insulin sensitivity is enhanced by cgmp- mediated mapk … · 2014-01-23 · insulin signalling....
TRANSCRIPT

INSULIN SENSITIVITY IS ENHANCED BY CGMP-MEDIATED MAPK INHIBITION IN RAT ADIPOCYTES
by
Garry Robert Thomas
A thesis submitted in conformity with the requirementsfor the degree of Master of ScienceGraduate Department of Physiology
University of Toronto
© Copyright by Garry Robert Thomas (2009)

ii
ABSTRACT
Garry Robert Thomas
Master of Science (2009)
Graduate Department of Physiology
University of Toronto
INSULIN SENSITIVITY IS ENHANCED BY cGMP-MEDIATED MAPK INHIBITIONIN RAT ADIPOCYTES
Bradykinin (BK) acts through eNOS to reduce MAPK-mediated feedback inhibition of
insulin signalling. Preliminary data suggests that the sGC-cGMP-PKG pathway, a prominent NO
target, is involved. Our present study aimed to support the role of this pathway with atrial natriuretic
peptide (ANP), which uses a receptor associated GC (NPR-A) to generate cGMP.
We found that treating adipocytes with ANP mimicked BK effects on insulin-stimulated
glucose uptake, Tyr-IRS-1 and Akt/PKB phosphorylation, as well as JNK and ERK1/2 inhibition.
These outcomes depended on GC-cGMP-PKG signalling since A71915 (NPR-A antagonist), and
KT-5823 (PKG inhibitor), completely abrogated them, while zaprinast (phosphodiesterase inhibitor),
prolonged ANP actions. Furthermore, decreased MAPK phosphorylation was independent of
upstream kinase activity, suggesting that MAPK phosphatases may be involved.
These data indicate that BK and ANP act through the GC-cGMP-PKG pathway to potentiate
insulin signalling via attenuated feedback inhibition. Stimulating the GC-cGMP-PKG pathway may,
therefore, be a promising therapy for T2DM.

iii
ACKNOWLEDGMENTS
First and foremost, I would like to thank my supervisor, Dr. I. George Fantus, for his
guidance, support, and confidence during my two years as a Master of Science student. His
constant motivation has allowed me to reach new heights of achievement and his efforts have
strengthened my knowledge and interest in the field of diabetes. Furthermore, as a future
physician, Dr. Fantus has served as a role model. His professionalism, knowledge, and balance
of both research and clinical responsibilities are attributes I hope to also achieve in my
professional career.
I would like to thank my supervisory committee members, Dr. John Floras and Dr. Adria
Giacca, for their valuable insight and direction. The success of my project could not have been
possible without their critical evaluations and expertise.
I would like to thank past and present members of the Fantus lab for their support and
assistance: Svetlana Altamentova, Yael Babichev, Howard Goldberg, Huogen Lu, Elodie
Masson, Zhiwen Yu, and Ling Xia. I would also like to offer a very special thanks to Kristin M.
Beard for setting the foundation for this project and demonstrating many of the techniques to me.
My work would likely not be at this stage without her earlier efforts to perfect the protocols.
Lastly, I am thankful to my family and girlfriend, Laura Voicu, for their patience and
tireless efforts to provide moral support.

iv
TABLE OF CONTENTS
ABSTRACT…………………………………………………………………………………ACKNOWLEDGMENTS…………………………………………………………………...TABLE OF CONTENTS……………………………………………………………………LIST OF TABLES………………………………………………….......................................LIST OF FIGURES………………………………………………………………………….ABBREVIATIONS………………………………………………………………………….
iiiiiivviiviiix
CHAPTER 1 BACKGROUND, RATIONALE AND HYPOTHESIS
1.1 Diabetes Mellitus...........................................................................................................1.2 Insulin…………………………………………………………………………….........
1.2.1 Properties of Insulin…………………………………………………………...1.2.2 Physiological Action of Insulin………………………………………………..1.2.3 The Insulin Signalling Pathway…………………………………………….....
1.2.3.1 The Insulin Receptor………………………………………………..1.2.3.2 Insulin Receptor Substrates…………………………………………1.2.3.3 PI3K Signalling Pathway…………………………………………...
1.2.3.3.1 PI3K…………………………………………………….1.2.3.3.2 Akt/PKB………………………………………………...1.2.3.3.3 GLUT4………………………………………………….
1.2.3.4 MAPK Signalling Pathway................................................................1.2.3.4.1 ERK..................................................................................1.2.3.4.2 JNK..................................................................................1.2.3.4.3 p38 Kinases......................................................................
1.2.4 Signalling Defects and Insulin Resistance........................................................1.2.5 Hypertension and Insulin Resistance................................................................
1.3 Blood Pressure Regulation............................................................................................1.3.1 Renin-Angiotensin System (RAS)....................................................................
1.3.1.1 Angotensin II (AngII)........................................................................1.3.1.2 AngII Receptors & Signalling............................................................
1.3.2 Kallikrein-Kinin System (KKS)........................................................................1.3.2.1 Bradykinin (BK).................................................................................1.3.2.2 BK Receptors & Signalling................................................................
1.4 Antihypertensive Therapy and Insulin Sensitivity.......................................................1.5 Nitric Oxide (NO)........................................................................................................
1.5.1 Nitric Oxide Synthase (NOS)...........................................................................1.5.2 NO and Insulin Sensitivity...............................................................................
1.6 Guanylate Cyclase-cGMP-Protein Kinase G Signalling Pathway...............................1.6.1 Guanylate Cyclase (GC)..................................................................................
1.6.1.1 Soluble Guanylate Cyclase (sGC).....................................................1.6.1.2 Particulate Guanylate Cyclase (GC).................................................
1.6.2 cGMP...............................................................................................................1.6.3 Protein Kinase G (PKG)...................................................................................
1
2223456889101112131414151616171718191920212222232323242526

v
1.7 Bradykinin Acts Through sGC-cGMP-PKG to Potentiate Insulin Signalling.............1.8 Natriuretic Peptides (NP).............................................................................................
1.8.1 Atrial Natriuretic Peptide (ANP)....................................................................1.8.2 B-type Natriuretic Peptide (BNP)...................................................................1.8.3 C-type Natriuretic Peptide (CNP)...................................................................
1.9 RATIONALE.............................................................................................................1.10 HYPOTHESES...........................................................................................................
CHAPTER 2 MATERIALS & METHODS
2.1 Materials……………………………………………………………………………....2.2 Animals……………………………………………………………………………….2.3 Isolation of Rat Adipocytes…………………………………………………...............2.4 Immunoblotting.............................................................................................................
2.4.1 Whole Cell Lysate Preparation............................................................................2.4.2 Cell Transfection and Immunoprecipitation........................................................2.4.3 Immunoblotting Procedure...................................................................................
2.5 Assay Kits.....................................................................................................................2.5.1 Quantification of cGMP Concentration (cGMP ELISA).....................................2.5.2 Quantification of Glycerol Production (Glycerol Assay)....................................
2.6 Glucose Uptake..............................................................................................................2.6.1 [3H]-2-Deoxy-D-Glucose Uptake Assay .............................................................2.6.2 Zaprinast Experiment...........................................................................................
2.7 Statistical Analysis.........................................................................................................
CHAPTER 3 RESULTS
3.1 Summary......................................................................................................................3.2 Results..........................................................................................................................
3.2.1 Natriuretic Peptides Enhance Insulin-Stimulated Glucose Uptake in RatAdipocytes..........................................................................................................
3.2.2 ANP Uses GC-cGMP-PKG Signalling to Enhance Insulin-StimulatedGlucose Uptake...................................................................................................3.2.2.1 ANP Requires the NPR-A to Enhance Insulin-Stimulated
Glucose Uptake......................................................................................3.2.2.2 ANP Requires cGMP to Enhance Insulin-Stimulated
Glucose Uptake......................................................................................3.2.2.3 ANP Requires PKG to Enhance Insulin-Stimulated
Glucose Uptake......................................................................................3.3 ANP Potentiates the Insulin Signalling Pathway........................................................3.4 ANP Decreases Insulin-Stimulated MAPK Phosphorylation.....................................3.5 ANP Reduces MAPK Phosphorylation Independent of Upstream
MAPK Kinases............................................................................................................
27293030313233
34
3535363737383939394040404141
42
4344
44
46
46
48
505154
57

vi
CHAPTER 4 DISCUSSION
4.1 INTRODUCTION......................................................................................................4.2 Natriuretic Peptides Enhance Insulin-Stimulated Glucose Uptake in
Rat Adipocytes...........................................................................................................4.3 ANP Acts Through the NPR-A(GC)-cGMP-PKG Signalling Pathway to
Enhance Insulin-Stimulated 2-DG Uptake in Rat Adipocytes...................................4.3.1 ANP Requires the NPR-A to Enhance Insulin-Stimulated Glucose Uptake....4.3.2 ANP Requires cGMP to Enhance Insulin-Stimulated Glucose Uptake............4.3.3 ANP Requires PKG to Enhance Insulin-Stimulated Glucose Uptake..............
4.4 ANP Potentiates the Insulin Signalling Pathway in Rat Adipocytes.........................4.5 ANP Reduces MAPK Phosphorylation in Insulin-Stimulated Rat Adipocytes.........4.6 ANP and the Regulators of MAPK Activity.............................................................
4.6.1 ANP Reduces MAPK Phosphorylation Independent of UpstreamMAPK Kinases..................................................................................................
4.6.2 ANP May Reduce MAPK Phosphorylation via MAPK PhosphataseUpregulation......................................................................................................
4.7 Caveats and Limitations of the Study........................................................................4.8 ANP-mediated Lipolysis – An Insulin Antagonizing Function?...............................
4.8.1 ANP and Insulin Resistance............................................................................4.8.1.1 ANP Signalling Enhances Mitochondrial Function.............................4.8.1.2 ANP Signalling Decreases Inflammation............................................
4.10 CONCLUSION......................................................................................................
CHAPTER 5 FUTURE DIRECTIONS
5.1 The Role of PKG1/cGKI in BK- and ANP-Mediated Enhanced Insulin Signalling5.2 The Role of MKPs in BK- and ANP-Mediated Enhanced Insulin Sensitivity.........5.3 The in vitro Effect of BK and ANP on Insulin Sensitivity in Skeletal Muscle........5.4 The in vivo Effect of ANP on Insulin Sensitivity in “Normal” Rodents..................5.5 The Effect of ANP on Insulin Resistance in Rodents...............................................
CHAPTER 6 REFERENCES
CHAPTER 7 APPENDIX
7.1 BK Acts through PKG1 to Decrease Insulin-stimulated JNK1 phosphorylation....7.2 ANP Binding to the NPR-A Stimulates Lipolysis in Rat Adipocytes......................
59
60
61
64646566676870
71
72737477787980
81
8283838484
86
118
119120

vii
LIST OF TABLES
Table 4.1 MKP Characteristics......................................................................................
LIST OF FIGURES
CHAPTER 1
Figure 1.1 Structure of Proinsulin.................................................................................Figure 1.2 The Insulin Signalling Pathway in Adipocytes...........................................Figure 1.3 Tyrosine Phosphorylation of the Insulin Receptor............................................Figure 1.4 IRS-1 Activity Regulation and Downstream Targets..................................Figure 1.5 MAPK Signalling Cascades........................................................................Figure 1.6 Major Mechanisms of Systemic Blood Pressure Regulation......................Figure 1.7 AngII Amino Acid Sequence.......................................................................Figure 1.8 BK Amino Acid Sequence...........................................................................Figure 1.9 Structure of soluble Guanylate Cyclase.......................................................Figure 1.10 Structure of Particulate Guanylate Cyclase.................................................Figure 1.11 cGMP Structure...........................................................................................Figure 1.12 cGMP Inducers and Targets........................................................................Figure 1.13 Mechanism of BK-mediated Enhanced Insulin Sensitivity.........................Figure 1.14 Natriuretic Peptide Structures......................................................................Figure 1.15 Characteristics of Natriuretic Peptide Receptors.........................................Figure 1.16 Proposed Mechanism of ANP-mediated Enhanced Insulin Sensitivity......
CHAPTER 2
Figure 2.1 Procedure for Isolating Rat Adipocytes (Epididymal)..........................................Figure 2.2 Complete Experiment Outline....................................................................Figure 2.3 Procedure for Transfecting and Immunoprecipitating Rat Adipocytes...............Figure 2.4 Procedure for [3H]-2-Deoxy-D-Glucose Uptake Assay........................................
CHAPTER 3
Figure 3.1 Natriuretic Peptides Mimic the Bradykinin Effect of EnhancedInsulin-Stimulated 2-DG Uptake in Rat Adipocytes...........................................
Figure 3.2 ANP Requires the NPR-A to Enhance Insulin-Stimulated2-DG Uptake in Rat Adipocytes.......................................................................
Figure 3.3 ANP Requires cGMP to Enhance Insulin-Stimulated 2-DGUptake in Rat Adipocytes...................................................................................
Figure 3.4 ANP Requires PKG to Enhance Insulin-Stimulated 2-DG Uptakein Rat Adipocytes................................................................................................
Figure 3.5 ANP Potentiates the Insulin Signalling Pathway via PKG in RatAdipocytes...........................................................................................................
73
3457121617192324252528292933
36373840
45
47
50
51
54

viii
Figure 3.6 ANP Reduces MAPK Phosphorylation via PKG in Insulin-Stimulated Rat Adipocytes...........................................................................
Figure 3.7 ANP Reduces MAPK Phosphorylation Indpendent of UpstreamMAPK Kinases....................................................................................................
CHAPTER 4
Figure 4.1 Regulation of Lipolysis in Adipocytes......................................................Figure 4.2 ANP-mediated Enhanced Insulin Sensitivity............................................
CHAPTER 7
Figure 7.1 BK Acts through PKG1 to Decrease Insulin-stimulated JNK1Phosphorylation.........................................................................................
Figure 7.2 ANP Promotes Lipolysis in Rat Adipocytes…………………………….
57
58
7680
119120

ix
ABBREVIATIONS
aa:ACE:
AC:ADH:AdV:AGT:
Ala:AngI:
AngII:APS:ANP:
aPKC:ARB:
ASK1:AT1 receptor:AT2 receptor:
ATF2:ATP:AU:
B1 receptor:B2 receptor:
BAD:BCL-2:
BNP:BH4:BK:
BSA:Ca2+:
cGMP:CHF:
CO:CPT-cGMP:
cNOS:CNP:cGK:
cGMP:CO:
COX IV:CVD:
Cyt C:DAG:
DM:DMEM:
DN:
Amino AcidAngiotensin Converting EnzymeAdenylate CyclaseAnti-diuretic HormoneAdenovirusAngiotensinogenAlanineAngiotensin IAngiotensin IIAdapter Protein with a PH and SH2 DomainAtrial Natriuretic PeptideAtypical Protein Kinase CAngiotensin Receptor BlockerApoptosis Signal-Regulating Kinase 1Angiotensin II Type 1 receptorAngiotensin II Type 2 receptorActivating Transcription Factor 2Adenosine TriphosphateArbitrary UnitsType 1 receptorBradykinin Type 2 receptorBcl2-Associated Death PromoterB-cell CLL/lymphoma 2B-type Natriuretic Peptide5,6,7,8-tetrahydrobiopterinBradykininBovine Serum AlbuminCalciumcyclic Guanosine MonophosphateCongestive Heart FailureCarbon Monoxide8-(4-Chlorophenylthio)-guanosine 3′,5′-cyclic monophosphateconstitutive NOSC-type Natriuretic PeptidecGMP-dependent Protein Kinase OR Protein Kinase G(PKG)cyclic Guanosine MonophosphateCarbon MonoxideCytochrome Oxidase Complex IVCardiovascular DiseaseCytochrome CDiacylglycerolDiabetes MellitusDulbecco’s Modified Eagle MediumDominant Negative

x
DNA:DOCK1/2:
EDTA:EGF:
EGTA:eNOS:ERK:ET-I:FAD:FFA:
FMN:Gab1/2:
GC:GDP:GEF:
GLUT:GTP:
GLP-1:GLUT:
Gly:Grb2:GTP:HA:
HEK:HFD:HGO:
HMW:HSL:
HT:IGF-1:IGF-2:
IL-1:IL-6:
iNOS:IP3:IR:
IRS:JNK:
K+
K+ATP
KKP:KO:
KRBH:Leu:
LMW:L-NAME:
Deoxyribonucleic AcidDedicator of cytokinesis 1/2Ethylenediamine Tetraacetic AcidEpidermal Growth FactorEthylene Glycol Tetraacetic AcidEndothelial Nitric Oxide SynthaseExtracellular Signal-Regulated KinaseEndothelin-IFlavin Adenine DinucleotideFree Fatty AcidFlavin MononucleotideGrb2-associated binder 1Guanylate CyclaseGuanosine DiphosphateGuanine Nucleotide Exchange FactorGlucose TransporterGuanosine TriphosphateGlucagon-Like Peptide-1Glucose TransporterGlycineGrowth Factor Receptor Bound Protein-2Guanosine TriphosphateHydroxylamineHuman Embryonic KidneyHigh Fat DietHepatic Glucose OutputHigh Molecular WeightHormone Sensitive LipaseHypertensionInsulin-like Growth Factor-1Insulin-like Growth Factor-2Interleukin-1Interleukin-6Inducible Nitric Oxide SynthaseInositol TriphosphateInsulin ReceptorInsulin Receptor Substratec-Jun-N-terminal KinasePotassiumATP-dependent K+ channelsKallikrein Kinin PathwayKnock outKrebs Ringer BufferLeucineLow Molecular WeightN (G)-nitro-L-arginine methyl ester

xi
LY2934002:Lys:
MAPK:MAPKK:
MAPKKK:MCP-1:
Mg2+
MI:MKP:
MLK3:Mn2+
mRNA:mTOR:
mw:NADPH:
Na+
NaCl:NEFA:
NEP:NF-κB:
nM:nNOS:
NO:NOS:NPR:ODQ:
PAGE:PCR:PDE:
PDGF:PDK:
PGC1-:PH:PI:
PI3K:PLC:PKB:PKC:
PM:PMSF:PP2A:PP2C:PPAR:
Pro:PTEN:PTIO:
2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-oneLysineMitogen Activated Protein KinaseMAPK KinaseMAPK Kinase KinaseMonocyte Chemoattractant Protein-1MagnesiumMyocardial InfarctionMAPK PhosphataseMixed Lineage Kinase 3ManganeseMessenger RNAMammalian Target of RapamycinMolecular WeightNicotinaminde Adenine Dinucleotide PhosphateSodiumSodium ChlorideNon-Esterified (free) Fatty AcidsNeutral EndopeptidaseNuclear Factor-κBNanomolarneuronal Nitric Oxide SynthaseNitric OxideNitric Oxide SynthaseNatriuretic Peptide Receptor1H-[1,2,4] Oxadiazolo-[4,3-a] quinoxalin-1-onePolyacrylamide Gel ElectrophoresisPolymerase Chain ReactionPhosphodiesterasePlatelet-Derived Growth FactorProtein Dependent KinasePeroxisome-Proliferator-Activated Receptor Co-Activator 1Pleckstrin HomologyPhosphatidylinositolPhosphatidylinositol-3 KinasePhospholipase CProtein Kinase B or AktProtein Kinase CPlasma MembranePhenylmethanesulphonylfluorideProtein Phosphatase 2AProtein Phosphatase 2CPeroxisome-Proliferator Activated ReceptorProlinePhosphatase and Tensin Homologue2-[4-carboxyphenyl]-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide

xii
PTP:RAS:
Rictor:RNA:ROS:RTK:
Ser:SDS-PAGE:
sGC:SH3:Shc:
SHIP2:SHP2:
siRNA:SKM:SNP:SOS:STZ:
t1/2:T1DM:T2DM:
Tak1:TG:
TGF-β:Thr:
TNFα:Tris-HCl:
TTBS:Tyr:
μl:μm:μM:UV:
VSMC:WAT:YC-1:ZDF:
Protein Tyrosine PhosphataseRenin Angiotensin SystemRapamycin Insensitive Companion of mTORRibonucleic AcidReactive Oxygen SpeciesReceptor Tyrosine KinaseSerineSodium Dodecyl Sulphate-Polyacrilamide Gel ElectrophoresisSoluble Guanylate CyclaseSrc Homology 3Src-Homology 2-Containing ProteinType II SH2-domain-containing Inositol-5-PhosphataseSrc Homology 2-Containing Tyrosine Phosphatasesmall-interfering RNASkeletal MuscleSodium NitroprussideSon of SevenlessStreptozotocinhalf-lifeType 1 Diabetes MellitusType 2 Diabetes MellitusTGF-beta activated kinase 1TriglycerideTransforming Growth Factor-βThreonineTumour Necrosis Factor-α2-Amino-2-(hydroxymethyl)-1,3-propanediol, hydrochlorideTris buffered saline with Tween-20TyrosineMicrolitreMicrometreMicromolarUltravioletVascular smooth muscle cellsWhite Adipose Tissue5-[1-(phenylmethyl)-1H-indazol-3-yl]-2-furanmethanolZucker Diabetic Fatty

1
CHAPTER 1
BACKGROUND, RATIONALE, HYPOTHESIS

2
1.1 Diabetes Mellitus
Diabetes Mellitus (DM) is a metabolic disorder characterized by hyperglycemia
resulting from the absence of insulin or a disruption of its functions[1]. Both genetic and
environmental factors can predispose individuals to DM and according to the International
Diabetes Federation, the number of worldwide cases will double to 500 million in the next 30
years[2]. If not treated, DM can increase the risk of macrovascular disorders (e.g:
cardiovascular disease (CVD)), as well microvascular disease (e.g: chronic renal failure, nerve
and retinal damage) [3]. DM has been subdivided into two general types (T1DM, T2DM) based
on the pathogenesis and current treatment of the disorder[1]. T1DM accounts for 10% of DM
and usually results from the autoimmune destruction of insulin producing pancreatic β-cells[4].
T2DM, in the early stages, however, is characterized by insulin resistance and compensatory
hyperinsulinemia[5]. As the disease progresses, however, β-cell dysfunction occurs, resulting in
a relatively insulin deficient phenotype[6]. In T1DM, since the body remains sensitive to
insulin, the most common treatment is lifelong insulin replacement by injection or pump[7].
Treatment for early T2DM often involves diet adjustments along with increased exercise and
weight loss to enhance insulin sensitivity in early stages of the disease[5]. This is followed by
anti-diabetic drugs including secretagogues (sulfonylureas), sensitizers (thiazolidinediones), or
incretin mimetics (GLP-1 analogues) for more advanced T2DM[8,9,10]. Finally, if these
medications fail due to further β-cell impairment, insulin therapy is required[5].
1.2 INSULIN
1.2.1 Properties of Insulin
Insulin is a 51-amino acid (aa) anabolic hormone, secreted by pancreatic β-cells of
mammals to promote a shift from the fed to fasted metabolic state[11]. Preproinsulin, the
precursor of insulin, is coded by the INS gene of chromosome 11 in humans, found exclusively

3
in β-cells and possibly the brain[12]. Pre-proinsulin is converted to proinsulin through the
removal of the N-terminal signal sequence[13]. Insulin is then generated by the action of
proteolytic prohormone convertases (PC) which excise the central c-peptide region,
leaving behind the C- and N-terminal ends joined
by disulphide bonds[13]. β-cells are able to
respond to circulating glucose levels in order to
regulate the release of insulin. Essentially, high
blood glucose results in greater β-cell glucose
uptake and thus, ATP production[14]. ATP can
Figure 1.1 Structure of Proinsulin.Proinsulin is converted to insulin by excisingthe central “C-peptide”, allowing for the A (C-terminal) and B (N-terminal) segments to joinvia disulphide bonds
then inactivate ATP-dependent K+ channels (K+ATP), leading to cellular depolarization, and
insulin secretion[14,15]. Physiologic plasma insulin levels are highest during the postprandial
period, often in the range of 300-600 pmol/L[16]. It has a half-life (t1/2) of less than 1h
following secretion, and is eliminated by hepatocyte insulin degrading enzymes (IDE) or via
insulin-receptor complex endocytosis[17]. Although receptors for insulin are found at many
sites throughout the body, three tissues have been classically identified as the major peripheral
targets for insulin signalling: liver, skeletal muscle (SKM), and adipose tissue[18].
1.2.2 Physiological Action of Insulin
Insulin regulates glucose homeostasis via several mechanisms. In the liver, it reduces
glucose production by inactivating glycogen phosphorylase and glucose-6-phosphatase which
are responsible for glycogenolysis and gluconeogenesis, respectively[19,20]. It also promotes
hepatic glycogen production through the activation of glycogen synthase and glucose
metabolism via phosphofructokinase[21]. In the SKM and adipose tissue, insulin promotes
glucose uptake by increasing the translocation of GLUT4, the predominant insulin-sensitive

4
glucose transporter in these tissues, to the plasma membrane (PM)[22]. Other features of insulin
signalling in these tissues include lipid storage through fatty acid synthase (FAS) activation
while inhibiting cAMP-mediated lipolysis in adipocytes[23,24], and promoting protein
synthesis in SKM by increasing amino acid uptake and ribsome content [25]. Insulin resistance
is characterized by an attenuation of these functions and will be further discussed.
1.2.3 The Insulin Signalling Pathway
Insulin signalling is accomplished through a series of phosphorylation events following
receptor binding, which ultimately results in a plethora of cellular processes[26]. Two major
targets of insulin signalling are i) the phosphatidylinositol 3-kinase (PI3K) pathway, which
accounts for most of insulin’s metabolic effects, and ii) the mitogen activated protein kinase
(MAPK) pathway, which has a fundamental role in cell growth and differentiation[26].
Figure 1.2 The Insulin Signalling Pathway in Adipocytes. Insulinhas two major signalling pathways, the a) PI3K Pathway and b) MAPK PathwayInsulin binds to the IR to activates IRS-1. IRS-1 can, in turn, activate PI3K, leadingto the conversion of PIP2 into PIP3, which can stimulate PDK and then Akt/PKB.Akt/PKB signalling can mediate a variety of effects, including fat, glycogen, andprotein synthesis, and glucose uptake via GLUT4 translocation to the PM. Insulin canalso act through IRS1 or Shc to activate Grb2, which in turn, activates SOS, RAS,then the MAPKKK, Raf. This will then stimulate MEK1/2 which can activate theMAPK, ERK. Insulin can also activate the MAPK, JNK, but the mechanism is notwell established.
Abbreviations
4EBP= 4E Binding ProteinAkt/PKB= Protein Kinase BBad=Bcl2-associated death promoterERK1/2= Extracellular signal-
regulated kinases 1/2GLUT4= Glucose transporter-4Grb2= growth factor receptor-bound
protein-2GSK3=Glycogen Synthase Kinase 3IR= Insulin ReceptorIRS-1=InsulinReceptor Substrate-1JNK= c-jun N-Terminal KinaseMEK1/2=MAPK kinase 1/2PDK= Phosphoinositide-Dependent
KinasePI3K= Phosphoinositide-3 KinasePIP2= phosphatidylinositol-4,5-
bisphosphatePIP3= phosphatidylinositol-3,4,5-
trisphosphatePKC= Protein Kinase CRaf=(MAPK kinase kinase)Shc=Src Homology 2 Domain
ContainingSOS=Sons of Sevenless

5
1.2.3.1 The Insulin Receptor (IR)
Insulin binds to the IR, a member of the transmembrane receptor tyrosine kinase (RTK)
family. It is a heterotetramer, where all four subunits are coded by the INSR gene of chromosome
19 in humans[27]. There are 2 α-subunits (135 kDa) which serve as extracellular ligand binding
domains and 2 β-subunits (95 kDa) which have both
transmembrane and catalytic cytosolic regions[28].
Upon insulin binding to the α-subunits, the
transmembrane domains of the β-subunits migrate
laterally until receptor dimerization occurs[29]. This
activates the tyrosine (Tyr) kinase activity of the β-
subunits. Each β-subunit transfers phosphate groups
from ATP to selected Tyr residues of the opposite β-
subunit, resulting in IR autophosphorylation [29]. An
NPXpY-recognition motif is then established, allowing
for the IR to bind to and phosphorylate Tyr residues on
Figure 1.3 Tyrosine Phosphorylationof the Insulin Receptor. Insulin bindingto the IR results in receptor activation by βsubunit autophosphorylation at tyrosineresidues.
a variety of intracellular scaffold peptides including Src homology 2 domain-containing
proteins (Shc), adapter protein with PH and SH2 domains (APS), Grb2-associated binder 1/2
(Gab1/2), dedicator of cytokinesis 1/2 (Dock1/2), casitas B-lineage lymphoma (cbl), and insulin
receptor substrate (IRS) proteins[30-32]. Once the cytosolic region of the IR β-subunits have
become autophosphorylated, the presence of insulin is no longer necessary for continued
signalling. Thus, in order to terminate insulin signalling, enhanced phosphotyrosine phosphatase
or serine (Ser)/threonine (Thr) kinase activity in the cell is required, both of which can result in
reduced Tyr phosphorylation of the IR[34,35].

6
Although the type 1 IGF receptor (IGF1R) and the IR are distinct, they are both
members of the same RTK family, and thus, share many structural and functional properties.
For this reason, both insulin and insulin-like growth factors (IGF) can bind to either receptor,
though the receptors have the greatest affinity for their respective ligands[33].
1.2.3.2 Insulin Receptor Substrates (IRS)
IRS proteins play a critical role in cellular processes including growth, differentiation,
proliferation, and metabolism[36]. They serve as an interface between growth factor RTKs and
intracellular signalling molecules containing Src homology 2 (SH2) domains[37]. Although
there are several potential targets for the IR, the IRS proteins make up the largest group with six
identified isoforms[38]. Insulin signalling in adipose and SKM predominantly occurs through
IRS-1, while IRS-2 is more critical in the liver and pancreas. IRS-3 and -4 are more limited in
expression, and may be found in adipose and embryonic tissue, respectively[38]. The other IRS
isoforms are not well characterized.
All IRS isoforms contain both an N-terminal pleckstrin homology (PH) domain,
important for membrane phospholipid binding, and a phosphotyrosine binding (PTB) domain
involved in docking on the IR β-subunit bearing the NPXpY recognition sequence[39].
Interactions at both PH and PTB domains promote Tyr phosphorylation in the C-terminal region
of the IRS, allowing it to subsequently interact with specific SH2 domain containing proteins
including PI3K regulatory subunits, growth factor receptor-bound protein-2 (Grb-2), and SH2
domain-containing tyrosine phosphatases (SHP-2)[40]. There are at least 18 distinct Tyr
phosphorylation sites on IRS-1 which can activate SH2 domain containing proteins. For
example, using rat numbering, the phosphorylation of Tyr608 and Tyr628 of IRS1 produces the
major docking sites for PI3K. Tyr phosphorylation of IRS-1 has been known to facilitate and
prolong insulin signalling, but it has been identified more recently that there are at least 50

7
potential Ser phosphorylation sites which may reduce the capacity of IRS-1 to dock on the IR,
thus resulting in reduced activation and signal transduction[40].
Figure 1.4 IRS-1 Activity Regulation and Downstream Targets. Tyrosine phosphorylation enhancesIRS-1signalling while serine phosphorylation attenuates it.
Abbreviations: Y=Tyrosine; S=Serine. PI3K= Phosphoinositide-3 Kinase; Grb= growth factor receptor-bound protein; SHP-2=SH2 domain-containing tyrosine phosphatases; JNK=c-jun-N-Terminal Kinase IKK= IκB kinase; PKCθ= Protein Kinase C (θisoform) GSK3β=Glycogen Synthase 3 β; ERK= Extracellular Signal-Related Kinases 1/2 ; mTOR=Mammalian Target ofRapamycin; AMPK=AMP-activated Protein Kinase
Evidence for the role of Ser phosphorylation first became apparent when okadaic acid, a
Ser phosphatase inhibitor, markedly reduced glucose uptake in insulin-stimulated SKM and
adipose tissue[41]. It was later identified that nearly 60% of individuals with T2DM and insulin
resistance have elevated IRS Ser phosphorylation. Thus, Tyr and Ser phosphorylation have
positive and negative effects on IRS-1 activity, respectively[40]. In fact, many factors
associated with insulin resistance including angiotensin II, endothelin-1, tumor necrosis factor-α
(TNF-α), and free fatty acids (FFA) can activate Ser/Thr kinases that phosphorylate IRS1[42-
44]. Interestingly, insulin signalling leads to the activation of MAPKs, including extracellular
signal-regulated kinases (ERK) and c-Jun-N-terminal kinase (JNK), which can phosphorylate
rodent IRS-1 Ser612 (human:Ser616) and Ser307 (human:Ser312), respectively[45,46]. More
recently, other Ser sites on IRS-1 which have lead to insulin resistance in rodent studies and are
targets of ERK or JNK include Ser632 and Ser302[47,48]. This mode of signalling has been
thought of as a mechanism of negative feedback regulation of insulin actions, and will be
further discussed.

