cd40: an upstream master switch for endothelial cell
TRANSCRIPT

HEMOSTASIS, THROMBOSIS, AND VASCULAR BIOLOGY
CD40: an upstream master switch for endothelial cell activation uncovered byRNAi-coupled transcriptional profilingRaquel Pluvinet,1 Rut Olivar,1 Jerzy Krupinski,2 Inmaculada Herrero-Fresneda,3 Anna Luque,2 Joan Torras,3
Josep M. Cruzado,3 Josep M. Grinyo,3 Lauro Sumoy,4 and Josep M. Aran1
1Medical and Molecular Genetics Center, Institut d’Investigacio Biomedica de Bellvitge (IDIBELL), Hospital Duran i Reynals, L’Hospitalet de Llobregat,Barcelona; 2Department of Neurology, Stroke Unit and 3Laboratory of Nephrology, Medicine Department, Hospital Universitari de Bellvitge, IDIBELL, L’Hospitaletde Llobregat, Barcelona; and 4Bioinformatics and Genomics Program, Centre de Regulacio Genomica (CRG)–Universitat Pompeu Fabra (UPF), Barcelona,Spain
The CD40-CD154 dyad seems to play aprominent role fostering the immune-inflammatory response triggered by endo-thelial cell (EC)–T-cell communication. Todelineate comprehensively the involve-ment of CD40 (TNFRSF5) in EC activation,we combined RNAi-mediated CD40 knock-down with comparative genome-widetranscriptional profiling of ECs interact-ing with (CD154�) T cells. We report theinitiation of a profound stress responsein ECs upon CD40-CD154 engagementthrough early up-regulation of, amongothers, the major proinflammatory NF-�B
and MAPK/SAPK pathways and their as-sociated transcription factors. Moreover,we have identified novel genes regulatedthrough the CD40-CD154 interaction, andpathways previously unrecognized tobe induced by CD40 signaling in ECs.Thus, we document a significant down-regulation of endothelial APLN by CD40-CD154 interaction, TNF�/IFN� exposure,and in immune-inflammatory patholo-gies, which could lead to hemodynamicdysfunction. Conversely, CD40-mediatedup-regulation of the viral immune surveil-lance system, notably TLR3, IFIH1, RIG-I,
and RNASEL, establishes a reverse linkfrom adaptive to innate immunity in ECs.Moreover, systematic enrichment analy-sis substantiates endothelial CD40 in-volvement in the transcriptional regula-tion of gene networks associated withadhesion and motility, immunity, cell fatecontrol, hemostasis, and metabolism. Ourstudy also highlights the anti-inflamma-tory potential of RNAi-mediated CD40 in-hibition, and the relevance of CD40 signal-ing for therapeutic intervention. (Blood.2008;112:3624-3637)
Introduction
It has been postulated that CD40, the tumor necrosis factorreceptor superfamily member 5 (TNFRSF5), when interactingwith its cognate ligand CD154 (CD40L; TNFSF5), plays aprominent role in the development of immune-inflammatoryconditions such as cardiovascular disorders, autoimmune dis-eases, and organ rejection.1
Indeed, CD154 has a restricted pattern of expression, beingup-regulated mainly in activated CD4� T cells and platelets. Incontrast, CD40 is expressed at relatively low abundance in mostresting cell types, showing the highest levels in B cells anddendritic cells, where it contributes to their maturation.2,3 Forexample, in B cells, where it was first discovered and has beenmore thoroughly characterized, CD40 engagement triggers thebinding of different members of the family of tumor necrosis factorreceptor-associated factors (TRAFs) to its intracellular domain,mediating activation of multiple signaling pathways that regulateB-cell survival, proliferation, and differentiation; immunoglobulinisotype switching; development of the germinal center; and thehumoral memory response.4
In ECs, the CD40-CD154 interaction triggers proinflammatorycytokine and chemokine production, matrix metalloproteinase, andtissue factor expression, procoagulant activity, angiogenesis, and ahallmark increase in expression of the leukocyte adhesion mol-ecules E-selectin (SELE, CD62E), vascular cell adhesion molecule
1 (VCAM1, CD106), and intercellular adhesion molecule 1(ICAM1, CD54).5 All these events modulate an essential step in theimmune-inflammatory process: leukocyte recruitment, adhesion,rolling, and extravasation to the injured or stressed tissue. Thus,interfering CD40-CD154 interaction represents a suitable therapeu-tic option. This has been demonstrated using blocking antibodiesagainst CD154 in a variety of animal models of immune-inflammatory disorders and several clinical trials, although severethromboembolic events have aroused in some of these studies.6-8
Alternatively, we have recently shown that genetic knockdown ofCD40 expression through RNAi prevents EC adhesion moleculeexpression and leukocyte adhesion on ECs.9 Nevertheless, ourunderstanding of the CD40-CD154 action is not yet complete topredict the full outcome and side effects of such therapeuticapproaches because CD40 has a remarkable variety of functions indifferent cell types. Thus, much remains to be learned about themolecular pathways that operate in ECs stimulated by interactionwith CD154 displayed on the surface of activated T lymphocytes.
In this report, we combine RNAi-mediated CD40 gene knock-down and genome-wide transcriptional profiling to comprehen-sively delineate the molecular signature of CD40 in ECs triggeredby CD154� T lymphocytes. This approach may be exploited to(1) study the temporal expression pattern of genes regulated byCD40 signaling, (2) identify novel genes and/or pathways not
Submitted March 3, 2008; accepted July 24, 2008. Prepublished online asBlood First Edition paper, July 31, 2008; DOI 10.1182/blood-2008-03-143305.
An Inside Blood analysis of this article appears at the front of this issue.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page chargepayment. Therefore, and solely to indicate this fact, this article is herebymarked ‘‘advertisement’’ in accordance with 18 USC section 1734.
© 2008 by The American Society of Hematology
3624 BLOOD, 1 NOVEMBER 2008 � VOLUME 112, NUMBER 9
For personal use only.on January 3, 2019. by guest www.bloodjournal.orgFrom

previously associated with CD40 action on ECs, and (3) establishcandidate targets, involved in immune-inflammatory processesthrough the CD40 pathway, for diagnosis/prognosis or potentialtherapeutic intervention.
Accordingly, our results indicate that CD40 signaling leads torobust down-regulation of the vasoactive peptide apelin by ECs,and suggest that the altered vascular homeostasis elicited by theloss of this cytokine under proinflammatory stimuli may contributeto endothelial dysfunction. Conversely, we propose a key role forCD40 triggering antiviral surveillance in ECs when challengedwith activated T cells. Furthermore, gene set enrichment analysis ofthe dynamically regulated genes identifies sequential geneticprograms characterizing CD40-induced endothelial activation.
Methods
Cell culture and siRNA transfection
Primary human umbilical vein ECs (HUVECs) representing a pool of atleast 3 different umbilical cord veins (Advancell, Barcelona, Spain) weremaintained in endothelial growth medium EGM-Bullet kit (Clonetics-Cambrex, Walkersville, MD). Third-passage primary ECs isolated fromnormal human carotid arteries (HCtAECs) were cultured in EC GrowthMedium II (Cell Applications, San Diego, CA). All endothelial cultureswere grown at 37°C in a 5% CO2 atmosphere. The siRNA duplex targetinghuman CD40 (siRNA-2) and its corresponding mismatched siRNA control(msiRNA-2) were chemically synthesized (QIAGEN, Hilden, Germany).Specific sequences and characterization of their silencing effect in HUVECshave already been described.9 Incorporation of the siRNAs into exponen-tially growing HUVECs (passages 3-4) was achieved by electroporationwith an ECM 830 Electropulse Generator System (BTX, San Diego, CA).For details, see Document S1 (available on the Blood website; see theSupplemental Materials link at the top of the online article).
siRNA-transfected HUVECs were stimulated by coincubation withJurkat D1.1 (CD154�) cells10 (ATCC, Manassas, VA; CRL-10915,1:10 ratio) at 4, 10, or 16 hours prior to analysis. At 48 hours aftertransfection, cocultures were extensively washed with PBS to release theattached Jurkat D1.1 cells from the HUVEC monolayer. Jurkat D1.1stimulation of HCtAECs was performed analogously to that of HUVECs.Residual Jurkat D1.1 contamination of HUVEC and HCtAEC monolayerswas estimated to be less than 0.5% by flow cytometry (data not shown).
Gene expression profiling
The RNA samples from treated HUVECs were extracted using the RNeasyRNA Isolation kit (Qiagen) and incubated with RNase-free DNase I(Ambion, Austin, TX) according to the manufacturer’s protocol. RNAquantification and quality assessment were performed as described inDocument S1.
RNA samples (500 ng total RNA) were reverse transcribed, amplified,labeled by in vitro transcription using the Low Input Linear AmplificationKit (5184-3523; Agilent, Wilmington, DE), and hybridized following themanufacturer’s instructions. The whole-genome oligonucleotide microar-rays used (G4112A; Agilent) contain 44 290 spots with 60-mer probes,41 675 of which represent in single or multiple copies a total of37 312 human transcripts.
Two biologic replicate experiments were performed, each comparingsiRNA-2–transfected with msiRNA-2–transfected HUVECs. Each experi-mental pair of labeled samples was cohybridized on 2 separate microarrayswith dye swapping to correct for dye bias effects. Thus, 4 microarrayhybridizations were processed for each of 3 EC-D1.1 Jurkat cell coincuba-tion time points, totaling 12 array data sets. Additional details on scanningand data analysis can be found in Document S1. Differentially expressedgenes were chosen, unless otherwise stated, using as cutoff criteria aBayesian statistic percentile (B rank) above 95% and absolute fold change(FC) greater than 1.2 for at least 1 of the 3 time points. The entire dataset for
all microarray experiments has been deposited in NCBI Gene ExpressionOmnibus (GEO, http://www.ncbi.nlm.nih.gov/geo) and is accessible throughGEO Series accession number GSE10601.11
Microarray data analysis
Significance analysis of microarrays (SAM) and gene ontology (GO)functional classification were carried out using the TMEV software (TIGRMultiple Experiment Viewer; The Institute for Genomic Research, Rock-ville, MD; http://www.tm4.org/mev.html).12 Gene set enrichment analysiswas performed with the GSEA software developed at the MassachusettsInstitute of Technology (MIT, Cambridge, MA)13 using the functional setC2, which includes 522 gene sets corresponding to specific metabolic andsignaling pathways from the Molecular Signature Database MSigDB 1.0.14
Either the expression intensity values or the log2 ratios from the whole-genome unfiltered microarray data were included to assess biologicfunctions or processes differentially regulated. We compared differencesbetween msiRNA-2 and siRNA-2 treatment, or between 2 given stimulationtimes. The permutation number was fixed at 1000, and the significance atfalse discovery rate q-value (FDR q-val) was less than 0.25.
