departamento de química inorgánica y analítica...
TRANSCRIPT

Universidad de Chile
Facultad de Ciencias Químicas y Farmacéuticas
Departamento de Química Inorgánica y Analítica
DETERMINACIÓN DE METABOLITOS ALQUILFOSFATOS DE PESTICIDAS
ORGANOFOSFORADOS UTILIZANDO DERIVATIZACIÓN ASISTIDA POR MICROONDAS EN
SOLUCIONES ACUOSAS Y ORINA HUMANA
TESIS ENTREGADA A LA UNIVERSIDAD DE CHILE PARA OPTAR AL
GRADO DE MAGISTER EN QUÍMICA
GILDA PAOLA ALVIAL ARAVENA
DIRECTORA DE TESIS:
DRA. MARÍA ESTRELLA BÁEZ CONTRERAS
SANTIAGO-CHILE
2008

ii
INDICE
Resumen …................................................................................................................................................ 2
Summary ……………............................................................................................................................... 5
Introducción ............................................................................................................................................. 8
Determinación de metabolitos de pesticidas organofosforados en orina .......................................... 11
Extracción de metabolitos de pesticidas organofosforados desde orina y limpieza del extracto .... 17
Derivatización de metabolitos de pesticidas organofosforados en orina ……................................... 18
Detección de pesticidas organofosforados y sus metabolitos en orina ……….................................. 21
Hipótesis ………….................................................................................................................................. 25
Objetivo general ……………………..................................................................................................... 25
Objetivos específicos …………………….............................................................................................. 25
Materiales y Métodos ……………............................................................................................... 26
Instrumentos ………………................................................................................................................... 26
Cromatógrafos de gases ............................................................................................................ 26
Horno de microondas …………………………........................................................................ 26
Otros equipos y materiales ……………………....................................................................... 27
Reactivos y soluciones ………………………….................................................................................... 28
Estándares de alquilfosfatos ……………………………......................................................... 28

iii
Reactivo derivatizante ……………………............................................................................... 28
Estándar interno ……………………….................................................................................... 28
Solventes ……………………..................................................................................................... 28
Sales ……………….................................................................................................................... 29
Soluciones madre ………………............................................................................................... 29
Soluciones intermedia de trabajo para los diseños experimentales ……….......................... 29
Métodos ............................................................................................................................................... 30
Metodología de derivatización de referencia Hardt y Angerer ( 2000) ............................... 30
Curva de calibración preliminar ............................................................................................. 31
Comportamiento energético del disolvente sometido a microondas .................................... 32
Etapa de derivatización utilizando la energía de microondas: Identificación de los
parámetros significativos para la reacción. Diseño Experimental Exploratorio ............................. 33
Optimización del programa cromatográfico para la determinación conjunta de los seis
metabolitos en estudio ............................................................................................................................ 35
Comparación de los métodos de Hardt y Angerer y Oglobline y colaboradores …............ 36
Diseño Experimental de Optimización para microondas ...................................................... 36
Curva de calibración utilizando microonda convencional ………………............................ 37
Diseño Experimental de Verificación ...................................................................................... 38
Preparación de curvas de calibración para los seis metabolitos en estudio utilizando las
condiciones obtenidas en el Diseño Experimental de Verificación .................................................... 39

iv
Estudio de cinética y de estabilidad en el tiempo a bajas concentraciones de seis metabolitos
de alquilfosfatos ………………………………...................................................................................... 39
Optimización de respuesta a bajas concentraciones para alquilfosfatos usando sistema de
microondas instrumental ....................................................................................................................... 40
Optimización del modo splitless de inyección usando dimetil y dietil fosfato como
compuestos modelo ................................................................................................................................ 41
Ensayo preliminar aplicando la metodología de Hardt y Angerer (2001) a una matriz de
orina ………………………………………………………………………………………………….…. 43
Obtención de los parámetros de calidad en la etapa de extracción y derivatización de los 6
metabolitos dialquilfosfatos como estándares puros y en matriz acuosa y orina ............................ 44
Calibración Instrumental ......................................................................................................... 44
Linealidad, intervalo de trabajo y precisión ........................................................................... 45
Límite de detección (LOD) y Límite de cuantificación (LOQ) ............................................. 45
Fortificación en medio acuoso y en matriz orina de compuestos dialquílicos a baja
concentración y extracción de los metabolitos ..................................................................................... 45
Resultados y Discusión ................................................................................................................ 47
Metodología de derivatización de referencia de Hardt y Angerer (2000) ............................ 47
Curva de calibración preliminar ............................................................................................. 48
Optimización del método cromatográfico para la determinación simultánea de los seis
metabolitos a partir de sus tiempos de retención ................................................................................ 50
Programas térmicos .................................................................................................................. 51
Comparación de métodos de referencia .................................................................................. 55

v
Ensayo preliminar para aplicación de la energía de microondas ........................................ 56
Etapa de derivatización utilizando la energía de microondas. Identificación de los
parámetros significativos para la reacción. Diseño experimental exploratorio ............................... 57
Optimización de la etapa de derivatización ............................................................................ 63
Curvas de calibración obtenidas con microondas .................................................................. 65
Diseño Experimental de verificación ....................................................................................... 66
Preparación de curvas de calibración para los seis metabolitos en estudio utilizando las
condiciones obtenidas en el Diseño Experimental de Verificación .................................................... 69
Estudio de Cinética y de Estabilidad en el tiempo a bajas concentraciones de los metabolitos
.................................................................................................................................................................... 71
Optimización de respuesta a bajas concentraciones para alquilfosfatos usando sistema de
microondas instrumental ....................................................................................................................... 73
Optimización del modo de inyección usando dimetil fosfato como compuesto modelo ...... 76
Comportamiento de los seis metabolitos en mezcla ............................................................... 78
Ensayo preliminar aplicando la metodología de Hardt y Angerer a una matriz de orina.... 79
Obtención de los parámetros de calidad en la etapa de extracción y derivatización de los 6
metabolitos dialquilfosfatos como estándares puros, en matriz acuosa y orina ............................... 83
Conclusiones ........................................................................................................................................... 89
Bibliografía ............................................................................................................................................. 92

vi
INDICE DE TABLAS
Tabla 1: Pesticidas organofosforados y alquilmetabolitos generados a partir de estos......................13
Tabla 2: Concentraciones y alícuotas de estándares de dialquilfosfato para estudio preliminar del
método de Hardt y Angerer…………………………………………………………………………..…32
Tabla 3: Diseño experimental exploratorio: factores y niveles empleados…………………………..34
Tabla 4: Matriz de diseño experimental exploratorio………………………………………………...34
Tabla 5: Segundo diseño experimental: factores y niveles empleados………………………….........36
Tabla 6: Matriz del segundo diseño…………………………………………………………………….37
Tabla 7: Diseño experimental de verificación: factores y niveles empleados………….……….…….38
Tabla 8: Matriz del diseño experimental de verificación………………………………….………….38
Tabla 9: Estudio de tiempo de reacción a 350 W……………………………………………………...40
Tabla 10: Estudio de potencias de reacción a 2 minutos……………………………………………...41
Tabla 11: Condiciones experimentales para la reacción de derivatización en mezcla………………41
Tabla 12: Temperatura inicial del programa térmico………………………………………………...42
Tabla 13: Tiempo de splitless…………………………………………………………………………....43
Tabla 14: Programa térmico inicial…………………………………………………………………….47
Tabla 15: Concentraciones y razones de área obtenidas para la curva de calibración……….……..48
Tabla 16: Resumen de las características obtenidas para la primera curva de calibración….……..50

vii
Tabla 17: Programas térmicos, modo de inyección y detector empleads para la optimizar la
separación de los seis dialquilfosfatos…………………………………………………………………..51
Tabla 18: Tiempos de retención para los seis dialquilfosfatos y para los compuestos usados como
estándar interno en cada programa térmico…………………………………………………………...52
Tabla 19: Tiempos de retención y ancho de pico a mitad de altura obtenida para los seis
dialquilfosfatos y clorpirifos como estándar interno…………………………………………………..52
Tabla 20: Programa térmico optimizado en sistema Varian con flujo constante de 2 mL/min y
detector NPD……………………………………………………………………………………………...54
Tabla 21: Razones de área obtenidas con método de derivatización de Oglobline usando el
programa térmico optimizado…………………………………………………………………………..55
Tabla 22: Razones de área obtenidas con método de derivatización de Hardt & Angerer usando el
programa térmico optimizado……………………………………………………………………...…...55
Tabla 23: Combinación de potencia, tiempo aplicado y temperatura alcanzada por los solventes
acetonitrilo y agua……………………………………………………………………………………..…56
Tabla 24: Resultados de matriz del diseño experimental exploratorio……………………………....58
Tabla 25: Razones de área obtenidas con las distintas condiciones de derivatización aplicadas…..61
Tabla 26: Resultados de matriz del diseño experimental de optimización……………………….….63
Tabla 27: Resultados de matriz del diseño experimental de verificación………………..…….…….67
Tabla 28: Programa térmico optimizado modificado para el estudio de cinética y estabilidad en el
tiempo de metabolitos oxigenados a bajas concentraciones…………………………………………...71
Tabla 29: Programa térmico optimizado para maximizar la respuesta de dimetilfosfato……….....77

viii
Tabla 30: Condiciones experimentales optimizadas para maximizar la respuesta de
dimetilfosfato……………………………………………………………………………………………..77
Tabla 31: Razones de área de los 6 metabolitos en mezcla de 100 μg/L……………………..………78
Tabla 32: Niveles de concentración y valores de razón de áreas usados en matriz de orina…….…80
Tabla 33: Curva de calibración con estándares puros en acetonitrilo………………………….……81
Tabla 34: Resumen de los datos de calidad para curvas de calibración…………………………......82
Tabla 35: Parámetros de calidad obtenidos en la curva de calibración instrumental en
acetonitrilo…………………………………..……………………………………………………………85
Tabla 36: Parámetros de calidad obtenidos en fortificaciones acuosas……………………….….….85
Tabla 37: Parámetros de calidad obtenidos en fortificaciones de orina………...……………...……86
Tabla 38: Datos de calidad obtenidos del método de extracción y derivatización……….……..…....86
Tabla 39: Porcentajes de recuperación en medio acuoso y en orina……………………..….……….87
Tabla 40: Resumen de parámetros de calidad obtenidos en muestra de orina fortificada con los seis
alquilfosfatos a 0,100 μg/mL …………………………….…...…………………………………………88

ix
INDICE DE FIGURAS
Figura 1: Formas tio y oxo de paratión………………………………………………….........................9
Figura 2: Estructuras de metabolitos más comunes de los pesticidas organofosforados…..….….....12
Figura 3: Estructuras de metabolitos distintos a alquilfosfatos que han sido investigados como
indicadores de exposición……………………………………………………………………………..…15
Figura 4: Esquema de la reacción de derivatización de un alquilfosfato con pentafluorobencil
bromuro (PFBBr)………………………………………………………………………………………...20
Figura 5: Resumen del método de referencia desarrollado por Hardt y Angerer…………………..31
Figura 6: Ordenamiento de viales en horno de microondas convencional……………………….…..32
Figura 7: Curva de calibración para dietilfosfato (DEP)…………….………………………….…….49
Figura 8: Curva de calibración para dietiltiofosfato (DETP) y dietiditiofosfato (DEDTP)…..….….49
Figura 9: Cromatograma obtenido para los seis metabolitos en sistema HP 5890………………….53
Figura 10: Cromatograma obtenido para los seis metabolitos en sistema Varian 3800…………….54
Figura 11: Comparación gráfica de razones de área obtenida con los métodos de referencia…...…56
Figura 12: Gráfico de Pareto para DEP…………………………………………………………….….59
Figura 13: Gráfico de Pareto para DETP…………………………………………………………...….59
Figura 14: Gráfico de Pareto para DEDTP…………………………………………………………….59
Figura 15: Razones de área a 70 W y concentraciones de 100 y 300 μg/L para dietilfosfatos ……...60
Figura 16: Razones de área a potencia intermedia y concentraciones de 100 y 300 μg/L para
dietilfosfatos ……………………………………………………………………………………………...60

x
Figura 17: Razones de área a potencia “descongelar” y concentraciones de 100 y 300 μg/L para
dietilfosfatos ……………………………………………………………………………………………...60
Figura 18: Gráfico de Pareto con factores e interacciones para derivatización de DMP y DEP..….64
Figura 19: efecto del tiempo y la potencia en las razones de área en el diseño experimental de
Optimización……………………….………………………………………………………….…………64
Figura 20: Curvas de calibración obtenidas con microondas convencional y sistema Varian 3800
con detección por NPD ……………………………………………………….………………………….65
Figura 21: Efecto del tiempo y la potencia en las razones de área en el diseño experimental de
verificación………………………………………………………………………………………………..68
Figura 22: Curvas de calibración obtenidas con microondas convencional y sistema HP 5890 con
detección por FPD ……………………………………………...…………….………………………….70
Figura 23: Efecto del catalizador sobre las señales de metabolitos alquilfosfato……………….……72
Figura 24: Gráfico de respuesta en función del tiempo de aplicación de microondas………………73
Figura 25: Gráfico de respuesta en función de la potencia de microondas aplicada………………..74
Figura 26: Gráfico de respuesta para 100 μg/L a distintos tiempos de inyección splitless…………76
Figura 27: Gráfico de respuesta para 100 μg/L a distintas temperaturas iniciales del programa
térmico………………………………………………………………………………………….…………78
Figura 28: Razón de área de alquilfosfatos a potencia fija y a distintos tiempos de aplicación….…79
Figura 29: Curvas de calibración obtenidas en matriz de orina concentraciones elevadas………....81
Figura 30: Curvas de calibración obtenidas en matriz de orina concentraciones bajas….………....84

2
RESUMEN
El contar con métodos analíticos rápidos y validados para la determinación de compuestos que
sirvan como indicadores de exposición por pesticidas es de gran importancia para realizar estudios en
población y evaluar el impacto de éstos sobre la salud humana.
Dentro de los pesticidas, los compuestos organofosforados son muy utilizados en la agricultura
intensiva de numerosos cultivos siendo los dialquilfosfatos dimetilfosfato (DMP), dietilfosfato (DEP),
dimetiltiofosfato (DMTP), dietiltiofosfato (DETP), dimetilditiofosfato (DMDTP) y dietilditiofosfato
(DEDTP) metabolitos comunes a la gran mayoría de los pesticidas pertenecientes a esta familia, por lo
tanto pueden ser usados como indicadores de exposición tanto en población laboralmente expuesta como
en población general, si es que se cuenta con la suficiente sensibilidad del método analítico. Uno de los
inconvenientes de estas moléculas es su polaridad por lo cual es necesario derivatizarlas para su
determinación por cromatografía de gases (GC) o por cromatografía líquida (HPLC) lo cual, en la
mayoría de los casos provoca que los métodos empleados considerando el procesamiento de la muestra y
cuantificación cromatográfica sean bastante largos.
En los últimos años se están aplicando diferentes tecnologías tales como aquéllas basadas en la
energía de microondas y ultrasonido para acelerar el proceso analítico y la investigación que se desarrolla
está orientada fundamentalmente a optimizar las etapas de extracción, limpieza de los extractos y
derivatización de compuestos. La energía de microondas permite acelerar procesos fisicoquímicos
involucrados en la extracción de analitos y también ejerce efectos que favorecen la cinética de algunas
reacciones, permitiendo de esa forma acortar los tiempos de realización del proceso analítico total.
Como objetivo de este trabajo se planteó el desarrollo de un método que permitiera tanto una
extracción como una concentración eficiente y que a la vez fuera simple, rápido y de bajo costo utilizando
la energía de microondas en la etapa de derivatización para la determinación de metabolitos
dialquilfosfato en muestras de orina humana.
El método desarrollado se basa en una extracción líquido-líquido de los metabolitos, una
concentración previa del extracto, la derivatización de los metabolitos utilizando PFBBr asistida por

3
microondas y posterior concentración de los derivados formados y, finalmente, la determinación de éstos
por cromatografía de gases. Los parámetros que influyen sobre la reacción de derivatización asistida por
microondas fueron definidos a través de diseños experimentales de “screening” (o exploratorios) y de
optimización para la obtención de las mas altas señales cromatográficas. Los resultados fueron
comparados con los de un método de referencia en el que se utiliza un sistema de calentamiento
convencional, obteniéndose rendimientos similares para la reacción de derivatización.
Factores tales como potencia y tiempo de calentamiento por microondas influyeron
significativamente en la reacción y los compuestos mas afectados fueron DMP y DEP. Con la
optimización de los parámetros instrumentales para la aplicación del método desarrollado en muestras, el
tiempo total de análisis fue reducido en un 99% si se compara con la metodología de referencia, desde 15
horas a 4 minutos en la reacción de derivatización, representando gran utilidad para el aumento de la
productividad en un laboratorio de control. El coeficiente de determinación para los 6 metabolitos en
orina fue mayor que 0,98 y los límites de detección varían de 11,8 a 18,5 µg/L en matriz de orina siendo
los límites más altos para los metabolitos oxigenados (DMP y DEP). El método desarrollado se aplicó a
soluciones acuosas y muestras de orina humana, la recuperación en muestras de orina fortificada varía
entre 20 a 55 % para un nivel de concentración de 50 µg/L mientras que el coeficiente de variación
porcentual es inferior al 10% para todos los alquilmetabolitos. Debido a la alta polaridad de este tipo de
compuestos se comprobó que es necesario para mejorar la recuperación de los metabolitos (DEP y DMP)
realizar un estudio exhaustivo a la etapa de extracción en una matriz de orina probando nuevas mezclas de
solventes que permitan mejorar la recuperación de los compuestos más desfavorables.
Se establecieron las funciones de calibración para el estudio de validación del método,
obteniéndose un intervalo dinámico entre 75 y 100 µg/L y todos los coeficientes de determinación (R2)
fueron superiores a 0.98. Se realizó un estudio comparativo de las pendientes de las curvas de calibración
obtenidas en agua y orina, obteniéndose valores similares para ambas, para los seis metabolitos estudiados
y representando aproximadamente entre un 50 y 60% de los correspondientes valores obtenidos en
solvente puro (acetonitrilo).

4
Los resultados de los estudios de recuperación desde muestras fortificadas de agua y orina a un
nivel de concentración de 50 µg/L, expresados como porcentaje, fueron relativamente bajos, entre un 20
% para los metabolitos oxigenados (DMP y DEP) y un 50 % para aquéllos azufrados (DETP, DEDTP,
DMTP, DEDTP). Las mas bajas recuperaciones fueron atribuidas a los pasos de extracción y
concentración de los extractos. Los límites de detección (LOD) y cuantificación (LOQ) obtenidos con
este proceso analítico fueron entre 11 - 19 µg/L y 39 - 62 µg/L, respectivamente. Estos pueden ser
considerados altos si el propósito del análisis es la determinación de metabolitos en muestras de población
no expuesta ocupacionalmente, no obstante éstos son concordantes con aquéllos descritos en la literatura
en donde se han empleados similares métodos de detección (NPD and FPD).
La precisión total (repetitividad) del método fue evaluada mediante la fortificación simultánea de
cuatro muestras de orina a un nivel de 100 µg/L para cada metabolito, obteniéndose valores bajo el 10 %
en todos los casos.
Teniendo en cuenta la polaridad de los compuestos en estudio y las bajas recuperaciones
obtenidas se deberán realizar estudios mas exhaustivos de las etapas de extracción y concentración de
éstos, mediante el uso de otros solventes o mezclas, sin embargo debe destacarse que es posible la
determinación de los seis metabolitos en orina donde existe un efecto de matriz que favorece la
recuperación.

5
SUMMARY
The development and validation of fast and reproducible analytical methods for the determination of
compounds that can be used as biomarkers of exposure to pesticides is required to carry out population
studies and to establish the impact of this kind of compounds on human health.
Organophosphorus pesticides are widely used in the intensive agriculture and the dialkylphosphates O,O-
Dimethyl phosphate (DMP), O,O-Diethylphosphate (DEP), O,O-Dimethyl phosphorothionate (DMTP),
O,O-Diethyl phosphorothionate (DETP), O,O-Dimethyl phosphorodithionate (DMDTP), O,O-Diethyl
phosphorodithionate (DEDTP) are their principal and common metabolites. They can be used as
biomarkers of exposure in different kinds of populations (occupationally and non-occupationally
exposed) if the analytical method has enough sensitivity. Because of its high polarity a derivatization step
previous to GC or HPLC determination is needed even though if mass spectrometry detection be
employed. Almost all the analytical processes developed for the determination of these compounds are
quite long and the whole analysis of samples is carried out in the range of days.
Recently, different technologies such as ultrasound and microwave energies are being employed to
accelerate the analytical process, and current research is oriented fundamentally to the extraction, clean up
and derivatization steps. Microwaves energy can be used for increasing the speed of the physicochemical
processes involved in the extraction of target analytes from different class of samples, with a considerable
time reduction of the total analysis. On the other hand, microwave energy is also a good tool to increase
the kinetic of several specific reactions.
The general objective of this work was to develop an analytical method that both provide an efficient
extraction and pre-concentration steps, and allow the simple, rapid and low-cost determination of
dialkylmetabolites of organophosphorus pesticides in human urine samples. A more specific approach
was to use the microwave energy to assist the derivatization step of these metabolites to be used in the GC
determination.
The developed method was based in a preliminary liquid-liquid extraction of metabolites from urine
samples, a subsequent concentration step, the derivatization assisted by microwaves with PFBBr as the

6
derivatizing reagent, the concentration of the resulting derivatives, and finally, the determination of
them by GC with NPD and FPD detection.
Parameters affecting the derivatization reaction were established through experimental screening designs
and later optimized to obtain the highest chromatographic signal. Results were compared with those
obtained through a reference method where the reaction is carried out by a conventional heating system.
Factors such as power and time of heating by microwaves influenced significantly the derivatization
reaction and the most affected compounds were DMP and DEP. With the optimization of microwave
instrumental parameters for the application of the developed method to samples, the total time was
reduced in 99% as compared with the reference methodology, from 15 hours to 4 minutes, being very
useful for enhance the productivity of a control laboratory.
The calibration functions for the validation study were established, so, the linear dynamic range was 75 to
250 ug/L, and all the determination coefficients (R2) were better than 0,98. A comparative study related to
the slopes of the calibration curves obtained in aqueous and urine matrix was done. We found that values
for aqueous and urine calibration solutions were very similar for the six metabolites under study, and
represented approximately a 50 to 60% of the corresponding values obtained in pure solvent
(acetonitrile).
The recovery results from water and urine fortified samples, at a 50 µg/L level and expressed as
percentage, were relatively low, between 20% for oxygenated dialkylmetabolites (DMP, DEP) and 50%
for the sulfur containing metabolites (DETP,DEDTP,DMTP,DEDTP), the last findings verifying that the
performance of the whole analytical method is decreased by the extraction and concentration steps. The
detection (LOD) and quantification (LOQ) limits obtained with this analytical process were between 11 -
19 µg/L and 39 - 62 µg/L, respectively. They can be considered high if the purpose of the analysis is the
determination of metabolites in non-occupationally exposed population but these results are in accordance
with the reports of works using similar detection systems (NPD and FPD). The whole precision
(repeatability) of the method was evaluated by fortifying human urine samples with 100 µg/L of each
metabolite in quadruplicate and the RSD values were below 10% in all cases.

7
Because of the high polarity of compounds, and taking into account the results obtained from the recovery
experiments, it is necessary to develop more exhaustive experiments related to the extraction step with
different mixtures of solvents for the enhancement of these recoveries, especially in the case of
oxygenated dialkylmetabolites (DMP, DEP); however, we can emphasize the fact that the analytical
determination of all six metabolites in urine samples is possible under the optimized conditions found for
the derivatization step obtained in the present study.

8
Introducción
Desde la década de 1940 el control de las diversas plagas se ha basado esencialmente en la
utilización masiva de los plaguicidas sintéticos. Si bien esto ha traído beneficios a la población, el uso de
estos productos tóxicos ha provocado importantes problemas en la salud, así como la contaminación
creciente del medio ambiente.
Dentro de los plaguicidas de origen sintético se encuentran los plaguicidas organofosforados. Las
propiedades tóxicas de los fosfatos orgánicos fueron reconocidas por primera vez en 1932 cuando los
investigadores Lang y Krueger observaron envenenamiento en ratas, sin embargo fue Schrader el primero
en descubrir las propiedades insecticidas de estos compuestos y desarrollar las primeras formulaciones
cuyos componentes activos fueron moléculas como el Schradan y el tetraetil pirofosfato (TEPP), este
último el primer producto comercial [1].
Los plaguicidas organofosforados son muy usados en nuestro país, están incorporados en
actividades de salud pública, en la agroindustria y de manera preponderante en el sector agrícola sobre
todo desde que se prohibiera el uso de pesticidas organoclorados. Las importaciones de plaguicidas para
uso agrícola alcanzaron entre los años 2000-2001 a 36.279 toneladas [2], de las cuales el 48%
correspondió a pesticidas organofosforados, si bien es cierto que en los últimos 4 años se ha producido un
cambio en las proporciones con un importante incremento en el uso de piretroides y la incorporación de
nuevos compuestos. Si a esto se suma la libre venta y circulación de estos productos, el fácil acceso a
ellos, la falta de conocimiento y capacitación en su manejo, nos encontramos con una población expuesta
a niveles importantes de plaguicidas.
Como población expuesta se destacan los habitantes de las zonas rurales, siendo los trabajadores
agrícolas uno de los grupos de alto riesgo. Entre los meses de noviembre de 2001 y enero de 2002 el
promedio de trabajadores agrícolas fue de 756.700 personas [3] y la cantidad de cultivos durante el año
fue de 1.788.498,7 hectáreas [4].
Al observar algunos indicadores de exposición encontramos que Chile utilizó un promedio
nacional de 2 kilos de plaguicidas/hectárea cultivada, con una carga promedio anual de 2,3 kilos de

9
plaguicidas por habitante [5], esto implica un valor casi 4 veces mayor a la media mundial estimada por la
OMS de 0,6 kilo/año/habitante.
Los plaguicidas organofosforados son usados en la agricultura como insecticidas, acaricidas y
fungicidas [6]. Estos compuestos en general son liposolubles y no ionizables por lo que son rápidamente
absorbidos por inhalación o ingestión, la absorción por la piel en general es lenta. Después de la
absorción, son rápidamente acumulados en el tejido adiposo, hígado, riñones, glándulas salivales y
pueden atravesar la barrera hematoencefálica en la mayoría de los casos. Los pesticidas con la estructura
fosforotioatos (P=S) (por ejemplo diazinon, parathion) son más lipofílicos que los fosfatos (P=O) como
ejemplo dichlorvos, por lo que se acumulan más en el tejido adiposo y pueden reaparecer los signos
clínicos de intoxicación después de la recuperación.[7].
Los compuestos que contienen el enlace P=O son activos biológicamente mientras que los
compuestos con la estructura P=S necesitan una etapa de bioactivación para generar los análogos tipo
fosfato denominados oxon (presentan la estructura P=O) este proceso ocurre por un mecanismo de
desulfuración oxidativa el cual es mediado por las isoformas CYP3A4 y CYP2D6 de la enzima citocromo
P450 principalmente en los microsomas del hígado [8]. En la figura 1 se muestra un esquema con las
formas tio y oxo de un pesticida organofosforado.
Figura 1. Formas tio y oxo de Paratión.
O2N O P OC2H5
S
OC2H5
O2N O P OC2H5
O
OC2H5
Paratión (forma tio)Paratión (forma oxo)
Bioactivación

10
El efecto tóxico de los pesticidas organofosforados, específicamente los derivados tipo oxon,
ocurre por la fosforilación de un grupo hidroxilo en un aminoácido de serina presente en el dominio que
une el sustrato de la enzima acetilcolinesterasa (EC 3.1.1.7). Esto ocurre en la sangre, cerebro y otros
tejidos [9] en forma dependiente del tiempo [7]. En las primeras 2 a 6 horas de exposición y dependiendo
de la dosis absorbida, se observa una disminución de la actividad enzimática en la sangre, al aumentar el
tiempo desde la absorción los efectos se detectan en cerebro e hígado. La inhibición de la enzima genera
una acumulación del neurotransmisor acetilcolina en los ganglios autonómicos, terminaciones nerviosas
post ganglionares y en las placas motoras.
La reactivación de la enzima alquilfosforilada depende de la estructura del compuesto
organofosforado unido, si la enzima es inhibida por pesticidas que contengan 2 grupos metilos unidos al
átomo de fósforo como por ejemplo diclorvos, dimetoato o malatión, entonces la reactivación será
espontánea y prácticamente no se necesitará ningún tratamiento. Si por el contrario se ha usado pesticidas
que presenten 2 grupos etilo como sustituyentes del átomo de fósforo como por ejemplo clorpirifos,
diazinon o paration, la reactivación de la enzima prácticamente no ocurre y se necesita tratamiento
médico y seguimiento en el tiempo de la actividad enzimática ya que se necesita esperar el recambio de la
enzima inactivada.
Como indicador biológico de exposición ocupacional o accidental a pesticidas organofosforados
se utiliza la determinación de la actividad de las enzimas acetilcolinesterasa plasmática (ChE) y
acetilcolinesterasa eritrocitaria [10]. Esto se utiliza como un indicador del efecto de los pesticidas
organofosforados absorbidos sobre el sistema nervioso central y en las uniones neuromusculares [11].
Habitualmente se realiza la determinación utilizando el método de Ellman [12].
La determinación de la actividad de la acetilcolinesterasa es un bioindicador muy utilizado y
efectivo en el caso de niveles altos de exposición, sin embargo es ampliamente reconocido que es un
parámetro relativamente insensible para estimar la dosis absorbida de pesticidas organofosforados [13];
por ejemplo se ha descrito que es necesario al menos un 15% de disminución en la actividad normal de la
acetilcolinesterasa plasmática o eritrocitaria para visualizar un efecto debido a la exposición a pesticidas

11
organofosforados. Diversas organizaciones internacionales recomiendan como valor límite de monitoreo
biológico una disminución de 30% en la actividad normal del individuo monitoreado para considerar que
existe una sobre exposición a pesticidas organofosforados. En general el valor normal de actividad
enzimática cuando es determinado con el método de Ellman en el caso de hombres es de 4,01 ± 0.65
UI/mL para acetilcolinesterasa eritrocitaria y de 3,03 ±0,66 para la acetilcolinesterasa plasmática, en el
caso de las mujeres los valores son un poco menores 3,03 ± 0,61 UI/mL para la acetilcolinesterasa
eritrocitaria y 3,03 ± 0,68 UI/mL para la acetilcolinesterasa plasmática. Para el caso del monitoreo
biológico es necesario contar con el valor de referencia para el individuo cuando no está expuesto, este
valor es contrastado con el valor obtenido al final de la jornada laboral para saber si ha ocurrido una sobre
exposición a estos compuestos, se obliga entonces a contar con a lo menos dos muestras de sangre para
confirmar la contaminación, lo cual en algunos casos es considerado como un método muy invasivo [11]
ya que considera un muestreo periódico de sangre. Además la disminución de la acetilcolinesterasa no es
un parámetro conveniente para establecer la exposición ocupacional en los niveles comunes de trabajo y a
su vez para valorar la exposición de la población general [14] debido a la variabilidad individual. Como
consecuencia de lo anterior es que surge la necesidad de la determinación de metabolitos específicos y/o
generales de pesticidas organofosforados ya que estos compuestos son excretados y pueden ayudar en la
interpretación del monitoreo biológico si se cuenta con buenos métodos analíticos, ya que se requiere una
buena sensibilidad y límite de detección por las bajas concentraciones en que se encuentran en los fluidos
biológicos.
Determinación de metabolitos de pesticidas organofosforados en orina
Una aproximación complementaria para evaluar la exposición a pesticidas organofosforados se
basa en la determinación de los metabolitos de estos pesticidas en la orina. El análisis de estas moléculas
puede realizarse de dos formas, la primera es a través del análisis de metabolitos específicos para cada
pesticida en particular y la segunda es el análisis de dialquil fosfatos que son comunes para muchos
pesticidas organofosforados.

