cgmp basic requirement. cgmp basic requirements quality system definitions regulations requirements
TRANSCRIPT

cGMP BASIC REQUIREMENT

cGMP Basic Requirements
Quality System
Definitions
Regulations
Requirements
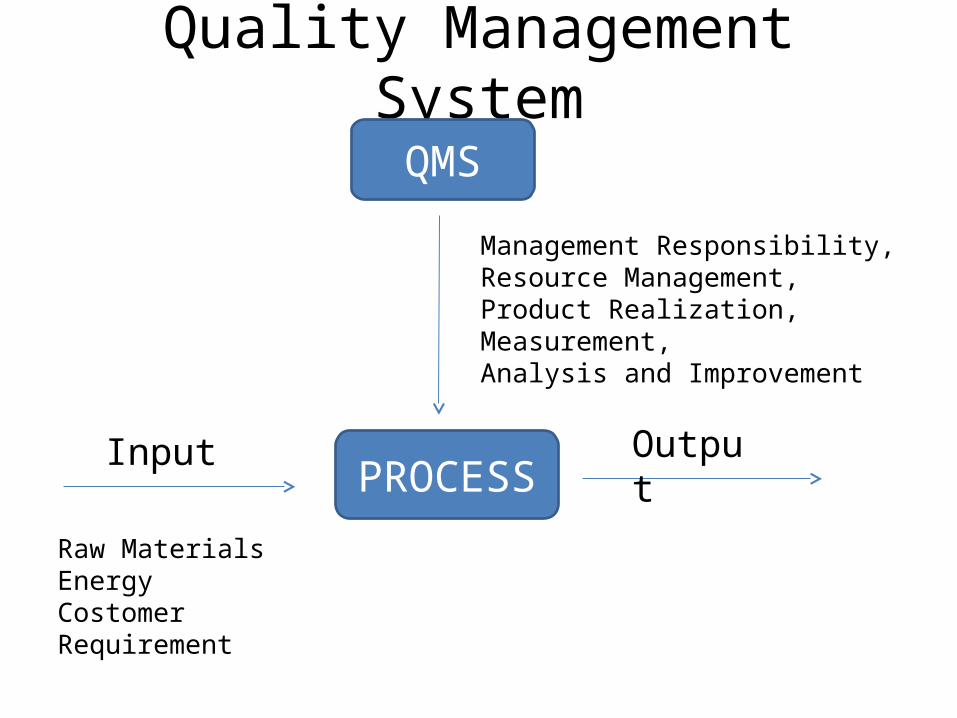
Quality Management System
QMS
PROCESSInput Output
Management Responsibility, Resource Management, Product Realization,Measurement,Analysis and Improvement
Raw MaterialsEnergyCostomer Requirement
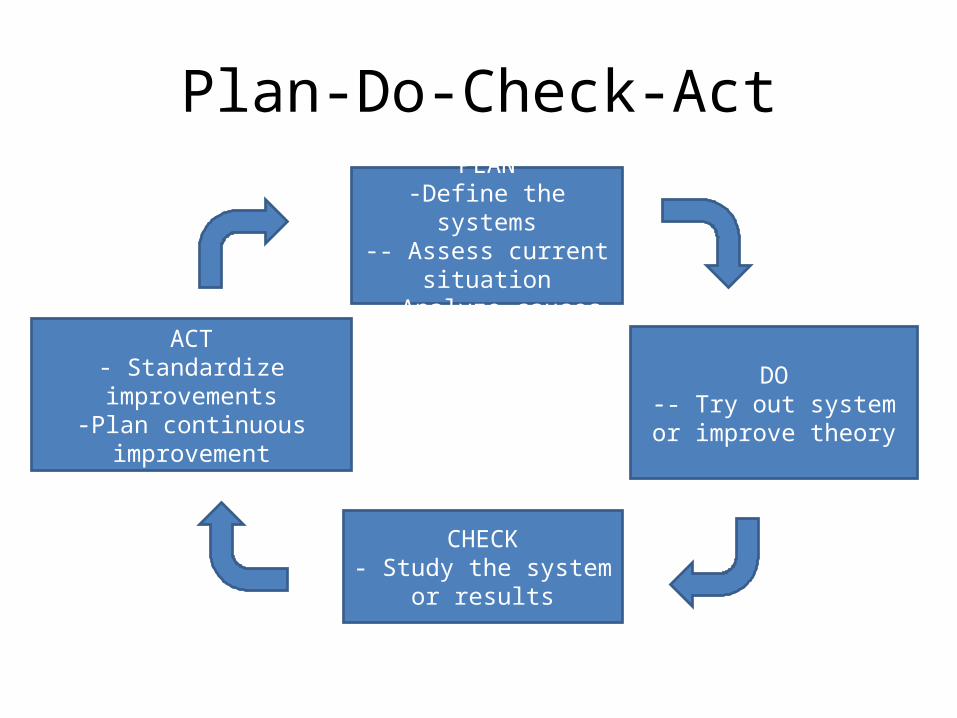
Plan-Do-Check-Act
PLAN-Define the systems
-- Assess current situation- Analyze causes
DO-- Try out system or
improve theory
CHECK- Study the system or
results
ACT- Standardize improvements
-Plan continuous improvement

