bai giang cnsh ung dung
TRANSCRIPT

TRƯỜNG ĐẠI HỌC NÔNG LÂM HUẾ
DỰ ÁN HỢP TÁC VIỆT NAM – HÀ LAN
BÀI GIẢNG
CÔNG NGHỆ SINH HỌC ỨNG DỤNG
Người biên soạn: TS. Trần Thị Lệ
Huế, 08/2009

5
BÀI I CÔNG NGHỆ DNA TÁI TỔ HỢP
I. Khái niệm 1. Ý nghĩa của tạo dòng DNA
Trước đây khoảng hơn một trăm năm Gregor Mendel đã đưa ra những định luật cho phép giải thích sự di truyền những đặc điểm sinh học. Cơ sở của những định luật này là mỗi một đặc điểm di truyền của sinh vật được điều khiển bởi 1 yếu tố, được gọi là gene, tồn tại ở đâu đó trong tế bào. Sự khám phá lại những định luật này của Mendel vào năm 1900 là sự khai sinh của di truyền học, ngành khoa học này có mục đich là hiểu bản chất của gene và giải thích những tác động của nó.
Công nghệ DNA tái tổ hợp là một tập hợp các kỹ thuật phân tử để định vị, phân lập, biến đổi và nghiên cứu các đoạn DNA. Thuật ngữ tái tổ hợp được dùng thường xuyên do mục tiêu của nó là phối hợp DNA từ hai nguồn xa nhau. Ví dụ: các gene từ hai nguồn vi khuẩn khác nhau có thể được liên kết lại, hoặc một gene người có thể được đưa vào nhiễm sắc thể vi khuẩn. Công nghệ DNA tái tổ hợp (thường được gọi là công nghệ di truyền) hiện nay bao gồm một mạng lưới các kỹ thuật phân tử được dùng để phân tích, biến đổi và tái tổ hợp hầu như mọi trình tự DNA. 2. Tạo dòng DNA là gì? Về nguyên tắc một thí nghiệm tạo dòng DNA gồm những bước cơ bản sau đây:
1. Tinh sạch DNA 2. Cắt phân tử DNA 3. Xác định độ lớn của các đoạn DNA 4. Đưa các đoạn DNA vào vectơ tạo DNA tái tổ hợp 5. Đưa DNA tái tổ hợp vào tế bào chủ 6. Xác định những tế bào chủ chứa phân tử tái tổ hợp DNA
3. Tại sao sự tạo dòng DNA quan trọng như vậy ? Ý nghĩa của tạo dòng đối với nghiên cứu và công nghệ sinh học
Từ hình 1.1 ta nhận thấy việc tạo dòng gene là một quá trình tương đối đơn giản. Tại sao phương pháp này trong sinh học lại có ý nghĩa lớn như vậy ? Câu trả lời là: Trước hết việc tạo dòng gene là tách một gene ra khỏi tất cả những gene khác ở dạng tinh khiết, mà những gene này thường tồn tại với nhau trong một tế bào.
Sẽ hiểu chính xác hơn khi quan sát thí nghiệm tạo dòng ở hình 1.2. Đoạn DNA được tạo dòng có thể thuộc một hỗn hợp gồm nhiều đoạn khác nhau, mà mỗi đoạn mang một gene hoặc một phần của gene. Hỗn hợp này có thể là toàn bộ hệ gene của một sinh vật (ví dụ hệ gene của người). Sau đó mỗi một đoạn được đưa vào một phân tử vector phù hợp và tạo nên một tập hợp các phân tử DNA tái tổ hợp, trong đó một phân tử mang gene cần tìm. Thông thường mỗi tế bào chủ chỉ tiếp nhận một phân tử DNA tái tổ hợp duy nhất. Như vậy, khi một gene nào đó được tách ra từ nhiều gene khác thì người ta có thể nghiên cứu đặc điểm của nó chính xác hơn.
Quyết định sự thành công của thí nghiệm tạo dòng là liệu người ta có phân biệt được dòng quan tâm từ nhiều dòng khác nhau không? Ví dụ khi quan sát hệ

6
gene của vi khuẩn E.coli có khoảng 2000 gene, có thể tìm ra một gene trong điều kiện có nhiều dòng không (Hình 1.3)?
Kỹ thuật gene đã mở ra một loạt cơ hội mà trước đây không thể có được. Vấn đề cơ bản là các gene có kích thước quá nhỏ và có hàng ngàn gene ở trong mỗi tế bào. Thậm chí khi quan sát trên kính hiển vi mạnh nhất, thì DNA xuất hiện như là một sợi dây bé xíu, không thể thấy các nucleotide và không có một dấu hiệu nào để nhận biết chỗ bắt đầu và kết thúc của một gene.
Hình 1.1. Các bước cơ bản của tạo dòng DNA
1. Thiết kế phân tử DNA tái tổ hợp
Vector Đoạn DNA ngoại lai
Phân tử DNA tái tổ hợp
2. Đưa vào tế bào chủ Vi khuẩn
3. Nhân phân tử DNA tái tổ hợp
Vi khuẩn chứa phân tử DNA tái tổ hợp
4. Nhân tế bào chủ
5. Xuất hiện một dòng qua nhiều lần phân chia tế bào
Các dòng vi khuẩn phát triển trên môi trường agar

7
Hình 1.2. Bằng việc tạo dòng người ta tạo ra dạng đồng nhất chứa các đoạn DNA khác nhau
Vector
Các đoạn DNA
Các phân tử DNA tái tổ hợp
Mỗi phân tử chứa một đoạn DNA khác nhau
Đưa vào vi khuẩn
Nuôi trên môi trường agar
Mỗi một dòng chứa một phân tử DNA tái tổ hợp khác nhau với nhiều bản sao

8
Để minh họa vấn đề này chúng ta hãy xem xét một ví dụ đặc trưng về di truyền phân tử như sau: Giả thiết chúng ta muốn phân lập một gene của người và đưa nó vào vi khuẩn để sản xuất một lượng lớn các protein người. Vấn đề đầu tiên là phải tìm được gene mong muốn. Geneome đơn bội của người chứa khoảng 3,3 tỷ cặp base. Giả sử gene mà chúng ta muốn phân lập dài 3.000 bp. Như vậy gene đích của chúng ta chỉ chiếm một phần triệu của geneome, vì thế để tìm kiếm gene trong một hệ gene đồ sộ là khó khăn hơn nhiều so với việc tìm kiếm một cây kim trong một đống cỏ khô. Và nếu chúng ta có thể định vị gene, thì tách nó ra khỏi geneome như thế nào? Không có forcep đủ nhỏ để gắp một đoạn DNA, và cũng không có một cái kéo cơ học nào đủ nhỏ để cắt ra khỏi hệ gene một đoạn gene riêng biệt.
Hình 1.3. Vấn đề khó khăn khi chọn lọc
Một đoạn rất nhỏ từ geneome của E. coli
Gene muốn tạo dòng
Những đoạn DNA với những gene khác nhau, trong đó có trpA mà người ta muốn tạo dòng
Làm thế nào mà người ta tìm ra được 1 gene hoặc xác định nó?

9
Công nghệ DNA tái tổ hợp đã biến đổi sâu sắc phương thức nghiên cứu
gene. Trước đây thông tin về cấu trúc và tổ chức của gene thu được bằng cách kiểm tra biểu hiện kiểu hình, ngày nay những kỹ thuật mới đã tạo ra khả năng tự đọc các trình tự nucleotide. Trước đây các nhà di truyền phải chờ đợi sự xuất hiện các đột biên ngẫu nhiên hoặc cảm ứng để phân tích hiệu quả của sự sai khác di truyền, ngày nay họ có thể tạo ra đột biến ở các điểm nhất định một cách chính xác và xem chúng thay đổi kiểu hình như thế nào.
Công nghệ DNA tái tổ hợp đã cung cấp thông tin mới về cấu trúc và chức năng của gene và đã thay đổi nhiều khái niệm cơ bản của di truyền học. Ví dụ: trong khi mã di truyền được xem là rất phổ biến, thì bây giờ chúng ta còn biết rằng các mã không phổ biến cũng tồn tại trong DNA ty thể. Trước đây chúng ta cho rằng tổ chức gene eukaryote giống với prokaryote, nhưng bây giờ đã chứng minh được nhiều gene eukaryote bị gián đoạn bởi các intron. Ngày nay, chúng ta đã biết đầy đủ hơn về các quá trình tái bản, phiên mã, dịch mã, biến đổi RNA (RNA processing) và điều hòa gene thông qua việc sử dụng các kỹ thuật DNA tái tổ hợp. Các kỹ thuật này cũng được dùng trong nhiều trong nhiều lĩnh vực khác, như hóa sinh học, vi sinh vật học, sinh học phát triển, sinh học thần kinh, tiến hóa và sinh thái học.
Công nghệ DNA tái tổ hợp cũng được ứng dụng để tạo ra nhiều sản phẩm thương mại: thuốc, hormone, enzyme và các giống cây trồng, vật nuôi. Một nền công nghiệp hoàn toàn mới, công nghệ sinh học, đã phát triển chung quanh việc sử dụng các kỹ thuật này để tạo ra các sản phẩm mới. Trong y học, kỹ thuật DNA tái tổ hợp được dùng để thăm dò bản chất của ung thư, chẩn đoán các bệnh di truyền và nhiễm trùng, sản xuất thuốc và điều trị các rối loạn di truyền.
Nếu chúng ta thành công trong việc định vị và phân lập gene mong muốn, thì bước tiếp theo là đưa nó vào tế bào vi khuẩn. Các đoạn DNA mạch thẳng sẽ bị thoái biến nhanh bởi vi khuẩn và để tồn tại và tái bản được gene phải được chèn vào trong một dạng ổn định.
Khi biến nạp DNA tái tổ hợp thành công vào vi khuẩn còn phải đảm bảo rằng gene được phiên mã và dịch mã. Sự biểu hiện của gene là một quá trình phức tạp đòi hỏi một số trình tự DNA khác nằm ở ngoài gene. Tất cả những trình tự này phải hiện diện trong các hướng, ở các vị trí thích hợp của chúng để sản xuất protein.
Phản ứng chuỗi polymerase

10
Hình 1.4. Bằng phản ứng chuỗi polymerase người ta có hàng triệu bản sao từ một gene
Cuối cùng, các phương pháp được sử dụng để chuyển gene có hiệu quả rất thấp, trong hàng triệu tế bào chỉ có một số tế bào được biến nạp thành công và biểu hiện gene. Vì vậy, chúng ta có phương pháp để phát hiện được tế bào mang DNA tái tổ hợp mong muốn.
Trong toàn bộ nghiên cứu sinh học chỉ có rất ít lĩnh vực không bị tác động bởi tạo dòng DNA, PCR và kỹ thuật tái tổ hợp DNA. Đã từ lâu con người đã sử dụng vi sinh vật như vi khuẩn để sản xuất các hợp chất có ích, ví dụ thuốc kháng sinh penicillin, được tạo thành từ loài nấm có tên là Penicillium và streptomycin, một sản phẩm của vi khuẩn Streptomyces griseus. Tạo dòng DNA đã đưa lại cho sinh học một cuộc cách mạng, vì chúng mở ra một khả năng sản xuất protein của động vật có vú trong tế bào vi khuẩn. Các gene tạo dòng có một đặc điểm quan trọng là có thể biểu hiện trong một loài sinh vật không có quan hệ họ hàng gì với loài của nó. Như vậy, người ta có thể tạo dòng gene động vật trong vi khuẩn (Hình 1.5). Điều này có ý nghĩa rất lớn, người ta có thể tách các gene điều khiển tổng hợp chất kích thích và hocmon từ một cơ thể tồn tại trong tự nhiên. Sự tách chiết một chất từ sinh vật nguyên thuỷ thường đắt và khó khăn, nhưng khi đưa gene tương ứng vào vi khuẩn hoặc sinh vật khác thì sẽ thu được sản phẩm dễ dàng và với khối lượng lớn. Với phương pháp này người ta đã thành công trong nhiều trường hợp, cho đến nay có ý nghĩa nhất là sản xuất insulin bằng công nghệ gene.
Tế bào động vật
Gene mã hóa cho một protein động vật
Vector cùng với gene động Nhiễm sắc thể
Vi khuẩn biến đổi gene
mRNA

11
Hình 1.5. Phương pháp để sản xuất một protein động vật trong vi khuẩn
Những nghiên cứu về y học và nông nghiệp nhờ tạo dòng DNA đã có những bước tiến quan trọng. Nhờ tạo dòng DNA mà người ta đã phát triển những chất tiêm phòng để bảo vệ cơ thể chống lại bệnh tật. Nhiều bệnh di truyền ngày nay có thể chẩn đoán ở giai đoạn bào thai và những nghiên cứu xác định được bệnh ở giai đoạn sớm nhất, như ung thư vú và những bệnh hiểm nghèo, có hy vọng chữa trị được. II. Các enzyme tác động lên DNA
Khả năng thay đổi DNA trong ống nghiệm là dựa trên những nghiên cứu về sự tổng hợp và biến đổi DNA trong tế bào sống. Người ta đã phát hiện những enzyme có trong tế bào có thể cắt và nối DNA và đã phân lập được những enzyme này. Với chúng ngày nay người ta có thể tạo ra các phân tử DNA tái tổ hợp. Đặc điểm của những enzyme này và việc sử dụng chúng vào thí nghiệm tạo dòng sẽ đề cập chi tiết sau đây.
Chỗ cắt
Chỗ cắt
Liên kết hydro
Nucleotide Liên kết phosphodiester
a. Một exonuclease
Chỗ cắt
b. Một endonuclease

12
Hình 1.6. Các phản ứng do hai loại nuclease xúc tác
a. Exonuclease tách các nucleotide từ các đầu cuối phân tử DNA
b. Endonuclease cắt liên kết phosphodiester từ bên trong Khi đã có DNA tinh khiết, bước tiếp theo của thí nghiệm tạo dòng là thiết kế
phân tử DNA tái tổ hợp. Vector tạo dòng cũng như DNA phải được cắt ở những vị trí xác định và bằng cách xác định để nối lại với nhau. Cắt và nối là hai ví dụ về phương pháp thay đổi DNA. Trong những năm qua một loạt các phương pháp được phát triển để biến đổi các phân tử DNA không những chỉ cắt, nối mà có thể làm ngắn, nối dài, gắn vào hoặc tách những nhóm hoá học xác định nào đó. Tất cả những biến đổi này có thể thực hiện được trong ống nghiệm, và tạo nên nền tảng không chỉ đối với tạo dòng mà còn đối với những nghiên cứu cơ bản về hoá sinh DNA, cấu trúc của gene và điều khiển sự biểu hiện gene.
Trong tế bào những enzyme này tham gia vào các quá trình sống, như tái sinh, sao chép, phân giải (ví dụ DNA của virus), sửa chữa DNA đột biến và kết hợp các phân tử DNA khác nhau. Nhiều loại enzyme này được tách từ dịch tế bào và chúng có thể thực hiện chức năng trong điều kiện nhân tạo. Các phản ứng enzyme thường rất đơn giản, nhưng đòi hỏi enzyme phải ở dạng tinh khiết.
Enzyme được chia làm 5 nhóm lớn dựa trên phản ứng mà enzyme xúc tác. 1. Nuclease là những enzyme cắt, làm ngắn, phân giải các phân tử nucleic
acid 2. Ligase nối các phân tử nucleic acid 3. Polymerase sản sinh các bản sao của phân tử nucleic acid 4. Các enzyme biến đổi bằng cách tách hoặc gắn những nhóm hoá học 5. Topoisomerase: làm xuất hiện hoặc mất cấu trúc xoắn của DNA dạng
vòng. Trước khi đi vào các nhóm enzyme cụ thể cần phải nêu lên hai điểm: Thứ nhất phần lớn các enzyme được xếp vào một nhóm, tuy nhiên một số
enzyme có thể thuộc vào hai hay nhiều nhóm. Vi dụ các polymerase, chúng vừa có thể tạo nên phân tử DNA mới và vừa có hoạt tính của nuclease là phân giải DNA.
Thứ hai là cần chú ý rằng, bên cạnh những enzyme làm thay đổi DNA, người ta cũng biết nhiều enzyme tương tự tác dụng lên RNA. Ví dụ enzyme ribonuclease, sử dụng để phân giải RNA trong dịch tế bào khi muốn thu nhận DNA tinh sạch. Ở đây trước hết đề cập đến enzyme tác động lên DNA. 1. Nuclease
Cũng theo tiêu chuẩn này người ta phân biệt các endonuclease. Endonuclease S1 (từ nấm Aspergillus oryzae) chỉ tác động lên từng sợi đơn (Hình 1.8a), trong khi đó deoxyribonuclease I (DNase I, từ dạ cỏ bò), cắt cả sợi đơn và sợi kép (Hình 1.8b). DNase I tác động lên DNA ở các liên kết phosphodiester bên trong. Khi xử lý với enzyme càng lâu thì xuất hiện một hỗn hợp các mononucleotide và các oligonucleotide rất ngắn.

13
Hình 1.7. Các loại exonuclease khác nhau xúc tác cho các phản ứng
a. Bal31 tách các nucleotide từ hai đầu của DNA sợi kép b. Exonuclease III tách nucleotide chỉ đầu cuối 3’
Ngược lại, có một nhóm đặc enzyme biệt hơn, được gọi là enzyme hạn chế
(restriction endonuclease), cắt DNA sợi kép chỉ ở một số vị trí xác định (Hình 1.8c). Những enzyme này sẽ được mô tả chi tiết ở phần sau.
a. Bal 31
b. Exonuclease III

14
2. Ligase Trong tế bào DNA-ligase sửa chữa những chỗ đứt trên sợi đơn của phân tử
DNA sợi kép, những chỗ đứt này có thể xuất hiện khi tái sinh DNA. Ngoài ra DNA-ligase của hầu hết các sinh vật có thể nối hai đoạn DNA sợi kép lại với nhau. 3. Polymerase
a. Nuclease S1
b. DNase I
c. Endonuclease
Không có sự cắt tiếp theo
Lỗ hổng trên sợi đơn

15
Hình 1.8. Các phản ứng được xúc tác bởi nhiều loại endonuclease khác nhau
a. Nuclease I chỉ cắt các sợi đơn và các lổ hổng trên sợi đơn của DNA sợi kép
b. DNAase I cắt DNA cả sợi đơn và kép c. Một endonuclease cắt sợi kép chỉ ở một số vị trí nhất định
DNA-polymerase là những enzyme tổng hợp sợi DNA mới nhờ có sợi DNA
hoặc RNA khuôn mẫu (template) (Hình 1.10a). Phần lớn các polymerase chỉ hoạt động khi trong khuôn có một đoạn sợi kép, đoạn này được gọi là primer (mồi) bắt đầu cho phản ứng tổng hợp. Có ba loại DNA-polymerase được sử dụng trong công nghệ gene: DNA-polymerase I, được tách ra từ E. coli. Enzyme này gắn vào một đoạn đơn ngắn là điểm đứt trên 1 sợi DNA trong DNA sợi đôi (nick) của phân tử DNA sợi kép và tổng hợp nên một sợi hoàn toàn mới. DNA-polymerase I cũng là một ví dụ cho enzym có hai chức năng: tổng hợp và phân giải DNA.
a. Sữa chữa chỗ đứt trên sợi đơn
Chỗ đứt trên sợi đơn
DNA-ligase
b. Gắn kết hai phân tử
DNA-ligase

16
Hình 1.9. Hai loại phản ứng được xúc tác bởi DNA-ligase
a. Sửa chữa liên kết phosphodiester bị thiếu trên một sợi đơn b. Gắn hai sợi kép lại với nhau
Hoạt tính polymerase và nuclease của DNA polymerase I được điều khiển
bởi những vùng khác nhau của phân tử enzyme. Hoạt tính nuclease nằm ở 323 gốc amino acid đầu tiên của chuỗi polypeptide. Nếu cắt đi phần này thì enzyme bị thay đổi, hoạt tính polymerase vẫn còn, nhưng hoạt tính phân giải DNA bị mất. Enzyme bị biến đổi này được gọi là đoạn Klenow. Đoạn Klenow có thể tổng hợp sợi bổ sung từ DNA khuôn, tuy nhiên sự tổng hợp không tiếp tục khi chỗ đứt trên sợi đơn được lấp đầy (Hình 1.10c). Ứng dụng quan trọng nhất là đoạn Klenow là xác định trình tự DNA.
Một loại DNA-polymerase thứ ba là reverse transcriptase, enzyme tham gia vào sự tái sinh của nhiều virus. Đặc tính duy nhất của nó là sử dụng RNA làm khuôn (Hình 1.10d). Khả năng tổng hợp nên sợi DNA bổ sung từ RNA khuôn của enzyme có ý nghĩa trung tâm trong kỹ thuật tạo dòng cDNA.
a. Phản ứng cơ bản
Sợi khuôn Đoạn mồi Sợi mới được tổng hợp
b. DNA-polymerase I
Sợi khuôn
Lỗ hổng trên sợi đơn (nick) Những nucleotide này được thay thế bằng những nucleotide mới
c. Đoạn Klenow
Những nucleotide này không được thay thế
Chỉ có lỗ hổng được lấp đầy
d. Reverse transcriptase

17
Hình 1.10. Các phản ứng được xúc tác bởi DNA-polymerase a. Phản ứng cơ bản: Một sợi DNA mới được tổng hợp theo hướng 5’
3’ b. DNA-polymerase I trước hết lấp những lỗ hổng trên sợi đơn và sau đó
tổng hợp sợi mới trên cơ sở phân giải sợi cũ c. Đoạn Klenow chỉ lấp những lỗ hổng d. Enzyme Reverse Transcriptase sử dụng RNA làm khuôn
4. Các enzyme biến đổi DNA
Có nhiều enzyme làm thay đổi DNA bằng cách gắn vào hoặc tách ra các nhóm hoá học. Quan trọng nhất là những enzyme sau đây: 1. Phosphatase kiềm (từ E. coli hoặc ruột bê) tách nhóm phosphate của đầu 5’ của
phân tử DNA (Hình 1.11a). 2. Polynucleotide kinase (từ E. coli nhiễm phage T4) có tác dụng ngược với
phosphatase kiềm: gắn nhóm phosphate vào đầu 5’ tự do (Hình 1.11b). Enzyme terminal deoxynucleotide transferase gắn một hoặc nhiều deoxynucleotide vào đầu cuối 3’ của phân tử DNA.
a. Phosphatase kiềm
b. Polynucleotide transfrease
c. Terminal deoxynucleotide transferase

18
Hình 1.11. Các phản ứng được xúc tác bởi các enzyme biến đổi DNA a. Phosphatase kiềm tách nhóm phosphate ở đầu 5’ b. Polynucleotide kinase gắn nhóm phosphate ở đầu 5’ c. Terminal deoxynucleotide transferase gắn các nucleotide vào đầu 3’ của
sợi đơn (1) hoặc sợi kép (2). 5. Topoisomerase
Topoisomerase là enzyme có khả năng thay đổi hình dạng của DNA vòng, đồng hoá trị (DNA plasmid), bằng cách làm mất hoặc xuất hiện xoắn ở mức cao hơn. Đối với nghiên cứu sự tái sinh DNA thì những enzyme này quan trọng, nhưng với công nghệ gene cho đến nay chưa có ứng dụng thực sự nào. III. Enzym hạn chế: Restriction endonuclease
Khi tạo dòng điều cần thiết là cắt các phân tử DNA chính xác và luôn luôn cùng cách. Vector phải được cắt ở một vị trí duy nhất để mở vòng và chèn đoạn DNA ngoại lai vào. Phân tử DNA bị cắt nhiều lần, thành hai hoặc nhiều đoạn riêng lẻ thì không thể dùng làm vector tạo dòng. Vậy vector phải được thiết kế như thế nào và cần những endonuclease nào để thực hiện sự thay đổi này?
a. Phân tử vector
Mở vector ở vị trí cắt

19
Hình 1.12. Tạ i sao ở thí nghiệm tạo dòng người ta phải cắt thật chính xác Thường người ta phải cắt DNA khi tạo dòng (Hình 1.12 b), vì phần lớn
những vector tạo dòng tiếp nhận đoạn DNA có độ lớn xác định. Ví dụ các vector trên cơ sở M13 chỉ tạo dòng có hiệu quả khi đoạn DNA nhỏ hơn 3 kb.
Với các enzyme hạn chế tinh khiết người ta mới có thể cắt chính xác các phân tử DNA. Nhờ sự phát hiện những enzyme này mà Arber, Smith và Nathans nhận được giải thưởng Nobel vào năm 1978, là một trong những điều kiện quan trọng nhất đối với sự phát triển của công nghệ gene. 1. Phát hiện và kiểu tác động của enzyme hạn chế
Quan sát đầu tiên dẫn đến sự phát hiện ra enzyme hạn chế đã bắt đầu từ những năm đầu những năm năm mươi. Người ta thấy một số loài vi khuẩn miễn dịch đối với một số phage, hiện tượng này được gọi là hạn chế ký chủ. Cơ chế của quá trình này không phức tạp lắm, tuy nhiên mãi 20 năm sau người ta mới hiểu một cách đầy đủ. Nguyên nhân của hạn chế là enzyme do vi khuẩn sản sinh ra, phân giải DNA của phage trước khi chúng tự tái sinh và tổng hợp nên hạt phage mới (Hình 1.13a).
a. Giới hạn của DNA phage
Phage đẩy DNA vào tế bào vi

20
Hình 1.13 Chức năng của endonuclease trong tế bào
DNA của phage (a) bị phân giải nhưng DNA của vi khuẩn (b) thì không Tuy nhiên, DNA của vi khuẩn không bị phân giải vì các base nitrogene ở
trình tự nhận biết đã bị methyl hóa nên enzyme không còn nhận ra được vị trí cắt (Hình 1.13b). Những enzyme phân giải này được gọi là enzym hạn chế (restriction endonuclease). Chúng có mặt ở tất cả các loài vi khuẩn. Hiện nay, đã có hơn 1200 enzyme thuộc loại này được xác định. Người ta biết có ba loại enzyme hạn chế khác nhau. Loại I và III có ý nghĩa rất giới hạn đối với công nghệ sinh học. Ngược lại loại II là những enzyme cắt rất quan trọng đối với tạo dòng DNA. 2. Enzyme hạn chế loại II cắt DNA ở những trình tự hoàn toàn xác định
Enzyme hạn chế loại II đặc biệt đã trở thành một công cụ rất hữu ích đối với các nhà sinh học phân tử. Những enzyme này gắn vào DNA ở bất kỳ vị trí nào và di chuyển dọc theo sợi DNA cho đến khi gặp trình tự nhận biết. Khi enzyme gắn vào trình tự nhận biết cấu trúc của enzyme thay đổi tạo điều kiện cho phản ứng cắt. Các vị trí nhận biết này có kích thước khác nhau tuỳ thuộc loại enzyme: Nhiều enzyme

21
hạn chế có trình tự nhận biết 6 nucleotide, một số khác có 4, 5, 8 nucleotide, hoặc cũng có thể là 20. Ví dụ Sau3A (từ Staphylococcus aureus dòng 3A) có trình tự nhận biết là GATC và AluI (từ Arthrobacter luteus) là trình tự AGCT. Ngoài ra còn có những enzyme nhận biết các trình tự thoái hoá, nghĩa là chúng có thể cắt DNA ở nhiều trình tự tương tự nhau. Ví dụ HinfI (từ Haemophulus influenzae dòng Rf) nhận biết trình tự GANTC và có khả năng cắt ở các trình tự sau: GAATC, GATTC, GACTC và GAGTC.
Mặc dù trình tự nhận biết của các enzyme hạn chế là khác nhau, nhưng khi cắt so le chúng đều tạo ra trình tự palindrome giống nhau khi đọc theo hướng 5' đến 3' trên mỗi sợi đơn. Một ví dụ về trình tự palindrome được tạo bởi enzyme EcoRI là AATT.
Hình 1.14 Enzym EcoRI
Các trình tự nhận biết của một số enzyme hạn chế thường được sử dụng trình bày ở bảng 1.1
EcoRI nhận biết trình tự 6 nucleotide là GAATTC đọc từ đầu 5' đến 3'. Trình tự bổ sung (trên sợi DNA bổ sung) khi đọc từ 5' đến 3' cũng là GAATTC. Trên hình 1.14 biểu diễn enzyme EcoRI gắn và cắt đối xứng ở cùng điểm bên trong trình tự (giữa G và A khi đọc từ 5' đến 3') trên cả hai sợi, ở đây ta có thể nhận ra trình tự palindrome.

22
Bảng 1.1 Một số enzymne hạn chế thường được sử dụng
Enzyme Nguồn vi sinh vật Trình tự nhận biết Loại đầu EcoRI
BamHI
BglII
PvuI
PvuII
HindIII
Hinfl
Sau3A
AluI
TaqI
HaeIII
Notl
Escherichia coli
Bacillus amyloliquefaciens
Bacillus globigii
Proteus vulgaris
Proteus vulgaris
Haemophilus influenzae Rd
Haemophilus influenzae Rf
Staphylococcus aureus
Arthrobacter luteus
Thermus aquaticus
Haemophilus aegyptius
Nocardia otitidis-caviarum
5’-G AATTC-3’
3”-CTTAA G-5’
5’-G GATCC-3’
3’-CCTAG G5’
5’-A GATCT-3’
3’-TCTAG A-5’
CGATCG
CAGCTG
AAGCTT
GANTC
GATC
AGCT
TCGA
GGCC
GCGGCCGC
Dính
Dính
Dính
Dính
Đầu bằng
Dính
Dính
Dính
Đầu bằng
Dính
Đầu bằng
Dính

23
3. Đầu bằng và đầu dính
Đối với thí nghiệm tạo dòng điều quan trọng là đoạn cắt được tạo ra từ enzyme hạn chế như thế nào. Nhiều enzyme cắt đơn giản hai sợi DNA ở giữa trình tự nhận biết (Hình 1.15a), vì vậy xuất hiện đầu bằng (blunt ends), ví dụ enzyme PvuII và AluI.
Hình 1.15. Các đầu cuối xuất hiện do DNA được cắt bằng những endonuclease khác nhau
a. Đầu bằng được tạo ra bởi AluI
a. Tạo ra các đầu bằng
b. Tạo ra các đầu dính
Các đầu bằng
Các đầu dính c. Các endonuclease khác nhau tạo ra các đầu dính giống nhau
BamHI
BglII
Sau3A
EcoRI
AluI
“N”=A, G, C hoặc T

24
b. Đầu dính được tạo ra bởi EcoRI c. Các đầu dính giống nhau được tạo ra bởi BamHI, BglII và
Sau3A Điểm quan trọng là khi cắt tạo nên những “đầu dính”, vì chúng dễ dàng gắn
lại với nhau theo nguyên tắc bổ sung. Những enzyme này cắt hai sợi DNA không ở cùng một vị trí mà thường dịch
chuyển 2 hoặc 4 nucleotide, vì vậy những đoạn DNA xuất hiện có đầu cuối là sợi đơn nhô ra (Hình 1.15b). Những enzyme hạn chế với các trình tự nhận biết khác nhau có thể tạo ra những đầu dính giống nhau. Ví dụ, BamHI (trình tự nhận biết là GGATCC) và BglII (trình tự nhận biết AGATCT). Khi cắt bằng hai enzyme này đều xuất hiện đầu dính có trình tự GATC (Hình 1.15c). Đầu dính như vậy cũng được tạo ra bởi Sau3AI, enzyme có trình tự nhận biết 4 nucleotide GATC. Các đoạn DNA xuất hiện khi cắt bằng những enzyme này có thể gắn lại được với nhau, vì các đầu dính này bổ sung cho nhau. 4. Số vị trí nhận biết của enzyme hạn chế trong phân tử DNA
Chúng ta có thể ước lượng được tần suất cắt DNA khi biết được kích thước của vị trí cắt. Có 4 loại nucleotide cấu tạo nên DNA nên khả năng cắt xảy ra ở bất kỳ một loại nucleotide nào là 1/4. Trong trường hợp kích thước của vị trí cắt có 4 nucleotide thì xác suất cắt xảy ra ở bất kỳ vị trí nào đó là 1/4 x 1/4 x 1/4 x 1/4 = 1/256, có nghĩa là cứ trung bình 256 bp có một lần cắt. Bằng cách tính tương tự có thể dự đoán được số đoạn DNA tạo ra nếu biết kích thước của trình tự nhận biết. Điều này có ý nghĩa đối với lập bản đồ gene và kỹ thuật di truyền.
Tuy nhiên, trong thực tế không hoàn toàn chính xác như vậy. Ví dụ DNA của phage có chiều dài 49 kb, theo tính toán có chứa khoảng 12 chỗ cắt cho một enzyme có trình tự nhận biết là 6 nucleotide, nhưng thực tế số lượng vị trí cắt ít hơn (ví dụ có 6 vị trí cắt đối với BglII, 5 với BamHI và chỉ 2 với SalI).
BglII: có 6 vị trí cắt
a. Các vị trí cắt của một số enzyme trên DNA
BamHI: có 5 vị trí cắt
SalI: có 2 vị trí cắt
b. Độ lớn của các đoạn DNA
BglII
BamHI

25
Hình 1.16. Cắt DNA phage bằng các enzyme hạn chế khác nhau a. Vị trí nhận biết của BglII, BamHI và SalI b. Các đoạn thu được khi cắt bằng các endonuclease. Các con số biểu
diễn độ lớn của các đoạn DNA bằng cặp base (bp)
Ngoài ra, các vị trí cắt phân bố không đồng đều trên phân tử DNA, vì nếu phân bố đồng đều thì các đoạn tạo ra có độ lớn phải bằng nhau. Hình 1.16b cho thấy độ lớn các đoạn thu được khi cắt phage bằng BglII, BamHI và SalI. Trong nhiều trường hợp các đoạn thu được có độ lớn phân tán rất rộng. Điều này nói lên rằng các nucleotide sắp xếp hoàn toàn ngẫu nhiên trên DNA.
Từ hình 1.16 với phương pháp toán học người ta có thể hình dung có bao nhiêu vị trí cắt có thể chờ đợi ở một phân tử DNA, nhưng bức tranh thực tế chỉ có được khi phân tích thực nghiệm.
5. Quá trình cắt bằng enzyme hạn chế trong phòng thí nghiệm
Ví dụ cắt -DNA (nồng độ 125 g/ml) bằng BglII. Trước hết cho DNA vào eppendorf, lượng bao nhiêu là tùy thuộc vào mục đích của từng loại thí nghiệm, ví dụ 2 g DNA tube (có trong 16 l, Hình 1.17). Thành phần được cho vào tiếp theo là dung dịch đệm. Đệm được cung cấp trên thị trường có nồng độ cao hơn 10 lần so với nồng độ phản ứng, vì vậy phải được pha loãng 10 lần. Ví dụ thể tích cuối cùng của hỗn hợp phản ứng là 20 l, thì chỉ cần 2 l dung dịch đệm BglII. Enzyme là thành phần cuối cùng được cho vào hỗn hợp phản ứng. Enzyme ở dạng tinh khiết với nồng độ đã biết do nhà sản xuất cung cấp. Nồng độ enzyme phải được tính chính xác để đảm bảo hoạt tính enzyme là cao nhất. Một đơn vị enzyme là lượng enzyme cần để cắt 1 g DNA trong thời gian 1 giờ, ví dụ có 2g DNA thì cần 2 đơn vị BglII. BglII trên thị trường có nồng độ 4 đơn vị/l nên chỉ cần 0,5 l cho thí nghiệm này. Để có thể tích cuối cùng là 20l cần thêm 1,5 l nước cất (Hình 1.17c).
Phần lớn các enzyme hạn chế hoạt động tốt ở pH 7,4, nhưng các loại enzyme khác nhau cần nồng độ ion khác nhau (được tạo ra bởi NaCl) và nồng độ Mg2+ (tất cả các enzyme hạn chế loại II cần Mg2+). Ngoài ra, cần cho thêm chất khử dithiothreitol để tăng sự bền vững và giảm sự bất hoạt của enzyme. Điều quan trọng là phải tạo điều kiện phản ứng chính xác cho mỗi loại enzyme. Nồng độ NaCl và

26
Mg2+ không chính xác không những làm giảm hoạt tính của enzyme mà còn thay đổi tính đặc hiệu của chúng là cắt DNA ở những vị trí không đặc hiệu. Thành phần dung dịch đệm thích hợp cho BglII được trình bày ở bảng 1.2.
Bảng 1.2 Thành phần đệm của enzyme BglII có nồng độ cao gấp 10 lần
Thành phần Nồng độ (mM) Tris-HCl, pH 7,4 MgCl2 NaCl Dithiothreitol
500 100 500 10
Yếu tố cuối cùng cần chú ý là nhiệt độ ủ. Hầu hết enzyme hạn chế có hoạt
tính cao nhất ở nhiệt độ 37 0C, nhưng một số khác có nhiệt độ tối thích cao hơn. Ví dụ TaqI thu được từ vi khuẩn Thermus aquaticus thích nghi ở nhiệt độ cao như suối nước nóng. Khi cắt bằng enzyme này người ta ủ ở nhiệt độ 650C mới đạt được hoạt tính cực đại.
Sau thời gian ủ 1 giờ phản ứng xảy ra hoàn toàn (Hình 1.17d). Nếu muốn sử dụng các đoạn DNA này cho thí nghiệm tạo dòng thì phải khử hoạt enzyme bằng cách nào đó. Có nhiều phương pháp làm bất hoạt enzyme. Trong nhiều trường hợp chỉ cần ủ trong thời gian ngắn ở nhiệt độ 700C hoặc chiết bằng phenol hoặc cho vào hỗn hợp phản ứng EDTA, chất này kết hợp với Mg2+ làm cho enzyme mất hoạt tính (Hình 1.17c).
Cho vào 2 l đệm BglII Cho vào 1,5 l H2O và 0,5 l BglII
2 g -DNA (16 l)
Ủ: 1 giờ ở 37oC
DNA của phage được cắt ra
Cho thêm phenol hoặc EDTA hoặc gia nhiệt 70oC trong 15 phút
(a) (b) (c)
(d)
(e)

27
Hình 1.17 Tiến trình cắt bằng enzyme giới hạn trong phòng thí nghiệm
6. Phân tích kết quả của phản ứng cắt bằng enzyme hạn chế Phản ứng cắt làm xuất hiện một số đoạn DNA, độ lớn của từng đoạn phụ thuộc vào vị trí các điểm cắt trong phân tử DNA gốc (Hình 1.16). Khi tạo dòng người ta cần một phương pháp để xác định số lượng và độ lớn của các đoạn DNA. Nói chung, phân tử DNA được cắt hay không, có thể dễ dàng nhận biết bằng cách xác định độ nhớt của dung dịch. Các phân tử DNA lớn làm cho dung dịch đặc quánh hơn phân tử DNA nhỏ. Tuy nhiên để xác định chính xác số lượng và độ lớn của sản phẩm là khó khăn và mất nhiều thời gian. Vào đầu những năm 70 vấn đề này đã được giải quyết nhờ phương pháp điện di. 6.1. Sự tách các phân tử bằng điện di
a. Điện di thường
DNA Đệm
Điện di
DNA di chuyển về cực dương nhưng quá trình tách theo độ lớn
b. Điện di trên gel

28
Hình 1.18. Điện di thường (a) không tách được các đoạn DNA có độ lớn khác nhau, điện di trên gel (b) thì tách được
Cũng như protein và nhiều phân tử khác, DNA mang điện và tích điện âm. Vì vậy, chúng di chuyển về cực dương khi ở trong điện trường (Hình 1.18a). Tốc độ di chuyển của chúng phụ thuộc vào hai đặc điểm: hình dạng và điện tích của phân tử. Phần lớn các phân tử DNA có cùng hình dạng và điện tích thì tỷ lệ với độ lớn. Bằng điện di đơn giản người ta không thể tách các đoạn DNA có độ lớn khác nhau. Khi cho hỗn hợp các đoạn DNA điện di trong gel được tạo nên từ agarose hoặc polyacrylamide, các phân tử DNA phải di chuyển qua hệ thống lỗ rây để đi đến cực dương. Phân tử DNA càng nhỏ, thì di chuyển càng nhanh. Nhờ vậy các phân tử DNA được tách ra trong điện di theo độ lớn của nó (Hình 1.18b).

29
Trong thực tế nồng độ agarose được xác định phụ thuộc vào độ lớn của các phân tử DNA. Ví dụ, gel có độ dày 0,5 cm từ 0,5% agarose, thì lỗ có kích thước tương đối lớn dùng để tách các phân tử DNA có độ lớn từ 1-30 kb, và phân biệt được rõ ràng khi phân tử có độ lớn 10-12 kb. Gel mỏng nhất (0,3 mm) được tạo nên từ 40% polyacrylamide dùng để tách các phân tử DNA ngắn hơn 300 bp. Những gel này cho phép phân biệt được các phân tử có độ dài khác nhau một nucleotide. 6.2. Sự hiển thị của phân tử DNA trong gel
Phương pháp đơn giản nhất để hiển thị kết quả điện di là nhuộm gel với
ethidium bromide (EtBr, Hình 1.19). Sau khi nhuộm EtBr và quan sát gel dưới ánh sáng tử ngoại có thể nhận biết vị trí các vạch tương ứng với độ lớn của các đoạn DNA với điều kiện là DNA phải đạt được một lượng nhất định.
Tuy nhiên, nhuộm gel bằng EtBt có nhược điểm là độ nhạy có giới hạn. Khi một vạch có lượng DNA ít hơn 25 ng thì không nhận ra được. Vì vậy, cần phải có phương pháp phát hiện nhạy hơn. Phóng xạ tự ghi giải quyết được vấn đề này. Trước khi điện di DNA được đánh dấu phóng xạ, rồi hiển thị chúng bằng cách đặt một bản phim lên gel. DNA phóng xạ “chiếu sáng” phim, nhờ vậy có thể nhận ra được các vạch (Hình 1.20).
Để đánh dấu DNA người ta đưa vào phân tử DNA các nucleotide chứa đồng vị phosphore phóng xạ 32P (Hình 1.21a). Có nhiều phương pháp đánh dấu nhưng thông dụng nhất là phương pháp nick-translation và gắn nucleotide vào đầu cuối.
Nick-translation được thực hiện bởi DNA-polymerase. Trong mọi trường hợp, ngay cả khi người ta tinh sạch rất cẩn thận mẫu DNA bao giờ cũng chứa một số phân tử với những chỗ gãy trên một sợi (nicks). DNA-polymerase I có khả năng gắn các nucleotide vào một sợi và xúc tác cho phản ứng được gọi là thay thế sợi (Hình 1.10b).
Ngâm gel trong dung dịch
Các giếng để cho mẫu vào
Gel agarose
Khung gel bằng chất tổng hợp cho tia cực tím đi qua

30
Hình 1.19. Các vạch DNA được hiển thị trong gel agarose sau khi nhuộm EtBr và quan sát dưới tia cực tím.

31
Tấm kính
Gel agarose được làm khô
Bản phim được đặt ltrực tiếp lên gel
Chiếu sáng (12 đến 100 giờ) để tráng phim
Phóng xạ tự ghi Các vạch DNA đã nhuộm đen phim

32
Hình 1.20. Hiển thị các vạch DNA bằng đánh dấu phóng xạ tự ghi
Với nick-translation người ta có thể đánh dấu được bất kỳ phân tử DNA nào, nhưng trong một số trường hợp dẫn đến sự phân cắt protein.
Một phương pháp khác là lấp đầy đầu cuối. Tuy nhiên với cách này chỉ đánh dấu được những phân tử có đầu dính. Enzyme được sử dụng là đoạn Klenow, lấp đầy các đầu dính bằng cách tổng hợp một sợi bổ sung (Hình 1.21). Nếu đưa vào phản ứng này các nucleotide đã dánh dấu phóng xạ thì thực hiện được sự đánh dấu DNA.
Với nick-translation và lấp đầy đầu cuối người ta có thể phát hiện bằng phóng xạ tự ghi các vạch chứa một lượng DNA rất nhỏ (2 ng).
a. [ -32P] dATP
32P phóng xạ
a. Đánh dấu bằng nick-translation
+ 32P + dATP
Những đoạn đánh dấu
DNA-polymerase I
Những chỗ đứt trên sợi đơn
Đoạn Klenow
Những đầu được đánh Nhứng đầu dính (EcoRI)
c. Đánh dấu bằn việc lấp đầy những đầu cuối
+ 32P + dATP

33
Hình 1.21. Đánh dấu phóng xạ DNA a. Cấu trúc của dATP b. Đánh dấu DNA bằng nick-translation c. Đánh dấu DNA bằng lấp đầy các đầu cuối
6.3. Đánh giá độ lớn của các đoạn DNA
Trong điện di các phân tử DNA được tách ra dựa trên độ lớn của nó: phân tử nhỏ nhất di chuyển nhanh nhất và phân tử lớn nhất di chuyển chậm nhất đến điện cực dương. Nếu có nhiều đoạn DNA có độ lớn khác nhau (ví dụ sau phản ứng cắt bằng enzyme hạn chế thành công), thì xuất hiện trong gel rất nhiều vạch. Vậy làm thế nào để biết được độ lớn của các vạch?
Mối quan hệ toán học giữa vận tốc di chuyển và khối lượng phân tử được biểu diễn theo công thức sau: D = a - b (log M)
Ở đây D là khoảng cách di chuyển của DNA, M là khối lượng phân tử, a và b là hằng số phụ thuộc vào điều kiện điện di.
Người ta cho DNA size marker (chuẩn kích thước của DNA), hỗn hợp các đoạn DNA có độ lớn đã biết, cùng chạy trong gel. Marker thường sử dụng là DNA của phage được cắt bằng HindIII , gồm 8 đoạn: đoạn nhỏ nhất có độ dài 125 bp và đoạn lớn nhất là hơn 23 kb. So sánh vị trí của các đoạn DNA trong mẫu nghiên cứu với các vạch.
a. Ước lượng độ lớn bằng mắt thường
Độ lớn chưa biết
HiindIII
khoảng 5000 bp
khoảng 3200 bp
khoảng 2000 bp
b. Ước lượng chính xác bằng đồ thị

34
Hình 1.22. Xác định độ lớn của các đoạn DNA trên gel agarose
a. Ước lượng bằng mắt thường b. Xác định độ lớn DNA chính xác hơn bằng đường chuẩn dựa
trên khoảng cách di chuyển của chúng trên gel. 6.4. Lập bản đồ giới hạn phân tử DNA
Như đã đề cập là làm thế nào để xác định được số lượng và độ lớn của các đoạn DNA được cắt bằng enzyme hạn chế? Bước tiếp theo trong phân tích là thiết kế bản đồ, trong đó chỉ ra vị trí cắt tương đối của các enzyme. Bản đồ giới hạn giúp cho việc lựa chọn enzyme phù hợp cho phản ứng cắt mong muốn.
Để thiết kế bản đồ phải tiến hành cắt phân tử DNA bằng một loạt các enzyme. Bằng điện di người ta biết được số lượng và độ lớn của các đoạn được tạo ra khi sử dụng các enzyme cắt riêng lẻ (Hình 1.23). Thêm vào những phát hiện này người ta cần một loạt các cắt đôi, nghĩa là đồng thời sử dụng hai loại enzyme. Nếu hai loại enzyme này có cùng yêu cầu về pH và nồng độ Mg2+ thì có thể để chúng cùng cắt trong một phản ứng. Ngược lại, thì cho hai phản ứng xảy ra kế tiếp nhau để có điều kiện phù hợp với từng loại enzyme.
Qua việc so sánh kết quả của phản ứng cắt đơn và đôi người ta có thể xác định được nhiều hay tất cả vị trí các điểm cắt (Hình 1.23). Trong những trường hợp còn chưa rõ ràng thì để phản ứng cắt xảy ra từng phần, nghĩa là chọn điều kiện phản ứng để chỉ một phần các vị trí được cắt. Sự cắt từng phần đạt được bằng cách ủ thời gian ngắn, hoặc ủ ở nhiệt độ thấp (ví dụ 40C), điều này làm giảm hoạt tính của enzyme. Kết quả của phản ứng cắt từng phần là các vạch phức tạp ở gel điện di. Bên cạnh những đoạn bình thường nhận được khi cắt hoàn toàn, còn xuất hiện thêm những đoạn khác. Biết được độ lớn của nó, người ta có thể biết được những vị trí nào ở cạnh nhau trong phân tử không được cắt (Hình 1.23).

35
Phản ứng cắt đơn và đôi Enzyme Số lượng đoạn Độ lớn (kb)
Xbal 2 24,0 24,5 Xhol 2 15,0 33,5
Kpnl 3 1,5 17,0 30,0 Xbal + Xhol 3 9,0 15,0 24,5
Xbal + Kpnl 4 1,5 6,0 17,0 24,0
Nhận xét: 1. Vì DNA ở dạng thẳng, có một vị trí cắt cho enzyme Xbal và Xhol, 2 vị trí cắt cho Kpnl 2. Vị trí nhận biết của Xbal và Xhol được sắp xếp như sau:
Các đoạn của Xbal 24 24,5
Các đoạn của Xbal và Xhol 9 15,5 24,5
Các đoạn của Xhol 15,0 33,5 Xhol Xbal
Khả năng duy nhất là 15,0 9,0 24,5
Cắt từng phần
Enzyme Độ lớn các đoạn (kb) KpnI, những điều kiện giới hạn 1,5 17,0 18,5 30,0 31,5 48,5
Nhận xét:
Đoạn có độ lớn 48,5 kb là DNA của phage không bị cắt Các đoạn 1,5, 17,0 và 30,0 kb là sản phẩm được cắt hoàn toàn
Các đoạn 18,5 và 31,5 kb là sản phẩm bị cắt từng phần
Các KpnI Vị trí cắt của Kpnl phải như sau:
XhoI XbaI KpnI Vị trí cắt của 3 enzyme như sau: 15,0 9,0 6,0 1,5 17,0

36
Hình 1.23. Lập bản đồ giới hạn. Ví dụ chỉ ra cách xác định vị trí cắt của Xbal, Xhol và các KpnI trên DNA của phage
IV. Gắn các phân tử DNA (ligation) Bước cuối cùng trong việc thiết kế một phân tử DNA tái tổ hợp là nối các
đoạn DNA lại với nhau (Hình 1.24). Enzyme xúc tác cho phản ứng này là DNA-ligase.
Hình 1.24. Nối kết (ligation): Bước cuối cùng khi thiết kế phân tử DNA tái tổ hợp
1. Tác dụng của DNA-ligase
Trong các tế bào sống đều có mặt DNA-ligase, nhưng enzyme được sử dụng trong công nghệ gene có nguồn gốc từ vi khuẩn E. coli nhiễm phage T4.
Phân tử DNA tái tổ hợp
Gene
DNA-ligase Vector
Gene tạo dòng

37
Trong tế bào ligase có nhiều chức năng quan trọng: DNA-ligase sửa chữa các chỗ đứt ở trên sợi đơn của DNA sợi kép (Hình
1.9a). Ở các vị trí đứt này là do thiếu liên kết phosphodiester giữa hai nucleotide ở cạnh nhau (ngược lại với những chỗ đứt mà ở đó một hay nhiều nucleotide bị mất).
DNA-ligase còn nối các phân tử DNA khác nhau hoặc hai đầu của một phân tử. Phản ứng hoá học ở sự sửa chữa các chỗ đứt cũng giống phản ứng nối, chỉ khác là ở phản ứng nối hai liên kết phosphotdiester giữa hai sợi được tạo nên (Hình 1.25a). 2. Các đầu dính tăng hiệu quả phản ứng gắn
Ở hình 1.25a hai đoạn DNA đầu bằng được nối lại với nhau. Tuy nhiên phản ứng này đạt hiệu quả rất thấp. Để phản ứng nối các đầu bằng có hiệu quả thì nồng độ DNA phải cao. Việc nối kết dễ dàng hơn khi thực hiện với các đầu dính. Các đầu dính phù hợp có thể liên kết với nhau bằng liên kết hydrogene giữa các cặp base bổ sung, sau đó liên kết bền vững được tạo nên dưới tác dụng của ligase. Khi các liên kết phosphodiester không được tạo nên thì những đầu cuối lại tách ra trở lại. Tuy nhiên, nhờ sự bắt cặp của các base mà hiệu quả của phản ứng nối được tăng lên, vì nhờ các liên kết hydrogene mà các đầu này duy trì tiếp xúc với nhau ở thời gian dài hơn.
DNA-ligase gắn các chỗ đứt trên sợi đơn lại với nhau
Trạng thái trung gian của cặp đôi base
Chỗ đứt trên sợi đơn
b. Gắn các đầu dính
a. Gắn các đầu bằng

38
Hình 1.25. Các phản ứng khác nhau được xúc tác bởi DNA-ligase a. Gắn các đầu bằng b. Gắn các đầu dính
3. Tạo đầu dính từ phân tử DNA đầu bằng
Như vậy muốn nối các phân tử DNA lại với nhau trong thí nghiệm tạo dòng là phải tạo ra các đầu dính. Để có được các đầu dính bổ sung cho nhau người ta sử dụng cùng một enzyme để mở vectơ và cắt đoạn DNA tạo dòng. Tuy nhiên, điều này không phải luôn luôn xảy ra. Thường là vector có các đầu dính, trong khi đó đoạn DNA tạo dòng có đầu bằng. Có ba phương pháp để tạo nên DNA đầu dính từ các đoạn đầu bằng. a. Gắn đoạn nối (linker) vào đầu bằng
Để thực hiện phương pháp đầu tiên là tạo nên đoạn nối, đoạn DNA nhỏ có trình tự đã biết, được tổng hợp nhân tạo. Một phần nối điển hình được nêu trong hình 1.26a. Đoạn nối này có đầu bằng và trong đó chứa một vị trí cắt cho BamHI. Nhờ DNA-ligase mà người ta có thể gắn đoạn nối này với phân tử DNA lớn hơn có đầu bằng. Mặc dù ở đây là nối các đoạn DNA có đầu bằng nhưng phản ứng xảy ra rất hiệu quả, vì có thể tạo ra một lượng lớn các oligonucleotide tổng hợp, nghĩa là phản ứng nối được thực hiện ở nồng độ DNA cao. Linker được cắt
ra
Đầu đính (BamHI)
BamHI
Linker gắn vào
Linker
DNA-ligase
Phân tử DNA đầu bằng
b. Sử dụng linker
a. Một linker điển hình

39
Hình 1.26. Linker và sử dụng linker a. Cấu trúc của một linker điển hình b. Gắn một linker vào phân tử có đầu bằng
Ở đầu cuối mỗi phân tử DNA có thể gắn nhiều linker, làm xuất hiện cấu trúc
chuỗi (Hình 1.26b). Bằng việc xử lý với enzyme hạn chế (ví dụ BamHI) chuỗi này được cắt ở những vị trí nhận biết tạo nên các đoạn chứa đầu dính BamHI. Như vậy đoạn DNA tạo dòng được biến đổi cho phù hợp với vector cũng được mở vòng bằng BamHI. b. Đầu gắn (adapter)
Sẽ xảy ra điều gì khi sử dụng linker với đoạn DNA tạo dòng chứa một hay nhiều vị trí nhận biết của BamHI? Ở phản ứng cắt cần thiết với linker thì phân tử DNA cũng bị cắt thành nhiều đoạn (Hình 1.27). Những đoạn này có các đầu dính mong muốn, nhưng không có lợi, vì gene đã bị cắt nhỏ. Hình 1.27. Một vấn đề xảy ra khi sử dụng linker: So sánh tình trạng ở đây với
kết quả mong muốn khi cắt bằng BamHI ở hình 1.26b.
BamHI
Các vị trí cắt của BamHI bên trong phân tử

40
Phương pháp thứ hai là gắn với một adapter. Adapter cũng như linker là
olygonucleotide tổng hợp, ngắn, có một đầu bằng và một đầu dính (Hình 1.28a). Để đạt được mục đích là đoạn DNA tạo dòng có được đầu dính thì đầu bằng của adapter phải được nối với một đầu của phân tử DNA. Phương pháp này đơn giản hơn so với gắn linker, nhưng cũng gặp khó khăn, vì các đầu dính của các adapter gắn lại với nhau, do các cặp base bổ sung, tạo nên dimer (Hình 1.28b).
Lời giải cho vấn đề nằm ở cấu trúc hoá học ở đầu cuối của adaptor. Thông thường hai đầu của một sợi polynucleotide có cấu tạo hoá học khác nhau (Hình 1.29a). Đầu cuối được gọi là 5 mang một gốc phosphate (5-P), một đầu khác được gọi là 3 mang một nhóm hydroxyl (3-OH). Trong DNA hai sợi nằm đối song (Hình 1.29b), như vậy ở mỗi đầu của phân tử có một đầu cuối 5 và 3. Phản ứng nối thường xảy ra giữa 5-P và 3-OH (Hình 1.29c).

