cloning of total mrna populations from adult and embryonic mice
TRANSCRIPT

ARCHIVES OF BIOCHEMISTRY AND BIOPHYSICS
Vol. 223, No. 1, May, pp. 140-148, 1983
Cloning of Total mRNA Populations from Adult and Embryonic Mice
MICHAEL J. TOCCI,’ KENNETH A. FLEMING,2 AND JOHN J. MONAHAN
Roche In-stitute of Molecular Biology, Department of Cell Biology, Nutley, New Jersey 07110
Received November 18, 1982
Total clone banks of cDNAs synthesized from poly(A)-RNA obtained from three stages of the developing mouse were constructed. The stages chosen were 13-day-old embryo, neonatal, and fully grown adult. To have as complete a bank as possible, large numbers of individual clones were generated -400,000 for the 13th day embryo and neonatal mouse and -610,000 for the adult bank. In each case the clone bank was constructed by inserting double stranded cDNA into the PstI site of pBR322 by the “G-C tailing” method. Sequences cloned in this way could be separated from the plasmid host DNA by treatment of the resultant total chimeric plasmid population with PstI. Aliquots of the cloned cDNA material were labeled with 32P by “nick translation” using Escherichia coli DNA polymerase I for the preparation of hybridization probes. Back- hybridization of these probes to the total clone banks allowed the determination of the sequence diversity among the above three very different developmental stages. The use of such clone banks should allow the identification of developmental stage specific mRNAs.
Very little is known about how growth and development is controlled at the mo- lecular level in developing animals. The continuously changing nature (as the em- bryo develops) of the source material often has made even superficial molecular anal- ysis difficult. Studies in the past were often confined to the measurements of rates of RNA synthesis during embryogenesis, the content of RNA/cell, or the effects of RNA inhibitors on the process of embryogene- sis. Recently, some progress has been made in the isolation and analysis of poly(A)- RNA from early mouse embryos using classical techniques (l-5). Quantitative as well as qualitative differences were seen as the embryo developed. However, fur-
’ Present address: Department of Biochemical Ge- netics, P.O. Box 2000, Merck Sharpe & Dohme Re- search Laboratories, Rahway, New Jersey 07065.
’ Present address: University of Oxford, Depart- ment of Pathology, John Radcliffe Hospital, Head- ington, Oxford, 0X3, 9DU, England.
3 To whom all correspondence should be addressed.
ther progress along these lines will prob- ably be slow because of the limited supply of starting material and the monumental amount of work involved in carrying out such studies.
The application of recombinant DNA techniques has brought new hope to the possibility of unraveling the complex pro- cess of multigene expression during em- bryogenesis. With this technology specific RNA species can be obtained and at least some attempt can be directed toward searching for nearby regulatory DNA se- quences. Perhaps equally significant, clon- ing methodology provides an essentially unlimited supply of cloned DNA from a small supply of initial starting material. For a study of mammalian embryogenesis and development often only minute quan- tities of material can be isolated practi- cally. We have chosen the mouse embryo as a model system for studying the mo- lecular biology of embryogenesis. It is well characterized in terms of the systems morphological development, embryogene-
0003-9861/83 $3.00 Copyright 0 1983 by Academic Press. Inc. All rights of reproduction in any form reserved
140

EMBRYONIC AND ADULT RNA CLONE BANKS 141
sis can proceed to a reasonably advanced stage in vitro (7-lo), and a number of in- teresting developmental mutants exist (10-11).
MATERIAL AND METHODS
Animals. Embryos and neonatal mice were ob- tained from SWR/J X SJ/L matings (The Jackson Laboratory, Bar Harbor, Me.). Embryos (13th day) were dissected from adult female mice, trimmed free of extraembryonic membranes, rinsed twice in phos- phate-buffered saline, and frozen immediately in liq- uid nitrogen. Neonatal mice were obtained on the morning of their birth and treated similarly. All an- imals were stored at -79°C until used.