QMS and cGMP in Biopharmaceutical Co.
Senior Management Responsibility
To archive the quality objectives, a well-establish and documented system of Quality assurance (QA)
incorporating Good Manufacturing Practice (GMP) should be followed)
- Quality - Safety - Efficacy

Quality Assurance and Quality Control

What QA systems should includes?1. GMP and GLP are taken into account during designing and developing
products.2. All production and control operations are clearly specified and
documented.3. Key personnel responsibilities are clearly defined.4. All arrangements for procurement and use of correct starting materials
and packaging materials are made.5. In process checks and validations are carried out in a defined manner.6. The final product is manufactured, packed and checked as per defined
procedures.7. Regulatory aspects and internal requirements for the final product are
fulfilled.8. Storage, handling and distribution procedures for the final product are
followed to ensure maintenance of quality throughout shelf life.9. Self-inspection procedures are defined to regularly monitor the
effectiveness of the quality assurance system.10. Corrective actions11. Statistical process control

What QC systems should includes?1. Appropriate procedures, training personnel and adequate facilities for sampling,
inspection and testing of starting materials, intermediate, bulk and finished products.
2. Validated test methods
3. Maintenance of records to demonstrate that all procedures have been carried out.
4. Certification of starting materials to be of specified quality and purity, and their storage and adequate labeling before use in final product.
5. Release of batch of product ONLY after certification by qualified person that it meet required criteria or specifications.
6. Maintain sufficient samples of starting materials and final product for future examination, if necessary.
7. Recording and investigation of out of specification results, changes, incidents and deviations.

cGMP for Biohharmaceutical Industries
Is it part of QA or QC?
GMP is that part of quality assurance which ensures that products are consistemly produced and controlled to be quality standards appropriate to their intended use and as required by the marketing authorization.
GMP rules are directed primarily to diminishing the risks, inherent in any biopharmaceutical production, that cannot be prevented completely through testing the final products.
Risk are:1. Cross contamination (in particular by unexpected
contaminants)2. Mix-ups (confusion) such as false labeling.

cGMP Guidelines for Biopharmaceuticals1. All manufacturing process are clearly defined, systematically
reviewed in the light of experience, and shown to be capable of consistently manufacturing biopharmaceutical products of the required quality and specifications.
2. All steps of manufacturing processes and any significant changes made to the process are validated.
3. All necessary facilities are provided, including:– Appropriate qualified and trained personnel– Adequate premises and space– Suitable equipment and services– Correct materials, containers and labels– Approved procedures and instructions– Suitable storage and transport– Adequate personnel, laboratories and equipment for in-process
control under the responsibility of the production management.

