theoretical studies of unusually short bond lengths in oxirane · pdf file ·...
TRANSCRIPT

Theoretical studies of unusually short bond lengths inoxirane and derivativesq
M. HoÃÁ 1, W.A. Szarek, V.H. Smith Jr.*
Department of Chemistry, Queen's University, Kingston, Ont., Canada K7L 3N6
Received 1 June 2000; accepted 28 June 2000
Abstract
The p-Complex-Back-Donation model (p-cbd) has been used together with the topological analysis of the ab initio and semi-
empirical densities to investigate bond lengths in the oxirane molecule and derivatives. Both models offer similar conclusions.
The shortenings of the C±C bond in the oxirane ring and of the neighbouring C±C bond arise mainly from the substituent
groups. However, neither the p-cbd model nor the atoms in molecules model offers a satisfactory explanation for the elongation
of one of the C±O bonds. q 2001 Elsevier Science B.V. All rights reserved.
Keywords: Oxirane molecule; p-Complex-Back-Donation model; Oxirane derivatives; Three-membered ring model; Charge density analysis;
Walsh orbitals; Molecular orbitals; Atoms in molecules
1. Introduction
The X-ray structure of 1,2-Anhydro-3,4:5,6-di-O-
isopropylidene-1-C-nitro-d-mannitol (1) has been
determined by Szarek et al. [1] (see Fig. 1). Their
result shows that the bond lengths of C(1)±C(2) (in
the oxirane ring) and C(2)±C(3) are unusually short.
The C(1)±C(2) bond distance is 1.441(4) AÊ , a value
which is unusually small compared to that of a typical
C±C bond length of oxirane of 1.462(3) AÊ [2±4]. The
observed C±C bond is also shorter than the compar-
able bond of oxirane derivatives such as 2-(¯uoro-
methyl)-2-((p-tolysul®nyl)methyl)oxirane (1.453 AÊ ) [5],
(1aa, 2b, 2ab, 4a, 7a, 7ab, 8b, 8aa)-octahydro-4,7-
epoxy-2,8-methanooxireno [h][3] benzoxepin-3(4H)-one
(1.462 AÊ ) [6] and 1,2:5,6-dianhydrogalactitol
(1.452(5) AÊ ) [7], and is similar to that estimated for
tetra¯uorooxirane [8] and that of methyl 3,4-anhydro-
1,6-bis-O-(p-tolylsulfonyl)-b-d-tagatofurano-
side (1.45(1) AÊ ) [9]. The C(2)±C(3) bond distance is
1.484(4) AÊ ; this is shorter than the corresponding C±
C bonds of certain isopropylidene substituents of
carbohydrates [10±13].
Saebo and Kavana [14] optimized three confor-
mers of epi¯uorohydrin at the HF/6-311Gpp level
and reported an average bond length of 1.453 AÊ
for the C±C bond in the oxirane ring, 1.402 AÊ for
the C±O bond, and 1.499 AÊ for the neighbouring
C±C bond.
In this paper, we applied two approaches in an
attempt to elucidate the unusually short bonds that
occur in oxirane and its derivatives.
Journal of Molecular Structure (Theochem) 537 (2001) 253±264
0166-1280/01/$ - see front matter q 2001 Elsevier Science B.V. All rights reserved.
PII: S0166-1280(00)00682-5
www.elsevier.nl/locate/theochem
q Dedicated to Professor SerafõÂn Fraga on the occasion of his 70th
birthday.
* Corresponding author.
E-mail address: [email protected] (V.H. Smith Jr.).1 Present address: Universidad AutoÂnoma del Edo de Morelos,
Centro de Investigaciones QuõÂmicas, Av. Universidad No. 1001
Col. Chamilpa. Cuernavaca, Mor. C. P. 62210, MexõÂco.

2. Results and discussions
The chemistry and structure of three-membered rings
(3MRs) including ethylene oxide have been the subject
of many theoretical and experimental studies [15±25].
Among these studies, there are two theoretical models,
which have proved to be satisfactory in explaining the
behaviour of the 3MR and oxirane in particular. The p-
complex-back-donation (p-cbd) model, ®rst proposed
by Dewar and Ford [26], and later expanded by others
[8,27±33], has been used to rationalize the substituent
effect on the 3MR. The second model, based on the
theory of atoms in molecules [34], was applied to
3MRs by Cremer and Kraka [19,20].
2.1. The p -complex-back-donation model
In the Molecular Orbital framework, one thinks of
MOs as the result of the mutual overlap of several
atomic orbitals. Since atomic and molecular orbitals
in principle are the same, as each represents an orbital
space that accommodates two electrons, it is possible
that MOs can also overlap with different AOs to form
new MOs. While s MOs are unable to overlap
ef®ciently with other AOs for steric reasons; the
same is not true for the p MOs. This idea is the essen-
tial one behind the p-cbd model. This model consid-
ers the 3MR as being composed of two parts: the
ole®n fragment (basal) and the acceptor (apical) (see
Fig. 2). The ole®n can use its ®lled p MO to form a
dative bond with the acceptor. The acceptor, on the
other hand, has ®lled p or d AOs that can be used to
form a reverse dative bond with the vacant (antibond-
ing) pp MO of the ole®n. Thus, the formation of the
3MR can be thought of as the interaction between two
opposed dative bonds resulting in an increase in the
overall bonding between the ole®n fragment and
the acceptor.
