nanoparticle depot for intraperitoneal …
TRANSCRIPT

Purdue UniversityPurdue e-Pubs
Open Access Dissertations Theses and Dissertations
January 2016
NANOPARTICLE DEPOT FORINTRAPERITONEAL CHEMOTHERAPY OFOVARIAN CANCERBo SunPurdue University
Follow this and additional works at: https://docs.lib.purdue.edu/open_access_dissertations
This document has been made available through Purdue e-Pubs, a service of the Purdue University Libraries. Please contact [email protected] foradditional information.
Recommended CitationSun, Bo, "NANOPARTICLE DEPOT FOR INTRAPERITONEAL CHEMOTHERAPY OF OVARIAN CANCER" (2016). OpenAccess Dissertations. 1480.https://docs.lib.purdue.edu/open_access_dissertations/1480

Graduate School Form 30 Updated
PURDUE UNIVERSITY GRADUATE SCHOOL
Thesis/Dissertation Acceptance
This is to certify that the thesis/dissertation prepared
By
Entitled
For the degree of
Is approved by the final examining committee:
To the best of my knowledge and as understood by the student in the Thesis/Dissertation Agreement, Publication Delay, and Certification Disclaimer (Graduate School Form 32), this thesis/dissertation adheres to the provisions of Purdue University’s “Policy of Integrity in Research” and the use of copyright material.
Approved by Major Professor(s):
Approved by:
Head of the Departmental Graduate Program Date
Bo Sun
NANOPARTICLE DEPOT FOR INTRAPERITONEAL CHEMOTHERAPY OF OVARIAN CANCER
Doctor of Philosophy
Yoon Yeo
Chair
Tonglei Li
Rodolfo Pinal
Kinam Park
Yoon Yeo
Lynne Taylor 11/25/2015

i
NANOPARTICLE DEPOT FOR INTRAPERITONEAL CHEMOTHERAPY OF
OVARIAN CANCER
A Dissertation
Submitted to the Faculty
of
Purdue University
by
Bo Sun
In Partial Fulfillment of the
Requirements for the Degree
of
Doctor of Philosophy
May 2016
Purdue University
West Lafayette, Indiana

ii
ACKNOWLEDGEMENTS
First and foremost, I would like to express my sincere gratitude to my advisor, Dr.
Yoon Yeo. I do not know where I would be if she did not offer me the opportunity to join
her research team five years ago. Her pure passion to science, insightful suggestion, and
constant support set an example of a true scientist and a knowledgeable mentor. I really
appreciate the great patience and valuable resources that she has provided in the past five
years for me to grow. Additionally, I would like to thank my committee members, Drs.
Kinam Park, Tonglei Li, and Rodolfo Pinal for sharing their knowledge in research and
advice in career with me.
My gratitude goes to all the current members and alumni of Dr. Yeo’s lab. Your
outstanding work established the foundation of this lab, letting me see forward on the
shoulders of giants. Special thanks to Drs. Eun Jung Cho, Kyung-Oh Doh and Hillary
Holback who trained me during my first couple of years in the lab.
I am also very grateful to our collaborators, Benjamin Ramsey, Sandra
Torregrosa-Allen and Bennett Elzey and undergraduate students, Alice Chang, Ji Ae Lee
and Olivia Rivera, who have made great contribution to my work.
Hail Purdue and Boiler up!

iii
TABLE OF CONTENTS
Page
ABSTRACT ....................................................................................................................... ix
CHAPTER 1. INTRODUCTION .................................................................................... 1
1.1 Background ............................................................................................................... 1
1.2 Materials for IP Delivery Systems ............................................................................ 2
1.2.1 Requirements for IP Drug Carriers .................................................................. 2
1.2.2 Biomaterials for IP Drug Delivery .................................................................. 4
1.2.2.1 Natural Polymers ........................................................................................ 4
1.2.2.2 Synthetic Polymers ..................................................................................... 5
1.3 Dosage Forms for IP Drug Delivery ......................................................................... 5
1.3.1 Solutions .......................................................................................................... 6
1.3.2 Micro- or Nanoparticles ................................................................................... 7
1.3.3 Implantable Depots ........................................................................................ 11
1.3.4 Injectable Hydrogels ...................................................................................... 13
1.4 Challenges in IP Chemotherapy .............................................................................. 15
1.4.1 Drug Release in Peritoneal Environment ...................................................... 16
1.4.2 Tumor Specificity and Penetration ................................................................ 18
1.4.3 Tissue Responses to Carrier Materials .......................................................... 21

iv
Page
1.4.3.1 Effects of Carrier Materials on Peritoneal Tissues ................................... 21
1.4.3.2 Effects of Carrier Materials on Tumors .................................................... 22
1.5 Conclusion ............................................................................................................... 23
1.6 References ............................................................................................................... 25
CHAPTER 2. DEVELOPMENT AND CHARACTERIZATION OF
NANOPARTICLE DEPOT FOR INTRAPERITONEAL CHEMOTHERAPY ............. 41
2.1 Introduction ............................................................................................................. 41
2.2 Materials and Methods ............................................................................................ 43
2.2.1 Materials ........................................................................................................ 43
2.2.2 Preparation and Characterization of PTX Nanocrystals (PNC) .................... 44
2.2.3 Synthesis of Gel Precursors (HA-ADH, HA-CHO, CMC-CHO) ................. 45
2.2.4 Synthesis of Hydrophobically Modified Gel Precursor (EtCA-CMC-CHO) 45
2.2.4.1 Synthesis of 5β-cholanic Acid Methyl Ester (MeCA) (Scheme 1) .......... 45
2.2.4.2 Synthesis of Aminoethyl 5β-cholanoamide (EtCA) (Scheme 1) ............. 45
2.2.4.3 Synthesis of EtCA-CMC-CHO Conjugate (Scheme 2) ........................... 46
2.2.5 Preparation of Gel formulations .................................................................... 47
2.2.6 Evaluation of Gel Precursors and Crosslinked Gels ...................................... 48
2.2.6.1 Hydrophilicity of Gel Precursors .............................................................. 48
2.2.6.2 Toxicity of EtCA and EtCA-CMC-CHO ................................................. 48
2.2.6.3 Toxicity of Gels ........................................................................................ 49
2.2.7 PTX Solubility and Stability in Various Media ............................................. 49

v
Page
2.2.7.1 PTX Solubility in PBS, PBS Containing Serum, or PBS Containing
Tween 80 ................................................................................................................. 49
2.2.7.2 PTX Stability in PBS Containing Tween 80 or PBS Containing Serum .. 50
2.2.8 Dissolution Kinetics of PNC ......................................................................... 50
2.2.9 PTX Release Kinetics of HA Gel Formulations ............................................ 52
2.2.9.1 In Tween/PBS under Sink Condition ....................................................... 52
2.2.9.2 In Tween/PBS under Non-Sink Condition ............................................... 53
2.2.9.3 In FBS/PBS or Tween/PBS containing Hyaluronidase under Non-Sink
Condition ................................................................................................................. 54
2.2.10 HPLC Analysis of PTX ............................................................................... 54
2.3 Results and Discussions .......................................................................................... 55
2.3.1 Characterization of PNC and PPT ................................................................. 55
2.3.2 Synthesis and Characterization of EtCA-CMC-CHO ................................... 57
2.3.2.1 Hydrophobicity of Gel Precursors ............................................................ 58
2.3.2.2 Toxicity of EtCA and EtCA-CMC-CHO Conjugate ................................ 58
2.3.3 PTX Solubility and Stability in Various Media ............................................. 60
2.3.3.1 PTX Solubility in PBS, 50-FBS/PBS, or Tween/PBS .............................. 60
2.3.3.2 PNC Stability in PBS Containing Serum ................................................. 61
2.3.4 Dissolution Kinetics of PNC and PPT ........................................................... 62
2.3.4.1 Dissolution Kinetics of PNC and PPT with Dialysis Method .................. 62
2.3.4.2 Limitations of Current Dissolution Kinetics Study Methods and Potential
Alternatives ............................................................................................................. 64

vi
Page
2.3.5 PTX Release Kinetics from HA Gel Formulations ....................................... 65
2.3.5.1 In Tween/PBS under Sink Condition ....................................................... 66
2.3.5.2 In Tween/PBS under Non-Sink Condition ............................................... 66
2.3.5.3 In FBS/PBS or Tween/PBS containing Hyaluronidase under Non-Sink
Condition ................................................................................................................. 68
2.3.6 PTX Release Kinetics from HA-EtCA-CMC Gel ......................................... 70
2.4 Conclusions ............................................................................................................. 71
2.5 References ............................................................................................................... 73
CHAPTER 3. BIOACTIVITY EVALUATION OF NANOPARTICLE DEPOT ........ 79
3.1 Introduction ............................................................................................................. 79
3.2 Materials and Methods ............................................................................................ 80
3.2.1 Materials ........................................................................................................ 80
3.2.2 Determination of IC50 of Taxol, PPT and PNC on SKOV3 Cell Line ......... 80
3.2.3 Cellular Retention and Cytotoxicity of PTX ................................................. 81
3.2.4 Determination of the Maximum Tolerated Doses of Treatments .................. 83
3.2.5 In-vivo Efficacy Studies ................................................................................ 84
3.2.6 Statistical Analysis ......................................................................................... 84
3.3 Results and Discussion ............................................................................................ 85
3.3.1 IC50 of PTX in the Form of Taxol, PPT or PNC ........................................... 85
3.3.2 Cellular Retention and Cytotoxicity of PPT and PNC .................................. 86
3.3.3 Cytotoxicity of PPT-gel and PNC-gel ........................................................... 87
3.3.4 Maximum Tolerated Doses of PTX Treatments ............................................ 89

vii
Page
3.3.5 Anti-tumor Effects of PPT-gel and PNC-gel ................................................. 89
3.4 Conclusion ............................................................................................................... 94
3.5 References ............................................................................................................... 95
CHAPTER 4. ALBUMIN-STABILIZED PACLITAXEL NANOCRYSTALS ........... 97
4.1 Literature Review .................................................................................................... 97
4.1.1 Production of Nanocrystals ............................................................................ 98
4.1.1.1 Bottom-up Technologies ........................................................................ 100
4.1.1.2 Top-down Technologies ......................................................................... 101
4.1.1.3 Combined Technologies ......................................................................... 102
4.1.1.4 Nanocrystal Stabilization ........................................................................ 103
4.1.2 Remaining Challenges in Nanocrystal Development for Parental
Applications ............................................................................................................. 104
4.1.2.1 Instability during Storage ....................................................................... 104
4.1.2.2 Instability during Applications ............................................................... 105
4.1.2.3 Lack of Target Specificity ...................................................................... 107
4.2 Introduction ........................................................................................................... 108
4.3 Materials and Methods .......................................................................................... 109
4.3.1 Materials ...................................................................................................... 109
4.3.2 Preparation of Albumin-stabilized PNC ...................................................... 110
4.3.2.1 Crystallization in Matrix (Cim) .............................................................. 110
4.3.2.2 Nonsolvent and Temperature-Induced Crystallization ........................... 111
4.3.3 Characterization of Nanocrystals ................................................................. 112

viii
Page
4.4 Results and Discussions ........................................................................................ 112
4.4.1 Nanocrystal Morphology ............................................................................. 112
4.4.2 Nanocrystal Size .......................................................................................... 113
4.4.3 Albumin content .......................................................................................... 115
4.4.4 Proposed Role of Albumin in Cim-alb ........................................................ 115
4.5 Conclusion ............................................................................................................. 116
4.6 References ............................................................................................................. 117
CHAPTER 5. CONCLUSION ..................................................................................... 125
VITA ............................................................................................................................... 128

ix
ABSTRACT
Sun, Bo. Ph.D., Purdue University, May 2016. Nanoparticle Depot for Intraperitoneal
Chemotherapy of Ovarian Cancer. Major Professor: Yoon Yeo.
Intraperitoneal (IP) chemotherapy is a promising post-surgical therapy of ovarian
cancer, with the full potential yet to be proven. To facilitate IP chemotherapy of ovarian
cancer, we have developed a nanoparticle depot for IP chemotherapy consisted of
paclitaxel (PTX) nanocrystals (PNC) and hyaluronic acid-based hydrogel (HA gel). PNC
with a size of ~310 nm was produced by nonsolvent and temperature-induced
crystallization. Dissolution kinetics of PNC could be determined by the light scattering
method rather than the dialysis method due to drug reprecipitation caused by diffusion
barrier. PTX release profiles from PNC-gel and PTX precipitate-gel (PPT-gel) were
estimated in both sink- and non-sink conditions, where the latter simulated the peritoneal
environment. In-vitro release kinetics studies did not reveal any difference between PNC-
gel and PPT-gel, partly due to the centrifugation-related artifacts.
In cellular toxicity test and maximum tolerated dose assessment, PNC-gel
provided more efficient killing effect and greater toxicity than PPT-gel, which contained
larger PTX particles, indicating a greater dissolution rate of PNC due to the small size. A
single IP administration of PNC-gel extended the survival of mice with IP tumors
significantly better than the same dose Taxol, due to the local depot effect, whereas PPT-

x
gel was not superior to Taxol in survival extension. While the cell toxicity test and in-
vivo results consistently point to the beneficial effect of particle size reduction, in-vitro
drug release kinetics did not predict the difference between PPT- and PNC-gels,
suggesting the limitation of current release study methods.
For PNC-gel to serve the cancer patients to its full potential, the compatibility
between PNC and HA gel could be optimized by incorporating hydrophobic domains in
the hydrogel and introducing surface stabilizer on PNC to achieve a well-controlled
release. Aminoethyl 5β-cholanoamide (EtCA) was conjugated to one of the gel
precursors, and the in-vitro PTX release was enhanced by the inclusion of EtCA.
However, it was not pursued in the subsequent studies due to the unexpected toxicity of
EtCA. Albumin-stabilized PNC with a sub-200 nm particle size were prepared using a
method involving incipient crystallization in polymer matrix and subsequent surface
stabilization with albumin. The function and quantitation of surface stabilizers need
further investigation. In-vitro dissolution test and bioactivity evaluation of Cim-alb
remains to be performed to test the contribution of small size to enhancing local
availability of PTX.

1
CHAPTER 1. INTRODUCTION1
1.1 Background
The mainstay of current peritoneal malignancy treatment is surgical debulking of
visible tumors and post-surgical chemotherapy to remove residual microscopic tumors [1-
3]. Recently, intraperitoneal (IP) chemotherapy has been pursued in post-surgical
management of peritoneal malignancies, due to the promise of a high local concentration
and a longer half-life of a drug in the peritoneal cavity, which provides a unique
opportunity for the locoregional treatment of the IP malignances [4-6]. Part of the IP-
administered drugs are absorbed to systemic circulation, but it occurs at a slower rate
than those administered intravenously [7-9]; therefore, IP dosage forms can also serve as
a depot for sustained systemic drug delivery. IP chemotherapy has proven significantly
more effective than intravenous (IV) therapy in several clinical studies [1, 3, 10].
Accordingly, the National Cancer Institute issued a clinical alert to recommend IP
chemotherapy for stage III patients with optimally debulked ovarian cancer in 2006 [11].
On the other hand, several challenges remain to be overcome before IP
chemotherapy to make a standard protocol for post-surgical management of peritoneal
1 The content of this chapter has been previously published in Ceeln, W.P. and Levine, E.A., eds.,
Intraperitoneal Cancer Therapy: Principles and Practice. CRC Press/Taylor & Francis Group, Boca Raton,
FL, 2016.

2
malignancies. For example, IP-administered drugs show limited penetration into tumors
[12], thus necessitating the use of high IP doses. The high IP doses in turn account for
increased toxicities such as myelotoxicity, neurotoxicity, nephrotoxicity, nausea,
vomiting, and abdominal pain [11, 13, 14]. Cumulative toxicities reduce options for
subsequent rounds of therapy [15]. Moreover, complications related to IP administration,
such as discomfort due to prolonged infusion, catheter implantation, and peritoneal
adhesion, result in poor quality of life and, thus, high rate of dropout prior to the
completion of planned treatment [1, 14].
While the long list of challenges seems discouraging, this leaves the formulation
scientists with several questions: Can drug delivery systems help overcome any of these
problems? What are the unique requirements for IP drug delivery? What needs to be
done and what has been done to improve drug delivery to the peritoneal cavity? In this
chapter, we intend to address these questions by reviewing recent literature concerning IP
drug delivery. We will discuss experimental approaches to improve the effectiveness of
IP chemotherapy, focusing on the biomaterials used as drug carriers and various dosage
forms that have been reported to date. The chapter will conclude with a discussion of
remaining challenges and future perspectives.
1.2 Materials for IP Delivery Systems
1.2.1 Requirements for IP Drug Carriers
Typical first-line chemotherapeutic agents such as paclitaxel (PTX), docetaxel
(DTX) and cisplatin are low molecular weight drugs (<20 kDa), which are absorbed

3
through the peritoneal capillaries and enter the systemic circulation in a few hours [7-9].
The short residence time not only compromises the effectiveness of local chemotherapy
but also requires frequent or continuous dosing, culminating in complications related to
catheters and infection [16]. For the delivery of low molecular weight drugs, it is
therefore important to attenuate fast systemic absorption and maintain a high local
concentration of a drug. Ideally, the IP drug carriers should provide a sustained drug
release to maintain the local drug concentration within an effective range over several
weeks. The sustained local delivery of chemotherapy is also found beneficial for avoiding
tumor repopulation, which can occur during drug-free cycles in conventional intermittent
therapy [17]. In addition, it is desirable that the carriers are degraded into molecules that
are readily absorbed and cleared from the body by the time the loaded drug is exhausted
so that surgical removal of empty carriers may not be necessary.
While the primary goal of the IP delivery system is to remain in the peritoneal
cavity and provide a local reservoir of a drug for a prolonged period, such an effort often
faces a challenge due to the sensitivity of the peritoneal cavity to foreign materials. The
peritoneal cavity is responsible for protecting the body from breaches in the integrity of
the gut; therefore, it is armed with powerful innate and adaptive immune mechanisms
[18]. When confronted by an insult, peritoneal mesothelial cells, polymorphonuclear
neutrophils, and the resident peritoneal-associated lymphoid tissues interact with one
another via chemical signaling to produce inflammatory responses to the foreign
materials [18]. Due to this sensitivity, some biomaterials typically considered
biocompatible are found to induce significant inflammatory responses such as peritoneal

4
adhesions [19, 20]. Therefore, in designing an IP drug delivery system, it is necessary to
apply more stringent criteria for the selection of biomaterials for formulations.
1.2.2 Biomaterials for IP Drug Delivery
A list of polymers used for other biomedical applications is a good starting point
for selection of drug carrier materials. In particular, biomaterials used for peritoneal
adhesion prevention are great candidates for IP drug delivery. Several natural and
synthetic polymers have been used clinically and experimentally as physical barrier
devices, as reviewed elsewhere in detail [21].
1.2.2.1 Natural Polymers
Polysaccharides such as hyaluronic acid (HA) [22-26], cellulose derivatives [26-
28], dextran [29-31], and chitosan [20, 32, 33] have been explored for IP application. The
popularity of these polysaccharides stems from the biocompatibility proven in various
biomedical applications. The polysaccharides are chemically modified into reactive
precursors, which can form crosslinkable hydrogels upon application [20, 34, 35]. For
example, HA is modified into two types of precursors - one with adipic dihydrazide and
the other oxidized to have aldehydes, which instantly form a hydrogel upon contact [25].
The polysaccharide-based hydrogels are enzymatically degraded; therefore, the
degradation rate can be controlled by combining polymers with different enzyme
susceptibility. For example, the degradation rate of HA gel in the peritoneal cavity was
extended by replacing one of the gel precursors with cellulose derivatives, which were

5
not degraded by human enzymes [35]. For the delivery of hydrophobic drugs, it is likely
necessary to balance the hydrophobicity and hydrophilicity of the polymer [36]. For this
purpose, it is conceivable to modify the polymer with hydrophobic moieties to increase
the compatibility between drugs and polymers [37, 38]. Chitosan has also been widely
explored as a local depot of chemotherapeutic agents, where various stimuli (light, pH,
temperature) are employed as a trigger to form a hydrogel in-situ [39].
1.2.2.2 Synthetic Polymers
Synthetic polymers used for the prevention of peritoneal adhesion and IP
chemotherapy include polylactic acid (PLA), poly(lactide-co-glycolide) (PLGA),
polyethylene glycol (PEG), poly(Ɛ-caprolactone) (PCL) and their block co-polymers [26,
40-44]. Most of these polymers are commercially available at reasonable prices. In
particular, PLGA is widely used as a drug carrier [45] or a device [46, 47] because of its
track record in the approved products and the well-known biodegradability and
biocompatibility [21]. An advantage of synthetic polymers over natural polymers is that it
is relatively easier to control the molecular weight, monomer composition, and structure
of the polymer; therefore, there is greater flexibility in delivering various types of drugs
[43, 44].
1.3 Dosage Forms for IP Drug Delivery
The most common form of IP chemotherapy is the repurposed IV solutions. To
increase the drug retention in the peritoneal cavity, viscous polymer solutions, micro- or

6
nanoparticle formulations, implantable polymeric depots, and hydrogel-based systems
have been explored [21, 34, 48, 49].
1.3.1 Solutions
PTX is poorly water-soluble and, thus, requires a solubility enhancer. An equal
parts mixture of ethanol and Cremophor EL (polyethoxylated castor oil) is used to
solubilize PTX in Taxol® [50, 51]. Alternative solubilization strategies are pursued to
reduce toxicities related to Cremophor EL. For example, PTX and randomly methylated-
β-cyclodextrin (RAME-β-CD) form water-soluble inclusion complexes, which does not
precipitate upon dilution and stay stable after 24 h storage at ambient temperature or 2 h
at 41.5 °C [52]. When used for hyperthermic peritoneal perfusion, PTX/RAME-β-CD
complexes showed 40-fold higher plasma concentration than Taxol in a rat model [53]
and delayed the growth of peritoneal carcinomatosis [54]. The authors argued that
PTX/RAME-β-CD complexes had a greater ability to penetrate into IP tumors than Taxol,
which entrapped PTX in surfactant micelles and thus limited direct tumor exposure of the
drug [53].
A viscous solution of hydroxyethyl starch was used for the IP delivery of PTX [55]
and DTX [56]. Sprague Dawley rats were administered IP with the taxane compounds
using 6% hydroxyethyl starch (hetastarch) or 1.5% dextrose peritoneal dialysis solution
as a carrier. Fluid clearance and mean taxane concentrations in plasma were lower when
the drugs were delivered with hetastarch solution than with the peritoneal dialysis
solution. Importantly, the total amount of drug remaining in the peritoneal cavity was

7
significantly higher with hetastarch solution [55, 56]. These studies demonstrate that
hetastarch solution helped retain taxane compounds in the peritoneal cavity and reduce
systemic exposure to the drugs.
1.3.2 Micro- or Nanoparticles
A tumor penetrating microparticles (TPM) loaded with PTX was designed for IP
drug delivery [49]. The TPM system consisted of two types of particles. One was priming
TPM, which encapsulated PTX in a low molecular weight PLGA (LA:GA=50:50) and
released the drug rapidly to “prime” the tumors: i.e., to expand the interstitial space via
apoptosis induction and enhance the penetration of the second type of particles into
tumors. The second component called sustaining TPM encapsulated PTX in a high
molecular weight PLGA (LA:GA=75:25) and, thus, provided sustained drug release to
kill tumor cells. TPMs were able to retain in the peritoneal cavity for a longer time,
achieve greater therapeutic effect and lower toxicity than Taxol [49]. The tumor-priming
technology was later used to promote the delivery of survivin siRNA, which targeted a
gene encoding survivin, an anti-apoptotic protein associated with metastases and poor
prognosis of patients with gastric and colorectal cancers [57]. Here, siRNA was
encapsulated in PEGylated cationic liposomes (PCat) were administered IP after TPM
treatment of IP tumors. The combination of PTX-loaded TPM and PCat-siSurvivin was
more effective than each treatment in suppressing tumor growth due to the synergy of the
two treatment: TPM enhancing the penetration of PCat-siSurvivin into peritoneal tumors

8
and PCat-siSurvivin reducing survivin expression and augmenting TPM-induced anti-
proliferation and apoptosis [57].
A nanocrystal (NC) form (sub-micron drug particles stabilized by surfactants
and/or polymers) of PTX has been used in conjunction with hyperthermia for IP
chemotherapy [58]. PTX NCs were produced using Pluronic F127 as a stabilizer and
administered to rats bearing peritoneal tumors via IP perfusion at 41.5°C for 45 min. The
PTX NCs showed similar anti-tumor activity as Taxol with relatively low apparent
toxicity [58]. Unlike Taxol, the blood level of PTX continued to increase even after the
discontinuation of NC HIPEC, which suggests the long-term residence of NCs in the
peritoneal cavity due to their mucoadhesive properties [58].
Polymeric micelles have been used for delivery of multiple drugs to IP tumors
[42]. A combination of three drugs with distinct mechanisms of action-PTX as a
cytotoxic drug, cyclopamine (CYP) as an inhibitor of hedgehog signaling to reverse
taxane resistance, and gossypol (GSP) as a proapoptotic compound-was encapsulated in
poly(ethylene glycol)-block-poly(ε-caprolactone) (PEG-b-PCL) micelles for co-delivery
to tumors. Simultaneous encapsulation of three drugs in a single polymeric micelle was
possible as they have similarly poor water solubility [42]. The 3-drug loaded micelles
were able to disaggregate three dimensional spheroids of ES-2 human ovarian cancer
cells, whereas micelles with a single-drug or 2-drug combination showed negligible
effects on tumor spheroids. In mouse models of IP xenograft, the IP administered 3-drug
micelles reduced tumor burden to a greater extent than PTX alone and vehicles controls
[42]. Following debulking and induction of apoptosis of IP tumors with the 3-drug
micelles, secondary micelles with a near-infrared (NIR) fluorescence probe, DiR, was

9
systemically administered to visualize and help remove residual tumors (Fig. 1) [59].
Here the secondary micelles were conjugated with a peptide having high affinity for
apoptotic tissues (externalized phosphatidylserine) so that the secondary micelles could
specifically visualize apoptotic tumors [59]. A similar approach was used for systemic
administration of drug combinations and visualization of residual tumors [60, 61].
Figure 1. Schematic illustration of two-step strategy for neoadjuvant therapy, apoptosis-
targeted optical imaging and intraoperative surgical guidance, enabled by a tandem of
PEG-b-PCL micelles. (Reprinted from Cho, H. and Kwon, G.S., PLos One, 9,e89968,
2014, per Creative Commons Attribution (CC BY) license.)
While the particle systems in the above examples are intended for local drug
delivery, they also provide a useful tool for lymphatic or systemic drug delivery. Particles
administrated in the peritoneal cavity are drained into lymphatic circulation (Fig. 2) [48,
62]. Within the lymphatic system, particles smaller than 50 nm pass through lymph nodes
and reach systemic circulation via thoracic lymph ducts [62]. Kohane et al reported that
even larger PLGA nanoparticles (265 nm) entered systemic circulation from the
peritoneal cavity in less than 2 days, ending up in the spleen and the liver [40]. On the
other hand, larger particles (>500 nm) are trapped in lymph nodes. Therefore, depending

10
on the particle size, nanoparticles can serve as a sustained systemic delivery system or for
targeting cancer cells spreading via lymphatics.
Conversely, microparticles (1-100 µm) are retained longer in the peritoneal cavity
and, thus, have a greater potential to enhance local availability of a drug in the peritoneal
cavity and attenuate systemic drug absorption than nanoparticles [48, 49] (Fig. 2).
However, the long-term residence of microparticles in the peritoneal cavity can cause
inflammatory tissue responses and peritoneal adhesions [40], which may offset the
benefits of the localized medicine. For this reason, a combination of nanoparticles and
hydrogels was proposed as an alternative option. Here, nanoparticles served as a
sustained drug delivery system and hydrogels prevented premature clearance of
nanoparticles from the peritoneal cavity [22]. Additionally, the hydrogel prevented the
retained polymeric nanoparticles from causing adhesions [22].

