novel mechanism of brain soluble epoxide hydrolase
TRANSCRIPT

©2005 FASEB
The FASEB Journal express article 10.1096/fj.04-3128fje. Published online January 19, 2005.
Novel mechanism of brain soluble epoxide hydrolase-mediated blood pressure regulation in the spontaneously hypertensive rat Kathleen W. Sellers,* Chengwen Sun,* Carlos Diez-Freire,* Hidefumi Waki,† Christophe Morisseau,‡ John R. Falck,§ Bruce D. Hammock,‡ Julian F. Paton,† and Mohan K. Raizada* *Department of Physiology and Functional Genomics, University of Florida College of Medicine and McKnight Brain Institute, Gainesville, Florida; †Department of Physiology, School of Medical Sciences, University of Bristol, Bristol, United Kingdom; ‡Department of Entomology and University of California–Davis Cancer Center, Davis, CA; §Departments of Biochemistry and Pharmacology, University of Texas Southwestern Medical Center, Dallas, Texas
Corresponding author: Mohan K. Raizada, Department of Physiology and Functional Genomics College of Medicine University of Florida McKnight Brain Institute Gainesville, FL 32610. E-mail: [email protected]
ABSTRACT
The role of soluble epoxide hydrolase (sEH) in the central control of blood pressure (BP) has not been elucidated in spite of peripheral sEH overexpression being linked to hypertension. Thus, our objective was to investigate the involvement of brain sEH in BP control. sEH expression in the hypothalamus and brain stem, two cardioregulatory brain areas, was increased in the spontaneously hypertensive rat (SHR) compared to the Wistar Kyoto (WKY) rat. Inhibition of the enzyme by intracerebroventricular (icv) delivery of AUDA further increased both BP and heart rate (HR) by 32 ± 6 mmHg and 54 ± 10 bpm, respectively, (P<0.05) in the SHR. Analysis of waveform telemetry data revealed a decrease in spontaneous baroreceptor reflex gain following sEH inhibition, indicating the sustained increase in BP may be due to a decrease in baroreceptor reflex function. The hypertensive effect of sEH inhibition is likely a result of an increase in epoxyeicosatrienoic acid (EET)-mediated generation of ROS. This view is supported by the following: 1) Inhibition of EET formation attenuates AUDA-induced increase in BP; 2) delivery of an EET agonist increases BP and HR in the WKY rat, and 3) inhibition of NAD(P)H oxidase by gp91ds-tat prevents AUDA-induced increases in BP and HR. Finally, electrophysiological studies demonstrate that AUDA increased neuronal firing rate exclusively in the SHR, an effect completely abolished by gp91ds-tat. These observations suggest that EETs and sEH inhibition are involved in increasing BP in the SHR. We suggest that an increased expression of sEH is a futile central nervous system response in protection against hypertension.
Key words: brain stem • baroreceptor reflex • central nervous system
Page 1 of 17(page number not for citation purposes)

eripheral soluble epoxide hydrolase (sEH) is a key alpha/beta hydrolase fold family enzyme that has been implicated in the control of cardiovascular functions by regulating the tissue levels of epoxyeicosatrienoic acids (EETs), dihydroxyeicosatrienoic acids
(DHETs), and other epoxy lipids (1–5). It is generally believed that EETs are vasodilatory, while their metabolism by sEH converts them into less potent vasodilatory metabolites (6–8). Thus, by regulating the balance between EET and DHET levels, sEH assists in maintaining normal vascular tone in the periphery. The significance of sEH in normal cardiovascular physiology is supported by evidence that the expression of this enzyme is significantly increased in both genetic and experimental rat models of hypertension (2, 3). Furthermore, inhibition of its activity by pharmacological agents decreased high BP and reverses hypertension (2). Additionally, disruption of the sEH gene (EPHX2) is associated with significantly lower systolic BP in the EPHX2-null mice compared to wild-type mice (1). In spite of this correlation of an increased peripheral sEH expression to hypertension, there are numerous reports that do not support this view. They indicate that EPHX2 gene polymorphisms may be more prevalent in hypertension but are not linked to systemic control of high BP (9, 10).
Our research group has been interested in elucidating the role of the central nervous system in the control of cardiovascular function. We had hypothesized that dysregulated expression of one or more genes in the cardioregulatory brain nuclei may be key in development of hypertension. Using microarray technology, the gene that showed the most profound increase in its expression was sEH in the hypothalamus and brain stem of the spontaneously hypertensive rat (SHR) vs. the Wistar Kyoto (WKY) rat brain. Interestingly, our initial observations indicated that inhibition of central sEH causes an increase in BP. This was contrary to the peripheral enzyme where sEH inhibition attenuated high BP. This led us to propose the following hypothesis on the role of central sEH in the control of cardiovascular function.
We hypothesized that fatty acid epoxides such as EETs in the brain are involved in an increase in BP via both a depression of the baroreceptor reflex and central neural pathways regulating the set-point of arterial pressure that involve reactive oxygen species (ROS). The role of ROS is derived from recent evidence linking it both to cytochrome P450 member 2C9 in the vasculature and to the central regulation of hypertension (11, 12). Such an increase in BP is contrary to the role of EETs in the peripheral vascular bed. This hypothesis would suggest that inhibition of sEH would increase EETs, which would lead to increased production of ROS that mediates sympatho-regulation of high BP. Thus, the present study was undertaken to support or refute this hypothesis.
P
sEHArachidonic Acid EETs DHETs
ROS Baroreceptor reflex BP
Page 2 of 17(page number not for citation purposes)

MATERIALS AND METHODS
Animals
Male SHR and WKY rats, weighing 220-250 g at the beginning of the study, were ordered from Charles River Laboratories (Wilmington, MA). Rats were housed individually and kept on 12:12 light-dark cycle in a climate-controlled room. Rat chow (Harlan Tekland, Madison, WI) and water were provided ad libitum. All animal protocols were approved by the Institutional Animal Care and Use Committee of the University of Florida.
ICV cannulation
Rats were anesthetized with inhaled isofluorane (3%) in all surgical procedures. A stereotaxic frame was used to position a 22-gauge guide cannula (PlasticsOne, Roanoke VA) into the right lateral ventricle (AP: 1.3 mm, ML: 1.5 mm, 4.5 mm below skull) of male SHR and WKY rats. The cannula was secured to the skull with dental resin assisted by 3 3/32 screws secured in the skull. Appropriate drugs (1-2 µl) were injected into the right lateral ventricle of unrestrained rats using a 28-gauge internal cannula, 4.5 mm. Rats were allowed to recover for 1 wk following all surgeries.
Abdominal aorta cannulation
Radiotelemetric pressure transducers (Data Sciences International, Arden Hills, MN) consisting of a fluid-filled catheter attached to a PA-C40 transmitter were implanted into the abdominal aorta. Before implantation, the aorta was clamped proximally and the catheter inserted and secured with medical adhesive. During experiments, blood pressure (BP) and heart rate (HR) were recorded every minute for an average of 10 s.
In vivo drug delivery
All drugs were delivered via the icv cannula into the right lateral ventricle of unrestrained rats. When testing the mechanism of sEH inhibition requiring two drugs, the first drug was injected 1 h before the second, and the dummy cannula was replaced following injection. sEH inhibitors N,N—dicyclohexyurea (DCU) and AUDA (0.8-60 ng, 2 µl vol) dissolved in 10% DMSO/90% aCSF and aCSF, respectively, were used to inhibit central sEH. MS-PPOH (680 ng, 2 µl, dissolved in aCSF) was used to block the formation of EETs. 11-NODA (100 ng, 1 µl, dissolved in aCSF), an EET agonist, was provided by JR Falck. gp91ds-tat and its scrambled peptide used as a control (100 ng each, 2 µl, dissolved in aCSF) were synthesized by the Temple University Protein Synthesis Core facility (Philadelphia, PA) and used to inhibit the action of NAD(P)H oxidase (13).
Evaluation of baroreceptor reflex function
To evaluate baroreceptor reflex function, the gain was determined from spontaneous changes in systolic blood pressure (SBP) and pulse interval (PI) using a time series method for the rat, as described by us (14, 15). Positive slope values were used only to avoid contaminating baroreceptor reflex data with nonbaroreceptor-mediated changes in PI.
Page 3 of 17(page number not for citation purposes)

Analysis of HR variability
HR variability was analyzed using a fast Fourier transformation (FFT). For each 10 min recording period, the beat-to-beat pulse interval data were converted into datum points every 100 ms using a spline interpolation, and power spectral density was computed using FFT algorithm and averaged. It is generally accepted that the high-frequency (HF) component of heart rate variability is mediated by cardiac parasympathetic tone (16).