It has been shown [35] that the electronegativity of the
apical group will affect the degree of back donation. If
the apical group is very electronegative, due to the
difference in the energy between thepp MO of the ethy-
lene fragment and the ®lled AO of the apical fragment,
the back donation will be negligible and the predomi-
nant effect will be the electron transfer from thepMO to
the empty AO of the apical group. The resulting struc-
ture will behave as a simple p-complex. If the apical
group is weakly electronegative, the back donation will
M. HoÃÁ et al. / Journal of Molecular Structure (Theochem) 537 (2001) 253±264254
Fig. 1. 1,2-Anhydro-3,4:5,6-di-O-isopropylidene-1-C-nitro-d-mannitol (1).

be more signi®cant; the result will be the conventional
3MR.
Dewar and Ford [26] showed that it is arbitrary to
decide whether a compound is a p-complex or a 3MR,
and that the distinction is based entirely on the suitability
for the interpretation of the experimental data. One
should note, however, that, in examining thep-complex
character according to this model, the essential criterion
is not the electron density associated with the apical
group but rather the ratio of electron transfer in the
forward and reverse directions. Therefore, any change
in the geometry of the molecule should be the result of
both p-donation and the back-donation changes.
Hoffmann and coworkers [27±29] and Allen and
coworkers [8,30±32] extended this model using the
Walsh orbital scheme [36], which deduces the mole-
cular geometry based on the symmetries and energy
orders of the AOs, to explain the geometric behaviour
of cyclopropane and of some other heterocyclic
compounds, although they did not treat oxirane and
derivatives systematically. Analysis based on this
scheme classi®es the substituents into four groups:
s-withdrawing, s-donating, p-withdrawing, and p-
donating, depending on the symmetry and relative
energy levels of the interacting MOs belonging to
these substituents. One still has to determine which
MOs of the 3MR will be affected by the substituents.
The interaction of an electron-donor MO from the
substituent with a bonding MO of the 3MR will
increase the bonding characteristic, and, therefore,
will shorten the bond. On the other hand, if the
same substituent interacts with an antibonding orbital
of a particular bond, it will enhance the antibonding
characteristic of that bond, and, as a result, lengthen it.
The electron-withdrawing substituents also work in an
analogous fashion.
The determination of which MO will be affected by
the substituent is a non-trivial task. For large mole-
cules, there are many more MOs involved in the
Walsh orbital picture, and, furthermore, these MOs
are much closer together energetically and the extent
of mixing of orbitals having the same symmetry and
compatible energy is enhanced greatly [37]. As will
be shown below, the matter is further complicated by
the fact that calculations at different levels of theory
can reverse the order of close-lying MOs, particularly
the HOMO±LUMO levels.
An HF/STO-3G calculation by McAlduff and Houk
[38] for the oxirane molecule using an experimentally
determined structure shows that the ®rst four
HOMO ionization potentials are as follows:
2b1 , 4a1 , 2b2 . 1a2. Our result, also at the same
level of theory using the same geometry, shows that
the order is 4a1 , 2b1 , 1a2 . 2b2. This order is
consistent with that of Pople and coworkers [39]
M. HoÃÁ et al. / Journal of Molecular Structure (Theochem) 537 (2001) 253±264 255
Fig. 2. The MO model of the three-membered ring according to
Dewar and Ford [26].
Table 1
Ionization potential order of oxirane at the HF level using different
basis sets
Basis set Ionization potential order Reference
STO-3G 2b1 , 4a1 , 2b2 , 1a2 McAlduff and Houk [38]
STO-3G 4a1 , 2b1 , 2b2 , 1a2 This work
6-31Gp 4a1 , 2b1 , 2b2 , 1a2 Pople et al. [39]
6-31Gpp 4a1 , 2b1 , 2b2 , 1a2 This work
DZ 2b1� 4a1 , 2b2 , 1a2 Basch et al. [40]
Expt. 2b1 , 4a1 , 2b2 , 1a2 Basch et al. [40]
Expt. 2b1 , 4a1 , 2b2� 1a2 Bieri et al. [44],
Potts et al. [45]

HF/6-31Gp calculation and our HF/6-31Gpp calcula-
tion. An earlier Hartree±Fock calculation using a
Double Zeta basis set by Basch et al., [40] predicted
an almost degenerate MO: 2b1� 4a1 , 2b2 , 1a2 (see
Table 1). Basch et al. removed the degeneracy by
further modifying the calculation, yielding the MO
order 2b1 , 4a1 , 2b2 , 1a2.
Within the Hartree±Fock scheme, Koopmans'
theorem [41] is employed to approximate the ioniza-
tion energies of the molecules as the negative of the
SCF orbital eigenvalues. The differences between
Koopmans' theorem ionization potentials and the
experimentally derived vertical ionization potentials
are due to the frozen orbital approximation. This
approximation does not allow the ionized cation to
stabilize itself by reorganizing its MOs after losing
the electron. Since orbital relaxation is inherent in an
ionization process, the MO levels predicted by Koop-
mans' theorem may not be the same as those predicted
by experimentally derived photoelectron spectra.
Basch et al. also used photoelectron data to support
their assignment of the MO levels. In subsequent
investigations [38,42,43] on ethylene oxide and deri-
vatives, interpretations were made assuming that
Basch et al.'s assignments were correct. However,
further experimental data from Bieri et al. [44] and
Potts et al. [45] show a different ionization potential
order for oxirane: 2b1 , 4a1 , 1a2� 2b2.
To determine which MOs will interact with the
substituent is even more complicated. For cyclopro-
pane, Clark et al. [21] argue that the Walsh orbital
having the largest coef®cient is the one most affected
by the electronegativity of the substituent, an aspect
which was not discussed in other similar studies. Even
though symmetry and energy compatibility are the
aspects that determine the interactions between
MOs, the ®nal assignment should come from the
experimental data.