11
Figure 2. A model of kinetic processes during IP administration of nano- or
microparticles. Nanoparticles are drained through the lymphatic system, reaching
systemic circulation or trapped in the lymph nodes depending on the size. Microparticles
tend to stay in the peritoneal cavity. Free drug released from particles are absorbed
directly to IP tissues or systemic circulation. Consequently, microparticles can maintain a
higher drug concentration in the peritoneal cavity than nanoparticles, whereas
nanoparticles yield a higher rate and extent of systemic absorption than microparticles.
(Modified from Tsai, M. et al., Pharm. Res., 24, 1691, 2007.)
1.3.3 Implantable Depots
Chitosan-phospholipid implantable formulations have been developed for
sustained and localized delivery of taxane compounds to ovarian tumors in the peritoneal
cavity [63-65]. The films were composed of chitosan and egg phosphatidylcholine (ePC)
(Chitosan-ePC film) blended with PLA-b-PEG [66], PLA-b-PEG/PLA [67], or PLGA
nanoparticles [68] containing PTX. PTX release from the nanoparticle/chitosan-ePC
films was evaluated in lysozyme-containing PBS [66], cell culture medium [68], or
ascitic fluid [67]. PTX was released over three months with no significant initial burst
Nano- or microparticles
Free drug
Peritoneal cavity Plasma
Free drug
Lymph node
Tissue
Lymphatic drainage Nanoparticles
Free drug
Absorption through peritoneumD
ose
rec
ove
red
Pla
sma
leve
l
Time Time
Nanoparticles
Microparticles Nanoparticles
Microparticles
[Drug]-time profile in peritoneal lavage sample
[Drug]-time profile in Plasma
Elimination

12
release as the chitosan-ePC matrix swelled and degraded by lysozyme [66-68]. Animals
implanted with chitosan-ePC films IP showed no signs of fibrous encapsulation or
inflammation around the implant area over 2-4 weeks, indicating good biocompatibility
of the implants [68]. The maximum tolerable dose (MTD) of PTX was 280 mg/kg/week
when delivered with the nanoparticle/chitosan-ePC film, much higher than Taxol with the
MTD of 20 mg/kg/week [64]. The PTX/nanoparticle/chitosan-ePC film significantly
enhanced the anti-tumor efficacy as compared to Taxol at total dose of 60 mg/kg [64, 65].
It is noticeable that continuous sustained delivery of PTX by PTX/nanoparticle/chitosan-
ePC film (60 mg/kg) was superior to intermittent Taxol administration (20 mg/kg q7d 3
schedule) at an equivalent dose and administration route (IP) in suppressing tumor
growth and extending animal survival [65]. Moreover, intermittent Taxol administration
resulted in a significant increase of tumor proliferation indices as compared with non-
treated controls, which indicates potential repopulation of tumors during treatment-free
intervals [65]. In contrast, PTX/nanoparticle/chitosan-ePC film is thought to increase
tumor responsiveness to the treatment by continuously exposing the tumors to high
concentration of PTX by localizing the drug source in the peritoneal cavity [65]. In
addition, PTX/nanoparticle/chitosan-ePC film did not induce the expression of multidrug
resistance gene (MDR1) in-vivo, whereas intermittent Taxol administration caused
significant MDR1 expression [69]. These results demonstrate the benefits of local
sustained drug delivery to IP tumors.

13
1.3.4 Injectable Hydrogels
The implantable films are an effective way of localizing chemotherapy in the
peritoneal cavity, but the fact that it requires surgical implantation poses challenges in
administration and patient compliance [70]. Therefore, the implantable film was replaced
with an injectable depot (PoLigel), composed of a water-soluble chitosan derivative, ePC,
and a fatty acid analog [70]. This mixture formed a gel at physiological temperature via
hydrophobic interaction between water-soluble chitosan and acyl chains of ePC,
reinforced by their interactions with a fatty acid analog [70]. Biocompatibility of the
PoLigel depended on the fatty acid analog–lauric aldehyde was better tolerated than
lauric chloride when injected subcutaneously in mice and observed over 4 weeks [71].
PoLigel was used for sustained and localized delivery of DTX to murine models of
SKOV3 ovarian cancer [63, 72]. DTX loaded in PoLigel (DTX/PoLigel) showed
constant release kinetics over 2 weeks with a minimal initial burst release in 0.01M PBS
(pH 7.4) containing lysozyme and albumin [63]. DTX/PoLigel IP administrated to mice
maintained constant DTX levels in plasma and peritoneal tissues over 2 weeks without
causing significant toxicity or inflammation [63]. Tumor burden was reduced by >70% in
DTX/PoLigel-treated group as compared with saline control [63]. Similar to the
PTX/nanoparticle/chitosan-ePC film [65], DTX/PoLigel achieved a greater anti-tumor
effect than intermittent Taxol injections, which was partly attributed to antiangiogenic
effect of local DTX, continuously released by PoLigel [73]. In addition to local tumors,
the IP administrated DTX/PoLigel was capable of delivering DTX to subcutaneous
tumors distant from the peritoneal cavity [72]. This result demonstrates that IP sustained

14
and localized delivery platform can treat not only local tumors but also metastasis distal
to the site of administration via the systemically absorbed drug. The PoLigel was also
used for co-delivery of DTX and cepharanthine, where the latter inhibited multidrug
resistance (MDR) due to drug efflux transporters and helped manage refractory ovarian
cancer with the MDR phenotype [74].
Another type of injectable gel used for IP chemotherapy is a thermosensitive
hydrogel based on PLGA-b-PEG-b-PLGA tri-block copolymers [75]. These polymers are
soluble in water, form a free-flowing solution at low temperature (e.g., 4°C), but
spontaneously gels at body temperature to create a water-insoluble gel called ReGel® [75].
ReGel® (23w/w%) injected subcutaneously into rats maintained the gel structure for 2
weeks but degraded into a viscous liquid and gradually resorbed in the next 2 weeks. The
hydrophobic segments (PLGA) formed hydrophobic regions in the gel, in which poorly
water-soluble drugs could be encapsulated. ReGel® loaded with PTX (OncoGel™)
released ~40% of the loaded PTX in the first 10 days and continuously released the
remaining payload in the next 40 days. In subcutaneous injection and the FDA Modified
Biocompatibility Tests, ReGel caused no signs of inflammation and significant toxicities,
confirming its biocompatibility [75]. OncoGel has been evaluated in local therapy of
solid tumors through preclinical and clinical studies and considered a promising adjuvant
therapy for ovarian cancer [76]. ReGel® was recently evaluated as a carrier of a three-
drug combination (PTX and two protein inhibitors) for IP chemotherapy [77]. The three-
drug loaded ReGel (Triogel) IP injected as a free-flowing solution formed a depot at body
temperature and released drugs at an equal rate according to gel erosion. The IP
administered Triogel showed greater antitumor efficacy and lower systemic toxicity than

15
IP or IV administrated PEG-b-PLA micelles with the same payloads in ES-2 ovarian
cancer xengraft model [77]. These results demonstrated the potential of Triogel as a
multi-drug carrier for IP chemotherapy of ovarian cancer.
While the PoLigel or ReGel form gels through non-covalent interactions between
polymer chains, polymers with functional groups that can react in-situ and form covalent
crosslinking to make hydrogels are also used for IP drug delivery [78]. The HA
derivatives that formed in-situ hydrogels via hydrazone bond (Section 2.2.1 ) were
evaluated for the prevention of post-surgical adhesion as a physical adhesion barrier
and/or drug carrier, showing excellent biocompatibility in the peritoneal cavity and
effectiveness in adhesion prevention [22, 34, 79, 80]. Accordingly, the in-situ
crosslinkable HA hydrogels have also been evaluated as an IP drug delivery system for
local therapy of cancers in the peritoneal cavity [36, 81].
1.4 Challenges in IP Chemotherapy
The above examples illustrate that various drug delivery systems deliver standard
anti-cancer drugs to the peritoneal cavity and help manage peritoneal malignancies by
providing sustained drug release and maintaining high local drug concentration. However,
several challenges remain to be addressed before IP chemotherapy becomes a standard
therapeutic option in the clinic.

16
1.4.1 Drug Release in Peritoneal Environment
The volume of peritoneal fluid in a healthy adult is about 50 mL with a turnover
rate of 4-5 mL/h, whereas blood volume is ~5 L [82]. The protein content of peritoneal
fluid is 25% of that in the blood [83]. Although malignancies may raise the protein level
and accelerate the turnover in the ascites, the relatively small volume of the peritoneal
fluid poses challenges to the IP delivery of poorly water-soluble drugs. Our previous
effort to deliver PTX with HA-based hydrogel provides an example of such a challenge
[36]. Here, PTX was mixed in HA hydrogel precursor solutions in the form of Taxol or
concentrated DMSO solution and administered IP to tumor-bearing mice so that the
precursors could form a hydrogel in-situ [36]. Upon dilution in the gel precursor solutions,
the concentrated PTX/DMSO solution started to form micrometer-scale precipitates
(PPTs), whereas Taxol maintained the micelle size (14 nm) [36]. The large particle size
of the former and HA hydrogel helped maintain PTX in the peritoneal cavity over 2
weeks. However, the prolonged retention of PPT-hydrogel did not translate to an
improved anti-tumor effect, due to the large size of PPTs and, thus, limited dissolution of
PTX [36]. This result indicates that hydrogel as a delivery medium helps localize drug in
the peritoneal cavity but does not prevent precipitation of poorly water-soluble drug,
limiting its availability to the local tumors.
A potential solution to this problem may be to reduce the particle size to facilitate
drug dissolution; however, it should not be as small as Taxol micelles as they are not
retained in the hydrogel [36]. Therefore, the challenge is to produce drug particles with
an optimal size, large enough to remain in the gel but small enough to allow continuous

17
and unhindered drug dissolution. There are several methods to produce drug particles in a
specific size range [84]. For example, nanocrystallization is a technique to produce
crystalline particles of poorly water-soluble drugs in the nanometer range (i.e., NCs). Due
to the size and, thus, the high surface area to volume ratio, NCs can increase the
dissolution rate of drug particles [85]. The unique advantage of NCs is that they are
mainly composed of drug molecules and create little concern for the safety of excipients
[86]. A minimal amount of surface stabilizers are however needed to prevent aggregation
of NCs during their lifetime [87]. For example, ionic surfactants such as sodium cholate,
sodium deoxycholate, and sodium lauryl sulfate are used to stabilize NCs via electrostatic
repulsion. Alternatively, NCs are stabilized with amphiphilic polymers that establish a
steric barrier against aggregation [86]. Another strategy is to use proteins as stabilizers
[88]. Serum proteins, such as human serum albumin (HSA), can serve as a stabilizer due
to their ability to adsorb onto hydrophobic surfaces and therefore provides steric
hindrance to NC aggregation and growth [89, 90]. Moreover, serum proteins can interact
with cell membrane to facilitate cellular uptake of anticancer drugs in tumors [91]. We
have recently used PTX NCs stabilized with HSA for IP delivery with hydrogels (Fig. 3)
and observed that the NC-hydrogel hybrid system had a superior anti-tumor activity than
Taxol at an equivalent total dose in a murine model of IP tumors.

18
Figure 3. Schematic illustration of an injectable depot system, composed of an in-situ
crosslinkable hydrogel and albumin-stabilized PTX NCs.
1.4.2 Tumor Specificity and Penetration
Reduced blood flow, increased interstitial fluid pressure, and high collagen
density interfere with drug transport into solid tumors [92]. In addition, tumor hyaluronan
[93] and stromal cells [94] in the tumor extracellular matrix aggravate the difficulty in
drug penetration into tumors. While the tumor penetration is a general issue in
chemotherapy of solid tumors [95], IP delivery faces a greater challenge than IV or
intratumoral administrations in tumor penetration because IP administrated drugs mostly
approach the tumor tissue from the periphery rather than the interior of the tumors (Fig.
4). In-vitro studies using multicellular layers [96-99] and tumor spheroids [100, 101]
have shown that limited drug penetration results in constant tumor exposure to a sub-
optimal level of drug, making an important mechanism for tumor resistance against
chemotherapy. An approach to address this challenge is to loosen up the tumor matrix
prior to chemotherapy to alleviate the drug penetration barrier [102, 103]. This strategy
involved two-step chemotherapy with an interval, where the first treatment with pro-
apoptotic agent like PTX induced reduction in cell density of solid tumors and allowed
In-situ cross-linkable hydrogel
PTX NCs
Paclitaxel Albumin

19
the subsequent dose to reach the interior of the tumors. This study provided a proof of
concept for the development of TPM for IP delivery of anticancer drugs or siRNA to
peritoneal malignances (Section 3.2) [49, 57]. Manipulating tumor microenvironment is
another way of improving delivery and intratumoral distribution of a drug or a
nanoparticulate drug delivery system [104]. For example, local mild hyperthermia was
applied to tumors to improve vasculature permeability, perfusion, and interstitial fluid
flow [105]. Local hyperthermia at 41°C increased tumor vasculature permeability and
allowed liposomes (~85 nm) to extravasate into the interstitial space (Fig. 5) [105]. This
effect may explain the increasing popularity and positive clinical outcomes of IP
chemotherapy combined with hyperthermia (hyperthermic intraperitoneal chemotherapy,
HIPEC) [106-113]. Recently, several new approaches have been proposed to improve
drug delivery to the interior of solid tumors [114, 115]. These approaches facilitate drug
penetration into tumors by enhancing interactions between drug carriers and tumor
stroma [116], preventing intracellular sequestration of a drug [117], or addressing
hypoxic region of tumors prior to standard chemotherapy [118]. Although these studies
have not been presented in the context of IP chemotherapy, it is worthwhile to explore
their applicability in the treatment of peritoneal malignances.

20
Figure 4. Schematic illustration of poor penetration of IP administered drug. Drug
approaching tumors from the peritoneal cavity has a limited depth of penetration, which
aggravates with increasing carrier size.
Nanoparticles
Released drug
Organ stroma
Basement membrane
Cancer cells
Organ epithelium

21
Figure 5. (a) Liposome (red) extravasation through tumor vasculature (green) with or
without mild hyperthermia for 1 h. (b) Quantification of liposome extravasation through
tumor vasculature in 4 tumor models under local mild hyperthermia at 41 °C for 1 h. NEF,
normalized extravascular fluorescence. Bar, 200 μm. Reprinted with permission from Li,
L., Koning, G.A., et al. J. Control. Release, 130-137, Copyright 2013 Elsevier.)
1.4.3 Tissue Responses to Carrier Materials
1.4.3.1 Effects of Carrier Materials on Peritoneal Tissues
As mentioned in section 2.1, careful selection of biomaterials as a drug carrier is
particularly important for IP application because of the high sensitivity of the peritoneal
cavity to foreign insults. Biomaterials generally considered biocompatible or wildly used

22
in other biomedical applications have caused inflammatory responses and peritoneal
adhesion upon IP application. For example, PLGA microparticles (5 µm) induced
adhesions 2 weeks after IP injection in mice [40]. This problem worsened with increasing
molecular weight of the polymer [40]. A photo-crosslinkable chitosan derivative, which
showed no attractive interactions or proliferative effect on mesothelial cells and
macrophages in-vitro, caused extensive and persistent peritoneal adhesions in animals
receiving it as a hydrogel in the peritoneal cavity [20]. It was later learned that the parent
chitosan had the same effect in-vivo [20]. Both chitosans had significant pro-
inflammatory properties, which might have been tolerated in other locations but not in the
peritoneal cavity.
1.4.3.2 Effects of Carrier Materials on Tumors
Drug carriers, once they are proven biocompatible, are often used under an
assumption that they have no other roles than delivering the payload to the body. Our
recent studies suggest otherwise. We have delivered platinum into the peritoneal cavity
using HA nanoparticles and hydrogels IP in mice for local chemotherapy of ovarian
cancer. We find that they do not show a greater anti-tumor efficacy than platinum
solution but rather cause a slight increase in tumor burdens at later time points, which
suggests a potential involvement of empty carriers and degradation products in the
growth of residual tumors. This hypothesis is not groundless, given various biological
roles of HA. HA is an indispensable extracellular matrix macromolecule for cell
migration, differentiation and proliferation [119]. Endogenous HA is implicated in the
invasion and growth of several tumor cells [119-122]. CD44, a well-known receptor of

23
HA, is found at the surface of various tumor cells and positively correlated with poor
prognosis of ovarian cancer patients [123]. HA concentration in tumor stroma is
considered an essential indicator of tumor aggressiveness and overall survival [124, 125].
However, the role of exogenous HA in tumor invasion and growth remains controversial.
Picaud et al reported that HA-carboxymethyl cellulose (CMC) membrane had no effect
on the proliferation of tumor cells in-vitro and in-vivo [126]. Similarly, HA-based anti-
adhesion membrane was found to have no influence on metastasis of colon cancer in a
human xenograft/nude mouse model [127]. On the other hand, Tan et al reported that HA
solution promoted proliferation and motility of colorectal tumor cell lines and increased
peritoneal tumor load as compared with non-treated control group, confirming its positive
role on metastatic potential of colorectal tumors [128]. Although the exact effect of HA-
based biomaterials on IP tumors and its role remain to be investigated, the mixed results
beg a question – if the drug carrier is indeed biologically inert and does no harm after
when it remains in the body after complete exhaustion of the payload. This means that in
designing a new drug carrier one should consider not only the biocompatibility of the
material but also its biological effects on residual tumors, which may not be readily
predicted from routine toxicity testing.
1.5 Conclusion
The premise of IP chemotherapy in the treatment of malignant diseases confined
in the peritoneal cavity lies in the theoretical potential for increased exposure of the
tumors to anti-cancer drugs and improved toxicity to local tumors [12]. Although the

24
proof of concept has been demonstrated in several clinical trials, current practice of IP
chemotherapy leaves plenty of room for improvement in delivery methods. Solutions,
micro- or nanoparticles, implantable depots, and in-situ crosslinkable hydrogels have
been evaluated in the context of IP chemotherapy, achieving varying levels of success in
preclinical studies. Due to the unique biological environment of the peritoneal cavity,
several challenges remain to be overcome before the IP drug delivery systems can benefit
patients to the full potential. Despite the needs and gravity of the challenges, there are
surprisingly few players in the field of IP drug delivery systems. It means that this is a
prime time to explore opportunities in IP drug delivery.

25
1.6 References
1. Armstrong, D.K., et al., Intraperitoneal Cisplatin and Paclitaxel in Ovarian
Cancer. New Eng J Med, 2006. 354(1): p. 34-43.
2. Ozols, R.F., et al., Phase III trial of carboplatin and paclitaxel compared with
cisplatin and paclitaxel in patients with optimally resected stage III ovarian
cancer: a Gynecologic Oncology Group study. J Clin Oncol, 2003. 21(17): p.
3194-3200.
3. Alberts, D.S., et al., Intraperitoneal Cisplatin plus Intravenous
Cyclophosphamide versus Intravenous Cisplatin plus Intravenous
Cyclophosphamide for Stage III Ovarian Cancer. New Eng J Med, 1996. 335(26):
p. 1950-1955.
4. Markman, M., Intraperitoneal drug delivery of antineoplastics. Drugs, 2001.
61(8): p. 1057-1065.
5. Markman, M., et al., Phase I trial of intraperitoneal taxol: a Gynecoloic
Oncology Group study. J Clin Oncol, 1992. 10(9): p. 1485-1491.
6. Dedrick, R.L., et al., Pharmacokinetic rationale for peritoneal drug
administration in the treatment of ovarian cancer. Cancer Treat Rep, 1978. 62(1):
p. 1-11.
7. Marchettini, P., et al., Docetaxel: pharmacokinetics and tissue levels after
intraperitoneal and intravenous administration in a rat model. Cancer Chemother
Pharmacol, 2002. 49(6): p. 499-503.

26
8. Nemes, K.B., et al., Oral, intraperitoneal and intravenous pharmacokinetics of
deramciclane and its N-desmethyl metabolite in the rat. J Pharm Pharmacol, 2000.
52(1): p. 47-51.
9. Krasner, C.N., et al., Case 11-2006. New Eng J Med, 2006. 354(15): p. 1615-
1625.
10. Markman, M., et al., Phase III Trial of Standard-Dose Intravenous Cisplatin Plus
Paclitaxel Versus Moderately High-Dose Carboplatin Followed by Intravenous
Paclitaxel and Intraperitoneal Cisplatin in Small-Volume Stage III Ovarian
Carcinoma: An Intergroup Study of the Gynecologic Oncology Group,
Southwestern Oncology Group, and Eastern Cooperative Oncology Group. J
Clinl Oncol, 2001. 19(4): p. 1001-1007.
11. NCI, NCI Clinical Announcement on Intraperitoneal Therapy for Ovarian Cancer.
2006: p. http://ctep.cancer.gov/highlights/docs/clin_annc_010506.pdf.
12. Markman, M., Intraperitoneal antineoplastic drug delivery: rationale and results.
The Lancet Oncol, 2003. 4(5): p. 277-283.
13. Zeimet, A., et al., Pros and Cons of Intraperitoneal Chemotherapy in the
Treatment of Epithelial Ovarian Cancer. Anticancer Res, 2009. 29(7): p. 2803-
2808.
14. Wenzel, L.B., et al., Health-Related Quality of Life During and After
Intraperitoneal Versus Intravenous Chemotherapy for Optimally Debulked
Ovarian Cancer: A Gynecologic Oncology Group Study. J Clin Oncol, 2007.
25(4): p. 437-443.

