apoptosis, retinitis pigmentosa, and degeneration
TRANSCRIPT

REVIEW / SYNTHESE
Apoptosis, retinitis pigmentosa, and degeneration
PAUL WONG National Eye Institute, National Institutes of Health, 9000 Rockville Pike, Bethesda, MD 20892, U.S.A.
Received November 14, 1994
WONG, P. 1994. Apoptosis, retinitis pigmentosa, and degeneration. Biochem. Cell Biol. 72: 489-498. The mechanism of photoreceptor cell death in different inherited retinal degenerations is not fully understood.
Mutations in a number of different genes (such as rhodopsin, the beta subunit of cGMP phosphodiesterase, and peripherin) have been identified as the primary genetic lesion in different forms of human retinitis pigmentosa, one of the most common causes of inherited blindness. In all cases the manifestation of the disorder regardless of the specific primary genetic lesion is similar, resulting in photoreceptor cell degeneration and blindness. A recent hypothesis is that the active photoreceptor cell death, which is characteristic of these genetically distinct disorders, is mediated by a common induction of apoptosis. In the present review, the current evidence for active cell death during retinal cell death in several different rodent models of retinitis pigmentosa and retinal degeneration is examined.
Key words: retinal degeneration, apoptosis, retinitis pigmentosa, clusterin, DNA fragmentation.
WONG, P. 1994. Apoptosis, retinitis pigmentosa, and degeneration. Biochem. Cell Biol. 72 : 489-498. Le mCcanisme de la mort des photorCcepteurs au cours de diffkrentes dCgCnCrescences rktiniennes hCrCditaires n'est
pas complbtement compris. Des mutations de certains g h e s (tels ceux de la rhodopsine, de la pkriphkrine et de la sous-unit6 p de la phosphodiestCrase du GMPc) ont CtC identifikes comme Ctant les lCsions gCnCtiques primaires dam diffkrentes formes de rCtinite pigmentaire humaine, une des causes les plus frCquentes de cCcitC hCrCditaire. Dans tous les cas, la maladie se manifeste de la m&me f a ~ o n , quelle que soit la lCsion gCnCtique primaire spkcifique : il y a dCgCnCrescence des photorCcepteurs et la cCcitC s'ensuit. Une hypothkse rCcente veut que la mort programmCe des photorkcepteurs, caractCristique de ces affections d'origines gCnCtiques distinctes, rksulte de I'induction de I'apoptose dans tous les cas. Dans cet article, nous passons en revue les preuves actuelles de la mort des cellules rCtiniennes par apoptose dans diffkrents modbles expkrimentaux de rCtinite pigmentaire et de dCgCnCrescence retinienne chez les rongeurs.
Mots cle's : dCgCnCrescence rktinienne, apoptose, retinitis pigmentaire, clusterine, fragmentation d'ADN. [Traduit par la rCdaction]
Introduction The retina represents the neural network that underlies
the initial steps of the visual process. The retina is located between the vitreous and the choroid layer of the eye. The tissue consists of a number of well-defined cell layers (Fig. 1). Within the outer segment of the photoreceptor cell are the membrane discs in which the initial detection of light takes place. Steps in phototransduction are known in some detail and have been reviewed previously (Baylor 1987).
Blindness is one of the primary health problems in the world. Statistics, released recently, by "Research to Prevent Blindness" (New York) revealed that one in every three Americans is affected by some form of eye disease, that 80 million individuals suffer from potential blinding eye diseases, and that seven million suffer directly from visual loss caused by retinal degeneration. The most common cause of inherited blindness is retinitis pigmentosa, a disorder which affects 1 in 3500 Americans and an estimated 1.5 million people worldwide (Steele 1994). Patients with RP become blind as early as age 30 and are usually legally blind by age 60 (Berson 1993).
RP is not a single disease but a heterogeneous group of human genetic disorders. All are characterized by a pro- gressive degeneration and loss of retinal cells leading to blindness (Berson 1993). RP is genetically heterogeneous
ABBREVIATIONS: RP, retinitis pigmentosa; TUNEL, terminal dUTP nick end labelling; P-PDE, P-subunit of cGMP phospho- diesterase; RPE, retinal pigment epithelial; bp, base pair(s); kDa, kilodalton(s). Prinled in Canada 1 Impr~rnC au Canada
and can be inherited as a dominant, recessive, or X-linked trait. Genetic loci underlying different forms of RP have been localized to a number of chromosomes including 1, 3, 4, 6, 7, 8, 11, 14, 16, 19, and X. Symptoms of these disorders include impaired adaptation, night blindness, and difficulty with midperipheral visual field in adolescence, progressing to a loss of farperipheral field and eventually to a loss of central vision.
The dilemma in understanding RP has been the question as to how so many different and diverse primary genetic lesions cause the same clinical manifestation that charac- terizes RP. It has been suggested that apoptosis is the bio- logical mechanism which underlies this (Adler 1986). In the last 2 years a number of research groups have provided evidence in animal models of RP to support this hypothesis. In the current review, this data together with new data will be discussed to define the type of apoptosis that may underlie photoreceptor degeneration.
Apoptosis during retinal development Apoptosis is thought to be a fundamental active cellular
process of cell deletion that together with proliferation underlies cell number and modification of cell number. There are three distinct aspects to apoptosis: (i) the initial cause that places a given cell in an apoptotic state (the trigger), (ii) the actual mechanism of apoptotic cell deletion, and (iii) the cellular response of other cells in the tissue that are not undergoing apoptosis.
Apoptosis is a normal and essential process during devel- opment and extensive cell death has long been known to
Bio
chem
. Cel
l Bio
l. D
ownl
oade
d fr
om w
ww
.nrc
rese
arch
pres
s.co
m b
y C
ON
CO
RD
IA U
NIV
on
12/0
9/14
For
pers
onal
use
onl
y.

BIOCHEM. CELL BIOL. VOL. 72, 1994
(A) (B) (C)
RETINA ROD OUTER
PHOTORECEPTOR SEGMENT
FIG. 1. The retina (modified after Steele 1994). (A) The retina consists of a number of defined cell layers: the ganglion cell layer (GCL), the inner nuclear layer (INL), and the outer nuclear layer (ONL), and the retinal pigment epithelial cell layer (RPEL). Specific cells that are present in the retina include the photoreceptor cells (P), horizontal cells (H), Mueller cells (M), bipolar cells (B), amacrine cells (A), and ganglion cells (G). (B) The rod photoreceptor cell can be divided into the nuclear region (N), the inner segment (IS), and the outer segment regions (0s ) . (C) The outer segments of the rod photoreceptor cell houses over 1000 disc- like structures. Each disc consists of a lipid bilayer whose integrity is maintained by proteins such as peripherin (p). The major integral membrane protein of the disc is rhodopsin. Phototransduction is initiated when a photon of light is absorbed by rhodopsin and causes it to be activated. Activated rhodopsin in turn activates transducin, which in turn stimulates cGMP phosphodiesterase. Activated cGMP phosphodiesterase then cleaves cGMP at the site of cGMP-gated cation channels (*) and causes the channels to close. This results in the hyperpolarization of the photoreceptor cell and the release of the neurotransmitter glutamate to the neighboring bipolar neuron, thereby initiating the signal that is sent to the brain. Termination of the response centers on the phosphorylation of the opsin and its association with arrestin, which inhibits the interaction of rhodopsin with transducin. i, the inactive state; a, the active state.
be necessary for normal retinal development (Glucksmann 1940). Although reports of cell death in the developing retina hae been reported (Silver and Hughes 1973; Rager and Rager 1978; Hughes and McLoon 1979; Sengelaub and Finlay 1982), the association between developmental retinal cell death and apoptosis was not made until 1984 (Young 1984). Cell death in the developing retina involves individual cells. This death is characterized by pyknosis, i.e., conden- sation of nuclear chromatin with morphological evidence of fragmentation and no inflammation (Young 1984), char- acteristics which all conform to the hallmarks of classical apoptosis (Kerr et al. 1972; Wyllie 1980).