2.1.1. Oxirane and derivatives
The p-acceptor substituents, such as CH21 CN,
NO2, Li, BeH, and BH2, having low-lying MOs will
withdraw charge from the 2b2 HOMO of the oxirane
molecule. This MO is bonding with respect to C±C
and antibonding for C±O (see Fig. 3). Thus, removing
charge from this MO will shorten the C±O bond and
lengthen the C±C bond. The experimentally deter-
mined geometry of tetracyanoethylene oxide [46]
shows that the C±C bond of tetracyanoethylene
oxide is 0.024 AÊ longer than that of oxirane, and
that the C±O bonds are 0.012 AÊ shorter than the C±
O bonds of oxirane.
M. HoÃÁ et al. / Journal of Molecular Structure (Theochem) 537 (2001) 253±264256
Fig. 3. The HOMO 2b2 of oxirane.

In the case of the s-acceptor substituents, the main
interaction could come from the 1a2 orbital of oxirane.
This MO has a large coef®cient on the C atom and
show C±X bonding characteristics (see Fig. 4). The
2b2 MO also has C±X bonding characteristics, but
with a smaller coef®cient at the C atom (see Fig. 3).
The 1a2 MO has antibonding characteristics for the
C±C bond; thus, s-acceptor substituents, such as F,
should shorten the C±C bond. The experimentally
determined structure of cis-1,2 di¯uorooxirane [47]
exhibits a shortening of the C±C bond by 0.021 AÊ
compared to that of oxirane. The shortening of the
C±O bonds by 0.027 AÊ in cis-1,2 di¯uorooxirane is
probably due to the contribution of the C±O antibond-
ing 2b2 MO mentioned above. Other s-acceptor
substituents are Cl, OH, and NH3. According to
Clark et al. [21] the CH3 group can also be considered
as a s-acceptor group. A study of the crystal structure
of methyloxirane [48] indicates that the CH3 substitu-
ent indeed shortens both vicinal C±O and C±C bonds
of the oxirane ring in the same manner as ¯uorine
does. By interpreting photoelectron spectra, McAlduff
and Houk [38] also con®rmed that the interaction
between an oxirane ring and alkyl substituents arises
mainly from the 1a2 and 2b2 MOs.
Strong p-donor substituents can transfer charge to
the low-lying 4b1 LUMO of oxirane. This MO has
antibonding characteristics for both the C±O and
C±C bonds (see Fig. 5). Thus p-donor substituents
such as CH22 and O2 would lengthen all three bonds
in the 3MR. s-Donor substituents (e.g. Li) can also
interact with the 4b1 MO of oxirane. Obviously, those
substituents have strong C±X bond characteristics,
and can interact in a manner similar to those of the
s-acceptor substituents. The situation becomes more
complicated, and requires more calculations and
experimental data, if one wishes to assign the proper
contributions of particular MOs. Currently, there have
been few investigations about the effect of s-donor
substituents on 3MR system.
M. HoÃÁ et al. / Journal of Molecular Structure (Theochem) 537 (2001) 253±264 257
Fig. 4. The 1a2 Molecular Orbital of oxirane.
Fig. 5. The LUMO 4b1 of oxirane.

2.1.2. 1,2-Anhydro-3,4:5,6-di-O-isopropylidene-1-C-
nitro-d-mannitol (1)
Following the Walsh orbital picture discussed
above, one can look at (1) as an oxirane derivative
having two substituent groups: the NO2 group and one
comprised of two O-isopropylidene rings. Both
groups will affect the geometry of the oxirane ring
in (1) in a different way. NO2, being a p-acceptor
substituent, will shorten the vicinal C±O bond and
lengthen the C±C bond. The fragment containing
the two isopropylidene rings is a s-acceptor substitu-
ent and thus would shorten both vicinal C±C and C±O
bonds. The combined effect is that the C±O bond
adjacent to the NO2 group in (1) is shortened by
0.048 AÊ compared to that in oxirane. The C±O bond
adjacent to the di-O-isopropylidene fragment on the
other hand is lengthened by 0.017 AÊ , an observation
which contradicts the predictions made above. A possi-
ble explanation is that the interaction of the 2b2 MO
which is responsible for the shortening of this C±O
bond could be diminished by the perpendicular orien-
tation of the 3,4-O-isopropylidene ring. Furthermore,
CNDO calculations by Furman and Meleshevich [49]
on the effect of an NO2 group on oxirane show weak-
ening (therefore lengthening) of this C±O bond.
2.2. The Laplacian of the charge density model
The p-cbd model for oxirane can be viewed from a
different perspective. While the Walsh orbital scheme
is convenient computationally and visually, the theory
of atoms in molecules [34] provides a more quantita-
tive approach for the oxirane ring. From the p-cbd
model, one can see that, for the dative donation,
there is a charge build-up along the C2 axis of oxirane.
Therefore, the resulting bond path would be from the
apical oxygen to the midpoint of the basal ethylene.
According to the catastrophe theory as applied to the
topological approach (see, for example, chapter 4 of
Ref. [34]), such a structure is known as the T-structure
and is topologically unstable. Any in®nitesimal
change in the symmetry of the molecule will alter
its geometry signi®cantly.
For the back donation, the charge build-up from the
occupied p orbital of oxygen to the empty antibonding
MO of ethylene will result in two convex bond paths
from oxygen to the carbon atoms. It is now possible to
determine the predominant donation based solely on
the molecular path of the molecule. If the apical group
is more electronegative than the basal group, a
concave molecular path will occur. A convex mole-
cular path exhibits a strong dative donation in which
the apical group is less electronegative than the basal
group. This approach is a quanti®cation of the intui-
tive orbital picture proposed by Dewar and Ford.
Cremer and Kraka [19,20] studied a series of
heterocyclic 3MRs to support this model. From Fig.