Isolation of poly(A)-RNA. Total RNA was isolated from la-day-old and neonatal embryos as follows: ap- proximately 20 g of tissue were homogenized in 200 ml of 20 mM Tris-HCl, pH ‘7.6, 1 mM EDTA, 2.0% SDS,’ and 0.5 mg/mi proteinase K (E. Merck, Darm- stadt) which had been preincubated at 45°C for 20 min. The material was homogenized for 60 s in a Po- lytron homogenizer (Brinkman, Westbury, N. Y.) in- cubated at 45°C for 1 h and then extracted with an equal volume of buffer-saturated phenol. The aqueous phase was ethanol-precipitated, the nucleic acid pel- let dissolved in 10 IIIM Tris-HCI, pH 7.6,5 mM EDTA, 0.01% SDS, and dry CsCl was added until a final density of 1.70 g/cm3 was achieved. Total RNA was pelleted by centrifugation at 30,000 rpm for 24 h at 10°C in a Beckman 70Ti rotor. After centrifugation, the supernatant was decanted, the RNA pellets dis- solved in water and the solution made up to 0.5 M
KCI, 20 mM Tris-HC1, pH 7.6, and 5 mM EDTA. Poly(A)-RNA was isolated by two cycles of oligo(dT)- cellulose column chromatography as described by Aviv and Leder (13). The poly(A)-RNA was then ethanol- precipitated and stored in water at -7O’C. Samples of RNA were monitored throughout the purification for degradation and DNA contamination by electro- phoresis in 1.5% agarose gels containing 10 mM
CHsHgOH as described by Bailey and Davidson (14). Total RNA from adult mice was isolated by phenol
extraction (15) and purified as described above. Synthesis qf‘ single-stranded complementary DNA
Iss-cDNA). The method for synthesizing ss-cDNA has been described previously (16). In general -7-15% of the poly(A)-RNA was converted to ss-cDNA in
4 Abbreviations used: AMV-RT, avian myelobas- tosis virus reverse transcriptase; TDT, terminal deoxynucleotidyl transferase; CFU, colony forming units; ss-cDNA, single-stranded cDNA; SDS, sodium dodecyl sulfate; ds-cDNA, double-stranded cDNA; NTBP, nucleotide base pairs, Tc’ tetracycline-resis- tant; Amp”, ampicillin-sensitive.
these experiments. For cloning purposes it was usu- ally necessary to generate -10 fig of ss-cDNA.
Synthesis of double-stranded cDNA (ds-cDNA). Af- ter hydrolysis of poly(A)-RNA with 0.3 M NaOH, the ss-cDNA was converted to double-stranded material using reverse transcriptase (17). Approximately 40- 60% of the ss-cDNA was coverted to ds-cDNA as monitored by S,-nuclease sensitivity.
Removal of hairpins from d.wDNA by S,-nuclease. Hairpins were cleaved from ds-cDNA preparations by digestion with S,-nuclease. The conditions used were critical. For optimum yields, the reaction was carried out in 20 mM NaOAc, 300 mM NaCI, 4.5 mM
ZnClz, pH 4.5. Digestion was for 20-30 min at room temperature using 100 units of S1 nuclease (Sigma Chemical Co., St. Louis, MO.), per microgram of ds- cDNA at a concentration of 1200 units/ml. The re- action mixture was diluted twofold with 0.1 M EDTA, phenol-extracted, and the aqueous phase precipitated with ethanol. Under these conditions 35-40% of the ds-cDNA was resistant to Si nuclease digestion. Sam- ples of ds-cDNA were assayed on 1.5% agarose-10 mM CH3HgOH gels before and after S1 digestion to determine the size of the cDNA and to ascertain whether or not the hairpins had been cleaved (16). ds-cDNA treated in this manner generally ranged from 200 to 1800 NTBP, with the average size being approximately 600 NTBP in length.
PuriJication of ds-cDNA by sucrose gradient cen- trifugation. Prior to “dCTP-tailing,” it was necessary to remove various small oligonucleotides from cDNA preparations by sucrose gradient centrifugation. Ap- proximately 1-5 Kg of S,-cleaved ds-cDNA in water was layered onto a 5-25% neutral sucrose gradient and centrifuged in a Beckman SW-40 rotor at 35,000 rpm for 20 h at 5°C. After centrifugation the gra- dients were collected and fractions containing ds- cDNA greater than 300 NTBP were pooled. These fractions were precipitated with ethanol at least twice. After precipitation the cDNA pellet was dissolved in water.
Terminal addition of dCTP to ds-cDNA. ds-cDNA was tailed with dCTP using calf thymus terminal deoxynucleotidyl transferase (TdT). The reaction mixture contained 100 PM dCTP, 1 mM CoCl,, 0.2 M K+ cacodylate, pH 6.9, and lo-20 Kg/ml ds-cDNA. Reaction mixtures were preheated to 37”C, 10 units of TdT per Fg of ds-cDNA was then added and the mixture incubated at 37°C for 3-5 min. The reaction mixture was then heated at 60°C for 2 min and frozen at ~70°C. Under these conditions 20-30 dC residues are added per end as determined by the incorporation of a-%PdCTP into acid-precipitable material.