The most used marked of apoptosis is the physical detec- tion of DNA fragmentation. DNA nucleosome ladder for- mation has been considered to be a hallmark of active cell death in many systems and is thought to mark the commit- ment step of the process (Wyllie et al. 1981; Kyprianou et al. 1988; McConkey et al. 1988; Bursch et al. 1990). Briefly, DNA fragmentation is believed to begin with the system- atic fragmentation of genomic DNA into fragments of 300 and 50 kb in size, fragments which can be seen after pulse field electrophoresis (Walker et al. 1993; Brown et al. 1993). In many but not all apoptotic systems, a c a 2 + - and M ~ ' + - dependent endonuclease further fragments the DNA by cleavage at linker DNA sites between nucleosomes, giving rise to fragments that are multimers of 180 bp in size (Arends et al. 1990). Upon electrophoresis these fragments give rise
to a DNA ladder. In addition, DNA fragments can be visu- alized in situ by TUNEL, i.e., end labeling of the 3'-OH DNA ends generated by the fragmentation and the subsequent in situ detection of this labeling (Gavrieli et al. 1992).
Confirmation of the morphological features of apoptosis during retinal development was provided recently by the observation of DNA fragmentation during ganglion cell development in the chick retina (Ilschner and Waring 1992). In addition the morphological profile of pyknotic cells in the developing postnatal mouse retina observed by Young (1984) has been confirmed by the in situ detection of DNA fragmentation over the same developmental period (Chang et al. 1993; Fig. 2). Detection of pyknotic cells, as well as TUNEL-labeled nuclei, occurred primarily in the inner nuclear layer. Isolation of DNA from retinas enucleated from mice 1 and 3 weeks of age (a time in which the retina has reached maturity) and subsequent electrophoresis reveals the presence of a DNA ladder in only the 1-week-old retina (Fig. 3). Detection of DNA fragmentation, therefore, seems to correlate well with the detection of morphological features of apoptosis in the retina.
Disease states and apoptosis Apoptosis itself is not a disease process. However, the
induction of massive apoptosis may lead to dystrophic states. During development, cell death is believed to be a functional process in which specific cells are sacrificed for the benefit
Bio
chem
. Cel
l Bio
l. D
ownl
oade
d fr
om w
ww
.nrc
rese
arch
pres
s.co
m b
y C
ON
CO
RD
IA U
NIV
on
12/0
9/14
For
pers
onal
use
onl
y.

Z l8 6 10 12 15 20 6 10 72 15 20 6 10 12 15 20 DAYS
FIG. 2. Pyknotic cells and DNA fragmentation during postnatiil retinal development. (A) Summary of data presented in Young (1984) showing the number o f pykno~ic cells in the three major retinal cell layers. The numbers used for ONL include both the counlq o f pyknotic cel ls in the outer and inner rod photoreceptor cells during the period of postnatal development. The majority of pyknosis occurs in the INL within thc first 10 days. (B) Summary of the data presented by Portera-Cailliau et al. (1994) showing the number of cells with TUNEL-stained nuclei in the thrcc major retinal cell layers during retinal postnatal development. The majority of the TUNEL labeling occurs in the inner nuclear cell layer within the first 10 days.
of the entire organism (Glucksmann 1951). Likewise then, in retinal degeneration models in a number of different animals, a given tissue, the random formation of a defective somatic including the mouse, rat, cat, dog, chicken, and horse. The cell serves no functional purpose. In this case, it is thought array of genetic lesions and abnormalities inherent to these that the cell will decide to activate the apoptotic mechanism animal models has been recently reviewed (Voaden 1991). and remove itself from the tissue so that it does not impart The present discussion is restricted to a few well-studied a sustained detrimental effect on the tissue, i.e., commit rodent models of retinal degeneration in which there is some suicide (Fig. 4). In the case of a genetic lesion and the cell support for tissue regression mediated by apoptosis. types that require the expression of the normal gene product, this gives rise to primarily dysfunctional cells in that tissue, leading to a population of cells that are abnormal and by default undergo apoptosis (mass suicide). This scenario leads directly to tissue regression and in the case of diseases like RP to retinal degeneration, visual cell loss, and blindness. In the case of the photoreceptor cell, mutations in genes under- lying the phototransduction cascade, the ability of opsin to respond to light, the integrity of the photoreceptor cell struc- ture, and it's ability to interact with either retinal pigment epithelial cells or bipolar cells, as well as a great many other things, could lead to an abnormal or dysfunctional photoreceptor cell. In the case of RP, it is therefore not unexpected that many different primary defects should lead to a common manifestation of visual cell loss and blindness.
Visual cell loss and DNA fragmentation in the rd mouse Two of the better studied rodent models of RP include
the rd (retinal degeneration; Tansley 1951), and the Rds (retinal degeneration slow, Van Nie et al. 1978; Sanyal et al. 1980) mice. In both models the primary lesion underly- ing the specific genetic abnormality is known. In the rd mutant, the gene encoding P-PDE is the affected gene (Bowes et al. 1990, 1993; Pittler and Baehr 1991). cGMP phos- phodiesterase is a key enzyme in phototransduction; when activated it mediates the cleaving of cGMP and causes the cGMP-dependent cation channels in the plasma membrane in the outer segment of the photoreceptor cell to close (Fig. 1) . cGMP phosphodiesterase consists of a tetramer made up of two subunits (a and p) that have catalytic activity and two y subunits that have inhibitory activitv (reviewed in Farber et 'al. 1994). Mutations in the &PDE gene are known
Model systems of RP to underlie cases of autosomal recessive RP in humans The direct study of human RP is slow owing to the diffi- (McLaughlin et al. 1993).
culty in obtaining the relevant tissues for biological study. As In the rd retina, just after postnatal day 8, there is an a consequence, animal models of retinal degeneration and RP abnormal retardation in growth of photoreceptor inner seg- have been used to gain an insight into photoreceptor cell ments and a delay in the separation of the outer and inner degeneration. At present there exists a number of distinct nuclear layers (Caley et al. 1972; Sanyal and Bal 1973). In
Bio
chem
. Cel
l Bio
l. D
ownl
oade
d fr
om w
ww
.nrc
rese
arch
pres
s.co
m b
y C
ON
CO
RD
IA U
NIV
on
12/0
9/14
For
pers
onal
use
onl
y.

BIOCHEM. CELL BIOL. VOL. 72, 1994
Age In Weeks
I 0
FIG. 3. Detection of DNA ladders from retinas from l-week- old mice, but not from retinas from 3-week-old mice.