Real-time RT-PCR
The RNAqueous-4PCR kit was used to isolate DNA-free RNA from tissue samples (Ambion, Austin TX). One-step real-time RT-PCR was performed on 50 ng RNA per sample. Briefly, samples were combined with sEH-specific primers and probe (900 nM forward: 5’-GATTCTCATCAAGTGGCTGAAGAC-3’; 900 nM reverse: 3’-GGACACGCCACTGGCTAAAT-5’) and probe (250 nM: 5’-CCAGAACCCATCGGTGACCTCCAA-3’) and TaqMan One-Step RT-PCR Master Mix and Multiscribe as described by Applied Biosystems (Foster City CA). Primers and probe to 18S were used as an internal control. Reactions were carried out in the ABI PRISM 7000 sequence detector, and the standard curve method was used to calculate relative expression levels.
Western blot analysis
Total cell lysate (TCL) was isolated according to the company’s research applications instructions (Santa Cruz Biotechnology, Santa Cruz, CA). Twenty µg of protein from either neuronal cultures or brain tissue were run on a 10% SDS-PAGE gel and transferred onto a nitrocellulose membrane. Following 3 hr blocking with 5% milk in TBS-Tween (TBS-T), the membrane was probed with the sEH rabbit-anti mouse antibody (1:3000 in 1% milk/TBS-T) overnight. Specificity of this antibody has been previously established (1, 2). The membrane was rinsed 3 times and washed for 20 min in TBS-T and then incubated with anti-rabbit IgG HRP-conjugated secondary antibody (1:3000) for 1 hour. Following final washes, the membrane was incubated with chemiluminescent agent for 1 min and then exposed to film to visualize the bands.
Neuronal cells in primary culture
Primary neuronal cultures (90% neurons, 10% glia) from the hypothalamus and brain stem of 1-day-old SHR and WKY rats were prepared as previously described (17). The cultures were grown 11-14 days in Dulbecco’s Modified Eagle’s Media (DMEM) + 10% horse serum at 37°C, 10% CO2.
Neuronal firing recording
Spontaneous action potentials (APs) were recorded with the whole-cell voltage clamp configuration in current clamp mode. Neurons cultured from hypothalamus were bathed in Tyrodes’s solution containing (in mM): 140 NaCl, 5.4 KCl, 2.0 CaCl2, 2.0 MgCl2, 0.3 NaH2PO4, 10 HEPES, and 10 dextrose, pH adjusted to 7.4 with NaOH. The patch electrodes (resistances from 3–4 MOhms) were filled with an internal pipette solution containing (in mM): 140 KCl, 4 MgCl2, 4 ATP, 0.1 guanosine 5’-triphosphate, 10 dextrose, and 10 HEPES, pH adjusted to 7.2
Page 4 of 17(page number not for citation purposes)

with KOH. The resting membrane potential was defined as the potential within a 1 s time period during which there were no spontaneously firing APs. Neuronal firing rate was measured as the number of fully developed APs (depolarization beyond 0 mV) per second (Hz).
Statistical analysis
All data are expressed as means ± SE. Comparisons between experimental groups were analyzed using ANOVA (SPSS) and Student’s t test (Stat View).
RESULTS
sEH expression in the SHR
Profiling with microarray has previously identified that the expression of sEH is most profoundly increased in the brain of the SHR, compared with its WKY normotensive control. Thus, our first objective was to confirm this observation. Figure 1A shows that sEH mRNA levels were three- to six-fold higher in the brain stem and hypothalamus of SHR compared to WKY rats. The sEH protein levels were barely detectable in WKY rats, while they were dramatically increased in the SHR (Fig. 1B, C).
Effect of sEH inhibition on BP and HR
Intracerebroventricular administration of AUDA, a potent sEH inhibitor, caused a dose-dependent increase in both BP and HR in the SHR. A significant increase of 25 ± 0.9 mmHg in BP was seen with as low as 0.8 ng AUDA, and a dose of 15 ng AUDA increased BP by 32 ± 6 mmHg (Fig. 2A, D). This increase began to be expressed as early as 30 min, reached maximal levels in 3-4 h, and returned to basal levels in 7 h. The same concentrations of AUDA showed only modest increases in BP in the WKY rats (Fig. 2A). The increase in BP was associated with a significant increase in HR in both WKY and SHR by 46 ± 12 bpm and 54 ± 10 bpm, respectively (Fig. 2B, C, E). DCU, another selective but weaker sEH inhibitor, produced a similar increase in BP and HR in the SHR (data not shown). To study the mechanism by which these increases are induced in the SHR, baroreceptor gain and cardiac vagal tone were determined in response to sEH inhibition. Analysis of waveform telemetry data showed a 56 ± 5% decrease in baroreceptor reflex gain (P<0.05) and a 33 ± 8% decrease in HR analysis of PI by icv AUDA administration in the SHR (Fig. 3A, B). No significant changes were observed in the WKY rats. These data demonstrate that increases in BP are consistent with a decrease in baroreceptor reflex functions.
Increased EETs linked to increase in BP through ROS production
sEH inhibition has been shown to change the equilibrium between EETs and DHETs in favor of the EETs. This led us to hypothesize that EETs may be central in sEH inhibitor-induced increases in BP. This hypothesis was tested initially with the use of MS-PPOH, an inhibitor of the cytochrome P450 epoxygenase pathway that generates EETs and related PUFA epoxides (18). Figure 4 shows that central treatment with MS-PPOH attenuated AUDA-induced increase in BP by 65% in the SHR. MS-PPOH alone had no effect on BP. In addition, icv delivery of the EET agonist 11-NODA in the WKY rat resulted in an increase in BP and HR by 13 ± 2 mmHg and 61 ± 19 bpm, respectively (Fig. 5).
Page 5 of 17(page number not for citation purposes)

Next, we examined the role of ROS on sEH-inhibitor-mediated increase in BP in the SHR. Because cytochrome P450 member 2C9 generates both EETs and ROS, and an increase in central ROS is associated with an increase in BP, we argued that one would be able to prevent AUDA-induced increases in BP by inhibitors of ROS (11, 12). Fig. 6 shows that pretreatment with gp91ds-tat, a blocker of NAD(P)H oxidase, caused an 85% attenuation of BP and a 77% decrease in HR induced by AUDA. In contrast, gp91ds-tat alone had no effect on BP and HR (data not shown).
In vitro validation with neuronal cells in primary culture
Primary coculture from hypothalamus-brain stem of WKY and SHR have been used in the past to elucidate cellular and molecular mechanisms of physiological dysregulation in neural control of hypertension (19, 20). We used these cultures to validate in vivo data and determine the cellular basis of sEH inhibitor-induced increase in BP in the SHR. Western blot analysis demonstrated a four-fold increase in sEH protein levels in neuronal culture of SHR vs. WKY rats (Fig. 7A). The effect of AUDA on neuronal firing rate was studied to determine if inhibition of sEH would increase neuronal activity in the SHR. Figure 7B shows that 0.4 mg/ml AUDA resulted in a two-fold increase in neuronal firing rate exclusively in the SHR neurons. This AUDA-induced increase was attenuated 75% by pretreatment with 5 µM gp91ds-tat (Fig. 7B, C). These observations provide cellular insight into the effects of sEH on neuronal activity in the SHR.
DISCUSSION
Our results suggest a novel role for sEH and EETs in the brain of the SHR. Following central sEH inhibition, both BP and HR increase and are associated with a dampening of the baroreceptor reflex. We surmise that these effects of sEH inhibition occur through the accumulation of their substrates, the EETs, and other epoxy lipids. This conclusion is supported by the finding that the selective inhibitor of lipid epoxidase MS-PPOH attenuates the AUDA-induced increase in BP. In addition, administration of an EET agonist results in a transient increase in BP in the WKY rat. Thus, the role of central EETs appears to be distinct from that of its peripheral expression, which is characterized by its vasodilatory properties that are associated with a decrease in BP.
In contrast to the SHR, sEH inhibition had no significant effect on BP in the WKY rat. This observation is consistent with our Western blot data indicating the presence of little sEH protein in the WKY rat brain. HR, however, was increased in the WKY rat, though the duration of the effect was attenuated compared to that of the SHR. This finding may be related to differential levels of EETs that induce BP and HR-specific effects. Further studies are needed to resolve these and other possibilities.