Preparation of pBR322 DNA. The methods for pu- rifying and preparing dG-tailed, PstI cut pBR322 DNA have been described elsewhere (16). Briefly 200 Fg of pBR322 DNA were cut to completion with PstI re- striction endonuclease. The DNA was phenol-ex-

142 TOCCI, FLEMING, AND MONAHAN
tracted, ethanol-precipitated, and dissolved in water. Terminal addition of dGTP to P&I. pBR322 was
tailed with dGTP using TdT under the same reaction conditions described above, except that the dGTP concentration was increased to 1.0 mM and the re- action allowed to proceed for 2 h at 37°C. The DNA was then phenol-extracted, ethanol precipitated, and dissolved in water. The dG-tailed DNA was centri- fuged through a 5 to 25% neutral sucrose gradient using an SW40 rotor at 4°C and 38,000 rpm for 13 h. The gradient fractions were collected and assayed as previously described (16). Material with low trans- forming activity (i.e., linear-tailed DNA) was pooled, ethanol-precipitated, and used for transformation of E. coli.
Annealing dC-ds-cDNA and dG-pBR.322 to form chi- me& plasmids. ds-cDNA and pBR322 DNA were mixed such that the molar ratio of ends of the DNAs were approximately 5:l in 0.15 M NaCl, 10 mM Tris- HCl, pH 7.6, 1 mM EDTA, at a total DNA concen- tration of 4 fig/ml or less. The DNA was allowed to anneal at 58°C for at least 1 h in water and then held on ice (18).
Transfwmatimz of cells. Cells for transformation were prepared as described previously (18-19). Ap- proximately 1.5 ml of E. coli RR1 cells were grown to ODSN = 0.3, washed, and resuspended in approx- imately 6.5 ml of Ca++ medium. The competent cells were then mixed with 3.3 ml (-10 pg of DNA) of chimeric plasmid DNA, held on ice for 1 h with oc- casional shaking, heat-shocked for 3-4 min at 37”C, and l.O-ml aliquots plated onto large pans (-900 cm’) containing L media with 10 rg/ml tetracycline-HCl. The pans were then incubated at 37°C for 24-48 h. Recombinant clones were scraped off the agar surface into L broth containing 10 pg/ml each of tetracycline and streptomycin. The cells were washed once by cen- trifugation resuspended in 100 ml of L broth con- taining 5% (w/v) glycerol, and frozen in 2- to 3-ml, aliquots at ~70°C.
Transfomnation eficiency. The efficiency of trans- formation for these experiments ranged from 50,000 to 200,000 colony forming units (CFU)/Fg ds-cDNA. The average transformation efficiency was approxi- mately 100,000 CFU/gg ds-cDNA.
Ampli$cation and isolation of plasmid DNA. The method for isolation of pBR322 and recombinant pDNAs has been described before in detail else- where (16).
Restriction endonuclease analysis qf cloned mouse cDNA. All restriction digests were performed in 6 rnM Tris-HCI, pH 7.6, 6 mM p-mercaptoethanol, and 6 mM MgClz. The salt concentration of each digestion was adjusted with the appropriate salt based upon the supplier’s recommendations and the enzyme used. All reactions were incubated at 37°C and analyzed by agarose-gel electrophoresis as described else- where (16).
Purijication of cloned cDNA inserts. Plasmid DNAs from each clone bank (-500 pg) were digested to completion with PstI restriction endonuclease (0.1 pg of DNA). The DNA was then phenol-extracted, ethanol-precipitated, and stored in water. PstI di- gested pDNA was layered onto a 5-25s neutral su- crose gradient and centrifuged in an SW40 rotor at 38,000 rpm for 20 h at 4°C. The gradient was collected and those fractions containing cDNA inserts but not pBR322 DNA were pooled and ethanol-precipitated.
Nick translation of cloned-cDNA inserts. Purified PstI-digested insert DNA used for complexity anal- ysis was nick-translated as described by Rigby et ~1. using @P]dATP as label (20).
Complexity analysis of mouse embryo cDNA li- braries. Liquid hybridization was carried out at 68°C in sealed Reacti-vials (Pierce Chemical Co.). Linear- ized plasmid DNA (using EcoRI) at various concen- trations was mixed on ice with nick-translated probe (-5,000 cpm/vial) in 0.5 M NaCl, 0.1 M Hepes, pH 7.6, and 1 mM EDTA. The DNA was denatured on ice for 10 min by the addition of l/10 vol 1.0 M NaOH and then neutralized with l/10 ~011.0 M HCl. All reaction vials were then incubated for various times until the appropriate Cot values were obtained.
Cross-hybridization analysis. Cross-hybridization experiments were carried out by hybridizing either 32P-labeled ss-cDNA (prepared form embryo RNAs) or nick-translated cloned cDNA inserts (isolated from cDNA libraries) to poly(A)-RNA from different de- velopmental stages, or back to the pDNA plasmid DNA from the cloned cDNA libraries. Hybridizations were incubated to a Cot or Rot of approximately 2500 mol s liter-’ under the same reaction conditions as described above, except that for RNA hybridizations the denaturation step was omitted.