8
1.2.3.3 PI3K Signalling Pathway
1.2.3.3.1 PI3K
PI3K is a target of insulin signalling which can phosphorylate the 3’OH of a
phosphatidylinositol ring[49]. PI3K has been divided into three classes (I-III) where only class
I is able to convert phosphatidylinositol-4,5-bisphosphate (PIP2) to phosphatidylinositol-3,4,5-
trisphosphate (PIP3) on the cytosolic side of the PM[50]. Class I PI3K is a heterodimer
composed of a regulatory and catalytic subunit. There are five potential regulatory subunits:
p85α, p55α, p50α, p85β, and p55γ, all of which contain the SH2 domain, allowing for
interaction with an IRS protein[51]. In quiescent cells, the regulatory subunit maintains the
catalytic subunit in a low-activity state until the PI3K is directly stimulated by the
phosphotyrosine residues of activated growth factor receptors or adaptor proteins such as IRS.
There are three potential catalytic subunits: p110α, β, and δ. The first two catalytic isoforms are
known to be expressed in all cells while p110δ is generally present in leukocytes. The activated
PI3K converts the plasma membrane lipid PIP2 to PIP3[51]. This stimulates the translocation of
signalling proteins with PH domains such as phosphoinositide-dependent kinase 1 (PDK1) and
its effector, Akt/protein kinase B (PKB) to the cytosolic region of the PM where the activated
PI3K and resulting PIP3 are present [52]. The association of these proteins with PIP3 at the
membrane brings them into close proximity with each other, facilitating the phosphorylation of
Akt/PKB at Thr308 by PDK1[53]. This, however, leads to only partial activation of Akt/PKB.
Phosphorylation of Ser473 is also required which is provided by PDK-2, a complex of the
protein kinase mTOR (mammalian target of rapamycin) and the regulatory protein, rictor[54].
PI3K activity is attenuated by PIP3 phosphatases including phosphatase and tensin homolog
(PTEN), type II SH2-domain-containing Inositol-5-Phosphatase (SHIP2), or through the use of
pharmacological agents including wortmannin or LY294002[55,56]. Although inhibition of

9
PI3K antagonists may prove useful at enhancing insulin signalling and thus, improving T2DM,
one caveat is that it may reduce the tumor suppression capacity within the cell[55].
1.2.3.3.2 Akt/PKB
Akt/PKB, is a member of the Ser/Thr-specific protein kinase family known for its
capacity to mediate the mitogenic and anti-apoptotic effects of growth factors including platelet-
derived growth factor (PDGF), epidermal growth factor (EGF), IGF-1, and insulin[57]. There
are three Akt/PKB isoforms, Akt 1-3/PKBα,β,γ, with conserved domain structures consisting of
a PH domain, a central kinase domain, and a C-terminal regulatory domain[58,59,60]. Despite
this structural similarity, the isoforms appear to express different distribution patterns and
functions. Akt1/PKBα, present in every cell of the body, appears to be involved in cell survival
and growth by respectively inhibiting apoptosis while stimulating protein synthesis[61]. The
anti-apoptotic effect may, in part, be due to Akt1/PKBα phosphorylating the pro-apoptotic
protein, Bcl2-associated death promoter (BAD) causing it to dissociate from the B-cell
CLL/lymphoma 2 (BCL-2/BCL-X) complex, or the activation of NF-κB which may enhance
the expression of pro-survival genes[61,62]. Akt2/PKBβ, however, is generally found only in
insulin-sensitive tissues as it is essential for the metabolic effects of insulin signalling. In
adipose and SKM, insulin stimulated Akt2/PKBβ can mediate GLUT4 translocation to the PM
from intracellular stores via AS160[63,64]. Akt2/PKBβ can also promote glycogen synthesis
through the inhibition of glycogen synthase kinase 3 (GSK3)[57]. Akt3/PKBγ has a less well
defined function and is primarily located in the brain and gonads[65]. The unique function of
these isoforms was identified in mouse knockout (KO) studies, where Akt1/PKBα-/- animals
were smaller in size but with normal glucose metabolism, whereas Akt2/PKBβ -/- demonstrated
insulin resistance and a diabetic phenotype[66-68]. Similarly, knockdown studies using small-

10
interfering RNA (siRNA) in 3T3-L1 adipocytes have demonstrated that insulin sensitivity and
glucose disposal are compromised only when Akt2/PKBβ levels are reduced[69,70].
Although PI3K has been traditionally linked to Akt/PKB activation, more recent studies
indicate alternative mechanisms. Compounds which increase intracellular cAMP such as
forskolin, prostaglandin-E1, 8-bromo-cAMP, and the β-adrenergic agonist, isoproterenol, have
been shown to activate Akt/PKB through protein kinase A (PKA) in a variety of cell
types[71,72]. It has also been shown that Akt/PKB can be activated by Ca2+/calmodulin-
dependent kinase directly in vitro[73].
1.2.3.2.3 GLUT4
The facilitative glucose transporters (GLUT) assist in moving blood glucose into cells
along a concentration gradient. There are 13 known GLUTs derived from different genes, which
have been divided into three classes (I-III)[74,75]. The GLUTs have twelve membrane-
spanning α-helical loop regions with cytosolic N- and C-terminals, but they exhibit different
substrate specificities, kinetic properties, and/or tissue expression profiles[76]. It is important to
note that transport through GLUTs is bidirectional, stereoselective, and saturable. Of the three
classes of GLUTs, Class I (GLUT1-4) is most relevant to insulin signalling and diabetes[76].
GLUT1 is ubiquitously expressed, though it is most notably present in the brain and
erythrocytes[76,77]. It is predominantly located in the PM, accounting for basal glucose uptake,
and is upregulated during extreme conditions of glucose deprivation or hypoxia. GLUT2 is
expressed primarily in the liver, kidneys, and pancreatic β-cells. In the β-cells, GLUT2 plays a
critical role in the glucose-sensing mechanism. GLUT3 has a high affinity for glucose and is
located in sites where glucose demand is high such as in the brain. The insulin-responsive
GLUT4 is found in the heart, SKM, and adipose tissue, where it facilitates the reduction of
postprandial blood glucose[76].

11
GLUT4 translocates to the PM from intracellular stores in insulin-sensitive cells. In fact,
insulin treatment leads to an 8-fold increase in PM GLUT4 in adipocytes and a 2-fold increase
in SKM[78,79]. Furthermore, global or tissue-specific GLUT4-/- mouse models were smaller
and insulin resistant, and reduced GLUT4 has been associated with T2DM[81,82]. Given that
the long 40 h t1/2 for GLUT4, it is recycled several times before being degraded [80].
1.2.3.4 MAPK Signalling Pathway
The MAPK family is a group of Ser/Thr kinases which mediate a variety of cell
processes including differentiation, proliferation, and apoptosis[83]. The MAPK signalling
pathway is involved, not only in inducing specific cellular effects, but is also important for
signal amplification[84]. There are 3 major discrete groups of MAPKs: i) extracellular signal-
regulated kinases 1/2 (ERK1/2), ii) c-Jun N-terminal kinases 1-3 (JNK1-3), and iii) p38 kinase
(α-δ), each, with different stimuli, distributions, and cellular outcomes[85].
MAPKs are part of a three-tiered cascade consisting of a MAPK, a MAPK kinase
(MAPKK) which is either MKK or MEK, and a MAPK kinase kinase (MAPKKK)[86].
MAPKs are phosphorylated at Thr/Tyr residues by MAPKKs, which are themselves
phosphorylated at Ser/Thr residues by upstream MAPKKKs [87]. Scaffold proteins bind the
components of MAPK-signalling pathway to facilitate specific and efficient activation cascades.
MAPK activation, however, is not a simple switch in that both the duration and extent of
activation result in different cellular outcomes. In order to regulate the state of activation, cells
have a series of phosphatase enzymes which can dephosphorylate and thus, turn off MAPK
activity. These include Ser/Thr phosphatases such as PP2A and PP2Cα, and members of the
tyrosine phosphatase (PTP) gene superfamily, such as dual specificity phosphatases
(DSPs)[88,89,90]. There are 10 identified DSPs that display differences in tissue expression,

12
subcellular distribution, and specificity for MAPK family members[91]. The largest group of
DSPs are the MAPK phosphatases(MKPs). These will be further discussed.
Figure 1.5 MAPK Signalling Cascades. Growth factors and/or stressorsactivate MAPKKKs which, in turn, stimulate specified MAPKKs. This leads tothe activation of MAPK (ERK1/2, JNK1-3, p38α-δ), each of which has a uniquecellular outcome.
1.2.3.4.1 ERK
ERK, or the classical MAPK, is involved in regulating meiosis, proliferation, and cell
growth[92]. It is strongly activated by growth factors including EGF, PDGF, prolactin, IGF-1,
and insulin, as well as, to a lesser extent, cytokines and osmotic stress[93]. There are two
isoforms of ERK which have 83% amino acid sequence similarity and are expressed to various
extent in all tissues[94]. Following autophosphorylation of the IR, Grb-2 will dock on specific
phosphotyrosines of the activated IRS1 through its SH2 domain, and through its SH3 domain,
binds to “son of sevenless” (SOS), a guanine nucleotide exchange factor (GEF)[86,95]. This
results in an exchange of GDP for GTP in Ras, and, by an undefined mechanism, leads to the
activation of Raf, a MAPKKK. Raf will then specifically activate the MAPKK, MEK1/2, which

13
in turn, activates ERK1/2[86,95]. ERK1/2 can phosphorylate a wide range of substrates in all
cellular compartments, including various membrane proteins (CD120a, calnexin) and nuclear
substrates (SRC-1, Elk-1, MEF2, c-Fos, c-Myc)[96,97].
It is through ERK1/2 that many of the insulin- and IGF-mediated growth signals are
conveyed. However, ERK1/2 is associated with Ser phosphorylation of IRS and thus, decreased
insulin signalling[98,99]. It has been proposed that this acts as a form of negative feedback
regulation but if hyperactive, can promote states of insulin resistance and T2DM[100].
1.2.3.4.2 JNK
JNK is a member of the MAPK family involved in cell differentiation, proliferation, and
apoptosis[101]. It is activated primarily by stressful stimuli including inflammatory cytokines,
UV radiation, hypoxia, FFAs, heat, and osmotic shock[101]. In addition to this, a number of
groups have also reported that various growth factors including EGF, PDGF, and insulin are
able to activate JNK in certain cells, though the mechanism by which this is achieved is poorly
understood[102-104]. There are at least 10 JNK isoforms derived from alternative splicing of
three genes [105]. JNK1/2 is expressed in all cells of the body while JNK3 is predominantly
found in neurons and the testes[105].
The MAPKKs for JNK are MKK4/Sek1 and MKK7, each of which has different
properties[101]. MKK4, for instance, primarily responds to extracellular stress such as hypoxia
and may also activate p38, while MKK7 is predominantly activated by cytokines[106,107]. In
addition, MKK4 phosphorylates the Tyr residue of the JNK activation loop while MKK7
phosphorylates Thr[108]. The MAPKKs are activated by dual-specific phosphorylation at Ser
and Thr residues by a variety of MAPKKKs including MEKK1-4, apoptosis signal-regulating
kinase 1 (ASK1), and mixed lineage kinase 3 (MLK3). This diversity in MAPKKKs reflects the
variety in pathway stimulators[109].

14
Once activated, JNK phosphorylates and regulates the activity of a number of
transcription factors including c-Jun, and activating transcription factor 2 (ATF2) [101]. It can
also cause Ser phosphorylation of IRS-1 to decrease insulin signalling[104]. This will be further
discussed.
1.2.3.4.3 p38 Kinases
p38 kinase is a member of the MAPK family involved in proliferation and apoptosis
and is primarily activated by stressful stimuli including inflammatory cytokines, UV radiation,
FFAs, hypoxia, heat, and osmotic shock[112]. Four p38 isoforms have been identified: p38-
α(MAPK14), -β(MAPK11), -γ(MAPK12) and -δ(MAPK13). The first two are expressed
ubiquitously[113-115]. Activation of the p38 isoforms results from phosphorylation by the
MAPKKs, MKK3 and MKK6[116]. As discussed, MKK4/Sek1 may also activate p38,
suggesting that MKK4/Sek1 may be a site of integration for the two stress-activated MAPKs
[106]. MKK3 and MKK6 are activated by a wide range of MAPKKKs including MEKK 1-4,
MLK3, DLK, ASK1, and TGF-beta activated kinase 1 (Tak1)[117].
p38 kinase can phosphorylate a wide variety of cellular targets, including cytosolic
phospholipase A2(cPLA2), and the transcription factors ATF1/2, Sap-1, Elk-1, NF-κB, and the
tumor suppressor, p53[118]. Although p38 kinase can be activated by insulin in certain tissues,
and can induce Ser phosphorylation, one group has suggested that p38 decreases insulin
signalling actions by downregulating GLUT4 [119,120].
1.2.4 Signalling Defects and Insulin Resistance
Insulin resistance is the condition where the actions of insulin are, to some degree,
compromised. The etiology is broad and diverse, though many agree that increased non-
esterified (free) fatty acids (NEFA), inflammatory cytokines (TNF-α, IL-1), and decreased

15
adiponectin are all correlated with an increased development of insulin resistance and
T2DM[121,122]. Any impairment of the insulin signalling pathway can result in resistance. It
has been reported that reduced Tyr phosphorylation, amplified Ser phosphorylation, or
decreased expression of IR and IRS are associated with the insulin resistant
phenotype[123,124]. Interestingly, exposing adipocytes to stressors including TNF-α or FFA
have resulted in a hyperactivated cellular JNK status and compromised insulin signalling,
whereas the induction of MKPs against JNK lead to potentiated insulin signalling[125,126].
Furthermore, there have been human and rodent studies which indicate reduced activation and
localization of PI3K at the membrane, along with enhanced Ser phosphorylation of the PI3K
p85 subunit in insulin resistance[127,128]. Finally, there is also evidence that Akt2/PKBβ is
significantly reduced in insulin resistance, as is the expression and translocation of
GLUT4[129,130]. This topic will be further discussed.
1.2.5 Hypertension and Insulin Resistance
Insulin resistance is associated with dyslipidemia and hypertension (HT) in what is
known as the metabolic syndrome[131]. Depending on the population studied, approximately
40% of non-obese non-diabetic patients with HT are insulin-resistant, and this increases with
age[132]. Despite the frequency of co-existence, it is not fully understood how they develop
together. Insulin resistance of peripheral tissues, whether from obesity, genetics, or variety of
environmental factors, results in hyperglycemia and compensatory hyperinsulinemia[5].
Chronically elevated levels of insulin can increase sympathetic nervous system activity,
promote vascular growth, Na+ retention, and increase α1-adrenergic receptors[133,134]. These
outcomes can all increase blood pressure (BP), thereby promoting HT. It must be noted that
although insulin can signal for vasodilation through vascular endothelial cells, these sites are
also prone to insulin resistance[135]. Furthermore, hyperglycemia also appears to inhibit nitric

16
oxide (NO) production and alter ion transport in vascular smooth muscle cells (VSMCs),
favoring vasoconstriction, as well as VSMC growth, proliferation, and migration[136,137].
Although these theories make a strong case for insulin resistance promoting HT, the reverse can
also be indicated. Vasoconstriction and HT can reduce blood flow to peripheral tissues and thus,
decrease insulin delivery and action at these sites[138]. In addition, angiotensin-II (AngII), a
hormone involved in elevating BP, has been shown to increase MAPK activity, leading to
greater Ser phosphorylation of IRS-1, and thus, decreased insulin signalling[139,140].
1.3 Blood Pressure Regulation
Figure 1.6 Major Mechanisms of Systemic Blood Pressure Regulation. In response to low BP,the RAS produces angiotensin II to promote BP elevating processes in the body. As the BP becomes too high, theKKS produces bradykinin to reverse these effects. Natriuretic peptides can assist the KKS in reducing BP.
1.3.1 Renin-Angiotensin System (RAS)
The RAS is a regulatory system which responds to decreasing BP by elevating AngII
production. This process was traditionally viewed as a systemic response, but has since been
identified to exist locally, at sites including the heart, brain, kidney, and adipose
tissue[142,143]. Furthermore, the systemic RAS activity may be normal while local RAS is
hyperactive, accounting for site specific hypoperfusion[144].

17
When a low volume output is detected by the macula densa cells of the kidney, the
juxtaglomerular apparatus is activated and secretes renin into circulation[145]. Renin is an
enzyme which cleaves the liver-derived 452 aa peptide, angiotensinogen (AGT), creating the
decapeptide, angiotensin I (AngI). AngI does not appear to have any significant biological
activity. It exists solely as a precursor to the octapeptide, AngII, produced by the dipeptidase,
angiotensin-converting enzyme (ACE)[145]. There are two major anti-hypertensive medications
which interfere with the RAS, i) ACE inhibitors (ACEi) and ii) AngII-receptor blockers (ARB).
ACEis disrupt the formation of AngII by compromising the actions of ACE, while ARBs interfere
with the AngII interaction with its AT1 receptor[146,147]. These will be further discussed.
1.3.1.1 Angiotensin II (AngII)
AngII is an 8 aa peptide which is very effective at elevating BP. It is a potent
vasoconstrictor which can also elevate the local secretion of ET-I[148]. It aids in expanding
blood volume and thus, BP, by acting on the subfornical
organ of the brain to enhance thirst and by increasing the Figure 1.7 AngII Amino Acid Sequence
secretion of vasopressin (ADH) from the posterior pituitary[149]. Furthermore, it promotes
aldosterone secretion from the zona glomerulosa of the adrenal cortex to cause Na+ retention at
the kidney[150]. AngII has a t1/2 of 30s (systemic RAS) or as long as 30 min (local RAS) and is
degraded by angiotensinases to the 7 aa, AngIII, with only a fraction of its initial potency[154].
1.3.1.2 AngII Receptors & Signalling
AngII has at least four receptors, but only two subtypes, designated AT1 and AT2, have
been well characterized[151]. The AT1 is responsible for vasoconstriction, sympathetic
activation, stimulation of aldosterone release, and cellular growth[194]. It is primarily found in
the brain, adrenals, heart, vasculature and kidney, and to a lesser extent at sites including the
liver, lung, and adipose tissue. The AT2 is involved in mediating anti-proliferation, cellular

18
differentiation, apoptosis, and the anti-AT1 effect of vasodilatation. In adults, AT2 receptors are
present in brain, adrenal medulla, kidney, heart, reproductive tissues, and adipose tissue. Both
AT1 and AT2 are seven transmembrane G-protein linked receptors, both of which have a
comparable binding affinity for AngII[151,194].
Since AngII generally acts through the AT1 receptor to influence cardiovascular actions,
ARBs, such as losartan, irbesartan, and valsartan, specifically target it[155]. It has been reported
that this AT1 receptor antagonism leads to elevated AngII which may cause hyperstimulation of
AT2 receptors[156]. Whether this is advantageous or detrimental is controversial. Some believe
that since AT2 receptors can promote vasodilation, this is another mechanism by which ARBs
serve as an anti-hypertensive agent[156]. Interestingly, AT2 receptor activation is associated
with increased NO and cGMP which appears to be essential for kinin-induced vasodilation and
decreased BP that will be discussed[157]. Furthermore, it is worth noting the discovery of
ACE2, a peptidase which catalyzes the synthesis of Ang1-7 from AngII, or Ang1-9 from AngI
which is subsequently converted to Ang1-7 by ACE[158]. Ang1-7 can act through a unique G-
protein coupled receptor, Mas, to counteract the effects of AngII on peripheral vascular
resistance, vascular cell growth, and cardiac remodelling[158,159].
1.3.2 Kalikrein Kinin System (KKS)
The KKS is a regulatory system which responds to increasing BP by elevating the
production of kinins[160]. In humans, the two forms of liver-derived kininogens, high
molecular weight kininogen (HMWK; 626aa) and low molecular weight kininogen
(LMWK;409aa), are formed through alternative splicing[161]. Although HMWK is not
catalytically active, it can serve as a cofactor in the intrinsic pathway of blood coagulation. As
indicated in Figure 1.6, kininogens are converted to kinins through the enzymatic action of
kallikrein, a member of the Ser protease family. Kallikrein itself is derived from prekallikrein,

19
produced by the liver, following cleavage at its N-terminal by factor XII (Hageman
factor)[162]. Active tissue kallikrein acts on HMWK to release the nonapeptide, bradykinin
(BK), and LMWK to produce the decapeptide, kallidin[160]. Kallidin can be converted to BK
by a plasma aminopeptidase[163]. In humans, BK is predominantly generated in the plasma
while kallidin peptides are produced in tissues[160]. It is important to note that there is another
system which uses natriuretic peptides to reduce BP via a similar mechanism to the KKS[204].
This will be further discussed.
1.3.2.1 Bradykinin (BK)
BK is a 9-aa peptide derived from HMWK, or indirectly from LMWK[164]. It is
implicated in many cardiovascular processes including vasodilation, natriuresis, left ventricular
hypertrophy inhibition, and cardioprotection during ischemia-reperfusion[160].
The average plasma concentration of BK is 50 pM with a t1/2
of only 15-30s [164,167]. It is degraded by a group of Figure 1.8 BK Amino Acid Sequence
kininase enzymes including aminopeptidase P (APP), neutral endopeptidase (NEP),
carboxypeptidase M and N, and most importantly, ACE[164,167]. Interestingly, ACE appears
to have a higher affinity for BK than AngI, suggesting that its primary function is to degrade
BK rather than produce AngII[168,169].
1.3.2.2 BK Receptors & Signalling
BK effects in the body are mediated by the stimulation of two G-protein-coupled
receptors, B1 and B2. The B1 receptors are usually only present in the PM following tissue
injury or exposure to noxious stimuli and are responsible for BK inflammatory responses. The
B2 receptor, however, is constitutively present in the PM of a wide variety of tissues, and is
responsible for the majority of BK actions [164].

20
Once BK binds to the B2 receptor, the coupled Gq/11α activates the membrane-bound
enzyme, phospholipase C (PLC), which in turn, converts PIP2 into the second messenger,
inositol triphosphate (IP3)[165,166]. Activation of the caveolae IP3 receptors leads to the release
of sequestered Ca2+ into the cytosol. The Ca2+ interacts with calmodulin, forming a complex
which activates the NO generating enzyme, nitric oxide synthase (NOS). It is through NO-
signalling that BK mediates the majority of its effects, including vasodilation[165,166].
1.4 Antihypertensive Therapy and Insulin Sensitivity
Many studies have examined the effect of antihypertensive agents on diabetes, given the
close association between HT and insulin resistance. Thiazide diuretics and β-blockers have had
an adverse impact, increasing the risk for new-onset diabetes by suppressing insulin secretion
and/or action[170,171]. In contrast, ACEi therapy has been shown to increase insulin sensitivity
in both rodents and humans[172,173]. It also delayed the development of T2DM in susceptible
hypertensive male subjects[174]. It must be noted that ACEi therapy both decreases AngII
production and diminishes BK degradation. ACEi studies have described a 2-fold increase in
plasma BK levels and a marked improvement in its physiological actions [175,176]. The dual
function of ACE poses the question of whether decreased AngII or enhanced BK is responsible
for the documented improvement in insulin sensitivity. Initially, AngII appeared more
important, given that ARBs can also enhance insulin sensitivity. Also, AngII inhibition leads to
reduced negative regulation of insulin signalling through MAPK[139,177]. However, many
studies have suggested an essential role for BK. For example, Brown Norway Katholiek (BNK)
rats, which are kinin-deficient and thus, lack BK, are insulin resistant and do not respond to
ACEi treatment[178]. Another study demonstrated that the use of HOE-140, a B2-receptor
antagonist, removed the effects of ACEi on insulin sensitivity[179]. Chronic i.v. infusion of BK
into insulin resistant fa/fa rats also improved glucose tolerance[180]. Finally, ramipril, an ACEi,

21
was more effective at improving insulin sensitivity than losartan, an ARB, in the myocardium,
SKM, and adipose tissue[181]. Collectively, these results suggest that the enhanced insulin
sensitivity observed with ACEi treatment is largely associated with elevated BK. It is also worth
noting that components of the RAS such as ACE are upregulated during diabetes while KKS
components including BK are reduced in obesity and insulin resistance[182,183]. Furthermore,
as mentioned, some suggest that hyperactivation of AT2 receptors following AT1 blockade with
ARBs may mimic BK actions through NO signalling. This may, in part, account for the
observed insulin sensitivity observed with ARB treatment[157].
Although BK is important in enhancing insulin sensitivity, it is also a potent vasodilator
which can improve blood flow and thus, insulin delivery to peripheral tissues. To address
whether BK can influence insulin signalling independent of hemodynamics, a number of groups
have investigated tissue in isolation. It has been reported that BK has a direct effect of
enhancing insulin signalling and GLUT4-mediated glucose uptake in L6 myoblasts, 3T3-L1
adipose cells, and both rodent and dog SKM and adipocytes [184,185,186]. This effect is
mediated specifically through the NOS isoform, endothelial NOS (eNOS), since BK had no
effect on eNOS-/- mice. Furthermore, the role of NO in BK signalling was supported by the
observation that the NO donor, sodium nitroprusside (SNP), mimicked BK actions, while the
NO scavenger, PTIO, removed them[184, 186].
1.5 Nitric Oxide (NO)
NO is short-lived free radical messenger that is produced on demand and is essential for
many processes including neurotransmission, vascular tone regulation, immunomodulation, and
insulin sensitivity[187]. NO is produced by the oxidation of L-arginine, resulting in this free
radical along with the byproduct, L-citrulline. This process is catalyzed by cell-specific
isoforms of NOS, and requires several cofactors including nicotinamide adenine dinucleotide

22
phosphate (NADPH), flavin adenine dinucleotide (FAD), flavin mononucleotide (FMN),
tetrahydrobiopterin (BH4), and a heme moiety[187]. NO can proceed to react with several
hemoproteins, the most well-characterized being soluble guanylate cyclase (sGC) which is
responsible for generating the second messenger, cGMP[188,189]. The t1/2 of NO is 3-30 s,
being inactivated by the superoxide anion (O2-) or oxyhemoglobin[187].
1.5.1 Nitric Oxide Synthase (NOS)
There are three NOS isoforms: i) neuronal NOS (nNOS; NOSI), ii) inducible NOS
(iNOS; NOSII), and iii) endothelial NOS (eNOS;NOSIII)[190]. Furthermore, nNOS and eNOS
are grouped as constitutive NOS (cNOS) because they are continuously expressed, unlike iNOS,
which must be induced[190]. All three isoforms are active as a dimer and share C-terminal
reductase and N-terminal oxygenase domains[191]. A heme prosthetic group is present at the N-
terminal which is linked to a calmodulin-binding domain in cNOS. Calmodulin couples
intracellular Ca2+ levels to cNOS activity such that when calmodulin is bound to its respective
site, it allows electrons to flow from the reductase domain to the heme, facilitating NO
production[191]. When cNOS is active, it acutely generates NO at low levels (nM). iNOS,
however, is activated independently of Ca2+ and synthesizes NO for longer periods, and at much
higher levels (µM) in response to specific inducers including IL-l, TNF-α, and interferon-γ
[187].
1.5.2 NO and Insulin Sensitivity
Being an important signalling molecule, low level production of NO through cNOS has
been shown to have protective effects and, specifically through eNOS, can promote insulin
sensitivity in peripheral tissues[192]. Chronic and excessive NO production by iNOS, however,
has been deemed toxic, as NO can bind to O2- in the cell to generate the highly unstable radical,
peroxynitrite (ONOO-)[187]. In addition to causing cell damage through lipid peroxidation and

23
DNA fragmentation, this compound exhausts the reactive oxygen species (ROS) buffering
capacity of the cell, leaving it susceptible to stressors that promote insulin resistance [193].
Therefore, low concentrations of NO appear to promote insulin sensitivity while excess NO
compromises it.
1.6 Guanylate Cyclase-cGMP-Protein Kinase G Signalling Pathway
1.6.1 Guanylate Cyclase (GC)
GC is an enzyme that converts GTP to the second messenger cGMP[195]. There are two
forms which exist in the cell, i) soluble GC (sGC), which is present in the cytosol and activated
by NO or carbon monoxide (CO), and ii) particulate GC, which is PM receptor associated
[195,196,197]. The catalytic conversion of GTP to cGMP by GC is dependent on the presence
of divalent cations such as Mn2+ and/or Mg2+ [195].
1.6.1.1 Soluble Guanylate Cyclase (sGC)
sGC is a heme-containing heterodimer protein comprised of one α(1, 2) and one β(1, 2)
subunit[198]. It is found in the cytosolic fraction and is present in essentially all mammalian
Figure 1.9 Structure of soluble Guanylate CyclaseEach sGC subunit is composed of three domains, i) heme-binding, ii) dimerization, iii) catalytic. sGC exists as aheterodimer of α- and β-subunits. NO binds in the hemedomain which activates of catalytic domain, resulting inthe conversion of GTP to cGMP.
cells where it mediates many of the effects of
NO. The N-terminal of each subunit constitutes
a heme-binding domain which allows sGC to
preferentially bind to NO[199]. To facilitate the
interaction between NO and the heme moiety,
the iron of the metalloporphyrin must be
maintained in a reduced Fe2+ (ferrous) state and
so reducing agents such as thiols are required to
enhance the enzyme activity[198]. At the C-

24
terminus of each subunit is a catalytic domain which exhibits significant sequence homology
with that of the membrane GC[198]. Although both α and β subunits of the sGC have catalytic
domains, both must be present for the enzyme to be functional[200]. In between the heme-
binding and catalytic regions of the subunits is a dimerization domain. This region mediates
subunit association, forming the heterodimer required for enzyme activity[201]. Upon NO
interaction with the heme domain, a conformational change occurs in the enzyme which
exposes the catalytic site to GTP[198]. A number of pharmacological agents can influence the
activity of sGC. YC-1, a benzyl indazole derivative, for example, has been shown to sensitize
sGC and increase its maximal catalytic rate by binding to an allosteric site on the enzyme,
thereby reducing the dissociation of NO from the heme group[202]. ODQ has been used
extensively as a specific inhibitor for sGC because it causes oxidation of the heme domain at the
N-terminal, making it unsuitable for NO interaction[203].
1.6.1.2 Particulate Guanylate Cyclase (GC)
Seven membrane GCs have been identified (GC-A— G) which are homomeric in
structure and only partly homologous to sGC[197].
The membrane GCs consist of an extracellular
ligand binding domain, a short transmembrane
region, and an intracellular domain with the
catalytic GC region at the C-terminal. The
extracellular domain functions as a receptor for
specific ligands, though ligands have only been
identified for GC-A—C. GC-A and B are activated
by the natriuretic peptides (NP), while the ligands
Figure 1.10 Structure of ParticulateGuanylate Cyclase. Each particulate GCsubunit is composed of three main domains, i)ligand-binding, ii) dimerization iii) catalytic(GC). The extracellular domain functions as areceptor for natriuretic peptides and when bound,activates the cytosolic catalytic (GC) domainwhich converts GTP to cGMP.

25
for GC-C are heat-stable enterotoxins and guanylins[197]. GC-A appears to show the most
variety in distribution, being present in VSMCs, endothelium, central nervous system, kidney,
heart and adipose tissue, while GC-B is predominantly in fibroblasts, and GC-C in the intestinal
epithelium [204]. Although both sGC and membrane GC produce cGMP, it is interesting to note
that the ligands for these receptors are distinct, being small gaseous activators and peptides,
respectively. These receptors will be further discussed.
1.6.2 cGMP
cGMP is a cyclic nucleotide that is generated by GC following activation by NO or
Figure 1.11 cGMP Structure
natriuretic peptides [195,196]. The specificity of cGMP actions is
dictated by binding motifs on target proteins. These intracellular
targets include protein kinase G (PKG), cyclic nucleotide-gated
channels, and cGMP-binding phosphodiesterases (PDE)[205].
The former two are primarily responsible for the biological
effects of cGMP while PDEs are a group of enzymes which
deactivate cyclic nucleotides and thus, regulate the localization, duration, and amplitude of
cGMP signalling[205]. There is evidence, however,
that cGMP can also influence the activity of cAMP-
specific PDEs[334,335]. There are 11 PDE
families(1-11) which differ in tissue distribution,
substrate specificity, and aa sequence[206]. Some are
specific for cAMP (PDE4,7,8), others for cGMP
(PDE5,6,9), while the remainder can target both
cAMP and cGMP[206]. A variety of pharmacological
Figure 1.12 cGMP Inducers andTargets. NO and natriuretic peptides canelevate cGMP in cells via sGC and theparticulate (membrane) GC, respectively. Thereare three main targets for cGMP, i) PDE(degradation), ii) PKG, and iii) ion channels

26
PDE inhibitors have been developed in order to maintain or elevate intracellular cGMP levels.
Some examples of PDE5 selective inhibitors include Sildenafil (Viagra), vardenafil, and
tadalafil [207,208]. Zaprinast is a traditional PDE5 inhibitor which also appears to antagonize
PDE1 functions[209].
1.6.3 Protein Kinase G (PKG)
PKG, also known as cGMP-dependent protein kinase (cGK) is a member of the Ser/Thr
kinase family and is a major target of cGMP[210]. It is involved in a wide variety of processes
including vascular smooth muscle relaxation, cardiac remodelling, platelet aggregation, long
bone growth, and circadian rhythmicity[211]. There are two distinct PKG genes: i) PKG type I
(PKGI; cGKI), which is predominantly found in the cytoplasm, and ii) PKG type II (PKGII;
cGKII), which is anchored to the PM. Furthermore, alternative splicing of the PKGI N-terminal
results in the isoforms, PKGIα and PKGIβ[211]. Almost all cells contain at least one of three
PKGs. PKGI is abundant in smooth muscles, platelets, cerebellum, kidney, and to a lesser extent
at sites including cardiac muscle, vascular endothelium, and adipose tissue[212,213]. PKGII is
present in the lung, intestinal mucosa, chondrocytes, and the adrenal cortex[214,215].
All PKGs possess an N-terminal domain, a regulatory domain, and a catalytic C-
terminal domain[211]. The N-terminal domain mediates cell localization and maintains PKG
inactivity in the absence of cGMP. The regulatory domain has two distinct cGMP-binding sites,
which, upon interaction with cGMP, produces a conformational change leading to the catalytic
activation of PKG. The catalytic domain allows PKG to transfer the phosphate from ATP to
Ser/Thr residues of substrate proteins[211].
PKG has a wide range of intranuclear, membrane, and cytosolic targets[212-214]. Most
of the substrates are members of other signalling pathways, including G proteins, ion channels,
cytoskeleton-associated proteins, or transcription factors where PKG can influence gene

27
expression[211]. The function and targets of PKG signalling have been effectively studied
through the use of a selective pharmacological inhibitor, KT-5823, which interferes with the
ATP binding site of the catalytic domain[219]. Other studies have relied on genetic methods of
compromising PKG functions including KO mice or siRNA transfections[220,221].
1.7 Bradykinin Acts Through sGC-cGMP-PKG to Potentiate Insulin Signalling
BK activates eNOS, and through NO, improves insulin sensitivity in peripheral
tissues[184,186]. As described, the sGC-cGMP-PKG signalling pathway is a major target for
NO. Although the role of cGMP in BK actions has not been fully assessed, one study showed
that chronically treating diet-induced insulin resistant mice with the PDE5 inhibitor, sildenafil,
resulted in improved energy balance and insulin actions[222].
In a previous study, our lab examined the role of sGC-cGMP-PKG signalling in insulin
treated primary rat adipocytes. We observed that pre-treating the cells for 30 min with the sGC
activator, YC-1 (50 µM), or the cGMP analogue, CPT-cGMP (100µM) reproduced the 1h effect of
BK (1.0µM) to enhance insulin-stimulated glucose uptake, while pre-treatment with the sGC
inhibitor, ODQ (50 μM), or the selective PKG inhibitor, KT-5823 (100 nM), removed the BK
effects.
To confirm that BK acts through sGC-cGMP-PKG signalling to potentiate the insulin
signalling pathway, the activity status of pathway members was next assessed. Pre-treatment of
adipocytes with YC-1 or CPT-cGMP mimicked the BK effect of enhancing insulin-stimulated
Tyr-IRS-1 and Akt/PKB phosphorylation, while ODQ and KT5823 pre-treatment abrogated the
BK effects. However, when assessing the MAPKs, which are also targets of insulin signalling,
BK, along with its prospective mimetics, decreased insulin-stimulated JNK and ERK
phosphorylation. Although this outcome seems paradoxical, it ultimately led to the expansion of
our proposed mechanism for BK actions. After 40 min of insulin stimulation, as employed in
our studies, Ser phosphorylation of IRS-1 by MAPKs becomes significant, resulting in an

28
attenuation of the insulin signalling pathway[223]. By doing this, MAPKs serve as a negative
feedback switch for insulin actions. We reasoned that BK may reduce MAPK activity, and thus,
Ser phosphorylation of IRS-1, resulting in a potentiation of insulin signalling. To support this
theory, we observed Ser307 and Ser612 phosphorylation of IRS-1, the respective target sites for
JNK and ERK[186], and found that it was also significantly decreased in BK-treated insulin
stimulated cells. Interestingly, the JNK inhibitor, SP600125, but not the ERK inhibitor,
PD98059, was able to mimic the BK effects on glucose uptake in our rat adipocytes, suggesting
that the BK effects occur primarily through compromised JNK activity. To further support this,
we observed that JNK1-/- mice had significantly higher glucose uptake compared with controls,
irrespective of BK treatment[186].
Taken together, these results suggest that BK acts through the sGC-cGMP-PKG
signalling pathway to decrease the activation of JNK, leading to reduced IRS-1 Ser
phosphorylation and thus, potentiated
insulin actions. This would also lead one
to suspect that activating the membrane
GC, as in the case with natriuretic
peptides, would elevate cGMP and mimic
this mechanism of BK action. To support
this, natriuretic peptides have many
functions in common with BK in the body,
including the antagonism of RAS through
the promotion of natriuresis and
vasorelaxation[204,225].
Figure 1.13 Mechanism of BK-mediatedEnhanced Insulin Sensitivity. BK binds to the B2receptor of adipocytes, resulting in the activation of eNOS andNO production. NO activates the sGC-cGMP-PKG signallingpathway which, in turn, attenuates the activity of JNK, anegative regulator of insulin signalling. As a result of reducedinhibition, insulin actions in adipocytes are potentiated.