The mechanism linking central sEH inhibition to the increase in BP and HR appears to function through ROS (11). This hypothesis is supported by our data: 1) Pretreatment with gp91ds-tat, an NAD(P)H oxidase blocker prevents AUDA-induced increase in BP and HR, and 2) AUDA treatment increased neuronal firing rate, an effect attenuated by gp91ds-tat. Thus, we propose that an increase in EETs by inhibition of sEH leads to an increase in ROS in the brain and hence an increase in BP. Another possibility is that ROS stimulates EETs, which therefore leads to an
Page 6 of 17(page number not for citation purposes)

increase in BP. The increase in BP is associated with dampening of the baroreceptor reflex. Whether this is a causative mechanism is unknown, but it should be mentioned that recent data indicate that baroreceptor reflex plays a critical role in chronic and long-term control of BP (21, 22). Finally, our study suggests that overexpression of sEH in the brain may be a compensatory response, which fails to override hypertensive conditions in the SHR. This would be an important concept to prove with the use of many other experimental models of hypertension. The SHR was exclusively chosen for this study because it exhibits many similarities to primary human hypertension. For example, there is an early onset of high BP and a slow, progressive increase in peripheral resistance, vascular reactivity and sympathetic nervous system activity in both the SHR and primary human hypertension. Additionally, both have neurogenic components in the development and establishment of hypertension (23–27). Thus, these observations may have important implications in elucidating the mechanism of neuronal control of primary human hypertension.
This study altogether provides a novel mechanism by which sEH regulates designated cardiovascular activity in the brain. It also raises important questions: for example, in what cell type in the brain is sEH localized? We believe that it is the neuronal sEH that participates in EET-mediated BP control. This is supported by our findings that neuronal cells in primary culture from the prehypertensive SHR hypothalamus and brain stem demonstrate an increased expression of sEH similar to that seen in the adult SHR brain. In addition, AUDA treatment caused an exclusive increase in neuronal firing in neurons from the SHR hypothalamus and brain stem. Finally, astroglial cultures from the same brain areas showed no significant differences in the sEH expression between WKY and SHR (data not shown). Although these data implicate neuronal sEH, they do not eliminate the role of cerebrovascular sEH, which we will investigate further. It would be important to determine what nucleus (or nuclei) in the hypothalamus and brain stem is/are key in EETs-mediated regulation of BP and how sEH-EET-ROS-mediated signals are transmitted to the peripheral system that is translated into increases in HR and BR. We have developed a lentiviral vector-mediated gene delivery system in selective brain nuclei and transmitter-specific neurons in vivo in an attempt to answer these questions (28). Nonetheless, these studies establish that an increase in gene expression for sEH appears to be a protective mechanism against neural control of hypertension, an increase that fails to override the hypertensive dysregulatory pathways.
ACKNOWLEDGMENTS
Authors wish to thank Tony Cometa and Adam Mecca for their surgical assistance. In addition, we thank Ms. Nichole Herring for secretarial assistance and Ms. Fan Lin for preparation of neuronal cultures. This work was supported by NIH grants HL33610 and HL56921 (MKR); R37 ES02710, P30 ES05707, and P42 ES04699 (BDH & CM); and GM31278 and the Robert A. Welch Foundation (JRF). HW and JFRP were both supported by the British Heart Foundation. Ms. KW Sellers is a predoctoral fellow of the Florida-Puerto Rico affiliate of the AHA.
REFERENCES
1. Sinal, C. J., Miyata, M., Tohkin, M., Nagata, K., Bend, J. R., and Gonzalez, F. J. (2000) Targeted disruption of soluble epoxide hydrolase reveals a role in blood pressure regulation. J. Biol. Chem. 275, 40,504–40,510
Page 7 of 17(page number not for citation purposes)

2. Yu, Z., Xu, F., Huse, L. M., Morisseau, C., Draper, A. J., Newman, J. W., Parker, C., Graham, L., Engler, M. M., Hammock, B. D., et al. (2000) Soluble epoxide hydrolase regulates hydrolysis of vasoactive epoxyeicosatrienoic acids. Circ. Res. 87, 992–998
3. Imig, J. D., Zhao, X., Capdevila, J. H., Morisseau, C., and Hammock, B. D. (2002) Soluble epoxide hydrolase inhibition lowers arterial blood pressure in angiotensin II hypertension. Hypertension 39, 690–694
4. Grant, D. F., Storms, D. H., and Hammock, B. D. (1993) Molecular cloning and expression of murine liver soluble epoxide hydrolase. J. Biol. Chem. 268, 17,628–17,633
5. Roman, R. J. (2002) P-450 metabolites of arachidonic acid in the control of cardiovascular function. Physiol. Rev. 82, 131–185
6. Campbell, W. B., Gebremedhin, D., Pratt, P. F., and Harder, D. R. (1996) Identification of epoxyeicosatrienoic acids as endothelium-derived hyperpolarizing factors. Circ. Res. 78, 415–423
7. Campbell, W. B., Deeter, C., Gauthier, K. M., Ingraham, R. H., Falck, J. R., and Li, P. L. (2002) 14,15-Dihydroxyeicosatrienoic acid relaxes bovine coronary arteries by activation of K(Ca) channels. Am. J. Physiol. Heart Circ. Physiol. 282, H1656–H1664
8. Falck, J. R., Krishna, U. M., Reddy, Y. K., Kumar, P. S., Reddy, K. M., Hittner, S. B., Deeter, C., Sharma, K. K., Gauthier, K. M., and Campbell, W. B. (2003) Comparison of vasodilatory properties of 14,15-EET analogs: structural requirements for dilation. Am. J. Physiol. Heart Circ. Physiol. 284, H337–H349
9. Fornage, M., Hinojos, C. A., Nurowska, B. W., Boerwinkle, E., Hammock, B. D., Morisseau, C. H., and Doris, P. A. (2002) Polymorphism in soluble epoxide hydrolase and blood pressure in spontaneously hypertensive rats. Hypertension 40, 485–490
10. Okuda, T., Sumiya, T., Iwai, N., and Miyata, T. (2002) Difference of gene expression profiles in spontaneous hypertensive rats and Wistar-Kyoto rats from two sources. Biochem. Biophys. Res. Commun. 296, 537–543
11. Fleming, I., Michaelis, U. R., Bredenkotter, D., Fisslthaler, B., Dehghani, F., Brandes, R. P., and Busse, R. (2001) Endothelium-derived hyperpolarizing factor synthase (Cytochrome P450 2C9) is a functionally significant source of reactive oxygen species in coronary arteries. Circ. Res. 88, 44–51
12. Zimmerman, M. C., Lazartigues, E., Lang, J. A., Sinnayah, P., Ahmad, I. M., Spitz, D. R., and Davisson, R. L. (2002) Superoxide mediates the actions of angiotensin II in the central nervous system. Circ. Res. 91, 1038–1045
13. Rey, F. E., Cifuentes, M. E., Kiarash, A., Quinn, M. T., and Pagano, P. J. (2001) Novel competitive inhibitor of NAD(P)H oxidase assembly attenuates vascular O(2)(-) and systolic blood pressure in mice. Circ. Res. 89, 408–414
Page 8 of 17(page number not for citation purposes)

14. Waki, H., Kasparov, S., Wong, L. F., Murphy, D., Shimizu, T., and Paton, J. F. (2003) Chronic inhibition of endothelial nitric oxide synthase activity in nucleus tractus solitarii enhances baroreceptor reflex in conscious rats. J. Physiol. 546, 233–242
15. Oosting, J., Struijker-Boudier, H. A., and Janssen, B. J. (1997) Validation of a continuous baroreceptor reflex sensitivity index calculated from spontaneous fluctuations of blood pressure and pulse interval in rats. J. Hypertens. 15, 391–399
16. Pagani, M., Lombardi, F., Guzzetti, S., Rimoldi, O., Furlan, R., Pizzinelli, P., Sandrone, G., Malfatto, G., Dell'Orto, S., Piccaluga, E., et al. (1986) Power spectral analysis of heart rate and arterial pressure variabilities as a marker of sympatho-vagal interaction in man and conscious dog. Circ. Res. 59, 178–193
17. Raizada, M. K., Lu, D., and Sumners, C. (1995) AT1 receptors and angiotensin actions in the brain and neuronal cultures of normotensive and hypertensive rats. Adv. Exp. Med. Biol. 377, 331–348
18. Brand-Schieber, E., Falck, J. F., and Schwartzman, M. (2000) Selective inhibition of arachidonic acid epoxidation in vivo. J. Physiol. Pharmacol. 51, 655–672
19. Yang, H., Reaves, P. Y., Katovich, M. J., and Raizada, M. K. (2004) Decrease in hypothalamic gamma adducin in rat models of hypertension. Hypertension 43, 324–328
20. Sun, C., Du, J., Sumners, C., and Raizada, M. K. (2003) PI3-kinase inhibitors abolish the enhanced chronotropic effects of angiotensin II in spontaneously hypertensive rat brain neurons. J. Neurophysiol. 90, 3155–3160
21. Thrasher, T. N. (2002) Unloading arterial baroreceptors causes neurogenic hypertension. Am. J. Physiol. Regul. Integr. Comp. Physiol. 282, R1044–R1053
22. Lohmeier, T. E., Irwin, E. D., Rossing, M. A., Serdar, D. J., and Kieval, R. S. (2004) Prolonged activation of the baroreflex produces sustained hypotension. Hypertension 43, 306–311
23. Goldstein, D. S. (1983) Plasma catecholamines and essential hypertension. An analytical review. Hypertension 5, 86–99
24. Lindpaintner, K. (1994) What can the molecular genetics of hypertensive rats teach us about the genetics of hypertension in humans? Curr. Opin. Nephrol. Hypertens. 3, 30–38
25. Louis, W. J., and Howes, L. G. (1990) Genealogy of the spontaneously hypertensive rat and Wistar-Kyoto rat strains: implications for studies of inherited hypertension. J. Cardiovasc. Pharmacol. 16, Suppl 7, S1–S5
26. Trippodo, N. C., and Frohlich, E. D. (1981) Similarities of genetic (spontaneous) hypertension. Man and rat. Circ. Res. 48, 309–319
Page 9 of 17(page number not for citation purposes)

27. Frohlich, E. D. (1988) The first Irvine H. Page lecture. The mosaic of hypertension: past, present and future. J. Hypertens. Suppl. 6, S2–11
28. Coleman, J. E., Huentelman, M. J., Kasparov, S., Metcalfe, B. L., Paton, J. F., Katovich, M. J., Semple-Rowland, S. L., and Raizada, M. K. (2003) Efficient large-scale production and concentration of HIV-1-based lentiviral vectors for use in vivo. Physiol. Genomics 12, 221–228
Received September 15, 2004; accepted November 22, 2004.