S, n&ease assay. All hybridizations were assayed for S, resistant material in the following manner. To each hybridization reaction (-20 ~1) was added 225 ~1 of Si nuclease buffer containing 300 mM NaCl, 30 mM sodium acetate, 4.5 mM ZnCla, pH 4.6, and 5 ~1 (200 units) of S1 nuclease (Miles Biochemicals). Sam- ples were incubated at 37°C for 90 min and then as- sayed by binding to DE-81 filter discs (21).
RESULTS
The procedure we have used to synthe- size and clone cDNAs is simply an exten- sion of the methods used to clone unique mRNAs such as hemoglobin and ovalbu- min (22-24). For the isolation of RNA from 13-day-old embryos and neonatal mice the proteinase K digestion procedure gave good yields of poly(A)-RNA. However, with adult mice, yields were poor by this method. Instead, for adult tissue, a

EMBRYONIC AND ADULT RNA CLONE BANKS 143
A B
910
600 520 403
281
FIG. 1. Electrophoresis of total adult clone bank plasmid DNA after treatment with EcoRI and BglI. The large smear at position X in lane A, is due to the numerous inserts of cDNA that have been cloned into this region of the PBR322 plasmid. Lane B is the molecular weight marker of pBR322 DNA cut with AlUI.
multiple phenol extraction method (15) was used. In every case the yield and pu- rity of the poly(A)-RNA were carefully monitored by electrophoresis on methyl mercury hydroxide gels. Single-stranded cDNAs was made from poly(A)-RNA preparations using AMV reverse tran- scriptase as described under Materials and Methods. Usually, about 10% of the RNA could be converted into cDNA. Gel elec- trophoresis of the 32P-labeled cDNA was used to determine the size of the cDNA. In these studies, the cDNA ranged in size from 200 to 1200 nucleotides in length. Short cDNAs were removed at a later stage in the procedure (see below). Reverse tran- scriptase was then used to convert the ss- cDNA to its double-stranded equivalent. Approximately 50-60% of ss-cDNA was converted in these experiments.
The critical step in the procedure was cutting of the hairpin loops in the cDNA. The concentrations of both DNA and S1 enzyme were critical. Using the conditions described under Materials and Methods, up to 50% of the DNA was S1 resistant and suitable for cloning. As an assay for complete cutting, the DNA was analyzed on a denaturing gel. A twofold decrease in the overall molecular weight of a band was taken as complete S1 cutting (16). The DNA was then phenol-extracted, ethanol-pre- cipitated, dissolved in dHpO, and sedi- mented through a neutral sucrose gra- dient. Material 8s and larger was pooled, ethanol-precipitated twice, and tailed with dCTP using TdT. After the addition of ap- proximately 15-20 dC residues to the 3’- OH ends, the ds-cDNA was annealed with dG-tailed Pat1 cut pBR322 (16).
Suitable clone banks were made as de- scribed under Materials and Methods. The number of individual clones was counted and the fraction containing no inserts (background) was determined by plating aliquots of the cells on Tc and Amp plates. Only clones that were Tc’ Amp” were scored as containing inserts.
Figure 1 shows the pattern obtained when the total plasmid clone bank (in this case the adult bank) was digested with EcoRI and &II. The discrete fragment that would exist in unmodified pBR322 con- taining the PstI site was replaced by ma- terial that migrated as a broad smear (la- beled X in Fig. 1). Figure 2 shows the op- tical density scan of such digests from each of the three clone banks (13-day-old, neo- natal, and adult). The arrow points to the area containing the cDNA inserts. Curve D is the pattern seen for pBR322 digested with BglI and EcoRI. From the area under each relevant peak the fraction of insert cDNA sequences in each clone bank was determined.
To analyze the sequence complexity of each clone bank a small amount of each cDNA insert was labeled with 32P. The to- tal insert population was separated from the plasmid DNA after digestion with PstI by sucrose gradient centrifugation (16).
When the 32P-labeled inserts were hy- bridized with an excess of plasmid clone
144 TOCCI, FLEMING, AND MONAHAN
FIG. 2. Optical density profiles of photographs sim- ilar to the one in Fig. 6. The inserts position is in- dicated by the arrow. A, the profile for the adult clone bank. B, the profile for the neonatal clone bank. C, the profile for the 13-day-old embryo clone bank. D, the profile of pBR322 control cut with EcoRI and B&I.