12
Se utiliza como matriz la orina ya que es una de las principales vías de excreción de los pesticidas
organofosforados y de sus metabolitos, la identificación y cuantificación de la concentración de los
dialquil metabolitos en orina se viene usando como bioindicador desde la década de 1960. Los
compuestos más comunes de esta clase de analitos que se encuentran en esta matriz son: dimetilfosfato
(DMP), dietilfosfato (DEP), o,o-dimetiltiofosfato (DMTP), o,o-dietiltiofosfato (DETP), o,o-
dimetilditiofosfato (DMDTP) y o,o-dietilditiofosfato (DEDTP) [15].
P
O CH3
CH3OHO
O
P
O CH3
CH3OHO
S
P
O CH3
CH3OHS
S
Dimetil fosfato Dimetiltiofosfato Dimetilditiofosfato
(DMP) (DMTP) (DMDTP)
CH2CH3P
OOHO
O
CH2CH3
CH2CH3P
OOHOCH2CH3
S
CH2CH3P
OOCH2CH3
HS
S
Dietil fosfato Dietiltiofosfato Dietilditiofosfato
(DEP) (DETP) (DEDTP)
Figura 2. Estructura de metabolitos más comunes de los pesticidas organofosforados.
Estos metabolitos son los más comunes para una gran variedad de pesticidas órgano fosforados.
Ejemplo de ello se muestra en la tabla 1.
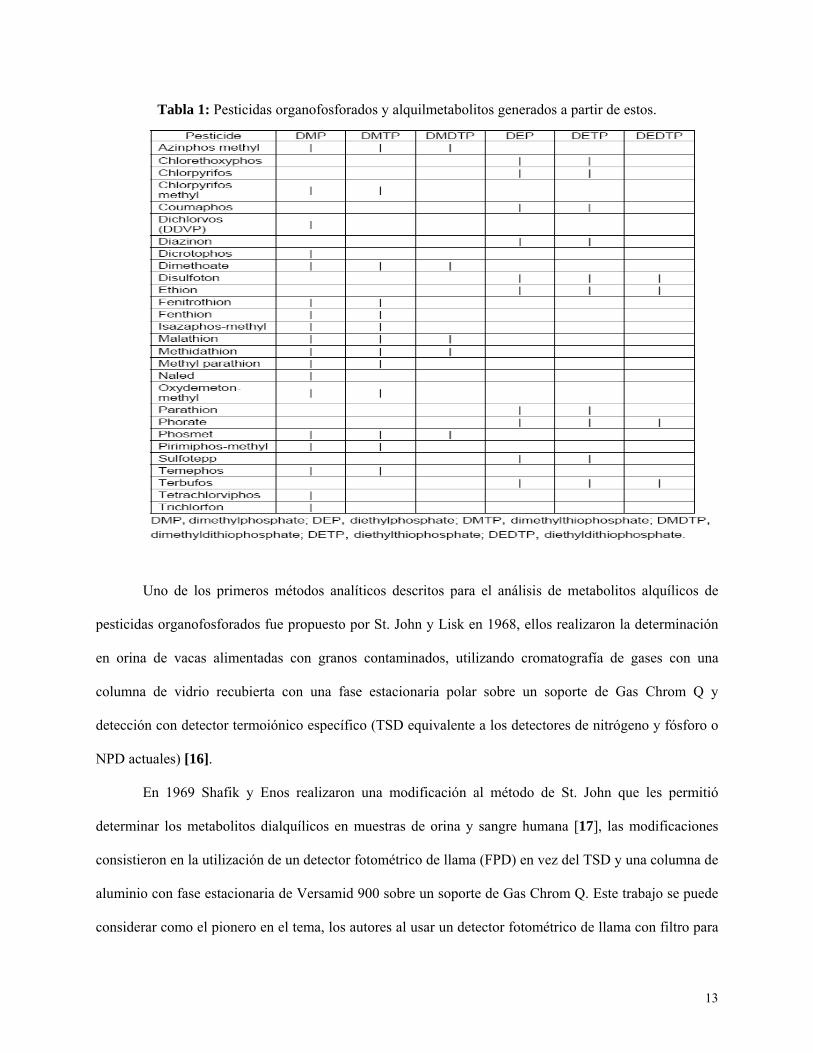
13
Tabla 1: Pesticidas organofosforados y alquilmetabolitos generados a partir de estos.
Uno de los primeros métodos analíticos descritos para el análisis de metabolitos alquílicos de
pesticidas organofosforados fue propuesto por St. John y Lisk en 1968, ellos realizaron la determinación
en orina de vacas alimentadas con granos contaminados, utilizando cromatografía de gases con una
columna de vidrio recubierta con una fase estacionaria polar sobre un soporte de Gas Chrom Q y
detección con detector termoiónico específico (TSD equivalente a los detectores de nitrógeno y fósforo o
NPD actuales) [16].
En 1969 Shafik y Enos realizaron una modificación al método de St. John que les permitió
determinar los metabolitos dialquílicos en muestras de orina y sangre humana [17], las modificaciones
consistieron en la utilización de un detector fotométrico de llama (FPD) en vez del TSD y una columna de
aluminio con fase estacionaria de Versamid 900 sobre un soporte de Gas Chrom Q. Este trabajo se puede
considerar como el pionero en el tema, los autores al usar un detector fotométrico de llama con filtro para

14
fósforo (FPD) pudieron cuantificar los seis metabolitos mencionados anteriormente, los límites de
detección variaron entre 10 μg/L para DEP y 200 μg/L para DMDTP.
Posteriormente en 1973 Shafik y col. [18], desarrollaron un nuevo método basado en el anterior
en el cual se mejoró la determinación cromatográfica al seleccionar una mejor fase estacionaria, se
reemplazó el Versamid 900 por la resina OV-210 al 5% sobre un soporte de Chromosorb W y también se
utilizó una mezcla de SE-30 al 4% con OV-210 al 6% sobre un soporte de Gas Chrom Q. De las dos
columnas estudiadas la primera, OV-210 al 5% sobre Chromosorb W, permitió reducir el tiempo de
cromatografía en forma significativa al compararlos con el método descrito por los mismos autores en el
año 1969.
Desde mediados de los años 70 hasta fines de los años 90 se han desarrollado varios métodos para
la determinación de estos analitos [13, 19-23]. Los principales esfuerzos se han realizado en lograr límites
de detección cada vez menores y en la robustez del proceso analítico para obtener métodos confiables y
poder cuantificar el nivel basal de la población no expuesta ocupacionalmente. Lo anterior se ha logrado
al introducir modificaciones en la derivatización al usar nuevos reactivos, también se ha investigado en la
optimización de la extracción usando nuevas tecnologías como la extracción en fase sólida y en los
procedimientos de limpieza de los extractos para disminuir las interferencias propias de la matriz; además
se ha aprovechado las nuevas tecnologías disponibles como la cromatografía líquida de alta resolución
acoplada a detección por espectrometría de masas (HPLC-MS) y HPLC acoplada a espectrometría de
masas en tandem (HPLC-MS/MS).
Los métodos analíticos más usados en la actualidad utilizan cromatografía de gases con detector
fotométrico de llama (FPD) [13] o espectrometría de masas (MS) [24, 25]. El desarrollo de métodos
analíticos para la determinación de estos compuestos continúa hasta nuestros días, es así como en el año
2002 se publicaron a lo menos 2 trabajos sobre el tema [26,27], en los cuales se combinan parte de los
procedimientos tradicionales con el uso de nuevas tecnologías. En el artículo de Bravo y col. [27] se
realiza la extracción de la muestra con acetonitrilo y luego una concentración del extracto por
evaporación, la derivatización se realiza con 1-cloro-3-yodo propano y la cuantificación se realiza con
espectrometría de masas-masas usando el método de dilución isotópica para la cuantificación en modo

15
SIM. En el método de Lin [26] se realiza la extracción de la muestra usando un disco de extracción con
una resina aniónica del tipo SAX, este disco es posteriormente derivatizado directamente en un vial con
ioduro de metilo y el extracto es cuantificado por interpolación en una curva de calibración, no se utiliza
ningún tipo de limpieza del extracto obtenido y se utiliza un detector del tipo FPD. La tendencia en los
últimos años para la determinación de los alquilfosfatos es utilizar cromatografía de gases con detección
de masas en tandem o GC-MS/MS [27,28] para la cuantificación con el objeto de lograr límites de
detección cada vez más bajos.
En el caso de la determinación de metabolitos específicos para pesticidas organofosforados, son
pocos los compuestos para los cuales se han desarrollado métodos analíticos, como ejemplo tenemos el
caso de 3,5,6-tricloro-2-piridinol (TCPyr) que es metabolito de clorpirifos y metil clorpirifos [29],
también se ha investigado en el análisis de p-nitrofenol (PNP) que es metabolito de paration y de metil
paration [30], en el caso de fenitrotion se utiliza el 3-metil-4-nitrofenol (MNP) [30] y en el caso de
malation se ha desarrollado la determinación en orina de ácido α-monocarboxílico y ácido dicarboxílico
de malation como metabolitos [31], también se ha descrito la determinación de pesticidas
organofosforados sin modificar en sangre o excretados por la orina, es el caso de acefato y metamidofos
[32]. En la figura 3 se muestran las estructuras de algunos de los compuestos antes señalados.
N Cl
Cl
OH
Cl NO2
OH
P
OO
O
NH2
CH3
CH3
3,5,6-tricloropiridinol paranitrofenol Metamidofos
(TCPyr) (PNP)
Figura 3. Estructura de metabolitos distintos a alquilfosfatos que han sido investigados como
indicadores de exposición

16
En general todos los procedimientos de determinación de metabolitos de pesticidas constan de las
etapas de extracción, limpieza del extracto, concentración, derivatización y detección. Se han realizado
esfuerzos para mejorar cada etapa del proceso analítico para alcanzar límites de detección lo más bajos
posibles con el fin de permitir el análisis de muestras de población no expuesta ocupacionalmente a estos
compuestos, principalmente por el hecho de que existe exposición crónica y en bajos niveles de
concentración como es el caso de los pesticidas de uso doméstico (casa y jardín) que pueden influir y
generar manifestaciones clínicas principalmente en mujeres embarazadas y niños pequeños además de la
exposición proveniente de la dieta. Otro enfoque en el cual se ha centrado la investigación está
relacionado con la reducción del tiempo de análisis para contar con una respuesta adecuada en el caso de
intoxicaciones agudas como es el caso de los intentos de suicidio en los cuales el contar con métodos
analíticos que permitan identificar los metabolitos genéricos de pesticidas organofosforados o algún
metabolito específico sería de gran utilidad en el tratamiento de estos pacientes en este caso no existe una
limitación en cuanto a la sensibilidad ya que por la naturaleza de la muestra se esperan altas
concentraciones.
Independiente del objetivo del método, sensibilidad o rapidez del análisis, los esfuerzos de
investigación y desarrollo se han centrado principalmente en la utilización de tecnologías más nuevas en
algunas de las etapas mencionadas anteriormente como por ejemplo extracción en fase sólida para la
etapa de extracción de los analitos desde la matriz y en la limpieza del extracto o combinaciones de
reactivos en la etapa de derivatización como por ejemplo reactivos silanizantes que permiten acelerar esta
etapa para algunos de estos analitos al reaccionar a temperatura ambiente y en tiempos cortos.
A continuación se presenta un breve resumen de los enfoques utilizados hasta la fecha en las
etapas de extracción, derivatización y detección en el ámbito del análisis de metabolitos alquilfosforados
de pesticidas organofosforados desde orina mediante cromatografía gaseosa.

17
Extracción de metabolitos de pesticidas organofosforados desde orina y limpieza del
extracto
En el caso de la determinación de los metabolitos alquílicos de pesticidas organofosforados la
técnica tradicional consiste en una extracción líquido-líquido, desde una alícuota acidificada de orina. En
1969 Shafik [17] utilizó una mezcla de acetonitrilo/dietiléter para la extracción, no se realizó una limpieza
del extracto antes de la concentración y derivatización, los autores realizaron la detección por FPD. En
1973 Shafik y sus colaboradores realizaron una modificación al método antes señalado, agregando una
etapa de limpieza para mejorar la detectabilidad de alquilfosfatos a niveles de concentración bajos en
orina humana y de rata [33]. La modificación consistió en la concentración de la mezcla de extracción y
su posterior paso a través de una columna de sílica gel activada para retener las impurezas que son
coextraidas en la mezcla de acetonitrilo/dietileter.
Posteriormente en 1976 Blair y col. [19] utilizaron como etapa previa a la extracción una
precipitación del fosfato inorgánico, que es interferente, con hidróxido de calcio y en la etapa de
extracción utilizaron una resina de intercambio iónico para luego extraer los metabolitos con etanol desde
este soporte. Un enfoque similar al antes señalado fue utilizado por Lores y col. un año después [34],
empleando acetona como solvente de extracción, este procedimiento fue levemente modificado en el año
1981 por Brokopp [21] al aumentar el volumen de la alícuota de orina y agregar un paso de centrifugación
previo a la extracción de la resina de intercambio iónico. En los métodos antes señalados se logró un bajo
porcentaje de recuperación y una pobre precisión analítica, sin embargo se estableció un antecedente
importante de la factibilidad del uso de resinas de intercambio iónico para la extracción de alquil fosfatos,
lo que derivó en el uso de otros tipos de resinas y soportes con capacidad de intercambio iónico para la
extracción de estos compuestos lo cual ha sido investigado incluso en los últimos años [26].
Un enfoque distinto de extracción fue utilizado por Fenske y Leffingwell [22] en el año 1989
ellos realizaron una destilación azeotrópica del extracto de orina con acetonitrilo como medio de
extracción, este procedimiento fue también utilizado en conjunto con la extracción liquido-líquido por
Hardt y Angerer en el año 2000 [25] y por Bravo y col. en el año 2002 [27].

18
La utilización de extracción en fase sólida fue ampliamente utilizada desde los años noventa por
ejemplo por los siguientes investigadores Park [24], Moate, [36], Lin, [26]. En 1992 Jauhiainen [35]
publicó un método donde se realiza la extracción de dimetilfosfato desde orina humana usando un
cartridge de extracción en fase sólida de carácter aniónico que retiene el metabolito para luego ser eluido
con acetonitrilo u otro solvente adecuado.
Otro método de extracción que ha sido utilizado recientemente por Oglobline en el año 2001[31]
es la liofilización de la muestra, esta consiste en el congelamiento de la muestra generalmente en baño de
alcohol a -20° C o a temperaturas menores, se prosigue con la aplicación de vacío sobre la orina con el fin
de deshidratar la muestra y generar un residuo sólido el cual es homogenizado para luego realizar la
derivatización de los alquilfosfatos en forma directa [28, 37] si bien este procedimiento reduce las etapas
de pretratamiento, es bastante largo ya que toma de 16 a 24 horas para ser completado.
Derivatización de metabolitos de pesticidas organofosforados en orina
Debido a su carácter iónico los alquil metabolitos de pesticidas organofosforados deben ser
derivatizados antes de que se produzca su separación y detección por cromatografía de gases. La
derivatización se realiza principalmente para convertir estos analitos en moléculas volátiles lo cual es
necesario para la determinación por cromatografía de gases.
En el trabajo original de Shafik [17] a fines de los años 60 se utilizó diazometano y diazoetano
como agentes derivatizantes, con esas reacciones se tiene el problema de que es difícil controlar la
volatilidad de los derivados lo que generó pérdida de los analitos en la determinación cromatográfica y la
reacción del fosfato inorgánico el cual es convertido en trimetilfosfato (TMP) o trietilfosfato (TEP) que
actúan como interferentes en la cuantificación de los otros metabolitos alquílicos. Posteriormente los
mismos autores en 1973 [18] mejoraron la reacción de derivatización al usar el compuesto diazopentano
como agente derivatizante lo que les permitió disminuir la volatilidad y aumentar la estabilidad de los
analitos investigados.

19
Otra alternativa de derivatización fue propuesta por Churchill y col. en el año 1978 [38] para
pesticidas organosfosforados y metabolitos alquílicos del tipo fosforotioatos, ellos usaron una solución
alcohólica de hidróxido de trimetilfenilamonio para realizar la derivatización en el cuerpo del inyector
siendo este trabajo una de las primeras alternativas a los diazoalcanos como agente derivatizante.
Un avance significativo en la reacción de derivatización fue publicado en 1979 por Takade y col.
[39] ellos utilizaron aril-alquil triacenos para generar derivados volátiles de los compuestos alquil fosfatos
antes señalados, este procedimiento por primera vez permitió la determinación de los seis metabolitos
principales a niveles de 10 μg/L.
En el mismo año, Daugthon y col. [40] publicaron la derivatización de estos metabolitos mediante
la reacción de bencilación, el reactivo utilizado por estos investigadores corresponde a benciltolil triaceno
el cual presenta un mejor rendimiento de derivatización y mejor respuesta si se quiere determinar estos
ésteres con un detector de ionización de llama (FID). Otra ventaja importante de este tipo de derivados es
que el grupo bencilo le otorga mejores propiedades de solubilidad a los derivados lo que permite utilizar
distintos tipos de solventes.
En 1981 Reid y Watts [20] publicaron un nuevo método analítico en el cual usaron bromuro de
pentafluorobencilo (PFBBr) como agente derivatizante, este compuesto posee fundamentalmente dos
problemas potenciales, como primer punto esta el hecho de que el PFBBr es un potente lacrimógeno, por
lo cual debe ser manejado siempre bajo campana, como segundo punto se tiene que en exceso puede
descomponerse a altas temperaturas generando halógenos libres que pueden dañar la columna y detector.
Lo anteriormente expuesto se puede evitar con un paso de limpieza del extracto después de realizada la
reacción de derivatización para eliminar el exceso de PFBBr. La principal ventaja de este reactivo es la
capacidad de generar principalmente un derivado en el caso de dimetiltiofosfato (DMTP) y de
dietiltiofosfato (DETP) para esto es necesario controlar muy bien las condiciones de reacción, mientras
que los agentes derivatizantes descritos anteriormente generan dos isómeros.
Se debe destacar además que los diazopentanos generalmente son explosivos y que en su síntesis
se utilizan reactivos cancerígenos, en el caso de los triacenos también se describen como compuestos
cancerígenos.

20
En los últimos años se han descrito otros reactivos para la reacción de derivatización de alquil
fosfatos como es el caso de la mezcla descrita en 1998 por Park [24] que consiste en hexametildisilazano,
trimetilclorosilano, piridina (2:1:10 v/v/v) la cual tiene el inconveniente de la utilización de piridina y fue
probada solo con algunos de los alquilfosfatos de interés. El año 2002 se publicó la utilización de ioduro
de metilo [26] y de 1-cloro-3-iodopropano [27] como agentes derivatizantes.
De todos los reactivos señalados el más común en la actualidad es el PFBBr, este compuesto ha
sido utilizado en los métodos que presentan los límites de detección menores y que han sido aplicados a
estudios epidemiológicos donde se han analizado una gran cantidad de muestras. En la figura 4 se muestra
un esquema de la reacción de derivatización de alquil fosfatos con PFBBr.
F
CH2Br
FF
F
F
+ P
O
ROHO
O
R
P
O
ROO
F
CH2
FF
F
FO
R
Figura 4. Esquema de la reacción de derivatización de un alquilfosfato con pentafluorobencil bromuro
(PFBBr).
Una de las principales limitantes de los métodos que utilizan PFBBr es el tiempo empleado en la
etapa de derivatización y el hecho de que en algunos de estos procedimientos se deben realizar dos
reacciones de derivatización para obtener en una reacción los metabolitos que contienen azufre en la
estructura y en la otra etapa los metabolitos oxigenados, las condiciones de reacción involucran la
aplicación de calor hasta alcanzar la temperatura de 90º C durante un periodo relativamente largo 2-3
horas para asegurar la reacción en el caso de los metabolitos oxigenados o, en otro método el tiempo
necesario es muy largo 15 horas a 40 ºC. Los métodos que mencionan tiempos de reacción de
derivatización largos y temperaturas altas son aquéllos para los compuestos no azufrados (DMP y DEP)
que son los que requieren mayor energía para la formación de sus derivados y por el contrario al emplear

21
altas temperaturas se pueden degradar los tio-compuestos (DMTP y DETP) dando lugar a la formación
de DMP Y DEP (Hardt y Angerer [25], Oglobline [28]).
Detección de pesticidas organofosforados y sus metabolitos en orina
Desde los primeros métodos desarrollados a fines de los 60 y comienzos de los 70 por Shafik
[17,18] se utilizó un detector fotométrico de llama (FPD) el cual presenta una respuesta específica para
azufre y/o fósforo, lo que permite alcanzar límites de detección muy bajos. También se ha descrito la
detección utilizando un detector de ionización de llama (FID) el cual es insensible a fósforo pero presenta
una buena respuesta a carbono, este detector fue utilizado para determinar los derivados con triacenos de
los alquil fosfatos a fines de los años 70 [40].
El detector termoiónico específico o sensible a nitrógeno y fósforo (NPD) también ha sido
utilizado para la determinación de estos compuestos [16] sin embargo es imprescindible realizar una
limpieza del extracto antes de la inyección para evitar problemas con la respuesta de este detector y los
problemas asociados de contaminación del mismo.
Cuando se utiliza compuestos halogenados en la derivatización se posibilita la detección mediante
un detector de captura de electrones (ECD), el detector antes mencionado presenta los mismos
inconvenientes que el NPD sobre todo cuando se utiliza (PFBBr) como agente derivatizante.
Los métodos más nuevos en los cuales el énfasis esta puesto en lograr los menores límites de
detección utilizan detección por espectrometría de masas (MS) [24, 25] en su modalidad de registro
específico de iones (SIM) o espectrometría de masas en tandem (MS/MS) [27, 28] lo que ha permitido
lograr métodos apropiados para la determinación de alquil fosfatos en población no expuesta
ocupacionalmente.
En general el detector más usado es el fotométrico de llama (FPD) por su sensibilidad y
especificidad mientras que en los últimos años se ha puesto más énfasis en la detección de estos
compuestos por espectrometría de masas. Por lo anterior si se cuenta con un buen procedimiento de

22
extracción, limpieza y derivatización es factible realizar la determinación de esta familia de compuestos
mediante cromatografía de gases independiente del tipo de detector empleado.
Después de la revisión bibliográfica descrita en los párrafos anteriores se muestra claramente que
la investigación en el desarrollo de métodos analíticos para la determinación de los dialquilfosfatos aún
continua vigente, los esfuerzos se concentran en lograr desarrollar metodologías analíticas confiables para
realizar los estudios en poblaciones que permitan evaluar principalmente la exposición no ocupacional a
los pesticidas organofosforados, el interés de esto consiste en lograr establecer valores de referencia
poblacionales para estos compuestos ya que son un buen bioindicador de la dosis interna de pesticidas
organofosforados, esto es la cantidad de compuesto absorbido, además no se conoce la formación de este
tipo de compuestos en forma endógena a partir de otros tipos de sustratos.
Existen muchos estudios sobre el tema de extracción de distintos compuestos orgánicos incluidos
los pesticidas organofosforados utilizando microondas en distintas matrices como alimentos, sedimentos y
agua [41] también se ha descrito la extracción de metidation desde muestras acuosas [42] en
concentraciones altas ya que el objetivo del estudio fue realizar pruebas de carácter fisicoquímico para
estudiar la adsorción y cinética de solubilización hacia la fase acuosa desde suelos, en este trabajo se
realizó la determinación directa sin una etapa de preconcentración. Si se considera que la matriz de orina
es básicamente una solución acuosa y que algunos pesticidas son excretados principalmente sin
modificación se tiene un antecedente sobre el tema de extracción mediante el uso de la tecnología de
microondas.
Una de las principales ventajas de usar un procedimiento de extracción asistido por microondas
consiste en la posibilidad de usar temperaturas y presiones elevadas lo que favorece la cinética en la
transferencia de masas desde la muestra hacia el solvente, lo anterior permite reducir el tiempo en la etapa
de extracción. El principio básico que permite acelerar el proceso de transferencia es la forma en la cual
se aumenta la energía del sistema, esto se debe al efecto directo de las microondas sobre las moléculas, se
produce por conducción iónica que genera una migración electroforética de los iones cuando se aplica un
campo electromagnético, la resistencia de la solución a este flujo genera fricción y como consecuencia se
calienta la solución. Otro mecanismo que permite un aumento de energía es la rotación de los dipolos,

23
esto se debe al realineamiento de ellos en el campo aplicado, en el caso de los sistemas comerciales que
utilizan una frecuencia de 2450 MHz la reorientación ocurre 4,9*109 veces por segundo y este
movimiento molecular forzado genera un aumento de la temperatura medida en el sistema.
La eficiencia en la extracción se debe al mecanismo de calentamiento de la solución, se han
descrito tres tipos distintos que pueden ser utilizados. El primer mecanismo es el uso de un solvente o
mezcla de solventes que absorban fuertemente las microondas aplicadas, otra forma es usar una mezcla de
solventes que absorban las microondas y que sean transparentes a ellas en distintas proporciones y
finalmente el tercer mecanismo corresponde al uso de un solvente que sea transparente a las microondas
esto se aplica en el caso de matrices que tengan una alta pérdida dieléctrica o alto contenido de agua. La
selección del mecanismo de calentamiento depende del sistema en estudio, matriz y analitos.
La derivatización asistida por microondas ha sido empleada para la extracción y derivatización in
situ de fenol y metil fenol en suelos [43] también se ha descrito la derivatización de compuestos
carbonílicos volátiles utilizando pentafluorobencil hidroxilamina [44], entre otras aplicaciones.
Recientemente se publicó un método que utiliza microondas para la síntesis de tiofosfatos a partir de dietil
fosfito y bromuros de alquilo en el cual no se utiliza solventes, los tiempos de reacción no superan los 5
minutos y los rendimientos son en todos los casos mayores al 75% [45].
Es importante destacar que no se ha encontrado en la literatura la aplicación de la tecnología de
microondas para la extracción de pesticidas organofosforados desde la matriz de orina, o para realizar la
reacción de derivatización de los metabolitos.
En la actualidad en nuestro país se realiza el monitoreo biológico de exposición a pesticidas
organofosforados principalmente con la determinación de la actividad de acetilcolinesterasa, no se realiza
la búsqueda de metabolitos generales como los alquil fosfatos o específicos como es el caso por ejemplo
del TCPyr o de los pesticidas intactos en caso de exposición ocupacional.
Existe preocupación a nivel de las autoridades de salud sobre el tema del monitoreo biológico en
poblaciones ocupacionalmente expuestas al uso de plaguicidas organofosforados y piretroides, por lo que
se postula que será de gran utilidad el contar con un método analítico para la determinación de los alquil
metabolitos de pesticidas organofosforados porque servirá para contrastar la información obtenida con el

24
análisis de colinesterasa en el caso de intoxicación o de exposición ocupacional y podría ser usado para el
estudio de la población general no expuesta ocupacionalmente si el método en cuestión logra tener la
suficiente sensibilidad.
Considerando lo anteriormente expuesto, las etapas analíticas que ameritan un esfuerzo de
investigación son principalmente los procedimientos de extracción, limpieza del extracto y derivatización.
En el presente trabajo de tesis se pretende estudiar la etapa de derivatización utilizando energía de
microondas tomando en consideración que se debe lograr un método idealmente muy sensible y rápido
que permita realizar en último caso estudios en poblaciones tanto ocupacionalmente expuestas como en
población general. Se utilizará como punto de partida métodos descritos en bibliografía como referencia
con los cuales se comparará la investigación propuesta.

25
HIPÓTESIS:
Es factible utilizar la tecnología de microondas para desarrollar un método analítico que
permita la determinación de metabolitos de pesticidas organofosforados desde orina humana de
manera de disminuir el tiempo total del proceso analítico sin pérdida de sensibilidad para los
compuestos en estudio.
OBJETIVO GENERAL:
Desarrollar un método que permita tanto una extracción como una concentración eficiente y que a
la vez sea simple, rápido y de bajo costo utilizando la energía de microondas en la etapa de derivatización
para la determinación de metabolitos dialquilfosfato en muestras de orina humana.
OBJETIVOS ESPECÍFICOS:
1.- Optimizar el protocolo de derivatización mediante microondas de los metabolitos alquilfosfato
para su determinación por cromatografía de gases.
2.- Establecer las condiciones analíticas para la identificación y cuantificación por cromatografía
de gases de los compuestos derivatizados.
3.- Establecer el mejor procedimiento de extracción y limpieza del extracto de alquilfosfatos
desde orina humana.
4.- Validar el método analítico optimizado para alquil metabolitos de pesticidas organofosforados.
5.- Aplicar el método analítico a muestras de orina de humana.

26
MATERIALES Y MÉTODOS
1.- INSTRUMENTOS
1.1.- Cromatógrafos de gases
Se utilizaron dos sistemas de cromatografía de gases, el primero consiste en un cromatógrafo
de gases marca Hewlett Packard (HP) modelo 5890, equipado con un inyector split-splitless programable,
detector fotométrico de llama (FPD), columna capilar Ultra 2 (crosslinked 5% Ph silicone de 25 m de
largo, 0,20 mm de diámetro y 0,33 μm de espesor de película y un sistema registrador HP modelo 5890.
Como gas portador se utilizó helio 4.5 (99,995% de pureza). El segundo cromatógrafo de gases
corresponde a un equipo marca Varian modelo 3800 equipado con un inyector de temperatura
programable (PTV) usado en modo splitless, detector termoiónico específico o NPD, columna DB-5 de
30 m de largo, 0,25 mm de diámetro y 0,25 µm de espesor de película; como gas portador se utilizó helio
99,995 % de pureza y para el registro de los cromatogramas se utilizó el software de control e integración
Star versión 5,0.
1.2.- Hornos de microondas
Se realizaron los estudios en dos tipos de hornos de microondas, uno convencional para
aplicaciones domésticas marca LG con capacidad para entregar 5 potencias prefijadas por el fabricante
(70 W, “descongelar”, 350 W, 500 W y “máximo”) ubicado en el Laboratorio de Química Analítica de
Residuos de Pesticidas y elementos traza de la Facultad de Ciencias Químicas y Farmacéuticas.
El otro sistema de microondas corresponde a un sistema especialmente diseñado para aplicaciones
de laboratorio marca Milestone modelo ETHOS 1600 ubicado en el Laboratorio de Salud Ambiental de la
Secretaría Ministerial de Salud Pública de la Región Metropolitana (SEREMI de Salud RM).

27
1.3.- Otros Equipos y Materiales
Estufas con control digital de temperatura, rotavapor marca Heindolph, microjeringas de 10, 25,
50, 100 y 500 µL, pipetas de vidrio con émbolo de 1, 2 y 5 mL, micropipetas de volumen variable de 100,
250, 1000 y 5000 µL, balanzas y campanas extractoras ubicadas en las dependencias del laboratorio de
Salud Ambiental del SEREMI de Salud RM.
Estufas, campanas de extracción, balanza, matraces aforados clase A con tapa de vidrio de 5 y 10
mL, refrigeradores y sistema de evaporación del Laboratorio de Química Analítica de Residuos de
Pesticidas y Elementos traza.

28
2.- REACTIVOS Y SOLUCIONES
2.1.- Estándares de alquilfosfatos
Se utilizaron sales con un 99 % de pureza para los siguientes compuestos: Dietilfosfato
(DEP); Dietiltiofosfato (DETP) y Dietilditiofosfato (DEDTP) marca Aldrich. Dimetilfosfato (DMP);
Dimetiltiofosfato (DMTP); y Dimetilditiofosfato (DMDTP) fueron adquiridos en Cerilliant (EEUU) como
soluciones metanólicas de 1000 µg/mL
2.2.- Reactivo derivatizante
El reactivo derivatizante utilizado fue pentafluorobencilbromuro (PFBBr) marca Aldrich
preparado por dilución en volumen en proporción 1 mL de PFBBr y 3 mL de acetonitrilo.
2.3.- Estándar Interno
Se utilizaron dos tipos de compuestos como estándar interno, estos son sales marca
Supelco de Trifenilfosfato (TFF) y clorpirifos (Supelco) los cuales fueron preparados por pesada y
posterior dilución en los solventes adecuados.
2.4.- Solventes
Los siguientes solventes fueron usados en el procedimiento analítico: Agua calidad
reactivo grado I (18 Mohm de resistividad) obtenida en un equipo Nanopure y MiliQ, Acetona grado
pesticida Merck y Acetonitrilo OmniSolv de Merck, hexano y dietiléter.

29
2.5.- Sales
Se utilizaron además carbonato de potasio y cloruro de sodio grado pesticida de Merck.
2.6.- Soluciones Madre
Se prepararon soluciones de DEP, DETP y DEDTP por disolución de las sales
correspondientes, pesando exactamente 3.0, 10.0 y 10.0 mg de cada uno de ellos por separado y llevados
a un volumen de 10 mL con acetonitrilo grado-HPLC, obteniéndose una concentración final de 300, 1000
y 1000 µg/mL (el resto, como se señaló anteriormente, fueron adquiridas como tales).
2.7.- Soluciones intermedias de trabajo para los diseños experimentales
Se prepararon soluciones individuales tomando 0,1 mL de cada una de las soluciones
patrón, diluyendo a 10 mL con acetonitrilo grado HPLC.

30
3.- MÉTODOS
3.1.- Metodología de derivatización de referencia Hardt y Angerer [25]
En este ensayo se prepararon 3 de los 6 dialquilfosfatos individualmente para el establecimiento
de los tiempos de retención utilizando un programa térmico desarrollado para análisis de pesticidas
organofosforados. La etapa de derivatización se realizó en tres viales distintos utilizando concentraciones
de 0.300 µg/mL para el DETP y DEDTP y de 0,900 µg/mL de DEP. Los estándares de 0,300 µg/mL de
DETP Y DEDTP se prepararon a partir de un estándar de trabajo de 10 µg/mL y el de 0,900 µg/mL de
DEP se preparó de uno de 3,00 µg/mL, luego se agregó reactivo derivatizante (PFBBr) en medio
acetonitrilo (150 µL) a 1000 µL de cada estándar y 50 mg de K2CO3 anhidro y se agregaron 350 µL de
AcN para completar el volumen de la reacción (1500 µL). Se llevó a la estufa a 40ºC por 15 horas para
lograr los derivados para posteriormente continuar con la metodología de extracción con hexano y
finalmente reconstituir con 1mL de AcN. Previa adición de 100 µL de patrón interno (TFF, 10µg/mL) se
inyectó 1 µL en el cromatógrafo gaseoso con detector FPD, en sistema “splitless”.
En la figura 5 se muestra un resumen del método utilizado como referencia en lo que corresponde
al método de derivatización y extracción. Cabe mencionar que en el método de referencia después del
último paso de concentración se llega a 150 μL como volumen final del que se inyecta 1 μL, a diferencia
de lo descrito previamente.

