current approaches for adme characterization of …...current approaches for adme characterization...
TRANSCRIPT

2016 IQ Webinar Series Presents:
Current Approaches for ADME Characterization of Antibody-Drug Conjugates
Sponsored by the IQ Drug Metabolism Leadership Group 1

Current Approaches for ADME Characterization of Antibody-Drug Conjugates
IQ- ADC ADME Working Group Feb 19, 2016
Team members: • Eugenia Kraynov (Team Lead) – Pfizer • Amrita Kamath – Genentech • Markus Walles – Novartis • Edit Tarcsa – Abbvie • Nagendra Chemuturi – Seattle Genetics • Antoine Deslandes – Sanofi • Ramaswamy Iyer – Bristol-Myers Squibb • Amita Datta-Mannan – Eli Lilly • Dan Rock – Amgen
• Priya Sriraman – Celgene • Michaela Bairlein – Bayer • Johnny Yang – Takeda • Matthew Barfield – GlaxoSmithKline • Guangqing Xiao – Biogen • Enrique Escandon – Merck • Weirong Wang – Jansen • David Moore – Roche
Current Approaches for ADME Characterization of Antibody-Drug Conjugates: An Industry White Paper. Kraynov et al, Drug Metabolism & Disposition, December 2015

3 Outline • Overview of ADC PK
– Nomenclature/Definitions – Mechanisms of disposition – Bioanalytical considerations – Factors impacting PK of ADCs
• Currents approaches to characterize ADC ADME
– In vitro and In vivo studies – Novel vs. previously used payloads
• Summary and Overall recommendations
Current Approaches for ADME Characterization of Antibody-Drug Conjugates: An Industry White Paper. Kraynov et al, Drug Metabolism & Disposition, December 2015
