chapter - iishodhganga.inflibnet.ac.in/bitstream/10603/24009/8/08... · 2018-07-09 · chapter - ii...
TRANSCRIPT

CHAPTER - II
ELECTRON TRANSFER REACTIONS OF ORGANIC CATIONS AND
RELATED SPECIES
I I ,1 Oxidation o f electron rich organic compounds to
stable rad ica l cations by DDQ in acid media*
11*2 Carbocations as electron transfer oxidants*
11*3 Electron Transfer reactions o f arene-diazonium
ions*

145
INTRODUCTION
Interaction o f two organic reagents, one
containing electron withdrawing groups such as C-0,
C«N, N»N# halogen, N02 or ca tion ic centres, and the other
electron rep e llin g ones e . g . , a lk y l, amino, alkoxy, and
anionic centres can lead to an in i t ia l step o f s in g le electron
transfer to form rad ica l cations- rad ica l anion pairs orkeep
rad ica l ion s-free rad ica ls in solution • Reports/pouring in
which suggest electron transfer steps in many o f these
reactions as a v iab le a ltern a tive to the conventional
mechanisms*
Highly cyanated unsaturated compounds such as
tetracyanoethylene (TCNE) and tetracyano-quinodimethane (TCMQ)
with weakly oxid is ing properties can undergo electron transfer
with eas ily ox id izab le compounds. For instance reaction o f
TCNE with metal hydrides proceeds by a rad ica l cation -rad ica l
anion pair from an in i t ia l charge-transfer complex1. Radical
processes have been invoked in the reaction o f TCMQ with some
anions such as sodium benzoate, sodium s a lic y la te and2
sodium acetate .
Peroxides reduce a host o f organic substrates by the
ET pathway. An ea r lie r example o f such a reaction is the
in teraction o f dibenzoylperoxide with M,N-dimethylaniline to3
generate the rad ica l cation o f the amine . A number o f other4 5compounds such as aromatic hydrocarbons , dimethoxybenzenes,
c 7 8anisplnacolone', diphenylhydroxyl amine and su lfid es have
been oxidised by the peroxides to the in i t ia l rad ica l cation.

Radical cations iso la ted in the so lid s ta te e f fe c t
oxidation o f a wide va rie ty o f compounds by the electron, . Q-14
transfer (ET) pathway . These reagents o f fe r a convenient
means o f generating rad ica l cations from organic substrates
fre e o f complications,such as ligand transfer, normally
encountered in oxidations with metal ions. The rad ica l cations
generated by th is method undergo a series o f bond scission
and bond formation analogous to the reactions encountered in
anodic processes.
Stable carbocations form charge transfer complexes
with donors e .g . , aromatic hydrocarbons and under certain
conditions complete ET from donor to acceptor can occur.
Certain o f the carbonium ions such as t r i t y l and i t s
deriva tives and tropylium ions are oxidants o f moderate
poten tia ls and many o f th e ir reactions involve electron
transfers. In many cases the rad ica l cations thus generated
have been id en tified by EPR spectroscopy^. Thus when t r i t y l
cation is mixed with perylene and tetracene in methylene
ch loride, the corresponding rad ica l cation is formed as
indicated by i t s EPR spectra (Eq,1 ). Tris-p-chlorophenyl
carbenium ion oxid ises pyrene and anthracene to the cation 15rad ica ls J•
146
(1 )Triphenyl~methyl cation accepts an e lectron from
ferrocene in nitromethane forming ferricin ium ions and the
triphenyl methyl rad ica l which reacts with oxygen to form
the peroxide1 (Scheme 11*13.

147
Ph3C
Fe4-
* Ph3C*
Ph3 COOCPhj \KU/
Scheme I I . 1
9-Methyl-9-fluorenyl trimethyl t in reacts with17 18triphenyl methyl sa lts by an ET process ’ (Scheme I I . 2)
+ Ph,CT +Ph,C*+Me,Sn+ 0 5
Scheme I I . 2
The fa te o f fre e rad ica ls formed on one electron
reduction o f the carbocations depends on the reaction1 Qconditions. In some cases dimer formation is observed (Eq.2).
(2 )Ph3C* Ph3C - CPh3
In the reaction o f tropylium sa lts with carbazole
evidence has been presented towards the formations o f20carbazole rad ica l cation . The ET reaction o f carbocations
21with o le fin s leads to polymerization • Several other reports
ex is t which describe the ro le o f stable carbocations as22-24electron acceptors •

148
Aryl diazonium ions have a high e lectron a f f in it y
and many o f i t s reactions with anions involve electron
transfer. For example Singh and coworkers have demonstrated25 26
that iodide and n it r i t e ions reduce various aryldiazonium
cations by a s ing le ET process to produce a ry l radicals which
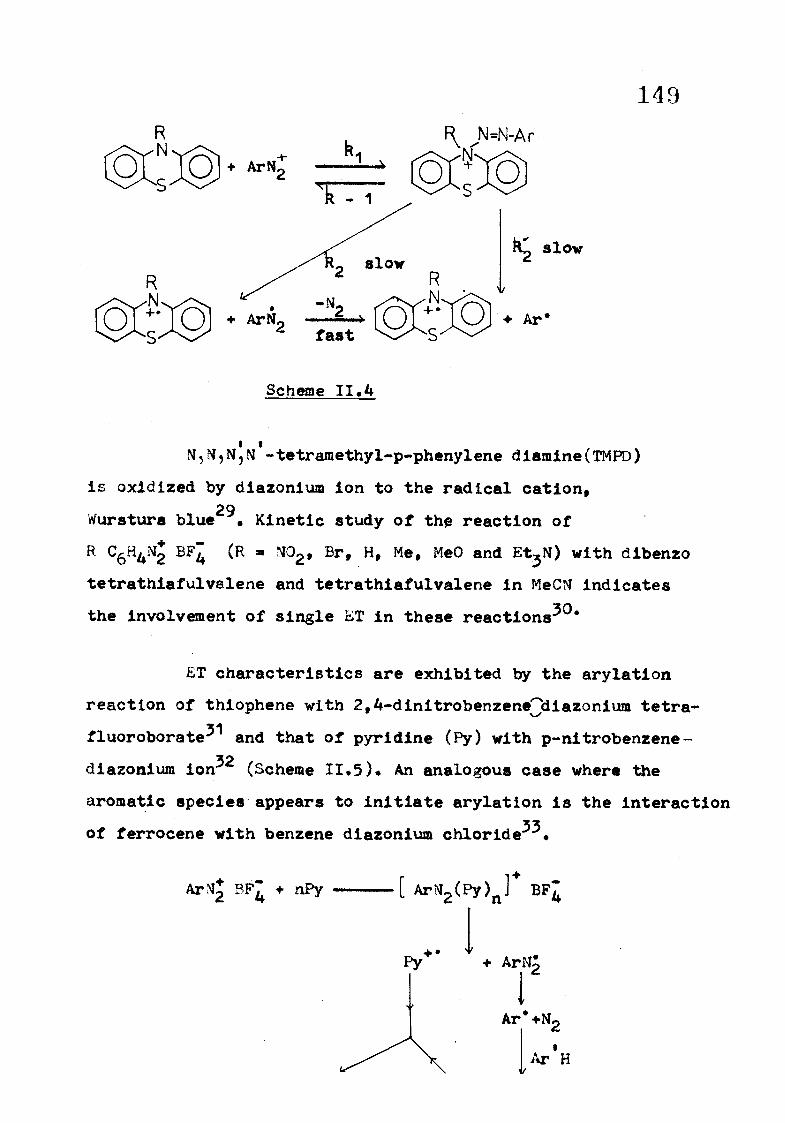
partic ipa te in a chain reaction (Scheme I I . 3 ) .
r^ n. n-
A « l7 MC>2
Scheme I I . 5
The formation o f azo compound in the reaction o f
diazonium cation with sodium phenoxide has been shown to
involve the in i t ia l generation o f rad ica ls followed by a27coupling reaction to form products •
In favourable circumstances, reactions o f diazonium
cations with certain neutral organic molecules lead to the
formation o f cation rad ical o f the substrates* Thus reactions
o f phenothiazlne and substituted phenothiazines with
4-methoxybenzenediazonium ion leads to the formation o fOQ
phenothiazlne radical cation (Scheme 11*4),,
149
Scheme I I . 4
N,M ,N 3N -tetraraethyl-p-phenylene diamine(TMPD)is oxidized by diazonium ion to the rad ica l cation,
29Wurstura blue • K inetic study o f the reaction o f
R BF (R * MOg, Brf H, Me, MeO and Et^N) with dibenzo
tetra th ia fu lva lene and tetra th ia fu lva lene in MeCN indicates
the involvement o f s ingle ET in these reactions^0,
ET characteristics are exhibited by the aryla tion
reaction o f thiophene with 2 ,4-dinitrobenzenejiiazonium te tra -
flu orobora te^ and that o f pyridine (Py) with p-nitrobenzene-32diazonium ion (Scheme I I . 5 ) . An analogous case where the
aromatic species appears to in it ia t e ary la tion is the in teraction
o f ferrocene with benzene diazonium c h lo r id e^ .
ArM+ BF ♦ nPy ---------[ A rN ^ P y jJ * BF^

Our quest fo r id en tify in g reagents that would
e f fe c t oxidation by the ET pathway led us to examine the
reactions o f some high poten tia l quinones such as
2 ,3-d ich loro-5 » 6-dicyano-p-benzoquinone (DDQ) with aromatic
hydrocarbons o f moderate poten tia ls that are known to y ie ld
stab le rad ica l cations chemically or a t an anode. In the
present in vestiga tion i t was found that in the presence o f
acid, aromatic and heterocyclic compounds read ily g ive up
an electron to DDQ generating the corresponding rad ica l
cations, the DDQ being converted to the dihydroquinone (DDQH .
Thus oxidations with DDQ provide an Instance o f acid induced
electron transfers taking place, and o ffe r newer avenues fo r
the generation o f stable rad ica l cations which can even be
iso la ted provided a judicious se lection o f solvent acid mixtures
i s made.
A number o f reports e x is t demonstrating the
involvement o f ET in the reaction o f stable carbocations with
many ea s ily ox id is ing molecules* Having a general in teres t in
ET reactions and in the use o f organic ET oxidants, I t was
considered worthwhile to in vestiga te the scope o f stable
carbocations such as t r i t y l hexachloroantimorate, tropylium
hexachloroantimonate and xanthylium perchlorate as electron
acceptors* In c iden ta lly , there are no reports on the ET
reactions o f xanthylium cation.
Stable solutions o f rad ica l cations were obtained
on treating the above mentioned carbocations with aromatic
and heterocyclic compounds o f moderate to low oxidation
poten tia ls such as phenothlazine, , N*,N*-tetramethyl-p-
150