29
1.8 Natriuretic Peptides (NP)
NPs are a family of structurally related but genetically distinct hormones that regulate
BP, pulmonary hypertension, ventricular hypertrophy, fat metabolism, and long bone
growth[204]. There are three general types of NPs in human, i) atrial natriuretic peptide (ANP;
28 aa), ii) B-type natriuretic peptide (BNP; 32 aa), and iii) C-type natriuretic peptide (CNP; 22
aa), all of which share a 17 aa ring structure formed by a disulphide bond[204]. NPs have three
Figure 1.14 Natriuretic Peptide Structures. There are three natriuretic peptides, i) ANP (28 aa),ii) BNP (32 aa), and iii) CNP (22 aa). All natriuretic peptides have a conserved 17 aa ring but unique N- and
C-terminals.
receptors(NPR), i) NPR-A (GC-A) with highest affinity for ANP and BNP, ii) NPR-B (GC-B)
with preference for CNP, and iii) NPR-C, also known as the “clearance receptor” [226,227].
Figure 1.15 Characteristics of NatriureticPeptide Receptors. There are three generalnatriuretic peptides receptors, i) NPR-A (GC-A), ii)NPR-B (GC-B), and iii) NPR-C. The first two are“biological” receptors with an associated cytosolic GC.The NPR-C is a “clearance” receptor, which is alsoassociated with an inhibitory G-protein.
NPR-A and NPR-B have a GC at the cytosolic
region of the receptor which can elevate
intracellular cGMP[204]. For this reason, they
are referred to as the “biological receptors”
since they have a signalling function. There is
growing evidence, however, for NPR-C acting
through an inhibitory Gi protein to decrease
adenylate cyclase (AC) activity and thus, cAMP
production[228,229]. This will be further
discussed. Although ANP preferentially binds to

30
the NPR-A, the ANP fragment known as C-ANP (aa 4-23) has the highest binding affinity for
the NPR-C, suggesting that it is the peptide terminal that dictates receptor affinities[230]. NPR-
A is highly expressed in the adrenal gland, brain, vascular smooth muscle, lung, kidney,
adipose, and heart, while the NPR-B is abundant in chondroctyes, brain, lung, and uterus, while
NPR-C is found in most tissues[204]. NPs are removed from circulation by the NPR-C or by
NEP, a zinc-dependent metalloprotease enzyme[231].
1.8.1 Atrial Natriuretic Peptide (ANP)
ANP is mainly produced and stored in the atria, and is present to a lesser extent in the
ventricles and kidney[232,233]. All NPs are synthesized as preprohormones. Human
preproANP is 151 aa which is cleaved at the N-terminus to yield the 126 aa proANP. The
mature 28 aa ANP is the C-terminal of the proANP, and is only produced when needed by the
Ser protease, corin[204,234]. The primary stimulus for ANP secretion is atrial wall stretch, but
it can also be released in response to increasing Na+, AngII, ET-I, arginine-vasopressin (AVP),
and of interest in the present context, insulin[204,235]. ANP has a t1/2 of 2-5 min in humans and
a physiological plasma concentration of approximately 10 fmol/mL, except during congestive
heart failure (CHF) where it is elevated 10- to 30-fold[236,237]. The effects of ANP in the body
are primarily cardioprotective, including enhanced natriuresis, vascular smooth muscle
relaxation, and decreased cardiac hypertrophy, thirst, and sympathetic tone[204]. Studies
involving the metabolic effects are limited, but it has been shown that ANP induces primate-
specific lipolysis, decreases inflammatory cytokine/adipokine production, and enhances energy
expenditure [238-240]. These actions will be further discussed.
1.8.2 B-type Natriuretic Peptide (BNP)
BNP was initially found in porcine brain but is mainly produced by cardiac ventricles
[241]. Human BNP is produced as a 134 aa preprohormone which is cleaved to the 108 aa pro-

31
BNP and stored in vesicles. When it is needed, an unknown protease produces the biologically
active BNP[204]. The length of BNP is species specific. Human, dog, and pig BNP are 32aa
while rat and mouse BNP are 45 aa [242,243]. BNP, just as ANP, binds to and activates the
NPR-A, although its affinity is believed to be as much as 10-fold less[244]. At the same time,
BNP also has a t1/2 of 20 min, likely due to a lesser affinity for the NPR-C compared with
ANP[245]. The physiological plasma concentration of BNP is approximately 1 fmol/mL though
this is markedly increased by 200- to 300-fold in CHF[204]. The actions of BNP are similar to
those of ANP which include increasing vasorelaxation and natriuresis [204].
1.8.3 C-type Natriuretic Peptide (CNP)
CNP is the most conserved NP and is highly expressed in the brain, chondrocytes, and
endothelial cells[246,247,248]. Unlike the other NPs, CNP is not stored in granules, and its
secretion is stimulated by exposure to TNF-α, IL-1, and sheer stress, but inhibited by
insulin[204]. Human proCNP, which is 103aa in length, is converted to the 22aa CNP through
the actions of the intracellular endopeptidase, furin. CNP has a t1/2 of approximately 20 min and
is normally present in plasma in the low fm/mL range. It is not elevated in CHF because it is not
involved in cardiovascular processes like other NPs, and is instead, essential for long bone
growth[204].

32
1.9 RATIONALE
The purpose of this study is to further elucidate the mechanism by which BK, acting
through NO, enhances insulin sensitivity in rat adipocytes. Preliminary data suggests that the
sGC-cGMP-PKG signalling pathway, a major target for NO, is involved, and to confirm this,
ANP, which increases intracellular cGMP, was selected for the present study. The long term
goals of our research program are to understand the mechanism of insulin signal transduction
and its regulation in normal physiology and disease states. Furthermore, the use of ANP in this
investigation may lead to the identification of novel therapeutic agents in the prevention of
T2DM and CVD.

33
1.10 HYPOTHESES
Figure 1.16. Proposed Mechanism of ANP-mediated EnhancedInsulin Sensitivity. Insulin binds to the IR, resulting in receptor activation throughautophosphorylation. The IR can then activate IRS-1, which can, in turn, stimulate thePI3K signalling pathway, resulting in Akt/PKB activation and eventual GLUT4translocation for glucose uptake. Insulin can also activate the MAPK (JNK,ERK)signalling pathway which can, in addition to other functions, mediate negative feedbackinhibition on insulin signalling. BK has previously been shown to bind to the B2receptor which, in turn, activates eNOS, resulting in NO production. A major target forNO is the sGC-cGMP-PKG signalling pathway. Preliminary data suggests that BK actsthrough this pathway to reduce MAPK activity, resulting in decreased feedbackinhibition of insulin signalling. It is proposed that ANP can mimic BK actions bybinding to the NPR-A, activating the receptor associated GC. This leads to elevatedintracellular cGMP which activates PKG, resulting in the attenuated MAPK activity,and thus, the potentiation of insulin actions in rat adipocytes.
Abbreviations
Akt/PKB=Protein Kinase BANP=Atrial Natriuretic
PeptideB2= B2-ReceptorBK=BradykinincGMP=cyclic GMPeNOS=Nitric Oxide SynthaseGLUT4=Glucose Transporter-4IR=Insulin ReceptorIRS-1=Insulin Receptor
Substrate-1JNK=c-Jun N-Terminal
KinaseNO=Nitric OxideNPR-A=Natriuretic peptide
Receptor-APDK= Phosphoinositide-
Dependent KinasePKG= Protein Kinase GPI3K=Phosphoinositide-3
KinasesGC=soluble Guanylate
Cyclase.
Two hypotheses will be tested in the present study:
1) Atrial natriuretic peptide, acting specifically through the NPR-A to elevate cGMP
and activate protein kinase G in primary rat adipocytes, will reproduce the effect of
bradykinin on enhanced insulin-stimulated 2-deoxy-D-glucose uptake, and Tyr-IRS-1,
Akt/PKB phosphorylation
2) Atrial natriuretic peptide enhances insulin actions, at least in part, by reducing the activity
of the negative regulator of insulin signalling, MAPK (JNK and ERK)

34
CHAPTER 2
MATERIALS & METHODS

35
2.1 Materials
Dulbecco’s modified Eagle’s medium (DMEM) was purchased from Invitrogen
(Burlington, ON), and type I collagenase was from Worthington (Lakewood, NJ). Bio-Rad
protein assay reagent was obtained from Bio-Rad (Hercules, CA), and the LumiGlo
chemiluminescent substrate kit was from KPL (Gaithersburg, MD). The Cyclic GMP EIA and
Glycerol Assay kits were purchased from Cayman Chemical Company (Ann Arbor, MI), and
the Deoxy-D-glucose,2-[3H(G)] was from Perkin Elmer (Boston, MA). Anti-Akt/PKB, anti-
phospho-Akt/PKB (Ser473), anti-ERK1/2, anti-phospho-ERK1/2 (Thr202/Tyr204), anti-JNK,
anti-phospho-JNK (Thr183/Tyr185), anti-MKK4/Sek1, anti-phospho-MKK4/Sek1 (Thr223),
anti-MKK7, anti-phospho-MKK7 (Ser271/Thr275), anti-MEK1/2, and anti-phospho-MEK1/2
(Ser221) antibodies were purchased from Cell Signaling Technology (Beverley, MA), and Anti-
IRS-1, anti-pY, and anti-FLAG-M2 antibodies were purchased from Upstate Chemical (Lake
Placid, NY). Goat anti-rabbit IgG-HRP secondary antibody and protein A/G PLUS-Agarose
beads were purchased from Santa Cruz Biotechnologies (Santa Cruz, CA). A71915 ( Arg6, β-
cyclohexyl-Ala8, Arg17, Cys18-Atrial Natriuretic Peptide(6-18) amide) was obtained from
Bachem Americas, Inc. (Torrance, CA), ODQ (1H-[1,2,4] Oxadiazolo-[4,3-a] quinoxalin-1-one)
from Cayman Chemical Company(Ann Arbor, MI), and Zaprinast (1,4-Dihydro-5-(2-
propoxyphenyl)-7H-1,2,3-triazolo(4,5-d)pyrimidin-7-one) from Calbiochem (San Deigo, CA).
All other chemicals were from Sigma-Aldrich (Oakville, ON).
2.2 Animals
Male Sprague-Dawley rats (150-180 g) were obtained from Charles River (St. Constant,
QC). Animals were given rodent chow and water ad libitum, exposed to 12 h light/dark cycles,
and acclimatized for 1 week prior to all experiments. Animal protocols were approved by the

36
Animal Care Committee of the Toronto General Hospital and all experiments were performed in
accordance with the Canadian Council of Animal Care guidelines.
2.3 Isolation of Rat Adipocytes
Figure 2.1 Procedure for Isolating Rat Adipocytes.
Male Sprague-Dawley rats were sacrificed and their epididymal fat pads were
transferred to 3% bovine serum albumin (BSA)-DMEM. Initial mechanical digestion was the
result of vigorous scissor cutting until the fat pads had been sectioned into fine pieces. Type I
collagenase was then added (1mg/ml) to provide chemical digestion and thus, further isolation
of the cells. The mixture was incubated in a water bath at 37˚C for 1h, shaking at 85 RPM. The
cells were then poured through a 20-micron mesh to remove any excess debris or undigested fat.
The filtrate was washed three times with 3% BSA-KRBH (Kreb’s Ringer Buffer: 118mM NaCl,
5 mM KCl, 1.2 mM MgSO4, 2.5 mM CaCl2, 1.2 mM KH2PO4, 5 mM NaHCO3, 30 mM HEPES,
1 mM pyruvate; pH 7.4), where centrifugation until 500 RPM at room temperature along with
the removal of the liquid phase was performed between washes. After the final wash, the liquid
phase was discarded and the isolated cells were used for experimentation.
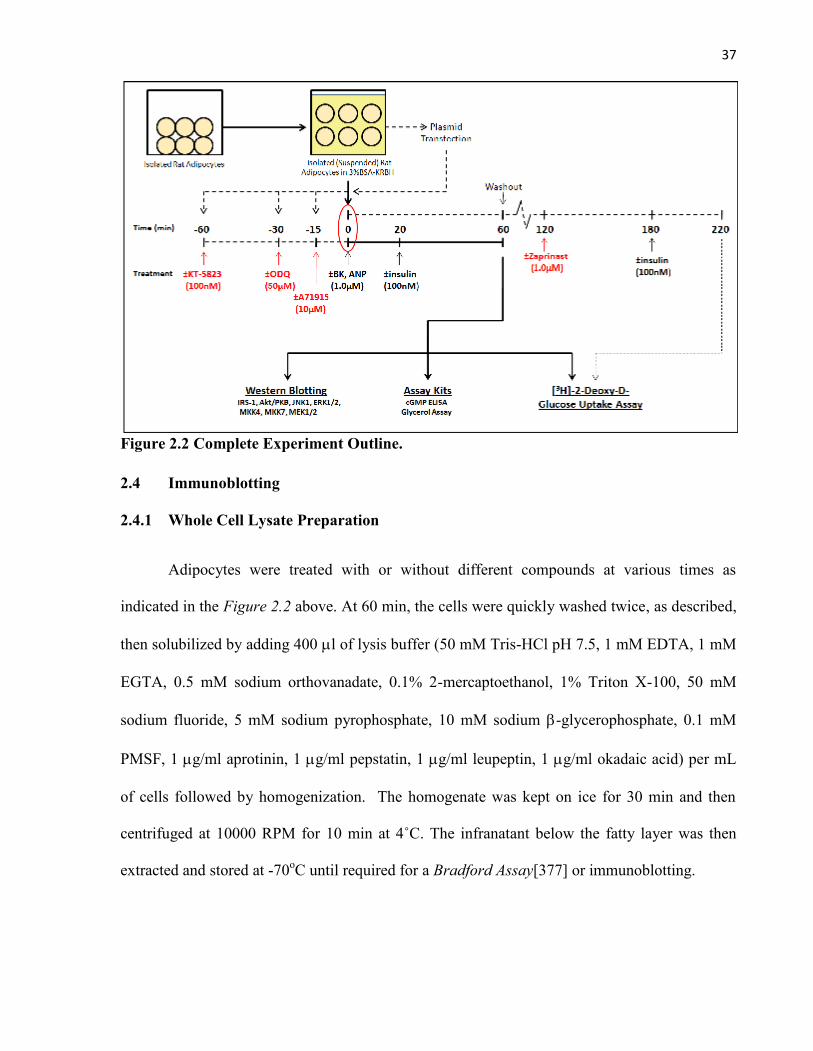
37
Figure 2.2 Complete Experiment Outline.
2.4 Immunoblotting
2.4.1 Whole Cell Lysate Preparation
Adipocytes were treated with or without different compounds at various times as
indicated in the Figure 2.2 above. At 60 min, the cells were quickly washed twice, as described,
then solubilized by adding 400 l of lysis buffer (50 mM Tris-HCl pH 7.5, 1 mM EDTA, 1 mM
EGTA, 0.5 mM sodium orthovanadate, 0.1% 2-mercaptoethanol, 1% Triton X-100, 50 mM
sodium fluoride, 5 mM sodium pyrophosphate, 10 mM sodium -glycerophosphate, 0.1 mM
PMSF, 1 g/ml aprotinin, 1 g/ml pepstatin, 1 g/ml leupeptin, 1 g/ml okadaic acid) per mL
of cells followed by homogenization. The homogenate was kept on ice for 30 min and then
centrifuged at 10000 RPM for 10 min at 4˚C. The infranatant below the fatty layer was then
extracted and stored at -70oC until required for a Bradford Assay[377] or immunoblotting.

38
2.4.2 Cell Transfection and Immunoprecipitation
Figure 2.3 Procedure for Transfecting and Immunoprecipitating Primary Rat Adipocytes
Adipocytes were co-transfected with the dominant negative (DN)-PKG or PC-DNA3
control plasmid and the wild-type(wt)-FLAG-JNK1 plasmid (20µg/mL/ plasmid) via
electroporation using the Bio-Rad Gene Pulser MXcell Electroporation system. The cells were
then washed two times, as described, and incubated for 18 h in 5% BSA-DMEM at 37˚C to
allow for plasmid expression. The adipocytes were then washed in 3%-BSA-KRBH and treated
with or without BK (1.0µM) for 1h with insulin (100nM) during the final 40 min. The technique
described in 2.4.1 Whole Cell Lysate Preparation for obtaining the infranatant was then
followed. The results from the Bradford assay helped determine the volume of cell lysate
required to obtain 1 g protein for each treatment group. The anti-FLAG-M2 antibody was then
used to immunoprecipitate the transfected FLAG-tagged JNK1 as the mixture was rotated for 18
h at 4oC. Each group was then treated with 30µL agarose beads and rotated for 2 h at 4oC. The
treatment groups were then carefully washed four times with ice-cold PBS (phosphate buffered
saline: 80.6mM sodium phosphate, 19.4mM potassium phosphate, 27mM KCl and 1.37M
NaCl), where centrifugation at 1000 RPM for 1 min at 4 oC and the removal of PBS took place
between washes. Immunoblotting was then performed.

39
2.4.3 Immunoblotting Procedure
Laemmli sample buffer with 0.006 % bromophenol blue and 10% 2-mercaptoethanol
was added to the protein equalized infranatant aliquots and boiled at 100˚C for 5 min. Each
western blot gel well was then loaded with 30µg of protein. Following separation by SDS-
PAGE (polyacrylamide gel electrophoresis), proteins were transferred onto nitrocellulose
membranes and blocked with 5% milk-TTBS (Tris-buffered saline with 0.1% Tween) for 1 h at
room temperature. Membranes were incubated overnight with primary antibodies at 4˚C
(1:1000), washed with TTBS, and then incubated with a horseradish peroxidase-conjugated
secondary antibody for 1 h at room temperature (1:5000). Membranes were then washed again
with TTBS, reacted with LumiGlo chemiluminescense, and exposed to film.
Equal amounts of protein were loaded onto separate gels, which were later probed for
phospho- and total proteins, respectively. We found this method to be more reproducible and
accurate in comparison to the stripping of membranes and reprobing for total proteins.
Appropriate controls were used to ensure accurate well loading.
2.5 Assay Kits
2.5.1 Quantification of cGMP Concentration (cGMP ELISA)
Adipocytes were treated with or without ANP (1.0 μM) for 1h, with ±insulin (100nM)
during the final 60 min as indicated in the Figure 2.2. The cells were then lysed, as described,
and the cytosolic cGMP concentration was determined using the EIA kit according to the
instructions of the manufacturer.

40
2.5.2 Quantification of Glycerol Production (Glycerol Assay)
Adipocytes were pretreated with or without A71915 (10 µM) followed by ±ANP (1.0
μM) for 1h as indicated in the Figure 2.2. Equal volumes of cell medium were then extracted
from each treatment group and the extracellular glycerol concentration was determined using
the assay kit according to the instructions of the manufacturer. All measurements were corrected
for the group lipocrits.
2.6 Glucose Uptake
2.6.1 [3H]-2-Deoxy-D-Glucose Uptake Assay
Figure 2.4 Procedure for [3H]-2-Deoxy-D-Glucose Uptake Assay
Adipocytes were treated with or without different compounds at various times as
indicated in the Figure 2.2 above. At 60 min, adipocytes were then exposed to Deoxy-D-
glucose,2-[3H(G)] (0.5mM at 1mCi/mL) for 3min. Ice-cold Phloretin (2’,4’,6’-Trihydroxy-3-p-
hydroxyphenylpropiophenone) (0.5mM) was then added to the cells to impair GLUT functions
and adipocytes were placed on ice[249]. The solution was then transferred to a microcentrifuge
tube containing di-isononyl phthalate and centrifuged at 10000 RPM for 1 min at 4˚C. The di-
isononyl phthalate separated the adipocytes from the liquid of the solution, facilitating the
transfer of only the fat layer to a vial containing 5mL of scintillation fluid. Vials were vortexed
and a scintillation counter was used to measure the [3H]-2-Deoxy-D-Glucose Uptake (2-DG

41
Uptake). The lipocrit was determined for each treatment group, and used as an accepted
correction factor for cell density[250].
2.6.2 Zaprinast Experiment
Adipocytes were treated with or without ANP (1.0 μM) for 1h as indicated in the Figure
2.2. This was followed by washing with 3%BSA-KRBH, as described, and incubation for 1 h at
37˚C. Adipocytes were then treated ±zaprinast (1.0μM) for 1h at 37˚C, followed by ±insulin
treatment (100nM) for 40 min. The remainder of the [3H]-2-Deoxy-D-Glucose Uptake Assay
protocol, as described, was then followed.
2.7 Statistical Analysis
The results are presented as means ± SEM. Two-way ANOVA was used for analysis,
followed by Tukey’s t-test for more than two treatments. Differences were deemed to be
significant at p<0.05.

42
CHAPTER 3
RESULTS

43
3.1 SUMMARY
We have demonstrated in rat adipocytes that BK, via NO signalling, activates the sGC-
cGMP-PKG pathway. This, in turn, leads to decreased MAPK activity and enhanced insulin
signalling. To support this finding, we used ANP, which increases intracellular cGMP, and
found that it, along with BNP, reproduced the effect of BK on enhanced insulin-stimulated
glucose uptake. ANP signalling was also dependent on GC-cGMP-PKG signalling since the use
of A71915, an NPR-A antagonist, and KT-5823, a PKG inhibitor, completely prevented the
ANP-mediated effects, while zaprinast, an agent which prevents cGMP degradation, maintained
ANP actions, 2h after washout. Similar to previous BK studies, ANP signalling through PKG
also significantly increased Tyr-IRS-1 and Akt/PKB phosphorylation while decreasing the JNK
and ERK1/2 phosphorylation in insulin-stimulated groups. Furthermore, the decreased MAPK
phosphorylation in ANP-treated insulin-stimulated groups was not the result of decreased
activation of MKK-4/7 or MEK1/2, the respective upstream MAPKKs for JNK and ERK1/2.

44
3.2 RESULTS
3.2.1 Natriuretic Peptides Enhance Insulin-Stimulated Glucose Uptake in Rat Adipocytes
We have previously shown that BK treated rat adipocytes potentiate insulin signalling
through the sGC-cGMP-PKG pathway [Data to be published]. To confirm this finding in the
present study, we selected ANP to elevate intracellular cGMP, and used the 2-Deoxy-D-glucose
(2-DG) uptake assay to assess the metabolic outcome of insulin signalling. Isolated rat
adipocytes were treated with ANP (0.01-1.0 M), BK (1.0µM), or vehicle for 1h, with ±insulin
(100nM) during the final 40 min. ANP and BK treatment had no effect on basal 2-DG uptake.
For insulin-stimulated groups, ANP produced a dose-dependent enhancement of 2-DG uptake
over the insulin control. This was significant at 0.1µM, and mimicked the effects of BK at the
same dose of 1.0µM (Relative arbitrary units (AU): control basal=0.16 0.05, insulin=1.0
0.00; 0.01µM ANP basal=0.21 0.06, insulin=1.10 0.14; 0.1µM ANP basal=0.12 0.04,
insulin=1.27 0.10; 1.0 µM ANP basal=0.19 0.04, insulin=1.56 0.09; 1.0 µM BK
basal=0.15 0.03, insulin=1.55 0.16 [Figure 3.1A]. Considering that BNP can also elevate
intracellular cGMP and has signalling outcomes in common with ANP, we examined whether it
could also replicate the effect of BK in rat adipocytes[251]. Isolated adipocyes were treated with
1.0µM ANP, BK, BNP, or vehicle for 1 h, with ±insulin(100nM) during the final 40 min. BNP
did not alter basal 2-DG uptake, but did significantly enhance insulin-stimulated 2-DG uptake,
mimicking the results for both ANP and BK (Relative AU: control basal=0.070.04,
insulin=1.000.0; BNP basal=0.13 0.04, insulin=1.400.15; ANP basal=0.100.03,
insulin=1.430.20; BK basal=0.140.06, insulin=1.410.04)
[Figure 3.1B].
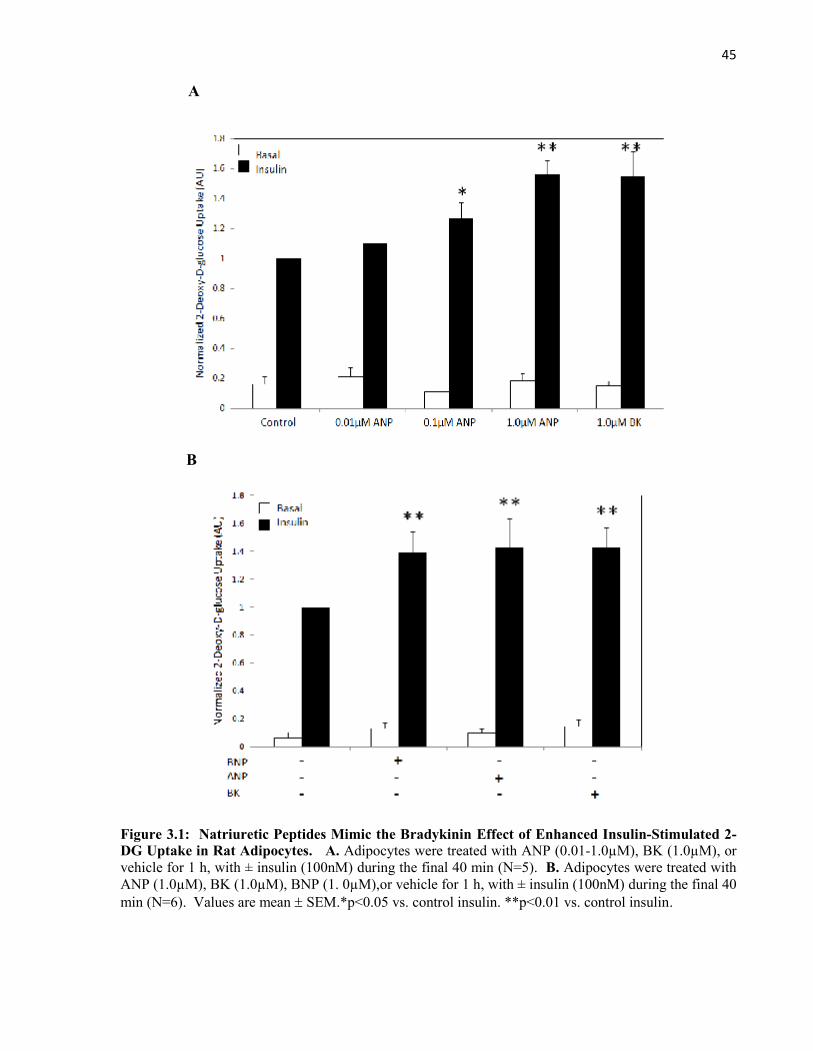
45
A
B
Figure 3.1: Natriuretic Peptides Mimic the Bradykinin Effect of Enhanced Insulin-Stimulated 2-DG Uptake in Rat Adipocytes. A. Adipocytes were treated with ANP (0.01-1.0µM), BK (1.0µM), orvehicle for 1 h, with ± insulin (100nM) during the final 40 min (N=5). B. Adipocytes were treated withANP (1.0µM), BK (1.0µM), BNP (1. 0µM),or vehicle for 1 h, with ± insulin (100nM) during the final 40min (N=6). Values are mean SEM.*p<0.05 vs. control insulin. **p<0.01 vs. control insulin.

46
3.2.2 ANP Uses GC-cGMP-PKG Signalling to Enhance Insulin-Stimulated Glucose Uptake
3.2.2.1 ANP Requires the NPR-A to Enhance Insulin-Stimulated Glucose Uptake
NPs can interact with two types of receptors in rat adipocytes: i) biological (NPR-A),
and ii) clearance (NPR-C), the latter which has been shown to also have a role in signalling[252].
To determine which receptor type is required for the ANP effects on insulin-stimulated 2-DG
uptake, we used A71915, a pharmacological competitive antagonist for NPR-A. Isolated adipocytes
were pre-treated with ±A71915 (10μM) for 15 min, followed by ANP (1.0µM) or vehicle for 1h,
with ±insulin (100nM) during the final 40 min. ANP treatment significantly improved insulin-
stimulated 2-DG uptake over the insulin control, but A71915 completely blocked these effects.
A71915 did not alter basal or vehicle-treated insulin-stimulated 2-DG uptake (Relative AU:
control basal= 0.06±0.02, insulin=1.0±0.0; ANP basal=0.05±0.02, insulin= 1.53±0.11; A71915
basal=0.06±0.02, insulin=0.93±0.06; ANP+A71915 basal=0.07±0.02; insulin=0.96±0.15)
[Figure 3.2A]. The NPR-A receptor has a functional GC in its cytosolic region which can
synthesize cGMP in the cell[204]. BK acts through sGC to produce cGMP which is responsible
for its effects on insulin-stimulated 2-DG uptake. Furthermore, NPR-C signalling has been
noted to activate eNOS in VSMCs, leading to NO generation and sGC activation[253,378]. To
further confirm that the ANP effects were strictly dependent on the NPR-A and its associated
GC, we used ODQ, a specific sGC inhibitor, which our lab previously used to remove the
effects of BK signalling [Data to be published]. Isolated adipocytes were pre-treated with
±ODQ (50µM) for 30 min, followed by ANP (1.0µM) or vehicle for 1h, with ±insulin (100nM)
during the final 40 min. ANP treatment significantly improved insulin-stimulated 2-DG uptake
over the insulin control, and this was not affected by ODQ treatment. ODQ did not alter basal or
vehicle-treated insulin-stimulated 2-DG uptake (Relative AU: control basal=0.23±0.05, insulin=
1.0±0.0; ANP basal=0.34±0.07,insulin=1.50±0.13; ODQ basal=0.24±0.06, insulin=0.93±0.09;
ANP+ODQ basal=0.33±0.06; insulin=1.42±0.17) [Figure 3.2B].
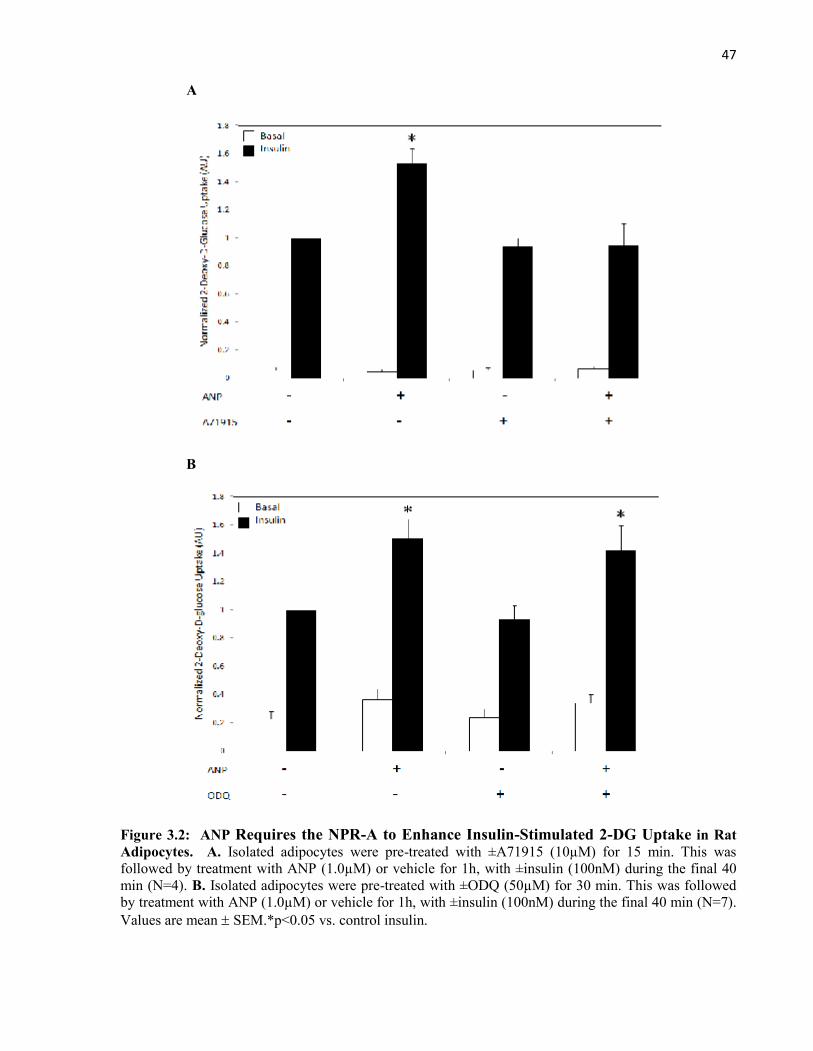
47
A
B
Figure 3.2: ANP Requires the NPR-A to Enhance Insulin-Stimulated 2-DG Uptake in RatAdipocytes. A. Isolated adipocytes were pre-treated with ±A71915 (10µM) for 15 min. This wasfollowed by treatment with ANP (1.0µM) or vehicle for 1h, with ±insulin (100nM) during the final 40min (N=4). B. Isolated adipocytes were pre-treated with ±ODQ (50µM) for 30 min. This was followedby treatment with ANP (1.0µM) or vehicle for 1h, with ±insulin (100nM) during the final 40 min (N=7).Values are mean SEM.*p<0.05 vs. control insulin.