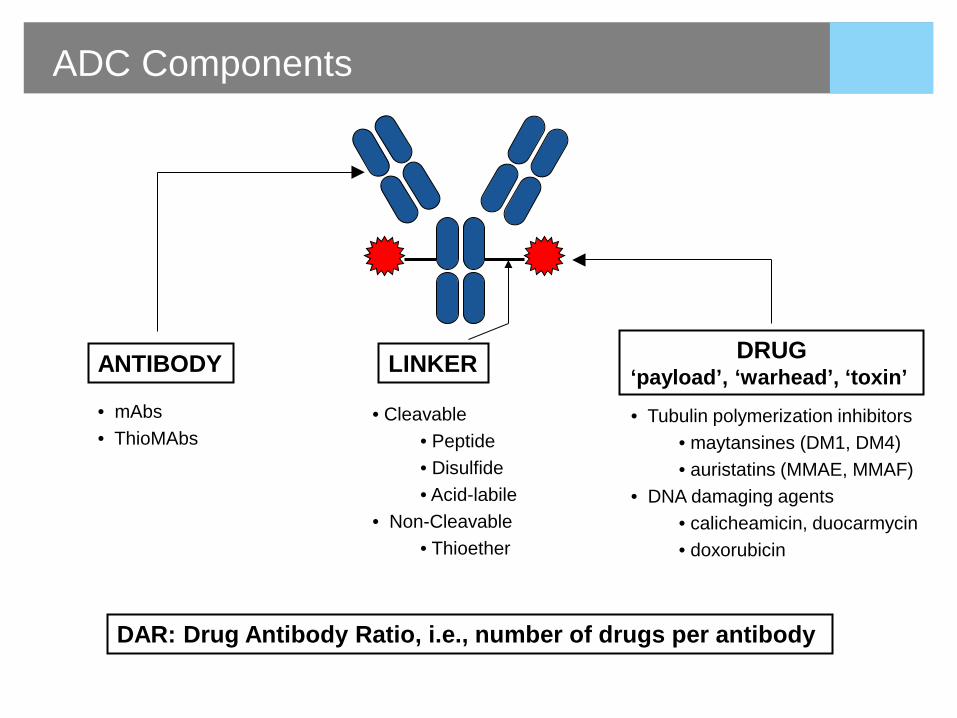
• mAbs • ThioMAbs
ADC Components
ANTIBODY LINKER DRUG ‘payload’, ‘warhead’, ‘toxin’
• Tubulin polymerization inhibitors • maytansines (DM1, DM4) • auristatins (MMAE, MMAF)
• DNA damaging agents • calicheamicin, duocarmycin • doxorubicin
DAR: Drug Antibody Ratio, i.e., number of drugs per antibody
• Cleavable • Peptide • Disulfide • Acid-labile
• Non-Cleavable • Thioether
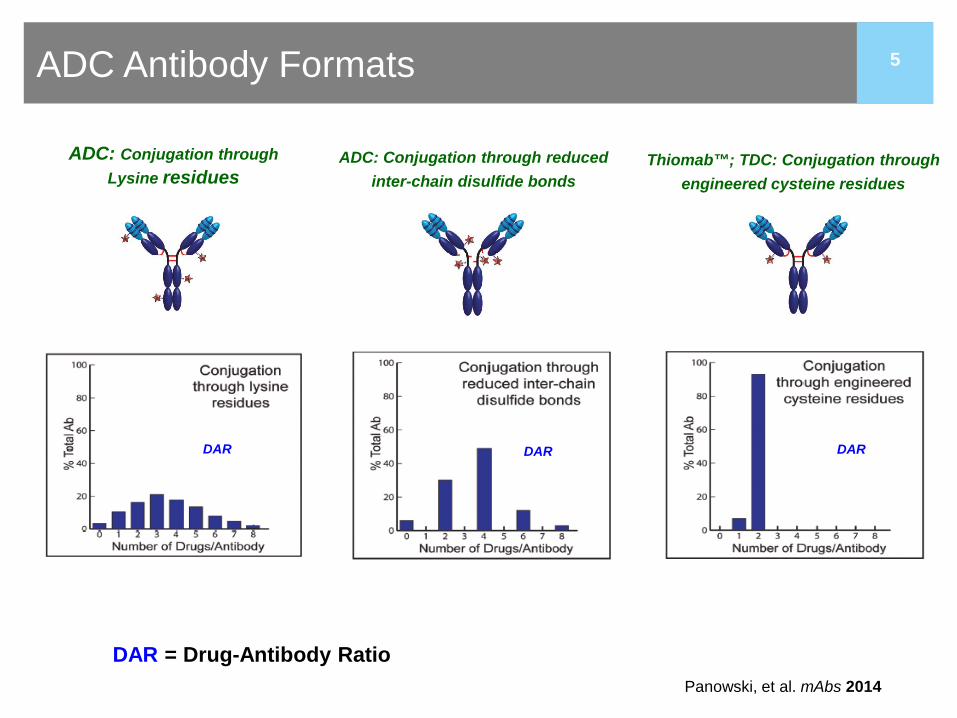
5 ADC Antibody Formats
ADC: Conjugation through Lysine residues
DAR = Drug-Antibody Ratio
DAR
ADC: Conjugation through reduced inter-chain disulfide bonds
DAR
Thiomab™; TDC: Conjugation through engineered cysteine residues
DAR
Panowski, et al. mAbs 2014
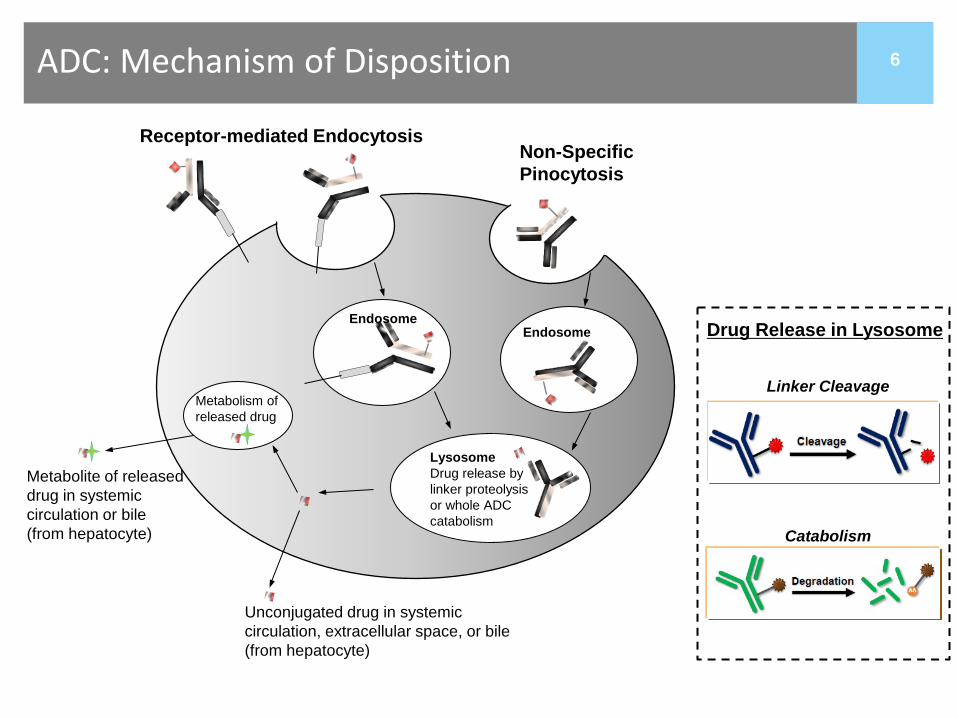
6
Receptor-mediated Endocytosis
Unconjugated drug in systemic circulation, extracellular space, or bile (from hepatocyte)
Metabolism of released drug
Metabolite of released drug in systemic circulation or bile (from hepatocyte)
Endosome Endosome
ADC: Mechanism of Disposition
Lysosome Drug release by linker proteolysis or whole ADC catabolism
Linker Cleavage
Catabolism
Drug Release in Lysosome
Non-Specific Pinocytosis
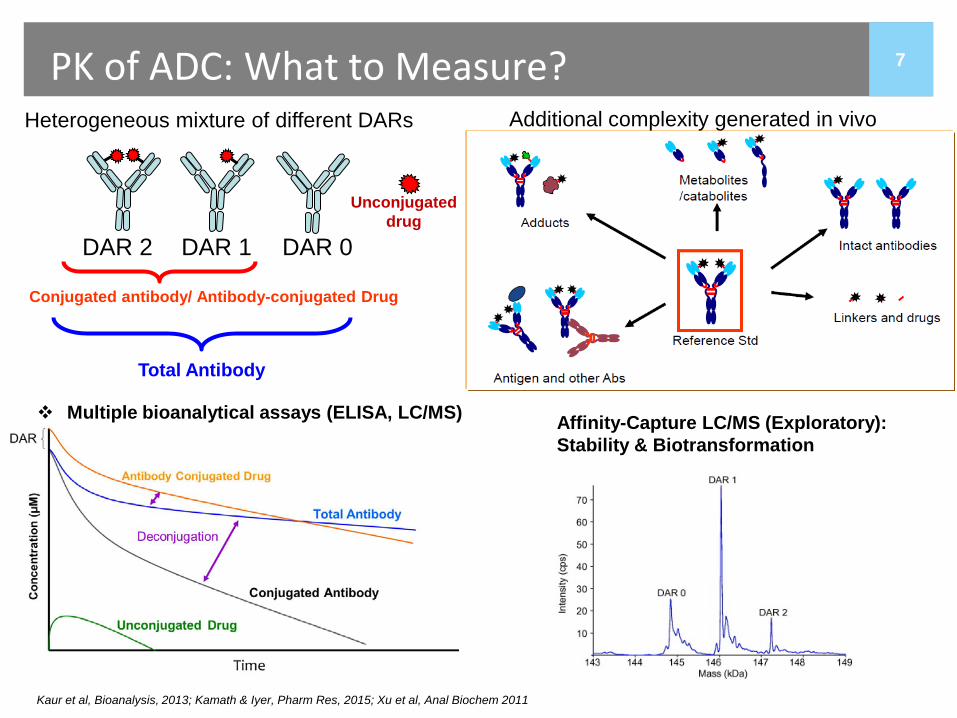
7 PK of ADC: What to Measure? Heterogeneous mixture of different DARs
DAR 0 DAR 1 DAR 2
Additional complexity generated in vivo
Conjugated antibody/ Antibody-conjugated Drug
Total Antibody
Unconjugated drug