Page 10 of 17(page number not for citation purposes)
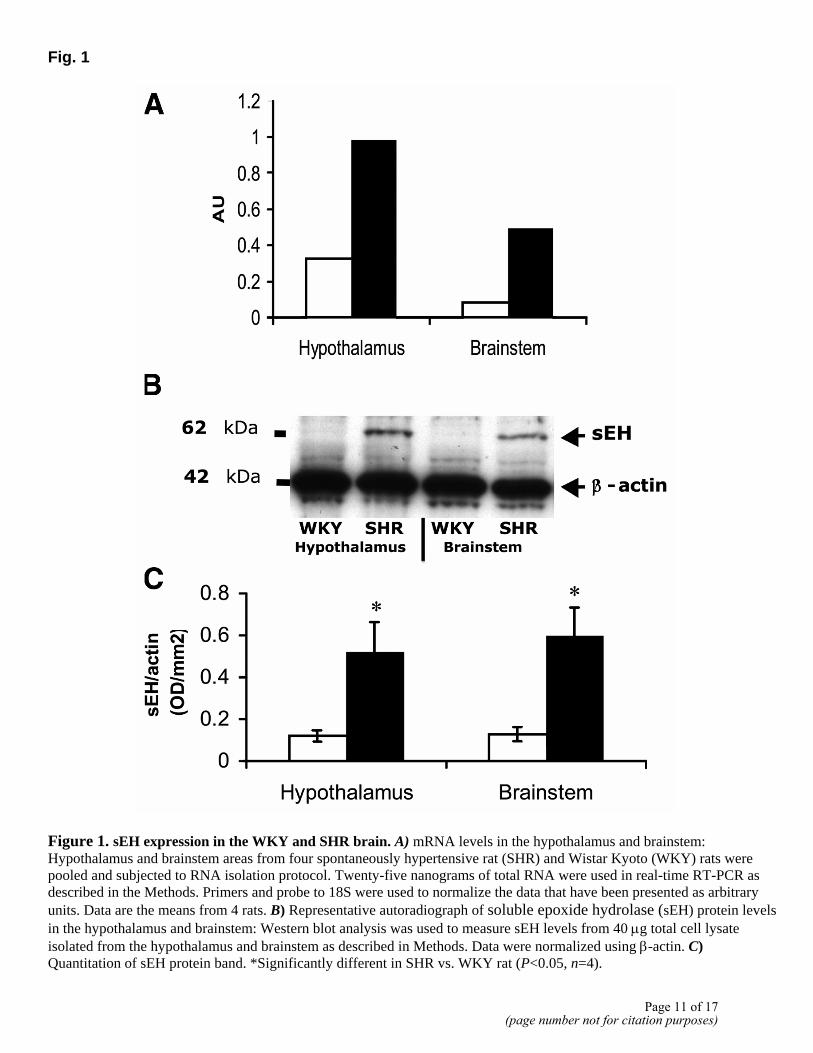
Fig. 1
Figure 1. sEH expression in the WKY and SHR brain. A) mRNA levels in the hypothalamus and brainstem: Hypothalamus and brainstem areas from four spontaneously hypertensive rat (SHR) and Wistar Kyoto (WKY) rats were pooled and subjected to RNA isolation protocol. Twenty-five nanograms of total RNA were used in real-time RT-PCR as described in the Methods. Primers and probe to 18S were used to normalize the data that have been presented as arbitrary units. Data are the means from 4 rats. B) Representative autoradiograph of soluble epoxide hydrolase (sEH) protein levels in the hypothalamus and brainstem: Western blot analysis was used to measure sEH levels from 40 µg total cell lysate isolated from the hypothalamus and brainstem as described in Methods. Data were normalized using β-actin. C) Quantitation of sEH protein band. *Significantly different in SHR vs. WKY rat (P<0.05, n=4).
Page 11 of 17(page number not for citation purposes)
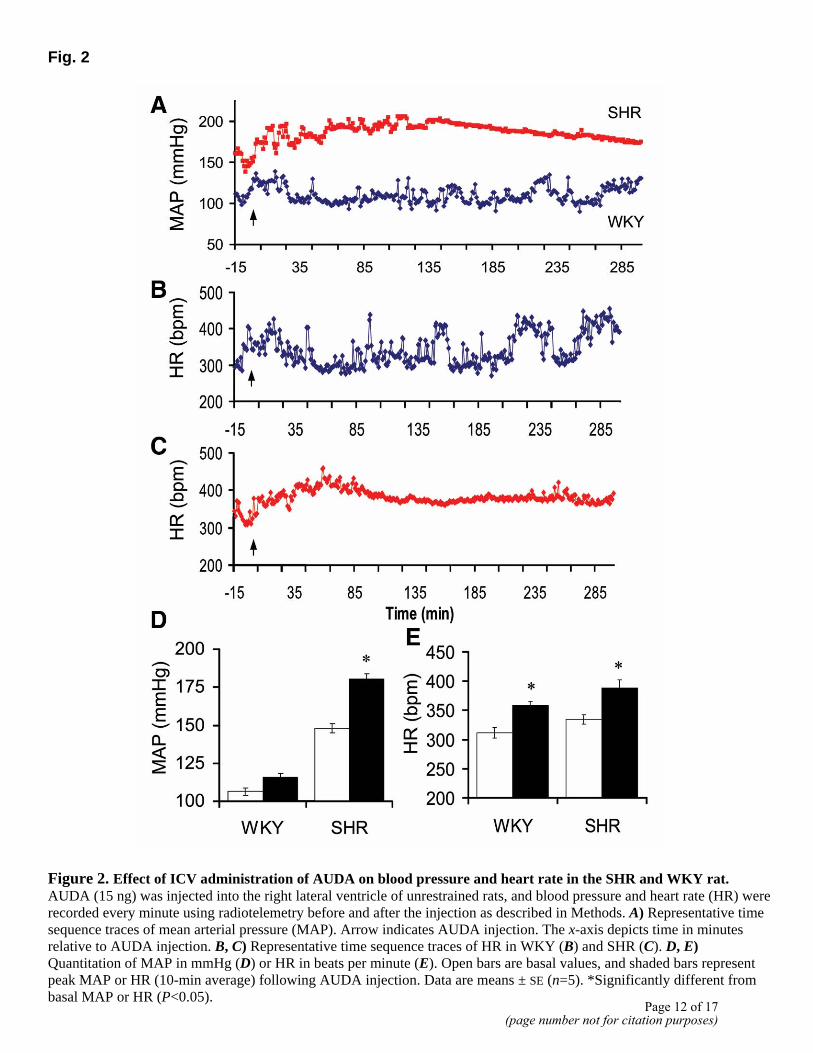
Fig. 2
Figure 2. Effect of ICV administration of AUDA on blood pressure and heart rate in the SHR and WKY rat. AUDA (15 ng) was injected into the right lateral ventricle of unrestrained rats, and blood pressure and heart rate (HR) were recorded every minute using radiotelemetry before and after the injection as described in Methods. A) Representative time sequence traces of mean arterial pressure (MAP). Arrow indicates AUDA injection. The x-axis depicts time in minutes relative to AUDA injection. B, C) Representative time sequence traces of HR in WKY (B) and SHR (C). D, E) Quantitation of MAP in mmHg (D) or HR in beats per minute (E). Open bars are basal values, and shaded bars represent peak MAP or HR (10-min average) following AUDA injection. Data are means ± SE (n=5). *Significantly different from basal MAP or HR (P<0.05).