48
3.2.2.2 ANP Requires cGMP to Enhance Insulin-Stimulated Glucose Uptake
Most of the reported ANP signalling effects require cGMP as the second messenger. To
confirm that exogenous ANP leads to the production of cGMP in adipocytes, a cGMP-specific
ELISA was performed. Isolated adipocytes were treated with ANP (1.0µM) or vehicle for 1 h,
with ±insulin (100nM) during the final 40 min. ANP treatment caused a significant increase in
the intracellular levels of cGMP in both basal and insulin-stimulated groups (Relative AU:
control basal=1.0±0.0, insulin=1.62±0.56, ANP=42.48±6.12, ANP+insulin=41.10±10.59)
[Figure 3.3A]. To examine the role of cGMP in ANP signalling, a series of experiments were
performed. First, it was important to determine the duration of ANP’s effects following a
washout. Isolated adipocytes were treated with ANP (1.0µM) or vehicle for 1h, washed out,
and incubated for 0, 1, or 2 h before treating with ±insulin for 40 min and performing a 2-DG
uptake assay. ANP-treated groups which were stimulated with insulin directly after washout
demonstrated a significant increase in insulin-stimulated 2-DG uptake over the insulin control,
but these effects were lost in cells incubated for 2 h prior to insulin treatment (Relative AU:
t=0: control basal=0.13±0.05, insulin=1.0±0.0; ANP basal=0.11±0.05, insulin=1.47±0.06. t=1:
control basal=0.16±0.05, insulin=1.0±0.0; ANP basal=0.20±0.04, insulin=1.26±0.09. t=2:
control basal=0.27±0.10, insulin=1.0±0.0; ANP basal=0.27±0.05, insulin=0.91±0.09) [Figure
3.3B]. Since PDE5 plays a major role in the degradation of cGMP in adipocytes, zaprinast, a
relatively specific PDE5 inhibitor, could then be used during the 2h following washout to
determine the role of cGMP in ANP-treated insulin stimulated 2-DG uptake[253,209].
Adipocytes were treated with ANP (1.0µM) or vehicle for 1h, washed out, and incubated for 2h,
with ±zaprinast (1.0µM) during the final hour. Cells were then treated with ±insulin for 40 min
and a 2-DG uptake assay was then performed. It was determined that the ANP effects on
insulin-stimulated glucose uptake were only significantly maintained in the presence of
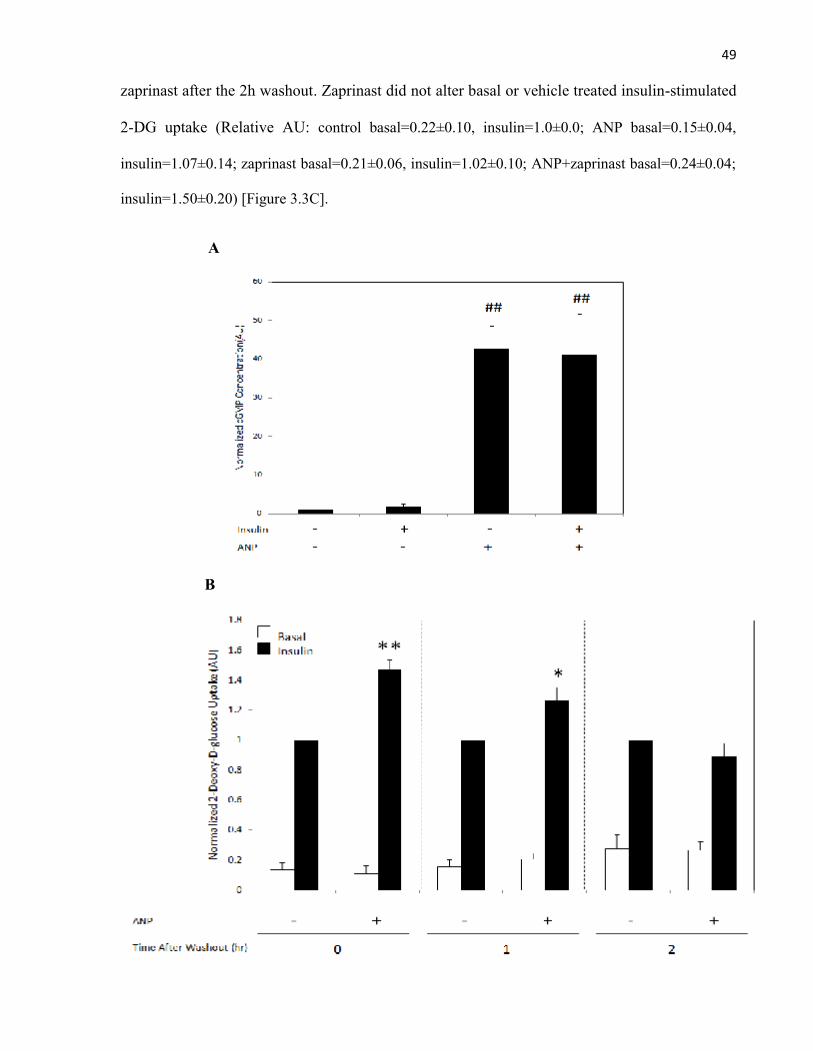
49
zaprinast after the 2h washout. Zaprinast did not alter basal or vehicle treated insulin-stimulated
2-DG uptake (Relative AU: control basal=0.22±0.10, insulin=1.0±0.0; ANP basal=0.15±0.04,
insulin=1.07±0.14; zaprinast basal=0.21±0.06, insulin=1.02±0.10; ANP+zaprinast basal=0.24±0.04;
insulin=1.50±0.20) [Figure 3.3C].
A
B
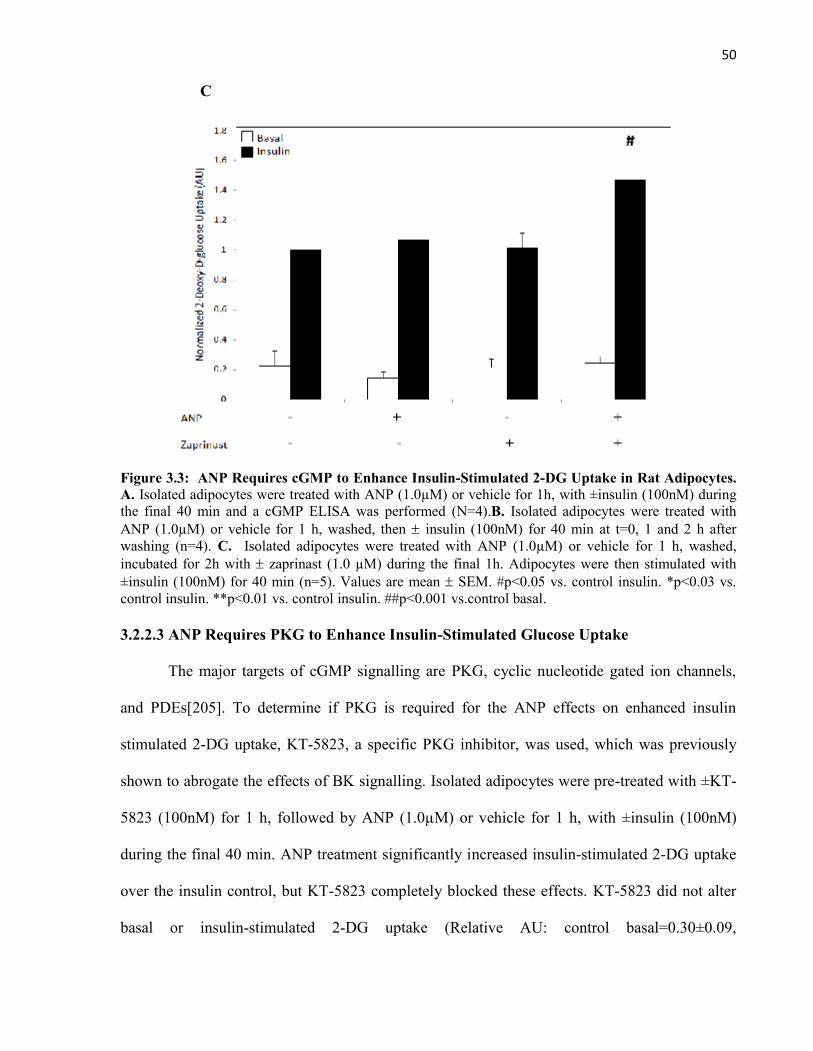
50
C
Figure 3.3: ANP Requires cGMP to Enhance Insulin-Stimulated 2-DG Uptake in Rat Adipocytes.A. Isolated adipocytes were treated with ANP (1.0µM) or vehicle for 1h, with ±insulin (100nM) duringthe final 40 min and a cGMP ELISA was performed (N=4).B. Isolated adipocytes were treated withANP (1.0µM) or vehicle for 1 h, washed, then insulin (100nM) for 40 min at t=0, 1 and 2 h afterwashing (n=4). C. Isolated adipocytes were treated with ANP (1.0µM) or vehicle for 1 h, washed,incubated for 2h with zaprinast (1.0 µM) during the final 1h. Adipocytes were then stimulated with±insulin (100nM) for 40 min (n=5). Values are mean SEM. #p<0.05 vs. control insulin. *p<0.03 vs.control insulin. **p<0.01 vs. control insulin. ##p<0.001 vs.control basal.
3.2.2.3 ANP Requires PKG to Enhance Insulin-Stimulated Glucose Uptake
The major targets of cGMP signalling are PKG, cyclic nucleotide gated ion channels,
and PDEs[205]. To determine if PKG is required for the ANP effects on enhanced insulin
stimulated 2-DG uptake, KT-5823, a specific PKG inhibitor, was used, which was previously
shown to abrogate the effects of BK signalling. Isolated adipocytes were pre-treated with ±KT-
5823 (100nM) for 1 h, followed by ANP (1.0µM) or vehicle for 1 h, with ±insulin (100nM)
during the final 40 min. ANP treatment significantly increased insulin-stimulated 2-DG uptake
over the insulin control, but KT-5823 completely blocked these effects. KT-5823 did not alter
basal or insulin-stimulated 2-DG uptake (Relative AU: control basal=0.30±0.09,
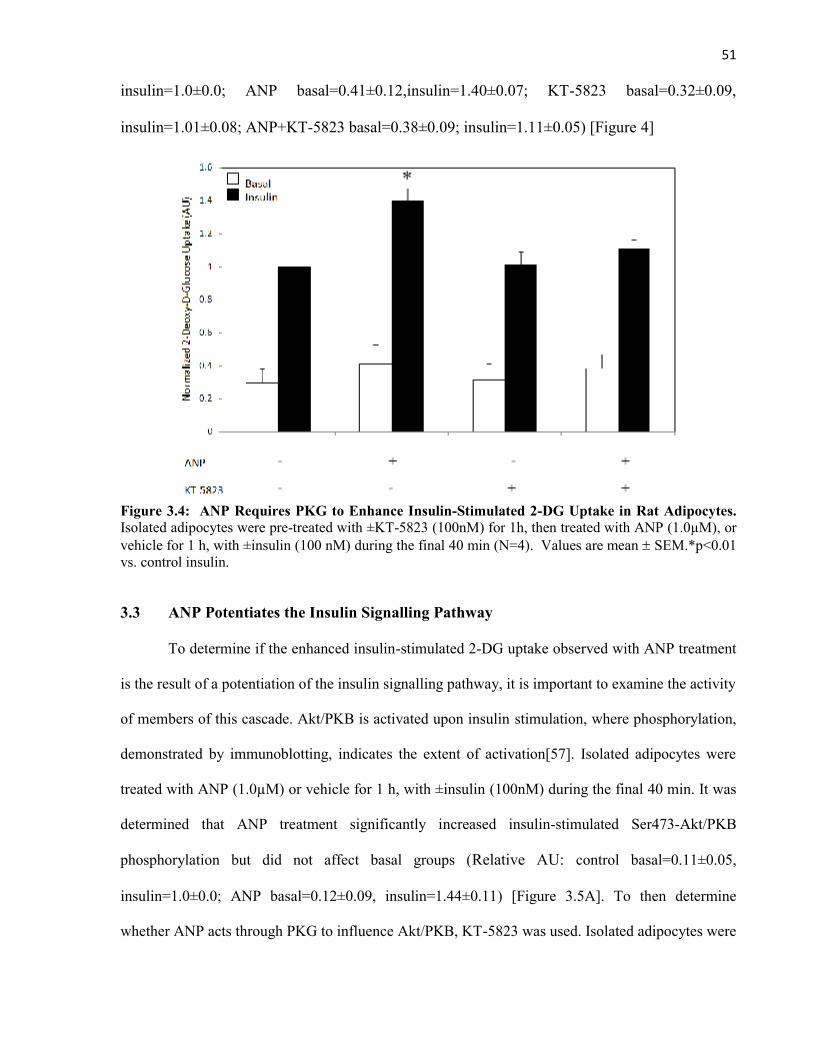
51
insulin=1.0±0.0; ANP basal=0.41±0.12,insulin=1.40±0.07; KT-5823 basal=0.32±0.09,
insulin=1.01±0.08; ANP+KT-5823 basal=0.38±0.09; insulin=1.11±0.05) [Figure 4]
Figure 3.4: ANP Requires PKG to Enhance Insulin-Stimulated 2-DG Uptake in Rat Adipocytes.Isolated adipocytes were pre-treated with ±KT-5823 (100nM) for 1h, then treated with ANP (1.0µM), orvehicle for 1 h, with ±insulin (100 nM) during the final 40 min (N=4). Values are mean SEM.*p<0.01vs. control insulin.
3.3 ANP Potentiates the Insulin Signalling Pathway
To determine if the enhanced insulin-stimulated 2-DG uptake observed with ANP treatment
is the result of a potentiation of the insulin signalling pathway, it is important to examine the activity
of members of this cascade. Akt/PKB is activated upon insulin stimulation, where phosphorylation,
demonstrated by immunoblotting, indicates the extent of activation[57]. Isolated adipocytes were
treated with ANP (1.0µM) or vehicle for 1 h, with ±insulin (100nM) during the final 40 min. It was
determined that ANP treatment significantly increased insulin-stimulated Ser473-Akt/PKB
phosphorylation but did not affect basal groups (Relative AU: control basal=0.11±0.05,
insulin=1.0±0.0; ANP basal=0.12±0.09, insulin=1.44±0.11) [Figure 3.5A]. To then determine
whether ANP acts through PKG to influence Akt/PKB, KT-5823 was used. Isolated adipocytes were
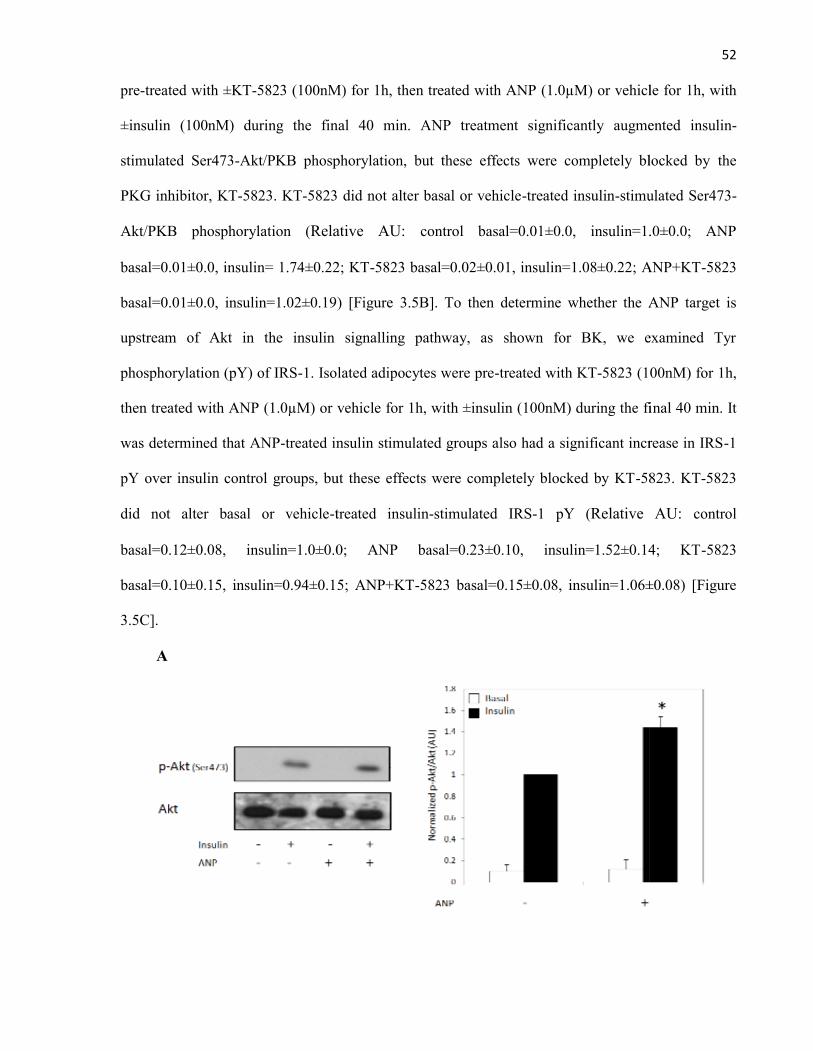
52
pre-treated with ±KT-5823 (100nM) for 1h, then treated with ANP (1.0µM) or vehicle for 1h, with
±insulin (100nM) during the final 40 min. ANP treatment significantly augmented insulin-
stimulated Ser473-Akt/PKB phosphorylation, but these effects were completely blocked by the
PKG inhibitor, KT-5823. KT-5823 did not alter basal or vehicle-treated insulin-stimulated Ser473-
Akt/PKB phosphorylation (Relative AU: control basal=0.01±0.0, insulin=1.0±0.0; ANP
basal=0.01±0.0, insulin= 1.74±0.22; KT-5823 basal=0.02±0.01, insulin=1.08±0.22; ANP+KT-5823
basal=0.01±0.0, insulin=1.02±0.19) [Figure 3.5B]. To then determine whether the ANP target is
upstream of Akt in the insulin signalling pathway, as shown for BK, we examined Tyr
phosphorylation (pY) of IRS-1. Isolated adipocytes were pre-treated with KT-5823 (100nM) for 1h,
then treated with ANP (1.0µM) or vehicle for 1h, with ±insulin (100nM) during the final 40 min. It
was determined that ANP-treated insulin stimulated groups also had a significant increase in IRS-1
pY over insulin control groups, but these effects were completely blocked by KT-5823. KT-5823
did not alter basal or vehicle-treated insulin-stimulated IRS-1 pY (Relative AU: control
basal=0.12±0.08, insulin=1.0±0.0; ANP basal=0.23±0.10, insulin=1.52±0.14; KT-5823
basal=0.10±0.15, insulin=0.94±0.15; ANP+KT-5823 basal=0.15±0.08, insulin=1.06±0.08) [Figure
3.5C].
A
52
pre-treated with ±KT-5823 (100nM) for 1h, then treated with ANP (1.0µM) or vehicle for 1h, with
±insulin (100nM) during the final 40 min. ANP treatment significantly augmented insulin-
stimulated Ser473-Akt/PKB phosphorylation, but these effects were completely blocked by the
PKG inhibitor, KT-5823. KT-5823 did not alter basal or vehicle-treated insulin-stimulated Ser473-
Akt/PKB phosphorylation (Relative AU: control basal=0.01±0.0, insulin=1.0±0.0; ANP
basal=0.01±0.0, insulin= 1.74±0.22; KT-5823 basal=0.02±0.01, insulin=1.08±0.22; ANP+KT-5823
basal=0.01±0.0, insulin=1.02±0.19) [Figure 3.5B]. To then determine whether the ANP target is
upstream of Akt in the insulin signalling pathway, as shown for BK, we examined Tyr
phosphorylation (pY) of IRS-1. Isolated adipocytes were pre-treated with KT-5823 (100nM) for 1h,
then treated with ANP (1.0µM) or vehicle for 1h, with ±insulin (100nM) during the final 40 min. It
was determined that ANP-treated insulin stimulated groups also had a significant increase in IRS-1
pY over insulin control groups, but these effects were completely blocked by KT-5823. KT-5823
did not alter basal or vehicle-treated insulin-stimulated IRS-1 pY (Relative AU: control
basal=0.12±0.08, insulin=1.0±0.0; ANP basal=0.23±0.10, insulin=1.52±0.14; KT-5823
basal=0.10±0.15, insulin=0.94±0.15; ANP+KT-5823 basal=0.15±0.08, insulin=1.06±0.08) [Figure
3.5C].
A
52
pre-treated with ±KT-5823 (100nM) for 1h, then treated with ANP (1.0µM) or vehicle for 1h, with
±insulin (100nM) during the final 40 min. ANP treatment significantly augmented insulin-
stimulated Ser473-Akt/PKB phosphorylation, but these effects were completely blocked by the
PKG inhibitor, KT-5823. KT-5823 did not alter basal or vehicle-treated insulin-stimulated Ser473-
Akt/PKB phosphorylation (Relative AU: control basal=0.01±0.0, insulin=1.0±0.0; ANP
basal=0.01±0.0, insulin= 1.74±0.22; KT-5823 basal=0.02±0.01, insulin=1.08±0.22; ANP+KT-5823
basal=0.01±0.0, insulin=1.02±0.19) [Figure 3.5B]. To then determine whether the ANP target is
upstream of Akt in the insulin signalling pathway, as shown for BK, we examined Tyr
phosphorylation (pY) of IRS-1. Isolated adipocytes were pre-treated with KT-5823 (100nM) for 1h,
then treated with ANP (1.0µM) or vehicle for 1h, with ±insulin (100nM) during the final 40 min. It
was determined that ANP-treated insulin stimulated groups also had a significant increase in IRS-1
pY over insulin control groups, but these effects were completely blocked by KT-5823. KT-5823
did not alter basal or vehicle-treated insulin-stimulated IRS-1 pY (Relative AU: control
basal=0.12±0.08, insulin=1.0±0.0; ANP basal=0.23±0.10, insulin=1.52±0.14; KT-5823
basal=0.10±0.15, insulin=0.94±0.15; ANP+KT-5823 basal=0.15±0.08, insulin=1.06±0.08) [Figure
3.5C].
A
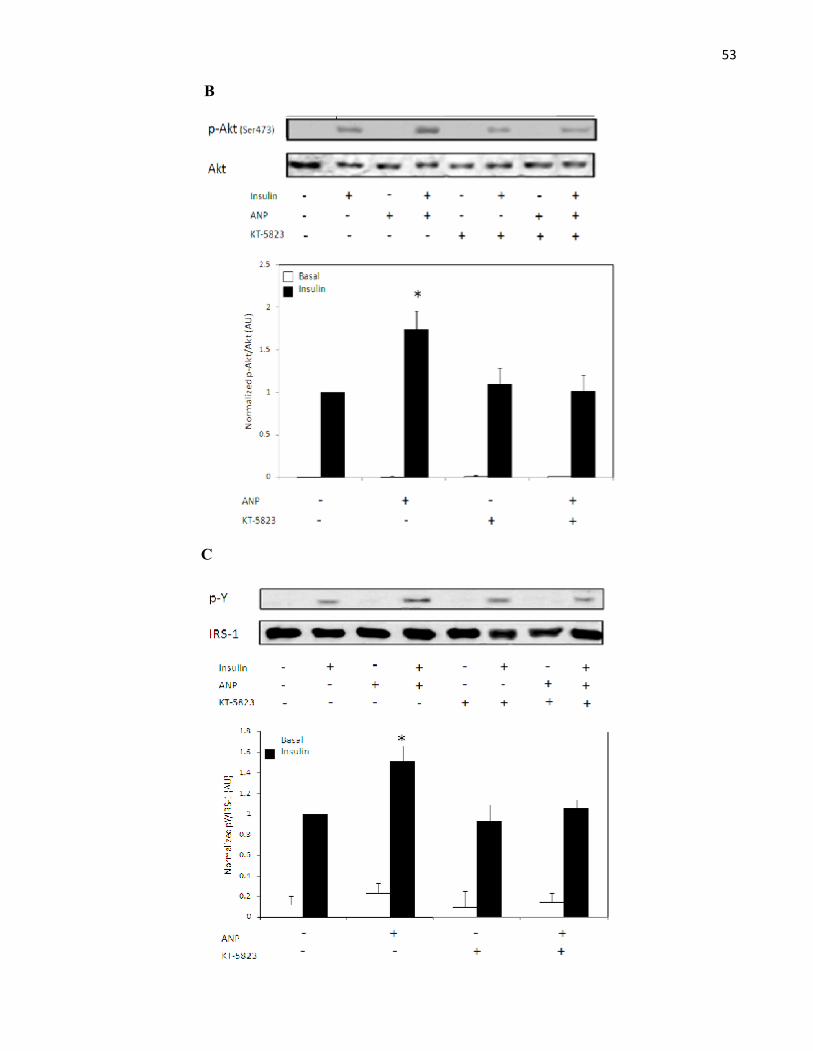
53
B
C
53
B
C
53
B
C

54
Figure 3.5: ANP Potentiates the Insulin Signalling Pathway via PKG in Rat Adipocytes. A.Isolated adipocytes were treated with ANP (1.0µM) or vehicle for 1 h, with ±insulin (100nM) during thefinal 40 min (N=3). B. & C. Adipocytes were pre-treated with ±KT-5823 (100nM) for 1h, followed byANP (1.0µM) or vehicle for 1 h, with ±insulin (100nM) during the final 40 min (N=4). Representativeimmunoblots of Akt/PKB, pSer473- Akt/PKB, IRS-1, and pY-IRS-1. The band intensities fromphospho- immunoblots were corrected for those of total protein. Values are mean SEM.*p<0.03 vs.control insulin.
3.4 ANP Decreases Insulin-Stimulated MAPK Phosphorylation
After 40 minutes of insulin stimulation, a negative feedback loop, mediated by members
of the MAPK family, is activated to turn off insulin signalling[223]. Furthermore, we previously
demonstrated that the BK effect on enhanced insulin signalling is through reduced MAPK
activity, principally, JNK. To determine if ANP mimics the mechanism of BK action, it was
essential to examine the phosphorylation status of MAPKs following treatment. Adipocytes
were treated with ANP (1.0µM) or vehicle for 1h, with ±insulin (100nM) during the final 40
min. ANP treatment led to a significant decrease in the phosphorylation state of both JNK and
ERK1/2 in insulin-stimulated adipocytes (Relative AU: JNK: control basal=0.64±0.16,
insulin=1.0±0.0; ANP basal=0.60±0.07, insulin=0.72±0.03. ERK1/2: control basal=0.64±0.10,
insulin=1.0±0.0; ANP basal=0.71±0.07, insulin=0.50±0.07) [Figure 3.6 A&B]. To then
determine if ANP acts through PKG to decrease the MAPK phosphorylation, KT-5823 was
used. Adipocytes were pre-treated with ±KT-5823 (100nM) for 1h, then treated with ANP
(1.0µM) or vehicle for 1h, with ±insulin (100nM) during the final 40 min. ANP treatment led to
a significant decrease in the phosphorylation state of both JNK and ERK1/2 in insulin-
stimulated adipocytes, but these effects were completely blocked with KT-5823. KT-5823 did
not alter basal or vehicle-treated insulin-stimulated MAPK phosphorylation (Relative AU: JNK:
control basal=0.61±0.18, insulin=1.0±0.0; ANP basal=0.39±0.02, insulin=0.46±0.05; KT-5823
basal=0.41±0.18, insulin=0.92±0.07; ANP+KT-5823 basal=0.54±0.06, insulin=1.01±0.13.
ERK1/2: control basal=0.61±0.18, insulin=1.0±0.0; ANP basal=0.72±0.10, insulin=0.69±0.09;
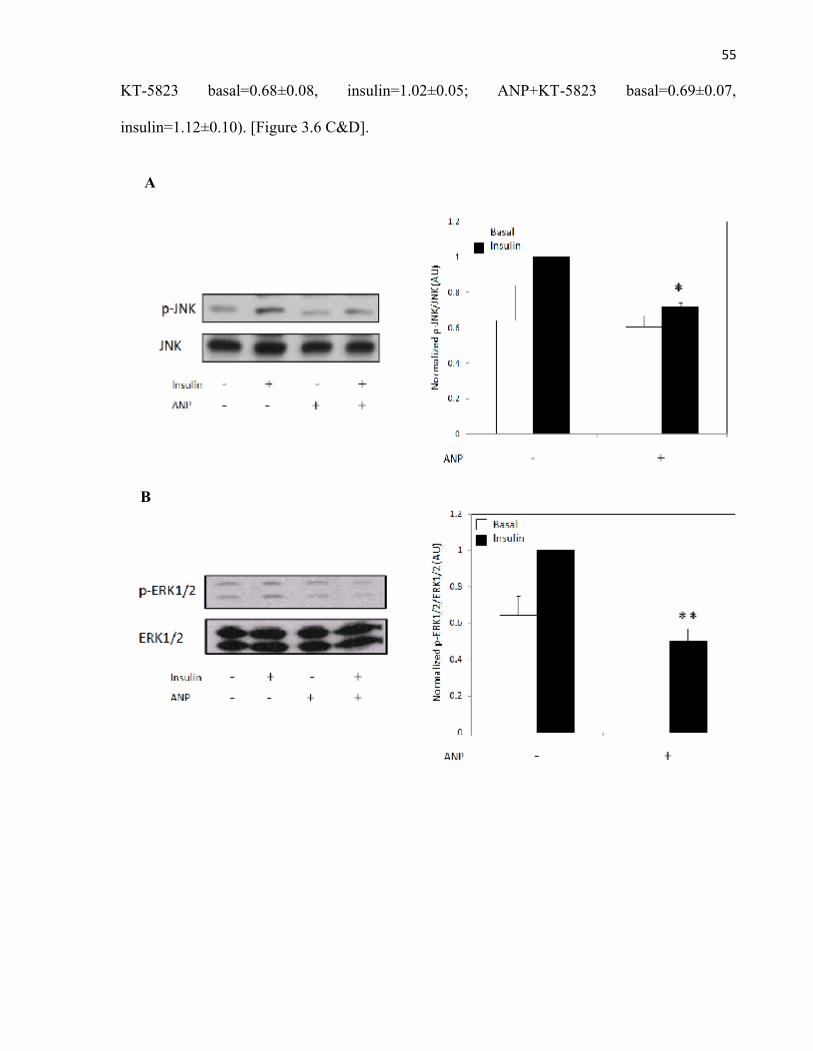
55
KT-5823 basal=0.68±0.08, insulin=1.02±0.05; ANP+KT-5823 basal=0.69±0.07,
insulin=1.12±0.10). [Figure 3.6 C&D].
A
B
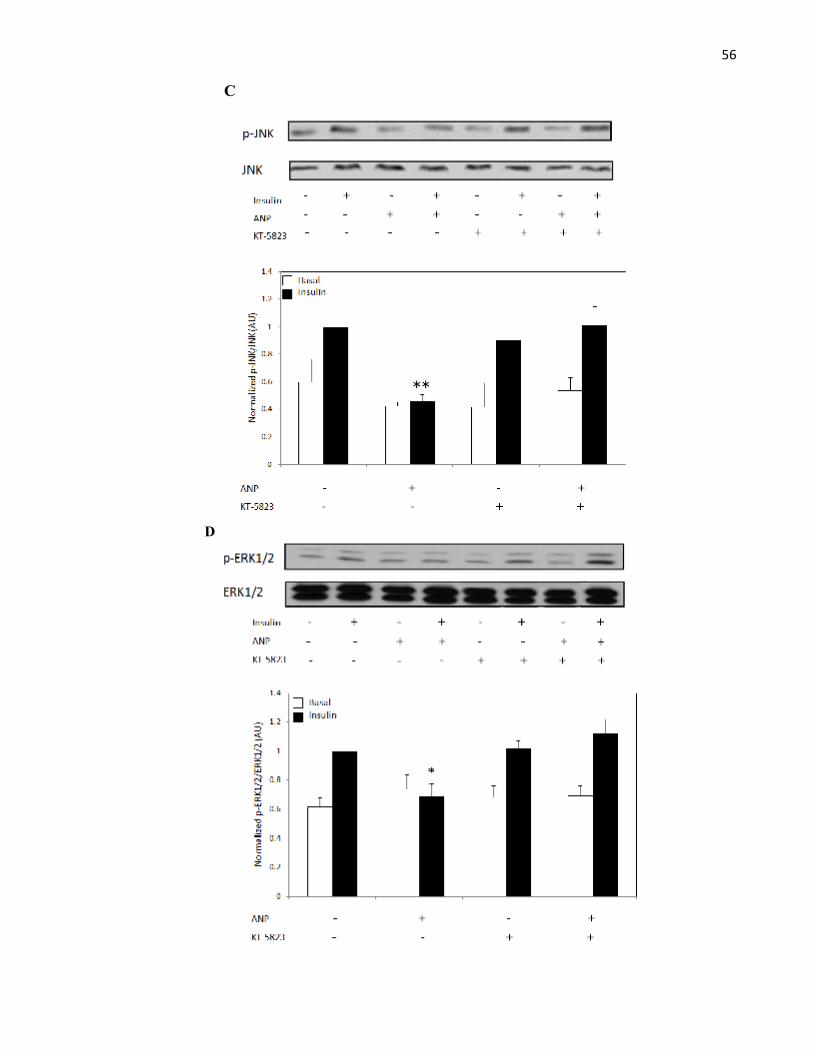
56
C
D

57
Figure 3.6: ANP Reduces MAPK Phosphorylation via PKG in Insulin-Stimulated Rat Adipocytes.A. & B. Isolated adipocytes were treated with ANP (1.0µM) or vehicle for 1 h, with ±insulin (100nM)during the final 40 min (N=4). C. & D. Isolated adipocytes were pre-treated with KT-5823 (100nM) for1h, followed by ANP (1.0µM) or vehicle for 1 h, with ±insulin (100nM) during the final 40 min (N=3-4). Representative immunoblots of JNK, p-JNK, ERK1/2, p-ERK1/2. The band intensities fromphospho- immunoblots were corrected for those of total protein. Values are mean SEM.*p<0.05 vs.control insulin. **p<0.01 vs. control insulin.
3.5 ANP Reduces MAPK Phosphorylation Independent of Upstream MAPK Kinases
MAPKs are phosphorylated by upstream MAPKKs[86]. MKK-4/7 and MEK1/2 are the
respective MAPKKs for JNK and ERK1/2[95,101]. To determine if the decreased
phosphorylation state of MAPKs with ANP treatment in insulin-stimulated cells is the result of
decreased upstream MAPKK activity, their activity level was assessed through immunoblotting.
Isolated adipocytes were treated with ANP (1.0µM) or vehicle for 1 h, with ±insulin (100nM)
during the final 40 min. It was determined that ANP did not influence MKK-4 or MEK1/2
phosphorylation in basal or insulin-stimulated groups. (Relative AU: MKK-4: control
basal=0.19±0.04, insulin=1.0±0.0; ANP basal=0.16±0.08, insulin=1.04±0.07. MEK1/2: control
basal=0.63±0.09, insulin=1.0±0.0; ANP basal=0.73±0.04, insulin=1.08±0.10) [Figure 3.7
A&B]. Although MKK7 protein was detected by immunoblotting, no phosphorylation was
detected for any of the treatment groups [data not shown].
A
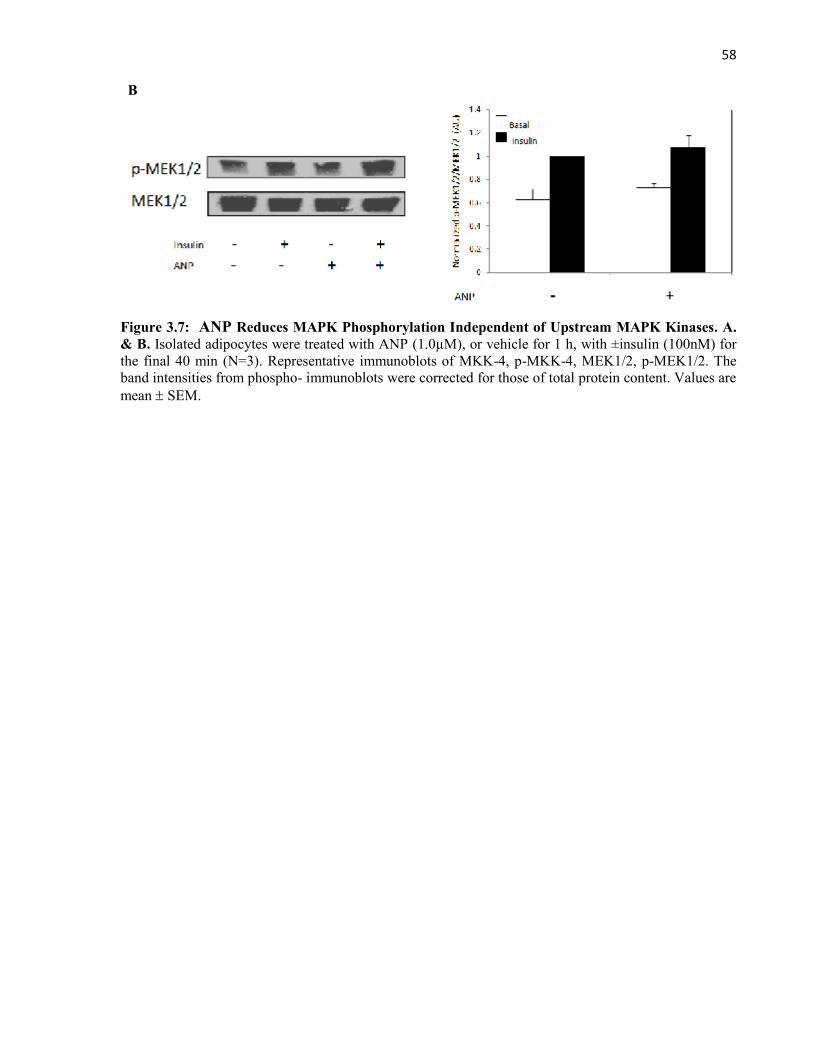
58
B
Figure 3.7: ANP Reduces MAPK Phosphorylation Independent of Upstream MAPK Kinases. A.& B. Isolated adipocytes were treated with ANP (1.0µM), or vehicle for 1 h, with ±insulin (100nM) forthe final 40 min (N=3). Representative immunoblots of MKK-4, p-MKK-4, MEK1/2, p-MEK1/2. Theband intensities from phospho- immunoblots were corrected for those of total protein content. Values aremean SEM.