27
15. Armstrong, D.K., Relapsed Ovarian Cancer: Challenges and Management
Strategies for a Chronic Disease. Oncologist, 2002. 7(suppl 5): p. 20-28.
16. Poveda, A., et al., Update in the management of ovarian and cervical carcinoma.
Clin Transl Oncol, 2007. 9(7): p. 443-451.
17. Vassileva, V., et al., Effects of sustained and intermittent paclitaxel therapy on
tumor repopulation in ovarian cancer. Mol Cancer Ther, 2008. 7(3): p. 630-637.
18. Hall, J.C., et al., The pathobiology of peritonitis. Gastroenterology, 1998. 114(1):
p. 185-196.
19. Dufrane, D., et al., The influence of implantation site on the biocompatibility and
survival of alginate encapsulated pig islets in rats. Biomaterials, 2006. 27(17): p.
3201-3208.
20. Yeo, Y., et al., Peritoneal application of chitosan and UV-cross-linkable chitosan.
J Biomed Mater Res A, 2006. 78A(4): p. 668-675.
21. Yeo, Y. et al., Polymers in the prevention of peritoneal adhesions. Eur J Pharm
Biopharm, 2008. 68(1): p. 57-66.
22. Yeo, Y., et al., In situ cross-linkable hyaluronan hydrogels containing polymeric
nanoparticles for preventing postsurgical adhesions. Ann Surg, 2007. 245(5): p.
819-824.
23. Burns, J.W., et al., Prevention of tissue injury and postsurgical adhesions by
precoating tissues with hyaluronic acid solutions. J Surg Res, 1995. 59(6): p. 644-
652.
24. Rodgers, K.E., et al., Reduction of adhesion formation with hyaluronic acid after
peritoneal surgery in rabbits. Fertil Steril, 1997. 67(3): p. 553-558.

28
25. Bulpitt, P., et al., New strategy for chemical modification of hyaluronic acid:
preparation of functionalized derivatives and their use in the formation of novel
biocompatible hydrogels. J Biomed Mater Res, 1999. 47(2): p. 152-169.
26. Park, S.N., et al., Preparation and characterization of biodegradable anti-
adhesive membrane for peritoneal wound healing. J Mater Sci Mater Med, 2007.
18(3): p. 475-482.
27. Leach, R.E., et al., Reduction of postsurgical adhesion formation in the rabbit
uterine horn model with use of hyaluronate/carboxymethylcellulose gel. Fertil
Steril, 1998. 69(3): p. 415-418.
28. Caicco, M.J., et al., Characterization of hyaluronan–methylcellulose hydrogels
for cell delivery to the injured spinal cord. J Biomed Mater Res A, 2012: p. 1472-
1477.
29. Hudson, S.P., et al., Injectable in situ cross-linking hydrogels for local antifungal
therapy. Biomaterials, 2010. 31(6): p. 1444-1452.
30. Ito, T., et al., Dextran-based in situ cross-linked injectable hydrogels to prevent
peritoneal adhesions. Biomaterials, 2007. 28(23): p. 3418-3426.
31. Patenaude, M., et al., Injectable, Mixed Natural-Synthetic Polymer Hydrogels
with Modular Properties. Biomacromolecules, 2012. 13(2): p. 369-378.
32. Risbud, M., et al, Chitosan-polyvinyl pyrrolidone hydrogel does not activate
macrophages: potentials for transplantation applications. Cell Transplant, 2001.
10(2): p. 195-202.
33. Prasitsilp, M., et al., Cellular responses to chitosan in vitro: the importance of
deacetylation. J Mater Sci Mater Med, 2000. 11(12): p. 773-778.

29
34. Yeo, Y., et al., In situ cross-linkable hyaluronic acid hydrogels prevent post-
operative abdominal adhesions in a rabbit model. Biomaterials, 2006. 27(27): p.
4698-4705.
35. Ito, T., et al., The prevention of peritoneal adhesions by in situ cross-linking
hydrogels of hyaluronic acid and cellulose derivatives. Biomaterials, 2007. 28(6):
p. 975-983.
36. Bajaj, G., et al., Hyaluronic acid-based hydrogel for regional delivery of
paclitaxel to intraperitoneal tumors. J Control Release, 2012. 158(3): p. 386-392.
37. Ilevbare, G.A., et al., Impact of Polymers on Crystal Growth Rate of Structurally
Diverse Compounds from Aqueous Solution. Mol Pharm, 2013. 10(6): p. 2381-
2393.
38. Kwon, S., et al., Physicochemical characteristics of self-assembled nanoparticles
based on glycol chitosan bearing 5 beta-cholanic acid. Langmuir, 2003. 19: p.
188-193.
39. Ta, H.T., et al., Injectable chitosan hydrogels for localised cancer therapy. J
Control Release, 2008. 126(3): p. 205-216.
40. Kohane, D.S., et al., Biodegradable polymeric microspheres and nanospheres for
drug delivery in the peritoneum. J Biomed Mater Res A, 2006. 77(2): p. 351-361.
41. Shin, H.C., et al., Pharmacokinetic study of 3-in-1 poly(ethylene glycol)-block-
poly(D, L-lactic acid) micelles carrying paclitaxel, 17-allylamino-17-
demethoxygeldanamycin, and rapamycin. J Control Release, 2012. 163(1): p. 93-
99.

30
42. Cho, H., et al., Poly(ethylene glycol)-block-poly(epsilon-caprolactone) micelles
for combination drug delivery: evaluation of paclitaxel, cyclopamine and
gossypol in intraperitoneal xenograft models of ovarian cancer. J Control Release,
2013. 166(1): p. 1-9.
43. Santovena, A., et al., Pharmacokinetics analysis of sustained release hGH
biodegradable implantable tablets using a mouse model of human ovarian cancer.
Int J Pharm, 2010. 388(1-2): p. 175-180.
44. Tang, Q., et al., Preparation of anti-tumor nanoparticle and its inhibition to
peritoneal dissemination of colon cancer. PLoS One, 2014. 9(6):p. e98455.
45. Wang, F., et al., A mechanistic model of controlled drug release from polymer
millirods: effects of excipients and complex binding. J Control Release, 2007.
119(1): p. 111-20.
46. Middleton, J.C., et al., Synthetic biodegradable polymers as orthopedic devices.
Biomaterials, 2000. 21(23): p. 2335-2346.
47. Wu, L. et al., In vitro degradation of three-dimensional porous poly(d,l-lactide-
co-glycolide) scaffolds for tissue engineering. Biomaterials, 2004. 25(27): p.
5821-5830.
48. Tsai, M., et al., Effects of Carrier on Disposition and Antitumor Activity of
Intraperitoneal Paclitaxel. Pharm Res, 2007. 24(9): p. 1691-1701.
49. Lu, Z., et al., Tumor-Penetrating Microparticles for Intraperitoneal Therapy of
Ovarian Cancer. J Pharmacol Exp Ther, 2008. 327(3): p. 673-682.
50. Trissel, L.A., et al., Pharmaceutical Properties of Paclitaxel and Their Effects on
Preparation and Administration. Pharmacotherapy, 1997. 17(2): p. 133-139.

31
51. Panchagnula, R., et al., Pharmaceutical aspects of paclitaxel. Int J Pharm, 1998.
172(1–2): p. 1-15.
52. Bouquet, W., et al., Paclitaxel/β-cyclodextrin complexes for hyperthermic
peritoneal perfusion – Formulation and stability. Eur J Pharm and Biopharm,
2007. 66(3): p. 391-397.
53. Bouquet, W., et al., In vivo toxicity and bioavailability of Taxol and a
paclitaxel/beta-cyclodextrin formulation in a rat model during HIPEC. Ann Surg
Oncol, 2010. 17(9): p. 2510-2517.
54. Bouquet, W., et al., Antitumour efficacy of two paclitaxel formulations for
hyperthermic intraperitoneal chemotherapy (HIPEC) in an in vivo rat model.
Pharm Res, 2011. 28(7): p. 1653-1660.
55. Mohamed, F., et al., Pharmacokinetics and tissue distribution of intraperitoneal
paclitaxel with different carrier solutions. Cancer Chemother Pharmacol, 2003.
52(5): p. 405-410.
56. Mohamed, F., et al., Pharmacokinetics and tissue distribution of intraperitoneal
docetaxel with different carrier solutions. J Surg Res, 2003. 113(1): p. 114-120.
57. Wang, J., et al., Tumor priming enhances siRNA delivery and transfection in
intraperitoneal tumors. J Control Release, 2014. 178(0): p. 79-85.
58. De Smet, L., et al., Development of a Nanocrystalline Paclitaxel Formulation for
Hipec Treatment. Pharm Res, 2012. 29(9): p: 2398-2406.
59. Cho, H., et al., Polymeric Micelles for Apoptosis-Targeted Optical Imaging of
Cancer and Intraoperative Surgical Guidance. PLoS One, 2014. 9(2): p. e89968.

32
60. Cho, H. et al., Polymeric Micelles for Neoadjuvant Cancer Therapy and Tumor-
Primed Optical Imaging. ACS Nano, 2011. 5(11): p. 8721-8729.
61. Cho, H., et al., In vivo cancer imaging by poly(ethylene glycol)-b-poly(ɛ-
caprolactone) micelles containing a near-infrared probe. Nanomedicine, 2012.
8(2): p. 228-236.
62. Hirano, K. et al., Lymphatic transport of liposome-encapsulated agents: effects of
liposome size following intraperitoneal administration. J Pharm Sci, 1985. 74(9):
p. 915-921.
63. Zahedi, P., et al., Chitosan–phospholipid blend for sustained and localized
delivery of docetaxel to the peritoneal cavity. Int J Pharm, 2009. 377(1–2): p. 76-
84.
64. Vassileva, V., et al., Novel biocompatible intraperitoneal drug delivery system
increases tolerability and therapeutic efficacy of paclitaxel in a human ovarian
cancer xenograft model. Cancer Chemother Pharmacol, 2007. 60(6): p. 907-914.
65. Vassileva, V., et al., Efficacy assessment of sustained intraperitoneal paclitaxel
therapy in a murine model of ovarian cancer using bioluminescent imaging. Br J
Cancer, 2008. 99(12): p. 2037-2043.
66. Grant, J., et al., Hybrid films from blends of chitosan and egg phosphatidylcholine
for localized delivery of paclitaxel. J Pharm Sci, 2005. 94(7): p. 1512-1527.
67. Lim Soo, P., et al., Drug release mechanism of paclitaxel from a chitosan–lipid
implant system: Effect of swelling, degradation and morphology. Eur J Pharm
Biopharm, 2008. 69(1): p. 149-157.

33
68. Ho, E.A., et al., In vitro and in vivo characterization of a novel biocompatible
polymer–lipid implant system for the sustained delivery of paclitaxel. J Control
Release, 2005. 104(1): p. 181-191.
69. Ho, E.A., et al., Impact of intraperitoneal, sustained delivery of paclitaxel on the
expression of P-glycoprotein in ovarian tumors. J Control Release, 2007. 117(1):
p. 20-27.
70. Grant, J., et al., Influence of molecular organization and interactions on drug
release for an injectable polymer-lipid blend. Int J Pharm, 2008. 360(1-2): p. 83-
90.
71. De Souza, R., et al., Biocompatibility of injectable chitosan–phospholipid implant
systems. Biomaterials, 2009. 30(23–24): p. 3818-3824.
72. Zahedi, P., et al., An injectable depot system for sustained intraperitoneal
chemotherapy of ovarian cancer results in favorable drug distribution at the
whole body, peritoneal and intratumoral levels. J Control Release, 2012. 158(3):
p. 379-385.
73. De Souza, R., et al., Continuous Docetaxel Chemotherapy Improves Therapeutic
Efficacy in Murine Models of Ovarian Cancer. Mol Cancer Ther, 2010. 9(6): p.
1820-1830.
74. Zahedi, P., et al., Combination Drug Delivery Strategy for the Treatment of
Multidrug Resistant Ovarian Cancer. Mol Pharm, 2010. 8(1): p. 260-269.
75. Zentner, G.M., et al., Biodegradable block copolymers for delivery of proteins
and water-insoluble drugs. J Control Release, 2001. 72(1–3): p. 203-215.

34
76. Elstad, N.L. et al., OncoGel (ReGel/paclitaxel)--clinical applications for a novel
paclitaxel delivery system. Adv Drug Deliv Rev, 2009. 61(10): p. 785-794.
77. Cho, H. et al., Thermosensitive poly-(d,l-lactide-co-glycolide)-block-poly(ethylene
glycol)-block-poly-(d,l-lactide-co-glycolide) hydrogels for multi-drug delivery. J
Drug Target, 2014. 22(7): p. 669-677.
78. Deligkaris, K., et al., Hydrogel-based devices for biomedical applications. Sens
Actuators B, 2010. 147(2): p. 765-774.
79. Yeo, Y., et al., Prevention of peritoneal adhesions with an in situ cross-linkable
hyaluronan hydrogel delivering budesonide. J Control Release, 2007. 120(3): p.
178-185.
80. Yeo, Y., et al., Peritoneal adhesion prevention with an in situ cross-linkable
hyaluronan gel containing tissue-type plasminogen activator in a rabbit repeated-
injury model. Biomaterials, 2007. 28(25): p. 3704-3713.
81. Emoto, S., et al., Intraperitoneal administration of cisplatin via an in situ cross-
linkable hyaluronic acid-based hydrogel for peritoneal dissemination of gastric
cancer. Surg Today, 2013: p. 1-8.
82. Sherwood, L., Human physiology : from cells to systems 2007, Australia; Belmont,
CA: Thomson/Brooks/Cole.
83. Watson, M.S., Oxford handbook of palliative care 2009, Oxford; New York:
Oxford University Press.
84. D'Addio, S.M. et al., Controlling drug nanoparticle formation by rapid
precipitation. Adv Drug Deliv Rev, 2011. 63(6): p. 417-426.

35
85. Patravale, V.B., et al., Nanosuspensions: a promising drug delivery strategy. J
Pharm Pharmacol, 2004. 56(7): p. 827-840.
86. Sun, B. et al., Nanocrystals for the parenteral delivery of poorly water-soluble
drugs. Curr Opin Solid State Mater Sci, 2012. 16(6): p: 295-301.
87. Van Eerdenbrugh, B., et al., Top-down production of drug nanocrystals:
Nanosuspension stabilization, miniaturization and transformation into solid
products. Int J Pharm, 2008. 364(1): p. 64-75.
88. Lu, Y., et al., Development and evaluation of transferrin-stabilized paclitaxel
nanocrystal formulation. J Control Release, 2013.
89. Jeyachandran, Y.L., et al., Quantitative and Qualitative Evaluation of
Adsorption/Desorption of Bovine Serum Albumin on Hydrophilic and
Hydrophobic Surfaces. Langmuir, 2009. 25(19): p. 11614-11620.
90. Seo, J., et al., Facile internalization of paclitaxel on titania nanoparticles in
human lung carcinoma cells after adsorption of serum proteins. J Nanopart Res,
2012. 14(10): p. 1-8.
91. Kratz, F. et al., Clinical impact of serum proteins on drug delivery. J Control
Release, 2012. 161(2): p. 429-445.
92. Torosean, S., et al., Nanoparticle uptake in tumors is mediated by the interplay of
vascular and collagen density with interstitial pressure. Nanomedicine, 2013.
9(2): p. 151-158.
93. Kohli, A.G., et al., Improving the distribution of Doxil(R) in the tumor matrix by
depletion of tumor hyaluronan. J Control Release, 2014. 20(14): p. 322-328.

36
94. Johansson, A. et al., Remodeling of tumor stroma and response to therapy.
Cancers, 2012. 4(2): p. 340-353.
95. Minchinton, A.I. et al., Drug penetration in solid tumours. Nat Rev Cancer, 2006.
6(8): p. 583-592.
96. Grantab, R., S. et al., The penetration of anticancer drugs through tumor tissue as
a function of cellular adhesion and packing density of tumor cells. Cancer Res,
2006. 66(2): p. 1033-1039.
97. Tannock, I.F., et al., Limited penetration of anticancer drugs through tumor tissue:
a potential cause of resistance of solid tumors to chemotherapy. Clin Cancer Res,
2002. 8(3): p. 878-884.
98. Kyle, A.H., et al., Limited tissue penetration of taxanes: a mechanism for
resistance in solid tumors. Clin Cancer Res, 2007. 13(9): p. 2804-2810.
99. Lee, J.H., et al., The distribution and retention of paclitaxel and doxorubicin in
multicellular layer cultures. Oncol Rep, 2012. 27(4): p. 995-1002.
100. Nicholson, K.M., et al., Influence of drug exposure parameters on the activity of
paclitaxel in multicellular spheroids. Eur J Cancer, 1997. 33(8): p. 1291-1298.
101. Ho, W.Y., et al., Development of multicellular tumor spheroid (MCTS) culture
from breast cancer cell and a high throughput screening method using the MTT
assay. PLoS One, 2012. 7(9): p. e44640.
102. Kuh, H.J., et al., Determinants of paclitaxel penetration and accumulation in
human solid tumor. J Pharmacol Exp Ther, 1999. 290(2): p. 871-880.

37
103. Jang, S.H., et al., Enhancement of paclitaxel delivery to solid tumors by
apoptosis-inducing pretreatment: effect of treatment schedule. J Pharmacol Exp
Ther, 2001. 296(3): p. 1035-1042.
104. Ishida, T. et al., Alteration of tumor microenvironment for improved delivery and
intratumor distribution of nanocarriers. Biol Pharm Bull, 2013. 36(5): p. 692-697.
105. Li, L., et al., Improved intratumoral nanoparticle extravasation and penetration
by mild hyperthermia. J Control Release, 2013. 167(2): p. 130-137.
106. Helm, J.H., et al., Cytoreductive Surgery and Hyperthermic Intraperitoneal
Chemotherapy for Malignant Peritoneal Mesothelioma: A Systematic Review and
Meta-analysis. Ann Surg Oncol, 2015. 22(5): p. 1686-1693.
107. Ihemelandu, C., L. et al., Iterative Cytoreductive Surgery and Hyperthermic
Intraperitoneal Chemotherapy for Recurrent or Progressive Diffuse Malignant
Peritoneal Mesothelioma: Clinicopathologic Characteristics and Survival
Outcome. Ann Surg Oncol, 2015. 22(5): p. 1680-1685.
108. Teo, M.C., et al., Colorectal peritoneal carcinomatosis treated with cytoreductive
surgery and hyperthermic intraperitoneal chemotherapy: The experience of a
tertiary Asian center. Asian J Surg, 2014. 21(14): p. 1-9.
109. Suidan, R.S., et al., A comparison of primary intraperitoneal chemotherapy to
consolidation intraperitoneal chemotherapy in optimally resected advanced
ovarian cancer. Gynecol Oncol, 2014. 17(14): p. 468-472.
110. Jarvinen, P., et al., Comparison of serial debulking and cytoreductive surgery with
hyperthermic intraperitoneal chemotherapy in pseudomyxoma peritonei of
appendiceal origin. Int J Colorectal Dis, 2014. 29(8): p. 999-1007.

38
111. Sammartino, P., et al., Long-term results after proactive management for
locoregional control in patients with colonic cancer at high risk of peritoneal
metastases. Int J Colorectal Dis, 2014. 29(9): p. 1081-1089.
112. Safra, T., et al., Cytoreduction surgery with hyperthermic intraperitoneal
chemotherapy in recurrent ovarian cancer improves progression-free survival,
especially in BRCA-positive patients-A case-control study. J Surg Oncol, 2014.
24(10): p. 23688.
113. Cui, H.B., et al., Effect of neoadjuvant chemotherapy combined with hyperthermic
intraperitoneal perfusion chemotherapy on advanced gastric cancer. Exp Ther
Med, 2014. 7(5): p. 1083-1088.
114. Saggar, J.K., et al., The tumor microenvironment and strategies to improve drug
distribution. Front Oncol, 2013. 3: p: 154.
115. Choi, I.K., et al., Strategies to increase drug penetration in solid tumors. Front
Oncol, 2013. 3: p: 193.
116. Sagnella, S.M., et al., Dextran-based Doxorubicin nanocarriers with improved
tumor penetration. Biomacromolecules, 2014. 15(1): p. 262-275.
117. Patel, K.J., et al., Use of the proton pump inhibitor pantoprazole to modify the
distribution and activity of doxorubicin: a potential strategy to improve the
therapy of solid tumors. Clin Cancer Res, 2013. 19(24): p. 6766-6776.
118. Saggar, J.K. et al., Activity of the hypoxia-activated pro-drug TH-302 in hypoxic
and perivascular regions of solid tumors and its potential to enhance therapeutic
effects of chemotherapy. Int J Cancer, 2014. 134(11): p. 2726-2734.

39
119. Zhang, L., et al, Hyaluronan on the surface of tumor cells is correlated with
metastatic behavior. Cancer Res, 1995. 55(2): p. 428-433.
120. Kimata, K., et al., Increased synthesis of hyaluronic acid by mouse mammary
carcinoma cell variants with high metastatic potential. Cancer Res, 1983. 43(3): p.
1347-1354.
121. Turley, E.A. et al., Glycosaminoglycan production by murine melanoma variants
in vivo and in vitro. Cancer Res, 1985. 45(10): p. 5098-5105.
122. McBride, W.H. et al., Hyaluronidase-sensitive halos around adherent cells. Their
role in blocking lymphocyte-mediated cytolysis. J Exp Med, 1979. 149(2): p. 507-
515.
123. Kayastha, S., et al., Expression of the hyaluronan receptor, CD44S, in epithelial
ovarian cancer is an independent predictor of survival. Clin Cancer Res, 1999.
5(5): p. 1073-1076.
124. Anttila, M.A., et al., High levels of stromal hyaluronan predict poor disease
outcome in epithelial ovarian cancer. Cancer Res, 2000. 60(1): p. 150-155.
125. Hiltunen, E.L., et al., Elevated hyaluronan concentration without hyaluronidase
activation in malignant epithelial ovarian tumors. Cancer Res, 2002. 62(22): p.
6410-6413.
126. Picaud, L., et al., Evaluation of the effects of hyaluronic acid-carboxymethyl
cellulose barrier on ovarian tumor progression. J Ovarian Res, 2014. 7(1): p. 40.
127. Hubbard, S.C. et al., Effects of a hyaluronan-based membrane (Seprafilm) on
intraperitoneally disseminated human colon cancer cell growth in a nude mouse
model. Dis Colon Rectum, 2002. 45(3): p. 334-341.

40
128. Tan, B., et al., Sodium hyaluronate enhances colorectal tumour cell metastatic
potential in vitro and in vivo. Br J Surg, 2001. 88(2): p. 246-250.

41
CHAPTER 2. DEVELOPMENT AND CHARACTERIZATION OF NANOPARTICLE DEPOT FOR INTRAPERITONEAL CHEMOTHERAPY2
2.1 Introduction
The potential advantages of IP chemotherapy have been discussed thoroughly.
However, frequently mentioned problems are the complications related to IP infusion,
including abdominal pain, intolerance to a high level of drug, and discomfort related to
the catheter implantation [1]. We speculate that these problems mainly stem from the
difficulty in controlling drug release. Small molecule drugs such as paclitaxel (PTX) or
docetaxel were cleared from the peritoneal cavity in less than one day [2-4]. The short IP
retention time requires frequent or continuous dosing, necessitating the use of large
volume of treatment and indwelling catheters. For IP chemotherapy to provide the
anticipated benefits, it is critical that the formulation control the drug release and avoid
burst initial release, followed by immediate absorption to the systemic circulation.
Considering this need, we previously used a hyaluronic acid (HA)-based hydrogel
(gel) as a carrier of PTX provided in the form of micrometer-scale precipitates (PPT) to
improve their IP retention and control the drug release [5]. The gel remained in the
peritoneal cavity and maintained the high level of local PTX concentration for 2 weeks.
2 The content of this chapter is adapted with permission from Sara A. Abouelmagd, Bo Sun, Alice C. Chang, et al., Release kinetics study of poorly water-soluble drugs from nanoparticles: Are we doing it right? Mol. Pharm. 2015, 12(3), p 997-1003. Copyright (2015) American Chemical Society.