41
Hình 1.28. Adapter và những khó khăn khi sử dụng a. Một adapter điển hình b. Adapter có thể nối lại với nhau tương tự linker c. Phân tử có đầu bằng sau khi gắn thêm adapter vẫn có đầu bằng, vì vậy cần phải cắt bằng một enzyme hạn chế
a. Một adapter điển hình
b. Adapter có thể nối lại với nhau
c. Phân tử DNA mới luôn luôn có 2 đầu bằng
Phân tử đầu bằng
Adaptor
DNA-ligase
Adapter nối lại với nhau
Đầu dính (BamHI)

42
Người ta tổng hợp adapter có đầu bằng ở dạng tự nhiên của nó, nhưng đầu dính ở 5 lại thiếu gốc phosphater, nghĩa là ở 5' mang nhóm OH (5-OH, Hình 1.30a). Ligase không thể tạo được liên kết phosphodieste giữa nhóm 3-OH và nhóm 5-OH. Vì vậy, các adapter không gắn lại với nha
Hình 1.29 Sự khác nhau giữa đầu 5’ và 3’
a. Cấu trúc của sợi polynucleotide: sự khác nhau giữa đầu 5’-phosphate và đầu 3’-OH
Một nucleotide Base
Base
Base
b. Trong DNA xoắn kép hai sợi polynucleotide nằm đối song
c. Sự gắn kết xảy ra giữa 5’-phosphate và 3’-OH

43
Adaptor có thể nối kết với các đoạn DNA tạo dòng nhờ ligase. Sau đó nhóm
OH ở C5' (5-OH) của đầu dính được chuyển sang dạng phosphate (5'-P) nhờ enzyme polynucleotide kinase sang dạng tự nhiên 5-P. Như vậy đoạn DNA tạo dòng có lại đầu dính bình thường để có thể nối kết với vector
Hình 1.30 Sử dụng adapter a. Cấu trúc của adapter có đầu 5’ đã biến đổi
a. Cấu trúc của một “đầu gắn” (adapter)
b. Gắn bằng adapter
Đầu 5’-OH được biến đổi
Phân tử có đầu bằng Adapter
DNA-ligase
Polynucleotide kinase
Đầu 5’-phosphate
44
b. Đoạn DNA tạo dòng đầu bằng đã biến thành đầu dính sau khi gắn thêm adapter
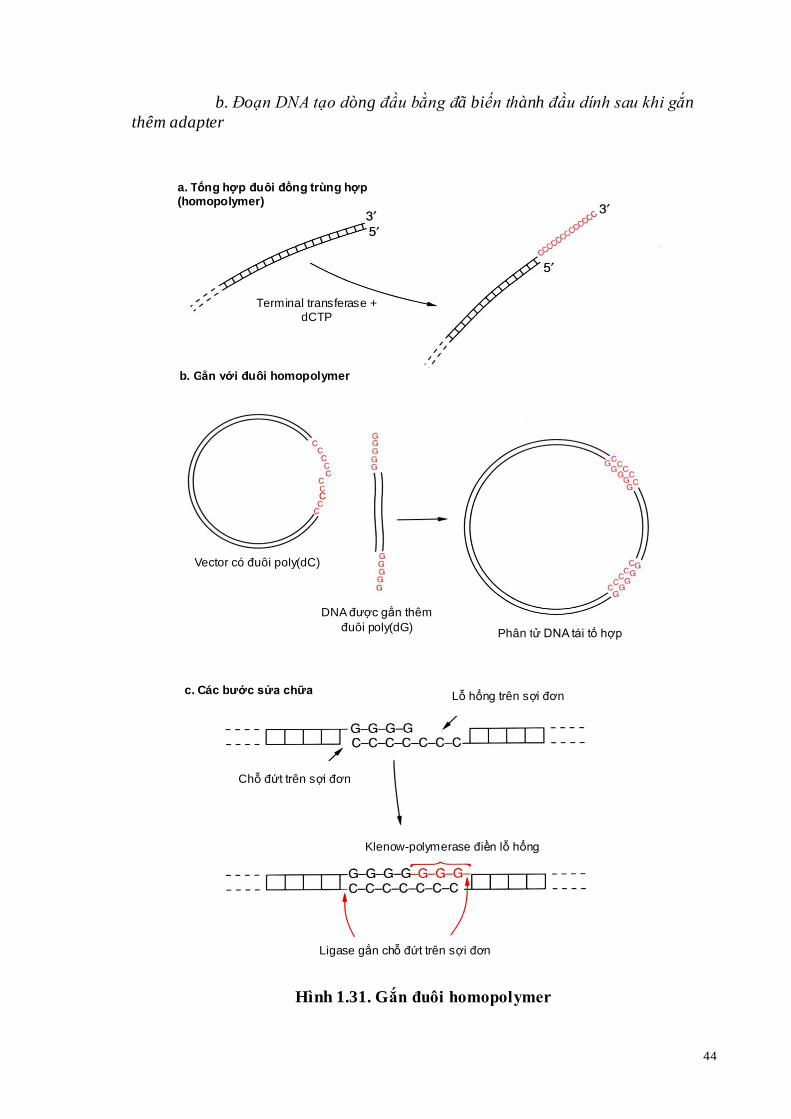
Hình 1.31. Gắn đuôi homopolymer
a. Tổng hợp đuôi đồng trùng hợp (homopolymer)
Terminal transferase + dCTP
b. Gắn với đuôi homopolymer
Vector có đuôi poly(dC)
DNA được gắn thêm đuôi poly(dG) Phân tử DNA tái tổ hợp
c. Các bước sửa chữa Lỗ hổng trên sợi đơn
Chỗ đứt trên sợi đơn
Klenow-polymerase điền lỗ hổng
Ligase gắn chỗ đứt trên sợi đơn

45
a. Tổng hợp đuôi homopolymer b. Tạo phân tử DNA tái tổ hợp từ một vector và đoạn DNA có gắn
đuôi c. Sửa chữa một phân tử DNA tái tổ hợp
c. Tạo đầu dính bằng việc gắn đuôi homopolymer
Một phương pháp hoàn toàn khác để tạo đầu dính ở phân tử DNA đầu bằng là gắn đuôi homopolymer (tailing). Homopolymer là một chuỗi polymer, được tạo nên từ nhiều đơn phân giống nhau, ví dụ chỉ gồm các nucleotide là deoxyguanosin, được gọi là poly deoxyguanosine hoặc poly (dG).
Khi gắn đuôi có sự tham gia của enzyme deoxynucleotide tranferase, các nucleoitde cùng loại lần lượt gắn vào đầu 3-OH của DNA sợi kép (Hình 1.31a).
Người ta thường gắn vào vector đuôi poly (dG) và đoạn DNA tạo dòng đuôi poly (dC). Trộn lẫn các phân tử này thì sự cặp đôi giữa các đuôi sẽ xảy ra theo nguyên tắc bổ sung.
Trong thực tế đuôi poly (dG) và poly (dC) có độ dài không chính xác bằng nhau, như vậy khi các đuôi này nối lại với nhau sẽ có những lỗ hổng trên sợi đơn (Hình 1.31c). Vì vậy, sự nối kết xảy ra hai bước: Trước hết đoạn Klenow lấp đầy các lỗ hổng và sau đó DNA-ligase tạo liên kết phosphodiester. Các đuôi homopolymer có chiều dài hơn 20 nucleotide gắn lại với nhau rất bền vững. V. Vector tạo dòng
Vấn đề trung tâm của thí nghiệm tạo dòng là vector tạo dòng. Một phân tử DNA được coi là vectơ tạo dòng khi có khả năng tồn tại và tái sinh trong tế bào chủ.
Tạo dòng DNA vẫn còn là một vấn đề mới mẽ, nhưng kỹ thuật này đã phát triển rất nhanh. Ngày nay, có nhiều loại vector tạo dòng khác nhau. Hầu hết chúng bắt nguồn từ các plasmid tồn tại trong tự nhiên hoặc virus, nhưng được biến đổi theo những cách khác nhau. Tuỳ theo mục đích tạo dòng mà lựa chọn vector cho phù hợp.
Sau đây sẽ mô tả các nhóm vector tạo dòng quan trọng nhất của E.coli. 1. Vector plasmid
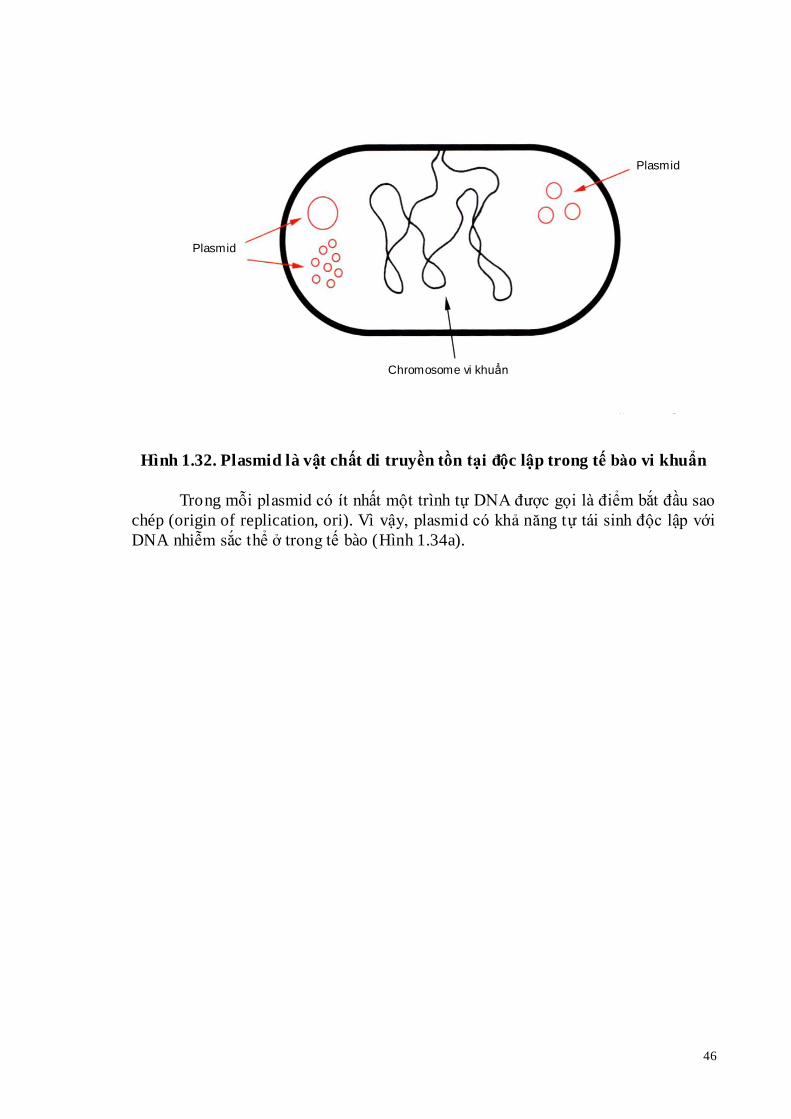
Plasmid là phân tử DNA dạng vòng, tồn tại độc lập trong tế bào vi khuẩn (Hình 1.32). Hầu hết plasmid chứa một hoặc hai gene mã hóa cho các đặc tính có lợi cho vi khuẩn. Ví dụ khả năng sống sót của vi khuẩn ở nồng độ gây chết của chất kháng sinh chloramphenicol hoặc ampicillin là nhờ plasmid mang các gene kháng kháng sinh. Các gene này được sử dụng làm gene chọn lọc (marker).
46
Hình 1.32. Plasmid là vật chất di truyền tồn tại độc lập trong tế bào vi khuẩn
Trong mỗi plasmid có ít nhất một trình tự DNA được gọi là điểm bắt đầu sao
chép (origin of replication, ori). Vì vậy, plasmid có khả năng tự tái sinh độc lập với DNA nhiễm sắc thể ở trong tế bào (Hình 1.34a).
Plasmid
Plasmid
Chromosome vi khuẩn

47
Hình 1.33. Kháng kháng sinh là đặc điểm chọn lọc của plasmid
Gene kháng ampicillin
Gene kháng tetracycline Gene kháng kanamycin
Một số tế bào E. coli, trong đó một số chứa RP4
Tế bào chứa plasmid
Tế bào không chứa plasmid
Dung dịch nuôi cấy không có kháng sinh
Dung dịch nuôi cấy chứa tetracilin (50 g/ml)
Tất cả các tế bào đều phát triển
Chỉ có tế bào chứa RP4 phát triển

48
RP4 mang các gene kháng ampicillin, tetracycline và kanamycin. Trong dung dịch nuôi cấy chứa một hoặc nhiều kháng sinh ở nồng độ gây độc, chỉ những tế bào
E.coli nào chứa RP4 (hoặc plasmid tương tự) thì mới sống sót.
Các plasmid nhỏ thường sử dụng những enzyme sao chép của tế bào để tái sinh. Ngược lại những plasmid lớn hơn, trong một số trường hợp chứa các gene mã hoá cho các enzyme đặc hiệu cho sự sao chép của plasmid.
Một số plasmid cũng có thể sao chép cùng với nhiễm sắc thể của vi khuẩn sau khi hoà nhập (Hình 1.34b). Những plasmid này được gọi là episome, có thể tồn tại bền vững qua nhiều lần phân chia tế bào, nhưng đến một lúc nào đó chúng như là yếu tố độc lập. Sự hoà nhập cũng là đặc điểm quan trọng của nhiễm sắc thể phage.
a. Plasmid không hòa nhập
Plasmid
Tế bào phân chia
Chromosome vi khuẩn
Chromosome kết hợp với plasmid Chromosome vi khuẩn Plasmid
Tế bào phân chia
b. Episome

49
Hình 1.34. Sự tái bản của plasmid không hòa nhập (a) và của episome (b)
Độ lớn và số lượng bản sao là hai đặc điểm quan trọng của plasmid tạo dòng. Các plasmid tồn tại trong tự nhiên có độ lớn từ 1 đến 250 kb (Bảng 1.3), Các vector tạo dòng nên có độ lớn nhỏ hơn 10 kb. Tuy nhiên các plasmid lớn hơn với những điều kiện nhất định cũng phù hợp cho mục đích tạo dòng.
Bảng 1.3 Độ lớn của một số plasmid quan trọng
Độ lớn Plasmid Dài (kb) Trọng lượng phân tử
(106 d*) Nguồn vi sinh vật
pBR345 0,7 0,46 E. coli p BR322 4,362 2,9 E. coli
CoIEI 6,4 4,2 E. coli RP4 54 36 Pseudomonas
F 95 63 E. coli TOL 117 78 Pseudomonas putida
pTiAch5 213 142 A. tume faciens d*: tương ứng 1,66018 x 10-27 kg
Số lượng bản sao là số lượng của mỗi loại plasmid tồn tại trong một tế bào vi khuẩn. Yếu tố nào ảnh hưởng đến số lượng bản sao, vẫn chưa được giải thích chính xác. Số lượng này biến động từ 1 (ở những plasmid lớn) đến 50 hoặc nhiều hơn. Nói chung, một vector tạo dòng nên có nhiều bản sao trong một tế bào, như vậy người ta có thể tách được phân tử DNA tái tổ hợp với một khối lượng lớn. 1.1. Vector tạo dòng pB322
Vector tạo dòng đơn giản nhất và được sử dụng nhiều nhất bắt nguồn từ plasmid của vi khuẩn, đặc biệt là E. coli. Đặc tính của các vector này là dễ làm sạch, biến nạp đạt hiệu quả cao, có các gene chọn lọc phù hợp và có thể tiếp nhận các đoạn DNA tạo dòng lớn (đến 8 kb). Một trong những vector tạo dòng đầu tiên được phát triển và hiện nay vẫn được sử dụng rộng rãi là pBR322. a. Cách gọi tên các vector tạo dòng plasmid Tên “pBR322” có nghĩa là:
- p là plasmid - BR: Tên viết tắt của người thiết kế ra plasmid: vector này được 2 nhà khoa
học là Bolivar và Rodriguez phát triển - 322: số để phân biệt plasmid này với plasmid khác (ví dụ pBR325, pBR327,
pBR28..) b. Ưu điểm của pBR322
Bản đồ vật lý và di truyền của pBR322 (Hình 1.35) đã nói lên tại sao plasmid này được sử dụng nhiều.

50
Hình 1.35. Bản đồ của pBR322 với vị trí của gene kháng ampicillin (ampR) và tetracycline (tetR), điểm khởi đầu sao chép (ori) và một số vị trí cắt quan trọng
của các enzyme hạn chế.
Ưu điểm đầu tiên của pBR322 là có độ lớn nhỏ (4.363 bp), có nghĩa là không những dễ dàng tinh sạch vector mà cả các phân tử DNA tái tổ hợp. Khi đoạn DNA tạo dòng dài 6 kp thì phân tử pBR322 tái tổ hợp vẫn còn có độ lớn phù hợp.
Đặc điểm thứ hai của pBR322 là mang 2 nhóm gene kháng kháng sinh. Người ta có thể sử dụng gene kháng ampicillin hoặc tetracyclline để chọn lọc tế bào chứa plasmid. Trong hai gene này có những vị trí cắt duy nhất của các enzyme hạn chế để tạo dòng. Nếu đưa một đoạn DNA ngoại lai vào vị trí cắt của PstI, PvuI hoặc ScaI thì gene ampR bị bất hoạt và khi sử dụng một trong 8 enzyme khác (ví dụ BamHI hoặc HindIII) thì khả năng kháng tetracyclline bị mất. Vì có nhiều vị trí cắt khác nhau trong vùng gene chọn lọc mà có thể tạo dòng trong pBR322 với nhiều đầu dính khác nhau.
Ưu điểm thứ ba của pBR322 là có số lượng bản sao lớn. Trong tế bào E. coli được biến nạp trung bình có 15 bản sao, số lượng này có thể tăng lên 1000 đến 3000 khi nhân plasmid có sử dụng chloramphenicol để kìm hãm tổng hợp protein. Nuôi cấy E. coli cung cấp DNA tái tổ hợp pBR322 với tỷ lệ cao. c. Nguồn gốc của pBR322
Vector tạo dòng pBR322 được thiết kế nhiều lần nên cuối cùng có được nhiều đặc điểm có lợi. Các bước tạo plasmid này được phác họa ở hình 1.36a. Có thể dễ dàng nhận ra rằng, để tạo ra plasmid này đã tiêu phí nhiều thời gian và phải có tay nghề cao. Hình 1.36b cho thấy plasmid pBR322 chứa các đoạn DNA từ 3 plasmid khác nhau tồn tại trong tự nhiên. Gene ampR có nguồn gốc từ plasmid R1 tìm thấy trong E. coli tự nhiên và gene tetR từ plasmid R6-5. Điểm khởi đầu sao chép của pBR322 để nhân bản vector trong tế bào chủ, có nguồn từ plasmid pMB1.

51
Hình 1.36 Nguồn gốc của pBR322 a. Các bước tạo plasmid b. Nguồn gốc của từng phần trong plasmid
1.2. pUC8: Plasmid có gene chọn lọc LacZ’ Vector này được nêu lên trong mối liên quan với sự xác định thể tái tổ hợp, ở
đây đoạn DNA chèn làm gián đoạn gene mã hoá cho -galactosidase. pUC8 cũng có nguồn gốc từ pBR322, điểm khởi đầu sao chép và gene ampR được giữ lại . Các vị trí chèn được tìm thấy ở pUC8 trong một đoạn ngắn của gene lacZ.
a. Các bước tạo pBR322
Ligation hai đoạn
b. Nguồn gốc của pBR322

52
Hình 1.37. Plasmid pUC8
Plasmid tái tổ hợp pUC8
a. Các vị trí cắt ở pUC8
b. Các vị trí cắt ở pUC10
c. Chuyển một đoạn DNA của pUC8 đến M13mp8
DNA mới Những vị trí cắt
Cắt bằng BamHI và EcoRI Gắn kết
DNA mới
Cắt bằng BamHI và EcoRI
Plassmid tái tổ hợp M13mp8

53
a. Các vị trí cắt trong gene LacZ’ của pUC8 b. Các vị trí cắt trong pUC10 c. Chuyển một đoạn DNA từ pUC8 đến M13mp8
pUC8 có ba ưu điểm quan trọng đã làm cho nó trở thành một trong những vector tạo dòng ưa thích nhất đối với E. coli. Ưu điểm thứ nhất là khi thiết kế plasmid đã tình cờ xuất hiện một đột biến ở điểm sao chép, nên không cần khuếch đại mà số lượng bản sao có thể đạt tới 500-700. Điều này có tác động rõ rệt lên hiệu quả khai thác DNA tạo dòng thu nhận được từ tế bào E. coli sau khi biến nạp pUC8 tái tổ hợp.
Ưu điểm thứ hai là có thể xác định các tế bào tái tổ hợp trong một bước duy nhất. Đơn giản là người ta nuôi chúng lên môi trường thạch có chứa X-gal. Ngược lại ở pBR322 việc xác định thể tái tổ hợp phải qua hai bước. Khuẩn lạc tái tổ hợp được xác định khi so sánh vị trí của chúng trên đĩa thạch có chứa kháng sinh này với đĩa nuôi chứa kháng sinh khác. Thí nghiệm tạo dòng với pUC8 tiết kiệm được một nửa thời gian so với pBR322.
Ưu điểm thứ ba ở chỗ là sự tập trung các vị trí cắt hạn chế. Bằng cách này
người ta có thể tạo dòng đoạn DNA với hai đầu dính khác nhau (ví dụ EcoRI và BamHI). Các vector pUC khác chứa tổ hợp các vị trí cắt hạn chế khác nhau cho phù hợp với sự đa dạng của các đọan DNA tạo dòng. Ngoài ra những vector này chứa tổ hợp các vị trí cắt hạn chế như loạt vector M13mp. Vì vậy, có thể đưa DNA trực tiếp vào vector M13mp sau khi tạo dòng trong vector pUC và sau đó có thể xác định trình tự hoặc đột biến gene in vitro (Hình 1.38c). 1.3. pGEM3Z: Sự phiên mã DNA tạo dòng in vitro
Tương tự pUC vectơ pGEM3Z mang gene ampR và LacZ. Trong gene LacZ có một nhóm các vị trí cắt hạn chế và gần như có cùng độ lớn

54
Hình 1.38. Plasmid pGEM3Z a. Bản đồ của vector b. Tổng hợp RNA in vitro.
Ngoài ra, ở pGEM3Z còn có 2 trình tự DNA ngắn là các vị trí nhận biết và
gắn của RNA-polymerase. Hai trình tự promoter này nằm về hai phía của các vị trí cắt, nơi mà các đoạn DNA tạo dòng được chèn vào. Khi đưa vào ống nghiệm pGEM3Z tái tổ hợp và RNA-polymerase thì xảy ra quá trình phiên mã làm xuất hiện các RNA của đoạn DNA tạo dòng (Hình 1.38b). Các RNA này có thể được sử dụng như là những mẫu dò hoặc trong nghiên cứu quá trình hoàn thiện RNA (RNA processing) và tổng hợp protein.
Ở đây không đề cập đến trình tự thông thường của hai promoter trong pGEM3Z và của các vector tương tự được nhận biết bởi polymerase của E. coli. Một promoter đặc hiệu cho RNA-polymerase của thực khuẩn thể T7, và promoter còn lại cho RNA-polymerase của phage SP6. Các RNA-polymerase này được tạo ra sau khi E. coli nhiễm một trong hai loại phage này và thực hiện phiên mã các gene
a. pGEM32
T7-promoter
T7-promoter
SP6-promoter b. Tổng hợp DNA in vitro
Đoạn DNA chèn vào
T7-DNA polymerase
Bản sao RNA

55
phage. Chúng được sử dụng cho phiên mã in vitro, vì rất hoạt động (chu kỳ sinh tan kéo dài chỉ 20 phút) và trong một phút có thể tạo ra 1-2 g RNA, lượng DNA nhiều hơn so với enzyme bình thường ở E. coli. 2. Vector tạo dòng trên cơ sở thực khuẩn thể M13
Một yêu cầu bắt buộc ở vector tạo dòng là khả năng sao chép trong tế bào chủ. Ở vector plasmid thì điều này đơn giản, vì ở chúng một đoạn trình tự DNA ngắn làm nhiệm vụ khởi đầu điểm sao chép và nó cung cấp phần lớn hoặc thậm chí tất cả các enzyme cần thiết cho sự sao chép trong tế bào chủ. Ở thực khuẩn thể M13 thì sự sao chép phức tạp hơn. Phân tử DNA của thực khuẩn thể nhìn chung mang nhiều gene hơn, trong đó có gene mã hóa cho phần vỏ và những enzyme sao chép đặc hiệu cho phage. Thay đổi hoặc loại trừ một trong những gene này làm ảnh hưởng hoặc phá huỷ khả năng sao chép của chúng. Vì vậy, ở sự thay đổi của DNA phage có giới hạn hơn và các vector tạo dòng phage nhìn chung chỉ khác biệt nhỏ so với phân tử ban đầu.
Khó khăn ở việc thiết kế vector tạo dòng phage rõ hơn khi quan sát M13. Hệ gene của M13 bình thường là 6,4 kb, phần lớn nhất trong đó chứa 10 gene nằm sát nhau (Hình 1.39) là không thể thiếu được cho toàn bộ quá trình sao chép phage. Chỉ có một trình tự duy nhất có 507 nucleotide là người ta chèn vào mà không làm cho các gene khác bị ảnh hưởng và trong vùng này điểm khởi đầu sao chép phải nguyên vẹn.
Như đã đề cập M13 có một ưu điểm rất hấp dẫn: với chúng người ta có thể thu được DNA tạo dòng ở dạng sợi đơn. Thực tế này là một kích thích để phát triển vector tạo dòng M13 2.1. Phát triển vector tạo dòng M13mp2
Bước đầu tiên ở sự thiết kế vector tạo dòng M13 là đưa gene LacZ’ vào. Như vậy, xuất hiện M13mp1 có khả năng tạo nên vết tan màu xanh trên môi trường thạch có chứa X-gal (Hình 1.40a).
M13mp1 chỉ chứa một vị trí sao chép đơn giản trong gene LacZ’, và có một hexanucleotide GGATTC ở gần điểm khởi đầu của gene , bằng việc thay đổi một nucleotide duy nhất sẽ xuất hiện trình tự GAATTC, là trình tự cắt của EcoRI. Sự thay đổi này thực hiện nhờ in vitro-mutagenease và kết quả là tạo ra vector M13mp2 (Hình 1.40b). Gene LacZ’ của M13mp2 thay đổi rất ít, tuy nhiên -galatosidase mà tế bào nhiễm bởi M13mp2 tạo ra là hoàn toàn hoạt động.
M13mp2 là vector tạo dòng đơn giản nhất của M13, người ta có thể đưa vào vị trí tạo dòng đoạn DNA có đầu dính của EcoRI và thể tái tổ hợp được nhận ra trên môi trường agar chứa X-gal là những vết tan trong suốt.
Hình 1.39. Hệ gene của M13 với các vị trí của gene I đến X

56
Hình 1.40 Cấu tạo của M13mp1 (a) và M13mp2 (b) từ geneom M13
M13mp7 - Nh ng v trí t o dòng i x ng
a. Cấu tạo của M13mp1 Cắt bằng enzyme giới hạn, nối (ligation)
b. Cấu tạo của M13mp2
Đột biến in vitro
Bắt đầu gene lacZ’ trong M13mp1
Bắt đầu gene lacZ’ trong M13mp2

57
Hình 1.41. Cấu tạo của M13mp7 a. Polylinker, b. Polylinker được đưa vào vị trí EcoRI của M13mp2
Bước tiếp theo để phát triển vector M13 người ta đưa vào trong gene LacZ’ các vị trí cắt bổ sung. Cho mục đích này người ta đưa vào polylinker là oligonucleotide ngắn, chứa các vị trí cắt và có các đầu dính cho EcoRI (Hình 1.42a). Polylinker được đưa vào vị trí của EcoRI của M13mp2 (Hình 1.42b) tạo nên M13mp7 (Hình 1.42b), một vector phát triển cao hơn với 4 vị trí tạo dòng (EcoRI, BamHI, SalI và PstI). Polylinker được đưa vào như thế nào đó để không làm hỏng gene LacZ’ và mặc dù -galatosidase bị thay đổi nhưng vẫn hoạt động.
a. Polylinker
b. Cấu tạo của M13mp7
Vị trí cắt giới hạn
EcoRI, ligase
Polylinker

58
Hình 1.42. Tạo dòng với M13mp7
Toàn bộ hoặc một polylinker a. Sự cắt giới hạn của M13mp7
Polylinker
Đầu dính
EcoRI, BamHI hoặc SalI
b. Nối với DNA mới: Các khả năng sản phẩm: 1. Đưa vào một DNA mới 2. Đưa vào một polylinker 3. Không đưa vào (tự đóng vòng)
c. Màu của vết tan trên agar có chưa X-gal
Gene LacZ’ bị gián đoạn không có -gal vết tan trong suốt
Gene LacZ’ gắn lại -gal vết tan màu xanh
Gene lacZ’ gắn lại -gal vết tan màu xanh

59
M13mp7 với vị trí tạo dòng đối xứng có ưu điểm lớn: người ta có cắt ra những đoạn DNA đã được đưa vào vị trí của BamHI, SalI hoặc PstI. (Hình 1.43). Chỉ
một số ít vector cho phép thu nhận lại DNA tạo dòng dễ dàng như vậy.
Khi xử lý M13mp7 với EcoRI, BamHI hoặc SalI thì polylinker bị cắt ra hoàn toàn hoặc một phần (Hình 1.42a). Ở sự gắn kết khi có những đoạn DNA mới có thể xảy ra 3 trường hợp (Hình 1.42b).
1. Đoạn DNA mới được chèn vào 2. Polylinker được chèn vào 3. Các đầu cuối của vector gắn lại với nhau mà không có đoạn DNA
Khi DNA ngoại lai được vào thì -galactosidase không được tổng hợp, nên vết tan tái tổ hợp trên môi trường agar có màu trong suốt (Hình 1.42c). Ngược lại, khi polylinker được tiếp nhận xuất hiện một thể mới của vector M13mp7 và tạo nên vết tan màu xanh. Nhưng điều gì xảy ra khi vector tự đóng lại mà không tiếp nhận DNA ngoại lai hoặc polylinker? Ở đây sự thiết kế polylinker có một vai trò quan trọng. Không phụ thuộc vào những vị trí cắt nào được sử dụng, sự tự đóng vòng dẫn đến gene mã hoá cho -galactosidase hoạt động (Hình 1.42c), tạo nên vết tan màu xanh. Rõ ràng rằng chỉ lựa chọn các phage có vết tan trong suốt.
Đoạn cắt bằng Sau3AI
Đầu cuối của Sau3AI (GATC) Cắt bằng BamHI
(đầu cuối: GATC)
Ligation, tạo dòng
1. Những vị trí nhận biết của BamHI không gắn kết lại 2. M13 chứa nhiều vị trí nhận biết của Sau3AI
Đoạn cắt bằng Sau3AI
Cắt bằng EcoRI
3. Thu nhận lại những đoạn khi cắt bằng EcoRI
Đầu cuối dính của EcoRI
Vector đươc đóng vòng

60
Hình 1.43. Thu nhận một đoạn DNA tạo dòng từ M13mp7 tái tổ hợp bằng việc cắt hạn chế ở các vị trí ngoài polylinker
3. Vector tạo dòng là phage
Bacteriophage, gọ i tắt là phage, là những virus chỉ nhân lên trong vi khuẩn nên được gọi là thực khuẩn thể.
Phage được chia làm 2 nhóm: Phage độc (virulence) và phage ôn hòa (temperaance)
Phage độc chỉ có cách phát triển duy nhất là làm tan tế bào chủ. Phage ôn hòa có hai cách phát triển: Phá vỡ tế bào hoặc không phá vỡ tế bào. Khi phage độc khi nhiễm vào tế bào chủ sẽ bơm DNA của nó vào bên trong
tế bào. Khi DNA trần đã xâm nhập vào trong tế bào, một loạt quá trình phiên mã xảy ra, sản sinh ra nhiều bản sao của hệ gene phage cũng như các protein cấu trúc. Sau đó DNA được đóng gói vào vỏ tạo phân tử phage hoàn chỉnh. Phage hoàn tất chu trình khi enzyme lysozyme được tạo ra tiêu hóa vách tế bào làm cho tế bào vi khuẩn bị vỡ ra. Phage độc luôn phá vỡ tế bào chủ trong quá trình sống tạo nên vệt tan hoàn toàn (vệt tan trong). Chu trình này được gọi là chu trình tan.
Phage ôn hòa khi nhiễm vào tế bào chủ chúng sẽ theo chu trình tan, tương tự phage độc, hoặc chu trình tiềm tan.
Phage thuộc nhóm phage ôn hòa, là DNA sợi đôi, ở dạng mạch thẳng, có chiều dài khoảng 49 kb, mã hóa cho 46 gene. Toàn bộ gene đã được giải trình tự, ở hai đầu có các đoạn ngắn (12 bp) sợi đơn bổ trợ cho nhau, được gọi là trình tự cos. Các đoạn này là những “đầu dính” tạo điều kiện cho hệ gene trở thành mạch vòng sau khi nhiễm vào vi khuẩn.
Khi phage nhiễm vào tế bào chủ, nếu tế bào chủ nghèo dinh dưỡng phage sẽ đi vào chu trình tiềm tan và ngược lại. Tuy nhiên nếu sử dụng làm vector thì phage phải đi vào chu trình tan mới có thể nhân dòng gene được.

61
Hình 1.44 DNA của phage λ ở dạng mạch thẳng và mạch vòng
Cấu tạo của phage λ gồm 1 đầu và 1 đuôi. Phần đầu có dạng nhiều mặt, chứa
DNA, đuôi cần cho sự gắn phage vào bề mặt của vi khuẩn và đưa DNA vào trong tế bào. Trước đây phage λ tự nhiên không được sử dụng làm vector vì các lý do:
- Độ lớn phân tử -DNA chỉ có thể được vượt quá 5%, tương ứng với đoạn DNA được đưa thêm vào là 3 kb. Những phân tử có độ dài tổng số vượt quá 52 kb sẽ không được gói ở trong cấu trúc đầu của . Độ lớn của đoạn DNA muốn tạo dòng trong vector không thay đổi là có giới hạn (Hình 1.45a).
- Hệ gene của là lớn nên chứa nhiều vị trí nhận biết của các enzyme hạn chế. Bằng sự cắt hạn chế người ta không thể cắt - DNA để đưa đoạn DNA mới vào vì phage sẽ bị cắt ra thành nhiều đoạn (Hình 1.45b).
Con đường để phát triển vector tạo dòng ở sự phát hiện là có thể cắt bỏ một đoạn lớn ở giữa phân tử -DNA mà không làm ảnh hưởng đến khả năng của phage lây nhiễm tế bào E. coli. Người ta cắt ra phần “không quan trọng”, ở hình 1.46 nó nằm giữa vị trí 20 đến 35, toàn bộ hoặc một phần đã giảm độ dài của -DNA đến 15 kb. Điều đó có nghĩa là bây giờ người ta có thể đưa vào đoạn DNA mới cho đến 18 kb.
a. Giới hạn độ lớn
Geneome của là 49 kb
Thể kết hợp có thể (> 52 kb)
DNA lạ > 3 kb
Quá lớn không thể gói được
b. Nhiều vị trí cắt hạn chế
Gói
EcoRI
EcoRI
Hỗn hợp các phân tử khác nhau
Ligation

62
Hình 1.45. Hai vấn đề phải được giải quyết trước khi phát triển hệ thống vector tạo dòng λ
a. Sự giới hạn độ lớn của hệ gene cần thiết cho đóng gói ở phần đầu
b. DNA của phage λ hầu như có tất cả các vị trí nhận biết cho endonuclease
Vùng “ không quan trọng” chứa phần lớn nhất của các gene tham gia vào sự
kết hợp và cắt ra của tiền phage từ chromosome của E. coli. Hệ gene được cắt ngắn vì vậy không có khả năng tiềm tan mà chỉ còn chu kỳ ly tan. Đó là đặc điểm có giá trị cho vector tạo dòng, vì bằng cách này vết tan tạo nên mà không cần lây nhiễm.
Hình 1.46. Bản đồ gene của phage λ. Vị trí của các gene quan trọng nhất
và chức năng của các nhóm gene Bằng sự chọn lọc tự nhiên có thể phân lập phage thiếu các vị trí giới hạn xác định.
Mặc dù hệ gene đã được làm ngắn vẫn còn chứa nhiều vị trí cắt của các enzyme hạn chế. Khi muốn loại bỏ một hoặc hai vị trí cắt người ta sử dụng kỷ thuật in vitro-mutagenease. Bằng cách này từ trình tự GAATTC là vị trí cắt của EcoRI, trình tự GGATTC được tạo ra và như vậy không còn được nhận biết bởi enzyme này nữa. Khi mới bắt đầu phát triển vector thì kỷ thuật này có ý nghĩa, nhưng ngày nay không còn là phương pháp hữu hiệu khi muốn thay đổi nhiều hơn vài vị trí cắt trong một phân tử.
Để thu được các chủng mà không có các vị trí cắt không cần thiết, người ta sử dụng chọn lọc tự nhiên. Cho mục đích này sử dụng một chủng E. coli là ký chủ tạo ra EcoRI. Phần lớn các phân tử DNA của phage xâm nhiễm vi khuẩn, bị phá huỷ bởi enzyme hạn chế. Tuy nhiên một số phage vẫn tồn tại và làm xuất hiện vết tan. Những phage này là thể đột biến, vì ở chúng ví trí cắt của EcoRI (hoặc nhiều hơn) tự nhiên mất đi. Nhiều chu kỳ nhiễm dạng này tạo nên những phân tử thiếu phần lớn hoặc tất cả các vị trí nhận biết của EcoRI (Hình 1.47).

63
Hình 1.47. Phân lập phage λ, ở chúng thiếu vị trí cắt của EcoRI nhờ chọn lọc tự nhiên
Tuy nhiên về sau người ta đã phát hiện ở giữa các DNA của phage có vùng
đệm (stuffer) hay còn gọ i là phần trung tâm có chiều dài khoảng 1/3 chiều dài của phage, vùng này không quan trọng và nếu cắt nó đi thì vẫn không ảnh hưởng đến khả năng sinh sản của phage trong tế bào vi khuẩn, đó là phần quy định khả năng đi vào chu trình tiềm tan của phage . Vì vậy, người ta đã cắt bỏ đoạn stuffer để lắp các đoạn gene ngoại lai vào. 3.1. Vector thay thế và vector xen đoạn
5 vị trí của EcoRI
-DNA bình thường
Lây nhiễm tế bào E. coli, những tế bào này sản sinh EcoRI
Vết tan xuất hiện do phage đột biến
Chỉ có 3 vị trí cắt cho EcoRI
Rất ít vết tan
Sự lây nhiễm mới với phage đột biến
Không có vị trí EcoRI
Chủng phage đột biến thứ hai
Có nhiều vết tan hơn

64
Sau khi giải quyết được khó khăn về lượng DNA giới hạn được gói và nhiều vị trí hạn chế, các vector tạo dòng khác nhau dựa trên phage được phát triển.
Các vector này được phân thành hai nhóm chính là vector xen đoạn và vector thay thế.
a. Vector xen đoạn Vector xen đoạn: Trong vector xen đoạn người ta loại bỏ một đoạn dài của
vùng bỏ được và gắn hai tay của phân tử phage lại với nhau. Trên vector xen đoạn chỉ có một vị trí cắt duy nhất cho phép cắt và chèn DNA mục tiêu vào.
Độ lớn của đoạn DNA ngoại lai phụ thuộc vào độ lớn của vùng loại bỏ (Hình 1.49a). Hai vector xen đoạn đặc biệt ưa thích là:
- gt10 (Hình 1.49b và Hình 1.50) có thể tiếp nhận DNA mới cho đến 8 kb ở vị trí cắt duy nhất EcoRI trong gene cI. Vì gene này bị bất hoạt bởi DNA đưa vào nên phân biệt thể tái tổ hợp có vết tan trong suốt với các vết tan đục.
- ZAPII (Hình 1.49c) cho phép đưa vào DNA có độ lớn 10kb vào một trong 6 vị trí hạn chế trong một polylinker. DNA làm bất hoạt gene LacZ’ trong vector, thể tái tổ hợp không tạo nên vết tan màu xanh mà là trong suốt.
b. Vector thay thế Vector thay thế: Vector thay thế chứa hai trình tự nhận biết của 2 enzyme cắt
hạn chế khác nhau. Khi sử dụng đồng thời 2 enzyme cắt hạn chế này, vector sẽ bị cắt làm 3 đoạn. Đoạn giữa có thể thay thế bằng DNA mục tiêu. Đoạn giữa thường chứa các vị trí cắt hạn chế khác nhau vì vậy người ta dùng các enzyme hạn chế để cắt nhỏ nó ra tránh trường hợp các đoạn vector tự gắn lại với nhau. Nhìn chung vector thay thế có thể tiếp nhận những đoạn DNA lớn hơn các vector xen đoạn. Khả năng tiếp nhận đoạn DNA ngoại lai của vector xen đoạn là 5 kb và của vector thay thế là 20-25 kb.
a. Thiết kế vector xen đoạn λ
Hình 1.48. Vector xen đoạn λ: P là polylinker trong gene LacZ’ của λZAPII, polylinker chứa vị trí cắt duy nhất cho Sacl, Notl, Spel, EcoRI và Xhol.
Vòng được cắt ra
Cắt, gắn
40 kb
41 kb
-DNA (49 kb)
Vector xen đoạn (35-40 kb)
b. gt10
c. -Charron 16

65
Hình 1.49. Vector xen đoạn λgt10
DNA ngoại lai, cho đến 23 kb
EcoRI, BamHI, SalI hoặc kết hợp
c. EMBL4
Không tái tổ hợp (35 kb)
Vị trí của EcoRI
b. WES.B’
Gắn với DNA ngoại lai
Nhỏ nhất
Lớn nhất
Tái tổ hợp (37-52 kb)
DNA mới
DNA ngoại lai
Đoạn giữa
Cắt bằng enzyme hạn chế
a. Tạo dòng với vector thay thế

66
Hình 1.50. Vector thay thế λ
Hình 1.51. Vector thay thế λEMBL3
3.2.Thí nghiệm tạo dòng với vector xen đoạn hoặc thay thế Những thí nghiệm tạo dòng với vector thực hiện theo nguyên tắc như với
plasmid. Phân tử được cắt bằng một enzyme hạn chế để đưa DNA tạo dòng vào và sau đó chuyển nhiễm chúng vào E. coli khả biến (Hình 1.51a). Với thí nghiệm loại này vector phải ở dạng vòng, nghĩa là các vị trí cos của nó được gắn lại nhờ những liên hydrogene.
Phương pháp chuyển nhiễm phục vụ cho nhiều mục đích, nhưng hiệu quả không cao. Người ta sẽ nhận được nhiều thể tái tổ hợp hơn khi thay đổi một hoặc hai điểm như sau: Điểm thứ nhất là làm sạch hai nhánh của vector. Khi xử lý vector ở dạng thẳng với enzyme hạn chế thì xuất hiện hai đoạn: nhánh phải và nhánh trái. Trong thực tế ở tất cả vector nhánh trái dài hơn nhánh phải, vì vậy có thể tách chúng ra khi ly tâm theo gradient saccharose. Để tạo nên phân tử tái tổ hợp người ta trộn DNA tạo dòng với mẫu có chứa nhánh phải và nhánh trái. Ở phản ứng gắn xuất hiện nhiều dạng phân tử, trong đó có sự lặp lại nhiều lần trình tự (concatemere) nhánh trái-đoạn DNA tạo dòng- nhánh phải (Hình 1.51b). Đoạn DNA có độ dài phù

67
hợp được đóng gói in vitro. Như vậy có thể sản xuất trong ống nghiệm các phage tái tổ hợp, sau đó lây nhiễm với E. coli. Với phương pháp này và đặc biệt với phương pháp gói in vitro người ta thu được một số lớn các vết tan tái tổ hợp.
Hình 1.51. Các phương pháp khác nhau khi tạo dòng với vector phage λ a. Dạng vòng của λ-DNA được sử dụng như là plasmid
a. Tạo dòng với -DNA vòng
DNA mới
b. Tạo dòng với -DNA thẳng
Thể tái tổ hợp
Phân tử tái tổ hợp Chuyển nhiễm E. coli
Lây nhiễm E. coli
Vector xen đoạn dạng vòng
Hỗn hợp gói in vitro

68
b. Với nhánh phải và nhánh trái của geneom λ và đóng gói in vitro người ta thu được một số lượng lớn hơn các vết tan tái tổ hợp. 4. Các vector lai plasmid/phage 4.1. Cosmid
Cosmid là vector kết hợp các thuộc tính của phage và plasmid: Chúng có chứa các vị trí cos của phage nên cho phép DNA có thể bọc gói trong phần đầu của phage, đồng thời chúng lại có một điểm tái bản của plasmid cho phép tự nhân lên như các plasmid trong vi khuẩn. Hầu như toàn bộ phần DNA của phage bị cắt bỏ nên cosmid có thể mang những đoạn DNA ngoại lai có kích thước khá lớn, khoảng 40 – 45 kb. Sau khi các đoạn DNA được ghép nối, các cosmid tái tổ hợp sẽ được bọc gói thành các phân tử phage. Các phage này không thể nhân được như phage bởi chúng hầu như không còn DNA của phage nhưng vẫn duy trì khả năng lây nhiễm vào tế bào vi khuẩn và có khả năng tự tái bản trong tế bào vi khuẩn do có mang điểm khởi đầu sao chép của plasmid.
Một cosmid về cơ bản là một plasmid có mang một vị trí cos (Hình 1.52a). Ngoài ra, nó phải có gene marker chọn lọc, ví dụ gene kháng ampicillin và một điểm bắt đầu sao chép plasmid, vì cosmid không chứa các -gene và vì vậy không tạo nên những vết tan. Trên môi trường chọn lọc xuất hiện nhiều dòng vi khuẩn, chính xác như ở vector plasmid.
a. Một cosmid điển hình
b. Tạo dòng với pJB8
pJB8 dạng vòng pJB8 dạng thẳng
Cắt bằng BamHI
Gói in vitro
Ligation
Các khuẩn lạc chứa phân tử pJB8 tái tổ hợp, dạng vòng
Lây nhiễm E. coli
Cosmid-DNA tái tổ hợp
-Partikel
DNA mới
Môi trường chứa ampicilin

69
Hình 1.52. Một cosmid điển hình và sử dụng nó khi tạo dòng với các
đoạn DNA lớn hơn Một thí nghiệm tạo dòng với cosmid xảy ra như sau (Hình 1.52b): Người ta
cắt cosmid ở một vị trí giới hạn duy nhất và đưa DNA ngoại lai vào. Đoạn này thường được tạo ra bằng cắt từng phần bằng một enzyme hạn chế, vì nếu phân giải hoàn toàn sẽ xuất hiện những phân tử mà tạo dòng trong cosmid là quá nhỏ. Phản ứng gắn kết được xảy ra làm xuất hiện các concatemere. Khi đoạn DNA tạo dòng có độ dài chính xác, các vị trí cos được cắt khi đóng gói in vitro và cosmid tái tổ hợp được đưa vào hạt phage. Với những hạt này người ta lây nhiễm tế bào E. coli, nhưng không tạo ra vết tan. Đưa những tế bào nhiễm này lên một môi trường chọn lọc và để cho những dòng kháng kháng sinh phát triển. Tất cả những dòng này là thể tái tổ hợp, vì những cosmid thẳng, không tái tổ hợp có kích thước nhỏ nên không được gói trong phage. 4.2. Phagemid
Phagemid là vector được cấu tạo nên trên cơ sở plasmid pBR322 gắn thêm đoạn khởi đầu sao chép (ori) của phage f1, một đoạn có chứa các promoter của phage T3, T7 và polylinker. Điển hình của phagemid là nhóm plasmid bluescript.
Hình 1.53. pBluescript SK
VI. Tạo dòng gene
Sau khi gắn một gene, một trình tự DNA cần thiết vào vector tạo dòng, tạo các phân tử DNA tái tổ hợp và đưa vào tế bào nhận (tế bào chủ), thì tiếp theo là tạo dòng gene nhằm thu một lượng lớn các bản sao của gene hoặc trình tự DNA cần thiết. Tạo dòng gene bao gồm quá trình tách và nhân dòng gene.
Có nhiều phương pháp tạo dòng gene khác nhau. Tạo dòng in vitro (in vitro genee cloning) là tách chiết các gene cần thiết từ bộ gene vi sinh vật, động vật, thực vật,... tạo vector tái tổ hợp và nhân các gene đó trong tế bào chủ.
Tạo dòng in silico là sự tổng hợp, phân tích các kết quả tạo dòng gene in vitro, rồi dựa vào cơ sở dữ liệu từ ngân hàng gene để lựa chọn các dòng gene ưu việt nhất, đưa vào ứng dụng trong thực tiễn sản xuất hoặc nghiên cứu.

70
Tạo dòng gene in vtro bao gồm tạo dòng gene đã biết và tạo dòng gene chưa biết. Tạo dòng gene đã biết là tạo dòng các gene đã biết kích thước hoặc vị trí của gene trong bộ gene. Tạo dòng gene chưa biết là nhân gene từ các sản phẩm của gene, chủ yếu là các loại mRNA. 1. Tạo dòng gene đã biết
Phân lập gene: Tách chiết các gene cần tạo dòng từ các mẫu nghiên cứu, nhân gene bằng máy PCR, sản phẩm PCR được điện di trên gel agar hoặc polyacrylamide, rồi dựa vào kích thước của gene và thang chuẩn DNA để tách ra được gene cần tìm.
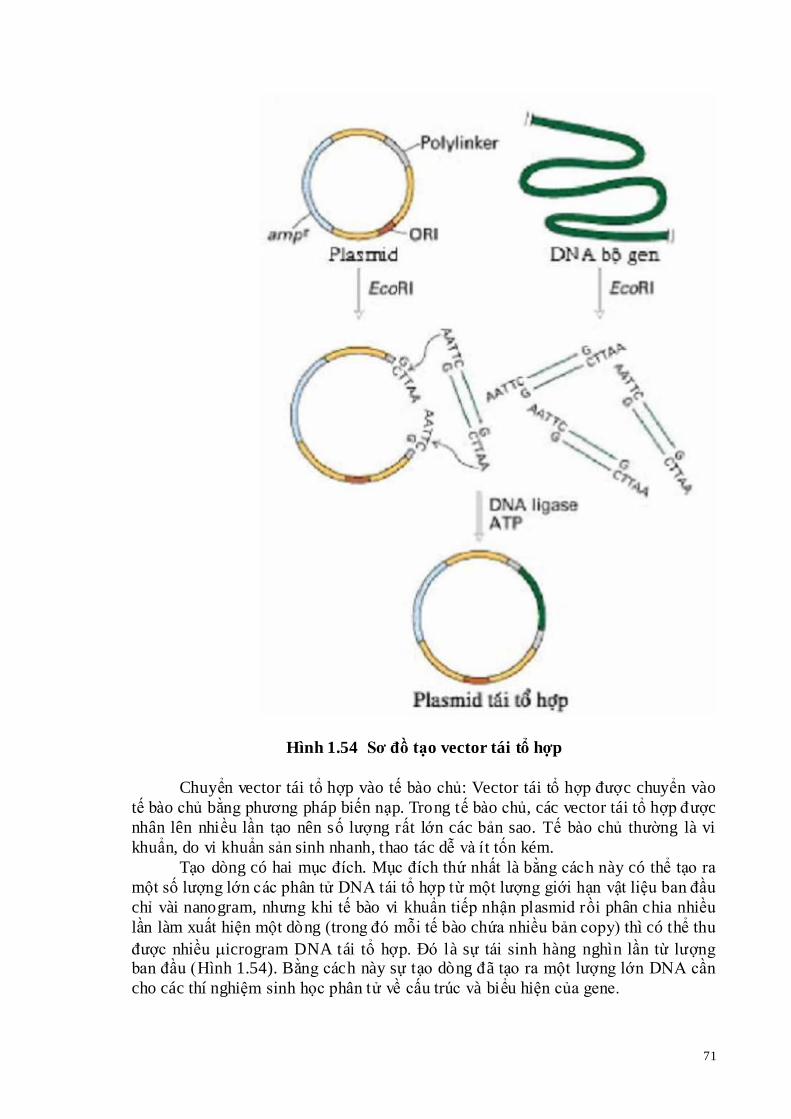
Tạo vector tái tổ hợp: Tùy theo mục đích tạo dòng để chọn vector tạo dòng phù hợp. Vector tạo dòng phải phù hợp với loại tế bào chủ và kích thước đoạn DNA cần tạo dòng (DNA chèn). Vector tạo dòng là plasmid phù hợp với tế bào chủ là tế bào vi khuẩn, có thể gắn đoạn DNA có kích thước 3-10 kb. Vector tạo dòng là phage cho gắn đoạn DNA từ 15 – 30kb, thích hợp với tế bào vi khuẩn, động vật, thực vật. Vector tạo dòng là cosmid có thể gắn các đoạn DNA lớn đến 45 kb. Sau khi chọn được vector tạo dòng phù hợp, trộn vector tạo dòng và các đoạn DNA chèn vào với nhau theo tỷ lệ nhất định, có sự tham gia của T4 DNA ligase. T4 DNA ligase xúc tác phản ứng gắn các đoạn DNA chèn với vector tạo dòng tạo nên vector tái tổ hợp.
71
Hình 1.54 Sơ đồ tạo vector tái tổ hợp
Chuyển vector tái tổ hợp vào tế bào chủ: Vector tái tổ hợp được chuyển vào tế bào chủ bằng phương pháp biến nạp. Trong tế bào chủ, các vector tái tổ hợp được nhân lên nhiều lần tạo nên số lượng rất lớn các bản sao. Tế bào chủ thường là vi khuẩn, do vi khuẩn sản sinh nhanh, thao tác dễ và ít tốn kém.
Tạo dòng có hai mục đích. Mục đích thứ nhất là bằng cách này có thể tạo ra một số lượng lớn các phân tử DNA tái tổ hợp từ một lượng giới hạn vật liệu ban đầu chỉ vài nanogram, nhưng khi tế bào vi khuẩn tiếp nhận plasmid rồi phân chia nhiều lần làm xuất hiện một dòng (trong đó mỗi tế bào chứa nhiều bản copy) thì có thể thu được nhiều icrogram DNA tái tổ hợp. Đó là sự tái sinh hàng nghìn lần từ lượng ban đầu (Hình 1.54). Bằng cách này sự tạo dòng đã tạo ra một lượng lớn DNA cần cho các thí nghiệm sinh học phân tử về cấu trúc và biểu hiện của gene.