Quantitative real-time RT-PCR
Confirmation of regulation was achieved by quantitative real-time reversetranscription–polymerase chain reaction (RT-qPCR) on selected genesusing SYBR Green and the Lightcycler technology (Roche MolecularBiochemicals, Indianapolis, IN). Reverse transcription reactions wereperformed with 2 �g total RNA using the Omniscript RT kit (Qiagen).Additional PCR amplification information is given in Document S1.Gene-specific primer pairs (Table S1) were designed using Oligo 4.0software (MBI, Cascade, CO) and selected to prevent primer-dimerformation.
Results were calculated as the normalized mRNA level ratio of eachgene over the housekeeping genes cyclophilin-A (CYPA) or 18S ribosomalRNA (LOC100008588), as indicated.
Northern blot analysis
For Northern blotting, 20 �g total RNA extracted from untreated orsiRNA-treated HUVECs further stimulated with the proinflammatorymediators (Jurkat D1.1 [CD154�] cells, or 100 U/mL TNF� � 1000 U/mLIFN�) was electrophoresed on 1.2% formaldehyde-agarose gels, transferredto a Hybond XL nylon membrane (Amersham Biosciences, ArlingtonHeights, IL), and immobilized by cross-linking with UV light (Stratalinker;Stratagene, La Jolla, CA). APLN-specific signals were detected usingprobes F3-R3 (244 bp) and F4-R4 (295 bp; Figure 6A, Document S1). Themembranes were stripped and rehybridized using a control glyceraldehyde-3-phosphate dehydrogenase (GAPDH) probe (Clontech BD Biosciences,Palo Alto, CA).
Western blot analysis
Immunodetection of CD40, apelin, and IFN� was carried out on thecorresponding cell extracts in radioimmunoprecipitation assay (RIPA)buffer as previously described.9 Total protein content of the supernatantswas measured using the BCA protein assay (Pierce, Rockford, IL). Totalprotein (40 �g from each sample) was separated by 10% SDS–polyacrylamide gel electrophoresis (PAGE) and electrophoretically trans-ferred to Hybond membranes (Amersham Biosciences). Human CD40expression was assessed with a mouse monoclonal antibody (H-10,1:1000;Santa Cruz Biotechnology, Santa Cruz, CA). Intracellular levels of apelinwere determined with a goat polyclonal antibody (S-20, 1:200; Santa CruzBiotechnology). Intracellular IFN� was measured with a rabbit antihumanIFN� polyclonal antibody (ab 9662; 1:1000; Abcam, Cambridge, MA). Thecorresponding anti-IgG HRP-conjugated secondary antibodies (Dako,Glostrup, Denmark) were used for enhanced chemiluminescence detection(ECL � Plus; Amersham Biosciences).
Membranes were reprobed with a rabbit polyclonal antiactin antibody(1:2000; Sigma-Aldrich, St Louis, MO) for normalization.
ROLE OF CD40 IN ENDOTHELIAL CELL ACTIVATION 3625BLOOD, 1 NOVEMBER 2008 � VOLUME 112, NUMBER 9
For personal use only.on January 3, 2019. by guest www.bloodjournal.orgFrom

MAPK phosphorylation
siRNA-transfected or untransfected HUVECs (2 � 106 cells) were cocul-tured with Jurkat D1.1 cells for 4, 10, and 16 hours prior to analysis. Toinvestigate phosphorylation of JNK, p38, and ERK1/2 MAPKs, cells wererecovered at 48 hours after transfection, washed with ice-cold PBS, andscraped into ice-cold lysis buffer (20 mM Tris-HCl pH 8.0; 150 mM NaCl;1% Triton X-100; 10 mM NaF; 40 mM �-glycerolphosphate; 2 mMNa3VO4) supplemented with Complete Protease Inhibitor Cocktail (RocheMolecular Biochemicals). After incubation for 15 minutes on ice, sampleswere centrifuged at 16 000g for 15 minutes at 4°C, and supernatants werecollected and processed as described in the previous section.
MAPK-specific phospho- and total anti–human antibodies (Cell Signal-ing, Beverly, MA) were used for probing Western blots. The Phospho-MAPK Family Antibody Sampler kit contains the rabbit polyclonal primaryantibodies Phospho-p44/42 MAPK (Thr202/Tyr204), Phospho-SAPK/JNK(Thr183/Tyr185), and Phospho-p38 MAPK (Thr180/Tyr182). The MAPKFamily Antibody Sampler kit contains the rabbit p44/42 MAPK (137F5)and SAPK/JNK (56G8) monoclonal antibodies and the rabbit p38 MAPKpolyclonal antibody.
Nuclear extractions and transcription factor activity assays
siRNA-transfected HUVECs were cocultured with Jurkat D1.1 cells for 4 hoursprior to analysis, at 48 hours after transfection. Nuclear extracts were preparedusing the Transfactor Extraction Kit (Clontech BD Biosciences) according to themanufacturer’s instructions. Protein concentration was determined with a Brad-ford-based assay (Bio-Rad Laboratories, Marnes-la-Coquette, France).
Detection of transcription factor activity was performed with 2 enzyme-linked immunosorbent assay (ELISA)–based colorimetric assays accordingto the supplier’s protocol: the BD Mercury Transfactor Profiling KitInflammation 1 (Clontech BD Biosciences) for transcription factors fromthe NF-�B family, and the TransAM AP-1 and MAPK family kits(ActiveMotif Europe, Rixensart, Belgium) for AP-1– and MAPK-dependent transcription factor families.
Human samples
Carotid arteries (n � 5) were obtained as vascular transplants from organdonors or postmortem autopsies. Carotid plaques were classified accordingto the American Heart Association (AHA; Dallas, TX) criteria with somemodifications.15 The study was approved by the local ethical committee,Comite Etico de Investigacion Clínica, from the Bellvitge University Hospital, inaccordance with institutional guidelines and with written informed consentobtained in accordance with the Declaration of Helsinki.
Experimental renal transplantation
Syngeneic renal transplantations between Wistar Agouti rats (n � 3) andallogeneic grafts using Wistar-Agouti rats as recipients of Brown-Norwaykidneys (n � 6) were performed as previously described.16 The animalswere maintained in accordance with the guidelines of the Committee onCare and Use of Laboratory Animals and Good Laboratory Practice fromthe Bellvitge Animal Care Facility (Barcelona, Spain).
Immunohistochemistry
Routine immunohistochemistry was performed on paraffin-processed sec-tions (Document S1) from human carotid samples using the rabbitpolyclonal antibodies anti–apelin-36 (human, 1:200; Phoenix Pharmaceuti-cals, Belmont, CA) and anti-CD40 (C-20, 1:200; Santa Cruz Biotechnol-ogy), and from rat kidneys using the rabbit polyclonal antibodies anti–apelin-36 (rat, mouse, 1:200; Phoenix Pharmaceuticals) and anti–PECAM-1 (M-20, 1:200; Santa Cruz Biotechnology).
Results
To infer the physiologic role of CD40 in the context ofT-lymphocyte–EC interaction, we sought to compare global
transcriptional profiles from Jurkat D1.1 (CD154�)–treatedHUVECs, in which CD40 expression was either knocked down(by siRNA-2) or unaffected (by msiRNA-2).9 Moreover, to gaininsight into the dynamics of CD40-mediated signal transductionin ECs, we extended our comparative transcriptomic analysis to4, 10, and 16 hours of CD40-CD154 interaction using thiscell-cell context.
As controls to assess the efficiency of CD40 gene silencingand the quality of the RNA prior to microarray processing,we quantified RNA levels of both CD40 and ICAM1,known to be up-regulated by CD40-CD154 interaction.Quantitative RT-PCR (CD40 and ICAM1) and Western immuno-blotting (CD40) confirmed efficient silencing of both thecostimulatory receptor as the primary target and the celladhesion molecule as a consequence of CD40 signal transduc-tion inhibition (Figure S1).
Microarray data mining and functional classification of CD40responsive genes in endothelial cells
We compared gene expression profiles from CD154�
T cell–stimulated ECs with or without RNAi-mediated CD40silencing. We considered genes as regulated when there wasboth B rank more than 95% and FC 1.2 or more in at least 1 ofthe 3 stimulation times (4, 10, and 16 hours). The analysisidentified 715 transcripts ( 2%) that were differentially regu-lated on the whole genome microarrays (3% of the HUVECtranscriptome). From the regulated genes, nearly 75% werefound up-regulated, whereas 25% were found down-regulated(Table S2). Moreover, the magnitude of differential generegulation was maximal at 4 hours. This suggests an earlymassive response of the EC upon CD40-CD154 signaling andconfirms the general role of CD40 as an EC “activator.”17
To explore the biologic functions affected by CD40 signaling inECs, we performed a GO overrepresentation analysis from the listof regulated genes with annotated functions. As a consequence ofCD40-CD154 engagement, most of the regulated genes belong tocategories directly or indirectly related to the immune-inflamma-tory process (Figure S2). Moreover, GO analysis confirms knownkey aspects of CD40-related EC activation early in the inflamma-tory process, such as the involvement of the NF-�B signaling, andthe induction of chemotactic signals allowing cross-talk with thecirculating leukocytes.1
A short list of up- and down-regulated annotated genes with aB rank more than 99% and FC 1.5 or more in at least 1 of the3 stimulation times is shown in Figure 1. The full list ofdifferentially expressed transcripts is accessible online (Table S2).We arranged representative annotated genes into selected func-tional classes according to their putative activities (Figure 2). Someof these genes had been already described as targets of CD40signaling, although we also identified many others previouslyunrecognized to be related to CD40 action in ECs (see DocumentS1, “Functional classification,” for details).