42
However, when tested in a murine model of IP SKOV-3 human ovarian cancer, the PPT-
gel had no more benefit than Taxol-gel, which did not retain PTX as well as PPT-gel did.
One of the possible explanations was that the dissolution of PPT was too slow to provide
an effective local drug concentration.
In the present study, we aim to overcome this challenge by designing a drug
delivery system that can localize PTX in the peritoneal cavity and provide sustained, yet
uninterrupted PTX release. PTX is used as a model drug because it is a first-line therapy
of ovarian cancer for both intravenous and intraperitoneal administration [6-9]. Besides,
PTX is a well-known poorly water-soluble drug, previously found to be challenging to
control the release in the peritoneal cavity with a limited amount of fluid.
PTX is formulated as nanocrystals (NCs), crystalline particles of poorly soluble
drugs in the nanometer range [10]. Due to the small size of NC and thus, the high surface
area to volume ratio, NC can increase the dissolution rate of drug particles [11].
Typically, NCs are produced by breaking down large particles (“top-down”) or
crystallizing from drug solutions (“bottom-up”) of the drug itself with an optional aid
with surfactants or polymeric stabilizers on the surface [10, 12]. Here, we produce PNC
by nonsolvent and temperature-induced crystallization [13] and evaluate its in-vitro
dissolution in comparison with PPT.
PNCs are then embedded in HA gel. IP-administered small molecule drugs could
be cleared out from peritoneal cavity in a few hours which compromised the
effectiveness of local chemotherapy [14-16]. In situ forming gel has been developed as a
standalone drug delivery system to handle the short residence time of chemotherapeutic

43
agents [5, 17-21]. HA gel is degradable in-vivo by prevalent hyaluronidase and highly
biocompatible [22-25]. In this study, we anticipate that HA gel will provide a local
reservoir of PNC and prolong the retention of PTX in the peritoneal cavity to provide
sustained local delivery of chemotherapy to ovarian tumors.
In order to predict PTX dissolution within the peritoneal cavity, it is necessary to
develop a dissolution method that accounts for biological and physicochemical factors
relevant to in-vivo conditions. Several attempts have been made to estimate the PTX
release profiles from gel formulations in conditions mimicking the peritoneal cavity with
a limited fluid volume and amphiphilic components.
2.2 Materials and Methods
2.2.1 Materials
Hyaluronic acid (HA, 20 kDa and 500 kDa) was purchased from Lifecore
Biomedical, LLC (Chaska, MN, USA). Paclitaxel (PTX) was a gift of Samyang Genex
Corp (Seoul, Korea). Carbamazepine was purchased from Enzo life sciences (Plymouth
Meeting, PA, USA). Cremophor ELP was a gift from BASF (New York, NY, USA).
Carboxyl methylcellulose sodium (CMC, 250 kDa) was purchased from Sigma-Aldrich.
Cell culture medium and supplements were purchased from Invitrogen (Carlsbad, CA,
USA). All other reagents were purchased from Sigma-Aldrich (St. Louis, MO, USA).

44
2.2.2 Preparation and Characterization of PTX Nanocrystals (PNC)
PTX nanocrystals (PNC) were prepared according to the published method [13].
Briefly, 4 mg/mL PTX/ethanol solution was added to 20 mL of deionized (DI) water and
stirred at 42 rcf for 10 min in a round-bottom flask immerged in a sonication bath filled
with ice water. The formed PNC was captured on a 100 nm polycarbonate membrane and
resuspended in DI water. Taxol and PTX precipitates (PPT) were prepared as described
previously [5]. Taxol was prepared by dluting “Taxol concentrate” (6 mg PTX dissolved
in 1 mL of 1:1 mixture of Cremophor ELP and ethanol). PPT was prepared by the
addition of 30 mg/mL of PTX-dimethyl sulfoxide (DMSO) solution to DI water without
low temperature, vigorous stirring and sonication. Particle size of PNC was measured
with a Zetasizer Nano-ZS90 (Malvern instruments, Westborough, MA, USA). The zeta
potential of PNC and PPT were measured with a Zeta sizer Nano-ZS90 in 1 mM
phosphate buffer (pH 7.4). The morphology of lyophilized PNC and PPT was visualized
with a FEI Nova nanoSEM field emission scanning electron microscopy (Hillsboro, OR,
USA). Freeze dried PNC or PPT was sputter-coated with platinum for 1 min and
observed with a high resolution through-the-lens detector under 5kV accelerating voltage
and spot size 3. PPT suspended in DI water was observed with Axio Imager 2 microscope
with polarized light (Carl Zeiss Microscopy GmbH, Germany). X-ray powder diffraction
(XPRD) patterns of lyophilized PNC and PPT were obtained on a Siemens D5000 X-ray
diffractometer with a Cu Kα radiation source (40kV, 40 mA). All the measurements were
conducted at room temperature over an angle (2θ) range of 5-40° with a step size of 0.02°
and a scan rate of 4°/min.

45
2.2.3 Synthesis of Gel Precursors (HA-ADH, HA-CHO, CMC-CHO)
In-situ crosslinkable HA derivatives, HA-adipic acid dihydrazide (HA-ADH) and
HA-aldehyde (HA-CHO), and a crosslinkable HA gel were prepared as described
previously [5, 26]. Briefly, HA-ADH was synthesized by conjugating adipic dihydrazide
to carboxyl groups in HA which was catalyzed by 1-ethyl-3-carbodiimide (EDC) and 1-
hydroxybenzotriazole (HOBt) at pH 6.8 and room temperature. HA-CHO was produced
by oxidizing HA with sodium periodate. HA-ADH and HA-CHO were purified by
dialysis, freeze dried and stored at 4°C until use. Carboxyl methylcellulose-aldehyde
(CMC-CHO) was synthesized in the same approach as HA-CHO.
2.2.4 Synthesis of Hydrophobically Modified Gel Precursor (EtCA-CMC-CHO)
2.2.4.1 Synthesis of 5β-cholanic Acid Methyl Ester (MeCA) (Scheme 1)
500 mg of 5β-cholanic acid (CA) (1.4 mmol) was dissolved in 50 mL of methanol
with 180 µL concentrated hydrochloride acid (4.9 µmol) at 60°C. The solution was
stirred for 6h under reflux. The solution was cooled to room temperature and added
dropwise into cold deionized water to obtain precipitates, which were further filtrated and
washed with cold methanol for 3 times and cold deionized water for 3 times. The product
was dried under vacuum at room temperature to obtain MeCA.
2.2.4.2 Synthesis of Aminoethyl 5β-cholanoamide (EtCA) (Scheme 1)
450 mg of MeCA (1.2 mmol) was dissolved in 8 mL of ethylene diamine (119.6
mmol) under stirring and reflux for 6h at 130 °C. The solution was cooled to room

46
temperature and added dropwise into cold deionized water to obtain precipitates, which
were further filtrated and washed with cold deionized water for 5 times. The final product
(EtCA) was dried under vacuum at room temperature and its chemical structure was
characterized with 1H NMR in CD3OD at a concentration of 10 mg/mL.
Scheme 1. Synthesis scheme of aminoethyl 5β-cholanoamide.
2.2.4.3 Synthesis of EtCA-CMC-CHO Conjugate (Scheme 2)
41 mg of CMC-CHO (100 µmol) was dissolved in 37 mL of methanol/water
(2v/1v) containing 20.6 mg of N, N’-Dicyclohexylcarbodiimide (DCC, 100 µmol) and
11.5 mg of N-Hydroxysuccinimide (NHS, 100 µmol) under stirring at room temperature
for 1h. After 4 or 12 mg of EtCA (10 or 30 µmol) in 1 mL of methanol was slowly added,
the reaction mixture was stirred for 12h. The resulting solution was dialyzed against
methanol/water (2v/1v) for 2 days and deionized water for another 2 days. EtCA0.1 or 0.3-
CMC-CHO was lyophilized and stored at -20°C until use.

47
Scheme 2. Synthesis scheme of EtCA-CMC-CHO conjugate.
2.2.5 Preparation of Gel formulations
Gels were prepared by suspending PNC or PPT in solutions of gel precursors (one
ADH derivative and one CHO derivative) in phosphate buffer saline (PBS, pH 7.4) and
extruding them through a common outlet using a double-barreled syringe. HA gel was
made of HA-ADH and HA-CHO; HA-CMC gel was made of HA-ADH and CMC-CHO.
The polymer concentration in gel was 40 mg/mL. When EtCA0.1-CMC-CHO was used,
37.5% (w/w) of HA-CHO was replaced with EtCA0.1-CMC-CHO. HA-EtCA0.1-CMC gel
was made of 40 mg/mL HA-ADH, 25 mg/mL CMC-CHO and 15 mg/mL EtCA0.1-CMC-
CHO.

48
2.2.6 Evaluation of Gel Precursors and Crosslinked Gels
2.2.6.1 Hydrophilicity of Gel Precursors
Contact angle is defined as the angle formed by the intersection of the liquid-solid
interface and the liquid-vapor interface [27]. The larger a contact angle is, the more
hydrophobic the surface is. The hydrophilicity of a specific material can be evaluated by
measuring the contact angle of water droplet placed on the thin film made of the material.
Four gel precursors, HA-CHO, CMC-CHO and EtCA0.1 or 0.3-CMC-CHO, were
dissolved in deionized water at 10 mg/mL. The solutions were casted on glass slides
respectively and dried at ambient temperature for 12h to form thin films. The contact
angle of water droplet on each polymeric film was measured with a contact angle
goniometer.
2.2.6.2 Toxicity of EtCA and EtCA-CMC-CHO
Toxicity of CA, MeCA, EtCA and EtCA0.1-CMC-CHO were studied with
NIH/3T3 fibroblast cells (ATCC). NIH/3T3 cells were maintained in Dulbecco's
Modified Eagle's Medium (DMEM) medium supplemented with 10% bovine calf serum
(BCS) and 1% penicillin-streptomycin. Cells were plated in a 96-well plate at a density of
8,000 cells per well with 0.2 mL of complete medium. After 24h incubation, CA, MeCA,
EtCA (in methanol) or EtCA0.1-CMC-CHO (in PBS) was added to each well at varied
concentrations and incubated with cells for 2 days. A control group was treated with an
equal volume of methanol or PBS. The cell viability was evaluated with the MTT (3-(4,
5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide) assay. The absorbance of the

49
solubilized formazan was measured with a SpectraMax M3 microplate reader (Molecular
Devices, Sunnyvale, CA, USA) at a wavelength of 562 nm. The measured absorbance
was normalized to the absorbance of the control group.
2.2.6.3 Toxicity of Gels
Toxicity of gels was evaluated with NIH/3T3 cells using Transwells. Cells were
plated in a 12-well plate at a density of 17,500 cells per wells with 1.5 mL DMEM
complete medium (cell density equal to 5,000 cells per well in 96-well plate). CMC-
CHO, HA-CMC gel or HA-EtCA0.1-CMC gel was added to the apical side of a Transwell
polycarbonate membrane insert (pore size = 0.4 µm), placed in the 12-well plate, and
incubated with cells for 2 days. Cell viability was evaluated with MTT assay as described
above.
2.2.7 PTX Solubility and Stability in Various Media
2.2.7.1 PTX Solubility in PBS, PBS Containing Serum, or PBS Containing Tween 80
PTX solubility in PBS, PBS containing 0.2 v/v% Tween 80 (Tween/PBS), and
PBS containing 50 v/v% FBS (50-FBS/PBS) were determined by incubating excess PTX
(0.6 – 2.4 mg) in 1 mL of each medium at 37 °C for 7 or 24 h with agitation. Samples
were centrifuged at 9300 rcf for 20 min to separate a supernatant. PTX dissolved in PBS
and Tween/PBS was analyzed with HPLC as described in Section 2.2.4. PTX dissolved
in 50-FBS/PBS was first extracted with ethyl acetate and reconstituted in the HPLC
mobile phase prior to HPLC analysis.

50
2.2.7.2 PTX Stability in PBS Containing Tween 80 or PBS Containing Serum
The stability of a drug was an important issue if it would be tested or delivered for
an extended period of time. PTX stability was first tested in a fully dissolved state (PTX
concentration below the solubility limit). 1.5 µg/mL of PTX solution in Tween/PBS was
prepared by diluting 10 mg/mL PTX-DMSO solution with Tween/PBS (final DMSO
concentration: 0.015%). The solution was divided into several 1 mL aliquots, and the
initial PTX concentration was determined with 7 of them. The remaining aliquots were
incubated at 37°C, and 3 aliquots were taken at predetermined time points and kept at -
80°C until HPLC analysis. The frozen samples were thawed and filtered through 0.45 µm
filters for HPLC analysis. The concentration of intact PTX at each time point was divided
by the original PTX concentration (1.5 µg/mL) and expressed as the percentage of
original PTX.
PTX stability in 50-FBS/PBS was tested in its solid form of PNC and PPT at a
concentration of 100 µg/mL (above solubility limit) and compared to that of Taxol. These
PTX formulations were incubated in 50-FBS/PBS at 37 °C for 10 days. PTX remained in
the suspensions was extracted with ethyl acetate and analyzed with HPLC.
2.2.8 Dissolution Kinetics of PNC
PTX dissolution was first tested with the dialysis method. Three milliliters of PBS
containing 200 µg PNC was injected into a dialysis cassette (MWCO= 3.5 kDa). The
cassette was placed in 200 mL PBS. Therefore, the initial PTX concentrations in cassette
and in the system were 66.7 µg/mL and 1 µg/mL, respectively. The cassette was

51
incubated in PBS at 37 °C on an orbital shaker for 10 days. At predetermined time point,
5 mL of the release medium was sampled and replaced with 5 mL of fresh PBS. PTX in
the release medium was extracted with ethyl acetate and analyzed as described in Section
2.2.4. At the completion of this study, dialysis cassettes were rinsed with PBS to collect
the remaining PTX. PTX leftover was analyzed in the same way as release samples.
To determine how quickly PTX precipitated in PBS at 37 °C, PTX solution in
PBS at a concentration of 20 µg/mL was prepared by diluting 1 mg/mL PTX stock
solution in Tween/PBS with PBS (Tween80 concentration in the diluted solution was
below the critical micelle concentration 0.0015%), aliquoted by 1 mL, and incubated at
37°C with shaking. At predetermined time points, 3 aliquots were taken and centrifuged
at 2095 rcf for 5 min. The supernatants were additionally centrifuged at 9300 rcf for 20
min and filtered with 0.45 µm PVDF syringe filters and directly analyzed with HPLC.
Dissolution kinetics of PPT and PNC was also tested in PBS containing 0.2%
Tween 80 (PBST) following a method based on light scattering [28]. PPT suspension was
prepared by diluting 30 mg/mL PTX in DMSO in PBS by 100 times. PNC suspension (1
mg/mL) was pre-diluted in 0.2% PBST by 10 times. Final dilution was performed
directly in a cuvette by adding 20 µL of pre-diluted PNC (equivalent to 2 µg of PTX) or
6.7 or 20 µL of PPT suspension (equivalent to 2 or 6 µg of PTX) to 1 mL of 0.2% PBST.
Their derived count rates were measured with a Zetasizer Nano-ZS90 from the time of
final dilution. Each measurement was done for 3 runs at 3s with a 30-sec interval and an
attenuator setting of 11. All measurements were performed at 25 °C.

52
2.2.9 PTX Release Kinetics of HA Gel Formulations
PTX release from gel formulations was studied in two ways: one in the sink
condition (total drug concentration being less than one third of the saturation solubility
[29]) and the other in a condition violating the sink condition to simulate the peritoneal
environment which has limited fluid available for drug dissolution. PNC-gel and PPT-gel
referred to PNC loaded in HA gel and PPT loaded in HA gel, respectively.
2.2.9.1 In Tween/PBS under Sink Condition
For the release kinetics study in the sink condition, 0.1 mL of PNC-gel or PPT-gel
containing 22 µg PTX was formed as described above and placed in a tube containing 20
mL of Tween/PBS (n=3). This condition made the initial PTX concentration in the
release medium 1.1 µg/mL, one third of the PTX solubility in Tween/PBS (3.3 µg/mL)
[30]. Thirty six identical samples were incubated at 37 °C on an orbital shaker. At
predetermined time points, 3 samples per group were taken and centrifuged at 2095 rcf or
10 min to separate the gels. The collected gels were freeze dried and incubated with 5 mL
mixture of acetonitrile/water (50/50) (ACN/water) at room temperature for 24 h. The
swollen gels were ground with a mortar and a pestle to release PTX into ACN/water. The
crushed gel suspension was filtered and analyzed with HPLC to determine the PTX
content.

53
2.2.9.2 In Tween/PBS under Non-Sink Condition
For a non-sink condition, 0.3 mL of PNC-gel or PPT-gel containing 200 µg PTX
was placed in a tube containing 20 mL of Tween/PBS (n=3). The initial PTX
concentration in the release medium was therefore 10 µg/mL, 3 times higher than the
PTX solubility in Tween/PBS [30]. All the samples were incubated at 37°C on an orbital
shaker. At predetermined time points, 3 mL release medium were sampled and
centrifuged at 9300 rcf for 10 min to separate a supernatant. Two milliliters of the
supernatant was taken for HPLC analysis, and the remaining 1 mL was combined with 2
mL of fresh Tween/PBS and returned to the sample tube for further incubation. At
completion of the study, the remaining mass in the sample container was freeze-dried and
reconstituted with 10 mL of acetonitrile to determine the PTX leftover.
PTX release from HA-EtCA0.1-CMC gel was also tested in Tween/PBS under
non-sink condition with minor differences. HA-EtCA0.1-CMC gel containing PPT was
formed as described in Section 2.2.5 by replacing 37.5% (w/w) of HA-CHO with
EtCA0.1-CMC-CHO. 0.3 mL of PPT-(HA-EtCA0.1-CMC) gel containing 200 µg of PPT
was placed in 20 mL of Tween/PBS. All the samples were incubated at 37 °C on an
orbital shaker. At predetermined time points, 5 mL release medium were sampled and
centrifuged at 2095 rcf for 10 min to separate a supernatant. Two milliliters of the
supernatant was taken for HPLC analysis, and the remaining 3 mL was combined with 2
mL of fresh Tween/PBS and returned to the sample container for further incubation. At
completion of this study, the remaining mass in the sample container was freeze-dried
and reconstituted with 10 mL of acetonitrile to determine the PTX leftover.

54
2.2.9.3 In FBS/PBS or Tween/PBS containing Hyaluronidase under Non-Sink
Condition
Hyaluronidase, a prevalent enzyme in the body, was added in the release medium
to simulate the degradation of HA gel in the peritoneal cavity. 0.3 mL of PNC-gel or
PPT-gel containing 200 µg PTX was placed in 20 mL of 25 v/v% FBS (25-FBS/PBS) or
Tween/PBS containing hyaluronidase (10 U/mL). The initial PTX concentration in the
release medium was therefore 10 µg/mL. All the samples were incubated at 37°C on an
orbital shaker. At predetermined time points, 5 mL release medium were sampled and
centrifuged at 2095 rcf for 10 min to separate a supernatant. Two milliliters of the
supernatant was taken for HPLC analysis, and the remaining 3 mL was combined with 2
mL of fresh 25-FBS/PBS and returned to the sample container for further incubation. At
the completion of the study, the remaining mass in the sample container was freeze-dried
and reconstituted with 10 mL of acetonitrile to determine the PTX leftover.
2.2.10 HPLC Analysis of PTX
PTX in PBS or PTX in Tween/PBS solution was directly analyzed with HPLC
after filtration with 0.45 µm PVDF syringe filter. PTX in PBS containing 50 or 25 v/v%
FBS was extracted with ethyl acetate prior to HPLC analysis. Briefly, 1 mL of PTX
solution in FBS medium with 10 µg of carbamazepine as an internal standard was mixed
with 3 mL ethyl acetate and shaken on a rotating shaker for 40 min. The mixture was then
centrifuged at 2095 rcf for 15 min to separate an organic layer, which was transferred to a
new glass vial and dried under vacuum. The dried sample was resuspended in the HPLC

55
mobile phase, filtered through 0.45 µm syringe filter, and analyzed by HPLC. A
calibration curve was drawn with PTX solutions in FBS medium in known
concentrations, treated in the same manner as the sample solutions. PTX was analyzed
with HPLC equipped with UV detector (1100 series, Agilent Technologies, Palo Alto,
CA) and an Ascentis C18 column (25 cm × 4.6 mm, particle size 5 µm) (Supelco, St.
Louis, MO, USA). The mobile phase was a mixture of acetonitrile and water (50:50) run
in the isocratic mode at a flow rate of 1 mL/min. PTX was detected at 227 nm.
2.3 Results and Discussions
2.3.1 Characterization of PNC and PPT
The properties of PNC and PPT are summarized in Table 1. PNC showed a rod-
shape with a length of 310 ± 86 nm and a width of 65 ± 10 nm under SEM (Fig. 6A). The
average diameter of PNC measured by Dynamic Light Scattering (DLS) was 258.0 ±
28.1 nm (average and standard deviation of 8 independently prepared batches) with
polydispersity index values ranging from 0.045 to 0.223, which indicated mid-range
polydispersity. On the contrary, PPT were needle-shaped crystals with a length of 11.5 ±
2.2 µm and a width of 2.0 ± 0.4 µm (Fig. 6B). Polarized light microscopy detected
clusters of PTX crystals, confirming the broad size distribution of PPT. Both PNC and
PPT showed weakly negative charges (PNC: -5.51 ± 0.42 mV; PPT: -3.08 ± 0.98 mV) at
pH 7.4. PNC and PPT exhibited sharp peaks typical of crystalline solids (Fig. 6C). The
crystal pattern of PNC was consistent with the result in the literature [13].

56
Figure 6. (A) Scanning electron micrograph of PNC. (B) Polarized light micrograph of PPT. (C) XPRD pattern of PNC and PPT.
Table 1. Summary of PNC and PPT properties PNC PPT Particle size Length1: 310 ± 86 nm;
Width1: 65 ± 10 nm z-average2: 258 ± 28.1 nm
Length3: 11.5 ± 2.2 µm; Width3: 2.0 ± 0.4 µm
Zeta potential4 -5.51 ± 0.42 mV -3.08 ± 0.98 mV 1Estimated by SEM; based on 50 measurements with ImageJ 2Measured by a zeta sizer (DLS); average and standard deviation of 8 independently prepared batches 3Estimated by light microscopy; based on 50 measurements with ImageJ 4Measured by a zeta sizer; average and standard deviation of 5 independently prepared batches

57
2.3.2 Synthesis and Characterization of EtCA-CMC-CHO
In an attempt to increase compatibility between PNC and the hydrogel carrier, 5β-
cholanic acid was introduced to one of the gel precursors. 5β-cholanic acid has been
conjugated to HA as EtCA to make an amphiphilic HA conjugate that formed self-
assembly nanoparticles for cancer therapy in the literature [31]. The EtCA synthesis was
adapted to produce EtCA-CMC-CHO. The chemical structure of EtCA was confirmed
with 1H NMR as shown in Fig. 7. The characteristic peaks of EtCA appeared from the
ethylene group (δ = 3.3 ppm [2H, –NHCH2–], δ = 2.7 ppm [2H,–CH2NH2]). EtCA0.1 or 0.3-
CMC-CHO was poorly soluble in water, which confirmed the conjugation of EtCA to
CMC-CHO. However, due to the low solubility in D2O and CD3OD, the chemical
structure of EtCA0.1 or 0.3-CMC-CHO was not successfully characterized with 1H NMR.
Figure 7. 1H NMR spectrum of EtCA (300 MHz, CD3OD). δ=3.30 (m, 2H), 2.86 (m, 2H), 0.97-0.94 (m, 6H), 0.68 (s, 3H).