151
phenylene diamine, tetra th ia fu lva lene e tc . Moreover
tropylium and t r i t y l hexachloroantimonate were found to e f fe c t
oxidation o f some nitrogen containing compounds such as
d iazoalkanes and hydrazones v ia the intermediacy o f rad ical
cations* A ll these resu lts lend credence to the view that ET
must be more prevelant than is recognised at present in the
reactions o f stable carbocations.
Reports presented in recent years regarding the
involvement o f ET in oxidation reactions o f diazonium sa lts
in cited the author to try unprecedented reactions that would
serve to exemplify novel ET processes. The substrates chosen
were diaryldiazomethanes, hydrazones o f aromatic ketones,
phosphazines and phosphoranes and oxidations were brought
about by p-nitrobenzenediazoniura tetra fluoroborate.
I I .1 Oxidation o f e lectron r ich organic compounds to
stable rad ica l cations by DPQ in acid media.
EXPERIMENTAL
Pu rifica tion o f reagents. The aromatic compounds were obtained
commercially and were pu rified by re c ry s ta llis a t io n . DDQ (BDH, Poole England) was pu rified by rec ry s ta llis a t io n from
chloroform (m.p. 217-219°). Dichloromethane and tr iflu o ro a ce tic
acid were purified as described previously (Chapter I ) .
EPR spectra were recorded on a Varian E-109
spectrometer in solutions deoxygenated by freeze-thaw technique
with in term ittant introduction o f n itrogen. UV-Vis spectra
were recorded on a P\.je lin'icom -SP 8100 scanning

sp ectrophotometer•
152
Experimental procedures; The oxidations were e ffec ted by
d isso lv ing the aromatic hydrocarbon ( ~ 10~* M) in CH2C12/TFA
(app. 9s1 v/v) and then adding so lid DDQ ( — lO ^So. Most o f
the reactions were carried out in a ir . Where EPR signals had
to be recorded, solutions were scrupulously degassed before
spectral measurement.
Iso la tion o f the stable t r la - (4-bromophenyl) amlnlum perchlorate
by_oxldatioo o f the amine w ith DDQ in ad d media.
To a solution o f tris(4-bromophenyl) amine
(1*3g, 2.7 mmol) in CH2C12(15 ml) was added O'OQ (0.6g, 2.6 mmol)
in MeCM-HClO (8s2 v/v) solvent mixtures (15 ml) while purging
with nitrogen. The reaction mixture developed a deep blue colour
almost instantaneously and on pouring the mixture into
scrupulously dried ether (100 ml) cooled previously to -10°C,
a blue p rec ip ita te o f the perchlorate sa lts o f the rad ica l
cation se tt led down which was f i l t e r e d and dried in a vacuum
desiccatdr ( m.p. 128°, l i t . m.p.34. 129°C, y ie ld 70%). A
major portion o f the f i l t r a t e was found to contain the
dihydroquinone, DDQHg.
RESULTS AND DISCUSSION;
We have observed that deep coloured solutions are
formed when electron donor molecules and high poten tia l
quinones, pa rticu la r ly DDQ are allowed to react in d ich loro-
methane or 1,2-dlchloroethane containing small amounts o f
tr if lu o ro a c e t ic acid (TFA). For example on adding a few drops

153
o f TFA to a mixture o f thianthrene and DDQ in CH2CI2 a t room
temperature (28°C), the solution developed a purple colour
almost instantaneously whose e lectron ic absorption spectrum
( X___ 545 nm) len t a convincing proof o f the thianthrenemaxrad ica l cation having formed.
S im ilarly , other electron r ich molecules o f a wide
range o f oxidation poten tia ls y ielded the corresponding rad ica l
cations whose e lectron ic absorption spectra matched very well
with those reported in lite ra tu re . Addition o f small amounts o f
triphenylphosphine, a p lausib le rad ica l cation scavenger ,
to each o f these solutions provided a more searching tes t in
that the rad ica l cations were reverted to parent molecules asTL-
marked by sudden discharge o f colours. In F i g u r e a r e shown
the EPR spectra o f 9,10-dimethylanthracene and phenothiazinen.
cation rad ica ls while F igu re^ presents the v is ib le absorption
spectra o f the rad ica l cations o f 9,10-dimethylanthracene,
ferrocene, biphenyl, te tra th ia fu lva lene and rubrene. Listed in
Table I I . 1 are the substrates oxid isable by DDQ in C^C l^/TFA
solu tion . The concentration o f TFA required to bring about
complete conversion o f the substrate to i t s rad ica l cation was
only 7-10^ (v/v) o f the solvent CH2C12.
1,2-Dichloroethane/TFA, dichloromethane/trichloroacetic
acid , nitromethane/HC10^, sulfolane/HClQ^ and MeCft/TFA systems
also worked but best resu lts were obtained with CH2C12/TFA
solvent mixtures.
The recogn ition o f ET having occurred in oxidations12with DDQ can be traced to 1#ed-with e t a l , who reported that
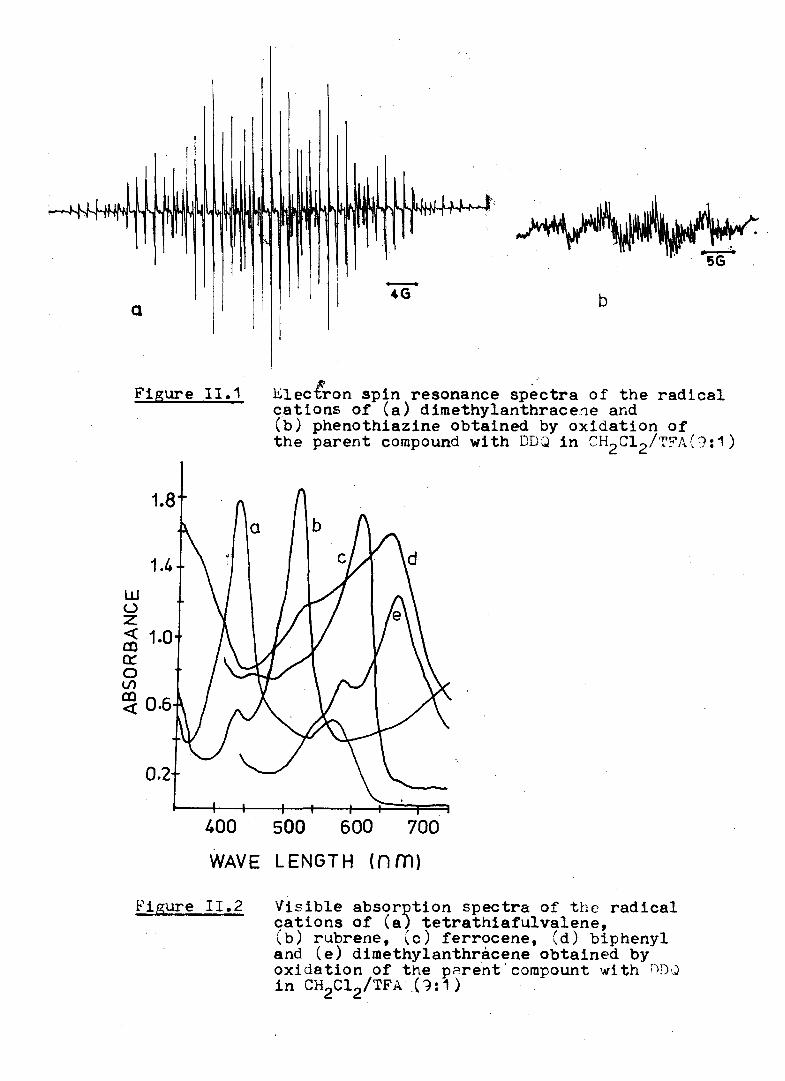
Figure I I . 1 Elec&on spin resonance spectra o f the radical cations o f (a ) dimethylanthracene and (b ) phenothiazine obtained by oxidation o f the parent compound with DDQ in CH2C12/TFA(9:1)
400 500 600 700
WAVE L E N G T H ( D m )
Figure I I . 2 V is ib le absorption spectra o f t h e radical cations o f (a ; te tra th ia fu lva lene,(b ) rubrene, (c ) ferrocene, (d ) biphenyl and (e ) dimethylanthracene obtained by oxidation o f the parent‘ compount with ODQ in CH2C12/TFA (9 :1 )

carbazole reacts with DDQ in acid medium to generate
carbazole rad ica l cation. The f i r s t in-depth in vestiga tion36has been carried out recently by Verhoeven et a l , who
proposed that protonation o f the oxidant, quinone (Q ), occurs
and i t is the protonated quinone (QH+, ) that in i t i a l l y accepts
an electron from the substrate (Eq«3)
Q + H+ --------------QH+V " 1 " 1 ...........
ArH ♦QH* -------------- * ArH+* + QH* . . .t--------------
ArH +QH* -------------- * ArH+* ♦ QH2 . . .T................
2ArH +Q+2H*------------- 2ArH** ♦ QH2 . . . (3 )
154
Our studies, wherein stoichiometry and products
o f the reaction were used to probe the mechanism, do as w ell
account fo r the «^uilibrium which is reminiscent o f the
electrochemical h a lf reaction (Eq.4)
Q ♦ 2H* +2e~--------- QH2 . . . (4 )
One may question, however, whether or not the ro le
o f acid is to form the protonated quinone (Eq.3a), that in
turn acts as one electron oxidant. We are o f the opinion
that the active species is not the protonated quinone, but
the quinone i t s e l f , whose ox id iz ing capacity is grea tly
enhanced in the presence o f strong acid. Reasons fo r th is
are as fo llo w s :(1 ) the e lectronegative ly substituted quinones,* » q
particu la r ly DDQ, possess low b a s ic ity * to be susceptible
to protonation by acids such as TFA. (2 ) I t was found that when
(3a)
(3b)
(3c)

an e x p e r im e n t p a r a l l e l t o t h a t o f E q ,3 was c a r r i e d o u t r e p l a c i n g
DDQ w i t h Pb02 » MnQg, o r ev en t h e weak o x i d a n t p - n i t r o b e n z e n e
d iazo n iu m t e t r a f l u o r o b o r a t e , i n w h ich t h e q u e s t i o n o f p r o t o n a t i o n
d o e s n o t a r i s e , an i d e n t i c a l se q u e n c e o f r e a c t i o n s o c c u r e d and
r a d i c a l c a t i o n s o f a l l t h e compounds l i s t e d i n T a b le I I . 1 w ere
fo rm ed .
T h is f a c t , th e n , s e r v e s t o e x c lu d e t h e p o s s i b i l i t y o f
t h e o x id a n t b e i n g p r o to n a t e d b u t a s s i g n s a d i f f e r e n t r o l e t o
t h e a c i d i n t h e o x i d a t i o n . A lth o u g h an e x p l i c i t answ er to
t h i s problem i s n o t known, i t i s r e a s o n a b le t o assume t h a t
t h e f u n c t i o n o f t h e a c i d i s t o prom ote t h e o x i d i z i n g power
o f t h e o x id a n t and th u s in d u c e e l e c t r o n t r a n s f e r s . I t i s
p e r t i n e n t t o m en tion h e re t h a t a c t i v a t i o n o f m e ta l o x id a n t s
i n TFA o r s t r o n g a c i d s h as b e e n o b s e r v e d w it h C o ( I I I ) , C e ( l V )
i i n ( l l l ) , C u ( I l i e t c ^ . Thus w h eth e r t h e o x i d a n t i s DDQ or
Pb02 , r a d i c a l c a t i o n s a r e form ed i n th e p r e s e n c e o f a c i d t h a t
prom otes t h e o x i d i s i n g power o f t h e o x id a n t ( E q . 5 ) .
ArH 4- DDQ + H* ---------- -*• ArH+* ♦ DDQH* . . . (5a)■I111"'...... ....
ArH ♦ DDQH* +H* ---------------- ArH+* + DDQHg . . . (5b)T.. .........
1 ArH + DDQ ♦ 2H*---------- *2ArH+* + DDQH ••• }t ...mmmmm.. .
Under a n h yd ro u s c o n d itio n s , s o l u t i o n s o f r a d i c a l c a t i o n s
formed b y DDQ o x i d a t i o n s ( T a b le I 1 . 1 ) were fou n d t o be s t a b l e
f o r l o n g p e r i o d s o f t im e (some o f them i n d e f i n i t e l y ) . I t was
v i s u a l i s e d t h a t r e a c t i o n s o f t h i s t y p e s h o u ld open up s im p le
p r e p a r a t i v e r o u t e s t o d i f f i c u l t y a c c e s s i b l e r a d i c a l c a t i o n s and
155