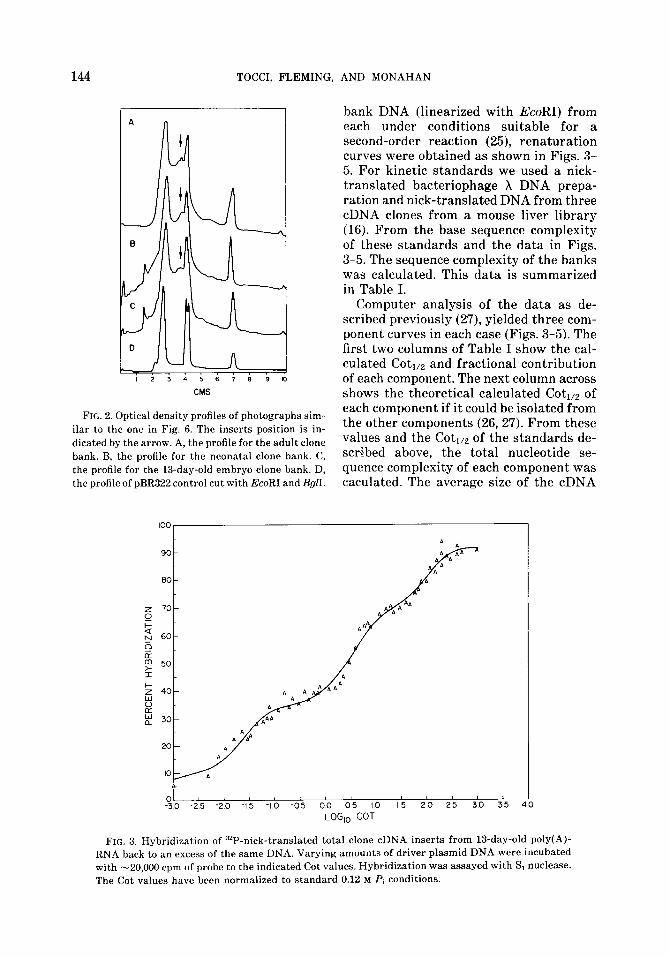
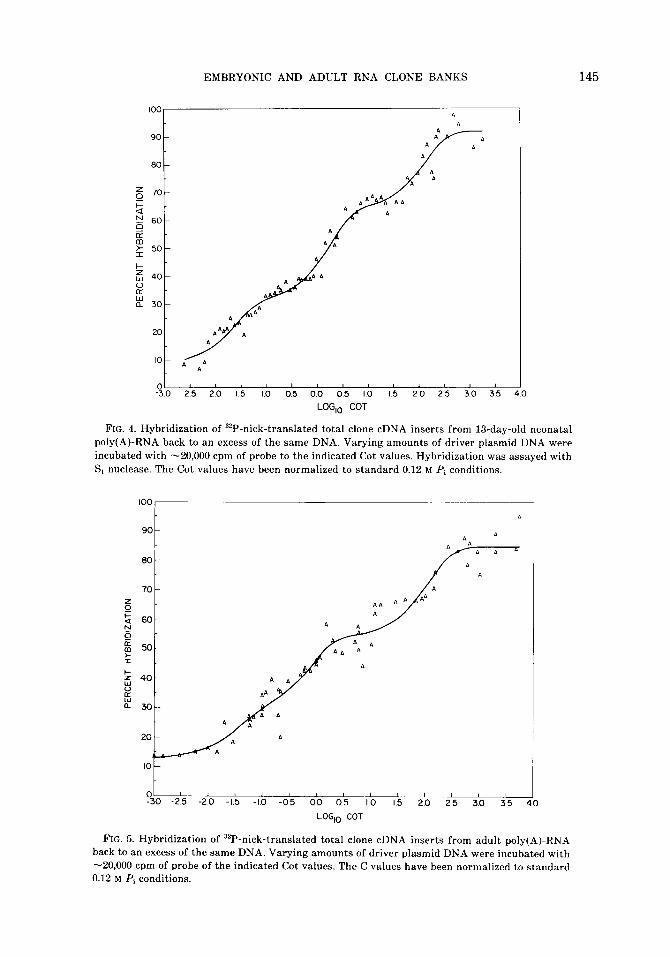
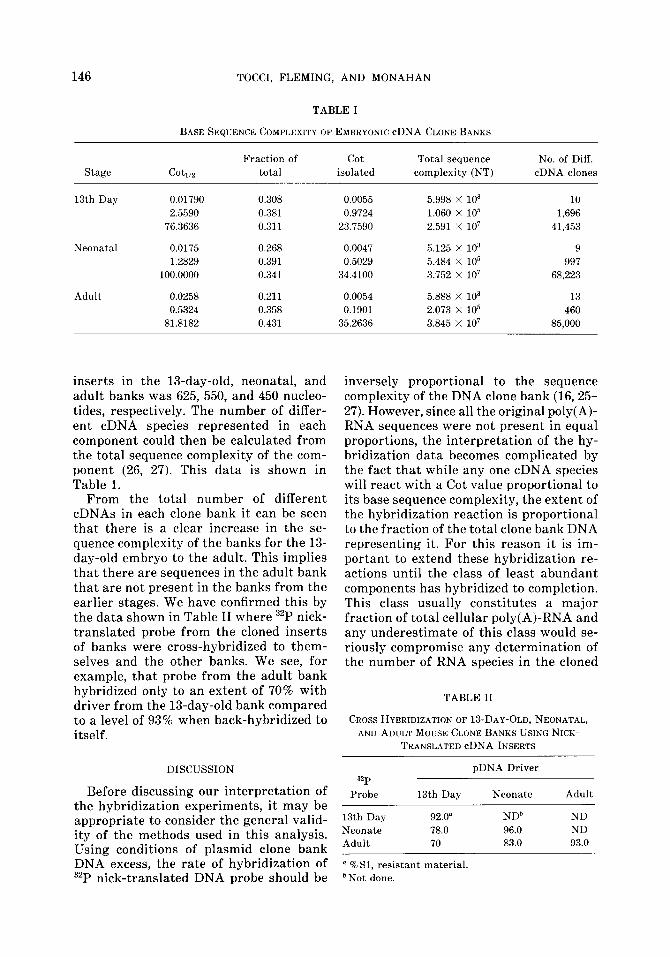
bank DNA (linearized with EcoRI) from each under conditions suitable for a second-order reaction (25), renaturation curves were obtained as shown in Figs. 3- 5. For kinetic standards we used a nick- translated bacteriophage X DNA prepa- ration and nick-translated DNA from three cDNA clones from a mouse liver library (16). From the base sequence complexity of these standards and the data in Figs. 3-5. The sequence complexity of the banks was calculated. This data is summarized in Table I.
Computer analysis of the data as de- scribed previously (27), yielded three com- ponent curves in each case (Figs. 3-5). The first two columns of Table I show the cal- culated Coti,z and fractional contribution of each component. The next column across shows the theoretical calculated Coti,a of each component if it could be isolated from the other components (26,27). From these values and the Coti,a of the standards de- scribed above, the total nucleotide se- quence complexity of each component was caculated. The average size of the cDNA
h
%I -25 -20 -15 -1 0 -05 00 05 IO 15 20 25 30 35 ‘ LOGlO COT
FIG. 3. Hybridization of 32P-nick-trans1ated total clone cDNA inserts from 13-day-old poly(A)- RNA back to an excess of the same DNA. Varying amounts of driver plasmid DNA were incubated with -20,000 cpm of probe to the indicated Cot values. Hybridization was assayed with Si nuclease. The Cot values have been normalized to standard 0.12 M Pi conditions.
EMBRYONIC AND ADULT RNA CLONE BANKS 145
I p3.0