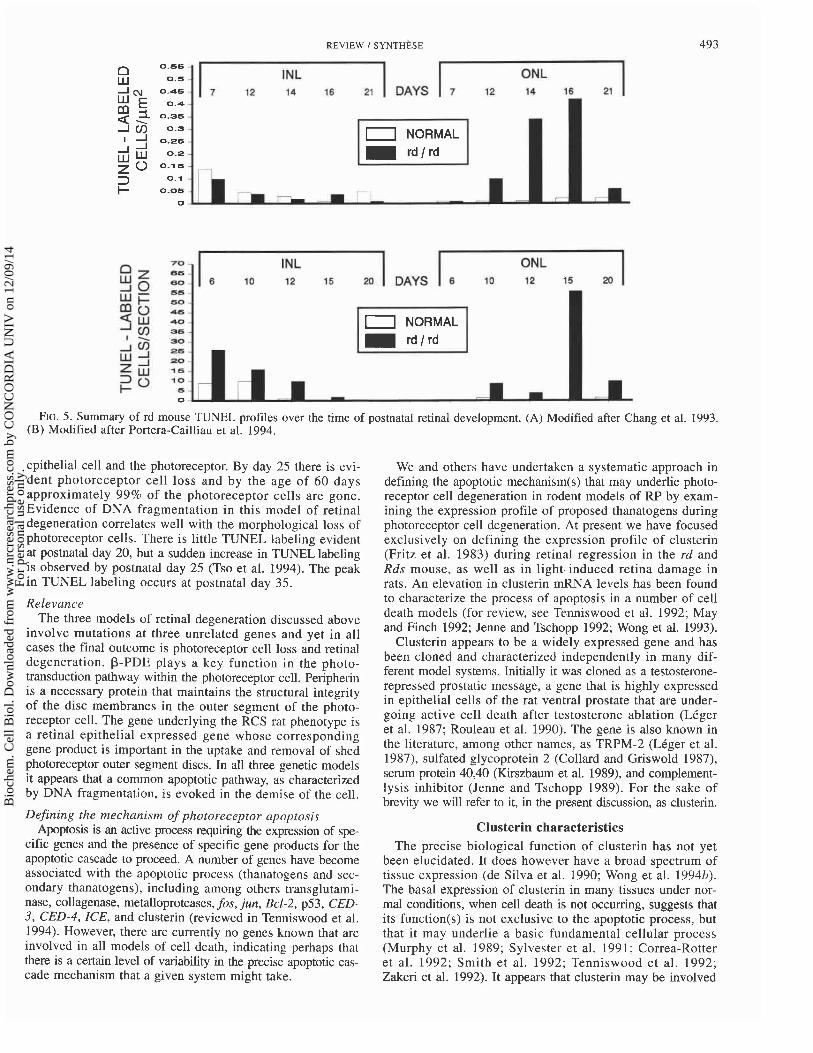
the rd mouse, photoreceptor nuclei begin to become pyknotic at about postnatal days 8-10. This is followed by rod photo- receptor cell loss within the next few days, resulting in rapid thinning of the outer nuclear layer (Tansley 1951). At post- natal day 14, approximately half of the photoreceptor cells are gone, and by postnatal day 21, only a single row of cone nuclei remains (Carter-Dawson et al. 1978). In the adult rodent, photoreceptor cells represent 90% of the retina (Treisman et al. 1988). Loss of these cells, therefore, rep- resents a substantial tissue regression. After loss of the photoreceptor cells, loss of other retinal neurons occurs with time (Ward 1982). A number of studies have surveyed the frequency of apoptotic cells over the degeneration profile of the rd mouse. In the two published reports, detection of DNA fragmentation was done in situ by TUNEL labeling (Chang et al. 1993; Portera-Cailliau et al. 1994). A summary of the results of both studies is provided in Fig. 5. Briefly, there is no detection of DNA fragmentation in the outer nuclear layer at any time in normal control mice during the period spanning the first 3 weeks of life. In contrast, retinas from rd mice show a small increase in TUNEL-labeled cells by postnatal days 10-12 and a peak in labeled cells by post- natal days 15-16, a time in which 50% of the photoreceptor cells have been lost.
Visual cell loss and DNA fragmentation in the Rds mouse In the Rds mutant, the primary lesion lies in the peripherid
Rds gene (Travis et al. 1989; Connell et al. 1991). Peripherin is believed to be a structural molecule that is essential for the
CELL DEFECT GENET^ DEFECT
c COMPROMISED ENVIRONMENT
( FUNCTION)
CELL DELE1 FIG. 4. Apoptotic model for retinal degenerative states.
integrity of the outer segment discs of the rod photoreceptor, although its exact biological function has not been elucidated (Molday 1994). Clinically, mutations in the peripherin gene have been identified in cases of autosomal dominant RP (Kajiwara et al. 1991) and macular dystrophy (Nichols et al. 1993).
Structurally, photoreceptor cells of Rds mice do not develop outer segments (Sanyal et al. 1980; Sanyal and Jansen 1981). The progression of photoreceptor cell death in the Rds mouse manifest later and more slowly than in the rd mouse (Sanyal et al. 1980). Loss of rod and cone nuclei starts after postnatal day 14 and their number is reduced to half by 3 months of age. In the case of the Rds mouse, the photoreceptor degen- eration continues to the end of the 1st year. By the end of this period there are very few photoreceptor cells remaining (Molday 1994). Evidence of DNA fragmentation has also been observed in the retinal developmental profile of the Rds mouse (Chang et al. 1993; Portera-Cailliau et al. 1994). In the Rds mouse there is a sudden peak in TUNEL-labeled nuclei at the end of postnatal week 3 (Chang et al. 1993), although an increase in labeling has been detected as soon as the end of postnatal week 2 (Portera-Cailliau et al. 1994). In either case, the detection of nuclear labeling correlates well to the known period of morphological Rds photoreceptor degeneration. It is not quite clear why there is such a marked peak in TUNEL-labeled cells at postnatal week 3, especially when morphologically photoreceptor cell loss is relatively slow. However, Western analysis of opsin levels, a marker of photoreceptor function, also shows a rapid decrease in levels starting between postnatal day 15 and 20 and continues to fall to undetectable levels by postnatal 30 (Usukura and Bok 1987).
Visual cell loss and DNA fragmentation in the RCS rat The RCS (Royal College of Surgeons) rat is characterized
by a form of photoreceptor degeneration (Dowling and Sidman 1962) that results from the inability of the retinal epithelial cells to phagocytose shedding rod outer segment discs (Bok and Hall 1971; LaVail 1981). The mutant gene that impairs phagocytosis in the RCS rat is specifically expressed in the RPE cells (Mullen and LaVail 1976). The precise affected gene has, however, not been characterized. The photoreceptor cells develop normally, reaching maturity at 17 days of age (Tso et al. 1994), and continue to appear morphologically normal at day 20. Shortly thereafter, excess rod photoreceptor outer segment membranes accumulate at their distal regions (Dowling and Sidman 1962), which is thought to impair the normal interaction between the retinal
Bio
chem
. Cel
l Bio
l. D
ownl
oade
d fr
om w
ww
.nrc
rese
arch
pres
s.co
m b
y C
ON
CO
RD
IA U
NIV
on
12/0
9/14
For
pers
onal
use
onl
y.
REVIEW I SYNTHESE
- -
70 INL ONL 6 10 12 15 M BAYS 6 10 12 15 20
FIG. 5. Summary of rd mouse TUNEL profiles over the time of postnatal retinal development. (A) Modified after Chang et al. 1993. (B) Modified after Portera-Cailliau et al. 1994.
- ONL
7 12 14 16 27
0.66
0.5 -
0.46
epithelial cell and the photoreceptor. By day 25 there is evi- dent photoreceptor cell loss and by the age of 60 days approximately 99% of the photoreceptor cells are gone. Evidence of DNA fragmentation in this model of retinal degeneration correlates well with the morphological loss of photoreceptor cells. There is little TUNEL labeling evident at postnatal day 20, but a sudden increase in TUNEL labeling is observed by postnatal day 25 (Tso et al. 1994). The peak in TUNEL labeling occurs at postnatal day 35.
Relevance The three models of retinal degeneration discussed above
involve mutations at three unrelated genes and yet in all cases the final outcome is photoreceptor cell loss and retinal degeneration. P-PDE plays a key function in the photo- transduction pathway within the photoreceptor cell. Peripherin is a necessary protein that maintains the structural integrity of the disc membranes in the outer segment of the photo- receptor cell. The gene underlying the RCS rat phenotype is a retinal epithelial expressed gene whose corresponding gene product is important in the uptake and removal of shed photoreceptor outer segment discs. In all three genetic models it appears that a common apoptotic pathway, as characterized by DNA fragmentation, is evoked in the demise of the cell.