31
Figura 5. Resumen del método desarrollado por Hardt y Angerer [25] usado como referencia en los
estudio iniciales.
3.2.-Curva de calibración preliminar
Una vez determinados los tiempos de retención, se procedió a preparar una solución mixta que
contenía 1µg/mL de DETP, DEDTP y de 3 µg/mL de DEP, desde donde se tomaron las respectivas
alícuotas para construir una curva de calibración de 5 niveles, como se indica en la tabla 2, según los
datos obtenidos las concentraciones de los compuestos se calcularon para un volumen final de 1 mL,
completándose el volumen por adición. La derivatización se realizó en viales con tapa rosca donde se
agregaron 150 µL de reactivo derivatizante (33 % v/v) y 50 mg de carbonato de potasio, procediendo de
igual forma que para la experiencia anterior.
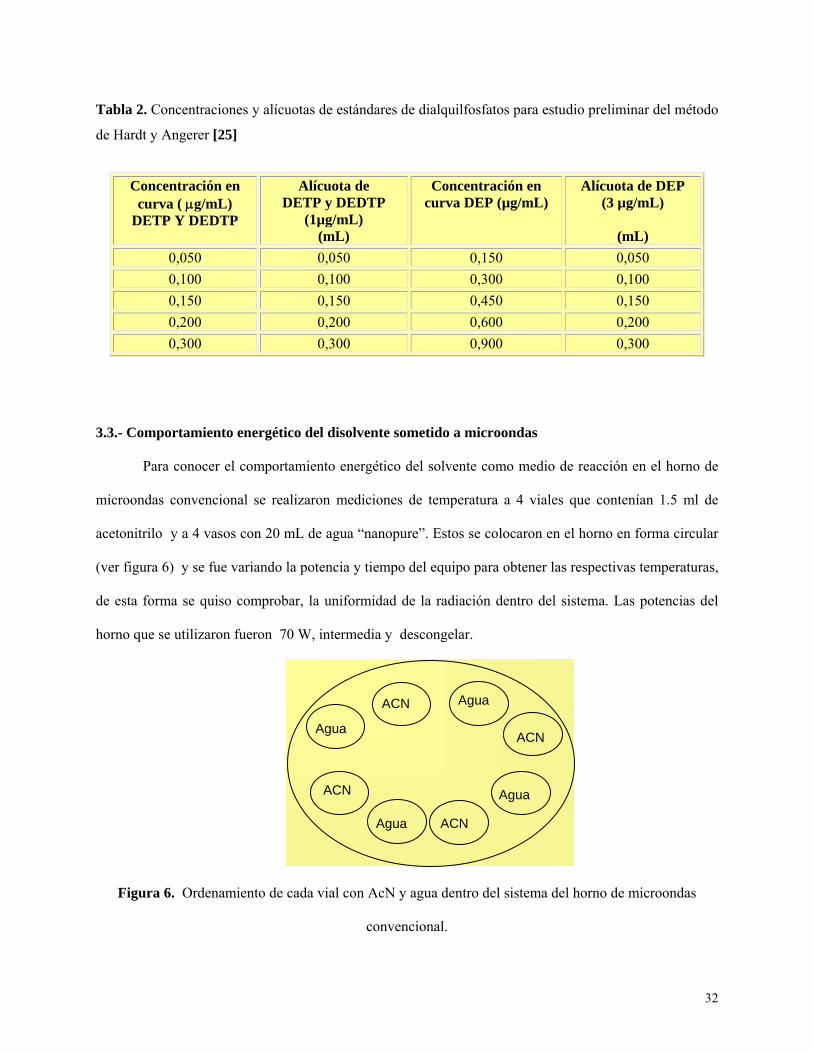
32
Tabla 2. Concentraciones y alícuotas de estándares de dialquilfosfatos para estudio preliminar del método
de Hardt y Angerer [25]
3.3.- Comportamiento energético del disolvente sometido a microondas
Para conocer el comportamiento energético del solvente como medio de reacción en el horno de
microondas convencional se realizaron mediciones de temperatura a 4 viales que contenían 1.5 ml de
acetonitrilo y a 4 vasos con 20 mL de agua “nanopure”. Estos se colocaron en el horno en forma circular
(ver figura 6) y se fue variando la potencia y tiempo del equipo para obtener las respectivas temperaturas,
de esta forma se quiso comprobar, la uniformidad de la radiación dentro del sistema. Las potencias del
horno que se utilizaron fueron 70 W, intermedia y descongelar.
Figura 6. Ordenamiento de cada vial con AcN y agua dentro del sistema del horno de microondas
convencional.
Concentración en curva ( μg/mL)
DETP Y DEDTP
Alícuota de DETP y DEDTP
(1µg/mL) (mL)
Concentración en curva DEP (µg/mL)
Alícuota de DEP (3 µg/mL)
(mL)
0,050 0,050 0,150 0,050 0,100 0,100 0,300 0,100 0,150 0,150 0,450 0,150 0,200 0,200 0,600 0,200 0,300 0,300 0,900 0,300
Agua
Agua
Agua
Agua ACN
ACN
ACN
ACN

33
De la misma solución mixta empleada para la curva de calibración se tomaron 4 alícuotas de
100 µL, las que se agregaron a sendos viales, dos de las cuales sirvieron para realizar la derivatización
según el método de referencia y las otras dos para derivatizar en el horno a una potencia de 70 W durante
5 minutos. Para asegurar la homogeneidad de la distribución de energía se utilizó el mismo ordenamiento
descrito en la figura 6. La determinación se realizó en idéntica forma que la descrita para el estudio de la
curva de calibración del método de referencia.
3.4.- Etapa de derivatización utilizando la energía de microondas: Identificación de los parámetros
significativos para la reacción. Diseño Experimental Exploratorio.
A través del diseño experimental se pretendió conocer aquellos factores que son significativos en
la obtención de los rendimientos de la etapa de derivatización expresados como relación de áreas entre la
señal de cada analito y el estándar interno (variable de respuesta). En primer lugar se elaboró un diseño
experimental exploratorio (Screening factor) con los metabolitos con grupos funcionales etilo (DEP,
DETP y DEDTP).
Para la realización de éstos estudios se prepararon tres estándares mixtos de 1 μg/mL, 1,5 μg/mL
y 2 μg/mL de DEP, DETP y DEDTP en acetonitrilo utilizándose la metodología de derivatización de
referencia.
Se estudiaron 5 factores mediante un diseño experimental factorial fraccionado (25-1), con cuatro
centros, para evaluar el error experimental (20 experimentos) en el horno de microondas convencional.
La elección de los factores y sus niveles se basó en las posibilidades relacionadas con el estudio
preliminar de comportamiento energético del solvente para la derivatización y en las posibles limitantes
propias de la reacción. Se estudiaron los siguientes factores: cantidad del reactivo derivatizante (33 % v/v
de PFBBr en acetonitrilo), concentración de los compuestos, cantidad de solvente (acetonitrilo), potencia
y tiempo. Los analitos se agregaron en mezcla en un volumen de 100 μL. Las alícuotas de acetonitrilo
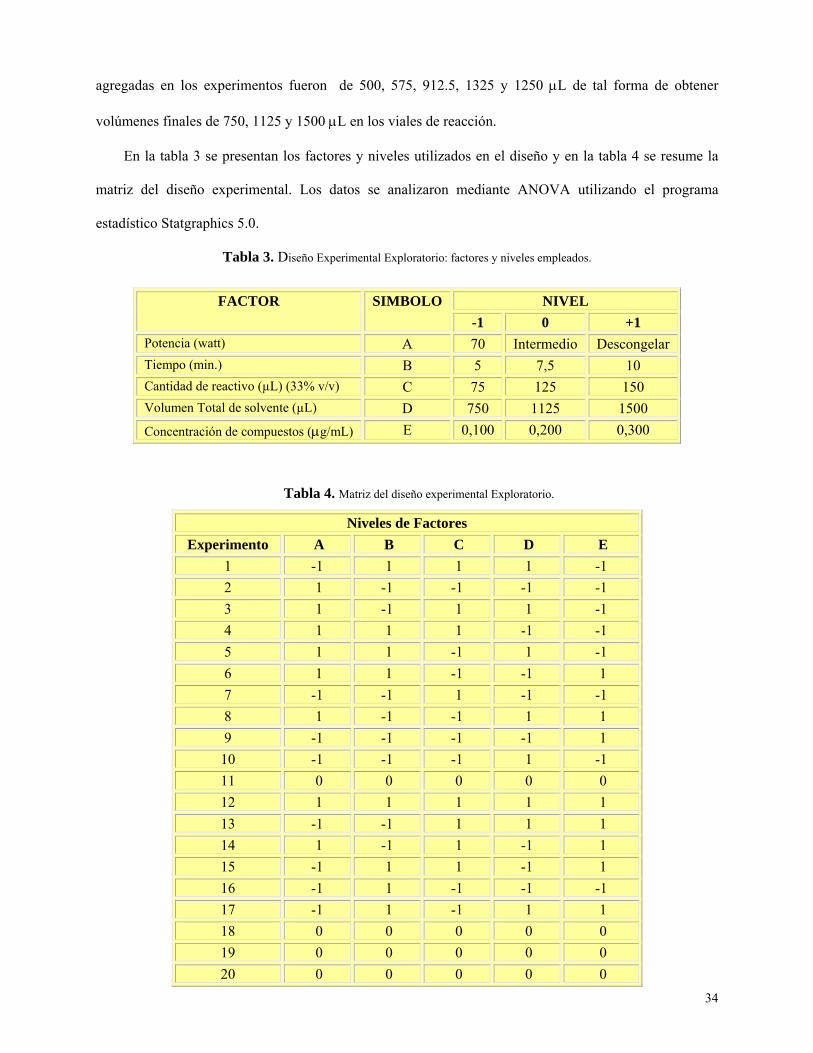
34
agregadas en los experimentos fueron de 500, 575, 912.5, 1325 y 1250 μL de tal forma de obtener
volúmenes finales de 750, 1125 y 1500 μL en los viales de reacción.
En la tabla 3 se presentan los factores y niveles utilizados en el diseño y en la tabla 4 se resume la
matriz del diseño experimental. Los datos se analizaron mediante ANOVA utilizando el programa
estadístico Statgraphics 5.0.
Tabla 3. Diseño Experimental Exploratorio: factores y niveles empleados.
Tabla 4. Matriz del diseño experimental Exploratorio.
FACTOR SIMBOLO NIVEL -1 0 +1
Potencia (watt) A 70 Intermedio DescongelarTiempo (min.) B 5 7,5 10 Cantidad de reactivo (µL) (33% v/v) C 75 125 150 Volumen Total de solvente (µL) D 750 1125 1500 Concentración de compuestos (μg/mL) E 0,100 0,200 0,300
Niveles de Factores Experimento A B C D E
1 -1 1 1 1 -1 2 1 -1 -1 -1 -1 3 1 -1 1 1 -1 4 1 1 1 -1 -1 5 1 1 -1 1 -1 6 1 1 -1 -1 1 7 -1 -1 1 -1 -1 8 1 -1 -1 1 1 9 -1 -1 -1 -1 1 10 -1 -1 -1 1 -1 11 0 0 0 0 0 12 1 1 1 1 1 13 -1 -1 1 1 1 14 1 -1 1 -1 1 15 -1 1 1 -1 1 16 -1 1 -1 -1 -1 17 -1 1 -1 1 1 18 0 0 0 0 0 19 0 0 0 0 0 20 0 0 0 0 0

35
3.5.- Optimización del programa cromatográfico para la determinación conjunta de los seis
metabolitos en estudio.
De acuerdo a los resultados obtenidos se procedió a incorporar los metabolitos con grupos
funcionales metilo para lo cual fue necesario replantear el método de separación cromatográfica, usándose
como punto de partida en esta etapa el método de derivatización y programa térmico de Oglobine y col.
[28] como referencia. Se optó por este método pues la derivatización se realiza de la misma forma que la
utilizada en el diseño anterior pero debido a la selectividad de los detectores empleados (FPD o NPD) no
fue necesaria la etapa de re-extracción desde una fase acuosa.
Se estudiaron por separado los 6 compuestos a una concentración de 1.00 μg/mL para DMP y
DEP y de 0,300 µg/mL para DMDTP, DETP y DEDTP a partir de los estándares de trabajo. Las
soluciones se prepararon en matraces aforados de 10 mL tomando las respectivas alícuotas de 1,00 mL de
DMP y de 0,300 mL para el resto de los metabolitos, de las cuales se tomaron 2 mL y se trasvasijó a un
vial de reacción con tapa, se agregó reactivo derivatizante (PFBBr al 33 % v/v) (100 µL) y 50 mg de
K2CO3 anhidro. Se llevó a la estufa a 60ºC por 4 horas para lograr los derivados. Previa la adición de 100
µL de patrón interno trifenil fosfato (TFF) de 10 µg/mL, se inyectó 1 µL en el cromatógrafo gaseoso con
detector FPD, en sistema “splitless”.
Así, se ensayaron varios programas térmicos para lograr la separación empleándose el índice de
separación (R) como una medida de la resolución de dos picos cromatográficos adyacentes.
R= (t2-t1)/1/2(w2+w1)
Donde:
w2 y w1=anchos de picos medidos a la mitad de la altura.
t2 y t1 =corresponden a los tiempos de retención de los dos picos

36
3.6.-Comparación de los métodos de Hardt y Angerer [25] y Oglobline y col. [28]
Una vez optimizados los programas térmicos se realizó el experimento de comparación entre los
métodos de derivatización descritos por Hardt yAngerer [25] y Oglobline y col. [28]. Se analizaron 4
réplicas de inyecciones de una mezcla de estándares a una concentración de 0,100 µg/mL con el método
de Oglobline y col. [28] y 4 réplicas de inyecciones de la misma concentración por el método de Hardt y
Angerer [25]. Se calcularon sus promedios y coeficientes de variación para analizar la variabilidad del
inyector del FPD.
3.7.- Diseño Experimental de Optimización para microondas
De acuerdo al análisis de los resultados del diseño exploratorio dos de los factores experimentales
potencia y tiempo, se sometieron a un diseño experimental factorial total con tres niveles y cuatro centros,
utilizando el sistema de microondas convencional, para confirmar si contribuyen significativamente en el
aumento de relaciones de áreas entre DEP, DETP, DEDTP, DMP, DMDTP y DMDTP y el estándar
interno. Las concentraciones usadas en este experimento fueron: 1,250 μg/mL para DMP; 0,750 μg/mL
para DEP y 0,250 μg/mL para DMTP, DMDTP; DETP y DEDTP.
Los factores y niveles empleados se encuentran en la tabla 5 y la correspondiente matriz de diseño
experimental en la tabla 6.
Tabla 5. Segundo Diseño Experimental: factores y niveles empleados.
FACTOR SIMBOLO NIVEL -1 0 +1
Potencia (W) A 350 425 500 Tiempo (min) B 5 7,5 10
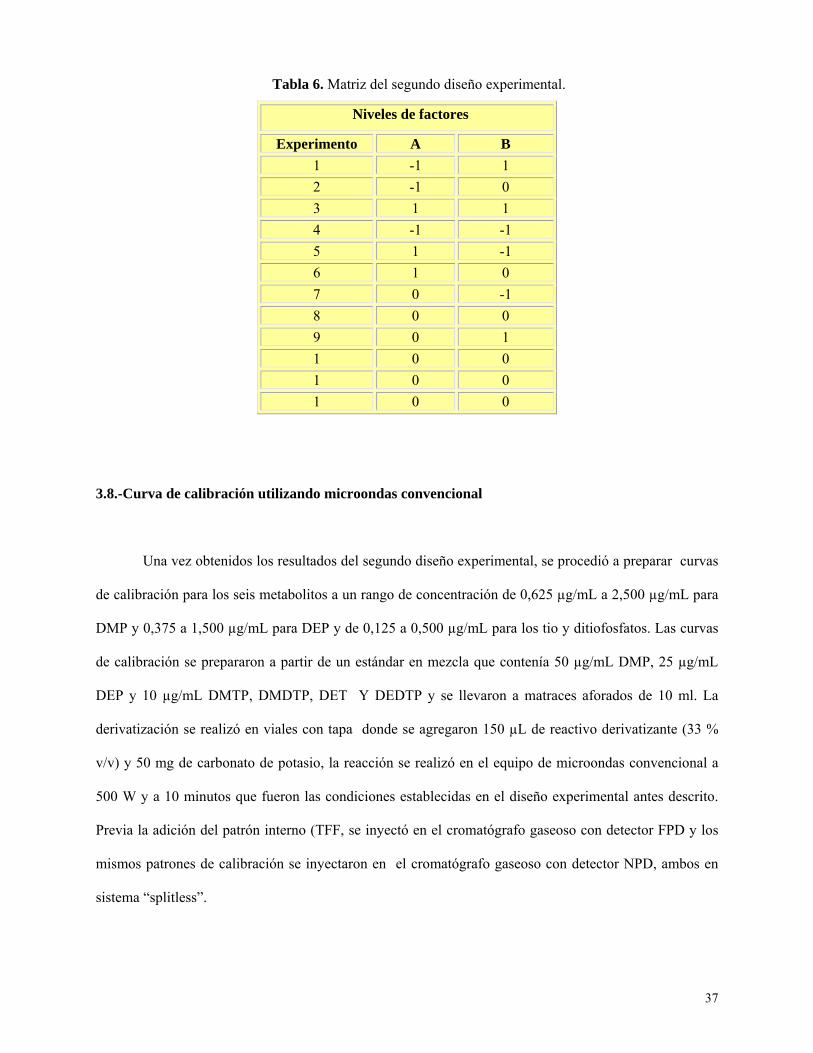
37
Tabla 6. Matriz del segundo diseño experimental.
Niveles de factores
Experimento A B 1 -1 1 2 -1 0 3 1 1 4 -1 -1 5 1 -1 6 1 0 7 0 -1 8 0 0 9 0 1 1 0 0 1 0 0 1 0 0
3.8.-Curva de calibración utilizando microondas convencional
Una vez obtenidos los resultados del segundo diseño experimental, se procedió a preparar curvas
de calibración para los seis metabolitos a un rango de concentración de 0,625 µg/mL a 2,500 µg/mL para
DMP y 0,375 a 1,500 µg/mL para DEP y de 0,125 a 0,500 µg/mL para los tio y ditiofosfatos. Las curvas
de calibración se prepararon a partir de un estándar en mezcla que contenía 50 µg/mL DMP, 25 µg/mL
DEP y 10 µg/mL DMTP, DMDTP, DET Y DEDTP y se llevaron a matraces aforados de 10 ml. La
derivatización se realizó en viales con tapa donde se agregaron 150 µL de reactivo derivatizante (33 %
v/v) y 50 mg de carbonato de potasio, la reacción se realizó en el equipo de microondas convencional a
500 W y a 10 minutos que fueron las condiciones establecidas en el diseño experimental antes descrito.
Previa la adición del patrón interno (TFF, se inyectó en el cromatógrafo gaseoso con detector FPD y los
mismos patrones de calibración se inyectaron en el cromatógrafo gaseoso con detector NPD, ambos en
sistema “splitless”.
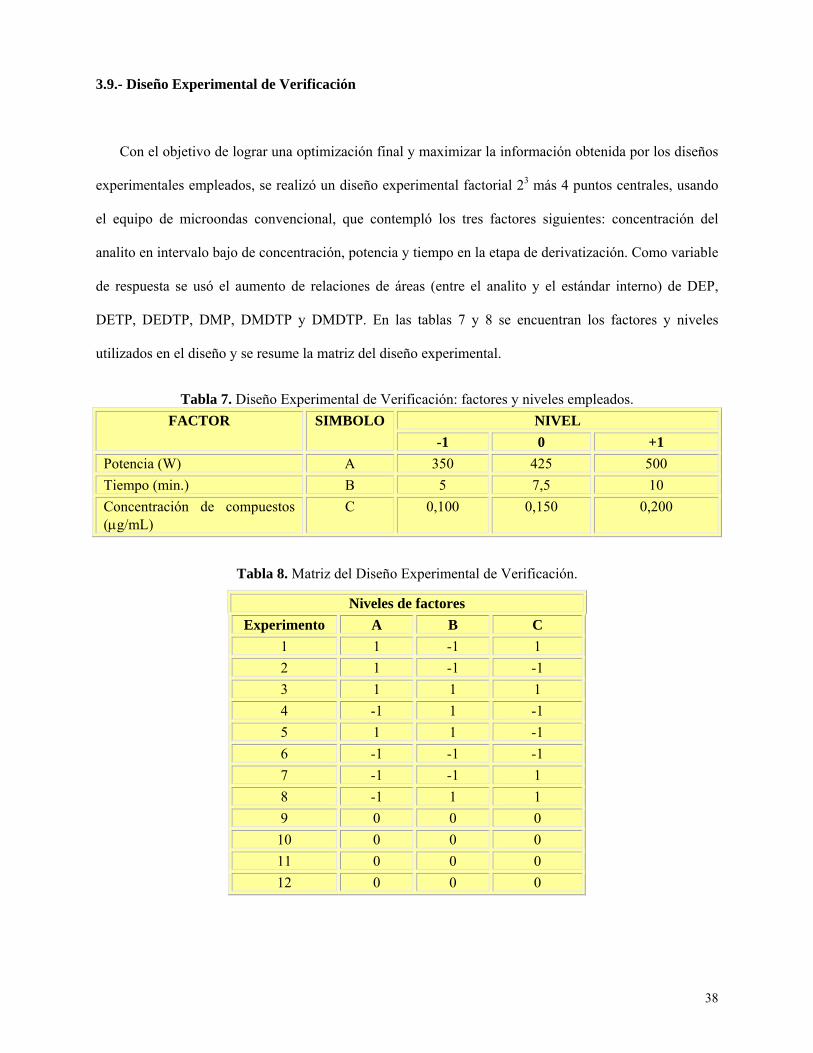
38
3.9.- Diseño Experimental de Verificación
Con el objetivo de lograr una optimización final y maximizar la información obtenida por los diseños
experimentales empleados, se realizó un diseño experimental factorial 23 más 4 puntos centrales, usando
el equipo de microondas convencional, que contempló los tres factores siguientes: concentración del
analito en intervalo bajo de concentración, potencia y tiempo en la etapa de derivatización. Como variable
de respuesta se usó el aumento de relaciones de áreas (entre el analito y el estándar interno) de DEP,
DETP, DEDTP, DMP, DMDTP y DMDTP. En las tablas 7 y 8 se encuentran los factores y niveles
utilizados en el diseño y se resume la matriz del diseño experimental.
Tabla 7. Diseño Experimental de Verificación: factores y niveles empleados.
FACTOR SIMBOLO NIVEL -1 0 +1
Potencia (W) A 350 425 500 Tiempo (min.) B 5 7,5 10 Concentración de compuestos (μg/mL)
C 0,100 0,150 0,200
Tabla 8. Matriz del Diseño Experimental de Verificación.
Niveles de factores Experimento A B C
1 1 -1 1 2 1 -1 -1 3 1 1 1 4 -1 1 -1 5 1 1 -1 6 -1 -1 -1 7 -1 -1 1 8 -1 1 1 9 0 0 0
10 0 0 0 11 0 0 0 12 0 0 0

39
3.10.-Preparación de curvas de calibración para los seis metabolitos en estudio utilizando las
condiciones obtenidas en el Diseño Experimental de Verificación.
Una vez analizado el tercer diseño se procedió a preparar curvas de calibración con seis
estándares preparados en duplicado con el fin de observar la linealidad de la respuesta para todos los
compuestos y a la vez lograr un mejor ajuste para el caso del DMP y DEP. Se prepararon las soluciones
en mezcla para los tío y di-tíosfosfatos en mezcla que fluctuaron entre 0,025 a 0,800 μg/mL en matraces
aforados de 10 mL a partir de los estándares de trabajo de 10 μg/mL y para el DMP y DEP
concentraciones de 0,200 a 0,8 μg/mL. Se derivatizó en el horno convencional a una potencia de 500W
durante 10 minutos para determinar finalmente la respuesta en el cromatógrafo gaseoso con detector FPD,
en sistema “splitless”.
3.11.- Estudio de cinética y de estabilidad en el tiempo a bajas concentraciones de los seis
metabolitos alquilfosfatos.
Este experimento se realizó con el fin de estudiar el efecto del catalizador (K2CO3) en la reacción
de derivatización y consistió en preparar en viales de reacción dos soluciones de concentraciones 0,050 y
0,100 µg/mL de la mezcla de los compuestos. En este ensayo los estándares permanecieron con el
catalizador durante diferentes tiempos de contacto antes de la inyección.
Las condiciones de trabajo para la reacción fueron las siguientes: para el horno de microondas
Potencia 500 W y tiempo 10 minutos y la cinética y estabilidad a temperatura ambiente.

40
3.12- Optimización de respuesta a bajas concentraciones para alquilfosfatos usando sistema de
microondas instrumental.
Al analizar los resultados del tercer diseño experimental no se obtuvieron resultados
significativos, por lo que se decidió trabajar a bajas concentraciones de metabolitos para estudiar distintos
tiempos y potencias aplicadas a la reacción de derivatización en un horno de microondas instrumental.
En esta etapa se prepararon los seis metabolitos en un vial de reacción, se utilizó acetonitrilo
como solvente para la reacción. Se realizaron dos experimentos, en el primero se prepararon soluciones
de 0,050 μg/mL y 0,100 μg/mL de la mezcla de metabolitos con el fin de evaluar el tiempo de reacción de
derivatización sobre la señal analítica de cada compuesto y en un segundo estudio se evaluó el efecto de
la potencia aplicada a la mezcla de analitos. Se utilizó el mismo procedimiento de derivatización de los
experimentos anteriores. Las tablas 9 y 10 muestran las condiciones estudiadas en estos experimentos.
Tabla 9. Estudio de tiempo de reacción a 350 W.
Experimento Tiempo de reacción(minutos)
1 2 2 4 3 6 4 8 5 10
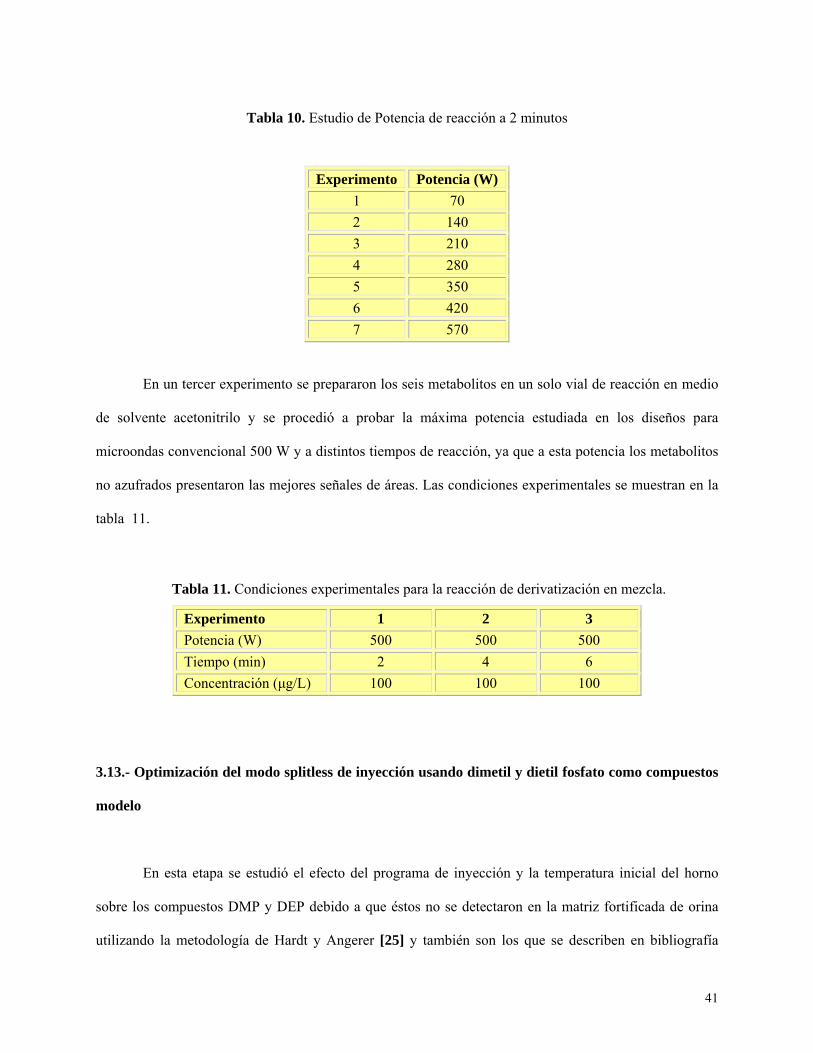
41
Tabla 10. Estudio de Potencia de reacción a 2 minutos
En un tercer experimento se prepararon los seis metabolitos en un solo vial de reacción en medio
de solvente acetonitrilo y se procedió a probar la máxima potencia estudiada en los diseños para
microondas convencional 500 W y a distintos tiempos de reacción, ya que a esta potencia los metabolitos
no azufrados presentaron las mejores señales de áreas. Las condiciones experimentales se muestran en la
tabla 11.
Tabla 11. Condiciones experimentales para la reacción de derivatización en mezcla.
Experimento 1 2 3 Potencia (W) 500 500 500 Tiempo (min) 2 4 6 Concentración (μg/L) 100 100 100
3.13.- Optimización del modo splitless de inyección usando dimetil y dietil fosfato como compuestos
modelo
En esta etapa se estudió el efecto del programa de inyección y la temperatura inicial del horno
sobre los compuestos DMP y DEP debido a que éstos no se detectaron en la matriz fortificada de orina
utilizando la metodología de Hardt y Angerer [25] y también son los que se describen en bibliografía
Experimento Potencia (W)1 70 2 140 3 210 4 280 5 350 6 420 7 570
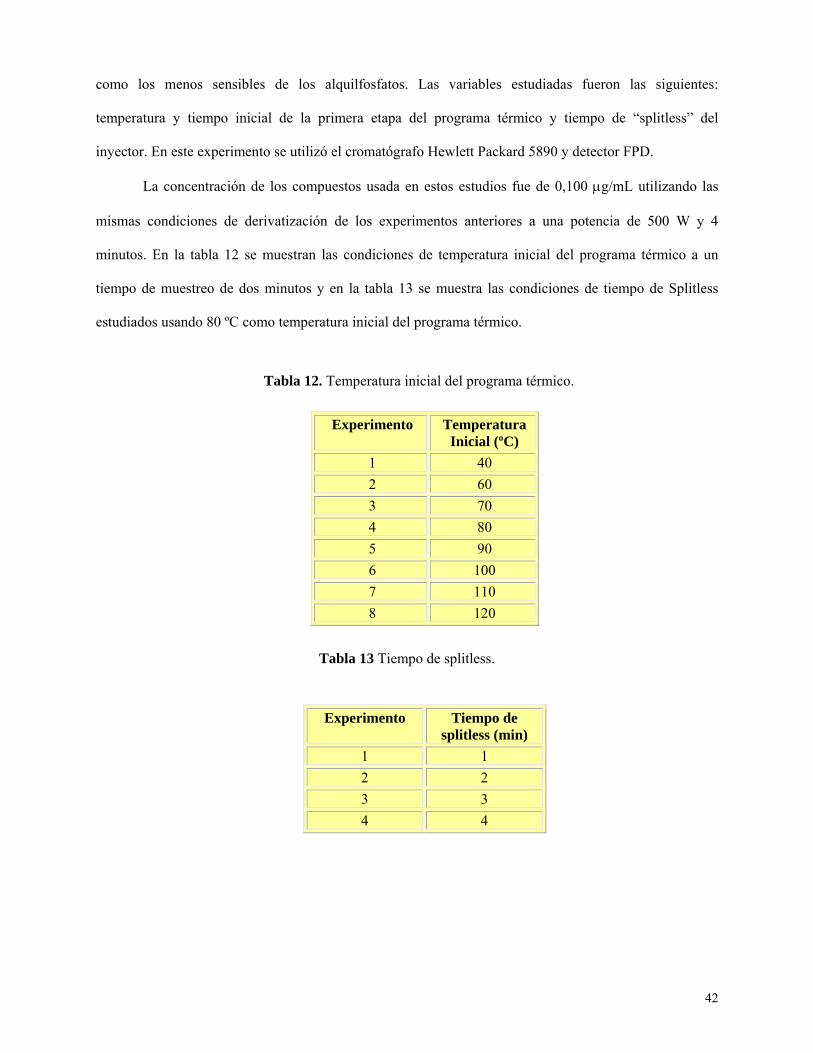
42
como los menos sensibles de los alquilfosfatos. Las variables estudiadas fueron las siguientes:
temperatura y tiempo inicial de la primera etapa del programa térmico y tiempo de “splitless” del
inyector. En este experimento se utilizó el cromatógrafo Hewlett Packard 5890 y detector FPD.
La concentración de los compuestos usada en estos estudios fue de 0,100 μg/mL utilizando las
mismas condiciones de derivatización de los experimentos anteriores a una potencia de 500 W y 4
minutos. En la tabla 12 se muestran las condiciones de temperatura inicial del programa térmico a un
tiempo de muestreo de dos minutos y en la tabla 13 se muestra las condiciones de tiempo de Splitless
estudiados usando 80 ºC como temperatura inicial del programa térmico.
Tabla 12. Temperatura inicial del programa térmico.
Tabla 13 Tiempo de splitless.
Experimento Temperatura Inicial (ºC)
1 40 2 60 3 70 4 80 5 90 6 100 7 110 8 120
Experimento Tiempo de splitless (min)
1 1 2 2 3 3 4 4

43
3.13.- Ensayo preliminar aplicando la metodología de Hardt y Angerer a una matriz de orina.
En este trabajo se probaron las etapas de extracción, derivatización, limpieza y concentración de
los seis metabolitos en una matriz de orina y en paralelo se preparó una curva de calibración con 4 niveles
de calibración, cuyas concentraciones fluctuaron entre 0 y 0,750 μg/mL. Los estándares puros se
disolvieron en acetonitrilo, luego se realizó la derivatización y extracción con agua y hexano, la fase
orgánica se trasvasijó a un vial de 10 mL para evaporar y concentrar los compuestos a un volumen final
de 150 μL.
El procedimiento utilizado para la orina consistió en adicionar distintas alícuotas de
concentraciones conocidas de los seis metabolitos a 5 ml de orina preparados a partir de un estándar de
trabajo de 1,00 μg/mL que contenía los seis compuestos en mezcla, luego se acidificó la muestra con 1 ml
de HCl 8M y se agregaron 4 gramos de NaCl, después se agregaron dos porciones de una mezcla de
solvente acetonitrilo / dietiléter (1:1v/v) y se agitó por 5 minutos. Una vez realizada la extracción se
separaron las fases por centrifugación a 2000 rpm durante 5 minutos. La fase orgánica se trasvasijó a un
vial de reacción de 10 mL y se procedió a evaporar el solvente con arrastre con nitrógeno hasta lograr
obtener un extracto totalmente seco. Luego se agregaron 1,5 ml de AcN 100 µL del reactivo derivatizante
y se llevó a reacción en el horno microondas instrumental, la potencia aplicada fue de 500 W y el tiempo
de reacción utilizado fue de 5 minutos. Una vez finalizada la reacción se dejaron los viales a temperatura
ambiente. Los viales de reacción se extrajeron con dos porciones de agua miliQ y 5 mL de hexano,
después de la extracción se redujo el volumen de la fase orgánica a 1 mL mediante evaporación por
arrastre de nitrógeno, posteriormente se agregó 150 µL de tolueno (que contenía el patrón interno) y se
continuó la evaporación hasta lograr reducir a un volumen final de 150 µL del extracto. Se inyectó 1 µL
en el cromatógrafo gaseoso con detector FPD siempre en la modalidad de “splitless”.