I 1 I 2.5 20 15 IO 0.5 0.0 05 IO I5 20 25 30 35 40
LOG,, COT
FIG. 4. Hybridization of 32P-nick-translated total clone cDNA inserts from 13-day-old neonatal poly(A)-RNA back to an excess of the same DNA. Varying amounts of driver plasmid DNA were incubated with -20,000 cpm of probe to the indicated Cot values. Hybridization was assayed with S, nuclease. The Cot values have been normalized to standard 0.12 M Pi conditions
n
t 01 I 1 1 I I -30 -25 -20 -15 -10 -05 00 05 IO 15 20 25 30 35 1
LOGlO COT
FIG. 5. Hybridization of “P-nick-translated total clone cDNA inserts from adult poly(A)-RNA back to an excess of the same DNA. Varying amounts of driver plasmid DNA were incubated with -20,000 cpm of probe of the indicated Cot values. The C values have been normalized to standard 0.12 M Pi conditions.
146 TOCCI, FLEMING, AND MONAHAN
TABLE I
BASE SEQUENCE COMPLEXITY OF EMBRYONIC cDNA CLONE BANKS
Stage cotm
Fraction of cot Total sequence No. of Diff.
total isolated complexity (NT) cDNA clones
13th Day 0.01790
2.5590
76.3636
0.308
0.381
0.311
0.0055
0.9724
23.7590
Neonatal 0.0175 0.268 0.0047
1.2829 0.391 0.5029
100.0000 0.341 34.4100
Adult 0.0258 0.211 0.0054
0.5324 0.358 0.1901
81.8182 0.431 35.2636
5.998 X lo3
1.060 X lo5
2.591 x lo7
5.125 X 10”
5.484 X lo5
3.752 X lo7
5.888 X lo3
2.073 X lo5 3.845 x lo1
10
1,696
41,453
9
997 68,223
13
460
85,000
inserts in the 13-day-old, neonatal, and adult banks was 625, 550, and 450 nucleo- tides, respectively. The number of differ- ent cDNA species represented in each component could then be calculated from the total sequence complexity of the com- ponent (26, 27). This data is shown in Table 1.