INL 7 12 14 16 21
0.4 -
0.35
0.3
0.26
0.2 -
0.1 6 -
0.1
0.06 - 7
0 rn r- -m I -
Defining the mechanism of photoreceptor apoptosis Apoptosis is an active process requiring the expression of spe-
cific genes and the presence of specific gene products for the apoptotic cascade to proceed. A number of genes have become associated with the apoptotic process (thanatogens and sec- ondary thanatogens), including among others transglutami- nase, collagenase, metalloproteases, fos, jun, Bcl-2, p53, CED- 3, CED-4, ICE, and clusterin (reviewed in Tenniswood et al. 1994). However, there are currently no genes known that are involved in all models of cell death, indicating perhaps that there is a certain level of variability in the precise apoptotic cas- cade mechanism that a given system might take.
DAYS
-
We and others have undertaken a systematic approach in defining the apoptotic mechanism(s) that may underlie photo- receptor cell degeneration in rodent models of RP by exam- ining the expression profile of proposed thanatogens during photoreceptor cell degeneration. At present we have focused exclusively on defining the expression profile of clusterin (Fritz et al. 1983) during retinal regression in the rd and Rds mouse, as well as in light-induced retina damage in rats. An elevation in clusterin mRNA levels has been found to characterize the process of apoptosis in a number of cell death models (for review, see Tenniswood et al. 1992; May and Finch 1992; Jenne and Tschopp 1992; Wong et al. 1993).
Clusterin appears to be a widely expressed gene and has been cloned and characterized independently in many dif- ferent model systems. Initially it was cloned as a testosterone- repressed prostatic message, a gene that is highly expressed in epithelial cells of the rat ventral prostate that are under- going active cell death after testosterone ablation (LCger et al. 1987; Rouleau et al. 1990). The gene is also known in the literature, among other names, as TRPM-2 (LCger et al. 1987), sulfated glycoprotein 2 (Collard and Griswold 1987), serum protein 40,40 (Kirszbaum et al. 1989), and complement- lysis inhibitor (Jenne and Tschopp 1989). For the sake of brevitv we will refer to it. in the mesent discussion. as clusterin.
Clusterin characteristics The precise biological function of clusterin has not yet
been elucidated. It does however have a broad spectrum of tissue expression (de Silva et al. 1990; Wong et al. 1994b). The basal expression of clusterin in many tissues under nor- mal conditions, when cell death is not occurring, suggests that its function(s) is not exclusive to the apoptotic process, but that it may underlie a basic fundamental cellular process (Murphy et al. 1989; Sylvester et al. 1991; Correa-Rotter et al. 1992; Smith et al. 1992; Tenniswood et al. 1992; Zakeri et al. 1992). It appears that clusterin may be involved
Bio
chem
. Cel
l Bio
l. D
ownl
oade
d fr
om w
ww
.nrc
rese
arch
pres
s.co
m b
y C
ON
CO
RD
IA U
NIV
on
12/0
9/14
For
pers
onal
use
onl
y.
BIOCHEM. CELL BIOL. VOL. 72. 1994
Clusterin (La w*
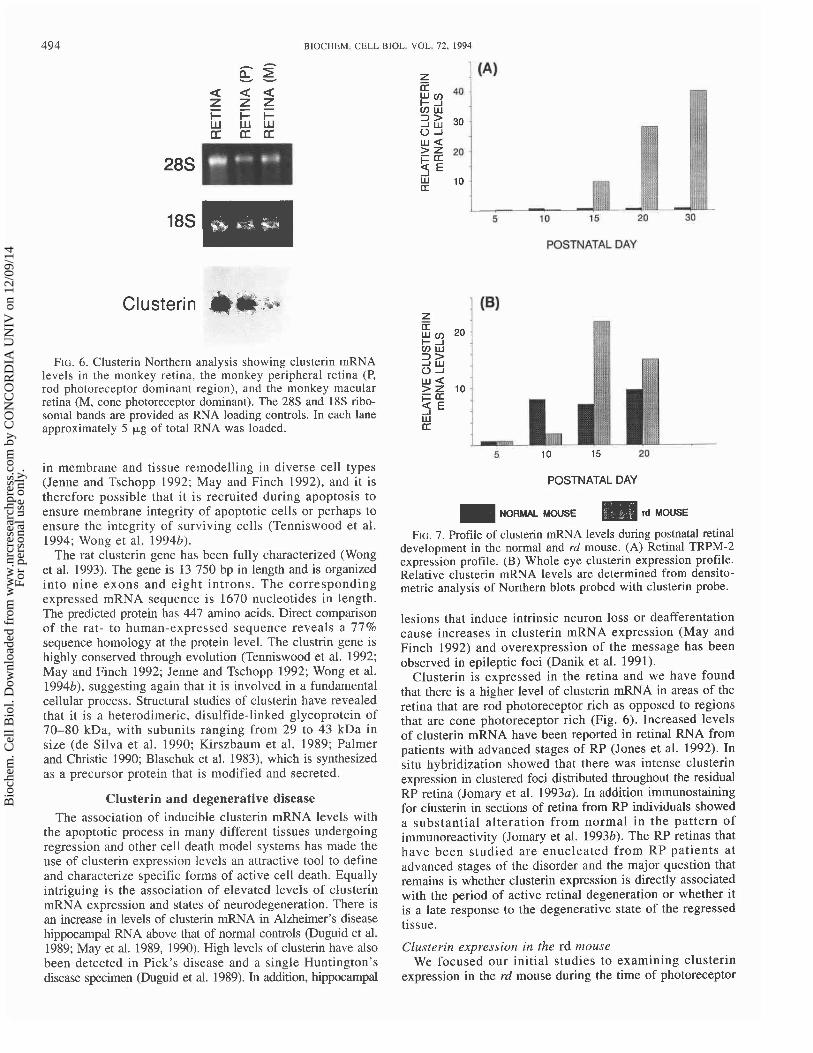
FIG. 6. Clusterin Northern analysis showing clusterin mRNA levels i n the monkey retina, the monkey peripheral retina (P, rod photoreceptor dominant region), and the monkey macular retina (M, cone photoreceptor dominant). The 28s and 18s ribo- somal bands are provided as RNA loading controls. In each lane approximately 5 pg of total RNA was loaded.
in membrane and tissue remodelling in diverse cell types (Jenne and Tschopp 1992; May and Finch 1992), and it is therefore possible that it is recruited during apoptosis to ensure membrane integrity of apoptotic cells or perhaps to ensure the integrity of surviving cells (Tenniswood et al. 1994; Wong et al. 1994b).
The rat clusterin gene has been fully characterized (Wong et al. 1993). The gene is 13 750 bp in length and is organized into nine exons and eight introns. The corresponding expressed mRNA sequence is 1670 nucleotides in length. The predicted protein has 447 amino acids. Direct comparison of the rat- to human-expressed sequence reveals a 77% sequence homology at the protein level. The clustrin gene is highly conserved through evolution (Tenniswood et al. 1992; May and Finch 1992; Jenne and Tschopp 1992; Wong et al. 1994b), suggesting again that it is involved in a fundamental cellular process. Structural studies of clusterin have revealed that it is a heterodimeric, disulfide-linked glycoprotein of 70-80 kDa, with subunits ranging from 29 to 43 kDa in size (de Silva et al. 1990; Kirszbaum et al. 1989; Palmer and Christie 1990; Blaschuk et al. 1983), which is synthesized as a precursor protein that is modified and secreted.