Multiple bioanalytical assays (ELISA, LC/MS)
Kaur et al, Bioanalysis, 2013; Kamath & Iyer, Pharm Res, 2015; Xu et al, Anal Biochem 2011
Affinity-Capture LC/MS (Exploratory): Stability & Biotransformation
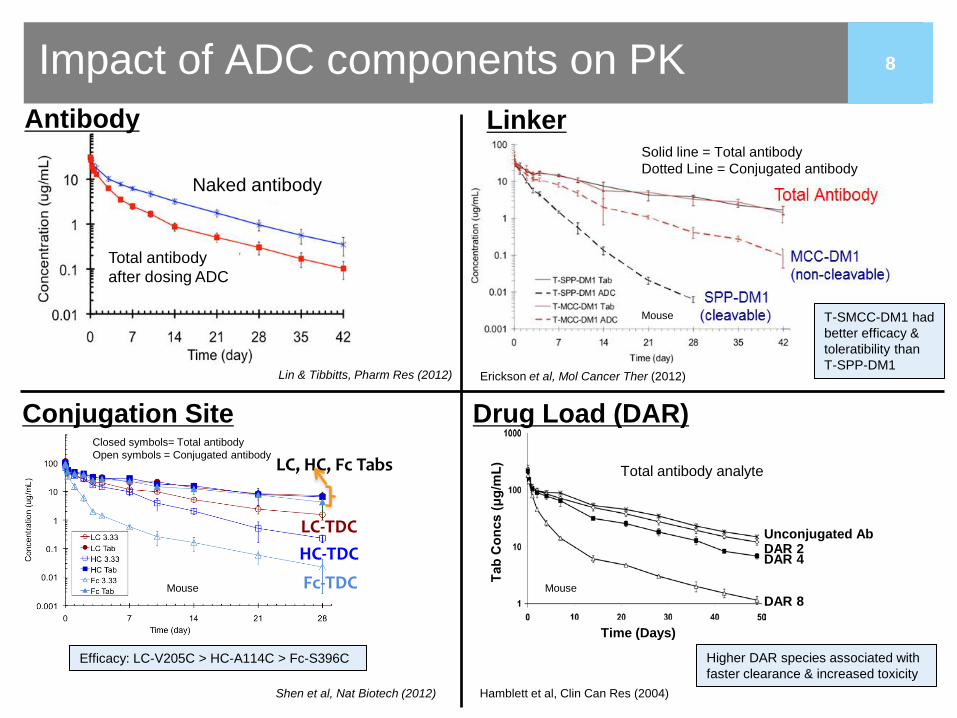
8
Unconjugated Ab DAR 2 DAR 4
DAR 8
Tab
Con
cs (μ
g/m
L)
Time (Days)
Fc-TDC
HC-TDC LC-TDC
LC, HC, Fc Tabs
Antibody
Impact of ADC components on PK
Conjugation Site Drug Load (DAR)
Linker
Lin & Tibbitts, Pharm Res (2012) Erickson et al, Mol Cancer Ther (2012)
Shen et al, Nat Biotech (2012) Hamblett et al, Clin Can Res (2004)
T-SMCC-DM1 had better efficacy & toleratibility than T-SPP-DM1
Efficacy: LC-V205C > HC-A114C > Fc-S396C Higher DAR species associated with faster clearance & increased toxicity
Solid line = Total antibody Dotted Line = Conjugated antibody
Mouse
Mouse Mouse
Total antibody after dosing ADC
Naked antibody
Closed symbols= Total antibody Open symbols = Conjugated antibody
Total antibody analyte
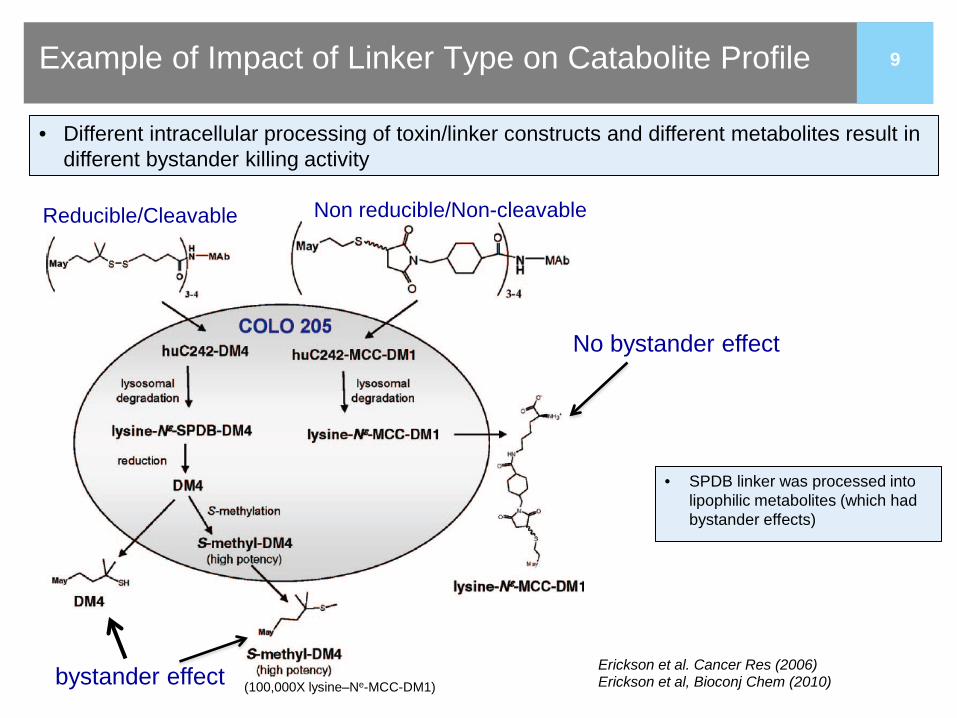
9 Example of Impact of Linker Type on Catabolite Profile
9 (100,000X lysine–Ne-MCC-DM1)
No bystander effect
Non reducible/Non-cleavable Reducible/Cleavable
bystander effect
• Different intracellular processing of toxin/linker constructs and different metabolites result in different bystander killing activity
• SPDB linker was processed into lipophilic metabolites (which had bystander effects)
Erickson et al. Cancer Res (2006) Erickson et al, Bioconj Chem (2010)