44
3.15.- Obtención de los parámetros de calidad en la etapa de extracción y derivatización de los 6
metabolitos dialquilfosfatos como estándares puros y en matriz acuosa y orina.
La obtención de los parámetros de calidad contempló principalmente evaluar intervalo lineal (R2)
y calcular el LOQ, LOD, porcentajes de recuperación y precisión obtenidos a partir de curvas de
calibración de los estándares puros, de una fortificación en medio acuoso y en orina. En este estudio se
utilizó el cromatógrafo Varian 3800 y detector NPD.
3.15.1.- Calibración Instrumental
Se determinó la función de calibración que describe la respuesta lineal del instrumento frente a la
concentración del analito utilizando como modelo una regresión lineal.
(Ec.1): y = a + bx
En que:
a: ordenada en el origen
b: pendiente de la regresión
A través de esta función de calibración se determinó la linealidad, intervalo de trabajo y se
determinaron los límites de detección, cuantificación y sensibilidad analítica.

45
3.15.2.- Linealidad, intervalo de trabajo y precisión.
Se escogió como intervalo de trabajo niveles de concentración de 0 a 0,250 µg/mL para todos los
metabolitos, se estableció como criterio de aceptación de linealidad mínimo un valor R2 mayor o igual a
0,98 (R > 0,99), la función de calibración se realizó con al menos 4 niveles de concentración.
La precisión fue evaluada analizando por cuadruplicado una concentración de 0,100 μg/mL de los
6 metabolitos preparados en acetonitrilo en forma simultánea.
3.15.3.- Límite de detección (LOD) y Límite de cuantificación (LOQ).
Se establecieron de acuerdo a las siguientes ecuaciones:
123 /
−−
⎟⎠⎞
⎜⎝⎛=
nn
mS
LOD XY
1210 /
−−
⎟⎠⎞
⎜⎝⎛=
nn
mS
LOQ XY
Donde Sy/x es el error estándar de la regresión, m la pendiente de la curva y n el número de pares de datos
utilizados.
3.15.4 Fortificación en medio acuoso y en matriz orina de compuestos dialquílicos a baja
concentración y extracción de los metabolitos.
Se preparó un estándar de trabajo de 10,0 µg/mL en acetonitrilo con los 6 metabolitos en mezcla
para obtener las siguientes concentraciones iniciales de 0,025 a 0,100 µg/mL.
Se tomaron 5 ml de agua milliQ o de orina y se trasvasijaron a un vial de reacción de 20 mL con tapa, se
adicionó una alícuota del estándar de trabajo, se agregaron 4 g de cloruro de sodio anhidro y luego se
adicionó 1 ml de HCL concentrado, la solución saturada se agitó, después se agregaron 5 mL de una
mezcla de acetonitrilo-dietileter (1:1 v/v) y se volvió a agitar por 5 minutos, después se centrifugó por 5

46
minutos a 2000 rpm. Se trasvasijó la fase orgánica a otro vial, repitiéndose la extracción por una segunda
vez, se juntaron los extractos, luego se evaporó suavemente en corriente de nitrógeno hasta llevar a
sequedad. Posteriormente, se realizó la derivatización en microondas instrumental a una potencia de 500
W por 4 minutos. La cuantificación se realizó por cromatografía gaseosa usando el detector NPD
optimizado en modo fósforo y a 2 minutos de tiempo de splitless.
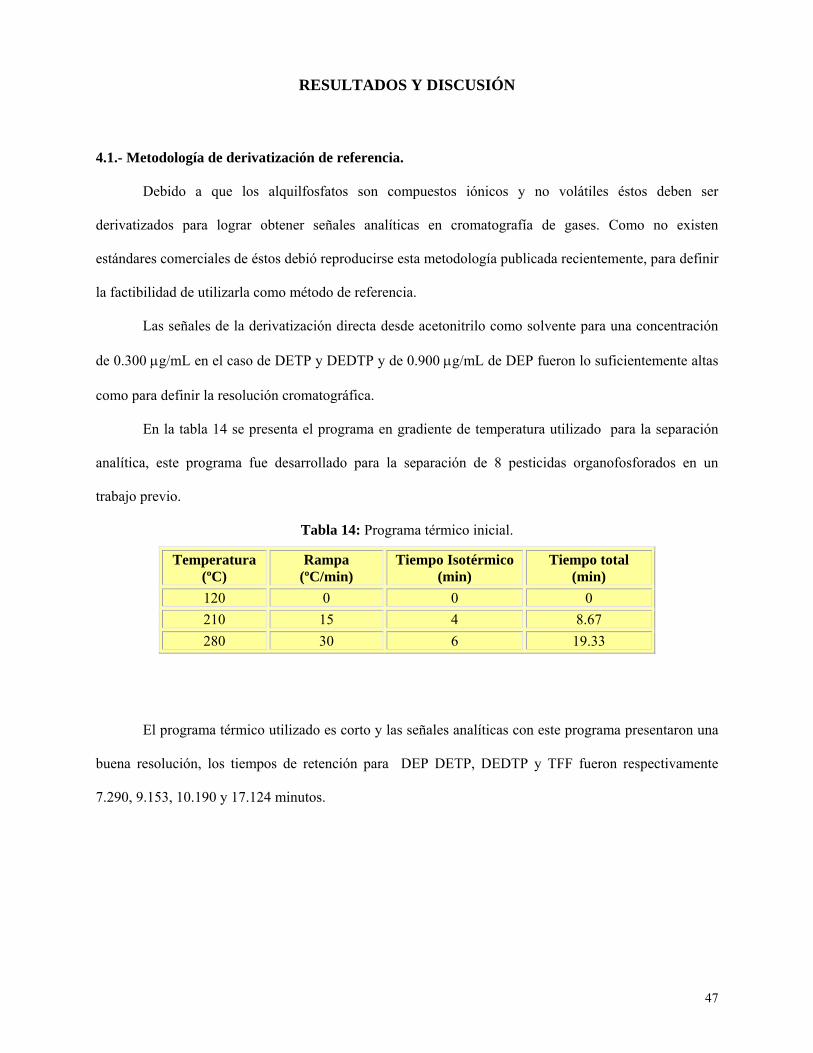
47
RESULTADOS Y DISCUSIÓN
4.1.- Metodología de derivatización de referencia.
Debido a que los alquilfosfatos son compuestos iónicos y no volátiles éstos deben ser
derivatizados para lograr obtener señales analíticas en cromatografía de gases. Como no existen
estándares comerciales de éstos debió reproducirse esta metodología publicada recientemente, para definir
la factibilidad de utilizarla como método de referencia.
Las señales de la derivatización directa desde acetonitrilo como solvente para una concentración
de 0.300 μg/mL en el caso de DETP y DEDTP y de 0.900 μg/mL de DEP fueron lo suficientemente altas
como para definir la resolución cromatográfica.
En la tabla 14 se presenta el programa en gradiente de temperatura utilizado para la separación
analítica, este programa fue desarrollado para la separación de 8 pesticidas organofosforados en un
trabajo previo.
Tabla 14: Programa térmico inicial.
Temperatura (ºC)
Rampa (ºC/min)
Tiempo Isotérmico (min)
Tiempo total (min)
120 0 0 0 210 15 4 8.67 280 30 6 19.33
El programa térmico utilizado es corto y las señales analíticas con este programa presentaron una
buena resolución, los tiempos de retención para DEP DETP, DEDTP y TFF fueron respectivamente
7.290, 9.153, 10.190 y 17.124 minutos.
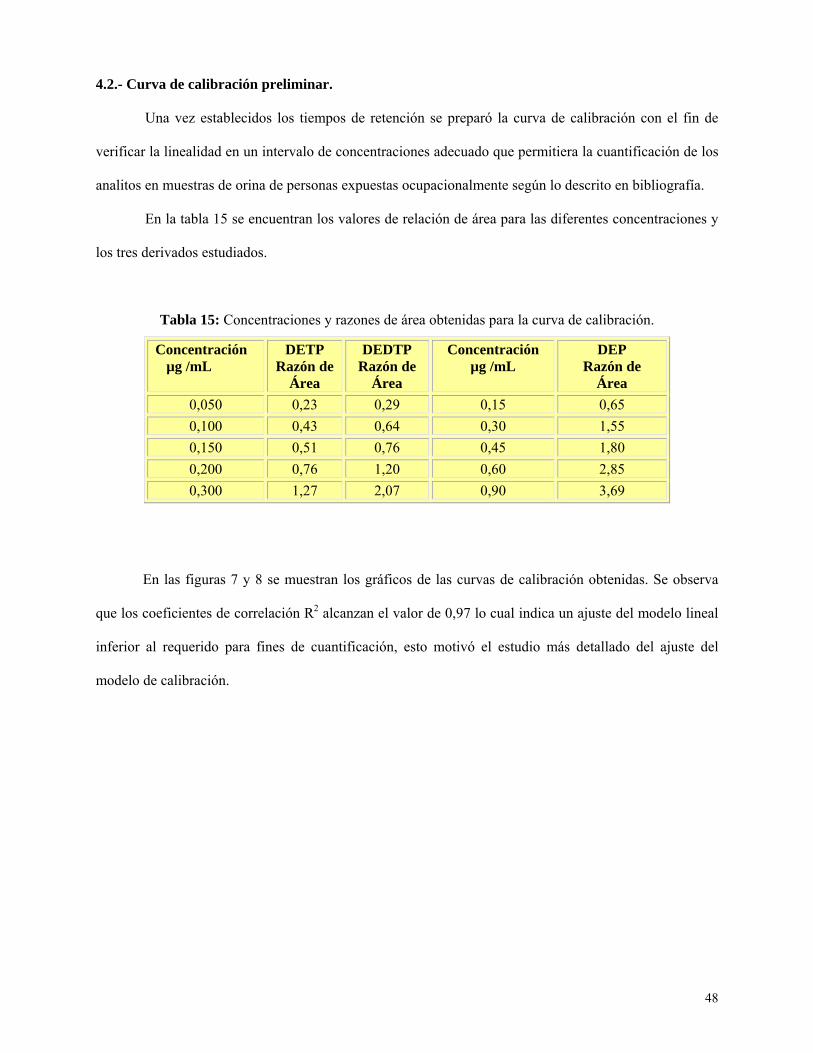
48
4.2.- Curva de calibración preliminar.
Una vez establecidos los tiempos de retención se preparó la curva de calibración con el fin de
verificar la linealidad en un intervalo de concentraciones adecuado que permitiera la cuantificación de los
analitos en muestras de orina de personas expuestas ocupacionalmente según lo descrito en bibliografía.
En la tabla 15 se encuentran los valores de relación de área para las diferentes concentraciones y
los tres derivados estudiados.
Tabla 15: Concentraciones y razones de área obtenidas para la curva de calibración.
Concentración µg /mL
DETP Razón de
Área
DEDTP Razón de
Área
Concentración µg /mL
DEP Razón de
Área 0,050 0,23 0,29 0,15 0,65 0,100 0,43 0,64 0,30 1,55 0,150 0,51 0,76 0,45 1,80 0,200 0,76 1,20 0,60 2,85 0,300 1,27 2,07 0,90 3,69
En las figuras 7 y 8 se muestran los gráficos de las curvas de calibración obtenidas. Se observa
que los coeficientes de correlación R2 alcanzan el valor de 0,97 lo cual indica un ajuste del modelo lineal
inferior al requerido para fines de cuantificación, esto motivó el estudio más detallado del ajuste del
modelo de calibración.
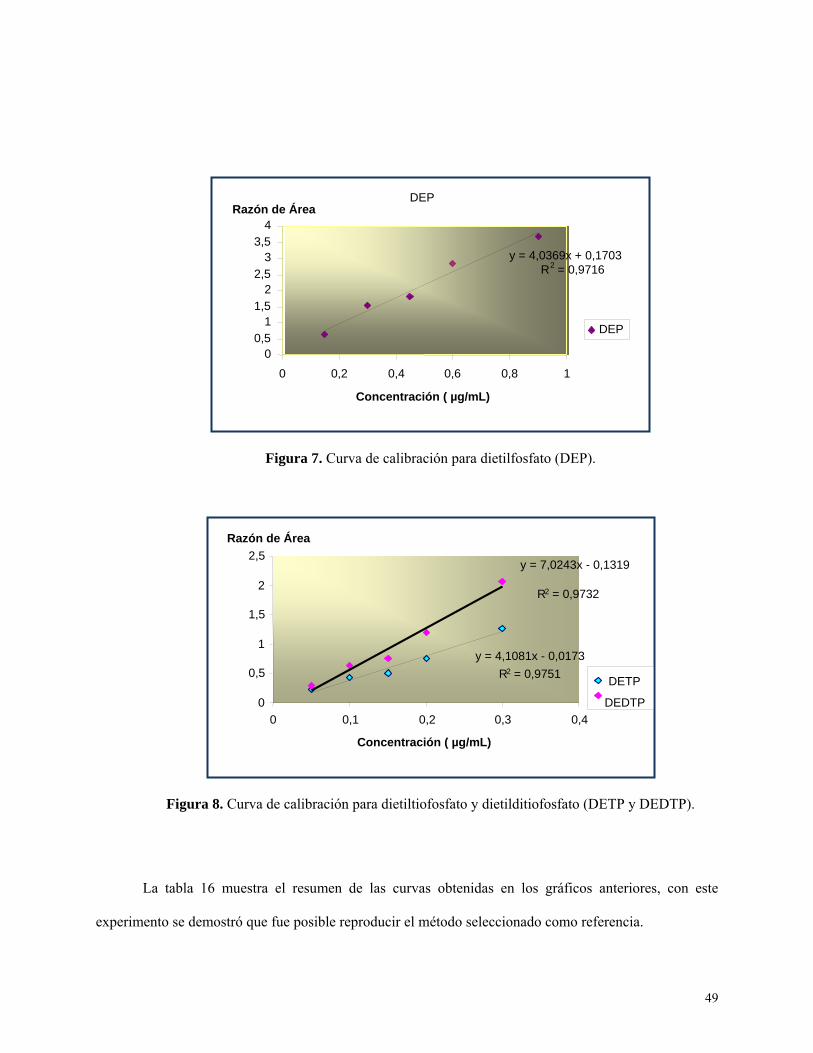
49
Figura 7. Curva de calibración para dietilfosfato (DEP).
Figura 8. Curva de calibración para dietiltiofosfato y dietilditiofosfato (DETP y DEDTP).
La tabla 16 muestra el resumen de las curvas obtenidas en los gráficos anteriores, con este
experimento se demostró que fue posible reproducir el método seleccionado como referencia.
y = 7,0243x - 0,1319
R2 = 0,9732
y = 4,1081x - 0,0173 R2 = 0,9751
0
0,5
1
1,5
2
2,5
0 0,1 0,2 0,3 0,4
Concentración ( µg/mL)
Razón de Área
DETP DEDTP
DEP
y = 4,0369x + 0,1703R2 = 0,9716
0 0,5
1 1,5
2 2,5
3 3,5
4
0 0,2 0,4 0,6 0,8 1
Concentración ( µg/mL)
Razón de Área
DEP
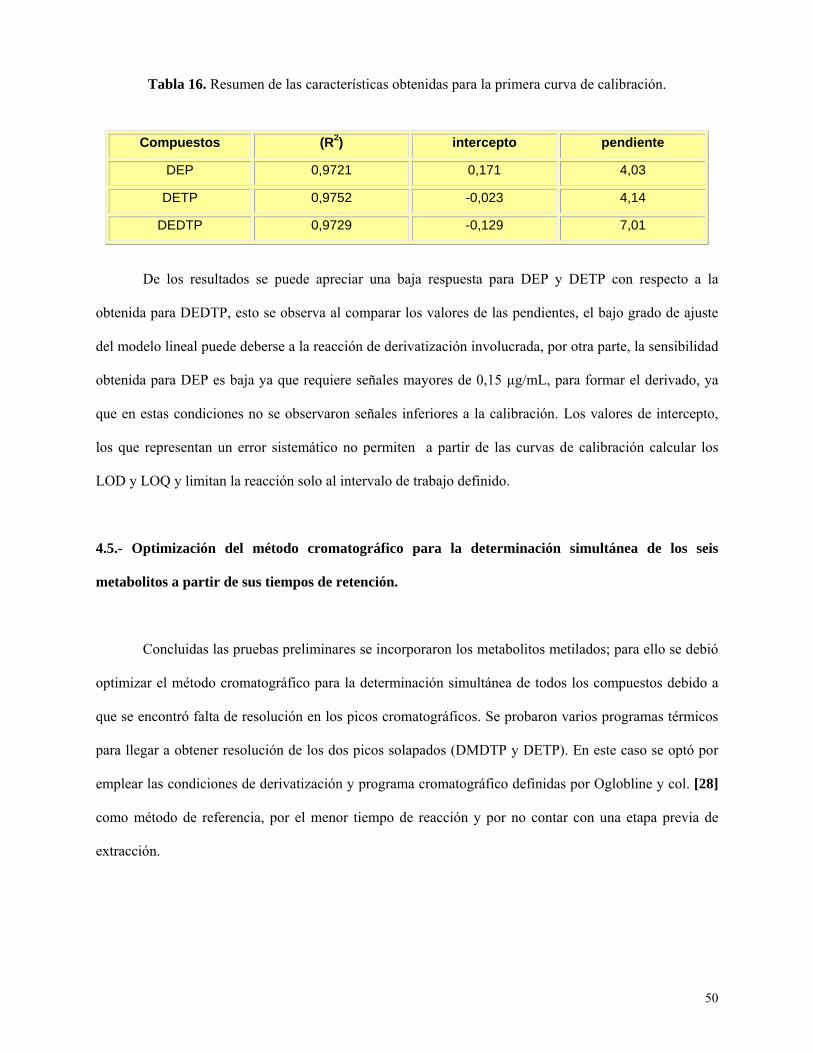
50
Tabla 16. Resumen de las características obtenidas para la primera curva de calibración.
De los resultados se puede apreciar una baja respuesta para DEP y DETP con respecto a la
obtenida para DEDTP, esto se observa al comparar los valores de las pendientes, el bajo grado de ajuste
del modelo lineal puede deberse a la reacción de derivatización involucrada, por otra parte, la sensibilidad
obtenida para DEP es baja ya que requiere señales mayores de 0,15 µg/mL, para formar el derivado, ya
que en estas condiciones no se observaron señales inferiores a la calibración. Los valores de intercepto,
los que representan un error sistemático no permiten a partir de las curvas de calibración calcular los
LOD y LOQ y limitan la reacción solo al intervalo de trabajo definido.
4.5.- Optimización del método cromatográfico para la determinación simultánea de los seis
metabolitos a partir de sus tiempos de retención.
Concluidas las pruebas preliminares se incorporaron los metabolitos metilados; para ello se debió
optimizar el método cromatográfico para la determinación simultánea de todos los compuestos debido a
que se encontró falta de resolución en los picos cromatográficos. Se probaron varios programas térmicos
para llegar a obtener resolución de los dos picos solapados (DMDTP y DETP). En este caso se optó por
emplear las condiciones de derivatización y programa cromatográfico definidas por Oglobline y col. [28]
como método de referencia, por el menor tiempo de reacción y por no contar con una etapa previa de
extracción.
Compuestos (R2) intercepto pendiente
DEP 0,9721 0,171 4,03
DETP 0,9752 -0,023 4,14
DEDTP 0,9729 -0,129 7,01
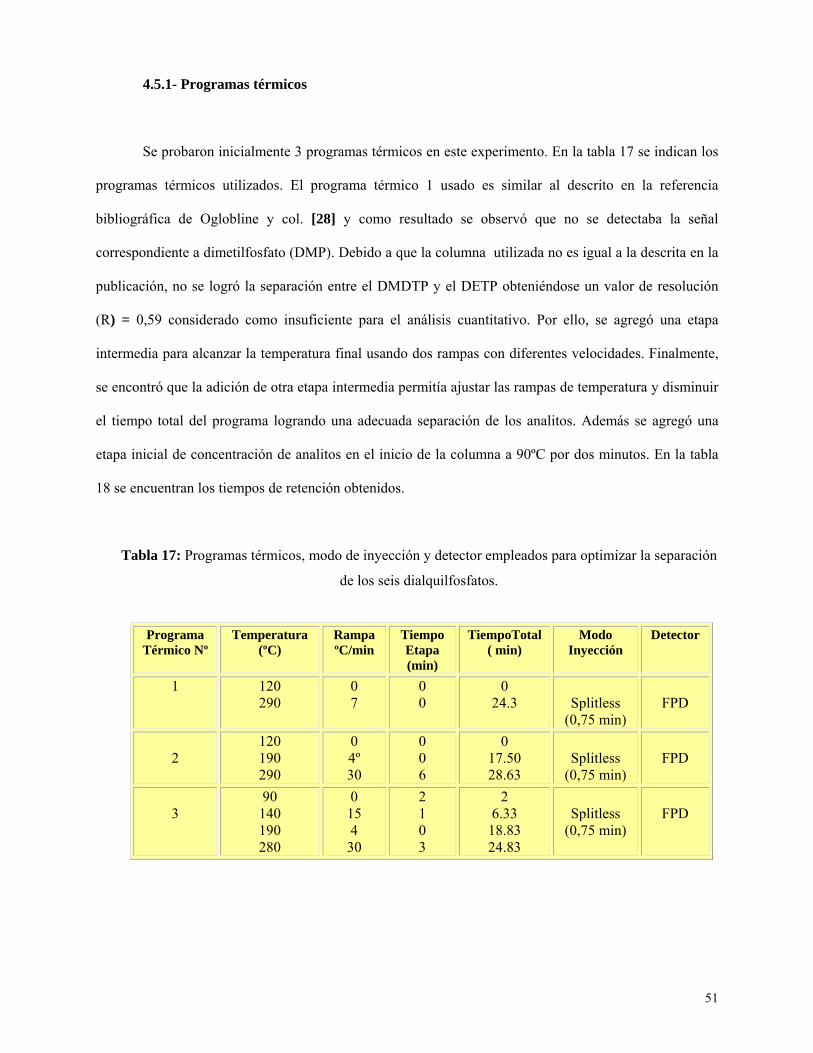
51
4.5.1- Programas térmicos
Se probaron inicialmente 3 programas térmicos en este experimento. En la tabla 17 se indican los
programas térmicos utilizados. El programa térmico 1 usado es similar al descrito en la referencia
bibliográfica de Oglobline y col. [28] y como resultado se observó que no se detectaba la señal
correspondiente a dimetilfosfato (DMP). Debido a que la columna utilizada no es igual a la descrita en la
publicación, no se logró la separación entre el DMDTP y el DETP obteniéndose un valor de resolución
(R) = 0,59 considerado como insuficiente para el análisis cuantitativo. Por ello, se agregó una etapa
intermedia para alcanzar la temperatura final usando dos rampas con diferentes velocidades. Finalmente,
se encontró que la adición de otra etapa intermedia permitía ajustar las rampas de temperatura y disminuir
el tiempo total del programa logrando una adecuada separación de los analitos. Además se agregó una
etapa inicial de concentración de analitos en el inicio de la columna a 90ºC por dos minutos. En la tabla
18 se encuentran los tiempos de retención obtenidos.
Tabla 17: Programas térmicos, modo de inyección y detector empleados para optimizar la separación
de los seis dialquilfosfatos.
Programa Térmico Nº
Temperatura (ºC)
Rampa ºC/min
Tiempo Etapa (min)
TiempoTotal ( min)
Modo Inyección
Detector
1 120 290
0 7
0 0
0 24.3
Splitless
(0,75 min)
FPD
2
120 190 290
0 4º 30
0 0 6
0 17.50 28.63
Splitless
(0,75 min)
FPD
3
90 140 190 280
0 15 4 30
2 1 0 3
2 6.33
18.83 24.83
Splitless
(0,75 min)
FPD
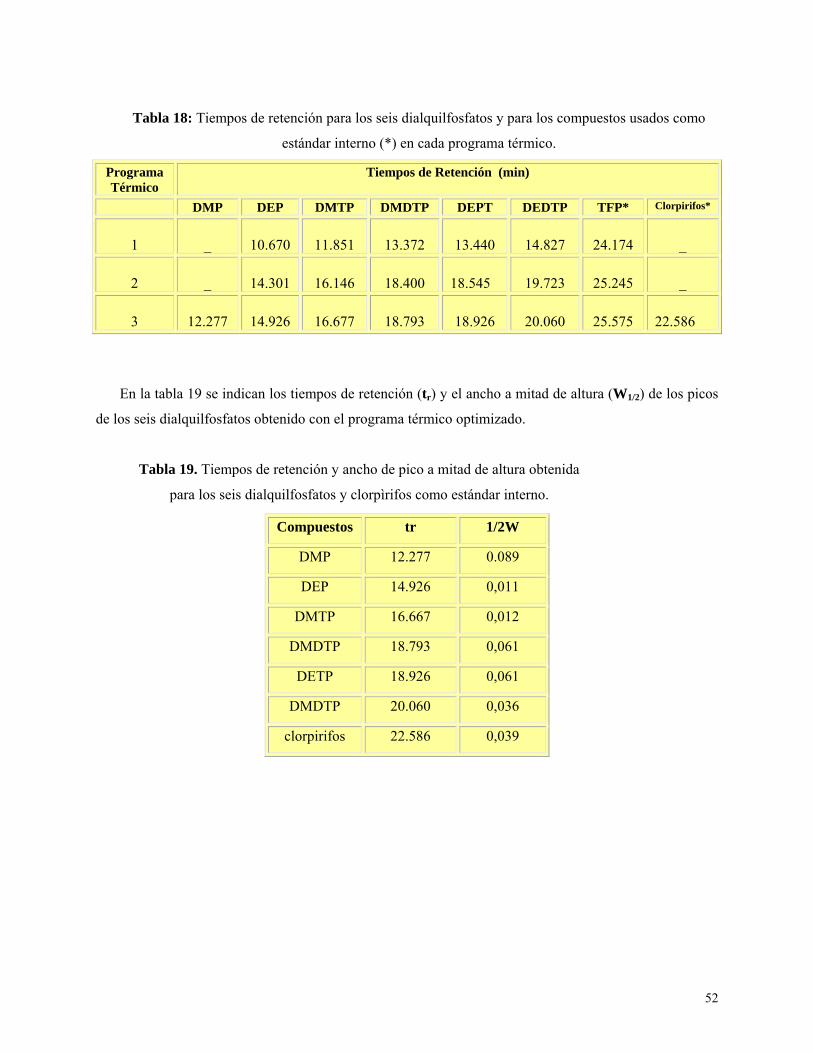
52
Tabla 18: Tiempos de retención para los seis dialquilfosfatos y para los compuestos usados como
estándar interno (*) en cada programa térmico.
Programa Térmico
Tiempos de Retención (min)
DMP DEP DMTP DMDTP DEPT DEDTP TFP* Clorpirifos*
1
_
10.670
11.851
13.372
13.440
14.827
24.174
_
2
_
14.301
16.146
18.400
18.545
19.723
25.245
_
3
12.277
14.926
16.677
18.793
18.926
20.060
25.575
22.586
En la tabla 19 se indican los tiempos de retención (tr) y el ancho a mitad de altura (W1/2) de los picos
de los seis dialquilfosfatos obtenido con el programa térmico optimizado.
Tabla 19. Tiempos de retención y ancho de pico a mitad de altura obtenida
para los seis dialquilfosfatos y clorpìrifos como estándar interno.
Compuestos tr 1/2W
DMP 12.277 0.089
DEP 14.926 0,011
DMTP 16.667 0,012
DMDTP 18.793 0,061
DETP 18.926 0,061
DMDTP 20.060 0,036
clorpirifos 22.586 0,039
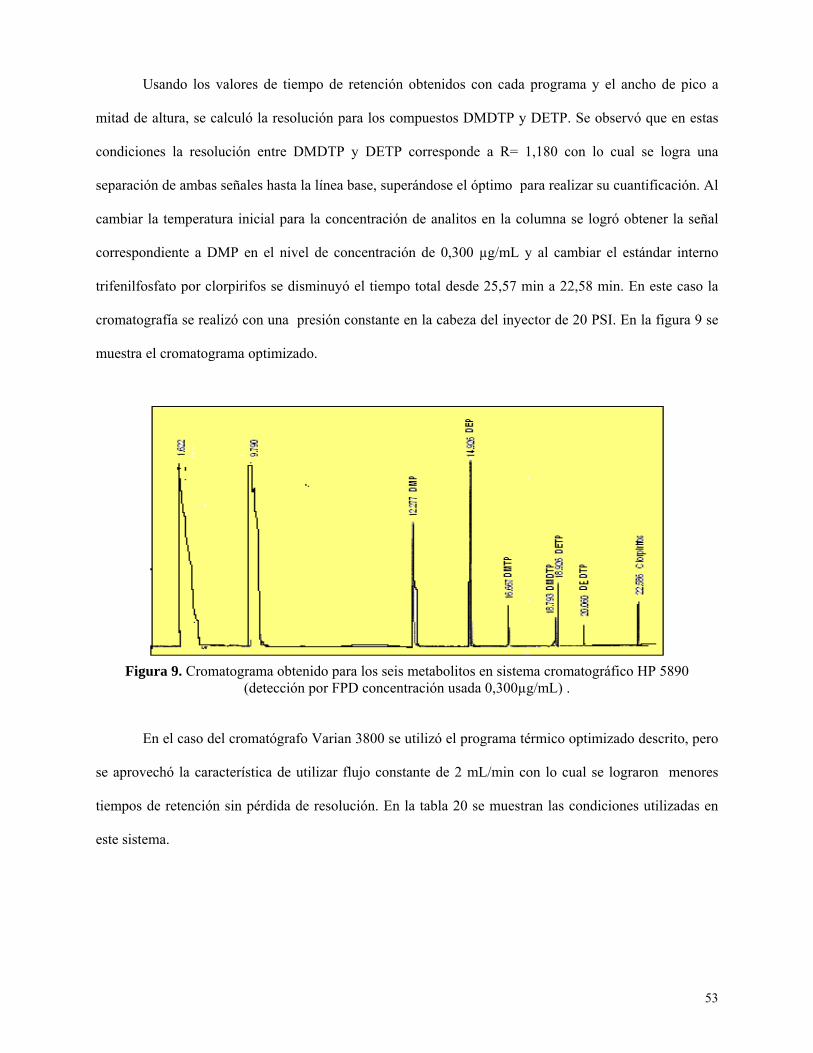
53
Usando los valores de tiempo de retención obtenidos con cada programa y el ancho de pico a
mitad de altura, se calculó la resolución para los compuestos DMDTP y DETP. Se observó que en estas
condiciones la resolución entre DMDTP y DETP corresponde a R= 1,180 con lo cual se logra una
separación de ambas señales hasta la línea base, superándose el óptimo para realizar su cuantificación. Al
cambiar la temperatura inicial para la concentración de analitos en la columna se logró obtener la señal
correspondiente a DMP en el nivel de concentración de 0,300 µg/mL y al cambiar el estándar interno
trifenilfosfato por clorpirifos se disminuyó el tiempo total desde 25,57 min a 22,58 min. En este caso la
cromatografía se realizó con una presión constante en la cabeza del inyector de 20 PSI. En la figura 9 se
muestra el cromatograma optimizado.
Figura 9. Cromatograma obtenido para los seis metabolitos en sistema cromatográfico HP 5890 (detección por FPD concentración usada 0,300µg/mL) .
En el caso del cromatógrafo Varian 3800 se utilizó el programa térmico optimizado descrito, pero
se aprovechó la característica de utilizar flujo constante de 2 mL/min con lo cual se lograron menores
tiempos de retención sin pérdida de resolución. En la tabla 20 se muestran las condiciones utilizadas en
este sistema.
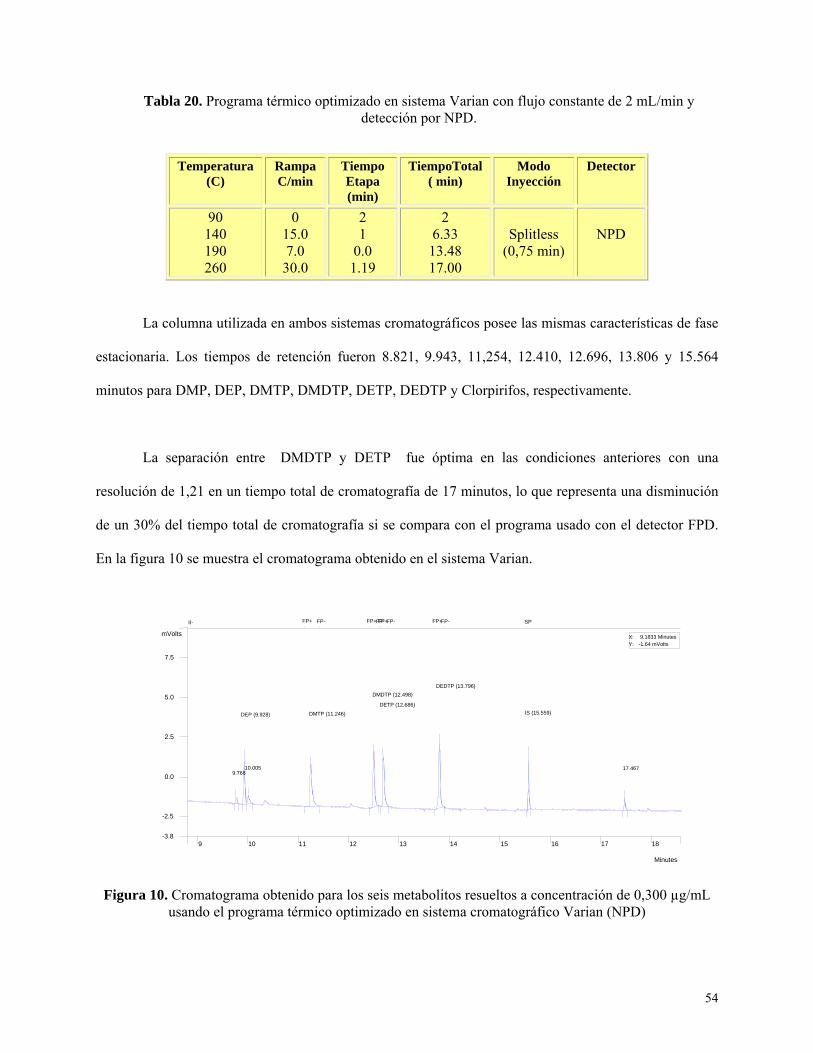
54
Tabla 20. Programa térmico optimizado en sistema Varian con flujo constante de 2 mL/min y
detección por NPD.
Temperatura (C)
Rampa C/min
Tiempo Etapa (min)
TiempoTotal ( min)
Modo Inyección
Detector
90 140 190 260
0 15.0 7.0
30.0
2 1
0.0 1.19
2 6.33 13.48 17.00
Splitless
(0,75 min)
NPD
La columna utilizada en ambos sistemas cromatográficos posee las mismas características de fase
estacionaria. Los tiempos de retención fueron 8.821, 9.943, 11,254, 12.410, 12.696, 13.806 y 15.564
minutos para DMP, DEP, DMTP, DMDTP, DETP, DEDTP y Clorpirifos, respectivamente.
La separación entre DMDTP y DETP fue óptima en las condiciones anteriores con una
resolución de 1,21 en un tiempo total de cromatografía de 17 minutos, lo que representa una disminución
de un 30% del tiempo total de cromatografía si se compara con el programa usado con el detector FPD.
En la figura 10 se muestra el cromatograma obtenido en el sistema Varian.
Figura 10. Cromatograma obtenido para los seis metabolitos resueltos a concentración de 0,300 µg/mL usando el programa térmico optimizado en sistema cromatográfico Varian (NPD)
9 10 11 12 13 14 15 16 17 18
Minutes
-3.8
-2.5
0.0
2.5
5.0
7.5
mVolts
9.768
DEP (9.928)
10.005
DMTP (11.246)
DMDTP (12.498)
DETP (12.686)
DEDTP (13.796)
IS (15.559)
17.467
X:Y:
9.1833 Minutes-1.64 mVolts
II- FP+ FP- FP+FP-FP+FP- FP+FP- SP
55
4.6- Comparación de métodos de referencia
Con los programas térmicos optimizados se realizó el experimento de comparación entre ambos
métodos de referencia. Se calcularon los promedios y coeficientes de variación para analizar la
variabilidad de la inyección en la detección por FPD para las 4 réplicas de la mezcla de estándares a una
concentración de 0.10 µg/mL, derivatizando por medio de ambos métodos. Los resultados se encuentran
en las tablas 21 y 22.
Tabla 21. Razones de área obtenidas con método de derivatización de Oglobline [28] usando el programa térmico optimizado.
Tabla 22. Razones de área obtenidas con método de derivatización de Hardt & Angerer [25] usando el programa térmico optimizado.
Al comparar ambos métodos de referencia se puede concluir que en el de Hardt y Angerer [25] se
obtienen las mejores respuestas para DEP con un promedio de relaciones de área de 2,20 y un 25 % más
de señal lo que indica que este compuesto necesita mayor tiempo y/o menor temperatura en la reacción de
derivatización. Pero para el caso del DMP no se detecta el derivado en ninguno de los métodos a la
Número de Replica
1 2 3 4 Promedio SD CV %
DMP 0 0 0 0 0 0 0 DEP 1.29 1.15 1.20 1.31 1.24 0.073 6.09
DMTP 1.32 1.4 1.27 1.47 1.36 0.089 6.56 DMDTP 1.44 1.40 1.36 1.51 1.43 0.063 4.45
DETP 1.54 1.51 1.43 1.56 1.51 0.057 3.82 DMDTP 1.44 1.40 1.36 1.43 1.42 0.052 3.69
Número de Réplica
1 2 3 4 Promedio SD CV %
DMP 0 0 0 0 0 0 0 DEP 2.25 2.00 2.25 2.36 2.20 1.35 6,10
DMTP 1.56 1.45 1.56 1.11 1.46 0,18 12,90 DMDTP 1.73 1.54 1.73 1.57 1.64 0,09 5,36
DETP 1.35 1.38 1.51 1.14 1.34 0,13 9,92 DMDTP 1.56 1.57 1.56 1.40 1.52 0,07 4,52
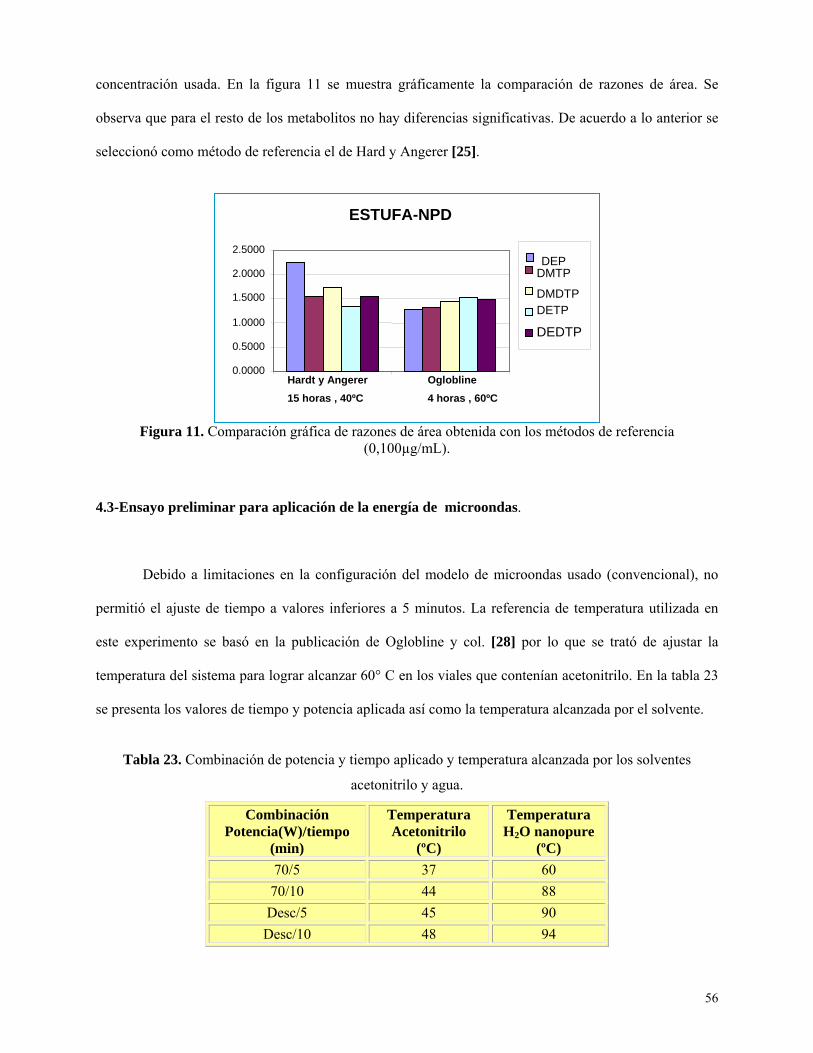
56
concentración usada. En la figura 11 se muestra gráficamente la comparación de razones de área. Se
observa que para el resto de los metabolitos no hay diferencias significativas. De acuerdo a lo anterior se
seleccionó como método de referencia el de Hard y Angerer [25].
Figura 11. Comparación gráfica de razones de área obtenida con los métodos de referencia
(0,100µg/mL).
4.3-Ensayo preliminar para aplicación de la energía de microondas.
Debido a limitaciones en la configuración del modelo de microondas usado (convencional), no
permitió el ajuste de tiempo a valores inferiores a 5 minutos. La referencia de temperatura utilizada en
este experimento se basó en la publicación de Oglobline y col. [28] por lo que se trató de ajustar la
temperatura del sistema para lograr alcanzar 60° C en los viales que contenían acetonitrilo. En la tabla 23
se presenta los valores de tiempo y potencia aplicada así como la temperatura alcanzada por el solvente.
Tabla 23. Combinación de potencia y tiempo aplicado y temperatura alcanzada por los solventes
acetonitrilo y agua.
Combinación Potencia(W)/tiempo
(min)
Temperatura Acetonitrilo
(ºC)
Temperatura H2O nanopure
(ºC) 70/5 37 60
70/10 44 88 Desc/5 45 90
Desc/10 48 94
ESTUFA-NPD
0.0000
0.5000
1.0000
1.5000
2.0000
2.5000
Hardt y Angerer15 horas , 40ºC
Oglobline4 horas , 60ºC
DEP
DMTP
DMDTP DETP
DEDTP