Page 12 of 17(page number not for citation purposes)
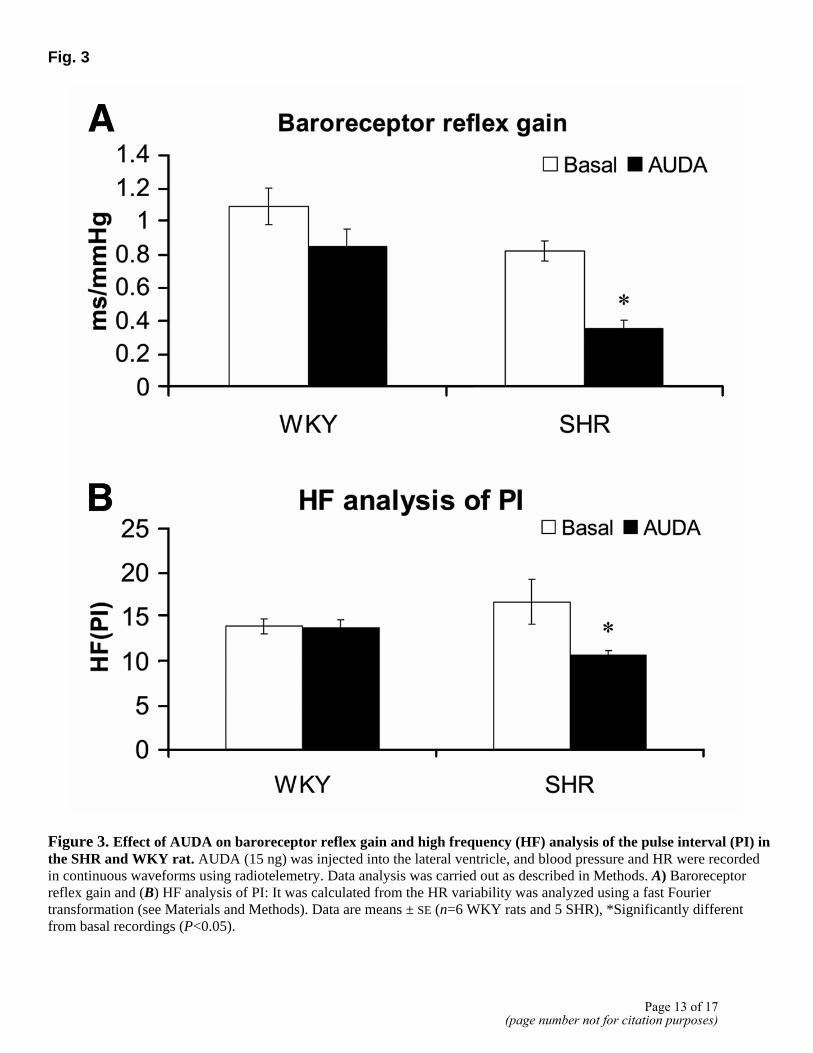
Fig. 3
Figure 3. Effect of AUDA on baroreceptor reflex gain and high frequency (HF) analysis of the pulse interval (PI) in the SHR and WKY rat. AUDA (15 ng) was injected into the lateral ventricle, and blood pressure and HR were recorded in continuous waveforms using radiotelemetry. Data analysis was carried out as described in Methods. A) Baroreceptor reflex gain and (B) HF analysis of PI: It was calculated from the HR variability was analyzed using a fast Fourier transformation (see Materials and Methods). Data are means ± SE (n=6 WKY rats and 5 SHR), *Significantly different from basal recordings (P<0.05).
Page 13 of 17(page number not for citation purposes)
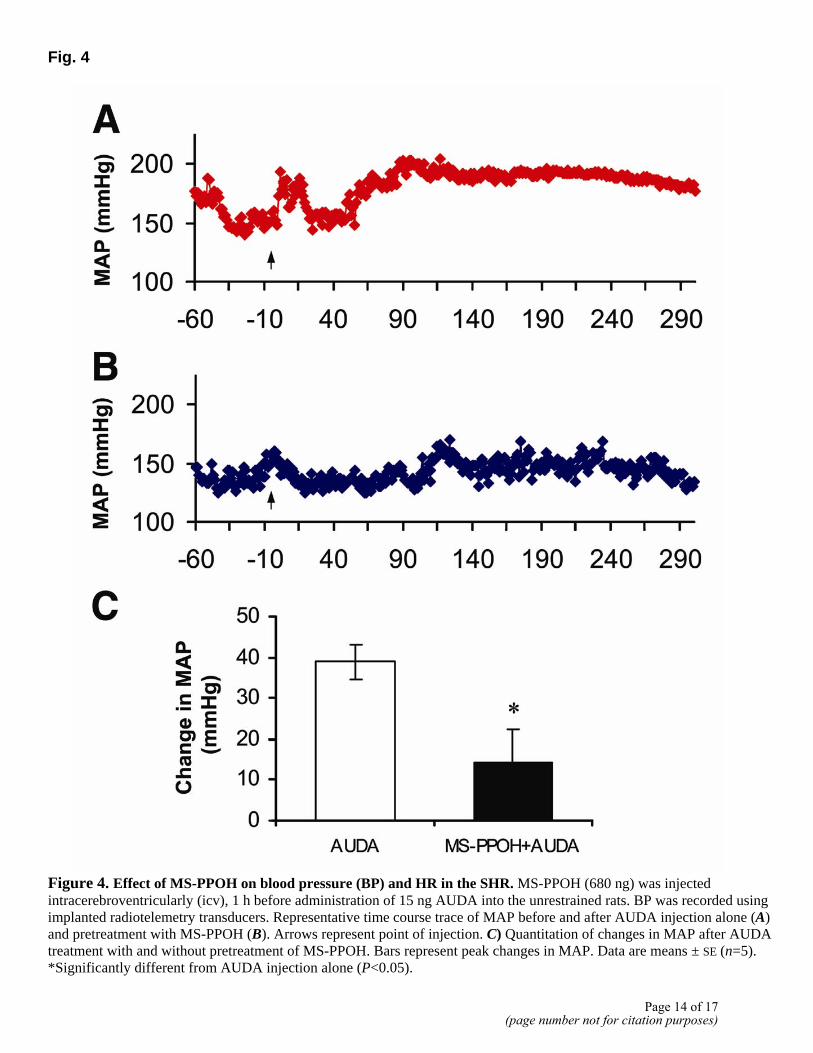
Fig. 4
Figure 4. Effect of MS-PPOH on blood pressure (BP) and HR in the SHR. MS-PPOH (680 ng) was injected intracerebroventricularly (icv), 1 h before administration of 15 ng AUDA into the unrestrained rats. BP was recorded using implanted radiotelemetry transducers. Representative time course trace of MAP before and after AUDA injection alone (A) and pretreatment with MS-PPOH (B). Arrows represent point of injection. C) Quantitation of changes in MAP after AUDA treatment with and without pretreatment of MS-PPOH. Bars represent peak changes in MAP. Data are means ± SE (n=5). *Significantly different from AUDA injection alone (P<0.05).