156
Table 11*1 A___ o f cation rad ica ls obtained by oxidation.............. . max *o f the parent compound with DDQ in acid media
at room temperature (28°C)
S. 'Jo Substrate* ObservedW ”
Reported\iax,nm
Ref.
1. Pyrene 505 505 402. Rubrene 525 b3. Fluorene 532 b4. Perylene 543 540 415. Acenaphthene 512 b6. Anthracene 715 715 407. 9,10-Dimethylanthracene 669.5 b8. 9,1O-Diphenylanthracene 727,652,593 724,653,596 369. Naphthalene 535 535 4010. Thianthrene 545 546 4211. Phenothiazine 513,439 515,437 4312. Tetrath iafu lvalene 578,434 580,435 4413. Biphenyl 658 660 4514. T r is - (4 -bromophenyl)amine 726 725 3415. Tetraphenylethylene 495 494 4616. M,N,M*, M1-Tetramethyl-p-
phenylene diamine 615,565 618,567 4717. Ferrocene 619.5 619 4818. 1 ,4-Dimethoxybenzene 472,437 475,440 49
b * not located in lite ra tu re .

also fa c i l i t a t e iso la tion o f stable rad ica l cation ?;alts.
Attempts in th is d irection proved rewarding whereby we could
is o la te perchlorate s a lt o f tris~(4~bromophenyl)amine
(.oee experimental) •
Thus oxidation with DDQ o ffe r newer avenues to the
formation o f rad ica l cations and as more work is done particulariy
in making a Judicious se lection o f solvent-acid mixtures,
oxidations o f th is type may rece ive wide-spread preference in
rad ica l cation formation and in reactions where rad ica l cations
intervene.
n .2 Carbocatlons as Electron.Transfer Oxidants
EXPERIMENTAL
The aromatic compounds were obtained commercially
and used as such. Hydrazones were prepared as described
previously (Chapter I ) . A ce ton itr ile was fra c t io n a lly d is t i l le d
from phosphorous pentoxide through a ten inch h e lix packed
column u n til the d is t i l la t e was free from hydrogen cyanide as
Indicated by the absence o f a p rec ip ita te with aqueous s i lv e r
n itra te . I t had a bo ilin g point o f 81°C and water content o f
0.02%.
.o£..,jyJB&P S2ffl.P.9¥Rfl,§,
a ) Diazodiphenvlmethanet To a mixture o f benzophenone hydrazone
(5.0. g ) and yellow mercuric oxide (10.0 g ) in petroleum ether
40-60° (25 m l), a few drops o f concentrated a lcoholic potassium
157

hycroxide were added. The mixture was covered with a s i lv e r f o i l
and l e f t s t ir r in g overnight. The resu lting purple solu tion was
f i l t e r e d through a sintered funnel and the solution concentrated
by using a rotary vacuum evaporator and then again f i l t e r e d .
C rysta llisa tion was then brought about by placing the solution
in a re fr ig e ra to r u n til needle-shaped crysta ls o f diazodiphenyl-
methane were obtained. These were dried in a vacuum desiccator
and stored in a brown b o ttle in the re fr ig e ra to r ,
\nax " 526 nm; 29-30°C, l i t m,p5° 29°
b) 9- D ia z o f lu o r e n e : F lu oren on e h yd razon e (4,7 g), y e llo w
m e rcu ric o x id e (1 , 5g ) a n d anhydrous sodium s u l f a t e (4 , 5g ) were
ground to g e th e r and suspended in d ry e th e r (50 ml ) , A s a tu r a te d
s o lu t io n o f KQH in m ethanol (1 m l) was th en added and th e
s o lu t io n s t i r r e d m a g n e t ic a lly f o r 1 h r . The s o lu t io n was
f i l t e r e d and th e r e s id u e washed w ith e th e r u n t i l th e w ashings
were no lo n g e r c o lo u r e d . The e th e r was removed to le a v e a red
s o l i d , w hich was r e c r y s t a l l i s e d from 40 - 60° p etro leu m
e th e r, m .p. 94- 95° •
Preparation o f stable carbocatlons
a) T r ity l hexachloroantlmonate: Antimony pentachloride
(BDH Poole, England) ( I 5 . 5g ) in carbon tetrach loride (75 ml)
wasaadded to a solution o f triphenylmethyl chloride (14 ,5g) in
carbon tetrach loride (100 m l), A yellow p rec ip ita te which formed
immediately was f i l t e r e d o f f , washed several times with carbon
te trach loride , and dissolved in a minimum quantity o f hot
dichloromethane. Pure triphenylmethyl hexachloroantlmonate
separated as orange crys ta ls . The product was stored under vacuuo.
158

m.p 230° | l i t m.p51 230°.
159
b) Tropyllum hexachloroantimonatet A solution o f cycloheptatrim e
(Fluka, Swiss) (13 ml) in GHgClg (50 ml) was added dropwise
during 30 min, to a solution o f triphenylmethyl hexachloroanti
monate (32g) in CH2C12 (250 ml) under nitrogen* The resultant
s lu rry was s tirred fo r a further 30 min a fte r which 100 ml o f
carbon tetrach loride was added* The mixture was ch illed in
ic e , f i l t e r e d and washed with cold carbon te trach loride (25 ml)*
The product was redissolved in CH2C12(150 m l), p recip ita ted with
carbon tetrach loride (150 ml) and r e f i lt e r e d to g ive white
tropyllum hexachloroantimonate*
m.p 283°; l i t m.p51 284°
c ) Xanthyllum perch lorate: Xanthene (1 ,82g), triphenylmethyl
perchlorate (3.43g) and acetic acid (100ml) were heated fo r
2 min. Xanthylium perchlorate began to separate out from the
b o ilin g solution as yellow p la tes . The product, f i l t e r e d from
the cooled solution was rec rys ta llis ed from acetic acid.
m.p 225°* l i t m.p52 225-226°C
II.2B Oxidation o f Dlazocompounds
a) Reaction ofdiazodlphenylmethane with t r i t y l hexachloroanti
monate
A solution o f dlazodiphenylmethane (1*5g, 7.7m mol)
in dry a c e to n itr ile (25 ffll) was taken in a 100 ml fla sk . To th is
was added an MeCN solu tion o f t r i t y l hexachloroantimonate
(2 .2g, 3.8 mmol) with constant s t ir r in g at room temperature. A

brisk evolution o f nitrogen ensued and the purple colour o f
the solution gradually disappeared. The reaction was over in
ten minutes• On allowing the reaction mixture to stand, a
mass o f shining white crysta ls se ttled down. These were f i l t e r e d ,
washed and id en tified as tetraphenylethylene (m.p 224°, l i t m .p^
224°) (67.5%). The f i l t r a t e was chromatographed on a s i l ic a
column. Thus benzhydrol (13»5%) and t r i t y l peroxide (traces )
were obtained. The compounds were characterised by mixed
melting points.
* ) Beastion o f dlazodljphenyliB ethane with tropyllum hexachloroanti-
On adding slowly a solution o f tropylium hexachloroanti-
monate (1.5g, 8 mmol) in MeCN to a solution o f diazodiphenylaiethan«
0 *5 g , 7*7 mmol) in 25 ml o f MeCN with constant s t ir r in g while
maintaining the temperature at 25°C, rapid expulsion o f nitrogen
gas occurred and the purple colour o f the diazoalkane disappeared
gradually. The solution on concentration yielded colourless
prisms, m.p 224° id en tified as tetraphenylethylene (70$), The
f i l t r a t e was found to contain benzhydrol (15%) and benzophenone
(4.5%).
c ) Reaction o f 9-diazofluorene with t r i t y l hexachloroantlmonatet
A solution o f t r i t y l hexachloroantlmonate (2.2g, 3.8rmnol
was added slow ly with s t ir r in g to a solution o f 9-diazofluorene
0 »5 g , 7.7 mmol) in dry a c e to n itr ile (25 ml) a t room temperature.
Brisk evolution o f nitrogen gas took place and the red colour o f
the solution turned yellow , the in tens ity o f which increased
with time. The reaction was over in 25 min. On completion, the
160

reaction mixture was allowed to stand whereupon orange crysta ls
deposited. These were f i l t e r e d , washed, rec ry s ta llized from
benzene-petrol and id en tified as b ifluorenylidene, m.p 191-192°,
l i t m.p^4 189-190° (66.5%)by comparisons (mixed melting point
and superimpossible IR spectrum) with authentic sample. The
f i l t r a t e on coluamc chromatography yielded fluorenone azine
(5*5%)» fluorenone (2.0%) and fluorenol (14.8%) also characterised
by comparisons with authentic samples.
d ) Reaction o£_fe jlazo flaargpg^vfith tropvllum hexachloroantlmonatg
To a solution o f 9-diazofluorene (1*0g, 5 mmol) in
dry a ce to n itr ile was added tropylium hexachloroantimonate
(1. Og, 2.5 mmol) in MeCM (10 m l). A brisk evolution o f nitrogen
ensued and the reaction was over in 15 minutes. Upon concentration
orange crysta ls o f b ifluorenylidene (68%) separated out and the
f i l t r a t e yielded fluorenol (15%)» fluorenone azine (6.2%) and
fluorenone (tra c e s ).
II.2C Oxidation o f hydrazones
a) ReactlOP-Of henaophenone hy dr azo ne with trlphenylaethyl
To a solution o f benzophenone hydrazone (1.0g, 5 mmol)
in dry a c e to n itr ile (20 ml) was added an MeCN solution (15 ml)
o f triphenylmethyl hexachloroantimonate (2.7g, 6 mmol). A brisk
evolution o f nitrogen resu lted . The reaction was carried out a t
room temperature and took about 20 minutes fo r completion. On
allowing the reaction mixture to stand fo r 4 hrs, colourless
needle shaped crysta ls appeared. These were f i l t e r e d and
161