59
CHAPTER 4
DISCUSSION

60
4.1 INTRODUCTION
BK is essential for the enhanced insulin sensitivity observed with ACEi treatment. This
has been strongly supported by studies demonstrating that kininogen-deficient rats are
unaffected by ACEi treatment and display insulin resistance[178], the insulin-sensitizing effect
of ACEi is lost when pretreating with the B2-receptor antagonist, HOE-140[179], and ACEi and
BK infusion into various insulin resistant rodent models improves glucose tolerance[180].
Relevant to this is that BK directly potentiates insulin signalling. Studies have shown that BK
can delay the dephosphorylation of the IR while enhancing PI3K activation and GLUT4
translocation in L6 muscle and 32D cells[184]. These studies were supported in dog SKM and
adipocytes, where BK treatment was accompanied by increased insulin-stimulated Tyr-IRS-1
and Akt/PKB phosphorylation[185]. We also demonstrated that BK enhances insulin signalling
in isolated rat adipocytes and this effect was dependent on eNOS activation and NO
signalling[186]. This latter finding has been confirmed by others[184,185], but the mechanism
downstream of NO remains unclear. A major intracellular target for NO is sGC, which has a
heme-binding domain relatively specific for NO[184, 199]. sGC activation leads to the
generation of the second messenger, cGMP, which can, in turn, activate PKG[195]. Despite the
prevalence of NO-cGMP signalling, its potential role in adipocyte insulin sensitivity has not
been effectively addressed. In a previous study, we demonstrated for the first time, the role of
the sGC-cGMP-PKG signalling pathway in adipocytes treated with BK following insulin
stimulation [Data to be published]. In an attempt to further support these observations, we
reasoned that if BK depended on cGMP signalling, a cGMP elevating agent should be able to
reproduce the actions of BK in our subject. One promising candidate for the present study was
ANP, a peptide which can bind to its biological NPR-A in adipocytes to elevate intracellular
cGMP[204]. Furthermore, kinins and NPs have many common signalling outcomes in the body.

61
Both BK and ANP, for example, signal through cGMP to induce vasodilation, diuresis,
natriuresis, and to provide protection from ischemia reperfusion injury[204,273,274,275]. For
these given reasons, it would be logical to suspect that ANP can also enhance insulin sensitivity
in isolated rat adipocytes.
4.2 Natriuretic Peptides Enhance Insulin-Stimulated Glucose Uptake in Rat Adipocytes
The literature pertaining to the metabolic effects of ANP is limited. In the present study,
we have demonstrated for the first time that ANP enhances insulin-stimulated 2-DG uptake in
primary rat adipocytes in a dose-dependent manner which mimics the extent of BK actions at
the same dose (1.0µM). This latter finding is particularly interesting, given that kinins and NPs
signal through distinct receptors with varying tissue distributions and densities[164,204]. Also,
while rat adipocytes have both the B2 and NPR-A receptors, they also have NPR-Cs which can
remove NPs from circulation[204,276]. Although it may be suggested that the cells have been
supersaturated with each peptide at 1.0 µM, it must be noted that in our previous studies, BK-
treated insulin-stimulated cells showed a similar dose-dependent response pattern[186]. These
results collectively suggest that either the i) BK is more readily degraded, given that there are
many more enzymes which can target it [164,167] or that ii) NPR-A signalling may have a
greater efficacy than B2 receptor signalling. This latter explanation may be supported by the
finding that ANP, but not the NO donor, SNP, induces the partial translocation of PKG to the
NPR-A where it acts as an NPR kinase to initiate a positive feed forward loop of activation and
thus, enhanced ANP signalling[277]. Although doses exceeding 1.0µM were not examined, they
would be of interest in order to determine if the ANP effects could further enhanced.

62
Although there is no reported evidence of ANP-mediated enhanced insulin signalling in
adipocytes, there may be indirect support. BK and NPs are predominantly degraded by ACE
and NEP, respectively[164,231]. Interestingly, the use of dual NEP/ACE inhibitors, such as
ompatrilat or mixanpril, produces significant improvements in myocardium, adipose, and SKM
insulin sensitivity which greatly exceeded that produced by ACEi treatment alone[278-280].
Although NEP can also degrade BK[164] it normally does not play a significant role[369-371],
and there are several other BK degrading enzymes including kininase I, aminopeptidase P, and
carboxypeptidases M and N [164,167]. These results may collectively suggest a potential role
for ANP in enhanced insulin sensitization.
Several groups have reported that ANP treatment of adult cardiomyocytes enhances
glucose uptake via GLUT4 during hypoxia[281,282]. Given that cardiomyocytes revert to
glucose metabolism from fatty-acid metabolism during hypoxia[282], this finding suggests of
specific action of ANP in glucose-dependent cells. Interestingly, adipocytes normally depend on
glucose metabolism[283], and if this observation in cardiomyocytes translates to other tissues, it
would support ANP-mediated enhanced glucose uptake in adipocytes. In addition to this,
hypoxia, physical activity, and insulin treatment all enhance glucose uptake in peripheral tissues
via GLUT4[282], and this may, in part, be related to elevations in ANP detected during
exercise, hyperglycemia/hyperinsulinemia, and hypoxia [284,285,286]. Insulin stimulates the
secretion of ANP. Although the elevated ANP may be a physiological response to Na+ retention
resulting from excessive insulin signalling in the kidney[287], it may be important for managing
hyperglycemia, whether by improving blood flow to peripheral tissues or via a direct
enhancement of insulin signalling, as we demonstrate in the present study. Interestingly, ANP
resistance/desensitization is also observed in T2DM, as is a increase in NPR-C during obesity,
which may explain why elevated ANP secretion during this disorder does not improve glucose

63
tolerance or the associated HT[286,346-348,376]. The potential role for ANP in systemic
insulin sensitivity will be further discussed.
To further demonstrate the effect of NPs on primary rat adipocytes, we used BNP,
which is known to act through the same receptors as ANP to mediate similar effects in the
body[204]. BNP produced a significant increase in insulin stimulated 2-DG uptake compared
with the insulin control and was also able to mimic the outcome of ANP and BK treatment at
the same dose (1.0µM). This latter finding was surprising, given that ANP has been reported to
have as much as 10-fold greater affinity for the NPR-A and BNP has been shown to be less
effective at promoting vasodilation in humans[204,288]. However, it must be noted that while
BNP has a lesser affinity for the NPR-A, it is also less prone to clearance through the NPR-C,
accounting for its significantly longer t1/2 and thus, possibly extended time to interact with the
NPR-A[204]. Although the use of a substantial dose of 1.0µM of each peptide may negate this
suggestion, it should also be indicated that NEP is present in these tissues and the density of
NPR-C exceeds that of NPR-A in rat adiocytes[239,290,375]. This may reduce the NPs to in
vivo levels of exposure within the 1 hr incubation period, making the results physiologically
relevant. Furthermore, it is important to recognize that signalling functions are tissue and
species specific. PC12 cells, for example, which are derived from a rat adrenal medulla
pheochromocytoma respond similarly to ANP and BNP, resulting in equivalent intracellular
cGMP production[289].
It is important to note that in these experiments, ANP, BNP, and BK enhanced 2-DG
uptake in insulin treated cells, but not in basal groups. This suggests that these peptides do not
have a direct effect on GLUT4 translocation in primary rat adipocytes and thus, potentiate
insulin signalling rather than acting synergistically with it. Although this effect of BK is
consistent with the observations of many others[184,185], one group reported an insulin-

64
independent action on GLUT4 translocation and glucose uptake[372]. In this study, however,
they used cells which overexpressed GLUT4myc and the human B2 receptor which may
account for the discrepancy in results.
4.3 ANP Acts Through the NPR-A(GC)-cGMP-PKG Signalling Pathway to EnhanceInsulin-Stimulated 2-DG Uptake in Rat Adipocytes
4.3.1 ANP Requires the NPR-A to Enhance Insulin-Stimulated Glucose Uptake
As discussed, rat adipocytes have two main types of NPRs, NPR-A and NPR-C[252].
The NPR-A is traditionally known as the “biological receptor” with an inherent GC which
generates cGMP, while the NPR-C is a “clearance receptor” which, along with NEP, is essential
for removing ANP from circulation[204]. More recently, however, a signalling capacity has
been ascribed to NPR-C[228,229]. In VSMCs and possibly adipocytes, NPR-C appears to be
associated with an inhibitory G-protein (Gi) which decreases adenylate cyclase (AC) activity
and thus, intracellular cAMP. Furthermore, signalling through the NPR-C has been shown to
activate NOS, generating NO, and may, in turn, activate sGC as seen in BK signalling[253].
The role of the NPR-C is also of particular importance in our study, given that the ratio of NPR-
C:NPR-A is much greater in rodents compared to humans[290]. The use of A71915, a selective
competitive antagonist for NPR-A, in the present study dismissed the role of the NPR-C,
however, in that pre-treating the rat adipocytes with it completely removed the ANP effects on
insulin-stimulated 2-DG uptake. To further confirm the role of the NPR-A and its associated
membrane GC, we used ODQ, a selective sGC inhibitor, and found that unlike with BK
treatment[Data not shown], it did not affect ANP treated insulin-stimulated 2-DG uptake.
Another study in guinea pig tracheal smooth muscle supports this observation in that ODQ

65
markedly inhibited the relaxant response to the NO donor, SNP, but did not hinder ANP-
mediated relaxation[291].
4.3.2 ANP Requires cGMP to Enhance Insulin-Stimulated Glucose Uptake
It has been reported that the ability of NO to improve 2-DG uptake via GLUT4 in SKM
is dependent upon the presence of sGC and cGMP [292-295]. Furthermore, insulin sensitivity is
mediated by NO/cGMP pathway activation in rat liver where it controls glucose output[296].
We have also previously demonstrated that the cGMP analog, CPT-cGMP, mimicked BK
actions on insulin-stimulated 2-DG uptake in primary rat adipocytes [Data to be published]
which was further supported in the present study with the cGMP generating agent, ANP[204].
One concern leading into this study was that cGMP signalling may be
compartmentalized, given that ANP acts through a membrane GC and BK through a cytosolic
sGC. This thinking was inspired by one study showing that the activation of the membrane GC
but not sGC had potent effects on Ca2+-ATPase pump activity, suggesting some distinct
functions[300]. Although our results do not disprove the existence of compartmentalization,
they do demonstrate that if it is present, it is not important in influencing the enhanced insulin
stimulated 2-DG uptake observed for both peptides. This is further supported by the
aforementioned effect of the cGMP analogue, CPT-cGMP, which does not localize to any
particular intracellular site.
There are three general intracellular targets for cGMP, i) PKG/cGK, ii) cyclic nucleotide
gated ion channels, and iii) PDEs, the latter which are involved in cAMP and/or cGMP
degradation[205]. It was determined that 2 h after a washout, the ANP-mediated effects on
insulin-stimulated 2-DG uptake were lost, and this is known to primarily result from cGMP-
specific PDE activity. We then pre-treated cells with zaprinast to inhibit PDE5A, a prominent
enzyme involved in adipocyte cGMP degradation[239]. This experiment was particularly

66
interesting, given that zaprinast can also potentiate ANP-mediated vasodilation[297]. We found
that zaprinast conserved the ANP effect on insulin-stimulated 2-DG uptake, 2 h after washout,
thus, further supporting the role of cGMP in our mechanism of enhanced insulin sensitivity. It
would be of interest to further support this finding with an assessment of cGMP levels following
zaprinast treatment. In a relevant study, administering the more potent PDE5A inhibitor,
sildenafil, to diet-induced insulin resistant C57BL/6J mice resulted in less weight gain
associated with increased energy expenditure and improved insulin sensitivity [222]. It is
interesting that in our study, zaprinast did not influence 2-DG uptake in the absence of ANP.
This may be the result of low basal cGMP levels which are unlikely to significantly improve
over 1h with PDE5A inhibition. Although PDE5A is important in cGMP degradation, it must be
noted that other cGMP targeting PDEs are present in adipocytes including PDE6, PDE9, and
several others which target both cGMP and cAMP[298]. In fact, there are reports of treating the
insulin resistance syndrome and T2DM with PDE9 inhibitors which have led to clinically
relevant improvements in BP, serum glucose, insulin, lipids, and procoagulant factors[299]. It
would be interesting to evaluate the effect of a combination of cGMP-specific PDE inhibitors
with ANP treatment.
4.3.3 ANP Requires PKG to Enhance Insulin-Stimulated Glucose Uptake
We have previously demonstrated that BK signals through PKG to enhance insulin-
stimulated 2-DG uptake. This finding was further supported in the present study with KT-5823,
a selective PKG inhibitor, which abolished the ANP effect on enhanced insulin actions. This
result is expected, given that many of the physiological outcomes of ANP signalling including
vasodilation and natriuresis are mediated through PKG[301,302]. Furthermore, PKG1 has also
been shown to play a functional role in GLUT4 translocation via actin and microtubule
cytoskeletal systems in human VSMCs[303].

67
There are three different PKG isoforms[211]. Although PKG1 is prominent in
adipocytes and suspected to be involved, KT-5823 does not distinguish between the
isoforms[254,255]. For this reason, the fact that KT-5823 blocks both ANP- and BK-mediated
enhanced insulin actions does not confirm that they act through the same isoform, though this
would be suspected, given that they mediate the same outcome and that PKGs tend to have
different substrates[212,214]. To further demonstrate the role of PKG isoforms in the signalling
pathway for both ANP and BK, adipocytes can be transfected with DN-PKG1 and examined for
changes. Figure 7.1 provides support for the role of PKG1 in the BK actions. This will be
further discussed.
4.4 ANP Potentiates the Insulin Signalling Pathway in Rat Adipocytes
As discussed, treatment of fa/fa rats with captopril, an ACEi, significantly enhanced the
insulin-stimulated Tyr phosphorylation of IR, IRS1, and PI3K(p85) in SKM and liver[304].
Furthermore, it has been shown that BK markedly improves insulin-stimulated Tyr-IRS-1 and
Akt/PKB phosphorylation in adipocytes [185,186].
In the present study, we found that ANP significantly enhanced insulin-stimulated Tyr-
IRS-1 and Akt/PKB phosphorylation and that this effect was dependent on PKG activity. This
potentiation of the insulin signalling pathway clearly supports our observation that ANP treated
adipocytes display enhanced insulin-stimulated 2-DG uptake in the absence of KT-5823
treatment. Although there are few studies addressing the metabolic actions of ANP with insulin,
it has been demonstrated that ANP and BK act through the GC-cGMP-PKG pathway to activate
PI3K and potentiate insulin signalling in VSMC[303,306]. It has also been observed that ANP
enhances Akt/PKB phosphorylation in cardiomyocytes in order to prevent apoptosis from

68
ischemia reperfusion, volume/pressure overload, hypoxia, hypoglycemia, and cardiotoxic
drugs[307,308]. Furthermore, many of these studies indicate that ANP affects the PI3K
signalling cascade, upstream of Akt/PKB [307,309] which we also saw through enhanced
insulin-stimulated Tyr-IRS-1 phosphorylation. This may provide indirect support for the role of
IRS-1 in the ANP-mediated regulation of insulin signalling. It is important to note, however,
that while many of these studies report a basal effect with ANP treatment, we only observed
changes in insulin-stimulated cells. This discrepancy may be attributed to the different cell types
examined or experimental conditions.
4.5 ANP Reduces MAPK Phosphorylation in Insulin-Stimulated Rat Adipocytes
MAPK-induced Ser-IRS-1 phosphorylation is believed to provide negative feedback
inhibition of insulin signalling[223]. Although this is a physiological process, hyperactive or
chronic MAPK phosphorylation promotes insulin resistance, and can be observed in T2DM and
obesity[310]. While it is well known that insulin activates ERK1/2 through raf signalling, the
role of insulin in JNK stimulation is poorly characterized given that JNK activators have
traditionally been thought of as stressors, including FFAs, inflammatory cytokines, and UV
radiation[93,101,223]. A variety of studies suggest that insulin can stimulate JNK through
PI3K, ras, or Shp2[311], though these pathways may converge, given that similar techniques
can inhibit both TNF-α- and insulin-mediated JNK phosphorylation[223]. It must be noted that
although IRS1 contains a JNK binding motif similar to JIP1/2, a direct interaction between JNK
and IRS1 does not appear to be involved in insulin-stimulated JNK activation[223].

69
In previous studies, we identified that BK decreased ERK1/2 and JNK phosphorylation
in insulin-treated rat adipocytes despite enhancing Akt/PKB phosphorylation and GLUT4
translocation. This outcome appeared paradoxical at first, but it eventually helped us identify the
BK mechanism of enhanced insulin sensitivity. Although there are several IRS-1 Ser residue
targets for MAPKs, the prominent sites for ERK1/2 and JNK are Ser612 and Ser307,
respectively[186]. Studies indicate that decreased phosphorylation at these sites results in
enhanced insulin-stimulated Tyr-IRS-1 and Akt/PKB phosphorylation, and glucose uptake, all
of which we observed with BK treatment[312]. To determine if the enhanced insulin sensitivity
is through decreased negative regulation of IRS-1, we previously examined Ser612 and Ser307
phosphorylation and found that BK decreased it in insulin-stimulated cells. Although factors
like IKKβ can also phosphorylate IRS-1 at Ser307[313], it did not appear to be important in
BK-mediated effects on IRS-1, and so we did not examine it further[186]. Furthermore,
although p38 is a member of the MAPK family which may participate in negative feedback
inhibition of insulin signalling, we did not observe p38 activation with insulin treatment in rat
adipocytes. Finally, it is worth noting that JNK1 but not JNK2-deficient animals are protected
from the development of obesity-induced insulin resistance [314] and so JNK1 was emphasized
in our investigations.
In the present study, ANP treatment led to a significant decrease in both ERK1/2 and
JNK1 phosphorylation in insulin-stimulated adipocytes and this outcome was dependent on
PKG activity. This observation is in agreement with our previous studies with BK and can
explain the enhanced insulin signalling and 2-DG uptake with ANP treatment. Similarly, ANP
has been shown to decrease PDGF- and AngII-induced ERK1/2 activation and ET-I-induced
JNK activation in rat mesangial cells via PKG[315,316]. The latter effect was reproduced using

70
8-bromo-cGMP and the NO donor, SNP, but the ANP actions were lost with HS-142-1, an
NPR-A antagonist, and KT-5823, a PKG inhibitor[316].
Basal MAPK activation was surprisingly elevated in the present study, which may be
due to the exposure of adipocytes to stressors such as type I collagenase and mild hypoxia
during incubation. ANP also tended to (non-significantly) reduce basal JNK phosophorylation
which may be due to a direct effect on MAPK or, in part, decreased pro-inflammatory cytokine
and adipokine secretion which has been reported with ANP-treatment in human adipose
tissue[239].
Although our past and present data collectively show that both ANP and BK reduce
MAPK phosphorylation in insulin treated adipocytes, we previously identified that the
inhibition of JNK with SP600125 mimicked the enhanced insulin-stimulated 2-DG uptake while
ERK1/2 inhibition with PD98059 did not[186]. We also observed that adipocytes from JNK-/-
mice behaved similarly to rat adipocytes treated with SP600125[186]. This finding led us to the
belief that JNK1 inhibition is primarily responsible for the observed effects of enhanced insulin
sensitivity with both BK and ANP treatment. Furthermore, the JNK target, Ser307-IRS-1, is
commonly hyperphosphorylated in states of insulin resistance, and can interfere with the IR-IRS
interaction given that it is located next to the IRS1 PTB binding domain[314]. It must be
acknowledged, however, that there are over 100 potential Ser phosphorylation sites in IRS1, and
studies of other kinases which phosphorylate IRS1 at such sites, including PKCζ, mTOR, and
AMPK, are warranted[223].
4.6 ANP and the Regulators of MAPK Activity
MAPK phosphorylation and activity is regulated by upstream kinases (MAPKK), and
downstream phosphatases[86,91]. It was of interest to identify how ANP decreases MAPK
phosporylation in insulin-stimulated cells, downstream of PKG.

71
4.6.1 ANP Reduces MAPK Phosphorylation Indpendent of Upstream MAPK Kinases
In the present study, we examined the activity of MKK-4/7 and MEK1/2, which are the
respective MAPKKs for JNK and ERK1/2. Although the insulin-mediated activation of ERK1/2
through MEK1/2 is well established, the role of MKK-4/7 is not as well defined. There are
some reports that insulin activates both MKK-4 and JNK in a dose dependent manner in rat
fibroblasts expressing human IRs, but there is no evidence for the role of MKK-7 [317]. This
experiment was of particular interest, given that another group observed that enhanced Akt/PKB
activity was concomitant with decreased MKK-4 and JNK phosphorylation in 293T cells [318].
In the present study, we observed that insulin stimulation resulted in MKK-4 phosphorylation
but that this was not altered with ANP treatment. A similar outcome was detected for MEK1/2,
while phosphorylation of MKK-7 was not observed in any of the treatment groups. These
observations suggest that the decreased MAPK phosphorylation with ANP treatment is not due
to decreased upstream MAPKK activity and may be due to enhanced phosphatase activity.
4.6.2 ANP May Reduce MAPK Phosphorylation via MAPK Phosphatase Upregulation
There are several intracellular negative regulators of MAPK signal intensity and
duration. The most robust and direct regulators are MKPs, a class of phosphatases with dual-
specificity activity against MAPK Thr/Tyr residues[91]. There is evidence that MKPs can
improve the insulin resistant phenotype. One recent study examined the effect of overexpressing
MKP4 in stress-induced (anisomycin, TNF-α) insulin resistant 3T3-L1 cells and insulin resistant
ob/ob mice[310]. MKP-4 inhibited ERK and JNK phosphorylation and, to a lesser extent, p38
phosphorylation, in 3T3-L1 cells. As a result, anisomycin- and TNF-α induced Ser307-IRS-1

72
phosphorylation was attenuated, with concomitant improvements in insulin-stimulated Tyr-IRS-
1 and Akt/PKB phosphorylation and glucose uptake. Furthermore, overexpressing MKP4 in
ob/ob mice liver resulted in decreased ERK and JNK phosphorylation, along with reduced fed/
fasting glycemia and improved glucose tolerance[310]. Interestingly, studies in other cell types
have demonstrated that PKG1 can induce the rapid expression of MKP genes[319]. ANP, acting
through PKG can elevate intracellular MKP-1, leading to decreased AngII- or TNF-α-induced
MAPK phosphorylation in rat mesangial and endothelial cells [320,321]. Furthermore, it was
found that MKP1-/- mice exhibited glucose intolerance and a 3-fold greater JNK activity than
control mice[322]. It is also worth noting that in VSMCs, both ANP and insulin are able to
elevate MKP-1 gene expression via PKG1 signalling within 30 min[323].
The fact that MKPs can target both ERK and JNK is of great interest given that both BK
and ANP decreased the phosphorylation status of these two MAPKs in insulin-stimulated
adipocytes. Furthermore, we previously identified that both BK and the NO donor,
hydroxylamine (HA), in combination with insulin significantly induced the expression of MKP5
mRNA after only 30 min. Although protein expression must still be confirmed, the rapid
induction of this MKP transcript provides more indirect evidence for the role of MKPs in our
acute studies. Finally, although the role of MKP5 in diabetes has yet to be examined, it should
be noted that it primarily targets JNK but can also prominently dephosphorylate ERK[Table
4.1].

73
Table 4.1 MKP Characteristics. The MKPs are coded by distinct genes,are expressed in different cellular compartments, and have different MAPKspecificities [373,374]
Although the hypothesis for the role of MKPs in BK- and ANP-mediated enhanced
insulin signalling is quite appealing, it does not rule out other potential phosphatases including
PP2A and PP2Cα [88,89]. Furthermore, although it has not been shown previously, ANP may
also interrupt MKK-4 and JNK interactions, possibly by disrupting the scaffold protein, JIP1/2.
Additionally, some of the actions of NO are reported to act through S-nitrosylation of proteins
which can suppress ERK through a thiol-redox mechanism[324]. These other possibilities
warrant further investigation. If, however, MKPs are implicated in the BK and ANP effects, this
would suggest that some actions of insulin, including growth, proliferation, and survival, which
are mediated through MAPK signalling would be disrupted with BK or ANP treatment while
the metabolic outcomes of insulin signalling, including glucose uptake, would be potentiated.
The long-term effects of this manipulation would also be worth investigating.
4.7 Caveats and Limitations of the Study
All experiments involved a 1 h incubation in a slightly hypoxic environment with type I
collagenase (1mg/mL), both of which can introduce stress to cells and artificially elevate basal
MAPK activity. Although efforts were made to minimize the influence of these factors, they
still may have had some effect on the results obtained. One other consideration is that while the

74
epididymal fat pads are selected for their relative purity of white adipocytes, there are still
resident macrophages and vascular cells which are present[325]. Although cellular digestion,
filtration, and washing techniques are fairly effective at isolating adipocytes, they are not
flawless, and so other cell types may, to a minor extent, contribute to the results. Furthermore,
although lipocrit measurements are accepted for cell number corrections during 2-DG uptakes,
the use of flow cytometry would likely yield more accurate estimations[250,326].
One obvious limitation of this study is that it strictly relies on pharmacological inhibitors
to examine the role of the GC-cGMP-PKG signalling pathway in ANP-mediated enhanced
insulin sensitivity. Genetic studies would provide more specific support for our hypothesis. The
present investigation also strictly examines the effects of ANP on insulin signalling in adipose
tissue, even though SKM is the predominant site for glucose disposal[259]. Adipocytes were
selected for this investigation in order to supplement the work previously established by our lab
with BK. Furthermore, adipose tissue is insulin-sensitive, relies on GLUT4 for insulin-
stimulated glucose uptake as does SKM, and is important in insulin sensitivity, given that
adipose-specific GLUT4 KO mice have systemic insulin resistance[18,327]. Another limitation
of our project is that our experiments were performed entirely in an in vitro context, and so in
vivo data will be required to support the relevance of our findings. We also only examined
young, lean (150-180g) male Sprague Dawley rats which are not insulin resistant compared
with older counterparts with more adipose tissue accumulation. Furthermore, since our rats are
“normal”, we do not know if our results will apply to the diabetic state. These are all interesting
studies which will be further discussed in Chapter 5 - Future Directions.
4.8 ANP-mediated Lipolysis – An Insulin Antagonizing Function?
ANP can act through the GC-cGMP-PKG signalling pathway to promote lipolysis, a
seemingly “anti-insulin” outcome, but this is only prominent in primates[328]. Some report that

75
the higher NPR-A:NPR-C ratio in primates accounts for this species specificity, suggesting that
ANP signalling is more potent in these organisms while it is more regulated and frequently
cleared in lower mammals[328]. However, as discussed, there is growing evidence for an NPR-
C signalling function. This receptor appears to be associated with a Gi protein whichleads to
reduced AC activation and thus, cAMP[252,329]. The ability of ANP to decrease intracellular
cAMP is of particular importance, given that cAMP signalling is responsible for catecholamine-
induced lipolysis and that it is through the activation of the cAMP-specific PDE3B that insulin
counters this outcome[330,331]. Thus, it would appear that ANP, signalling through the NPR-C,
acts synergistically with insulin to ensure anti-lipolysis in adipocytes. Interestingly, the ratio of
NPR-A:NPR-C increases during the fasting state[332], and so it would be appealing to examine
rodents under these experimental conditions for lipolysis. It is worth noting that some groups
have identified marginal lipolysis in rats following ANP treatment[252], which we also
observed in our model[Figure 7.2], while others report significant increases in cGMP without
concomitant lipolysis[333]. If NPR-A signalling results in lipolysis and NPR-C signalling
results in decreased lipolysis, albeit by a different mechanism, this may account for the overall
absence of lipolysis over basal measurements in rodents. Also, given the greater NPR-A:NPR-C
ratio in primates, it is reasonable to assume that there is greater cGMP production and less NPR-
C stimulation than in rodents.
The fact that cGMP can promote lipolysis but enhance adipocyte insulin sensitivity
seems paradoxical. One possibility is that ANP signalling can activate several pathways, some
with antagonistic functions. One group reported that ANP treatment of human adipocytes
resulted in decreased pro-inflammatory cytokine secretion and increased lipolysis, while
inhibition of this lipolysis promoted the release of anti-inflammatory cytokines[239]. This
would suggest that one arm of ANP signalling in adipocytes can antagonize the other. This

76
study will be further discussed. Another consideration is that increasing cGMP can activate
PDE2 to further degrade cAMP while excessive increases can impair the PDE3B, and thus,
insulin-mediated anti-
lipolysis [334,335]. The
lower NPR-A:NPR-C ratio
in rodents may, therefore,
guard against PDE3B
inhibition. It is also worth
noting that cGMP-mediated
lipolysis is not normally
antagonized by the anti-
lipolysis signalling of
insulin. This was
demonstrated by studies
showing that isoprotenol-
Figure 4.1 Regulation of Lipolysis in Adipocytes. Natriureticpeptides act through the NPR-A to generate cGMP which can stimulatelipolysis. ANP can also act through the NPR-C to decrease cAMP and inhibitan alternative mechanism of lipolysis. Insulin attenuates lipolysis by activating,PDE3B, a phosphodiesterase which specifically degrades cAMP.
mediated lipolysis was completely distinct from cGMP-stimulated lipolysis, and insulin
treatment was unable to influence the latter effect[252,336]. It may, therefore, be reasonable to
assume that only when there is excessive cGMP is the anti-lipolytic effect of insulin directly
antagonized. This paradoxical, concentration-dependent role for cGMP has been shown in
another context, where low cGMP has a stimulatory effect on ion channels leading to increased
intracellular Ca2+, an outcome which is associated with enhanced GLUT4-mediated glucose
uptake in 3T3-L1 adipocyte cells, while too much cGMP reversed this outcome [337,338].
Other paradoxical outcomes of ANP signalling have also been identified. While ANP can guard
against cardiomyocyte apoptosis in response to hypoxia, ischemic reperfusion injury, or volume

77
overload, the therapeutic window is relatively narrow, such that excessive signalling can
actually promote apoptosis[339]. Furthermore, a similar observation, albeit by a different
mechanism, is observed for NO, an upstream activator of sGC and cGMP production. When NO
is produced acutely and at low levels through eNOS, it promotes insulin sensitivity[192]. In
fact, eNOS-/- mice exhibit insulin resistance and T2DM[340]. However, chronic and excessive
NO production, via iNOS, appears to promote insulin resistance[187]. Disruption of the iNOS
gene has been shown to protect against obesity-induced insulin resistance in muscle and
improve whole-body insulin action and glucose tolerance[341,342].
4.8.1 ANP and Insulin Resistance
ANP is chronically elevated during CHF, which may result in excessive lipolysis and
FFAs in circulation[343,344]. It is known that FFAs can promote insulin resistance[345], but
whether ANP contributes to the insulin resistance in CHF or whether it acts as a compensatory
mechanism is unknown. Interestingly, insulin stimulates ANP secretion, and during insulin
resistance there is hyperinsulinemia and thus, elevated ANP secretion[235]. ANP has been
noted to inhibit pre-adipocyte proliferation and adipogenesis while reducing BP, but chronically
elevated ANP leads to receptor desensitization and the NPR-C is upregulated during obesity,
resulting in decreased ANP in circulation[346-348,376]. A very interesting observation was also
made in human adipocytes that ANP decreases the secretion of inflammatory
cytokines/adipokines which promote insulin resistance[239]. These observations collectively
suggest a link between inflammation, HT, insulin resistance, and obesity with less ANP or its
actions. In addition, despite increasing lipolysis in primates, ANP also appears to promote FFA
oxidation[240]. Thus, in contrast to insulin resistance caused by FFA release, in this case, ANP
actions would presumptively lead to rapid FFA consumption while maintaining a lean state and
insulin sensitivity.

78
4.8.1.1 ANP Signalling Enhances Mitochondrial Function
There is recent evidence that NO and cGMP signalling can critically regulate
mitochondrial mass and function[352]. This is achieved by inducing the expression of the
transcriptional co-activator, peroxisome-proliferator-activated receptor co-activator 1
(PGC1-) which can eventually lead to the expression of nuclear and mitochondrial genes, such
as cytochrome oxidase complex IV (COX IV) and cytochrome C (Cyt C), important for
mitochondrial oxidative phosphorylation [352]. Interestingly, eNOS∕ mice, which are insulin
resistant, display decreased energy expenditure, mild obesity, decreased mitochondria, PGC1-,
and COXIV and Cyt C protein in a number of tissues, indicating that this NO signal is of major
physiological importance [352]. In a follow-up study, this group showed that mice subjected to
a calorie restricted diet, which resulted in a higher NPR-A:NPR-C ratio, showed increased
cGMP, eNOS, and mitochondria in WAT and other organs, along with increased O2
consumption, ATP synthesis, and the expected changes in protein expression mentioned above
[353]. In this study, similar to effects of ANP, there was increased fat mobilization from WAT,
concomitant with increased mitochondrial fatty acid -oxidation, suggesting that the elevated
FFAs with ANP are easily metabolized and thus, do not promote insulin resistance.
Relevant to these data is the evidence that mitochondrial dysfunction, either genetic or
acquired, is a major contributor to insulin resistance in humans with T2DM and “pre-diabetes”
[354]. Even in lean, young non-diabetic insulin resistant individuals with a strong family
history of T2DM, there is a decrease in mitochondrial density in skeletal muscle [354]. The
cause of this decrease is not clear as some studies [355,356] but not all [357] found reduced
levels of PGC1- mRNA along with other regulators of mitochondrial biogenesis. Relevant
and interesting is a recent report that exposing C2C12 cultured mouse muscle cells to high

79
glucose and insulin caused mitochondrial dysfunction which was prevented by co-treatment
with cGMP [358].
4.8.1.2 ANP Signalling Causes Decreased Inflammation
Insulin resistance has also been closely linked with a chronic inflammatory state
[359,360]. Both adipocytes and infiltrating macrophages produce various inflammatory
adipokines and cytokines such as TNF-, IL-l, Il-6, MCP-1 and resistin, which are associated
with insulin resistance [361,362,363]. While AngII has pro-inflammatory actions [364], ANP
has been shown in macrophages to have anti-inflammatory effects [365]. For example, ANP can
inhibit iNOS expression in macrophages [366] and iNOS has been strongly implicated as a key
promoter of insulin resistance in muscle and liver [367]. ANP can also inhibit LPS-stimulation
of TNF- and NF-kB production [368]. As discussed, Moro et al. showed in human adipose
tissue that ANP, acting through GC-cGMP-PKG signalling, reduces the secretion of several
adipokines (leptin, RBP-4) from adipocytes and cytokines (IL-6, TNF-α, MCP-1, MIP-1β,
GRO-α) from macrophages[239]. These findings collectively suggest an anti-inflammatory
effect of ANP which may critical for circumventing insulin resistance.