cGMP Guidelines for Biopharmaceuticals (cont)
4. Instructions and procedures are written in clear language, specifically applicable to the facilities provided.
5. Operators are trained to carry out procedures correctly
6. Records are made (Manually, with signature or recording instruments) during manufacturing to show that all steps required by the defined procedures have been taken and the expected quantity and quality were reached. Any significant deviations are fully recorded and investigated.
7. Records covering manufacturing and distribution, which enable the complete history of a batch to be traced, are retained in a comprehensible form.
8. Proper storage and distribution of the products minimized any risk to their quality
9. A system is available to recall any batch or product from sale or supply
10. Complains about marketed products are examined, the causes of quality defects investigated, and appropriate measurements taken in respect of the defective product(s) and to prevent recurrence.

Information and material flow in pharmaceutical/ biopharmaceutical production process
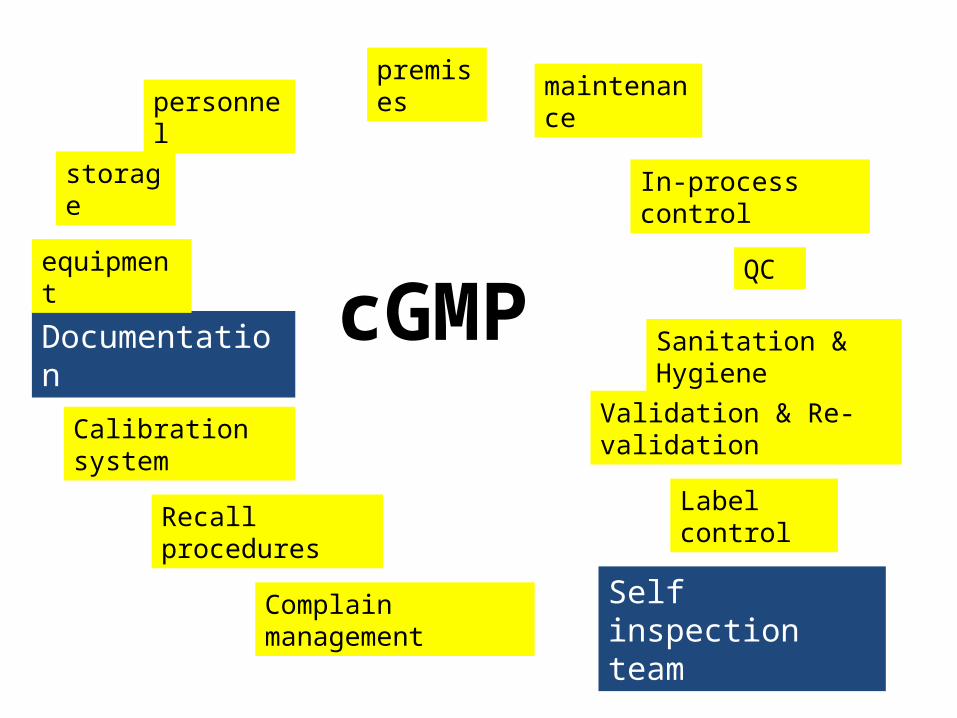
cGMP
premises maintenance
In-process control
QC
Sanitation & Hygiene
Validation & Re-validation
Self inspection teamComplain management
Recall procedures
Calibration system
Documentation
equipment
Label control
personnel
storage

The cGMP Regulations
• First issued: June 1963
• Current = Dynamic roles (standarsd evolve over time)
cGMP for finised Pharmaceuticals 21 CFR 210, 211
www.fda.gov/cder/dmpq

Examples of cGMP codes (FDA)
Building, Facilities, Equipments (21 CFR 211.42-72)
Equipment Identification (21 CFR 211.105)
Equipment cleaning and use (21 CFR 211.182)
Electronic records, electronic signature (21 CFR Part 11)
Process validation (General code of FDA process validation)
Rubber articles for repeated use (21 CFR 177.2600)

Buildings and Facilities

Ǿ 211.42 Design and Construction features
a) Any building or buildings used in the manufacture, processing, packing or holding of drug product shall be of suitable size, construction and location to facilities cleaning, maintenance, and proper operation
External Environment and Internal Environment