Validation of CD40-dependent endothelial cell activation
To confirm the array results, we performed RT-qPCR of a set of15 representative genes at 4 hours after stimulation, and from8 of them we also evaluated their behavior at 10 hours and16 hours after stimulation (Figure 3A). Among the up-regulatedgenes we selected chemokines (CCL2, IL8), transcription fac-tors (FOSB, NLF2 [and also NLF1; data not shown]), growthfactors (CSF2, VEGF), the adhesion molecule SELE, the
3626 PLUVINET et al BLOOD, 1 NOVEMBER 2008 � VOLUME 112, NUMBER 9
For personal use only.on January 3, 2019. by guest www.bloodjournal.orgFrom

antiapoptotic BIRC3, the antioxidant SOD2, the metalloproteaseMMP10, the regulator of calcineurin DSCR1, the complementfactor CFI, and the T-cell activation marker CD69. The down-regulated genes included the anticoagulant THBD and the cytokineIL11. Four of these genes (SOD2, DSCR1, IF, and IL11) showedsmall FC values ranging 1.2 to 1.3. Our RT-qPCR validation resultsare in full agreement with the microarray data, both in terms of up-
and down-regulated genes and in terms of early (FOSB, CSF2) andlate (MMP10, VEGF) regulated ones (Figure 3A). Overall, wefound correlation between the magnitude of change as measured bymicroarray hybridization and RT-qPCR (Figure 3B), confirmingprevious observations20 and suggesting that microarray-derivedlog2 ratio, though attenuated in high-density microarrays,21 accu-rately reflects gene expression changes.
Figure 1. Expression profile of selected CD40-regulated genes upon EC stimulation with Jurkat D1.1 (CD154�) T cells. Heat map and the corresponding list ofup-regulated (green) and down-regulated (red) genes fulfilling criteria of B rank 99 or more and mean FC 1.5 or more in at least 1 of the 3 stimulation times analyzed: 4 hours(columns 1-4), 10 hours (columns 5-8), and 16 hours (columns 9-12). Rows representing individual genes are ordered according to the mean FC across the 3 stimulation times.Unigene symbols18 and GenBank accession numbers19 were used.
ROLE OF CD40 IN ENDOTHELIAL CELL ACTIVATION 3627BLOOD, 1 NOVEMBER 2008 � VOLUME 112, NUMBER 9
For personal use only.on January 3, 2019. by guest www.bloodjournal.orgFrom
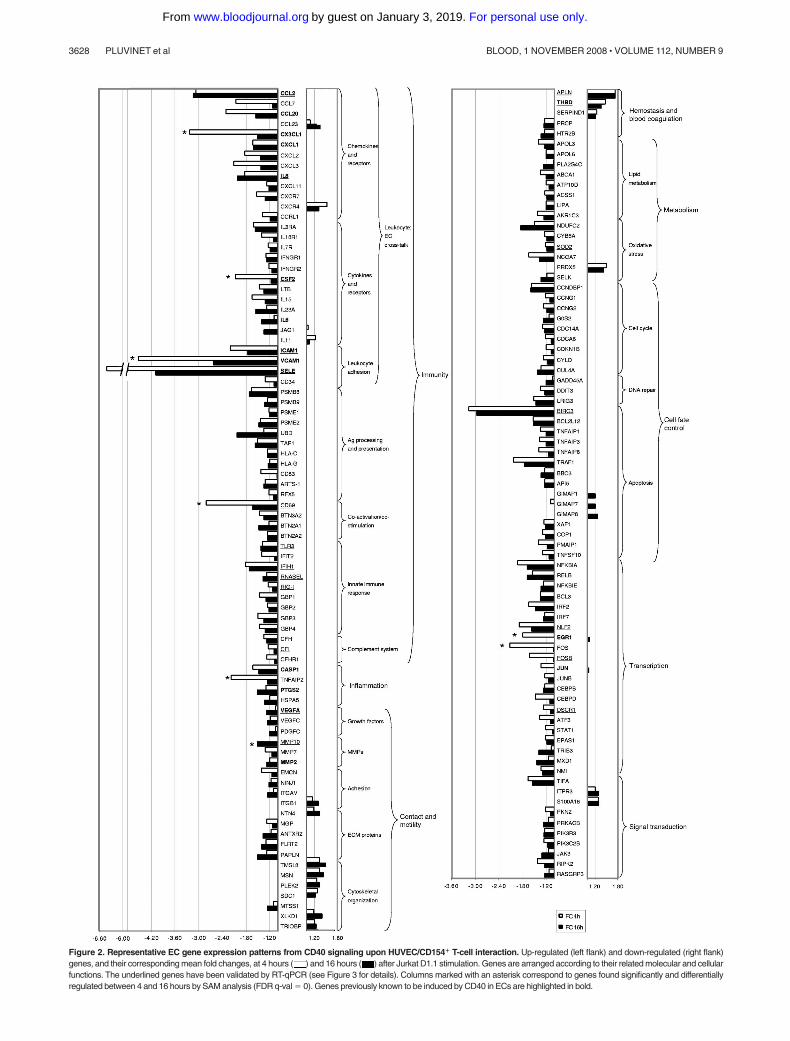
Figure 2. Representative EC gene expression patterns from CD40 signaling upon HUVEC/CD154� T-cell interaction. Up-regulated (left flank) and down-regulated (right flank)genes, and their corresponding mean fold changes, at 4 hours ( ) and 16 hours ( ) after Jurkat D1.1 stimulation. Genes are arranged according to their related molecular and cellularfunctions. The underlined genes have been validated by RT-qPCR (see Figure 3 for details). Columns marked with an asterisk correspond to genes found significantly and differentiallyregulated between 4 and 16 hours by SAM analysis (FDR q-val � 0). Genes previously known to be induced by CD40 in ECs are highlighted in bold.
3628 PLUVINET et al BLOOD, 1 NOVEMBER 2008 � VOLUME 112, NUMBER 9
For personal use only.on January 3, 2019. by guest www.bloodjournal.orgFrom

T cell–stimulated endothelial CD40 activates NF-�B and MAPKfamily transcription factors
To assess the functional implication of NF-�B and additional transcrip-tion factors in the early response to CD40-mediated EC induction, wemonitored their activity in nuclear extracts from Jurkat D1.1 (CD154�)–stimulated ECs pretreated with either siRNA-2 or msiRNA-2 at 4 hoursafter stimulation. Of 15 different transcription factors analyzed, signifi-cant differential DNA-binding activity could be monitored for theNF-�B family members NF-�B p50 and NF-�B p65, but not for c-Rel;for the AP-1 family members c-Fos, c-Jun, FosB, JunB, and JunD, butnot for Fra-1 and Fra-2; and for alternative MAPK-regulated factorssuch as ATF-2 and STAT-1, but not for CREB-1 (Figure 4). Thus,several important transcription factor gene families such as NF-�B,AP-1, and STAT operating simultaneously appear to be essentialmodulators of the early immune-inflammatory process in ECs throughthe CD40-CD154 dyad.
MAPK functional validation
Engagement of the CD40 receptor on ECs through specific TRAFadaptor molecules leads to the induction of different proinflamma-tory signaling cascades.5 In agreement with previous studies, majorsignal transduction pathways operating in ECs by CD154 stimula-tion include those of the mitogen-activated protein kinase (MAPK)subfamily. ERK1/2, JNK, and p38 are responsible for the phosphor-ylation of many of the active transcription factors identified in theprevious DNA-binding assays, including c-Fos, c-Jun, ATF-2, andSTAT-1. We wished to assess the activation status of thesepathways in ECs after coincubation for 4, 10, and 16 hours withJurkat D1.1 (CD154�) cells. Thus, upon Jurkat D1.1 stimulation,ECs presented induced phosphorylation of both ERK1/2 and JNKat 4 and 10 hours, whereas at 16 hours these kinases seemed to
return to basal phosphorylation levels. In contrast, p38 appeared tobe constitutively activated regardless of Jurkat D1.1 stimulation(Figure 5A). To confirm that MAPK induction was indeed CD40dependent, we assessed differential phosphorylation of ERK1/2,JNK, and p38 in protein extracts from either siRNA-2– ormsiRNA-2–pretreated ECs. RNAi-mediated silencing of CD40expression on treated cells paralleled inhibition of both total andphosphorylated ERK1/2 at 4 and 10 hours. JNK was also differen-tially phosphorylated, although total JNK remained constantthroughout the stimulation period. In contrast, no differentialregulation was apparent on either the total or the phosphorylatedforms of p38 MAPK (Figure 5B-C).
Figure 3. Validation of microarray results by RT-qPCR. (A) Relative mRNA levels corresponding torepresentative genes from msiRNA-2–treated ( )and siRNA-2–treated ( ) HUVECs stimulated with(CD154�) T cells for 4, 10, and 16 hours were assessedby real-time RT-qPCR using the housekeeping cyclophi-lin A gene (CYPA) as a control. Results are meanvalues plus or minus SEM from 2 independent experi-ments performed in triplicate. In those genes validatedat the 3 stimulation times, the comparative expressionkinetics between microarrays (f) and RT-qPCR (Œ) interms of mean fold change is shown. (B) Dot-plot of theexpression values in log2 ratio between microarrayhybridization (ordinate) and RT-qPCR (abscissa) fromall validated genes irrespective of the stimulation timeused. Linear regression demonstrates a significantcorrelation.
Figure 4. Transcription factor DNA-binding assays confirm that CD40-dependent EC activation by Jurkat D1.1 (CD154�) T cells is coordinatedthrough NF-�B, AP-1, and other MAPK-regulated activities. Nuclear extracts fromHUVECs transfected with siRNA-2 ( ) or msiRNA-2 ( ) and stimulated withJurkat D1.1 for 4 hours were analyzed through an ELISA-based assay (see “Nuclearextractions and transcription factor activity assays” for details). The histogramrepresents the relative DNA-binding levels of NF-�B (NF-�B p50, NF-�B p65, c-Rel),AP-1 (c-Jun, c-Fos, FosB, JunB, JunD, Fra-1, Fra-2), and several other MAPK-regulated (CREB-1, ATF-2, c-Myc, STAT-1, Mef-2) transcription factors. Data repre-sent the average plus or minus SEM from 3 different experiments (*P .05, vsmsiRNA-2).