80
4.10 CONCLUSION
Figure 4.2 ANP-mediated Enhanced Insulin Sensitivity. ANP binds to theNPR-A receptor, activating the receptor associated GC. This leads to elevatedintracellular cGMP, which activates PKG. PKG, in turn, attenuates the activity of MAPK,while potentiating the insulin signalling pathway (Tyr-IRS-1 and Akt/PKBphosphorylation) and insulin-mediated glucose uptake.
Abbreviations
Akt/PKB=Protein Kinase BANP=Atrial Natriuretic
PeptideB2= B2-ReceptorBK=BradykinincGMP=cyclic GMPeNOS=Nitric Oxide SynthaseERK=Extracellular Signal
Related KinaseGLUT4=Glucose
Transporter-4IR=Insulin ReceptorIRS-1=Insulin Receptor
Substrate-1JNK=c-Jun N-Terminal
KinaseMEK1/2=MAPK Kinase1/2MKK4=MAPK Kinase 4MKP=MAPK PhosphataseNO=Nitric OxideNPR-A=Natriuretic peptide
Receptor-APKG= Protein Kinase GPI3K=Phosphoinositide-3
KinasesGC=soluble Guanylate
Cyclase
In conclusion, ANP reproduced the BK effect of enhanced insulin-stimulated 2-DG
uptake in isolated rat adipocytes, and was dependent on NPR-A-cGMP-PKG signalling. ANP
was responsible for potentiating the insulin-stimulated PI3K pathway as indicated by enhanced
Tyr-IRS-1 and Akt/PKB phosphorylation while paradoxically, decreasing insulin-stimulated
MAPK(JNK, ERK1/2) phosphorylation. Interestingly, after 40 min of insulin stimulation,
MAPK activation has been shown to be associated with a negative feedback loop on insulin
signalling, mediated by increased Ser residue phosphorylation of IRS-1[312]. Thus, it is
possible that ANP, as demonstrated for BK, potentiates the insulin signalling pathway by
reducing its negative feedback regulation. ANP did not appear to influence the activity of
upstream MAPK kinases, suggesting that it may upregulate phosphatases to reduce MAPK
phosphorylation.

81
CHAPTER 5
FUTURE DIRECTIONS

82
5.1 The Role of PKG1/cGKI in BK- and ANP-Mediated Enhanced Insulin Signalling
In the present study, we have shown that ANP mimics BK actions by signalling through
PKG to enhance insulin-stimulated Tyr-IRS-1 and Akt/PKB phosphorylation while decreasing
JNK and ERK1/2 phosphorylation[Chapter 3]. The role of PKG was determined using KT-
5823, a pharmacological PKG inhibitor, but this agent does not differentiate between the three
PKG isoforms[254,255]. Furthermore, while we were certain to use concentrations which
provide specific inhibition of PKG, there is still the possibility of non-specific effects with a
pharmacological agent. For this reason, genetic evidence for the role of PKG1/cGKI will be
required.
The genetic approach will consist of using electroporation to co-transfect adipocytes
with the DN-PKG1 (provided by R. Pilz, UCSD) or empty pcDNA3 (control) vector, along with
the “outcome protein” plasmid, i.e. wt-IRS-1, wt-Akt/PKB, wt-JNK1 or wt-ERK (provided by
D. Bar-Sagi, NYU). This technique has been well established by our lab for adipocytes.
Furthermore, expressed proteins are myc- or flag-tagged so we can isolate them from
endogenous proteins by immunoprecipitation.
Co-transfected adipocytes will be treated with BK (1.0µM), ANP (1.0µM), or vehicle for 1
h, with insulin (100nM) during the final 40 min. This will be followed by immunoblotting for
the immunoprecipitated protein. It has already been determined that BK treatment decreases wt-
JNK1 phosphorylation in insulin-stimulated cells compared with insulin controls in the PC-
DNA3 groups, while these effects were lost in DN-PKG1 groups[Chapter 7]. We would
anticipate that DN-PKG1 would also remove the effects of BK and ANP on ERK and Akt/PKB
phosphorylation in insulin-stimulated groups. Our transfection efficiency using this technique is
25-50% with 2 vectors. As a back-up approach, we will use adenovirus transduction to express
the protein. Adenovirus use has the advantage of providing greater efficiency (>50%) [256].

83
5.2 The Role of MKPs in BK- and ANP-Mediated Enhanced Insulin Sensitivity
As described in Chapter 4, BK and ANP may upregulate MKP expression in order to
enhance insulin signalling. We have previously identified through quantitative real-time RT-
PCR, that BK and the NO donor, hydroxylamine (HA), significantly enhanced MKP5 mRNA
within 30 min of treatment. It will also be important to replicate this experiment with ANP.
Although we would expect ANP to also increase MKP5 mRNA, this may not be essential, as
other MKPs are capable of dephosphorylating JNK and ERK to a similar extent, and ANP has
been shown to elevate MKP1 in a variety of other cell types[257,258]. This will be followed by
immunoblotting for the expression of specific MKP isoforms identified through the quantitative
real-time RT-PCR experiments, where adipocytes will first be treated with BK (1.0µM), ANP
(1.0µM), or vehicle for 1h, with ±insulin (100nM) during the final 40 min. It is anticipated that
BK and ANP will result in the upregulation of one or more MKPs against JNK and ERK.
Finally, it would also be interesting to assess the role of the suspected MKPs through the use of
specific siRNA-mediated gene silencing. If MKP is important, the latter treatment would
remove the ANP and BK-mediated effects on 2-DG uptake as well as JNK, ERK, IRS-1, and
Akt/PKB phosphorylation in insulin stimulated groups.
5.3 The in vitro Effect of BK and ANP on Insulin Sensitivity in Skeletal Muscle
SKM is insulin-sensitive and plays a greater role in peripheral glucose uptake than
adipose tissue[259]. Also, given that there are limited data on ANP action in SKM, it would be
interesting to see if our result of enhanced acute insulin sensitivity in adipocytes with ANP
treatment would also apply to SKM. We would pursue in vitro studies in cell culture models,
i.e. L6 rat SKM cells, and ex vivo studies in EDL and soleus muscle. Cells would be treated
with BK (1.0µM), ANP (1.0µM), or vehicle for 1h, with ±insulin (100nM) during the final 40
min. Immunoblotting and 2-DG uptake assays would then be performed. Given that plasma

84
ANP and BK are markedly elevated during exercise when SKM energy demands are greatest,
and ANP promotes energy expenditure and glucose uptake by cardiomyocytes during hypoxia,
we believe that SKM insulin sensitivity will be enhanced with treatment[260,261,240].
5.4 The in vivo Effect of ANP on Insulin Sensitivity in “Normal” Rodents
It is important to determine whether ANP can enhance in vivo insulin sensitivity in order
to determine whether our results may have any translational importance. The euglycemic
hyperinsulinemic clamp technique is well established and can be used to determine the role of
ANP in mediating systemic insulin sensitivity.
The initial studies will be carried out in normal Wistar rats infused with ANP or vehicle
for 2h followed by 2h of insulin. In rats, an effective infusion rate was 50 pmol/kg min-1, which
resulted in an increase in urinary cGMP excretion and natriuresis[262]. The 2h
hyperinsulinemic-euglycemic clamp will be carried out as previously described [263,264] and
infusion rates would be adjusted to avoid hypotension and the activation of RAS[265-267].
These studies will reveal the effects of ANP on insulin-stimulated peripheral glucose disposal
(Rd), as well as on suppression of hepatic glucose output (HGO). We expect that ANP will
enhance insulin action.
To supplement these studies, mice with mutant nonfunctional NPR-C, and with a
targeted deletion of NPR-A are commercially available (Jackson) and can be used to determine
the effects of excessive and absent ANP-mediated cGMP signaling, respectively. We expect
that NPR-C knockouts will show enhanced insulin sensitivity, and these effects will be reversed
for NPR-A KOs.
5.5 The Effect of ANP on Insulin Resistance in Rodents
One major limitation of our study is that although BK and ANP appear to enhance
insulin sensitivity in rat adipocytes, this tissue was acquired from young and relatively lean

85
animals which are unlikely to show insulin resistance. Although ACEi and BK treatment has
been shown to improve insulin resistance or delay the onset of T2DM[172,173,180], we cannot
be certain that the GC-cGMP-PKG signalling pathway is involved in these effects without
testing under insulin resistant conditions. Thus, it would be interesting to evaluate whether BK
or ANP can reverse or prevent insulin resistance.
One in vitro method of inducing insulin resistance which we have extensive experience
with is treating primary adipocytes with D-glucose (20mM) and insulin (100nM) for 18 h [268].
Initial experiments would examine whether BK or ANP can prevent the development of insulin
resistance in this model by incubating the cells with BK or ANP during the 18 h, followed by
studies indicating whether treatment can improve it, using the same treatment protocol
established in the present study. In vivo methods of achieving insulin resistance in rodents
include i) glucose or FFA (intralipid + heparin) infusions, ii) providing high fat diets for
animals, or iii) using Zucker diabetic fatty (ZDF) insulin resistant rats for experiments [269-
272] . We can infuse rats with BK, ANP, or vehicle for 2 h (50 pmol/kg min-1), followed by a 2
h hyperinsulemic-euglycemic clamp to assess insulin sensitivity. Another option would be to
extract tissues, i.e. adipose tissue and SKM, from the insulin resistant rodents and treat cells
with BK (1.0µM), ANP (1.0µM), or vehicle for 1h, with ±insulin (100nM) during the final 40
min. This would be followed by 2-DG uptake assays and immunoblotting for insulin signalling
pathway proteins.

86
CHAPTER 6
REFERENCES

87
1) Chipkin SR, Klugh SA, Chasan-Taber L. Exercise and diabetes. Cardiol Clin. 2001: 19(3);489-505.
2) Alper J. New insights into type 2 diabetes. Science. 2000: 289(5476); 37–39.
3) Nathan DM. Long-term complications of diabetes mellitus. N Eng J Med. 1993: 328(23);1676-1685.
4) Foulis AK, McGill M, Farquarson MA. Insulinitis in type 1 (insulin-dependent) diabetesmellitus in man—macrophages, lymphocytes, and interferon-gamma containing cells. JPathol. 1991: 165; 97-103.
5) Stumvolt M, Goldstein BJ, Haeften TWV. Type 2 diabetes: principles of pathogenesis andtherapy. Lancet. 2005: 365; 1333-1346.
6) Schinner S, Scherbaum WA, Bornstein SR, Barthel A. Molecular mechanisms of insulinresistance. Diabet Med. 2005: 22(6); 674-682.
7) LeRoith D. Treatment of diabetes: a clinical update on insulin trials. Clin Cornerstone. 2007:8(2); 21-29.
8) Rendell M. The role of sulphonylureas in the management of type 2 diabetes mellitus. Drugs.2004: 64; 1339-1358.
9) Deacon CF. Therapeutic strategies based on glucagon-like peptide 1. Diabetes. 2004: 53;2181-2189.
10) Bailey CJ, Turner RC. Metformin. N Eng J Med. 1996: 334; 574-579.
11) Rinderknecht E, Humbel RE. The amino acid sequence of human insulin-like growth factorI and its structural homology with proinsulin. J Biol Chem. 1978: 253(8); 2769-2776.
12) Harper ME, Ullrich A, Saunders GF. Localization of the human insulin gene to the distalend of the short arm of chromosome 11. Proc Natl Acad Sci. 1981: 78(7); 4458-4460.
13) Steiner DF, Oyer PE. The biosynthesis of insulin and a probable precursor of insulin by ahuman islet cell adenoma. Proc Natl Acad Sci. 1967: 57(2); 473–480.
14) Zaitsev SV, Efanov AM, Efanova IB, Larsson O, Ostenson CG, Gold G, Berggren PO,Efendic S. Imidazoline compounds stimulate insulin release by inhibition of K (ATP)channels and interaction with the exocytotic machinery. Diabetes. 1996: 45(11); 1610-1618.
15. Ashcroft FM, Gribble F. ATP-sensitive K+ channels and insulin secretion: their role inHealth and disease. Diabetologia. 1999: 42(8); 903-919.

88
16. Raskin P, Rappaport EB, Cole ST, Yan Y, Patwardhan R, Freed MI. Rosiglitazone short-term monotherapy lowers fasting and post-prandial glucose in patients with type II diabetes.
Diabetologia. 2000: 43(3); 278-284.
17) Bergamini E, Cavallini G, Donati A, Gori Z. The role of autophagy in aging: Its essentialpart in the anti-aging mechanism of caloric restriction. Ann N Y Acad Sci. 2007: 1114;6978.
18) Lang CH, Dobrescu C, Bagby GJ. Tumor necrosis factor impairs insulin action onperipheral glucose disposal and hepatic glucose output. Endocrinology. 1992: 130; 43-52.
19) Rath VL, Ammirati M, Danley DE, Ekstrom JL, Gibbs EM, Hynes TR, Mathiowetz AM,McPherson RK, Olson TV, Treadway JL, Hoover DJ. Human liver glycogen phosphorylaseinhibitors bind at a new allosteric site. Chemistry & Biology. 2000: 7(9); 677-682.
20) Minassian C, Tarpin S, Mithieux G. Role of glucose-6 phosphatase, glucokinase, andglucose-6 phosphate in liver insulin resistance and its correction by metformin.Biochemical Pharmacology. 1998: 55(8); 1213-1219.
21) Choudhury J, Sanyal AJ. Insulin resistance and the pathogenesis of nonalcoholic fatty liverdisease. Clinics in Liver Disease. 2004: 8(3); 575-594.
22) Pessin JE, Thurmond DC, Elmendorf JS, Coker KJ, Okada S. Molecular basis of insulin-stimulated GLUT4 vesicle trafficking. J Biol Chem. 1999: 274(5); 2593-2596.
23) Wang D, Sul HS. Insulin stimulation of the fatty acid synthase promoter is mediated by thephosphatidylinositol 3-kinase pathway. J Biol Chem. 1998: 273(39); 25420-25426.
24) Löbbert RW, Winterpacht A, Seipel B, Zabel BU. Molecular cloning and chromosomalassignment of the human homologue of the rat cGMP-inhibited phosphodiesterase 1(PDE3A)—A gene involved in fat metabolism located at 11p15.1. Genomics. 1996: 37(2);211-218.
25) Hasselgren PO, Warner BW, James JH, Takehara H, Fischer JE. Effect of insulin on aminoacid uptake and protein turnover in skeletal muscle from septic rats. Evidence for insulinresistance of protein breakdown. Arch Surg. 1987: 122(2); 228-233.
26) Avruch J. Insulin signal transduction through protein kinase cascades. Mol Cell Biochem.1998: 182(1-2); 31-48.
27) Olson TS, Bamberger MJ, Lane MD. Post-translational changes in tertiary and quaternarystructure of the insulin proreceptor. Correlation with acquisition of function. J Biol Chem.1988: 263(15); 7342-7351.
28) Kasuga M, Karlsson FA, Kahn CR. Insulin stimulates the phosphorylation of the 95,000-dalton subunit of its own receptor. Science. 1982: 215(4529); 185-187.

89
29) White MF, Shoelson Se, Keutmann H, Kahn CR. A cascade of tyrosineautophosphorylation in the beta-subunit activates the phosphotransferase of the insulinreceptor. J Biol Chem. 1988: 263(6); 2969-2980.
30) Pawson T, Scott JD. Signaling through scaffold, anchoring, and adaptor proteins. Science.1997: 278; 2075-2080.
31) Nelms K, O'Neill TJ, Li S, Hubbard SR, Gustafson TA, Paul WE. Alternative splicing, genelocalization, and binding of SH2-B to the insulin receptor kinase domain. Mamm.Genome.1999: 10; 1160-1167.
32) Saltiel AR, Kahn CR. Insulin signalling and the regulation of glucose and lipid metabolism.Nature. 2001: 414; 799-806.
33) De Meyts P, Christoffersen CT, Urso B, Wallach B, Gronskov K, Yakushiji F, ShymkoRM. Role of the time factor in signaling specificity: application to mitogenic and metabolicsignaling by the insulin and insulin-like growth factor-I receptor tyrosine kinases.Metabolism. 1995: 44(10, Suppl. 4); 2-11.
34) Olichon-Berthe C, Hauguel -De Mouzon S, Péraldi P, Van Obberghen E, Le Marchand-Brustel Y. Insulin receptor dephosphorylation by phosphotyrosine phosphatases obtainedfrom insulin-resistant obese mice. Diabetologia. 1994: 37; 56-60.
35) Hotamisligil GS, Budavari A, Murray D, Spiegelman BM. Reduced tyrosine kinase activityof the insulin receptor in obesity-diabetes. Central role of tumor necrosis factor-alpha. JClin Invest. 1994: 94(4); 1543–1549.
36) Sadagurski M, Nofech-Mozes S, Weingarten G, White MF, Kadowaki T, Wertheimer E.Insulin receptor substrate 1 (IRS-1) plays a unique role in normal epidermal physiology. JCell Physio. 2007: 213(2); 519-527.
37) Myers MG, Backer JM, Sun XJ, Shoelson S, Hu P, Schlessinger J, Yoakim M,Schaffhausen B, White MF. IRS-1 activates phosphatidylinositol 3'-kinase by associatingwith src homology 2 domains of p85. Proc Natl Acad Sci. 1992: 89(21); 10350-10354.
38) Giovannone B, Scaldaferri ML, Federici M, Porzio O, Lauro D, Fusco A, Sbraccia P,Borboni P, Lauro R, Sesti G. Insulin receptor substrate (IRS) transduction system: distinctand overlapping signalling potential. Diabetes/Metab Res Rev. 2000: 16; 434-441.
39) Backer JM, Shoelson SE, Weiss MA, Hua QX, Cheatham RB, Haring E, Cahill DC, WhiteMF. The insulin receptor juxtamembrane region contains two independent tyrosine/beta-turn internalization signals. J Cell Biol. 1992: 118(4); 831-839.
40) Gual P, Le Marchand-Brustel Y, Tanti JF. Positive and negative regulation of insulinsignaling through IRS-1 phosphorylation. Biochimie. 2005: 87(1); 99-109.

90
41) Tanti JF, Gremeaux T, Van Obberghen E, Le Marchand-Brustel Y. Serine/threoninephosphorylation of insulin receptor substrate 1 modulates insulin receptor signaling, J BiolChem. 1994: 269(8); 6051–6057.
42) Kanety H, Feinstein R, Papa MZ, Hemi R, Karasik A. Tumor necrosis factor alpha-inducedphosphorylation of insulin receptor substrate-1 (IRS-1). Possible mechanism forsuppression of insulin stimulated tyrosine phosphorylation of IRS-1. J Biol Chem. 1995:270(40); 23780–23784.
43) Shulman GI. Cellular mechanisms of insulin resistance. J Clin Invest. 2000: 106(2); 171–176.
44) Haruta T, Uno T, Kawahara J, Takano A, Egawa K, Sharma PM, Olefsky JM, KoboyashiM. A rapamycin-sensitive pathway down-regulates insulin signalling via phosphorylationand proteasomal degradation of insulin receptor substrate-1. Mol Endocrinol. 2000: 14(6);783–794.
45) Aguirre V, Werner ED, Giraud J, Lee YH, Shoelson SE, White MF. Phosphorylation ofSER307 in IRS-1 blocks interactions with the insulin receptor and inhibits insulin action. JBiol Chem. 2002: 277(2); 1531-1537.
46) Andreozzi F, Laratta E, Procopio C, Hribal ML, Sciacqua A, Perticone M, Miele C,Perticone F, Sesti G. Interleukin-6 impairs the insulin signaling pathway, promotingproduction of nitric oxide in human umbilical vein endothelial cells. Mol Cell Biol. 2007:27(6); 2372–2383.
47) Bloch-Damti A, Potashnik R, Gual P, Le Marchand-Brustel Y, Tanti JF, Rudich A, BashanN. Differential effects of IRS1 phosphorylated on Ser307 or Ser632 in the induction ofinsulin resistance by oxidative stress. Diabetologia. 2006: 49(10); 2463-2473.
48) Werner ED, Lee J, Hansen L, Yuan M, Shoelson SE. Insulin resistance due tophosphorylation of insulin receptor substrate-1 at serine 302. J Biol Chem. 2004: 279(34);35298-35305.
49) Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002: 296(5573); 1655-1657.
50) Cohen P. The Croonian Lecture 1998. Identification of a protein kinase cascade of majorimportance in insulin signal transduction. Philos Trans R Soc Lond B Biol Sci. 1999:354(1382); 485-95.
51) Toker A. Phosphoinositides and signal transduction. Cell Mol Life Sci. 2002: 59(5); 761-779.
52) Vanhaesebroeck B, Alessi DR. The PI3K–PDK1 connection: more than just a road to PKB.Biochem J. 2000: 346; 561-576.

91
53) Flynn P, Wong M, Zavar M, Dean NM, Stokoe D. Inhibition of PDK-1 activity causes areduction in cell proliferation and survival. Current Biology. 2000: 10; 1439–1442.
54) Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation ofAkt/PKB by the rictor-mTOR complex. Science. 2005: 307(5712); 1098–1101.
55) Sansal I, Sellers WR. The biology and clinical relevance of the PTEN tumor suppressorpathway. J Clin Oncology. 2004: 22(14); 2954-296.
56) Walker E, Pacold M, Perisic O, Stephens L, Hawkins P, Wymann M, Williams R.Structural determinants of phosphoinositide 3-kinase inhibition by wortmannin, LY294002,quercetin, myricetin, and staurosporine. Molecular Cell. 2000: 6(4); 909-919.
57) Song G, Ouyang G, Bao S. The activation of Akt/PKB signaling pathway and cell survival.Cell Mol Med. 2005: 9(1): 59-71.
58) Laine J, Künstle G, Obata T, Noguchi M. Differential regulation of Akt kinase isoforms bythe members of the TCL1 oncogene family. J Biol Chem. 2002: 277(5); 3743-3751.
59) Franke TF, Sung-Il Y, Chan TO, Data K, Kazlauskas A, Morrison DK, Kapland R, TsichlisPN. The protein kinase encoded by the Akt proto-oncogene is a target of the PDGF-activated phosphatidylinositol 3-kinase. Cell. 1995: 81(5); 727-736.
60. Liu AX, Testa JR, Hamilton TC, Jove R, Nicosia SV, Cheng JQ. AKT2, a member of theprotein kinase B family, is activated by growth factors, v-Ha-ras, and v-src throughphosphatidylinositol 3-kinase in human ovarian epithelial cancer cells. Cancer Res. 1998:58(14); 2973-2977.
61) Lawlor MA, Alessi DR. PKB/Akt: a key mediator of cell proliferation, survival and insulinresponses? J Cell Science. 2001: 114; 2903-2910.
62) Hatano E, Brenner DA. American Akt protects mouse hepatocytes from TNF-α- and Fas-mediated apoptosis through NK-kB activation. Journal of Physiology Gastrointestinal andLiver Physiology. 2001: 44(6); G1357-G1368 (56 ref.)
63) Cho H, Mu J, Kim JK, Thorvaldsen JL, Chu Q, Crenshaw III EB, Kaestner KH, BartolomeiMS, Shulman GI, Birnbaum MJ. Insulin resistance and a diabetes mellitus-like syndrome inmice lacking the protein kinase Akt2 (PKBβ). Science. 2001: 292(5522); 1728–1731.
64) Mîinea CP, Sano H, Kane S, Sano E, Fukuda M, Peränen J, Lane WS, Lienhard GE.AS160, the Akt substrate regulating GLUT4 translocation, has a functional Rab GTPase-activating protein domain. Biochem J. 2005: 391(Pt 1); 87–93.

92
65) Easton RM, Cho H, Roovers K, Shineman DW, Mizrahi M, Forman MS, Lee VMY,Szabolcs M, de Jong R, Oltersdorf T, Ludwig T, Efstratiadis A, Birnbaum MJ. Role forAkt3/protein kinase B γ in attainment of normal brain size. Molecular and CellularBiology. 2005: 25(5); 1869-1878.
66) Cho H, Mu J, Kim JK, Thorvaldsen JL, Chu Q, Crenshaw III EB, Kaestner KH, BartolomeiMS, Shulman GI, Birnbaum MJ. Insulin resistance and a diabetes mellitus-like syndrome inmice lacking the protein kinase Akt2 (PKB beta). Science. 2001: 292(5522); 1728-1731.
67) Cho H, Thorvaldsen JL, Chu Q, Feng F, Birnbaum MJ. Akt1/PKBalpha is required fornormal growth but dispensable for maintenance of glucose homeostasis in mice. J BiolChem. 2001: 276(42); 38349-38352.
68) Chen WS, Xu P-Z, Gottlob K, Chen M-L, Sokol K, Shiyanova T, Roninson I, Weng W,Suzuki R, Tobe K, Kadowaki T, Hay N. Growth retardation and increased apoptosis inmice with homozygous disruption of the Akt1 gene. Genes Dev. 2001: 15(17); 2203-2208.
69) Katome T, Obata T, Matsushima R, Masuyama N, Cantley LC, Gotoh Y, Kishi K, ShiotaH, Ebina Y. Use of RNA interference-mediated gene silencing and adenoviraloverexpression to elucidate the roles of AKT/protein kinase B isoforms in insulin actions. JBiol Chem. 2003: 278(30); 28312-28123.
70) Jiang ZY, Zhou QL, Coleman KA, Chouinard M, Boese Q, Czech MP. Insulin signalingthrough Akt/protein kinase B analyzed by small interfering RNA-mediated gene silencing.Proc Natl Acad Sci. 2003: 100(13); 7569-7574.
71) Sable CL, Filippa N, Hemmings B, Van Obberghen E. cAMP stimulates protein kinase Bin a wortmannin-insensitive manner. FEBS Lett. 1997: 409; 253-257.
72) Filippa N, Sable CL, Filloux C, Hemmings BA, Van Obberghen E. Mechanism of proteinkinase B activation by cyclic AMP-dependent protein kinase. Mol Cell Biol. 1999: 19(7);4989-5000.
73) Perez-Garcia MJ, Cena V, de Pablo Y, Llovera M, Comella JX, Soler RM. Glial cell line-derived neurotrophic factor increases intracellular calcium concentration: Role ofcalcium/calmodulin in the activation of the phosphatidylinositol 3-kinase pathway. J BiolChem. 2004: 279(7); 6132-6142.
74) Gould GW, Holman GD. The glucose transporter family: structure, function and tissue-specific expression. Biochem J. 1993: 295(Pt 2); 329–341.
75) Wood IS, Trayhurn P. Glucose transporters (GLUT and SGLT): expanded families of sugartransport proteins. British J Nutrition. 2003: 89; 3-9.
76. Nishimura H., et al., Kinetics of GLUT1 and GLUT4 glucose transporters expressed inXenopus oocytes. J Biol Chem. 1993: 268(12); 8514-8520.

93
77) Pessin JE, Bell G. Mammalian facultative glucose transporter family: Structure andmolecular regulation. Annual Review of Physiology. 1992: 54; 911-930.
78) Cushman SW, Wardzala LJ. Potential mechanism of insulin action on glucose transport inthe isolated rat adipose cell. Apparent translocation of intracellular transport systems to theplasma membrane. J Biol Chem. 1980: 255(10); 4758-4762.
79) Klip A, Ramlal T, Young DA, Holloszy JO. Insulin-induced translocation of glucosetransporters in rat hindlimb muscles. FEBS Lett. 1987: 224(1); 224-230.
80) Ishiki M, Klip A. Minireview: recent developments in the regulation of glucose transporter-4 traffic: New signals, locations, and partners. Endocrinology. 2005: 146(12); 5071-5078.
81) Stenbit AE, Tsao T-S, Li J, Burcelin R, Geenen DL, Factor SM, Houseknecht K, Katz EB,Charron MJ. GLUT4 heterozygous knockout mice develop muscle insulin resistance anddiabetes. Nat Medicine. 1997: 3(10); 1096-1101.
82) Katz EB, Stenbit AE, Hatton K, Depinhot R, Charron MJ. Cardiac and adipose tissueabnormalities but not diabetes in mice deficient in GLUT4. Nature. 1995: 377(6545); 151-155.
83) Pearson G, Robinson F, Beers Gibson T, Xu BE, Karandikar M, Berman K, Cobb MH.Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions.Endocr Rev. 2001: 22(2); 153–183.
84) Schoeberl B, Eichler-Jonsson C, Gilles ED, Müller G. Computational modeling of thedynamics of the MAP kinase cascade activated by surface and internalized EGF receptors.Nature Biotechnology. 2002: 20; 370-375.
85) Johnson GL, Lapadat R. Mitogen-activated protein kinase pathways mediated by ERK,JNK, and p38 protein kinases. Science. 2002: 298(5600); 1911–1912.
86) Seger R, Krebs EG. The MAPK signaling cascade. FASEB. 1995: 9; 726-735.
87) Gotoh Y, Nishida E. Intracellular Signaling From Ras to Genes: Activation mechanism andfunction of the MAP kinase cascade. Molecular Reproduction and Development. 1995:42(4); 486-492.
88) Takekawa M, Maeda T, Saito H. Protein phosphatase 2C alpha inhibits the human stress-responsive p38 and JNK MAPK pathways. Embo J. 1998: 17(16); 4744-4752.
89) Zhou B, Wang Z-X, Zhao Y, Brautigan DL, Zhang Z-Y. The specificity of extracellularsignal-regulated kinase 2 dephosphorylation by protein phosphatases. J Biol Chem. 2002:277(35); 31818-31825.

94
90) Avdi NJ, Malcom KC, Nick JA, Worthen GS. A role for protein phosphatase-2A in p38mitogen-activated protein kinase-mediated regulation of the c-Jun NH(2)-terminal kinasepathway in human neutrophils. J Biol Chem. 2002: 277(43); 40687-40696.
91) Lang R, Hammer M, Mages J. DUSP meet immunology: Dual specificity MAPKphosphatases in control of the inflammatory response. The Journal of Immunology. 2006:177; 7497-7504.
92) Sharma G-D, He J, Bazan HEP. p38 and ERK1/2 coordinate cellular migration andproliferation in epithelial wound healing. J Biol Chem. 2003: 278(24); 21989-21997.
93) Miranti CK, Brugge JS. Sensing the environment: a historical perspective on integrin signaltransduction. Nature Cell Biology. 2002: 4; E83 - E90.
94) Boulton TG, Cobb MH. Identification of multiple extracellular signal-regulated kinases(ERKs) with antipeptide antibodies. Cell Regulation. 1991: 2; 357-571.
95) Kyriakis JM, App H, Zhang X-F, Banerjee P, Brautigan DL, Rapp UR, Avruch J. Raf-1activates MAP kinase-kinase. Nature. 1992: 358(6385); 417-421.
96) Feldman EL, Russell JW, Greene DA. Diabetic nephropathy in: Principles and practice ofendocrinology and metabolism. Becker KL (ed.). Philadephia PA: Lippincott, Williams &Wilkins, 2001: 1391-1399.
97) Babior BM. The NADPH oxidase of endothelial cells. IUBMB Life. 2000: 50(4-5); 267-269.
98) Fujishiro M, Gotoh Y, Katagiri, H, Sakoda H, Ogihara T, Anai M, Onishi Y, Ono H, AbeM, Shojima N, Fukushima Y, Kikuchi M, Oka Y, Asano T. Three mitogen-activatedprotein kinases inhibit insulin signaling by different mechanisms in 3T3-L1 adipocytes.Molecular Endocrinology. 2003: 17(3); 487–497.
99) Wang CN, O’Brien L, Brindley DN. Effects of cell-permeable ceramides and tumornecrosis factor-α on insulin signaling and glucose uptake in 3T3-L1 adipocytes. Diabetes.1998: 47; 24–31.
100) Paz K, Hemi R, LeRoith D, Karasik A, Elhanany E, Kanety H, Zick Y. A molecular basisfor insulin resistance. Elevated serine/threonine phosphorylation of IRS-1 and IRS-2inhibits their binding to the juxtamembrane region of the insulin receptor and impairs theirability to undergo insulin-induced tyrosine phosphorylation. J Biol Chem. 1997: 272(47);29911-29918.
101) Willoughby EA, Perkins GR, Collins MK, Whitmarsh AJ. The JNK-interacting protein-1scaffold protein targets MAPK phosphatase-7 to dephosphorylate JNK. J Biol Chem.2003: 278(12); 10731–10736.

95
102) Hurd C, Rozengurt E. Protein kinase D is sufficient to suppress EGF-induced c-Jun Ser 63phosphorylation. Biochemical and Biophysical Research Communications. 2001: 282(2);404-408.
103) Yu J, Liu X-W, Kim HRC. Platelet-derived growth factor (PDGF) receptor-α-activatedc-Jun NH2-terminal kinase-1 is critical for PDGF-induced p21WAF1/CIP1 promoter activityindependent of p53. J Biol Chem. 2003: 278(49); 49582-49588.
104) Aguirre V, Werner ED, Giraud J, Lee YH, Shoelson SE, White MF. Phosphorylation ofSer307 in insulin receptor substrate-1 blocks interactions with the insulin receptor andinhibits insulin action. J Biol Chem. 2002: 277(2); 1531-1537.
105) Gupta S, Barrett T, Whitmarsh AJ, Cavanagh J, Sluss HK, Derijard B, Davis RJ. Selectiveinteraction of JNK protein kinase isoforms with transcription factors. Embo J. 1996:15(11); 2760-2770.
106) Guan Z, Buckman SY, Pentland AP, Templeton DJ, Morrison AR. Induction ofcyclooxygenase-2 by the activated MEKK1 SEK1/MKK4 p38 mitogen-activatedprotein kinase pathway. J Biol Chem. 1998: 273(21); 12901-12908.
107) Tournier C, Dong C, Turner TK, Jones SN, Flavell RA, Davis RJ. MKK7 is an essentialcomponent of the JNK signal transduction pathway activated by proinflammatorycytokines. Genes Dev. 2001: 15(11); 1419-1426.
108) Lawler S, Fleming Y, Goedert M, Cohen P. Synergistic activation of SAPK1/JNK1 byTwo MAP kinase kinases in vitro. Curr Biol. 1998: 8(25); 1387-1390.
109) Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell. 2000: 103; 239–252.
110) Wellen KE, Gokhan S, Hotamisligil S. Obesity-induced inflammatory changes in adiposetissue. J Clin Invest. 2003: 112(12); 1785-1788.
111) Tuncman G, Hirosumi J, Solinas G, Chang L, Karin M, Hotamisligil GS. Functional invivo interactions between JNK1 and JNK2 isoforms in obesity and insulin resistance. ProcNatl Acad Sci. 2006: 103(28); 10741-10746.
112) Saklatvala J. The p38 MAP kinase pathway as a therapeutic target in inflammatorydisease. Curr Opin Pharmacol. 2004: 4(4); 372-377.
113) Han J, Lee JD, Bibbs L, Ulevitch RJ. A MAP kinase targeted by endotoxin andhyperosmolarity in mammalian cells. Science. 1994: 265(5173); 808-811.

96
114) Jiang Y, Chen C, Li Z, Guo W, Gegner JA, Lin S, Han J. Characterization of the structureand function of a new mitogen-activated protein kinase (p38 beta). J Biol Chem. 1996:271(30); 17920-17926.
115) Lechner C, Zahalka MA, Giot JF, Moller NP, Ullrich A. ERK6, a mitogen-activatedProtein kinase involved in C2C12 myoblast differentiation. Proc Natl Acad Sci.1996: 93(9); 4355-4359.
116) van der Houven van Oordt W, Diaz-Meco MT, Lozano J, Krainer AR, Moscat J, CáceresJF. The MKK(3/6)-p38-signaling cascade alters the subcellular distribution of hnRNP A1and modulates alternative splicing regulation. J Cell Biol. 2000: 149(2); 307-316.
117) Roux PP, Blenis J. ERK and p38 MAPK-activated protein kinases: a family of proteinkinases with diverse biological functions. Microbiology and Molecular Biology Reviews.2004: 68(2); 320-344.
118) Cook R, Wu CC, Kang YJ, Han J. The role of the p38 pathway in adaptive immunity.Immunology. 2007: 4(4); 253-259.
119) Alvaro C, Teruel T, Hernandez R, Lorenzo M. Tumor necrosis factor α produces insulinresistance in skeletal muscle by activation of inhibitor κB kinase in a p38 MAPK-dependent manner. J Biol Chem. 2004: 279(17); 17070-17078.
120. Carlson CJ, Koterski S, Sciotti RJ, Poccard GB. Enhanced basal activation of mitogen-activated protein kinases in adipocytes from type 2 diabetes: Potential role of p38 in thedownregulation of GLUT4 expression. Diabetes. 2003: 52(3); 634-641.
121) Dandona P, Aljada A, Bandyopadhyay A. Inflammation: the link between insulinresistance, obesity and diabetes. Trends in Immunology. 2004: 25(1); 4-7.
122) Yamauchi T, et al. The fat-derived hormone adiponectin reverses insulinresistance associated with both lipoatrophy and obesity. Nature Medicine. 2001: 7; 941-946.
123) Hotamisligil GS, Peraldi P, Budavari A, Ellis R, White MF, Spiegelman BM. IRS-1-mediated inhibition of insulin receptor tyrosine kinase activity in TNF-α- and obesity-induced insulin resistance. Science. 1996: 271(5249); 665–670.
124) Goodyear LJ, Giorgino F, Sherman LA, Carey J, Smith RJ, Dohm GL. Insulin receptorphosphorylation, insulin receptor substrate-1 phosphorylation, and phosphatidylinositol 3-kinase activity are decreased in intact skeletal muscle strips from obese subjects. J ClinInvest. 1995: 95(5); 2195–2204.
125) Borst SE. The role of TNF-α in insulin resistance. Endocrine. 2004: 23(2-3); 177-182.