Site preparation and Plant design
Site arrangement and over-all layout design (green spaces parking, traffic, recreation area, tanks, site utilities, etc..)
Water supply and waste management area (waste contractor!!)
Site security and access (fences, guard, cameras, etc..)
Utilities design, layout, backup (critical utilities backup)
Equipment-design, layout, spares, capacity
Safety (personnel and equipment), emergency services access.
External architecture should take in account the local environment (temperature, humidity, wind, etc..)
Ease of maintenance (service ducts, cat floor, etc..)
Project management (managers, consultants, etc..)
Validation Plans and an effective change control procedures. Provision of design and (as builds) drawing.
Contractor (Experienced contractor)

Ǿ 211.42 Design and Construction features
b) Any such building shall have adequate space for the orderly placement of equipment, drug product containers, closures, labeling, in-process materials, or drug products, and to prevent contamination. The flow of components, drug product containers, closures, labeling, in-process materials, and drug products through the buildings shall be designed to prevent contamination.

Ǿ 211.42 Design and Construction features
c) Operations shall be performed within specifically defined areas of adequate size. There shall be separate or defined areas or other such control systems for the firm’s operations as are necessary to prevent contamination or mix-ups during the course of the following procedures:1. Receipt, identification, storage and with folding from use of components, drug
product containers, closures and labeling, pending the appropriate sampling, testing, or examination by quality control unit before release for manufacturing or packaging.
2. Holding rejected components, drug product containers, closures and labeling before dispersion.
3. Storage of released components, drug product containers, closures and labeling4. Storage of in-process materials5. Manufacturing and processing operations6. Packaging and labeling operations7. Quarantine storage before release of drug products8. Storage of drug products after release9. Control and laboratory operation

Ǿ 211.42 Design and Construction features Part (c) cont
10. Aseptic processing, which include as appropriate:
i. Floors, walls, and ceilings of smooth, hard surfaces the easily cleanable
ii. Temperature and humidity controlsiii. An air supply filtered through high-efficiency particular air
filters under positive pressure, regardless of whether flow is laminar or non-laminar
iv. A system for monitoring environmental conditionsv. A system for cleaning and disinfecting the room and
equipment to produce aseptic conditionsvi. A system for maintaining any equipment used to control
the aseptic conditions.

Construction Materials
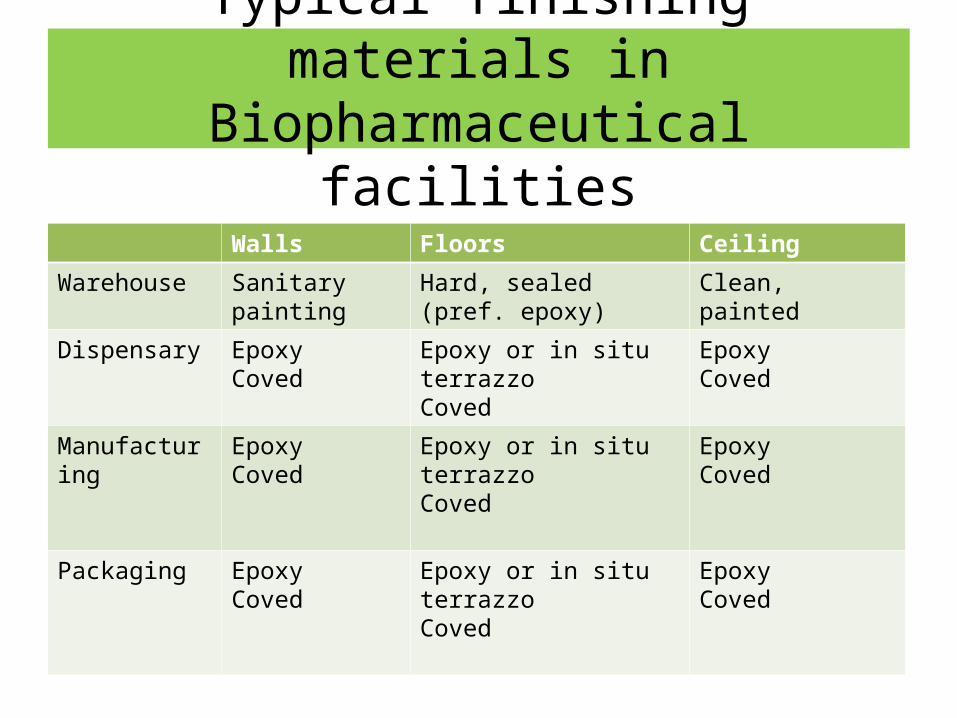
Typical finishing materials in Biopharmaceutical facilities
Walls Floors Ceiling
Warehouse Sanitary painting Hard, sealed(pref. epoxy)
Clean, painted
Dispensary Epoxy Coved
Epoxy or in situ terrazzoCoved
Epoxy Coved
Manufacturing Epoxy Coved
Epoxy or in situ terrazzoCoved
Epoxy Coved
Packaging Epoxy Coved
Epoxy or in situ terrazzoCoved
Epoxy Coved
Ǿ 211.42 Design and Construction Features
d) Operations relating to the manufacture, processing and packing of PENICILIN shall be performed in facilities separate from those used for other drug products for human use