58
2.3.2.1 Hydrophobicity of Gel Precursors
Contact angles of water droplet on the thin films of different gel precursors were
measured to compare their hydrophobicity. The contact angles of EtCA0.1-CMC-CHO
and EtCA0.3-CMC-CHO were similar but much higher than those of HA-CHO or CMC-
CHO, indicating the hydrophobicity imparted by EtCA (Table 2). EtCA0.1-CMC-CHO
was chosen for further studies and used as a partial replacement of HA-CHO, because it
was easier to dissolve in water than EtCA0.3-CMC-CHO.
Table 2. Contact angles of water droplet on gel precursor films. n=3 HA-CHO CMC-CHO EtCA0.1-CMC-
CHO EtCA0.3-CMC-CHO
Contact angle ( ) 33.0±2.0 38.8±1.4 63.2±7.3 67.2±2.7
2.3.2.2 Toxicity of EtCA and EtCA-CMC-CHO Conjugate
Quite unexpectedly, EtCA showed high toxicity on NIH/3T3 cells at all tested
concentrations (2-71 µg/mL) (Fig. 8A). CA or MeCA, precursors of EtCA, did not show
comparable toxicity, which indicated that the toxicity came from ethylene diamine
conjugation. Accordingly, EtCA0.1-CMC-CHO was toxic to NIH/3T3 cells at
concentrations ≥ 100µg/mL (equivalent to EtCA 9 µg/mL or above with a theoretical
EtCA content in EtCA0.1-CMC-CHO of 9.5%) (Fig. 8B and 8C). Toxicity of EtCA0.1-
CMC-CHO was further evaluated after gel formation. HA-EtCA0.1-CMC gel was placed
on the apical side of the insert and immersed in the culture medium, letting degradants or
molecules dissociated from the gel approach the cells through the pores of the membrane
insert. After 2 days incubation, cells treated with HA-EtCA0.1-CMC gel (equivalent to
67.5 µg/mL EtCA) were all dead (Table 3). CMC-CHO showed no toxicity on NIH/3T3
cells, and cells treated with HA-CMC gel maintained about 72% cell viability. Due to the

59
significant toxicity, EtCA0.1-CMC-HA gel containing 1.7% w/w of EtCA was not further
pursued in animal studies. Nevertheless, in-vitro release kinetics from PNC-loaded
EtCA0.1-CMC-HA gel was studied to examine the effect of hydrophobic moiety in the gel
on controlling the PTX release.
Figure 8. Toxicity of EtCA and EtCA0.1-CMC-CHO.
Table 3. Toxicity of gel precursors and gels with NIH/3T3 cells Samples CMC-CHO CMC-HA gel EtCA0.1-CMC-
HA gel Control
Sample preparation*
1.5 mg CMC-CHO in 0.2 mL PBS
0.2 mL gel consisting of 4 mg HA-ADH, 2.5 mg HA-CHO and 1.5 mg CMC-CHO
0.2 mL gel consisted of 4mg HA-ADH, 2.5 mg HA-CHO and 1.5 mg EtCA0.1-CMC-CHO
0.2 mL PBS
Cell viability% # 106.4 ± 4.9 71.6 ± 3.2 0 100% * Each sample (0.2 mL total) was placed in the apical side of a Transwell and supplemented with 0.3 mL DMEM prior to placement in the 12-well plate containing 1.5 mL medium and 17,500 cells. #: cell viability expressed as average ± standard deviation after treated with 3 independently and identically prepared gels.
25 50 100
250
500
750
A B C

60
2.3.3 PTX Solubility and Stability in Various Media
2.3.3.1 PTX Solubility in PBS, 50-FBS/PBS, or Tween/PBS
Prior to testing PNC dissolution kinetics and PTX release kinetics from gel
formulations, PTX solubility was evaluated in PBS, Tween/PBS, and 50-FBS/PBS after
incubation at 37 °C for 7 or 24h. PTX solubility in PBS was measured at ~0.2 µg/mL
with no specific trend according to the incubation time, although the values were variable
due to the sensitivity limitation of HPLC (Fig. 9A). PTX solubility in Tween/PBS was
measured to be 3.3 µg/mL irrespective of the incubation time (Fig. 9B). PTX solubility in
50-FBS/PBS was measured at 35 µg/mL after 7h incubation at 37 °C, much higher than
those in PBS or Tween/PBS, which confirmed the solubilizing effect of serum proteins
(Fig. 9C). Notably, PTX concentration measured after 24h incubation was 25 µg/mL,
28.6% lower than that after 7h. This difference is attributable to the instability of PTX in
serum, reported in our previous study [5] as well as in others. 38.5% of paclitaxel was
hydrolyzed to 7-epitaxol in cell culture medium containing FBS within 10h [32] and
35 % of paclitaxel would degrade in 24h at 37 °C in human plasma [33]. This indicates
that, if PTX release kinetics studies are performed in serum-containing medium and the
medium is not sampled and analyzed frequently, one may not recover 100% of PTX from
the formulation due to the degradation of released PTX. On the other hand, PTX was
relatively more stable in Tween/PBS as compared to 50-FBS/PBS, maintaining 94% of
the initial concentration for 1 day (Fig. 9D). This means that, as long as the medium is
sampled at least once a day, PTX stability in Tween/PBS is less likely to be a problem.

61
Figure 9. PTX solubility in (A) PBS, (B) Tween/PBS, or (C) 50-FBS/PBS. Blue square: measured after 7h; red triangle: measured after 24h. (D) Stability of 1.5 µg/mL PTX in Tween/PBS at 37 °C.
2.3.3.2 PNC Stability in PBS Containing Serum
PTX stability in serum-containing medium improved when it was incubated as
solid particles. PTX was least stable as Taxol and more stable as solid (PNC and PPT) in
25-FBS/PBS (Fig. 10). Between PNC and PPT, the recovery of PNC was relatively low,
likely due to the faster initial dissolution of PNC due to the small size. The improved
stability justifies the use of solid crystal forms for a long-term delivery of PTX.
A B
C D

62
Figure 10. Stability of PTX formulations (0.1 mg/mL) in 25-FBS/PBS for 10 days (n=6). *: p<0.05.
2.3.4 Dissolution Kinetics of PNC and PPT
2.3.4.1 Dissolution Kinetics of PNC and PPT with Dialysis Method
PTX dissolution kinetics from PNC or PPT was evaluated using the dialysis
method and PBS as a release medium. Here, the PNC equivalent to 200 µg PTX was
suspended in 3 mL of PBS, put in a dialysis cassette, and incubated in 200 mL of PBS.
Fig. 11A shows that PTX dissolution from the PNC was very slow, reaching less than
10% cumulative release in 10 days. Given that the PTX concentration in the dialysis
cassette was 66.7 µg/mL, far exceeding the solubility (0.2 µg/mL), and the dialysis
membrane could delay PTX diffusion into the release medium, we suspected that PTX
might have precipitated in the dialysis cassette. Indeed a significant fraction of PTX
remained in the dialysis cassette after 10 days. To estimate how quickly PTX precipitated
in a dialysis cassette, we prepared a 20 µg/mL PTX solution in PBS (<1/3 of the initial
concentration in a dialysis cassette) and sampled the solution at different time points to
quantify the dissolved PTX. PTX rapidly precipitated out in less than 30 min, leaving
Taxol PPT NC0
50
100
150
% P
TX
rec
ov
ery
**
*
P

63
PTX in solution only to the solubility level (Fig. 11B). This result suggests that even
though the small size of PNC had increased the dissolution rate of PTX, the dissolved
PTX might have undergone reprecipitation in the dialysis cassette. In other words, PTX
detected in the release medium did not necessarily reflect the dissolution of PNC but that
of PTX precipitates entrapped in the cassette. Since only the dissolved PTX could pass
the membrane and became diluted in the release medium, the PTX concentration in the
release medium was <0.1 µg/mL, much below the solubility limit, at any time point (Fig.
11C).
Drug dissolution or release kinetics of a particulate formulation is typically
studied by separating dissolution medium from the formulation by high speed
centrifugation or a dialysis bag and measuring the drug concentration in the sampled
medium. Neither is appropriate for estimating PTX dissolution from PNC or PPT:
centrifugation does not completely separate small particles from the suspension and
accelerates particle aggregation, and the dialysis method bears the risk of underestimating
drug release/dissolution due to drug adsorption to the membrane and/or reprecipitation of
dissolved drug in the bag [30].
Given these challenges, we resorted to an in-situ analytical technique based on
light scattering [28]. As expected, the derived count rate of PNC decreased from the
initial value of 214.3 kcps to 22.0 kcps (count rate of PBST) in 10 min, indicating rapid
dissolution of PNC in PBST. On the other hand, PPT showed an average count rate of
23.0 kcps from the beginning and did not change for 4 min, likely due to the small
number of PPT particles per volume and/or their instantaneous sedimentation. The count
rate was not much different (26 kcps) even when the concentration of PPT suspension

64
was tripled (6 µg/mL, above the PTX solubility in 0.2% PBST) (Fig. 11D). This indicates
that light scattering method was not suitable for large particles.
Figure 11. (A) Dissolution of PNC in PBS. (B) Kinetics of PTX precipitation in PBS. (C) PTX concentration in the sampled medium at each time point. Symbols indicate each replicate. (D) Derived count rate of PNC and PPT suspended in 0.2% PBST.
2.3.4.2 Limitations of Current Dissolution Kinetics Study Methods and Potential
Alternatives
Our release kinetics demonstrated a critical limitation of dialysis as a sampling
method in testing dissolution kinetics of particle formulations of PTX. Dialysis method is
often preferred to centrifugation because centrifugation makes it difficult to resuspend
nanoparticles for further incubation. However, the dialysis membrane itself could delay
A B
C D

65
the drug diffusion and involve drug adsorption, especially for poorly water-soluble drugs
like PTX, causing underestimation of dissolution kinetics.
Given these challenges, it is worthwhile to note several in-situ analytical
techniques developed to measure the dissolved drug directly and non-invasively. An
ultraviolet/visible (UV/Vis) fiber optic probe is a good example of real-time method in
dissolution test [34-36]. However, this technique is only applicable when excipients do
not interfere with analysis. Moreover, the utility of UV/Vis probe in the release kinetics
study of nanoparticle is still limited because of the light absorbing potential of
nanoparticles [37]. Electrochemical analytical methods were also explored but their
application was restricted to electroactive drugs [38, 39]. A method based on the light
scattering of micro/nano particles was implemented in determination of the dissolution
rate of nanoparticles [40, 41]. A linear decrease of scattering intensity is expected if
particles dissolves in the release medium with time. As indicated by our results, this
approach was not suitable for large particles with inadequate count rates and high
tendency of sedimentation. An alternative method could be using hydrogel to contain
drug particles, thereby separating them from receptor medium [42].
2.3.5 PTX Release Kinetics from HA Gel Formulations
Due to the limitations of the dialysis method mentioned above, gel formulations
were directly exposed to release media, and the released PTX was sampled by
centrifugation.

66
2.3.5.1 In Tween/PBS under Sink Condition
PNC and PPT were loaded in the HA gel, and the PTX release was tested in-vitro
using Tween/PBS as release medium. We first attempted to compare the dissolution rates
under a sink condition as defined by the United States Pharmacopeia (the volume of
medium at least three times that required to form a saturated solution of a drug [29]).
Since the highest PTX concentration (1.1 µg/mL) in the release medium fell far below the
limit of quantitation (3.8 µg/mL) which was calculated per the ICH guideline, the
released amount was indirectly determined by measuring the PTX amount remaining in
the gel at each time point and deducting it from the initial PTX amount. With this
method, up to 80% of PTX was found to be released from the gels in 7 days, with no
apparent difference between the two formulations (Fig. 12A).
2.3.5.2 In Tween/PBS under Non-Sink Condition
We next tested the dissolution rate in a non-sink condition to reflect the limited
fluid volume in the peritoneal cavity. The volume of peritoneal fluid in a healthy adult is
approximately 50 mL with a protein content 75% lower than that of the blood [44, 45].
The turnover rate of peritoneal fluid is 4-5 mL/h [44]. Although malignancies may
increase the protein level and accelerate the volume and the turnover rate of the ascites,
the small volume of the peritoneal fluid (compared to blood) is likely to challenge the
sink condition assumption. Therefore, we used 20 mL of release medium (Tween/PBS)
for PPT-gel and PNC-gel equivalent to 200 µg of PTX to create a release condition that
intentionally violated a sink condition. The release medium was directly analyzed at each
time point to determine the cumulative drug release. Under this non-sink condition,

67
32.6% and 30.6% of the total PTX were released from PPT-gel and PNC-gel,
respectively, in 12 days (Fig. 12B). 66.0% (PPT-gel) and 59.7% of PTX (PNC-gel) were
recovered in the remaining gel (Fig. 12C). The relatively slow release (compared to the
sink condition) suggests that the released PTX might have undergone reprecipitation in
the medium as the PTX concentration reached the saturation solubility. We expected that
the two gels would show different release kinetics at least initially but did not observe
any difference, most likely due to rapid reprecipitation that might have offset the effect of
particle size difference.
Figure 12. (A) In-vitro PTX release kinetics from HA-gel under sink condition (initial PTX concentration=1.1 µg/mL). (B) In-vitro PTX release kinetics from HA-gel under non-sink condition (initial PTX concentration=10 µg/mL). (C) Total PTX recovery from Tween/PBS medium. Data are expressed as averages and standard deviations of three independently and identically prepared samples.
The lack of difference in in-vitro release kinetics between the two gels may be
attributable to several reasons. First, both methods may involve the centrifugation-
induced artifacts. In the sink condition method, it is possible that the remaining PTX at
each time point might have been underestimated (i.e., drug release overestimated) due to
A B C

68
the centrifugation, which would have pressurized the gel and caused artificial release of
PPT and PNC loosely associated with the gel. In the non-sink method, the centrifugation
force applied to separate the released PTX from the gels might have contributed to
underestimation by accelerating the aggregation of reprecipitates. Second, the
invasiveness of Tween80 might also have masked the potential difference between two
gels. In this regard, it is worthwhile to reconsider the utility of in-vitro release kinetics
studies in predicting in-vivo outcomes. The release kinetics is largely affected by the
sampling method, concentration gradient between the formulation and release medium
(i.e., whether it satisfies the sink condition or not) and the choice of release medium [30].
However, the sampling method involves the aforementioned artifacts, and the sink
condition assumption is not necessarily applicable to drug release in the peritoneal cavity
due to the limited fluid volume and delay in systemic absorption of the dissolved drug.
Tween/PBS is widely used as a dissolution/release medium for poorly water-soluble drug
formulations to ensure the solubility [46-51]; however, its physiological relevance is not
clearly established, and the active role of Tween80 in drug release is seldom considered
in data interpretation.
2.3.5.3 In FBS/PBS or Tween/PBS containing Hyaluronidase under Non-Sink
Condition
In-vitro PTX release kinetics was studied with a medium simulating the peritoneal
cavity environment, which contains amphiphilic solutes (plasma proteins or Tween80)
and hyaluronidase: 25-FBS/PBS with 10 U/mL HAase or Tween/PBS with 10 U/mL
HAase. We were curious if HAse decelerated PTX release by degrading the gels and

69
facilitating the particle aggregation. The total PTX concentration in the media was kept at
10 µg/mL, above the PTX solubility in Tween/PBS (3.3 µg/mL). PTX solubility in 25-
FBS/PBS was not measured, but given that PTX solubility in 50-FBS/PBS was 35
µg/mL, so PTX solubility in 25-FBS/PBS was likely to be lower than 35 µg/mL. Thus,
both tests were done under a non-sink condition.
HA gel gradually degraded in 6 days in the presence of HAase. PPT-gel and
PNC-gel continuously released 35.2% and 29.5% over 10 days in 25-FBS/PBS,
respectively, similar to those tested without HAse (Fig. 13A). This indicates that the PTX
reprecipitation dominated the potential effect of HAse. The PTX recovery from PNC or
PPT-gel was much lower (< 60%) than that in Tween/PBS, probably due to the poor
stability of PTX in serum containing medium and inadequate sampling frequency after
the first day of the release kinetics study (Fig. 13B).
Similarly, PPT-gel and PNC-gel released 32.7% and 29.6% over 10 days in
Tween/PBS, respectively (Fig. 13C). Total PTX recovery from PNC or PPT-gel (~70%)
was higher than that in 25-FBS/PBS reflecting the stability profile (Fig. 13D). On the
other hand, this was lower than that in Tween/PBS without HAse (above 90%, Fig. 12C).
This difference is not due to the effect of HAse but more likely due to the sampling
condition. Here, the samples were centrifuged at 2095 rcf (as opposed to 9300 rcf in
section 1.2.9.2), which might have caused incomplete separation of supernatant and
subsequent loss of samples during each replacement with fresh medium.

70
Figure 13. (A) In-vitro PTX release kinetics performed in 25-FBS/PBS with 10U/mL HAase. (B) Total PTX recovery from 25-FBS/PBS medium (n=3). (C) In-vitro PTX release kinetics performed in Tween/PBS with 10U/mL HAase. (D) Total PTX recovery from Tween/PBS medium (n=3).
2.3.6 PTX Release Kinetics from HA-EtCA-CMC Gel
PTX release from HA-EtCA0.1-CMC gel was tested under non-sink condition.
Although reprecipitation of PTX is likely to have played a dominant role in PTX release
kinetics from the gel formulations in the non-sink condition, we speculated that the
segregation of PNC within the gel might further decelerate drug release during
incubation. We hypothesized that the addition of hydrophobic moieties like 5β-cholanic
PPT-gel
PNC-g
el
PPT-gel
PNC-g
el
A B
C D

71
acid would create domains for PNC to dwell in and prevent segregation of PNC in the gel
during the delivery period. As expected, HA- EtCA0.1-CMC gel increased to 31.8%, as
compared to 19.5% from HA gel in 10 days (Fig. 14A). Total PTX recovery was also
improved from 30.5% in HA gel to 45.2% in EtCA0.1-CMC-HA gel (Fig. 14B). This
result suggested that PTX release was facilitated by incorporating hydrophobic domains
to the gel.
Figure 14. (A) In-vitro PTX release from PNC containing HA gel or EtCA0.1-CMC-HA gel. (B) Total PTX recovery from Tween/PBS medium (n=3).
2.4 Conclusions
We have developed a nanoparticle depot for IP chemotherapy consisted of PNC
and HA gel. PNC with a size of ~310 nm was produced by nonsolvent and temperature
induced crystallization. To improve compatibility between hydrophobic PNC and
0.1
A B

72
hydrophilic HA gel, a hydrophobic moiety, EtCA, was conjugated on CMC-CHO and
employed as a partial replacement of HA-CHO. Dissolution kinetics of PNC could not be
determined by the dialysis method due to drug reprecipitation caused by diffusion barrier,
while fast dissolution of PNC was observed with light scattering method. PTX release
profiles from PNC-gel and PPT-gel were estimated in both sink- and non-sink conditions,
where the latter simulated the peritoneal environment. In-vitro release kinetics studies did
not reveal any difference between PNC-gel and PPT-gel, partly due to the centrifugation-
related artifacts. Despite the technical limitations, in-vitro release kinetics studies
detected enhancement of PTX release due to the inclusion of EtCA in the gel, indicating
that hydrophobic domain in the gel helped prevent segregation of PNC. However, due to
the unexpected toxicity of EtCA-CMC-CHO, EtCA-CMC-HA gel was not further
pursued in subsequent studies.

73
2.5 References
1. Markman, M., et al., Intraperitoneal Chemotherapy of Ovarian Cancer: A
Review, With a Focus on Practical Aspects of Treatment. J Clin Oncol, 2006.
24(6): p. 988-994.
2. Mohamed, F., et al., Pharmacokinetics and tissue distribution of intraperitoneal
paclitaxel with different carrier solutions. Cancer Chemother Pharmacol, 2003.
52(5): p. 405-410.
3. Mohamed, F., et al., Pharmacokinetics and tissue distribution of intraperitoneal
docetaxel with different carrier solutions. J Surg Res, 2003. 113(1): p. 114-120.
4. Tsai, M., et al., Effects of Carrier on Disposition and Antitumor Activity of
Intraperitoneal Paclitaxel. Pharm Res, 2007. 24(9): p. 1691-1701.
5. Bajaj, G., et al., Hyaluronic acid-based hydrogel for regional delivery of
paclitaxel to intraperitoneal tumors. J Control Release, 2012. 158(3): p. 386-392.
6. Armstrong, D.K., et al., Intraperitoneal Cisplatin and Paclitaxel in Ovarian
Cancer. New Eng J Med, 2006. 354(1): p. 34-43.
7. Hofstra, L.S., et al., Kinetic Modeling and Efficacy of Intraperitoneal Paclitaxel
Combined with Intravenous Cyclophosphamide and Carboplatin as First-Line
Treatment in Ovarian Cancer. Gynecol Oncol, 2002. 85(3): p. 517-523.
8. Chambers, S.K., et al., Phase I Trial of Intraperitoneal Pemetrexed, Cisplatin,
and Paclitaxel in Optimally Debulked Ovarian Cancer. Clin Cancer Res, 2012.
18(9): p. 2668-2678.

74
9. de Bree, E., et al., Intraperitoneal chemotherapy with taxanes for ovarian cancer
with peritoneal dissemination. Eur J Surg Oncol, 2006. 32(6): p. 666-670.
10. Sun, B., et al., Nanocrystals for the parenteral delivery of poorly water-soluble
drugs. Curr Opin Solid State Mater Sci, 2012. 16(6): p. 295-301.
11. Patravale, V.B., et al., Nanosuspensions: a promising drug delivery strategy. J
Pharm Pharmacol, 2004. 56(7): p. 827-840.
12. Müller, R.H., et al., State of the art of nanocrystals – Special features, production,
nanotoxicology aspects and intracellular delivery. Eur J Pharm and Biopharm,
2011. 78(1): p. 1-9.
13. Zhao, R., et al., Hybrid Nanocrystals: Achieving Concurrent Therapeutic and
Bioimaging Functionalities toward Solid Tumors. Mol Pharm, 2011. 8(5): p.
1985-1991.
14. Marchettini, P., et al., Docetaxel: pharmacokinetics and tissue levels after
intraperitoneal and intravenous administration in a rat model. Cancer Chemother
Pharmacol, 2002. 49(6): p. 499-503.
15. Nemes, K.B., et al., Oral, intraperitoneal and intravenous pharmacokinetics of
deramciclane and its N-desmethyl metabolite in the rat. J Pharm Pharmacol,
2000. 52(1): p. 47-51.
16. Krasner, C.N., et al., Case 11-2006. New Eng J Med, 2006. 354(15): p. 1615-
1625.
17. Zahedi, P., et al., Combination Drug Delivery Strategy for the Treatment of
Multidrug Resistant Ovarian Cancer. Mol Pharm, 2010. 8(1): p. 260-269.

75
18. De Souza, R., et al., Biocompatibility of injectable chitosan–phospholipid implant
systems. Biomaterials, 2009. 30(23–24): p. 3818-3824.
19. Zahedi, P., et al., Chitosan–phospholipid blend for sustained and localized
delivery of docetaxel to the peritoneal cavity. Int J Pharm, 2009. 377(1–2): p. 76-
84.
20. Yu, J., et al., The antitumor effect of a thermosensitive polymeric hydrogel
containing paclitaxel in a peritoneal carcinomatosis model. Invest New Drugs,
2012. 30(1): p. 1-7.
21. Wang, Y., et al., 5-FU-hydrogel inhibits colorectal peritoneal carcinomatosis and
tumor growth in mice. BMC Cancer, 2010. 10(402): p. 1471-2407.
22. Yeo, Y., et al., Prevention of peritoneal adhesions with an in situ cross-linkable
hyaluronan hydrogel delivering budesonide. J Control Release, 2007. 120(3): p.
178-185.
23. Yeo, Y., et al., Peritoneal adhesion prevention with an in situ cross-linkable
hyaluronan gel containing tissue-type plasminogen activator in a rabbit repeated-
injury model. Biomaterials, 2007. 28(25): p. 3704-3713.
24. Yeo, Y., et al., In situ cross-linkable hyaluronic acid hydrogels prevent post-
operative abdominal adhesions in a rabbit model. Biomaterials, 2006. 27(27): p.
4698-4705.
25. Yeo, Y., et al., In situ cross-linkable hyaluronan hydrogels containing polymeric
nanoparticles for preventing postsurgical adhesions. Ann Surg, 2007. 245(5): p.
819-824.

76
26. Bulpitt, P., et al New strategy for chemical modification of hyaluronic acid:
preparation of functionalized derivatives and their use in the formation of novel
biocompatible hydrogels. J Biomed Mater Res, 1999. 47(2): p. 152-169.
27. Yuan, Y., et al., Contact Angle and Wetting Properties, in Surface Science
Techniques, G. Bracco and B. Holst, Editors. 2013, Springer Berlin Heidelberg. p.
3-34.
28. Anhalt, K., et al., Development of a new method to assess nanocrystal dissolution
based on light scattering. Pharm Res, 2012. 29(10): p. 2887-2901.
29. The United States Pharmacopeia: The National Formulary (USP37/NF32).
General Chapters: <1092> The Dissolution Procedure: Development and
Validation2014, Rockville, MD: The United States Pharmacopeial Convention,
Inc.
30. Abouelmagd, S.A., et al., Release Kinetics Study of Poorly Water-Soluble Drugs
from Nanoparticles: Are We Doing It Right? Mol Pharm, 2015. 12(3): p. 997-
1003.
31. Choi, K.Y., et al., Self-assembled hyaluronic acid nanoparticles as a potential
drug carrier for cancer therapy: synthesis, characterization, and in vivo
biodistribution. J Mater Chem, 2009. 19(24): p. 4102-4107.
32. Ringel, I., et al., Taxol is converted to 7-epitaxol, a biologically active isomer, in
cell culture medium. J Pharmacol and Exp Ther, 1987. 242(2): p. 692-698.
33. Willey, T.A., et al., High-performance liquid chromatographic procedure for the
quantitative determination of paclitaxel (Taxol®) in human plasma. J Chromatogr
B, 1993. 621(2): p. 231-238.