57
Como resultado se obtuvo que en el caso del acetonitrilo las dos potencias usadas conducen a
diferencias de temperatura de escasa magnitud; en el caso del agua se observó una mayor diferencia de
temperatura la que depende del tiempo y de la potencia empleada. El agua posee un mayor coeficiente de
disipación de la energía de microondas debido a su mayor constante dieléctrica.
4.4.- Etapa de derivatización utilizando la energía de microondas. Identificación de los parámetros
significativos para la reacción. Diseño experimental exploratorio.
El primer diseño experimental exploratorio consideró los factores potencia, tiempo, cantidad de
reactivo derivatizante, volumen de solvente empleado y concentración de dialquilfosfato considerándose
en este experimento sólo los dietilderivados. A partir del análisis estadístico (ANOVA) se identificó la
potencia como único parámetro significativo para la obtención de buenos rendimientos en la reacción de
derivatización. Los gráficos estandarizados de Pareto indican que una mayor potencia conduce a un
mayor rendimiento. La tabla 24 muestra las razones de área obtenidas en la matriz del diseño
experimental exploratorio.
En las figuras 12, 13 y 14 se muestran dichos gráficos para los tres compuestos. En este tipo de gráfico se
puede deducir el sentido positivo o negativo de la influencia de cada factor y sus interacciones.
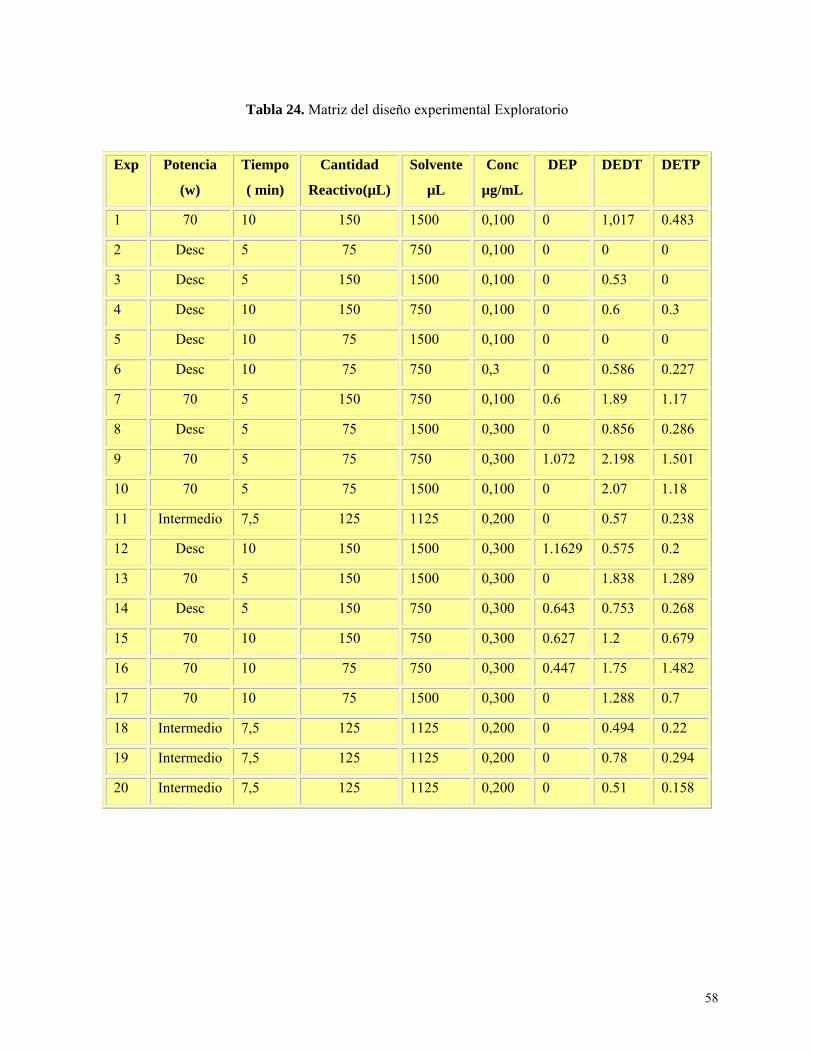
58
Tabla 24. Matriz del diseño experimental Exploratorio
Exp Potencia
(w)
Tiempo
( min)
Cantidad
Reactivo(µL)
Solvente
µL
Conc
µg/mL
DEP DEDT DETP
1 70 10 150 1500 0,100 0 1,017 0.483
2 Desc 5 75 750 0,100 0 0 0
3 Desc 5 150 1500 0,100 0 0.53 0
4 Desc 10 150 750 0,100 0 0.6 0.3
5 Desc 10 75 1500 0,100 0 0 0
6 Desc 10 75 750 0,3 0 0.586 0.227
7 70 5 150 750 0,100 0.6 1.89 1.17
8 Desc 5 75 1500 0,300 0 0.856 0.286
9 70 5 75 750 0,300 1.072 2.198 1.501
10 70 5 75 1500 0,100 0 2.07 1.18
11 Intermedio 7,5 125 1125 0,200 0 0.57 0.238
12 Desc 10 150 1500 0,300 1.1629 0.575 0.2
13 70 5 150 1500 0,300 0 1.838 1.289
14 Desc 5 150 750 0,300 0.643 0.753 0.268
15 70 10 150 750 0,300 0.627 1.2 0.679
16 70 10 75 750 0,300 0.447 1.75 1.482
17 70 10 75 1500 0,300 0 1.288 0.7
18 Intermedio 7,5 125 1125 0,200 0 0.494 0.22
19 Intermedio 7,5 125 1125 0,200 0 0.78 0.294
20 Intermedio 7,5 125 1125 0,200 0 0.51 0.158
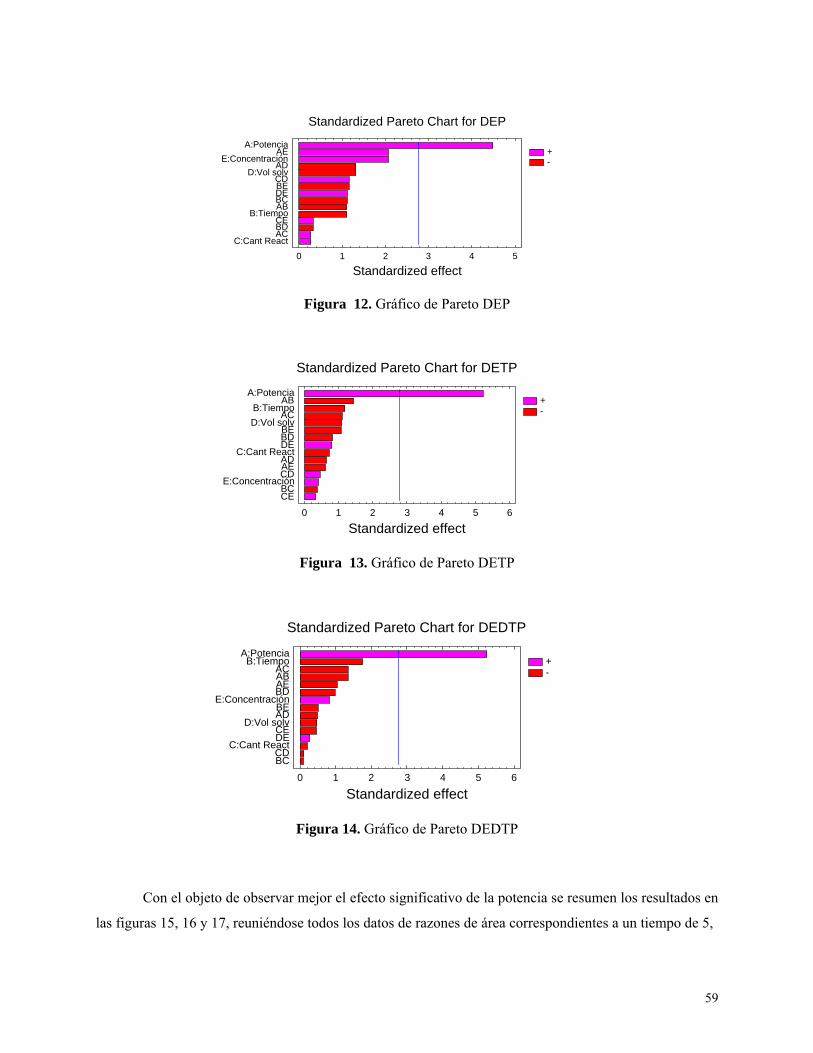
59
Standardized Pareto Chart for DEP
0 1 2 3 4 5
Standardized effect
C:Cant ReactACBDCE
B:TiempoABBCDEBECD
D:Vol solvAD
E:ConcentraciónAE
A:Potencia+-
Figura 12. Gráfico de Pareto DEP
Standardized Pareto Chart for DETP
0 1 2 3 4 5 6
Standardized effect
CEBC
E:ConcentraciónCDAEAD
C:Cant ReactDEBDBE
D:Vol solvAC
B:TiempoAB
A:Potencia+-
Figura 13. Gráfico de Pareto DETP
Standardized Pareto Chart for DEDTP
Standardized effect
+-
0 1 2 3 4 5 6
BCCD
C:Cant ReactDECE
D:Vol solvADBE
E:ConcentraciónBDAEABAC
B:TiempoA:Potencia
Figura 14. Gráfico de Pareto DEDTP
Con el objeto de observar mejor el efecto significativo de la potencia se resumen los resultados en
las figuras 15, 16 y 17, reuniéndose todos los datos de razones de área correspondientes a un tiempo de 5,
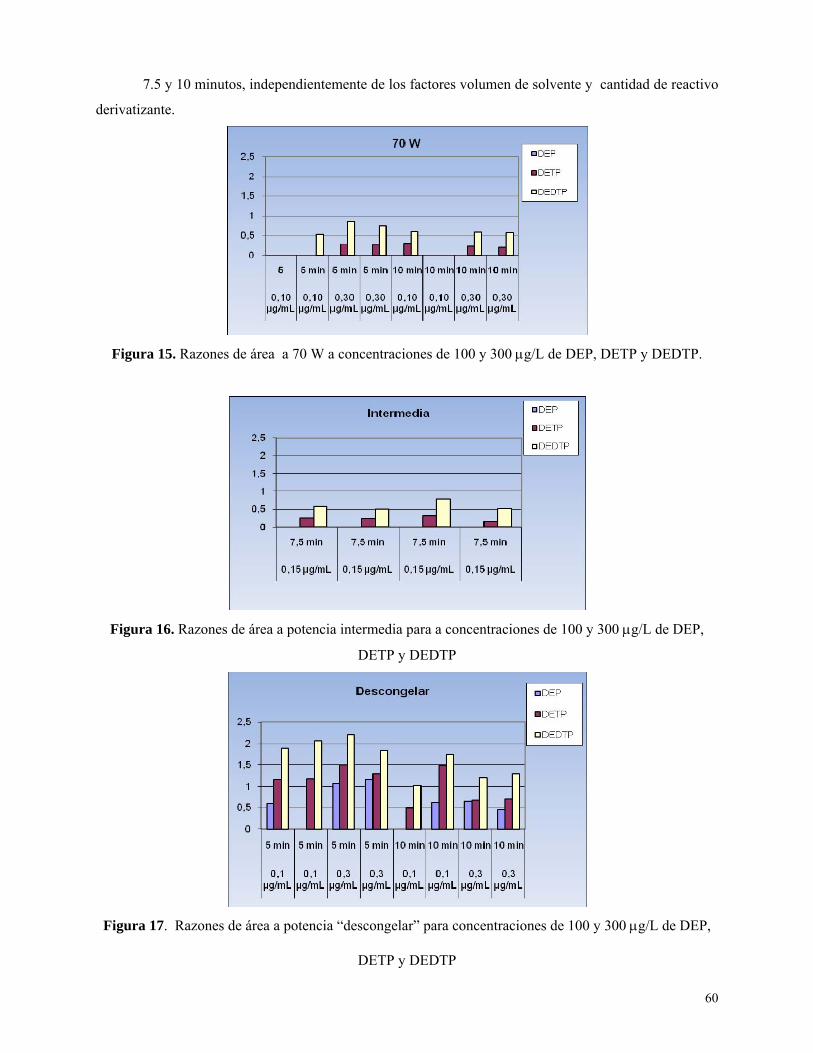
60
7.5 y 10 minutos, independientemente de los factores volumen de solvente y cantidad de reactivo
derivatizante.
Figura 15. Razones de área a 70 W a concentraciones de 100 y 300 μg/L de DEP, DETP y DEDTP.
Figura 16. Razones de área a potencia intermedia para a concentraciones de 100 y 300 μg/L de DEP,
DETP y DEDTP
Figura 17. Razones de área a potencia “descongelar” para concentraciones de 100 y 300 μg/L de DEP,
DETP y DEDTP

61
A 70 W y a potencia intermedia se obtuvieron bajas respuestas para DETP y DEDTP y no se
obtuvo respuesta para DEP. La potencia correspondiente a “descongelar” es la mayor y generó las
mejores respuestas para los tres compuestos. Independiente de la significancia estadística del factor
tiempo, es posible observar una tendencia negativa al aumentar el tiempo de reacción, sin embargo no se
observó en los cromatogramas signos de degradación a través de la aparición de nuevos picos. La
potencia “descongelar” usada por 5 minutos produce las mayores respuestas de área a ambas
concentraciones estudiadas.
A pesar de lo anterior se optó por utilizar en los experimentos siguientes la potencia de 70 W ya
que otorga un valor concreto que puede transferirse al sistema de microondas instrumental (Ethos 1600),
el tiempo seleccionado fue el necesario para alcanzar la temperatura de reacción de derivatización usada
en la referencia elegida (60°C). Las mediciones se hicieron en duplicado para cada compuesto por
separado, con el fin de establecer la posibilidad de degradaciones, que impidieran visualizar los
rendimientos reales en cada reacción; además se realizó la inyección en duplicado de cada solución de
0,300 μg/mL para la cuantificación. Se compararon los resultados con los obtenidos a través del método
de referencia antes elegido (tabla 25).
Tabla 25 Razones de área obtenidas con las distintas condiciones de derivatización aplicadas.
Método de derivatización aplicado
DEP Rel. Área 1
DEP Rel. Área 2
DETP Rel. Área 1
DETP Rel. Área 2
DEDTP Rel. Área 1
DEDTP Rel. Área 2
Método de microondas 70 W, 5 minutos
0.137 0.338 1.728 1.721 3.582 2.658
Método de referencia (Hardt and Angerer)
3.1556 3.228 1.961 1.641 2.694 2.666
Una segunda comparación se realizó entre la derivatización con microondas en la condición 70 W por 5
minutos contra la condición de derivatización usada por Hardt y Angerer [25]. Se encontró que para DEP
la derivatización en estufa entrega razones de área más favorables mientras que para los otros 4
metabolitos no se aprecia una diferencia significativa, para DMP tampoco se logró ver señal. Por lo

62
anterior se decidió utilizar un microondas instrumental que permite tener un mejor control de la potencia
utilizada. Con este sistema se procedió a realizar un diseño experimental de optimización que utilizó los
seis metabolitos en forma simultánea, se utilizó como factores de estudio la potencia y el tiempo.
Como resultado de la comparación se puede señalar que aunque es posible la obtención de señales
de los derivados mediante el uso de microondas, en el caso de DEP son mucho mas bajas que las del
método de referencia y de acuerdo a la experiencia anterior, sin embargo para los tio-fosfatos estas
señales alcanzan valores similares, aunque de menor reproducibilidad, en el caso de dietil-ditiofosfato la
relación de áreas es a lo menos similar lo cual indica que no se está perdiendo este compuesto por
descomposición oxidativa hacia dietilfosfato como está descrito en la bibliografía o si esto ocurre lo hace
en una porcentaje menor o similar a lo que ocurre en el método de referencia. Tanto el diseño exploratorio
como la comparación anterior indican la factibilidad de uso de microondas para lograr la derivatización
en un corto tiempo, con escaso grado de manipulación de las muestras y justifica la necesaria
optimización del método.
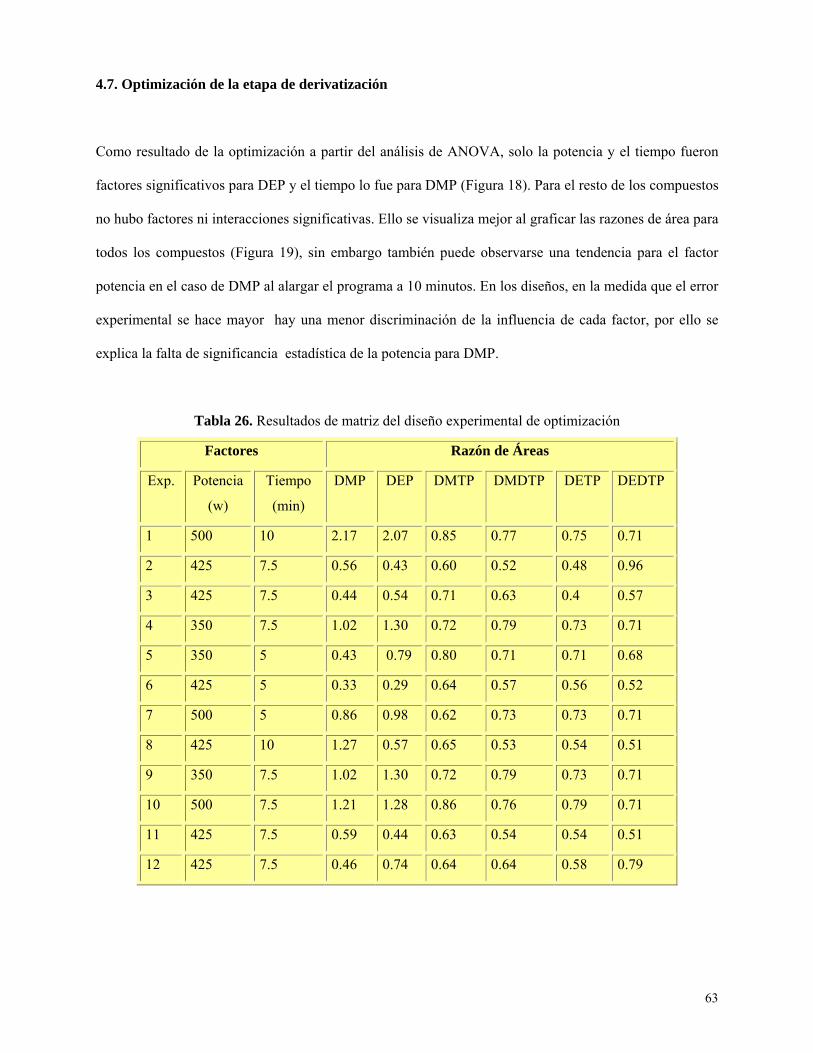
63
4.7. Optimización de la etapa de derivatización
Como resultado de la optimización a partir del análisis de ANOVA, solo la potencia y el tiempo fueron
factores significativos para DEP y el tiempo lo fue para DMP (Figura 18). Para el resto de los compuestos
no hubo factores ni interacciones significativas. Ello se visualiza mejor al graficar las razones de área para
todos los compuestos (Figura 19), sin embargo también puede observarse una tendencia para el factor
potencia en el caso de DMP al alargar el programa a 10 minutos. En los diseños, en la medida que el error
experimental se hace mayor hay una menor discriminación de la influencia de cada factor, por ello se
explica la falta de significancia estadística de la potencia para DMP.
Tabla 26. Resultados de matriz del diseño experimental de optimización
Factores Razón de Áreas
Exp. Potencia
(w)
Tiempo
(min)
DMP DEP DMTP DMDTP DETP DEDTP
1 500 10 2.17 2.07 0.85 0.77 0.75 0.71
2 425 7.5 0.56 0.43 0.60 0.52 0.48 0.96
3 425 7.5 0.44 0.54 0.71 0.63 0.4 0.57
4 350 7.5 1.02 1.30 0.72 0.79 0.73 0.71
5 350 5 0.43 0.79 0.80 0.71 0.71 0.68
6 425 5 0.33 0.29 0.64 0.57 0.56 0.52
7 500 5 0.86 0.98 0.62 0.73 0.73 0.71
8 425 10 1.27 0.57 0.65 0.53 0.54 0.51
9 350 7.5 1.02 1.30 0.72 0.79 0.73 0.71
10 500 7.5 1.21 1.28 0.86 0.76 0.79 0.71
11 425 7.5 0.59 0.44 0.63 0.54 0.54 0.51
12 425 7.5 0.46 0.74 0.64 0.64 0.58 0.79
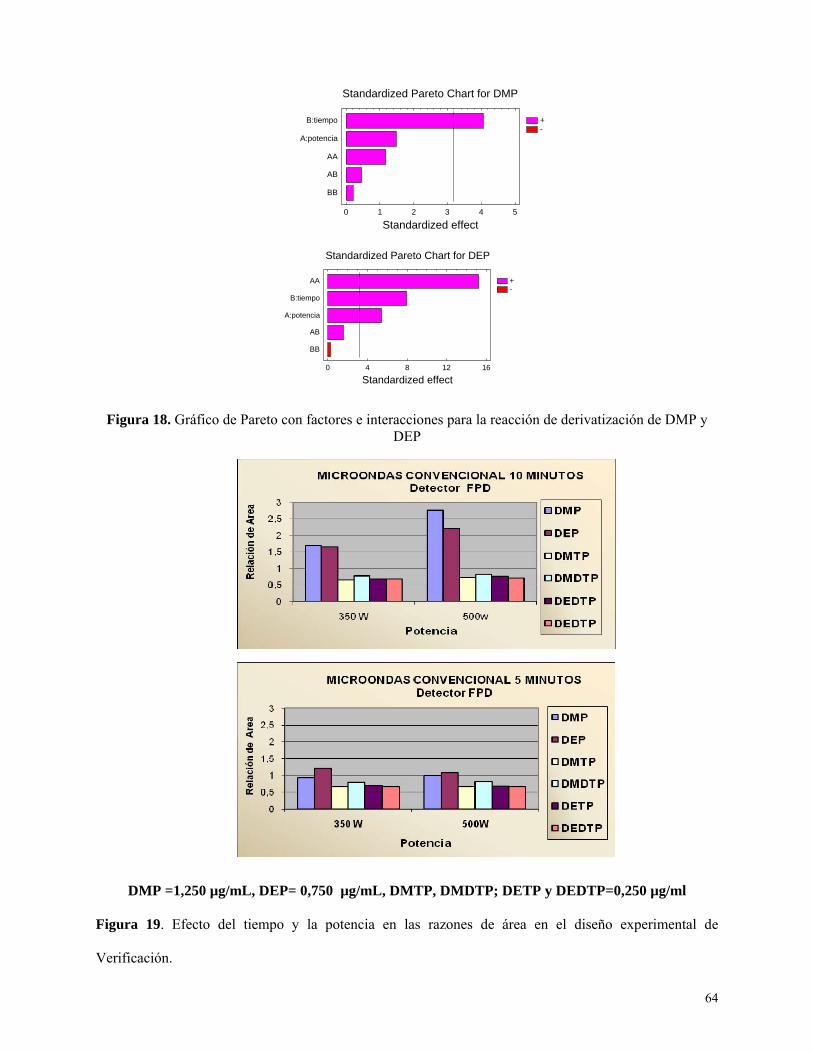
64
Standardized Pareto Chart for DMP
Standardized effect
+-
0 1 2 3 4 5
BB
AB
AA
A:potencia
B:tiempo
Standardized Pareto Chart for DEP
Standardized effect
+-
0 4 8 12 16
BB
AB
A:potencia
B:tiempo
AA
Figura 18. Gráfico de Pareto con factores e interacciones para la reacción de derivatización de DMP y DEP
DMP =1,250 µg/mL, DEP= 0,750 µg/mL, DMTP, DMDTP; DETP y DEDTP=0,250 µg/ml
Figura 19. Efecto del tiempo y la potencia en las razones de área en el diseño experimental de
Verificación.
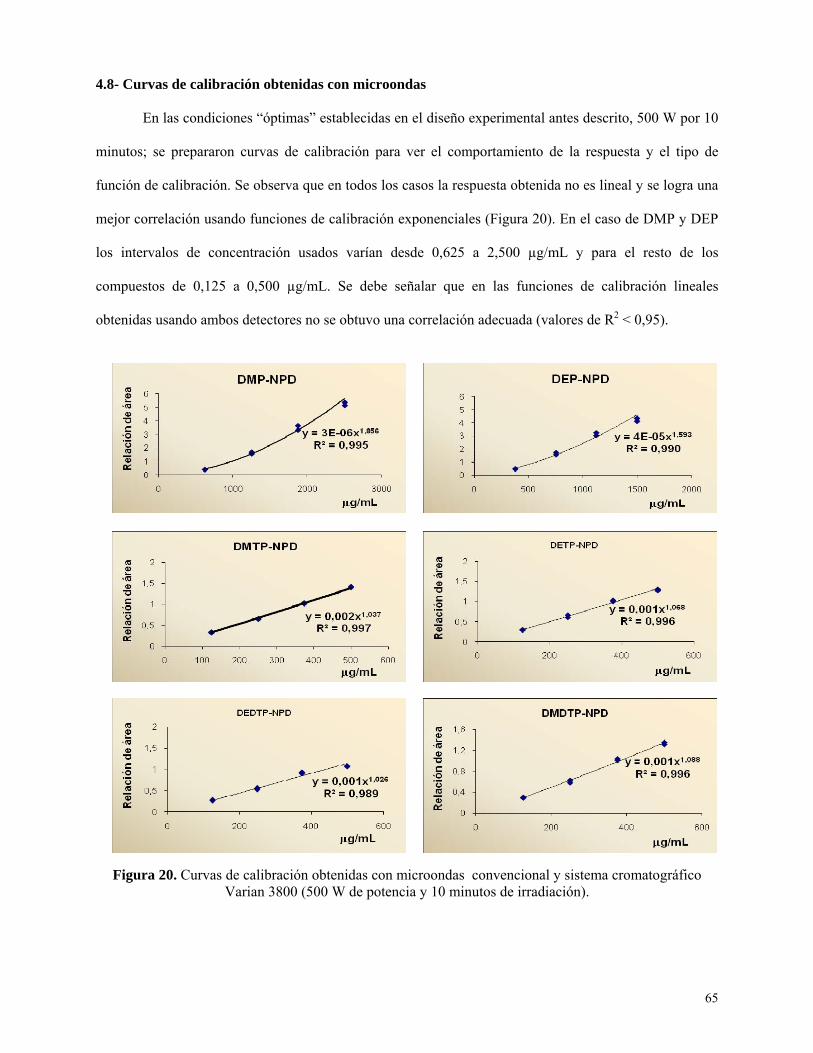
65
4.8- Curvas de calibración obtenidas con microondas
En las condiciones “óptimas” establecidas en el diseño experimental antes descrito, 500 W por 10
minutos; se prepararon curvas de calibración para ver el comportamiento de la respuesta y el tipo de
función de calibración. Se observa que en todos los casos la respuesta obtenida no es lineal y se logra una
mejor correlación usando funciones de calibración exponenciales (Figura 20). En el caso de DMP y DEP
los intervalos de concentración usados varían desde 0,625 a 2,500 µg/mL y para el resto de los
compuestos de 0,125 a 0,500 µg/mL. Se debe señalar que en las funciones de calibración lineales
obtenidas usando ambos detectores no se obtuvo una correlación adecuada (valores de R2 < 0,95).
Figura 20. Curvas de calibración obtenidas con microondas convencional y sistema cromatográfico Varian 3800 (500 W de potencia y 10 minutos de irradiación).