Ǿ 211.44 Lighting
Adequate lighting shall be provided in all area
The amount of light reaching the working surface of each area involved in the production chain should be defined (lux or foot-candles)
Normally, for public standard, a range of 30-50 candles is necessary to ensure worker comfort and ability to perform efficiently and effectively
One hundred (100) foot-candles is required in some area such as inspection and filling area
Lighting should measured periodically and the results recorded.
Routine replacement of light sources on some schedule to ensure that light levels do not drop below the established minimum.

Ǿ 211.46 Ventilation, air filtration, air heating and cooling
a) Adequate ventilation should be provided.
b) Equipment for adequate control over air pressure, micro-organisms, dust, humidity, and temperature shall be provided when appropriate for the manufacture, processing, packing, or holding of a drug product
c) Air filtration system, including pre-filters and particulate matter air filters, shall be used when appropriate on air supplies to production areas, measures shall be taken to control recirculation of dust from production. In areas where air contamination occurs during production, there shall be adequate exhaust systems adequate to control contaminants.
d) Air-handling systems for the manufacture, processing and packing of PENICILIN shall be completely separate from those for other drug, products for human use.

Consideration of Air-Handling System Placement of air inlet and outlet ports. These should be sited to minimize the entry of airbone
particles or odors from the surrounding areas. Outlets should not be sited near inlet.
Where recirculation of air acceptable, adequate precautions must be taken to ensure that particulates from a processing area are removed. This will usually require an alarm system or an automatic cutoff in the event that a filter develops a hole. Dust extraction systems should be provided, where appropriate, to further minimize this potential problem.
The degree of filtration and air volumes should be matched to the operations involved.
Temperature and humidity conditions should provide personnel comfort- to enhance performance
Temperature and humidity conditions should be within the optimal condition of equipment operation
Where differential pressures are required between adjacent areas, suitable monitoring equipment must be provided.
The siting of final air filters close to each room being serviced eliminates concerns regarding the possibility of small leaks in the air duct system. Air usually enters rooms near the ceiling and leaves from the opposite side near the floor.

Ǿ 211.48 Plumbing
a) Potable water shall be supplied under continuous positive pressure in a plumbing system free of defects that could contribute contamination to any drug product. Potable water shall meet the standard prescribed in the Environmental Protection Agency’s Primary Drinking Water Regulation set in 40 CFR Part 141. water not meeting such standards shall not be permitted in the potable water systems.
b) Drains shall be of adequate size and, where connected directly to a sewer, shall be provided with air break or other mechanical device to prevent back-siphonage