Page 14 of 17(page number not for citation purposes)
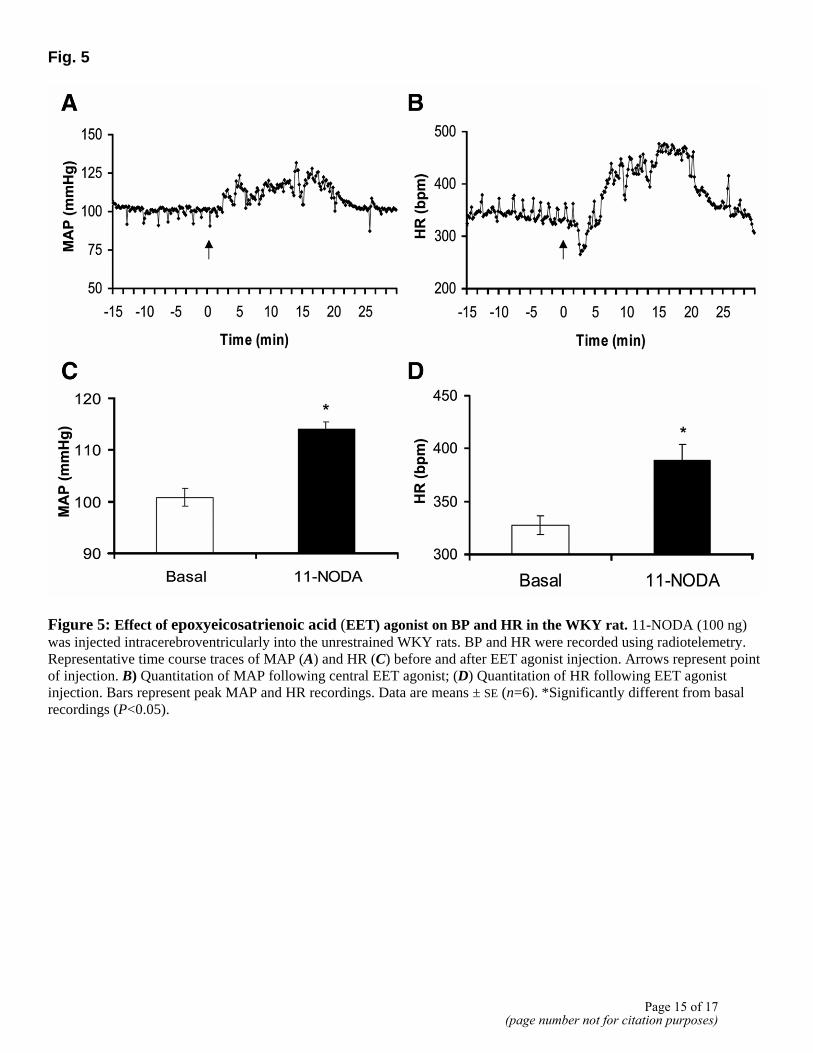
Fig. 5
Figure 5: Effect of epoxyeicosatrienoic acid (EET) agonist on BP and HR in the WKY rat. 11-NODA (100 ng) was injected intracerebroventricularly into the unrestrained WKY rats. BP and HR were recorded using radiotelemetry. Representative time course traces of MAP (A) and HR (C) before and after EET agonist injection. Arrows represent point of injection. B) Quantitation of MAP following central EET agonist; (D) Quantitation of HR following EET agonist injection. Bars represent peak MAP and HR recordings. Data are means ± SE (n=6). *Significantly different from basal recordings (P<0.05).
Page 15 of 17(page number not for citation purposes)
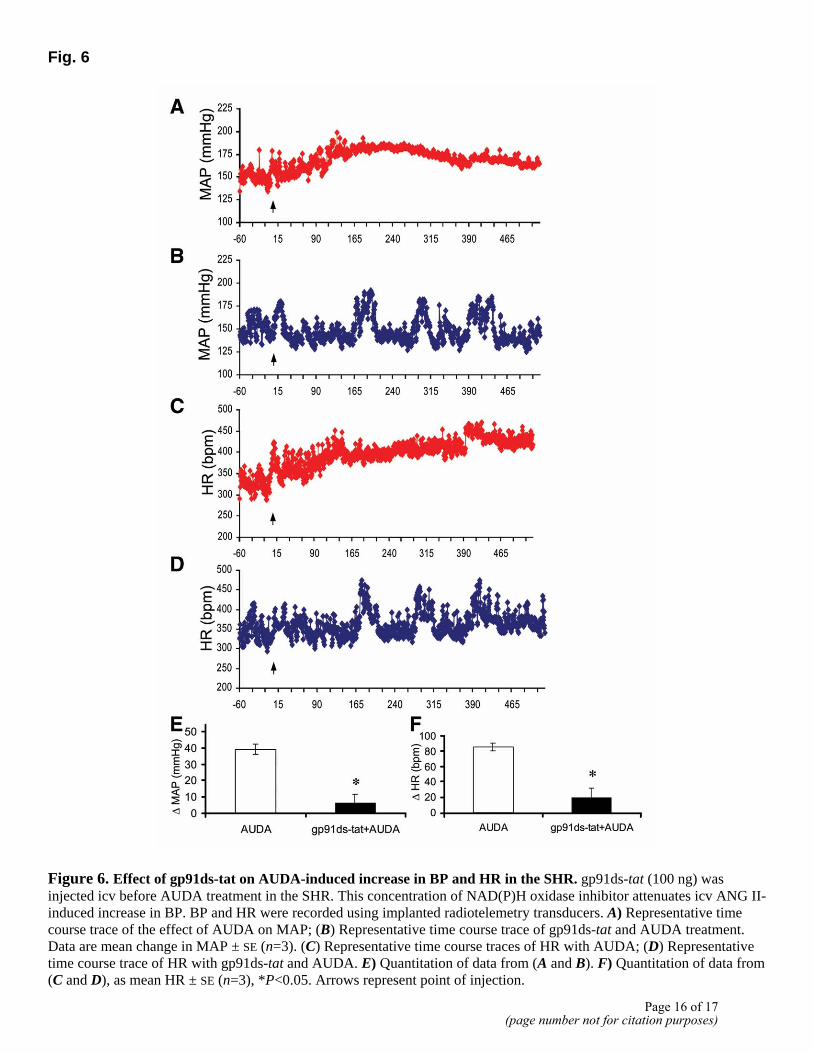
Fig. 6
Figure 6. Effect of gp91ds-tat on AUDA-induced increase in BP and HR in the SHR. gp91ds-tat (100 ng) was injected icv before AUDA treatment in the SHR. This concentration of NAD(P)H oxidase inhibitor attenuates icv ANG II-induced increase in BP. BP and HR were recorded using implanted radiotelemetry transducers. A) Representative time course trace of the effect of AUDA on MAP; (B) Representative time course trace of gp91ds-tat and AUDA treatment. Data are mean change in MAP ± SE (n=3). (C) Representative time course traces of HR with AUDA; (D) Representative time course trace of HR with gp91ds-tat and AUDA. E) Quantitation of data from (A and B). F) Quantitation of data from (C and D), as mean HR ± SE (n=3), *P<0.05. Arrows represent point of injection.
Page 16 of 17(page number not for citation purposes)
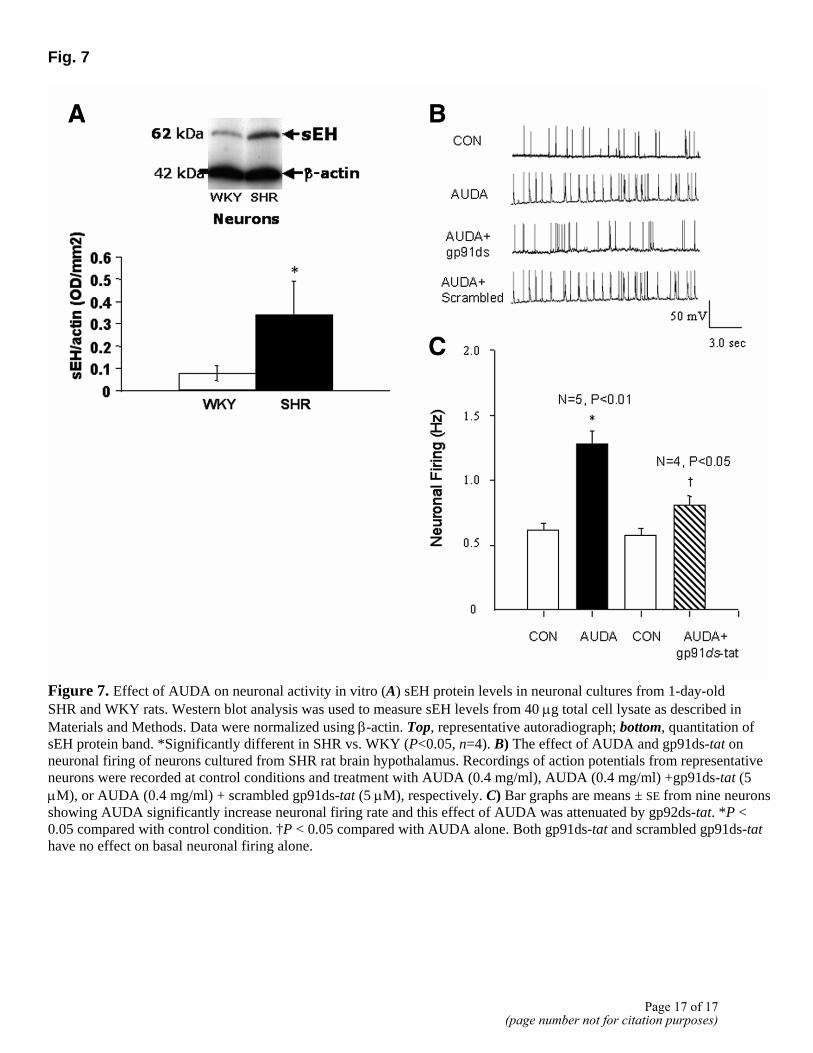
Fig. 7
Figure 7. Effect of AUDA on neuronal activity in vitro (A) sEH protein levels in neuronal cultures from 1-day-old SHR and WKY rats. Western blot analysis was used to measure sEH levels from 40 µg total cell lysate as described in Materials and Methods. Data were normalized using β-actin. Top, representative autoradiograph; bottom, quantitation of sEH protein band. *Significantly different in SHR vs. WKY (P<0.05, n=4). B) The effect of AUDA and gp91ds-tat on neuronal firing of neurons cultured from SHR rat brain hypothalamus. Recordings of action potentials from representative neurons were recorded at control conditions and treatment with AUDA (0.4 mg/ml), AUDA (0.4 mg/ml) +gp91ds-tat (5 µM), or AUDA (0.4 mg/ml) + scrambled gp91ds-tat (5 µM), respectively. C) Bar graphs are means ± SE from nine neurons showing AUDA significantly increase neuronal firing rate and this effect of AUDA was attenuated by gp92ds-tat. *P < 0.05 compared with control condition. †P < 0.05 compared with AUDA alone. Both gp91ds-tat and scrambled gp91ds-tat have no effect on basal neuronal firing alone.