66
Las funciones exponenciales no resultan adecuadas para la obtención de límites de detección y por otra
parte la no linealidad podría estar indicando degradación de los compuestos a bajas concentraciones,
especialmente para DMP y DEP. Se debe señalar además que el nivel de concentración empleado es alto
si se quiere monitorear personas no expuestas ocupacionalmente, por lo anterior se abordó un tercer
diseño experimental incorporando el factor concentración de cada metabolito, esta vez a bajos niveles
para estudiar la optimización de condiciones.
4.9.- Diseño Experimental de verificación.
En este estudio se utilizaron tres concentraciones bajas para realizar un tercer diseño experimental
de potencia y tiempo, donde las concentraciones usadas fueron 0,1 μg/mL, 0,15 μg/mL y 0,2 μg/mL. Las
condiciones de tiempo y potencia fueron iguales a las estudiadas en el experimento anterior.
A partir del análisis estadístico (ANOVA) se concluyó que ninguno de los factores era
significativo a estos niveles de concentración. Ello podría confirmar el efecto de degradación a bajos
niveles de concentración que oculten los efectos significativos anteriormente logrados.
Los resultados obtenidos en este diseño son muy opuestos a los resultados del segundo diseño
experimental que utilizó concentraciones mucho más altas en el caso de DMP y DEP, al realizar más
réplicas en algunos de los experimentos usados en el tercer diseño, se dejó en evidencia un aumento en la
dispersión de las señales. Esto se puede deber al hecho de que los sellos de los viales no resisten bien
tiempos mayores de 7,5 minutos de irradiación generando pérdidas que no fueron evidenciadas en el
segundo diseño experimental ya que las concentraciones son mucho más altas que las usadas en este
tercer diseño. Lo anterior puede ayudar a explicar los resultados obtenidos en los diagramas de Pareto
antes mostrados. Por lo antes señalado, se procedió a realizar un estudio en triplicado para ver el efecto de
la combinación potencia /tiempo que se resume en la tabla 27 y figura 21. En estos experimentos se
utilizó el sistema cromatográfico con detector FPD y se tomaron datos de viales que no sufrieron
pérdidas.
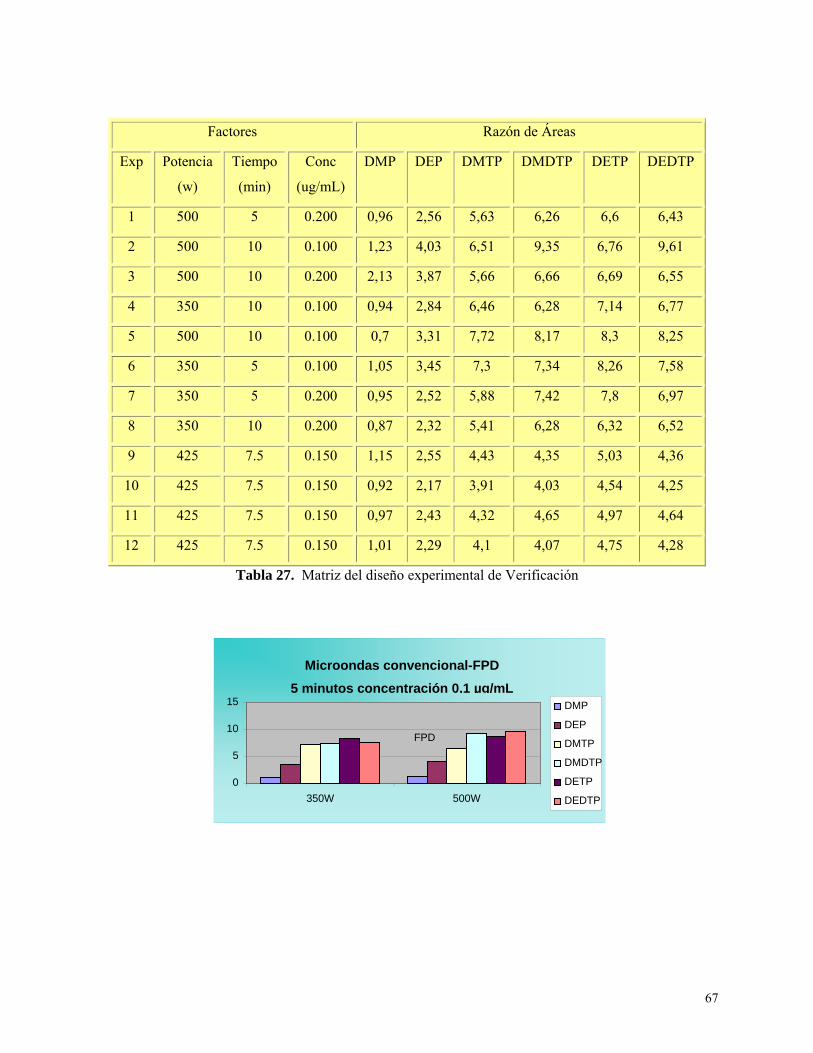
67
Tabla 27. Matriz del diseño experimental de Verificación
Factores Razón de Áreas
Exp Potencia
(w)
Tiempo
(min)
Conc
(ug/mL)
DMP DEP DMTP DMDTP DETP DEDTP
1 500 5 0.200 0,96 2,56 5,63 6,26 6,6 6,43
2 500 10 0.100 1,23 4,03 6,51 9,35 6,76 9,61
3 500 10 0.200 2,13 3,87 5,66 6,66 6,69 6,55
4 350 10 0.100 0,94 2,84 6,46 6,28 7,14 6,77
5 500 10 0.100 0,7 3,31 7,72 8,17 8,3 8,25
6 350 5 0.100 1,05 3,45 7,3 7,34 8,26 7,58
7 350 5 0.200 0,95 2,52 5,88 7,42 7,8 6,97
8 350 10 0.200 0,87 2,32 5,41 6,28 6,32 6,52
9 425 7.5 0.150 1,15 2,55 4,43 4,35 5,03 4,36
10 425 7.5 0.150 0,92 2,17 3,91 4,03 4,54 4,25
11 425 7.5 0.150 0,97 2,43 4,32 4,65 4,97 4,64
12 425 7.5 0.150 1,01 2,29 4,1 4,07 4,75 4,28
Microondas convencional-FPD 5 minutos concentración 0,1 µg/mL
0
5
10
15
350W 500W
DMP
DEP
DMTP
DMDTP
DETP
DEDTP
FPD
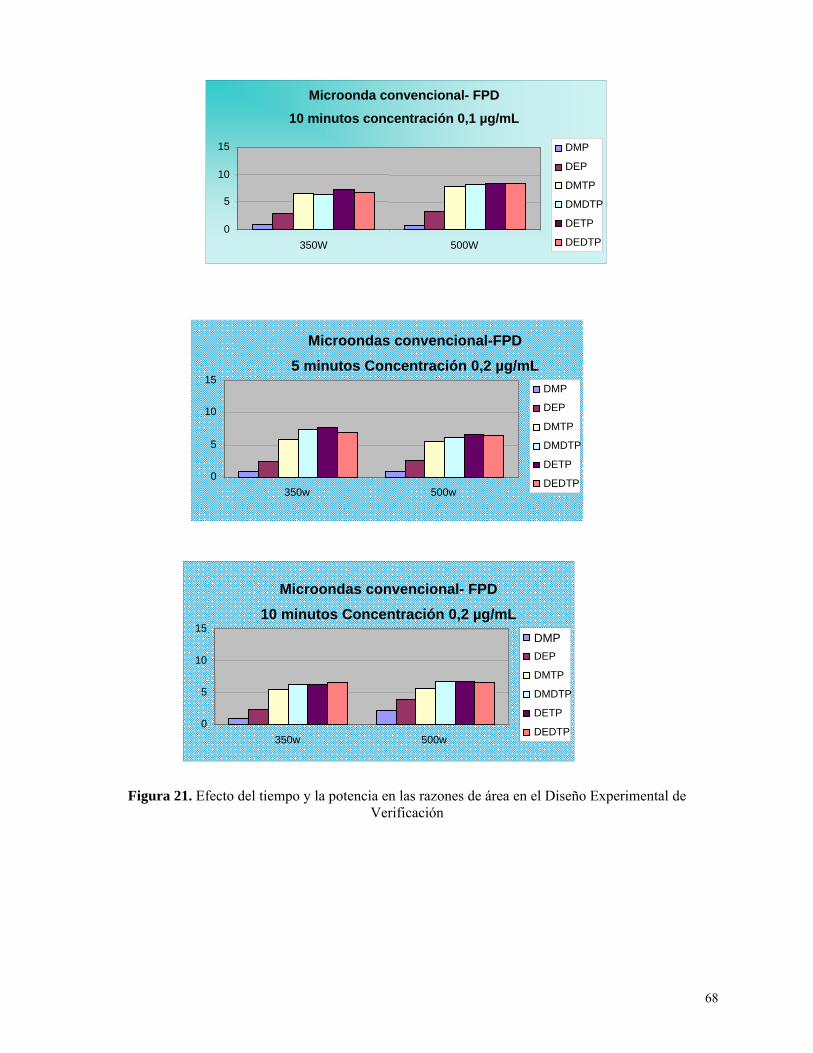
68
Figura 21. Efecto del tiempo y la potencia en las razones de área en el Diseño Experimental de Verificación
Microonda convencional- FPD
10 minutos concentración 0,1 µg/mL
0
5
10
15
350W 500W
DMP
DEP
DMTP
DMDTP
DETP
DEDTP
Microondas convencional-FPD 5 minutos Concentración 0,2 µg/mL
0
5
10
15
350w 500w
DMP
DEP
DMTP
DMDTP
DETP
DEDTP
Microondas convencional- FPD 10 minutos Concentración 0,2 µg/mL
0
5
10
15
350w 500w
DMPDEP
DMTP
DMDTP
DETP
DEDTP

69
Si se comparan las razones de área obtenidas en este experimento con el anterior se observa que
son mayores ya que en varios casos las razones de área superan el valor de 5 mientras que cuando las
concentraciones son mayores, como las usadas en el segundo diseño experimental, esta razón de áreas no
sobrepasa el valor de tres lo que significa que el método de las microondas es más favorable para
concentraciones bajas de analitos.
4.10.-Preparación de curvas de calibración para los seis metabolitos en estudio utilizando las
condiciones obtenidas en el Diseño Experimental de Verificación.
En las condiciones anteriores se repitieron las curvas de calibración usando un mayor número de
puntos de calibración en duplicado con el fin de establecer un mejor ajuste para el caso de DMP y DEP,
se usó un rango de concentración de 0,025 μg/mL a 0,8 μg/mL para ver el comportamiento de la
respuesta en detalle, en la figura 22 se muestran las curvas obtenidas.
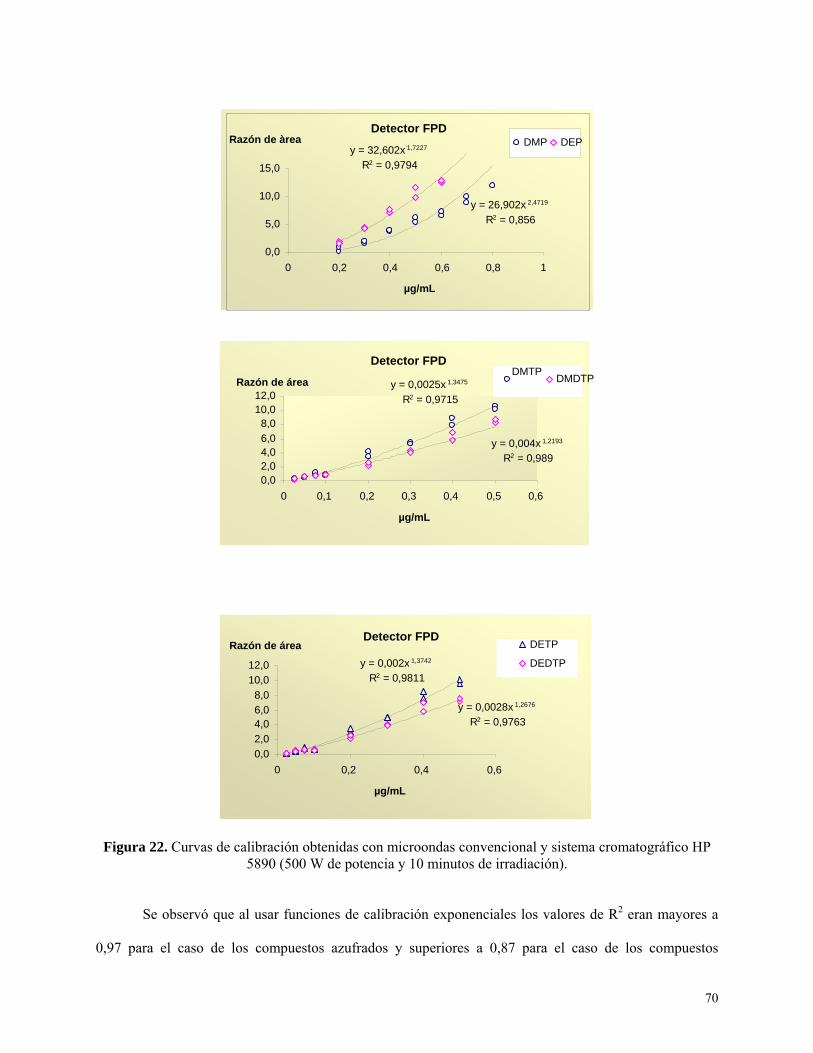
70
Figura 22. Curvas de calibración obtenidas con microondas convencional y sistema cromatográfico HP 5890 (500 W de potencia y 10 minutos de irradiación).
Se observó que al usar funciones de calibración exponenciales los valores de R2 eran mayores a
0,97 para el caso de los compuestos azufrados y superiores a 0,87 para el caso de los compuestos
Detector FPD
y = 32,602x 1,7227
R2 = 0,9794
y = 26,902x 2,4719
R2 = 0,856
0,0
5,0
10,0
15,0
0 0,2 0,4 0,6 0,8 1
µg/mL
Razón de àrea DMP DEP
Detector FPD
y = 0,0025x 1,3475
R2 = 0,9715
y = 0,004x 1,2193
R2 = 0,989
0,02,04,06,08,0
10,012,0
0 0,1 0,2 0,3 0,4 0,5 0,6
µg/mL
Razón de áreaDMTP
DMDTP
Detector FPD
y = 0,002x 1,3742
R2 = 0,9811
y = 0,0028x 1,2676
R2 = 0,9763
0,02,04,06,08,0
10,0 12,0
0 0,2 0,4 0,6
µg/mL
Razón de área DETP
DEDTP

71
oxigenados (DMP y DEP), sin embargo estos niveles de ajuste no son lo suficientemente buenos para
realizar determinaciones cuantitativas. Además, del análisis de las curvas obtenidas se puede apreciar que
el rango de concentraciones empleados es aún muy amplio por lo que se decidió realizar una optimización
a bajas concentraciones, se puso especial cuidado en la optimización de los metabolitos no azufrados ya
que son estos los que presentan menor sensibilidad en los estudios antes realizados.
4.11.-Estudio de Cinética y de Estabilidad en el tiempo a bajas concentraciones de los metabolitos.
Se exploró también el efecto de la permanencia del catalizador junto al extracto, ausente por
separación previa a la determinación cromatográfica en todas las pruebas anteriores, asumiendo que el
efecto de las microondas daría solo inicio a la reacción. Ello podría conducir a un método simple para la
determinación de todos los compuestos simultáneamente, fijando un tiempo de reacción posterior, para
dar comienzo a la determinación cromatográfica. El programa térmico para el análisis se cambió con el
objeto de visualizar una mejor respuesta a DMP. Para ello se realizaron pruebas preliminares que
indicaron esta posibilidad, la diferencia fundamental fue la velocidad de la rampa en la etapa 3 del
programa donde se disminuyó de 7º C/ min a 4º C/ min y el aumento del tiempo isotérmico en la etapa
final de 1,19 a 3 minutos, estos cambios permitieron obtener una señal de DMP bien definida. La tabla 28
muestra el programa optimizado modificado usado en este estudio.
Tabla 28. Programa térmico optimizado modificado para el estudio de cinética y estabilidad en el tiempo de metabolitos oxigenados a bajas concentraciones.
Las condiciones de trabajo para la reacción de derivatización fueron las siguientes: Potencia 500
W y tiempo 10 minutos la estabilidad se evaluó a temperatura ambiente. Se observó en algunos casos un
90ºC 0 2 min 140 ºC 15ºC 1min 190ºC 4ºC 0 min 260ºC 30ºC 3 min
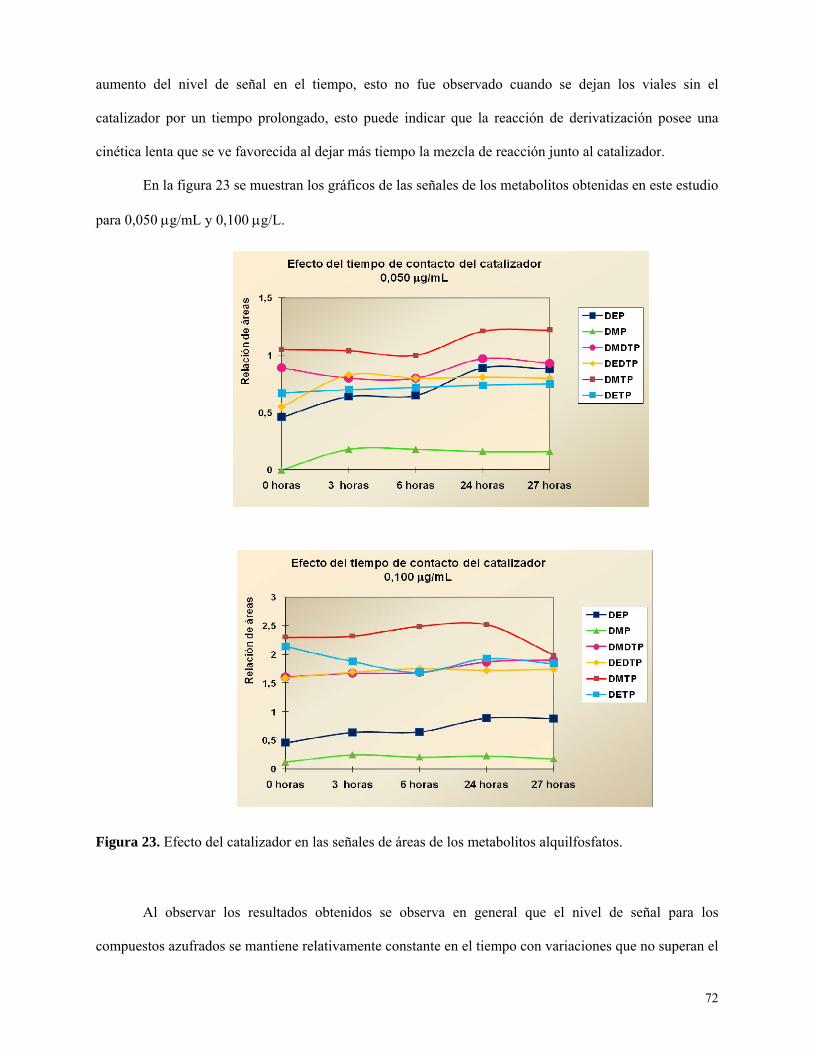
72
aumento del nivel de señal en el tiempo, esto no fue observado cuando se dejan los viales sin el
catalizador por un tiempo prolongado, esto puede indicar que la reacción de derivatización posee una
cinética lenta que se ve favorecida al dejar más tiempo la mezcla de reacción junto al catalizador.
En la figura 23 se muestran los gráficos de las señales de los metabolitos obtenidas en este estudio
para 0,050 μg/mL y 0,100 μg/L.
Figura 23. Efecto del catalizador en las señales de áreas de los metabolitos alquilfosfatos.
Al observar los resultados obtenidos se observa en general que el nivel de señal para los
compuestos azufrados se mantiene relativamente constante en el tiempo con variaciones que no superan el
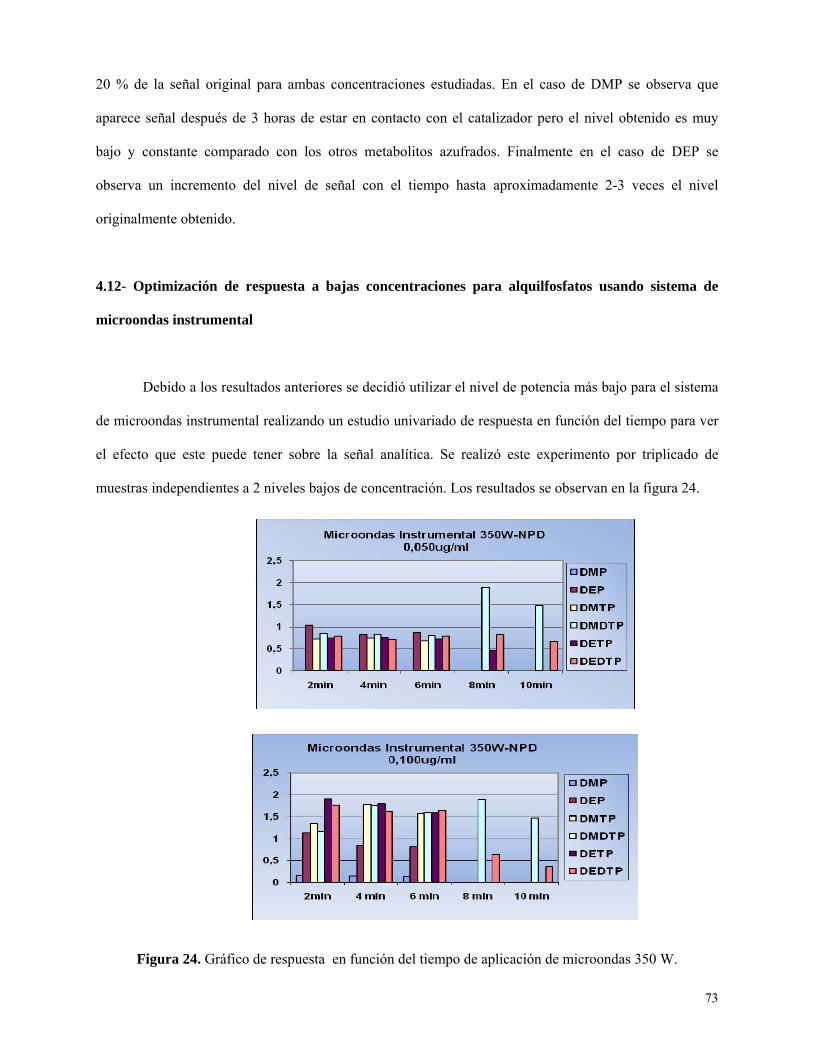
73
20 % de la señal original para ambas concentraciones estudiadas. En el caso de DMP se observa que
aparece señal después de 3 horas de estar en contacto con el catalizador pero el nivel obtenido es muy
bajo y constante comparado con los otros metabolitos azufrados. Finalmente en el caso de DEP se
observa un incremento del nivel de señal con el tiempo hasta aproximadamente 2-3 veces el nivel
originalmente obtenido.
4.12- Optimización de respuesta a bajas concentraciones para alquilfosfatos usando sistema de
microondas instrumental
Debido a los resultados anteriores se decidió utilizar el nivel de potencia más bajo para el sistema
de microondas instrumental realizando un estudio univariado de respuesta en función del tiempo para ver
el efecto que este puede tener sobre la señal analítica. Se realizó este experimento por triplicado de
muestras independientes a 2 niveles bajos de concentración. Los resultados se observan en la figura 24.
Figura 24. Gráfico de respuesta en función del tiempo de aplicación de microondas 350 W.
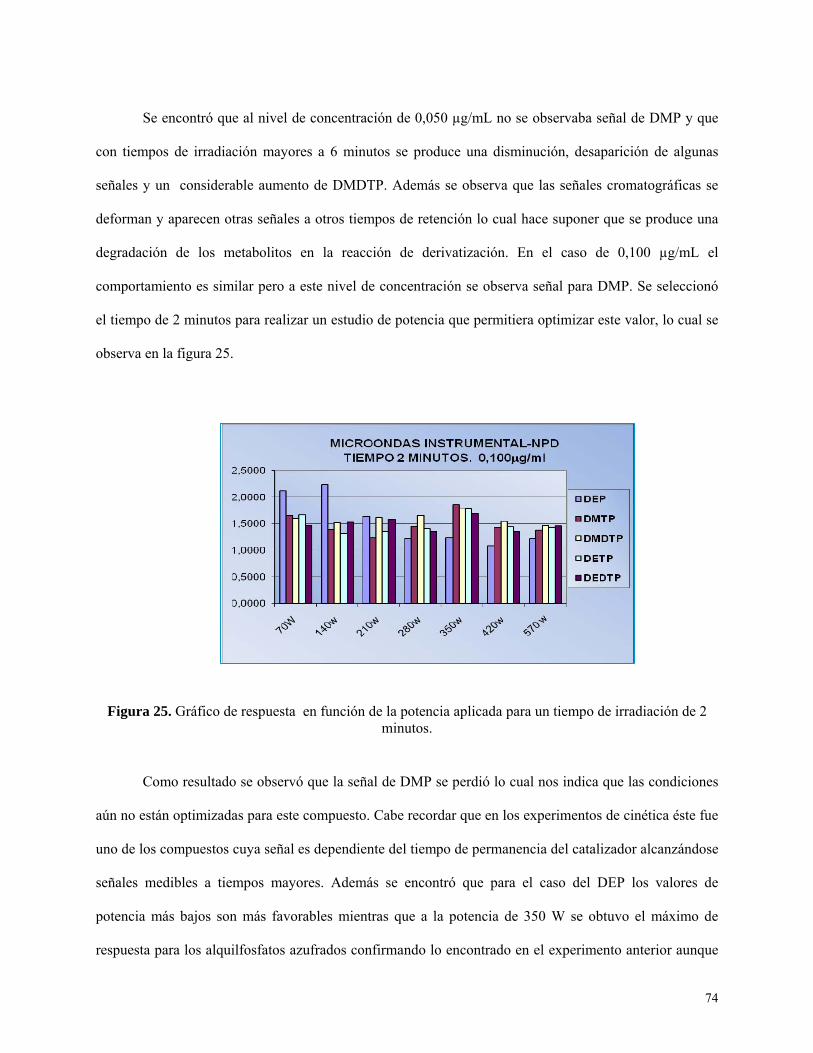
74
Se encontró que al nivel de concentración de 0,050 µg/mL no se observaba señal de DMP y que
con tiempos de irradiación mayores a 6 minutos se produce una disminución, desaparición de algunas
señales y un considerable aumento de DMDTP. Además se observa que las señales cromatográficas se
deforman y aparecen otras señales a otros tiempos de retención lo cual hace suponer que se produce una
degradación de los metabolitos en la reacción de derivatización. En el caso de 0,100 µg/mL el
comportamiento es similar pero a este nivel de concentración se observa señal para DMP. Se seleccionó
el tiempo de 2 minutos para realizar un estudio de potencia que permitiera optimizar este valor, lo cual se
observa en la figura 25.
Figura 25. Gráfico de respuesta en función de la potencia aplicada para un tiempo de irradiación de 2 minutos.
Como resultado se observó que la señal de DMP se perdió lo cual nos indica que las condiciones
aún no están optimizadas para este compuesto. Cabe recordar que en los experimentos de cinética éste fue
uno de los compuestos cuya señal es dependiente del tiempo de permanencia del catalizador alcanzándose
señales medibles a tiempos mayores. Además se encontró que para el caso del DEP los valores de
potencia más bajos son más favorables mientras que a la potencia de 350 W se obtuvo el máximo de
respuesta para los alquilfosfatos azufrados confirmando lo encontrado en el experimento anterior aunque