FDA usually not inquire documents that the potable water is meeting the standard if the manufacturer connects the potable waterline to a public supply that meet the standard
The water can lose quality in transmission through the public piping system and through the manufacturer’s system
If potable water is obtained from wells under the control of manufacturer, periodic testing is mandatory.
In case of providing potable water storage system, an automatic chlorination system should be installed, usually at 2-3ppm

Ǿ 211.50 Sewage and refuse
Sewage, trash, and other refuse in and from the building and immediate premises shall be disposed of in a safe and sanitary manner
Product disposal: any product requiring disposal should initially be separated from packaging. Disposal procedures should involve agents with a proven record of dealing with such sensitive material from plant to disposal.
Printed packaging disposal: labels, inserts and cartons poses usually no health risk. For public. However, this may rise the public concern that product may be associated with the packaging. Incineration of such materials is preferred.
General trash and sewage: an internal procedures should be established to ensure that product and packaging waste does not get intermixed. Containers used wiyhin the plant to accumulate waste materials should be clearly marked to denote their designated use.
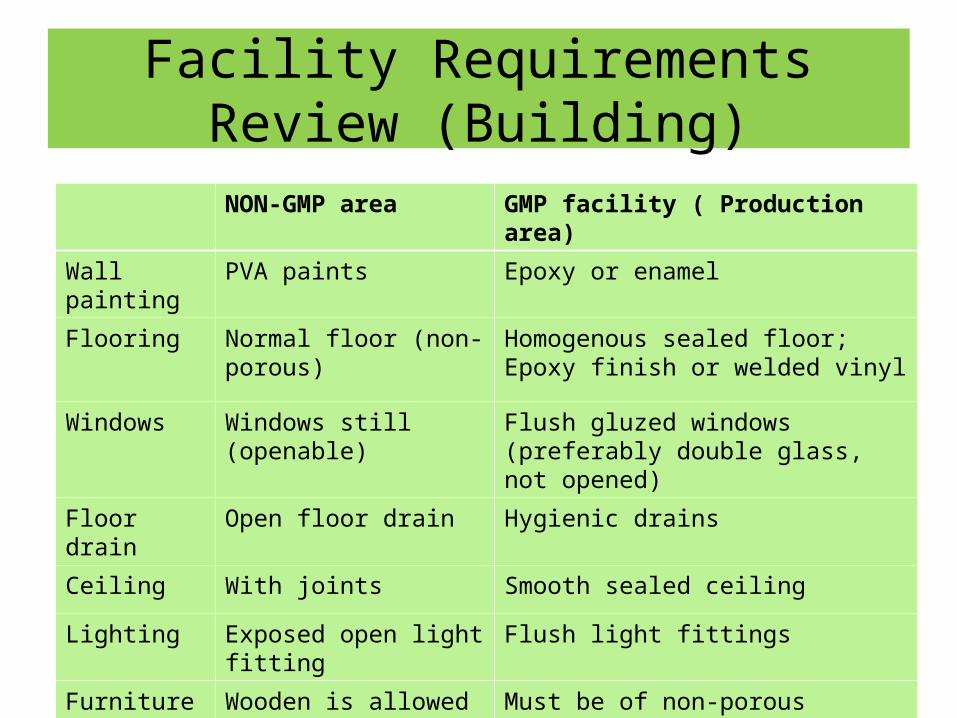
Facility Requirements Review (Building)
NON-GMP area GMP facility ( Production area)
Wall painting PVA paints Epoxy or enamel
Flooring Normal floor (non-porous) Homogenous sealed floor; Epoxy finish or welded vinyl
Windows Windows still (openable) Flush gluzed windows (preferably double glass, not opened)
Floor drain Open floor drain Hygienic drains
Ceiling With joints Smooth sealed ceiling
Lighting Exposed open light fitting Flush light fittings
Furniture Wooden is allowed Must be of non-porous materials (stainless steel or Melamine)