Clusterin and degenerative disease The association of inducible clusterin mRNA levels with
the apoptotic process in many different tissues undergoing regression and other cell death model systems has made the use of clusterin expression levels an attractive tool to define and characterize specific forms of active cell death. Equally intriguing is the association of elevated levels of clusterin mRNA expression and states of neurodegeneration. There is an increase in levels of clusterin mRNA in Alzheimer's disease hippocampal RNA above that of normal controls (Duguid et al. 1989; May et al. 1989, 1990). High levels of clusterin have also been detected in Pick's disease and a single Huntington's disease specimen (Duguid et al. 1989). In addition, hippocampal
-
5 10 15 20
POSTNATAL DAY
POSTNATAL DAY
FIG. 7. Profile of clusterin mRNA levels during postnatal retinal development in the normal and rd mouse. (A) Retinal TRPM-2 expression profile. (B) Whole eye clusterin expression profile. Relative clusterin mRNA levels are determined from densito- metric analysis of Northern blots probed with clusterin probe.
lesions that induce intrinsic neuron loss or deafferentation cause increases in clusterin mRNA expression (May and Finch 1992) and overexpression of the message has been observed in epileptic foci (Danik et al. 1991).
Clusterin is expressed in the retina and we have found that there is a higher level of clusterin mRNA in areas of the retina that are rod photoreceptor rich as opposed to regions that are cone photoreceptor rich (Fig. 6). Increased levels of clusterin mRNA have been reported in retinal RNA from patients with advanced stages of RP (Jones et al. 1992). In situ hybridization showed that there was intense clusterin expression in clustered foci distributed throughout the residual RP retina (Jomary et al. 1993~). In addition immunostaining for clusterin in sections of retina from RP individuals showed a substantial alteration from normal in the pattern of immunoreactivity (Jomary et al. 199317). The RP retinas that have been studied are enucleated from RP patients at advanced stages of the disorder and the major question that remains is whether clusterin expression is directly associated with the period of active retinal degeneration or whether it is a late response to the degenerative state of the regressed tissue.
Clusterin expression in the rd mouse We focused our initial studies to examining clusterin
expression in the rd mouse during the time of photoreceptor
Bio
chem
. Cel
l Bio
l. D
ownl
oade
d fr
om w
ww
.nrc
rese
arch
pres
s.co
m b
y C
ON
CO
RD
IA U
NIV
on
12/0
9/14
For
pers
onal
use
onl
y.
REVIEW I SYNTHESE 495
Postnatal Day
Clusterin
Retina
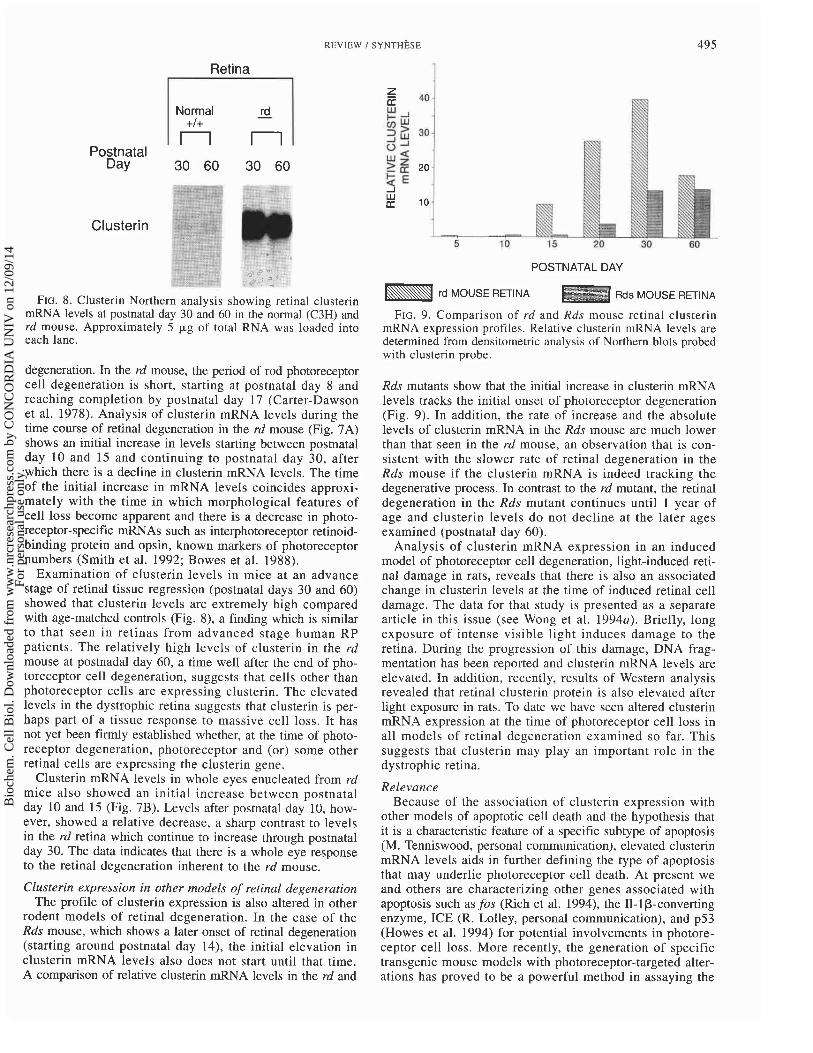
FIG. 8. Clusterin Northern analysis showing retinal clusterin mRNA levels at postnatal day 30 and 60 in the normal (C3H) and rd mouse. Approximately 5 pg of total RNA was loaded into each lane.
I I
degeneration. In the rd mouse, the period of rod photoreceptor cell degeneration is short, starting at postnatal day 8 and reaching completion by postnatal day 17 (Carter-Dawson et al. 1978). Analysis of clusterin mRNA levels during the time course of retinal degeneration in the rd mouse (Fig. 7A) shows an initial increase in levels starting between postnatal day 10 and 15 and continuing to postnatal day 30, after which there is a decline in clusterin mRNA levels. The time of the initial increase in mRNA levels coincides approxi- mately with the time in which morphological features of cell loss become apparent and there is a decrease in photo- receptor-specific mRNAs such as interphotoreceptor retinoid- binding protein and opsin, known markers of photoreceptor numbers (Smith et al. 1992; Bowes et al. 1988).
Examination of clusterin levels in mice at an advance stage of retinal tissue regression (postnatal days 30 and 60) showed that clusterin levels are extremely high compared with age-matched controls (Fig. 8), a finding which is similar to that seen in retinas from advanced stage human RP patients. The relatively high levels of clusterin in the rd mouse at postnadal day 60, a time well after the end of pho- toreceptor cell degeneration, suggests that cells other than photoreceptor cells are expressing clusterin. The elevated levels in the dystrophic retina suggests that clusterin is per- haps part of a tissue response to massive cell loss. It has not yet been firmly established whether, at the time of photo- receptor degeneration, photoreceptor and (or) some other retinal cells are expressing the clusterin gene.
Clusterin mRNA levels in whole eyes enucleated from rd mice also showed an initial increase between postnatal day 10 and 15 (Fig. 7B). Levels after postnatal day 10, how- ever, showed a relative decrease, a sharp contrast to levels in the rd retina which continue to increase through postnatal day 30. The data indicates that there is a whole eye response to the retinal degeneration inherent to the rd mouse.
Normal rd - +I+
n n
Clusterin expression in other models of retinal degeneration The profile of clusterin expression is also altered in other
rodent models of retinal degeneration. In the case of the Rds mouse, which shows a later onset of retinal degeneration (starting around postnatal day 14), the initial elevation in clusterin mRNA levels also does not start until that time. A comparison of relative clusterin mRNA levels in the rd and
POSTNATAL DAY
rd MOUSE RETINA Rds MOUSE RETINA
FIG. 9. Comparison of rd and Rds mouse retinal clusterin mRNA expression profiles. Relative clusterin mRNA levels are determined from densitometric analysis of Northern blots probed with clusterin probe.