75
exista una diferencia de tiempo de dos minutos siendo las razones de área similares. Después de este
estudio se cambió el tipo de vial y sello en que se realizó los siguientes experimentos, ya que en las
pruebas a mayor potencia se perdieron algunas muestras, por ello se utilizaron viales de micro reacción
con sellos resistentes a la presión generada dentro de éstos.
Se puede concluir con estos resultados que se pueden establecer condiciones instrumentales para
la aplicación de la energía de las microondas en la obtención de buenos rendimientos en la derivatización
de los derivados azufrados, pero que son menos favorables para DEP y no adecuadas para DMP. Con
todos los diseños experimentales aplicados para la etapa de derivatización se comprobó la mayor
eficiencia para los compuestos azufrados. Hay que aclarar que en todos los estudios los compuestos
fueron preparados en solventes y derivatizados directamente, sin la etapa previa de extracción, la que se
obvió para aislar alguna posible interferencia y pérdidas de los compuestos. Por otro lado, de la revisión
de datos de la bibliografía para todos los compuestos se analizaron muestras fortificadas en matriz de
orina, con concentraciones entre 0 y 0,5 µg/mL, pasando por todas las etapas de extracción y
concentración, existiendo por ende un factor de concentración muy considerable y a pesar de las posibles
pérdidas de los compuestos en las etapas previas y consecutivas se llevaba a cabo la determinación con la
obtención de considerables señales con la obtención de límites de detección del orden de los µg/L,
calculados sobre la base del nivel de ruido instrumental y consideradas todas las etapas de la
determinación. Así, los resultados obtenidos en el presente trabajo son altamente sensibles para todos los
compuestos, exceptuando DMP, el que de acuerdo a los datos bibliográficos también es el que presentó la
menor sensibilidad. Por otra parte, a pesar de que las condiciones cromatográficas fueron diferentes
(columnas, programas térmicos, etc.) a las usadas en los métodos de referencia se logró obtener una
excelente resolución y el mismo orden de elución de los 6 metabolitos.
En los experimentos con orina a bajas concentraciones se observó que la reacción de derivatización era
más favorable para los compuestos no azufrados (DMP y DEP) que en el caso de los estándares
preparados en ACN. Existe un efecto de matriz notable o bien la acidificación en la etapa de extracción y
la adición de NaCl pueden ser factores preponderantes para mejorar la eficiencia.
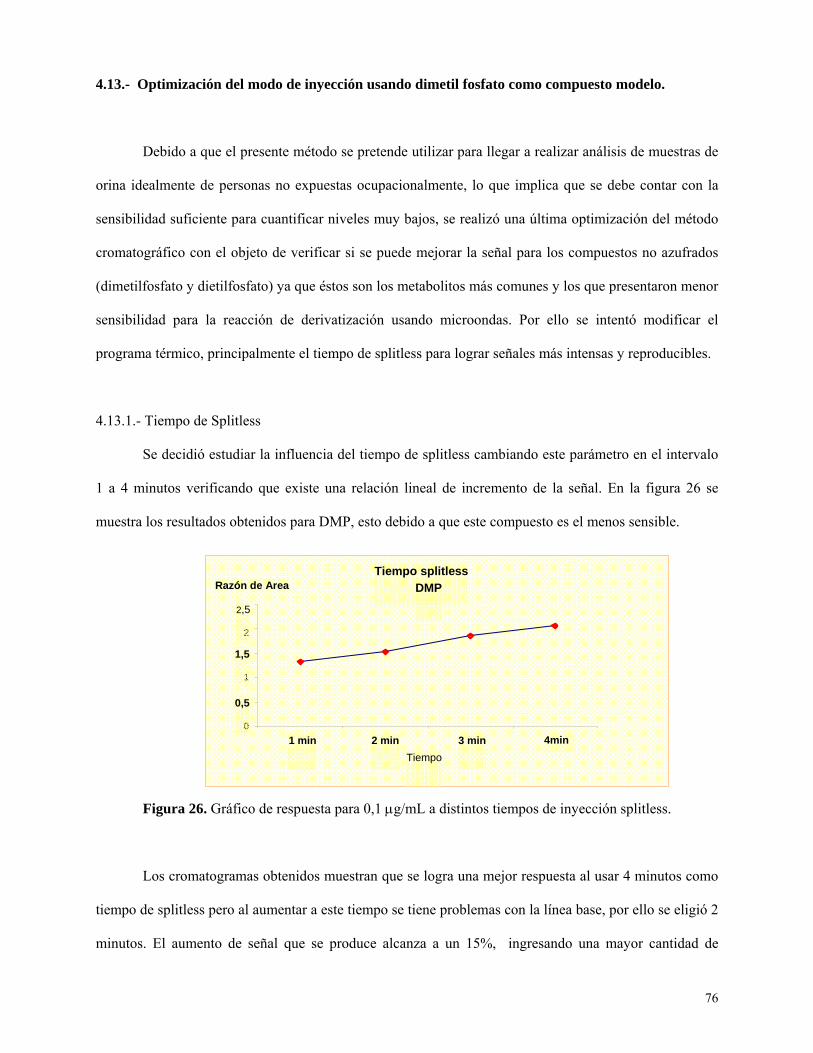
76
4.13.- Optimización del modo de inyección usando dimetil fosfato como compuesto modelo.
Debido a que el presente método se pretende utilizar para llegar a realizar análisis de muestras de
orina idealmente de personas no expuestas ocupacionalmente, lo que implica que se debe contar con la
sensibilidad suficiente para cuantificar niveles muy bajos, se realizó una última optimización del método
cromatográfico con el objeto de verificar si se puede mejorar la señal para los compuestos no azufrados
(dimetilfosfato y dietilfosfato) ya que éstos son los metabolitos más comunes y los que presentaron menor
sensibilidad para la reacción de derivatización usando microondas. Por ello se intentó modificar el
programa térmico, principalmente el tiempo de splitless para lograr señales más intensas y reproducibles.
4.13.1.- Tiempo de Splitless
Se decidió estudiar la influencia del tiempo de splitless cambiando este parámetro en el intervalo
1 a 4 minutos verificando que existe una relación lineal de incremento de la señal. En la figura 26 se
muestra los resultados obtenidos para DMP, esto debido a que este compuesto es el menos sensible.
Figura 26. Gráfico de respuesta para 0,1 μg/mL a distintos tiempos de inyección splitless.
Los cromatogramas obtenidos muestran que se logra una mejor respuesta al usar 4 minutos como
tiempo de splitless pero al aumentar a este tiempo se tiene problemas con la línea base, por ello se eligió 2
minutos. El aumento de señal que se produce alcanza a un 15%, ingresando una mayor cantidad de
Tiempo splitlessDMP
0
0,5
1
1,5
2
2,5
1 min 2 min 3 min 4min
Tiempo
Razón de Area
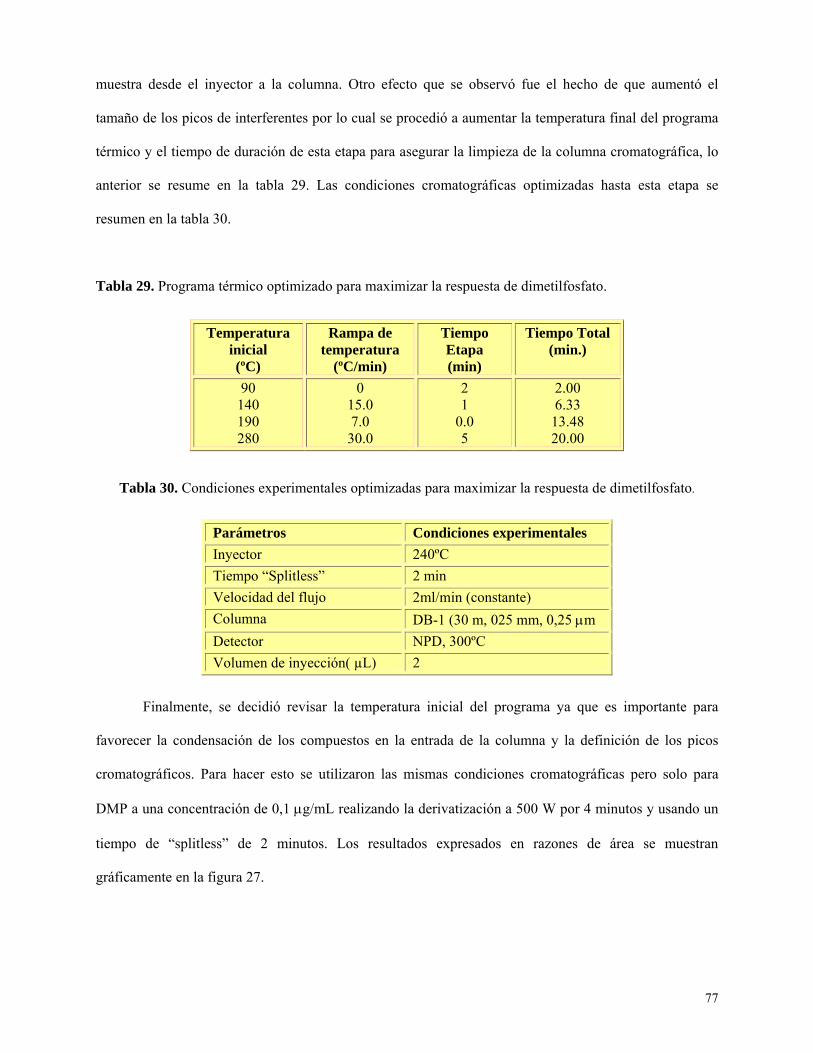
77
muestra desde el inyector a la columna. Otro efecto que se observó fue el hecho de que aumentó el
tamaño de los picos de interferentes por lo cual se procedió a aumentar la temperatura final del programa
térmico y el tiempo de duración de esta etapa para asegurar la limpieza de la columna cromatográfica, lo
anterior se resume en la tabla 29. Las condiciones cromatográficas optimizadas hasta esta etapa se
resumen en la tabla 30.
Tabla 29. Programa térmico optimizado para maximizar la respuesta de dimetilfosfato.
Tabla 30. Condiciones experimentales optimizadas para maximizar la respuesta de dimetilfosfato.
Finalmente, se decidió revisar la temperatura inicial del programa ya que es importante para
favorecer la condensación de los compuestos en la entrada de la columna y la definición de los picos
cromatográficos. Para hacer esto se utilizaron las mismas condiciones cromatográficas pero solo para
DMP a una concentración de 0,1 μg/mL realizando la derivatización a 500 W por 4 minutos y usando un
tiempo de “splitless” de 2 minutos. Los resultados expresados en razones de área se muestran
gráficamente en la figura 27.
Temperatura inicial (ºC)
Rampa de temperatura
(ºC/min)
Tiempo Etapa (min)
Tiempo Total (min.)
90 140 190 280
0 15.0 7.0
30.0
2 1
0.0 5
2.00 6.33
13.48 20.00
Parámetros Condiciones experimentales Inyector 240ºC Tiempo “Splitless” 2 min Velocidad del flujo 2ml/min (constante) Columna DB-1 (30 m, 025 mm, 0,25 μm Detector NPD, 300ºC Volumen de inyección( µL) 2
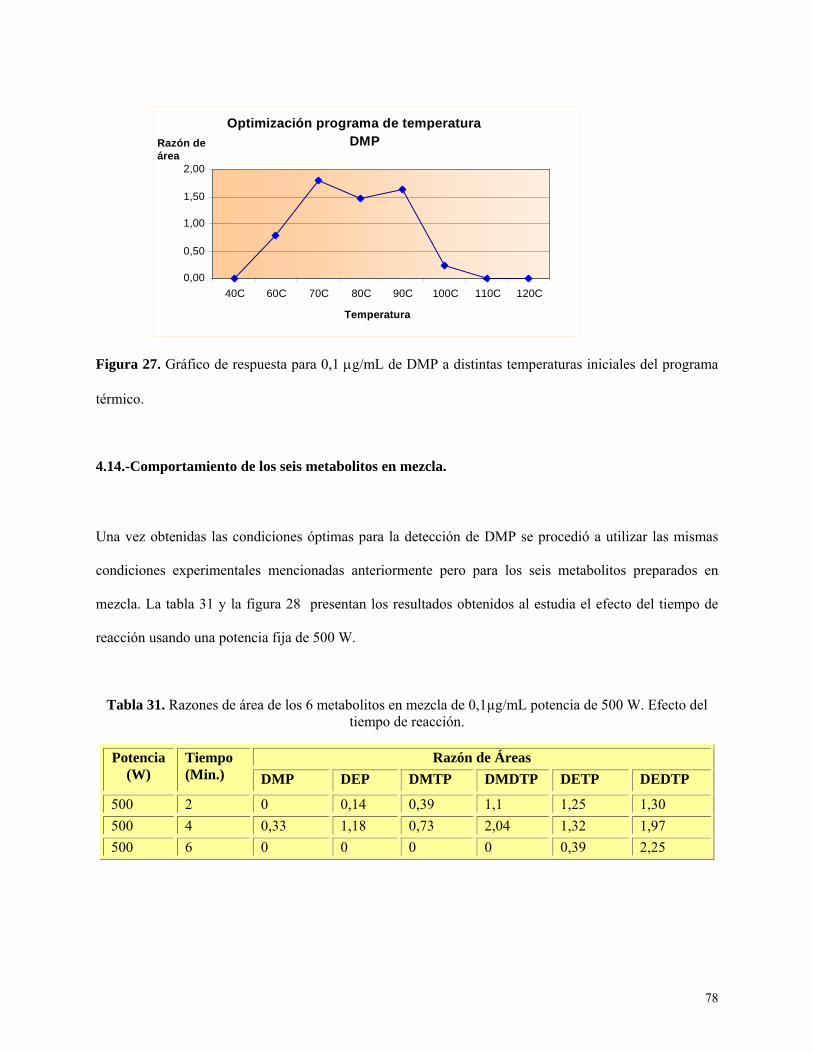
78
Figura 27. Gráfico de respuesta para 0,1 μg/mL de DMP a distintas temperaturas iniciales del programa
térmico.
4.14.-Comportamiento de los seis metabolitos en mezcla.
Una vez obtenidas las condiciones óptimas para la detección de DMP se procedió a utilizar las mismas
condiciones experimentales mencionadas anteriormente pero para los seis metabolitos preparados en
mezcla. La tabla 31 y la figura 28 presentan los resultados obtenidos al estudia el efecto del tiempo de
reacción usando una potencia fija de 500 W.
Tabla 31. Razones de área de los 6 metabolitos en mezcla de 0,1µg/mL potencia de 500 W. Efecto del tiempo de reacción.
Potencia(W)
Tiempo (Min.)
Razón de Áreas DMP DEP DMTP DMDTP DETP DEDTP
500 2 0 0,14 0,39 1,1 1,25 1,30 500 4 0,33 1,18 0,73 2,04 1,32 1,97 500 6 0 0 0 0 0,39 2,25
Optimización programa de temperatura
DMP
0,00
0,50
1,00
1,50
2,00
40C 60C 70C 80C 90C 100C 110C 120C
Temperatura
Razón de área

79
Figura 28. Razones de área de los metabolitos alquílicos a una potencia fija y a distintos tiempos de reacción.
Se logran las condiciones óptimas de derivatización a 500 W y 4 minutos ya que a mayor tiempo
de reacción los compuestos sufren degradación y los sellos de los viales no resisten las altas presiones
generadas a esa potencia.
Una vez seleccionadas las condiciones más favorables se procedió a realizar la validación de la
etapa de derivatización de los compuestos DMP, DEP, DMTP, DMDTP, DETP y DEDTP como
estándares puros y en matriz acuosa y orina.
4.15.- Ensayo preliminar aplicando la metodología de Hardt y Angerer a una matriz de orina.
Una vez establecidas las condiciones favorables para la reacción de derivatización de los
alquilmetabolitos a bajas concentraciones con énfasis en los metabolitos oxigenados, se procedió a
realizar la extracción desde una matriz de orina a la cual se adicionó una concentración conocida de estos
compuestos y se realizó la extracción siguiendo el procedimiento de referencia; la posterior derivatización
se realizó en el sistema de microondas instrumental utilizado en los estudios previos. En este estudio se
utilizó 3 concentraciones en duplicado, las concentraciones y razones de área obtenidas se muestran en la
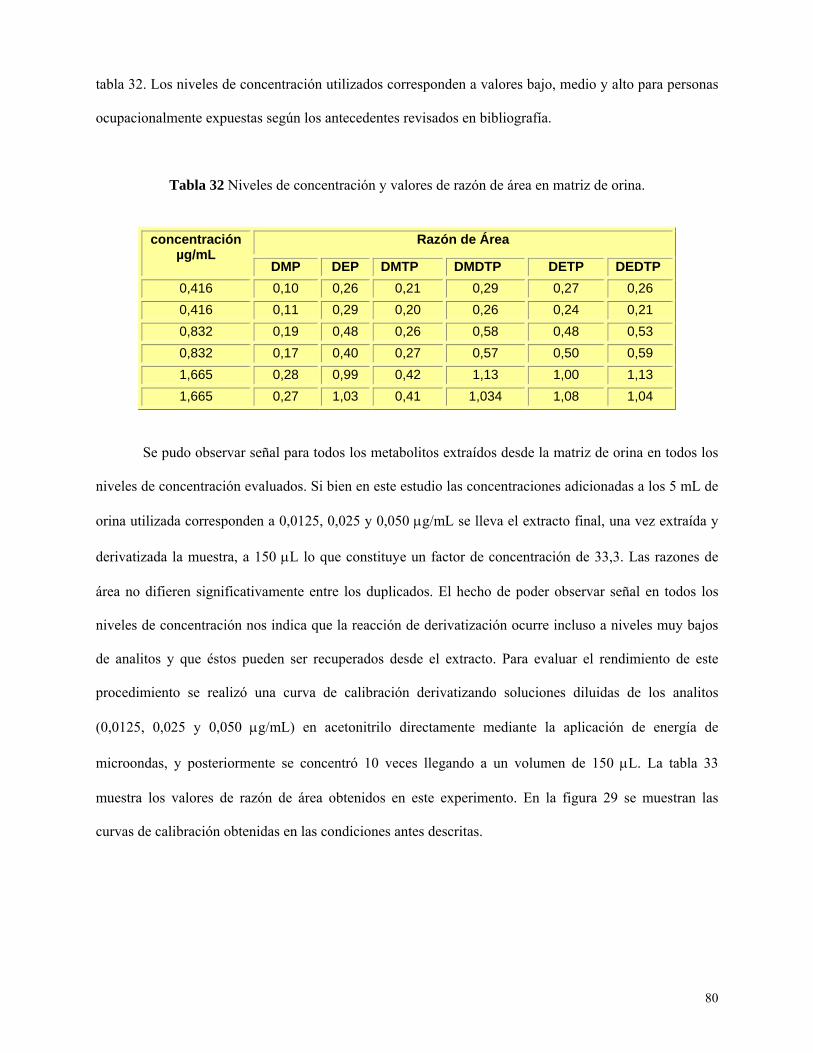
80
tabla 32. Los niveles de concentración utilizados corresponden a valores bajo, medio y alto para personas
ocupacionalmente expuestas según los antecedentes revisados en bibliografía.
Tabla 32 Niveles de concentración y valores de razón de área en matriz de orina.
concentración µg/mL
Razón de Área
DMP DEP DMTP DMDTP DETP DEDTP 0,416 0,10 0,26 0,21 0,29 0,27 0,26 0,416 0,11 0,29 0,20 0,26 0,24 0,21 0,832 0,19 0,48 0,26 0,58 0,48 0,53 0,832 0,17 0,40 0,27 0,57 0,50 0,59 1,665 0,28 0,99 0,42 1,13 1,00 1,13 1,665 0,27 1,03 0,41 1,034 1,08 1,04
Se pudo observar señal para todos los metabolitos extraídos desde la matriz de orina en todos los
niveles de concentración evaluados. Si bien en este estudio las concentraciones adicionadas a los 5 mL de
orina utilizada corresponden a 0,0125, 0,025 y 0,050 μg/mL se lleva el extracto final, una vez extraída y
derivatizada la muestra, a 150 μL lo que constituye un factor de concentración de 33,3. Las razones de
área no difieren significativamente entre los duplicados. El hecho de poder observar señal en todos los
niveles de concentración nos indica que la reacción de derivatización ocurre incluso a niveles muy bajos
de analitos y que éstos pueden ser recuperados desde el extracto. Para evaluar el rendimiento de este
procedimiento se realizó una curva de calibración derivatizando soluciones diluidas de los analitos
(0,0125, 0,025 y 0,050 μg/mL) en acetonitrilo directamente mediante la aplicación de energía de
microondas, y posteriormente se concentró 10 veces llegando a un volumen de 150 μL. La tabla 33
muestra los valores de razón de área obtenidos en este experimento. En la figura 29 se muestran las
curvas de calibración obtenidas en las condiciones antes descritas.
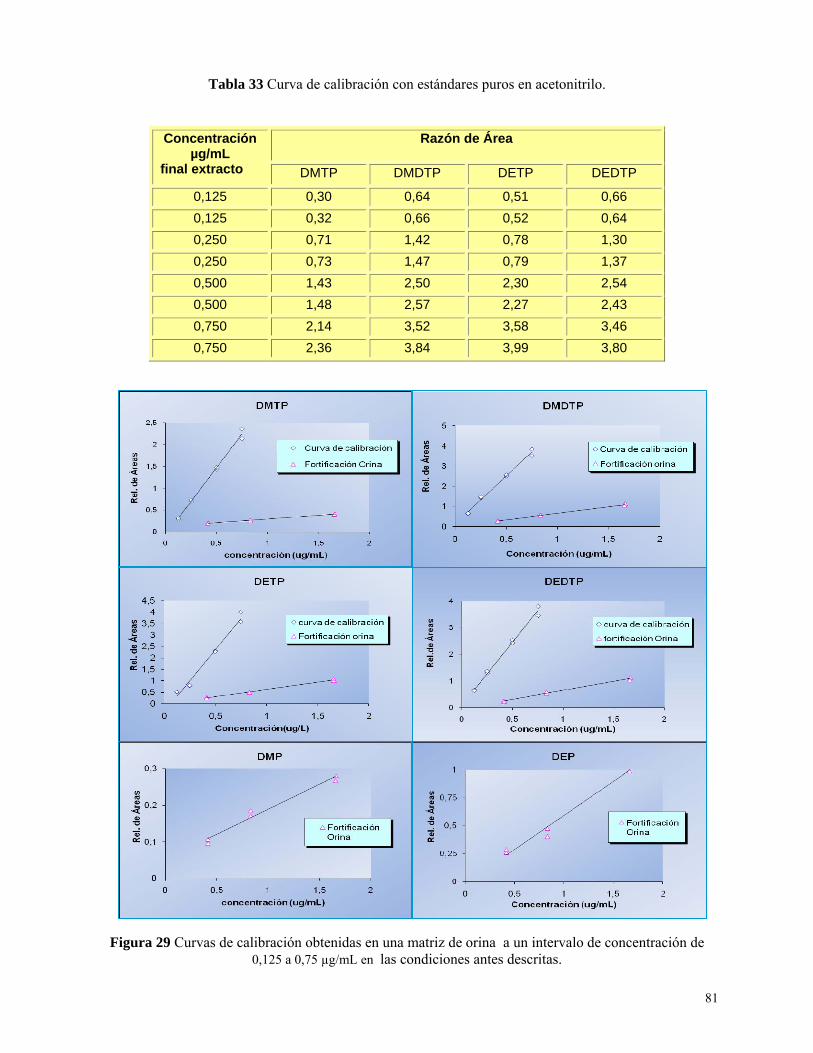
81
Tabla 33 Curva de calibración con estándares puros en acetonitrilo.
Concentración
µg/mL final extracto
Razón de Área
DMTP DMDTP DETP DEDTP
0,125 0,30 0,64 0,51 0,66 0,125 0,32 0,66 0,52 0,64 0,250 0,71 1,42 0,78 1,30 0,250 0,73 1,47 0,79 1,37 0,500 1,43 2,50 2,30 2,54 0,500 1,48 2,57 2,27 2,43 0,750 2,14 3,52 3,58 3,46 0,750 2,36 3,84 3,99 3,80
Figura 29 Curvas de calibración obtenidas en una matriz de orina a un intervalo de concentración de 0,125 a 0,75 µg/mL en las condiciones antes descritas.
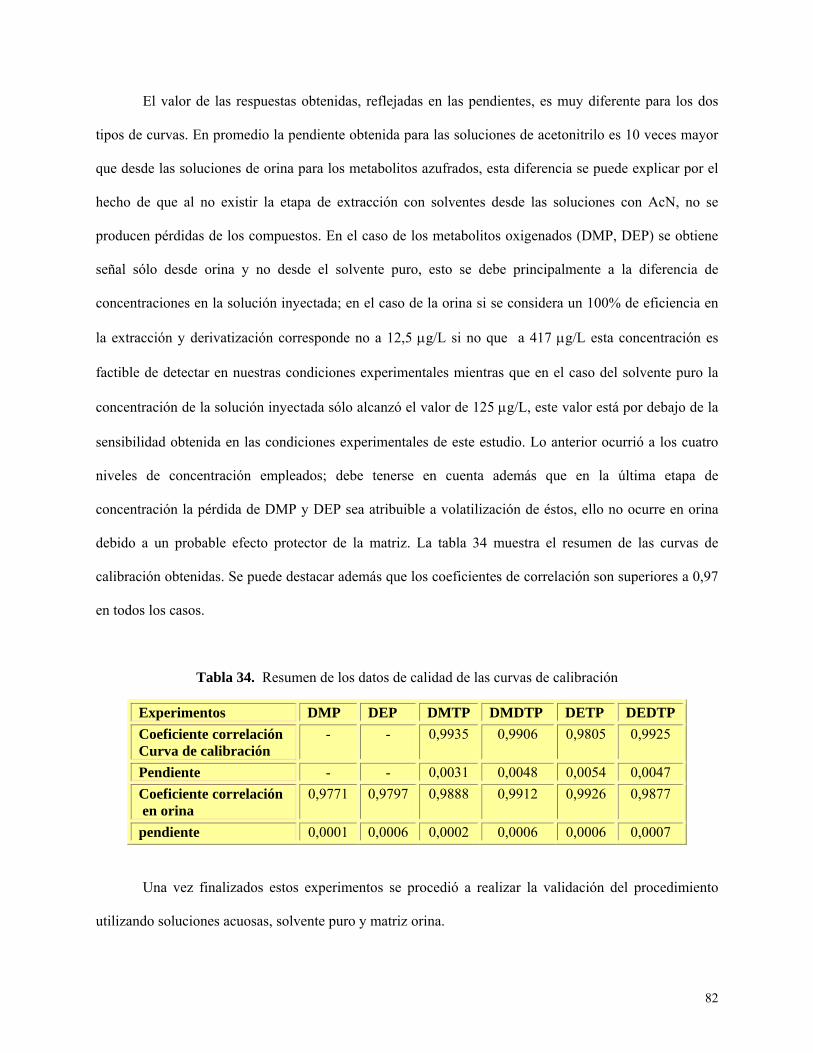
82
El valor de las respuestas obtenidas, reflejadas en las pendientes, es muy diferente para los dos
tipos de curvas. En promedio la pendiente obtenida para las soluciones de acetonitrilo es 10 veces mayor
que desde las soluciones de orina para los metabolitos azufrados, esta diferencia se puede explicar por el
hecho de que al no existir la etapa de extracción con solventes desde las soluciones con AcN, no se
producen pérdidas de los compuestos. En el caso de los metabolitos oxigenados (DMP, DEP) se obtiene
señal sólo desde orina y no desde el solvente puro, esto se debe principalmente a la diferencia de
concentraciones en la solución inyectada; en el caso de la orina si se considera un 100% de eficiencia en
la extracción y derivatización corresponde no a 12,5 μg/L si no que a 417 μg/L esta concentración es
factible de detectar en nuestras condiciones experimentales mientras que en el caso del solvente puro la
concentración de la solución inyectada sólo alcanzó el valor de 125 μg/L, este valor está por debajo de la
sensibilidad obtenida en las condiciones experimentales de este estudio. Lo anterior ocurrió a los cuatro
niveles de concentración empleados; debe tenerse en cuenta además que en la última etapa de
concentración la pérdida de DMP y DEP sea atribuible a volatilización de éstos, ello no ocurre en orina
debido a un probable efecto protector de la matriz. La tabla 34 muestra el resumen de las curvas de
calibración obtenidas. Se puede destacar además que los coeficientes de correlación son superiores a 0,97
en todos los casos.
Tabla 34. Resumen de los datos de calidad de las curvas de calibración
Experimentos DMP DEP DMTP DMDTP DETP DEDTPCoeficiente correlación Curva de calibración
- - 0,9935 0,9906 0,9805 0,9925
Pendiente - - 0,0031 0,0048 0,0054 0,0047 Coeficiente correlación en orina
0,9771 0,9797 0,9888 0,9912 0,9926 0,9877
pendiente 0,0001 0,0006 0,0002 0,0006 0,0006 0,0007
Una vez finalizados estos experimentos se procedió a realizar la validación del procedimiento
utilizando soluciones acuosas, solvente puro y matriz orina.