ROLE OF CD40 IN ENDOTHELIAL CELL ACTIVATION 3629BLOOD, 1 NOVEMBER 2008 � VOLUME 112, NUMBER 9
For personal use only.on January 3, 2019. by guest www.bloodjournal.orgFrom

The CD40-CD154 dyad and other proinflammatory stimulimediate apelin down-regulation in endothelial cells
The most down-regulated gene as a consequence of CD40signaling through EC–T-lymphocyte interaction was the AGTRL1ligand gene APLN, coding for apelin (Figure 1). Among themultiple physiologic functions that are being attributed to thisbioactive peptide, it has been suggested to have an importantrole in the regulation of cardiovascular function and fluidhomeostasis.22 Further validation by RT-qPCR confirmed APLNdown-regulation across the 3 stimulation times (Figure 6B). TheAPLN gene includes 2 polyadenylation signals at the 3�UTRleading to 2 alternative transcripts of 2.7 and 3.1 kb (Figure 6A).Northern analysis using 2 discriminating probes showed identi-cal CD40-mediated down-regulation of both APLN mRNAspecies in ECs (Figure 6C). Interestingly, not only CD40-CD154–mediated cell-cell interactions, but also other proinflammatorysoluble factors, such as TNF� plus IFN�, were able to signifi-cantly down-regulate APLN transcript levels. Moreover, West-ern analysis revealed that the 77 amino acid prepropeptideapelin is repressed upon CD40 signaling in HUVECs stimulatedby Jurkat D1.1 (CD154�) T cells (Figure 6D). Conversely,arterial ECs from different sources (eg, HCtAECs [Figure 6E]and human coronary artery endothelial cells [HCAECs; notshown]) demonstrated also transcriptional down-regulation ofAPLN upon proinflammatory stimulation. Furthermore, in agree-ment with previous reports,23,24 we found strong presence ofapelin in ECs lining the luminal surface of normal-looking bigarteries (internal and external carotid arteries and coronaryarteries). Carotid arteries with type I and II atheroma lesionsalso presented significant staining for apelin (Figure 6F, ECtA).In contrast, carotids (Figure 6F, ICtA) and coronary arteries (notshown) with type III to VI atheromas had weak or absent
staining for apelin in the intraluminal endothelium. Thus, inconcordance with the decreased APLN expression observed inHCtAECs challenged by proinflammatory stimuli, there was aninverse correlation between the degree of inflammation in theneointima from carotid arteries, and the apelin-36 peptide levelswithin ECs lining these large conduit vessels.
In the etiology of kidney acute rejection, allorecognition isthe main factor responsible for immune-inflammatory responseactivation. Thus, we investigated apelin regulation in a renaltransplant model involving vascular dysfunction. We havepreviously shown that renal insufficiency in this model involvesstrong CD40 up-regulation and a marked increase of the Th1proinflammatory cytokines TNF� and IFN�.16,25 In addition, wefound significant transcriptional down-regulation of APLN, anda marked decrease of apelin immunoreactivity in the microvas-cular and tubular cells from the rejecting kidneys (Figure S3).Taken together, these data indicate that vascular activationinduced by proinflammatory states is associated with repressionof apelin expression.
CD40 engagement through interaction with activated (CD154�)T cells induces the viral innate immune surveillance system inendothelial cells
Our transcriptional profiling data suggested that T-lymphocyte–ECcross-talk through CD40 signaling alerts the EC viral innateimmune surveillance system by up-regulating key sensors of viralinfection: TLR3, recognizing extracellular dsRNA; the RNAhelicases IFIH1 (MDA5) and RIG-I, sensing cytoplasmic dsRNA;the OAS/RNASEL axis, which mediates RNA degradation; and theIRF7 transcription factor, a common downstream regulator of theantiviral immune response. Moreover, important components ofIFN� action capable of stimulating innate cell-mediated immunity,such as both IFN� receptor subunits (IFNGR1 and IFNGR2) andits signaling intermediate STAT1, were also found up-regulatedupon CD40 engagement (Figure 2).
Again, RT-qPCR was used to validate CD40-dependenttranscriptional up-regulation of the main arms of the EC innateresponse to dsRNA: TLR3, IFIH1, RIG-I, and RNASEL. Todiscriminate any potential sequence-dependent immunostimula-tory effects caused by the introduction of our siRNAs into theEC, independent of CD40 action, we normalized the expressionvalues obtained from Jurkat D1.1–stimulated HUVECs withrespect to unstimulated HUVECs, both subject to the samesiRNA treatment (Figure 7A).
Furthermore, we confirmed the functionality of the EC antiviralsurveillance system by demonstrating CD40-dependent up-regulation of2 downstream effectors of TLR3/MDA5/RIG-I signaling, IL8 andIFN�, following dsRNA analog challenging. We measured both cyto-kines by RT-qPCR in ECs upon poly (I:C) treatment, at 4 and 16 hoursafter Jurkat D1.1 (CD154�) stimulation (Figure 7B). IFN� was alsoup-regulated at the protein level (Figure 7C).
Toward a systematic integrated portrait of CD40 signaling inendothelial cells: gene set enrichment analysis
To gain additional insight into the signaling cascades and themolecular and cellular functions directly or indirectly influencedby CD40 in ECs as a consequence of their interaction withCD154-expressing T lymphocytes, we undertook a threshold-free gene set enrichment analysis (GSEA) of our RNAi-coupledtranscriptional profiling data. The analysis was performed foreach of the 3 stimulation times: 4, 10, and 16 hours. Moreover,
Figure 5. CD40-dependent EC activation by Jurkat D1.1 (CD154�) T cellsinduces MAPK and SAPK signaling pathways. (A) HUVECs were either leftuntreated (lane 1) or coincubated with Jurkat D1.1 (CD154�) cells for the indicatedtimes (lanes 2-4). Cell extracts (40 �g total protein/sample) were separated by 10%SDS-PAGE, subjected to Western blotting, and reacted with anti–phospho ERK1/2,anti–phospho JNK, or anti–phospho p38 antibodies. (B,C) HUVECs were transfectedwith siRNA-2 or msiRNA-2 and further stimulated with Jurkat D1.1 (CD154�) cells forthe indicated times. Western blots were performed as in panel A, and probed withanti-CD40 MoAb (B), or with antibodies detecting either total or phosphorylatedERK1/2, JNK, and p38 isoforms (C). The bottom bands of the panels indicate �-actinprotein levels used to normalize for equal loading of the gel lanes. All blots shown arerepresentative from 3 different experiments.
3630 PLUVINET et al BLOOD, 1 NOVEMBER 2008 � VOLUME 112, NUMBER 9
For personal use only.on January 3, 2019. by guest www.bloodjournal.orgFrom

to further investigate the dynamics of CD40-dependent ECactivation, we also mined for gene sets differentially regulatedbetween 4 and 16 hours of cell-associated CD154 stimulation.
Figure 8 displays a list of relevant, significantly regulated (FDRq-val 0.25) functional gene sets: (1) at 4 hours after stimulation,(2) at 16 hours after stimulation, and (3) comparing the response
Figure 6. The endogenous vasoactive peptide apelin is strongly down-regulated through CD154 and other proinflammatory stimuli in ECs. (A) Schematicrepresentation of the APLN full transcript (GenBank accession no. NM 017413) showing the coding sequence and the relative positions of the 2 polyadenylation siteswithin the 3� noncoding sequence of the gene. The cDNA fragments (F3-R3 probe and F4-R4 probe) used to distinguish the alternative transcripts are also shown.(B) Relative APLN mRNA levels from msiRNA-2–treated (black columns) and siRNA-2–treated (white columns) HUVECs stimulated with Jurkat D1.1 (CD154�) cells for 4, 10, and16 hours were assessed by real-time RT-qPCR using the housekeeping cyclophilin A gene (CYPA) as a control. Results are mean values plus or minus SEM from 2 independentexperiments performed in triplicate. (C) Northern blot analysis of total RNA isolated from HUVECs either unstimulated, or stimulated with Jurkat D1.1 (CD154�) cells or with TNF�(100 U/mL) plus IFN� (1000 U/mL) for 4 hours (left panels).Alternatively, HUVECs were treated with siRNA-2 or msiRNA-2 and further stimulated with Jurkat D1.1 (CD154�) T cells (rightpanels). All Northern blots were hybridized with the APLN-specific F3-R3 or F4-R4 probes as indicated, and a G3PDH probe was used for normalization. (D) At the protein level, cellextracts (40 �g of total protein/sample) from HUVECs either untransfected and unstimulated (lane 4), transfected with msiRNA-2, transfected with siRNA-2, or untransfected, and furtherstimulated for 4 hours with Jurkat D1.1 (CD154�) T cells (lanes 1, 2, and 3, respectively) were fractionated by 10% SDS-PAGE and probed with a 77–amino acid preproapelin-specificantibody.The housekeeping �-actin protein levels were assessed for lane normalization. (E) RelativeAPLN mRNAlevels from HCtAECs either unstimulated, or stimulated with Jurkat D1.1(CD154�) cells or with TNF� (100 U/mL) plus IFN� (1000 U/mL) for 4 hours, were assessed by real-time RT-qPCR using the housekeeping cyclophilinAgene (CYPA) as a control. Resultsare mean values plus or minus SEM from 2 independent experiments performed in triplicate. (F) Apelin-36 immunostaining in human carotid arteries. A representative section from thecarotid bifurcation showing the internal (ICtA) and external (ECtA) carotid arteries (central image: �20). In the ICtA with advanced atheroma (type V lesion) there was almost absentapelin-36 immunostaining in the intraluminal ECs (➞ ). In the normal-looking ECtA, strong positive intraluminal EC immunostaining for apelin-36 was evident (➞ ). Both ICtA and ECtApresented significant endothelial CD40 immunoreactivity (➤ ). Peripheral images, �400. The sections were computerized as color-encoded digitized images using a Nikon Eclipse 80imicroscope equipped with a CFI Plan Fluor 4�/0.13 NA and 40�/0.75 NA air objectives, a Nikon DS-2Mv camera, and the DS-U2 image processing unit and analyzing software NIKONNIS-Elements version 3.21 (all Nikon, Melville, NY).