77
34. Aldridge, P.K., et al., A robotic dissolution system with on-line fiber-optic UV
analysis. J Pharm Sci, 1995. 84(8): p. 909-914.
35. Chen, C.-S., et al., A Drug Dissolution Monitor Employing Multiple Fiber Optic
Probes and a UV/Visible Diode Array Spectrophotometer. Pharm Res, 1994.
11(7): p. 979-983.
36. Alonzo, D., et al., Understanding the Behavior of Amorphous Pharmaceutical
Systems during Dissolution. Pharm Res, 2010. 27(4): p. 608-618.
37. Van Eerdenbrugh, B., et al., Influence of Particle Size on the Ultraviolet Spectrum
of Particulate-Containing Solutions: Implications for In-Situ Concentration
Monitoring Using UV/Vis Fiber-Optic Probes. Pharm Res, 2011. 28(7): p. 1643-
1652.
38. Rosenblatt, K.M., et al., Drug release from differently structured
monoolein/poloxamer nanodispersions studied with differential pulse
polarography and ultrafiltration at low pressure. J Pharm Sci, 2007. 96(6): p.
1564-1575.
39. Charalampopoulos, N., et al., Differential pulse polarography: a suitable
technique for monitoring drug release from polymeric nanoparticle dispersions.
Anal Chim Acta, 2003. 491(1): p. 57-62.
40. Crisp, M.T., et al., Turbidimetric measurement and prediction of dissolution rates
of poorly soluble drug nanocrystals. J Control Release, 2007. 117(3): p. 351-359.
41. Anhalt, K., et al., Development of a New Method to Assess Nanocrystal
Dissolution Based on Light Scattering. Pharm Res, 2012. 29(10): p. 2887-2901.

78
42. Peschka, R., et al., A simple in vitro model to study the release kinetics of
liposome encapsulated material. J Control Release, 1998. 56(1-3): p. 41-51.
43. The United States Pharmacopeia: The National Formulary 574
(USP37/NF32); The United States Pharmacopeial Convention, Inc.: 575
Rockville, MD, 2014.
44. Sherwood, L., Human physiology : from cells to systems 2007, Australia;
Belmont, CA: Thomson/Brooks/Cole.
45. Watson, M.S., Oxford handbook of palliative care 2009, Oxford; New York:
Oxford University Press.
46. Kilfoyle, B.E., et al., Development of paclitaxel-TyroSpheres for topical skin
treatment. J Control Release, 2012. 163(1): p. 18-24.
47. Yang, T., et al., Enhanced solubility and stability of PEGylated liposomal
paclitaxel: In vitro and in vivo evaluation. Int J Pharm, 2007. 338(1–2): p. 317-
326.
48. Cha, E.-J., et al., Stabilized polymeric micelles by electrostatic interactions for
drug delivery system. Eur J Pharm Sci, 2009. 38(4): p. 341-346.
49. Gullotti, E., et al., Beyond the imaging: Limitations of cellular uptake study in the
evaluation of nanoparticles. J Control Release, 2012. 164(2): p. 170-176.
50. Amoozgar, Z., et al., Low Molecular-Weight Chitosan as a pH-Sensitive Stealth
Coating for Tumor-Specific Drug Delivery. Mol Pharm, 2012. 9(5): p. 1262-1270.
51. Gullotti, E., et al., Polydopamine-Based Surface Modification for the
Development of Peritumorally Activatable Nanoparticles. Pharm Res, 2013.
30(8): p. 1956-1967.

79
CHAPTER 3. BIOACTIVITY EVALUATION OF NANOPARTICLE DEPOT
3.1 Introduction
Although the difference in in-vitro release profile between PNC-gel and PPT-gel
was not identified, it is still possible that PNC-gel can demonstrate potential advantages
over PPT-gel in a more biologically relevant experimental setting or in-vivo because the
in-vitro release kinetics does not always correlate well with the in-vivo outcomes. In this
chapter, the half maximal inhibitory concentrations (IC50) of PTX in the form of PNC or
PPT were measured with different cell viability assays on SKOV3 human ovarian cancer
cells. Low IC50 value could be attributed to fast drug release as well as sensitivity of the
cells to the drug. In addition, Maximum tolerated doses (MTD) of PPT-gel and PNC-gel
were estimated using healthy mice to determine the PTX dose in in-vivo efficacy study
and observe the difference in drug release rate in-vivo. In anti-tumor efficacy study, PNC
was administered IP to nude mice bearing peritoneal ovarian tumor xenografts with the
HA gel as a carrier to retain the drug in the peritoneal cavity. Progression of tumor
burden after single treatment of PNC-gel was monitored over 10 weeks via non-invasive
whole body imaging, and the outcome was compared with that of PPT-gel. We expect
that PNC can show higher cytotoxicity than PPT in both in-vitro and in-vivo bioactivity
studies

80
3.2 Materials and Methods
3.2.1 Materials
Hyaluronic acid (HA, 20 kDa and 500 kDa) was purchased from Lifecore
Biomedical, LLC (Chaska, MN, USA). Paclitaxel (PTX) was a gift of Samyang Genex
Corp (Seoul, Korea). Carbamazepine was purchased from Enzo life sciences (Plymouth
Meeting, PA, USA). Cremophor ELP was a gift from BASF (New York, NY, USA). D-
Luciferin potassium salt was purchased from Gold Biotechnology (St. Louis, MO, USA).
Geneticin® selective antibiotic (G418 sulfate, 50 mg/mL) was purchased from Life
technologies (Grand Island, NY, USA). Cell culture medium and supplements were
purchased from Invitrogen (Carlsbad, CA, USA). Micro BCA Protein Assay Kit was
purchased from Life technologies (Grand Island, NY, USA). CellTiter-Glo® 2.0 Assay
Kit was purchased from Promega (Madison, WI, USA). All other reagents were
purchased from Sigma-Aldrich (St. Louis, MO, USA).
3.2.2 Determination of IC50 of Taxol, PPT and PNC on SKOV3 Cell Line
IC50 of PNC on SKOV3 cell line was determined with luminescent cell viability
assay (CellTiter-Glo® 2.0 Assay) which determines the number of viable cells by
quantitating the amount of ATP from metabolically active cells. SKOV3 human ovarian
cancer cells (ATCC) were maintained in RPMI-1640 medium supplemented with 10%
fetal bovine serum (FBS) and 1% penicillin-streptomycin. Cells were plated in a 96-well
plate at a density of 5,000 cells per well with 0.2 mL of complete medium. After 24h

81
incubation, Taxol, PPT or PNC suspended in 22 µL PBS was added to each well and the
final PTX concentration ranged from 0.05 to 100 nM. Control groups were treated with
22 µL PBS or Cremophor ELP in PBS at the same concentrations as Taxol treated group.
Cells were exposed to treatments for 24h and allowed to recover for 2 days after
replacing the treatments with fresh medium. One hundred microliters of CellTiter-Glo
Reagent was added to each well after removal of 100 µL of medium on the top. The plate
was placed on an orbital shaker for 2 minutes to induce cell lysis and incubated at room
temperature for 10 minutes to stabilize the luminescent signal. The luminescence of each
treatment group was measured with a SpectraMax M3 microplate reader (Molecular
Devices, Sunnyvale, CA, USA) and normalized to the signal of PBS control group.
3.2.3 Cellular Retention and Cytotoxicity of PTX
SKOV3 human ovarian cancer cells (ATCC) were maintained in RPMI-1640
medium supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-
streptomycin. Cells were plated in a 12-well plate at a density of 200,000 cells per well
with 0.8 mL of complete medium. After 24h incubation, PNC or PPT suspended in 88 µL
PBS was added to each well to make the final PTX concentration 6 µg/mL (7 µM). A
control group was treated with 88 µL PBS. Cells were incubated with the treatments for
3h and washed twice with 0.5 mL complete medium after removal of the treatments. The
treated cells were incubated for two days in fresh medium and evaluated with the MTT
(3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide) assay. The absorbance
of the solubilized formazan was measured with a SpectraMax M3 microplate reader

82
(Molecular Devices, Sunnyvale, CA, USA) at a wavelength of 562 nm. The measured
absorbance was normalized to the absorbance of the control group.
To determine PTX retained in the cells after each treatment, SKOV3 cells were
treated in the same way as described above. Immediately after removing treatments, 1
mL of sterile water was added to each well, frozen and thawed once to lyse the cells. The
cell lysate was analyzed with HPLC (see section 2.2.5) and Micro BCA assay to
determine the concentrations of PTX and protein, respectively.
PTX in the cell lysate was analyzed after ethyl acetate extraction. Briefly, 2 mL of
the cell lysate was spiked with 10 µg/mL carbamazepine as an internal standard, mixed
with 6 mL ethyl acetate, and agitated on a rotating shaker for 40 min. The mixture was
then centrifuged at 3724 rcf for 15 min to separate an organic layer, which was
transferred to a new glass vial and dried under vacuum. The dried sample was dissolved
in HPLC mobile phase, filtered through 0.45 µm syringe filter, and analyzed with HPLC.
A calibration curve was drawn with PTX in FBS medium in known concentrations,
treated in the same manner as the sample solutions. PTX was analyzed with HPLC
equipped with UV detector (1100 series, Agilent Technologies, Palo Alto, CA) and an
Ascentis C18 column (25 cm × 4.6 mm, particle size 5 µm) (Supelco, St. Louis, MO,
USA). The mobile phase was a mixture of acetonitrile and water (50:50), run in the
isocratic mode at a flow rate of 1 mL/min. PTX was detected at 227 nm.
In a separate experiment, SKOV3 cells were plated in a 12-well plate at a density
of 60,000 cells per well with 1.5 mL of complete medium. After 24h incubation, a
Transwell insert was suspended in each well as a container of PTX treatment. The
Transwell membrane was perforated with fifteen 21-gauge needle holes to facilitate the

83
transport of released PTX (either as free molecules or particles) to the underlying cell
layer. 0.1 mL of PNC-gel, PPT-gel, PNC, or PPT suspension was placed in each well
along with 0.4 mL PBS, making the final PTX concentration in the well 30 µg/mL (35
µM). A control group was treated with 0.1 mL of PBS. Cells were incubated with the
treatments for 3h, 24h or 48h, removed of the treatments, washed twice, and incubated
for additional two days in complete medium prior to the MTT assay. The solubility of
PTX in the culture medium containing 10% FBS was determined in the same method as
PBS containing 50 v/v% FBS described in Chapter 2.
3.2.4 Determination of the Maximum Tolerated Doses of Treatments
All animal procedures were approved by Purdue Animal Care and Use
Committee, in conformity with the NIH guidelines for the care and use of laboratory
animals. The MTD of each treatment was determined according to the method published
by the National Cancer Institute’s Developmental Therapeutics Program [1]. Healthy
female Balb/c wild-type mice (8-10 week old, ~20g, Harlan Laboratories, Indianapolis,
IN, USA) were randomly assigned to Taxol, PPT-gel, and PNC-gel groups and given a
single IP injection of each formulation at different dose levels (one mouse per dose). The
mice were observed over a period of 2 weeks after the injection. The highest dose
tolerated without >20% weight loss or other signs of significant toxicity was designated
as the MTD of each treatment. This experiment was repeated at least three times to
confirm the reproducibility.

84
3.2.5 In-vivo Efficacy Studies
A mouse model of IP tumor was prepared as described in our previous study [2].
Luciferase-expressing SKOV3 cells (SKOV3-luc, donated by Prof. Glen Kwon at
University of Wisconsin-Madison) [3] were maintained in a complete RPMI-1640
medium containing 500 µg/mL G418 sulfate. 107 cells were suspended in 1 mL RPMI-
1640 medium and IP injected to a female Balb/c nude mouse (8-10 week old, ~20g,
Harlan Laboratories). Tumor growth was monitored every week by measuring the
bioluminescence with the IVIS Lumina II whole body imaging system (Caliper Life
Science, Hopkinton, MA, USA) [4]. When the radiance of tumors reached 105-106
p/s/cm2/sr, which usually took one or two weeks, animals were evenly assigned to 5
treatment groups (n=3 per group per study): PBS, HA gel, Taxol, PNC-gel and PPT-gel
(equivalent to 30 mg/kg of PTX). One milliliter of each treatment was IP injected through
a catheter, and the skin was sealed with GLUture topical tissue adhesive (Abbott Park,
IL, USA). Bioluminescence of IP tumor was measured weekly over 10 weeks. Animals
were observed every 3 days for weight change and the signs of pain until they reached the
criteria of sacrifice, which include the loss of appetite and/or body weight, bloated
abdomen and signs of respiratory distress, according to the approved animal procedures.
3.2.6 Statistical Analysis
All in-vitro data were analyzed using GraphPad Prism 6 (La Jolla, CA), with
unpaired t test to determine the difference of means between two groups. The comparison
of survival curves was conducted with Log-rank (Mantel-Cox) test built in GraphPad

85
Prism 6. A value of p< 0.05 was considered statistically significant. For the tumor growth
study, analysis of variance (ANOVA) was performed using IBM SPSS Statistics 23
(Armonk, NY, USA) to test the differences between the treatment groups in the change of
tumor burden over time.
3.3 Results and Discussion
3.3.1 IC50 of PTX in the Form of Taxol, PPT or PNC
The growth profile of a certain cell line plays an important role in the
determination of IC50 of a treatment. PTX interferes with the normal depolymerization of
microtubules during cell proliferation [5, 6]. The population doubling time of SKOV3
cell line was approximately 35h, which means that SKOV3 cells need at least 1.5 days to
show their response to PTX. These indicate that it is necessary to leave SKOV3 cells in
fresh medium for at least 1.5 days to observe the cytotoxic effect of PTX. However, this
recovery time cannot be too long because challenged cells may become viable as before
if the treatment discontinues, resulting in underestimation of PTX cytotoxicity. On the
other hand, complete medium contains 10% FBS, which can solubilize PTX; thus,
relatively long incubation time of PNC or PPT in the culture medium may diminish the
potential difference in the release rate between PPT and PNC. Given these constraints,
cells were exposed to each treatment for only one day, a time frame speculated to be
short enough to observe differential PTX release from PPT and PNC, and incubated for 2
more days in treatment-free medium to develop responses to the treatments. This
experiment was repeated twice, and IC50 value was obtained with nonlinear regression

86
using GraphPad Prism 6. Although the apparent IC50 values increased in the order of
Taxol, PNC, and PPT, reflecting the expected difference in dissolution rate of PTX, they
did not show statistical difference, probably due to the variation of cell response to PTX
(Fig. 15). Therefore, alternative study was carried out to directly quantitate the amount of
PTX retained with cells.
Figure 15. IC50 of PTX on SKOV3 cell line in the form of Taxol, PPT and PNC (n=3).
3.3.2 Cellular Retention and Cytotoxicity of PPT and PNC
The cytotoxicity of PPT and PNC were compared as an indirect indicator of drug
dissolution profiles. SKOV3 cells were incubated with PPT and PNC for 3h, and the level
of PTX retained with cells were determined. Much less PTX was detected in the SKOV3
cells treated with PNC than those with PPT (Fig. 16A). The relatively high level of PTX
remaining in PPT-treated cells may be explained by the large size of PPT, which was not
readily removed from cells by simple washing. Surprisingly, despite the low level of PTX
retained with the cells, PNC showed higher cytotoxicity than PPT (Fig. 16B). The cell
IC5
0 o
f P
TX
(n
M)
Taxo
l
PP
T
PN
C
0
5
1 0
1 5
2 0

87
fraction affected by the unit amount of PTX was 3.3 times greater for PNC than PPT
(Fig. 16C), which indicates that PTX provided as PNC was much more efficient in
killing SKOV-3 cells than PPT. These results suggest that PNC made PTX better
available than PPT via the small size that facilitated the drug dissolution. In addition, the
small size may have helped PNC to enter cells. A recent study supports that drug
nanocrystals could be endocytosed as solid particles [7].
3.3.3 Cytotoxicity of PPT-gel and PNC-gel
Acknowledging aforementioned limitations of in-vitro release kinetics tests, we
measured the cytotoxicity of PPT-gel and PNC-gel varying the exposure time to predict
their in-vivo effects (Fig.16D). The gel was contained in a perforated Transwell insert to
avoid direct contact with the cell layer that might limit oxygen supply. Free particles
were supplied in the same manner to mimic a situation where a degrading gel was no
longer able to retain the particles. Therefore, the tests with gels and free particles
represented the initial and later phase of delivery, respectively. With 3h exposure, PNC-
gel was found to be more toxic than PPT-gel, indicating faster dissolution of PNC than
PPT. This difference disappeared upon longer incubation (24h and 48h), which may be
explained by the reprecipitation of released PTX exceeding the saturation solubility of
PTX in the culture medium (1.4 µg/mL), as predicted from the release kinetics results. In
contrast, free PNC showed consistently high cytotoxicity as compared to PPT, indicating
that at least part of PNC were endocytosed by the cells as solid particles before they
underwent dissolution and reprecipitation in the medium. This result suggests that PNC-

88
gel would achieve greater anti-tumor effect than PPT-gel in-vivo, as the endocytosis of
released PNC offset the effect of reprecipitation of dissolved PTX.
Figure 16. (A) PTX retention normalized with total protein content. Micro BCA assay
was performed to determine the total protein content in the cell lysate as an estimate of
cell population. The PTX to protein ratio reflects PTX retained by each cell. (B)
Cytotoxicity of PPT and PNC (equvilant to 7 µM PTX) to SKOV3 cells after 3h of direct
exposure followed by incubation in drug-free medium. (C) Fraction of cells affected by
unit amount of PTX. Data are expressed as averages and standard deviations of three
measurements of a representative batch. Cytotoxicity of (D) PNC-gel and PPT-gel and
(E) PPT and PNC (equivalent to 35 µM PTX) to SKOV3 cells after 3h, 24h or 48 h
exposure followed by incubation in drug-free medium. Data are expressed as averages
and standard deviations of three measurements of a representative batch. *: p<0.05 by t-
test.
PT
X (
g)/
pro
tein
(
g)
PP
T
PN
C
0 .0 0
0 .0 5
0 .1 0
0 .1 5
Ce
ll v
iab
ilit
y (
%)
PP
T
PN
C
0
2 0
4 0
6 0
De
ad
ce
lls
(%
) /
PT
X (
g)
PP
T
PN
C
0
2
4
6
8
A B C
**
*
3h
24h
48h
0
2 0
4 0
6 0
8 0
1 0 0
Ce
ll v
iab
ilit
y (
%)
P N C -g e l
P P T -g e l
3h
24h
48h
0
2 0
4 0
6 0
8 0
1 0 0
Ce
ll v
iab
ilit
y (
%)
P P T
P N C
D E
*
**
*

89
3.3.4 Maximum Tolerated Doses of PTX Treatments
MTDs of PNC-gel, PPT-gel, and Taxol were determined using healthy Balb/c
mice for two purposes: (i) to determine the maximum PTX dose to administer in the anti-
tumor efficacy study and (ii) to observe the difference in drug release rate in-vivo.
Specifically, we hypothesized that the gel more rapidly releasing PTX would have a
lower MTD value. Mice did not survive a single administration at dose higher than 60
mg/kg. On the other hand, PPT-gel and PNC-gel were tolerated much higher doses: 120
mg/kg and 90 mg/kg, respectively. This result first confirms the benefit of the gel
formulations free of Cremophor EL, which is associated with several adverse effects such
as severe anaphylactoid hypersensitivity reactions, abnormal lipoprotein pattern and
peripheral neuropathy [8]. Interestingly, PNC-gel showed lower MTD than PPT-gel,
despite the identical composition. The difference between PNC-gel and PPT-gel suggests
that PNC-gel might have released a greater amount of PTX than PPT-gel in the given
time. This result is consistent with the cellular PTX retention and cytotoxicity results.
3.3.5 Anti-tumor Effects of PPT-gel and PNC-gel
The anti-tumor efficacy of PPT- and PNC-gels was studied using a mouse model
of IP tumor. Once the tumor reached a certain size as indicated by the bioluminescence
signals, treatments (Taxol, PPT-gel and PNC-gel) equivalent to 30 mg/kg PTX as well as
vehicle controls were administered IP once. A single administration regimen was chosen
to compare the duration of the therapeutic effects of the treatments (Fig. 17A). The tumor
burden was monitored up to 14 weeks until the animals reach a humane endpoint (Fig.

90
17B). Animals treated with PBS or HA gel vehicle reached the endpoint in <7 weeks
with median survival periods of 37 days. All animals receiving PTX-treatments survived
longer than the vehicle control groups with notable difference according to the treatment.
Tumor growth in animals treated with Taxol was initially delayed but resumed after 3
weeks to reach the endpoint in <9 weeks with a median survival time of 56 days. This
result may be explained by the rapid clearance of PTX from the peritoneal cavity [2].
Animals treated with PPT-gel showed a similar survival curve (median survival time of
51 days) as Taxol-treated ones. On the other hand, mice treated with PNC-gel showed a
significant extension of the survival period (67 days), which clearly contrasted with
Taxol-treated groups (p<0.05: PNC-gel vs. Taxol, Log rank Mantel-Cox test) (Fig. 18A).
Both PPT- and PNC-gel-treated animals showed minimal increase in tumor signals in
surviving animals until 7 weeks post-treatment (Fig. 18B). However, it was difficult to
obtain meaningful statistical analysis from averages of the bioluminescence values, due
to the attrition of animals over the course of survival period and large variations in tumor
growth. Thus, the change of tumor burden (tumor burden at each time point-initial tumor
burden) was plotted with respect to the survival time for each mouse, and the area under
the curve over time (AUC/time) was calculated as an average tumor burden for the mouse
during the survival period. The mice treated with PNC-gel or PPT-gel showed the lowest
median AUC/time value among the treatment (Fig. 19). The AUC/time value of Taxol
group was not significantly different from PBS or HA gel control groups. Both PPT-gel
and PNC-gel showed significantly lower AUC/time values than Taxol group, but the
difference between PNC-gel and Taxol groups was more significant than that of PPT-gel
and Taxol groups (p: 0.0012 vs. 0.0306).

91
Figure 17. (A) Treatment schedule. (B) Representative whole body bioluminescence
imaging of animals administered with different treatments. The most representative
animal in each group is presented.
a
b
\
\
Weekly monitoring of tumor growth
0 1w 2w 3w 4w 5w 6-14w
Tumor injection
Single IP administration(PBS, HA gel, Taxol, PPT-gel, PNC-gel at 30 mg/kg)
-1 or 2w
B
A

92
Figure 18. (A) Kaplan-Meier analysis for survival time post tumor cells inoculation. n=9
per group. *: p<0.05, PNC-gel vs. Taxol by Log-rank Mantel-Cox test. (B)
Bioluminescence of IP tumors in animals treated with PBS, HA gel, Taxol, PPT-gel, and
PNC-gel. Data are expressed as median with ranges of surviving animals. The box
extends from the 25th to 75th percentiles, the line in the middle of the box indicates the
median, and the whiskers go down to the smallest value and up to the largest.
0 2 4 6 8 0 2 4 6 8 0 2 4 6 8 0 2 4 6 8 0 2 4 6 8
0
5 .01 0 6
1 .01 0 7
1 .51 0 7
2 .01 0 7
T im e (w e e k s )
Av
g R
ad
ian
ce
[p
/s/c
m²/
sr] P B S c o n tro l
H A g e l c o n tro l
T a x o l
P P T g e l
P N C g e l
c
d
0 5 0 1 0 0
0
5 0
1 0 0
T im e (d a y s )
Pe
rc
en
t s
urv
iva
l
P B S c o n tro l
H A g e l c o n tro l
T a x o l
P P T g e l
P N C g e l
*
Overall survival
Tumor radiance in surviving animalsB
A

93
Figure 19. AUC of tumor burden change over survival time of animals treated with PBS,
HA gel, Taxol, PNC-gel and PPT-gel. *: p<0.05, PPT-gel vs. Taxol, **: p<0.05, PNC-gel
vs. Taxol by Tukey’s test.
Taken together, the in-vivo studies demonstrated that a single dose PNC-gel was
significantly better in delaying tumor progression as compared to Taxol, while PPT-gel
was not different from Taxol in the median survival time. This result is consistent with
the in-vitro cellular toxicity test and MTD values, which indicated greater dissolution and
cellular uptake of PNC than PPT. Based on the depot effect of HA gel, PNC could have a
prolonged effect on IP tumors than Taxol. It remains to be investigated how the
prolonged local delivery improved the efficacy of PTX. A potential mechanism may
involve the tumor priming effect by initial PTX exposure, which suppresses stroma
expansion [9], thereby facilitating the transport of subsequently released PTX. A
remaining challenge is to improve the specificity of local delivery of PTX. The current
PNC-gel does not have a mechanism to distinguish tumors vs. normal tissues; thus, some
toxicity to adjacent normal tissues is likely unavoidable (although no abnormalities were
PB
S c
on
tro
l
HA
gel co
ntr
ol
Taxo
l
PP
T-g
el
PN
C-g
el
-4 .01 0 6
-2 .01 0 6
0
2 .01 0 6
4 .01 0 6
6 .01 0 6
AU
C/t
ime
AUC/time
* **

94
observed on macroscopic level in this study). This may be achieved by surface
modification of PNC with cell-specific ligands.
3.4 Conclusion
PTX-gel formulation comprising PNC and HA gel was developed for IP
chemotherapy of ovarian cancer. In cellular toxicity test and MTD assessment, PNC-gel
provided more efficient killing effect and greater toxicity than PPT-gel, which contained
larger PTX particles, indicating a greater dissolution rate of PNC due to the small size. A
single IP administration of PNC-gel extended the survival of mice with IP tumors
significantly better than the same dose Taxol, due to the local depot effect, whereas PPT-
gel was not superior to Taxol in survival extension. While the cell toxicity test and in-
vivo results consistently point to the beneficial effect of particle size reduction, in-vitro
drug release kinetics did not predict the difference between PPT- and PNC-gels,
suggesting the limitation of current release study methods.