72
Chức năng thứ hai là tinh sạch DNA. Khi tạo dòng bên cạnh các phân tử tái tổ hợp mong muốn hỗn hợp phản ứng gắn (ligation) còn chứa các phân tử sau:
1. Các phân tử vector 2. Các đoạn DNA tạo dòng 3. Các phân tử vector đóng vòng trở lại, nhưng không tiếp nhận DNA mới (tự
đóng vòng) 4. Phân tử DNA tái tổ hợp nhưng đã kết hợp với đoạn DNA không mong muốn.
Chọn và sàng lọc các thể tái tổ hợp: Trong thí nghiệm tạo vector tái tổ hợp, ví dụ plasmid tái tổ hợp, hỗn hợp
vector và một lượng lớn các phân tử DNA được cắt cùng một enzyme hạn chế được gắn với nhau bằng enzyme DNA ligase. Kết quả tạo một hỗn hợp có plasmid tái tổ hợp lẫn các plasmid không mang gene được chuyển vào vi khuẩn. Sau đó, tất cả các tế bào vi khuẩn đó được chuyển lên môi trường dinh dưỡng để phát triển thành các dòng, tức là khuẩn lạc vi khuẩn. Do thí nghiệm tiến hành với một hỗn hợp plasmid không đồng nhất như vậy nên các dòng vi khuẩn mọc lên có 3 loại:
- Tế bào vi khuẩn không nhận được plasmid - Tế bào vi khuẩn nhận được plasmid không mang gene ngoại lai mong muốn - Tế bào vi khuẩn nhận được đúng plasmid tái tổ hợp
Có nhiều phương pháp khác nhau để xác định dòng mong muốn, nhưng phương pháp được sử dụng nhiều nhất là xác định trực tiếp gene mong muốn. Đây là phương pháp lới dụng các chỉ thị (marker) trên vector tạo dòng để chọn lọc các dòng mang vector tái tổ hợp. Hình 1.55. Bằng cách tạo dòng người ta thu được một lượng lớn DNA tái tổ hợp
Tế bào chứa nhiều bản sao của phân tử DNA tái tổ hợp
Phân chia: Tạo ra một dòng Thu được:
nhiều g DNA tái tổ hợp
Đưa vào nuôi trong 500 ml môi trường lỏng, ủ trong 18 giờ
Thu được: nhiều mg DNA tái tổ hợp

73
Các phân tử vector và DNA tạo dòng, nếu được tế bào vi khuẩn tiếp nhận chúng chỉ tái bản trong trường hợp ngoại lệ và thường bị phân giải bởi enzyme của tế bào chủ. Ngược lại, các phân tử vector tự đóng vòng hoặc những plasmid kết hợp với đoạn DNA không mong muốn thì tái bản giống phân tử tái tổ hợp mong muốn (Hình 1.56b). Các dòng khác nhau chứa các phân tử DNA khác nhau. Chỉ có một số ít tế bào vi khuẩn chứa cấu trúc mong muốn. Vậy làm thế nào để xác định được chúng?Trong phần này sẽ đề cập các phương pháp đưa plasmid và phage (phân tử tái tổ hợp) vào tế bào vi khuẩn và chọn lọc chúng.
a. Sản phẩm của ligation Các đoạn DNA
gene
Vector tạo dòng
gene
DNA tái tổ hợp mong muốn
Vector được mở vòng
DNA tái tổ hợp “sai”
b. Tất cả DNA dạng vòng được tạo dòng
Tế bào với vector tự đóng vòng
Tế bào với DNA tái tổ hợp “sai”
Tế bào với phân tử mong muốn
Dòng với vector tự đóng vòng
Dòng với DNA “sai”
Dòng với vector mong muốn

74
Hình 1.56. Tạo dòng là quá trình tinh sạch DNA. Từ một hỗn hợp các phân tử DNA khác nhau người ta thu được nhiều dòng mà mỗi dòng chỉ chứa một
phân tử duy nhất với nhiều bản copy 1.1. Biến nạp (transformation): Sự tiếp nhận DNA của tế bào vi khuẩn
Phần lớn các loài vi khuẩn có thể tiếp nhận các phân tử DNA từ dung dịch nuôi cấy. Những phân tử DNA này thường bị phân giải trong tế bào. Khi phân tử DNA là một plasmid thì điểm bắt đầu sao chép của nó được nhận biết bởi tế bào ký chủ.
Sự tiếp nhận và tồn tại của plasmid trong tế bào được nhận biết nhờ sự biểu hiện của các gene nằm trên plasmid đó. Các tế bào E. coli thường nhạy cảm với tác dụng kìm hãm sinh trưởng của kháng sinh như ampicillin và tetracyclline. Các tế bào vi khuẩn chứa plasmid pBR322, một trong những vector tạo dòng đầu tiên, được phát triển trong những năm 70, có khả năng kháng kháng sinh. Nguyên nhân là có hai nhóm gene nằm trên plasmid đó: Một gene mã hoá -lactamase, biến đổi ampicillin thành một dạng không độc đối và gene của nhóm thứ hai làm cho tetracyclline trở thành vô hại. Các tế bào đã tiếp nhận pBR322, biến đổi từ dạng nhạy cảm với ampicillin và tetracyclline (ampstets) sang dạng kháng (amprtetr). a. Không phải tất cả tế bào vi khuẩn tiếp nhận DNA với hiệu quả như nhau
Trong tự nhiên sự biến nạp có thể không phải là con đường quan trọng để đưa thông tin di truyền vào vi khuẩn. Trong phòng thí nghiệm chỉ có một số ít loại (Bacillus và Streptococcus) biến nạp được thực hiện dễ dàng.
Phần lớn những loài vi khuẩn, trong đó có E. coli tiếp nhận DNA trong điều kiện bình thường rất giới hạn. Để tăng khả năng biến nạp của những loài này, người ta phải xử lý bằng phương pháp vật lý và hoá học. Những tế bào vi khuẩn được xử lý này gọi là tế bào khả biến. b.Tạo tế bào khả biến
Các tiến bộ quan trọng trong kỹ thuật tái tổ hợp DNA cũng là bước phát triển quan trọng liên quan đến sự biến nạp vào đầu những năm 70. Lúc đó người ta thấy rằng, tế bào E.coli được xử lý bằng dung dịch muối lạnh tiếp nhận DNA nhiều hơn những tế bào không được xử lý. Người ta thường sử dụng dung dịch CaCl2 50 mM, nhưng các muối khác cũng có cùng chức năng đặc biệt rubidiumchlorid.
Tại sao việc xử lý này có tác dụng, vẫn chưa được giải thích một cách chi tiết. Có thể CaCl2 làm tăng khả năng kết tủa DNA ở bên ngoài tế bào, hoặc có thể muối là nguyên nhân làm thay đổi thành tế bào làm cho DNA bám tốt hơn. Xử lý bằng CaCl2 chỉ có tác dụng tăng sự bám DNA ở phía bên ngoài tế bào. Sự di chuyển thực chất của DNA vào tế bào khả biến được thực hiện do việc tăng nhiệt độ lên 420C trong thời gian ngắn. Tuy nhiên, người ta vẫn chưa biết chính xác, tại sao thay đổi nhiệt độ đột ngột lại gây ra tác dụng này.
1.2. Chọn lọc các tế bào biến nạp

75
Hình 1.57. Tiếp nhận và biến nạp DNA của tế bào vi khuẩn khả biến Sự biến nạp của các tế bào khả biến là một quá trình không có hiệu quả, cả
khi người ta chuẩn bị các tế bào này cẩn thận. Khi biến nạp 1 ng pUC8 có thể thu được 1000 đến 10 000 tế bào được biến nạp, nhưng điều đó nói lên rằng chỉ 0,1 % phân tử được tế bào tiếp nhận. 10 000 tế bào được biến nạp cũng chỉ một phần rất nhỏ trong dung dịch tế bào khả biến. Vì vậy, phải tìm ra cách để phân biệt tế bào vi khuẩn tiếp nhận plasmid với hàng ngàn tế bào không biến nạp.
Nhằm mục đích này người ta sử dụng gene chọn lọc có trong plasmid. Gene chọn lọc đơn giản là gene tạo cho tế bào biến nạp có một đặc tính mới, ví dụ gene kháng ampicillin trong pBR322. Các tế bào E. coli đã tiếp nhận plasmid amprterr có thể tạo khuẩn lạc trên dung dịch thạch chứa ampicillin và tetracycline. Các tế bào không biến nạp không có khả năng này. Bằng cách này người ta phân biệt một cách dễ dàng các tế bào được biến nạp và không biến nạp.
Phần lớn các vector tạo dòng plasmid mang ít nhất một gene làm cho ký chủ có khả năng kháng một kháng sinh. Để chọn lọc các tế bào này người ta đưa chúng lên môi trường thạch chứa kháng sinh đã đề cập. Cũng nên biết rằng tính kháng không phải đơn giản xuất hiện ngay khi có mặt của plasmid trong tế bào. Gene
Vi khuẩn bình thường
Xử lý bằng CaCl2
Plasmid gắn phía ngoài tế bào
Tế bào khả biến
42oC trong 2 phút Plasmid được đưa vào trong tế bào
Tế bào biến nạp

76
kháng phải biểu hiện, nghĩa là enzyme được tổng hợp. Vì vậy, sự biểu hiện của gene biến nạp sớm nhất cũng sau một vài phút. Do vậy không nên nuôi cấy vi khuẩn trên môi trường chọn lọc ngay sau khi xử lý schock nhiệt mà nên giữ chúng trong dung dịch lỏng có thể tích nhỏ không có kháng sinh trong thời gian ngắn. Bằng cách này sự tái sinh và biểu hiện của gene có thể xảy ra và khi đưa tế bào này tiếp cận với kháng sinh trên môi trường, chúng đã tạo nên đủ enzyme để sống sót (Hình 1.58).
Hình 1.58. Chọn lọc tế bào chứa plasmid pBR322 bằng cách nuôi chúng trên môi trường chứa ampicillin và/ hoặc tetracycline
1.3. Xác định dạng tái tổ hợp
Bằng việc nuôi cấy trên môi trường chọn lọc người ta có thể phân biệt được những tế bào được biến nạp và không biến nạp. Tiếp theo phải khẳng định được tế bào biến nạp nào đã tiếp nhận phân tử DNA tái tổ hợp và những tế bào nào chỉ chứa vector tự đóng vòng. Ở các vector tạo dòng khi đoạn DNA ngoại lai chèn vào thì
Agar với 40 g/ml ampicillin, 15 g/ml tetracylline
Sống sót và tạo khuẩn lạc
Tế bào E. coli không chứa plasmid
Không sóng sót

77
gene chọn lọc bị gián đoạn (tách thành hai phần). Các thể tái tổ hợp được phân biệt, do các tế bào này không còn có đặc tính mới do gene mã hóa đưa lại. Nguyên lý của bất hoạt do bị chèn được làm sáng tỏ ở thí nghiệm tạo dòng với vector pBR322.
Hình 1.59. Biểu hiện kiểu hình. Thể biến nạp sống sót tốt hơn sau khi ủ một giờ ở 370C, so với trước khi nuôi trên môi trường chọn lọc, vì sau đó vi khuẩn mới
bắt đầu tổng hợp enzyme. a. Chọn lọc các biến nạp chứa pBR322: bất hoạt do DNA chèn vào gene kháng kháng sinh
Plasmid pBR322 có nhiều chỗ cắt duy nhất cho các enzyme hạn chế, ở đó đoạn DNA được chèn vào. Ví dụ BamHI cắt plasmid chỉ ở một vị trí bên trong nhóm gene kháng tetracyclline. Một đoạn DNA ngoại lai được chèn vào vị trí cắt của BamHI (Hình 1.61b) làm cho tế bào không còn tính kháng tetracyclline, vì gene kháng này đã bị gián đoạn. Các tế bào chứa plasmid tái tổ hợp này vẫn còn kháng ampicillin, nhưng mẫn cảm với tetracyclline (amprtets).
Việc tìm kiếm các thể tái tổ pBR322 thực hiện như sau: Sau khi biến nạp (gene kháng tetracycline bị bất hoạt do chèn DNA lạ vào) người ta nuôi các tế bào này trên môi trường thạch chứa ampicillin và ủ chúng cho đến khi xuất hiện các khuẩn lạc (Hình 1.62a). Các khuẩn lạc này bao gồm cả tế bào biến nạp (chứa phân tử pBR322 tái tổ hợp) và không biến nạp (chứa plasmid không thay đổi). Để xác
Protein (enzyme) làm bất hoạt kháng sinh
Plasmid
Ngay sau khi biến nạp
Sau một giờ ủ ở nhiệt độ 37oC

78
định các khuẩn lạc tái tổ hợp người ta chuyển các khuẩn lạc này lên môi trường thạch chứa tetracyclline theo đúng trật tự cũ (Hình 1.61b). Sau khi ủ chỉ một số khuẩn lạc gốc phát triển (Hình 1.62c). Những khuẩn lạc phát triển bình thường không chứa plasmid tái tổ hợp (đoạn DNA ngoại lai không được chèn vào, vì ở chúng nhóm gene kháng tetracylin vẫn còn nguyên (amprtetr). Ngược lại những khuẩn không phát triển trên môi trường này là các tế bào chứa plasmid tái tổ hợp (amprtets). Biết được chính xác vị trí của nó trên đĩa chứa ampicillin, người ta có thể dễ dàng tách chúng ra và tiếp tục sử dụng.
Hình 1.60. Bất hoạt do chèn DNA ngoại lai vào a. Vector bình thường chứa một gene chọn lọc, đưa lại cho tế bào chủ
một đặc tính xác định nào đó. b. DNA ngoại lai được chèn vào gene chọn lọc làm gene gián đoạn, vì
vậy tế bào chủ không còn đặc tính đó nữa.
a. Vector
Sản phẩm gene
Gene đích được hoạt hóa
Không có sản phẩm gene
Gene đích bị gián đoạn khi đưa DNA vào
b. Vector tái tổ hợp

79
Hình 1.61. Vector tạo dòng pBR322 a. Vector pBR322, b. Phân tử pBR322 tái tổ hợp, đoạn DNA ngoại lai được
chèn vào vị trí cắt của BamHI
b. Sự bất hoạt do DNA chèn vào không phải luôn luôn là gene kháng kháng sinh
Các gene kháng kháng sinh là phương tiện thuận lợi để xác định thể tái tổ
hợp, nhưng ở một số vector tạo dòng plasmid người ta còn sử dụng một hệ thống khác, ví dụ pUC8 (Hình 1.37a). Plasmid này mang một gene kháng ampicillin và một gene LacZ mã hoá một phần enzyme -galactosidase. Khi tạo dòng với pUC8 thì gene LacZ bị bất hoạt, vì vậy ở thể biến nạp không tổng hợp được -galactosidase (Hình 1.37b).
a. Vector
b. pBR322 tái tổ hợp
Gene kháng ampicillin BamHI
Gene kháng tetracylline
DNA ngoại lai được chèn vào vị trí cắt của BamHI
ampR tetS
ampR tetR

80
-galactosidase là enzyme phân giải lactose thành glucose và galactose. Bình thường enzyme này được mã hoá bởi gene LacZ có trong nhiễm sắc thể của E.coli. Một số chủng E.coli có gene LacZ thay đổi, gene này thiếu một đoạn lớn, gọi là gene LacZ mã hoá cho một phần (-peptide) enzyme -galactosidase. Những thể đột biến này có thể tạo nên enzyme khi chúng mang một plasmid, ví dụ như pUC8 chứa phần thiếu LacZ của gene.

81
Hình 1.62. Tìm kiếm thể biến nạp pBR322, ở đây gene kháng tetracycline bị bất hoạt do chèn DNA ngoại lai vào
a. Tế bào biến nạp được nuôi trên môi trường thạch chứa ampicillin: Tất cả các tế bào đều tạo khuẩn lạc
b. Các khuẩn lạc này được chuyển sang môi trường chứa tetracycline. Những khuẩn lạc phát triển trên môi trường này là những thể không biến nạp (amR tetR)
a. Khuẩn lạc trên môi trường chứa ampicillin
b. Chuyển khuẩn lạc từ đĩa agar này lên đĩa agar khác, trên đó khuẩn lạc tiếp tục phát triển theo đúng vị trí ban đầu
Khối gỗ bọc nhung
Tiếp xúc trên bề mặt
Tế bào gắn khối gỗ Tiếp xúc trên bề mặt
Khuẩn lạc tetR
Khuẩn lạc trên môi trường chứa ampicillin
Môi trường chứa tetracylline
c. Khuẩn lạc ampR tetR phát triển trên môi trường chứa tetracyclline
Vị trí của một thể biến nạp ampR tetS
ampR tetR
: không có thể biến nạp

82
c. Các thể biến nạp (ampR tetS) không phát triển, nhưng người ta có thể biết được vị trí của nó trên đĩa chứa ampicillin
Ở một thí nghiệm tạo dòng với pUC8 người ta chọn lọc các tế bào sau khi biến nạp trên môi trường chứa ampicillin và kiểm tra hoạt tính của -galactosidase để xác định thể biến nạp. Tế bào chứa plasmid pUC8 kháng ampicillin và có thể tổng hợp -galactosidase. Thể biến nạp cũng kháng ampicillin nhưng không tạo ra -galactosidase.
Hình 1.63. Vector tạo dòng pUC18: a. Vector pUC18 b. Phân tử pUC18 tái tổ hợp, đoạn DNA ngoại lai được chèn vào vị trí
cắt của BamHI
ampR -gal+
BamHI gene LacZ’
a. pUC18
b. Phân tử pUC18 tái tổ hợp
Gene kháng ampicillin
DNA ngoại lai được chèn vào vị trí cắt của BamHI
ampR -gal-

83
Thí nghiệm không kiểm tra lactose có được phân giải thành glucose và galactose hay không mà kiểm tra một phản ứng được xúc tác bởi enzyme. Ở đây sử dụng một chất tương tự lactose là X-gal (5-brom-4-chlor-3-indolyl-D-galactopyranoside), chất này được phân giải thành một sản phẩm có màu xanh tối nhờ enzyme -galactosidase. Người ta cho X-gal vào agar và một chất cảm ứng enzyme, isopropythiogalactoside (IPTG), các khuẩn lạc không chứa thể tái tổ hợp thì có màu xanh. Ở thể biến nạp gene LacZ bị gián đoạn nên không tạo được -galactosidase và khuẩn lạc có màu trắng.
2. Tạo dòng gene chưa biết
Khi chưa biết kích thước hoặc vị trí của gene trong bộ gene thì nhân dòng gene thường sử dụng các sản phẩm của gene, chủ yếu là các loại mRNA.
Tạo dòng gene chưa biết từ mRNA là một quá trình phức tạp, có thể thực hiện theo các bước sau:
- Tách chiết mRNA - Tạo DNA bổ sung, cDNA (complemantary DNA) - Tạo vector tái tổ hợp - Biến nạp vector tái tổ hợp vào tế bào nhận - Chọn lọc

84
Hình 1.64. Nguyên tắc bất hoạt gene LacZ’ của pUC8 bằng việc chèn DNA ngoại lai
a. Gene của vi khuẩn và plasmid bổ sung cho nhau tạo nên β-galactosidase hoạt động
b. Để tìm kiếm thể tái tổ hợp người ta nuôi vi khuẩn trên môi trường thạch chứa X-gal và IPTG
Nói chung các bước tiến hành về cơ bản cũng như tạo dòng gene đã biết. Các
kỹ thuật chi tiết sẽ trình bày ở chương sau.
a. Ý nghĩa của gene: LacZ’
b. Tìm tế bào biến nạp pUC8 tái tổ hợp
E. coli Lac Z’
Phân tử enzyme không hoàn chỉnh
+ pUC18
pUC18
Phân tử -galactose hoàn chỉnh
Một phần của -galactosidase được mã hóa bởi gene của vi khuẩn
Một phần của -galactosidase được mã hóa bởi gene của plasmid
Một phần của -galactosidase hoàn chỉnh
Agar + X-gal + IPTG
Khuẩn lạc màu xanh: tế bào không biến nạp pUC8 tái tổ hợp
Khuẩn lạc màu xanh: tế bào không biến nạp
Khuẩn lạc màu xanh: -galactosidase được tạo nên X-gal sản phẩm màu xanh
Khuẩn lạc màu trắng: -galactosidase không được tạo nên X-gal Không có sản phẩm màu xanh

85
Tài liệu tham khảo/đọc thê 1. Nguyễn Hoàng Lộc, Trần Thị Lệ, Hà Thị Minh Thi, Giáo trình Sinh học phân
tử, NXB Đại học Huế, 2007. 2. Cherfas, J. Man made Life. Oxford (Blackwell) 1982, Eine Geschichte der
fruehen Jahre in der Genetechnik, 3. Felsenfeld, G. DNA. In: Spektrum der Wissenschaft 12 (1985) S. 50 -63,
Kurzer Ueberblick ueber die wichtigsten Eigeneschaften der DNA 4. Brown, T. A. (Hrsg.), The Encyclopedia of Molecular Biology, Oxford
(Blackwell) 1994, Ein ausgezeichnetes Nachschlagewerk. 5. Beebee, T.; Burke, J, Genee Structure and Transcription.2, Aufl. Oxford
(IRL Press at Oxford University Press) 1992. 6. Dale, J. W. Molecular geneetics of Bacteria, 2. Aufl. Chicheester (Wiley)
1994. 7. Marmur, J. A Procedure for the Isolation of Deoxyribonucleic Acid from
Micro-organisms, In: Journal of Molecular Biology 3 (1961) 8. Rogers, S.O.; Bendich, A. J. Extraction of DNA from Milligram Amounts of
Fresh, Herbarium and Mummified Plant Tissues, In: Plant Molecular Biology (1985).
9. Birnboim, H. C.; Doly, J. A Rapid Alkaline Extraction Procedure for Screening Recombinant Plasmid DNA, In: Nucleic Acid Research 7 (1979)
10. Smith, H. O.; Wilcox, K. W. A Restriction Enzyme from Haemophilus influenzae, In: Jounal of molecular Biology 51 (1970)
11. Roberts, R. J.; Macelis, D. Restriction Enzymes and Methylase, In: Nucleic Acid research 21 (1993).
12. Rothstein, R. J. et al. Synthetic Adaptors for Cloning DNA, In: methods in Enzymology 68 (1979).
13. Hohn, B.; Murray, K. Packaging Recombinant DNA Molecules into Bacteriophage Particles in Vitro, In: Proceeding of the NationalAcadamy of Sciences, USA 74 (1977).
14. Bolivar, F.et al. Construction and Characterisation of New Cloning Vectors, II. Amulti-Purpose Cloning System. In: Genee 2 (1977).
15. Leder, P. et al. EK2 Derivatives of Bacteriophage Lambda Useful in Cloning of DNA from Higher Organisms, The gtWES System. In: Science 196 (1977)

86

70
Chương 2
CÁC KỸ THUẬT CHÍNH SỬ DỤNG TRONG PHÂN TÍCH NUCLEIC ACID
I. Phương pháp tách chiết DNA
Trong công nghệ gene luôn luôn yêu cầu làm sạch ít nhất là 3 loại DNA. Thứ nhất là DNA tổng số của vi khuẩn, thực vật và động vật mà người ta muốn nghiên cứu.Loại DNA thứ hai là plasmid: Theo nguyên tắc như tinh sạch DNA tổng số, tuy nhiên có sự khác nhau là phải tách DNA-plasmid ra khỏ i DNA nhiễm sắc thể, mà chúng chỉ chiếm phần nhỏ DNA của tế bào. Cuối cùng là DNA của phage, khi muốn sử dụng chúng làm vector tạo dòng. Nói chung DNA phage không được tách từ tế bào bị nhiễm mà từ các hạt phage. Vì vậy, phải sử dụng các phương pháp đặc biệt hơn để loại phần vỏ của phage. Trường hợp ngoại lệ M13 sợi kép thì được tách như plasmid của tế bào E.coli.
1. Phương pháp tách DNA tổng số
Để tách chiết được DNA tổng số từ tế bào vi khuẩn cần thực hiện 4 bước sau đây (Hình 2.1):
1. Nuôi cấy và thu hoạch vi khuẩn 2. Phá vỡ tế bào để giải phóng phần bên trong. 3. Dịch tế bào được xử lý để loại bỏ các thành phần của tế bào trừ
DNA. 4. Cô đặc dung dịch DNA thu được.
Hình 2.1 Các bước cơ bản để thu được DNA tổng số từ tế bào vi khuẩn
1. Vi khuẩn được nuôi và thu hoạch 2. Vi khuẩn được tách ra và phá vỡ tế bào, làm xuất hiện dịch tế bào
Ly tâm
Vi khuẩn nuôi trong dung dịch Vi khuẩn kết tủa Dịch tế bào
4. Làm tăng nồng độ DNA DNA sạch
3. DNA sạch thu được từ dịch tế bào

71
1.1. Nuôi và thu hoạch vi khuẩn Vi khuẩn được nuôi dễ dàng trong môi trường lỏng. Dung dịch nuôi cấy là
hỗn hợp các chất cần thiết đảm bảo cho vi khuẩn phát triển và phân chia có hiệu quả. Có hai loại môi trường nuôi cấy thường được sử dụng là M9 và LB (Bảng 2.1). M9 là hỗn hợp các chất vô cơ cần thiết cho sự sống như N, Mg, Ca và đường. Tùy từng loài vi khuẩn mà người ta bổ sung vào M9 các nguyên tố vi lượng và vitamin. Ngược lại LB là môi trường mà người ta không biết chính xác chất nào và với lượng chất bao nhiêu chứa ở trong đó. Ví dụ, trypton và dịch chiết nấm men là hỗn hợp phức tạp chứa các chất chưa biết.
Bảng 2.1 Thành phần của hai môi trường nuôi cấy vi khuẩn
Thành phần Nồng độ (g/l) 1. Môi trường M9 Na2HPO4 KH2PO4 NaCl NH4Cl MgSO4 Glucose CaCl2 2. Môi trường LB (Luria-Bertani) Trypton Dịch chiết nấm men NaCl
6,0 3,0 0,5 1,0 0,5 2,0
0,015
10 5 10
Trypton cung cấp các amino acid cơ bản và các peptide nhỏ. Dịch chiết từ
nấm men (dạng bột khô, một phần là những tế bào nấm men được phân giải) cung cấp N, đường, các hợp chất vô cơ và hữu cơ. Môi trường hỗn hợp như LB không cần cho thêm những chất bổ sung. Các loài vi khuẩn khác nhau phát triển tốt trên môi trường này.
Khi chỉ muốn tách DNA từ dung dịch vi khuẩn nuôi cấy, thì môi trường LB là phù hợp. Trong môi trường LB, ở nhiệt độ 370C, trên máy lắc với tốc độ 150-250 vòng/phút, tế bào vi khuẩn phân chia trong khoảng 20 phút cho đến khi dung dịch nuôi cấy đạt được mật độ cực đại khoảng 2-3 x 109 tế bào/ml. Để theo dõi sự phát triển của vi khuẩn nuôi cấy, người ta đo mật độ quang (OD) ở 600 nm (Hình 2.2). Ở độ dài bước sóng này 1 đơn vị OD tương ứng với khoảng 0,8 x 109 tế bào vi khuẩn/ml.
Để thu vi khuẩn phải ly tâm dung dịch nuôi cấy ở số vòng quay thấp, vi khuẩn sẽ đọng lại ở đáy ống nghiệm (Hình 2.3). Bằng cách này 1 lít dung dịch vi khuẩn nuôi cấy có thể cô đặc lại trong 10 ml hoặc ít hơn.
Để phá vỡ tế bào vi khuẩn có thể dùng phương pháp vật lý hoặc phương pháp hoá học, trong đó phương pháp hóa học được sử dụng phổ biến hơn. Đối với tế bào vi khuẩn người ta thường sử dụng các hóa chất như lysozyme kết hợp với ethylene diamine tetraacetic acid (EDTA) để phá hủy màng tế bào. Lysozyme sẽ phân hủy màng tế bào giải phóng các thành phần bên trong tế bào

72
(dịch tế bào), trong khi đó EDTA tách ion Mg, ion này rất quan trọng trong việc duy trì cấu trúc của vỏ tế bào, ngoài ra nó còn kìm hãm enzyme phân giải DNA. Dưới những điều kiện xác định lysozyme làm yếu màng tế bào và EDTA làm cho tế bào bị vỡ ra, nhưng thường thì người ta cho thêm một hoá chất như sodiumdecysulfate (SDS). 1.2. Xử lý dịch tế bào
Hình 2.2. Xác định số lượng vi khuẩn bằng đo mật độ quang ở máy quang phổ kế
a. Đo mật độ quang
b. Xác định số lượng tế bào nhờ đường chuẩn
Ánh sáng đi qua
Máy đo
Dung dịch vi khuẩn trong một curvet có độ dày 1 cm
Ánh sáng có bước sóng 600 nm
Mật độ quang ở 600 nm
Số lượng tế bào (x 109)/ml

73
a. Cho mẫu vào cuvet thủy tinh và chiếu ánh sáng có bước sóng 600 nm. Đo lượng ánh sáng đi qua dung dịch vi khuẩn và mật độ quang (OD) được tính là
OD = -log10 (cường độ ánh sáng đi qua/ cường độ ánh sáng chiếu vào dung dịch) b. Biết được số lượng tế bào tương ứng với mỗi giá trị OD nhờ một đường chuẩn,
đường chuẩn này được xây dựng từ các giá trị OD của dung dịch vi khuẩn ở các nồng độ đã biết. Đố i với E.coli 1 đơn vị OD = 0,8 x 109 tế bào/ml
Chất này làm tăng quá trình hoà tan, vì chúng tách ra những phân tử lipid ra
khỏi màng tế bào. Tuy nhiên, trong một số trường hợp vẫn phải sử dụng đến các phương pháp vật lý như sử dụng máy nghiền hay cối và chày sứ. Nhược điểm của phương pháp này là có nguy cơ làm đứt gãy cơ học các phân tử DNA lớn và điều có thể gây trở ngại cho quá trình xử lý tiếp theo.
Sau khi tế bào phân giải, người ta phải tách những phần tế bào không hoà tan ra khỏi dịch tế bào, ví dụ các mảnh vỡ màng tế bào không bị phân giải và các bào quan như ty thể, không bào bằng ly tâm (Hình 2.4b), kết quả cuối cùng là thu được phần dịch trong suốt nằm ở phía trên.
Hình 2.3. Thu sinh khối vi khuẩn bằng ly tâm
Máy ly tâm
Ly tâm 10 phút ở 800 rpm
Dung dịch vi khuẩn
Sinh khối vi khuẩn

74
Hình 2.4. Thu nhận dịch tế bào a. Ly tan tế bào, b. Ly tâm dịch tế bào để tách ra những phần không tan
1.3. Tách DNA từ dịch tế bào
Để thu được DNA tinh sạch người ta cần loại bỏ những thành phần tạp nhiễm mà quan trọng nhất là protein. Bước này được thực hiện bằng cách tách chiết một hoặc vài lần với tác nhân tách chiết, thường là phenol, chloroform, hoặc một hỗn hợp phenol và chloroform cùng thể tích.
Phenol là chất làm biến tính protein mạnh nhưng không hòa tan nucleic
acid. Chloroform thường đi kèm với phenol có tác dụng bổ sung và loại bỏ hoàn
toàn dấu vết của phenol. Vì vậy, người ta hay sử dụng hỗn hợp phenol và chloroform để loại bỏ protein.
Như vậy, việc loại bỏ thành phần tạp nhiễm và tách chiết DNA dựa trên sự hòa tan khác nhau của DNA và protein trong 2 pha không hòa tan phenol và chloroform/nước.
a. Ly tan tế bào
Phá vỡ thành tế bào
Phá vỡ màng tế bào
Dịch tế bào
b. Ly tâm để tách ra phần không hòa tan
DNA, RNA và protein
Chất cặn tế bào
Ly tâm Dịch tế bào

75
Hình 2.5. Tách protein bằng dung dịch phenol
Các phân tử protein kết tủa trắng ở ranh giới giữa pha lỏng và pha hữu cơ (Hình 2.5). Phần dịch chứa nucleic acid được tách ra bằng micropipette.
Một số dịch tế bào chứa nhiều protein thì một lần chiết bằng phenol là không đủ để làm sạch nucleic acid. Để giải quyết vấn đề này phải xử lý với phenol nhiều lần, nhưng mỗi lần trộn lẫn và ly tâm một số các phân tử DNA bị gãy. Thay thế cho xử lý bằng phenol nhiều lần là xử lý bằng protease (pronase hoặc protease K). Những enzyme này phân giải chuỗi peptide thành các phân tử nhỏ hơn nên dễ dàng tách ra bằng phenol.
Một phần RNA, đặc biệt là mRNA cũng được tách ra khi xử lý bằng phenol, nhưng phần lớn RNA cùng với DNA ở trong dung dịch. Một phương pháp có hiệu qủa duy nhất để tách chúng là xử lý với ribonuclease, enzyme này phân giải RNA rất nhanh thành các ribonucleotide. 1.4. Làm giàu DNA
Ở quá trình tinh sạch nếu thu được dung dịch DNA có nồng độ cao thì không cần làm đặc lại. Nếu dung dịch có nồng độ DNA thấp thì sử dụng các phương pháp sau đây để làm tăng nồng độ DNA.
Phương pháp được sử dụng nhiều nhất là kết tủa bằng ethanol ở nhiệt độ dưới -200C. Khi dung dịch DNA có nồng độ cao có thể tạo kết tủa trên bề mặt với ethanol. Khi nhúng đũa thuỷ tinh qua ethanol vào dung dịch DNA và lấy đũa ra thì các phân tử DNA bám chặt vào đũa, vì vậy có thể lấy DNA từ dung dịch ra ở dạng sợi dài (Hình 2.6a). Ngược lại nếu nồng độ DNA thấp có thể tách kết tủa này (DNA) bằng cách ly tâm (Hình 2.6b) và sau đó hoà tan trở lại với một lượng nước hoặc đệm thích hợp. Sự kết tủa bằng ethanol có ưu điểm là các đoạn ngắn và monomer xuất hiện khi xử lý bằng ribonuclease ở lại trong dung dịch nên chúng được tách ra ở giai đoạn này.
Dịch tế bào
Trộn với phenol Tách các lớp băng ly tâm
Pha lỏng (DNA và RNA)
Lớp mặt giới hạn (protein kết tủa)
Phenol

76
Hình 2.6. Thu nhận DNA bằng kết tủa với ethanol a. Dung dịch DNA có nồng độ cao được phủ bằng ethanol tuyệt đối. Bằng đũa thủy
tinh có thể lấy ra các sợi DNA b. Với dung dịch DNA có nồng độ thấp hơn thì kết tủa bằng ethanol tuyệt đối với tỷ
lệ 2,5 thể tích so với thể tích dung dịch DNA, sau đó thu kết tủa bằng ly tâm 1.5. Định lượng và đánh giá độ tinh sạch của nucleic acid tách chiết
Đối với thí nghiệm tạo dòng, điều quan trọng là phải biết chính xác lượng nucleic acid trong dung dịch. Để xác định độ tinh sạch cũng như hàm lượng nucleic acid thu được sau tách chiết, người ta sử dụng phương pháp đo quang phổ. Phương pháp này dựa trên khả năng hấp thụ ánh sáng khác nhau của các base.
Giá trị hấp thụ (absorbance, ký hiệu A) là mật độ quang (optical density) tại bước sóng 260nm (ký hiệu là OD260nm) cho phép xác định nồng độ nucleic acid trong dung dịch.
Dung dịch DNA mạch kép có giá trị mật độ quang OD = 1.0 ở bước sóng 260nm tương ứng với nồng độ là 50 mg/ml.
Dung dich DNA mạch đơn có giá trị mật độ quang OD =1.0 ở bước sóng 260nm tương ứng với nồng độ là 33 mg/ml.
Dung dịch RNA có giá trị mật độ quang OD = 1.0 ở bước sóng 260nm tương ứng với nồng độ là 40 mg/ml.
Ethanol
Đũa thủy tinh
Sợi DNA
Dung dịch DNA
Ethanol
DNA kết tủa bằng ly tâm

77
Như vậy, nồng độ (mg/ml) của dung dịch nucleic acid được tính bằng công thức sau:
Nồng độ dung dịch = A260nm x Φ Ứng với DNA mạch kép, Φ = 50 mg/ml Ứng với DNA mạch đơn, Φ = 33 mg/ml Ứng với RNA, Φ = 40 mg/ml
Dung dịch nucleic acid tách chiết từ tế bào thường lẫn tạp protein. Do vậy, khi đo nồng độ dung dịch bằng quang phổ kế, tỷ lệ giữa A260nm/A280nm cho biết độ tinh sạch của dung dịch nucleic acid. Dung dịch này đạt yêu cầu tinh sạch khi tỷ lệ A260nm/A280nm nằm trong khoảng 1.8 đến 2.0.
Trong phương pháp định lượng nucleic acid bằng mật độ quang, nồng độ của dung dịch cần định lượng phải nằm trong khoảng xác định. Trong khoảng này, mật độ quang tương quan tuyến tính với nồng độ của dung dịch cần định lượng.
Sau khi xác định được nồng độ của dung dịch nucleic acid thì ta có thể lấy bất kỳ lượng DNA hay RNA nào, kể cả nanogram hay picogram bằng cách pha loãng dung dịch gốc với độ pha loãng thích hợp. 1.6. Tách chiết DNA tổng số từ các sinh vật khác
Cơ sở của các bước làm sạch DNA tổng số từ tế bào động vật, thực vật là như nhau, tuy nhiên cũng có những biến đổi cho phù hợp với đặc điểm của từng loại tế bào.
Sự khác biệt lớn nhất là ở sự phá vỡ tế bào. Các hoá chất sử dụng để hoà tan tế bào vi khuẩn thường không có tác dụng đối với tế bào các sinh vật khác, ví dụ lysozyme không có tác dụng ở tế bào thực vật. Để phân giải thành tế bào có những enzyme đặc hiệu, nhưng thường phương pháp vật lý phù hợp hơn, ví dụ phá huỷ vật liệu làm lạnh với cối và chày sứ. Ngược lại tế bào động vật không có thành tế bào nên có thể hoà tan chúng đơn giản bằng hoá chất.
Đối với vi khuẩn, các chất quan trọng nhất có trong dịch tế bào là protein, DNA và RNA. Bằng phương pháp xử lý phenol và/hoặc xử lý bằng protease và sau đó tách RNA bằng ribonuclease người ta có được mẫu chứa DNA đủ sạch. Ở các mô thực vật thường chứa một lượng lớn carbohydrate, chất này không tách ra được khi xử lý bằng phenol. Trong trường hợp này người ta phải sử dụng một hoá chất có tên là cetyltrimethylammoniumbromide (CTAB), chất này kết hợp với nucleic acid thành một phức không hoà tan. Khi cho CTAB vào dịch tế bào thực vật, xảy ra kết tủa với nucleic acid, trong khi đó, carbohydrate, protein và các tạp chất khác ở lại trong dung dịch (Hình 2.7). Người ta tách kết tủa này bằng ly tâm và hoà tan trở lại trong NaCl 1M. Sau đó có thể nâng nồng độ nucleic acid lên bằng kết tủa ethanol và tách RNA bằng xử lý với ribinuclease.

78
Hình 2.7. Tinh sạch DNA thực vật bằng phương pháp CTAB
Phương pháp thứ hai là dựa vào đặc tính phân tử nucleic acid là mang điện tích âm tương đối cao. Vì vậy, nucleic acid kết hợp với bề mặt tích điện dương, ví dụ các hạt ở trong chất nhựa trao đổi anion (Hình 2.8a). Người ta có thể cho chất nhựa trao đổi này vào dịch chiết của tế bào tương tự như CTAB, nhưng đơn giản hơn là sử dụng cột sắc ký. Chất nhựa trao đổi này được nhồi đầy cột và cho dịch chiết tế bào đi qua (Hình 2.8b). Acid nucleic gắn với chất nhựa trao đổi và được giữ lại trong cột, trong khi đó các chất khác không tích điện và tích điện dương thì được đi qua. Sau khi rửa các chất không cần thiết người ta thu lại nucleic acid bằng cách rửa cột với dung dịch muối có nồng độ cao, muối này làm cho tác dụng tương hỗ tĩnh điện giữa nucleic acid và chất trao đổi không bền vững. 2. Tách chiết DNA plasmid
Tinh sạch DNA plasmid từ dung dịch vi khuẩn nuôi cấy được thực hiện như tinh sạch DNA tổng số. Sự khác biệt quan trọng giữa tinh sạch DNA plasmid và DNA tổng số của tế bào là phải tách ra lượng lớn DNA nhiễm sắc thể của vi khuẩn, mà chúng cùng tồn tại trong tế bào.
Ly tâm CTAB Carbohydrate, protein, v.v.
Dịch tế bào Phức hệ kết tủa gồm acid nucleic và CTAB
Bỏ phần dung dịch, giữ lại phần kết tủa
DNA sạch
Phức hệ kết tủa gồm nucleic acid và CTAB
Kết tủa được hòa tan trong NaCl 1M
Kết tủa bằng ethanol, xử lý bằng ribonuclease
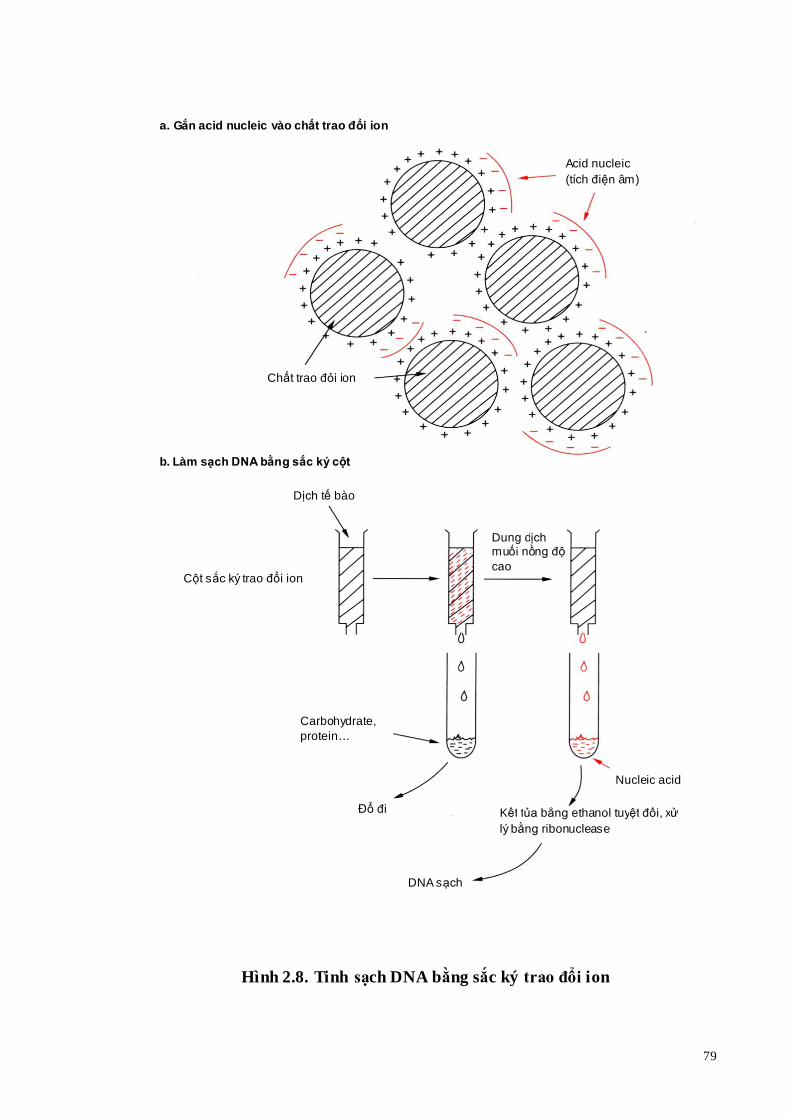
79
Hình 2.8. Tinh sạch DNA bằng sắc ký trao đổi ion
Carbohydrate, protein…
Đổ đi
DNA sạch
Nucleic acid
Kết tủa bằng ethanol tuyệt đối, xử lý bằng ribonuclease
Dung dịch muối nồng độ cao
Cột sắc ký trao đổi ion
Dịch tế bào
b. Làm sạch DNA bằng sắc ký cột
Chất trao đỏi ion
Acid nucleic (tích điện âm)
a. Gắn acid nucleic vào chất trao đổi ion

80
a. Nucleic acid tích điện âm liên kết với bề mặt tích điện dương của các hạt trao đổi,
b. DNA được tách ra từ cột sắc ký
Có nhiều phương pháp để tinh sạch DNA plasmid. Có thể sử dụng riêng lẻ hoặc kết hợp các phương pháp này sẽ thu được DNA plasmid tinh khiết.
Phương pháp này dựa trên những đặc điểm vật lý khác nhau của DNA plasmid và DNA nhiễm sắc thể. Sự khác biệt lớn nhất là độ lớn. Plasmid lớn nhất cũng chỉ đạt đến 8% độ dài của nhiễm sắc thể E.coli và phần lớn plasmid là nhỏ hơn. Các phương pháp phân biệt được phân tử DNA lớn và nhỏ là phương pháp làm sạch DNA plasmid rất có hiệu quả.
Ngoài độ lớn thì người ta còn phân biệt DNA plasmid và của vi khuẩn là nhờ cấu trúc của chúng. Các DNA có hình dạng nhất định trong không gian. Plasmid cũng như nhiễm sắc thể vi khuẩn có dạng vòng, nhưng ở quá trình tinh sạch thì DNA nhiễm sắc thể luôn luôn bị gãy, xuất hiện các đoạn thẳng. Với phương pháp này dạng vòng và dạng thẳng được tách ra nên người ta thu được plasmid rất sạch. 2.1. Tách DNA dựa trên độ lớn
Sự phân đoạn theo độ lớn được thực hiện trong khi tạo ra dịch chiết tế bào. Hoà tan tế bào thận trọng trong những điều kiện xác định, thì chỉ xảy ra sự gảy rất nhỏ đối với DNA nhiễm sắc thể. Các đoạn DNA xuất hiện ở đây vẫn còn rất dài và lớn hơn rất nhiều so với plasmid. Chúng được tách ra cùng với phần tạp chất bằng cách ly tâm, cùng với mảnh vỡ của tế bào. Khi phá vỡ tế bào người ta phải rất thận trọng, để DNA nhiễm sắc thể không bị gảy thành đoạn. Xử lý bằng lysozyme và EDTA khi có mặt của saccharose sẽ ngăn cản sự vỡ tế bào. Bằng phương pháp này DNA của vi khuẩn bị gãy rất ít và sau khi ly tâm người ta thu được một dung dịch trong suốt hầu như chỉ chứa DNA plasmid.
Một lượng nhỏ DNA nhiễm sắc thể vẫn còn tồn tại trong dung dịch trong suốt này. Và các plasmid có phân tử lớn cũng có thể lắng kết với các mảnh vỡ của tế bào. Vì vậy sự phân đoạn theo độ lớn chưa đủ, người ta còn sử dụng các phương pháp khác nhằm loại bỏ DNA nhiễm sắc thể. 2.2. Tách DNA dựa vào hình dạng
Có sự khác nhau về hình dạng giữa plasmid và DNA nhiễm sắc thể của vi khuẩn: Plasmid có cấu trúc dạng vòng, nhưng không hoàn toàn chính xác như vậy, vì trong thực tế có hai dạng hoàn toàn khác nhau. Phần lớn plasmid ở trong tế bào ở dạng siêu xoắn (supercoils) (Hình 2.10a). Dạng siêu xoắn xuất hiện trong quá trình sao chép được thực hiện bởi enzyme topoisomerase. Dạng này được duy trì khi hai sợi polynucleotide không bị tổn hại. Vì vậy, người ta gọi vòng DNA là vòng đóng đồng hoá trị (covalently closed-circular DNA, ccc). Nếu có một chỗ đứt trên một trong hai sợi thì sợi kép trở lại trạng thái duỗi xoắn bình thường, và plasmid có dạng hình thứ hai được gọi là dạng vòng mở (open-circular, Hình 2.9b).
Dạng siêu xoắn có ý nghĩa đối với việc tinh sạch plasmid. Có hai phương pháp làm sạch DNA plasmid từ dịch thô của tế bào. Trong thực tế người ta thu được kết qủa tốt nếu đầu tiên tạo ra được dịch tế bào trong suốt.
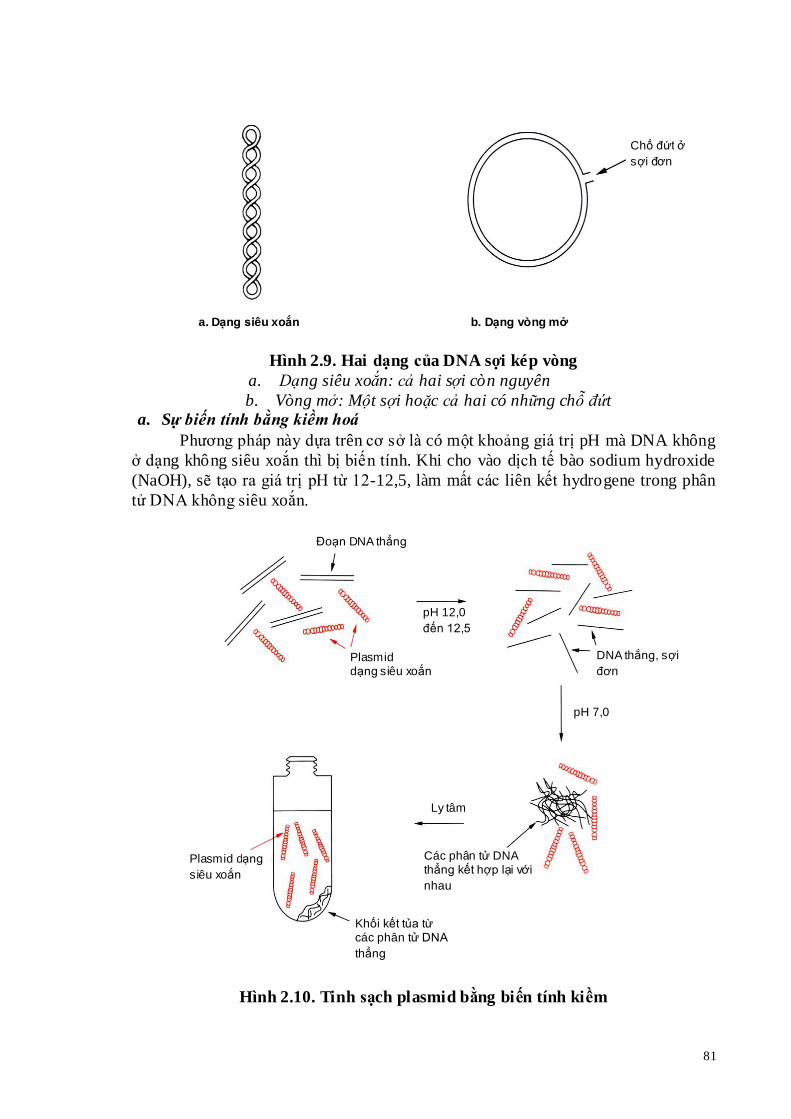
81
Hình 2.9. Hai dạng của DNA sợi kép vòng a. Dạng siêu xoắn: cả hai sợi còn nguyên
b. Vòng mở: Một sợi hoặc cả hai có những chỗ đứt a. Sự biến tính bằng kiềm hoá
Phương pháp này dựa trên cơ sở là có một khoảng giá trị pH mà DNA không ở dạng không siêu xoắn thì bị biến tính. Khi cho vào dịch tế bào sodium hydroxide (NaOH), sẽ tạo ra giá trị pH từ 12-12,5, làm mất các liên kết hydrogene trong phân tử DNA không siêu xoắn.
Hình 2.10. Tinh sạch plasmid bằng biến tính kiềm
Chổ đứt ở sợi đơn
a. Dạng siêu xoắn b. Dạng vòng mở
Đoạn DNA thẳng
pH 12,0 đến 12,5
DNA thẳng, sợi đơn
pH 7,0
Ly tâm
Plasmid dạng siêu xoắn
Plasmid dạng siêu xoắn
Các phân tử DNA thẳng kết hợp lại với nhau
Khối kết tủa từ các phân tử DNA thẳng