ROLE OF CD40 IN ENDOTHELIAL CELL ACTIVATION 3631BLOOD, 1 NOVEMBER 2008 � VOLUME 112, NUMBER 9
For personal use only.on January 3, 2019. by guest www.bloodjournal.orgFrom

between 4 and 16 hours after CD40-mediated EC activation.Overall, these results confirm the important role of the CD40receptor as a mediator of EC activation upon interaction withactivated T lymphocytes (Document S1, “GSEA”).
Discussion
In the present study, we report a key role of CD40 signaling inT cell–dependent transcriptional activation of ECs. Moreover, wefurther dissect the molecular pathways defining the physiologicbehavior of endothelial CD40 in the immune-inflammatory processthrough genome-wide expression profiling at 3 different timepoints to obtain “snapshots” of the course of transcriptionalalterations. We demonstrate that the activated T lymphocyte is astrong enough stimulus to induce EC activation by direct interac-tion through the CD40-CD154 dyad.
Both activated platelets and activated CD4� T cells are themajor source of CD154. However, it has been suggested thatalthough CD154� platelets contribute significantly to the recruit-ment of inflammatory cells to damaged endothelium in vivo, due tothe short half-life of platelet CD154, the chronic CD154-driveninflammatory component can be sustained only by activated CD4�
T cells.26 Consequently, we undertook the study of the CD40-CD154 interaction using as experimental paradigm EC activated byT-lymphocyte coculture, to assess exclusively the relevance and
consequences of CD40 engagement in ECs. This has been achievedby combining RNAi-mediated CD40 knockdown9 and comparativewhole-genome microarrays. The gene silencing specificity ofoptimized siRNAs has been confirmed in several expressionprofiling studies,27,28 which allows confident investigation of genefunction on genome-wide scale.29
CD40 signaling is a complex process where different pathwaysexert overlapping independent control of gene expression modules,as already reported in B lymphocytes30-32 and dendritic cells.21 Ourresults corroborate the role of CD40 as an activator in ECs and infera substantial perturbation of the EC transcriptome as a consequenceof CD40 engagement. By gene ontology analysis, the main specificcategories overrepresented, inflammatory and immune response,support the validity of our data. To further explore the biologicmechanisms behind CD40-mediated EC activation we have usedGSEA.13 This knowledge-based high-content data interpretationtool has proven especially suitable to detect biologic processesaffected by networks of genes and not apparent at the level ofindividual gene analysis.
According to our microarray results, within the first 4 hours ofT cell–mediated, CD40-dependent activation of resting ECs, anengagement of the stress response program takes place in thesecells, leading to the activation of multiple transcriptional programs.Our HUVEC profiling study may not fully reflect the heterogeneityof microvascular and macrovascular ECs from distinct bodycompartments.33,34 Nevertheless, the achievement of an EC-activated state through CD40 resembles the condition ensued whenactivated immune cells, inflammatory cytokines, and other acute-phase reactants interact with the vascular bed. This turns out in aprothrombotic, proadhesive, and proinflammatory phenotype typi-cal of pathologic conditions related to the endothelium includingsepsis, solid organ rejection, and atherosclerosis.35 Indeed, signifi-cant transcriptional profile similarities between ECs activated byCD154� T cells and by inflammatory cytokines, particularlyTNF-� and IL-1,36-39 support the proinflammatory effects of theCD40-CD154 dyad in ECs.
CD40 signaling through TRAF effectors regulates key transcrip-tion factors involved in the immune-inflammatory process such asNF-�B, AP-1, and NFAT families.40 Accordingly, we report theearly induction of the NF-�B and AP-1 families, a common featureof EC activation through proinflammatory mediators. Our DNAbinding results confirm CD40-mediated activation of the majorproteins involved in the canonical pathway (p50, p65/RelA).Moreover, our microarray data also suggest engagement of thenoncanonical pathway (through Rel B up-regulation), and perhapsof alternative pathways, such as that leading to a transcriptionalmodule formed by a p50 homodimer interacting with the IkB-likecoactivator Bcl341 (also induced by CD40). Similarly, the extensiveearly induction of central AP-1 components (c-Fos, FosB, c-Jun,JunB, JunD, ATF-2, ATF-3) substantiates the importance of thesetranscription factors for CD40-mediated transcriptional up-regulation of a diverse range of genes involved in EC proliferation,differentiation, and response to cell damage. We also foundup-regulation of many other transcription factors, CD40 effectors,and regulators (Document S1, “CD40 signaling: pathway inductionand transcription factor activation”). This highlights the complex-ity of multiple signaling pathways that cooperate in CD40-regulated gene expression in ECs. In addition, we document for thefirst time a strong CD40-dependent induction of the novel NLFtranscription factor family (NLF1 and NLF2) in ECs, which under
Figure 7. CD40 engagement through CD154� T cells induces the antiviral innateimmune surveillance system in ECs. (A) Validation of CD40-dependent inductionof key antiviral innate immune response genes by RT-qPCR. Relative mRNA levelscorresponding to TLR3, IFIH1, RIG-I, and RNASEL from msiRNA-2–treated (f) andsiRNA-2–treated (�) HUVECs stimulated with (CD154�) T cells for 4 hours (IFIH1,RIG-I, RNASEL), or 4, 10, and 16 hours (TLR3), were assessed by real-timeRT-qPCR using CYPA as a control, and normalized from the corresponding valuesobtained from unstimulated, msiRNA-2–treated, or siRNA-2–treated HUVECs. Re-sults are mean values plus or minus SEM from 2 independent experiments performedin triplicate. (B) Functional validation of the antiviral surveillance pathways induced byCD40 signaling in HUVECs. msiRNA-2–treated (f) and siRNA-2–treated HUVECswere stimulated with Jurkat D1.1 (CD154�) T cells for 4 or 16 hours and eitheranalyzed or further challenged with poly (I:C) (25 �g/mL) for 6 hours and analyzed.Relative mRNA levels of the proinflammatory chemokine IL8 and the typeI IFN� were assessed by real-time RT-qPCR using the housekeeping CYPA as acontrol. Results are mean values plus or minus SEM from 3 independent experimentsperformed in triplicate. (C) The relative amount of synthesized IFN� was alsoevaluated at the protein level by immunoblotting using an anti-IFN� polyclonalantibody, in fractionated cell extracts from msiRNA-2–treated (lanes 2 and 4) andsiRNA-2–treated (lanes 1 and 3) HUVECs stimulated with Jurkat D1.1 (CD154�)T cells for 4 and 16 hours, and further challenged with poly (I:C) (25 �g/mL) for6 hours. �-actin protein levels were assessed for normalization. A representative blotfrom 3 independent experiments is shown.
3632 PLUVINET et al BLOOD, 1 NOVEMBER 2008 � VOLUME 112, NUMBER 9
For personal use only.on January 3, 2019. by guest www.bloodjournal.orgFrom

h61svh4h61h4teSeneGnoitcnuF
Rank NES FDR q-val
Rank NES FDR q-val
Rank NES FDR q-val
Signal Transduction
NFKB_INDUCED (see Figure S4) 1 2,23 0,018 2 2,07 0,014 13 1,79 0,044 NFKB_PATHWAY 6 2,02 0,020 30 1,69 0,032 P38MAPK_PATHWAY 36 1,67 0,060 56 1,45 0,118 CA_NF_AT_SIGNALLING 37 1,67 0,060 28 1,70 0,030 RELAPATHWAY 45 1,60 0,080 26 1,71 0,030 ST_JNK_MAPK_PATHWAY 48 1,59 0,079 52 1,46 0,115 MAPK_PATHWAY 53 1,56 0,090 ST_ERK1_ERK2_MAPK_PATHWAY 57 1,54 0,097 46 1,50 0,101 MAPK_CASCADE 58 1,54 0,095 AKT_PATHWAY 88 1,39 0,156 ST_P38_MAPK_PATHWAY 54 1,45 0,121
Transcription Factors
FETAL_LIVER_HS_ENRICHED_TF_JP 9 1,97 0,020 58 1,44 0,121 29 1,70 0,060 HUMAN_CD34_ENRICHED_TF_JP 16 1,83 0,029 17 1,76 0,049 HEMO_TF_LIST_JP 38 1,67 0,059 68 1,33 0,211 72 1,49 0,119 CR_TRANSCRIPTION_FACTORS 56 1,54 0,098 43 1,53 0,084
HOXA9 DOWNREG_BY_HOXA9 67 1,51 0,104 73 1,30 0,225 HOXA9_DOWN 72 1,48 0,117 31 1,67 0,035
Nuclear RAR_UP 42 1,63 0,072 receptors PPARA_PATHWAY 50 1,56 0,091
Stress and Inflammation
STRESS_PATHWAY 11 1,96 0,020 34 1,65 0,040 INFLAM_PATHWAY 31 1,72 0,047 25 1,71 0,029 INFLAMMATORY_RESPONSE_PATHWAY 57 1,45 0,116 EICOSANOID_SYNTHESIS 66 1,34 0,201 GPCRPATHWAY 40 1,65 0,062 55 1,56 0,092 SIG_CD40PATHWAYMAP 63 1,53 0,098 53 1,46 0,114 GPCRS_CLASS_B_SECRETIN-LIKE 86 1,40 0,153 PROTEASOME_PATHWAY 101 1,31 0,210
Immunity CR_IMMUNE_FUNCTION 7 1,98 0,021 71 1,31 0,214 49 1,58 0,089 CYTOKINE_PATHWAY 29 1,73 0,048 15 1,78 0,024 IL17PATHWAY 30 1,72 0,048 24 1,72 0,027 STEMPATHWAY 64 1,52 0,098 37 1,62 0,049 TH1TH2PATHWAY 19 1,76 0,025 91 1,38 0,165 IL6_PATHWAY 49 1,47 0,111
Contact and Motility
Adhesion CR_CAM (see Figure S4) 8 1,97 0,021 1 2,14 0,014 CELL_ADHESION_MOLECULE_ACTIVITY 34 1,71 0,049 42 1,55 0,075 64 1,53 0,106 CELL_ADHESION 90 1,38 0,164 32 1,67 0,036
Cytoskeleton ACTINYPATHWAY 25 1,71 0,064 MCALPAIN_PATHWAY 1 -1,98 0,095 75 1,49 0,117 INTEGRIN_PATHWAY 2 -1,92 0,075 ECM_PATHWAY 1 -1,86 0,176 4 -1,87 0,061 83 1,41 0,180 RASPATHWAY 7 -1,75 0,083 RHO_PATHWAY 13 -1,63 0,128 ST_INTEGRIN_SIGNALING_PATHWAY 86 1,38 0,209
Growth MET_PATHWAY 3 -1,92 0,059 14 1,78 0,047 Factors NGFPATHWAY 5 -1,77 0,084 7 1,87 0,025
EGFPATHWAY 9 -1,71 0,092 38 1,65 0,066 PDGFPATHWAY 15 -1,63 0,116 70 1,50 0,117 ERK5_PATHWAY 18 -1,60 0,129 76 1,49 0,116 IGF1PATHWAY 22 -1,51 0,208 VEGFPATHWAY 24 -1,51 0,200 TGF_BETA_SIGNALING_PATHWAY 65 1,52 0,099 18 1,75 0,052
Chemotaxis SIG_CHEMOTAXIS 30 -1,47 0,207 CXCR4PATHWAY 14 -1,63 0,121
Vesicle NDKDYNAMIN_PATHWAY 17 -1,60 0,130 71 1,50 0,117 transport CR_TRANSPORT_OF_VESICLES 26 -1,51 0,192 35 1,66 0,070
52 1,58 0,086
Figure 8. Representative gene sets induced upon CD40 signaling in ECs challenged by interaction with Jurkat D1.1 (CD154�) T cells. GSEA lists of selected inducedgene sets (see “Microarray data analysis”) arranged according to their related molecular and cellular functions. Comparison of misRNA-2–treated versus siRNA-2–treatedHUVEC transcriptional profiles at 4 hours and 16 hours after Jurkat D1.1 (CD154�) T-cell stimulation. A complementary list of CD40-dependent differentially regulated genesets between 4 hours and 16 hours (4 h vs 16 h) is also given. For each functional gene set, its corresponding normalized enrichment score (NES) and FDR q-val is shown.Gene sets are ranked according to their FDR q-val. Positive NES values indicate up-regulated gene sets; negative NES values indicate down-regulated gene sets.