6 one can see the changes of the molecular paths with
respect to the p-complex character of the molecules.
Beryllocyclopropyne (BeC2, structure n) possesses
a T-structure, since the main charge build-up comes
from the 2s orbital of the Be atom to the s bonding
orbital of the basal fragment [50]. Due to its extreme
electronegativity, the ¯uoroethyl cation (structure 1)
shows a very concave molecular path, which is almost
a T-structure. Protonation of oxygen in oxirane makes
it even more electronegative, as can be seen by the
molecular path of the protonated oxirane (k) being
M. HoÃÁ et al. / Journal of Molecular Structure (Theochem) 537 (2001) 253±264258
Fig. 6. Changes in molecular paths with respect to the p-complex
character of the molecule (after Cremer and Kraka [19]).

more concave than that of the neutral oxirane mole-
cule (j). The importance of the back donation is
demonstrated clearly in oxirane, cyclopropane (i),and, particularly, beryllocyclopropane (m) where the
back donation has outweighed the dative donation due
to the electropositivity of the beryllium atom.
The effect of the substituent group on the oxirane
ring can be investigated using the Laplacian of the
charge density. According to topological de®nitions,
the integration of the Laplacian of the charge density
over the subspace V , de®ned by the zero-¯ux surface
of r (r), vanishes:ZV7 2r�~r� d~r �
I~7r�~r�´~n dS � 0: �1�
Any changes in the Laplacian of r are accompanied
by other changes within the subspace V boundary in
order to satisfy Eq. (1). The physical interpretation of
this equation can be explained by the example of an
A±X molecule. If X is more electronegative than A,
there will be a shift of the location of the bond critical
point toward A due to the fact that the valence sphere
of A is being pulled toward X. This deformation
causes a decrease in the charge concentration of A
in the direction of X; thus, Eq. (1) indicates that
there must be an increase in the charge concentration
at A in the opposite direction.
An increase of the charge concentration in the inter-
nuclear region shields the nuclei from repelling each
other, and, hence, a shortened bond results. Similarly,
a decrease in the charge concentration in this region
will increase the nuclear±nuclear repulsion, causing a
bond elongation.
The Laplacian of the charge density indicates the
locally electron-depleted and locally electron-concen-
trated areas on the valence sphere of the substituents.
The effect of these electronic holes and electronic
lumps allows one to distinguish the substituents as
s-attractor, s-repeller, p-attractor, or p-repeller.
The s-attractor substituents, which transmit their
effect through the bond path, withdraw electrons
from the 3MR, and thereby create an electronic hole
in the adjacent carbon atom. According to Eq. (1)
there will be electronic lumps toward the direction
of the oxygen and the distal carbon atoms. These
electronic lumps will deshield the vicinal carbon
from the oxygen atom and the distal carbon. Conse-
quently, shortening of the vicinal bond occurs. The
electronic lumps also become staggered in order to
avoid each other, thus making the distal bond longer.
F, OH and NH2 are substituents that have s-attracting
ability, even though they also have stronger p-repel-
lent effect.
In the same manner, a s-repeller such as Li and
BeH donates electrons to the adjacent carbon of the
3MR, thereby forming an electronic lump between
itself and this carbon atom. Elongation of the vicinal
M. HoÃÁ et al. / Journal of Molecular Structure (Theochem) 537 (2001) 253±264 259
Fig. 7. Model derivatives of (1).

bonds occurs, due to the electron holes, toward the
oxygen and the distal carbon atoms. The distal C±O
bond contracts because of the reduction of the
nuclear±nuclear repulsion.
The p-attractors possess electronic holes in their
valence spheres and the effects are transmitted
through space rather than through the bond paths.
CN, NO2, and phenyl act as p-attractors and cause a
decrease of the charge density in the vicinal bond
region, and, hence, shorten the distal bond and stretch
the vicinal bonds.
Strong p-repellers such as F, OH and NH2 have the
reverse effect on the 3MR, namely contraction of the
vicinal bonds and expanding of the distal one.
2.2.1. 1,2-Anhydro-3,4:5,6-di-O-isopropylidene-1-C-
nitro-d-mannitol (1)
To study the effect of the substituents on compound
(1), its structure and six more derivatives were opti-
mized using the AM1 method [51]. The starting
geometry of (1) was taken from the X-ray structure
[1]. In order to study the effect of the substituents, the
NO2 and isopropylidene groups were also replaced or
removed, and the structures were optimized indepen-
dently (Fig. 7). Missing experimental parameters were
replaced mainly with data from Pople et al. [52].
The results from the AM1 optimized geometry
show good agreement with those from the crystal
structural determination (see Tables 2±5). For the
most part, the optimized bond lengths agree within
2% with the experimental ones. The discrepancies
of the C(1)±C(2) and C(2)±C(3) bond lengths are
reasonable within the AM1 approximation. Bond
lengths involving the methyl groups, for example,
C(7)±C(9) and C(10)±C(11), give large deviations.
However, the similar type of bond in C(7)±C(8) and
C(10)±C(12) shows better agreement. In general, the
C±O bond lengths are reproduced satisfactorily.