Page 17 of 17(page number not for citation purposes)

The FASEB Journal • FJ Express
Novel mechanism of brain soluble epoxidehydrolase-mediated blood pressure regulationin the spontaneously hypertensive rat
Kathleen W. Sellers,* Chengwen Sun,* Carlos Diez-Freire,* Hidefumi Waki,†
Christophe Morisseau,‡ John R. Falck,§ Bruce D. Hammock,‡ Julian F. Paton,†
and Mohan K. Raizada*,1
*Department of Physiology and Functional Genomics, University of Florida College of Medicine andMcKnight Brain Institute, Gainesville, Florida, USA; †Department of Physiology, School of MedicalSciences, University of Bristol, Bristol, UK; ‡Department of Entomology and U.C.D. Cancer Center,University of California Davis, California, USA; and §Departments of Biochemistry andPharmacology, University of Texas Southwestern Medical Center, Dallas, Texas, USA
To read the full text of this article, go to http://www.fasebj.org/cgi/doi/10.1096/fj.04-3128fje;doi: 10.1096/fj.04-3128fje
SPECIFIC AIMS
Gene profiling using microarray technology revealedthat soluble epoxide hydrolase (sEH) mRNA levelswere elevated in the spontaneously hypertensive rat(SHR) vs. the Wistar Kyoto (WKY) rat brain. PeripheralsEH overexpression has been linked to animal modelsof hypertension, and inhibition of sEH activity bypharmacological agents decreased high blood pressure(BP). Whereas sEH in the vasculature is proposed toexert its actions by converting vasodilatory epoxyeico-satrienoic acids (EETs) to their relatively inactive diolforms, the role of this enzyme in the brain is unknown.Thus, our objective was to investigate the involvementof brain sEH in BP control.
PRINCIPAL FINDINGS
1. sEH is up-regulated in the hypothalamus andbrainstem of the SHR
Our first objective was to validate sEH expressionchanges identified in microarray analysis. Real-timeRT-PCR and Western blot analysis revealed that sEHexpression was several fold higher in the brainstem andhypothalamus of adult SHR than in WKY rats. Thisincrease seems to be genetically linked, as neuronalcultures from prehypertensive rats exhibited a similarincrease in expression. Thus, sEH is overexpressed incardiovascularly relevant brain regions of the SHR.
2. Central sEH inhibition increases BP and heart ratein the SHR
Intraperitoneal delivery of pharmacological inhibitorsto sEH results in a depressor effect in the SHR. We
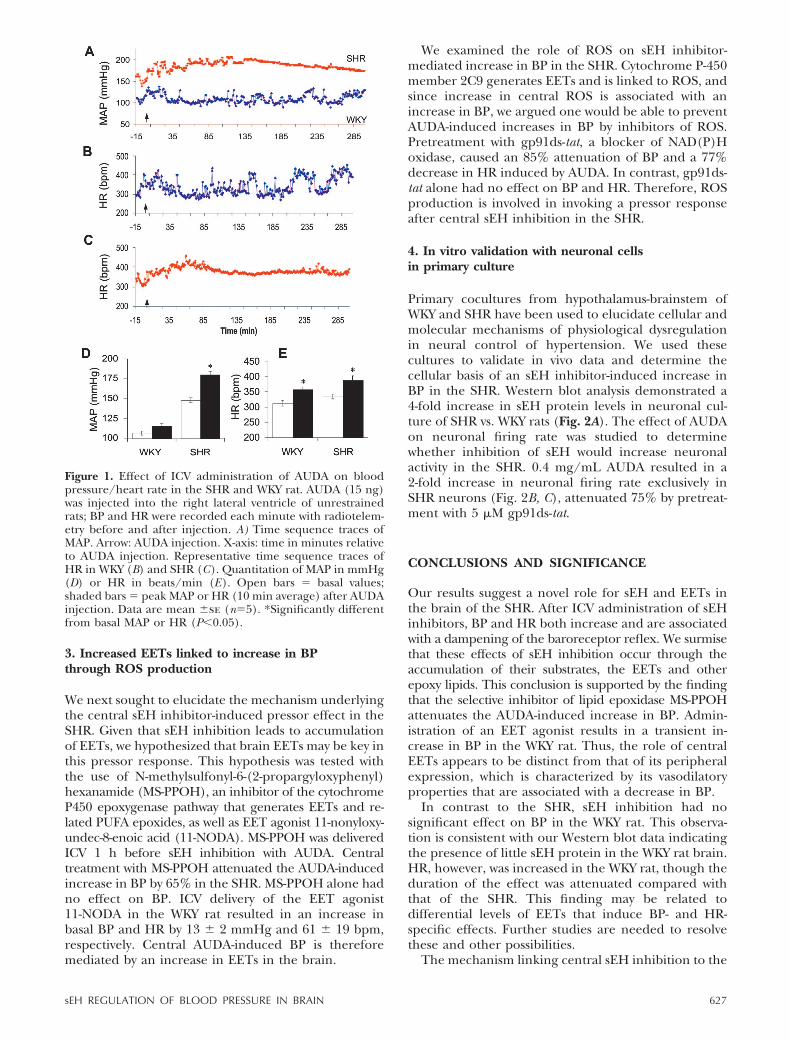
sought to determine what effect central inhibition ofsEH had on blood pressure in SHR and WKY rats. Ratswere implanted with a radiotelemetric pressure trans-ducer (Data Sciences International, Arden Hills, MN,USA) in the abdominal aorta to transmit blood pres-sure and heart rate (HR) data every minute for analysis.sEH inhibitors were delivered ICV through a cannulainserted into the right lateral ventricle. ICV administra-tion of adamantyl dodecanoic acid urea (AUDA), apotent sEH inhibitor, caused a dose-dependent in-crease in BP and HR in the SHR (Fig. 1). An increaseof 25 � 0.9 mmHg in BP was seen with as little as 0.8 ngAUDA; a dose of 15 ng AUDA increased BP by 32 � 6mmHg. This increase was observed as early as 30 minpost-AUDA administration, reached maximal levels in3–4 h, and returned to basal levels in 7 h. The increasein BP was associated with increases in HR in WKY andSHR by 46 � 12 bpm and 54 � 10 bpm, respectively.Central sEH inhibition did not alter BP in the WKY rat(Fig. 1).
To study the mechanism by which BP and HR areconcomitantly increased in SHR, spontaneous barore-ceptor reflex gain was determined from spontaneouschanges in systolic blood pressure (SBP) and pulseinterval (PI) using a time series method. Analysis ofwave form telemetry data showed a 56 � 5% decreasein baroreceptor reflex gain. High-frequency analysis ofPI as an indicator of cardiac vagal tone decreased 33 �8% after ICV AUDA administration in the SHR. Thesedata demonstrate that increases in BP are consistentwith a decrease in baroreceptor reflex functions.