162o 5*5 oId en tified as benzophenone azine, m.p, 162 , l i t ®.p 163
by mixed melting poin t. The mother liquor gave more o f the
azine (to ta l y ie ld 65%), tetraphenylethylene (5%) and t r i t y l
peroxide (62%) m.p.185°, l i t m.p56 185-186°.
b) Reaction o f benzophenone hydrazone with troplium
hexachloroantimonate
Benzophenone hydrazone O.Og, 5 mmol) was dissolved
in dry a c e to n itr ile (25 ml) and tropylium hexachloroantimonate
(2 .0g, 5 mmol) in MeCN (15 ml) was added a t once. A brisk
evolution o f nitrogen ensued which continued throughout the
reaction . Towards completion, colourless needle shaped crysta ls
deposited. These were f i l t e r e d and id en tified as benzophenone
azine (63.6%). The f i l t r a t e was found to contain more o f the
azine, trace amount o f tetraphenylethylene and b itrop y l (61.3%)
(m.p 61°, l i t m.p'5 61°).
c ) Beas-tlon o f fluorenone hydrazone with t riphenylmethyl
hexachloroant imonate
The addition o f an MeCN solution (15 ml) o f
triphenylmethyl hexachloroantimonate (2,7g , 5 mmol) to fluorenone
hydrazone (1.0g, 5 mmol) in MeCN (25 ml) was marked by a slow
evolution o f n itrogen. The reaction carried out at 25°,
completed within 30 minutes by which time a red c ry s ta llin e
p rec ip ita te se ttled down. This was separated and id en tified as
fluorenone azine fup 268°, l i t m.p^8 270° by mixed melting
point and superimposible IR spectrum with that o f the authentic
sample. The f i l t r a t e indicated the presence o f more azine
( to ta l y ie ld 68.2%), b ifluorenylidene (traces ) and t r i t y l

163
peroxide.
d ) Reaction o f fluorenone hvdrazone with tropyllum
hexachloroantlmonate
When a solution o f tropyllum hexachloroantlmonate
(2 .0g, 5 mmol) was added to fluorenone hydrazone (1.0g, 5 mmol)
in a c e to n itr ile (25 ml) a slow evolution o f nitrogen could be
observed which continued throughout the reaction carried out
a t room temperature. A fter 30 minutes a red crop o f fluorenone
azine settled down. TLC examination o f the mother liquor showed
the presence o f b ifluorenylidene (tra ces ) and b itropy l
(not determined).
RESULTS AND DISCUSSIONi
IX*2A Formation o f stable rad ica l cations by oxidation with
carbo cations
On adding t r i t y l hexachloroantlmonate, tropyllum
hexachloroantlmonate and xanthyllum perchlorate (10" "S i) to a
solution o f te tra th ia fu lva lene (1 ), phenothiazine (2 ),
ft,M,N*, M*-tetramethyl-p-phenylene diamine (3 ) and ferrocene (4 )
(10“ %I) in a c e to n itr ile , deep coloured solutions were formed
(red in the case o f 1, reddish yellow with 2., blue with 2. and
green with 4 ). These deep coloured solutions were found to be
EPFi active . The ESR spectra recorded at room temperature in
degassed solutions were w ell resolved and matched reasonably59w ell with those reported in lite ra tu re . UV-Vis scanning o f
the solutions gave peaks sim ilar to those reported fo r the


165
ma^or product o f the reaction alongwith benzhydrol and traces
o f t r i t y l peroxide.
Likewise the addition o f tropylium hexachloroantimonate
to a solution o f PhgC^ in MeCN resu lts in a rapid decomposition
o f the diazoalkane as seen in the change o f colour o f the
solution from dark pink to colourless. Nitrogen is evolved
throughout the reaction which ends up in the formation o f
tetraphenylethylene as the p rinc ipa l product.
In an analogous manner, 9-diazofluorene reacts with
t r i t y l and tropylium hexachloroantimonate to e f fe c t a rapid
decomposition o f the diazoalkane to b ifluorenylidene and small
amounts o f fluorenone azine, fluorenone and flu oren o l.
The iso la tion o f t r i t y l peroxide in trace amounts in
the reaction o f PhgCN with t r i t y l hexachloroantimonate is
suggestive,o f the reaction to invo lve ET process. The in i t ia l
step o f the reaction presumably involves the Ph2CN2 or F1N2
cation rad ica l (5 ) (Scheme I I . 6) as a resu lt o f s in g le ET
from the parent diazoalkane molecule. Such intermediates havegO 4
been proposed in the electrochemical and C u ( I I )J induced
decomposition o f PhgCM .
In the absence o f any nucleophile, the rad ica l
cation (5 ) reacts with the parent molecule in a ra te lim itin g
step as shown in Scheme I I . 6. Such a coupling would e ither
occur at the diazocarbon atom or a t the terminal nitrogen o f
the reactant with the subsequent loss o f nitrogen to y ie ld
the rad ica l cations o f the o le f in (6 ) and the azine (7 ).

The emergence o f dimeric o le f in (8 ) as the principal
product o f the reaction suggests that the major portion o f the
reaction proceeds v ia pathway (a ) that involves the rad ica l
cation ( 5 ) as chain ca rr ie r .
Traces o f water present in the solvent MeCN are
expected to attack the principa l intermediate ( 5 ) , in it ia t in g
a pathway that Involves rad ica l species such as 10 and V\_ as
formulated in Scheme I I . 6. This explains the formation o f
products obtained in small amounts, such as alcohol ( 12 ) and
ketone (14 ). The explanation receives strong support from the
fa c t that when water is d e lib era te ly added to the system, products
such as T2 and 14 particu la rly the alcohol, emerge in good
y ie ld s at the expense o f the o le f in .
166
(a )-2 N ,
R2C*”C+R2(6)
R2™2
r2c- cr2» r2c. n2( 8 ) (5 )
■* R2C- f,2 ‘ - ... t *2 C0H2 r(5 )
R2C-N2
(10),-H
T b T-Mr
R~C - OH (11 )
R2C#-N-N-C'>R2(7 ) F^CHOH
R2C»^2 R2C-0H
r 0c«n-m«cr0 2 (9 ) +•
r2c« n+-(5 )
R2C-0H (13)
Disproportion* -ation
R-CHOH R5C-O (12) (14)
R-j - t r i t y l or tropyllum cation .
R2 » diphenyl or fluorenyl residue
Scheme I I .6

1G7
II.2C Oxidation o f hydrazones
An examination o f products formed in the reaction o f
benzophenone and fluorenone hydrazone with carbocations ( t r i t y l
hexachloroantimonate and tropylium hexachloroantimonate) revealed
that the corresponding azines were formed in appreciable y ie ld s .
The emergence o f t r i t y l peroxide in the oxidations with t r i t y l
cation and that o f b itropy l in reactions with tropylium cation
indicates that the reaction proceeds by an ET mechanism,
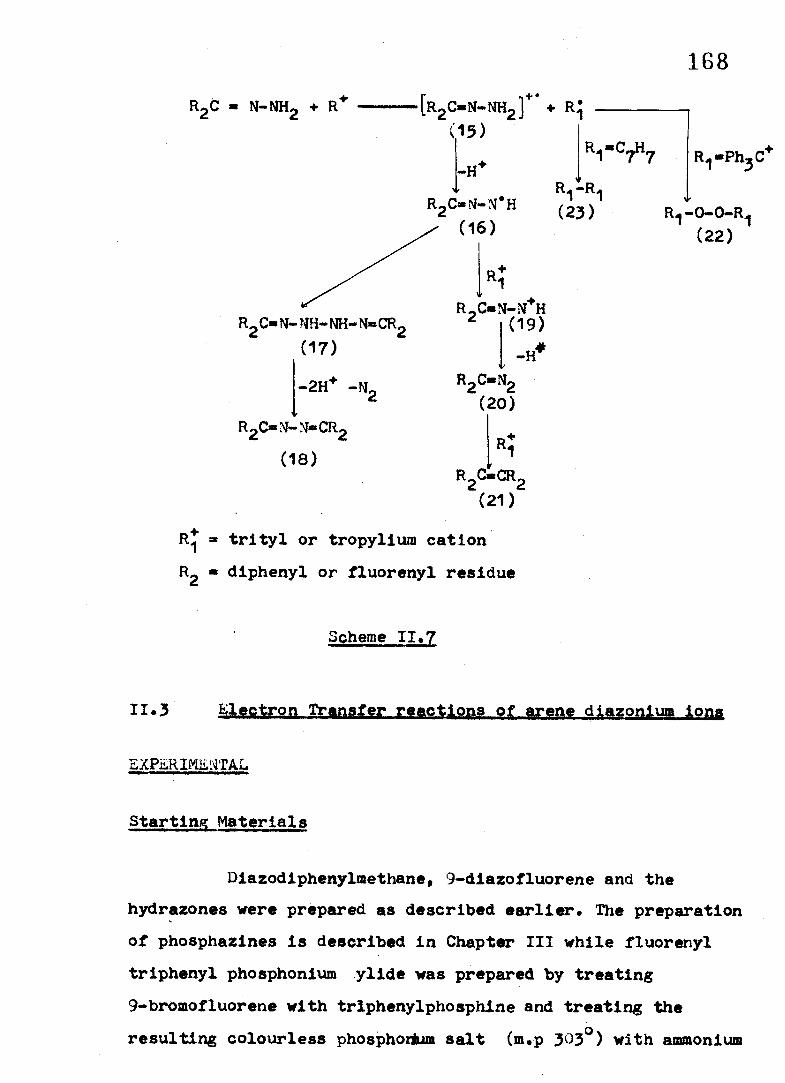
A single ET from the hydrazone to the organic cation
should in p rin c ip le generate the cation rad ica l o f the hydrazone.
A ready loss o f proton from the rad ica l cation (15) would
generate the rad ica l (16 ), as shown In Scheme I I . 7, The rad ica l
(16) can partake in two competing reactions, one involving
dimerisation with a subsequent oxidation o f the dimer ( 1 7 ) ,
coupled with nitrogen loss, to y ie ld the product azine (18 ). The
second pathway involves a further oxidation o f the rad ica l (16 ),
followed by a loss o f proton, to form the dlazoalkane (20 ). The
diazoalkan* is subject to oxidation by the carbenium ion to
y ie ld the o le f in (21) by a mechanism as suggested in the
preceeding section .
Thus a close s im ila r ity ex is ts in the lead dioxide/TFA
(Section I-2A ) and carbocation in it ia te d decomposition o f the
hydrazones. The reaction is yet another demonstration o f the
ET attribu tes o f the stable carbocations. The work suggests that
the involvement o f rad ica ls in the reactions o f carbocations
must be more wide-spread than is generally recognised.
168
R2c « N-NH2 ♦ R
R
r2c- n- mh- nh- n« cr2(17)
-2H+ -N„
R2C«M-M«CR2
(18)
•[r2C-M-NH2]+* + R* (15)
- Hi R*.—R*r2c- m- n*h (2 3 ) '
(16)
R-OH-N H2 (19)
-H*
R2C- » 2 ( 20)
"1R2<5.CR2
(21)
R ^ » P h j C
R1-0-0-R1(22)
* t r i t y l or tropylium cation
R2 « diphenyl or fluorenyl residue
Scheme I I . 7
11*3 Electron Transfer,reactions o f arene d iazonluia long
EXPERIMENTAL
Starting M aterials
Diazodiphenylmethane, 9-diazofluorene and the
hydrazones were prepared as described e a r l ie r . The preparation
o f phosphazines is described in Chapter I I I while fluorenyl
triphenyl phosphonium y lid e was prepared by trea ting
9-brdmofluorene with trlphenylphosphine and treating the
resu lting colourless phosphoriim sa lt (m.p 303° ) with ammonium