95
3.5 References
1. Lengyel, E., et al., Expression of latent matrix metalloproteinase 9 (mmp-9)
predicts survival in advanced ovarian cancer, Gynecol. Oncol. 2001. 82(2): p.
291-298.
2. Bajaj, G., et al., Hyaluronic acid-based hydrogel for regional delivery of
paclitaxel to intraperitoneal tumors. J Control Release, 2012. 158(3): p. 386-392.
3. Cho, H., et al., Poly(ethylene glycol)-block-poly(ε-caprolactone) micelles for
combination drug delivery: Evaluation of paclitaxel, cyclopamine and gossypol in
intraperitoneal xenograft models of ovarian cancer. J Control Release, 2013.
166(1): p. 1-9.
4. Cho, E.J., et al., Intraperitoneal delivery of platinum with in-situ crosslinkable
hyaluronic acid gel for local therapy of ovarian cancer. Biomaterials, 2015. 37: p.
312-319.
5. Horwitz, S.B., Taxol (paclitaxel): mechanisms of action. Ann Oncol, 1994. 5(6):
p. S3-6.
6. http://physics.cancer.gov/docs/bioresource/ovary/NCI-PBCF-HTB77_SK-OV-
3_SOP-508.pdf.
7. Chen, Y. et al., Cellular Uptake Mechanism of Paclitaxel Nanocrystals
Determined by Confocal Imaging and Kinetic Measurement. AAPS J, 2015.
17(5): p. 1126-1134.
8. Gelderblom, H., et al., Cremophor EL: the drawbacks and advantages of vehicle
selection for drug formulation. Eur J Cancer, 2001. 37(13): p. 1590-1598.

96
9. Lu, Z., et al., Tumor-Penetrating Microparticles for Intraperitoneal Therapy of
Ovarian Cancer. J Pharmacol Exp Ther, 2008. 327(3): p. 673-682.

97
CHAPTER 4. ALBUMIN-STABILIZED PACLITAXEL NANOCRYSTALS
4.1 Literature Review3
Approximately 40% of active pharmaceutical ingredients in the discovery stage
have poor water-solubility [1]. In order to attain adequate bioavailability of poorly
soluble drugs, special formulation strategies are employed to increase their dissolution in
aqueous medium [2]. Traditionally, organic solvents are used as co-solvents [3] or a part
of emulsion [4] to formulate the poorly soluble drugs as aqueous dosage forms.
Alternatively, the drug is fitted in cyclodextrins, which have a hydrophobic interior and a
hydrophilic exterior, and made soluble in water [5]. Another way of enhancing the
solubility of poorly soluble drugs is to produce nanoparticulate formulations, such as
liposomes [6], micelles [7], nanoemulsion [8], solid lipid nanoparticles [9], and
polymeric nanoparticles [10]. However, relatively low drug loading efficiency, concerns
for the safety of excipients, and complicated manufacturing process are noted as potential
disadvantages of these strategies.
Nanocrystallization is a technique to produce crystalline particles of poorly
soluble drugs in the nanometer range (i.e., nanocrystals). Due to the size and, thus, the
high surface area to volume ratio, nanocrystals can increase the saturation solubility of a
3 Reprinted from Current Opinion in Solid State & Materials Science, 16(6), Bo Sun, Yoon Yeo,
Nanocrystals for the parenteral delivery of poorly water-soluble drugs, p 295-301, Copyright (2015), with
permission from Elsevier.

98
drug and the dissolution rate of drug particles [11]. Nanocrystals have gained increasing
interest in the pharmaceutical industry because of the simple structures and compositions.
They have been explored for a variety of therapeutic applications including oral [12],
dermal [13], pulmonary [14], systemic administration [15], as well as targeted drug
delivery [16] and intraperitoneal chemotherapy [17]. The objective of this section is to
review the production of nanocrystals and discuss the remaining challenges in the
development of nanocrystal products.
4.1.1 Production of Nanocrystals
Nanocrystals of poorly soluble drugs can be created by “top-down” or “bottom-
up” technologies (Fig. 20), or combinations of the two [18]. Nanocrystals are produced
from the drug itself, with surfactants or polymeric stabilizers on the surface; thus, the
drug content in nanocrystals approaches 100% [18]. The production of nanocrystals is
relatively easy to scale up and transfer to industry as compared to other formulations on
the market, such as liposomes [19, 20] and albumin-based nanoparticles [21]. Several
nanocrystal products, produced by wet milling and high-pressure homogenization, have
been approved by the US Food and Drug Administration as oral products (Table 4).
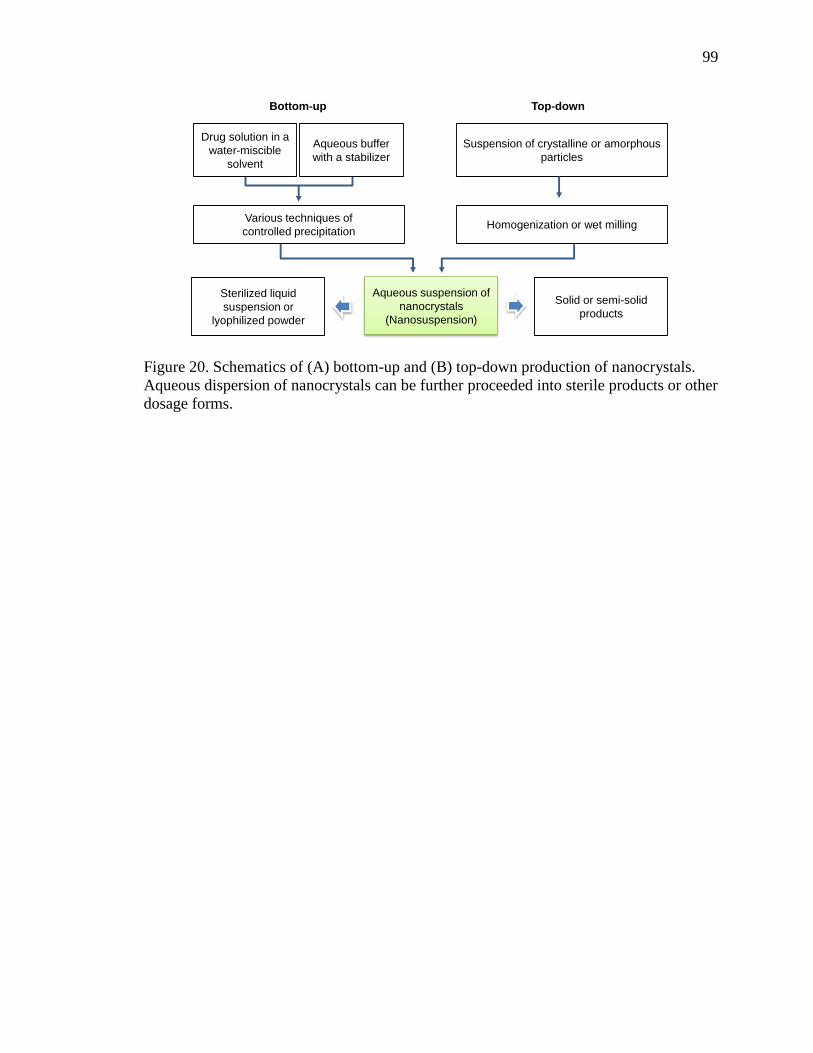
99
Figure 20. Schematics of (A) bottom-up and (B) top-down production of nanocrystals.
Aqueous dispersion of nanocrystals can be further proceeded into sterile products or other
dosage forms.
Suspension of crystalline or amorphous
particles
Homogenization or wet millingVarious techniques of
controlled precipitation
Aqueous suspension of
nanocrystals
(Nanosuspension)
Sterilized liquid
suspension or
lyophilized powder
Solid or semi-solid
products
Top-downBottom-up
Drug solution in a
water-miscible
solvent
Aqueous buffer
with a stabilizer

100
Table 4. Examples of nanocrystal products for oral administration approved by the FDA
[22, 23].
Trade name (drug) Manufacturing
techniques
Indications Company
Rapamune® (Sirolimus) Top-down, wet milling Immunosuppres
sive
Wyeth
Pharmaceuticals
Emend® (Aprepitant) Top-down, wet milling Antiemetic Merck & Co.
Tricor® (Fenofibrate) Top-down, wet milling Hypercholestero
lemia
Abbott
Laboratories
Triglide® (Fenofibrate) Top-down, high-pressure
homogenization
Hypercholestero
lemia
Skye Pharma
Megace ES® (Megestrol
acetate)
Top-down, wet milling Antianorexic Par
Pharmaceutical
Avinza® (Morphine sulfate) Top-down, wet milling Psychostimulant
drug
King
Pharmaceuticals
Focalin® XR (Dexmethyl-
phenidate HCl)
Top-down, wet milling Attention deficit
hyperactivity
disorder
Novartis
Ritalin® LA
(Methylphenidate HCl)
Top-down, wet milling Attention deficit
hyperactivity
disorder
Novartis
Zanaflex CapsulesTM
(Tizanidine HCl)
Top-down, wet milling Muscle relaxant Acorda
4.1.1.1 Bottom-up Technologies
The bottom-up approach refers to methods that create small drug particles from
drug molecules dissolved in an organic solution. Small drug particles are formed as drug
molecules precipitate from solution in the presence of an agent and/or a condition that
induces nucleation of the molecules. For example, a non-solvent, which is miscible with
the solvent but does not dissolve the drug, is used to induce the nanocrystal formation, in
conjunction with various methods to mix the drug solution with non-solvents such as
rotation, liquid jets, or multi-inlet vortex mixing [24]. Alternatively, supercritical fluid,

101
ultrasonic waves, or controlled solvent evaporation are employed to induce drug
precipitation. These technologies are discussed in detail in a recent review article [24].
Particles produced by the bottom-up approach can be crystalline or amorphous.
Amorphous nanoparticles produced by a technique called NanomorphTM achieve higher
saturation solubility and faster dissolution rate than nanocrystals [25, 26]. However, they
are prone to partial or complete re-crystallization, which may lead to decreased
bioavailability. Due to the stability and consistent performance, nanocrystals are usually
favored over amorphous particles. On the other hand, the production of nanocrystals
within a desired size range depends critically on precise control of the precipitation and
prevention of the crystal growth during the production [18]. The complexity of the
process control and potential risk of residual organic solvents have discouraged the
development of commercial products [22]. Recently, spray-drying [27] and freeze-drying
[28, 29] processes have been used to achieve continuous control of the crystallization at
large scales.
4.1.1.2 Top-down Technologies
Top-down approach is based on two basic size reduction methods: wet milling
[30] and high-pressure homogenization [31]. The wet milling process applies shear stress
on large drug particles by grinding an aqueous suspension that contains a drug and a
surface stabilizer using beads or pearls in a milling chamber [32]. The outcome of the
milling process is determined by the hardness of the drug, energy input, milling time, and
stabilizer concentration [32]. Microfluidization and piston-gap homogenization are
examples of high-pressure homogenization [18]. Microfluidization is based on the jet

102
stream principle, where the size diminution is achieved by collision of two fluid streams
of particle suspension in a Y-type chamber under high pressure [22, 31]. The piston-gap
homogenizer forces a particle suspension to pass a small gap (~5 µm) under pressure.
The high shear forces, turbulent flow, and cavitation generated during this passage can
reduce the particle size to the nanometer range [22, 31]. The performance of this process
depends on the number of cycles, power density, and temperature [31]. These techniques
are widely used in industry.
Compared to bottom-up methods, top-down methods require higher energy
consumption and a longer operation time. The risk of contamination due to the erosion of
milling beads is also a disadvantage of wet milling [33]. Moreover, the high-energy
process may induce phase transition of a drug, which may compromise the in-vivo
performance of the products [34].
4.1.1.3 Combined Technologies
A pre-treatment step (bottom-up) and particle size reduction step (top-down) may
be combined. For example, precipitates are first obtained from anti-solvent precipitation,
spray-drying, or lyophilization (pre-treatment step), followed by high-pressure
homogenization (particle-size reduction) [18, 22]. One of the roles of the high-pressure
homogenization step is to anneal the initial precipitates, which are often
thermodynamically unstable, into an ordered crystal structure [22]. Two well-established
techniques, NANOEDGETM and smartcrystals®, have been discussed elsewhere in detail
[18, 22].

103
4.1.1.4 Nanocrystal Stabilization
Due to the high surface energy generated by nanonization, surface stabilizers are
needed to prevent aggregation and precipitation of nanocrystals. Examples of stabilizing
systems are summarized in a recent review article [23]. For drug nanocrystals with no
surface charge, anionic surfactants such as sodium cholate, sodium deoxycholate, and
sodium lauryl sulfate are often used to keep the particles separated via electrostatic
repulsion. Another way of stabilizing nanocrystals is to apply polymeric stabilizers on
their surface and establish a steric barrier against aggregation. Polymers used for this
purpose are derivatives of cellulose, polyvinyl alcohol, polyvinyl pyrrolidone,
Polysorbates (polyoxyethylene sorbitan fatty acid esters), and Pluronics (or Poloxamers,
triblock-copolymers of polyoxyethylene and polyoxypropylene) [35, 36]. Some of the
stabilizers, such as arginine, amphiphilic amino acid copolymers, and vitamin E
polyethylene glycol succinate (TPGS), are biologically active and provide additional
functions to the nanocrystals [30]. For example, TPGS, an effective P-glycoprotein
inhibitor [37], enables paclitaxel (PTX) nanocrystals to overcome multidrug resistance
[38]. The effectiveness of PTX nanocrystals stabilized with TPGS was demonstrated in a
nude mouse model bearing multidrug-resistant NCI/ADR-RES human ovarian cancer
cells [38].
The effectiveness of a nanocrystal stabilizer depends on its affinity for a drug, the
concentration, and the stabilizer to drug ratio in suspension. Relatively hydrophobic
stabilizers have higher affinity for drug crystals and a greater stabilizing effect [39].
There is a positive correlation between particle size and the hydrophilic lipophilic balance
(HLB) value of a non-ionic surfactant in bottom-up approaches; thus, the HLB value can

104
be a useful guideline for the selection of a stabilizer [40]. Typically, a surfactant with a
low HLB value (lipophilic surfactant) is a good stabilizer of hydrophobic nanocrystals.
Adequate surface coverage by stabilizers, irrespective of their mechanisms, is critical to
the stabilization of nanocrystals. However, it does not necessarily mean that the stability
increases in proportion to the concentration of a stabilizer. When the concentration of a
surfactant exceeds the critical micelle concentration (CMC), the excessive surfactant has
a negative effect on the stability of the nanocrystal suspension (nanosuspension) because
micelle formation begins to compete with adsorption to the nanocrystal surface [41-43].
4.1.2 Remaining Challenges in Nanocrystal Development for Parental Applications
4.1.2.1 Instability during Storage
Ostwald ripening refers to a phenomenon that small particles gradually dissolve
and redeposit on the surface of larger particles over time (Fig. 21) [44]. It occurs when
the particle size in a dispersion system is heterogeneous and the dispersed phase (drug)
has a limited solubility in the medium (water) [45], which, unfortunately, are the
conditions frequently encountered in pharmaceutical suspensions [46, 47]. Ostwald
ripening leads to the particle size growth and physical instability of a dispersion system
during storage; therefore, there is a strong need for preventing this process.

105
Figure 21. Schematic representation of Ostwald ripening in nanosuspension.
Surfactants and polymers are commonly used as stabilizers to delay detachment
and attachment of drug molecules at the surface of dispersed particles [45]. Polymers are
believed to be more effective than small molecular-weight surfactants because they tend
to adhere to the nanocrystal surface less dynamically than surfactants [48]. Another way
of preventing particle size growth is to produce uniform nanocrystals, thus eliminating
one of the conditions for Ostwald ripening. Optimizing the process parameters, such as
the number of high-pressure homogenization cycles [49], milling time [34, 50] and
milling speed [51], can eliminate large particles and obtain narrow size distribution.
Processing nanosuspensions into solid products is another way of avoiding dynamic
changes in the medium and thus Ostwald ripening [23, 52].
4.1.2.2 Instability during Applications
Due to the high surface area to volume ratio, complete dissolution of nanocrystals
can occur quickly (in less than an hour) as long as a sink condition is maintained (Table.
5). On the other hand, in locations with a limited volume of fluid, such as the peritoneal
cavity, stomach, or the lungs, nanocrystals may be exposed to a non-sink condition for an
Nanocrystals with different
particle sizes
Drug molecules gradually
transfer from small particles
to large particles.
Large particles grow
continuously at the expense of
small particles and dominate
the system.

106
extended period of time. This may limit the dissolution rate of the nanocrystals and
provide an opportunity for sustained drug release [16]. However, with the gradual surface
erosion and loss of stabilizing agents, the nanocrystals may become increasingly unstable
and, thus, undergo agglomeration and Ostwald ripening over time. When this occurs,
drug dissolution slows down significantly, to an extent that the drug is no longer
bioavailable. We have experienced a consequence of particle agglomeration in an animal
model of IP tumors [53]: we produced PTX particles with precipitation followed by
sonication and administered the particles IP into mice bearing ovarian tumors in the
peritoneal cavity, using an in-situ crosslinkable hyaluronic acid-based hydrogel as a
carrier [53]. The PTX precipitates delivered with the hydrogel were best retained in the
peritoneal cavity as compared to other formulations, which included multiple injections
of Taxol, a bolus injection of Taxol, PTX precipitates alone, and Taxol delivered with the
hydrogel. Despite the prolonged IP retention, the anti-tumor effect of hydrogel-embedded
PTX precipitates was not significantly different from the others, which were cleared from
the peritoneal cavity much earlier. One of the possible explanations is the agglomeration
of PTX precipitates, which led to incomplete dissolution of PTX [53].

107
Table 5. Examples of nanocrystal dissolution in a sink condition.
Drug Particle size (nm) Dissolution rate Reference
Oridonin 322.7 98% dissolved in 24 min [41]
Asulacrine d(v; 0.5)* 133 ± 20 42% dissolved in 6 h [15]
Celecoxib d(v; 0.5) 360 91.8% dissolved in 50 min [54]
Meloxicam d(v; 0.5) 530 ± 110 100% dissolved in 10 min [55]
Artemisinin 100-360 75.9% dissolved in 4 h [56]
Nitrendipine 209 ± 9 90% dissolved in 2 min [57]
Quercetin 213.6 ± 29.3 73.2% dissolved in 20 min [58]
Itraconazole ~300 85% dissolved in 90 min [12]
Camptothecin 200-700 50% dissolved in 2 h [16]
*Note: size of the particles for which 50% of the same volume contains particles smaller
than d (v; 0.5).
4.1.2.3 Lack of Target Specificity
The surface of nanocrystals is decorated with specific ligands for target-specific
delivery. However, continuous surface erosion poses a challenge to the longevity of the
targeting effect. In this regard, it is worthwhile to note a recent approach to produce a co-
crystal of a drug and functional molecules [59]. Here, Li et al. produced hybrid crystals
by co-crystallization of PTX and fluorescent dyes, where guest dye molecules were
integrated in PTX nanocrystals [59]. Since the dye was embedded throughout the matrix,
the PTX-dye hybrid nanocrystals could be located via real-time imaging during their
lifetime in the body. The same principle is applicable to producing drug-ligand hybrid
nanocrystals with the prolonged target specificity.

108
4.2 Introduction
While the design of nanoparticulate formulations has become increasingly
sophisticated, structurally and conceptually simple nanocrystals have a unique advantage
with respect to the development of commercial products. Nanocrystals can greatly
enhance the dissolution rate of poorly soluble drugs. The large contact area of
nanocrystals can allow for a greater interaction with tissue or cell surfaces and enhance
drug absorption. Several oral nanocrystal products are available on the market and dermal
and IV products are actively explored. However, the potential of nanocrystals has not
been thoroughly investigated for different applications such as targeted or local drug
delivery. For example, nanocrystals can be combined with implantable delivery systems
to attain a higher local concentration for a prolonged period of time. This requires
nanocrystals to maintain the small size during long-term delivery without using toxic
surfactants as surface stabilizers. Current methods of nanocrystal production do not
adequately address the challenges in development of such products, and new approaches
to engineer nanocrystals are strongly awaited.
To overcome the limitations in current nanocrystal production for local therapy,
we developed a method to make PTX nanocrystals smaller than 200 nm with albumin as
a surface stabilizer. Albumin was chosen because of its ability of adsorbing onto
hydrophobic surfaces and therefore provides steric hindrance to nanocrystal aggregation
and growth [60, 61]. Moreover, serum proteins have the ability to bind certain membrane
proteins which may facilitate targeting of anticancer drugs to the tumors [62, 63]. This
method involves crystallization in a matrix of surfactant, such as Pluronic F127. The

109
presence of polymer matrix limits the growth of crystals; thus, this method can form
relatively small particle size (<200 nm). The incipient nanocrystals (iCim) are harvested
by sonication and hydration and stabilized with surface modifiers such as albumin.
Pluronic F127 is replaced by the surface modifiers with higher affinity for nanocrystals
and mostly absent in the final product. Due to the small size and the lack of polymeric
surfactant, it is expected to provide faster dissolution of a drug and greater safety than
other nanocrystal formulations.
4.3 Materials and Methods
4.3.1 Materials
Paclitaxel (PTX) was a gift of Samyang Genex Corp (Seoul, Korea). Pluronic
F127 was a gift from BASF (New York, NY, USA). Albumin from human serum (alb, 66
kDa, ≥ 96%) was purchased from Sigma-Aldrich (St. Louis, MO, USA). Reagents for
sodium dodecyl sulfate polyacrylamide (SDS-PAGE) gel electrophoresis were purchased
from Bio-Rad (Hercules, CA, USA). Micro BCA Protein Assay Kit was purchased from
Life technologies (Grand Island, NY, USA). All other reagents were purchased from
Sigma-Aldrich (St. Louis, MO, USA).

110
4.3.2 Preparation of Albumin-stabilized PNC
4.3.2.1 Crystallization in Matrix (Cim)
Crystallization in Matrix consists of two steps: first, incipient crystallization in
matrix (iCim), adapted from the literature [64]; second, surface stabilization of iCim.
Incipient Crystalliation in Matrix (iCim): PTX was crystallized in the polymeric
matrix and recovered by sonication and hydration (Fig. 22). Briefly, 6 mg PTX and 24
mg Pluronic F127 (1/4, w/w) were fully dissolved in 3 mL of chloroform in a round
bottom flask. Chloroform was evaporated with rotary evaporator at 40 ℃ for 2 hours to
form a thin film on the wall of the flask. Six milliliters of DI water was added to hydrate
the film under stirring at room temperature for 1hour. The hydrated suspension was
sonicated with ice-water bath to help reduce the particle size. The settings of the probe
sonicator were: power = 40%, time = 15 min and pulse = 1s/1s.
Surface Stabilization of iCim: iCim was stabilized with albumin. iCim suspension
(PTX concentration = 1 mg/mL) was mixed with an equal volume of aqueous albumin
aqueous solution (8 mg/mL) and incubated at room temperature for 3 hours on a rotating
rocker. The mixture was centrifuged at 4 ℃ at 104 rpm (9300 rcf) for 10 min to remove
Pluronic F127 and unadsorbed albumin. The pellet was re-suspended with 0.5 mg/mL
albumin solution, and the surface stabilized nanocrystals were collected by centrifugation
at 4 ℃ at 104 rpm (9300 rcf) for 10 min. This nanocrystal formulation was called Cim-
alb.