1 Correspondence: Department of Physiology and Func-tional Genomics, College of Medicine, University of Florida,McKnight Brain Institute, Gainesville, FL 32610, USA. E-mail:[email protected].
626 0892-6638/05/0019-0626 © FASEB
3. Increased EETs linked to increase in BPthrough ROS production
We next sought to elucidate the mechanism underlyingthe central sEH inhibitor-induced pressor effect in theSHR. Given that sEH inhibition leads to accumulationof EETs, we hypothesized that brain EETs may be key inthis pressor response. This hypothesis was tested withthe use of N-methylsulfonyl-6-(2-propargyloxyphenyl)hexanamide (MS-PPOH), an inhibitor of the cytochromeP450 epoxygenase pathway that generates EETs and re-lated PUFA epoxides, as well as EET agonist 11-nonyloxy-undec-8-enoic acid (11-NODA). MS-PPOH was deliveredICV 1 h before sEH inhibition with AUDA. Centraltreatment with MS-PPOH attenuated the AUDA-inducedincrease in BP by 65% in the SHR. MS-PPOH alone hadno effect on BP. ICV delivery of the EET agonist11-NODA in the WKY rat resulted in an increase inbasal BP and HR by 13 � 2 mmHg and 61 � 19 bpm,respectively. Central AUDA-induced BP is thereforemediated by an increase in EETs in the brain.
We examined the role of ROS on sEH inhibitor-mediated increase in BP in the SHR. Cytochrome P-450member 2C9 generates EETs and is linked to ROS, andsince increase in central ROS is associated with anincrease in BP, we argued one would be able to preventAUDA-induced increases in BP by inhibitors of ROS.Pretreatment with gp91ds-tat, a blocker of NAD(P)Hoxidase, caused an 85% attenuation of BP and a 77%decrease in HR induced by AUDA. In contrast, gp91ds-tat alone had no effect on BP and HR. Therefore, ROSproduction is involved in invoking a pressor responseafter central sEH inhibition in the SHR.
4. In vitro validation with neuronal cellsin primary culture
Primary cocultures from hypothalamus-brainstem ofWKY and SHR have been used to elucidate cellular andmolecular mechanisms of physiological dysregulationin neural control of hypertension. We used thesecultures to validate in vivo data and determine thecellular basis of an sEH inhibitor-induced increase inBP in the SHR. Western blot analysis demonstrated a4-fold increase in sEH protein levels in neuronal cul-ture of SHR vs. WKY rats (Fig. 2A). The effect of AUDAon neuronal firing rate was studied to determinewhether inhibition of sEH would increase neuronalactivity in the SHR. 0.4 mg/mL AUDA resulted in a2-fold increase in neuronal firing rate exclusively inSHR neurons (Fig. 2B, C), attenuated 75% by pretreat-ment with 5 �M gp91ds-tat.
CONCLUSIONS AND SIGNIFICANCE
Our results suggest a novel role for sEH and EETs inthe brain of the SHR. After ICV administration of sEHinhibitors, BP and HR both increase and are associatedwith a dampening of the baroreceptor reflex. We surmisethat these effects of sEH inhibition occur through theaccumulation of their substrates, the EETs and otherepoxy lipids. This conclusion is supported by the findingthat the selective inhibitor of lipid epoxidase MS-PPOHattenuates the AUDA-induced increase in BP. Admin-istration of an EET agonist results in a transient in-crease in BP in the WKY rat. Thus, the role of centralEETs appears to be distinct from that of its peripheralexpression, which is characterized by its vasodilatoryproperties that are associated with a decrease in BP.
In contrast to the SHR, sEH inhibition had nosignificant effect on BP in the WKY rat. This observa-tion is consistent with our Western blot data indicatingthe presence of little sEH protein in the WKY rat brain.HR, however, was increased in the WKY rat, though theduration of the effect was attenuated compared withthat of the SHR. This finding may be related todifferential levels of EETs that induce BP- and HR-specific effects. Further studies are needed to resolvethese and other possibilities.
The mechanism linking central sEH inhibition to the
Figure 1. Effect of ICV administration of AUDA on bloodpressure/heart rate in the SHR and WKY rat. AUDA (15 ng)was injected into the right lateral ventricle of unrestrainedrats; BP and HR were recorded each minute with radiotelem-etry before and after injection. A) Time sequence traces ofMAP. Arrow: AUDA injection. X-axis: time in minutes relativeto AUDA injection. Representative time sequence traces ofHR in WKY (B) and SHR (C). Quantitation of MAP in mmHg(D) or HR in beats/min (E). Open bars � basal values;shaded bars � peak MAP or HR (10 min average) after AUDAinjection. Data are mean �se (n�5). *Significantly differentfrom basal MAP or HR (P�0.05).
627sEH REGULATION OF BLOOD PRESSURE IN BRAIN
increase in BP and HR appears to function throughROS. This hypothesis is supported by our data: 1)pretreatment with gp91ds-tat, an NAD(P)H oxidaseblocker prevents AUDA-induced increase in BP andHR, and 2) AUDA treatment increased neuronal firingrate, an effect attenuated by gp91ds-tat. Thus, we pro-pose that an increase in EETs by inhibition of sEH leadsto an increase in ROS in the brain and hence an increasein BP. An alternate possibility is that ROS stimulates EETs,which leads to an increase in BP. The increase in BP isassociated with dampening of the baroreceptor reflex.Whether this is a causative mechanism is unknown, butrecent data indicates that baroreceptor reflex plays acritical role in chronic and long-term control of BP.Finally, our study suggests that overexpression of sEH inthe brain may be a compensatory response that fails tooverride hypertensive conditions in the SHR. This
would be important to prove with the use of otherexperimental models of hypertension. The SHR wasexclusively chosen for this study because it exhibitsmany similarities to primary human hypertension.
This study provides a novel mechanism by which sEHregulates designated cardiovascular activity in the brainand raises important questions. For example, in whatcell type in the brain is sEH localized? We believe it isthe neuronal sEH that participates in EET-mediated BPcontrol. This is supported by our findings that neuronalcells in primary culture from the prehypertensive SHRhypothalamus and brainstem demonstrate increasedexpression of sEH similar to that seen in the adult SHRbrain. AUDA treatment caused an exclusive increase inneuronal firing in neurons from the SHR hypothala-mus and brainstem. Finally, astroglial cultures from thesame brain areas showed no significant differences inthe sEH expression between WKY and SHR. Whilethese data implicate neuronal sEH, they do not elimi-nate the role of cerebrovascular sEH, which we willinvestigate. It is important to determine what nucleus(or nuclei) in the hypothalamus and brainstem is/arekey in EETs-mediated regulation of BP and how sEH-EET-ROS-mediated signals are transmitted to the pe-ripheral system, which is translated into increases in HRand BR. We have developed a lentiviral vector-mediatedgene delivery system in selective brain nuclei andtransmitter-specific neurons in vivo in an attempt toanswer these questions. Nonetheless, these studies es-tablish that an increase gene expression for sEH ap-pears to be a protective mechanism against neuralcontrol of hypertension, an increase that fails to over-ride the hypertensive dysregulatory pathways.
Figure 2. Effect of AUDA onneuronal activity in vitro. A)sEH protein levels in neuronalcultures from 1-day-old SHRand WKY rats. Top: representa-tive audioradiograph; bottom:quantitation of sEH proteinband. *Significantly different inSHR vs. WKY (P�0.05, n�4). B)Effect of AUDA and gp91ds-taton neuronal firing of neuronscultured from SHR rat brainhypothalamus. Recordings ofaction potentials from represen-tative neurons were recorded atcontrol conditions and treat-ment with AUDA (0.4 mg/mL),AUDA � gp91ds-tat (5 �M), orAUDA � scrambled gp91ds-tat(5 �M). C) Bar graphs: means �se from 9 neurons showing thatAUDA significantly increaseneuronal firing rate; this effectwas attenuated by gp92ds-tat. *P� 0.05 compared with controlcondition. †P � 0.05 vs. AUDA.
Figure 3. Proposed mechanism of central sEH on bloodpressure regulation in the SHR. An increase in central EETsleads to an increase in blood pressure via ROS mediation. Wepropose that EETs induce the production of ROS, includingNAD(P)H oxidase. While high sEH expression in the SHRconverts EETs, inhibition of this enzyme activates this cas-cade, leading to a further increase in blood pressure.
628 Vol. 19 April 2005 SELLERS ET AL.The FASEB Journal