10 ADC PK/ADME: Key Assessments Types of Assessments • Linker stability
– What is optimal stability for efficacy/toxicity? – Should be stable in circulation, but promptly
release the drug in the target cells
• Catabolite/Metabolite ID – What is released? Is it active? – Does it accumulate in tissues/tumor? – How it is eliminated? – Is it clinically relevant?
• In vivo exposure (efficacy & tox)
– Exposure-response analysis – What is the driver of efficacy/toxicity? – Which analyte correlates with activity? – Plasma conc? Or tissue concs?
• Other assessments
– Drug-drug interactions – Immunogenicity
ADC Stability • In Vitro Study
– Plasma or Serum from human and efficacy & tox species – Incubation conditions
– 37ºC at pH 7.4 for 96 hours – ADC concentration around observed/predicted Cmax in
animal species or human – Typical analytes: Tab, conjugate, released drug, DAR – Can help optimize the combination of mAb, linker & drug
In Vivo PK Choice of animal species for ADC, same general principles as mAb • Ideal if cross-reactive in animal species (i.e., binding species)
– If not cross-reactive, PK & toxicity may not be reflective of humans. May still provide some information on non-specific disposition of ADCs and on potential drug-related metabolites
• Important to choose species that has similar in vivo fate/ deconjugation mechanism as in humans
• PK characterization at doses low enough to evaluate target mediated clearance and high enough to understand toxicokinetics
Typical PK analytes: Tab, conjugate, released drug, DAR