Ǿ 211.52 Washing and toilet facilities
Adequate washing facilities shall be provided, including hot and cold water, soap or detergent, air driers or single-service towels, and
clean toilet facilities easily accessible to working areas.
Separates toilet facility for each sex except where individual locked toilet rooms are available (the number is based on the number users)
Suggested additional emphasis.
Eating facility: Eating, drinking are permitted in separate area and well segregated from all production areas. Prominent sign indicating this role at the entrance of production areas. Enforcement procedures against violation are taken by management
In production area: Tissues and loosed disposal containers are readily available.
Lavoratories and lockers: Adequate number for personnel employed, hot shower facility, disinfectant soaps, adequate ash and waste receptacles are provided, periodic cleaning of area during each shift, complete daily cleaning using disinfectant, specific rest area for female employees should provided, areas separated from all aseptic spaces by an air lock.

Ǿ 211.56 Sanitization
a) Any building used in the manufacture, processing, packing, or holding of a drug product shall be maintained in a clean and sanitary condition. Any such building shall be free of infestation by rodents, birds, insects, and other vermin (other than laboratory animals). Trash and organic waste matter shall be held and disposed of in a timely and sanitary manner.
b) There shall be written procedures assigning responsibility for sanitization and describing in sufficient detail the cleaning schedules, methods, equipment and materials to be used in cleaning the building and facilities; such written procedures should be followed.
c) There shall be written procedure for use of suitable rodenticides, insecticides, fungicides, fumigitation agents, and cleaning and sanitization agents. Such written, procedures shall be designed procedures should be designed to prevent the contamination of equipment, components, drug product containers, closures, packaging, labeling materials, or drug product and shall be followed. Rodenticides, insecticides, fungicides shall not be used unless registered and used in accordance with the Federal Insecticide, Fungicide, and Rodenticide Act (7 U.S.C. 135).
d) Sanitation procedures shall apply to work performed by contractors or temporary employees as well as work performed by full-time employees during the ordinary course of operations.

Ǿ 211.58 Maintenance
Any building used in the manufacture, processing, packing or holding of a drug product shall be maintained
in a good state of repair
Deterioration of buildings not only presents poor image of the facility, but also can influence product quality. Cracks in ceiling, hole in wall or floor crack is potential source of insects, microbial contaminations.
Water leakage can cause significant damage for materials and equipment, give rise to electrical failure and fires and result in damage to the basic structure of the building.
Holes in the roof or near the tops of building proved ready access to birds, which may then be encourages to nest within the building.

Equipment
Ǿ 211.62 Equipment design, size and location
Equipment used in the manufacture, processing, packing, or holding of a drug product shall be of appropriate design, adequate size, and suitably located to facilitate operations for its intended use and for its cleaning and maintenance.

Ǿ 211.65 Equipment construction
Equipment shall be constructed so that surfaces that contact components, in-process materials, or drug products shall not be reactive, additive, or absorptive so as to alter the safety, identify, strength, quality, or purity of the drug product beyond that official or other established requirements.
Any substance required for operation, such as lubricants or coolants, shall not come into contact with components, drug product containers, closures, in-process materials, or drug products so as to alter the safety, identity strength, quality, or purity of the drug product beyond the official or other established requirements.

Ǿ 211.67 Equipment cleaning and maintenance a) Equipment and utensils shall be cleaned, maintained and sanitized at appropriate
intervals to prevent malfunctions or contamination that would alter the safety, identity, strength, quality, or purity of the drug product beyond the official or other established requirements.
b) Written procedures shall be established and followed for cleaning and maintenance of equipment, including utensils, used in the manufacture, processing, packing, or holding of a drug product. These Procedure shall include, but not necessarily limited to the following:1) Assignment of responsibility for cleaning and maintaining equipment.2) Maintenance and cleaning schedule, including, where appropriate, sanitizing schedule.3) A description in sufficient detail of the methods, equipment and materials used in cleaning and maintenance operations, and the methods of dissembling equipment as necessary to assure proper cleaning and maintenance.4) Removal of obliteration of previous batch identification5) Protection of clean equipment from contamination prior to use6) inspection of equipment for cleanliness immediately before use.
c) Records shall be kept of maintenance, cleaning, sanitizing and inspection as specified in 211.180 and 211.182