Rds mutants show that the initial increase in clusterin mRNA levels tracks the initial onset of photoreceptor degeneration (Fig. 9). In addition, the rate of increase and the absolute levels of clusterin mRNA in the Rds mouse are much lower than that seen in the rd mouse, an observation that is con- sistent with the slower rate of retinal degeneration in the Rds mouse if the clusterin mRNA is indeed tracking the degenerative process. In contrast to the rd mutant, the retinal degeneration in the Rds mutant continues until 1 year of age and clusterin levels do not decline at the later ages examined (postnatal day 60).
Analysis of clusterin mRNA expression in an induced model of photoreceptor cell degeneration, light-induced reti- nal damage in rats, reveals that there is also an associated change in clusterin levels at the time of induced retinal cell damage. The data for that study is presented as a separate article in this issue (see Wong et al. 1994~) . Briefly, long exposure of intense visible light induces damage to the retina. During the progression of this damage, DNA frag- mentation has been reported and clusterin mRNA levels are elevated. In addition, recently, results of Western analysis revealed that retinal clusterin protein is also elevated after light exposure in rats. To date we have seen altered clusterin mRNA expression at the time of photoreceptor cell loss in all models of retinal degeneration examined so far. This suggests that clusterin may play an important role in the dystrophic retina.
Relevance Because of the association of clusterin expression with
other models of apoptotic cell death and the hypothesis that it is a characteristic feature of a specific subtype of apoptosis (M. Tenniswood, personal communication), elevated clusterin mRNA levels aids in further defining the type of apoptosis that may underlie photoreceptor cell death. At present we and others are characterizing other genes associated with apoptosis such as fos (Rich et al. 1994), the 11-lp-converting enzyme, ICE (R. Lolley, personal communication), and p53 (Howes et al. 1994) for potential involvements in photore- ceptor cell loss. More recently, the generation of specific transgenic mouse models with photoreceptor-targeted alter- ations has proved to be a powerful method in assaying the
Bio
chem
. Cel
l Bio
l. D
ownl
oade
d fr
om w
ww
.nrc
rese
arch
pres
s.co
m b
y C
ON
CO
RD
IA U
NIV
on
12/0
9/14
For
pers
onal
use
onl
y.

496 BIOCHEM. CELL BIOL. VOL. 72. 1994
cause and effects of specific genes with respect to photo- receptor cell loss (Al-Ubaidi et al. 1992; Howes et al. 1994). Data from these studies should further delineate the factors involved in apoptosis and apoptosis-related responses to photoreceptor cell death.
Summary The study of the mechanism and effects of apoptosis in
retinal and photoreceptor cell loss is at its infancy. The existing data suggests that apoptosis may be a common pathway that leads to retinal degeneration. It would appear, however, that there are many potential genetic and non- genetic triggers that can lead to this mechanism of photo- receptor cell loss. In all retinal degeneration models that we have examined for clusterin expression, it appears that an elevation in clusterin levels is part of the response to photo- receptor cell loss. The further understanding of both the tis- sue response to photoreceptor cell loss, as well as the mech- anistic apoptotic cascade of events that underlies the process in these animal models, should provide further insight into human RP and potential treatments.
Acknowledgements I thank the collaborative efforts of Sylvia Smith (Medical
College of Georgia, Georgia), Deborah Farber (Jules Stein Eye Insti tute, Calif .) , Dan Organisciak (Wright Sta te University, Ohio) and Theo van Veen (University of Goteborg, Sweden) during the course of these studies as well as the RP Foundation-Fighting Blindness (Canada) for their support. I am also deeply indebted to Gerald Chader (National Eye Institute, NIH, MD) and Martin Tenniswood (W. Alton Jones Cell Science Center, Lake Placid, N.Y.) for their continued support and encouragment.
Adler, R. 1986. Trophic interactions in retinal development and in retinal degenerations. in vivo and in vitro studies. In The retina, a model for cell biology studies. Part I. Edited by R. Adler and D. Farber. Academic Press, Orlando, Fla. pp. 111-150.
Al-Ubaidi, M.R., Hollyfield, J.G., Overbeek, P.A., and Baehr, W. 1992. Photoreceptor degeneration induced by the expression of simian virus 40 large tumor antigen in the retina of trans- genic mice. Proc. Natl. Acad. Sci. U.S.A. 89: 1194-1198.
Arends, M.J., Morris, R.G., and Wyllie, A.H. 1990. Apoptosis: the role of the endonuclease. Am. J. Pathol. 136: 593-608.
Baylor, D. 1987. Photoreceptor signals and vision. Invest. Ophthalmol. Visual Sci. 28: 34-49.
Berson, E.L. 1993. Retinitis pigmentosa. Invest. Ophthalmol. Visual Sci. 34: 1659-1676.
Blaschuk, O.L., Burdzy, K., and Fritz, I.B. 1983. Purification and characterization of a cell-aggregating factor (clusterin), the major glycoprotein in ram rete testis fluid. J. Biol. Chem. 258: 77 14-7720.
Bok, D., and Hall, M. 1971. The role of the pigment epithelium in the etiology of inherited retinal dystrophy in the rat. J. Cell Biol. 49: 664-682.
Bowes, C., van Veen, T., and Farber, D.B. 1988. Opsin, G-protein and 48-kDa protein in normal and rd mouse retinas: develop- mental expression of mRNAs and proteins and lightidark cycling of mRNAs. Exp. Eye Res. 47: 369-390.
Bowes, C., Li, T., Danciger, M., Baxter, L., Applebury, M., and Farber, D. 1990. Retinal degeneration in the rd mouse is caused by a defect in the P-subunit of rod cGMP-phosphodiesterase. Nature (London), 347: 677-680.
Bowes, C., Li, T., Frankel, W.N., Danciger, M., Coffin, J.M.,
Applebury, M.A., and Farber, D.B. 1993. Localization of a retroviral element within the rd gene coding for the P-subunit of cGMP-phosphodiesterase. Proc. Natl. Acad. Sci. U.S.A. 90: 2955-2959.
Brown, D.G., Sun, X.-M., and Cohen, G.M. 1993. Dexamethasone- induced apoptosis involves cleavage of DNA to large frag- ments prior to internucleosomal fragmentation. J. Biol. Chem. 268: 3037-3039.
Bursch, W., Kleine, L., and Tenniswood, M.P. 1990. The bio- chemistry of cell death by apoptosis. Biochem. Cell Biol. 68: 1071-1074.
Caley, D.W., Johnson, C., and Leibelt, R.A. 1972. The postnatal development of the retina in the normal and rodless CBA mouse: a light and electron microscopic study. Am. J. Anat. 133: 179-212.
Carter-Dawson, L.D., LaVail, M.M., and Sidman, R.L. 1978. Differential effect of the rd mutation on rods and cones in the mouse retina. Invest. Ophthalmol. Visual Sci. 17: 489-498.
Chang, G.Q., Hao, Y., and Wong, F. 1993. Apoptosis: final com- mon pathway of photoreceptor death in rd, rds and rhodopsin mutant mice. Neuron, 11: 595-605.
Collard, M.W., and Griswold, M.D. 1987. Biosynthesis and mol- ecular cloning of sulfated glycoprotein 2 secreted by rat sertoli cells. Biochemistry, 26: 3297-3303.
Connell, G., Bascom, R., Molday, L., Reid, D., McInnes, R., and Molday, R. 1991. Photoreceptor peripherin is the normal product of the gene responsible for retinal degeneration in the rds mouse. Proc. Natl. Acad. Sci. U.S.A. 88: 723-726.