82
Sợi kép được duỗi xoắn và hai sợi polynucleotide tách nhau ra (Hình 2.10). Sau khi cho thêm acid vào, các sợi DNA biến tính kết hợp lại với nhau thành một khối phức tạp, không tan và dễ dàng được tách ra khi ly tâm. Trong dung dịch chỉ còn lại plasmid. Phương pháp này còn có một ưu điểm khác nữa: Dưới những điều kiện xác định, đặc biệt khi phân giải tế bào bằng sodium doecylsulfate và tiếp theo là phản ứng trung hoà với sodiumacetate thì phần lớn protein và RNA không hoà tan, có thể loại chúng ra bằng cách ly tâm. Khi đã biến tính DNA bằng kiềm hoá thì việc xử lý bằng phenol và bằng ribonuclease trong nhiều trường hợp là không cần thiết. b. Ly tâm gradient mật độ -Ethidium bromide – Caesium chloride
Gradient mật độ xuất hiện khi thực hiện ly tâm dung dịch muối caesium chloride (CsCl) ở vận tốc rất cao (Hình 2.11) vì một lực ly tâm mạnh kéo ion caesium và chloride xuống đáy của ống ly tâm. Tác động đi xuống này chống lại sự khuếch tán tạo ra một gradient nồng độ: dung dịch CsCl ở đáy ống ly tâm luôn trở nên đậm đặc hơn.
Các đại phân tử tìm thấy khi ly tâm trong dung dịch CsCl ở những vùng (vạch) xác định của gradient (Hình 2.11b). Mỗi loại phân tử tập hợp chính xác tại vị trí xác định phụ thuộc vào tốc độ di chuyển. Đối với DNA mật độ nằm khoảng 1,7 g/cm3, vì vậy chúng di chuyển trong gradient đến vị trí mà ở đó tỷ trọng của dung dịch CsCl cũng đạt đến giá trị này. Ngược lại, phân tử protein có tỷ trọng di chuyển nhỏ hơn và nên tập trung ở phần trên của ống ly tâm. RNA thì nằm ở đáy ống (Hình 2.11b). Với ly tâm gradient mật độ người ta tách được protein, DNA, RNA ra khỏi nhau. Ở sự tinh sạch DNA đây là một sự thay thế cho xử lý bằng phenol và ribonuclease.
Hình 2.11. Ly tâm theo gradient tỷ trọng CsCl a. Một gradient tỷ trọng xuất hiện khi ly tâm tốc độ cao. b. Sự phân tách protein, DNA và RNA theo gradient tỷ trọng
Nồng độ CsCl tăng lên
Protein
DNA
Tỷ trọng của dung dịch CsCl (g/cm3)
RNA
a b
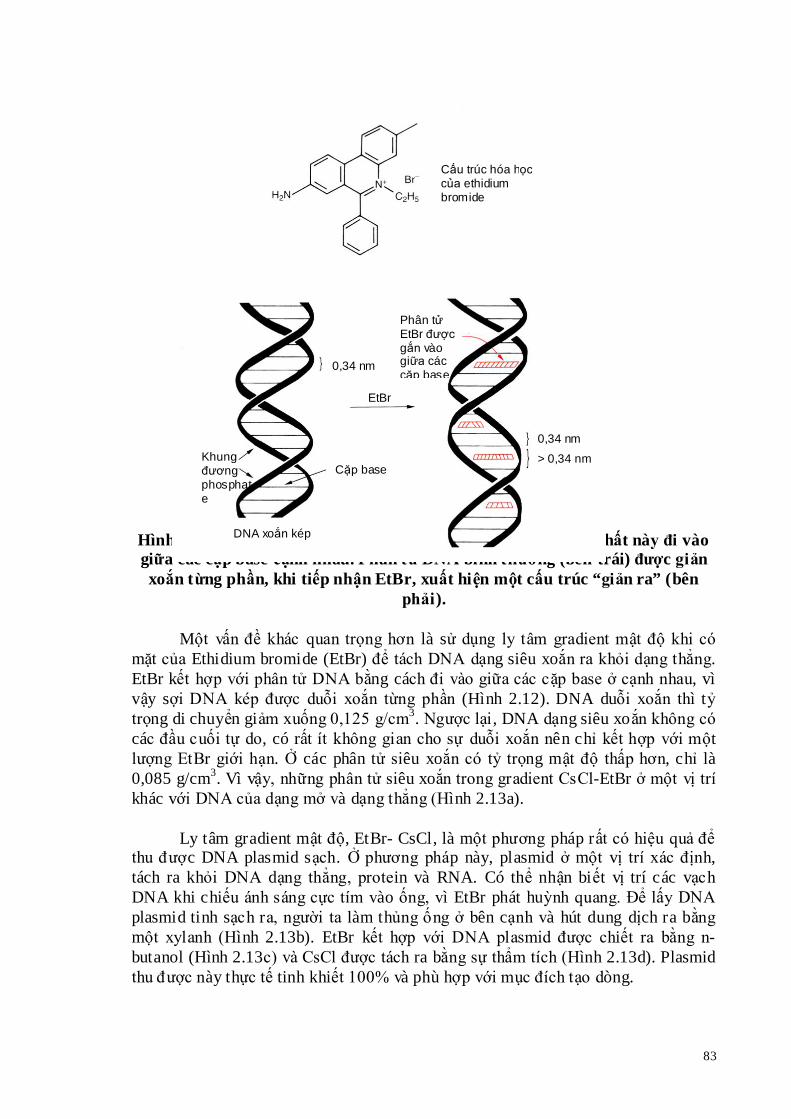
83
Hình 2.12. Giản xoắn từng phần của DNA xoắn kép bởi EtBr, chất này đi vào giữa các cặp base cạnh nhau. Phân tử DNA bình thường (bên trái) được giản xoắn từng phần, khi tiếp nhận EtBr, xuất hiện một cấu trúc “giản ra” (bên
phải). Một vấn đề khác quan trọng hơn là sử dụng ly tâm gradient mật độ khi có
mặt của Ethidium bromide (EtBr) để tách DNA dạng siêu xoắn ra khỏi dạng thẳng. EtBr kết hợp với phân tử DNA bằng cách đi vào giữa các cặp base ở cạnh nhau, vì vậy sợi DNA kép được duỗi xoắn từng phần (Hình 2.12). DNA duỗi xoắn thì tỷ trọng di chuyển giảm xuống 0,125 g/cm3. Ngược lại , DNA dạng siêu xoắn không có các đầu cuối tự do, có rất ít không gian cho sự duỗi xoắn nên chỉ kết hợp với một lượng EtBr giới hạn. Ở các phân tử siêu xoắn có tỷ trọng mật độ thấp hơn, chỉ là 0,085 g/cm3. Vì vậy, những phân tử siêu xoắn trong gradient CsCl-EtBr ở một vị trí khác với DNA của dạng mở và dạng thẳng (Hình 2.13a).
Ly tâm gradient mật độ, EtBr- CsCl, là một phương pháp rất có hiệu quả để
thu được DNA plasmid sạch. Ở phương pháp này, plasmid ở một vị trí xác định, tách ra khỏi DNA dạng thẳng, protein và RNA. Có thể nhận biết vị trí các vạch DNA khi chiếu ánh sáng cực tím vào ống, vì EtBr phát huỳnh quang. Để lấy DNA plasmid tinh sạch ra, người ta làm thủng ống ở bên cạnh và hút dung dịch ra bằng một xylanh (Hình 2.13b). EtBr kết hợp với DNA plasmid được chiết ra bằng n-butanol (Hình 2.13c) và CsCl được tách ra bằng sự thẩm tích (Hình 2.13d). Plasmid thu được này thực tế tinh khiết 100% và phù hợp với mục đích tạo dòng.
Cấu trúc hóa học của ethidium bromide
Phân tử EtBr được gắn vào giữa các cặp base
Khung đương phosphate
DNA xoắn kép
Cặp base
EtBr
0,34 nm
0,34 nm
> 0,34 nm

84
Hình 2.13. Làm sạch plasmid bằng ly tâm gradient tỷ trọng EtBr-CsCl 2.3. Nhân plasmid
Khi plasmid chỉ chiếm một phần nhỏ trong DNA tổng số của tế bào thì việc tách DNA plasmid khó khăn. Hiệu quả thu hồi DNA của một dung dịch vi khuẩn thường rất thấp. Một khả năng để nâng cao nó là nhân plasmid.
Nhân có mục đích là tăng số lượng bản sao của một plasmid. Loại plasmid có nhiều bản sao (số lượng bản sao là 20 hoặc lớn hơn) là đặc tính có lợi, nó có thể tự tái sinh mà không tổng hợp protein. Ngược lại nhiễm sắc thể của vi khuẩn không tái sinh dưới điều kiện này. Người ta lợi dụng sự khác này khi nuôi cấy dung dịch vi khuẩn. Khi mật độ tế bào đủ cao, thì cho vào một chất kìm hãm sự tổng hợp protein ví dụ chloramphenicol và tiếp tục nuôi cấy thêm 12 giờ nữa. Trong thời gian này, plasmid tiếp tục tái sinh, mặc dù sự tái sinh nhiễm sắc thể và sự phân chia tế bào bị phong tỏa (Hình 2.14). Kết quả là số lượng bản sao của plasmid có thể tăng lên
Protein
DNA dạng thẳng và dạng mở vòng
DNA siêu xoắn
RNA
Lấy DNA siêu xoắn ra bằng một mũi kim gắn syringe
a. Gradient tỷ trọng EtBr-CsCl b. Lấy ra vạch DNA
Đệm
Dung dịch DNA đi vào ống thẩm tích
Cho n-butanol vào để phân tách hai pha
EtBr trong pha chất hữu cơ
DNA trong pha lỏng
CsCl khuếch tán vào dung dịch đệm
d. Tách CsCl bằng thẩm tích c. Tách EtBr bằng n-butanol

85
hàng ngàn. Sự nhân này cũng là một phương pháp có hiệu quả để tăng sự khai thác ở plasmid có nhiều bản sao.
Hình 2.14. Khuếch đại plasmid
3. Tinh sạch DNA của phage
Sự khác biệt quan trọng nhất trong việc tinh sạch DNA của phage và plasmid và tổng số là ở phage thì người ta không đi từ dịch chiết tế bào. Nguyên nhân là có thể thu được một lượng lớn hạt phage từ dung dịch nuôi cấy vi khuẩn bị nhiễm phage. Khi ly tâm dung dịch này, các tế bào vi khuẩn lắng xuống, trong khi đó các hạt phage ở lại trong dung dịch (Hình 2.15). Tiếp theo là tách phage từ dung dịch này và tách DNA của nó trong một bước duy nhất là tách ra vỏ protein.
Về tổng thể thì quá trình này hầu như đơn giản hơn tách DNA plasmid hoặc DNA tổng số. Tuy nhiên ở sự tinh sạch này để có đủ một lượng DNA cần phải có dung dịch có số lượng phage cao (phagetiter: phage chuẩn độ là số lượng phage/ml dung dịch). Trong thực tế nồng độ phage cao nhất mà người ta mong đợi đối với phage là 1010/ml. Nhưng 1010 phage chỉ cho khoảng 500 ng (10-9 g) DNA. Vì vậy cần một thể tích lớn (500-1000 ml) khi muốn có một lượng DNA cần thiết. 3 1. Nuôi cấy dung dịch để đạt được phagetiter cao
Phage λ tồn tại trong tự nhiên chủ yếu ở dạng tiềm tan, vì vậy dung dịch vi khuẩn bị nhiễm phage là phần lớn gồm những tế bào mà DNA phage hợp nhất vào DNA của vi khuẩn. Trường hợp này số lượng phage bên ngoài những tế bào này rất thấp.
Nhiều bản sao plasmid (số lượng bản sao >20)
DNA vi khuẩn
Nuôi cấy khi có chloramphenicol
Có hàng nghìn plasmid

86
Hình 2.15. Thu dung dịch phage từ dung dịch vi khuẩn nhiễm Để đạt được hiệu quả khai thác cao, người ta phải tác động vào dung dịch để
tất cả tế bào ở pha sinh tan của chu kỳ xâm nhiễm, sau đó tế bào vi khuẩn chết giải phóng ra các hạt . Sự tác động này bình thường khó điều khiển, nhưng phần lớn các chủng phage sử dụng trong phòng thí nghiệm mang một gene cI, một đột biến mẫn cảm với nhiệt độ. Gene cI thuộc vào nhóm gene đảm bảo phage ở trạng thái hợp nhất. Gene này bị bất hoạt do đột biến nên phage chuyển sang dạng sinh tan. Ở đột biến cIts, gene cI hoạt động ở nhiệt độ 300C, phage ở trạng thái tiềm tan (lysogene). Ngược lại, ở nhiệt độ 420C phage λ chuyển sang trạng thái sinh tan. Có thể kích thích dung dịch tế bào E. coli lây nhiễm cIts tạo ra sản phẩm là phage tự do bằng cách tăng nhiệt độ từ 30 0C lên 420C (Hình 2.16).
Môi trường nuối cấy bị nhiễm (vi khuẩn và phage tự do)
Ly tâm
Dung dịch phage
Vi khuẩn kết tủa
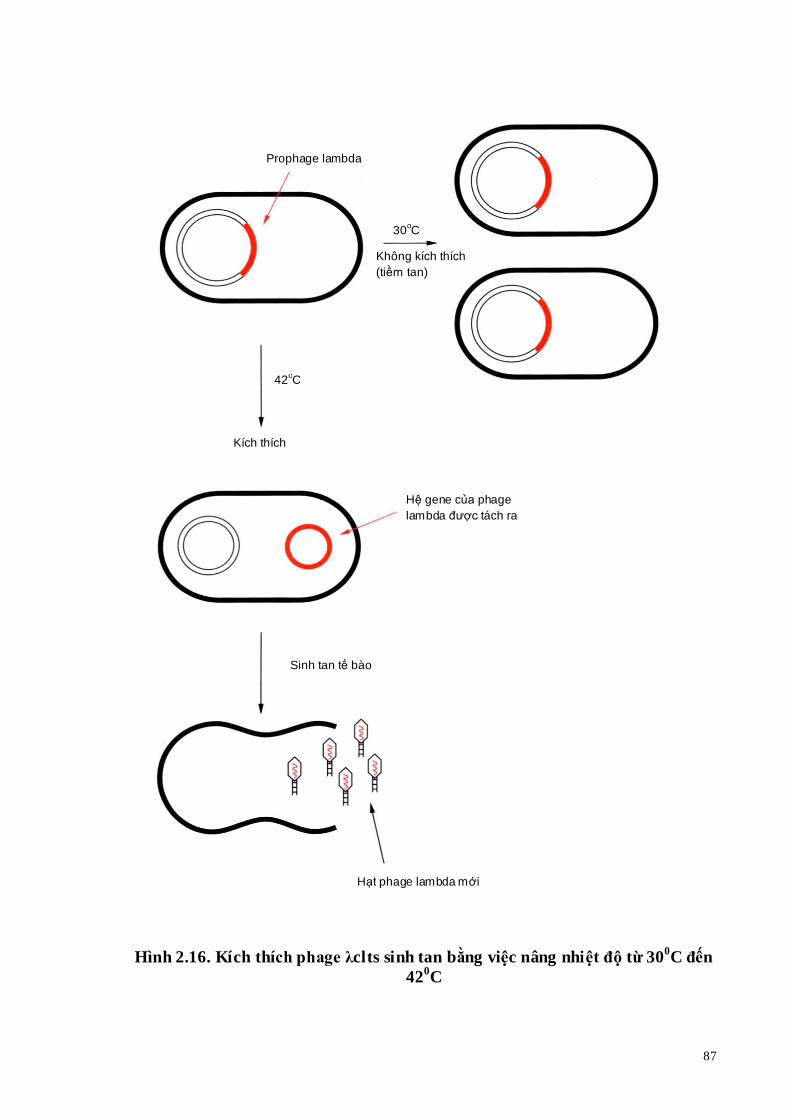
87
Hình 2.16. Kích thích phage λclts sinh tan bằng việc nâng nhiệt độ từ 300C đến 420C
Sinh tan tế bào
Hạt phage lambda mới
Kích thích
42oC
Hệ gene của phage lambda được tách ra
Prophage lambda
Không kích thích (tiềm tan)
30oC

88
Hình 2.17. Tạo một sự cân bằng chính xác giữa tuổi của dung dịch tế bào và số lượng phage cho vào khi chuẩn bị mẫu phage không tiềm tan
a. Mật độ vi khuẩn quá thấp
b. Mật độ vi khuẩn quá cao
Vi khuẩn
Bổ sung thêm phage
Cho vào phage
Tất cả tế bào bị sinh tan nhanh Số lượng phage/ml thấp
Dung dịch tế bào không bao giờ sinh tan hoàn toàn Số lượng phage/ml thấp
Tế bào trong dung dịch tiếp tục phát triển, tuy nhiên cuối cùng cũng bị sinh tan Số lượng phage/ml cao
c. Mật độ vi khuẩn chính xác

89
Hình 2.18. Thu hoạch phage bằng kết tủa với polyethyleneglycol (PEG) 3.2. Tinh sạch -phage không tiềm tan
Phần lớn chủng là tiềm tan nhưng nhiều vector tạo dòng có nguồn gốc từ phage này được biến đổi bằng cách loại gene cI và các gene khác làm mất vĩnh viễn trạng thái tiềm tan. Những phage này không những không có khả năng hợp nhất vào hệ gene của vi khuẩn, mà chỉ nhiễm các tế bào này ở chu kỳ sinh tan.
Phagetiter của các loại phage này phụ thuộc vào cách chúng được nuôi, đặc biệt là cách lây nhiễm tế bào. Cho phage vào dung dịch nuôi cấy trước khi tế bào đạt đến tốc độ tái sinh cực đại, thì tất cả những tế bào này sẽ được phân giải rất nhanh, lúc này nồng độ phage có giá trị thấp (Hình 2.17a). Ngược lại khi mật độ tế bào đã cao mới cho phage vào, thì dung dịch nuôi cấy không bao giờ được sinh tan hoàn toàn và nồng độ phage cũng thấp (Hình 2.17b). Trường hợp lý tưởng là tuổi của dung dịch nuôi cấy và lượng phage cho vào cùng xác định, tiếp tục nuôi cấy, ở đây tất cả tế bào bị nhiễm và sinh tan (Hình 2.17c). 3.3. Thu phage từ dung dịch nhiễm vi khuẩn
Phage có kích thước nhỏ nên chúng chỉ kết lắng khi ly tâm với tốc độ rất cao. Vì vậy, để thu hoạch chúng người ta thường kết tủa với polyethyleneglycol (PEG). Hợp chất này là một polymer có chuỗi dài, hấp thu nước khi có mặt của các muối, vì vậy nó kết hợp với những phân tử lớn như hạt phage. Kết lắng này được tách ra nhờ ly tâm và được hoà tan trở lại trong một thể tích thích hợp (Hình 2.18).
Dung dịch phage Phage được kết tủa
Kết tủa từ phage và từ phần không tan của tế bào
Hòa tan kết tủa
Ly tâm Bổ sung PEG + NaCl

90
Hình 2.19. Tinh sạch phage λ bằng ly tâm tỷ trọng CsCl 3.4. Tinh sạch DNA từ hạt phage
Kết tủa PEG thường chứa một lượng nhất định các chất cặn, có thể DNA không mong muốn. Các tạp chất này được tách ra khỏi phage bằng ly tâm gradient mật độ CsCl. Những hạt phage tạo nên vạch trong CsCl-gradient ở 1,45-1,5 g/cm3 (Hình 2.19) và có thể lấy chúng ra một cách chính xác từ ống ly tâm, như đã được mô tả (Hình 2.13). Tiếp theo tách CsCl bằng sự thẩm tích, như vậy chỉ còn lại phage tinh khiết, có thể xử lý với phenol hoặc protease để phân giải vỏ protein của phage và thu được DNA tinh khiết. 3.5. M13 được tinh sạch dễ dàng
Những khác nhau trong chu kỳ xâm nhiễm giữa phage M13 và đối với các nhà sinh học phân tử chủ yếu là ưu điểm khi tinh sạch M13-DNA. Trước hết M13 ở dạng sao chép sợi kép, ở dạng này thể hiện như là plasmid có nhiều bản sao, rất dễ phân lập theo phương pháp tiêu chuẩn đối với plasmid. Người ta tạo ra một dịch tế bào từ những tế bào bị nhiễm M13 và tách dạng M13 sợi kép bằng ly tâm gradient mật độ EtBr-CsCl của DNA vi khuẩn.
Phage
Tỷ trọng của dung dịch CsCl (g/cm3)

91
Hình 2.20. Thu nhận DNA của M13 từ dung dịch vi khuẩn nhiễm Tuy nhiên, người ta cần hệ gene của M13 ở dạng mạch đơn, dạng này tìm
thấy ở bên ngoài tế bào. Ở đây M13 có ưu thế lớn so với phage , là dễ dàng thu được phagetiter cao (số lượng phage/ml dung dịch). Vì các tế bào bị nhiễm thường xuyên giải phóng ra hạt M13 vào dung dịch, ở đây tế bào không phân giải. Trong thực tế người ta dễ dàng đạt được nồng độ 1012 hạt phage hoặc cao hơn/ml. Ở titer cao người ta thu được một lượng đáng kể M13-DNA sợi đơn trong một thể tích nhỏ 5 ml hoặc ít hơn. Vì tế bào nhiễm không bị phân giải, nên không gặp khó khăn đối với mảnh vụn của tế bào. Để tách DNA của M13 sợi đơn người ta phải nuôi dung dịch vi khuẩn bị nhiễm trong một thể tích nhỏ và ly tâm, phage kết tủa với PEG, vỏ protein của phage được tách bằng xử lý phenol, và nâng nồng độ DNA được giải phóng ra bằng kết tủa phenol.
a. Dung dịch tế nhiễm b. Tách tế bào bằng ly tâm c. Cho PEG vào dung
dịch tế bào và ly tâm
g. M13-DNA trong thể tích nhỏ
f. Lấy pha lỏng (chứa M13-DNA) cho vào ethanol và ly tâm
e. Cho phenol vào để tách ra protein võ
d. Hòa tan phage vào dung dịch đệm
M13-DNA
Protein
Phenol
M13-DNA
Phage M13 kết tủa
Dung dịch phage M13
Tế bào kết tủa

92
II. Kỹ thuật lai nucleic acid 1. Kỹ thuật Southern blot
Nguyên lý: Dựa vào sự tương tác của các cặp base theo nguyên tắc bổ sung (A-T, G-C), các đoạn polynucleotide đơn có cấu trúc bổ sung có thể bắt cặp với nhau tạo nên các phân tử lai.
Điều kiện: Để tiến hành phản ứng lai cần tạo điều kiện nhiệt độ cao hoặc hóa chất gây
biến tính để tách các phân tử DNA sợi kép thành 2 sợi đơn. Nhờ vậy các sợi polynucleotide có trình tự bổ sung dễ dàng bắt cặp với nhau. Để phát hiện phản ứng lai người ta phải sử dụng mẫu dò.
Mẫu dò DNA là oligonucleotide được đánh dấu và nó bổ sung với trình tự gene quan tâm. Thông qua phương pháp lai, các mẫu dò sẽ bắt cặp với DNA quan tâm. Dựa trên sự phát hiện mẫu dò trên bản lai, người ta xác định được sự hiện diện của gene quan tâm.
Theo phương pháp này đoạn DNA nghiên cứu được cắt bằng một enzyme hạn chế, kết quả cho ra một hỗn hợp các đoạn cắt DNA có kích thước khác nhau. Tiến hành điện di các đoạn thu được trên gel agarose. Sau đó biến tính các vạch DNA trên gel để tạo các sợi đơn. Tiếp đó, các đoạn này được chuyển lên màng lai bằng phương pháp thấm truyền (blot). Màng lai có cấu tạo là nitrocellulose hoặc màng nylon có chức năng giữ các đoạn DNA được chuyển lên từ gel. Trên màng lai, các đoạn này chính là bản sao của các đoạn DNA trên gel. Màng lai được lai với mẫu dò phóng xạ và được phát hiện theo cách tương tự như màng lai vi khuẩn hay vết tan.
Có nhiều phương pháp để xác định vị trí của gene tạo dòng trong phân tử DNA. Sử dụng phương pháp nào là tùy thuộc vào độ lớn của phân tử DNA. Các phân tử nhỏ như plasmid và chromosome của phage thì sử dụng phương pháp khác với các gene có trong những phân tử DNA rất lớn của sinh vật nhân chuẩn. 1.1. Định vị của gene tạo dòng trên phân tử DNA nhỏ
Như ví dụ ở phần trước, gene kháng kanamycin trong plasmid R6-5 là một đọan được cắt bằng EcoRI và tạo dòng trong plasmid pBR322. Tuy nhiên, người ta muốn biết gene kháng đó định vị trên đoạn nào trong 13 đoạn được cắt bằng EcoRI. Qua đó có thể định vị tương đối gene trên bản đồ của R6-5.
Trước hết phải tách các đoạn DNA của R6-5 được cắt bằng EcoRI trên gel agarose. (Hình 2.21a). Các đoạn này được đưa vào vector pBR322 mang gene kháng kanamycin và sử dụng làm mẫu dò cho các đoạn cắt hạn chế. Có thể nhận biết được các đoạn này ở trên gel, nhưng thường thì kết quả thu nhận này không được tốt vì các chất trong gel dẫn đến sự lai không đặc hiệu. Vì vậy, phải chuyển các vạch này lên màng nitrocellulose hoặc nylon cho thấy kết quả lai rõ ràng hơn.
Để chuyển những đoạn DNA từ agarose lên màng lai người ta sử dụng phương pháp được đề nghị bởi E.M Southern (1975), vì vậy phương pháp này được gọi là Southern blot. Màng lai được đặt trực tiếp lên gel và sau đó để dung dịch đệm đi qua, DNA đi theo dung dịch đệm từ gel lên màng và được giữ lại ở đây. Có thể thực hiện công việc này bằng máy nhưng nhiều nhà sinh học phân tử vẫn sử dụng dụng cụ tự làm với nhiều cho chồng giấy (Hình 2.21b). Phương pháp này cũng được sử dụng đối với RNA (Northern blot) hoặc protein (Western blot).

93
Hình 2.21. Phương pháp Southern blot
Các đoạn của R6-5 cắt bằng EcoRI
a. Điện di DNA của R6-5 cắt bằng EcoRI
b. Southern blot
Chồng giấy thấm
Màng nitrocellulose hoặc nylon Gel
Giấy hút
Đệm
Giá đỡ
c. Kết quả lai
Tín hiệu positive: đoạn số 6
Đoạn số 6: vị trí của gene KanR
d. Vị trí của đoạn trên bản đồ cắt hạn chế R6-5
Vị trí nhận biết của EcoRI

94
Bằng Southern blot người ta có màng với các vạch DNA là bản sao của gel
agarose. Sau đó, cho mẫu dò đánh dấu vào để xác định phản ứng lai và vị trí lai bằng phóng xạ tự ghi (hoặc một hệ thống đánh dấu không phóng xạ) (Hình 2.21c). Nhờ vậy người ta biết được vị trí của gene kháng kanamycin trên bản đồ plasmid R6-5.
Hình 2.22. Có thể xác định được vị trí của gene tạo dòng bằng Southern blot
Với phương pháp lai Southern người ta biết được chính xác vị trí của gene tạo dòng trong phân tử DNA tái tổ hợp. Quan trọng là khi đoạn DNA tạo dòng tương đối lớn (ở vector cosmid cho đến 40 kb), mà gene cần tìm có thể chỉ là 1 kb. Đoạn DNA tạo dòng có thể mang nhiều gene khác ngoài gene mà người ta quan tâm. Vì vậy, để xác định được một dòng từ thư viện hệ gene phải thực hiện phân
R6-5
Phân tử DNA tái tổ hợp-gene đó ở đâu?
Lai Southern
Vector
Đoạn cắt cùng với gene

95
tích Southern blot với phân tử DNA tái tổ hợp, nhằm xác định chính xác vị trí của gene quan tâm trong đoạn tạo dòng (Hình 2.22). a. Ứng dụng kỹ thuật Southern blot
Kỹ thuật Southern blot là kỹ thuật đơn giản nhưng được ứng dụng rất nhiều và là phương pháp rất có giá trị trong phân tích gene. Đặc biệt, đây là phương pháp chuẩn được áp dụng trong việc đánh giá tính đa hình của các đoạn DNA đã được xử lý bằng enzyme hạn chế, được gọ i là RFLP (Restriction Fragment Length Polymorphisms- Sự đa hình chiều dài các đoạn hạn chế).
RFLP là sự khác biệt về chiều dài của các đoạn DNA được cắt đặc hiệu bằng enzyme hạn chế. RFLP được tạo ra khi DNA có nhiều điểm nhận biết của enzyme hạn chế. Vì vậy, việc xen đoạn cũng làm thay đổi vị trí tương đối của các điểm cắt hạn chế. b. Nghiên cứu cấu trúc của gene tạo dòng
Khi nghiên cứu cấu trúc của gene hoặc hệ gene mà chưa biết được trình tự nucleotide thì việc sử dụng các phương pháp gián tiếp cũng thu được những hiểu biết có giá trị. Các phương pháp này có ưu điểm là tiến hành tương đối nhanh và có thể so sánh giữa nhiều mẫu DNA khác nhau. Hai phương pháp được đề cập ở đây: phân tích đa hình độ dài các đoạn hạn chế (RFLPs) và DNA-fingerprinting.
Đoạn DNA không đột biến
Đột biến điểm ở vị trí cắt thứ 3
Loại bỏ một đoạn nhỏ trong đoạn 2-3
Loại bỏ một đoạn trong đó có cả vị trí 3

96
Hình 2.23. Ba đột biến và biểu hiện của chúng lên bản đồ hạn chế của phân tử DNA
- Phân tích đa hình chiều dài các đoạn hạn chế (RFLP)
Hai phân tử DNA rất giống nhau và có trình tự nucleotide chỉ khác nhau rất ít được nhận biết khi so sánh bản đồ hạn chế của chúng. Ba khả năng (có thể còn có những khả năng khác) được thể hiện ở hình 2.23. Cả ba trường hợp đều được chứng minh bằng đa hình độ dài các đoạn hạn chế khi hiển thị những đoạn cần nghiên cứu bằng điện di trên gel agarose.
Nhưng ở đây có một vấn đề: Các đoạn hạn chế của hầu hết sinh vật không thể nhận biết được bằng điện di. Ví dụ khi cắt DNA của người bằng EcoRI xuất hiện khoảng 700 000 đoạn, ở trên gel agarose tạo nên một “đám”, trong một hỗn hợp thì hoàn toàn không thể nhận biết một vạch duy nhất thay đổi.

97
Hình 2.24. Tì m đa dạng chiều dài các đoạn cắt hạn chế trong một mẫu dò
DNA người
Ở đây một phương pháp biến đổi từ Southern blot được sử dụng. Sản phẩm của phản ứng cắt cho chạy điện di bình thường, nhưng không hiển thị bằng nhuộm ethidiumbromid, mà chuyển “đám” vạch này lên màng nitrocellulose hoặc màng nylon bằng Southern blot (Hình 2.24). Sử dụng mẫu dò là đoạn DNA chứa vùng nghi vấn và thực hiện phản ứng lai. Khi mẫu dò lai với đoạn DNA thì được “đánh dấu” trong phóng xạ tự ghi. So sánh độ dài của nó với mẫu gene bình thường (không đột biến) và có thể nhận biết RFLP rõ ràng. - DNA-fingerprinting
Phóng xạ tự ghi: RFLP trong mẫu được nghiên cứu (*)
Lai Southern
Mấu dò
Cấu trúc gene thường (đối chứng)
Agarose gel
Giếng 1 = Marker về độ lớn Giếng 2 = DNA người, mẫu nghiên cứu Giếng 3 = DNA người, mẫu đối chứng

98
DNA-Fingerprinting là một dạng đặc biệt của phân tích RFLP, với phương pháp này người ta có thể tạo ra một bức tranh về cấu trúc của một hệ gene. Ở DNA-fingerprinting không chỉ tập trung vào RFLP mà đồng thời chứng minh một số lớn sự sai khác. Trong trường hợp này người ta sử dụng mẫu dò là một hỗn hợp các phân tử DNA đặc trưng cho các phần khác nhau của hệ gene, là những vị trí chứa các trình tự lặp lại (trình tự ngắn, khoảng 64 bp) (Hình 2.25a). Hai đặc điểm của trình tự lặp lại kế tiếp nhau (tandem) tạo cơ sở cho DNA-fingerprinting:
- Trình tự tương tự hoặc giống với trình tự nằm ở nhiều vị trí trong geneome (Hình 2.25b). Như vậy người ta có thể chứng minh đồng thời một loạt RFLPs khi sử dụng 1 mẫu dò duy nhất.
- Số lượng trình tự lặp lại ở các vị trí biến động rất lớn. Sự sắp xếp chỉ ra RFLPs với một tần suất lớn. Ở DNA-fingerprinting người ta tiến hành như ở phân tích RFLP bình
thường. DNA hệ gene được cắt bằng một enzyme hạn chế và được tách bằng điện di, sau đó được chuyển lên màng và lai với mẫu dò. Sự khác nhau ở chỗ phóng xạ tự ghi không phải là một mô hình đơn giản gồm 4 hoặc 5 vạch như ở hình 2.24. Người ta thu được sự sắp xếp các vạch phức tạp hơn vì mẫu dò lai với trình tự lặp lại ở nhiều vị trí của hệ gene. Mẫu vạch của hai người là khác nhau vì sự biến động mạnh của sự sắp xếp trình tự lặp lại với xác xuất cao (ở đây là trường hợp sinh đôi cùng trứng).
DNA-fingerprinting được sử dụng phổ biến trong pháp y học toà án. Sử dụng chính xác các mẫu dò, xác suất hai người không phải là sinh đôi cùng trứng có mô hình vạch DNA giống nhau chỉ khoảng một phần triệu. Một bị cáo có DNA-fingerprinting trùng với mô hình từ máu hoặc tóc ở hiện trường, thì sự nghi ngờ là cao nhất. Sự chứng minh mối quan hệ mẹ con hoặc bố con cũng sử dụng DNA-fingerprinting.
DNA-fingerprinting là đặc trưng cho mỗi người, nó chỉ ra sự giống nhau tương ứng với bố và mẹ (Hình 2.26). Khi so sánh DNA-fingerprinting người ta có thể khẳng định, hai người đó có quan hệ họ hàng với nhau hay không? Điều này cũng có ý nghĩa quan trọng trong nghiên cứu các động vật có giá trị như ngựa đua, quần thể chim và các quần thể động vật khác.

99
Hình 2.25. DNA- fingerprinting
a. Sự lặp lại các trình tự lặp
b. Những đơn vị lặp lại giống nhau có thể ở các vị trí khác nhau trong geneome
c. Kết quả phóng xạ tự ghi
Giếng 1 và 2: DNA của hai người khác nhau

100
Hình 2.26. DNA-fingerprint của bố mẹ và con cái tương tự nhau.
Con gái di truyền các vạch từ bố hoặc mẹ Sự sai khác về kích thước của các đoạn cắt thu được khi cắt DNA của nhân,
cơ quan tử hay toàn bộ cơ thể bằng enzyme hạn chế được gọi là “sự đa hình chiều dài của các đoạn hạn chế: Restriction fragment length polymorphisms: RFLP”.
Các đoạn DNA (cắt bằng một loại enzyme hạn chế) với kích thước hay chiều dài khác nhau có thể được dùng như các dấu hiệu di truyền để xem xét các mẫu nhiễm sắc thể.
Sự nhận biết các đọan cắt được thực hiện nhờ kỹ thuật lai với mẫu dò DNA (các mẫu DNA tương tự đã được chuẩn bị trước).
Có thể sử dụng nhiều enzyme hạn chế để cắt một DNA (mỗi lần cắt sử dụng một loại enzyme), sau đó phân tích các mẫu DNA thu được của từng loại enzyme mà thiết lập nên bản đồ enzyme hạn chế “ Restriction enzyme map” cho một phân tử DNA nghiên cứu.
Các DNA tương đối nhỏ như DNA lục lạp thường bị cắt thành vài chục đoạn khi sử dụng enzyme cắt phổ biến EcoRI. Các DNA của lục lạp nếu khác nhau về trình tự base sẽ cho các đoạn cắt có kích thước khác nhau, dễ dàng được nhận biết. Nhiều nghiên cứu đã sử dụng tính đa hình của các đoạn cắt của DNA lục lạp để nghiên cứu mối quan hệ huyết thống giữa các nhóm thực vật khác nhau.
Tuy nhiên, việc xem xét tính đa hình các đoạn cắt của DNA lục lạp trong công tác giống cây trồng là rất hạn chế, vì các gene quy định các tính trạng nông nghiệp quan trọng chủ yếu định vị trong nhiễm sắc thể của nhân.
Việc phân tích tính đa hình của các đoạn cắt DNA nhân phức tạp hơn do độ phức tạp của DNA nhân lớn hơn rất nhiều. Khi cắt DNA nhân bằng enzyme hạn chế đặc hiệu sẽ tạo ra hàng triệu đoạn cắt xếp liên tục nhau theo kích thước trên điện di. Nếu nhuộm màu các đoạn cắt này và xem xét dưới ánh sáng tử ngoại thì không thể thấy sự phân cách giữa các đoạn cắt, điện di đồ như một dãi mây. Để giải quyết vấn
Giếng 1: Mẹ Giếng 2: Con gái Giếng 3: Bố

101
đề này, người ta không thể so sánh sự sai khác của toàn bộ các đoạn cắt có mặt trên điện di đồ mà chỉ so sánh sự sai khác của một trình tự nucleotide bất kỳ nào đó trên các băng điện di.
Một đoạn DNA nào đó lấy từ thư viện hệ gene sẽ được sử dụng làm mẫu dò để phát hiện ra trình tự nucleotide tương ứng có mặt trên các băng điện di. Tiến hành lai Southern blot, so sánh sự hiện diện của các vị trí lai sẽ phát hiện được tính đa hình của các đoạn cắt. Nếu sử dụng mẫu dò là gene cần tìm ta có thể phát hiện được gene bằng kỹ thuật trên.
Kỹ thuật này được ứng dụng để phát hiện được sự có mặt của gene cần tìm trong hệ gene sinh vật nghiên cứu, phát hiện sự đa dạng DNA của các cá thể khác nhau, các đột biến, các thể lai soma qua dung hợp tế bào trần và lập bản đồ giới hạn của một gene. 1.2. Định vị gene tạo dòng trên phân tử DNA lớn
Lai Southern blot chỉ có ý nghĩa khi đã có bản đồ hạn chế của phân tử DNA, vì vậy phương pháp này phần lớn phù hợp với plasmid, phage và virus. Các phân tử DNA lớn hơn người ta không thể định vị gene tạo dòng. Nếu phân tử lớn hơn 250 kb việc lập bản đồ hạn chế rất phức tạp, có thể dễ dàng tưởng tượng được khi quan sát hình 1.23. Vì vậy, người ta sử dụng các phương pháp khác để nhận biết vị trí của những gene tạo dòng trên các phân tử DNA của nhiễm sắc thể. Sau đây là các khả năng để xác định vị trí của gene tạo dòng trên phân tử DNA có kích thước lớn. a. Tách nhiễm sắc thể bằng điện di
Câu hỏi đặt ra đầu tiên là: Gene quan tâm thuộc nhiễm sắc thể nào? Ở một số sinh vật có thể khẳng định được nhờ phân tích Southern blot đặc biệt, ở đây không sử dụng enzyme hạn chế mà là các nhiễm sắc thể nguyên vẹn được tách bằng phương pháp điện di mới.
Ở điện di thường điện trường chạy dọc theo chiều dài của gel và các phân tử DNA di chuyển theo đường thẳng đến điện cực dương (Hình 2.27a). Các phân tử DNA có độ lớn khác nhau di chuyển với vận tốc khác nhau qua hệ thống lỗ rây của gel. Bằng cách này người ta chỉ có thể tách được các phân tử có độ lớn khác nhau trong giới hạn nhất định, vì các phân tử DNA lớn thì sự khác nhau về tốc độ di chuyển rất nhỏ (Hình 2.27b). Trong thực tế các phân tử lớn hơn 50 kb thì không thể tách được bằng điện di bình thường.
Có thể vượt qua sự giới hạn của phương pháp này khi người ta tạo ra một điện trường phức tạp hơn. Có nhiều phương pháp khác nhau, nhưng được sử dụng nhiều nhất là điện di thay đổi điện trường vuông góc (orthogonal field alternation gel electrophoresis, OFAGE). Điện trường không chạy dọc theo gel mà thay đổi nhanh giữa hai điện cực tới và lui và mỗi chuyển động lệch 45 độ theo hướng di chuyển (Hình 2.28a). Trong điều kiện này các phân tử DNA phải thay đổi hướng thường xuyên theo điện trường.

102
Hình 2.27. Điện di thường và giới hạn của nó Khi hướng điện trường thay đổi thường xuyên thì các phân tử DNA chuyển
động trong gel không theo đường thẳng (Hình 2.28a). Ở đây các phân tử DNA phải quay 900 theo hướng thay đổi của điện trường, trước khi chúng tiếp tục di chuyển. Sự chuyển động này được quyết định: Phân tử DNA nhỏ thích nghi với hướng di chuyển mới nhanh hơn phân tử lớn, vì vậy những phân tử nhỏ di chuyển nhanh hơn đến cuối gel. Quá trình này đã làm cho khả năng phân biệt của gel tăng lên, nhờ đó người ta có thể tách những phân tử gồm hàng nghìn kilobase như nhiễm sắc thể của sinh vật nhân chuẩn nấm men và nấm sợi.
Bằng phương pháp OFAGE và một số phương pháp tương tự có thể làm sạch DNA của từng nhiễm sắc thể từ gel và tạo ra các thư viện gene đặc trưng nhiễm sắc thể. Mỗi một thư viện chỉ chứa các gene của một nhiễm sắc thể, chúng nhỏ hơn và dễ thao tác hơn so với một thư viện hệ gene đầy đủ. Ngoài ra có thể chuyển các nhiễm sắc thể lên màng nitrocellulose hoặc nylon bằng Southern blot và sau đó phân tích phản ứng lai. Bằng cách này có thể xác định được nhiễm sắc thể chứa gene quan tâm.
a. Điện di agarose gel khởi đầu
b. Ảnh hưởng của độ dài DNA đến tốc độ dịch chuyển
Không phân biệt rõ các phân tử DNA ngoài một độ lớn xác định
Phân biệt tốt ở các phân tử DNA trong vùng độ lớn này
Khoảng cách di chuyển trong x giờ
llog
der m
olec
ular

103
Hình 2.28. Điện di thay đổi điện trường vuông góc
b. Lai in-situ có thể làm hiển thị vị trí của gene tạo dòng trên nhiễm sắc thể sinh vật nhân chuẩn Khả năng sử dụng OFAGE và phương pháp điện di mới giới hạn ở những
sinh vật nhân chuẩn bậc thấp có nhiễm sắc thể tương đối nhỏ. Sự tách các nhiễm sắc thể lớn hơn ( 50 000 kb) của động vật có vú và của những sinh vật bậc cao khác nằm ngoài khả năng của phương pháp này. Để định vị gene trên các phân tử DNA
a. Điện di điện trường vuông góc (OFAGE)
b. Tách nhiễm sắc thể nấm men bằng OFAGE
Số lượng nhiễm sắc thể
700 kb
500 kb
200 kb

104
lớn, có phương pháp phù hợp hơn lai in situ. Phương pháp này có ưu điểm là
biết được vị trí của gene tạo dòng trên nhiễm sắc thể.
Hình 2.29. Nhiễm sắc thể và lai in situ
Những vết chỉ ra vị trí của gene tạo dòng trên nhiễm sắc thể
Tế bào đã được cố định trên kính
bổ sung mẫu dò, phóng xạ tự ghi
Nhiễm sắc thể tự phát triển
Các vạch màu có thể nhận ra
10 m
a. Nhiễm sắc thể người giai đoạn kỳ giữa
b. Lai in situ

105
Phương pháp in situ có nguồn gốc từ kính hiển vi thường, với chúng người ta
có thể quan sát nhiễm sắc thể trong tế bào trong khi phân chia (Hình 2.29a). Ở nhiều loài sinh vật người ta có thể phân biệt dễ dàng hình dạng và các vạch khi sử dụng các phương pháp nhuộm khác nhau. Với phương pháp in situ có thể quan sát được vị trí gene tạo dòng trên nhiễm sắc thể dưới kính hiển vi.
Các tế bào được xử lý bằng các chất cố định để gắn chúng vào vật mang rồi ủ với ribunuclease và NaOH để phân giải RNA và biến tính DNA. Ở đây các liên kết hydrogene giữa các base nitrogene bị đứt và nhiễm sắc thể được duỗi ra ở một mức độ nhất định (Hình 2.29b). Gene tạo dòng được đánh dấu và cho vào hỗn hợp nhiễm sắc thể. Phản ứng lai xảy ra nếu gene tạo dòng tìm thấy đoạn gene bổ sung trên nhiễm sắc thể và nhận ra vệt đen trên phóng xạ tự ghi. Vị trí vệt đen cho biết vị trí gene tạo dòng ở trên nhiễm sắc thể. Lai in situ với mẫu dò đánh dấu phóng xạ là phương pháp khó nhưng đã định vị được một loạt gene trong tế bào chất của người.
Thay thế cho phương pháp phóng xạ là gắn mẫu dò với chất phát quang và kết quả lai được quan sát trực tiếp bằng kính hiển vi đặc biệt. Với các chất màu phát quang khác nhau người ta chứng minh được sự hiện diện đồng thời hai hoặc nhiều gene, vì các tín hiệu lai phân biệt được ở màu sắc. Phương pháp phát quang trong lai in situ được gọi là FISH, được sử dụng thường xuyên khi gắn với mẫu dò. Phương pháp này đặc biệt có lợi cho nghiên cứu tế bào, trong đó đã có sự sắp xếp nhiễm sắc thể. Sự sao chép nhiễm sắc thể hoặc sự giải mã các bước riêng rẽ trên các nhiễm sắc thể khác nhau được phân tích bằng phương pháp FISH nhanh hơn nhiều so với các phương pháp khác.
c. Sự di chuyển gene từ nhiễm sắc thể này đến nhiễm sắc thể khác
Vấn đề đặt ra là làm thế nào để nhận biết vị trí của một gene tạo dòng trên nhiễm sắc thể? Người ta thường đứng trước vấn đề ngược lại là đã biết vị trí của một gene, nhưng không có mẫu dò tương ứng, như vậy không thể tách gene ra được từ thư viện hệ gene. Trong trường hợp này có thể xác định dòng mong muốn bằng sự di chuyển nhiễm sắc thể (chromosome walking). Nhưng với điều kiện là: người ta đã có một dòng cho một gene thứ hai, được biết ở mức độ nào đó là ở gần gene cần tìm trên nhiễm sắc thể.
Như vậy cần có hai thư viện dòng được tạo ra bằng hai enzyme hạn chế khác
nhau, ví dụ enzyme EcoRI và BamHI. Các đoạn trong những dòng của hai thư viện trùm lên nhau (Hình 2.30a). Người ta lấy dòng với gene đã biết từ thư viện A và sử dụng nó là mẫu dò đối với thư viện B (Hình 2.30b). Ở một dòng từ thư viện B (hoặc ở nhiều thư viện khác) người ta nhận được một tín hiệu lai, vì đoạn tạo dòng này trùm lên với đoạn trong mẫu dò. Một dòng được tìm như vậy từ thư viện B bây giờ được sử dụng là mẫu dò cho thư viện A (Hình 2.30c). Phản ứng lai xảy ra với dòng thứ nhất và có thể với một số khác. Bằng cách lặp lại nhiều lần dần dần người ta nhận được bản đồ hạn chế của các đoạn trùm lên nhau và cuối cùng tiến đến được gene cần tìm (Hình 2.30d).
Sự di chuyển trên nhiễm sắc thể là một phương pháp khó và tốn nhiều thời gian. Sự di chuyển dài nhất mà đến nay đạt được là 200-250 kb
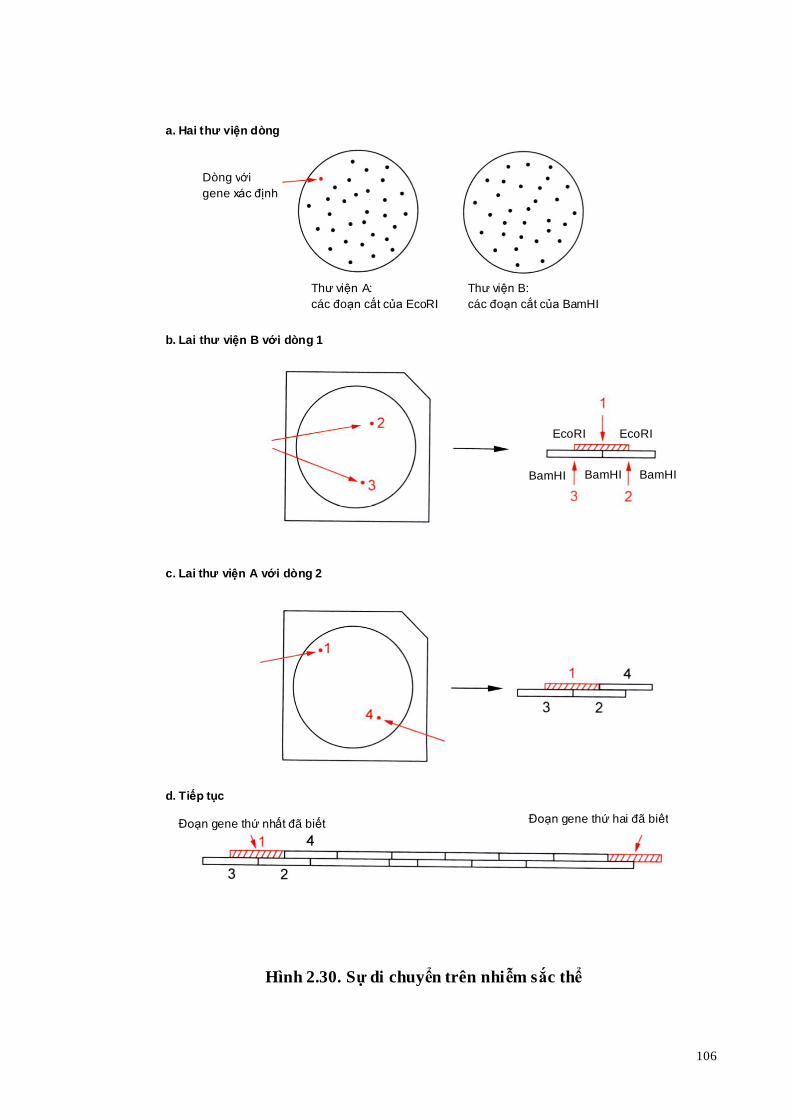
106
Hình 2.30. Sự di chuyển trên nhiễm sắc thể
a. Hai thư viện dòng
Dòng với gene xác định
Thư viện A: các đoạn cắt của EcoRI
Thư viện B: các đoạn cắt của BamHI
b. Lai thư viện B với dòng 1
BamHI
EcoRI
c. Lai thư viện A với dòng 2
…………..
d. Tiếp tục
Đoạn gene thứ nhất đã biết Đoạn gene thứ hai đã biết
BamHI BamHI
EcoRI

107
d. Phương pháp xác định một dòng Khi tạo được một thư viện hợp lý, người ta có thể sử dụng nhiều phương
pháp để tìm ra dòng mong muốn. Một số phương pháp dựa trên sự có mặt của sản phẩm gene, nhưng đơn giản hơn là xác định chính xác sự hiển diện của DNA tái tổ hợp bằng phương pháp lai acid nucleic.
Một phương pháp tạo dòng cDNA, xem chi tiết ở phần viết
Hình 2.31. Lai nucleic acid a. Phân tử lai không bền vững từ hai sợi DNA không bổ sung b. Phân tử lai bền vững từ hai sợi bổ sung c. Lai DNA-RNA: Một gene có thể lai với bản sao của nó
DNA RNA
c. Lai DNA-RNA
Những đoạn ngắn không cặp bổ sung không làm ảnh hưởng đến độ bền vững chung
b. Dạng lai bền
a. Dạng lai không bền

108
Hai sợi đơn nucleic acid thường có khả năng cặp đôi lại với nhau. Phần lớn là cấu trúc lai không bền vững, vì giữa hai sợi này chỉ một số liên kết hydrogene được tạo thành (Hình 2.31a). Tuy nhiên, ở các trình tự polynucleotide bổ sung thì xuất hiện nhiều liên kết giữa các base và tạo nên sợi kép bền vững (Hình 2.31b). Phản ứng lai không chỉ xảy ra giữa các phân tử DNA sợi đơn mà còn xảy ra giữa các sợi đơn RNA với nhau hoặc giữa một sợi DNA và RNA (Hình 2.31c).
Bằng phản ứng lai nucleic acid người ta có thể chứng minh được sự có mặt của một dòng tái tổ hợp xác định khi mẫu dò (probe) RNA hoặc DNA bổ sung cho gene tìm kiếm.
Mẫu dò DNA sợi kép
Biến tính bằng nhiệt
DNA sợi đơn
Đưa vào các nucleotide có 6 cặp base với trình tự ngẫu nhiên
Một số hexane tạo liên kết base
Bổ sung Klenow-polymerase và dNTPs, trong đó một loại được đánh dấu
DNA đánh dấu