The bond angles show an average deviation of
2.5%. The largest deviation comes from the angle
M. HoÃÁ et al. / Journal of Molecular Structure (Theochem) 537 (2001) 253±264260
Table 2
Bond lengths of AM1 optimized structure of (1)
Bond Distance (AÊ ) Bond Distance (AÊ ) Bond Distance (AÊ )
O(1)±C(1) 1.425 O(5)±C(5) 1.430 C(4)±C(5) 1.533
O(1)±C(2) 1.442 O(5)±C(10) 1.438 C(5)±C(6) 1.531
O(1N)±N 1.199 O(6)±C(6) 1.428 C(7)±C(8) 1.522
O(2N)±N 1.198 O(6)±C(10) 1.431 C(7)±C(9) 1.522
O(3)±C(3) 1.431 N±1C(1) 1.515 C(10)±C(11) 1.522
O(3)±C(7) 1.435 C(1)±C(2) 1.498 C(10)±C(12) 1.520
O(4)±C(4) 1.430 C(2)±C(3) 1.507
O(4)±C(7) 1.434 C(3)±C(4) 1.539
Table 3
Bond angles of AM1 optimized structure of (1)
Bonds Angle (8) Bonds Angle (8) Bonds Angle (8)
C(1)±O(1)±C(2) 62.968 O(1)±C(2)±C(3) 116.053 O(3)±C(7)±O(4) 105.788
C(3)±O(3)±C(7) 110.957 C(1)±C(2)±C(3) 120.954 O(3)±C(7)±C(8) 109.153
C(4)±O(4)±C(7) 110.888 O(3)±C(3)±C(2) 110.148 O(3)±C(7)±C(9) 109.853
C(5)±O(5)±C(10) 110.933 O(3)±C(3)±C(4) 105.305 O(4)±C(7)±C(8) 108.813
C(6)±O(6)±C(10) 110.431 C(2)±C(3)±C(4) 112.323 O(4)±C(7)±C(9) 110.700
O(1N)±N±O(2N) 123.232 O(4)±C(4)±C(3) 104.988 C(8)±C(7)±C(9) 112.311
O(1N)±N±C(1) 117.175 O(4)±C(4)±C(5) 109.150 O(5)±C(10)±O(6) 105.843
O(2N)±N±C(1) 119.563 C(3)±C(4)±C(5) 112.446 O(5)±C(10)±C(11) 109.332
O(1)±C(1)±N 116.766 O(5)±C(5)±C(4) 108.479 O(5)±C(10)±C(12) 109.384
O(1)±C(1)±C(2) 59.084 O(5)±C(5)±C(6) 105.141 O(6)±C(10)±C(11) 108.998
N±C(1)±C(2) 120.266 C(4)±C(5)±C(6) 112.561 O(6)±C(10)±C(12) 110.630
O(1)±C(2)±C(1) 57.948 O(6)±C(6)±C(5) 105.124 C(11)±C(10)±C(12) 112.434

C(6)±O(6)±C(10) of the isopropylidene ring; surpris-
ingly, the angles C(3)±O(3)±C(7) and C(5)±O(5)±
C(7) are accurately reproduced. These results indicate
a distortion of one side of each of the isopropylidene
rings. The dihedral angles show the largest deviations.
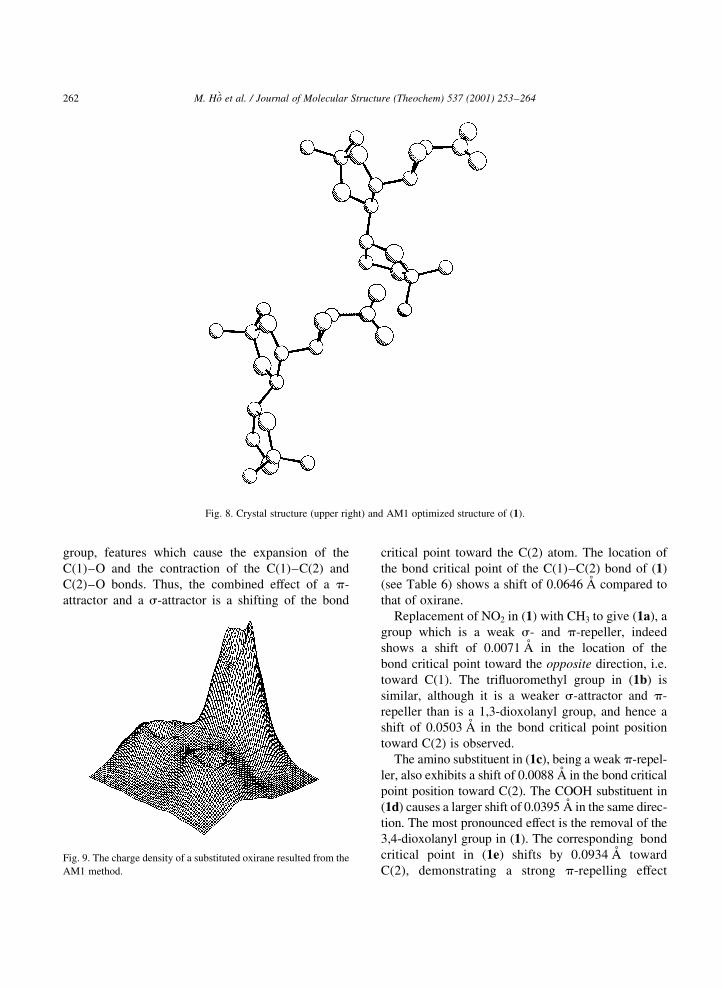
However, from Fig. 8 one can see that the optimized
structure of (1) is less-closely packed than is the crys-
tal one. This deviation could come from the fact that
(1) is optimized in the gas phase, an approach which
involves a structure, which has none of the intermo-
lecular forces present in a crystal structure.
The topological properties of these molecules were
obtained using the semi-empirical wavefunctions
produced with the AM1 method. The bond critical
points of the oxirane moiety of (1) and derivatives
were used to analyse the effects of the NO2 group
and the fragment containing the two isopropylidene
groups. It should be noted that, in this method, energy
contributions arising from core electrons and their
interactions with valence electrons are represented
only in a parameterized form. Consequently, no expli-
cit charge distribution associated with these electrons
can be given. The AM1 wavefunctions, therefore,
contain only valence orbitals resulting in profound
consequences for their topological analysis (see Fig.