97
126) Xu H, Dembski M, Yang QID, Moriarty A, Tayber O, Chen H, Kapeller R, Tartaglia LA.Dual specificity mitogen-activated protein (MAP) kinase phosphatase-4 plays a potentialrole in insulin resistance. J Biol Chem. 2003: 278(32); 30187-30192.
127) Yu C, Chen Y, Cline GW, Zhang D, Zong H, Wang Y, Bergeron R, Kim JK, CushmanSW, Cooney GJ, Atcheson B, White MF, Kraegen EW, Shulman GI. Mechanism bywhich fatty acids inhibit insulin activation of insulin receptor substrate-1 (IRS-1)-associated phosphatidylinositol 3-kinase activity in muscle. J Biol Chem. 2002: 277(52);50230-50236.
128) Zick Y. Insulin resistance: a phosphorylation-based uncoupling of insulin signalling.Trends in Cell Biology. 2001: 11(11); 437-441.
129) Shao J, Yamashita H, Qiao L, Friedman JE. Decreased Akt kinase activity and insulinresistance in C57BL/KsJ-Leprdb/db mice. J Endocrin. 2000: 167; 107-115.
130) Rosenbaum D, Haber RS, Dunaif A. Insulin resistance in polycystic ovary syndrome:decreased expression of GLUT-4 glucose transporters in adipocytes. Am J Physio Endo &Metab. 1993: 264(2); E197-E202.
131) Alexander CM, Landsman PB, Teutsch SM, Haffner SM. NCEP-defined metabolicsyndrome, diabetes, and prevalence of coronary heart disease among NHANES IIIparticipants age 50 years and older. Diabetes. 2003: 52(5); 1210-1214.
132) Hypertension in diabetes study (HDS): I. Prevalence of hypertension in newly presentingtype 2 diabetic patients and the association with risk factors for cardiovascular anddiabetic complications. J Hypertens. 1993: 11(3); 309-317.
133) Reaven GM, Lithell H, Landsberg L. Hypertension and associated metabolicabnormalities--the role of insulin resistance and the sympathoadrenal system. N Eng JMed. 1996: 334(6); 374-381.
134) Landsberg L. Insulin resistance and hypertension. Clin Exp Hypertens. 1999: 21(5-6);885-894.
135) Baron AD. Hemodynamic actions of insulin. Am J Physiol Endocrinol Metab. 1994: 267;E187-E202.
136) Xiang L, Naik JS, Abram SR, Hester RL. Chronic hyperglycemia impairs functionalvasodilation via increasing thromboxane-receptor-mediated vasoconstriction. Am JPhysiol Heart Circ Physiol. 2007: 292; H231-H236.
137) Dzau VJ, Braun-Dullaeus RC, Sedding DG. Vascular proliferation and atherosclerosis:new perspectives and therapeutic strategies. Nat Med. 2002: 8(11); 1249-1256.
138) Julius, S. The hemodynamic link between insulin resistance and hypertension. JHypertens. 1991: 9(11); 983-986.

98
139) Izawa Y, et al. ERK1/2 activation by angiotensin II inhibits insulin-induced glucoseuptake in vascular smooth muscle cells. Exp Cell Res. 2005: 308(2); 291-299.
140) Zhou M-S, Hernandez Schulman I, Raij L. Role of angiotensin II and oxidative stress invascular insulin resistance linked to hypertension. Am J Physiol Heart Circ Physiol. 2009:296; H833-H839.
141) Wei Y, Sowers JR, Clark SE, Li W, Ferrario CM, Stump CS. Angiotensin II-inducedskeletal muscle insulin resistance mediated by NF-κB activation via NADPH oxidase. AmJ Physiol Endocrinol Metab. 2009: 294; E345-E351.
142) Fleming I, Kohlstedt K, Busse R. The tissue renin-angiotensin system and intracellularsignalling. Curr Opin Nephrol Hypertens. 2006: 15(1); 8-13.
143) Goossens GH, Blaak EE, van Baak MA. Possible involvement of the adipose tissue renin-angiotensin system in the pathophysiology of obesity and obesity-related disorders. ObesRev. 2003: 4(1); 43-55.
144) Ogawa T, Linz W, Schölkens BA, de Bold AJ. Variable renal atrial natriuretic factor geneexpression in hypertension. Hypertension. 1999: 33; 1342-1347.
145) Mercure C, Ramla D, Garcia R, Thibault G, Deschepper CF, Reudelhuber TL. Evidencefor intracellular generation of angiotensin II in rat juxtaglomerular cells. FEBS Lett. 1998:422(3); 395-399.
146) Flather M, Yusuf S, Køber L, Pfeffer M, Hall A, Murray G, Torp-Pedersen C, Ball S,Pogue J, Moyé L. Long-term ACE-inhibitor therapy in patients with heart failure or left-ventricular dysfunction: a systematic overview of data from individual patients. Lancet.2000: 355(9215); 1575-158.
147) Taal MW, Brenner BM. Combination ACEI and ARB therapy: Additional benefit inrenoprotection? Current Opinion in Nephrology and Hypertension. 2002: 11(4); 377-381.
148) Munzel T, Keaney Jr. JF. Are ACE inhibitors a “magic bullet” against oxidative stress?Circulation. 2001: 104(13); 1571-1574.
149) Ehlers MR, Riordan JF. Angiotensin-converting enzyme: new concepts concerning itsbiological role. Biochemistry. 1989: 28(13); 5311-5318.
150) Ullian ME, Hutchison FN, Hazen-Martin DJ, Morinelli TA. Angiotensin II-aldosteroneinteractions on protein synthesis in vascular smooth muscle cells. Am J Physiol CellPhysiol. 1993: 264; C1525-C1531.
151) Bottari SP, de Gasparo M, Steckelings UM, Levens NR. Angiotensin II receptor subtypes:characterization, signalling mechanisms, and possible physiological implications. FrontNeuroendocrinol. 1993: 14(2); 123-171.

99
152) Kaschina E, Unger T. Angiotensin AT1/AT2 receptors: regulation, signalling andfunction. Blood Press. 2003: 12(2); 70-88.
153) Edwards RM, Stack EJ. Angiotensin II inhibits glomerular adenylate cyclase via theangiotensin II receptor subtype 1 (AT1). J Pharm and Experimental Therapeutics. 1993:266(2); 506-510.
154) Kats JP, de Lannowy LM; Jan Danser AH; van Meegen JR; Verdouw PD, SchalekampMADH. Angiotensin II type 1 (AT1) receptor–mediated accumulation of angiotensin II intissues and its intracellular half-life in vivo. Hypertension. 1997; 30; 42-49.
155) Schupp M, Janke J, Clasen R, Unger T, Kintscher U. Angiotensin type 1 receptor blockersinduce peroxisome proliferator–activated receptor-γ activity. Circulation. 2004: 109;2054-2057.
156) AbdAlla S, Lother H, Abdel-tawab Am, Quitterer U. The angiotensin II AT2 receptor is anAT1 receptor antagonist. J Biol Chem. 2001: 276(43); 39721-39726.
157) Hiyoshi H, Yayama K, Takano M, Okamoto H. Stimulation of cyclic GMP production viaAT2 and B2 receptors in the pressure-overloaded aorta after banding. Hypertension. 2004:43; 1258.
158) Ferrario CM. Angiotension-(1-7) and antihypertensive mechanisms. J Nephrol. 1998:11(6); 278-283.
159) Santos RAS, Simoes e Silva AC, Maric C. Angiotensin-(1–7) is an endogenous ligand forthe G protein-coupled receptor Mas. Proc Natl Acad Sci. 2003: 100(14); 8258-8263.
160) Sharma JN. Role of tissue kallikrein–kininogen–kinin pathways in the cardiovascularsystem. Archives of Medical Research. 2006: 37; 299–306.
161) Nagayasa T, Nagasawa S. Studies of human kininogen. Isolation, characterization, andcleavage by plasma kallikrein of high molecular weight (HMW) kininogen. J Biochem.1979: 85; 249–258.
162) Cochrane CG, Revak SD, Wuepper D. Activation of hageman factor in solid and fluidphase. A critical role of kallikrein. J Exp Med. 1973: 138; 1564–1583.
163) Kato H, Enjyoji K. Hydroxylated kininogens and kinins. In: Fritz H, Müller-Esterl W,Jochum M, Roscher A, Luppertz K (eds). Recent Progress on Kinins. Biochemistry andMolecular Biology of the Kallikrein–Kinin System. Birkhèuser-Verlag, Basel. 1992: 217–224.
164) Campbell DJ. The kallikrein-kinin system in humans. Clinical and ExperimentalPharmacology and Physiology. 2001: 28; 1060–1065.

100
165) Cruzblanca H, Koh D-S, Hille B. Bradykinin inhibits M current via phospholipase C andCa2+ release from IP3-sensitive Ca2+ stores in rat sympathetic neurons. Proc Natl Acad Sci.1998: 95(12); 7151-7156.
166) Kobayashi N, Honda T, Yoshida K, Nakano S, Ohno T, Tsubokou Y, Matsuoka H.Critical role of bradykinin-eNOS and oxidative stress-LOX-1 pathway in cardiovascularremodeling under chronic angiotensin-converting enzyme inhibition. Atherosclerosis.
2006: 187(1); 92-100.
167) Sharma JN. Involvement of the kinin-forming system in physiopathology of rheumatoidinflammation. Agents Actions. 1992: 38(III); 343–361.
168) Bunning P, Holmquist B, Riordan JF. Substrate specificity and kinetic characteristics ofangiotensin converting enzyme. Biochemistry. 1983: 22(1); 103-110.
169) Jaspard E, Alhenc-Gelas F. Catalytic properties of the two active sites of angiotensin I-converting enzyme on the cell surface. Biochem Biophys Res Commun. 1995: 211(2);528-534.
170) Ostman J. Beta-adrenergic blockade and diabetes mellitus. A review. Acta Med ScandSuppl. 1983: 672; 69-77.
171) Furman BL. Impairment of glucose tolerance produced by diuretics and other drugs.Pharmacol Ther. 1981: 12(3); 613-649.
172) Harada N, Takishita E, Ishimura N, Minami A, Sakamoto S, Nakaya Y. Combined effectof ACE inhibitor and exercise training on insulin resistance in type 2 diabetic rats. LifeSciences. 2002: 70(15); 1811-1820.
173) Heinemann L, Heise T, Ampudia J, Sawicki P, Sindelka G, Brunner G, Starke AA.Four week administration of an ACE inhibitor and a cardioselective beta-blocker inhealthy volunteers: no influence on insulin sensitivity. Euro J ClinicalInvestigation. 1995: 25(8); 595-600.
174) Weinsaft JW, O'Rourke MF, Nichols WW, Sharma AM, Pischon T, Engeli S, Gavras H,Yusuf S, Francis GS. Effect of ramipril on cardiovascular events in high-risk patients. NEng J Med. 2000: 343; 64-66.
175) Campbell DJ, Dixon B, Kladis A, Kemme M, Santamaria JD. Activation of the kallikrein-kinin system by cardiopulmonary bypass in humans. Am J Physiol Regul Integr CompPhysiol. 2001: 281(4); R1059-70.
176) Tom B, de Vries R, Saxena PR, Jan Danser AH. Bradykinin potentiation by angiotensin-(1-7) and ACE inhibitors correlates with ACE C- and N-domain blockade. Hypertension.2001: 38(1); 95-99.

101
177) Goldman EL. ARB increases insulin sensitivity in hypertensives. Internal Medicine News.2006: 39(17); 40.
178) Damas J, Bourdon V, Lefebvre PJ. Insulin sensitivity, clearance and release in kininogen-deficient rats. Exp Physiol. 1999: 84(3); 549-557.
179) Kudoh A, Matsuki A. Effects of angiotensin-converting enzyme inhibitors on glucoseuptake. Hypertension. 2000: 36(2); 239-244.
180) Henriksen EJ, Jacob S, Augustin HJ, Dietze GJ. Glucose transport activity in insulin-resistant rat muscle. Effects of angiotensin-converting enzyme inhibitors and bradykininantagonism. Diabetes. 1996: 45(Suppl 1); S125-128.
181) Wang CH, Leung N, Lapointe N, Szeto L, Uffelman KD, Giacca A, Rouleau JL, LewisGF. Vasopeptidase inhibitor omapatrilat induces profound insulin sensitization andincreases myocardial glucose uptake in Zucker fatty rats: Studies comparing avasopeptidase inhibitor, angiotensin-converting enzyme inhibitor, and angiotensin II typeI receptor blocker. Circulation. 2003: 107(14); 1923-1929.
182) Engeli S, Bohnke J, Gorzelniak K, Janke J, Schling P, Bader M, Luft FC, Sharma AM.Weight loss and the renin-angiotensin-aldosterone system. Hypertension. 2005: 45(3);356-362.
183) Sharma JN, Uma K, Yusof AP. Left ventricular hypertrophy and its relation to the cardiackinin-forming system in hypertensive and diabetic rats. Int J Cardiol. 1998: 63(3); 229-235.
184) Miyata T, Taguchi T, Uehara M, Isami S, Kishikawa H, Kaneko K, Araki E, Shichiri M.Bradykinin potentiates insulin-stimulated glucose uptake and enhances insulin signalthrough the bradykinin B2 receptor in dog skeletal muscle and rat L6 myoblasts. Euro JEndo. 1998: 138; 344–352.
185) Isami S, Kishikawa H, Araki E, Uehara M, Kaneko K, Shirotani T, Todaka M, Ura S,Motoyoshi S, Matsumoto K, Miyamura N, Shichiri M. Bradykinin enhances GLUT4translocation through the increase of insulin receptor tyrosine kinase in primaryadipocytes: evidence that bradykinin stimulates the insulin signalling pathway.Diabetologia. 1996: 39; 412-420.
186) Beard KM, Lu H, Ho K, Fantus IG. Bradykinin augments insulin-stimulated glucosetransport in rat adipocytes via endothelial nitric oxide synthase–mediated inhibition of JunNH2-terminal kinase. Diabetes. 2006: 55; 2678-2687.
187) Schroeder RA, Kuo PC. Nitric oxide: Physiology and pharmacology. Anesthesia &Analgesia. 1995: 81; 3052-9.
188) Nathan C. Nitric oxide as a secretory product of mammalian calls. FASEB. 1992: 6; 3051-64.

102
189) Henry Y, Lepoivre M, Drapier JC, Ducrocq C, Boucher JL, Guissani A. EPRcharacterization of molecular targets for NO in mammalian cells and organelles. FASEB.1993: 7; 1124-34.
190) Michel T, Feron O. Perspective series: Nitric oxide and nitric oxide synthases. J ClinInvest. 1997: 100(9); 2146–2152.
191) Marlettaz MA. Nitric oxide synthase structure and mechanism. J Biol Chem. 1993:268(17); 12231-12234.
192) Young ME, Radda GK, Leighton B. Nitric oxide stimulates glucose transport andmetabolism in rat skeletal muscle in vitro. Biochem. J. 1997: 322; 223-228.
193) Dallaire P, Marette A. Obesity-linked insulin resistance: Is nitric oxide the missing link?Canad J Diabetes. 2004: 28(1); 59-66.
194) Dinh DT, Frauman AG, Johnston CI, Fabiani ME. Angiotensin receptors: distribution,signalling and function. Clinical Science. 2001: 100; 481–492.
195) Denninger JW, Marletta MA. Guanylate cyclase and the .NO/cGMP signaling pathway.Biochim Biophys Acta. 1999: 1411(2-3); 334-350.
196) Kharitonov VG, Sharma VS, Pilz RB, Magde D, Koesling D. Basis of guanylate cyclaseactivation by carbon monoxide.. Proc Natl Acad Sci. 1995: 92(7); 2568–2571.
197) Garbem DL, Lowell DG. Guanylyl cyclase receptors. J Biol Chem. 1994: 269(49); 30741-30744.
198) Hobbs AJ. Soluble guanylate cyclase: the forgotten sibling. Trends PharmSciences. 1997: 18(4); 484-491.
199) Ignarro LJ, Degnan JN, Baricos WH, Kadowitz PJ, Wolin MS. Activation of purifiedguanylate cyclise by nitric oxide requires heme. Comparison of heme-deficient, heme-reconstituted and heme-containing forms of soluble enzyme from bovine lung. BiochemBiophys Acta. 1982: 718(1); 49-59.
200) Wedel B, Harteneck C, Foerster J, Friebe A, Schultz G, Koesling D. Functional domainsof soluble guanylyl cyclise. J Biol Chem. 1995: 270(42); 24871-24875.
201) Wilson EM, Chinkers M. Identification of sequences mediating guanylyl cyclisedimerization. Biochemistry. 1995: 34(14); 4696-4701.
202) Friebe A, Koesling D. Mechanism of YC-1-induced activation of soluble guanylylcyclase. Molecular Pharm. 1998: 53; 123–127.
203) Zhao Y, Brandish PE, DiValentin M, Schelvis JPM, Babcock GT, Marletta MA.Inhibition of soluble guanylate cyclase by ODQ. Biochemistry. 2000: 39(35); 10848-10854.

103
204) Potter LR, Abbey-Hosch S, Dickey DM. Natriuretic peptides, their receptors, and cyclicguanosine monophosphate-dependent signaling functions. Endocrine Reviews. 2006:27(1); 47–72.
205) Lohse MJ, Förstermann U, Schmidt HHHW. Pharmacology of NO:cGMP signaltransduction. Naunyn-Schmiedeberg’s Arch Pharmacol. 1998: 358; 111–112.
206) Omori K, Kotera J. Overview of PDEs and their regulation. Circ Res. 2007: 100; 309-327.
207) Gresser CH, Gleiter CH. Erectile dysfunction: Comparison of efficacy and side effects ofthe PDE-5 inhibitors sildenafil, vardenafil and tadalafil review of the literature. Eur J MedRes. 2002: 7; 435-446.
208) Jackson G. Vardenafil--PDE5 inhibitor number 3. Int J Clin Pract. 2004: 58(3); 230-239.
209) Ahmad M, Abdel-Wahab YH, Tate R, Flatt PR, Pyne NJ, Furman BL. Effect of type-selective inhibitors on cyclic nucleotide phosphodiesterase activity and insulin secretion inthe clonal insulin secreting cell line BRIN-BD11. British J Pharm. 2000: 129(6); 1228-1234.
210) Francis Sh, Corbin JD. Structure and function of cyclic nucleotide-dependent proteinkinases. Annu Rev Physiol. 1994: 56; 237-372.
211) Hofmann F, Feil R, Kleppisch T, Schlossmann J. Function of cGMP-dependent proteinkinases as revealed by gene deletion. Physiol Rev. 2006: 86; 1–23.
212) Feil S, Zimmermann P, Knorn A, Brummer S, Schlossmann J, Hofmann F, and Feil R.Distribution of cGMP-dependent protein kinase type I in the mouse CNS and eye.Neuroscience. 2005: 135(3); 863-868.
213) Keilbach A, Ruth P, and Hofmann F. Detection of cGMP dependent protein kinaseisozymes by specific antibodies. Eur J Biochem. 1992: 208; 467–473.
214) De Vente J, Asan E, Gambaryan S, Markerink-van Ittersum M, Axer H, Gallatz K,Lohmann SM, and Palkovits M. Localization of cGMP-dependent protein kinase type II inrat brain. Neuroscience. 2001: 108; 27–49.
215) el-Husseini AE, Bladen C, Vincent SR. Molecular characterization of a type II cyclicGMP-dependent protein kinase expressed in the rat brain. J Neurochem. 1995: 64; 2814–2817.
216) Gudi T, Lohmann SM, Pilz RB. Regulation of gene expression by cyclic GMP-dependentprotein kinase requires nuclear translocation of the kinase: identification of a nuclearlocalization signal. Molecular & Cellular Biology. 1997: 17(9); 5244-5254.

104
217) Jiang LH, Gawler DJ, Hodson N, Milligan CJ, Pearson HA, Porter V, Wray D. Regulationof cloned cardiac L-type calcium channels by cGMP-dependent protein kinase. J BiolChem. 2000: 275; 6135-6143.
218) Lincoln TM, Dey N, Sellak H. Invited review: cGMP-dependent protein kinase signalingmechanisms in smooth muscle: from the regulation of tone to gene expression. J AppliedPhysio. 2001: 91; 1421-1430.
219) Shen J, Harada N, Nakazawa H, Yamashita T. Involvement of the nitric oxide-cyclicGMP pathway and neuronal nitric oxide synthase in ATP-induced Ca2+ signalling incochlear inner hair cells. Euro J Neuroscience. 2005: 21(11); 2912-2922.
220) Schlossmann J, Feil R, Hofmann F. Insights into cGMP signalling derived from cGMPkinase knockout mice. Frontiers in Bioscience. 2005: 10; 1279-1289.
221) Das A, Xi L, Kukreja RC. Protein Kinase G-dependent Cardioprotective Mechanism ofPhosphodiesterase-5 Inhibition Involves Phosphorylation of ERK and GSK3β. Biol Chem.2008: 283(43); 29572-29585.
222) Ayala JE, Bracy DP, Julien BM, Rottman JN, Fueger PT, Wasserman DH. Chronictreatment with sildenafil improves energy balance and insulin action in high fat-fedconscious mice. Diabetes. 2007: 56(4); 1025-1033.
223) Lee YH, Giraud J, Davis RJ, White MF. c-jun N-terminal kinase (JNK) mediates feedbackinhibition of the insulin signaling cascade. J Biol Chem. 2003: 278(5); 2896-2902.
224) Beard KM, Lu H, Ho K, Fantus IG. Bradykinin augments insulin-stimulated glucosetransport in rat adipocytes via endothelial nitric oxide synthase–mediated inhibition of junNH2-terminal kinase. Diabetes. 2006: 55; 2678-2687.
225) Ren Y, Garvin J, Carretero OA. Mechanism involved in bradykinin-induced efferentarteriole dilation. Kidney International. 2002: 62; 544-549.
226) Koller KJ, Lowe DG, Bennett GL, Minamino N, Kangawa K, Matsuo H, Goeddel DV.Selective activation of the B natriuretic peptide receptor by C-type natriuretic peptide(CNP). Science. 1991: 252; 120–123.
227) Suga S, Nakao K, Hosoda K, Mukoyama M, Ogawa Y, Shirakami G, Arai H, Saito Y,Kambayashi Y, Inouye K, Imura H. Receptor selectivity of natriuretic peptide family,atrial natriuretic peptide, brain natriuretic peptide, and C-type natriuretic peptide.Endocrinology. 1992: 130; 229-239.
228) Pagano M, Anand-Srivastava MB. Cytoplasmic domain of natriuretic peptide receptor-C(NPR-C) constitutes Gi-activator sequences that inhibit adenylyl cyclase activity. J BiolChem. 2001: 276(25); 22064-70.

105
229) William M, Hamilton EJ, Garcia A, Bundgaard H, Chia KKM, Figtree GA, RasmussenHH. Natriuretic peptides stimulate the cardiac sodium pump via NPR-C-coupled NOSactivation. Am J Physiol Cell Physiol. 2008: 294; C1067-C1073.
230) Costa MA, Elesgaray R, Balaszczuk AM, Arranz C. Role of NPR-C natriuretic receptor innitric oxide system activation induced by atrial natriuretic peptide. Regul Pept. 2006:135(1-2); 63-68.
231) Chen HH, Lainchbury JG, Burnett JC. Natriuretic peptide receptors and neutralendopeptidase in mediating the renal actions of a new therapeutic synthetic natriureticpeptide dendroaspis natriuretic peptide. J Am Coll Cardiol. 2002: 40; 1186-1191.
232) de Bold AJ, de Bold ML, Sarda IR. Functional-morphological studies on in vitrocardionatrin release. J Hypertens Suppl. 1986: 4; S3–S7.
233) Edwards BS, Zimmerman RS, Schwab TR, Heublein DM, Burnett Jr JC. Atrial stretch,not pressure, is the principal determinant controlling the acute release of atrial natriureticfactor. Circ Res. 1988: 62; 191–195.
234) Yan W, Wu F, Morser J, Wu Q. Corin, a transmembrane cardiac serine protease, acts as apro-atrial natriuretic peptide-converting enzyme. Proc Natl Acad Sci. 2000: 97; 8525–8529.
235) Bai GY, Piao FL, Kim SY, Yuan K, Kim SZ, Kim SH. Augmentation of insulin-stimulated ANP release through tyrosine kinase and PI 3-kinase in diabetic rats. Peptides.2006: 27; 2756–2763.
236) Rosenzweig A, Seidman CE. Atrial natriuretic factor and related peptide hormones. AnnuRev Biochem. 1991: 60; 229-255.
237) Jarolim P. Serum biomarkers for heart failure. Cardiovascular Pathology. 2006: 15(3);144-149.
238) Sengenes C, Zakaroff-Girard A, Moulin A, Berlan M, Bouloumie A, Lafontan M,Galitzky J. Natriuretic peptide-dependent lipolysis in fat cell is a primate specificity. Am JPhysiol Regul Integr Comp Physiol. 2002: 283(1); R257-R265.
239) Moro C, Klimcakova E, Lolmède K, Berlan M, Lafontan M, Stich V, Bouloumié A,Galitzky J, Arner P, Langin D. Atrial natriuretic peptide inhibits the production ofadipokines and cytokines linked to inflammation and insulin resistance in humansubcutaneous adipose tissue. Diabetologia. 2007: 50; 1038–1047.
240) Birkenfeld AL, Budziarek P, Boschmann M, Moro C, Adams F, Franke G, Berlan M,Marques MA, Sweep FCGJ, Luft FC, Lafontan M, Jordan J. Atrial natriuretic peptideinduces postprandial lipid oxidation in man. Diabetes. 2008: 57(12); 3199-3204.
241) Sudoh T, Kangawa K, Minamino N, Matsuo H. A new natriuretic peptide in porcine brain.Nature. 1988: 332; 78–81.

106
242) Seilhamer JJ, Arfsten A, Miller JA, Lundquist P, Scarborough RM, Lewicki JA, PorterJG. Human and canine gene homologs of porcine brain natriuretic peptide. BiochemBiophys Res Commun. 1989: 165; 650–658.
243) Ogawa Y, Itoh H, Tamura N, Suga S, Yoshimasa T, Uehira M, Matsuda S, Shiono S,Nishimoto H, Nakao K. Molecular cloning of the complementary DNA and gene thatencode mouse brain natriuretic peptide and generation of transgenic mice that overexpressthe brain natriuretic peptide gene. J Clin Invest. 1994: 93; 1911–1921.
244) Hirose M, Furukawa Y, Miyashita Y, Kurogouchi F, Nakajima K, Tsuboi M, Chiba S.CNP causes receptor-mediated positive dromotropic effects in anesthetized dog hearts.Am J Physiol Heart Circ Physiol. 1998: 275; H717-H720.
245) Budeus M, Salibassoglu E, Schymura AM, Reinsch N, Wieneke H, Sack S, Erbel R.Effect of induced ventricular fibrillation and shock delivery on brain natriuretic peptidemeasured serially following a predischarge ICD test. Indian Pacing Electrophysiol J.2007: 7(4); 195–203.
246) Hagiwara H, Sakaguchi H, Itakura M, Yoshimoto T, Furuya M, Tanaka S, Hirose S.Autocrine regulation of rat chondrocyte proliferation by natriuretic peptide C and itsreceptor, natriuretic peptide receptor-B. J Biol Chem. 1994: 269; 10729–10733.
247) Hagiwara H, Sakaguchi H, Lodhi KM, Suda K, Hirose S. Subtype switching of natriureticpeptide receptors in rat chondrocytes during in vitro culture. J Biochem. 1994: 116(3);606–609.
248) Suga S, Nakao K, Itoh H, Komatsu Y, Ogawa Y, Hama N, Imura H. Endothelialproduction of C-type natriuretic peptide and its marked augmentation by transforminggrowth factorβ: Possible existence of “vascular natriuretic peptide system.” J Clin Invest.1992: 90; 1145–1149.
249) Teerijoki H, Krasnov A, Pitkänen TI, Mölsä H. Cloning and characterization of glucosetransporter in teleost fish rainbow trout (Oncorhynchus mykiss). Biochimica et BiophysicaActa (BBA) - Gene Structure and Expression. 2000: 1494(3); 290-294.
250) Fine JB, DiGirolamo M. A simple method to predict cellular density in adipocytemetabolic incubations. International Journal of Obesity. 1997: 21(9); 764-768.
251) Wijdicks EFM, Schievnik WI, Burnett JC. Natriuretic peptide system and endothelin inaneurysmal subarachnoid hemorrhage. Journal of Neurosurgery. 1997: 87(2); 275-280.
252) Nishikimi T, Iemura-Inaba C, Akimoto K, Ishikawa K, Koshikawa S, Matsuoka H.Stimulatory and Inhibitory Regulation of Lipolysis by the NPR-A/cGMP/PKG and NPR-C/G(i) Pathways in Rat Cultured Adipocytes. Regul Pept. 2009: 153(1-3); 56-63.

107
253) William M, Hamilton EJ, Garcia A, Bundgaard H, Chia KKM, Figtree GA, RasmussenHH. Natriuretic peptides stimulate the cardiac sodium pump via NPR-C-coupled NOSactivation. Am J Physiol Cell Physiol. 2008: 294; C1067-C1073.
253) Moro C, Klimcakova E, Lafontan M, Berlan M, Galitzky J. Phosphodiesterase-5A andneutral endopeptidase activities in human adipocytes do not control atrial natriureticpeptide-mediated lipolysis. British J Pharm. 2007: 152(7); 1102–1110.
254) Serulle Y, Zhang S, Ninan I, Puzzo D, McCarthy M, Khatri L, Arancio O, Ziff EB. Anovel GluR1-cGKII interaction regulates AMPA receptor trafficking. Neuron. 2007:56(4); 670–688.
255) Burkhardt M, Glazova M, Gambaryan S, Vollkommer T, Butt E, Bader B, Heermeier K,Lincoln TM, Walter U, Palmetshofer A. KT5823 inhibits cGMP-dependent protein kinaseactivity in vitro but not in intact human platelets and rat mesangial cells. J Biol Chem.2000: 275(43); 33536-41.
256) Daval M, Diot-Dupuy F, Bazin R, Hainault I, Viollet B, Vaulont S, Hajduch E, Ferre, P,Foufelle F. Anti-lipolytic action of AMP-activated protein kinase in rodent adipocytes. JBiol Chem. 2005: 280; 25250-25257.
257) Chuang S-M, Liou G-Y, Yang J-L. Activation of JNK, p38 and ERK mitogen-activatedprotein kinases by chromium(VI) is mediated through oxidative stress but does not affectcytotoxicity. Carcinogenesis. 2000: 21(8); 1491-1500.
258) Fürst R, Brueckl C, Kuebler WM, Zahler S, Krötz F, Görlach A, Vollmar AM, KiemerAK. Atrial natriuretic peptide induces mitogen-activated protein kinase phosphatase-1 inhuman endothelial cells via Rac1 and NAD(P)H oxidase/Nox2-activation. Circ Res. 2005:96(1); 43-53.
259) Abel ED, Peroni O, Kim JK, Kim YB, Boss O, Hadro E, Minnemann T, Shulman GI,Kahn BB. Adipose-selective targeting of the GLUT4 gene impairs insulin action inmuscle and liver. Nature. 2001: 409; 729-733.
260) Kato M, Kinugawa T, Omodani H, Osaki S, Ogino K, Hisatome I, Miyakoda H, FujimotoY. Augmented response in plasma atrial natriuretic peptide to dynamic exercise in patientswith congestive heart failure. Jpn Circ J. 1996: 60(12); 909-916.
261) Cugno M, Agostoni P, Mari D, Meroni PL, Gregorini L, Bussotti M, Anguissola GB,Donatelli F, Nussberger J. Impaired bradykinin response to ischaemia and exercise inpatients with mild congestive heart failure during angiotensin- converting enzymetreatment. Relationships with endothelial function, coagulation and inflammation. BritishJ Haematology. 2005: 130(1); 113-120.

108
262) Beltowski J, Jamroz-Wisniewska A, Borkowska E, Marciniak A. Phosphodiesterase 5inhibitor ameliorates renal resistance to atrial natriuretic peptide associated with obesityand hyperleptinemia. Arch Med Res. 2006: 37; 307-315.
263) Haber A, Gupta N, Yu Z, Goh T, Bogdanovic E, Giacca A, Fantus IG. N-acetylcysteine(NAC) prevents hyperglycemia-induced insulin resistance in vivo: possible role ofoxidative stress. Am J Physiol Endocrinol Metab. 2003: 4; E744-753.
264) Lam TK, Yoshii H, Haber CA, Bogdanovic E, Lam L, Fantus IG, Giacca A. Free fattyacid-induced hepatic insulin resistance: a potential role for protein kinase C-delta. Am JPhysiol Endocrinol Metab. 2002: 283; E682-E691.
265) Beltowski J, Jamroz-Wisniewska A, Borkowska E, Marciniak A. Phosphodiesterase 5inhibitor ameliorates renal resistance to atrial natriuretic peptide associated with obesityand hyperleptinemia. Arch Med Res. 2006: 37; 307-315.
266) Suwa M, Seino Y, Nomachi Y, Matsuki S, Funahashi K. Multicenter prospectiveinvestigation on efficacy and safety of carperitide for acute heart failure in the “realworld” of therapy. Circ J. 2005: 69; 283-290.
267) Abraham WT, Adams KF, Fonarow GC, Costanzo MR, Berkowitz RL, LeJemtel TH,Cheng ML, Wynne J, the ADHERE Scientific Advisory Committee and Investigators, theADHERE Study Group. In-hospital mortality in patients with acute decompensated heartfailure requiring intravenous vasoactive medications: An analysis from the acutedecompensated Heart Failure National Registry (ADHERE). J Am Coll Cardiol. 2005:46; 57-64.
268) Garvey WT, Olefsky JM, Marshall S. Glucose and insulin co-regulate the glucosetransport system in primary cultured adipocytes. A new mechanism of insulin resistance.J Biol Chem. 1987: 262(1); 189-197.
269) Amery C, Round R, Smith J, Nattrass M. Elevation of plasma fatty acids by ten-hourintralipid infusion has no effect on basal or glucose-stimulated insulin secretion in normalman. Metabolism. 2000: 49(4); 450-454.
270) Hoy AJ, Bruce CR, Cederberg A, Turner N, James DE, Cooney GJ, Kraegen EW.Glucose infusion causes insulin resistance in skeletal muscle of rats without changes inAkt and AS160 phosphorylation. Am J Physiol Endocrinol Metab. 2007: 293; E1358-E1364.
271) Wilkes JJ, Bonen A, Bell RC. A modified high-fat diet induces insulin resistance in ratskeletal muscle but not adipocytes. Am J Physiol Endocrinol Metab. 1998: 275; E679-E686.
272) Leonard BL, Watson RN, Loomes KM, Phillips AR, Cooper GJ. Insulin resistance in theZucker diabetic fatty rat: a metabolic characterisation of obese and lean phenotypes. ActaDiabetol. 2005: 42(4); 162-170.