109
Hình 2.32. Đánh dấu DNA bằng phương pháp dùng mồi ngẫu nhiên. Hỗn hợp hexane (oligonucleotide gồm 6 nucleotide với trình tự ngẫu nhiên) gồm nhiều
loại để ít nhất một số phân tử có thể gắn vào mẫu dò e. Ứng dụng lai acid nucleic
Một dòng tái tổ hợp nào đó được xác định với lai khuẩn lạc hoặc lai dòng khi có mẫu dò tương ứng. Mẫu dò phải có trình tự bổ sung ít nhất với một phần của gene tạo dòng. Nếu gene đó chưa biết (trong trường hợp thí nghiệm sẽ tìm ra 1 dòng), người ta phải tìm một mẫu dò khác.
Một mẫu dò như thế nào là phụ thuộc vào thực tế những gì mà người ta đã biết về gene cần tìm. Ba khả năng được giải thích ở đây:
1. Gene tìm kiếm biểu hiện ở phạm vi lớn trong một loại tế bào, từ loại tế bào này người ta đã tạo ra một thư viện cDNA.
2. Trình tự amino acid của protein, sản phẩm của gene cần tìm đã biết hoàn toàn hoặc từng phần.
3. Gene đó thuộc một gene họ hàng. - Thí nghiệm về tần suất xuất hiện khi phân tích thư viện cDNA
Để chứng minh dòng gliadin người ta sử dụng các cDNA từ thư viện làm mẫu dò, với chúng người ta thí nghiệm với tất cả những dòng khác có trong thư viện (Hình 2.34). Người ta chọn ra một dòng tuỳ ý, tinh sạch DNA tái tổ hợp, đánh dấu chúng và sử dụng làm mẫu dò cho tất cả các dòng khác. Phương pháp này được lặp lại với nhiều dòng khác, cho đến khi tìm được một dòng đó là mẫu dò lai với phần lớn các dòng cDNA. cDNA thường gặp này được cho là dòng gliadin, bằng những thí nghiệm tiếp theo (xác định trình tự DNA và phân lập sản phẩm giải mã) người ta có thể khẳng định kết quả này

110
Hình 2.33. Hai phương pháp đánh dấu DNA mẫu dò không dùng phóng xạ
a. Đánh dấu với một nucleotide được biotin hóa
Mẫu dò DNA Biotin-dUTP
Lai
Nick-translation được lấp đầy đầu cuối hoặc đưa vào primer ngẫu nhiên
Chứng minh avidin và fluorescens marker được gắn kết
b. Đánh dấu enzyme peroxidase của giống củ cải cay
Peroxidase của củ cải cay + Glutaraldehyde
Mẫu dò DNA sợi đơn
Sự phát quang hóa học
Cho vào luminol
Lai

111
Hình 2.34. Lai với một thư viện để xác định dòng phổ biến
- Mẫu dò oligonucleotide đối với những gene có sản phẩm giải mã xác định Sản phẩm của một gene tạo dòng là một protein thường được phân tích. Đặc
biệt là phương pháp xác định trình tự amino acid đã được biết từ hơn 30 năm nay. Khi biết trình tự amino acid của một protein, người ta có thể thiết kế trình tự nucleotide của gene mã hoá cho protein đó nhờ mã di truyền. Một trình tự nucleotide chỉ là một sự gần đúng, vì chỉ có methionine và tryptophan được xác định rõ ràng từ một codon xác định. Những amino acid còn lại được xác định ít nhất bởi hai cođon khác nhau. Trong nhiều trường hợp các codon khác nhau mã hóa cho một amino acid. Ví dụ alanine được mã hoá bởi GCA, GCT, GCG và GCC, nghĩa là người ta có thể đoán chắc chắn được 2 trong 3 nucleotide mã hoá cho alanine.
Ví dụ dự đoán cho protein là cytochrome c, có vai trò quan trọng trong chuỗi hô hấp của sinh vật hiếu khí. Protein của cytochrome c từ nấm men được xác định trình tự năm 1963. Từ acid amin thứ 59 có trình tự trp-asp-glu-asn-asn-met. Theo mã di truyền này thì trình tự nucleotide như sau: TGG-GAT-GAA-AAT-AAT-ATG. Có thể có 16 kiểu trình tự mã di truyền khác nhau, nhưng 14 trong 18 nucleotide là chắc chắn. Nhiều năm trước đây đường hướng này ít được các nhà khoa học chú ý. Tuy nhiên ngày nay các oligonucleotide ngắn với trình tự xác định
Thư viện dòng
Mẫu dò A Mẫu dò B Lai dòng
Phóng xạ tự ghi Mẫu dò A tương ứng với dòng hiếm
Mẫu dò B tương ứng với dòng phổ biến

112
được tổng hợp trong phòng thí nghiệm, vì vậy có thể thiết kế mẫu dò oligonucleotide theo trình tự nucleotide phỏng đoán. Với mẫu dò này người ta xác định được gene mã hoá cho protein đó. Trong trường hợp cytochrome c của nấm men người ta tổng hợp 16 loại oligonucleotide riêng rẽ hoặc hỗn hợp và dùng chúng làm mẫu dò cho thư viện hệ gene hoặc thư viện dòng của nấm men. Mẫu dò có sự đồng nhất về trình tự với một phần gene của cytochrome c đủ để tạo ra tín hiệu lai. Khi có nhiều dòng lai có tín hiệu dương tính, người ta có thể lặp lại với một mẫu dò tương ứng với một đoạn khác của gene cytochrome c. Đoạn protein được sử dụng để suy diễn trình tự nucleic acid phải được lựa chọn rất cẩn thận: Ví dụ trình tự amino acid: ser-glu-tyr-leu-thr-asn, kế tiếp trình tự trên có thể được mã hoá bởi hàng nghìn kiểu trình tự oligonucleotide và vì vậy không thích hợp cho việc sản xuất mẫu dò tổng hợp. 2. Kỹ thuật Northern blot
Phương pháp lai Northern blot là phương pháp lai RNA của tế bào với mẫu dò cDNA. Về nguyên lý và các bước tiến hành tương tự phương pháp lai Southern blot.Phương pháp lai Northern blot có ý nghĩa trong việc nghiên cứu sự hiện diện sản phẩm phiên mã trong qúa trình biểu hiện của các đoạn DNA tạo dòng. 3.Kỹ thuật Western blot 3.1. Điện di SDS-PAGE
Thư viên gene từ DNA của nấm men
Oligonucleotide, đánh dấu đầu cuối, tổng hợp
Lai dòng
Phóng xạ tự ghi
Oligonucleotide lai với dòng cytochrome c
Dòng cytochrome c phỏng đoán
Tính hiệu không đặc hiệu ?
Lai với một nucleotide từ đó được biến đổi
Chứng minh xác định dòng cytochrome c

113
Hình 2.35. Xác định một dòng gene cytochrome c của nấm men bằng một nucleotide tổng hợp
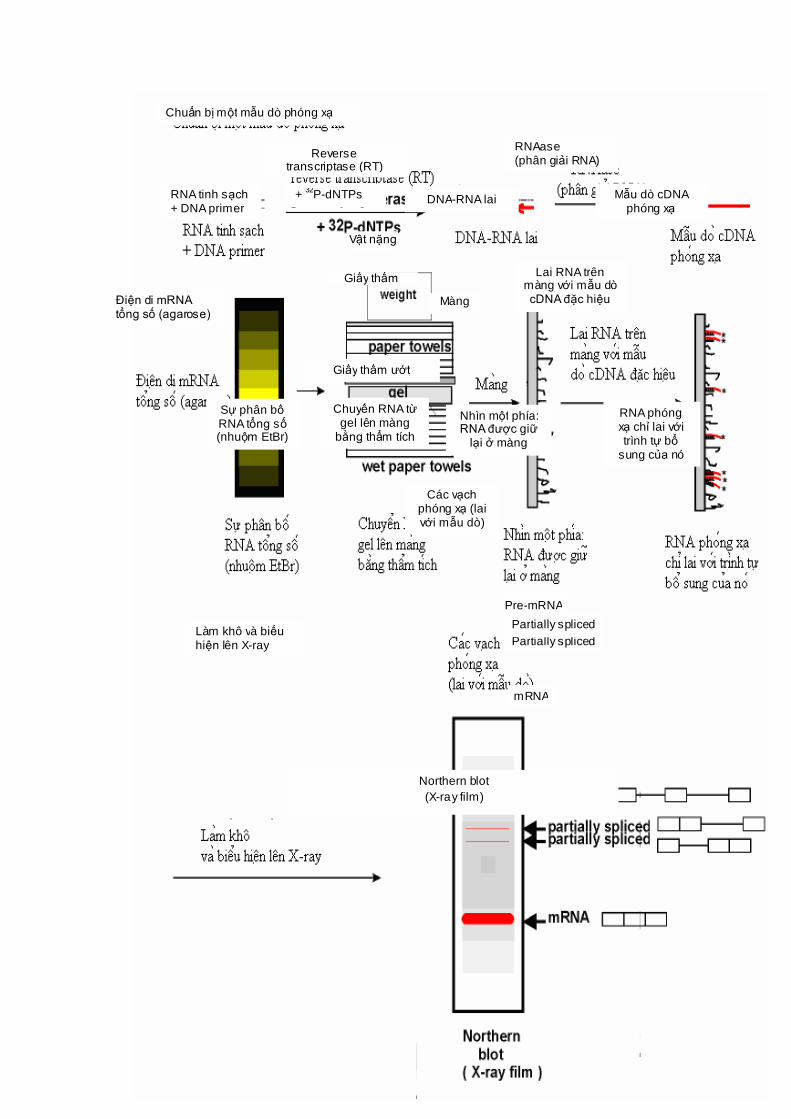
114
Partially spliced
Northern blot (X-ray film)
mRNA
Làm khô và biểu hiện lên X-ray
Các vạch phóng xạ (lai với mẫu dò)
Pre-mRNA Partially spliced
Sự phân bố RNA tổng số (nhuộm EtBr)
Chuyển RNA từ gel lên màng
bằng thẩm tích
Nhìn một phía: RNA được giữ
lại ở màng
RNA phóng xạ chỉ lai với trình tự bổ
sung của nó
Giấy thấm ướt
Giấy thấm
Vật nặng
Điện di mRNA tổng số (agarose)
Màng
Lai RNA trên màng với mẫu dò cDNA đặc hiệu
Mẫu dò cDNA phóng xạ
DNA-RNA lai
RNAase (phân giải RNA)
Chuẩn bị một mẫu dò phóng xạ
RNA tinh sạch + DNA primer
+ 32P-dNTPs
Reverse transcriptase (RT)

115
Hình 2.36. Kỹ thuật Northern blot Điện di trên gel polyacrylamide với sự có mặt của SDS cho phép phân ly các
phân tử protein có trọng lượng khác nhau. SDS có điện tích âm rất lớn và có khả năng liên kết với mạch peptide. Như vậy, số lượng SDS tương tác với protein tỷ lệ với kích thước phân tử protein và điện tích của SDS bám vào có thể làm bất cứ phân tử protein nào cũng chuyển động trong điện trường từ cực âm sang cực dương. Do đó, bằng phương pháp điện di, có thể phân tách riêng biệt các phân tử protein có trọng lượng phân tử khác nhau. Ngoài ra, có thể điện di các protein tùy theo điểm đẳng điện (isoelectric focusing) của chúng. Phương pháp này được gọi là điện di đẳng điện. Trong dung dịch đệm có pH biến thiên liên tục (gradient pH), các protein sẽ phân ly đến vị trí tương thích với điểm đẳng điện của chúng. Hai phương pháp điện di theo trọng lượng phân tử và điểm đẳng điện có thể kết hợp với nhau tạo nên kỹ thuật điện di 2 chiều (2-D electrophoresis). Điện di protein trên gel polyacrylamide cho phép phân đoạn, xác định trọng lượng phân tử và phân lập protein. Ngoài ra, trọng lượng protein còn được xác định chính xác bằng phương pháp sắc ký khối phổ. 3.2. Phản ứng lai kháng nguyên-kháng thể
Phản ứng kháng nguyên-kháng thể có tính đặc hiệu rất cao. Vì vậy, có thể áp dụng phản ứng này để phát hiện sự có mặt và tinh sạch protein. Kháng thể (antibody) được sản xuất khi đưa kháng nguyên vào thỏ và được tinh sạch từ máu thỏ sau khi gây nhiễm.
Những kháng thể tạo ra bằng cách này là những kháng thể đa dòng (polyclonal antibodies-do các tế bào lympho khác nhau tiết ra), do đó chúng có khả năng nhận biết một số kháng nguyên. Ngược lại, kháng thể đơn dòng (monoclonal antibodies) chỉ tương tác với một kháng nguyên nhất định.
Kháng thể được đánh dấu bằng enzyme (hoặc bằng chất phát huỳnh quang) để phát hiện protein đặc hiệu (thường được thẩm tích lên màng nitrocellulose sau khi chạy điện di SDS-PAGE, và cố định ở đó) thông qua kỹ thuật Western blot (hoặc immunoblot) (Hình 2.37). Cơ chế của phản ứng lai kháng nguyên-kháng thể được trình bày ở hình 2.38. Sau khi protein trên màng lai gắn với kháng thể thứ nhất đặc hiệu và tiếp đến là kháng thể thứ hai có đánh dấu enzyme (alkaline phosphatase, horse-radish peroxidase…) thì phức hợp này sẽ được liên kết với cơ chất để tạo màu. Sự hiện diện của protein ngoại lai (sản phẩm dịch mã của gene ngoại lai được chuyển vào tế bào vật chủ) sẽ được phát hiện nhờ sự xuất hiện màu của phản ứng lai.

116
Hình 2.37. Sơ đồ kỹ thuật Western blot
Sự phân bố của protein đặc hiệu trong tế bào và tổ chức mô cũng có thể phát hiện bằng kỹ thuật lai in situ (in situ hybridization) với nguyên tắc tương tự Western blot. Ngoài ra, kháng thể được sử dụng để tinh sạch protein đặc hiệu bằng kết tủa miễn dịch hoặc sắc ký ái lực (affinity chromatography). Kháng thể đánh dấu được dùng để định lượng kháng nguyên trong kỹ thuật xét nghiệm hấp thụ miễn dịch liên kết enzyme (enzyme-linked immunosorbent assay) gọi tắt là ELISA.
Hình 2.38. Phương pháp ELISA
Ủ và rửa màng lai
Western blot
Kết quả
Protein gel
Kháng thể Chất đánh dấu
Enzyme
Cơ chất
Sản phẩm
Chỗ gắn
Kháng nguyên
(antigene)
Kháng thể thứ 2
hoặc kháng nguyên
Mẫu
Gắn Rửa Đánh dấu
Đọc

117
- Các bước của phương pháp ELISA
Kháng nguyên (antigene) HIV được gắn trên bề mặt đĩa ELISA
Nếu huyết thanh của bệnh nhân chứa kháng thể (antibody)của HIV thì kháng thể này sẽ gắn với kháng nguyên HIV trên đĩa.
Kháng thể thứ hai đã được gắn với một enzyme được đưa vào sẽ gắn với kháng thể người
Gene tạo màu hoặc cơ chất sẽ thay đổi màu khi bị phân giải bởi enzyme gắn với kháng thể thứ hai
Phản ứng ELISA dương tính
Phản ứng ELISA âm tính
ELISA sử dụng một kháng thể đánh dấu với enzyme peroxidase (horseradish). Trong khi enzyme hoặc là kháng thể gắn cơ chất, cả hai vẫn duy trì hoạt tính sinh học. Sự thay đổi hoạt tính của enzyme là kết quả của phản ứng enzyme – kháng thể – kháng nguyên, tỷ lệ với nồng độ của kháng nguyên và được đo bằng quang phổ kế.
a. Phương pháp xác định gene tạo dòng bằng chứng minh sản phẩm gene Nhìn chung lai nucleic acid là một phương pháp ưu thế để xác định thể tái tổ
hợp trong thư viện dòng. Phương pháp này đơn giản, có thể kiểm tra 10 000 thể tái tổ hợp trong một thí nghiệm duy nhất. Tuy nhiên, người ta cần một mẫu dò mà ít nhất nó bổ sung từng phần với gene quan tâm.
Khác với lai nucleic acid người ta chứng minh đoạn DNA tạo dòng bằng phương pháp miễn dịch, nghĩa là xác định protein được mã hoá bởi gene tạo dòng. Phương pháp miễn dịch cần điều kiện là gene tạo dòng phải biểu hiện và dĩ nhiên là protein này không có trong tế bào chủ.
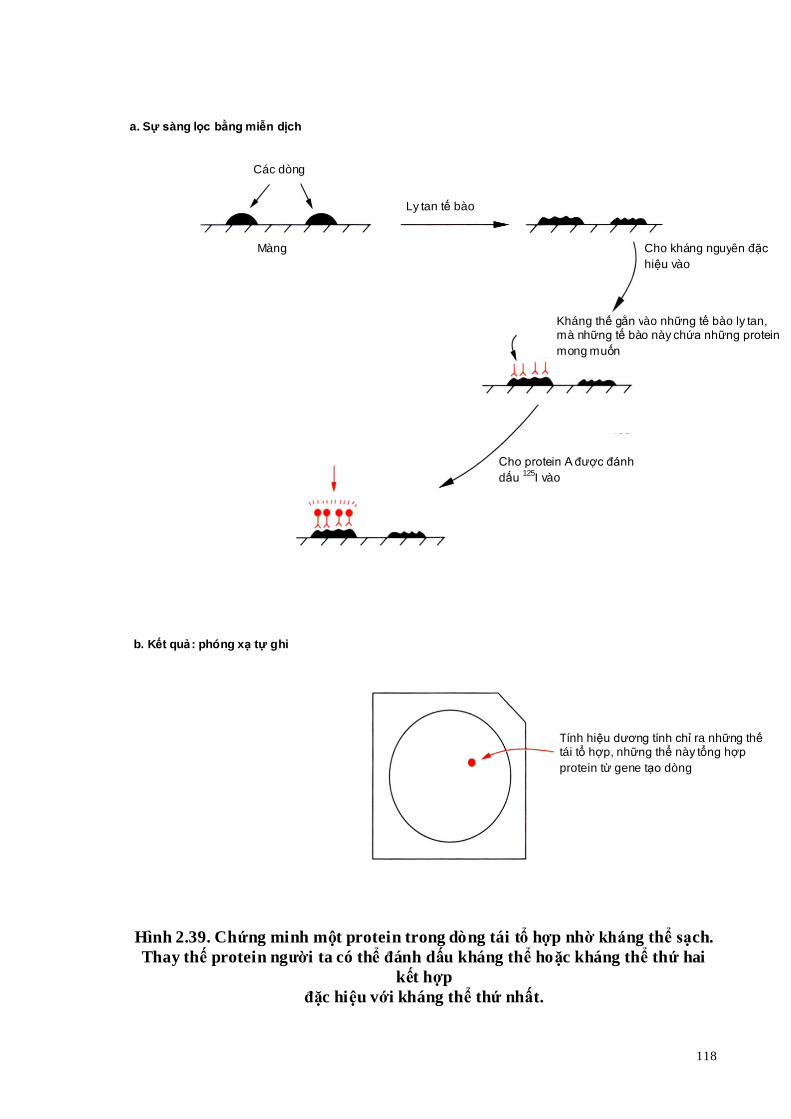
118
Hình 2.39. Chứng minh một protein trong dòng tái tổ hợp nhờ kháng thể sạch. Thay thế protein người ta có thể đánh dấu kháng thể hoặc kháng thể thứ hai
kết hợp đặc hiệu với kháng thể thứ nhất.
a. Sự sàng lọc bằng miễn dịch
b. Kết quả : phóng xạ tự ghi
Các dòng
Màng
Ly tan tế bào
Cho kháng nguyên đặc hiệu vào
Kháng thể gắn vào những tế bào ly tan, mà những tế bào này chứa những protein mong muốn
Cho protein A được đánh dấu 125I vào
Tính hiệu dương tính chỉ ra những thể tái tổ hợp, những thể này tổng hợp protein từ gene tạo dòng

119
b. Cần kháng thể cho phương pháp chứng minh miễn dịch Người ta tiêm một protein tinh sạch vào máu một con thỏ, sau đó hệ thống
miễn dịch tạo nên một kháng thể kết hợp với protein lạ và tham gia vào sự phân giải protein. Đây là một cơ chế tự vệ trong tự nhiên, nhờ vậy động vật chống lại được vi khuẩn, virus và những tác nhân khác xâm nhập.
Sau khi tiếp xúc với protein lạ sau một vài ngày nồng độ kháng thể ở trong máu động vật có nồng độ cao đến nỗi người ta có thể tinh sạch một lượng kháng thể. Người ta không cần giết thỏ vì trong 10 ml máu thu được một lượng lớn kháng thể. Kháng thể tinh sạch chỉ kết hợp với protein mà đã tiêm vào cho thỏ. c. Chứng minh một protein trong dòng tái tổ hợp với kháng thể tinh sạch Có nhiều phương pháp kiểm tra miễn dịch, trong đó có phương pháp lai dòng trực tiếp, ly tan tế bào và cho vào dung dịch chứa kháng thể đặc hiệu (Hình 2.39a). Ở đây hoặc là đánh dấu kháng thể hoặc rửa màng với dung dịch protein A, một protein vi khuẩn gắn đặc hiệu vào kháng thể (Hình 2.39b). Ở phương pháp đánh dấu phóng xạ có thể chứng minh được dòng gắn vào chất đánh dấu bằng phóng xạ tự ghi. Phương pháp không phóng xạ là tín hiệu phát quang và phát sáng hoá học III. Kỹ thuật xác định trình tự DNA
Vấn đề quan trọng nhất trong sinh học phân tử là xác định trình tự nucleotide trên DNA. Phương pháp xác định trình tự đã có từ hơn 30 năm nay, nhưng phương pháp nhanh và có hiệu quả có từ cuối những năm 70. Gần như cùng một lúc hai phương pháp khác nhau được phát triển: Phương pháp enzyme hay dideoxy được thực hiện bởi Sanger và Coulson ở Anh và phương pháp hoá học bởi Maxam và Gilbert ở Mỹ. Hai phương pháp này có nguyên tắc khác nhau, nhưng được sử dụng như nhau. Bằng 2 phương pháp này người ta có thể xác định trình tự nhiều kb với thời gian ít nhất. Ngày nay trình tự DNA là thông tin đầu tiên, cơ bản nhất mà người ta thu được từ gene tạo dòng. 1. Phương pháp Sanger-Coulson (phương pháp enzyme hay dideoxy)
Phương pháp này do Frederick Sanger đề xuất. Trong phương pháp này, DNA xác định trình tự phải chuyển thành dạng sợi
đơn và sợi đơn sẽ được sử dụng như một sợi khuôn để tổng hợp sợi mới. Điều đặc biệt là trong quá trình tổng hợp này tác giả sử dụng các nhân tố kết thúc chuỗi đặc hiệu – đó là các dideoxynucleotide triphosphate (ddNTP). Các phân tử này vẫn gắn bình thường vào chuỗi DNA đang tổng hợp nhưng lại không gắn được với nucleotide tiếp theo. Vì vậy khi đưa 4 loại dNTP (A, G, T, C) vào 4 phản ứng (mỗi hỗn hợp phản ứng một loại ddNTP), đồng thời vẫn bổ sung 4 loại dNTP bình thường thì sẽ thu được một loạt các đoạn DNA được kết thúc tại một loại nucleotide đặc thù.
Phương pháp enzyme cần DNA sợi đơn, vì vậy người ta tạo dòng đoạn DNA cần xác định trình tự trong vector M13. Ở phương pháp này thực chất là một sợi DNA thứ hai được tổng hợp bằng enzyme bổ sung cho sợi đã có sẵn. Các bước cụ thể như sau: a. Mồi (primer)

120
Bước đầu tiên của phương pháp enzyme là cho mồi (một đoạn nucleotide ngắn) lai với phân tử M13 tái tổ hợp (Hình 2.40a). Đoạn mồi này là điểm khởi đầu cho tổng hợp sợi bổ sung, phản ứng được thực hiện bởi đoạn Klenow hoặc T7-DNA-polymerase. Mồi gắn vào một đoạn của polylinker trong vector. b. Tổng hợp sợi bổ sung
Phản ứng xảy ra khi có enzyme và 4 deoxyribonucleotide triphosphate, ngoài ra còn cho vào hỗn hợp phản ứng một loại nucleotide đã được biến đổi, đó là dideoxynucleic (ví dụ dideoxy-ATP). Vì nucleotide này thiếu nhóm OH ở vị trí carbon 3 của đường deoxyribose (Hình 2.40b). Nhóm này cần cho việc gắn vào của nucleotide tiếp theo. Vì vậy chuỗi luôn luôn kết thúc ở vị trí mà dideoxynucleotide được gắn vào.
Khi trong hỗn hợp phản ứng có mặt của ddATP thì chuỗi sẽ kết thúc ở các vị trí đối diện với thymine ở trên sợi khuôn (Hình 2.40c). Tỷ lệ nồng độ giữa dATP và ddATP phải phù hợp để các sợi được tổng hợp dài hơn trước khi ddATP đi vào. Kết quả là một tập hợp các sợi có độ dài khác nhau nhưng tất cả các sợi đều kết thúc chuỗi bằng dideoxy-ATP.

121
Hình 2.40. Xác định trình tự bằng phương pháp enzyme c. Thực hiện bốn phản ứng riêng rẽ người ta thu được 4 họ các đoạn xếp
Người ta để 4 phản ứng tổng hợp sợi xảy ra song song với nhau. Bên cạnh phản ứng với ddATP còn có phản ứng với ddTTP, ddGTP và ddCTP. Bằng cách
Những đoạn nhỏ nhỏ
Dideoxy-ATP
primer
Tất cả những sợi mới đều kết thúc với dideoxy-ATP
a. Sự gắn (lai) của primer
b. Dideoxy-ATP
c. Tổng hợp sợi
d. Kết quả : Phóng xạ tự
Vị trí, ở đây nhóm OH của dNTP được thay thế bằng 1 H
DNA-polymerase dATP, dTTP, dGTP, dCTP, Dideoxy-ATP
Gene được đưa vào vector M13mp
primer

122
này người ta nhận được 4 họ polynucleotide mới tổng hợp, trong đó một họ có tất cả các sợi kết thúc với ddATP, họ thứ hai kết thúc với ddTTP...
Bước tiếp theo các sợi của từng họ được tách ra để xác định chiều dài bằng phương pháp điện di. Để tách các sợi có chiều dài chỉ khác nhau một nucleotide người ta sử dụng gel polyacrylamide rất mỏng (độ dày 0,5 mm). Gel có chứa urea để làm biến tính DNA và tách sợi mới tổng hợp ra khỏ i sợi khuôn. Ngoài ra phải thực hiện điện di ở điện thế cao và nhiệt độ 600C hoặc cao hơn để đảm bảo các sợi đơn không hồi tính.
Vì mỗi vạch trên gel chứa rất ít DNA nên phải sử dụng phóng xạ tự ghi để hiển thị kết quả (Hình 2.40d). Để đánh dấu những sợi mới tổng hợp người ta cho vào hỗn hợp phản ứng một deoxyribonucleotide đánh dấu phóng xạ (32P- hoặc 35 S-ATP).
Hình 2.41. Đọc trình tự của phóng xạ tự ghi bằng phương pháp enzyme. Các đoạn xuất hiện trong 4 giếng, mà ở mỗi giếng trong quá trình tổng hợp sợi có mặt một ddNTPs. Để đọc trình tự người ta chú ý đoạn đó nằm trong giếng nào, bắt
đầu với giếng di chuyển xa nhất rồi đọc dần lên phía trên. d. Đọc trình tự trên phóng xạ tự ghi
Để đọc trình tự trước hết xác định vạch nào di chuyển xa nhất là sợi DNA ngắn nhất, vì nó kết thúc chuỗi khi dideoxyribonucleotide gắn vào vị trí thứ nhất
Dideoxy-NTP
Hướng điện di

123
của sợi khuôn. Nhìn vào điện di đồ người ta khẳng định ở giếng nào trên gel có vạch xuất hiện, vi dụ ở giếng A, nucleotide thứ nhất của trình tự là A. (Hình 2.41).
Vạch di chuyển nhanh thứ hai tương ứng với phân tử DNA dài hơn phân tử DNA thứ nhất một nucleotide. Cũng như vậy người ta xác định ở đây là giếng T, nucleotide thứ hai là T và trình tự là AT.
Lặp lại như vậy cho đến hết chiều dài của phóng xạ tự ghi, cho đến khi các vạch nằm gần nhau đến nỗi mà người ta không phân biệt được. Nói chung với phóng xạ tự ghi có thể xác định được trình tự khoảng 400 nucleotide. 2. Phương pháp Maxam-Gilbert (Phương pháp hoá học)
Năm 1977 Allan Maxam và Walter Gibbert ở Đại học Haward đã đề xuất một phương pháp để xác định trình tự DNA.
Phương pháp này được tiến hành như sau: Đoạn DNA cần xác định trình tự được đánh dấu bởi 32P tại đầu 5’ của mỗi sợi. Sau đó, các đoạn DNA này được biến tính tạo thành các sợi đơn và được tinh sạch, rồi chia ra thành 4 nhóm để thực hiện phản ứng. Tiếp theo là thực hiện xử lý hóa học đối với các base trên sợi DNA để gây biến tính đối với 1 hoặc 2 base. Để biến đổi base G người ta sử dụng dimethylsulfate và dùng pH thấp (pH = 2) để biến đổi base tại A và G. Hydrazin được sử dụng để biến đổi base C và T và xử lý kết hợp hydrazin cùng với nồng độ muối cao để biến đổi base C.
Sau đó các base bị biến đổi như trên được cắt bởi piperidin, vì chất này có tác dụng loại bỏ các base tại các vị trí biến đổi. Kết quả thu được là một loạt các đoạn đánh dấu có chiều dài khác nhau trên gel polyacrylamide. Sự phân bố của các đoạn này trên điện di đồ tương ứng với kích thước của chúng và phân biệt được các đoạn có chiều dài hơn kém nhau 1 nucleotide. Từ đó có thể xác định được trình tự nucleitide trên sợi đơn.
Có nhiều biến đổi trong phương pháp Maxam-Gilbert, phân biệt ở chỗ là cách tạo ra DNA và những hoá chất được sử dụng để cắt. Phần lớn những hoá chất này rất độc đối với người, vì vậy phải hết sức chú ý.
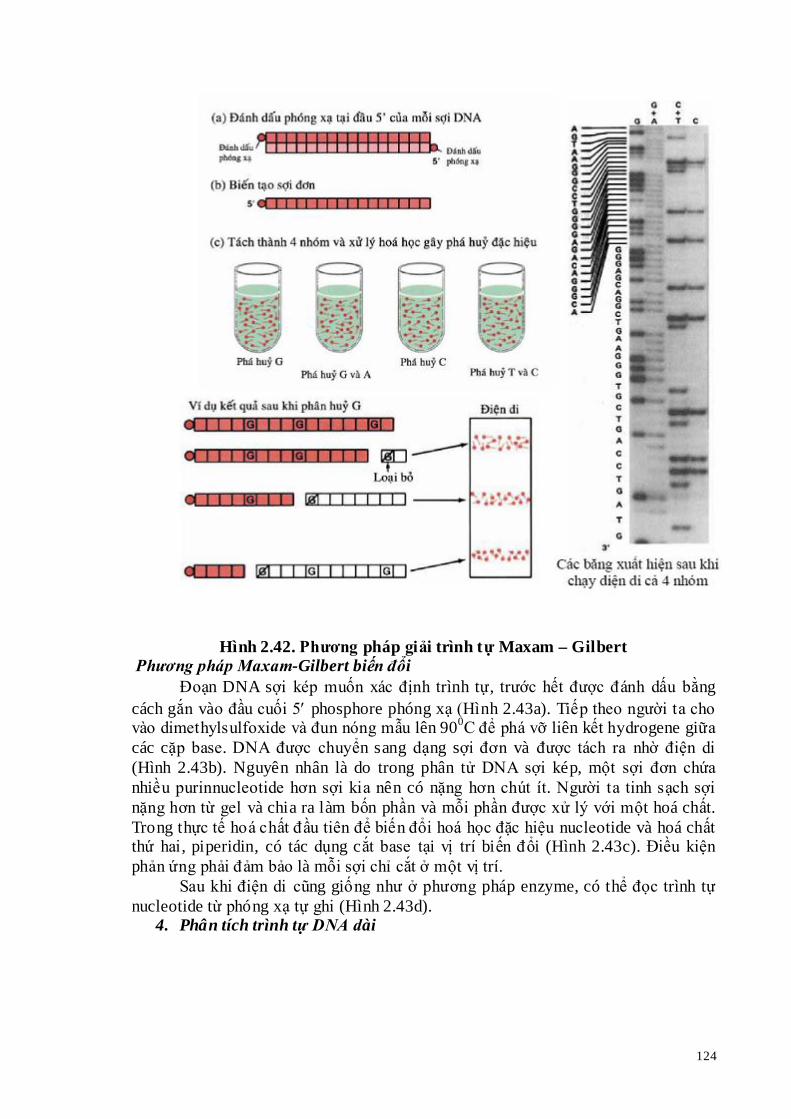
124
Hình 2.42. Phương pháp giải trình tự Maxam – Gilbert
Phương pháp Maxam-Gilbert biến đổi Đoạn DNA sợi kép muốn xác định trình tự, trước hết được đánh dấu bằng
cách gắn vào đầu cuối 5 phosphore phóng xạ (Hình 2.43a). Tiếp theo người ta cho vào dimethylsulfoxide và đun nóng mẫu lên 900C để phá vỡ liên kết hydrogene giữa các cặp base. DNA được chuyển sang dạng sợi đơn và được tách ra nhờ điện di (Hình 2.43b). Nguyên nhân là do trong phân tử DNA sợi kép, một sợi đơn chứa nhiều purinnucleotide hơn sợi kia nên có nặng hơn chút ít. Người ta tinh sạch sợi nặng hơn từ gel và chia ra làm bốn phần và mỗi phần được xử lý với một hoá chất. Trong thực tế hoá chất đầu tiên để biến đổi hoá học đặc hiệu nucleotide và hoá chất thứ hai, piperidin, có tác dụng cắt base tại vị trí biến đổi (Hình 2.43c). Điều kiện phản ứng phải đảm bảo là mỗi sợi chỉ cắt ở một vị trí.
Sau khi điện di cũng giống như ở phương pháp enzyme, có thể đọc trình tự nucleotide từ phóng xạ tự ghi (Hình 2.43d).
4. Phân tích trình tự DNA dài

125
Hình 2.43. Phân tích trình tự DNA bằng phương pháp hóa học
a. Đánh dấu và tách sợi
b. Tách các đoạn nặng và nhẹ
c. Tách các sợi
d. Kết quả : Phóng xạ tự ghi
Các phản ứng tách
Làm sạch một sợi đơn
Những sợi đơn được đánh dấu
nặng
nhẹ
Các đoạn cắt giới hạn
Polynucleotide kinase
32P-dATP Đầu 5’ đánh dấu
DMSO 90oC

126
Bằng phương pháp Sanger-Coulson hoặc Maxam-Gilbert, người ta chỉ đọc được trình tự khoảng 400 nucleotide. Nhưng phần lớn gene là dài hơn, vậy làm thế nào để phân tích trình tự có độ dài nhiều kilobase? Câu trả lời là phân tích trình tự DNA với các đoạn có độ dài khác nhau có nguồn gốc từ phân tử DNA lớn cần xác định trình tự (Hình 2.44). Những đoạn này nên chùm lên nhau, nên trình tự các đoạn DNA riêng rẽ sẽ có phần như nhau. Vùng chùm lên nhau có thể xác định bằng mắt thường hoặc bằng sự trợ giúp của máy tính mà dần dần toàn bộ trình tự được xác định.
Hình 2.44. Xác định trình tự DNA dài với nhiều đoạn ngắn có phần gối lên nhau
Gene
200 bp Phân tử DNA lớn
Tạo ra những đoạn có phần chồng lên nhau
Đưa vào vector M13
Phân tích trình tự những đoạn riêng lẻ
Trình tự những phần gối lên nhau
Trình tự phân tích trên trình tự dài được xác định

127
Để tạo ra những đoạn trùm lên nhau người ta cắt phân tử DNA bằng hai loại
enzyme hạn chế khác nhau, ví dụ một nhóm các đoạn xuất hiện do cắt bằng Sau3A và nhóm thứ hai cắt bằng AluI (Hình 2.45). Tuy nhiên, phương pháp này cũng có nhược điểm là vị trí cắt có thể không nằm ở chỗ thích hợp và một số đoạn có thể quá dài. Vì vậy, người ta phải sử dụng 4 hoặc 5 loại enzyme hạn chế để lấp tất cả những lỗ hổng ở trong toàn bộ trình tự.
Hình 2.45. Xác định trình tự đoạn DNA lớn bằng cách xác định trình tự các đoạn cắt hạn chế trùm lên nhau
4. Các khả năng xác định trình tự DNA
Lần đầu tiên, vào năm 1975 người ta đã xác định trình tự toàn bộ nhiễm sắc thể phage X174 (5 386 bp) và sau đó là trình tự của virus SV40 (5 243 bp) năm 1977, và của plasmid pBR322 (4 363 bp) năm 1978. Trình tự của các phân tử DNA lớn hơn dần dần cũng được công bố. Nhóm Sanger công bố trình tự gene ty thể người (16,6 kb) vào năm 1981 và của phage (49 kb) vào năm 1982. Nhiệm vụ quan trọng nhất hiện nay là dự án xác định trình tự toàn bộ hệ gene của một sinh vât. Trình tự hoàn chỉnh của nhiễm sắc thể nấm men đã được xác định và trình tự hệ gene người đã được công bố. Sắp xếp lại, đánh giá và hiểu được kết quả trình tự phong phú này chắc chắn là thách thức quan trọng nhất của 10 năm đầu thế kỷ 21 của các nhà sinh học phân tử. 5. Giải trình tự gene bằng phương pháp tự động
Nguyên tắc chung của máy giải trình tự gene tự động dựa trên cơ sở của phương pháp Sanger – Coulson. Ở đây quy trình tổng hợp được thực hiện nhờ máy PCR với các hóa chất tiêu chuẩn và sự hỗ trợ của phần mềm xử lý số liệu. Máy đọc trình tự trên cả 2 mạch đơn nên kết quả rất chính xác.
Phân tử DNA với những vị trí nhận biết Sau3A-(S) và Alul-(A)
Trình tự của những đoạn được cắt ra bằng Alul
Trình tự của những đoạn được cắt ra bằng Sau3A
Những đoạn quá lớn mà người ta không thể xác định được trình tự
200 bp

128
Việc giải trình tự DNA có thể cung cấp nhiều thông tin hữu ích về cấu trúc và tổ chức của các gene. Ngày nay máy tính là phương tiện lý tưởng để tiến hành nhanh và chính xác công việc này. Mặc dù vậy, vẫn cần sự hỗ trợ của các bằng chứng thực nghiệm về cấu trúc và chức năng của gene. Những thực nghiệm đó khẳng định chức năng mà phương pháp giải trình tự đã tiên đoán, nhưng đôi khi cũng nhận được những thông tin mới.
Phản ứng xác định trình tự Chu kỳ nhiệt của phản ứng xác định trình tự có 3 bước chính (giống PCR)
được lặp lại từ 30 đến 40 chu kỳ. - Biến tính ở 940C
Trong quá trình biến tính, sợi kép DNA chuyển thành sợi đơn, tất cả các phản ứng enzyme ở chu kỳ trước bị dừng. - Gắn mồi ở nhiệt độ 500C
Trong phản ứng xác định trình tự chỉ có một mồi được sử dụng nên chỉ có một sợi được làm khuôn (trong PCR: hai mồi được sử dụng, nên hai sợi đều làm khuôn). Primer chuyển động nhẹ (chuyển động Brown) nên liên kết hydrogene thường xuyên được tạo nên và mất đi giữa primer và sợi khuôn. Các liên kết ổn định và tồn tại lâu hơn là nhờ mồi gắn chính xác vào sợi khuôn. Trên đoạn DNA sợi kép ngắn này (khuôn và primer) enzyme polymerase gắn vào và bắt đầu phản ứng tổng hợp sợi bổ sung. Khi các liên kết phosphodiester giữa các base được tạo nên thì liên kết hydrogene giữa khuôn và primer không bị gãy nữa. - Kéo dài mồi ở nhiệt độ 600C
Đây là nhiệt độ thích hợp cho polymerase (bình thường là 720C, nhưng vì phải gắn ddNTPs đánh dấu huỳnh quang nên nhiệt độ được hạ thấp để có thời gian gắn với phân tử “lạ”). Các primer đã gắn với khuôn có lực ion mạnh hơn lực phá vỡ liên kết. Các primer không gắn được chính xác sẽ được tách ra và không kéo dài được. Các base bổ sung với khuôn được gắn với primer ở đầu 3’(gắn dNTPs hoặc ddNTPs từ 5’ đến 3’, đọc khuôn từ 3’ đến 5’). Khi ddNTP được gắn vào phản ứng kéo dài bị dừng lại.Vì ddNTPs được đánh dấu huỳnh quang nên phát hiện được màu của base cuối bằng máy xác định trình tự tự động. - Tách các phân tử
Sau phản ứng tổng hợp, hỗn hợp các sợi có độ dài khác nhau và đều kết thúc bằng một dNTP được đánh dấu huỳnh quang, phải được tách ra bằng điện di trên gel polyacrylamide.

129
Hình 2.46. Các bước khác nhau trong xác định trình tự
Vì chỉ có một mồi được sử dụng nên chỉ một sợi được copy, số lượng sợi này tăng lên theo đường thẳng. Vì vậy, sẽ có một số lượng lớn bản copy gene trong hỗn hợp để xác định trình tự. Có khoảng 1000 bản copy đoạn DNA khi bắt đầu, sau một chu kỳ sẽ có 2000 copy: trong đó 1000 bản khuôn ban đầu và 1000 bản bổ sung được đánh dấu huỳnh quang ở base cuối cùng, sau 2 chu kỳ sẽ có 2000 sợi bổ sung,
Có 30 chu kỳ gồm 3 bước
Bước 1: Biến tính 1 phút, 94oC
Bước 2: Gắn mồi 15 giây, 50oC
Bước 2: Kéo dài 4 phút, 60oC
Hỗn hợp dNTP’s
và ddNTP’s
1 primer !!!

130
sau 3 chu kỳ sẽ có 3000 sơi bổ sung và cứ tiếp tục như vậy.
Hình 2.47. Số lượng sợi DNA được khuếch đại theo đường thẳng trong giải trình tự
Hình 2.48. Tách các phân tử bằng điện di
6 phân tử khuôn Chu kỳ 1
6 sợi bổ sung Chu kỳ 2
12 sợi bổ sung
30 chu kỳ 180 sợi bổ sung
Hỗn hợp các sợi với độ dài khác nhau có đầu cuối là 1 ddNTP đánh dấu
Sản phẩm PCR Primer chỉ gắn với 1 sợi Khi gắn ddNTP đánh dấu huỳnh quang (bổ sung với base trên khuôn) sự kéo dài sợi dừng lại
Acrylamide gel
Phát hiện (detector)
t = 0 t = 2 giờ t = 1 giờ

131
- Phát hiện bằng máy xác định trình tự tự động Đoạn được đánh dấu huỳnh quang di chuyển dọc theo gel, đi qua chùm laser
ở cuối gel. Khi chùm laser hướng vào nucleotide đánh dấu huỳnh quang, chúng phát ra ánh sáng màu. Ánh sáng được thu lại vào các kính đi vào máy quang phổ. Mỗi một base có một màu riêng, nhờ vậy máy xác định trình tự có thể phát hiện trật tự base của gene. - Lắp ráp các trình tự của một gene
Để có được kết quả chính xác trình tự của gene phải được xác định cả hai hướng. Vì vậy phải thực hiện phản ứng tổng hợp với cả mồi ngược và xuôi.
Khi tất cả các đoạn được giải trình tự, một chương trình máy tính lắp ráp các phân lại với nhau để được trình tự gene hoàn chỉnh.
Hình 2.49. Lắp trình tự của một gene
IV. Kỹ thuật PCR 1. Nguyên tắc
Kỹ thuật PCR (phản ứng trùng hợp, phản ứng chuỗi polymerase) là kỹ thuật tổng hợp nhân tạo các đoạn DNA với tốc độ nhanh và chính xác được thực hiện trong máy chu trình nhiệt (máy PCR).
PCR là một kỹ thuật được sử dụng phổ biến trong công nghệ sinh học hiện đại và đã đóng góp rất lớn cho những tiến bộ về sinh học phân tử, đánh dấu một bước tiến vô cùng quan trọng tương đương với việc khám phá ra các enzyme hạn chế và kỹ thuật Southern blot (phân tích DNA).
PCR là khuếch đại một đoạn nào đó trong phân tử DNA. Người ta có thể chọn bất kỳ đoạn nào với điều kiện là trình tự ở hai đầu cuối đã biết. Điều này rất cần vì để bắt đầu PCR mỗi đoạn ngắn oligonucleotide phải lai với đầu một sợi đơn của phân tử DNA. Các đoạn oligonucleotide này là mồi cho phản ứng tổng hợp DNA và giới hạn đoạn cần khuếch đại.
Base 0 Base 600 Base 1200 Base 1750
mồi xuôi
mồi ngược

132
Hình 2.50. Tổng quát về phản ứng PCR
PCR dựa trên cơ sở phản ứng kéo dài mồi nhờ enzyme Taq polymerase để
khuếch đại in vitro các đoạn DNA đặc trưng. PCR cho phép khuếch đại theo hàm mũ lên đến hàng triệu lần các đoạn DNA có chiều dài từ 200-3.000 bp. Đoạn DNA được khuếch đại (DNA đích) được nhận dạng nhờ cặp mồi đặc trưng (oligonucleotide) thường có chiều dài khoảng 20 nucleotide.
Kỹ thuật PCR sử dụng các đặc điểm của quá trình sao chép DNA: Các liên kết hydroen ổn định cấu trúc xoắn và gắn 2 sợi với nhau phải được
phá vỡ để tách thành 2 sợi đơn. Sợi mới được tổng hợp theo hướng 5’ → 3’ nhờ hoạt động của DNA polymerase. Phải có đoạn mồi và có đủ 4 loại nucleotide triphosphate (dNTP).
Hình 2.51. Máy PCR 2. Kỹ thuật PCR 2.1. Thành phần phản ứng
DNA khuôn là đoạn DNA chứa vùng cần khuếch đại. Mồi là đoạn nucleotide ngắn có trình tự bổ sung với 2 đầu trình tự sợi khuôn. Mồi ở bên trái tác động lên sợi DNA theo hướng 3’→5’ được gọi là mồi thuận (forward primer-ký hiệu là F).
Đoạn DNA chứa 3 gene, chỉ cần khuếch đại gene B
genee A genee B genee C
Cần phải có 2 mồi để lai với mỗi đầu của gene B
Phản ứng PCR
Chỉ có gene B được khuếch đại, sau đó được tinh sách cho phân tích tiếp theo

133
Mồi ở bên phải tác động lên sợi DNA theo hướng 5’→3’ được gọi là mồi ngược (reverse primer-ký hiệu R).
Polymerase chịu nhiệt là loại DNA polymerase không bị biến tính ở nhiệt độ cao, thường dùng Taq polymerase chiết xuất từ vi khuẩn Thermus aquaticus sống ở suối nước nóng.
Các loại nucleotide triphosphate (dNTP) Dung dịch đệm thích hợp và MgCl2 Nước tinh khiết (không chứa DNAase, Rnase) Nguyên tắc của PCR được trình bày ở hình 2.52. Theo đó, từ chu kỳ thứ hai
Taq polymerase bắt đầu tạo ra các đoạn DNA có chiều dài xác định. PCR thường tiến hành khoảng 25-35 chu kỳ, qua đó từ 10
-6 µg DNA ban đầu có thể khuếch đại
(amplification) lên tới trên 1 µg (khoảng 2 kb).
Hình 2.52.Khuếch đại lũy thừa đoạn DNA bằng phản ứng PCR
Thí nghiệm PCR về nguyên tắc là rất đơn giản, nhưng để đạt kết quả tốt phải thực hiện rất cẩn thận. Trình tự của đoạn mồi cũng quan trọng như đảm bảo đúng nhiệt độ trong từng giai đoạn của chu kỳ phản ứng. Cuối cùng là những vấn đề quan trọng khi sử dụng sản phẩm khuếch đại PCR. 2.2. Các bước của phản ứng PCR
Chu kỳ thứ 35
236 = 68 tỷ bản sao
Gene cần khuếch đại
DNA khuôn

134
Hình 2.53. Mỗi chu kỳ PCR bao gồm ba giai đoạn có nhiệt độ khác nhau
Phản ứng PCR gồm nhiều chu kỳ lặp lại, mỗi chu kỳ gồm 3 bước: - Biến tính (denaturation) DNA là tách DNA sợi đôi thành 2 sợi đơn bằng
nhiệt. Các phân tử DNA bị biến tính ở nhiệt độ cao hơn nhiệt độ nóng chảy (Tm:
Phản ứng PCR được thực hiện 25 - 35 chu kỳ, mỗi chu kỳ gồm 3 bước
Bước 1: Biến tính 1 phút ở 94oC
Bước 2: Gắn mồi 45 giây ở 54oC
Mồi xuôi và ngược
Bước 3: Kéo dài phân tử
2 phút ở 72oC chỉ dNTP’s

135
temperature melting), nên bước này phải tiến hành ở nhiệt độ cao hơn nhiệt độ nóng chảy của DNA khuôn, thường là 94-95
oC từ 30 giây đến 1 phút.
- Bắt cặp (lai) (annealing). Mồi bắt cặp với DNA khuôn ở các vị trí có trình tự bổ sung. Nhiệt độ hạ thấp dưới Tm của các mồi để mồi bắt cặp với khuôn. Nhiệt độ thường sử dụng trong khoảng 60-70
oC, tùy thuộc vào độ lớn và Tm của các mồi,
thời gian từ 30 giây đến 1 phút. - Tổng hợp (kéo dài) (extension): Sợi mới được kéo dài từ mồi, bước này xảy
ra ở nhiệt độ 72oC, thời gian từ 30 giây cho đến vài chục phút, tùy thuộc vào kích
thước đoạn DNA cần khuếch đại.
Hình 2.54. Một chu kỳ nhiệt điển hình của PCR
Cứ như vậy, phản ứng PCR xảy ra trong 25 – 35 chu kỳ. Sau chu kỳ cuối
nhiệt độ được duy trì ở 72oC trong vòng 5 – 10 phút sao cho toàn bộ DNA có trong
phản ứng xoắn lại tạo nên sản phẩm PCR. Sự tổng hợp DNA trong phản ứng PCR tăng theo cấp số 2n.
Trong 3 bước trên thì gắn mồi là quan trọng, ảnh hưởng tới tính đặc hiệu của phản ứng. Nếu nhiệt độ cao quá sẽ không xảy ra phản ứng lai, mồi và khuôn đứng riêng lẽ (Hình 2.55a). Nếu nhiệt độ thấp quá, các thể lai không đặc hiệu bền vững, trong đó không phải tất cả các cặp base bắt cặp chính xác (Hình 2.55b). Khi các cặp base bắt cặp không tương ứng thì số lượng vị trí lai với mồi tăng lên rất nhiều và dẫn đến nhân những đoạn không mong muốn. Nhiệt độ lai lý tưởng một mặt phải thấp để xảy ra phản ứng lai giữa mồi và sợi khuôn, mặt khác phải cao để không tạo
Biến tính 1 phút)
Kéo dài chuỗi (2 phút)
Gắn mồi (1 phút)
Thời gian (phút)
Nhi
ệt đ
ộ (o C
)

136
nên thể lai không đặc hiệu (Hình 2.55c). Có thể xác định nhiệt độ gắn mồi (Tm) bằng thực nghiệm, nhưng phần lớn tính theo công thức đơn giản sau đây:
Tm = 4 x (G + C) + 2 x (A + T) 0C G + C là số lượng nucleotide G và C, A + T là số lượng nucleotide A và T
trong trình tự mồi. Để xác định nhiệt độ lai cho phản ứng PCR người ta phải tính Tm cho hai mồi và nhiệt độ này thấp hơn Tm 1 đến 20C. Nên thiết kế 2 mồi có cùng Tm.
a. Nhiệt độ lai quá cao
b. Nhiệt độ lai quá thấp
c. Nhiệt độ gắn mồi chính thức
Mồi và khuôn không gắn vào nhau
Mồi gắn không đúng vị trí Không phải tất cả các cặp base đã cặp đôi đúng
Mồi chỉ gắn đúng vào vị trí mong muốn

137
Hình 2.55. Nhiệt độ có ảnh hưởng lớn đến phản ứng lai giữa mồi và DNA khuôn
2.3. Thiết kế mồi oligonucleotide cho phản ứng PCR
Mồi quyết định sự thành công hay thất bại của phản ứng PCR. Mồi là các oligonucleotide tổng hợp (synthetic oligonucleotide) có khoảng 10-20 nucleotide. Nếu biết trình tự của đoạn gene cần khuếch đại thì có thể tổng hợp nhân tạo các mồi tương ứng để thực hiện PCR và tách chúng ra bằng kỹ thuật điện di. Đoạn DNA muốn khuếch đại không nên dài hơn 3 kb, trường hợp lý tưởng độ lớn của chúng nên nhỏ hơn 1kb. Bằng kỹ thuật PCR thông thường có thể khuếch đại các đoạn DNA có độ lớn đến 10 kb, nhưng khi độ dài càng lớn thì hiệu quả phản ứng càng thấp. Có thể khuếch đại đoạn DNA dài (cho đến 40 kb) nhưng phải sử dụng phương pháp đặc biệt hơn. Độ dài của mồi ảnh hưởng đến sự gắn vào DNA nhanh hay chậm. Hiệu quả của phản ứng PCR là số lượng bản sao được tạo trong một thí nghiệm sẽ giảm khi mồi có độ dài lớn, vì với thời gian cho phép trong chu kỳ phản ứng lai xảy ra không đầy đủ. Trong thực tế không có mồi nào dài hơn 30 nucleotide được sử dụng.
Hình 2.56. Kết quả phản ứng PCR với mồi thiết kế đúng và không đúng: Đường 1: Đoạn có độ lớn mong muốn được khuếch đại khi mồi được thiết kế
đúng Đường 2: Không có sản phẩm khuếch đại, ở đây có thể mồi không gắn được
với DNA khuôn. Đường 3: Đoạn DNA có độ lớn không mong muốn được khuếch đại Đường 4: Đoạn DNA mong muốn và hai đoạn không mong muốn được khuếch
đại Như vậy ở đường 3 và 4 mồi đã gắn với DNA khuôn không đúng vị trí mong
muốn
DNA size marker

138
Hình 2.57. Một cặp mồi khuếch đại gene α-globin người, các vạch đen biểu diễn các đoạn intron, các vạch trắng là các đoạn exon
3. Phân tích sản phẩm PCR
Có nhiều phương pháp được phát triển để phân tích sản phẩm PCR. Quan trọng nhất là ba phương pháp sau đây:
- Điện di trên gel agarose hoặc polyacrylamide - Phân tích trình tự trực tiếp sản phẩm PCR - Các phân tích liên quan tới nhân dòng sản phẩm PCR
3.1. Điện di sản phẩm PCR
Kiểm tra kết quả phản ứng PCR phần lớn bằng cách điện di một phần sản phẩm PCR. Sau khi nhuộm màu với ethidiumbromid, vạch DNA được hiển thị. Nếu thiếu vạch mong đợi và xuất hiện nhiều vạch khác thì phải thực hiện lại phản ứng.
Trong một số trường hợp, điện di không những khẳng định được kết quả phản ứng PCR, mà còn thu được các thông tin khác. Ví dụ có thể khẳng định trong đoạn DNA được khuếch đại có tồn tại các vị trí cắt của enzyme hạn chế hay không? Nhằm mục đích này sản phẩm PCR được xử lý bằng một enzyme hạn chế và sau đó thực hiện điện di. Dựa trên độ dài của sản phẩm điện di, có thể khẳng định trong vùng khuếch đại có chứa đoạn chèn hoặc loại bỏ đi một đoạn không? (Hình 2.59). Trong cả hai trường hợp đề cập đến một dạng biến đổi của RFLP ở phản ứng PCR.
Gene mã hóa cho 1-globin ở người

139
Hình 2.58. Độ dài của mồi quyết định kết quả phản ứng PCR
a. PCR với khuôn là DNA người và mồi gồm 8 nucleotide
Các vị trí lai
Nhiều sản phẩm PCR được tạo nên từ nhiều cặp mồi
b. PCR với khuôn là DNA người và primer gồm 17 nucleotide
Chỉ đoạn mong muốn được nhân lên
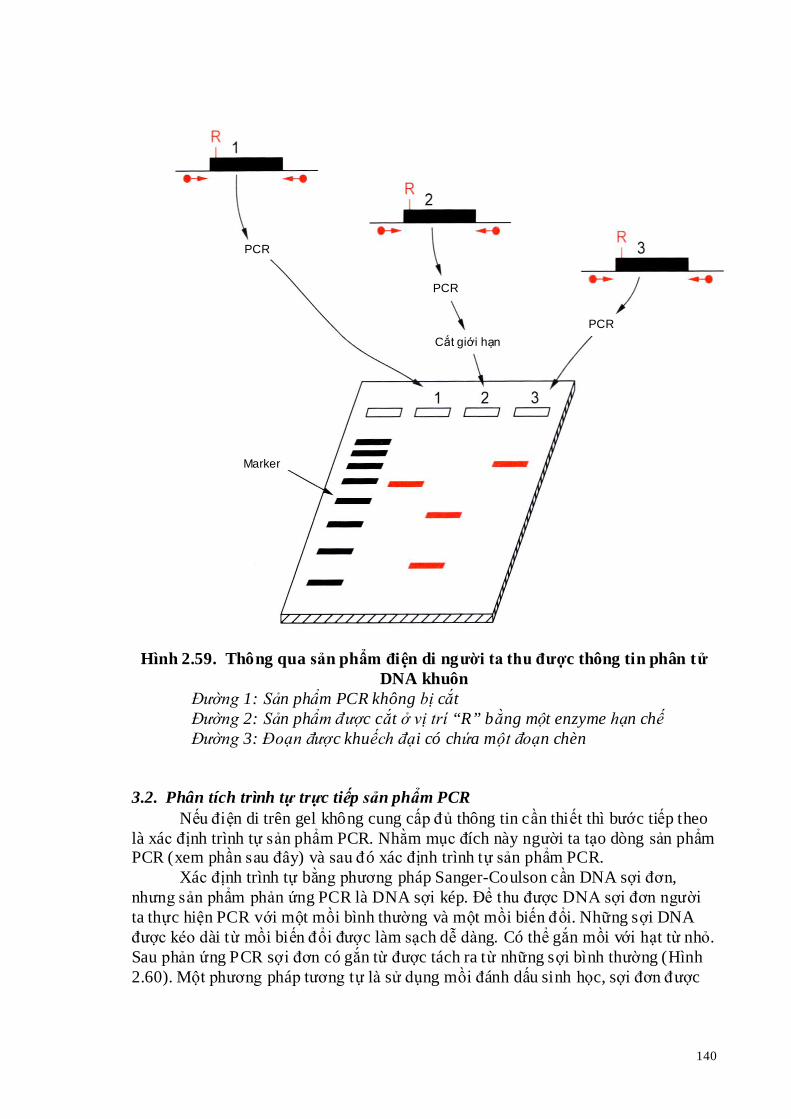
140
Hình 2.59. Thông qua sản phẩm điện di người ta thu được thông tin phân tử DNA khuôn
Đường 1: Sản phẩm PCR không bị cắt Đường 2: Sản phẩm được cắt ở vị trí “R” bằng một enzyme hạn chế Đường 3: Đoạn được khuếch đại có chứa một đoạn chèn
3.2. Phân tích trình tự trực tiếp sản phẩm PCR
Nếu điện di trên gel không cung cấp đủ thông tin cần thiết thì bước tiếp theo là xác định trình tự sản phẩm PCR. Nhằm mục đích này người ta tạo dòng sản phẩm PCR (xem phần sau đây) và sau đó xác định trình tự sản phẩm PCR.
Xác định trình tự bằng phương pháp Sanger-Coulson cần DNA sợi đơn, nhưng sản phẩm phản ứng PCR là DNA sợi kép. Để thu được DNA sợi đơn người ta thực hiện PCR với một mồi bình thường và một mồi biến đổi. Những sợi DNA được kéo dài từ mồi biến đổi được làm sạch dễ dàng. Có thể gắn mồi với hạt từ nhỏ. Sau phản ứng PCR sợi đơn có gắn từ được tách ra từ những sợi bình thường (Hình 2.60). Một phương pháp tương tự là sử dụng mồi đánh dấu sinh học, sợi đơn được
Cắt giới hạn
Marker
PCR
PCR
PCR

141
tách ra là do đã gắn với avidin, một protein có ái lực rất lớn với biotin. Sợi đơn được tạo dòng trong vector M13 ở cạnh polylinker.