hydroxide, when yellow le a f l ik e crysta ls o f the y lid e separated
out. R ecrysta llisa tion from MeCN yielded pure fluorenyl triphenyl
phosphonium y lid e , m.p 253 ( l i t m.p 253 • p-Nitrobenzene-
diazonium tetrafluoroborate^3 and 4 ,4~dini trobiphenyl^4 were
prepared according to la id down procedures.
II.3A RgfteMon_ofL arene diazonium ion with aliphatic dlazocompounds
a) Baasiifln af.Prnltrobeaaenedlazonium tetrafluoroborate withdlazodiphenvlmethane.
A solution o f p-nitrobenzened lazonium tetra fluoroborate
(1 .Og, 45 mmol) was added with constant s t ir r in g , to a solution
o f diazodiphenylmethane (3.88g, 20 mmol) in a c e to n itr ile placed
In a 100 ml round bottomed fla sk while maintaining the temperature
a t 15°C. The solution assumed a transient green colouration
followed by a brisk evolution o f nitrogen and the reaction was
complete in 5 min. On concentration, colourless crystals
separated out. R ecrysta llisa tion from benzene-absolute efchanol
gaye colourless prisms id en tified as tetraphenylethylene,
m.p 224° ( l i t m.p'*3 224°) in Ca. 68% y ie ld . A fter the removal o f
tetraphenylethylene, the remaining solution was chromatographed
over an alumina column. Elution with petroleum ether gave a
liqu id which on d is t i l la t io n in a bulb gave nitrobenzene at a
bath temperature o f 90° ( l i t b.p 85°) (8% y ie ld ) . F'urther
e lu tion with pet-ether/benzene (1*1 ) gave 4 ,4*-d in itrob iphenylf
m.p 238°, l i t m.p64 238-239° (14%).
In a separate experiment, allowing equimolar quantities
o f p-nitrobenzenediazonium tetrafluoroborate (4.38g, 20 mmol)
169

170
in MeCN and diazodiphenylmethane in a c e to n itr ile (3#88g,20 mmol)
to react* a transient green colour again appeared followed by a
vigorous evolution o f n itrogen. The yellow solution on standing
overnight deposited yellow needle-Shaped crys ta ls , id en tified
as p-nitrophenylazodlphenylmethanol. The f i l t r a t e was found to
contain nitrobenzene and 4 ,4-d in itrobiphenyl. When the same
reaction was carried out In the presence o f water (0.5M), an
almost instantaneous expulsion o f nitrogen occurred and a
thick yellow p rec ip ita te o f p-nitrophenylazodiphenylmethanol
separated out (80%), The compound, m.p 150° was characterised
by IR, NMR & mass spectrometry.
b ) RgflC-t.iPtv, o f-^ltrobenzjtn.edlazonlum. te tra flu o rob ora tew ith
9-dlazofluorene
The addition o f an MeCN solution o f p-nitrobenzenedia-
zonium tetra fluoroborate (1.06g, 4.5 mmol) to 9-diazofluorene
(3*88g, 20 mmol) in MeCN was marked by a brisk evolution o f
n itrogen. The reaction was over in two minutes. On completion,
the reaction mixture was allowed to stand whereupon reddish-
orange c rys ta ls , id en tified as b ifluorenylidene (m.p 190°, l i t
m.p'*** 189-190°) were obtained in Ca 60% y ie ld . The f i l t r a t e
revealed the presence o f nitrobenzene and 4,4*-d in itrob iphenyl.
In a d iffe ren t experiment, equimolar amounts o f
p-nitrobenzenediazonium tetra fluoroborate (3*43g# 14.5 mmol)
and 9-dlazofluorene (2.98g, 15»5 mmol) in MeCM were allowed to
react a t 15°C. Nitrogen was evolved and a dark yellow precipitali
se ttled down. This was rec rys ta llis ed from CHCl and id en tified
by IR, NMR and mass spectra as fluorenone-p-nitrophenylhydrazone

(m.p 269°, l i t m.p65 269°) (70#). The f i l t r a t e was
chromatographed over an alumina column to g ive nitrobenzene
k , 4-dinitrobiphenyl and traces o f b ifluorenylidene•
R eac tion o f arenedlazonlum ions with hydrazones
a ) Reaction ofp-nitrobenzenedlazonlum tetra fluoroborate
with benzophenone hydrazone
In a round bottomed fla sk was placed an MeCN solution
o f tbenzophenone hydrazone (2 .0gt 10 mmol). To th is was added
p-nltrobenzenediazonium tetra fluoroborate (2.32g, 10 mmol) in
25 ml o f MeCN with constant s t ir r in g . A brisk evolu tion o f
n itrogen ensued and the solution turned yellow . A fte r allowing
to stand fo r sometime, colourless crysta ls deposited. These
were separated and washed with MeCN. R ecrysta llisa tion from
ethanol gave f in e needle shaped crysta ls (m.p 162°) id en tified
as benzophenone azine ( l i t m .p^ 163°), The f i l t r a t e was
chromatographed on an alumina column. Elution with petroleum
ether (40-60°) gave tetraphenylethylene m.p 224°, l i t ra.p^
224°) (12%), nitrobenzene (a t bath temperature 90° and 10mm
pressure) (18%), p-nitrophenylazodiphenylmethanol (m.p 150°,8%)
while use was made o f pet.ether/benzene mixtures to elu te
4 ,4-din itrobiphenyl (m.p 237°, l i t m.p64 238-239°, 10%).
Fluorenone hydrazone (2 .0g, 10 mmol) in MeCN (25ml)
was allowed to react with p-nitrobenzenediazonium te tra flu o ro
borate {2,32g, 10 mmol) a t 25°C. The reaction was marked by a
slow evolution o f nitrogen which continued throughout the
171

172
reaction . The yellow solution turned red and a dark red mass
se ttled down. This was separated and rec rys ta llis ed to g iv e
brigh t red crysta ls o f fluorenone azine (m.p 269°, l i t m.p^8
269°) (80% y ie ld ) . The f i l t r a t e was concentrated and
chromatographed on an alumina column. Thus nitrobenzene and
4 ,4 , -d in itrobiphenyl were obtained.
Reaction with benzaldehyde hvdrazAne
To a solution o f benzaldehyde hydrazone (1*5g»
7.5 mmol) in 25®1 o f MeCN was added a solu tion o f p-nitrobenzene
diazonium tetra fluoroborate (2.9g, 7.5 mmol) with constant s
s t ir r in g a t room temperature. A brisk evolution o f nitrogen
was fo llow ed by turning o f the solution to bright ye llow . A fter
the usual work-up and chromatographic separation o f the product
mixture on an alumina column benzaldehyde azine (m.p 92°, l i t
m .p^ 93°) (55%), nitrobenzene and 4,4-din itrobiphenyl were
obtained.
I I . 3C Reaction of^arenedlazonlum ion with phosphazlnes and
phosphoranes.
a) RaagJtlQa o f p-Mtrobenzenediazonlum tetra fluoroborate with
dlRheaylmethvlene^trlphenvl phosphazlne
On adding a solu tion o f p-nitrobenzenediazonium
tetra fluoroborate (0 .8g, 3.3 mmol) in MeCN (10 ml) to
diphenylmethylene triphenylphosphazine (1.5g» 3.3 mmol) in
CH2Cl2 (20 ml) a brisk evolution o f nitrogen commenced and the
solution darkened to bright yellow . Upon completion, the

reaction mixture was concentrated and chromatographed on an
alumina column. Elution with petroleum ether followed by
evaporation o f the solvent gave f i r s t a colourless so lid (m.p224°
id en tified as tetraphenylethylene ( l i t m*p^ 224°) (35%)
followed by a liqu id product which on d is t i l la t io n in a bulb
gave nitrobenzene at a bath temperature o f 90° (18%). Further
e lu tion w ith the same solvent yielded a yellow compound
(m.p 150° ) (30%) which was id en tified as p-nitrophenylazodiphenyl
methanol. Use o f pet.ether/chloroform mixtures fa c i l i ta te d the
iso la tion o f 4 ,4-dinitrobiphenyl (m.p 239°) (10.5%) fo llowed
by triphenylphosphine oxide, m.p 150°, l i t m .p^ 150-151°.
b ) R eaction w ith flu oren v lld en e trlphenvlphosphazlne
p-Mitrobenzenediazonium tetra fluoroborate (1 .0g,
4.2 mmol) in a c e to n itr ile (10ml) was added to fluorenylidene
triphenylphosphazine (2 .0g, 4.4 mmol) in CHgCl,, (20 ml) a t
room temperature. A brisk evolution o f nitrogen ensued, the
solution turning to dark yellow . Upon completion, the reaction
mixture was concentrated and chromatographed on an alumina
column. Bifluorrenylidene (m.p 190°, l i t m .p^ 189-190°) was
iso la ted f i r s t (38*») fo llowed by nitrobenzene (20%), fluorenone-
p-nitrophenyl hydrazone (35%), 4 ,4 '-d in itrobiphenyl (12%) and
triphenylphosphene oxide (75%)* A ll the compounds were
characterised by a comparison (mixed melting point and
superimposible 3R spectra) with authentic samples.
c ) Reaction with fluorenvl triphenvlphosphonlum v lid e
A solution o f p-nitrobenzenediazonium tetra fluoroborate
(0.5g, 2.1 mmol) in 10ml o f MeCN was added to fluorenyl
173

174
triphenylphosphonlum y lid e (1.0g, 2.3 mmol) in CH^Cl^ (15ml)
a t room temperature with constant s t ir r in g . A v io le t coloured
solution was formed which decomposed a fte r sometime to y ie ld
a yellow solu tion . The reaction mixture was allowed to
concentrate and on standing a dark yellow p rec ip ita te
(m.p 260°) separated out. This was f i l t e r e d , washed with MeCN
and rec rys ta llis ed from CHCl and id en tified as fluorenone-p-
nitrophenyl hydrazone (46^). The f i l t r a t e yielded
triphenylphosphine oxide (48%) and nitrobenzene (6%).
RESULTS AND DISCUSSION
II.3 A iAlBfraUfl
dlazocompounds
On adding p-nitrobenzenedlazonium tetra fluoroborate
to Ph2CN2 at 15°C, a transient green colour was formed followed
by brisk evolitton o f nitrogen gas. The reaction ended up in
the formation o f tetraphenylethylene as the major product
(6Q% y ie ld ) .
Likewise, the reaction o f 9-diazofluorene with
p-nitrobenzenediazonium tetra fluoroborate produced a brisk
evolu tion o f nitrogen and yielded b ifluorenylidene in Ca.60%
y ie ld .
In a separate experiment, u t i l iz in g equlmolar amounts
o f PhgCNg and P"^°2C6H4N2 * reac^ on was again marked by
the appearance o f transient green colour and a rapid evolution
o f n itrogen . TLC examination o f the reaction mixture revealed