9). We have addressed this question by studying the
topological properties of various valence type wave-
functions [53]. We found that there are problems for
only a few systems, mostly of a p-bonded nature,
having short bond lengths and large differences in
atomic charges. In these systems, the bond critical
points could not be located. The bond critical point
by de®nition is the minimum in the charge density
along the internuclear path. The short bond lengths
plus the high atomic charges of the atoms suppress
these minima. This feature is not due to the inability of
AM1 to calculate the charge density in the bonding
region of these molecules, or to the direct contribution
of the core orbitals to the charge density in this region.
In fact, we have shown [53] that topological proper-
ties of AM1 wavefunctions are consistent with those
of the Hartree±Fock calculations at the split-valence
(6-31G) basis level. Furthermore, the missing core
orbitals can be replaced with those from Near
Hartree±Fock atomic wave functions [54] of the
corresponding atoms. For the molecules studied
here, this implementation was not necessary.
NO2 is a p-attractor group, a feature which shortens
the C(2)±O bond and lengthens the C(1)±O and
C(1)±C(2) bonds. A 1,3-dioxolanyl group can be
considered both as a s-attractor and a p-repeller
M. HoÃÁ et al. / Journal of Molecular Structure (Theochem) 537 (2001) 253±264 261
Table 4
Difference in bond lengths (%) between optimized structure of (1)
and crystal structure
Bond Bond Bond
O(1)±C(1) 2.66 O(5)±C(5) 21.22 C(4)±C(5) 1.93
O(1)±C(2) 20.74 O(5)±C(10) 1.30 C(5)±C(6) 0.54
O(1N)±N 21.42 O(6)±C(6) 22.07 C(7)±C(8) 1.37
O(2N)±N 2.00 O(6)±C(10) 20.32 C(7)±C(9) 2.05
O(3)±C(3) 20.23 N±C(1) 1.82 C(10)±C(11) 2.83
O(3)±C(7) 0.86 C(1)±C(2) 3.92 C(10)±C(12) 0.26
O(4)±C(4) 0.41 C(2)±C(3) 1.58
O(4)±C(7) 0.84 C(3)±C(4) 20.20
Table 5
Difference in bond angles (%) between optimized structure of (1) and crystal structure
Bonds Bonds Bonds
C(1)±O(1)±C(2) 3.4 O(1)±C(2)±C(3) 21.3 O(3)±C(7)±O(4) 20.1
C(3)±O(3)±C(7) 1.3 C(1)±C(2)±C(3) 21.6 O(3)±C(7)±C(8) 0.4
C(4)±O(4)±C(7) 3.3 O(3)±C(3)±C(2) 20.5 O(3)±C(7)±C(9) 20.1
C(5)±O(5)±C(10) 0.7 O(3)±C(3)±C(4) 1.5 O(4)±C(7)±C(8) 1.0
C(6)±O(6)±C(10) 5.0 C(2)±C(3)±C(4) 20.3 O(4)±C(7)±C(9) 0.4
O(1N)±N±O(2N) 22.4 O(4)±C(4)±C(3) 2.7 C(8)±C(7)±C(9) 1.3
O(1N)±N±C(1) 1.0 O(4)±C(4)±C(5) 1.3 O(5)±C(10)±O(6) 0.4
O(2N)±N±C(1) 1.5 C(3)±C(4)±C(5) 21.7 O(5)±C(10)±C(11) 20.8
O(1)±C(1)±N 1.6 O(5)±C(5)±C(4) 1.1 O(5)±C(10)±C(12) 0.9
O(1)±C(1)±C(2) 24.4 O(5)±C(5)±C(6) 0.7 O(6)±C(10)±C(11) 1.1
N±C(1)±C(2) 1.0 C(4)±C(5)±C(6) 0.0 O(6)±C(10)±C(12) 20.2
O(1)±C(2)±C(1) 1.1 O(6)±C(6)±C(5) 2.9 C(11)±C(10)±C(12) 21.3
group, features which cause the expansion of the
C(1)±O and the contraction of the C(1)±C(2) and
C(2)±O bonds. Thus, the combined effect of a p-
attractor and a s-attractor is a shifting of the bond
critical point toward the C(2) atom. The location of
the bond critical point of the C(1)±C(2) bond of (1)
(see Table 6) shows a shift of 0.0646 AÊ compared to
that of oxirane.
Replacement of NO2 in (1) with CH3 to give (1a), a
group which is a weak s- and p-repeller, indeed
shows a shift of 0.0071 AÊ in the location of the
bond critical point toward the opposite direction, i.e.
toward C(1). The tri¯uoromethyl group in (1b) is
similar, although it is a weaker s-attractor and p-
repeller than is a 1,3-dioxolanyl group, and hence a
shift of 0.0503 AÊ in the bond critical point position
toward C(2) is observed.
The amino substituent in (1c), being a weak p-repel-
ler, also exhibits a shift of 0.0088 AÊ in the bond critical
point position toward C(2). The COOH substituent in
(1d) causes a larger shift of 0.0395 AÊ in the same direc-
tion. The most pronounced effect is the removal of the
3,4-dioxolanyl group in (1). The corresponding bond
critical point in (1e) shifts by 0.0934 AÊ toward
C(2), demonstrating a strong p-repelling effect
M. HoÃÁ et al. / Journal of Molecular Structure (Theochem) 537 (2001) 253±264262
Fig. 8. Crystal structure (upper right) and AM1 optimized structure of (1).
Fig. 9. The charge density of a substituted oxirane resulted from the
AM1 method.