ROLE OF CD40 IN ENDOTHELIAL CELL ACTIVATION 3633BLOOD, 1 NOVEMBER 2008 � VOLUME 112, NUMBER 9
For personal use only.on January 3, 2019. by guest www.bloodjournal.orgFrom

proinflammatory conditions has been suggested to play an impor-tant role regulating architecture and cell adhesion allowing in-creased vascular permeability.42
Our data imply the CD40-dependent induction of pathwaysregulating cell fate decisions (PI-3K/AKT, JAK/STAT), and theearly and transient activation of major MAPK pathways. Erk1/2and JNK have been functionally confirmed, analogously to thatdescribed for monocytes.43
All these events shape the transcriptional cascade elicited as aconsequence of CD40-mediated EC–T-cell cross-talk, affectingfundamental EC processes such as chemotaxis, adhesion, tissueremodeling, carbohydrate and lipid metabolism, oxidative stress,and angiogenesis (Document S1).
Conversely, the fundamental contribution of CD40 as T-cellcostimulatory molecule has been widely recognized.44 Moreover, ithas been recently suggested than ECs themselves play a far more
h61svh4h61h4teSeneGnoitcnuFRank NES FDR
q-val Rank NES FDR
q-val Rank NES FDR
q-val
Hemostasis and Blood coagulation
INTRINSICPATHWAY 51 1,58 0,087
Surveillance systems
IL1RPATHWAY 2 2,12 0,018 8 1,96 0,014 TOLL_PATHWAY 3 2,07 0,018 20 1,75 0,026 NTHIPATHWAY 19 1,81 0,034 33 1,66 0,036 IL12PATHWAY 21 1,78 0,037 TIDPATHWAY 33 1,71 0,049 DRUG_RESISTANCE_AND_METABOLISM 46 1,59 0,080 67 1,51 0,115 FCER1_PATHWAY 83 1,42 0,146 16 -1,62 0,113 11 1,81 0,033 FASPATHWAY 98 1,32 0,203
Cell fate control
EMT_UP 17 1,83 0,029 27 1,71 0,029
Cell cycle ATRBRCAPATHWAY 21 -1,57 0,142 CELL_CYCLE 27 -1,51 0,186 SA_G1_AND_S_PHASES 23 -1,51 0,202 94 1,33 0,242 G1PATHWAY 43 -1,40 0,242 22 1,73 0,064 G2PATHWAY 45 -1,39 0,246 58 1,55 0,099 CELL_CYCLE_CHECKPOINT 31 1,67 0,071 CELL_CYCLE_REGULATOR 47 1,59 0,084 CR_CELL_CYCLE 3 2,00 0,010
DNA repair ATM_PATHWAY 10 1,96 0,020 59 1,54 0,101 DNA_DAMAGE_SIGNALLING 62 1,38 0,166 CR_REPAIR 54 1,57 0,088
Apoptosis TNF_AND_FAS_NETWORK 15 1,84 0,027 5 2,00 0,014 ST_TUMOR_NECROSIS_FACTOR_PATHWAY 18 1,82 0,032 6 1,98 0,014 DEATHPATHWAY 39 1,66 0,062 9 1,93 0,016 MITOCHONDRIA_PATHWAY 43 1,61 0,078 11 1,90 0,017 CASPASE_PATHWAY 61 1,53 0,096 12 1,84 0,022 SA_CASPASE_CASCADE 81 1,44 0,131 3 2,06 0,014
Metabolism
Carbohydrate GLUCOSE_DOWN 89 1,39 0,157 6 1,88 0,026 GLUCOSE_UP 45 1,51 0,098 GLYCOGEN_METABOLISM 62 1,54 0,102 INSULIN_PATHWAY 80 1,45 0,130 35 -1,45 0,208 INSULIN_2F_UP 105 1,29 0,229
Lipid MAP00650_BUTANOATE_METABOLISM 46 1,60 0,075 FATTY_ACID_METABOLISM 60 1,53 0,097 MAP00120_BILE_ACID_BIOSYNTHESIS 95 1,34 0,199 81 1,42 0,172 FATTY_ACID_SYNTHESIS 19 -1,58 0,139 MAP00071_FATTY_ACID_METABOLISM 40 1,63 0,067
Amino acid LEU_UP 38 1,61 0,050 GLUT_UP 39 1,59 0,060 MAP00260_GLYCINE_SERINE_AND_THREONINE_METABOLISM 75 1,29 0,231 MAP00251_GLUTAMATE_METABOLISM 29 -1,48 0,201 MAP00410_BETA_ALANINE_METABOLISM 19 1,75 0,049 MAP00280_VALINE_LEUCINE_AND_ISOLEUCINE_DEGRADATION 2 2,04 0,010 MAP00640_PROPANOATE_METABOLISM (see Figure S4) 1 2,06 0,010 MAP00340_HISTIDINE_METABOLISM 12 1,81 0,035
Krebs cycle TCA 43 1,61 0,073 MAP00620_PYRUVATE_METABOLISM 49 1,58 0,080 10 1,85 0,021
Nucleic acids MAP00240_PYRIMIDINE_METABOLISM 28 -1,49 0,197 34 1,66 0,071 MAP00230_PURINE_METABOLISM 68 1,50 0,118
Figure 8 (continued)
3634 PLUVINET et al BLOOD, 1 NOVEMBER 2008 � VOLUME 112, NUMBER 9
For personal use only.on January 3, 2019. by guest www.bloodjournal.orgFrom

direct role in immunity.45 Our results indicate that CD40 signalingin ECs leads to transcriptional induction of several immunity-related molecules and actively contributes to the transformation ofECs to antigen-presenting cells (Document S1, “Immunity”). Thus,CD40-mediated leukocyte–endothelial cell interactions appear toinfluence immune cell function through antigen presentation anddirectly affect adaptive immunity, inducing transendothelial migra-tion and/or polarization of different T-cell subsets.
In addition, several important components of the innate immunesystem were found induced in ECs after CD40-mediated EC–T-lymphocyte interaction (Figure 2; Document S1, “Immunity”).Remarkably, as a part of the EC stress response, we show that oneof the “alert” mechanisms most relevantly induced upon CD40signaling was the viral immune surveillance system. Thus, TLR3,recognizing the viral replication intermediate dsRNA insideintracellular organelles such as endosomes, was one of the mosthighly up-regulated pattern recognition receptors (PRRs) found inECs (implied by functional annotation and GSEA, and confirmedby RT-qPCR). Moreover, additional PRRs, such as the cytosolicCARD-helicases RIG-I and MDA546,47 were also found signifi-cantly induced upon EC CD40 engagement (also validated byRT-qPCR), along with common downstream effectors such asseveral interferon regulatory factors (IRFs)48 (Figure 2). Weverified the functionality of this induced viral recognition systemby challenging CD40-activated ECs with the synthetic dsRNAanalog poly (I:C), which, upon activation of their downstreamsignaling cascades, led to transcriptional up-regulation and produc-tion of IFN�, and NF-�B– and AP-1–dependent proinflammatorycytokines (IL8).46
Moreover, CD40 action also induced the nonredundant type II(immune) IFN system in ECs by up-regulating both IFN� receptorsubunits and the regulatory STAT1 transcription factor49 (Figure 2),adapting the endothelium to the paracrine antiviral and antimicro-bial IFN� action from the surrounding immune cells.