175
the presence o f tetraphenylethylene in only trace amounts* The
reaction mixture was allowed to stand overnight when a yellow
c ry s ta llin e p rec ip ita te appeared, which was separated and
id en tif ie d as p-nitrophenylazodiphenylmethanol.
On repeating the above experiment in MeCN to which
preadditions o f water had been made (0.5M), the reaction was
again marked by a brisk evolution o f nitrogen and a fte r
10 minutes a thick yellow p rec ip ita te separated out. This was
id en tif ie d as p-nitrophenylazodiphenylmethanol (80%). Besides
th is , small amounts o f tetraphenylethylene, nitrobenzene and
4 ,4*-din itrobiphenyl were also obtained.
On the other hand, 9-diazofluorene reacted with
p-nitrobenzenediazonium tetra fluoroborate to y ie ld a dark yellow
p rec ip ita te id en tified as fluoren©ne-p-nitrophenylhydrazone.
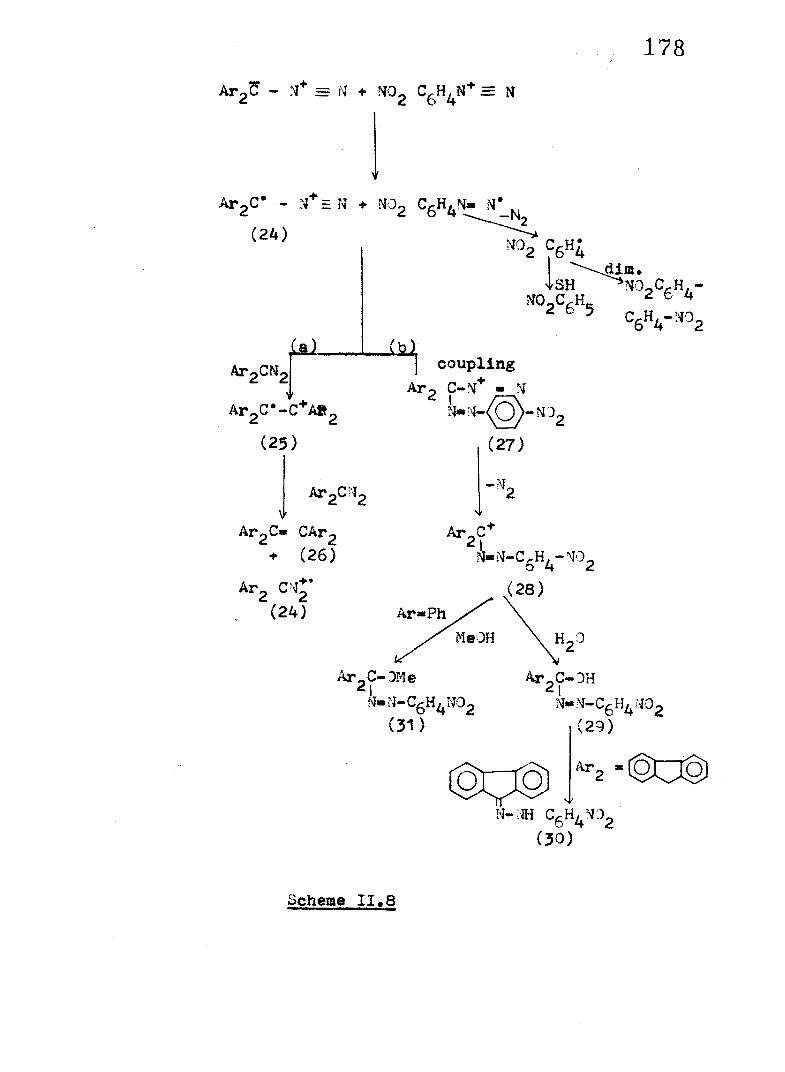
A mechanistic in terpreta tion , formulated in Scheme I I .
8, Is proposed on the basis o f the aforementioned resu lts . The
f i r s t step o f the reaction may be considered to involve electron
tran sfer to generate the rad ical cation o f the diazo compound.
I t is known that diazoalkanes l ik e PhgCNg and Fl?^ are in i t ia l l yfiooxidised to the corresponding cation rad ica ls at an anode or
in chemical oxidation with C u (ll) perchlorate^1.
I f we are r ig h t in assuming that the key step in the
reaction o f arenediazonium ion with diazoalkane molecule en ta ils
diazoalkane cation rad ica l formation, i t implies that the
diazonium ion must possess the a b i l i t y to tr ig g e r an ET reaction .
Although not many examples are known, there are some references
in the lite ra tu re that subscribe to the ro le o f diazonium cations

178
as weak ET oxidants* For example, reaction o f phenothiazine and
i t s methylated d eriva tive toy arened iazonium ions has been shown2Bto y ie ld the corresponding phenothiazine cation rad ica l •
Likewise, reaction o f tetra th ia fu lva lene with several p-substitu
ted arenediazonium ions are reported to involve s ingle electron
transfers^0* Arylation o f pyridine^2 and thiophene^1 proceeds
by the Intermediacy o f rad ica l cations* Other references are
ava ilab le in lite ra tu re which demonstrate the ET character o f29 32arenediazonium ions • Moreover, i t was observed by us that in
a c id ic medium, i f e* in the presence o f TFA, the arenediazonium
ion atta ins s u ff ic ie n t poten tia l to oxid ise aromatic and
heterocyclic compounds o f moderately high oxidation poten tia ls
such as thianthrene, t r i s - (-4 bromophenyl) amine,
9,10-diphenylanthracene, perylene, tetraphenylethylene e tc . to
the corresponding rad ica l cations.
Returning to our discussion o f the oxidation o f
diazoalkanes with N02 18 obv*ous that once the
diazoalkane cation rad ical is formed, i t should partic ipa te in
the reaction co-ordinate in very much the same way as discussed
in the preceeding section and outlined in pathway (a ) o f
Scheme I I .8.
With the use o f equimolar amounts o f the
diazocompound and p-^O,, the reaction assumes new
dimensions. In such a s ituation the d iazorad ical cations are
qu an tita tive ly generated in the in i t ia l fa s t ET step o f the
reaction . There are p ra c tica lly no diazoalkane molecules l e f t
fo r the cation rad ica l to couple w ith. Hie probable fa te o f
cation rad ica ls in th is situation is a coupling reaction with

the diazenyl rad ica ls to y ie ld the carbocation (28) a fte r the
loss o f n itrogen from the diazonium intermediate (27) as shown
in pathway (b ) o f Scheme I I . 8. The carbonium ion (28) is
bound to undergo a fa s t s o lv o ly t ic reaction to y ie ld the
product (29) provided water is present in the solvent system.
Thus when delibera te additions o f water are made 29 emerges as
the sole product o f the reaction . Even in dried MeCN presence
o f trace amounts o f water cannot be r ll led out. Further proof
o f the intermediacy o f the carbonium ion was obtained when the
reaction o f Ph2CM2 with p-tf02 was carried out in the
presence o f MeOH wherein the methoxy d er iva tive (31) emerged as
the principa l product.
The formation o f fluorenone-p-nitrophenyl hydrazone
(30) In the reaction o f 9-diazofluorene with arenediazonlum
Ion probably resu lts from a further redox reaction o f68p-nitrophenylazo-9-fluorenol formed in i t ia l ly .
Small amounts o f 4 ,4 , -d ln itrobiphenyl and nitrobenzene
which were formed are considered to be the products o f side
reaction o f the a ry l rad ica l derived from the diazenyl rad ica l.
These are the usual reactions involv ing dim erisation to the
biphenyl and hydrogen atom abstraction to y ie ld nitrobenzene.
177
178
Ar2£ - M* ~ Nf ♦ m 2 C6H4N+ = N
Ar2 C* - + N02 C6h4 ^ N*_n
(2 4 )
L§1
Ar2CN2
Ar2C*-C+A»2
mo2 c 6h;
I si4iau
'SH ^NO,C,H. -M02C6H5
5 c6h 4 - mo2
c o u p li n g
A r2 C-N* - M
(2 5 )
Ar2C»J2
Ar2C» CAr2 ♦ (26)
Ar2 CM*’ (24)
( 2 7 )
^ 2 °iU m- c 6ha - no2
Aj~ C-OMe 21 A r oC-0H 2 1N-M-C6H4N02
( 3 1 )
N«N-C6H4 i\T02
(2 9)
N-MH C6H4M02(30)
Scheme 11*8

179
^ 3B Reaction with hydrazones
The reaction of aldehyde hydrazones with diazonium
(Scheme II.9)
To our knowledge no report exists on the reaction of benzophenone or fluorenone hydrazone with diazoniuin salts.
On adding an MeC'J solution of p-nitrobenzenediazonium
evolution of nitrogen took place and the solution turned yellow. After allowing to stand colourless crystals identified as benzophenone azine appeared in Ca. 55% yield together with tetraphenyl ethylene (12%), p-nitrophenylazodiphenylmethanol (8%). Products of dediazonation of N O ^ - s u c h as nitrobenzene (18^) and 4,4*-dinitroblphenyl (v^10%) wfere also obtained.
In a similar manner fluorenone hydrazone reacted with yield fluorenone azine (Ca.80%) as the main
69cations is known to yield forraazans (Scheme II.9) via the intermediate formation of bis-arylazocompound (32).
Phft.2 N* NPh Ph* Ph C - M-M*"""\ \Nf - N... H
IPh
PhCH« 4-MHPh * PhCHNPh
(32)
tetrafluoroborate to benzophenone hydrazone at 25°, a brisk
product.
An analogous reaction occurred when under identical
benzaldehyde azine emerged as the principal product of the reaction.