142
PCR
Các hạt từ
Mồi cho sợi dưới
Mồi cho sợi trên
DNA khuôn
Biến tính
Tách ra các sợi đánh dấu từ

143
Hình 2.60. Phương pháp tinh sạch sợi đơn từ sản phẩm PCR sợi kép Một mồi được đánh dấu bằng cách gắn với hạt từ nhỏ. Sau khi kết thúc PCR
người ta biến tính sản phẩm và tách ra được các sợi đã đánh dấu 3.3. Tạo dòng sản phẩm PCR
Khi muốn có thêm thông tin về sản phẩm PCR thì người ta đưa sản phẩm này vào vector tạo dòng và nghiên cứu bằng phương pháp tiêu chuẩn để phân tích DNA tạo dòng. Tuy nhiên cũng gặp khó khăn vì sản phẩm PCR có đầu bằng, vì vậy có thể gắn adapter vào để tạo nên đầu dính. - Gắn các sản phẩm PCR
Trong một số trường hợp có thể thay thế phương pháp tạo dòng truyền thống bằng kỹ thuật PCR, vì PCR cho phép sản xuất ra một lượng lớn đoạn DNA mong muốn và sau đó có thể tạo dòng đoạn DNA này trong các vector plasmid hoặc phage thích hợp. Phương thức tạo dòng cho các sản phẩm PCR hoàn toàn giống tạo dòng các đoạn DNA thu được từ những thao tác DNA truyền thống, và có thể tạo dòng với đầu bằng hoặc đầu dính. DNA polymerase ổn nhiệt như Taq polymerase đã khuếch đại các sản phẩm PCR có gốc A lồi ra ở đầu 3’. Như vậy, có thể tạo dòng sản phẩm PCR vào trong các dT vector (được gọi là tạo dòng dA:dT). Điều này cho thấy việc bổ sung các gốc A vào đầu cuối có thể giúp gắn thành công sản phẩm PCR với vector đã được chuẩn bị với gốc T lồi ra (Hình 2.61). Phản ứng được xúc tác bởi DNA ligase, như trong một phản ứng gắn truyền thống.
Hình 2.61. Tạo dòng các sản phẩm PCR bằng phương thức tạo dòng dA:dT.

144
Người ta cũng có thể tạo dòng đầu dính với các sản phẩm PCR. Trong trường hợp này các oligonucleotide primer được thiết kế với một vị trí cắt hạn chế được kết hợp chặt chẽ trong chúng. Do sự bổ trợ của các primer cần thiết là tuyệt đối ở đầu 3’, nên thông thường đầu 5’ của primer là vùng định vị của vị trí cắt hạn chế. Điều này cần phải được thiết kế với sự chú ý thận trọng do hiệu suất cắt DNA bằng một enzyme hạn chế nhất định sẽ giảm nếu các nucleotide bổ sung thêm cho sự nhận biết của enzyme lại thiếu ở đầu 5’. Trong trường hợp này các phản ứng cắt và gắn giống như các phản ứng truyền thống.
4. Những khó khăn do sự khiếm khuyết của Taq-polymerase Các DNA-polymerase thỉnh thoảng gắn sai một nucleotide vào sợi DNA
đang tổng hợp. Tuy nhiên chúng cũng có khả năng sửa chữa các lỗi này, nghĩa là nó quay lại để cắt nucleotide đã gắn sai và gắn vào nucleotide đúng. Đặc tính có “chức năng sửa chữa” phụ thuộc vào hoạt tính của exonuclease theo hướng 3’ đến 5’ của polymerase. Tuy nhiên Taq-polymerase thiếu chức năng sửa chữa này. Theo ước đoán cứ khoảng 10.000 nucleotide thì có một lỗi, như vậy sau 30 chu kỳ số lỗi có thể lên đến 300bp. 5. Ứng dụng PCR
PCR là một phương pháp rất hữu dụng, mặc dù nó chỉ khuếch đại một đoạn DNA và ở đây biểu hiện nhược điểm là phải biết được trình tự hai đầu của đoạn DNA. PCR có ý nghĩa rất lớn trong sinh học phân tử. Dưới đây sẽ trình bày một số ứng dụng của kỹ thuật này. a. PCR có thể phân tích một lượng DNA rất nhỏ

145
Hình 2.62. So sánh phương pháp nguyên thủy (a) và phương pháp dựa trên PCR để xác định trình tự DNA người
Với kỹ thuật PCR, một phân tử DNA duy nhất có thể làm khuôn cho phản ứng khuếch đại. Điều này thể hiện rõ khi khuếch đại DNA từ một tế bào tinh trùng, nó chỉ chứa một bản đơn bội duy nhất của hệ gene người. Điều này có ý nghĩa rất lớn vì với lượng DNA như vậy các phương pháp phân tích truyền thống không thể thực hiện được. Đây cũng là điểm quan trọng trong pháp y, người ta có thể xác định thủ phạm nhờ sợi tóc và vệt máu còn sót lại trên hiện trường. PCR được sử dụng cả với xương và trong một số trường hợp những phần còn lại của người chết, mà bằng phương pháp truyền thống không thể thực hiện được.
Một khả năng mới mở ra đối với khảo cổ học là nhờ độ nhạy cực kỳ của PCR. Người ta có thể xác định được trình tự nucleotide của các vệt DNA trong nguyên liệu hóa thạch. Với phân tích DNA mối quan hệ họ hàng của con người trong quá khứ được nghiên cứu, trong đó DNA được khuếch đại từ xương, xác ướp hoặc xác ở đầm lầy và sau đó được xác định trình tự. Về lịch sử trái đất người ta xác định trình tự của DNA được khuếch đại từ các mẫu thực vật và động vật hóa thạch. b. PCR tham gia vào chẩn đoán lâm sàng
Trong phạm vi lớn phân tích RFLP nguyên thủy có thể tìm được các đột biến
dẫn đến các bệnh di truyền. Tuy nhiên nó chỉ được sử dụng trong trường hợp đột biến là do thay đổi độ dài của một đoạn cắt hạn chế. Trong nhiều trường hợp độ dài các đoạn không thay đổi do đột biến thì chỉ chứng minh được bằng việc xác định trình tự đoạn gene đó. Theo phương pháp nguyên thủy, người ta phải thiết lập một thư viện hệ gene cho từng người, rồi từ đó tách ra dòng chứa gene đột biến và xác định trình tự (Hình 2.62a). Cách này có thể thực hiện được nhưng rất tốn thời gian.
Xác định trình tự gene
Tạo dòng để tạo ra đoạn ngắn cho việc xác định trình tự
Tinh sạch DNA
Lai để xác định dòng mong muốn
Xác định trình tự gene
Tạo một thư viện hệ gene
Mẫu DNA
PCR

146
PCR đưa ra một phương pháp thay thế là thực hiện khuếch đại vùng nghi vấn từ hệ gene và phân tích trình tự trực tiếp, từ đó người ta sẽ thu được thông tin về trình tự đó (Hình 2.62b).
Sự nhận biết nhanh những đột biến không những quan trọng đối với chẩn đoán bệnh mà còn có ý nghĩa đối với các bệnh di truyền.
Độ nhạy của phản ứng PCR là cơ hội để chẩn đoán các bệnh khác, ví dụ bằng việc khuếch đại DNA virus sự chẩn đoán bệnh được sớm trước hàng tuần, thậm chí hàng tháng trước khi triệu chứng bệnh xuất hiện. Điều này có ý nghĩa rất lớn trong điều trị bệnh, đặc biệt dạng ung thư, có nhiều triển vọng hơn khi bệnh mới bắt đầu.
Hình 2.63. Phản ứng RT-PCR c. PCR khuếch đại RNA
Khả năng của PCR không giới hạn ở sự khuếch đại khuôn DNA, mà còn khuếch đại được các phân tử RNA với enzyme phiên mã ngược reverse transcriptase thành thể lai DNA-RNA, sau đó nhờ enzyme Taq-polymerase mà cDNA được tổng hợp (Hình 2.63).
RNA
Reverse transcriptase
Thể lai DNA/RNA
Phân giải sợi RNA
PCR như bình thường
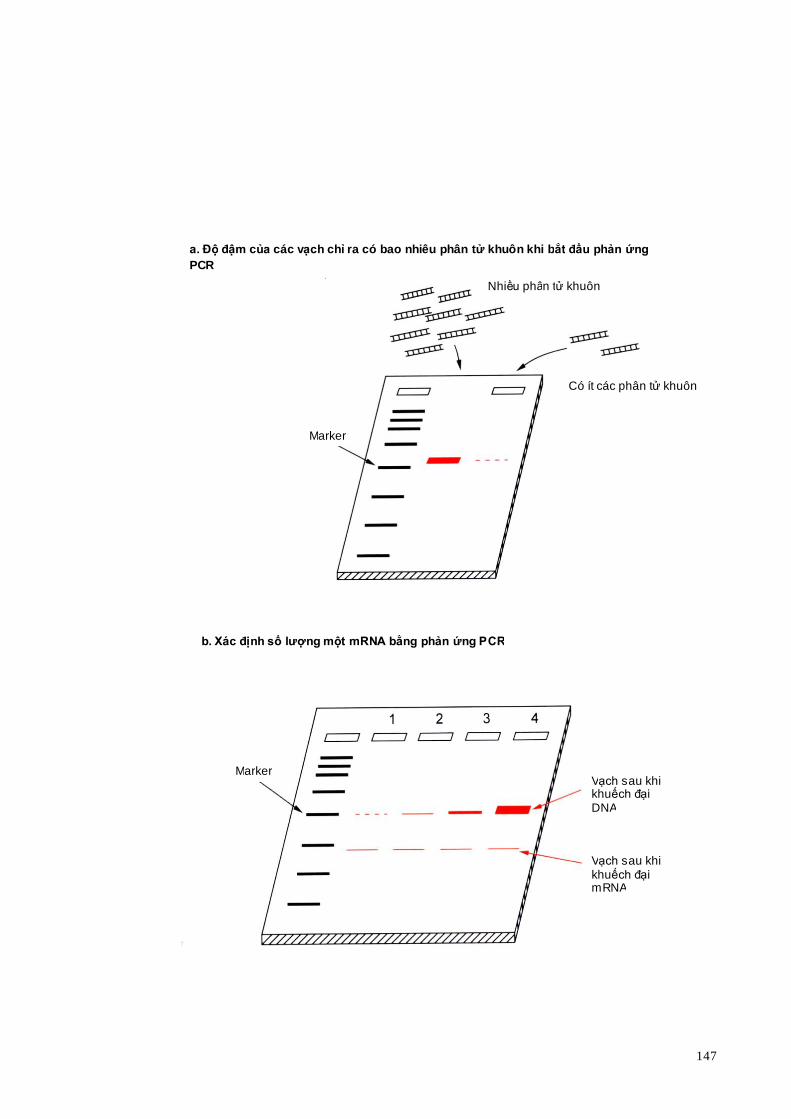
147
Vạch sau khi khuếch đại mRNA
Vạch sau khi khuếch đại DNA
Marker
b. Xác định số lượng một mRNA bằng phản ứng PCR
a. Độ đậm của các vạch chỉ ra có bao nhiêu phân tử khuôn khi bắt đầu phản ứng PCR
Marker
Có ít các phân tử khuôn
Nhiều phân tử khuôn

148
Hình 2.64. Xác định số lượng sản phẩm phản ứng RT-PCR. Ở b các phản ứng PCR được thực hiện với cùng một lượng RNA và lượng DNA
ngày càng tăng. DNA chứa một gene, được phiên mã thành mRNA và đoạn được khuếch đại chứa một intron trong gene. Sản phẩm PCR là DNA tạo nên dài hơn mRNA của nó. Các vạch DNA và RNA trên giếng 2 chỉ một nồng độ tương tự.
Nghĩa là PCR chứa dạng DNA và RNA của trình tự nghiên cứu gần như cùng số lượng bản copy
Một lĩnh vực sử dụng hữu ích được gọi là RT-PCR là xác định tỷ lệ khối lượng RNA trong các mô khác nhau hoặc trong một mô ở các thời điểm khác nhau. Có thể nói rằng lượng mRNA trong một tế bào phản ánh hoạt động của các gene đó, người ta có thể chứng minh được những thay đổi hoạt động gene bằng cách xác định số lượng mRNA. Có thể thực hiện định lượng này bằng Northern blot, nhưng chỉ với loại mRNA tồn tại với lượng tương đối lớn. Những loại RNA hiếm không chứng minh được bằng phương pháp lai. PCR cho phép mở rộng khả năng nghiên cứu biểu hiện của các gene ít hoạt động mà chúng thường đặc biệt thú vị .
Phương pháp PCR để định lượng đi từ nhận thức là lượng sản phẩm PCR tỷ lệ với lượng RNA (hoặc DNA) khuôn khi bắt đầu phản ứng. Sản phẩm của phản ứng được thực hiện điện di trên gel agarose và có thể ước lượng nồng độ của mRNA có mặt khi so sánh với độ đậm của các vạch chuẩn (Hình 2.64b). Có thể ước lượng chính xác khi phản ứng đối chứng chạy trong cùng một ống nghiệm với cùng một mồi như ở RT-PCR. Điều này được chỉ ra khi RNA phiên mã từ gene có một hoặc nhiều intron, như vậy đoạn DNA khuôn được khuếch đại có kích thước dài hơn sản phẩm bắt nguồn từ RNA và sẽ di chuyển trên gel ở các vị trí khác nhau. d. PCR so sánh các hệ gene khác nhau
Ở phương pháp đa hình DNA khuếch đại ngẫu nhiên (random amplified polymorphic DNA analysis, RADP) người ta thu được một hỗn hợp các đoạn có kích thước khác nhau được khuếch đại khi sử dụng các mồi ngẫu nhiên. Đây là phương pháp hữu dụng để nghiên cứu hệ tộc phát sinh học, lĩnh vực khoa học liên quan đến lịch sử tiến hóa và nguồn gốc của các loài. Mẫu vạch thu được của các mồi ngẫu nhiên khi điện di phản ánh cấu trúc tổng thể của DNA khuôn. PCR với các mồi ngẫu nhiên có thể chứng minh được sự khác nhau trong hệ gene của hai loài sinh vật. Ở hai cá thể có họ hàng gần nhau mô hình các vạch giống nhau nhiều hơn so với các cá thể có họ hàng xa nhau. V. Các kỹ thuật xác định tính đa hình DNA dựa trên PCR 1. Kỹ thuật RAPD: Đa hình các đoạn DNA khuếch đại ngẫu nhiên.
Phương pháp RADP được William, Welsh, McClelland đề xuất năm 1990. Phương pháp RAPD thực chất là quá trình nhân bản các đọan DNA bằng kỹ thuật PCR sử dụng các mồi được thiết kế ngẫu nhiên. Các mồi này sẽ bắt cặp một cách ngẫu nhiên vào DNA khuôn ở vị trí bất kỳ nào mà tại đó có trình tự bổ sung với nó.

149
Hình 2.65. Phản ứng RADP Mũi tên chỉ nhiều bản copy của 1 mồi (tất cả mồi có cùng trình tự). Hướng của mũi tên chỉ hướng DNA sẽ kéo dài Số vị trí có mặt trên khuôn DNA mà mồi gắn vào. Các mồi gắn vào vị trí 1, 2 và 3 ở sợi DNA dưới và gắn vào vị trí 4, 5 và 6 ở sợi DNA trên.
Tuy nhiên, trong phân tích RAPD, đoạn được khuếch đại chưa biết trình tự. Nhà khoa học thiết kế mồi với trình tự ngẫu nhiên gồm 10 nucleotide (có thể do máy tính tạo ra ngẫu nhiên), sau đó tổng hợp mồi, thực hiện phản ứng PCR và điện di trên gel agarose để xác định các đoạn DNA được khuếch đại khi có mặt của mồi ngẫu nhiên.
Để đoạn DNA được khuếch đại ngẫu nhiên các mồi phải gắn vào khuôn theo hướng ngược nhau và trong một khoảng cách phù hợp.
Hình 2.65 giới thiệu tổng quát phản ứng RADP. Đoạn DNA được sử dụng để làm khuôn trong phản ứng PCR có nhiều vị trí gắn cho một mồi ngẫu nhiên. Trong đó sản phẩm A được tạo nên là do khuếch đại trình tự nằm giữa vị trí 2 và 5, sản phẩm B là do khuếch đại trình tự nằm giữa vị trí 3 và 6. Trình tự giữa vị trí 1 và 4 không được khuếch đại vì đoạn này có độ dài quá lớn.
Ngoài ra, sản phẩm cũng không được tạo nên giữa các mồi ở vị trí 4 và 2, 5 và 3 vì các mồi này không định hướng đối diện nhau.
Phương pháp RADP được sử dụng nhiều để tìm kiếm sự khác nhau giữa các hệ gene. Giả sử có sự thay đổi trình tự ở một vị trí nào đó trên DNA nên mồi không có khả năng gắn vào thì đoạn đó sẽ không được khuếch đại và trên điện di đồ sẽ không có mặt của sản phẩm đó. Thậm chí sự thay thế một base trong trình tự nhận biết sẽ ngăn cản sự gắn mồi và khuếch đại đoạn DNA đó. Kết quả là sẽ có một sự đa dạng mà trong đó các vạch xuất hiện hoặc không.
Có khoảng 1 – 20 vị trí gắn ở trong hệ gene của một thực vật điển hình tùy thuộc vào sự lựa chọn của mồi. Khuếch đại sẽ cho 3-4 vạch phân biệt được bằng điện di trên gel agarose. Ví dụ một mồi ngẫu nhiên gồm 10 nucleotide thì xác suất bắt gặp 1 trình tự bổ sung với nó trên mạch DNA (được cấu tạo từ 4 loại nucleotide) là 1/104 = 1/ 1
DNA khuôn
Sản phẩm A
Phản ứng PCR
Sản phẩm B

150
048 576. Một bộ gene đơn bội của lúa có kích thước 550 000 Kb, vậy khả năng mồi ngẫu nhiên (10 nucleotide) có thể có 550 000 000/ 1 048 576 = 524 vị trí gắn. Do mồi gắn với DNA ở hai điểm khác nhau (đối diện nhau) trên sợi DNA nên sẽ có 262 đoạn DNA khác nhau được nhân.
Phương pháp này có ưu điểm là phát hiện tính đa dạng đáng tin (mất đoạn nhiễm sắc thể, thêm, bớt nucleotide, xen đoạn…) đều làm thay đổi kích thước đoạn nhân bản. Bộ mồi ngẫu nhiên nên có thể sử dụng cho các loài khác nhau. Phương pháp này tiến hành đơn giản hơn so với RFLP và thời gian ngắn hơn (2-4 giờ), ít tốn kém, phù hợp cho phân tích đa dạng di truyền và lập bản đồ gene sử dụng quần thể RIL (recombinant inbred line) và phân tích dòng gần đồng gene (near isogeneic line): các dòng chỉ khác nhau ở một tính trạng.
Tuy nhiên, phương pháp này có nhược điểm là rất nhạy cảm với yếu tố tham gia phản ứng, đặc biệt là nhiệt độ gắn mồi, vì vậy cần tuân thủ nghiêm ngặt quy trình, điều kiện thí nghiệm, lặp lại nhiều lần. Mặt khác, khoảng cách giữa 2 điểm gắn mồi cần thích hợp và không chắc chắn các đoạn có cùng kích thước từ 2 mẫu DNA khác nhau thực sự được tạo ra từ cùng một vị trí trên hệ gene. 2. Kỹ thuật AFLP (amplified fragment length polymorphism) – Sự đa hình chiều dài các phân đoạn DNA được khuếch đại
Phương pháp AFLP được Zabeau và Vos cùng cộng sự đề xuất vào những năm 1993-1995. Bản chất của phương pháp này là sử dụng kỹ thuật PCR để khuếch đại các đoạn DNA đã được cắt bởi enzyme hạn chế. Về nguyên tắc giống phương pháp RFLP, nhưng điểm khác biệt cơ bản là không tiến hành lai phân tử như RFLP, nên thực hiện nhanh hơn.
Khâu then chốt của kỹ thuật này là thiết kế các mồi đặc trưng. Để thiết kế được các mồi đặc trưng trước hết DNA của mẫu nghiên cứu được cắt bằng các enzyme hạn chế. Thường dùng đồng thời 2 enzyme hạn chế là EcoRI có trình tự nhận biết là 6 nucleotide (-GAATTC-/- CTTAAG-) và MseI có trình tự nhận biết là 4 nucleotide (-AATT-/-TTAA-). Kết quả sau khi xử lý với enzyme, DNA bị cắt thành nhiều đoạn có kích thước khác nhau nhưng trình tự nucleotide ở hai đầu đoạn cắt là giống nhau và xác định. Dựa vào trình tự đã biết ở hai đầu cắt để thiết kế các đoạn gắn (adapter) và gắn chúng vào các đầu đã cắt. Dựa vào trình tự adapter để thiết kế mồi PCR gồm 2 phần: phần trình tự bổ sung với adapter và phần bổ sung các nucleotide tùy ý, thường từ 1đến 3 nucleotide. Với mồi thiết kế như trên chỉ có các đọan DNA có trình tự 2 đầu gắn với mồi được khuếch đại. Đây là phương pháp kết hợp giữa RFLP và RAPD nên có nhiều ưu điểm: đơn giản, ổn định và dễ thiết kế mồi ở lượng lớn và khả năng ứng dụng rộng rãi.

151
Hình 2.66. Kỹ thuật AFLP

152
3. Kỹ thuật SSR (simple sequence reppeats): Đa hình đoạn trình tự lặp lại đơn giản
SSR là kỹ thuật khuếch đại các đoạn lặp lai đơn giản (Simple Sequence Repeats), còn gọi là phương pháp vệ tinh hay tiểu vệ tinh (micro satellite).
Hệ gene sinh vật eukaryote có nhiều đoạn lặp lại, có kích thước khác nhau đặc trưng cho từng loài, từng giống. SSR gồm 2-5 nucleotide lặp lại nhiều lần: (TG)n hoặc (AAT)n. Ở lúa đã phát hiện các nhóm GA, GT, CAT, CTT, các đoạn này được gọi là SSR, STRs (short tandom repeats), SSLPs (single sequence length polymorphism) và gọi chung là microsatelite (vi vệ tinh). Các đoạn DNA nhắc lại này có trình tự hai đầu rất đặc trưng nên được sử dụng để thiết kế mồi cho PCR. Do có sự sai khác về kích thước giữa các đoạn lặp lại nên kỹ thuật SSR rất thích hợp cho nghiên cứu đa hình, lập bản đồ và phân lập gene.
Kỹ thuật SSR sử dụng các mồi đơn giản để khuếch đại các đoạn DNA lặp lại. Dựa vào sự khác nhau về độ dài và kích thước thu được các đoạn lặp lại của các giống khác nhau để xác định mức độ di truyền của các mẫu so sánh.
Kỹ thuật SSR đơn giản, không tốn kém, là chỉ thị đồng trội được sử dụng để phát hiện cá thể dị hợp tử.
Hình 2.67. Kỹ thuật SSR
VI. Thư viện hệ gene (geneomic library) và thư viện cDNA 1. Thư viện hệ gene ( geneomic library)
Thư viện hệ gene là một tập hợp tất cả các đoạn DNA được tạo ra từ phản ứng cắt hạn chế geneome trong bacteriophage vector, đại diện được cho toàn bộ thông tin di truyền của một hệ gene.

153
Các thư viện được dùng chủ yếu để sàng lọc theo các phương pháp khác nhau nhằm phân lập một, hoặc một số trình tự nucleotide quan tâm, hoặc để xác định vị trí và thứ tự của các trình tự trong geneome.
Hệ gene của sinh vật nào đó được cắt bằng một số enzyme cắt hạn chế tạo ra các đoạn DNA khác nhau, được gắn vào vector tạo dòng và nhân dòng trong vi khuẩn. Vậy thư viện hệ gene là tập hợp tất cả các dòng tái tổ hợp đại diện cho toàn bộ hệ gene của một sinh vật.
Một thư viện hệ gene là một tập hợp các dòng, số lượng rất lớn, trong đó có thể là mỗi gene của một sinh vật đều có mặt. Để tạo được thư viện hệ gene người ta phải tinh sạch toàn bộ DNA của tế bào, cắt từng phần bằng một enzyme hạn chế và tạo dòng các đoạn đó trong vector phù hợp, thường là phage biến đổi, cosmid hoặc có thể là YAC.
Như vậy để xây dựng một thư viện hệ gene gồm các bước như sau: - Tách chiết DNA tổng số của tế bào - Tạo đoạn chèn (DNA insert) bằng các enzyme cắt hạn chế
thích hợp (thường dùng BamHI và Sau3AI), cắt hệ gene thành từng đoạn có kích thước xác định và xử lý các đầu cắt để chúng không tự nối lại với nhau.
- Tạo vector tái tổ hợp - Biến nạp vector tái tổ hợp vào tế bào nhận - Chọn lọc các thể tái tổ hợp.
Thư viện hệ gene có nhiều ứng dụng, chẳng hạn để lập bản đồ vật lý (physical mapping) và xác định các gene gây bệnh hoặc các chuỗi DNA quan tâm cho những phân tích khác.
Tạo ra các dòng mang các đoạn DNA khác nhau nhưng gối lên nhau có nhiều thuận lợi và thư viện có thể được sử dụng trong kỹ thuật chromosome walking. Chromosome walking thường được thực hiện với thư viện của cosmid, phage hoặc YAC.
Các thư viện hệ gene cũng cần thiết cho việc xác định các gene gây bệnh bằng cách tạo dòng chức năng (functional cloning). Theo hướng này, thông tin về chức năng của gene được khai thác để phân lập gene mong muốn từ thư viện. Một oligonucleotide có trình tự dựa trên chuỗi amino acid từng phần được dùng như là một probe (mẫu dò) để phân lập dòng cDNA bằng cách sàng lọc thư viện cDNA. Dòng cDNA này sau đó có thể được dùng để sàng lọc thư viện hệ gene nhằm phân lập các dòng hệ gene và cho phép quan sát đặc điểm của hệ gene hoàn chỉnh.
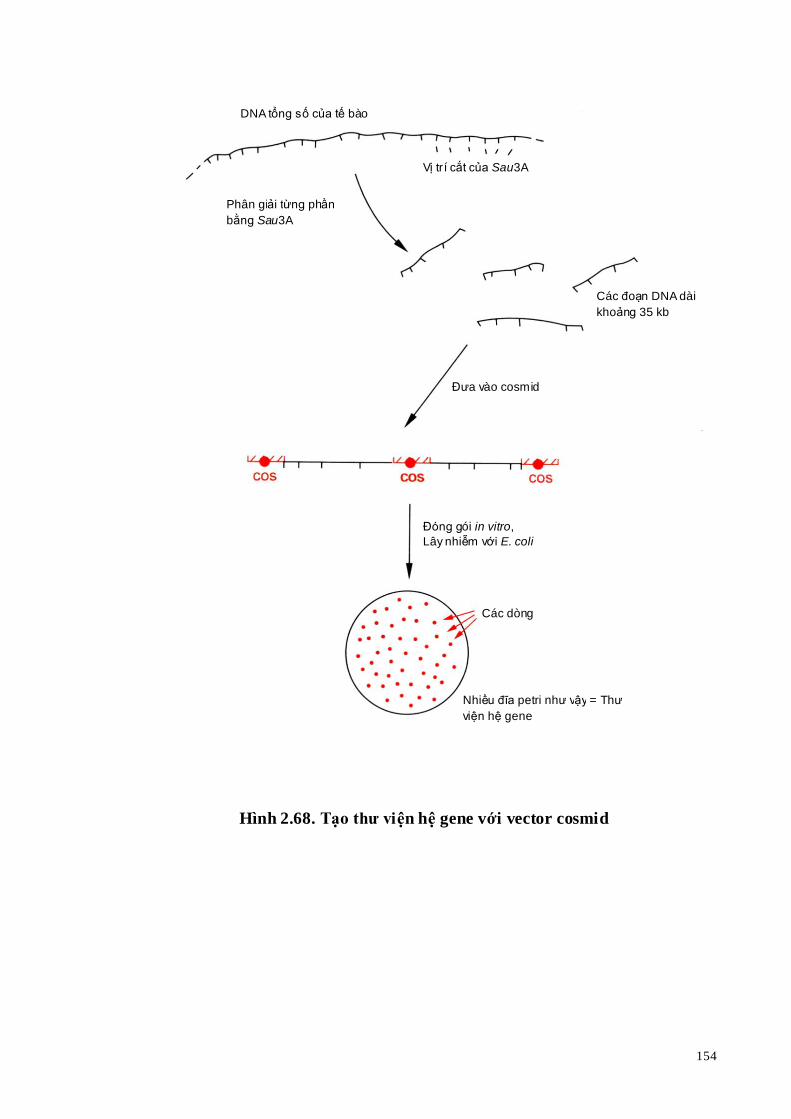
154
Hình 2.68. Tạo thư viện hệ gene với vector cosmid
DNA tổng số của tế bào
Vị trí cắt của Sau3A
Phân giải từng phần bằng Sau3A
Các đoạn DNA dài khoảng 35 kb
Đưa vào cosmid
Đóng gói in vitro, Lây nhiễm với E. coli
Các dòng
Nhiều đĩa petri như vậy = Thư viện hệ gene

155
DNA
Phân đoạn theo kích thước (~40kb) bằng BamHI
T4 DNA ligase
ori cos cos Tetr
50kb
cos
cos
Tetr
ori
BamHI
Scal
Scal
BamHI
Đóng gói phage in vitro
Đầu
DNA vector được đóng gói 50 kb
Đuôi
Sợi đuôi
Đầu cos
Plasmid
Tetr
ori cos
Xâm nhiễm

156
Hình 2.69. Tạo dòng trong cosmid. Hai vị trí cos gần vị trí cắt hạn chế ScaI và BamHI. DNA nguồn bị cắt bởi BamHI để phân đoạn có kích thước khoảng 40
kb. Phân tử plasmid DNA được cắt bởi BamHI và ScaI. Hai mẫu DNA này được trộn vào nhau và được gắn bằng T4 DNA ligase. Sau khi gắn xong, những phân tử này được đóng gói trong phần đầu của phage , và những phần tử có
thể lây nhiễm sẽ được hình thành sau khi tạo đuôi. 2. Thư viện cDNA
Thư viện cDNA (complementary DNA) là tập hợp các dòng DNA được tạo ra từ mRNA của một tế bào hoặc một mô cụ thể trong bacteriophage vector, đại diện cho thông tin di truyền mà các tế bào đó biểu hiện.
Sử dụng thư viện cDNA có hai ưu điểm sau: - Các dòng cDNA chứa trình tự mã hóa liên tục của một gene. Nhiều gene ở
eukaryote là gián đoạn, chứa nhiều đoạn intron. Sau khi cắt và nối lại, các đoạn intron của tiền mRNA đã bị loại và mRNA (mature mRNA) có trình tự mã hoá liên tục được tạo thành. Do cDNA được tạo nên từ mRNA nên các dòng cDNA có thể tổng hợp protein cần thiết với số lượng lớn như mong muốn.
- Nhiều protein được tổng hợp với số lượng lớn do những tế bào chuyên hóa và ở trong các tế bào này mRNA của protein đó có tỷ lệ cao và thư viện cDNA được tạo ra từ các tế bào này sẽ có nhiều cDNA mã hóa cho các protein tương ứng. Sự dồi dào một vài loại cDNA nào đó làm giảm nhẹ đáng kể việc xác định đúng dòng mong muốn từ thư viện hệ gene.
Để tạo nên một thư viện hệ gene vi khuẩn, nấm men và các loài nấm khác không cần nhiều dòng. Nhưng ở thực vật và động vật thư viện hệ gene gồm có nhiều dòng, nên việc xác định một dòng là rất khó khăn. Vì vậy, ở những sinh vật này tiện lợi hơn là tạo thư viện không phải cho toàn bộ sinh vật đó mà là cho một loại tế bào nhất định. 2.1. Không phải tất cả các gene được biểu hiện cùng lúc

157
.
Hình 2.70. Trong các loại tế bào khác nhau các gene được hoạt hóa khác nhau
Đặc tính của phần lớn sinh vật đa bào là sự phân hoá tế bào riêng biệt. Ví dụ
ở người có một số lượng lớn các loại tế bào khác nhau: tế bào não, tế bào máu, tế bào gan…. Tất nhiên mỗi tế bào chứa lượng thông tin di truyền như nhau, nhưng trong từng loại riêng biệt những nhóm gene khác nhau được hoạt hóa (Hình 2.69).
Thực tế là trong mỗi loại tế bào chỉ có một số lượng nhỏ gene biểu hiện, lợi dụng đặc điểm này để tạo thư viện cDNA. Chỉ những gene hoạt động mới được phiên mã sang mRNA, và khi sử dung chúng làm nguyên liệu thì những dòng tương ứng chỉ đại diện cho một số gene của tế bào.
Loại tế bào A
Loại tế bào B
mRNA Protein
Gene không hoạt động
mRNA Protein
Gene không hoạt động
mRNA Protein
Protein mRNA

158
Đặc biệt có lợi cho việc tạo thư viện cDNA là gene cần tìm trong tế bào nào đó được biểu hiện mạnh mẽ. Ví dụ gene gliadin mã hóa cho protein quan trọng trong hạt lúa mỳ. Trong các tế bào này có 30% mRNA mã hoá cho gliadin. Khi tạo dòng mRNA từ hạt lúa mỳ người ta nhận được nhiều dòng mã hoá cho gliadin 2.2. mRNA được tạo dòng là cDNA
Vì mRNA không gắn được với vector tạo dòng nên phải chuyển sang DNA, nghĩa là tổng hợp cDNA.
Enzyme quan trọng cho quá trình này là reverse transcriptase, tổng hợp sợi DNA bổ sung cho mRNA (Hình 2.71a). Khi tổng hợp xong sợi DNA thứ nhất, sợi RNA của phân tử lai được phân giải từng phần bằng enzyme ribonuclease H (Hình 2.71b). Các đoạn RNA còn lại có chức năng là mồi cho enzyme DNA-polymerase I để tổng hợp nên sợi thứ hai của cDNA (Hình 2.71c). Kết quả tạo nên cDNA sợi kép, gắn được vào vector tạo dòng (Hình 2.71d).
Dòng cDNA đại diện cho mRNA có nguồn gốc trong mẫu ban đầu. Thư viện dòng cDNA của hạt lúa mỳ phần lớn đại diện cho mRNA gliadin (Hình 2.71e). Bên cạnh đó tồn tại những dòng khác, nhưng để tìm kiếm cDNA của gliadin bằng cách này đơn giản hơn từ thư viện hệ gene của lúa mỳ.
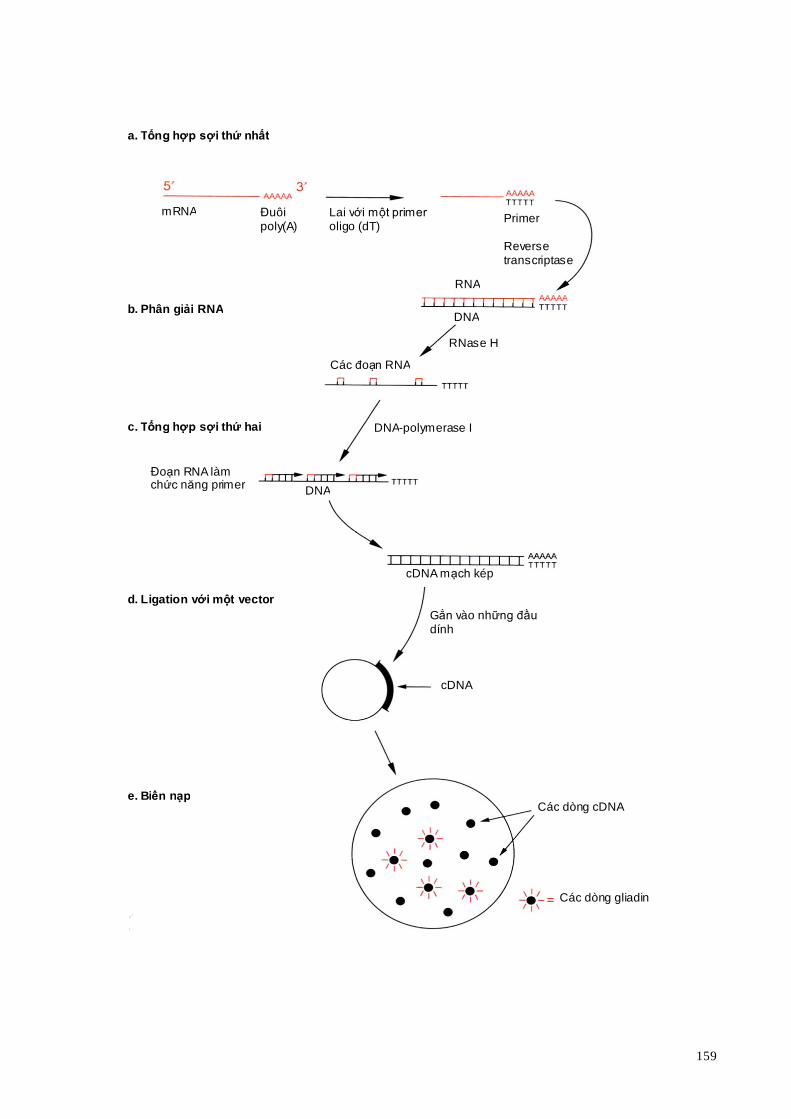
159
mRNA
a. Tổng hợp sợi thứ nhất
Đuôi poly(A)
Lai với một primer oligo (dT)
Primer
Reverse transcriptase
RNA
DNA
RNase H
Các đoạn RNA
DNA-polymerase I
DNA
b. Phân giải RNA
c. Tổng hợp sợi thứ hai
Đoạn RNA làm chức năng primer
cDNA mạch kép
Gắn vào những đầu dính
d. Ligation với một vector
e. Biến nạp
cDNA
Các dòng cDNA
Các dòng gliadin

160
Hình 2.71. Phương pháp tạo dòng cDNA Phương pháp tổng hợp gene từ mRNA ngày càng được phát triển và ứng
dụng nhiều trong lĩnh vực sinh học phân tử. Để xây dựng thư viện cDNA gồm các bước sau: - Tách chiết mRNA từ RNA tổng số của một phận cơ thể sinh vật.
- Tổng hợp DNA bổ sung (cDNA) - Tạo vector tái tổ hợp
- Biến nạp vector tái tổ hợp vào tế bào chủ, thường là tế bào vi khuẩn. Tập hợp tất cả các dòng vi khuẩn mang các cDNA khác nhau tạo nên thư
viện cDNA. Thư viện cDNA được sử dụng để nghiên cứu sự biểu hiện của gene, quá trình điều hòa hoạt động và sự tương tác giữa các gene…
Hình 2.72. Kỹ thuật tạo thư viện cDNA

161
Các linker chứa một hoặc nhiều vị trí cắt hạn chế cho phép gắn cDNA với các plasmid vector hoặc bacteriophage λ vector. Sợi đôi cDNA được xử lý với DNA polymerase (ví dụ: đoạn Klenow của DNA polymerase I của E. coli), enzyme này loại bỏ đầu tận cùng 3’ sợi đơn so le bằng hoạt tính exonuclease 3’→ 5’ và lấp đầy các đầu tận cùng 3’-OH bị khuyết bằng hoạt tính trùng hợp (polymerization). Sự phối hợp của các hoạt tính này đã tạo ra các phân tử DNA đầu bằng, sau đó các cDNA này được ủ với các phân tử linker có mặt của enzyme gắn là DNA ligase.

162
Hình 2.73. Các phân tử DNA sợi đôi mang các đầu dính nhân tạo được cắt hạn chế ở vị trí nhận biết trong linker, tinh sạch, và sau đó gắn với vector
cũng được cắt bằng enzyme hạn chế tương ứng tạo ra các đầu dính tương đồng với các đầu của linker. Có thể hạn chế sự tái tạo lại vòng của plasmid vector
(không tái tổ hợp) bằng cách xử lý các vector được cắt bằng alkaline phosphatase trước khi thực hiện phản ứng gắn với cDNA.

163
Tài liệu tham khảo/đọc thêm 1. Nguyễn Quang Thạch (Chủ biên), Nguyễn Thị Lý Anh, Nguyễn Thị
Phương Thảo. Giáo trình Công nghệ Sinh học Nông nghiệp. NXB Nông nghiệp. 2005
2. Southern, E. M. Detection of Specific Sequences Among DNA Fragments Separated by Gel Electrophoresis. In: Journal of Molecular Biology 98 (1975).
1. Sanger, F. et al. DNA Sequencing with Chain-Terminating Inhibitors. In: Proceedings of the National Academy of Sciences, USA 74 (1977).
2. Maxam, A.; Gilbert, W. A New Method of Sequencing DNA. In: Proceeding of the National Academy of Sciences, USA 74 (1977).
3. Leitch, A. R. et al. In situ-Hybridisierung. Heidelberg (Spektrum Akademischer Verlag) 1994.
4. Brown, T. A. DNA Sequencing: The Basics. Oxford (Oxford University Press) 1994
5. Galas, D. J.; Schmitz, A. DNAase Footprinting: A Simple Method for the Detection of Protein-DNA Binding Specificity. In: Nucleic Acid Research 5 (1978).
6. Newton, C. R.; Graham, A. PCR. Heidelberg (Spektrum Akademischer Verlag) 1994

145
Bài 3
CÔNG NGHỆ CHUYỂN GENE VÀO TẾ BÀO THỰC VẬT
I. Giới thiệu chung Chuyển gene vào thực vật thành công lần đầu tiên vào những năm 1960, mặc
dù còn thiếu marker và công cụ phân tử để khẳng định sự hợp nhất của gene được chuyển và sự biểu hiện của chúng. Cho đến cuối những năm 1970, quá trình này mới được sáng tỏ nhờ làm rõ cơ chế hình thành khối u (crown gall).
Chuyển gene vào thực vật là một trong những lĩnh vực phát triển nhanh nhất của công nghệ sinh học phân tử. Kỹ thuật chuyển gene là một công cụ có tiềm năng trong việc giải quyết một số vấn đề hiện đại của sinh lý học, hóa sinh học, sinh học phát triển và di truyền học.
Để chuyển gene từ một cơ thể này sang cơ thể khác, các nhà sinh học phân tử sử dụng vector. Vector là các vật mang (carriers) được sử dụng để chuyển gene vào tế bào chủ mới, chúng có thể vào tế bào, duy trì và biểu hiện gene ngoại lai. Vector được sử dụng để chuyển gene bao gồm virus, plasmid, gene nhảy (transposon). Các gene cũng có thể đưa vào nhờ phương pháp hóa chất, xung điện, vi tiêm… Nguyên tắc cơ bản cho các kỹ thuật trên là giống nhau cho động vật, thực vật, vi sinh vật, nhưng có những biến đổi đặc trưng phù hợp cho từng nhóm.
Có một số yếu tố sinh học ảnh hưởng đến quá trình chuyển gene. Không phải tất cả các tế bào trong cơ thể đều có tính toàn thể, các cây khác nhau có phản ứng không giống nhau với sự xâm nhập của một gene lạ. Một loại cây nào đó có thể được biến nạp khi nó có khả năng tái sinh và chấp nhận sự biến nạp. Mô tế bào là một tập hợp của nhiều tế bào có phản ứng khác nhau khi gặp một yếu tố tác động đến. Một số ít tế bào trong cây có khả năng tái sinh và chấp nhận sự biến nạp. Những tế bào khác chỉ có một trong hai khả năng ấy. Một số lớn các tế bào trong cơ thể có khả năng điều chỉnh được khả năng đó, một số khác ngược lại không thể làm được. Tương quan giữa các quần thể tế bào kiểu như vậy phụ thuộc vào loài, kiểu gene, cơ quan, thậm chí tùy từng vùng trong một cơ quan.
Một trong các cơ chế có thể chuyển các tế bào từ trạng thái tiềm ẩn sang trạng thái thực sự có khả năng tái sinh là sự xuất hiện thương tổn trên cơ thể. Phản ứng với sự thương tổn có lẽ là cơ sở sinh học cho sự tăng sinh và tái sinh của các tế bào soma. Tuy nhiên phản ứng này khác nhau giữa các loài cây, cũng như giữa các nhóm tế bào trong cùng một cây. Nói chung các cây hòa thảo và các cây họ đậu phản ứng này xảy ra rất yếu. Thành tế bào ngăn cản sự xâm nhập của DNA. Vì thế, gene chỉ có thể được đưa vào thành tế bào thông qua Agrobacterium, virus, bằng phương pháp vi tiêm hay bằng súng bắn gene. Việc chuyển gene chỉ thành công khi đưa được gene vào nhóm tế bào có khả năng tái sinh và tiếp nhận gene lạ. Tuy nhiên, khả năng biến nạp không có liên quan chặt chẽ với khả năng biểu hiện của gene được biến nạp. DNA không phải của virus có thể liên kết với hệ gene của vật chủ và không di chuyển từ tế bào này qua tế bào khác. Trong khi đó các DNA của virus có thể không liên kết với hệ gene của vật chủ, thậm chí cả khi có rất nhiều bản sao trong tế bào chủ. DNA, RNA của virus di chuyển từ tế bào này sang tế bào khác và có thể lan truyền khắp trong cây trừ vùng mô phân sinh.

146
Mục đích chính của một quy trình chuyển gene là đưa một cách ổn định một đoạn DNA vào hệ gene nhân của các tế bào có khả năng phát triển thành một cây biến nạp. Ở nhiều loài thực vật, sự xác định kiểu tế bào nào trong cây có khả năng tiếp nhận sự biến nạp là điều khó khăn. Hạt phấn hay tế bào trứng sau khi được biến nạp có thể được dùng để tạo ra cây biến nạp hoàn toàn thông qua quá trình thụ tinh bình thường. Hạt phấn thường được coi là đối tượng lý tưởng để gây biến nạp. Trong khi đó, việc biến nạp gene vào hợp tử in vivo hay in vitro lại gặp nhiều khó khăn. Trong trường hợp này, thường phải kết hợp với kỹ thuật cứu phôi. Việc biến nạp đối với các tế bào đơn của các mô phức tạp như phôi hay mô phân sinh thường cho ra những cây khảm.
Tính toàn thể của tế bào thực vật đã tạo điều kiện cho sự tái sinh cây hoàn chỉnh in vitro qua quá trình phát sinh cơ quan (hình thành chồi) hay phát sinh phôi. Các chồi bất định hay các phôi được hình thành từ các tế bào đơn được hoạt hóa là những bộ phận dễ dàng tiếp nhận sự biến nạp và có khả năng cho những cây biến nạp hoàn chỉnh (không có tính khảm)
Kể từ năm 1984, khi người ta bắt đầu tạo được cây chuyển gene, đến nay kỹ thuật này đã có những bước tiến rất lớn. Những thành công của nó không chỉ giới hạn ở những cây mang tính chất mô hình như thuốc lá và các cây họ cà, mà còn đạt những kết quả ứng dụng ở một số cây trồng quan trọng. Vì vậy, chúng ta có cơ sở để hy vọng rằng trong tương lai kỹ thuật gene sẽ trở thành một công cụ không thể thiếu được của chọn giống thực vật. II. Phương pháp chuyển gene gián tiếp 1. Hệ thống Agrobacterium
Vào năm 1907, Smith và Townsend chứng minh rằng vi khuẩn đất Gram âm Agrobacterium tumefaciens, một thành viên của họ vi khuẩn thật Rhizobiaceae, là vi khuẩn gây ra khối u ở thực vật. Sự tạo thành khối u xảy ra do sự nhiễm vi khuẩn thường ở vết thương của nhiều cây hai lá mầm và một số cây một lá mầm
Agrobacterium tumefaciens và Agrobacterium rhizogenes là hai loài vi khuẩn gây bệnh cho thực vật được sử dụng như các vector tự nhiên để mang các gene ngoại lai vào mô và tế bào thực vật. A. tumefaciens có chứa một plasmid lớn có kích thước khoảng 200 kb gọi là Ti-plasmid (tumor inducing plasmid) chính là tác nhân truyền bệnh cho cây (Hình 3.1).