Other IFN-inducible antiviral pathways that we found transcrip-tionally induced in ECs upon CD40 engagement include thedsRNA-dependent, 2�-5� oligoadenylate synthetase/RNase L sys-tem mediating RNA degradation,50 and several guanylate-bindingproteins (GBP1-4).51 The global induction of the antiviral sensingnetwork in ECs when interacting with activated T lymphocyteshighlights the important contribution of CD40 preserving thevasculature from the harmful consequences and the spread ofsystemic viral infections in the host (Figure S5). Relatedly,repression of genes encoding receptors involved in viral entry, suchas FAM89B and CXCR4, a chemokine coreceptor of HIV, was alsoevident. Taken together, these results highlight the key role ofCD40 in instructing the immune surveillance systems in ECsactivated through T-lymphocyte interaction.
Thus, endothelial CD40 appears to play a fundamental roleregulating both the adaptive and the innate immunity. Conse-quently, CD40-mediated induction of the viral immune surveil-lance system might also participate in the mechanisms underlyingautoimmune disorders that are often associated with overproduc-tion of type I IFNs52,53 and, perhaps, other chronic inflammatoryprocesses.54
Furthermore, upon activation, ECs transform to a vasopressive,procoagulant state characteristic of the inflammatory process. Ourresults indicate that CD40 also influences such processes byregulating expression of genes involved in maintenance of vascularhomeostasis, in the control of blood pressure, and in thrombinbinding and inhibition (Document S1, “Hemostasis and bloodcoagulation”).
Interestingly, our study infers that endothelial APLN, whichis expressed predominantly in normal endothelia,55,56 wassignificantly down-regulated not only through the CD40-CD154dyad, but also by other proinflammatory stimuli. This previouslyunrecognized feature of EC activation was functionally vali-dated both in vitro and in vivo. Atherosclerotic plaques areinfiltrated by activated macrophages, T and B lymphocytes,plasma cells, and mast cells releasing inflammatory molecules,which amplify the severity of the disease. Moreover, CD40-CD154 has proven a crucial mediator not only in the initialevents of atherogenesis but also during the evolution ofestablished atheroma.57 Thus, we found an inverse correlationbetween the pathologic degree of stenotic plaque from humancarotid arteries and the level of their endothelial apelin expres-sion revealed by immunohistochemistry, substantiating therelevance of this vasoactive peptide in vascular immune-inflammatory processes. Several studies have indicated thatapelin is an arterial and venous dilator through a NOS-dependent mechanism, causing NO release from ECs.58,59 Arecent report highlights the complex and fine regulation of renalhemodynamic functions exerted by apelin through actions onpreglomerular and postglomerular microvasculature and alsoprobably on tubular functions.60 Accordingly, our results sug-gest that endothelial apelin down-regulation might also consti-tute a major factor in the hypertension and deregulation of bodyfluid homeostasis at the peripheral level associated with renaltransplant rejection.
Given the central role of ECs in the immune-inflammatoryprocess and the prominent position of CD40 regulating severalmediators and signaling pathways that contribute to immunityand inflammation, it seems reasonable to consider the CD40-CD154 dyad as a relevant therapeutic target for anti-inflamma-tory action. Indeed, preclinical assessment of the efficacy ofblocking the CD40-CD154 interaction is either ongoing inseveral autoimmune disease models, such as multiple sclerosis61
and lupus erythematosus,62 or is being considered for cardiovas-cular disease63,64 and to achieve immunologic tolerance intransplantation.65 Our comparative transcriptional profiling studyhighlights the specificity and efficiency of the siRNA anti-CD40modulating CD40 expression and function in ECs, whichsupports its anti-inflammatory potential.9 Nevertheless, ongoingwork using animal models of immune-inflammatory diseaseswith established CD40 relevance will prove the overall therapeu-tic effectiveness of this RNAi-based reagent. Moreover, thedetailed knowledge of the molecular pathways affected byCD40 action in ECs should contribute to dissect potentialtargets downstream of CD40 signaling relevant to immune-inflammatory processes.
Acknowledgments
We thank Juanjo Lozano and David Otero from the MicroarrayCore Facility (CRG-UPF), and Nuria Bolanos from the Laboratoryof Nephrology (CSUB-IDIBELL) for their expert technicalassistance.
This work was supported entirely by the Ministerio de Sanidady Consumo (Madrid, Spain) through grants 03/0516 and 05/1018from the Fondo de Investigaciones Sanitarias (FIS), from the Redde Centros del Instituto de Salud Carlos III (Refs C03/03 andC03/07), and by an ISCIII fellowship to R.P. (BF02/9166). I.H.-F.
ROLE OF CD40 IN ENDOTHELIAL CELL ACTIVATION 3635BLOOD, 1 NOVEMBER 2008 � VOLUME 112, NUMBER 9
For personal use only.on January 3, 2019. by guest www.bloodjournal.orgFrom

was supported by FIS/ISCIII. J.M.A. is sponsored by the Research-ers Stabilization Program from the National Health System (SNS).
Authorship
Contribution: R.P., R.O., J.K., I.H.-F., and A.L. performed the research;J.T., J.M.C., and J.M.G. contributed essential reagents and providedgeneral support; R.P. and L.S. performed bioinformatic analyses; R.P.
analyzed the data and wrote the paper; and J.M.A. designed the research,analyzed the data, and wrote the paper.
Conflict-of-interest disclosure: The authors declare no compet-ing financial interests.
Correspondence: Josep M. Aran, Medical and Molecular Genet-ics Center, Institut de Investigacio Biomedica de Bellvitge (IDI-BELL), Hospital Duran i Reynals, Gran Via s/n km 2.7, 08907L’Hospitalet de Llobregat, Barcelona, Spain; e-mail: [email protected].
References
1. Schonbeck U, Libby P. The CD40/CD154 recep-tor/ligand dyad. Cell Mol Life Sci. 2001;58:4-43.
2. Banchereau J, Bazan F, Blanchard D, et al. TheCD40 antigen and its ligand. Annu Rev Immunol.1994;12:881-922.
3. van Kooten C, Banchereau J. CD40-CD40 ligand.J Leukoc Biol. 2000;67:2-17.
4. Pullen SS, Dang TT, Crute JJ, Kehry MR. CD40signaling through tumor necrosis factor receptor-associated factors (TRAFs): binding site specific-ity and activation of downstream pathways bydistinct TRAFs. J Biol Chem. 1999;274:14246-14254.
5. Schonbeck U, Libby P. CD40 signaling andplaque instability. Circ Res. 2001;89:1092-1103.
6. Chess L. Blockade of the CD40L/CD40 pathway.In: Austen KF, Burakoff SJ, Rosen FS, Strom TB,eds. Therapeutic Immunology. 2nd ed. 2001:441-456.
7. Boumpas DT, Furie R, Manzi S, et al. A shortcourse of BG9588 (anti-CD40 ligand antibody)improves serologic activity and decreases hema-turia in patients with proliferative lupus glomerulo-nephritis. Arthritis Rheum. 2003;48:719-727.
8. Kawai T, Andrews D, Colvin RB, Sachs DH,Cosimi AB. Thromboembolic complications aftertreatment with monoclonal antibody againstCD40 ligand [letter]. Nat Med. 2000;6:114.
9. Pluvinet R, Petriz J, Torras J, et al. RNAi-mediated silencing of CD40 prevents leukocyteadhesion on CD154-activated endothelial cells.Blood. 2004;104:3642-3646.
10. Lederman S, Yellin MJ, Krichevsky A, Belko J,Lee JJ, Chess L. Identification of a novel surfaceprotein on activated CD4� T cells that inducescontact-dependent B cell differentiation (help). JExp Med. 1992;175:1091-1101.
11. National Center for Biotechnology Information.Gene Expression Omnibus (GEO). http://www.ncbi.nlm.nih.gov/geo. Accessed February 21,2008.
12. Saeed AI, Sharov V, White J, et al. TM4: a free,open-source system for microarray data manage-ment and analysis. Biotechniques. 2003;34:374-378.
13. Subramanian A, Tamayo P, Mootha VK, et al.Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wideexpression profiles. Proc Natl Acad Sci U S A.2005;102:15545-15550.
14. Massachusetts Institute of Technology. MolecularSignatures Database (MSigDB). http://www.broad.mit.edu/gsea/msigdb/index.jsp. AccessedOctober 3, 2006.
15. Stary HC, Chandler AB, Dinsmore RE, et al. Adefinition of advanced types of atheroscleroticlesions and a histological classification of athero-sclerosis: a report from the Committee on Vascu-lar Lesions of the Council on Arteriosclerosis,American Heart Association. Circulation. 1995;92:1355-1374.
16. Herrero-Fresneda I, Franquesa M, Torras J, et al.Role of cold ischemia in acute rejection: charac-terization of a humoral-like acute rejection in ex-perimental renal transplantation. Transplant Proc.2005;37:3712-3715.
17. Phipps RP. Atherosclerosis: the emerging roleof inflammation and the CD40-CD40 ligand sys-tem. Proc Natl Acad Sci U S A. 2000;97:6930-6932.
18. National Center for Biotechnology Information.UniGene. http://www.ncbi.nlm.nih.gov/sites/entrez?db�unigene. Accessed June 11, 2007.
19. National Center for Biotechnology Information.GenBank. http://www.ncbi.nlm.nih.gov/Genbank.Accessed June 11, 2007.
20. Barrans JD, Allen PD, Stamatiou D, Dzau VJ,Liew CC. Global gene expression profiling of end-stage dilated cardiomyopathy using a human car-diovascular-based cDNA microarray. Am JPathol. 2002;160:2035-2043.
21. Tureci O, Bian H, Nestle FO, et al. Cascades oftranscriptional induction during dendritic cellmaturation revealed by genome-wide expressionanalysis. FASEB J. 2003;17:836-847.
22. Kleinz MJ, Davenport AP. Emerging roles of ape-lin in biology and medicine. Pharmacol Ther.2005;107:198-211.
23. Lee DK, Cheng R, Nguyen T, et al. Characteriza-tion of apelin, the ligand for the APJ receptor.J Neurochem. 2000;74:34-41.
24. Medhurst AD, Jennings CA, Robbins MJ, et al.Pharmacological and immunohistochemical char-acterization of the APJ receptor and its endoge-nous ligand apelin. J Neurochem. 2003;84:1162-1172.
25. Herrero-Fresneda I, Gulías O, Franquesa M, etal. Local gene therapy with anti-CD40 siRNA plusrapamycin prolongs survival and decreases post-transplant renal acute rejection [abstract]. TransplInt. 2007;20:s2. Abstract O230.