83
4.16.- Obtención de los parámetros de calidad en la etapa de extracción y derivatización de los 6
metabolitos dialquilfosfatos como estándares puros, en matriz acuosa y orina.
Se realizaron curvas de calibración por duplicado a concentraciones de 75 a 250 μg/L para los 6
compuestos en mezcla preparado en acetonitrilo. Luego en esta misma etapa se realizaron fortificaciones
en una matriz acuosa y en orina a niveles de concentración de 25 a 100 μg/L.
Las condiciones de trabajo para la reacción de derivatización fueron las siguientes: Potencia 500
W y Tiempo 4 minutos y estabilidad a temperatura ambiente. En el caso de acetonitrilo no se realizó una
concentración de la muestra mientras que las soluciones acuosas y de orina se extrajeron y pre-
concentraron 2,5 veces previo a la derivatización, obteniéndose las mismas concentraciones finales que
aquéllas de las curvas de calibración en acetonitrilo, En la figura 30 se muestran las curvas obtenidas en
las tres condiciones.
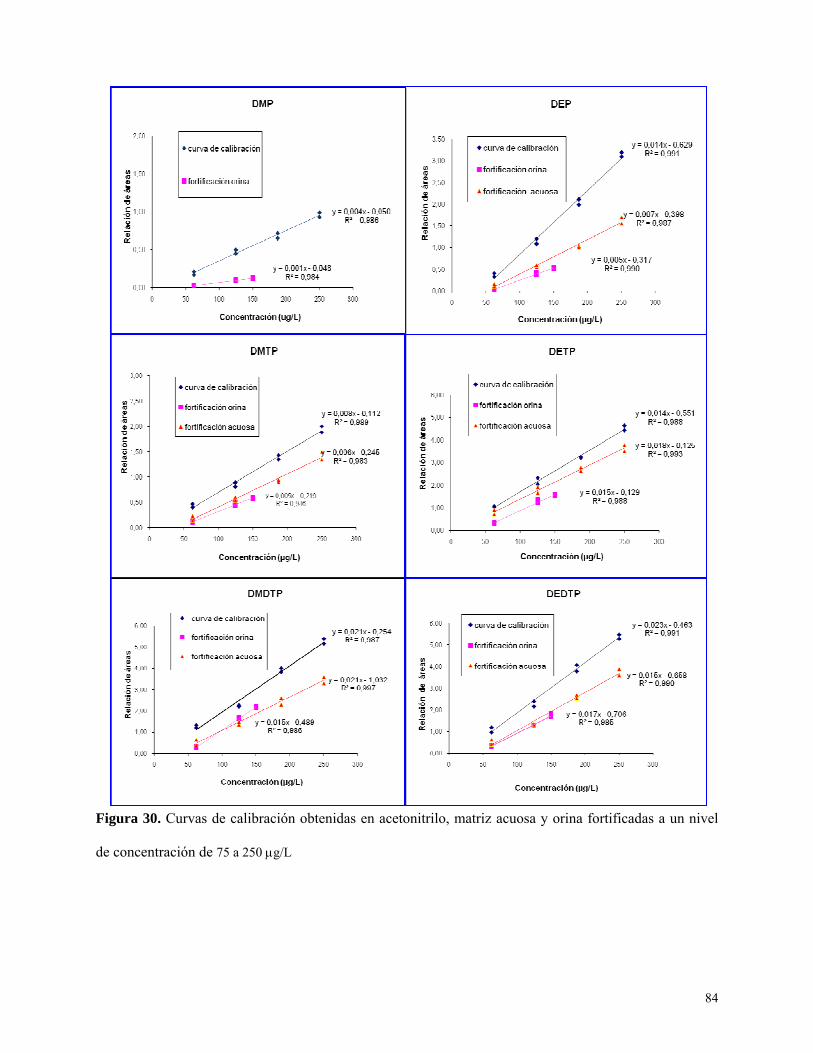
84
Figura 30. Curvas de calibración obtenidas en acetonitrilo, matriz acuosa y orina fortificadas a un nivel
de concentración de 75 a 250 μg/L
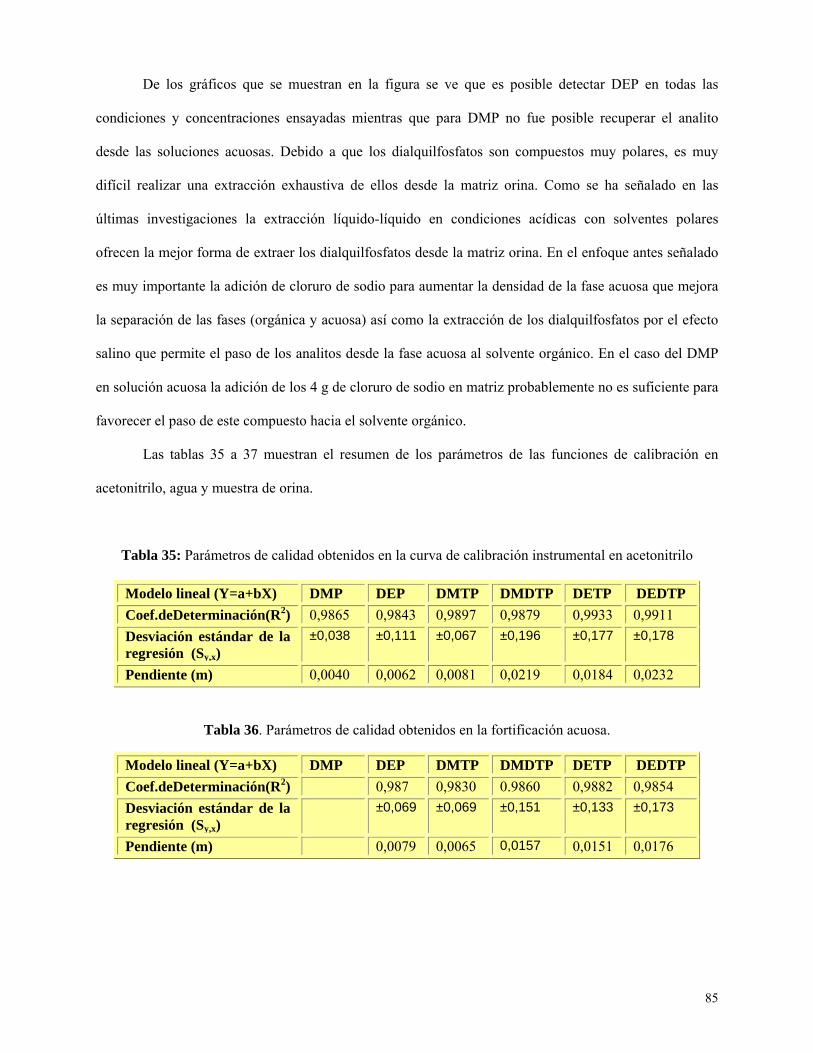
85
De los gráficos que se muestran en la figura se ve que es posible detectar DEP en todas las
condiciones y concentraciones ensayadas mientras que para DMP no fue posible recuperar el analito
desde las soluciones acuosas. Debido a que los dialquilfosfatos son compuestos muy polares, es muy
difícil realizar una extracción exhaustiva de ellos desde la matriz orina. Como se ha señalado en las
últimas investigaciones la extracción líquido-líquido en condiciones acídicas con solventes polares
ofrecen la mejor forma de extraer los dialquilfosfatos desde la matriz orina. En el enfoque antes señalado
es muy importante la adición de cloruro de sodio para aumentar la densidad de la fase acuosa que mejora
la separación de las fases (orgánica y acuosa) así como la extracción de los dialquilfosfatos por el efecto
salino que permite el paso de los analitos desde la fase acuosa al solvente orgánico. En el caso del DMP
en solución acuosa la adición de los 4 g de cloruro de sodio en matriz probablemente no es suficiente para
favorecer el paso de este compuesto hacia el solvente orgánico.
Las tablas 35 a 37 muestran el resumen de los parámetros de las funciones de calibración en
acetonitrilo, agua y muestra de orina.
Tabla 35: Parámetros de calidad obtenidos en la curva de calibración instrumental en acetonitrilo
Modelo lineal (Y=a+bX) DMP DEP DMTP DMDTP DETP DEDTP Coef.deDeterminación(R2) 0,9865 0,9843 0,9897 0,9879 0,9933 0,9911 Desviación estándar de la regresión (Sy,x)
±0,038 ±0,111 ±0,067 ±0,196 ±0,177 ±0,178
Pendiente (m) 0,0040 0,0062 0,0081 0,0219 0,0184 0,0232
Tabla 36. Parámetros de calidad obtenidos en la fortificación acuosa.
Modelo lineal (Y=a+bX) DMP DEP DMTP DMDTP DETP DEDTP Coef.deDeterminación(R2) 0,987 0,9830 0.9860 0,9882 0,9854 Desviación estándar de la regresión (Sy,x)
±0,069 ±0,069 ±0,151 ±0,133 ±0,173
Pendiente (m) 0,0079 0,0065 0,0157 0,0151 0,0176
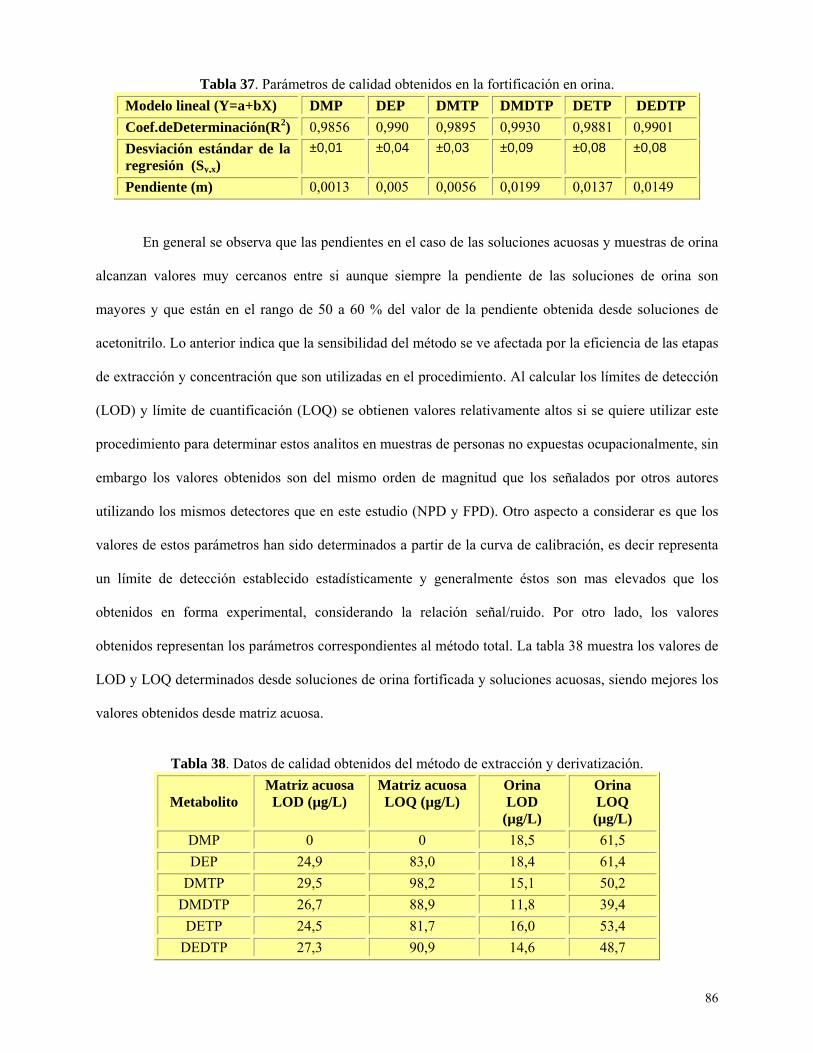
86
Tabla 37. Parámetros de calidad obtenidos en la fortificación en orina. Modelo lineal (Y=a+bX) DMP DEP DMTP DMDTP DETP DEDTP Coef.deDeterminación(R2) 0,9856 0,990 0,9895 0,9930 0,9881 0,9901 Desviación estándar de la regresión (Sy,x)
±0,01 ±0,04 ±0,03 ±0,09 ±0,08 ±0,08
Pendiente (m) 0,0013 0,005 0,0056 0,0199 0,0137 0,0149
En general se observa que las pendientes en el caso de las soluciones acuosas y muestras de orina
alcanzan valores muy cercanos entre si aunque siempre la pendiente de las soluciones de orina son
mayores y que están en el rango de 50 a 60 % del valor de la pendiente obtenida desde soluciones de
acetonitrilo. Lo anterior indica que la sensibilidad del método se ve afectada por la eficiencia de las etapas
de extracción y concentración que son utilizadas en el procedimiento. Al calcular los límites de detección
(LOD) y límite de cuantificación (LOQ) se obtienen valores relativamente altos si se quiere utilizar este
procedimiento para determinar estos analitos en muestras de personas no expuestas ocupacionalmente, sin
embargo los valores obtenidos son del mismo orden de magnitud que los señalados por otros autores
utilizando los mismos detectores que en este estudio (NPD y FPD). Otro aspecto a considerar es que los
valores de estos parámetros han sido determinados a partir de la curva de calibración, es decir representa
un límite de detección establecido estadísticamente y generalmente éstos son mas elevados que los
obtenidos en forma experimental, considerando la relación señal/ruido. Por otro lado, los valores
obtenidos representan los parámetros correspondientes al método total. La tabla 38 muestra los valores de
LOD y LOQ determinados desde soluciones de orina fortificada y soluciones acuosas, siendo mejores los
valores obtenidos desde matriz acuosa.
Tabla 38. Datos de calidad obtenidos del método de extracción y derivatización.
Metabolito
Matriz acuosa LOD (µg/L)
Matriz acuosa LOQ (µg/L)
Orina LOD
(µg/L)
Orina LOQ (µg/L)
DMP 0 0 18,5 61,5 DEP 24,9 83,0 18,4 61,4
DMTP 29,5 98,2 15,1 50,2 DMDTP 26,7 88,9 11,8 39,4
DETP 24,5 81,7 16,0 53,4 DEDTP 27,3 90,9 14,6 48,7
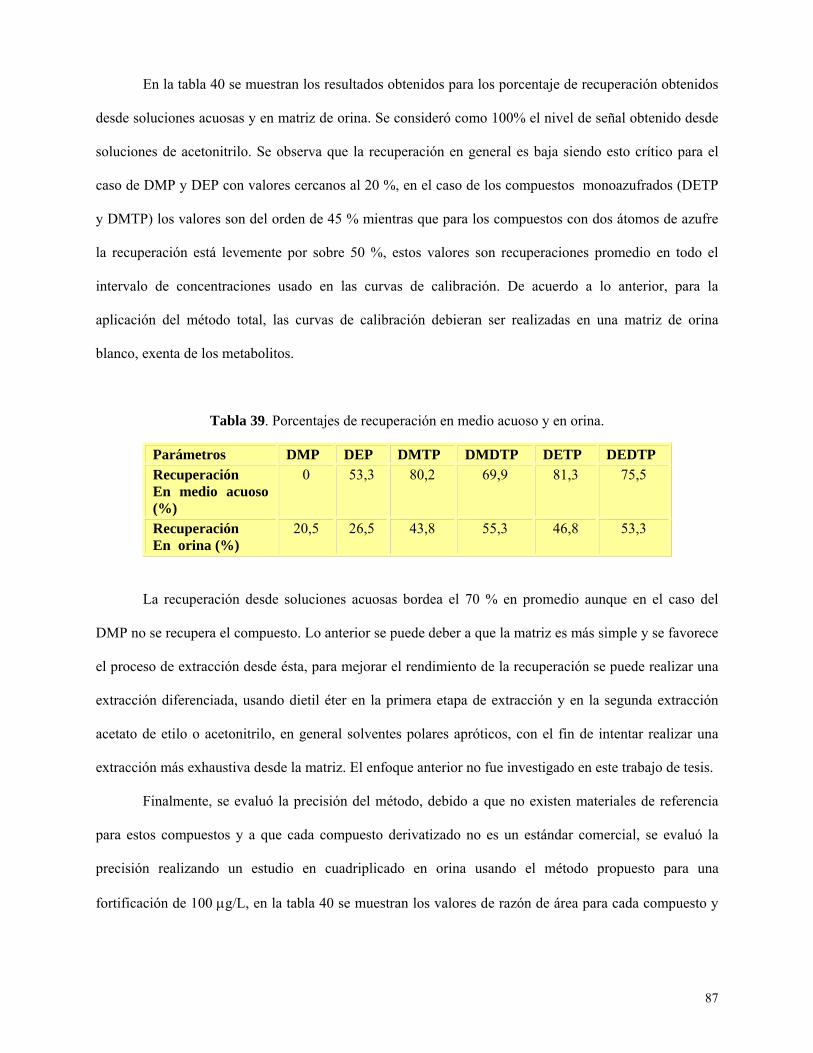
87
En la tabla 40 se muestran los resultados obtenidos para los porcentaje de recuperación obtenidos
desde soluciones acuosas y en matriz de orina. Se consideró como 100% el nivel de señal obtenido desde
soluciones de acetonitrilo. Se observa que la recuperación en general es baja siendo esto crítico para el
caso de DMP y DEP con valores cercanos al 20 %, en el caso de los compuestos monoazufrados (DETP
y DMTP) los valores son del orden de 45 % mientras que para los compuestos con dos átomos de azufre
la recuperación está levemente por sobre 50 %, estos valores son recuperaciones promedio en todo el
intervalo de concentraciones usado en las curvas de calibración. De acuerdo a lo anterior, para la
aplicación del método total, las curvas de calibración debieran ser realizadas en una matriz de orina
blanco, exenta de los metabolitos.
Tabla 39. Porcentajes de recuperación en medio acuoso y en orina.
Parámetros DMP DEP DMTP DMDTP DETP DEDTP Recuperación En medio acuoso (%)
0 53,3 80,2 69,9 81,3 75,5
Recuperación En orina (%)
20,5 26,5 43,8 55,3 46,8 53,3
La recuperación desde soluciones acuosas bordea el 70 % en promedio aunque en el caso del
DMP no se recupera el compuesto. Lo anterior se puede deber a que la matriz es más simple y se favorece
el proceso de extracción desde ésta, para mejorar el rendimiento de la recuperación se puede realizar una
extracción diferenciada, usando dietil éter en la primera etapa de extracción y en la segunda extracción
acetato de etilo o acetonitrilo, en general solventes polares apróticos, con el fin de intentar realizar una
extracción más exhaustiva desde la matriz. El enfoque anterior no fue investigado en este trabajo de tesis.
Finalmente, se evaluó la precisión del método, debido a que no existen materiales de referencia
para estos compuestos y a que cada compuesto derivatizado no es un estándar comercial, se evaluó la
precisión realizando un estudio en cuadriplicado en orina usando el método propuesto para una
fortificación de 100 μg/L, en la tabla 40 se muestran los valores de razón de área para cada compuesto y
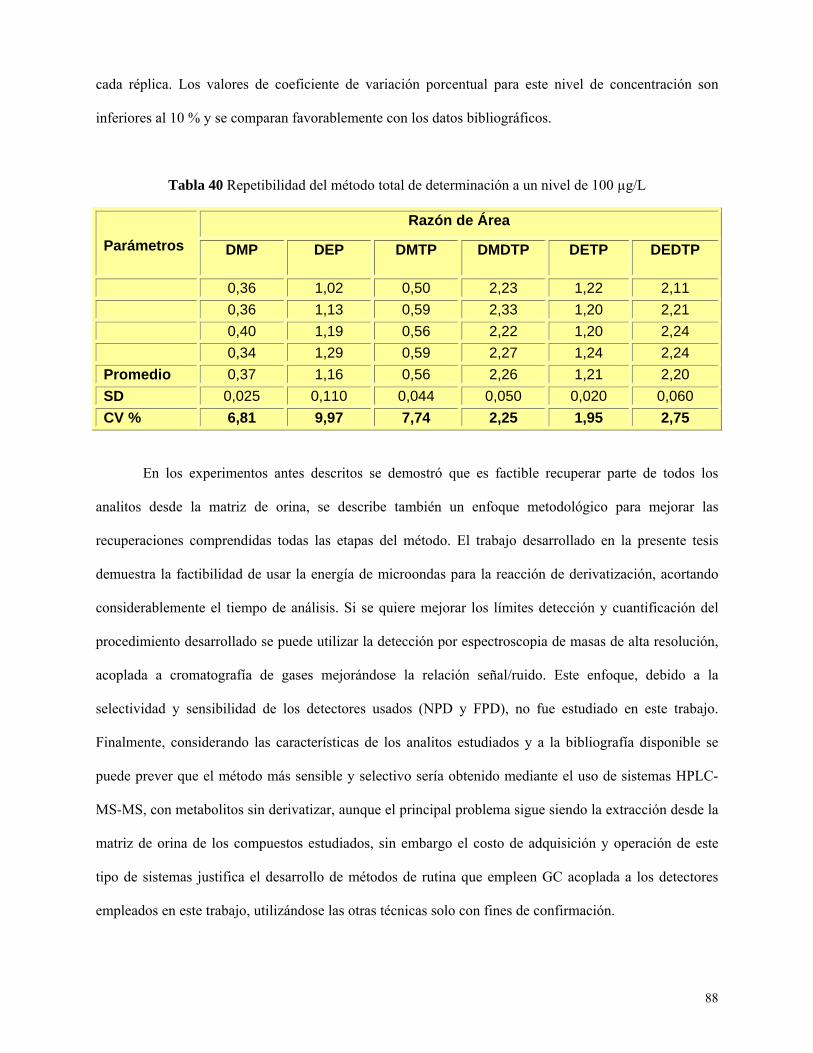
88
cada réplica. Los valores de coeficiente de variación porcentual para este nivel de concentración son
inferiores al 10 % y se comparan favorablemente con los datos bibliográficos.
Tabla 40 Repetibilidad del método total de determinación a un nivel de 100 µg/L
Parámetros
Razón de Área
DMP DEP
DMTP
DMDTP
DETP
DEDTP
0,36 1,02 0,50 2,23 1,22 2,11 0,36 1,13 0,59 2,33 1,20 2,21 0,40 1,19 0,56 2,22 1,20 2,24 0,34 1,29 0,59 2,27 1,24 2,24 Promedio 0,37 1,16 0,56 2,26 1,21 2,20 SD 0,025 0,110 0,044 0,050 0,020 0,060 CV % 6,81 9,97 7,74 2,25 1,95 2,75
En los experimentos antes descritos se demostró que es factible recuperar parte de todos los
analitos desde la matriz de orina, se describe también un enfoque metodológico para mejorar las
recuperaciones comprendidas todas las etapas del método. El trabajo desarrollado en la presente tesis
demuestra la factibilidad de usar la energía de microondas para la reacción de derivatización, acortando
considerablemente el tiempo de análisis. Si se quiere mejorar los límites detección y cuantificación del
procedimiento desarrollado se puede utilizar la detección por espectroscopia de masas de alta resolución,
acoplada a cromatografía de gases mejorándose la relación señal/ruido. Este enfoque, debido a la
selectividad y sensibilidad de los detectores usados (NPD y FPD), no fue estudiado en este trabajo.
Finalmente, considerando las características de los analitos estudiados y a la bibliografía disponible se
puede prever que el método más sensible y selectivo sería obtenido mediante el uso de sistemas HPLC-
MS-MS, con metabolitos sin derivatizar, aunque el principal problema sigue siendo la extracción desde la
matriz de orina de los compuestos estudiados, sin embargo el costo de adquisición y operación de este
tipo de sistemas justifica el desarrollo de métodos de rutina que empleen GC acoplada a los detectores
empleados en este trabajo, utilizándose las otras técnicas solo con fines de confirmación.

89
CONCLUSIONES
1.- Se reprodujo con éxito un método analítico para ser usado como referencia optimizando el programa
térmico para la separación cromatográfica. Del estudio de tres programas térmicos en dos sistemas
cromatográficos con distintos detectores (NPD y FPD) se logró una buena separación de los seis
compuestos con resolución superior a 1 en todos los casos y un tiempo total de análisis inferior a 23
minutos.
2.- Mediante la optimización del tiempo de “splitless” en el sistema cromatográfico se logró un aumento
de un 15 % en la señal de DMP y DEP lo cual permite mejorar la sensibilidad y los límites de detección y
cuantificación de estos analitos.
3.- Se demostró la factibilidad de aplicar energía de microondas para realizar la derivatización de los
metabolitos dialquilfosfato (DMP, DEP, DMTP, DETP, DMDTP, DEDTP) de pesticidas
organofosforados en solución acuosa, solvente puro y extraídos desde orina humana.
4.- Los estudios de “screening” arrojaron como único factor significativo la potencia empleada. Lo
anterior se verificó especialmente en el caso de los dialquilmetabolitos oxigenados (DMP, DEP) con una
mayor señal al usar valores de potencia más altos. En el caso de los dialquilmetabolitos azufrados
(DMTP, DETP, DMDTP, DEDTP) no se observaron grandes diferencias en el nivel de señales obtenidas
al ser sometidos a potencias y tiempos distintos. Al incrementar en exceso los valores de la potencia
aplicada se produce la degradación de estos compuestos. El comportamiento diferente para los
metabolitos oxigenados y azufrados es consistente con lo señalado en la bibliografía siendo las
condiciones óptimas para la reacción de derivatización 500 W de potencia y 4 minutos como tiempo de
reacción.

90
5.- Con la aplicación y optimización de la energía de microondas en la derivatización se reduce en un 99
% el tiempo total de esta etapa si se compara con el método usado como referencia pasando de 15 horas
a 4 minutos; lo anterior beneficia la productividad de un laboratorio que requiera realizar este análisis
pasando el proceso analítico total del rango de días a horas.
6.- Con las condiciones de derivatización optimizadas se procedió a realizar el estudio de validación,
estableciendo las funciones de calibración, las que resultaron ser funciones lineales en el intervalo de
concentración de 75 a 250 ug/L, con coeficientes de determinación para los seis metabolitos superiores a
0,98. Al comparar las pendientes en general se observó que los valores obtenidos en soluciones acuosas y
muestras de orina fueron muy cercanos entre si y fluctuaron en el rango de 50 a 60 % del valor de
pendiente obtenido desde soluciones de solvente puro (acetonitrilo).
7.- Los valores de recuperación, expresados como porcentaje, obtenidos desde muestras fortificadas de
agua y orina, en general son bajos y fluctúan desde un 20% para los dialquilmetabolitos oxigenados
(DMP y DEP) hasta 50% para los dialquilmetabolitos azufrados; lo anterior significa que el método se ve
afectado por la eficiencia de las etapas de extracción y concentración.
8.- Los límites de detección (LOD) y cuantificación (LOQ) obtenidos con este proceso analítico varían
entre 11 a 19 µg/L (LOD) y 39 a 62 µg/L (LOQ) y se pueden considerar altos si el objetivo es determinar
los niveles de estos compuestos en población ocupacionalmente no expuesta, sin embargo los resultados
obtenidos son del mismo orden de magnitud de lo señalado en la literatura utilizando similares sistemas
de detección (NPD y FPD). La precisión del método (repetitividad), evaluada por cuadruplicado en
muestras de orina fortificadas a 100 µg/L, presenta valores de coeficiente de variación porcentual
inferiores a 10% en todos los casos.
9.- Debido a la alta polaridad de este tipo de compuestos y a los resultados obtenidos en el estudio de
recuperación, es necesario realizar estudios más exhaustivos de la etapa de extracción probando nuevas

91
mezclas de solventes que permitan mejorar la recuperación de los analitos, especialmente de los
dialquilmetabolitos oxigenados (DMP, DEP); es necesario destacar que la extracción de los seis
metabolitos es factible desde la matriz orina.

92
BIBLIOGRAFÍA
[1] Rose R., Hodgson E., Roe M., Pesticides chapter 28, 663-697 in Toxicology , Marquadt Ed. 1999.
[2] Fuente Aduanas 2002
[3] Fuente INE 2002
[4] Fuente INE 2002
[5] Situación epidemiológica de las intoxicaciones agudas por plaguicidas, Chile año 1998, REVEP,
Departamento de Epidemiología, Ministerio de Salud , Gobierno de Chile.
[6] C.R. Worthing and R.J. Hance, The Pesticide Manual-A World Compendium, 9th Ed. The British
Crop Protection Council, Surrey, U.K. 1991.
[7] J.A. Vale, Toxicokinetics and Toxicodinamic Aspects of Organophosphorus (OP) Insecticide
Poisoning, Toxicol. Lett.102-103, 649-652(1998).
[8] Sams C., Mason H., Rawbone R., Evidence for the activation of organophosphate pesticides by
cytochromes P450 3A4 and 2D6 in human liver microsomes, Toxicol. Lett, 116, 217-221(2000).
[9] G.B. Koelle, Pharmacology of Organophosphates. J. Appl. Toxicol.. 14:105-109(1994).
[10] M. J. Coyle, J. A. Lowe and K. T. Maddy, biological Monitoring of agricultural workers exposed to
pesticides:I. Cholinesterase activity determination, J. Occup. Med. 28:619-627(1986).
[11] Cocker J., Mason H.J., Garfitt S.J., Jones K., Biological monitoring of exposure to
organophosphate pesticides, Toxicol. Lett. 134, 97-103(2002).
[12] Ellman G.L., Courtney K.D, Andres V., Featherstone R.M., Biochem. Pharmacol.1961, 7, .88.
[13] Nutley B., Cocker J., Biological monitoring of workers occupationally exposed to
organophosphorus pesticides, Pesticide Science. 38, 315-322(1993).

93
[14] He F., Biological Monitoring of occupational pesticides exposure, Int. Arch. Occup. Environ.
Health 65:S69-S76 (1993).
[15] World Health Organization, Environmental Health Criteria 63:Organophosphorus Insecticides,
Geneva, Switzerland, 1986.
[16] St. John Jr. L. E, D.J. Lisk., .Determination of hydrolytic metabolites of organophosphorus
insecticides in cow urine using improved thermoionic detector, J. Agric. Food Chem. 16, 48-49
(1968)
[17] Shafik M.T., Enos H.F., determination of metabolic and hydrolytic products of organophosphorus
pesticides chemicals in human blood and urine, J. Agric. Food Chem. 17, 1186-1189 (1969).
[18] Shafik MT., Bradway D.E., Enos H.F., Yobs A.R., Human exposure to organophosphorus
pesticides. A modified procedure for the gas-liquid chromatographic analysis of alkyl phosphate
metabolites in urine, J. Agric. Food Chem. 21, 625-629 (1973).
[19] Blair D., Roderick H.R., An improved method for the determination of urinary dimethyl phosphate,
J. Agric. Food Chem. Vol. 24, Nº 6, 1221-1223 (1976).
[20] Reid S.J., Watts R.R., A method for the determination of dialkyl phosphate residues in urine, J.
Anal. Toxicol. Vol. 5, issue 3, 126-132 (1981).
[21] Brokopp C.D., Wyatt J.L., Gabica J., Dialkyl phosphates in urine samples from pesticide
formulators exposed to disulfoton and phorate, Bull. of Environ. Contam. Toxicol. Vol. 26, 524-
529, (1981).
[22] Fenske R.A., Leffinwell J.T., Method for the determination of dialkyl phosphate metabolise in
urine for studies of human exposure to malathion, J. Agric. Food Chem. Vol. 37, 995-998 (1989).
[23] Aprea C., Sciarra G., LunghiniL., Analytical method for the determination of urinary
alkylphosphates in subjects occupationally exponed to organophosphorous pesticides and in the
general population, J. Anal. Toxicol. vol. 20, 559-563 (1996)

94
[24] Park S.S., Pyo H., Lee K.J., Park S.J., Park T.K., The analysis of common metabolites of
organophosphorus pesticides in urine by gas chromatography/mass spectrometry, Bulletin of the
Korean Chemical Society, vol. 19, Nº1, 45-50, (1998).
[25] Hardt J., Angerer J., Determination of dialkyl phosphates in human urine using gas
chromatography-mass spectrometry, J. Anal. Toxicol. Vol. 24, 678-684, (2000).
[26] Lin W.C., Kuei C.H., Wu H.C., Yang C.C., Chang H.Y., Method for the determination of Dialkyl
phosphates in Urine by strong Anion exchange disk Extraction and in-vial derivatization, J. Anal.
Toxicol., Vol. 26, 176-180, (2002).
[27] Bravo R., Driskell W.J., Whitehead R.D., Needham L.L., Barr D.B., Quantitation of Dialkyl
Phosphate Metabolites of Organophosphate Pesticides in Human urine Using GC-MS-MS with
Isotopic Internal Standars, J. Anal. Toxicol. Vol. 26, 245-252, (2002).
[28] Oglobline A.N., Elimelakh H., Tattam B., Geyer R., O`donnell G.E., Holder G., Negative ion
chemical ionization GC/MS-MS analysis of dialkylphosphate metabolites of organophosphate
pesticides in urine of non-occupationally exposed subjects, The Analyst, vol. 126, pp. 1037-1041,
(2001)
[29] Koch H.M., Angerer J., Analysis of 3,5,6-trichloro-2-pyridinol in urine samples from the general
population using gas chromatography-mass spectrometry after steam distilation and solid-phase
extraction, J. Chromatogr. B, vol. 759, 43-49, (2001).
[30] Shafik T.M., Sullivan H.C., Enos H.R., Multiresidue procedure for halo and nitrophenols
measurement of exposure to biodegradable pesticides yielding these compounds as metabolites, J.
Agric. Food Chem. vol. 21, Nº 2, pp.295-298 (1973).
[31] Bradway D.E., Shafik T.M., Malathion exposure studies determination of mono- and dicarboxilic
acid sand alkyl phosphates in urine, J. Agric. Food Chem., Vol. 25, Nº 6, 1342-1344 (1977).
[32] Maroni M., Catenacci G., Galli D., Cavallo D., RAvazzani G., Biological monitoring of human
exposure to acephate Arch. Environ. Contam. Toxicol., Vol.19, Issue 5, 782-788, (1990).

95
[33] Shafik M.T., Bradway D., Enos H.F., A clean up procedure for the determination of low levels of
alkyl phosphates, thiophosphates and dithiophosphates in rat and human urine, J. Agric. Food
Chem., Vol,19, issue 5, 885-889, (1971).
[34] Lores E.M., Bradway D.E., extraction and recovery of organophosphorus metabolites from urine
using an anion exchange resin, J. Agric. Food Chem., Vol. 25, Issue 1, pp. 75-79, (1977).
[35] Jauhiainen A., Kangas J., Laitinen S., Savolainen K., Biological Monitoring of workers exposed to
Mevinphos in greenhouses, Bull. Environ. Contam. Toxicol. Vol.49, 37-43, (1992).
[36] Moate T.F., Lu C., Fenske R.A., Hahne R.M.A., Kalman D.A., Improved cleanup and
determination of dialkyl phosphates in the urine of children exposed to organophosphorus
insecticides, J. .Anal. Toxicol., Vol. 23, 230-236, (1999).
[37] Oglobine A.N., O´Donnell G.E., Geyer R., Holder G.M., Tattam B., Routine gas chromatographic
determination of dialkylphosphate metabolites in the urine of workers occupationally exposed to
organophosphorus insecticides, J. Anal. Toxicol., Vol. 25, 153-157, (2001).
[38] Churchill F.C., Ku D.N., Miles J.W., Gas-liquid chromatographic inlet block derivatization of
organophosphorus pesticides and related dialkyl phosphorotioates, J. Agric. Food Chem., Vol. 26,
Issue 5, 1108-1112 (1978).
[39] Takade D.Y., Reynolds J.M., Nelson J.H., 1-4(nitrobenzyl)-3-(4-tolyl)triazene as derivatizing
reagent for the analysis of urinary dialkyl phosphate metabolites of organophosphorus pesticides by
gas chromatography, J. Agric. Food Chem. Vol. 27, Issue 4, 746-753 (1979).
[40] Daughton C.D., Cook A.M., Alexander M., Gas chromatographic determination of phosphorus-
containing pesticide metabolites via benzylation, Analytical Chemistry, Vol.51, Issue 12, 1949-
1953, (1979)
[41] Sparr Eskisson C., Björklund E., Analytical-scale microwave-assisted extraction, J. Chromatogr. A,
vol. 902, 227-250 (2000).
[42] Sánchez L., Mingorance M.D., Peña A, Microwave assisted process for methidathion in water
samples for physicochemical studies, Analyst Vol. 125, 1199-1203, (2000).

96
[43] Llompart M.P., Lorenzo R.A., Cela R., Paré J.R., Bélanger J., Li K., Phenol and methylphenol
isomers determination in soils by in-situ microwave-assisted extraction and derivatization, J.
Chromatogr. A, Vol. 757, pp. 153-164, (1997).
[44] Strassnig S., Wenzl T., Lankmayr E.P., Microwave-assisted derivatization of volatile carbonyl
compounds with O-(2,3,4,5,6-pentafluorobenzyl)hydroxylamine, J. Chromatogr. A, Vol. 891, 267-
273 (2000).
[45] Kobaudin B., A simple and new method for the synthesis of thiophosphates, Tetrahedron Lett. Vol
43, Issue 48, 8713-8714 (2002).