111
4.3.2.2 Nonsolvent and Temperature-Induced Crystallization
Control nanocrystals were prepared by the bottom-up method based on
nonsolvent and temperature-induced crystallization [59] (Section 2.2.2) and referred to as
PNC. To further stabilize PNC, PNC suspension (1 mg/mL) was mixed with an equal
volume of aqueous albumin solution (2 mg/mL) and incubated at room temperature for
1.5 hours on a rotating rocker. The albumin-coated PNC (PNC-alb) was collected by
centrifugation at 4 ℃ at 104 rpm (9300 rcf) for 15 min and washed with deionized (DI)
water twice.
Figure 22. Diagram of preparation of Cim-alb.
PTX and F127 in
chloroform
Evaporate at 40⁰C
for 2 h
A dry film of PTX and
F127 forms on the flask
wall
Hydrate the film at R.T.
for 1 h
Ice-water bath
Probe sonication
for size reduction
-F127
-PTX particles
-PTX nanocrystals
Mixing PNC-
F127 with HSA
solution
Centrifugation
to collect
particles
-Albumin
Cim-alb

112
4.3.3 Characterization of Nanocrystals
The particle sizes of iCim, Cim-alb, PNC, and PNC-alb were measured in DI
water with a Zetasizer Nano-ZS90 (Malvern instruments, Westborough, MA, USA). The
morphology of lyophilized Cim-alb and PNC-alb was visualized with a FEI Nova
nanoSEM field emission scanning electron microscopy (Hillsboro, OR, USA). Cim-alb
and PNC-alb were sputter-coated with platinum for 1 min and observed with a high
resolution through-the-lens detector under 5kV accelerating voltage and spot size 3. The
protein content of Cim-alb or PNC-alb was determined with Micro BCA protein assay or
gel electrophoresis.
4.4 Results and Discussions
4.4.1 Nanocrystal Morphology
PNC-alb showed a rod-shape with a length of ~400 nm and a width of ~150 nm
under SEM (Fig. 23A). Cim-alb showed a similar shape as PNC with a length of ~500
nm and the width of ~80 nm under SEM (Fig. 23B).

113
Figure 23. Scanning electron micrograph of lyophilized (A) PNC-alb and (B) Cim-alb.
Scale bar: 500 nm.
4.4.2 Nanocrystal Size
The size difference visualized in SEM was reflected in DLS measurement. The
average diameter of iCim measured by DLS was 157.1 nm. With surface stabilization
with albumin, Cim-alb had an average diameter of 186.4 nm (Table 6). The initial size
increase was induced by centrifugation, but Cim-alb withstood subsequent centrifugation
with no further size increase. The average diameter of PNC was 301.3 nm. With albumin
stabilization the particle size was 335.5 nm.
A B

114
Table 6. Particle size of albumin-stabilized PNC and intermediate products.
Size (nm)* Polydispersity index range
Min. Max.
iCim (n=8) 157.1 ± 14.6 0.079 0.226
Cim-alb after first centrifugation
(n=9)
188.7 ± 24.6 0.052 0.313
Cim-alb after second centrifugation
(n=2)
186.4 ± 18.1 0.126 0.209
PNC (n=5) 301.3 ± 17.9 0.049 0.362
PNC-alb (n=7) 335.5 ± 19.7 0.152 0.372
*: average diameter and standard deviation of n batches prepared independently and
identically.
The protective effect of albumin was evident when PNC and PNC-alb were
subjected to repeated centrifugation at 4 ℃ at 104 rpm (9300 rcf) for 10 min: bare PNC
aggregated to form ~3 µm particles, while PNC-alb was kept at ~330 nm (Fig. 24). This
observation proved the effectiveness of albumin as a surface stabilizer to PNC.
Figure 24. Particle size of PNC and PNC-alb after repeated centrifugations.
pa
rti
cle
siz
e (
d.
nm
)
Or i
gin
al
1st
cen
trif
ug
at i
on
2n
d c
en
trif
ug
at i
on
3rd
cen
trif
ug
at i
on
4th
cen
trif
ug
at i
on
0
1 0 0 0
2 0 0 0
3 0 0 0
4 0 0 0
5 0 0 0P N C -a lb P N C

115
4.4.3 Albumin content
The albumin contents in Cim-alb and PNC-alb were determined using standard
Micro BCA protein assay. Four percent SDS aqueous solution was used to strip off the
albumin from the nanocrystal surface. The weight ratio between PTX and albumin was
8.21/1 (PTX loading % = 89.1%). However, PTX can interfere with BCA assay because
of the secondary amino group in the structure. SDS-PAGE gel electrophoresis was
recommended to determine the albumin content though the influence of PTX could be
deducted in the BCA assay. The albumin content in iCim-alb was measured based on the
correlation between albumin concentration and band intensity in SDS-PAGE gel
electrophoresis. The weight ratio between PTX and albumin in iCim-alb was 6.3/1 (PTX
loading % =85%) (With courtesy of Joonyoung Park).
4.4.4 Proposed Role of Albumin in Cim-alb
Cim-alb maintained original particle size after repeated centrifugation, whereas
iCim (before albumin protection) could not be resuspended. This indicates albumin was
present on the nanocrystal surface and prevented hydrophobic aggregation of the
particles. The quantity of remaining Pluronic F127 after albumin adsorption has not been
directly measured. Given that albumin and PTX account for 13.5% and 85%,
respectively, the remaining Pluronic F127 is likely to be <2% (With courtesy of
Joonyoung Park). There are two possible scenarios in albumin coverage of the
nanocrystal surface: first, albumin added onto Pluronic F127 surface; second, albumin
replacing Pluronic F127 on nanocrystal surface. Considering the low Pluronic F127

116
content in Cim-alb and improved size stability, second scenario should be the case though
direct evident was still needed.
4.5 Conclusion
Albumin-stabilized nanocrystals with a sub-200 nm particle size were prepared
using a method involving incipient crystallization in polymer matrix and subsequent
surface stabilization with albumin. The presence of polymeric matrix limited the growth
of crystals; thus, PTX could form nanocrystals at ~160 nm. The surface stabilized
nanocrystals (Cim-alb) maintained particle size below 200 nm. Due to the reduced
particle size and the lack of a surfactant, Cim-alb is expected to provide faster PTX
dissolution and greater safety than conventional nanocrystal formulations. The function
and quantitation of surface stabilizers need further investigation. In-vitro dissolution test
and bioactivity evaluation of Cim-alb remains to be performed to test the contribution of
small size to enhancing local availability of PTX.

117
4.6 References
1. Merisko-Liversidge, E.M. et al., Drug Nanoparticles: Formulating Poorly Water-
Soluble Compounds. Toxicol Pathol, 2008. 36(1): p. 43-48.
2. Rabinow, B.E., Nanosuspensions in drug delivery. Nat Rev Drug Discov, 2004.
3(9): p. 785-796.
3. Squillante, I., Emilio et al., Solid Dispersions: Revival with Greater Possibilities
and Applications in Oral Drug Delivery. Crit Rev Ther Drug Carrier Syst, 2003.
20(2&3): p. 34.
4. He, S., et al., A Cremophor-Free Self-Microemulsified Delivery System for
Intravenous Injection of Teniposide: Evaluation In Vitro and In Vivo. AAPS
PharmSciTech, 2012. 13(3): p. 846-852.
5. Bouquet, W., et al., Paclitaxel/β-cyclodextrin complexes for hyperthermic
peritoneal perfusion – Formulation and stability. Eur J Pharm Biopharm, 2007.
66(3): p. 391-397.
6. Chen, H., et al., Lactoferrin-modified procationic liposomes as a novel drug
carrier for brain delivery. Eur J Pharm Sci, 2010. 40(2): p. 94-102.
7. Shao, K., et al., Angiopep-2 modified PE-PEG based polymeric micelles for
amphotericin B delivery targeted to the brain. J Control Release, 2010. 147(1): p.
118-126.
8. Gong, Y., et al., An Excellent Delivery System for Improving the Oral
Bioavailability of Natural Vitamin E in Rats. AAPS PharmSciTech, 2012. 13(3):
p. 961-966.

118
9. Dodiya, S.S., et al., Solid lipid nanoparticles and nanosuspension formulation of
Saquinavir: preparation, characterization, pharmacokinetics and biodistribution
studies. J Microencapsul, 2011. 28(6): p. 515-527.
10. Gullotti, E. et al., Beyond the imaging: Limitations of cellular uptake study in the
evaluation of nanoparticles. J Control Release, 2012 164(2): p 170-176.
11. Patravale, V.B., et al., Nanosuspensions: a promising drug delivery strategy. J
Pharm Pharmacol, 2004. 56(7): p. 827-840.
12. Sun, W., et al., Nanonization of itraconazole by high pressure homogenization:
Stabilizer optimization and effect of particle size on oral absorption. J Pharm Sci,
2011. 100(8): p. 3365-3373.
13. Mitri, K., et al., Lutein nanocrystals as antioxidant formulation for oral and
dermal delivery. Int J Pharm, 2011. 420(1): p. 141-146.
14. Zhang, J., et al., Enhanced bioavailability after oral and pulmonary
administration of baicalein nanocrystal. Int J Pharm, 2011. 420(1): p. 180-188.
15. Ganta, S., et al., Formulation and pharmacokinetic evaluation of an asulacrine
nanocrystalline suspension for intravenous delivery. Int J Pharm, 2009. 367(1–2):
p. 179-186.
16. Zhang, H., et al., Preparation and antitumor study of camptothecin nanocrystals.
Int J Pharm, 2011. 415(1–2): p. 293-300.
17. De Smet, L., et al., Development of a Nanocrystalline Paclitaxel Formulation for
Hipec Treatment. Pharm Res, 2012. 29(9): p. 1-9.

119
18. Müller, R.H., et al., State of the art of nanocrystals – Special features, production,
nanotoxicology aspects and intracellular delivery. Eur J Pharm Biopharm, 2011.
78(1): p. 1-9.
19. Barenholz, Y., Doxil® — The first FDA-approved nano-drug: Lessons learned. J
Control Release, 2012. 160(2): p. 117-134.
20. Xiong, S., et al., Preparation, therapeutic efficacy and intratumoral localization
of targeted daunorubicin liposomes conjugating folate-PEG-CHEMS. Biomed
Pharmacother, 2011. 65(1): p. 2-8.
21. Montero, A.J., et al., Nab-paclitaxel in the treatment of metastatic breast cancer:
a comprehensive review. Expert Rev Clin Pharmacol, 2011. 4(3): p. 329-334.
22. Shegokar, R. et al., Nanocrystals: Industrially feasible multifunctional
formulation technology for poorly soluble actives. Int J Pharm, 2010. 399(1–2): p.
129-139.
23. Van Eerdenbrugh, B., et al., Top-down production of drug nanocrystals:
Nanosuspension stabilization, miniaturization and transformation into solid
products. Int J Pharm, 2008. 364(1): p. 64-75.
24. Chan, H.-K. et al., Production methods for nanodrug particles using the bottom-
up approach. Adv Drug Deliv Rev, 2011. 63(6): p. 406-416.
25. Lindfors, L., et al., Amorphous Drug Nanosuspensions. 3. Particle Dissolution
and Crystal Growth. Langmuir, 2007. 23(19): p. 9866-9874.
26. Thombre, A.G., et al., In vitro and in vivo characterization of amorphous,
nanocrystalline, and crystalline ziprasidone formulations. Int J Pharm, 2012.
428(1–2): p. 8-17.

120
27. Hu, J., et al., Continuous and scalable process for water-redispersible
nanoformulation of poorly aqueous soluble APIs by antisolvent precipitation and
spray-drying. Int J Pharm, 2011. 404(1–2): p. 198-204.
28. de Waard, H., et al., Preparation of drug nanocrystals by controlled
crystallization: Application of a 3-way nozzle to prevent premature crystallization
for large scale production. Eur J Pharm Sci, 2009. 38(3): p. 224-229.
29. de Waard, H., et al., A novel bottom–up process to produce drug nanocrystals:
Controlled crystallization during freeze-drying. J Control Release, 2008. 128(2):
p. 179-183.
30. Merisko-Liversidge, et al., Nanosizing for oral and parenteral drug delivery: A
perspective on formulating poorly-water soluble compounds using wet media
milling technology. Adv Drug Deliv Rev, 2011. 63(6): p. 427-440.
31. Keck, C.M. et al., Drug nanocrystals of poorly soluble drugs produced by high
pressure homogenisation. Eur J Pharm Biopharm, 2006. 62(1): p. 3-16.
32. Merisko-Liversidge, et al., Nanosizing: a formulation approach for poorly-water-
soluble compounds. Eur J Pharm Sci, 2003. 18(2): p. 113-120.
33. Juhnke, M., et al., Generation of wear during the production of drug
nanosuspensions by wet media milling. Eur J Pharm Biopharm, 2012. 81(1): p.
214-222.
34. Begat, P., et al., The effect of mechanical processing on surface stability of
pharmaceutical powders: Visualization by atomic force microscopy. J Pharm Sci,
2003. 92(3): p. 611-620.

121
35. Raghavan, S.L., et al., Crystallization of hydrocortisone acetate: influence of
polymers. Int J Pharm, 2001. 212(2): p. 213-221.
36. Raghavan, S.L., et al., Formation and stabilisation of triclosan colloidal
suspensions using supersaturated systems. Int J Pharm, 2003. 261(1–2): p. 153-
158.
37. Dintaman, J.M. et al., Inhibition of P-Glycoprotein by D-α-Tocopheryl
Polyethylene Glycol 1000 Succinate (TPGS). Pharm Res, 1999. 16(10): p. 1550-
1556.
38. Liu, Y., et al., Paclitaxel Nanocrystals for Overcoming Multidrug Resistance in
Cancer. Mol Pharm, 2010. 7(3): p. 863-869.
39. Van Eerdenbrugh, B., et al., A screening study of surface stabilization during the
production of drug nanocrystals. J Pharm Sci, 2009. 98(6): p. 2091-2103.
40. Verma, S., et al., A comparative study of top-down and bottom-up approaches for
the preparation of micro/nanosuspensions. Int J Pharm, 2009. 380(1–2): p. 216-
222.
41. Gao, L., et al., Preparation and Characterization of an Oridonin Nanosuspension
for Solubility and Dissolution Velocity Enhancement. Drug Dev Ind Pharm, 2007.
33(12): p. 1332-1339.
42. Deng, J., et al., Understanding the structure and stability of paclitaxel
nanocrystals. Int J Pharm, 2010. 390(2): p. 242-249.
43. Deng, Z., et al., Understanding a relaxation behavior in a nanoparticle
suspension for drug delivery applications. Int J Pharm, 2008. 351(1–2): p. 236-
243.

122
44. Yao, J.H., et al., Theory and simulation of Ostwald ripening. Physical Review B,
1993. 47(21): p. 14110-14125.
45. Verma, S., et al., Physical stability of nanosuspensions: Investigation of the role
of stabilizers on Ostwald ripening. Int J Pharm, 2011. 406(1–2): p. 145-152.
46. Lindfors, L., et al., Amorphous Drug Nanosuspensions. 1. Inhibition of Ostwald
Ripening. Langmuir, 2005. 22(3): p. 906-910.
47. Meinders, M.B.J. et al., The role of interfacial rheological properties on Ostwald
ripening in emulsions. Adv Colloid Interfa Sci, 2004. 108(1): p. 119-126.
48. Walstra, P., Formation of emulsions, in Encyclopedia of Emulsion Technology:
Volume 1 - Basic Theory, P. Becher, Editor 1983, Marcel Dekker: New York. p.
57-128.
49. Gao, Y., et al., Preparation, characterization, pharmacokinetics, and tissue
distribution of curcumin nanosuspension with TPGS as stabilizer. Drug Dev Ind
Pharm, 2010. 36(10): p. 1225-1234.
50. Ali, H.S.M., et al., Hydrocortisone nanosuspensions for ophthalmic delivery: A
comparative study between microfluidic nanoprecipitation and wet milling. J
Control Release, 2011. 149(2): p. 175-181.
51. Singare, D.S., et al., Optimization of formulation and process variable of
nanosuspension: An industrial perspective. Int J Pharm, 2010. 402(1–2): p. 213-
220.
52. Van Eerdenbrugh, B., et al., Drying of crystalline drug nanosuspensions—The
importance of surface hydrophobicity on dissolution behavior upon redispersion.
Eur J Pharm Sci, 2008. 35(1–2): p. 127-135.

123
53. Bajaj, G., et al., Hyaluronic acid-based hydrogel for regional delivery of
paclitaxel to intraperitoneal tumors. J Control Release, 2012. 158(3): p. 386-392.
54. Dolenc, A., et al., Advantages of celecoxib nanosuspension formulation and
transformation into tablets. Int J Pharm, 2009. 376(1–2): p. 204-212.
55. Ambrus, R., et al., Investigation of preparation parameters to improve the
dissolution of poorly water-soluble meloxicam. Int J Pharm, 2009. 381(2): p. 153-
159.
56. Kakran, M., et al., Fabrication of drug nanoparticles by evaporative precipitation
of nanosuspension. Int J Pharm, 2010. 383(1–2): p. 285-292.
57. Xia, D., et al., Preparation of stable nitrendipine nanosuspensions using the
precipitation–ultrasonication method for enhancement of dissolution and oral
bioavailability. Eur J Pharm Sci, 2010. 40(4): p. 325-334.
58. Gao, L., et al., Preparation of a chemically stable quercetin formulation using
nanosuspension technology. Int J Pharm, 2011. 404(1–2): p. 231-237.
59. Zhao, R., et al., Hybrid Nanocrystals: Achieving Concurrent Therapeutic and
Bioimaging Functionalities toward Solid Tumors. Mol Pharm, 2011. 8(5): p.
1985-1991.
60. Jeyachandran, Y.L., et al., Quantitative and Qualitative Evaluation of
Adsorption/Desorption of Bovine Serum Albumin on Hydrophilic and
Hydrophobic Surfaces. Langmuir, 2009. 25(19): p. 11614-11620.
61. Seo, J., et al., Facile internalization of paclitaxel on titania nanoparticles in
human lung carcinoma cells after adsorption of serum proteins. J Nanopart Res,
2012. 14(10): p. 1-8.

124
62. Kratz, F. et al., Clinical impact of serum proteins on drug delivery. J Control
Release, 2012. 161(2): p. 429-45.
63. Zhang, J.-Y., et al., Preparation of the albumin nanoparticle system loaded with
both paclitaxel and sorafenib and its evaluation in vitro and in vivo. J
Microencapsul, 2011. 28(6): p. 528-536.
64. Liu, F., et al., Targeted cancer therapy with novel high drug-loading
nanocrystals. J Pharm Sci, 2010. 99(8): p. 3542-3551.

125
CHAPTER 5. CONCLUSION
PTX was delivered as PNC via an in-situ crosslinkable HA gel, aiming to increase
the retention and chemotherapeutic effect in the peritoneal cavity. Several attempts were
made to evaluate the difference in dissolution rate between PNCs and uncontrolled
precipitates (PPTs) using in-vitro conditions reflecting the peritoneal cavity, such as non-
sink condition and amphiphilic components. The expected difference between PNC and
PPT or gels containing the two was not clearly seen in the in-vitro release studies
(Chapter 2). Nevertheless, PNC was more efficient in killing SKOV3 cells than PPT, and
PNC-gel showed some advantage in anti-tumor efficacy on tumor bearing mice over
PPT-gel. However, the heterogeneity of tumor growth and decreasing number of animals
following the tumor relapse made it difficult to statistically differentiate PNC-gel from
PPT-gel based on bioluminiescence imaging (Chapter 3).
Although the cytotoxicity result and in-vivo anti-tumor effects demonstrate the
PNC-gel demonstrated its effectiveness as an IP chemotherapy delivery system, the small
extent of improvement over PPT-gel suggest that PTX dissolution was still limited in the
non-sink condition of the peritoneal cavity. PNC-gel can be further improved at least in
two aspects. First, it is possible that PNC may have segregated from the hydrogel matrix
and formed aggregates during the prolonged incubation in hydrogel, further delaying the
PTX release. To prevent PNC segregation, HA gel may be modified to improve the

126
compatibility with PNC. To this end, HA gel conjugated with a derivative of 5β-cholanic
acid was synthesized, which would form hydrophobic domains to accommodate
hydrophobic PNC (Chapter 2). The hydrophobically modified hydrogel improved PTX
release, confirming that compatibility between PNC and hydrogel is an important factor
in local delivery of PTX. The current form of the hydrophobically modified hydrogel
will not be pursued due to the unexpected toxicity of the gel precursor (EtCA-CMC-
CHO), but it will be worthwhile to synthesize an alternative hydrophobitized gel
precursor with low toxicity. Second, PNC size may be further reduced to improve
dissolution of PTX. PNC produced with nonsolvent and temperature induced
crystallization method, used for the in-vivo study, and had an average diameter of ~ 260
nm. Albumin-stabilization helped prevent aggregation of PNC but did not produce
smaller PNC. A new method of producing smaller PNC has been developed, based on
crystallization in surfactant matrix and surface stabilization with albumin (Chapter 4).
This method involves forming sub-200 nm PNC with Pluronic F127 and replacing it with
albumin for further stabilization. The function and quantitation of surface stabilizers on
Cim-alb remain to be investigated.
The future delivery systems may be built with hydrophobically modified
hydrogels and/or Cim-alb NCs. Ultimately, their effectiveness in reducing IP tumor
burdens needs to be tested in-vivo, but mechanistic understanding of the contributions of
each component will depend on well-controlled in-vitro studies. This study identified the
limitations of current in-vitro drug release kinetics in predicting the in-vivo outcome of
different formulations. Ideally in-vitro release kinetics should not involve artifacts due to
sampling procedures, such as centrifugation and dialysis, but should be easy to accurately

127
analyze. In this regard, it is worthwhile to note a promising method based on the linearity
between light scattering intensity and the concentration of partially dissolved particles. A
new release/dissolution kinetics test method will be highly beneficial with the advent of
new drug delivery systems, in particular those including poorly water-soluble drugs for
local application to sites with limited body fluid, such as the lungs and the peritoneal
cavity.

127
VITA

128
VITA
Bo Sun, Ph.D.
Education
2011-2016 Ph.D. in Pharmaceutics (Advisor: Dr. Yoon Yeo)
Dept. of Industrial and Physical Pharmacy, College of Pharmacy, Purdue
University, West Lafayette, IN, USA
2008-2011 M.S. in Pharmaceutics (Advisor: Dr. Jianping Zhou)
China Pharmaceutical University, Nanjing, China
2002-2006 B.S. in Pharmaceutics
China Pharmaceutical University, Nanjing, China
Research Experience
8/2011-5/2016 Graduate Research Assistant
Dept. of Industrial and Physical Pharmacy, College of Pharmacy,
Purdue University, West Lafayette, IN, USA
Ph.D. Dissertation: Nanoparticle Depot for Intraperitoneal
Chemotherapy of Ovarian Cancer
9/2008-5/2011 Graduate Student
Dept. of Pharmaceutics, College of Pharmacy, China Pharmaceutical

129
University, Nanjing, China
Master’s Theses: The Study of Selective Cell Penetrating Peptide on
Tumor Cells and Its Combined Nanocarriers
Professional Experience
6/2015-8/2015 Internship at Genentech
Manager: Jane Li, Senior Scientist
Dept. of Small Molecule Analytical Chemistry and Quality Control,
Genentech, South San Francisco, USA.
6/2006-2/2007 Production Management Engineer
Sino-American Tianjin Smith Kline & French Laboratories, Ltd.,
Tianjin, China.
Publications
1. Sun, B., Taha, M.S., Ramsey, B., Torregrosa-Allen, S., Elzey, B.D., Yeo, Y.
Intraperitoneal Chemotherapy of Ovarian Cancer by Hydrogel Depot of Paclitaxel
Nanocrystals. (Manuscript under 2nd review)
2. Sun, B. and Yeo, Y. Biomaterials and Drug Delivery Systems for Intraperitoneal
Chemotherapy. In: Wim Ceelen and Edward Levine (Eds.) Intraperitoneal Cancer
Therapy: Principles and Practice. CRC press (Taylor and Francis). 2015.
3. Abouelmagd, S.A.*, Sun, B.*, Chang, A.C., Ku, Y.J., Yeo, Y. Release Kinetics Study
of Poorly Water-Soluble Drugs from Nanoparticles: Are We Doing It Right? Molecular
Pharmaceutics. (2015) 12(3): 997-1003. (*: co-first authors)

130
4. Cho, E.J.*, Sun, B.*, Doh, K.O., Wilson, E.M., Torregrosa-Allen, S., Elzey, B.D.,
Yeo, Y. Intraperitoneal Delivery of Platinum with in-Situ Crosslinkable Hyaluronic
Acid Gel for Local Therapy of Ovarian Cancer. Biomaterials. (2015) 37: 312-319. (*:
co-first authors)
5. Sun, B., Yeo, Y. Nanocrystals for the parenteral delivery of poorly water-soluble
drugs. Current Opinion in Solid State and Materials Science. (2012) 16(6): 295-301.
Awards and Honors
2015 O'Malley Scholarship, College of Pharmacy, Purdue University
2015 Travel award from the Purdue University Center for Cancer Research Hasson
Graduate Travel Fund
2014 Ronald W. Dollens Graduate Scholarship in the Life Sciences, College of
Pharmacy, Purdue University.
2013 Purdue Research Foundation grant for proposal entitled “Nanoparticle depot for
intraperitoneal chemotherapy of ovarian cancer”.
2005 Scholarship from Suzhou Capsugel Ltd., China
2004 Third-class scholarship at China Pharmaceutical University, Nanjing, China