11 Is there a single in vitro system that can be used for characterization of both ADC and drug?
Hepatocytes Contain all relevant microsomal and cytosolic
enzymes Target protein is not expressed Drug may have limited permeability.
Liver microsomes Contain most relevant drug metabolizing enzymes Not confounded by drug permeability or uptake Lack the lysosomal enzymes responsible for
release of drug from ADC molecule
Liver S9 fraction Contains the same drug metabolizing enzymes
as hepatocytes. Does not rely on drug permeability Transporter independent Can be used at either pH 7.4 (to study
metabolism of the drug) or acidified to mimic lysosomal degradation of an ADC. Lysosomes
Mimic ADC degradation in the cell Artificial system which does not contain drug-
metabolizing enzymes Uptake of ADC might be limited
Cancer cells Selection of cell line would depends on target
expression, Limited by drug permeability Drug metabolizing enzymes expressed by
cancer cells are found in the live Have been shown to up-regulate Phase II
enzymes and down regulate Phase I enzymes as compared to the liver
Plasma Contains proteases

12 Is there a single in vitro system that can be used for characterization of both ADC and drug?
Liver microsomes Contain most relevant drug metabolizing enzymes Not confounded by drug permeability or uptake Lack the lysosomal enzymes responsible for
release of drug from ADC molecule
Liver S9 fraction Contains the same drug metabolizing enzymes
as hepatocytes. Does not rely on drug permeability Transporter independent Can be used at either pH 7.4 (to study
metabolism of the drug) or acidified to mimic lysosomal degradation of an ADC.
Plasma Contains proteases
Drug’s metabolic stability, reaction phenotyping, CYP/UGT inhibition
Characterization of drug-containing species released from an ADC. Identification of metabolites formed from the drug
Assessment of linker stability in the systemic circulation. PPB of released drug
In general, it is recommended that understanding of the linker and drug chemical structures and potential reactions that they can undergo, be taken into consideration when selecting the in vitro test system and the most straightforward (or simplest) system is used.