From the total number of different cDNAs in each clone bank it can be seen that there is a clear increase in the se- quence complexity of the banks for the 13- day-old embryo to the adult. This implies that there are sequences in the adult bank that are not present in the banks from the earlier stages. We have confirmed this by the data shown in Table II where 32P nick- translated probe from the cloned inserts of banks were cross-hybridized to them- selves and the other banks. We see, for example, that probe from the adult bank hybridized only to an extent of 70% with driver from the 13-day-old bank compared to a level of 93% when back-hybridized to itself.
inversely proportional to the sequence complexity of the DNA clone bank (16,25- 27). However, since all the original poly(A)- RNA sequences were not present in equal proportions, the interpretation of the hy- bridization data becomes complicated by the fact that while any one cDNA species will react with a Cot value proportional to its base sequence complexity, the extent of the hybridization reaction is proportional to the fraction of the total clone bank DNA representing it. For this reason it is im- portant to extend these hybridization re- actions until the class of least abundant components has hybridized to completion. This class usually constitutes a major fraction of total cellular poly(A)-RNA and any underestimate of this class would se- riously compromise any determination of the number of RNA species in the cloned
TABLE II
CROSS HYBRIDIZATION OF I3-DAY-OLD, NEONATAL, AND ADULT MOUSE CLONE BANKS USING NICK-
TRANSLATED cDNA INSERTS
DISCUSSION
Before discussing our interpretation of the hybridization experiments, it may be appropriate to consider the general valid- ity of the methods used in this analysis. Using conditions of plasmid clone bank DNA excess, the rate of hybridization of 32P nick-translated DNA probe should be bNot done.
pDNA Driver
=P Probe 13th Day Neonate Adult
13th Day 92.0” NDb ND
Neonate 78.0 96.0 ND
Adult 70 83.0 93.0
u. % Sl, resistant material.

DNA population. Fortunately, with plas- quences. For example, we would like to mid DNA sequences there is no problem identify those clones corresponding to the in obtaining enough DNA to drive these new mRNAs that are produced as the reactions essentially to completion. Any mouse embryo progresses from the 13-day remaining unhybridized sequences would stage to the adult. From the data in Table by definition be extremely rare, and would II, we see that hybridization of a nick- be present at a frequency of less than 1 translated adult cDNA probe to 13-day-old molecule per -100 cells (25, 27). clone bank material yields a value of only
The data in Table I, summarize our ob- 70% (compared to 93% when the probe is servations of the sequence complexity of hybridized to its own adult clone bank the DNA in the 13th day, neonatal, and DNA). The remaining -20% of unhybrid- adult mouse banks. As can be clearly seen ized DNA has provided us with suitable from column five of Table I, there is a pro- radioactive probes to identify what we call gressive increase in the number of differ- “difference clones.” There are clones cor- ent sequences in the banks from 13-day- responding to new mRNAs that appear as old embryo to the adult, the adult total the embryo develops. Such “difference complexity being almost twice that of t,he clones” sequences are presently being uti- 13-day-old embryo. The number of se- lized in assays involving in vitro hybrid- quences in the class of most abundant se- ization and autoradiography on tissue of quences is similar in all three cases. It is the developing embryo. tempting to speculate that these corre- spond to structural proteins that are pro- duced by many cells such as actin, tubu-
ACKNOWLEDGMENTS
lin, myosin, collagen, laminin, and fibro- We thank Carolyn Fimiani for her excellent tech-
nectin. nical help during the course of this work and Dr.
In the case of the intermediate abun- Michael I. Sherman and Jay Haron for many valuable
dance class there is an almost fourfold re- discussions.
duction in the number of different se- quences in between the 13-day-old embryo REFERENCES
and the adult. The significance of this is not clear to us. It should be stressed that
1. LETTEY, I. L., STULL, G. B., ANL) BRINSTEK, R. L. (1978) Dev. Bid. 64, 140-148.
what we have analyzed is the sequence 2. BACHX~AROVA, R. (1981) Dew. Bid. 86, 384-392. complexity of the clone banks and not the 3. SCHIJLTZ, G. A., AND CHIJRCH, R. B. (1975) in The originating poly(A)-RNA starting mate- Biochemistry of Animal Development (Weber,
rials. It is conceivable that the cloning pro- R., ed.), Vol. 3, pp. 47-90, Academic Press, New
cess has discriminated among some cDNA York.