180
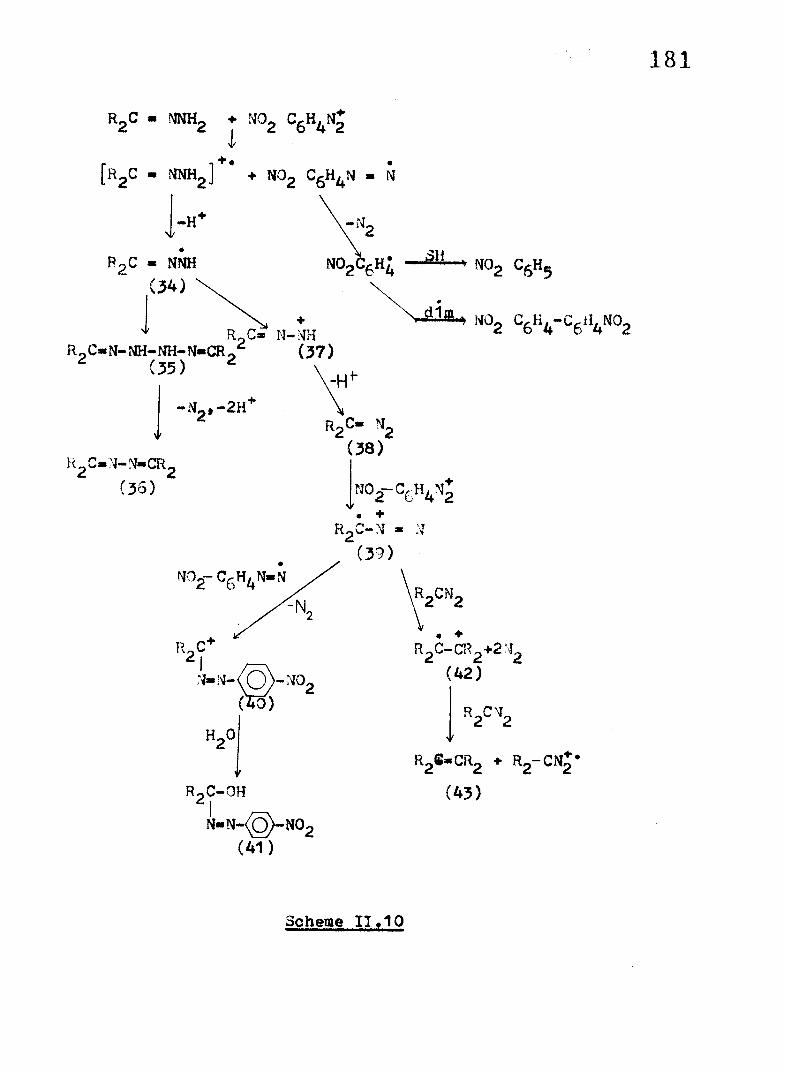
On the basis of pattern of products obtained in the reaction of these hydrazones with p-nitrobenzenediazonium tetrafluoroborate, a mechanistic interpretation is outlined inScheme 11.10.
The diazonium cation believably acts as an ET oxidant, generating the cation radicals of the hydrazones in an initial ET step. The radical cation (33) once formed would most readily lose a proton to form a radical (34). Dimerisation of the radical would yield the dimer (35), which being unstable can easily undergo oxidation to yield the azine as th« principal product of the reaction.
An alternative pathway of consumption of the radical(34) involves further oxidation followed by deprotonation to form the diazoalkane (38), the follow-up reaction of which involves a chain conducive to the formation of the olefin (43)*
The formation of p-nitrophenylazodiphenylmethanol in the reaction of Ph2O N M H 2 with f l O ^ - s u g g e s t s the presence of diazenyl radicals and Ph2CN2 radical cations in the system which partake in a coupling reaction followed by loss of ^ and hydrolysis of the resulting carbonium ion (40) to form the azo product (41) (as detailed in section II.2B)
The diazenyl radical on nitrogen loss would yield the p-nitrophenyl radical (44) which is expected to abstract hydrogen atom from the solvent to form nitrobenzene (45) or dimerise to4 ,4-dinitrobiphenyl (46).
jj»N-<g>-N02(41)
r2c-oh (43)
Scheme 11.10

182
B ftftgttoaof arertedlazonluro Ions with phosphazlnes and phosphorane.
To our knowledge no report exists on the reaction of diazonium salts with fluorenylldene triphenylphosphazlne and diphenyliaethylene triphenylphosphazlne, although coupling products have been reported in the reaction of phosphoranes with diazonium ion70.
The r e s u l t s o b t a i n e d i n t h e p r e s e n t s t u d y c o n c e r n i n g
the r e a c t i o n o f U02 w i t h Ph2 C«^-N«PPh3 and F l - N - N - P P h y
and F l - P P h j , a r e suggestive o f an e l e c t r o n t r a n s f e r mechanism
b e i n g o p e r a t i v e h e r e a l s o .
On adding MeCM solution of m 2 to Ph2C«N-N»PPh3i n CH2 C 12 at 25° , a brisk evolution of nitrogen gas occurredand the solution turned bright yellow. On work-up for products, tetraphenylethylene (40&), p-nitrophenylazodiphenylmethanol (25%), triphenylphosphine oxide (69^), together with products of dediazonation of X02 nitrobenzene (18%) and4,4*-dinitrobiphenyl (10.5%) were obtained.
Likewise, Fl«?J-M«PPh3 reacted with M02 to yieldMfluorenylldene (38&) fluorenone-p-nitrophenlhydrazone (33*5%), rh^P-0 (75#), nitrobenzene (12#) and 4,4*-dinitrobiphenyl (8$).
The reaction of Fl-PPhj with NX, CgH^N2 proceeded through the formation of a violet coloured intermediate which soon decomposed to yield fluorenone-p-nitrophenylhydrazone (46%), Ph^P-0 (48%) and nitrobenzene (6%).

183
On the basis o f our resu lts obtained in the reaction
o f Pi^ON-N-PPhj and Fl-M-N-PPh^ with NOg a mechanistic
in terpretation outlined in Scheme 11,11 is proposed*
The oxidation o f Fl*N-N-PPh, and Ph0C»N-N«PPh, withD Z 3SbCl,j and tris(-4-bromophenyl) aminium s a lt proceeds by the
in i t ia l generation o f the rad ica l cation and y ie ld s the dimeric
o le fin s and Ph^P-0 in good y ie ld s* The intermediacy o f rad ica l
cations in these reactions has been confirmed by EPR
spectroscopy and cy c lic voltammetric studies (see Chapter I I I ) .
Considering the a b i l i t y o f diazonium ion to act as
an ET oxidant and keeping in mind the moderate oxidation
poten tia l o f the phosphazines (see Chapter I I I ) and also the
fa c t that the dimeric o le fin s are produced in substantial
amounts, we propose that the reactions under discussion proceed
by an in i t ia l ET step to generate the cation rad ica l o f the
phosphazine (47) and the d iazenyl rad ica l.
The rad ica l cation once formed su ffers a homolytic
cleavage o f the C-M and M-P bonds to form the carbene (48 ),
nitrogen and the cation rad ica l o f PPhj(49) which on in teraction71with oxygen y ie ld s the oxide by a known mechanism .
Dimerisation o f the carbene forms the o le f in (50 ).
An a lte rn a tive pathway also seems to be operative.
In the solution are present the rad ica l cations (47) and the
diazenyl rad ica ls . A coupling o f the two followed by expulsion
o f PPh and nitrogen would lead to the carbonium ion (53) which
on so lv o ly t ic reaction with water, generally present in MeCN
despite scrupulous drying, y ie ld s the azo product (55 ). PPh on
reaction with excess would form the oxide, PPh^-0, as
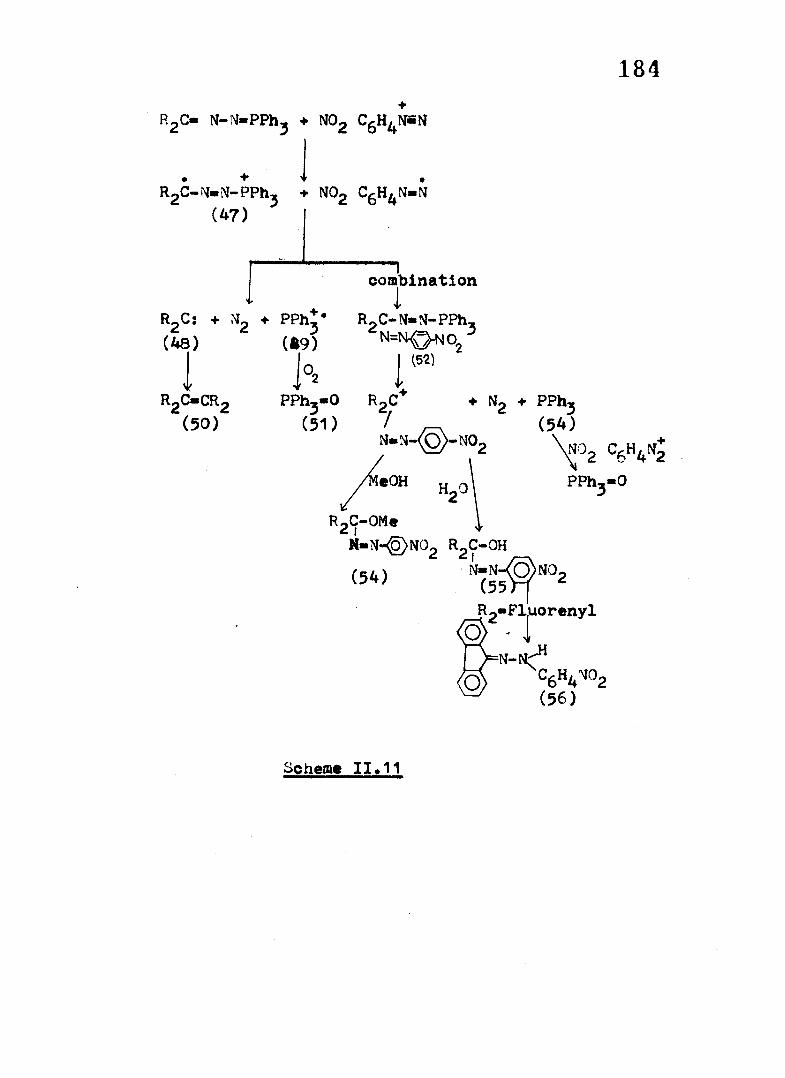
184
R20 N-N«PPh3 ♦ N02 CgH^NSN
R2C-N«N-PPh3 N02 CgH^N-N(47)
combinationI
R0Cs + N- ♦ PPhI* R0C-M«ft-PPh,2 2 ? 2n=n^ o, 3(48)
R2C-CR2
(*9 )
1(52)
(50)PPh3-0 R2C
(51) /♦ Nf2 * PPh-j
A t
N-N-<O>-N0(54)
\N°2 c6H4*2eOH
R-C-OMe 2 i
h20' PPh3-0
N-M-©>N0p RpC-OHn« n- (Q )no,(54) (5 5 r fR2»Fluorenyl
d> * IN-N;J i
\C6H4*°2(56)
Scheme 11.11