147
Hình 3.1. Mô hình đơn giản biểu thị các vùng trên Ti-plasmid của Agrobacterium tumefaciens
A. tumefaciens tạo khối u ở nhiều loại cây 2 lá mầm như cà chua, thuốc
lá, khoai tây, đậu,... Khi cây bị nhiễm A. tumefaciens qua các vết thương, biểu hiện rõ nhất là các khối u được hình thành ở ngay chỗ lây nhiễm (Hình 3.2).
Tạo khối u Tổng hợp nopaline
Sử dụng nopaline
Điểm xuất phát sao chép
Các chức năng chuyển T-DNA

148
Hìn
h 3.
2. A
grob
acter
ium
gây
khố
i u ở
thực
vật

149
Sự hình thành khối u sau đó có thể tiếp tục mà không cần phải có sự hiện diện của vi khuẩn. Khả năng này có được do A. tumefaciens đã chuyển một đoạn DNA của Ti-plasmid là T-DNA xâm nhập vào hệ gene của cây bị bệnh.
2. Ti-plasmid là nhân tố gây biến nạp tự nhiên ở thực vật
Sự có mặt tiếp tục của Agrobacterium không được yêu cầu cho sự duy trì tế bào thực vật ở trạng thái biến nạp của chúng. Rõ ràng nguyên lý kích thích tạo khối u được chuyển từ vi khuẩn sang tế bào thực vật qua vết thương. Zaenen và cộng sự (1973) lần đầu tiên phát hiện các chủng A. tumefaciens độc chứa những plasmid và thí nghiệm liên quan đến sự chuyển những plasmid như thế giữa những chủng sử dụng octopine và nopaline làm hình thành ngay tính độc. Khả năng sử dụng và kích thích tổng hợp opine là những đặc tính tự nhiên của plasmid.
Ti-plasmid đặc trưng cho kiểu opine mà nó tổng hợp ở trong mô thực vật được biến nạp và opine được sử dụng bởi vi khuẩn. Plasmid trong nhóm octopine liên hệ chặt chẽ với mỗi plasmid trong nhóm nopaline, trong khi những plasmid trong nhóm nopaline gồm nhiều loại hơn. Giữa các nhóm có 4 vùng tương đồng bao gồm các gene chịu trách nhiệm trực tiếp cho sự tạo thành khối u (Drummon và Chilton 1978, Engler và cs 1981) (Hình 3.3).
Hình 3.3. Bản đồ gene của Ti-plasmid
Phần lớn các loại Ti-plasmid gồm 4 vùng A, B, C, D. Vùng A luôn luôn được truyền sang tế bào thực vật nên được gọi là T-DNA
(Transferred DNA), có chứa 2 hệ gene: Hệ gene gây khối u onc (oncogene), chứa ít nhất 3 gene chịu trách nhiệm tổng hợp các hormone auxin và cytokinin và do đó có liên quan đến việc sản sinh ra và duy trì sự phân chia tế bào liên tục. Thứ hai là hệ gene mã hóa cho một số enzyme điều khiển quá trình tổng hợp các dẫn xuất của các amino acid hay đường gọi là opine. Hai loại enzyme đã được nghiên cứu kỹ nhất là octopine synthetase và nopaline synthetase.

150
Vùng B có liên quan đến sự tái sinh.Vùng C có liên quan đến sự tiếp hợp. Vùng D là vùng độc (virulence region) có chứa các gene vir, giữ vai trò quan trọng trong việc chuyển T-DNA vào hệ gene nhân của tế bào thực vật.
Ngoài ra mỗi Ti-plasmid còn chứa các gene nằm ngoài vùng T-DNA mã hóa cho các enzyme phân giải opine để làm nguồn dinh dưỡng carbon và nitrogen cho vi khuẩn. 3. T-DNA và các trật tự biên 25 bp
DNA của Ti-plasmid hoàn chỉnh không được tìm thấy trong tế bào khối u, nhưng một đoạn nhỏ đặc trưng của plasmid được tìm thấy gắn vào DNA nhân thực vật ở vị trí dường như ngẫu nhiên. Đoạn DNA này được gọi là T-DNA (transferred DNA), mang các gene liên quan với sự sinh trưởng không được điều hòa và cả khả năng tổng hợp opine ở mô thực vật được biến nạp. Tuy nhiên, những gene này không mẫn cảm với sự chuyển, nên có thể thay thế bằng DNA ngoại lai. Cấu trúc và tổ chức của trình tự T-DNA plasmid nopaline thường đơn giản chẳng hạn chỉ có một đoạn gắn vào đơn lẻ. Ngược lại đoạn T-DNA octopine gồm có 2 đoạn: TL (mang những gene yêu cầu cho sự tạo khối u) và TR (mang những gene tổng hợp opine). Cả hai đoạn được chuyển vào genome thực vật một cách độc lập và có thể có nhiều bản sao.
Vùng T-DNA có kích thước từ 10 kb đến 20 kb, nằm giữa hai trật tự 25 bp lặp lại không hoàn chỉnh gọi là bờ trái (left border – LB) và bờ phải (right border – RB) được bảo tồn ở plasmid octopine và nopaline. Toàn bộ phần giới hạn giữa hai đoạn biên này đều được chuyển nguyên vẹn sang tế bào thực vật. LB và RB là những yếu tố cần thiết để định hướng cho sự chuyển DNA. Việc mất đi 6 bp đầu tiên hoặc 10 bp cuối cùng trong số 25 bp đều ngăn cản sự chuyển của T-DNA. Quá trình chuyển của T-DNA được bắt đầu từ vị trí RB và kết thúc ở vị trí LB. Sự định hướng này là rất quan trọng nếu không việc chuyển sẽ kém hiệu quả. Trong các gene được mã hóa ở vùng T-DNA có 3 gene rất quan trọng trong việc phát triển khối u: một gene mã hóa cho việc chuyển enzyme AMP-isopentonyl transferase và 2 gene mã hóa cho enzyme tham gia vào sinh tổng hợp auxin (tryptophane-2 mono oxygenase và acetamide hydrolase). Sự hoạt động của các gene này tạo điều kiện cho sự phát triển của tế bào thực vật có sự phụ thuộc vào auxin và cytokinin. Đặc điểm này được sử dụng để chọn lọc các tế bào được biến nạp từ phần lớn các tế bào không được biến nạp. 4. Vùng vir
Các gene chịu trách nhiệm chuyển T-DNA được định vị trên một phần tách biệt của Ti-plasmid gọi là vùng vir (virulence). Chính hoạt động của vùng vir đóng vai trò quan trọng cho việc chuyển T-DNA sang tế bào thực vật. Nó có kích thước khoảng 40 kb và gồm 6 operon: virA, B, D và G là hoàn toàn cần thiết cho việc tạo ra độc tính, còn virC và E có liên quan với việc hình thành khối u. Trừ virG và A, các operon khác đều là đa cistron. Vir A và virG được biểu hiện cơ bản ở nồng độ thấp và điều khiển hoạt tính của các gene vir khác. Nói chung, gene virA biểu hiện ở mọi điều kiện, gene virC biểu hiện thấp ở các tế bào sinh dưỡng nhưng thể hiện rất mạnh ở các dịch chiết từ các mô bị tổn thương. Hoạt động của các operon này có liên quan chặt chẽ với sự có mặt của các hợp chất phenol được tiết ra từ vết thương
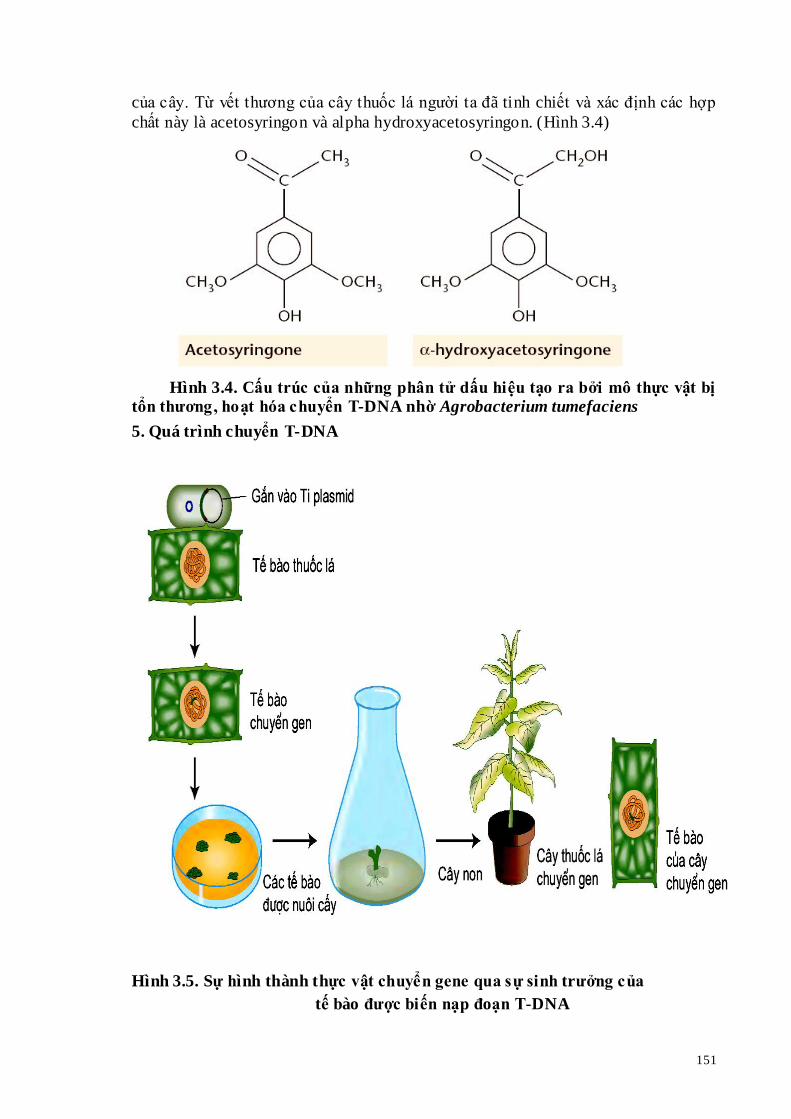
151
của cây. Từ vết thương của cây thuốc lá người ta đã tinh chiết và xác định các hợp chất này là acetosyringon và alpha hydroxyacetosyringon. (Hình 3.4)
Hình 3.4. Cấu trúc của những phân tử dấu hiệu tạo ra bởi mô thực vật bị
tổn thương, hoạt hóa chuyển T-DNA nhờ Agrobacterium tumefaciens 5. Quá trình chuyển T-DNA
Hình 3.5. Sự hình thành thực vật chuyển gene qua sự sinh trưởng của tế bào được biến nạp đoạn T-DNA

152
Đoạn T-DNA muốn được chuyển, trước tiên nó cần phải được hoạt hóa nhờ hoạt động của các gene vir. Hoạt động này xảy ra khi Agrobacterium bắt đầu tiếp
xúc với hợp chất chứa phenol được chiết ra từ vết thương của cây hoặc từ các tế bào nuôi cấy hoặc protoplast.
Đầu tiên là sự hoạt động của các sản phẩm được tạo ra từ gene virA (protein virA). Protein virA là kinase mở rộng màng tế bào vi khuẩn. Protein này nằm ở màng trong của tế bào Agrobacterium, đóng vai trò như là một chất thụ cảm đối với acetosyringon có trong môi trường. Tín hiệu này sẽ được truyền dẫn qua sự hoạt hóa (có lẽ bằng việc phosphoryl hóa) gene virG. Protein virG sau đó lại gắn vào gene chỉ huy nằm sát gene khởi động thuộc các gene vir khác. Bằng cách như vậy, protein của gene virG đã được hoạt hóa làm tăng quá trình phiên mã của chính nó, đồng thời làm giảm quá trình này của các operon B, C, D và E. Thêm vào virG, các gene xa hơn trên nhiễm sắc thể cũng mã hóa cho các nhân tố phiên mã điều hòa biểu hiện của gene vir.
Trong tiến trình chuyển T-DNA, ở giai đoạn đầu xuất hiện các vết cắt Ti-plasmid tại hai trật tự biên 25 bp, ở vị trí base thứ ba và thứ tư của mạch đơn phía dưới mới được tổng hợp. Từ đó, một mạch T-DNA được giải phóng ra khỏi Ti-plasmid và thay vào đó là một mạch đơn mới được tổng hợp theo hướng 5’-3’, bắt đầu từ chỗ đứt của trật tự biên phải. Điều này giải thích tại sao trật tự biên phải lại cần thiết cho sự chuyển T-DNA. Operon D chủ yếu mã hóa cho một loại endonuclease tạo ra các vết cắt nói trên tại hai trật tự biên. Sự hình thành mạch đơn T-DNA được tăng cường nhờ sự tương tác giữa protein virD2 với một hoặc hai protein do gene virC mã hóa. Protein gene virE2 gắn vào mạch đơn T-DNA sau khi được cắt ra làm cho nó ổn định trong quá trình chuyển vào nhân tế bào thực vật. Phức hợp protein-mạch đơn T-DNA là cấu trúc cần thiết cho việc chuyển T-DNA sang tế bào thực vật và nó phải được sản sinh ra từ Agrobacterium. Gene virB mã hóa cho ít nhất 11 protein để hình thành nên một cầu nối tế bào chất (pilus), cho phức hợp trên chuyển từ Agrobacterium sang tế bào thực vật.
Các nghiên cứu gần đây đã đề nghị là kiểu T-DNA trung gian (T-DNA intermediate) được tạo thành (sợi đơn hoặc sợi đôi) phụ thuộc vào kiểu Ti-plasmid. Kiểu T-DNA sợi đôi thích hợp với plasmid nopaline (nơi mà T-DNA là một yếu tố đơn) và kiểu T-DNA sợi đơn thích hợp với plasmid octopine và succinopine, nơi mà T-DNA được chia ra thành những phần không kề nhau (Steck 1999). Sợi T-DNA được bao bọc với virE2 – một loại protein bám sợi đơn. Toàn bộ phức hợp được chuyển qua cầu tế bào chất pilus vào tế bào thực vật. Protein virD2 bảo vệ T-DNA chống lại nuclease và gắn vào genome thực vật. Protein có 2 tín hiệu định vị nhân phân biệt, với tín hiệu đầu tận cùng C (C-terminal signal) đóng vai trò chủ yếu trong xác đinh T-DNA (Tinland và cs. 1992). Trong nhân, T-DNA được gắn vào genome qua quá trình tái tổ hợp. 6. Các dẫn xuất Ti-plasmid như là vector thực vật a. Vector đồng xâm nhập (cointegrate vector)
Mặc dù các dạng biến đổi của Ti-plasmid hoang dại có thể được sử dụng cho biến nạp ở thực vật nhưng chúng không thuận lợi như vector thực nghiệm do chúng có kích thước lớn và không có vị trí cắt hạn chế thống nhất trong vùng T (T-region). Vấn đề này được khắc phục nhờ cấu trúc vector đồng xâm nhập.

153
Hình 3.6. Chức năng tái tổ hợp của Ti-plasmid đã được biến đổi nhờ sự tạo đồng xâm nhập
T-DNA phân lập từ Ti-plasmid của bố mẹ được tạo dòng trong vector plasmid E. coli, tạo ra vector trung gian (intermediate vector) (Matzke và Chilton 1981). Những vector này không có khả năng sao chép trong A. tumefaciens và cũng thiếu chức năng tiếp hợp. Việc chuyển gene được thực hiện nhờ giao phối ba bố mẹ (Triparental mating), trong đó 3 chủng vi khuẩn được phối hợp với nhau:
- Chủng E. coli mang helper plasmid giúp tiếp hợp hoàn toàn.

154
- Chủng E. coli mang vector trung gian tái tổ hợp. - A. tumefaciens thể nhận mang Ti-plasmid. Sự tiếp hợp giữa 2 chủng E. coli giúp chuyển helper plasmid sang chủng vi
khuẩn mang vector trung gian tái tổ hợp. Chỉ có vector trung gian tái tổ hợp được chuyển vào Agrobacterium. Tái tổ hợp tương đồng giữa trình tự T-DNA của Ti-plasmid và vector trung gian, kết quả tạo vector đồng xâm nhiễm. Từ đó T-DNA tái tổ hợp được chuyển vào genome thực vật (Hình 3.6). Plasmid pGV 3850 được dùng như là một thể nhận của vector trung gian vì các chuỗi pBR322 trong vùng T-DNA của nó tương đồng với hầu hết plasmid trung gian pBR322 gốc (Hình 3.7). Trong hệ thống vector đồng xâm nhiễm, sự duy trì của T-DNA tái tổ hợp phụ thuộc vào sự tái tổ hợp. Nó được tăng cường nếu mở rộng vùng tương đồng chung giữa 2 plasmid.
Hình 3.7. Cấu trúc của Ti-plasmid pGV3850, trong đó T-DNA bị cắt bỏ
b. Vector hai nguồn (binary vector)
Mặc dù vector trung gian được sử dụng rộng rãi, nhưng sự đồng xâm nhiễm lớn là không cần thiết cho biến nạp. Các gene vir của Ti-plasmid có chức năng trans và có thể hoạt động trên bất kỳ T-DNA nào có mặt trong cùng tế bào.Vì vậy, gene vir và T-DNA bị biến đổi có chứa gene cần chuyển được cung cấp bới các plasmid riêng rẽ. Đây là nguyên lý của hệ thống binary vector.
T-DNA có thể được tạo dòng trong một plasmid nhỏ của E. coli, gọi là mini-Ti hoặc micro-Ti, và đưa vào chủng Agrobacterium mang Ti-plasmid. Chức năng vir giúp chuyển T-DNA tái tổ hợp. T-DNA plasmid có thể được đưa vào

155
Agrobacterium bằng giao phối ba bố mẹ hoặc bằng phương thức biến nạp đơn giản như điện biến nạp. 7. Phương thức truyền gene vào thực vật nhờ Agrobacterium
Tế bào thực vật bị thương tổn là nơi chuyển gene T-DNA. Trong các mô được sử dụng để chuyển gene, lá là nguồn tốt nhất cho tái sinh (Hình 3.8).
Những tế bào ở mép lá bắt đầu tái sinh và khi những đĩa lá được nuôi cấy trên môi trường chứa Agrobacterium, những tế bào này rất hiệu quả cho tác nhân chuyển gene. Kỹ thuật đĩa lá được sử dụng hiệu quả cho chuyển gene vào thực vật nhờ sử dụng Ti-plasmid của Agrobacterium.
Agrobacterium được chứng minh là một công cụ hữu ích cho việc chuyển gene vào cây trồng, nhưng phạm vi của nó bị hạn chế đối với những cây trồng có thể được nhiễm bằng vi khuẩn. Thực vật một lá mầm như lúa mì, lúa mạch, lúa và ngô là những mục tiêu cho thao tác di truyền nhưng lại kháng với việc nhiễm Agrobacterium. Lý do cho điều này là chưa rõ, có lẽ sự cảm ứng phân bào và là hệ quả của sự sinh tổng hợp DNA, xảy ra ở vết thương của cây hai lá mầm đã giúp cho quá trình hợp nhất. Sự thương tổn của cây một lá mầm đã dẫn đến kết quả sự hóa gỗ. Sự biến nạp vào lúa qua trung gian Agrobacterium có thể xảy ra với tần số cao nếu acetosyringone, một hợp chất phenolic được tạo ra nhờ mô thực vật hai lá mầm bị thương, được tính đến trong phản ứng biến nạp. Nhiều loại cây một lá mầm khác cũng được biến nạp thành công nhờ sử dụng Agrobacterium cùng với hóa chất hoặc sử dụng phôi của thực vật mà trong đó sự phân chia tế bào xảy ra nhanh chóng sẽ cho phép T-DNA gắn vào.

156
Hình 3.8. Tái sinh cây từ đĩa lá nhiễm Agrobacterium
Tạo chồi
Tạo rễ
Chuyển cây ra đất

157
III. Phương pháp chuyển gene trực tiếp 1. Chuyển gene vào protoplast
Hầu hết các tế bào thực vật được bao bọc bởi thành cellulose nên khó để hấp thụ các phân tử DNA ngoài vào tế bào. Tuy nhiên, thành tế bào có thể làm tan nhờ các xử lý tế bào mô. Kết quả thu được các protoplast chỉ còn màng sinh chất rất thuận lợi cho các thao tác thí nghiệm.
Protoplast có thể được tạo thành từ nuôi cấy huyền phù, mô callus hoặc mô nguyên vẹn như lá, thành tế bào của chúng bị phá vỡ do tác động cơ học hoặc tốt nhất là xử lý bằng enzyme phân giải cellulose và phân giải pectin. Xử lý mô tế bào lần lượt với enzyme pectinase và cellulase, pectinase làm bẻ gãy các liên kết giữa các tế bào tạo các tế bào đơn và cellulase sẽ tiêu hóa thành tế bào. Sau khi xử lý bằng enzyme, dịch huyền phù protoplast được chọn lọc bằng ly tâm, được rửa trong môi trường không có enzyme và tách những tế bào nguyên vẹn khỏi các mảnh tế bào.
Sau khi trải trên môi trường dinh dưỡng, protoplast sẽ tổng hợp nên thành tế bào mới trong khoảng 5 - 10 ngày sau khi trải qua sự phân chia tế bào và dưới điều kiện thích hợp, chúng có thể tái sinh thành cây mới (Hình 3.9). Protoplast có thể hấp thụ các đại phân tử như DNA và chúng có khả năng tái sinh thành cây hoàn chỉnh thông qua sự tạo thành callus. Ở giai đoạn protoplast, tế bào có thể hấp thu DNA bằng hóa chất, điện biến nạp hoặc bằng con đường tạo liposome. DNA xâm nhiễm vào tế bào bằng các con đường này không có khả năng sao chép độc lập. Tuy nhiên, nó có thể được gắn vào một nhiễm sắc thể nào đó của cây trồng một cách ngẫu nhiên nhờ quá trình tái tổ hợp không tương đồng. Nếu DNA có chứa gene marker như gene kháng kanamycine, có thể chọn lọc được các thể biến nạp.

158
.
Hinh 3.9. Tái sinh cây từ protoplast Mặc dầu protoplast đã được sử dụng thành công cho nhiều loài, nhưng hầu hết
các cây nông nghiệp quan trọng như ngũ cốc là rất khó tái sinh từ protoplast. Gần đây đã thu được một vài thành công trong sinh trưởng ở lúa và ngô từ
protoplast của nuôi cấy tế bào phát sinh phôi. Phôi thực vật sinh trưởng in vitro là nguồn tế bào quan trọng cho nuôi cấy. Các cây lúa và ngô hữu thụ được tạo thành từ những tế bào được thao tác di truyền thu được từ nuôi cấy dịch huyền phù phát sinh phôi 2. Chuyển nạp DNA ngoại lai vào tế bào bằng xung điện
Phương pháp điện xung hay điện biến nạp (electroporation) có liên quan với kỹ thuật sử dụng điện trường cường độ cao, ngắn để gây ra tính thấm thuận nghịch qua lớp lipid kép của màng tế bào. Xung điện làm mở rộng lỗ màng và làm mỏng màng sinh chất. Kết quả sự tạo thành tạm thời các lỗ sẽ cho phép các đại phân tử khác nhau (DNA, RNA, kháng thể, …) chuyển qua màng.

159
Tế bào được đặt ở trong một thiết bị có hiệu điện thế cao khoảng 500 V/cm với khoảng thời gian 4-5 phần nghìn giây, xung điện tạo các lỗ tạm thời (cỡ 30 nm) ở trên màng tế bào và DNA có thể xâm nhập vào chất nguyên sinh.
Điện biến nạp là phương pháp thuận tiện cho chuyển gene. Trong hầu hết các trường hợp, phương pháp này hiệu quả hơn các phương pháp khác được thiết kế cho cùng mục đích như phương pháp bắn gene. Hơn nữa, khi sử dụng phương pháp này không bị giới hạn bởi phổ vật chủ như các hệ thống dựa trên cơ sở sinh học như hệ thống Agrobacterium tumefaciens và cũng không gặp vấn đề về độc tố như khi sử dụng phương thức dựa trên polyethylene glycol. Ngoài ra, điện biến nạp gắn liền với phép thử biểu hiện nhanh, cho phép phát hiện sự tái sinh của các sản phẩm gene trong vài giờ sau khi đưa DNA vào. Khác với kiểu biến nạp gene ổn định, phải mất thời gian hàng tháng để tái sinh thể biến nạp và phải chấp nhận những thay đổi lớn không thể điều chỉnh được trong biểu hiện của gene vì “hiệu ứng vị trí” (positional effects). Điện biến nạp trên cơ sở hệ thống chuyển gene có một số biến đổi quan trọng gồm phương pháp chuẩn bị protoplast, cường độ và thời gian xung điện, nồng độ ion, thành phần của đệm điện biến nạp, độ tinh sạch của DNA,…Việc phân tích những biến đổi này với mục đích là xác định và tối ưu hóa các thông số cần thiết để tăng tần số biểu hiện trong quần thể các protoplast thuốc lá. Bằng kỹ thuật xung điện, Molina (1992) đã tạo được giống mía chuyển gene. Nhiều thực vật khác cũng đã được tạo ra bằng kỹ thuật chuyển gene nhờ xung điện.
3. Chuyển gene bằng phương pháp bắn gene
Đây là phương pháp hiện đang được sử dụng phổ biến tại các phòng thí nghiệm công nghệ sinh học thực vật ở trong nước và trên thế giới. Phương pháp này được Sanford (Cornell University, USA) đề xuất lần đầu tiên vào năm 1987. Nguyên tắc của phương pháp này là sử dụng viên đạn có kích thước hiển vi (1-4 µm), có tỷ trọng cao để đạt gia tốc (khoảng 250 m/s) đủ để xuyên qua vách tế bào và màng sinh chất, đưa lớp DNA bọc ngoài tiếp cận với bộ máy di truyền của tế bào.
Hạt tungsten hoặc vàng có đường kính 1-1,5 µm được dùng làm vi đạn (microprojectile). Vi đạn được trộn với DNA theo một tỷ lệ thích hợp cùng với các chất phụ gia và sau khi kết tủa DNA bao quanh vi đạn, hỗn hợp được làm khô trên một đĩa kim loại mỏng kích thước 0,5-0,8 cm. Đĩa kim loại được gắn vào đầu một viên đạn lớn (macroprojectile) vừa khít với nòng súng. Thường đạn lớn làm bằng nhựa hoặc bông nén hay các vật liệu nhẹ. Khi bắn, áp suất hơi đẩy viên đạn lớn đi với tốc độ cao. Ra khỏi đầu nòng, một lưới thép mịn cản viên đạn lớn lại, nhưng các vi đạn vẫn tiếp tục quỹ đạo với gia tốc lớn đến đích và xuyên vào tế bào. Một tỷ lệ nhất định DNA ngoại lai hội nhập với DNA tế bào và biểu hiện, thực hiện quá trình biến nạp gene (Hình 3.10).

160
Hình 3.10. Sơ đồ súng bắn gene Trong hệ thống kiểm tra ban đầu, tế bào biểu bì hành nguyên vẹn đã được thực
hiện bằng phương pháp dội bom với hạt tungsten được bọc bằng RNA của virus khảm thuốc lá (tobacco mosaic virus). Ba ngày sau khi tiến hành dội bom, có khoảng 40% tế bào biểu bì hành biến nạp có bằng chứng về sự sao chép của virus khảm thuốc lá (Sanford và cs 1987). Một plasmid chứa gene thông báo cat, dưới sự điều khiển của promoter CaMV 35S đã được kiểm tra và xác định được DNA phát tán bởi cùng một phương pháp. Phân tích mô biểu bì hành 3 ngày sau dội bom cho thấy hoạt tính tạm thời của enzyme chloramphenicol transacetylase ở mức độ cao.
Sự biến nạp bền vững từ nhiều loài thực vật đã đạt được những thành tựu nhanh chóng từ sau thí nghiệm đầu tiên này như biến nạp vào đậu tương (Christou và cs 1988), vào cà chua (Klein và cs 1988b), vào ngô (Klein và cs 1988a). Trong mỗi trường hợp, gene nptII được sử dụng như là một marker chọn lọc và sự biến nạp đã được khẳng định bởi sự sống sót của các callus này trên môi trường bổ sung kanamycin.
Cây đậu tương chuyển gene đã được tạo ra từ mô phân sinh đỉnh được phân lập từ hạt chín (McCabe và cs 1988). Trong thí nghiệm này, marker sàng lọc là gene gusA đã được biến nạp bằng dội bom và cây chuyển gene được phục hồi sau khi sàng lọc nhờ hoạt tính của -glucoronidase (GUS). Các thành công sớm khác có thể kể là ở bông, đu đủ, ngô và thuốc lá (Finer và McMullen 1990, Fitch và cs 1990, Fromm và cs 1990, Kamm và cs 1990, Tomes và cs 1990).
So với biến nạp vào protoplast, chuyển gene bằng phương pháp bắn gene đơn giản hơn và ít phụ thuộc vào kiểu gene. Nó đã trở thành phương pháp được sử dụng rộng rãi để biến nạp gene vào những loài thực vật một lá mầm.

161
Hình 3.11. Các thành phần của hệ thống dội bom Biolistic®PDS-1000 Hiện nay có nhiều kiểu súng bắn gene được chế tạo ngày càng hiện đại và hiệu
quả hơn nhờ sử dụng luồng khí helium áp lực cao. Có thể không dùng viên đạn lớn mà dùng bộ phận nén khí mạnh tạo áp lực đẩy vi đạn như súng bắn gene.
Thiết bị được sử dụng rộng rãi nhất cho biến nạp gene vào thực vật là hệ thống Biolistic PDS-100 của Biorad (Hình 3.11).
Trong hệ thống này, dòng khí helium áp lực cao được giải phóng qua một nắp đậy, đẩy giá gắn viên đạn lớn (macrocarrier) mang hàng triệu viên đạn nhỏ (microcarrier) bằng kim loại gắn DNA (Hình 3.12). Bộ phận sàng làm dừng sẽ giữ viên đạn lớn lại, các viên đạn nhỏ tiếp tục phóng về hướng tế bào đích và xâm nhập vào tế bào

162
Hình 3.12. Quá trình dội bom sinh học
Vì bản chất vật lý và tính đơn giản của phương pháp, quá trình chuyển gene bằng dội bom được sử dụng để bắn các vật liệu vào nhiều tế bào và mô của các cơ thể khác nhau. Ở thực vật, phương pháp này được ứng dụng đề nghiên cứu biểu hiện của gene được chuyển, sản xuất các thực vật biến đổi di truyền, ủ thực vật với mầm bệnh virus… Những năm gần đây nhiều tác giả đã sử dụng súng bắn gene để chuyển vào callus các plasmid mang các gene chống rầy, chống nấm và đã thu được nhiều dòng lúa chuyển gene có các đặc tính tốt.
4. Chuyển nạp DNA ngoại lai vào tế bào bằng kỹ thuật vi tiêm (microinjection) Vi tiêm là kỹ thuật sử dụng phổ biến trong công nghệ tế bào động vật (animal cell biotechnology). Ở thực vật, tiêm DNA vào thực vật cũng có thể là phương pháp trực tiếp để tạo ra cây chuyển gene. Trên hiển vi trường, DNA plasmid có thể được tiêm vào protoplast và thực hiện biến nạp gene thành công ở khá nhiều đối tượng thực vật (Hình 3.13). Tuy nhiên, kỹ thuật này hiện nay ít được các phòng thí nghiệm sử dụng, vì không hiệu quả, thao tác vi tiêm dưới kính hiển vi đòi hỏi thiết bị vi thao tác (micromanipulator) cực nhạy, thiết bị kéo và mài kim tiêm từ các ống thủy tinh (puller) rất đắt tiền. Ngoài ra, nó còn đòi hỏi kỹ năng thao tác và sự kiên nhẫn cao của kỹ thuật viên.

163
Hình 3.13. Chuyển nạp DNA ngoại lai vào tế bào bằng kỹ thuật vi tiêm

164
Bài 4 ỨNG DỤNG CỦA CÔNG NGHỆ CHUYỂN GEN VÀO TẾ BÀO THỰC VẬT 1. Chuyển gene kháng thuốc diệt cỏ
Tính đề kháng thuốc diệt cỏ, côn trùng và bệnh có sự di truyền đơn gene. Đa phần các thực vật chuyển gene được nhận các gene đề kháng thuốc diệt cỏ (56%). Cho đến cuối năm 1992, có 489 trường hợp được thử nghiệm ra đồng ruộng, gồm các cây cải dầu, bắp, củ cải đường và đậu nành. Điều đặc biệt là kỹ thuật di truyền đã chuyển các gene đề kháng căn cứ vào phương thức tác động của các chất diệt cỏ chứ không phải chỉ căn cứ vào kiểu hình như trước đây. Kỹ thuật di truyền cho phép can thiệp vào trao đổi chất của thực vật tạo nên tính chất mới cho giống.
Trong số các thực vật thử nghiệm có 78% đề kháng glyphosate hoặc glufosinate.
Glyphosate là thành phần có hoạt tính trong thuốc diệt cỏ được bán rộng rãi nhất trên thế giới. Glyphosate ức chế sự tạo thành amino acid thơm ở cả thực vật và vi khuẩn. Hai phương pháp được sử dụng để thao tác với tính kháng để khi sử dụng chất diệt cỏ trong điều khiển cỏ dại mà không gây hại đến cây trồng. Thứ nhất là các protein mục tiêu của chất diệt cỏ - EPSPase (5-enolpyruvylshikinic acid-3- phosphate synthetase) được sản xuất thừa để sự kháng xảy ra như là hệ quả của việc có nhiều enzyme có sẵn đối với tế bào. Phương pháp thứ hai kết quả từ sự biểu hiện của thể đột biển EPSPase, mà nó là kháng được với chất diệt cỏ bên trong tế bào. Cây trồng kháng glyphosate chẳng hạn như cây “Roundup Ready” của công ty Monsanto có sản lượng tăng do kết quả của sự loại trừ cỏ dại. Tuy nhiên, các ảnh hưởng bất lợi của sự sử dụng chất diệt cỏ tăng lên và tiềm năng của sự chuyển gene kháng chất diệt cỏ sang những loài thực vật khác vẫn còn những liên quan chưa được biết đến.
Gene đích của glyphosate có mặt trong vi khuẩn Salmonella typhimurium. Dạng kháng của gene thu được nhờ các đột biến Salmonella sinh trưởng trong môi trường có mặt glyphosate. Gene sau đó được tạo dòng trong E. coli và được tạo dòng lại trong T-DNA của Agrobacterium. Các cây trồng được biến nạp gene kháng glyphosate như ngô, bông, thuốc lá... đã kháng được với các chất diệt cỏ. Ở ruộng các cây trồng này, có thể phun glyphosate ở bất kỳ giai đoạn sinh trưởng nào của cây trồng, cỏ dại sẽ bị tiêu diệt nhưng không ảnh hưởng đến cây trồng.
2. Chuyển gene kháng sâu bệnh
Số lượng các thử nghiệm xếp thứ hai là các thực vật kháng bệnh và côn trùng: - Kháng virus (15%): sự đề kháng thông qua protein vỏ virus được thử nghiệm
ở các cây: khoai tây, cà chua, củ cải đường, thuốc lá và một số cây khác. - Kháng bệnh vi khuẩn (4%): chủ yếu thực hiện ở khoai tây. - Kháng côn trùng (10%): được thực hiện nhờ gene tạo -endotoxin từ vi
khuẩn Bacillus thuringiensis. Gene này được chuyển vào các cây trồng: khoai tây, bông vải, bắp, cà chua và thuốc lá.

165
Cây trồng được thao tác di truyền để biểu hiện gene độc tố trừ sâu (insecticidal toxin gene) của Bacillus thuringiensis để khi côn trùng tấn công và ăn những cây này sẽ bị giết chết. Bacillus thuringiensis là vi khuẩn tạo bào tử Gram dương, chúng tổng hợp một tinh thể lớn trong tế bào chất chứa độc tố trừ sâu. Các chủng vi khuẩn khác nhau tạo ra các độc tố có ảnh hưởng chống những loài sâu khác nhau. Các tinh thể được gắn kết thành protein có trọng lượng phân tử khoảng 130 kDa. Chúng được tạo ra như là một tiền độc tố, phải được hoạt hóa trước khi có ảnh hưởng. Protein tinh thể là không hòa tan vì vậy nó tương đối an toàn với người, động vật bậc cao và hầu hết côn trùng. Tuy nhiên, protein được hoà tan trong điều kiện pH cao (pH>9,5), đây là điều kiện được tìm thấy phổ biến ở ruột giữa (mid-gut) của ấu trùng sâu bọ cánh vảy (Lepidopteran larvae) cả sâu bướm (moth) và bướm (butterfly). Một khi nó được hoà tan ở trong ruột của côn trùng, protoxin sẽ bị cắt bởi protease của ruột tạo ra toxin có hoạt tính, được gọi là -endotoxin, có trọng lượng khoảng 60 kDa, sẽ gắn vào tế bào biểu mô (epithelial cell) của ruột giữa, tạo ra các lỗ trên màng tế bào và dẫn đến sự cân bằng của các ion. Kết quả, ruột sẽ nhanh chóng bị giữ cố định, có sự dung giải tế bào biểu mô, ấu trùng ngừng ăn và pH của ruột bị giảm xuống nhờ sự cân bằng với pH của máu. pH thấp hơn này sẽ làm cho bào tử của vi khuẩn nẩy mầm và vi khuẩn tràn vào tế bào chủ, gây chết do nhiễm trùng máu. Nhiều loại cây trồng được thao tác di truyền chứa bản sao của gene cry1Ac của vi khuẩn Bacillus thuringiensis mã hóa cho protoxin. Ngoài ra, gene được biểu hiện ở mức độ cao ở lục lạp của cà chua, kết quả là cây này kháng được một loạt côn trùng gây hại. Phương pháp này đã thu được thành công lớn. nhưng có bất lợi là sự tiếp xúc liên tục của côn trùng với độc tố sẽ chọn lọc cho sự phát triển của tính kháng độc tố.
3. Chuyển gene gây chín chậm- DR (delayed ripening )
Hình 3.14. Quả cà chua chín chậm

166
Chín là một giai đoạn bình thường trong quá trình trưởng thành của rau và quả. Quá trình chỉ xảy ra mạnh mẽ vài ngày trước khi rau và quả có thể ăn được. Sự chín không thể tránh khỏi này gây thiệt hại không nhỏ cho cả nông dân và người tiêu dùng.
Các nhà khoa học đã nghiên cứu để tìm ra phương pháp làm chậm quá trình chín của quả giúp nông dân có thể chủ động tiêu thụ nông sản và đảm bảo cho người tiêu dùng có thể sử dụng những sản phẩm còn tươi.
Sự chín là một phần quan trọng của sản xuất trái cây thương mại. Trái cây chưa chín hoặc quá chín đều kém hấp dẫn với người tiêu dùng. Sự vận chuyển của nhiều loại quả chín mềm hoàn toàn có thể dẫn đến kết quả là sự hư hỏng của chúng. Điều này liên quan đặc biệt với sự vận chuyển các loại quả như cà chua, là loại quả mà có bất kỳ sự hư hại nào có thể làm cho không thể bán được quả. Các sản phẩm protein của một số gene điều khiển quá trình chín chậm đã được nghiên cứu. Một trong số chúng mã hóa cho enzyme polygalacturonase, có liên quan với sự làm hỏng chậm thành phần acid polygalacturonic của thành tế bào trong vỏ quả. Ảnh hưởng của nó gây mềm dần quả, làm cho quả có thể ăn được. Tuy nhiên, kéo dài hơn ảnh hưởng của enzyme có thể gây ảnh hưởng lên thành tế bào, làm mềm quả và quả sẽ trở nên quá chín. Vì vậy nếu các ảnh hưởng của enzyme có thể bị làm dừng thì quả sẽ chín chậm hơn và kết quả cà chua có thể ở trên cây lâu hơn để tích luỹ mùi vị nhiều hơn. Cà chua là đối tượng đã được sử dụng trong kỹ thuật gene để biểu hiện enzyme polygalacturonase kém. Điều này có thể đạt được qua việc chèn vào một trình tự antisense với vùng 5’ của gene polygalacturonase vào bộ gene của cà chua. Sự biểu hiện của trình tự antisense nhờ promoter 35S của virus khảm cây cải hoa (cauliflower mosaic virus) và toàn bộ cấu trúc này đã được đưa vào tế bào cà chua nhờ sử dụng Agrobacterium. Cây cà chua chuyển gene được biểu hiện giảm mức độ hoạt động của gene polygalacturonase (giảm 6%) so với đối chứng hoang dại của chúng. Nhờ vậy quả được bảo quản trong thời gian dài hơn trước khi bắt đầu hư hỏng.
Antisense RNA cũng có thể được sử dụng để làm giảm sự biểu hiện của một vài gene được yêu cầu cho sinh tổng hợp ethylene, kết quả cũng làm chậm sự chín của quả.
Ethylene là một hormone thực vật tự nhiên liên quan đến quá trình sinh trưởng, phát triển, chín và sự lão hóa của thực vật. Người ta cho rằng phytohormone này thúc đẩy quá trình chín của rất nhiều loài quả như: chuối, dứa, cà chua, xoài, dưa hấu và đu đủ. Nó được tạo ra ở nhiều nồng độ khác nhau phụ thuộc vào nhiều loại quả. Nhưng khi nồng độ ethylene đạt từ 0,1 đến 1,0 ppm (phần triệu) thì bắt đầu xảy ra quá trình chín của quả vùng nhiệt đới. Quả vùng nhiệt đới thường được thu hoạch một lần khi chúng đạt được độ trưởng thành nhất định, và sau đó chúng chín rất nhanh trong khi vận chuyển và bảo quản. Các loại quả nhiệt đới quan trọng như: chuối, xoài, dứa, đu đủ và ổi là những ví dụ. Các loại quả không thuộc vùng nhiệt đới thì không chín ngay sau khi thu hoạch. Do đó, để đạt được độ chín và tăng hương vị của quả, thì những quả như: dâu tây và cam thường được thu hoạch một lần sau khi chúng đã chín hoàn toàn.
Ở cà chua, mất khoảng 45 đến 55 ngày để quả trưởng thành hoàn toàn, sau đó nó bắt đầu quá trình chín. Sản phẩm ethylene trong quả là tín hiệu cho hoạt động

167
của nhiều loại enzyme khác nhau dẫn đến những thay đổi sinh lý như: quả thay đổi màu sắc từ xanh sang đỏ, mềm và có mùi vị khác nhau.
Thông thường nông dân thu hoạch khi quả còn xanh, sau khi vận chuyển đến
những nơi cần thiết, quả được làm chín bằng phương pháp phun khí ethylene. Đối với những chuyến hàng vận chuyển dài ngày, quả được làm lạnh để giảm thiệt hại và làm chậm quá trình chín.Tuy nhiên, trên thực tế, có một vài trở ngại sau khi thu hoạch. Quả được thu hoạch trước khi chín có thể giảm mùi vị và chất lượng và quả được vận chuyển trong thời gian dài dưới điều kiện bảo quản lạnh cũng có khuynh hướng giảm chất lượng.
Vì vậy, các nhà khoa học có thể sử dụng một vài phương pháp để điều khiển quá trình chín chậm bằng công nghệ biến đổi gene, điều khiển sự tổng hợp ethylene.
Lượng ethylene tạo ra có thể được điều khiển bằng cách “đóng” hoặc làm giảm sự tạo ethylene trong quả, theo một số cách sau đây:
+ Ức chế sự biểu hiện của gene ACC (1-aminocyclopropane-1-carbonxylic acid) synthase. ACC synthase là enzyme chịu trách nhiệm chuyển hóa S-adenosylmethiomine (SAM) thành ACC; từ bước thứ hai tới bước cuối cùng trong quá trình sinh tổng hợp ethylene. Sự biểu hiện của enzyme bị cản trở khi một antisense hoặc một đoạn của bản sao gene synthase được chuyển vào trong genome của thực vật.
+ Chuyển gene ACC diaminase. Gene mã hóa cho enzyme này nhận được từ một vi khuẩn đất (Pseudomonas chlororaphis) không gây bệnh. Vi khuẩn này có khả năng chuyển hóa ACC thành một phân tử khác, nhờ vậy làm giảm lượng ACC có thể nhận được để tạo ethylene.
+ Chuyển gene SAM hydrolase. Phương pháp này cũng tương tự như ACC diaminase, bằng cách giảm tiền chất của ethylene. Trong trường hợp này, SAM được chuyển hóa thành homoserine. Gene mã hóa cho enzyme này được phân lập từ thể thực khuẩn E. coli T3.
+ Ức chế sự biểu hiện của gene ACC oxidase. ACC oxidase là enzyme xúc tác cho sự oxi hóa ACC thành ethylene, bước cuối cùng trong con đường sinh tổng hợp ethylene. Thông qua công nghệ antisense, giảm sự điều khiển gene ACC oxidase dẫn đến ức chế sự hình thành ethylene, do đó làm chậm sự chín của quả.
- Điều khiển việc nhận ethylene: Khi ethylene có tín hiệu bắt đầu cho việc chín quả, việc làm chậm quá trình chín có thể đạt được bằng cách biến đổi thụ thể

168
của ethylene. Gene ETR1 là một ví dụ, nó mã hóa cho protein liên kết ethylene. Ở thực vật ETR1 được biến đổi thì mất khả năng phản ứng với ethylene.
- Ức chế hoạt tính của polygalacturonase: Enzyme polygalatuonae (PG) chịu trách nhiệm cho việc phân giải pectin, chất duy trì độ cứng thành tế bào thực vật. Việc phân giải pectin xảy ra lúc bắt đầu quá trình chín, làm cho mềm quả. Để tạo ra quả DR bằng phương pháp này, các nhà khoa học đã chuyển gene antisene hoặc một đoạn bản sao của gene PG vào trong geome của thực vật dẫn đến sự ức chế tạo ra enzyme PG.
Cây trồng biến đổi gene lần đầu tiên được chấp nhận trên thị trường là cây cà chua Flavr-Savr TM được tạo ra bởi công ty Calgene, Hoa Kỳ vào năm 1994. Sau khi tiến hành nghiên cứu công nghệ DR và các sản phẩm của nó, các cơ quan quản lý của Hoa Kỳ đã kết luận rằng công nghệ DR là an toàn, cà chua rạo ra nhờ công nghệ này có thành phần dinh dưỡng giống như cà chua thông thường và không có sự sai khác về mức độ dị ứng cũng như độc tố so với quả bình thường. Ngoài ra, các thử nghiệm đồng ruộng cho thấy cà chua DR không gây ra bất kỳ một sự đe dọa nào tới những thực vật và những sinh vật có ích khác.
Sau đó, những cây cà chua DR khác cũng được chấp nhận ở Hoa Kỳ, Canada và Mexico. Năm 1996, các nhà quản lý an toàn thực phẩm ở Anh đã đưa ra lời tuyên bố đồng ý cho triển khai cà chua DR của công ty Zeneca Seeds, nhưng hiện nay sản phẩm này không được bán trên thị trường.
Những lợi ích của công nghệ DR: + Thời hạn sử dụng sản phẩm tăng lên mang lại lợi ích cho cả người sản xuất
và người tiêu dùng. + Bảo đảm về chất lượng rau quả trên thị trường. Nông dân có thể chờ rau quả
trưởng thành đầy đủ mới thu hoạch. Người tiêu dùng có thể mua được hàng hóa tốt với giá tiền phù hợp.
+ Nông dân yên tâm khi vận chuyển sản phẩm của mình trong một khoảng thời gian dài mà không cần phải bảo quản lạnh.
+ Giảm thiệt hại sau thu hoạch. Quả DR có độ cứng cao hơn quả thông thường do đó nó không bị dập trong quá trình vận chuyển và thời gian lưu thông trên thị trường lâu hơn.
+ Kéo dài thời hạn sử dụng nhưng rau quả vẫn tươi ngon.
4. Chuyển gene tạo hoa có màu sắc và kiểu hoa mới Hoa hồng xanh có thể được coi là chén thánh (Holy Grail) của những nhà lai
tạo hoa hồng kể từ năm 1840. Khi đó hiệp hội làm vườn của Anh và Bỉ đã treo giải thưởng 500.000 francs cho người đầu tiên tạo được hoa hồng màu xanh.
Các nhà di truyền học phân tử của công ty Florigene và Suntory đã đoạt được giải thưởng này. Yếu tố tạo ra màu xanh trên hoa hồng chính là gene delphinidin đã được các nhà di truyền học của công ty Florigene tạo dòng từ loài hoa păng-xê (Viola x wittrockiana) để tổng hợp trực tiếp màu xanh hoa hồng. Yếu tố vay mượn chính là gene iris nhằm tạo ra enzyme dihydroflavonol reductase (DFR), enzyme này sẽ hoàn thành chu trình phản ứng tổng hợp delphinidin trên hoa hồng. Yếu tố mới chính là một gene nhân tạo. Gene này được tạo ra bởi nhóm các nhà di truyền

169
học của công ty Suntory bằng một kỹ thuật mới là RNA interference, viết tắt là RNAi. Kỹ thuật này được tư vấn bởi viện CSIRO nhằm mục đích tắt sự hoạt động của gene hình thành màu đỏ trong hoa hồng. Chính gene này đã đánh bại những nổ lực của nhóm nghiên cứu Florigene nhằm làm hoạt hóa chu trình delphinidin trong hoa hồng gần cả một thập kỷ nay. Chính vì thế mà các nhà khoa học của Suntory đã tạo ra một gene “silence” để vượt qua sự khó khăn này bằng kỹ thuật RNAi. Kỹ thuật RNAi là một hướng rất mới trong nghiên cứu y sinh khoảng 25 năm trở lại đây.
Đội ngũ nghiên cứu của Peter Waterhouse (Viện CSIRO, Canberra, Úc) đã đi tiên phong trong việc sử dụng kỹ thuật RNAi cho việc khám phá và ứng dụng các chức năng của gene trong cây trồng. Mặc dù lúc đầu hoa hồng có màu hoa cà nhạt nhưng nó là hoa hồng đầu tiên trên thế giới có khả năng di truyền tạo ra những hoa hồng xanh thực sự làm cầu nối cho việc hình thành phổ màu trên hoa hồng từ màu xanh tái chuyển sang màu xanh vùng Địa Trung Hải hay thậm chí là màu xanh nước biển. Như vậy có thể nói rằng đây là loài hoa mang tình thương mại đầu tiên trên thế giới được hình thành bằng kỹ thuật RNAi. Hoa hồng xanh của Florigene là sự báo trước một tương lai tươi sáng cho các nhà nhân giống cây trồng trong thế kỷ 21.
Phương pháp tạo ra hoa hồng xanh: Trong cây trồng, anthocyanin được coi là sắc tố chủ đạo trên hoa, trái và các
mô tế bào khác. Thông thường các màu chính của hoa bắt nguồn từ anthocyanin với sự có mặt của một ít các chất carotenoid màu vàng. Ngoài ra, anthocyanin dihydrokaempferol (DHK) lại là một enzyme chi phối cho cả 3 chu trình hình thành sắc tố trên cây trồng bao gồm: cyanidin, pelargonidin và delphinidin. Gene cyanidin mã hóa một enzyme làm thay đổi enzyme DHK nhằm hình thành chu trình cyanidin dẫn đến biểu hiện các màu đỏ, hồng hay màu tím hoa cà. Trong khi đó gene delphinidin không hiện diện trong cây hoa hồng sẽ mã hóa một enzyme khá tương đồng cho việc thay đổi enzyme DHK nhằm hình thành sự tổng hợp màu theo chu trình delphinidin. Enzyme khác là dihydroflavinol reductase (DFR) sẽ hỗ trợ các màu chỉ chị trong cả ba chu trình trên (hình 3.15). Enzyme này rất quan trọng vì không có nó sẽ không thể tạo màu trên các cánh hoa. Chính vì vậy mà các đột biến gene DFR đều cho ra những hoa có màu trắng. Trong hoa hồng không có gene delphinidin để hình thành màu theo chu trình của nó. Chu trình delphinidin có thể hình thành màu đỏ hoặc xanh trên hoa dưới sự tác động của DRF và pH.
170
Hình 3.15. Sơ đồ chu trình tổng hợp anthocyanin Sơ đồ chỉ ra vai trò của dihydrokaempferol và ba nhánh chu trình hình thành
nên các màu khác nhau. Khung màu đỏ là chu trình delphinidin góp phần hình thành màu xanh trên hoa hồng.
Trong suốt thế kỷ 20, các nhà lai tạo hoa hồng đã tạo ra một loạt màu hoa lạ như hoa hồng màu lilac hay hoa hồng xám được coi là bước đệm để tạo ra hoa hồng xanh. Tuy nhiên chúng là những biến thể hiếm từ chu trình cyanidin. Bơi vậy chúng ta có thể hiểu rằng tại sao việc lai tạo truyền thống không thể tạo ra hoa hồng xanh như mong muốn bởi vì hoa hồng về mặt di truyền không có gene để tạo ra chu trình delphinidin. Chính vì thế các nhà khoa học Florigene đã đi một bước rất dài bằng việc tạo dòng gene delphinidin từ loài hoa dã yến thảo vào năm 1991. Vào khoảng giữa thập niên 1990 các nhà khoa học đã có những kỹ thuật hoàn hảo cho việc lai tạo hoa hồng và tạo hoa từ các dòng tế bào nuôi cấy mô. Cũng trong khoảng thời này, công ty Florigene đã có giống hoa hồng đỏ thẩm đầu tiên được tạo ra từ gene delphinidin có tên gọi là “Cardinal”. Việc kết hợp gene cyanidin và gene
delphinidin 3-glucoside pelargonidin 3-glucoside cyanidin 3-glucoside
dihydroquercetin dihydrokaempherol dihydromyricetin
naringenin
tetrahydroxychalcone

171
delphinidin đã tạo ra một giống hoa hồng màu đỏ tía rất ấn tượng. Dĩ nhiên nó không phải màu xanh nhưng về mặt kỹ thuật đó là một bước tiến rất lớn.
Chính vì thế để tạo ra một bông hồng màu xanh, các nhà nghiên cứu Florigene cần một loại bông hồng trắng trong đó gene DFR đã bị bất hoạt. Vào năm 2001, Waterhouse đã thảo luận việc sử dụng kỹ thuật RNAi nhằm ức chế một gene mong muốn để sau đó có thể thay thế bằng một gene khác. Do đó, các nhà khoa học của công ty Florigene ngay lập tức nhận ra được lợi ích của việc dùng kỹ thuật RNAi nhằm ức chế hoạt động của gene DFR trong hoa hồng đỏ dẫn đến ức chế chu trình cyanidin và sau đó chuyển gene delphinidin với một gene DFR hoàn toàn mới nhằm hoàn chỉnh chu trình tổng hợp delphinidin trong hoa hồng. Cùng lúc đó các nhà nghiên cứu của công ty Suntory (Nhật Bản) cũng có cùng ý tưởng bằng cách dùng kỹ thuật RNAi để ức chế gene DFR sau đó họ tạo dòng một gene delphinidin mới từ loài hoa păng-xê (pansy) và gene DFR từ hoa iris. Các gene DFR của hoa hồng và iris khá tương tự nhau và chia sẽ nhiều đoạn mã DNA nhưng kỹ thuật RNAi cũng rất tinh tế bởi vì nó có thể ức chế gene DFR của hoa hồng mà không ảnh hưởng đến gene DFR của hoa iris bằng việc tạo ra một cấu trúc ức chế gene có tác dụng tạo ra các phân tử RNA mạch kép (dsRNA) có vòng kẹp tóc (hairpin dsRNA) với trình tự tương đồng với gene DFR của hoa hồng.
Để tạo ra hoa hồng xanh, các nhà khoa học của Suntory đã sử dụng một bộ 3 gene. Một gene nhân tạo được dùng cho kỹ thuật RNAi nhằm ức chế gene DFR của hoa hồng làm cho hoa hồng không biểu hiện màu. Sau đó chuyển gene delphinidin từ loài hoa păng-xê và gene DFR từ loài hoa iris sẽ tạo ra hoa hồng có hàm lượng delphinidin rất cao trong cánh hoa. Tuy nhiên cũng phải lưu ý một yếu tố ảnh hưởng đến màu xanh trên cánh hoa đó chính là độ pH tế bào và đó là một trong những lý do chính là tại sao các loài hoa có cùng chu trình anthocyanin nhưng lại có màu khác nhau. Khi nồng độ pH tế bào mang tính kiềm thì sắc tố của anthocyanin thường trở nên xanh hơn. pH của đất không ảnh hưởng hay ảnh hưởng rất ít đến pH tế bào cánh hoa. Nồng độ pH tế bào cánh hoa thường mang tính di truyền. Cánh hoa hồng thông thường có nồng độ pH khoảng 4,5. Chính vì vậy để tạo ra các cánh hoa hồng có nồng độ pH thấp thì rất hạn chế. Các nhà khoa học sử dụng biện pháp ức chế gene bằng kỹ thuật RNAi nhằm xác định những gene ảnh hưởng đến tính acid của cánh hoa hay điều chỉnh màu của cánh hoa theo những hướng khác.
Hoa hồng xanh là một trong những sản phẩm được tạo ra từ việc ứng dụng kỹ thuật RNAi. Đây là một trong hàng loạt ứng dụng của RNAi trong nghiên cứu y sinh và là công cụ rất hữu ích cho việc tìm hiểu và khám phá các chức năng bí ẩn của các gene trong thời đại nghiên cứu hậu genome (post-genomic era).
Ngoài ra, kỹ thuật di truyền còn được ứng dụng trong tạo ra hoa có kiểu dáng mới. Gene pttknI được phân lập từ vùng thượng tầng của cây dương lai (hybrid aspen) đã được đưa vào Petunia hybrida Vilm sử dụng phương pháp biến nạp đĩa lá qua trung gian Agrobacterium. Hàng loạt kiểu hình đã được quan sát thấy ở cây Petunia chuyển gene, gồm sự hình thành cụm hoa lệch vị trí trên mặt bên trục của tràng hoa và cánh hoa nhỏ trên mặt xa trục của tràng hoa, rút ngắn gân giữa của tràng hoa, tạo thành các nốt giống khối u dọc theo gân giữa trên mặt xa trục và các thùy của mép tràng hoa bị phân chia và có sự biến đổi màu của cánh hoa.

