugation-8,500×g for 20 minutes lysed in 20 ml of TEMK buffer (4 mM Tris–HCl,pH 8.0+0 mM EDTA+6.6 mM 2-mercaptoethanol+25 mMKCl),
Incubation at 37°C for 1 hour
centrifugation-20,000×g for 15 minutes supernatants containing protoplast extracts stored at −80°C.
1.0 μg plasmid DNA+25% protoplast lysate+20 μl of 1× CpaAI reaction buffer (6 mMTris–HCl,pH 7.4+6 mM MgCl2+6 mM 2-mercaptoethanol).
At 37°C for 2–4 hours

DNA Isolation and manipulation• Plasmid DNA was extracted and purified from C.pasteurianum
•
• 3–9 ml-late-exponential phase cells were collected by centrifugation and washed twice in KET buffer(0.5 M KCl+0.1 M EDTA+0.05 M Tris–HCl, pH 8.0)+200 μl SET buffer (25% sucrose, 0.05 M EDTA,and 0.05 M Tris–HCl, pH 8.0)+5 mg/ml lysozyme
• Incubated anaerobically at 37°C for 20mins
• RNase A in 100 μg/ml and cell lysis and plasmid purification+400 μl of alk.SDS sol II.
• Colony PCR of C.pasteurianum by suspending single colonies in 50 μl colony lysisbuffer (20 mM Tris–HCl,pH 8.0+2 mM EDTA +1% Triton X-100).
• Microwave Heating for 2 minutes at maximum power setting+1μl of the cell suspension+9μl PCR containing Std Taq DNA Polymerase. An initial denaturation of 5 minutes at 95°C was employed to further cell lysis.

Vector construction• Plasmid pFnuDIIMKn was derived from pFnuDIIM to allow
methylation of E. coli-C. pasteurianum shuttle vectors.
• Firstly FRT-kan-FRT PCR cassette was amplified from plasmid pKD4 and inserted into the MCS of BlpI/XhoI-digested pET-20b(+) to generate pETKnFRT.
• Then FRT-kan-FRT cassette was digested out of pETKnFRTusing ScaI and EcoRI and subcloned into the corresponding restriction sites within the catP gene of pFnuDIIM to yield pFnuDIIMKn.

Preparation of electrocompetent cells andelectrotransformation
• Seed culture preparation-inoculation of 20 ml of reduced 2×YTG+0.2 ml of a thawed glycerol stock-20-2-dilution..
• overnight growth at 37°C
• 1 ml culture transferred to 125 ml Erlenmeyer flask(20 ml of reduced 2×YTG). Cells grown to exponential phase Filter sterilized solutions of sucrose and glycine 0.4 M and 1.25%.
• Growth until culture attained an OD600 of 0.6-0.8 (approx2–3 h) and 20 ml culture transferred to 50 ml pre-chilled, screw-cap centrifuge tube.
At this point, all manipulations at 4°C
• Cells removed from anaerobic chamber and collected by centrifugation at 8,500×g-20 minutes.Pellet returned to anaerobic chamber and washed in 5 ml of filter sterilized SMP buffer (270 mMsucrose+1 mM MgCl2+5 mM sodium phosphate-pH 6.5) centrifugation cell pellet resuspended in 0.6 ml SMP buffer.

Preparation of electrocompetent cells andelectrotransformation
• For transfer of plasmids to C.pas, E. coli-C.pas shuttle vectors co-transformed with pFnuDIIMKn into E. coli ER1821 to methylate external cytosine residue.
• Plasmid mixtures isolated and 0.5 μg 20 μl of 2 mMTris–HCl,pH 8.0 580 μl of C. pas competent cells.
• cell-DNA mixture transferred to pre-chilled electroporation cuvette with 0.4 cm gap+30 μl of cold 96% ethanol.
• Incubation on ice for 5 minutes• Decay pulse using Gene Pulser 1.8 kV, 25 μF, at time constant of 12–14
ms.
• Following pulse delivery,cuvette flooded with 1 ml 2×YTG medium+0.2 M sucrose+9 ml of the same medium.Recovery cultures incubated for 4–6 hours prior to plating 50–250 μl aliquots onto 2×YTG agar plates(15 μg/ml thiamphenicol+20 μg/ml erythromycin). Plates were incubated for 2–4 days under secondary containment within 3.4 L Anaerobic Jars each with a 3.5 L Anaerobic Gas Generating sachet.

References• 1. Lee SY, Park JH, Jang SH, Nielsen LK, Kim J, Jung KS: Fermentative
butanol production by clostridia. Biotechnol Bioeng 2008, 101:209–228.
• 2. Zheng YN, Li LZ, Xian M, Ma YJ, Yang JM, Xu X, He DZ: Problems with the microbial production of butanol. J Ind Microbiol Biotechnol2009,36:1127–1138.
• 3. Pfromm PH, Amanor-Boadu V, Nelson R, Vadlani P, Madl R: Bio-butanol vs.bio-ethanol: A technical and economic assessment for corn and switchgrass fermented by yeast or Clostridium acetobutylicum. Biomass Bioenerg 2010, 34:515–524.
• 4.Jensen TO, Kvist T, Mikkelsen MJ, Christensen PV, Westermann P: Fermentation of crude glycerol from biodiesel production by Clostridium pasteurianum. J Ind Microbiol Biotechnol 2012, 39:709–717.
• 5. da Silva GP, Mack M, Contiero J: Glycerol: A promising and abundant carbon source for industrial microbiology. Biotechnol Adv 2009, 27:30–39.