185
was found to be the case when a sample o f PPh^ was separately
treated with N02
In the case o f Fl«N-M»PPhj, the formation o f the
hydrazone ( 56 ) may be due to the further redox reaction o f the
in i t ia l l y formed p-nitrophenylazo-9-fluorenol (see section II.2 B )
Evidence fo r the intermediacy o f carbo cation (53) was
obtained when the reaction o f Ph^ON-N-PPh^ with MC CgH N was
carried out in the presence o f methanol wherein the methoxy
product, p-nitrophenylazodiphenylmethoxymethane (54) m.p 135° ,
could be iso la ted in 20% y ie ld .
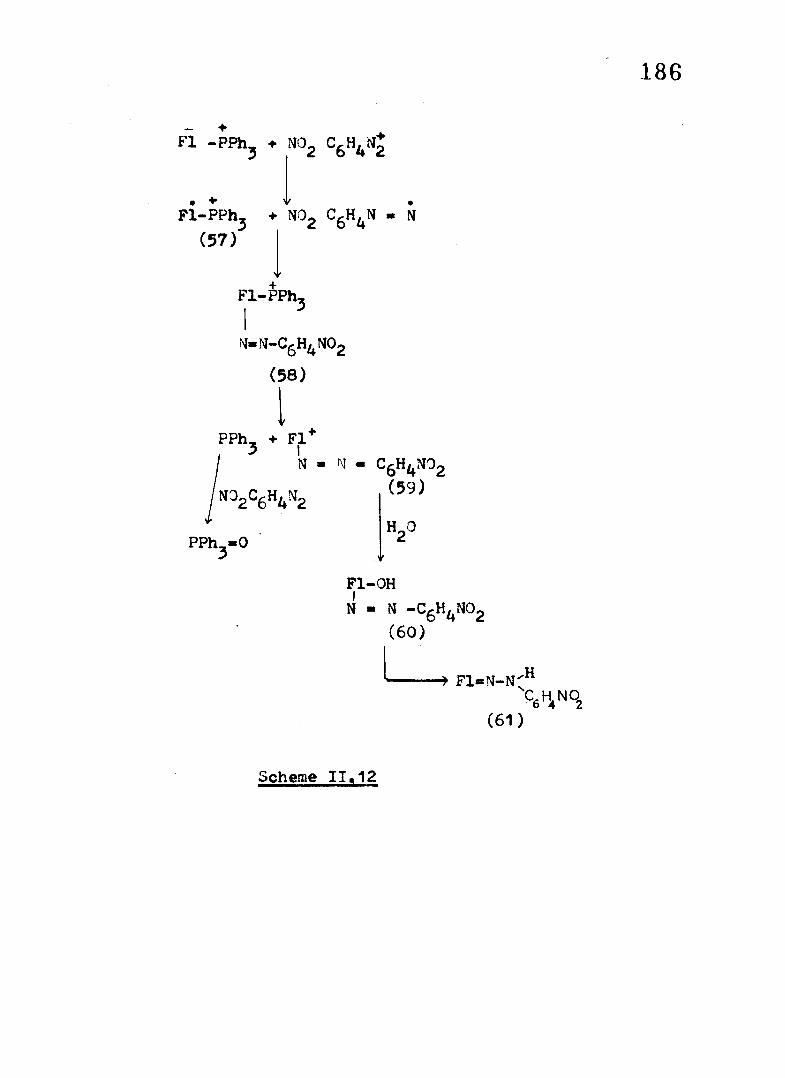
Fluorenyl triphenylphosphorane has a very low
oxidation poten tia l (0.05V) and an ET reaction with ArM is- + 72
quite conceivable. Fl-PPh^ has been shown to be oxidised by
chemical and electrochemical means to i t s rad ica l cation . The
transient v io le t colour that develops in these reactions may be
due to the formation o f these paramagnetic species. A coupling . +
o f Fl-PPhj (57) with the diazenyl rad ica l followed by expulsion
o f PPhj would generate the carbocation (59 ). This on hydrolysis68and a further redox reaction leads to the formation o f
fluorenone-p-nitrophenylhydrazone (61) Scheme 11.12 .
186
FI -PPhj ♦ N02 C6Ha N*
• +Fl-PPhj + N02 C6H4N
(57)
Fl-PPh,
N
n- n- c6h4no2
(58)
PPh, + F l+ 3 IN - M « C6H ft02
N32C6H4M2
PPh3«0
(59)
h2o
FI-OHN ■ N -C6H/fN02
(60)
(61)
Scheme 11,12

REFERENCES187
1. K linger R J, Mochida K and Kochi J K, J Araer Chem Soc,101 (1979) 6636.
2. Farcasiu M and Russell C S, J Org Chem, 41_ (1976) 571.
3. Horner L and Schwenk E, Justus L ieb igs Ann Chem, 566 (1950) 69.
4a. Zupancic J J, Horn K A and Schuster G B, J Amer Chem Soc,102 (1980) 5279.
b. Dixon B G and Schuster G B, J Amer Chera Soc, 103 (1981) 3068.
5a. Walling C and Zhao C, Tetrahedron, £8 (1982) 1105.
b. Zhaq C, E l'Ta liw a i G M and Walling C, J Org Chem, 48 (1983) 4908.
6. Kateuchi K, Murai 0, Matsui S, Inoue T, Kitagawa T and Okamato K, J Chem Soc Perkin I I , (1983) 1301.
7. Chalfond C R and Perkin M J, J Chem Soc Bt (1971) 245.
8. Pryor W A & Henrickson W H Jr, J Amer Chem Soc, 105 (1983) 7114.
9. Beresford P, Lambert M C and Ledwith A, J Chem Soc C,(1970) 2508.
10. Ledwith A, ACC Chem Res, £ (1972) 133.
11. Kricka L J & Ledwith A, J Chem Soc Perkin I , (1973) 294.
12. Beresford P, I le a D H, Kricka L J and Ledwith A, J Chem Soc Perkin I , (1974) 276.
13* B ell F A, Beresford P, Kricka L J and Ledwith A, j Chem Soc Perkin I , (1974) 1788.
14. Nelson S F, Weissman " R, Hintz P J, Olp D and Fahey M J,J Amer Chem Soc, £6 (1974) 2916.
15* Dauben H J and Wilson J D, J Chem Soc Chem Comaun, (1968) 1629.
16. Hawthrone M F, J Org Chem, 21, (1956) 363#
17. Kashin A N, Bumagin N A. Beletskaya I P and Reutov 0 A, JOrg Chem USSR, 1*[ (1979) 204.
18. Kashin A M, Bumagin N A, Beletskaya I P and Reutov 0 A,Dokl Akad Nauk SSSR, 244 (1979) 98.
19. Murray R W and Kalpan M L, J Org Chem £1 (1968) 962.

20. Ledwith A & Sambi M, «J Chem Soc Chem Commun, (1965) 64.
21. Bawn CEH, Fitzsimmons C and Led with A. Proc Chem Soc, (1964) 391.
22. B ilev itch K A, Bubnov N N and Okhlobystin 0 Yu, Tetrahedron Lett, (1968) 3465.
23. Singh H and Tedder J M, J Chem Soc Chem Commun, (1981)70.
24. Okamoto K, Komatsu K, Mural 0 and Sakaguchl 0,Tetrahedron L e tt, (1972) 4989.
25. Singh P R and Kumar R, Aust J Chem, 2£ (1972) 2133.
26. Singh P R and Kumar R, Tetrahedron Lett, (1982) 5191.
27. Bubnov M N, B ilev itch K A, Polyakova L A and Okhlobystin0 Yu, J Chem Soc Chem Commun, (1972) 1058.
28. Bisson J M, Hanson P and Salocum D, J Chem Soc Perkin I I ,(1978) 1331.
29». B ilev itch K A, Bubnov N M e t a l . Dokl Akad Nauk SSSR, (1970) 583.
b. B ilev itch K A et a l . , Teor-i-k KSP Khia, 8 (1972) 256.
30. Kampars, V, Bumbure G, Kokars V and Neilands 0, Zh Obshch Khim £0 (1980) 2057.
31. Bartie MG, Machie R K and Tedder J M, J Chem Soc Chem Commun, (1974) 271.
32. Elofson R M, Gadallah F F and Schulz K F, J Org Chem,26 (1971) 1526.
33. Broadhead G D and Panson P L, J Chem Soc, (1955) 367.
34. B e ll F A, Ledvrith A and Sherrington D C, J Chem Soc C, (1969) 2719.
35* Handoo K L and Gadru K, Indian J Chem, (1985)* In press.
36. Sep W J, Verhoeven J W and DeBoer Th J, Tetrahedron, 35(1979) 2161.
37. Paul M A and Long F A, Chem Rev, £Z (1957) 1.
38. Nepras M, Kratochvil V, T itz M, Novak A and S lavik V, C o llect Chech Chem Commun, (1973) 1003.
39a. Kochi J K, Tang R T and Bernath T, J Amer Chem Soc,(1973) 7114.
b. Bard A J, Ledwith A and Shine H J, A d v , Phys Org Chem,11 (1976) 155.
188

c ) Heiba E I , Dessan R M and Koehl Jr W J, J Amer Chem Soc,21 (1969) 138, 6830;
Helba E I and Dessau R M, J Amer Chem Soc, 22 (1971) 995.
40. Das M and Basu 3, Spectrodhira Acta, (1961) 897.
41. Hoijtink G J and We land W P, Rec Trav Chitn, JS (1957)836.
42. Pava A, Sago P B and Calvin M, J Amer Chem Soc, 79 (1957)1078.
43* Dwivedi P C, Rao K G, Bhatt 3 M and Rao CMR, SpectrochimActa, 21A (1975) 129.
44. Coffen D L, Chambers J Q, Williams DR, Garrett P E andCanfield N D, J Amer Cheai Soc, 92. (1971) 2258.
45. Sehested K and Hart H J, J Phys Chera, 22 ( 1975) 1639.
46. Suzuki H, Koyano K. Shida T and Kira A, Bull Chem Soc Jpn, 22 (1979) 2794.
47. Foster R and Thomson T J, Trans Faraday Soc, *>8 (1962)560.
48. Wilkinson G, Rosenblum M, Whiting M C and Woodward R B,J Amer Chem Soc, £4 (1952) 2125.
49. Zarubin M Ya, Kutnevich A M and Rudenko A P, Zh Org Khim, 11 (1975) 1284.
50. Roberts J D and Wantabe W, J Amer Chera Soc, 72(1950)4869.
51. Cowell C W, Ledwith A, White A C and Woods H J, J Chem Soc B, (1970) 227.
52. Bonthrone w and Reid D H, J Chem Soc, (1959) 2773.
53. Schlenk and Bergmann, Anaalen, 463 (1928) 15*
54. Ingold C K and Jessop J A, J Chem Soc, (1929) 2357.
55. Szmant H H and McGinnis C, J Amer Chem Soc, (1950)2890.
56. Gomberg M, J Amer Chem Soc, 22 (1900) 757.
57. Doering W E and Knox L H, J Amer Chem Soc, 22 ( 1957) 352.
189

190
58. Pinck L A and H ilbert G Ef J Amer Chem Soc, 68 (1946)867.
59a. B illon J P, Cauquis G and Combrisson J, J China Phys, (1964) 374.
b. Wudl F, Smith G M and Hufnagel E J, J Chem Soc Chem Commun, (1970) 1453.
c. Bolton J R» Carrington A and Santos-Veiga J D, Mol Phys,2(1962) 615.
60. Jugelt W and Pragst F, Tetrahedron 24 (1968) 5123IJugelt W and Pragst F, Angew Chem Int Ed Engl,2 (1968) 290.
61. Bethell D, Handoo K L , Fairhurst S A and S u tc lif fe L H,J Chem Soc Perkin 11,(1979) 707.
62. Pinck L A and Guido H E, J Amer Chem Soc, 69 (1947) 723.
63. Starkley E B, Org. Synth, Coll Vol I I , (1943) 225.
64. B e ll F and Kenyon J, J Chem Soc, (1926) 2767.
65. Vogel A I , A Text Book o f P ractica l Organic Chemistry, Longmans, 4th ELBS Ed, page 1196.
66. Pearson D E, Carter K N and Greer C M, J Amer Chem Soc, 75 (1953) 5905.
67. Blake C A, Brown K B and Coleman C F, U S Atomic Energy Coma, 0RNL-1964 (1955)106pj CA 50* 15320.
68. Huisgen V R and Koch H J, Ann Chem, $9± (1955) 200.
69. Organic reactions, Ed B la tt AH , Boekethelde V, Cope C A, David D Y, McGraw F C and Niemann C,John Wiley & Sons,New York, (1959), Vo l.10, Chap 1.
70. Mareki G, Tetrahedron Lett, (1961) 807*
71. P litsyna 0 A, Pudeeva M E and Reutov 0 Af Dokl Akad Nauk SSSR, 2§S, (1965) 838.
72. Koul A, M.Phil Thesis, University o f Kashmir (1985).