26. Buchner K, Henn V, Grafe M, de Boer OJ, BeckerAE, Kroczek RA. CD40 ligand is selectively ex-pressed on CD4� T cells and platelets: implica-tions for CD40-CD40L signalling in atherosclero-sis. J Pathol. 2003;201:288-295.
27. Chi JT, Chang HY, Wang NN, Chang DS, DunphyN, Brown PO. Genomewide view of gene silenc-ing by small interfering RNAs. Proc Natl Acad SciU S A. 2003;100:6343-6346.
28. Semizarov D, Frost L, Sarthy A, Kroeger P,Halbert DN, Fesik SW. Specificity of short inter-fering RNA determined through gene expressionsignatures. Proc Natl Acad Sci U S A. 2003;100:6347-6352.
29. Semizarov D, Kroeger P, Fesik S. siRNA-mediated gene silencing: a global genome view.Nucleic Acids Res. 2004;32:3836-3845.
30. Dadgostar H, Zarnegar B, Hoffmann A, et al.Cooperation of multiple signaling pathways inCD40-regulated gene expression in B lympho-cytes. Proc Natl Acad Sci U S A. 2002;99:1497-1502.
31. Gricks CS, Zahrieh D, Zauls AJ, et al. Differentialregulation of gene expression following CD40activation of leukemic compared to healthy Bcells. Blood. 2004;104:4002-4009.
32. Basso K, Klein U, Niu H, et al. Tracking CD40 sig-naling during germinal center development.Blood. 2004;104:4088-4096.
33. Lang I, Pabst MA, Hiden U, et al. Heterogeneity
of microvascular endothelial cells isolated fromhuman term placenta and macrovascular umbili-cal vein endothelial cells. Eur J Cell Biol. 2003;82:163-173.
34. Ribatti D, Nico B, Vacca A, Roncali L, DammaccoF. Endothelial cell heterogeneity and organ speci-ficity. J Hematother Stem Cell Res. 2002;11:81-90.
35. Hansson GK. Inflammation, atherosclerosis, andcoronary artery disease. N Engl J Med. 2005;352:1685-1695.
36. Horrevoets AJ, Fontijn RD, van Zonneveld AJ, deVries CJ, ten Cate JW, Pannekoek H. Vascularendothelial genes that are responsive to tumornecrosis factor-alpha in vitro are expressed inatherosclerotic lesions, including inhibitor of apo-ptosis protein-1, stannin, and two novel genes.Blood. 1999;93:3418-3431.
37. Zhou J, Jin Y, Gao Y, et al. Genomic-scale analy-sis of gene expression profiles in TNF-alphatreated human umbilical vein endothelial cells.Inflamm Res. 2002;51:332-341.
38. Viemann D, Goebeler M, Schmid S, et al. Tran-scriptional profiling of IKK2/NF-kappa B- and p38MAP kinase-dependent gene expression in TNF-alpha-stimulated primary human endothelial cells.Blood. 2004;103:3365-3373.
39. Sana TR, Janatpour MJ, Sathe M, McEvoy LM,McClanahan TK. Microarray analysis of primaryendothelial cells challenged with different inflam-matory and immune cytokines. Cytokine. 2005;29:256-269.
40. Francis DA, Karras JG, Ke XY, Sen R, RothsteinTL. Induction of the transcription factors NF-kappa B, AP-1 and NF-AT during B cell stimula-tion through the CD40 receptor. Int Immunol.1995;7:151-161.
41. Bates PW, Miyamoto S. Expanded nuclear rolesfor IkappaBs. Sci STKE. 2004 Oct 12;2004(254):pe48.
42. Warton K, Foster NC, Gold WA, Stanley KK.A novel gene family induced by acute inflamma-tion in endothelial cells. Gene. 2004;342:85-95.
43. Pearson LL, Castle BE, Kehry MR. CD40-mediated signaling in monocytic cells: up-regulation of tumor necrosis factor receptor-associated factor mRNAs and activation ofmitogen-activated protein kinase signaling path-ways. Int Immunol. 2001;13:273-283.
44. Grewal IS, Flavell RA. CD40 and CD154 in cell-mediated immunity. Annu Rev Immunol. 1998;16:111-135.
45. Marelli-Berg FM, Jarmin SJ. Antigen presentationby the endothelium: a green light for antigen-specific T cell trafficking? Immunol Lett. 2004;93:109-113.
46. Meylan E, Tschopp J. Toll-like receptors and RNAhelicases: two parallel ways to trigger antiviralresponses. Mol Cell. 2006;22:561-569.
47. Kawai T, Akira S. Innate immune recognition ofviral infection. Nat Immunol. 2006;7:131-137.
48. Honda K, Takaoka A, Taniguchi T. Type I inter-feron [corrected] gene induction by the interferonregulatory factor family of transcription factors.Immunity. 2006;25:349-360.
3636 PLUVINET et al BLOOD, 1 NOVEMBER 2008 � VOLUME 112, NUMBER 9
For personal use only.on January 3, 2019. by guest www.bloodjournal.orgFrom

49. Samuel CE. Antiviral actions of interferons. ClinMicrobiol Rev. 2001;14:778-809.
50. Silverman RH. Viral encounters with 2�,5�-oligoadenylate synthetase and RNase L duringthe interferon antiviral response. J Virol. 2007;81:12720-12729.
51. Degrandi D, Konermann C, Beuter-Gunia C, et al.Extensive characterization of IFN-inducedGTPases mGBP1 to mGBP10 involved in hostdefense. J Immunol. 2007;179:7729-7740.
52. Theofilopoulos AN, Baccala R, Beutler B, KonoDH. Type I interferons (alpha/beta) in immunityand autoimmunity. Annu Rev Immunol. 2005;23:307-336.
53. Baccala R, Hoebe K, Kono DH, Beutler B,Theofilopoulos AN. TLR-dependent and TLR-in-dependent pathways of type I interferon inductionin systemic autoimmunity. Nat Med. 2007;13:543-551.
54. Andreakos E, Foxwell B, Feldmann M. Is target-ing Toll-like receptors and their signaling pathwaya useful therapeutic approach to modulating cyto-kine-driven inflammation? Immunol Rev. 2004;202:250-265.
55. Kleinz MJ, Davenport AP. Immunocytochemicallocalization of the endogenous vasoactive pep-tide apelin to human vascular and endocardialendothelial cells. Regul Pept. 2004;118:119-125.
56. Kleinz MJ, Skepper JN, Davenport AP. Immuno-cytochemical localisation of the apelin receptor,APJ, to human cardiomyocytes, vascular smoothmuscle and endothelial cells. Regul Pept. 2005;126:233-240.
57. Schonbeck U, Sukhova GK, Shimizu K, Mach F,Libby P. Inhibition of CD40 signaling limits evo-lution of established atherosclerosis in mice.Proc Natl Acad Sci U S A. 2000;97:7458-7463.
58. Ishida J, Hashimoto T, Hashimoto Y, et al. Regu-latory roles for APJ, a seven-transmembrane re-ceptor related to angiotensin-type 1 receptor inblood pressure in vivo. J Biol Chem. 2004;279:26274-26279.
59. Tatemoto K, Takayama K, Zou MX, et al. Thenovel peptide apelin lowers blood pressure via anitric oxide-dependent mechanism. Regul Pept.2001;99:87-92.
60. Hus-Citharel A, Bouby N, Frugiere A, Bodineau L,Gasc JM, Llorens-Cortes C. Effect of apelin onglomerular hemodynamic function in the rat kid-ney. Kidney Int. 2008;74:486-494.
61. ’t Hart BA, Hintzen RQ, Laman JD. Preclinicalassessment of therapeutic antibodies against hu-man CD40 and human interleukin-12/23p40 in anonhuman primate model of multiple sclerosis.Neurodegener Dis. 2008;5:38-52.
62. Haubitz M. Exploring new territory: the move to-wards individualised treatment. Lupus. 2007;16:227-231.
63. Lutgens E, Lievens D, Beckers L, Donners M,Daemen M. CD40 and its ligand in atherosclero-sis. Trends Cardiovasc Med. 2007;17:118-123.
64. Vishnevetsky D, Kiyanista VA, Gandhi PJ. CD40ligand: a novel target in the fight against cardio-vascular disease. Ann Pharmacother. 2004;38:1500-1508.
65. Burkhart C, Heusser C, Morris RE, et al. Pharma-codynamics in the development of new immuno-suppressive drugs. Ther Drug Monit. 2004;26:588-592.
ROLE OF CD40 IN ENDOTHELIAL CELL ACTIVATION 3637BLOOD, 1 NOVEMBER 2008 � VOLUME 112, NUMBER 9
For personal use only.on January 3, 2019. by guest www.bloodjournal.orgFrom

online July 31, 2008 originally publisheddoi:10.1182/blood-2008-03-143305
2008 112: 3624-3637
Torras, Josep M. Cruzado, Josep M. Grinyó, Lauro Sumoy and Josep M. AranRaquel Pluvinet, Rut Olivar, Jerzy Krupinski, Inmaculada Herrero-Fresneda, Anna Luque, Joan uncovered by RNAi-coupled transcriptional profilingCD40: an upstream master switch for endothelial cell activation
http://www.bloodjournal.org/content/112/9/3624.full.htmlUpdated information and services can be found at:
(2485 articles)Hemostasis, Thrombosis, and Vascular Biology Articles on similar topics can be found in the following Blood collections
http://www.bloodjournal.org/site/misc/rights.xhtml#repub_requestsInformation about reproducing this article in parts or in its entirety may be found online at:
http://www.bloodjournal.org/site/misc/rights.xhtml#reprintsInformation about ordering reprints may be found online at:
http://www.bloodjournal.org/site/subscriptions/index.xhtmlInformation about subscriptions and ASH membership may be found online at:
Copyright 2011 by The American Society of Hematology; all rights reserved.of Hematology, 2021 L St, NW, Suite 900, Washington DC 20036.Blood (print ISSN 0006-4971, online ISSN 1528-0020), is published weekly by the American Society
For personal use only.on January 3, 2019. by guest www.bloodjournal.orgFrom