Correa-Rotter, R., Hostetter, T.H., Manival, J.C., Eddy, A.A., and Rosenberg, M.E. 1992. Intrarenal distribution of clusterin following reduction of renal mass. Kidney Int. 41: 938-950.
Danik, M., Chabot, J.-G., Mercier, C., Benabid, A.-L., Chauvin, C., Quirion, R., and Suh, M. 1991. Human gliomas and epileptic foci express high levels of a mRNA related to rat testicular sulfated glycoprotein 2, a purported marker of cell death. Proc. Natl. Acad. Sci. U.S.A. 88: 8577-8581.
de Silva, H.V., Harmony, J.A.K., Stuart, W.D., Gil, C.M., and Robbins, J. 1990. Apolipoprotein J: structure and tissue dis- tribution. Biochemistry, 29: 5380-5389.
Dowling, J., and Sidman, R. 1962. Inherited retinal dystrophy in rat. J. Cell Biol. 14: 73-109.
Duguid, J.R., Bohmont, C.W., Liu, N., and Tourtellotte, W.W. 1989. Changes in brain gene expression shared by scrapie and Alzheimer disease. Proc. Natl. Acad. Sci. U.S.A. 86: 7260-7264.
Farber, D.B., Flannery, J.G., and Bowes-Rickman, C. 1994. The rd mouse story: seventy years of research on an animal model of inherited retinal degeneration. Progress in Retinal and Eye Research, 13: 3 1-64.
Fritz, I.B., Burdzy, K., Setchell, B., and Blaschuk, 0 . 1983. Ram rete testis fluid contains a protein (clusterin) which influences cell-cell interactions in vitro. Biol. Reprod. 28: 1173-1 188.
Gavrieli, Y., Sherman, Y., and Ben-Sasson, S.A. 1992. Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J. Cell Biol. 119: 493-501.
Glucksmann, A. 1940. Development and differentiation of the tadpole eye. Br. J. Ophthalmol. 24: 153-178.
Glucksmann, A. 1951. Cell death in normal vertebrate ontogeny. Biol. Rev. 26: 59-86.
Howes, K.A., Ransom, N., Papermaster, D.S., Lasudry, J.G.H., Albert, D., and Windle, J.J. 1994. Apoptosis or retinoblastoma: alternative fates of photoreceptor expressing the HPV-16 E7 gene in the presence or absence of p53. Genes Dev. 8: 1300-1310.
Hughes, W.F., and McLoon, S.C. 1979. Ganglionic cell death during normal retinal development in the chick: comparisons with cell death induced by target field destruction. Exp. Neurol. 66: 587-601.
Bio
chem
. Cel
l Bio
l. D
ownl
oade
d fr
om w
ww
.nrc
rese
arch
pres
s.co
m b
y C
ON
CO
RD
IA U
NIV
on
12/0
9/14
For
pers
onal
use
onl
y.

REVIEW I SYNTHESE 497
Ilschner, S.U., and Waring, P. 1992. Fragmentation of DNA in the Murphy, B.F., Saunders, J.R., O'Bryan, M.K., Kirszbaum, L., retina of chicken embryos coincides with retinal ganglion cell Walker, I.D., and d'Apice, A.J. 1989. SP-40,40 is an inhibitor death. Biochem. Biophys. Res. Commun. 183: 1056-1061. of C5b-6-initiated haemolysis. Int. Immunol. 1: 551-554.
Jenne, D.E., and Tschopp, J. 1989. Molecular structure and func- Nichols, B., Schiffield, V., Vandenburgh, K., Drack, A., Kimura, tional characterization of a human complement cytolysis A., and Stone, E. 1993. Butterfly-shaped pigment dystrophy of inhibitor found in blood and seminal plasma: identity to sulfated the fovea caused by a point mutation in codon 167 of the rds glycoprotein 2, a constituent of rat testis fluid. Proc. Natl. gene. Nature Genet. 3: 202-207. Acad. Sci. U.S.A. 86: 7123-7127. Palmer, D.J., and Christie, D.L. 1990. The primary structure of
Jenne, D.E., and Tschopp, J. 1992. Clusterin: the intriguing glycoprotein 111 from bovine adrenal medullary chromaffin guises of a widely expressed glycoprotein. Trends Biochem. Sci. granules. J. Biol. Chem. 265: 6617-6623. 17: 154-159. Pittler, S.J., and Baehr, W. 1991. Identification of a nonsense
Jomary, C., Murphy, B.F., Neal, M.J., and Jones, S.E. 1993a. mutation in the rod photoreceptor cGMP phosphodiesterase Abnormal distribution of retinal clusterin in retinitis pigmentosa. p-subunit gene of the rd mouse. Proc. Natl. Acad. Sci. U.S.A. Mol. Brain Res. 20: 274-278. 88: 8322-8326.
Jomary, C., Neal, M.J., and Jones, S.E. 1993b. Comparison of Portera-Cailliau, C., Sung, C.-H., Nathans, J., and Adler, R. clusterin gene expression in normal and dystrophic human 1994. Apoptotic photoreceptor cell death in mouse models of retinas. Mol. Brain Res. 20: 279-284. retinitis pigmentosa. Proc. Natl. Acad. Sci. U.S.A. 91: 974-978.
Jones, S.E., Meerabux, D.A., Yeats, D.A., and Neal, M. 1992. Rager, C., and Rager, U. 1978. Systems-matching by degeneration. Analysis of differentially expressed genes in retinitis pigmentosa Exp. Brain Res. 33: 65-78. retinas. Altered clusterin mRNA. FEBS Lett. 300: 279-282. Rich, K.A., Ahan, Y., and Blanks, J.C. 1994. Aberrant expression
Kajiwara, K., Hahn, L., Mukai, S., Travis, G., Berson, E., and of c-fos accompanies photoreceptor cell death in the rd mouse. Dryja, T. 1991. Mutations in the human retinal degeneration Invest. Ophthalmol. Visual Sci. 35: 1833. slow gene in autosomal dominant retinitis pigmentosa. Nature Rouleau, M. LCger, J., and Tenniswood, M. 1990. Ductal hetero- (London), 354: 480-482. geneity of cytokeratins, gene expression, and cell death in the
Kerr, J.F.R., Wyllie, A.H., and Currie, A.R. 1972. Apoptosis: a rat ventral prostate. Mol. Endocrinol. 4: 2003-2013. basic biological phenomenon with wide ranging implications Sanyal, S., and Bal, A.K. 1973. Comparative light and electron- in tissue kinetics. Br. J. Cancer, 26: 239-257. microscopic study of the retinal histogenesis in normal and
Kirszbaum, L., Sharpe, J.A., Murphy, B., d'Apice, A.J., Classon, rd mutant mice. Z. Anat. Entwicklungsgesch. 142: 219-238. B., Hudson, P., and Walker, I.D. 1989. Molecular cloning and Sanyal, S., and Jansen, H.G. 1981. Absence of receptor outer characterization of the novel, human complement-associated segments in the retina of rds mutant mice. Neurosci. Lett. 21: protein, SP-40,40: a link between the complement and repro- 23-26. ductive systems. EMBO J. 8: 711-718. Sanyal, S., DeRuiter, A., and Hawkins, R. 1980. Development
Kyprianou, N., English, H.F., and Isaacs, J.T. 1988. Activation of and degeneration of retina in rds mutant mice: light microscopy. a c a 2 + - ~ g ~ + - d e p e n d e n t endonuclease as an early event in J. Comp. Neurol. 194: 193-207. castration-induced prostatic cell death. Prostate (N.Y.), 13: Sengelaub, D.R., and Finlay, B.L. 1982. Cell death in the mam- 103-117. malian visual system during normal development. I. Retinal
LaVail, M.M. 1981. Analysis of neurological mutants with inher- ganglion cells. J. Comp. Neurol. 204: 311-317. ited retinal degeneration. Invest. Ophthalmol. Visual Sci. 21: Silver, J., and Hughes, A.F.W. 1973. The role of cell death during 638-657. morphogenesis of the mammalian eye. J. Morphol. 140: 159-170.