13
In most cases, systemic concentrations of released drug are extremely low, therefore, risk of ADC being a DDI perpetrator can be considered minimal.
o In a clinical DDI study, ADCETRIS® (vc-MMAE ADC) did not affect the PK of midazolam.
Assessment of DDI potential.
Probability of released drug to be a DDI victim exists and impact can be high due to the drug’s narrow therapeutic margin.
DDI risk assessment for a novel drug used in an ADC needs to be performed during development to determine if formal clinical studies should be conducted in accordance with the FDA and EMA guidelines.
Studies to assess transporter-mediated DDI may be valuable at later stages of the development.
Han & Zhao, DMD , 2014
o No profound changes in clinical PK of ADCETRIS® was observed when coadministered with rifampicin or ketoconazole. However, exposure of released MMAE was reduced by ~46 % and increased by ~34 % by coadministration of rifampicin and ketoconazole, respectively.
o Kadcyla® (DM1-containing ADC) label contains a caution that coadministration with strong CYP3A4 inhibitors should be avoided due to the potential for an increase in DM1 exposure and toxicity.
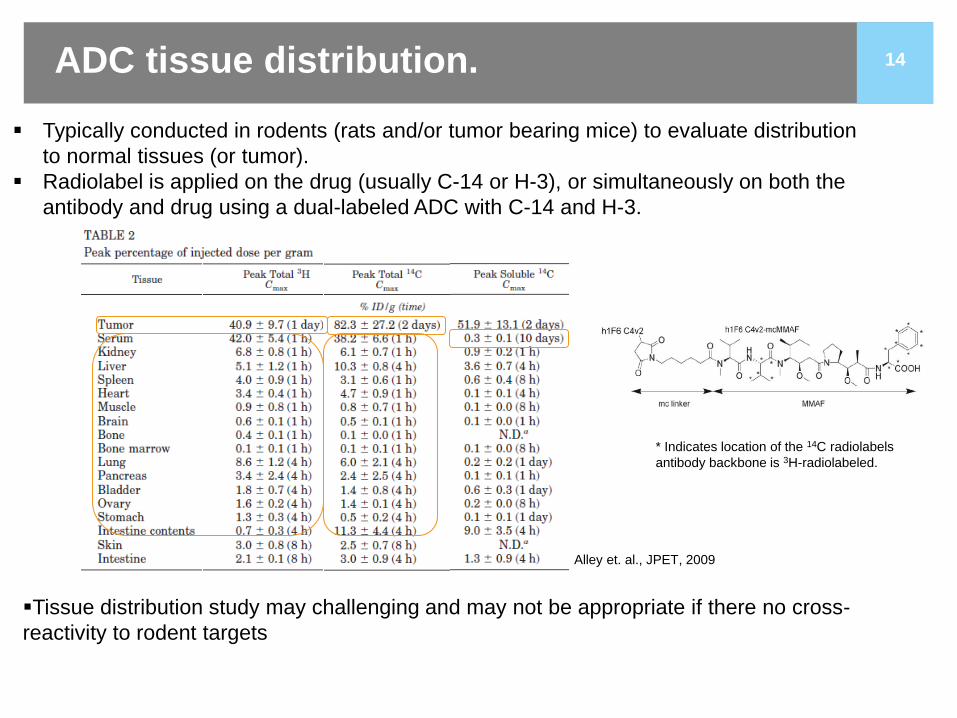
14
Typically conducted in rodents (rats and/or tumor bearing mice) to evaluate distribution to normal tissues (or tumor).
Radiolabel is applied on the drug (usually C-14 or H-3), or simultaneously on both the antibody and drug using a dual-labeled ADC with C-14 and H-3.
ADC tissue distribution.
Tissue distribution study may challenging and may not be appropriate if there no cross-reactivity to rodent targets
Alley et. al., JPET, 2009
* Indicates location of the 14C radiolabels antibody backbone is 3H-radiolabeled.