whereby the density is pushed towards the C(1)
and O(1) atoms by the electron lumps in the
1,3-dioxolane ring. Removing the second O-
isopropylidene group to give (1f) shows a similar
effect but to a lesser extent (0.0597 AÊ ). Similar
behaviour is observed for the C(2)±C(3) bond,
again showing the signi®cant effect of
the fragment containing the two isopropylidene
rings.
Comparing the topological changes in the C(1)±O,
C(2)±O, and C±C bonds one can see that the NO2
group has more effect on the C(1)±O and C(2)±O
bonds. However, changes in the bond critical point
of C(1)±O and C(2)±O are not as signi®cant. The
NO2 group in¯uences the 72r (r) of C(1)±O and
C(1)±C(2) mostly, and changes in the C(2)±O bond
come mainly from the immediate O-isopropylidene
group.
3. Conclusions
Using both the p-cbd model with the Walsh orbitals
scheme and the Laplacian of the charge density, the
structure of (1) has been analyzed to explain the beha-
viour of the oxirane ring and the C(2)±C(3) bond. The
®rst model requires little computation and relies on
experimental data. The theory of atoms in molecules
also allows one to use the inexpensive AM1 method.
Both models come to the same conclusion. The short-
enings of the C(1)±C(2) and C(2)±C(3) bonds arose
mainly from the two O-isopropylidene groups,
whereas the NO2 is responsible for the contraction
of the C(1)±O bond. Neither the p-cbd model nor
the Laplacian of the charge density model offers a
satisfactory explanation for the elongation of the
C(2)±O bond.
In larger molecules, the Walsh picture becomes
increasingly complicated and thus more MO inter-
actions should be considered. This aspect, however,
makes the model lose its attractive features of
being simple and convenient. The Laplacian
model on the other hand does not really differenti-
ate between various 3MR compounds in claiming
that the substituents will in¯uence all 3MRs in the
same fashion. From the molecular orbital point of
view, this claim is not the case for cyclopropane
and oxirane. The experimental structures of simple
derivatives of these two compounds do not support
the model completely. A systematic study of the
Laplacian and p-cbd together with experimentally
determined structures and spectra should minimize
these defects.
Acknowledgements
This research was supported in part by the Natural
Sciences and Engineering Research Council of
Canada (NSERCC). We would like to thank Dr Hart-
mut L. Schmider for valuable discussions.
M. HoÃÁ et al. / Journal of Molecular Structure (Theochem) 537 (2001) 253±264 263
Table 6
Topological properties of (1) and derivatives (from top to bottom:
(1), (1a)±(1f)) from AM1 wavefunctions
Bond Location of r b (AÊ ) r b (ea023) 72r b (ea0
25)
C(1)±C(2) 0.8006 0.2239 20.4703
C(1)±O 0.5439 0.2637 20.3911
C(2)±O 0.5457 0.2516 20.3376
C(2)±C(3) 0.7961 0.2397 20.5787
C(1)±C(2) 0.7755 0.2289 20.5024
C(1)±O 0.5474 0.2566 20.3429
C(2)±O 0.5421 0.2528 20.3407
C(2)±C(3) 0.8008 0.2400 20.5759
C(1)±C(2) 0.7448 0.2274 20.5028
C(1)±O 0.5564 0.2412 20.2522
C(2)±O 0.5457 0.2576 20.3627
C(2)±C(3) 0.7771 0.2408 20.5856
C(1)±C(2) 0.7289 0.2302 20.5090
C(1)±O 0.5435 0.2530 20.3402
C(2)±O 0.5462 0.2571 20.3516
C(2)±C(3) 0.7693 0.2409 20.5888
C(1)±C(2) 0.7862 0.2298 20.5034
C(1)±O 0.5449 0.2638 20.3871
C(2)±O 0.5466 0.2537 20.3412
C(2)±C(3) 0.7929 0.2400 20.5812
C(1)±C(2) 0.8294 0.2215 20.4403
C(1)±O 0.5456 0.2641 20.3845
C(2)±O 0.5420 0.2487 20.3277
C(2)±C(3) 0.8090 0.2453 20.5981
C(1)±C(2) 0.7975 0.2243 20.4735
C(1)±O 0.5428 0.2637 20.3929
C(2)±O 0.5466 0.2514 20.3320
C(2)±C(3) 0.8075 0.2389 20.5646

References
[1] W.A. Szarek, G.W. Hay, R.K. Sood, K. Trouton, S. Fortier,
Can. J. Chem. 66 (1988) 1600.
[2] G.L. Cunningham, A.W. Boyd, R.J. Meyers, W.D. Gwinn,
W.I. Le Van, J. Chem. Phys. 19 (1951) 676.
[3] T.E. Turner, J.A. Howe, J. Chem. Phys. 24 (1956) 924.
[4] C. Hirose, Bull. Chem. Soc. Jpn 47 (1974) 1311.
[5] G. Fabrizi, W. Fedeli, Acta Crystallogr., Sect. C 48 (1991)
1131.
[6] T.T. Stevenson, M.G. Essig, F. Sha®zadeh, L.H. Jensen, R.E.
Stenkamp, Carbohydr. Res. 118 (1983) 261.
[7] M. Csugler, K. Simon, L. Institoris, I. Vidra, I. CsoÈregh,
Carbohydr. Res. 108 (1982) 173.
[8] C.A. Deakyne, J.P. Cravero, W.S. Hobson, J. Phys. Chem. 88
(1984) 5975.
[9] R.D. Guthrie, I.D. Jenkins, R. Yamasaki, B.W. Skelton, A.H.
White, J. Chem. Soc., Perkin Trans. 1 (1981) 2328.