Ǿ 211.68 Automatic, mechanical, and electronic equipment
a) Automatic, mechanical or electrical equipment or other types of equipment, including computers, or related systems that will perform a function satisfactorily, may be used in manufacture, processing, packing, and holding of a drug product. If such equipment is so used, it shall be routinely calibrated, inspected, or checked according to a written program designed to assure proper performance. Written records of those calibration checks and inspections shall be maintained.

b) Appropriate controls shall be exercised over a computer or related systems to assure that changes in master production and control records or other records are instituted only by authorized personnel. Input and output from the computer or related system of formulas or other records data shall be checked for accuracy. The degree and frequency of input/output verification shall be maintained except where certain data, such as calculations performed in connection with laboratory analysis, are eliminated by computerization or other automated processes. In such instances a written record of the program shall be maintained along with appropriate validation data. Hard copy or alternative systems, such as duplicate, stapes, or microfilm, designed to assure that backup data are exact and complete and that it is secure from alteration, inadvertent erasures, or loss shall be maintained
Ǿ 211.68 Automatic, mechanical, and electronic equipment

Ǿ 211.70 Filters
Filters for liquid filtration used in manufacture, processing, or packing of injectible drug products intended for human use shall not release fibers into such products. Fiber-releasing filters may not be used in the manufacture, processing, or packing of these injectable drug products without the use of such filters. If use of a fiber-releasing filter in necessary, an additional non-fiber releasing filter of 0.22 micron maximum mean porosity (0.45 micron if the manufacturing conditions so dictate) shall subsequently be used to reduce the content of particles in the injectable drug product.
Use of an asbestos-containing filter, with or without subsequent use of a specific non-fiber releasing filter, is permissible only upon submission of proof to the appropriate bureau of the Food and Drug Administration that use of a non-fiber-releasing filter will, or is likely to, compromise the safety or effectiveness of the injectible drug product

Filters in Biopharmaceuticals Factory
Air filters HEPA / ULPA filters- pre-filters/filter- Microbiological Filters(Filter defect cause contamination)
-Air filter for operation (pneumatic valves)(Filter defect is destructive for the valve system)
- Air filter for process (aeration and transfer)(Filter defect cause contamination)

Filters in Biopharmaceuticals Factory (cont.)
A- For Cooling line
I. Prefilter
II. Filter
(Filter defect is destructive for heat exchanger and valve on the cooling line)
B- For Process water
I. Water filtration before distillation
II. Medium filtration (sterilization)
III. WFI for buffer and product formulation (Pyrogen free)
(Filter defect directly affecting process and product Quality, depending on the filter position)
C- For media preparation
I. Microbiological filter
(Filter defect cause direct contamination)
43
Liquid filters

- Process steam: (heating, sterilization of double jacketed vessels
(Filter defect make damage in heat exchangers and also for steam valves)
- Sterilization steam (direct steam injection): (for sterilization of empty vessels such as for media
transfer tanks and holding tanks), or SIP equipments
(Filter defect make damage for filter and direct contamination of products by foreign particle!)
44
Filters in Biopharmaceuticals Factory (cont.)
(Filtering steam from solid particles)Steam filters

- Process steam: (heating, sterilization of double jacketed vesels)
(Filter defect make damage in heat exchangers and steam valves)
- Sterilization steam (direct steam injection) (for sterilization of empty vessels such as for media transfer
tanks and holding tanks), or SIP equipments.
(Filters defect make valve damage and direct contamination of products by foreign particle!)
45
Filters in Biopharmaceuticals Factory (cont.)
(Filtering steam from solid particles)Steam filters

• Used for protein separation based on membrane molecular weight cut-off. Different filter systems are usually applied. The common used are 1000/10000/50000/100000 Daltons
(Filters defect cause improper protein separation and leakage and loss of total protein production)
46
Filters in Biopharmaceuticals Factory (cont.)
Ultra filtration system (protein concentrator)

Filters in Interferons production Platform
47