15
Currently, a human ADME study using radiolabeled material is not recommended. o For the cytotoxic/genotoxic drugs typically used in oncology ADCs, dosing of ADCs in
healthy volunteers is not appropriate. Evaluation would have to be conducted in cancer patients.
o Due to typically long ADC half-life, patients would have to be sequestered for prolonged periods of time (3-4 weeks) with little to no benefit to the patient, which would not be ethical.
o An ADME study of shorter duration may not be adequate and can result in incomplete mass balance data.
o Identification of the circulating products of further metabolism of the drug may be challenging due to typically very low concentrations of those products.
ADME (mass balance) evaluation.
An animal (rodent) ADME study using an ADC with radiolabel on the drug may be considered.
o Matrices to be collected: serum/plasma, bile, urine, and feces. o Since most of the ADCs do not cross react with rodent targets, this evaluation would
primarily address nonspecific uptake and degradation pathways and may not necessarily represent the disposition of ADC in humans.
o Due to the long half-life of ADCs the study duration would need to be extended in order to achieve good recovery of radioactivity and mass balance.

16 In vitro and in vivo studies for characterization of ADC ADME
Molecule ADME data
ADC* In vitro stability in plasma or serum from animals and humans.
ADC* PK in pharmacology and toxicology species
ADC**
ADC
Drug
Drug
Animal (rodent) ADME: PK, excretion, and metabolism
Identification of circulating metabolites formed from the released drug in patients
Rodent PK
Plasma protein binding across species
Drug
Drug
In vitro characterization of metabolites formed from the released drug (safety species and human)
Reaction phenotyping
Drug
Drug
Passive/active (uptake or efflux) transport (as substrate)
CYP inhibition and induction
* Analytes that could be measured as appropriate include Tab, ADC, unconjugated drug ** This evaluation is recommended to be conducted with an ADC bearing a radiolabel on the drug

17 What should be done for novel ADCs with previously characterized drugs?
• Drugs or linker-drugs previously tested in the clinic – Now conjugated to different mAbs to form new ADCs – Using novel linker or novel conjugation chemistry
• Existing ADME information usually available
– Published reports or filings – Internal unpublished data – Need to only generate key data specific to the novel ADC
• Additional ADME evaluation
– Plasma stability of the ADC – Major released drug-containing species – Major ADC clearance mechanisms – Confirm that projected human PK properties support intended dose and frequency of
administration.