109
273) Carey RM, Park J. Role of Angiotensin Type 2 Receptors in Vasodilation of Resistanceand Capacitance Vessels. Hypertension. 2006: 48; 824-825.
274) Lahera V, Navarro J, Biondi ML, Ruilope LM, Romero JC. Exogenous cGMP preventsdecrease in diuresis and natriuresis induced by inhibition of NO synthesis. Am J PhysiolRenal Physiol. 1993: 264; F344-F347.
275) Feng J, Li H, Rosenkranz ER. Bradykinin protects the rabbit heart after cardioplegicischemia via NO-dependent pathways. Ann Thorac Surg. 2000: 70; 2119-2124.
276) Duka I, Shenouda S, Johns C, Kintsurashvili E, Gavras I, Gavras H. Role of the B2Receptor of Bradykinin in Insulin Sensitivity. Hypertension. 2001: 38; 1355.
277) Airhart N, Yang Y-F, Roberts Jr CT, Silberbach M. ANP Induces Natriuretic PeptideReceptor-PKG Interaction. J Biol Chem. 2003: 278(40); 38693-38698.
278) Wang C-H, Leung N, Lapointe N, Szeto L, Uffelman KD, Giacca A, Rouleau JL, LewisGF. Vasopeptidase Inhibitor Omapatrilat Induces Profound Insulin Sensitization andIncreases Myocardial Glucose Uptake in Zucker Fatty Rats. Circulation. 2003:107; 1923.
279) Arbin V, Claperon N, Fournie-Zaluski M-C, Roques BP, Peyroux J. Effects of dualangiotensin-converting enzyme and neutral endopeptidase 24-11 chronic inhibition bymixanpril on insulin sensitivity in lean and obese Zucker rats. J Cardio Pharm. 2003:41(2); 254-264.
280) Arbin V, Claperon N, Fournié-Zaluski MC, Roques BP, Peyroux J. Acute effect of thedual angiotensin-converting enzyme and neutral endopeptidase 24-11 inhibitor mixanprilon insulin sensitivity in obese Zucker rat. British J Pharm. 2001: 133(4); 495-502.
281) Kudoh A, Katagai H, Takazawa T. Atrial Natriuretic Peptide Increases Glucose UptakeDuring Hypoxia in Cardiomyocytes. J Cardio Pharm. 2002: 40; 601–610.
282) Sosa V, Carbó R, Guarner V. Participation of glucose transporters on atrial natriureticpeptide-induced glucose uptake by adult and neonatal cardiomyocytes underoxygenation and hypoxia. Euro J Pharm. 2007: 568; 83–88.
283) Leturque A, Guerre-Millo M, Lavau M, Girard J. Effect of insulin on glucose metabolismin adipocytes from virgin and late-pregnant rats. Biochem J. 1984: 224(2); 685–688.
284) Kato M, Kinugawa T, Omodani H, Osaki S, Ogino K, Hisatome I, Miyakoda H, FujimotoY. Augmented response in plasma atrial natriuretic peptide to dynamic exercise in patientswith congestive heart failure. Jpn Circ J. 1996: 60(12); 909-916.
285) Ortola FV, Ballermann BJ, Anderson S, Mendez RE, Brenner BM. Elevated plasma atrialnatriuretic peptide levels in diabetic rats. Potential mediator of hyperfiltration. J ClinInvest. 1987: 80(3); 670–674.

110
286) Bai GY, Piao FL, Kim SY, Yuan K, Kim SZ, Kim SH. Augmentation of insulin-stimulated ANP release through tyrosine kinase and PI 3-kinase in diabetic rats. Peptides.2006: 27(11); 2756-2763.
287) Trevisan R, Fioretto P, Semplicini A, Opocher G, Mantero F, Rocco S, Remuzzi G,Morocutti A, Zanette G, Donadon V. Role of insulin and atrial natriuretic peptide insodium retention in insulin-treated IDDM patients during isotonic volume expansion.Diabetes. 1990: 39(3); 289-298.
288) Nakamura M, Arakawa N, Yoshida H, Makita S, Niinuma H, Hiramori K. Vasodilatoryeffects of B-type natriuretic peptide are impaired in patients with chronic heart failure. AmHeart J. 1998: 135(3); 414-420.
289) Takekoshi K, Ishii K, Isobe K, Nomura F, Nammoku T, Nakai T. Effects of natriureticpeptides (ANP, BNP, CNP) on catecholamine synthesis and TH mRNA levels in PC12cells. Life Sciences. 2000: 66(22); PL303-PL311.
290) Sengenès C, Zakaroff-Girard A, Moulin A, Berlan M, Bouloumié A, Lafontan M,Galitzky J. Natriuretic peptide-dependent lipolysis in fat cells is a primate specificity. AmJ Physiol Regul Integr Comp Physiol. 2002: 283; R257-R265.
291) Devillier P, Corompt E, Bréant D, Caron F, Bessard G. Relaxation and modulation ofcyclic AMP production in response to atrial natriuretic peptides in guinea pig trachealsmooth muscle. European Journal of Pharmacology. 2001: 430(2-3); 325-333.
292) Etgen GJ Jr., Fryburg DA, Gibbs EM. Nitric oxide stimulates skeletal muscle glucosetransport through a calcium/contraction- and phosphatidylinositol-3-kinase-independentpathway. Diabetes. 1997: 46(11); 1915-1919.
293) Roberts CK, Barnard RJ, Scheck SH, Balon TW. Exercise-stimulated glucose transport inskeletal muscle is nitric oxide dependent. Am J Physiol. 1997: 273(1 Pt 1); E220-225.
294) Fryer LG, Hajduch E, Rencurel F, Salt IP, Hundal HS, Hardie DG, Carling D. Activationof glucose transport by AMP-activated protein kinase via stimulation of nitric oxidesynthase. Diabetes. 2000: 49(12); 1978-1985.
295) Li J, Hu X, Selvakumar P, Russell RR III, Cushman SW, Holman GD, Young LH. Role ofthe nitric oxide pathway in AMPK-mediated glucose uptake and GLUT4 translocation inheart muscle. Am J Physiol Endocrinol Metab. 2004: 287(5); E834-841.
296) Guarino MP, Correia NC, Lautt WW, Macedo MP. Insulin sensitivity is mediated by theactivation of the ACh/NO/cGMP pathway in rat liver. Am J Physiol Gastrointest LiverPhysiol. 2004: 287; G527-G532.

111
297) Merkel L. Zaprinast: A cGMP-Selective Phosphodiesterase Inhibitor. CardiovascularDrug Reviews. 1993: 11(4); 501-515
298) Snyder PB, Esselstyn JM, Loughney K, Wolda SL, Florio VA. The role of cyclicnucleotide phosphodiesterases in the regulation of adipocyte lipolysis. Journal of LipidResearch. 2005: 46; 494-503.
299) US Patent 6967204 - Treatment of insulin resistance syndrome and type 2 diabetes withPDE9 inhibitors. http://www.freepatentsonline.com/6967204.html
300) Su J, Scholz PM, Weiss HR. Differential Effects of cGMP Produced by Soluble andParticulate Guanylyl Cyclase on Mouse Ventricular Myocytes. Experimental Biology andMedicine. 2005: 230; 242-250.
301) Eklof A-C, Holthack U, Svennilson J, Fienberg A, Greengard P, Aperia A. Increasedblood pressure and loss of anp-induced natriuresis in mice lacking DARPP-32 gene.Clinical and experimental hypertension. 2001: 23(6); 449-460.
302) Tanaka Y, Tang G, Takizawa K, Otsuka K, Eghbali M, Song M, Nishimaru K, ShigenobuK, Koike K, Stefani E, Toro L. Kv channels contribute to NO- and ANP-inducedrelaxation of a rat conduit artery. J. Pharmacol Exp Ther. 2006: 317(1); 341-354.
303) Bergandi L, Silvagno F, Russo I, Riganti C, Anfossi G, Aldieri E, Ghigo D, Trovati M,Bosia A. Insulin stimulates glucose transport via nitric oxide/cyclic GMP pathway inhuman vascular smooth muscle cells. Arteriosclerosis, Thrombosis, and Vascular Biology.2003: 23; 2215-2221.
304) Carvalho CR, Thirone AC, Gontijo JA, Velloso LA, Saad MJ. Effect of captopril,losartan, and bradykinin on early steps of insulin action. Diabetes. 1997: 46(12);1950-1957.
305) Kishi K, Muromoto Y, Miyata I, Hagi A, Hayashi H, Ebina Y. Bradykinin directlytriggers GLUT4 translocation via an insulin-independent pathway. Diabetes. 1998: 47(4);550-558.
306) Murohara T, Kugiyama K, Yasue H. Interactions of Nitrovasodilators, Atrial NatriureticPeptide and Endothelium-Derived Nitric Oxide. J Vasc Res. 1996: 33; 78-85.
307) Tsujita Y, Muraski J, Shiraishi I, Kato T, Kajstura J, Anversa P, Sussman MA. Nucleartargeting of Akt antagonizes aspects of cardiomyocyte hypertrophy. Proc Natl Acad Sci.2006: 103(32); 11946–11951.
308) Wang R, Brattain MG. AKT can be activated in the nucleus. Cellular Signalling. 2006:18(10); 1722-1731.

112
309) Grutzner U, Keller M, Bach M, Kiemer AK, Meissner H, Bilzer M, Zahler S,Gerbes AL, Vollmar AM. PI 3-kinase pathway is responsible for antiapoptotic effects ofatrial natriuretic peptide in rat liver transplantation. World J Gastroenterol. 2006: 12(7);1049-1055.
310) Emanuelli B, Eberlé D, Suzuki R, Kahn CR. Overexpression of the dual-specificityphosphatase MKP-4/DUSP-9 protects against stress-induced insulin resistance. Proc NatlAcad Sci. 2008: 105(9); 3545-3550.
311) Fukunaga K, Noguchi T, Takeda H, Matozaki T, Hayashi Y, Itoh H, Kasuga M.Requirement for protein-tyrosine phosphatase SHP-2 in insulin-induced activation of c-Jun NH(2)-terminal kinase. J.Biol.Chem. 2000: 275; 5208-5213.
312) Aguirre V, Werner ED, Giraud J, Lee YH, Shoelson SE, White MF. Phosphorylation ofSer307 in Insulin Receptor Substrate-1 Blocks Interactions with the Insulin Receptor andInhibits Insulin Action. J Biol Chem. 2002: 277(2); 1531-1537.
313) Bikman BT, Donghai A, Cortright RN, Neufer PD, Kane DA, Anderson EJ, Woodlief TL,Price JW, Dohm GL. A novel mechanism for metformin in improving insulin signaling inskeletal muscle. The FASEB Journal. 2008: 22; 959.6
314) Hirosumi J, Tuncman G, Chang L, Görgün CZ, Uysal KT, Maeda K, Karin M,Hotamisligil GS. A central role for JNK in obesity and insulin resistance. Nature. 2002:420; 333-336.
315) MacKenzie CJ, Wakefield JM, Cairns F, Dominiczak AF, Gould GW. Regulation ofglucose transport in aortic smooth muscle cells by cAMP and cGMP. Biochem J. 2001:353(Pt 3); 513–519.
316) Isono M, Haneda M, Maeda S, Omatsu-Kanbe M, Kikkawa R. Atrial natriuretic peptideinhibits endothelin-1-induced activation of JNK in glomerular mesangial cells. KidneyInternational (1998) 53, 1133–1142.
317) Morino K, Maegawa H, Fujita T, Takahara N, Egawa K, Kashiwagi A, Kikkawa R.Insulin-Induced c-Jun N-Terminal Kinase Activation Is Negatively Regulated by ProteinKinase C δ. Endocrinology. 2001: 142(6); 2669-2676.
318) Park HS, Kim MS, Huh SH, Park J, Chung J, Kang SS, Choi EJ. Akt (protein kinase B)negatively regulates SEK1 by means of protein phosphorylation. J Biol Chem. 2002:277(4); 2573-2578.
319) Fürst R, Brueckl C, Kuebler WM, Zahler S, Krötz F, Görlach A, Vollmar AM, KiemerAK. Atrial natriuretic peptide induces mitogen-activated protein kinase phosphatase-1 inhuman endothelial cells via Rac1 and NAD(P)H oxidase/Nox2-activation. Circ Res. 2005:96(1); 43-53.

113
320) Hannken T, Schroeder R, Stahl RA, Wolf G. Atrial natriuretic peptide attenuates ANG II-induced hypertrophy of renal tubular cells. Am J Physiol Renal Physiol. 2001: 281(1);F81-90.
321) Kiemer AK, Weber NC, Fürst R, Bildner N, Kulhanek-Heinze S, Vollmar AM. Inhibitionof p38 MAPK activation via induction of MKP-1: atrial natriuretic peptide reduces TNF-alpha-induced actin polymerization and endothelial permeability. Circ Res. 2002: 90(8);874-881.
322) Wu JJ, Roth RJ, Anderson EJ, Hong EG, Lee MK, Choi CS, Neufer PD, Shulman GI,Kim JK, Bennett AM. Mice lacking MAP kinase phosphatase-1 have enhanced MAPkinase activity and resistance to diet-induced obesity. Cell Metab. 2006: 4(1); 61-73.
323) Sugimoto T, Haneda M, Togawa M, Isono M, Shikano T, Araki S, Nakagawa T,Kashiwagi A, Guan K-L, Kikkawa R. Atrial Natriuretic Peptide Induces the Expression ofMKP-1, a Mitogen-activated Protein Kinase Phosphatase, in Glomerular Mesangial Cells.American Society for Biochemistry and Molecular Biology. 1996: 271(1); 544-547.
324) Kim TW, Lee C-H, Choi C-Y, Kwon NS, Baek KJ, Kim Y-G, Yun H-Y. Nitric oxidemediates membrane depolarization-promoted survival of rat neuronal PC12 cells.Neurosci Lett. 2003: 344(3); 209-211.
325) Nomiyama T, Perez-Tilve D, Ogawa D, Gizard F, Zhao Y, Heywood EB, Jones KL,Kawamori R, Cassis LA, Tschöp MH, Bruemmer D. Osteopontin mediates obesity-induced adipose tissue macrophage infiltration and insulin resistance in mice. J ClinInvest. 2007: 117(10); 2877-2888.
326) Mou X, Moran MA, Stepanauskas R, González JM, Hodson RE. Flow-Cytometric CellSorting and Subsequent Molecular Analyses for Culture-Independent Identification ofBacterioplankton Involved in Dimethylsulfoniopropionate Transformations. Appl EnvironMicrobiol. 2005: 71(3); 1405–1416.
327) Minokoshi Y, Kahn CR, Kahn BB. Tissue-specific ablation of the GLUT4 glucosetransporter or the insulin receptor challenges assumptions about insulin action and glucosehomeostasis. J Biol Chem. 2003: 278; 33609-33612.
328) Yu J, Jeong YJ, Kwon K-B, Kim S-Z, Kim SH, Park J-W, Yu H-C, Park B-H. AComparison of the Lipolytic Activity of Different Natriuretic Peptides on HumanAdipocytes. International Journal of Peptide Research and Therapeutics. 2008: 14(2);167-172.
329) Woodard GE, Li X, Rosado JA. Water deprivation enhances the inhibitory effect ofnatriuretic peptides on cAMP synthesis in rat renal glomeruli. Am J Physiol RenalPhysiol. 2004: 287: F418-F426.

114
330) Riviere D, Crampes F, Beauville M, Garrigues M. Lipolytic response of fat cells tocatecholamines in sedentary and exercise-trained women. Journal of Applied Physiology.1989: 66(1); 330-335.
331) Mei J, Holst LS, Landström TR, Holm C, Brindley D, Manganiello V, Degerman E. C(2)-ceramide influences the expression and insulin-mediated regulation of cyclic nucleotidephosphodiesterase 3B and lipolysis in 3T3-L1 adipocytes. Diabetes. 2002: 51(3); 631-637.
332) Sarzani R, Paci VM, Zingaretti CM, Pierleoni C, Cinti S, Cola G, Rappelli A, Dessì-Fulgheri P. Fasting inhibits natriuretic peptides clearance receptor expression in ratadipose tissue. Journal of hypertension. 1995: 13(11); 1241-1246.
333) Jeandel L, Okamura H, Belles-Isles M, Chabot J-G, Dihl F, Morel G, Kelly PA, Heisler S.Immunocytochemical localization, binding, and effects of atrial natriuretic peptide in ratadipocytes. Mol Cell Endocrinol. 1989: 62; 69-78.
334) Gustafssoni ÅB, Brunton LL. Attenuation of cAMP accumulation in adult rat cardiacfibroblasts by IL-1_ and NO: role of cGMP-stimulated PDE2. Am J Physiol Cell Physiol.2002: 283; C463–C471.
335) Degerman E, Belfrage P, Manganiello VC. Structure, Localization, and Regulation ofcGMP-inhibited Phosphodiesterase (PDE3). Journal of Biol Chem. 1997: 272(11); 6823-6826.
336) Moro C, Galitzky J, Sengenes C, Crampes F, Lafontan M, Berlan M. Functional andpharmacological characterization of the natriuretic peptide-dependent lipolytic pathway inhuman fat cells. J Pharmacol Exp Ther. 2004: 308(3); 984-992.
337) Vandecasteele G, Verde I, Rucker-Martin C, Donzeau-Gouge P, Fischmeister R. CyclicGMP regulation of the L-type Ca2+ channel current in human atrial myocytes. Journal ofPhysiology. 2001: 533: 2; 329–340.
338) Whitehead JP, Molero JC, Clark S, Martin S, Meneilly G, James DE. The Role of Ca2+ inInsulin-stimulated Glucose Transport in 3T3-L1 Cells. J Biol Chem. 2001: 276; 27816-27824.
339) Tsujita Y, Muraski J, Shiraishi I, Kato T, Kajstura J, Anversa P, Sussman MA. Nucleartargeting of Akt antagonizes aspects of cardiomyocyte hypertrophy. Proc Natl Acad SciUSA. 2006: 103(32); 11946–11951.
340) Duplain H, Burcelin R, Sartori C, Cook S, Egli M, Lepori M, Vollenweider P, PedrazziniT, Nicod P, Thorens B, Scherrer U. Insulin Resistance, Hyperlipidemia, and Hypertensionin Mice Lacking Endothelial Nitric Oxide Synthase. Circulation. 2001: 104; 342.
341) Perreault M, Marette A. Targeted disruption of inducible nitric oxide synthase protectsagainst obesity-linked insulin resistance in muscle. Nature Medicine. 2001: 7; 1138-1143.

115
342) Marette A. Mediators of cytokine-induced insulin resistance in obesity and otherinflammatory settings. Current Opinion in Clinical Nutrition and Metabolic Care. 2002:5(4); 377-383.
343) McGrath MF, Kuroski de Bold ML, de Bold AJ. The Endocrine function of the heart.Trends Endocrinol Metab. 2005: 16; 469-477.
344) Lommi J, Kupari M, Yki-Jarvinen H. Free fatty acid kinetics and oxidation in congestiveheart failure. Am J Cardiol. 1998: 181; 45-50.
345) Boden G. Free fatty acids (FFA), a link between obesity and insulin resistance. FrontBiosci. 1998: 3; 169-175.
346) Chinkers M, Garbers DL. The protein kinase domain of the ANP receptor is required forsignalling. Science. 1989: 245(4924); 1392-1394.
347) Dessi-Fulgheri P, Sarzani R, Tamburrini P, Moraca A, Espinosa E, Cola G, GiantomassiL, Rappelli A. Plasma atrial natriuretic peptide and natriuretic peptide receptor geneexpression in adipose tissue of normotensive and hypertensive obese patients. JHypertens. 1997: 15; 1596-16l9.
348) Sarzani R, Marcucci P, Salvi F, Bordicchia M, Espinosa E, Mucci L, Lorenzetti B,Minardi D, Muzzonigro G, Dessi-Fulgheri P, Rappelli A. Agniotension II stimulates andatrial natriuretic peptide inhibits human visceral adipocyte growth. Internat J Obesity.2007: 10; 1-9.
349) Matsukawa N, Grzesik WJ, Takahashi N, Pandey KN, Pang S, Yamauchi M, Smithies O.The natriuretic peptide clearance receptor locally modulates the physiological effects ofthe natriuretic peptide system. Proc Natl Acad Sci. 1999: 96; 7403-7408.
350) Jaubert J, Jaubert F, Martin N, Washburn LL, Lee BK, Eicher EM, Guenet J-L. Three newallelic mouse mutations that cause skeletal overgrowth involve the natriuretic peptidereceptor C gene (Npr3). Proc Natl Acad Sci. 1999: 96; 10278-10283.
351) Lafontan M, Moro C, Sengenes C, Galitzky J, Crampes F, Berlan M. An unsuspectedmetabolic role for atrial natriuretic peptides: The control of lipolysis, lipid mobilization,and systemic nonesterified fatty acids levels in humans. Arterioscler Thromb Vasc Biol.2005: 25; 2032-2042.
352) Nisoli E, Clementi E, Paolucci C, Cozzi V, Tonello C, Sciorati C, Bracale R, Valerio A,Francolini M, Moncada S, Carruba MO. Mitochondrial biogenesis in mammals: The roleof endogenous nitric oxide. Science. 2003: 299; 896-899.

116
353) Nisoli E, Tonello C, Cardile A, Cozzi V, Bracale R, Tedesco L, Falcone S, Valerio A,Cantoni O, Clementi E, Moncada S, Carruba MO. Calorie restriction promotesmitochondrial biogenesis by inducing the expression of eNOS. Science. 2005: 310; 314-317.
354) Morino K, Petersen KF, Shulman GI. Molecular mechanisms of insulin resistance inhumans and their potential links with mitochondrial dysfunction. Diabetes. 2006;55(Suppl 2); S9-S15.
355) Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P,Carlsson E, Ridderstrale M, Laurila E, Houstis N, Daly M.J, Patterson N, Mesirov JP,Golub TR, Tamayo P, Spiegelman B, Landr ES, Hirschhorn JN, Altshuler D, Groop LC.PGC-1 alpha-responsive genes involved in oxidative phosphorylation are coordinatelydownregulated in human diabetes. Nat Genet. 2003: 34; 267-273.
356) Patti ME, Butte AJ, Crunkhorn S, Cusi K, Berria R, Kashyap S, Miyazaki Y, Kohane I,Costello M, Saccone R, Landaker EJ, Goldfine AB, Mun E, DeFronzo R, Finlayson J,Kahn CR, Mandarino LJ. Coordinated reduction of genes of oxidative metabolism inhumans with insulin resistance and diabetes: potential role of PGC1 and NRF1. Proc NatlAcad Sci. 2003: 100; 8466-8471.
357) Morino K, Petersen K/F, Dufour S, Befroy D, Frattini J, Shatzkes N, Neschen S, WhiteMF, Bilz S, Sono S, Pypaert M, Shulman GI. Reduced mitochondrial density andincreased IRS-1 serine phosphorylation in muscle of insulin-resistant offspring of type 2diabetic fparents. J Clin Invest. 2005: 115; 3587-3593.
358) Mitsuishi M, Miyashita K, Itoh H. cGMP rescues mitochondrial dysfunction induced byglucose and insulin in myocytes. Biochem Biophy Res Comm. 2008: 36l7; 840-845.
359) Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. J Clin Invest.2006: 116; 1793-1801.
360) Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006: 444; 860-867.
361) Kershaw EE, Flier JS. Adipose tissue as an endocrine organ. J Clin Endocrinol Metab.2004: 89; 2548-2556.
362) Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, Sole J, Nichols A, Ross JS,Tartaglia LA, Chen H. Chronic inflammation in fat plays a crucial role in the developmentof obesity- related insulin resistance. J Clin Invest. 2003: 112; 1821-1830.
363) Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW Jr. Obesity isassociated with macrophages accumulation in adipose tissue. J Clin Invest. 2003: 112;1796-1808.
364) Sachse A, Wolf G. Angiotensin II-induced reactive oxygen species and the kidney. J AmSoc Nephrol. 2007: 18; 2439-2446.

117
365) Vollmar AM. The role of atrial natriuretic peptide in the immune system. Peptides. 2005:26: 1086-1094.
366) Kiemer AK, Vollmar AM. Autocrine regulation of inducible nitric-oxide synthase inmacrophages by atrial natriuretic peptide. J Biol Chem. 1998: 273; 13444-13451.
367) Perreault M, Marette A. Targeted disruption of inducible nitric oxide synthase protectsagainst obesity-linked insulin resistance in muscle. Nat. Med. 2001: 7; 1138-1143.
368) Ladetzki-Baehs K, Keller M, Kiemer AK, Koch E, Zahler S, Wendel A, Wollmar AM.Atrial natriuretic peptide, a regulator of nuclear factor-kB activation in vivo. Endocrinol.2008: 148; 332-336.
369) Decarie A, et al., Serum interspecies differences in metabolic pathways of bradykinin and[des-Arg9]BK: influence of enalaprilat. Am J Physiol. 1996. 271: p. H1340-H1347.
370) Dumoulin MJ, et al., Metabolism of bradykinin by the rat coronary vascular bed.Cardiovasc Res, 1998. 38(1): p. 229-236.
371) Coutant KD, Corvaia N, Ryder NS, Bradykinin induces tyrosine phosphorylationof epidermal growth factor-receptor and focal adhesion proteins in human keratinocytes.Biochem Biophys Res Commun, 1995. 210(3): p. 774-780.
372) Kishi K, et al., Bradykinin directly triggers GLUT4 translocation via an insulin-independent pathway. Diabetes, 1998. 47(4): p. 550-558.
373) Theodosiou A, Ashworth A. MAP kinase phosphatases. Genome Biology2002, 3(7): 3009.1–3009.10.
374) Dickinson RJ, Keyse SM. Diverse physiological functions for dual-specificity MAPkinase phosphatases. Journal of Cell Science. 2006. 119, 4607-4615.
375) Moro C, Klimcakova E, Lafontan M, Berlan M, Galitzky J. Phosphodiesterase-5A andneutral endopeptidase activities in human adipocytes do not control atrial natriureticpeptide-mediated lipolysis. Brit J Pharm. 2009. 152(7): 1102-1110.
376) Solini A, DeFronzo RA. Insulin resistance, hypertension and cellular ion transportsystems. Acta Diabetologica. 1992. 29(3-4):196-200.
377) Bradford MM. A rapid and sensitive method for quantitation of microgramquantities of protein utilizing the principle of protein-dye-binding. AnalBiochem. 1976. 72:248-254.
378) Galligan J. Focus on: "G protein-dependent activation of smooth muscle eNOS vianatriuretic peptide clearance receptor". Am J Physiol Cell Physiol. 1998. 275:C1407-C1408.

118
CHAPTER 7
APPENDIX
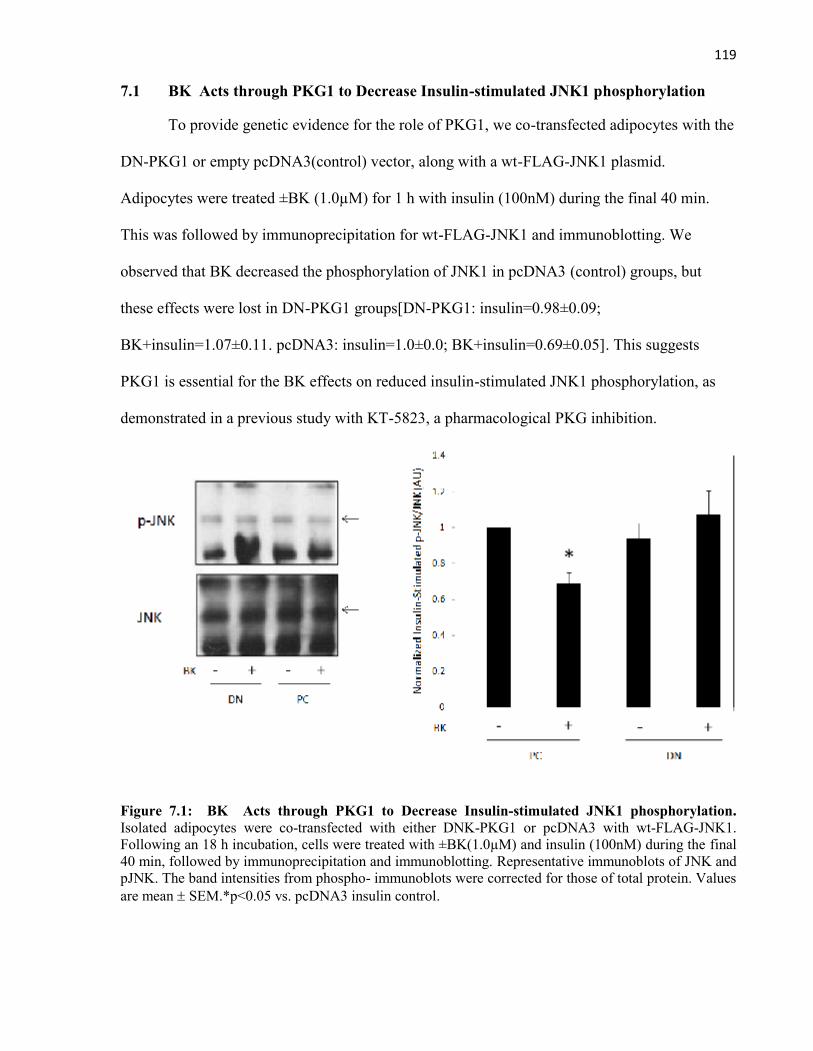
119
7.1 BK Acts through PKG1 to Decrease Insulin-stimulated JNK1 phosphorylation
To provide genetic evidence for the role of PKG1, we co-transfected adipocytes with the
DN-PKG1 or empty pcDNA3(control) vector, along with a wt-FLAG-JNK1 plasmid.
Adipocytes were treated ±BK (1.0µM) for 1 h with insulin (100nM) during the final 40 min.
This was followed by immunoprecipitation for wt-FLAG-JNK1 and immunoblotting. We
observed that BK decreased the phosphorylation of JNK1 in pcDNA3 (control) groups, but
these effects were lost in DN-PKG1 groups[DN-PKG1: insulin=0.98±0.09;
BK+insulin=1.07±0.11. pcDNA3: insulin=1.0±0.0; BK+insulin=0.69±0.05]. This suggests
PKG1 is essential for the BK effects on reduced insulin-stimulated JNK1 phosphorylation, as
demonstrated in a previous study with KT-5823, a pharmacological PKG inhibition.
Figure 7.1: BK Acts through PKG1 to Decrease Insulin-stimulated JNK1 phosphorylation.Isolated adipocytes were co-transfected with either DNK-PKG1 or pcDNA3 with wt-FLAG-JNK1.Following an 18 h incubation, cells were treated with ±BK(1.0µM) and insulin (100nM) during the final40 min, followed by immunoprecipitation and immunoblotting. Representative immunoblots of JNK andpJNK. The band intensities from phospho- immunoblots were corrected for those of total protein. Valuesare mean SEM.*p<0.05 vs. pcDNA3 insulin control.
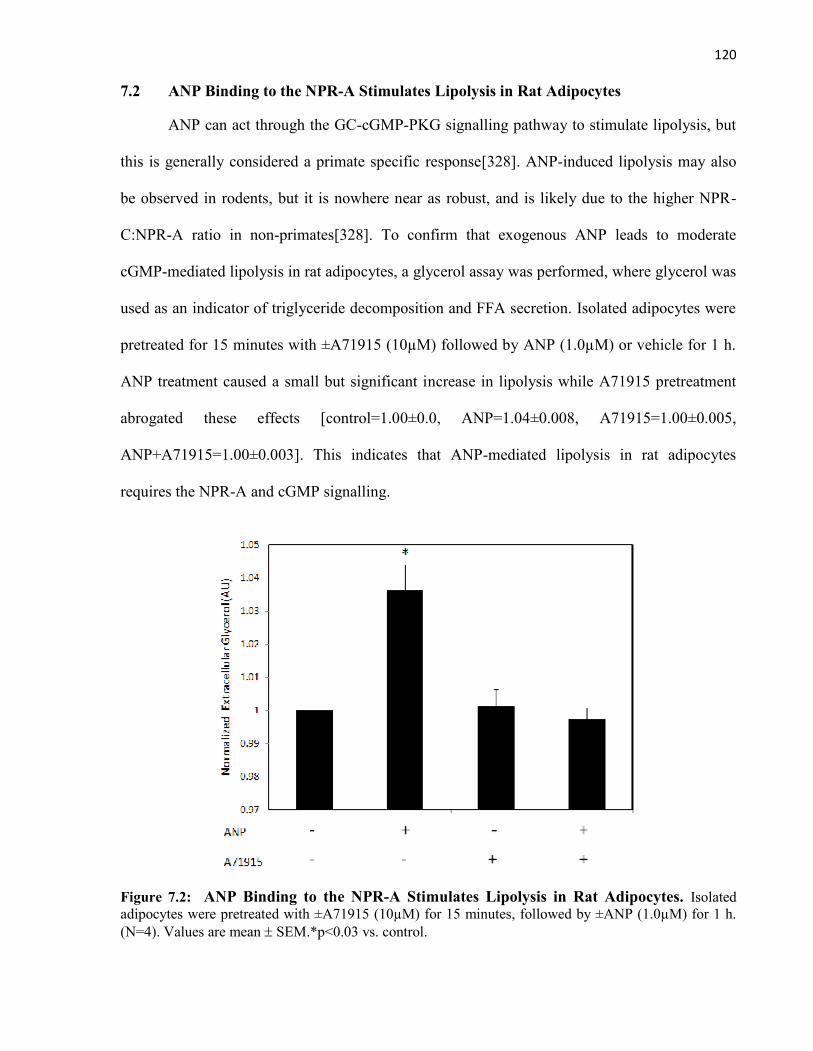
120
7.2 ANP Binding to the NPR-A Stimulates Lipolysis in Rat Adipocytes
ANP can act through the GC-cGMP-PKG signalling pathway to stimulate lipolysis, but
this is generally considered a primate specific response[328]. ANP-induced lipolysis may also
be observed in rodents, but it is nowhere near as robust, and is likely due to the higher NPR-
C:NPR-A ratio in non-primates[328]. To confirm that exogenous ANP leads to moderate
cGMP-mediated lipolysis in rat adipocytes, a glycerol assay was performed, where glycerol was
used as an indicator of triglyceride decomposition and FFA secretion. Isolated adipocytes were
pretreated for 15 minutes with ±A71915 (10µM) followed by ANP (1.0µM) or vehicle for 1 h.
ANP treatment caused a small but significant increase in lipolysis while A71915 pretreatment
abrogated these effects [control=1.00±0.0, ANP=1.04±0.008, A71915=1.00±0.005,
ANP+A71915=1.00±0.003]. This indicates that ANP-mediated lipolysis in rat adipocytes
requires the NPR-A and cGMP signalling.
Figure 7.2: ANP Binding to the NPR-A Stimulates Lipolysis in Rat Adipocytes. Isolatedadipocytes were pretreated with ±A71915 (10µM) for 15 minutes, followed by ±ANP (1.0µM) for 1 h.(N=4). Values are mean SEM.*p<0.03 vs. control.