LCger, J.G., Montpetit, M.L., and Tenniswood, M.P. 1987. Smith, S.B., Lee, L., Nickerson, J., Si, J.S., Chader, G.J., and Characterization and cloning of androgen-repressed mRNAs Wiggert, B. 1992. Synthesis and secretion of interphotoreceptor from rat ventral prostate. Biochem. Biophys. Res. Commun. retinoid-binding protein (IRBP) and developmental expression 147: 196-203. of IRBP mRNA in normal and rd mouse retinas. Exp. Eye
May, P.C., and Finch, C.E. 1992. Sulfated glycoprotein 2: new Res. 54: 957-964. relationships of this multifunctional protein to neurodegener- Steele, F.R. 1994. Shedding light on inherited blindness: the ation. Trends Neurosci. 15: 391-396. genetics of retinitis pigmentosa. J. Natl. Inst. Health Res. 6:
May, P.C., Johnson, S.A., Poirier, J., Lampert-Etchells, M.A., 58-62. and Finch, C.E. 1989. Altered gene expression in Alzheimer's Sylvester, S.R., Morales, C., Oko, R., and Griswold, M.D. 1991. disease brain tissue. Can. J. Neurol. Sci. 16: 473-476. Localization of sulfated glycoprotein-2 (clusterin) on sper-
May, P.C., Lampert-Etchelles, M.A., Johnson, S.A., Poirier, J., matozoa and in the reproductive tract of the male rat. Biol. Masters, J.N., and Finch, C.E. 1990. Dynamics of gene expres- Reprod. 45: 195-207. sion for a hippocampal glycoprotein elevated in Alzheimer's Tansley, K. 1951. Hereditary degeneration of the mouse retina. disease and in response to experimental lesions in the rat. Br. J. Ophthalmol. 35: 572-582. Neuron, 5: 831-839. Tenniswood, M., Guenette, R., Lakins, J., Mooibroek, M., Wong,
McConkey, D.J., Hartzell, P., Duddy, S.K., Hakansson, H., and P., and Welsh, J.-E. 1992. Active cell death in hormone- Orrenius, S. 1988. 2,3,7,8-Tetrachlorodibenzo-p-dioxin kills dependent tissues. Cancer Metastasis Rev. 11: 197-220. immature thymocytes by ca2+-mediated endonuclease activa- Tenniswood, M., Taillefer, D., Lakins, J., Guenette, R., Mooibroek, tion. Science (Washington, D.C.), 242: 256-259. M., Daehlin, L., and Welsh, J. 1994. Control of gene expression
McLaughlin, M., Sandberg, M., Berson, E., and Dryja, T. 1993. during apoptosis in hormone-dependent tissues. In Apoptosis Recessive mutations in the gene encoding the P-subunit of 11: the molecular basis of apoptosis in disease. Edited by L.D. rod phosphodiesterase in patients with retinitis pigmentosa. Tomei and F.O. Cope. Cold Spring Harbor Laboratory Press, Nature Genet. 4: 130-134. Plainveiw, N.Y. pp. 283-3 11.
Molday, R.S. 1994. Peripherinlrds and rom-I: molecular properties Travis, G., Brennan, M., Danielson, P., Kozak, C., and Sutcliffe, and role in photoreceptor cell degeneration. Progress in Retinal J. 1989. Identification of a photoreceptor-specific mRNA and Eye Research, 13: 271-299. encoded by the gene responsible for retinal degeneration slow
Mullen, R.J., and LaVail, M.M. 1976. Inherited retinal dystrophy: (rds). Nature (London), 338: 70-73. primary defect in pigment epithelium determined with exper- Treisman, J.E., Morabito, M.A., and Barnstable, C.J. 1988. Opsin imental rat chimeras. Science (Washington, D.C.), 192: expression in the rat retina is developmentally regulated by 799-801. transcription activation. Mol. Cell. Biol. 8: 1570-1579.
Bio
chem
. Cel
l Bio
l. D
ownl
oade
d fr
om w
ww
.nrc
rese
arch
pres
s.co
m b
y C
ON
CO
RD
IA U
NIV
on
12/0
9/14
For
pers
onal
use
onl
y.

498 BIOCHEM. CELL BIOL. VOL. 72. 1994
Tso, M.O.M., Zhang, C., Abler, AS . , Chang, C.-J., Wong, R., Wong, P., Kutty, R.K., Darrow, R.M., Shivaram, S., Kutty, G., Chang, G.-Q., and Lam, T.T. 1994. Apoptosis leads to photo- Fletcher, R.T., Wiggert, B., Chader, G., and Organisciak, D.T. receptor degeneration in inherited retinal dystrophy of RCS 1994a. Changes in clusterin expression associated with light- rats. Invest. Ophthalmol. Visual Res. 35: 2693-2699. induced retinal damage in rats. Biochem. Cell Biol. 72. This
Usukura, J., and Bok, D. 1987. Changes in the localization and issue. content of opsin during retinal development in the rds mutant Wong, P., Taillefer, D., Lakins, J., Pineault, J., Chader, G., and mouse: immunocytochemistry and immunoassay. Exp. Eye Tenniswood, M. 1994b. Molecular characterization of human Res. 45: 501-515. TRPM-2/clusterin, a gene associated with sperm maturation,
Van Nie, R., Ivanyi, D., and Demant, P. 1978. A new H-2 linked apoptosis and neurodegeneration. Eur. J. Biochem. 221: mutation, rds, causing retinal degeneration in the mouse. Tissue 917-925. Antigens, 12: 106-108. Wyllie, A.H. 1980. Glucocorticoid-induced thymocyte apoptosis
Voaden, M.J. 1991. Retinitis pigmentosa and its models. Progress is associated with endogenous endonuclease activation. Nature in Retinal and Eye Research, 10: 293-331. (London), 284: 555-556.
Walker, P.R., Kokileva, L., LeBlanc, J., and Sikorska, M. 1993. Wyllie, A.H. Beattie, G.J., and Hargreaves, A.D. 1981. Chromatin Detection of the initial stages of DNA fragmentation in apop- changes in apoptosis. Histochem. J. 13: 681-692. tosis. BioTechniques, 15: 1032-1047. Young, R. 1984. Cell death during differentiation of the retina in
Ward, R. 1982. Quantitative effects of retinal degeneration in the mouse. J. Comp. Neurol. 229: 362-373. mice. Rev. Can. Biol. Exp. 41: 115-119. Zakeri, A,, Curto, M., Hoover, D.M., Wightman, K., Engelhardt,
Wong, P., Pineault, J., Lakins, J., Taillefer, D., LCger, J.G., Wang, J., Smith, F.F., Kierszenbaum, A.L., Gleeson, T.G., and C., and Tenniswood, M.P. 1993. Genomic organization and Tenniswood, M. 1992. Developmental expression of the S35- expression of the rat TRPM-2 (clusterin) gene, a gene impli- S45lSGP-2lTRPM-2 gene in rat testis and epididymis. Mol. cated in apoptosis. J. Biol. Chem. 268: 5021-5031. Reprod. Dev. 33: 373-384.
Bio
chem
. Cel
l Bio
l. D
ownl
oade
d fr
om w
ww
.nrc
rese
arch
pres
s.co
m b
y C
ON
CO
RD
IA U
NIV
on
12/0
9/14
For
pers
onal
use
onl
y.