sequences causing them to move from one 4. CHURCH, R. B., AND BROWN, I. R. (1972) in Nucleic
abundance group to another (either up or Acid Hybridization in the Study of Cell Dif-
down). However, using a total mouse liver ferentiation (Ursprung, H., ed.), pp. 11-24,
poly(A)-RNA clone bank, we have previ- Springer, New York.
ously shown that if precautions are taken 5. GELDERMAN, A. G., RAKE, A. V., AND BKITTEN,
to make large scale plasmid DNA prepa- R. J. (1971) PTOC. Nat Acad Sci. USA 68, 17% 176.
rations from the initial stock of trans- 6. BROWN, I. R., AND CHIJRCH, R. B. (1972) Dev. Bid. formed cells (rather than serially passage 29, 73-84.
of cultures), the resulting complexity of 7. R~JGII, R. (1967) in The Mouse, pp. 44-102, Bur-
the cloned cDNA sequences bears a good gess, Minneapolis, Minn.
relationship to the original poly(A)-RNA 8. HXJ, Y. C. (1978) in Methods in Mammalian Re-
preparation (16). In any case, relatively production (Daniel, J. C., ed.), p. 229, Academic
minor distortions of a clone bank (com- Press, New York.
pared to the source material) are not of 9. SHERMAN, M. I. (1975) Di~erentiation 3, 51-67.
great concern to us, because our main use 10. RIZZINO, A., AND SHERMAN, M. I. (1979) Exp. Cell
of such banks will be as a repository form Res. 121, 221-223.
which to isolate cloned stage specific se- 11. SHERMAN, M. I., AND WUDL, L. R. (1976) in The
Cell Surface in Animal Embryoyenesis and
EMBRYONIC AND ADULT RNA CLONE BANKS 147

148 TOCCI, FLEMING, AND MONAHAN
Development (Poste, G., and Nicolson, G. L., eds.), p. 81, North-Holland, Amsterdam, Am- sterdam.
12. SILVER, L. M., ARTZT, K., AND BENNETT, D. (1979) Cell 17,275-284. WEITLAUNG, H. M., AND KIES- SLING, A. A., (1980) Den. BioL 77, 116-129.
13. AVIV, H., AND LEDER, P. (1972) Proc. Nut. Acad Sci. USA 69, 1408-1412.
14. BAILEY, J. M., AND DAVIDSON, N. (19’76) Anal Biochem. 70,75-85.
15. PENMAN, S. (1979) in Fundamental Techniques in Virology (Habel, K., and Salzman, N. P., eds.), pp. 35-48, Academic Press, New York.
16. NORGARD, M. V., TOCCI, M. J., AND MONAHAN, J. J. (1980) J. BioL Chem. 255, 7665-7672.
17. MONAHAN, J. J., HARRIS, S. E., AND O’MALLEY, B. W. (1976) J. BioL Chem. 251, 3738-3748.
18. PEACOCK, S. L., MCIVER, C. M., AND MONAHAN, J. J. (1981) B&hem. Biophys. Acta 655, 243- 250.
19. NORGARD, M. V., KEEM, K., AND MONAHAN, J. J. (1978) Gene 3, 279-292.
20.
21.
22.
23.
24.
25.
26.
27.
RIGBY, P. W. J., DIECKMANN, M., RHODES, C., AND BERG, P. (1977) J. Mol. BioL 113, 237-251.
MAXWELL, I. H., VANNESS, J., AND HAHN, W. E. (1978) NucL Acid Res. 5, 2033-2038.
ROUGEON, F., KOURILSKY, P., AND MACH, B. (1975) NucL Acid Res. 2, 2365-2377.
MANIATIS, T., KEE, S. G., EFSTRATIADIS, A., AND KAFATOS, F. C. (1976) Cell 8, 163-182.
MCREYNOLDS, L. A., MONAHAN, J. J., BENDURE, D. W., Woo, S. L. C., PADDOCK, G. V., SALSER, W., DORSON, J., MOSES, R. E., AND O’MALLEY, B. W. (1977) J. BioL Chem. 252, 1840-1843.
BRITTEN, R. J., AND DAVIDSON, E. H. (1976) Proc. Nat. Acad Sci. USA 73, 415-419.
BISHOP, J. O., MORTON, J. G., ROSBASH, M., AND RICHARDSON, M. (1974) Nature (London) 250, 199-203.
MONAHAN, J. J., HARRIS, S. E., AND O’MALLEY, B. W. (1977) in Receptors and Hormone Action (O’Malley, B. W., and Birnbaumer, L., eds.), Vol. 1, pp. 297-329.