[10] A. Ducruix, C. Pascard-Billy, Acta Crystallogr., Sect. B 33
(1977) 1384.
[11] A. Aubry, J. Protas, B. Duchaussoy, P.D. Cesare, B. Gross,
Acta Crystollogr., Sect. B 36 (1980) 187.
[12] V.J. James, J.D. Stevens, Carbohydr. Res. 82 (1980) 167.
[13] M. Argentini, R. Weinreich, R. Oberti, L. Ugaretti, J. Fluorine
Chem. 32 (1986) 239.
[14] S. Saebo, K. Kavana, Theochemistry 235 (1991) 447.
[15] A. de Meijere, Angew. Chem. 91 (1979) 867.
[16] R. Gleiter, Top. Curr. Chem. 86 (1979) 197.
[17] W.A. Lathan, L. Radom, P.C. Hariharan, W.J. Hehre, J.A.
Pople, Top. Curr. Chem. 40 (1973) 1.
[18] M.D. Newton, in: H.F. Schaefer (Ed.), Modern Theoretical
Chemistry, vol. 4, Plenum Press, New York, 1977, p. 223.
[19] D. Cremer, E. Kraka, J. Am. Chem. Soc. 107 (1985) 3800.
[20] D. Cremer, E. Kraka, J. Am. Chem. Soc. 107 (1985) 3811.
[21] T. Clark, G.W. Spitznagel, R. Klose, P.v.R. Schleyer, J. Am.
Chem. Soc. 106 (1984) 4412.
[22] E. Lewars, Chem. Rev. 83 (1983) 519.
[23] E. Lewars, Theochemistry 391 (1997) 39.
[24] G. Vacek, J.M. Galbraith, Y. Yamaguchi, H.F. Schaefer, R.H.
Nobes, A.P. Scott, L. Radom, J. Phys. Chem. 98 (1994) 8660.
[25] J.E. Fowler, J.M. Galbraith, G. Vacek, H.F. Schaefer, J. Am.
Chem. Soc. 116 (1994) 9311.
[26] M.J.S. Dewar, G.P. Ford, J. Am. Chem. Soc. 101 (1979) 783.
[27] R. Hoffmann, H. Fujimoto, J.R. Swenson, C.-C. Wan, J. Am.
Chem. Soc. 95 (1973) 7644.
[28] R. Hoffmann, Tetrahedron Lett. 33 (1970) 2907.
[29] R. Hoffmann, J. Am. Chem. Soc. 90 (1968) 1475.
[30] C. Liang, L.C. Allen, J. Am. Chem. Soc. 113 (1991) 1878.
[31] C.A. Deakyne, L.C. Allen, N.C. Craig, J. Am. Chem. Soc. 99
(1977) 3895.
[32] C.A. Deakyne, L.C. Allen, V.W. Laurie, J. Am. Chem. Soc. 99
(1977) 1343.
[33] M.-M. Rohmer, B. Roos, J. Am. Chem. Soc. 97 (1975) 2025.
[34] R.F.W. Bader, Atoms in Molecules: a Quantum Theory,
Oxford University Press, London, 1990.
[35] M.J.S. Dewar, Bull. Soc. Chim. Fr. (1951) C71.
[36] A.D. Walsh, Nature (London) 159 (1947) 165.
[37] B.J. Gimarc, Acc. Chem. Res. 7 (1974) 392.
[38] E.J. McAlduff, K.N. Houk, Can. J. Chem. 55 (1977) 318.
[39] W.A. Lathan, L. Radom, P.C. Hariharan, J.A. Pople, Fortschr.
Chem. Forsch. 40 (1973) 1.
[40] H. Basch, M.B. Brown, N.A. Kuebler, C. Baker, D.W. Turner,
J. Chem. Phys. 51 (1969) 52.
[41] T. Koopmans, Physica 1 (1934) 104.
[42] P.D. Mollere, K.N. Houk, J. Am. Chem. Soc. 99 (1976) 3226.
[43] A. Schweig, W. Thiel, Chem. Phys. Lett. 21 (1973) 541.
[44] G. Bieri, L. AÊ sbrink, W. von Niessen, J. Electron Spectrosc.
27 (1982) 129.
[45] A.W. Potts, T.A. Williams, W.C. Price, Faraday Discuss.
Chem. Soc. 54 (1972) 104.
[46] D.A. Mathews, J. Swanson, M.H. Mueller, G.D. Stucky, J.
Am. Chem. Soc. 93 (1971) 5945.
[47] C.W. Gillies, J. Mol. Spectrosc. 71 (1978) 85.
[48] R.A. Creswell, R.H. Schwendeman, J. Mol. Spectrosc. 64
(1977) 295.
[49] E.G. Furman, A.P. Meleshevich, Theor. Exp. Chem. 14 (1978)
84.
[50] W. Koch, G. Frenking, J. Gauss, D. Kremer, A. Sawayn,
P.v.R. Schleyer, J. Am. Chem. Soc. 108 (1986) 5732.
[51] M.J.S. Dewar, E. Zoebisch, E.F. Healy, J.J.P. Stewart, J. Am.
Chem. Soc. 107 (1985) 3902.
[52] W.J. Hehre, L. Radom, P.v.R. Schleyer, J.A. Pople, Ab initio
Molecular Orbital Theory, Wiley-Interscience, New York,
1986.
[53] M. HoÃÁ, H. Schmider, K.E. Edgecombe, V.H. Smith Jr., Int. J.
Quantum Chem. S28 (1994) 215.
[54] E. Clementi, C. Roetti, Atomic Data and Nuclear Data Tables
14 (1974) 177.
M. HoÃÁ et al. / Journal of Molecular Structure (Theochem) 537 (2001) 253±264264