18 Conclusions
ADME characterization for an ADC is a complex process as it needs to take into account both the mAb and small molecule components of this modality.
No standard “one size fits all” approach can be applied to all ADCs.
ADC ADME working group has evaluated advantages and disadvantages of the currently used experimental systems and strategies, and published white paper which provides guidance that should help investigators to develop successful novel ADCs with desirable ADME properties.
Since ADC technology is still evolving, the working group has proposed that this area of science is continuously monitored as it matures over the next several years and, if needed, currently used approaches are re-evaluated.

19 Acknowledgements
Colleagues from IQ member companies for their input and review of the white paper
IQ DMLG for recognizing the importance of this topic and for their guidance and support
• Marcel Hop (Genentech) • Volker Fischer (AbbVie) • Thomayant Prueksaritanont (Merck) • Sekhar Surapaneni (Celgene)

IQ Webinar Series: Current Approaches for ADME Characterization of Antibody-Drug Conjugates Q&A Session

Audience Q&A submissions • When testing metabolic pathways of an ADC with non cleavable
linker, what should be tested? Drug or linker-drug? Or both? • When testing metabolic pathways of an ADC with non cleavable
linker, what should be tested? Drug or linker-drug? Or both? • what about using Cyno PK for translation into the Clinic? • what are the advantages of having a smaller DAR ratio • Amrita, The data you showed for the difference in clearance of
DAR2,4,&8 were only for cysteine conjugated mAbs correct? This my not bee the same for lysine or other amino acid conjugated ADCs (with cysteine conjugated mAbs the mAb will have reduced covelent bonds between LC & HC which may impact the PK in a very differnt manner than ADCs conjugated through other amino acids.
21

Audience Q&A submissions • In the table, CYP inhibition studies were recommended, although
circulatory concentrations are expected to be very low. How about transporter inhibition studies?
• Is there any study looked at the impact of different PL on the profile of ADC?
• Have PBPK modelling been used by industries and accepted by regulatory to address ADC-DDIs
• Has ADME studies been conducted in tumor bearing animals? • For the rodent ADME study, would you consider doing them in
transgenic mice expressing the relevant DMEs for the toxin (e.g., CYP3A4) also bearing orthotopic human cell-based tumors that express the relevant target?
22

Audience Q&A submissions • Would you still run DAR assay for Thiomab ADC with 2 bound
payloads? • Generally at whats stage of development the DDI risk need to be
evaluated? • is there any update for ADC combinational therapy in immuno-
oncology field? • For in vitro assays, do you have any additional literature except for
apporved two ADCS? • For approved ADCs, how have other companies determined DAR?
Specifically, did they perform DAR investigations in the preclinical or clinical phase? If in clinical phase, were those investigations GLP or non-GLP?
23

Audience Q&A submissions • Can you comment on the status of LC/MS/MS vs. immunoassay for
measuring ADC and TAb, and what are your recommendations? • Can you comment on special considerations for ADME assessment
both preclinically and clinically for ADCs targeted for CNS malignancies? There are now ADCs being evaluated for CNS malignancies. What are the key attributes that maximize likelihood of success?
• What is the role of FCgamma receptor binding on toxicity of ADCs? • What species (ADC only or Total Antibody as well?) would you looks
at as the the predictor variable in clinical exposure-response analyses of ADCs (for efficacy)?
24

Audience Q&A submissions • Can you comment on the role/ utility of clinical imaging approaches
for assessing biodistribution/ target access in tumors as a component of POM ahead of Phase 2?
• What approach do you recommend for human PK predictions for ADCs?
• Is it necessary to repeat clinical CYP3A inhibitor or inducer DDI studies for new ADCs that have MMAE as a payload or can the results from brentuximab vedotin be extrapolated
25