172
5. Chuyển gene sản xuất protein động vật và người Trong số các ứng dụng quan trọng nhất của kỹ thuật di truyền là tạo ra một
lượng lớn các protein đặc biệt mà khó có thể thu được bằng các phương pháp khác. Chẳng hạn, các protein có mặt chỉ vài phân tử trong tế bào hoặc các protein chỉ được tạo ra ở một số ít tế bào hoặc chỉ được tạo ra ở tế bào người. Về nguyên tắc, có một trình tự DNA mã hóa cho một protein mong muốn được tạo dòng trong vector kề bên trình tự điều hoà. Phương pháp này thường được sử dụng với cDNA vì cDNA có chứa tất cả trình tự mã hóa nối nhau theo trật tự đúng.
Hiện nay, thực vật được xem là nguồn sản xuất các protein có giá trị như là các thuốc hay vaccine phòng bệnh cho người và động vật.
Thực vật sản xuất vaccine đã thu được kết quả đầu tiên ở khoai tây, tiếp theo là ở chuối và gần đây là ở cà chua. Người ăn chuối có vaccine sẽ khỏi phải tiêm vào cơ thể và một hướng phát triển mới hình thành là ăn để trị bệnh, còn gọi là dinh dưỡng dược (neutriceutical). Ví dụ gene mã hóa kháng nguyên bề mặt virus viêm gan B được chuyển vào cây chuối và cây khoai tây. Gene mã hóa các thành phần protein của độc tố dịch tả đã được đưa vào cây và bước đầu đã thu được sản phẩm như là một vaccine dịch tả. Các nhà khoa học cũng đã tạo ra được giống khoai tây chứa kháng nguyên vô hại từ E. coli là loại vi khuẩn gây bệnh tiêu chảy, ăn loại khoai tây này sẽ có khả năng miễn dịch. Theo cách này, tương lai các loại vaccine khác sẽ được đưa vào thức ăn cho người cũng như thức ăn gia súc.
Các nhà nghiên cứu đã tạo ra các thực vật chuyển gene sản xuất các protein trị liệu: cây cải dầu sản xuất enkephalin làm chất giảm đau, khoai tây tạo ra serum albumin để tăng máu. Vào năm 1996, các gene của globin máu đã được đưa vào cây thuốc lá. Các gene immunoglobin được biểu hiện ở thực vật có thể đạt 1% protein hoà tan của lá. Gần đây đã tạo được giống lúa biểu hiện các protein người là lysozyme và lactoferrin, hứa hẹn có thể sản xuất hàng tấn. Các thực vật có thể sẽ cung cấp nhiều protein quan trọng cho việc chữa trị các bệnh, kể cả máu.
Quá trình chuyển gene lipase acid vào cây cải dầu để tạo ra thuốc chữa bệnh nhầy nhớt và suyển nặng (mucavisoidose) mà trước đây không có cách nào cứu chữa. Ở những phòng thí nghiệm miền Đông nước Pháp của bang Biocem đã tạo ra những cây cải dầu và cây thuốc lá mang gene động vật đó là gene lipase acid của chó. Enzyme này có tác dụng làm cho trẻ con mắc chứng nhầy nhớt hen suyển có thể tiêu hóa chất béo dễ dàng. Tuy nhiên, không thể giết nhiều chó để đáp ứng nhu cầu chữa bệnh cho bệnh nhân, nên người ta đã nghĩ ra cách ghép gene này cho cây cải dầu để nó sản sinh ra protein enzyme lipase sử dụng cho điều trị. Sử dụng thử nghiệm ghép gene vào thực vật có nhiều ưu điểm so với sử dụng E. coli, do vi khuẩn không đủ enzyme để glucose hóa các protein có gốc glucosyl mà chỉ có ở động vật hoặc thực vật. Còn nếu chiết suất protein từ động vật thì lại hay gặp protein lạ, giá thành cao và phải có thiết bị phức tạp, ngoài ra phải chăm sóc động vật công phu và không an toàn. Trường hợp sử dụng thực vật sẽ giúp hạ giá thành và an toàn tối đa. Với thực vật, không có vấn đề bị nhiễm virus độc như các chất ly trích từ máu hay rau thai. Một số virus tần công thực vật thì vô hại đối với người và động vật.

173
6. Chuyển gene thay đổi protein Chuyển gene làm thay đổi protein là một bước phát triển lớn của thực vật
chuyển gene và đã thành công trong biến đổi hàng loạt tính chất như tăng amino acid có giá trị (lysine, methionine, cystein), tăng lượng vitamin, độ ngọt,...
Cây trồng là nguồn chủ yếu cung cấp protein cho người và vật nuôi. Các hạt, đặc biệt là ngũ cốc, rau đậu chứa một lượng lớn protein và là nguồn chủ yếu của protein ăn kiêng. So với thịt, protein hạt kinh tế hơn nhiều trong sản xuất, dự trữ và vận chuyển, nhưng ở protein hạt lại thiếu một vài amino acid cần thiết. Protein ngũ cốc có lysine và tryptophan thấp, protein của đậu hạt thiếu amino acid có sunfur như methionine và cystein. Nhìn chung các protein hạt thường không đầy đủ về dinh dưỡng. Các nhà di truyền học và các nhà trồng trọt trong thời gian qua đã có nhiều cố gằng để cải tiến chất lượng protein hạt. Những biến dị tạo hạt lúa mạch có lysine cao đã được xác định và phát triển, tuy nhiên chúng có năng suất thấp và nhạy cảm hơn với sâu hại và bệnh. Với những phát triển hiện nay trong công nghệ DNA tái tổ hợp có thể thay đổi thành phần amino acid của các protein hạt nhằm cải tiến chất lượng dinh dưỡng.
Mặc dù gạo được một nữa dân số thế giới ăn hàng ngày, nhưng nó lại thiếu một số chất dinh dưỡng và vitamine A. Khoảng 124 triệu trẻ em trên thế giới bị thiếu vitamine A và hậu quả là 1-2 triệu trẻ em chết hàng năm. Bằng biến nạp nhờ Agrobacterium, con người đã chuyển gene quy định toàn bộ chuỗi sinh tổng hợp beta-carotene (tiến chất vitamine A) vào lúa. Giống lúa này cho hạt gạo giàu beta-carotene có màu vàng, được gọi là lúa vàng (golden rice).
Giống lúa đã được chuyển 3 gene: gene mã hóa cho ferrilin (protein trữ sắt hàm lượng cao) từ đậu cô ve (Phaseolus vulgaris), gene mã hóa phytase và gene mã hóa metallothionein từ nấm Aspergillus fumigatus làm tăng hấp thu sắt và cho hạt gạo giàu sắt dễ hấp thụ.
Monellin là protein có trong một loại trái cây ở châu Phi, nó ngọt gấp 3.000 lần đường ăn khi so cùng khối lượng. Gene mã hóa monellin được biến đổi đã được chuyển thành công vào cà chua và rau xà lách.
Trong thời kỳ thiếu lương thực, thức ăn của động vật nhai lại trên đồng cỏ được cải thiện với các hạt rau quả có protein cao, tuy nhiên lại nghèo nguồn methionin. Các nổ lực được thực hiện để tạo ra loại rau quả giàu methionin nhờ sử dụng công nghệ biến đổi gene. Các nhà nghiên cứu đã xác định được protein ở hạt hướng dương có hàm lượng amino acid chứa sulfur cao khác thường. Một đặc tính nữa của protein này là chống lại sự bẻ gãy của hệ vi sinh vật trong dạ cỏ của động vật. Vì vậy nó đi qua phía dưới và vào ruột, nới có các enzyme tiêu hóa thuỷ phân nó thành các amino acid thành phần, bao gồm methionine và động vật sử dụng để tổng hợp protein. Các nhà nghiên cứu người Úc, đã chuyển gene mã hóa cho protein này vào cỏ lupin (một loại legume) và cho biểu hiện ở hạt. Kết quả làm tăng 100% hàm lượng methionine ở protein của hạt và khi sử dụng những hạt này làm thức ăn cho cừu, thì trọng lượng của chúng tăng 7% và sản lượng lông tăng 8% so với những cừu nuôi bằng hạt không biến đổi. Thành công này đã thúc đẩy các nghiên cứu chuyển gene tổng hợp protein hạt hướng dương có methionine cao vào lá các loại cỏ với mục đích cải thiện sự cân bằng các amino acid thiết yếu cho động vật ăn cỏ.

174
Một số nghiên cứu chuyển gene cải thiện chất lượng protein cũng đã được thực hiện. Ngô là nguồn thức ăn quan trọng cho người và động vật, nhưng nó chứa lượng lysin và tryptophan thấp. Đó là do zein là một protein dự trữ hạt của ngô, thiếu lysine và tryptophan. Wallace và cộng sự (1988) đã tạo ra các codon lysine và tryptophan ở những vị trí khác nhau trong đoạn mã hóa -zein 19 kDa bởi thay thế nucleotide và gài thêm oligonucleotide vào khung đọc mở 50 bp của protein vỏ của virus SV40 đã được gài vào đoạn zein như là đoạn thay đổi kiểm soát. Kết quả đã có thêm những gốc lysine đơn hoặc đa, những oligopeptid giàu tryptophan và lysine ngắn không làm ảnh hưởng đến sự dịch mã và tính bền của zein.
Ngoài ra, kỹ thuật di truyền còn có thể cải thiện thành phần protein của thực vật hay loại bỏ các độc tố của thực phẩm.
Việc nghiên cứu chuyển gene để cải tiến cả chất và lượng của các protein dự trữ trong hạt đã mở ra một số kết quả ban đầu và hy vọng trong tương lai cùng với các loại chuyển gene khác sẽ tạo ra vụ mùa bội thu các hạt ngũ cốc cả về chất lẫn lượng. 7. Chuyển gene tạo tính bất dục của cây trồng
Một ứng dụng thực tiễn khác của kỹ thuật di truyền là tạo ra cây trồng bất dục. So với các bố mẹ lai cùng dòng, những cây lai thường có nhiều đặc điểm ưu việt về nhiều chi tiết bao gồm sản lượng cao và tăng tính chống chịu bệnh. Bất dục đực có vai trò quan trọng trong tạo hạt ở con lai vì nó đẩy mạnh khả năng lai giữa các dòng với nhau. Bất dục bào chất đực (cytoplasmic male sterility) được sử dụng rộng rãi trong lai giống bắp. Các cây bất dục cũng có thể trở nên hữu thụ nhờ sự phục hồi hữu dục (fertility restore), nhờ vậy các dòng con lai có thể sinh sản được (Hình 3.16)

175
Hình 3.16. Thao tác di truyền với tính bất dục đực và phục hồi hữu dục (A) hạt phấn (PG) phát triển trong lớp mỏng tế bào tapel (B) Biểu hiện của nuclease RNA barnase dưới sự điều khiển của
vùng điều hoà đặc trưng tapetum của gene TA29, phá huỷ tapetum và làm cây bất dục đực
(C) Biểu hiện của barstar (chất ức chế của barsnar) trong các tế bào tapetal bất hoạt barsnase và phục hồi hữu dục (Courtesy of Robert B. Goldberg)
Hệ thống thao tác di truyền của tính bất dục đực và phục hồi hữu dục được áp dụng ở một số loài thực vật như cây cải dầu (Oilseed rape), cải bắp (Brassica napus). Đây là các cây trồng chủ yếu để chiết dầu thực vật (Hình 3.17). Tiến hành thao tác di truyền của tính bất dục đực và phục hồi hữu dục dựa trên sử dụng 2 gene. Cơ sở của tính bất dục đực là một nuclease ngoại bào được gọi là barnase, được tạo ra bởi vi khuẩn Bacillus amyloliquefaciens. Nuclease barnase là một độc tố rất có hiệu lực của tế bào.

176
Hình 3.17. Hoa (A, C và E) và lát cắt ngang bao phấn (B, D và F) của cây
cải dầu. Genotype: A và B: kiểu dại C và D: barsnase TA29 E và F: barnase TA29 và barstar TA29

177
Cây phục hồi hữu dục (fertility restoreed plants) E và F, được phân biệt với cây kiểu dại (A và B).
Phần chú thích trên hoa: A (anther): bao phấn; P (petal): cánh hoa; PI (Pistil): nhuỵ hoa; N
(nectary): tuyến mật ở hoa Phần chú thích qua lát cắt ngang: En (Endothecium): vách trong bao phấn E (epidermis): biểu bì PG (pollen grain): hạt phấn PS (pollen sac): túi phấn T (tapetum): Tế bào bao quanh bao phấn Trong việc sử dụng barnase để tạo tính bất dục, trình tự mã hóa của gene vi
khuẩn được dung hợp với trình tự điều hoà cuả gene TA29, có biểu hiện đặc trưng mô (tissue specific expression) ở lớp tế bào tapetal bao quanh bao phấn (pollen sac). Khi gene barnase TA29 nhân tạo được đưa vào genome nhờ sử dụng T-DNA từ Agrobacterium. Cây thu được bị bất dục đực vì sự phá huỷ RNA tế bào tapetal nhờ enzyme barnase và làm chết các tế bào bao quanh bao phấn ở tapetum.
Sự phục hồi hữu dục được tạo ra nhờ sử dụng một protein khác được gọi là barstar. Enzyme này cũng được tạo ra nhờ vi khuẩn Bacillus amyloliquefaciens. Barstar là một protein nội bào, bảo vệ chống lại hiệu quả gây chất của barsnase nhờ sự tạo thành phức hợp bất hoạt enzyme bền vững trong tế bào chất. Vì vậy, barstar là sự tự bảo vệ của tế bào vi khuẩn chống lại barnase. Những thực vật được chuyển gene barstar TA29 nhân tạo đều khoẻ mạnh và hữu thụ, chúng tạo ra barstar trong tapetum với sự biểu hiện cử vùng điều hoà TA29 đặc trưng tapetum. Genotype thu được tổ hợp của barnase TA29 với barstar TA29 là bất dục đực (Hình 3.10. C). Lý do cho sự phục hồi hữu dục là tổ hợp protein barnase TA29 với nuclease barnase và làm nó không có hiệu quả. Hình thái vĩ mô và hình thái vi mô của các genotype khác nhau của cây cải dầu được mô tả ở hình 3.11. Ở kiểu hình hoang dại (phần A và B), số lớn hạt phấn (pollen grain - PG) xuất hiện ở mặt cắt ngang qua nhị. Phần C và D cho thấy hoa và nhị của cây mang barsnar TA29, nhị nhỏ, teo lại và không có phấn hoa (pollen). Phần E và F cho thấy hoa và bao phấn của cây mang kiểu gene gồm barsnase TA29 có thêm barstar TA29, kiểu hình của nó phân biệt với cơ thể kiểu dại.
8. Chuyển gene giúp cây trồng chống điều kiện bất lợi
Stress vô sinh (abiotic stress) là nhân tố ngăn cản sử dụng các yếu tố môi trường nhất định cho trồng trọt và làm hạn chế sản lượng nông nghiệp toàn thế giới.
Stress vô sinh bao gồm nhiệt độ cao hoặc thấp, hạn, muối, lạnh, cường độ ánh sáng cao, mất cân bằng về dinh dưỡng ... Các nhân tố này có thể tác động độc lập hoặc tác động đồng thời. Các stress này dẫn đến stress thẩm thấu và stress oxy hóa ở mức độ tế bào bởi sự ức chế hấp thu và vận chuyển nước. Khí khổng bị đóng, giảm sự cung cấp CO2, tỷ lệ phản ứng sinh hóa thấp trong suốt thời gian khử nước (dehydrate) kéo dài. Cường độ ánh sáng cao, nhiệt độ cao hoặc thấp đều dẫn đến sản phẩm phản ứng qua trung gian oxy (ROI - reactive oxygen intermediate) cao trong chloroplast gây ra thiệt hại tế bào và ức chế sáng không thuận nghịch. Để

178
phản ứng lại stress khử nước và stress thẩm thấu, hàng loạt chất thẩm thấu (osmolyte) tương thích được tích luỹ để điều chỉnh thẩm thấu, giữ nước và bắt gốc tự do. Tương tự, sự biểu hiện quá mức của các enzyme nhất định như superoxide dismutase, ascorbate peroxidase và glutathione reductase được thực hiện để khử độc các gốc tự do và bắt các gốc tự do xuất hiện do stress oxy hóa.
- Tạo cây trồng chuyển gene chống stress thẩm thấu, sự khử nước và muối: Proline được xác định là một chất bảo vệ thẩm thấu hiệu lực và thích hợp được
tích lũy với nồng độ cao ở thực vật nước ngọt (glycophyte) và thực vật nước măn (halophyte) trong phản ứng với stress thẩm thấu như hạn và nồng độ muối cao.. Hai enzyme quan trọng cho sinh tổng hợp proline là A1-pyrroline-5-carboxylate synthetase (P5CS) và A1-pyrroline-5-carboxylate reductase (P5CR) đã được tạo dòng vào nhiều cây trồng và sự biểu hiện của chúng đã được nghiên cứu dưới những stress vô sinh khác nhau.
Một số thực vật chuyển gene biểu hiện mức độ cao mRNA P5CS cũng đã tích luỹ mức độ cao protein P5CS. Thực vật chuyển gene sản xuất proline nhiều hơn cây đối chứng từ 10 đến 18 lần.. Dưới điều kiện hạn, hàm lượng protein tăng từ 80 mg/g trọng lượng tươi của lá (trước stress) lên đến 3.000 mg/g trọng lượng tươi của lá (sau stress) ở cây đối chứng và đã tăng từ 1.000 mg/g đến 6.500 mg/g ở những dòng chuyển gene. Sự héo ở cây chuyển gene diễn ra ít gay gắt và chậm hơn so với cây đối chứng kiểu dại. Kết quả này đã chứng minh proline hoạt động như một chất bảo vệ thẩm thấu và sự tổng hợp nó tăng lên ở cây chuyển gene để chống lại stress hạn và muối.
Glycinebetaine, một amino acid có cấu trúc bậc bốn cũng là chất hòa tan khác trong thực vật và bảo vệ thực vật chống stress muối và lạnh. Một số cây như rau bina (spinach) và lúa mạch (barley), betain được tổng hợp từ choline nhờ sự oxy hóa choline tạo betain aldehyde nhờ sự xúc tác của enzyme choline mono-oxygenase và rồi tạo ra betain nhờ xúc tác của enzyme betain aldehyde dehydrogenase (được mã hóa do gene nhân). Các cây được biến nạp gene sinh trưởng chậm ở nồng độ 200 mM NaCl trong khi không một cây kiểu dại nào có thể sinh trưởng được. Sự tích lũy glycine betain qua biến đổi di truyền ở Arabidopsis làm tăng cường khả năng chống chịu của nó với stress muối và lạnh.
- Tạo cây trồng chuyển gene kháng kim loại nặng: Sự sinh trưởng, tạo thành sản phẩm và thậm chí cả sự gieo trồng của thực vật
bị giới hạn bới nồng độ của các ion vô cơ có trong đất như ion Na+ trong đất mặn, Al3+ và Mn2+ trong đất acid và các kim loại nặng như Cu, Zn, Pb, Ni, Cd, ... kết quả của khai khoáng, trong kỹ nghệ và trong các hoạt động của con người.Cây trồng kháng với độc tố Al. Sự loại thải và sự hấp thu Al từ mút rễ được nhận thấy là tương ứng với gia tăng khả năng giải phóng các acid hữu cơ như cỉtric acid mà chelate Al3+ bị thải ra ngoài màng sinh chất. Thuốc lá và đu đủ chuyển gene biểu hiện quá mức trong tế bào chất của chúng gene citrate synthetase (CSb) được tách từ Pseudomonas aeruginosa . các dòng thuốc lá này biểu hiện CSb cao gấp 10 lần nồng độ citrate trong mô rễ của chúng và một trong số các dòng đó giải phóng nồng độ citrate ngoại bào cao hơn 4 lần, ở cây đu đủ sự sản xuất citric acid cũng tăng lên

179
2-3 lần. Sự sản xuất citric acid tăng đã được chứng minh là kết quả chống chịu Al ở những loài này.
Trong phương pháp cải thiện cây trồng truyền thống, chủ yếu sử dụng phương pháp lai, dựa vào sự chọn lọc kiểu hình mong muốn, nhưng không thể điều khiển qua kết quả kiểu gene. Kỹ thuật tái tổ hợp DNA giúp trao đổi gene giữa các loài không tương hợp về mặt sinh sản dẫn đến sự bổ sung trong điều hoà ở thực vật chuyển gene.
Hầu hết cây trồng không được chọn giống theo hướng sống sót ở điều kiện môi trường khắc nghiệt. Vì vậy, khả năng sống sót của cây trồng dưới những điều kiện này bị hạn chế. Stress vô sinh ảnh hưởng đến sự sống sót, sự tổng hợp sinh khối và sản lượng của hầu hết cây trồng và có thể dẫn đến làm giảm sự nảy mầm của hạt, sự hình thành cây mầm, khả năng phát triển của hạt phấn. Sử dụng kỹ thuật di truyền có thể tạo cây trồng sống được ở điều kiện môi trường khắc nghiệt.

180
Tài liệu tham khảo
1. Anthony J. F. Griffiths, Susan R. Wessler, Richard C. Lewontin, William M. Gelbart, David T. Suzuki, Jeffrey H. Miller. 2004. An introduction to genetics analysis. W.H. Freeman Publishers.
2. Đái Duy Ban, Đái Hằng Nga. 1998. Tổng luận phân tích cây trồng chuyển gene - một hướng công nghệ cao của công nghệ sinh học phục vụ nông nghiệp và y dược. Trung tâm thông tin tư liệu, Trung tâm Khoa học Tự nhiên và Công nghệ Quốc gia, Hà Nội.
3. Harlt D.L., Jones E.W. 1998. Genetics - Principle and analysis. Jone and Bartlett Publshers, Toronto, Canada.
4. Phạm Thành Hổ. 2005. Nhập môn công nghệ sinh học. NXB Giáo dục. 5. Jamese Tavares, Phyllis B. Moses, Susannee Mason. 1987. Agricultural
biology. National academy Press. Washington D.C. 6. Leandro Pena. 2005. Transgenic plants method. Humana Press. Volume 286. 7. Nguyễn Hoàng Lộc. 1998. Bài giảng Công nghệ gene thực vật. Đại học Khoa
học Huế. 8. Maarten J. Chrispeels & David E. Sadava. 2003. Plants, Genes and Crop
Biotechnology. Jones and Bartlett Publishers. Sudbury, Massachsetts. 9. Maniatis T, Fritsch EF, Sambrook J. 1989. Molecular Cloning (A laboratory
manual). Cold Spring Habor Laboratory. USA. 10. Mark Tester and Antony Bacic. 2005. Abiotic stress tolerance in grasses, from
model plants to crop plants. Plant Physiology. American Society of plant Biologists.
11. Newton CR and Graham A. 1994. PCR. BIOS Scientific Publishers Limited. 12. Nigel Halford. 2006. Plant biotechnology. Current and future application of
genetically modified crops. John Wiley & Sons, Ltd. 13. Richard J. Reece. 2004. Analysis of genes and Genomes. John Wiley & Sons,
Ltd. 14. Tac Secretariat. 2001. Genetic engineering for abiotic stress tolerance in
plants. Food and agriculture organization of the United nations. 15. Khuất Hữu Thanh. 2005. Cơ sở di truyền phân tử và kỹ thuật gen. NXB Khoa
học và Kỹ thuật. 16. Lê Duy Thành. 2000. Cơ sở di truyền chọn giống thực vật. NXB Khoa học và
Kỹ thuật. 17. Xin HU, Qing-Feng WU, Ya-Hong XIE, Hong RU, Feng XIE, Xin-Yu Wang,
Chong-Ying Wang. 2005. Ectopic expression of the PttknI gene induces alterations in the morphology of the leave and flowers in Petunia hybrida Vilm.
18. Wiser MF. 2002. Lecture notes for method in cell. Spring 19. (http://www.florigene.com/research/research.php)

175
Bài 5 CÔNG NGHỆ NUÔI CẤY MÔ TẾ BÀO THỰC VẬT
I. GIỚI THIỆU CHUNG 1. Sơ lược lịch sử phát triển
Gottlied Haberlandt (1902), nhà thực vật học người Đức, đã đặt nền móng đầu tiên cho công nghệ nuôi cấy mô tế bào thực vật với giả thuyết tính toàn năng của tế bào trong cuốn ”Thực nghiệm nuôi cấy tế bào tách rời“. Tuy nhiên, những thí nghiệm đầu tiên của ông trên tế bào mô mềm, biểu bì đã không thành công do chúng không thể phân chia.
Năm 1922, Kotte là học trò của Haberlandt cùng với Robbins đã lặp lại thí nghiệm của Haberlandt với đỉnh sinh trưởng tách từ đầu rễ của một cây hòa thảo (cây ngô). Hai tác giả đã nuôi được đỉnh sinh trưởng này trong một thời gian ngắn trên môi trường lỏng có chứa đường glucose, muối khoáng và đã thu được hệ rễ nhỏ.
Năm 1934, bắt đầu giai đoạn thứ 2 của lịch sử nuôi cấy mô tế bào thực vật, khi White đã duy trì được sự sinh trưởng của đầu rễ cà chua (Lycopersicon esculentum) trong một thời gian dài trên môi trường lỏng chứa đường, một số muối khoáng và dịch chiết nấm men. Cũng trong giai đoạn này, vai trò của hàng loạt vitamin nhóm B trong việc thúc đẩy sinh trưởng trong nuôi cấy như thiamin (vitamin B1), pyridoxin (vitamin B6), nicotinic acid (vitamin B3 hay PP)... đã được phát hiện.
Nuôi cấy mô thực vật có nhiều thuận lợi hơn khi Went và Thimann tìm ra chất kích thích sinh trưởng đầu tiên là indole acetic acid(IAA). Trong thời gian này, nhiều chất kích thích sinh trưởng thuộc nhóm auxin được nghiên cứu và tổng hợp thành công như naphthalen acetic acid (NAA), 2,4-dichlorophenoxy acetic acid(2,4-D) ...
Van Overbeck (1941) đã chứng minh tác dụng tốt của nước dừa trong nuôi cấy phôi cây họ cà. Năm 1954, Skoog phát hiện chế phẩm phân hủy của tinh dịch cá bẹ có tác dụng kích thích rõ rệt trong nuôi cấy các mảnh mô thân cây thuốc lá. Chất này sau đó được tổng hợp thành công gọi là kinetin có tác dụng trong kích thích phân bào.
Việc phát hiện NAA, 2,4-D, kinetin cùng các loại vitamin và nước dừa là những bước tiến ý nghĩa trong giai đoạn 2 của nuôi cấy mô tế bào.
Skoog và Miler (1975) đã chứng minh sự biệt hóa rễ, chồi trong nghiên cứu nuôi cấy mô thuốc lá phụ thuộc vào nồng độ tương đối của auxin/cytokinin và cũng từ đó đưa ra quan niệm điều khiển hormone trong quá trình hình thành cơ quan của thực vật. Thành công này của Skoog và Miler dẫn đến nhiều phát hiện quan trọng khác và mở đầu cho giai đoạn 3 của nuôi cấy mô tế bào thực vật.
Khả năng nuôi cấy tế bào thực vật và tái tạo cây hoàn chỉnh từ tế bào mở ra những triển vọng cho chọn dòng đột biến, sản xuất các chất trao đổi thứ cấp.... Ngày nay, nuôi cấy mô tế bào thực vật được ứng dụng rộng rãi trong nhân giống nhiều loại thực vật, chọn dòng chống chịu, lai xa, chuyển gene vào cây trồng... Đấy chính là giai đoạn thứ 4 của nuôi cấy mô tế bào thực vật - giai đoạn nuôi cấy mô tế bào

176
thực vật được ứng dụng mạnh mẽ vào thực tiễn chọn tạo giống cây trồng, vào việc sản xuất các hợp chất thứ cấp, vào nghiên cứu lý luận di truyền của thực vật bậc cao.
Như vậy, có thể nói rằng với sự phát triển đa dạng của các hướng nghiên cứu, nuôi cấy mô và tế bào thực vật là một trong những lĩnh vực ứng dụng đạt nhiều thành công nổi bật của công nghệ sinh học thực vật. Bằng các kỹ thuật nuôi cấy các bộ phận tách rời của cơ thể thực vật trong điều kiện vô trùng và môi trường dinh dưỡng thích hợp, người ta đã nhân giống in vitro thành công nhiều loại cây trồng có giá trị, nuôi cấy đơn bội (1n) để tạo giòng thuần chủng, phục tráng giống cây trồng, dung hợp protoplast giúp mở rộng nguồn gene tạo ra nhiều loài thực vật mang tính trạng mới hữu ích, chọn dòng biến dị soma có khả năng chống chịu các điều kiện bất lợi của ngoại cảnh, sản xuất các cây sạch bệnh virus, chuyển gene vào cây trồng nhằm tạo ra những cây trồng mang các gene kháng sâu, kháng bệnh, kháng chất diệt cỏ...
2. Cơ sở khoa học của nuôi cấy mô tế bào thực vật 2.1. Tính toàn năng của tế bào (Topipotency)
Gottlied Haberlandt lần đầu tiên đưa ra giả thuyết về tính toàn năng của tế bào trong cuốn sách “Thực nghiệm về nuôi cấy mô tế bào tách rời”. Theo ông, tế bào bất kỳ của cơ thể sinh vật đa bào nào cũng đều có khả năng phát triển thành cơ thể hoàn chỉnh khi gặp điều kiện thuận lợi. Đó chính là tính toàn năng của tế bào.
Tính toàn năng của tế bào mà Haberlandt nêu ra chính là cơ sở lý luận của phương pháp nuôi cấy mô tế bào thực vật. Cho đến nay, người ta đã hoàn toàn chứng minh được khả năng tái sinh một cơ thể hoàn chỉnh từ một tế bào riêng lẻ.
2.2. Sự phản phân hóa và phân hóa của tế bào Cơ thể sinh vật trưởng thành gồm nhiều cơ quan có chức năng khác nhau
được hình thành từ nhiều loại tế bào. Tuy nhiên, tất cả các tế bào đó đều bắt nguồn từ một tế bào ban đầu (tế bào hợp tử). Ở giai đoạn đầu, tế bào hợp tử phân chia thành nhiều tế bào phôi sinh chưa mang chức năng riêng biệt (chuyên hóa). Sau đó, các tế bào phôi sinh này tiếp tục được biến đổi thành các tế bào chuyên hóa đặc hiệu cho các mô, cơ quan khác nhau.

177
Hình 4.1. Sự phân hóa mô nuôi cấy thành chồi, rễ, phôi Tuy nhiên, khi tế bào đã phân hóa thành các tế bào có chức năng chuyên biệt, chúng không hoàn toàn mất khả năng biến đổi của mình. Trong trường hợp cần thiết, ở điều kiện thích hợp, chúng có thể trở về dạng tế bào phôi sinh và phân chia mạnh mẽ. Quá trình đó gọi là quá trình phản phân hóa tế bào, ngược với quá trình phân hóa tế bào.
Trong nuôi cấy mô tế bào thực vật, mẫu vật ban đầu phải trải qua giai đoạn phản phân hóa để trở lại trạng thái phân sinh và tạo ra những mô, tế bào không phân hóa, sau đó các tế bào và mô này sẽ tạo thành cây hay cơ quan hoàn chỉnh thông qua quá trình tái phân hóa.
Như vậy, kỹ thuật nuôi cấy mô tế bào thực vật là quá trình điều khiển sự phát sinh hình thái của tế bào thực vật (khi nuôi cấy tách rời trong điều kiện nhân tạo vô trùng) một cách có định hướng dựa vào sự phân hóa và phản phân hóa của tế bào trên cơ sở tính toàn năng của tế bào thực vật.
3. Một số thuật ngữ và khái niệm cơ bản
- Nuôi cấy mô tế bào thực vật (plant cell and tisue culture) là khái niệm chung cho tất cả các quá trình nuôi cấy từ nguyên liệu thực vật trên môi trường dinh dưỡng nhân tạo trong điều kiện vô trùng.
- Thuật ngữ nhân giống in vitro (in vitro propagation) hay còn gọi là vi nhân giống (micropropagation) thường được sử dụng cho việc ứng dụng các kỹ thuật nuôi cấy mô để nhân giống thực vật, bắt đầu bằng nhiều bộ phận khác nhau của thực vật với kích thước nhỏ.
Trong thực tế, các nhà vi nhân giống thường dùng thuật ngữ nuôi cấy mô và nhân giống in vitro hoặc nuôi cấy in vitro thay đổi cho nhau để chỉ mọi phương thức nhân giống thực vật trong điều kiện vô trùng với các mục đích khác nhau.
- Nuôi cấy đỉnh phân sinh (meristem-tip culture): Phương thức nhân giống bằng cách dùng các phần rất nhỏ của đỉnh chồi (shoot tip) bao gồm mô phân sinh đỉnh riêng rẽ (single apical meristem) và mầm lá non (young leaf primordia) để kéo dài chồi (shoot elongation). Kiểu nuôi cấy này thường được sử dụng để làm sạch virus (virus-free) ở thực vật.

178
- Sinh sản chồi nách (axillary shoot proliferation): Kiểu sinh trưởng này sử dụng chồi của các điểm sinh trưởng bên và ngọn khi sự kéo dài của chồi tận cùng (elongation of terminal shoot) bị kìm hãm và sự sinh sản chồi nách được đẩy mạnh. Kiểu nuôi cấy này cho phép nhân nhanh được các chồi in vitro.
- Tạo chồi bất định (adventitious shoot induction): Mẫu vật được dùng trong loại nuôi cấy này là: đoạn thân, mảnh lá, cuống lá, vảy củ, đoạn mầm, các bộ phận của hoa, ...Chồi bất định được hình thành hoặc trực tiếp trên mẫu vật hoặc gián tiếp từ mô callus. Các mô callus này đuợc hình thành trên bề mặt vết cắt của mẫu vật.
- Phát sinh cơ quan (organogeneesis): Đây là quá trình tái sinh chồi hoặc/và rễ bất định từ các khối tế bào callus đã được cảm ứng tạo ra trước đó. Đối với mục đích nhân giống in vitro, nếu tái sinh được cây hoàn chỉnh trực tiếp từ mẫu nuôi cấy ban đầu thì không những nhanh chóng thu được cây với số lượng lớn mà các cây cũng khá đồng nhất về mặt di truyền. Do tế bào callus khi cấy chuyển nhiều lần sẽ không ổn định về mặt di truyền nên trong trường hợp phải tái sinh cây thông qua giai đoạn callus thì nhất thiết phải sử dụng callus vừa phát sinh.
- Phát sinh phôi vô tính (somatic embryogeneesis): Thuật ngữ này dùng để chỉ sự phát triển của các phôi hoàn chỉnh từ các tế bào sinh dưỡng được sản xuất từ các nguồn mẫu vật khác nhau sinh trưởng trong nuôi cấy in vitro. Phôi vô tính có cấu trúc tương tự phôi hữu tính của thực vật sinh trưởng trong điều kiện tự nhiên. Điểm khác nhau cơ bản giữa phôi hữu tính và phôi vô tính là phôi hữu tính luôn luôn có nội nhũ là cơ quan dự trữ năng lượng và chất dinh dưỡng phục vụ cho quá trình nảy mầm, còn ở phôi vô tính hoàn toàn không có nội nhũ.
Hình 4.2. Nuôi cấy mô tế bào thực vật
- Cảm ứng (induction): Một bộ phận, cấu trúc hay quá trình nào đó được tạo ra trong điều kiện in vitro dưới tác động của hormon.
- Cấy chuyển (subculture): Chuyển tế bào, mô hoặc mẫu vật nuôi cấy sang môi trường mới pha, kết hợp với việc tách nhỏ mô hoặc làm loãng mật độ tế bào.
- Chồi hoặc rễ bất định (adventitious shoots or roots): Chồi hoặc rễ phát sinh từ những vùng khác thường (không phải là hợp tử).

179
- Mẫu vật (explant): Mô được tách từ nguyên liệu ban đầu dùng để nuôi cấy. - Mô sẹo (callus): Khối mô thực vật gồm những tế bào không phân hóa, có
khả năng phân chia, được hình thành từ những tế bào đã phân hóa. - Phân hóa (differentiation): Quá trình chuyên hóa các tế bào về chức năng
và hình thái để tạo ra các mô, cơ quan và cơ thể hoàn chỉnh.
Hình 4.3. Mô sẹo (callus)
- Phát sinh hình thái (morphogeneesis): Hiện tượng phát sinh và phát triển
các cấu trúc như chồi, rễ, lá,... từ tế bào, mô sẹo hay mẫu vật nuôi cấy. Chủ yếu nói đến sự hình thành phôi hoặc rễ từ mô sẹo.
Hình 4.4. Phát sinh chồi từ mẫu cấy ban đầu
- Tái sinh (regeneeration): Hiện tượng tế bào hoặc mô nuôi cấy phân hóa thành mô, cơ quan hoặc cây hoàn chỉnh khi chịu sự kích thích.
- Tế bào trần (protoplast): Tế bào bị mất toàn bộ thành tế bào nhưng vẫn duy trì được sức sống và chức năng.
4. Môi trường dinh dưỡng

180
Môi trường dinh dưỡng là nhân tố quan trọng quyết định sự thành công của quá trình nuôi cấy. Hầu hết các môi trường dinh dưỡng nhân tạo được sử dụng để nuôi cấy mô và tế bào thực vật đều bao gồm các thành phần sau đây
4.1. Các nguyên tố muối khoáng
Nguyên tố đa lượng: nồng độ sử dụng lớn hơn 30 ppm (mg/l) quan trọng nhất là các nguyên tố N, S, P, K, Mg, Ca, Na, và một vài nguyên tố khác nhưng chưa rõ vai trò của chúng.
- Nitơ: được sử dụng ở dạng NO3- hoặc NH4
+ hầu hết các thực vật đều có khả năng khử nitrate thành ammonium nhờ hệ thống nitratreductase ammonium được thực vật đồng hóa trực tiếp để tổng hợp nên các sản phẩm hữu cơ.
Nếu chỉ dùng NH4+để thay thế toàn bộ NO3
- thì tế bào có thể giảm hoặc dừng hẳn sinh trưởng vì pH môi trường thay đổi (pH giảm). Khi pH môi trường giảm thì quá trình hấp thụ Fe sẽ kém đi.
- Lưu huỳnh: sử dụng chủ yếu là SO42-
- Phosphore: mô và tế bào nuôi cấy có nhu cầu sử dụng P rất cao, nó có tác dụng như một hệ thống đệm (buffer) làm ổn định pH môi trường trong quá trình nuôi cấy.
- Kali: thường dùng là KNO3, KCl.6H2O, KH2PO4 - Canci: sử dụng chủ yếu CaNO3.4H2O, CaCl2.6H2O, CaCl2.2H2O - Magie: sử dụng chủ yếu MgSO4.7H2O Nguyên tố vi lượng: nồng độ sử dụng nhỏ hơn 30 ppm. Gồm các nguyên tố
sau: Fe, B, Mn, I, Mo, Cu, Zn, Ni, Co. - Fe: thiếu Fe tế bào mất khả năng phân chia. Nhiều thí nghiệm cho thấy Fe
được dự trữ trong nhân rất nhiều. Thiếu Fe làm giảm lượng RNA và protein nhưng làm tăng DNA, amino acid tự do và làm giảm phân bào. Fe thường tạo phức hợp với các thành phần khác và khi pH môi trường thay đổi thì phức hợp này mất khả năng giải phóng Fe cho các nhu cầu trao đổi chất của tế bào. Fe thường được sử dụng ở dạng Na2-EDTA, các phức chất này Fe được giải phóng ra trong phạm vi pH khá rộng.
- Mn: thiếu Mn thì hàm lượng amino acid tự do và DNA tăng nhưng lượng RNA và quá trình tổng hợp protein giảm, dẫn đến hoạt động phân bào giảm.
- B: thiếu B trong môi trường gây nên những biểu hiện như thừa auxin vì B sẽ làm cho các chất ức chế enzymee auxinoxydase trong tế bào giảm. Mô nuôi cấy có biểu hiện mô sẹo hóa mạnh nhưng thường là mô sẹo xốp, mọng nước, khả năng tái sinh cây kém.
- Mo: là ion đóng vai trò cofector trong hệ thống nitrate reductase.
4.2. Nguồn carbon Mô và tế bào thực vật trong nuôi cấy in vitro sống chủ yếu theo phương thức
dị dưỡng mặc dù trong nhiều trường hợp chúng có thể sống bán dị dưỡng nhờ điều kiện ánh sáng nhân tạo và lục lạp có khả năng quang hợp. Vì vậy, đưa vào môi trường nuôi cấy nguồn carbon hữu cơ là điều kiện bắt buộc. Nguồn carbon hữu cơ thông dụng nhất là đường saccharose, với nồng độ thích hợp là 2-3%. Tùy theo mục đích và đối tượng nuôi cấy mà thay đổi chẳng hạn như chọn dòng: 0,2%; tạo củ in vitro: 9-12%; cảm ứng stress nước: 12%.

181
Glucose cũng được dùng để nuôi cấy protoplast, maltose được dùng để nuôi cấy bao phấn. Fructose, lactose, galactose cũng được thử nghiệm nhưng kém hiệu quả.
Đối với những loại tế bào có khả năng thủy phân nhờ có enzyme amylase thì có thể sử dụng nguồn carbon hữu cơ là polysaccharide như tinh bột, pectin, dextrin, …
Các loại rượu: glycerol, mannitol, sorbitol, … được sử dụng trong nuôi cấy dịch huyền phù và nuôi cấy protoplast với mục đích là ổn định áp suất thẩm thấu.
4.3. Vitamin
Thông thường các loại tế bào nuôi cấy có khả năng tồng hợp vitamin nhưng không đủ về lượng nên phải bổ sung các vitamin nhóm B.
- Vitamin B1 (Thiamine HCl) là chất bổ sung rất cần cho môi trường nuôi cấy. Khi khử trùng ở nhiệt độ cao vitamin B1 thường bị nhiệt phân thành 2 cấu tử là pyrimidin và thiazol. Tuy nhiên tế bào nuôi cấy có khả năng tổng hợp vitamin từ hai cấu tử này, do đó không cần thay thế cách khử trùng khác.
- Vitamin B2 (Riboflavin) có thể tiệt trùng bằng nhiệt nhưng loại vitamin này dễ bị ánh sáng phân hủy.
- Vitamin B6 (Pyridoxin) là tiền chất của pyridoxal phosphate, là cofactor của các nhóm enzymee carboxylase và transminase. Khi ở nhiệt độ cao thì pyridoxin kết hợp với phosphat tạo thành pyridoxylphosphat.
- Myo-inositol (Bios I) có vai trò trong sinh tổng hợp thành tế bào. Inositol là chất bền vững ở nhiệt độ cao, thường sử dụng ở nồng độ 100ppm.
- Biotin (Bios II) cần thiết cho phân bào ở một số loại mô, chỉ sử dụng ở nồng độ rất thấp.
4.4. Các chất hữu cơ tự nhiên
- Nước dừa (cocconut milk): phân tích thành phần nước dừa từ non đến già cho thấy trong thành phần nước dừa có:
Amino acid tự do: 190,5-685 ppm khi hấp ở nhiệt độ cao còn 70 ppm. Các amino acid trong protein, các acid hữu cơ, đường, DNA, RNA. Ngoài ra
trong nước dừa còn có các chất quan trọng đối với nuôi cấy phân lập: cytokinin ở dạng glucoside, các chất có hoạt tính auxin.
- Dịch thủy phân casein: được sử dụng rộng rãi trong nuôi cấy vi sinh vật. Trong nuôi cấy mô tế bào thực vật được sử dụng như nguồn bổ sung amino acid.
- Dịch chiết nấm men: thành phần hóa học chủ yếu chứa đường và các amino acid. Dịch chiết nấm men có tác dụng đối với rễ rất tốt.

182
Bảng 4.1. Thành phần của một số môi trường thông dụng
Thành phần Môi trường
(mg/l) Murashige và Skoog
(MS)
Gamborg (B5)
Woody plant medium (WPM)
Nitsch và Nitsch
Khoáng đa lượng NH4NO3 1650 - 400 - NH4SO4 - 134 - - CaCl2.2H2O 332,2 150 96 166 Ca(NO3)2.4H2O - - 556 - MgSO4.7H2O 370 250 370 185
KNO3 1900 2500 - 950
K2SO4 - - 990 -
KH2PO4 170 - 170 68 NaH2PO4 - 130,5 - -
Khoáng vi lượng H3BO3 6,2 3,0 6,2 10 CoCl2.6H2O 0,025 0,025 - - CuSO4.5H2O 0,025 0,025 0,25 0,025 Na2EDTA 37,3 37,3 37,3 37,3 FeSO4.7H2O 27,8 27,8 27,8 27,8 MnSO4. H2O 16,9 10,0 22,3 18,9 KI 0,83 0,75 - - Na2MoO4.2H2O 0,25 0,25 0,25 0,25 ZnSO4.7H2O 8,6 2,0 8,6 10
Chất hữu cơ Myo-inositol 100 100 100 100 Glycine 2,0 - 2,0 2,0 Acid nicotinic 0,5 1,0 0,5 5 B1 0,5 0,1 0,5 0,5 B6 0,1 10,0 1,0 0,5 4.5. Các chất điều hòa sinh trưởng
Các chất điều hòa sinh trưởng thường được sử dụng ở nồng độ thấp (0,001-10 µM). Chúng kích thích sự hình thành và phát triển chồi, rễ và phôi vô tính trong nuôi cấy mô do khả năng kích thích sự kéo dài và phân chia tế bào. Mô nuôi cấy có thể tự tổng hợp các chất điều hòa sinh trưởng nhưng thông thường chúng ta phải bổ sung thêm các chất này vào môi trường nuôi cấy.
Nhóm chất điều hòa sinh trưởng thường được sử dụng trong nuôi cấy mô là auxin và cytokinin, ngoài ra còn có các nhóm chất như gibberllin, abscisic acid.
- Auxin là hormone sinh trưởng gồm có các chất như IAA (indolylacetic acid), NAA (naphthylacetic acid), 2,4-D (diclorophenoxy acetic acid), IBA (indolyl butyric acid)… trong đó IAA là auxin tự nhiên còn lại NAA, IBA, 2,4-D là auxin

183
nhân tạo. Các auxin nhân tạo có hoạt tính mạnh hơn. Auxin có vai trò quan trọng trong các quá trình phát triển, kéo dài tế bào, tạo ưu thế đỉnh, hình thành rễ bất định, tạo phôi vô tính. Thông thường, khi nồng độ auxin thấp sẽ kích thích sự tạo rễ, nồng độ auxin cao thì dẫn đến sự hình thành callus. Kinh nghiệm sử dụng auxin trong nuôi cấy mô là lúc đầu sử dụng nồng độ cao sau đó giảm dần để tránh tình trạng mô bị suy và nhiễm độc.
- Cytokinin là hormon phân bào do Skoog phát hiện ra đầu tiên năm 1950. Ngoài tác dụng kích thích quá trình phân bào, cytokinin còn có tác dụng thúc đẩy sự hình thành và phát triển chồi. Ở nồng độ cao (1-10 mg/l), cytokinin kích thích sự hình thành chồi bất định nhưng ức chế sự tạo rễ. Có các loại cytokinin sau:
Kinetin (6-furfuryl-aminopurin): nếu phối hợp với auxin với tỷ lệ nồng độ thích hợp thì sẽ có tác dụng kích thích phân chia tế bào.
Zeatin: được tách và tinh chế từ nội nhũ ở dạng sữa của hạt ngô. BAP (6-benzyl aminopurin): hoạt tính của BAP cao hơn nhiều so với zeatin.
BAP bền vững hơn dưới tác dụng của nhiệt độ. Ngoài ra còn có TDZ (thidiazuron), … - Gibberellin thường ít được sử dụng trong nuôi cấy mô. Trong nhóm này
GA3 thường được sử dụng nhất, nó có tác dụng kích thích sự phát triển của mô phân sinh lóng.
- Abscicic acid (ABA): thường được sử dụng trong nuôi cấy mô và tế bào thực vật với mục đích chọn dòng tế bào có khả năng chống chịu với điều kiện bất lợi của ngoại cảnh. ABA hoàn toàn không phải là chất gây đột biến mà là chất ức chế. Khi xử lý ABA lên mô lúa, mô này có thể chịu được mất nước đến 90% mà vẫn có khả năng phục hồi trên môi trường phục hồi. Kết quả chạy điện di cho thấy có xuất hiện protein lạ chứng tỏ có DNA lạ. Người ta chứng minh được rằng có gene (DNA) chịu mất nước nhưng promoter của nó không hoạt động, chính ABA đã xúc tác cho promoter này hoạt động.
5. pH của môi trường
Độ pH của môi trường ảnh hưởng trực tiếp đến khả năng thu nhận các chất dinh dưỡng từ môi trường vào tế bào. Vì vậy, trong từng trường hợp cụ thể phải điều chỉnh độ pH của môi trường về mức ổn định ban đầu. Trong các thí nghiệm nuôi cấy tế bào đơn hay tế bào trần thì việc điều chỉnh pH là bắt buộc.
6. Vô trùng mẫu cấy
Phương pháp phổ biến hiện nay là dùng các hóa chất có khả năng tiêu diệt vi sinh vật để vô trùng mẫu cấy. Hiệu quả diệt nấm khuẩn của các chất này phụ thuộc vào thời gian, nồng độ xử lý và khả năng xâm nhập của chúng vào các ngõ ngách trên bề mặt mẫu cấy. Các hóa chất được lựa chọn trong quá trình vô trùng mẫu cấy phải đảm bảo 2 thuộc tính: có khả năng diệt nấm khuẩn tốt và không có hoặc có mức độ độc thấp đối với mẫu thực vật. Để tăng khả năng diệt nấm khuẩn người ta thường lắc mẫu cấy với cồn 700 trong 30 giây đến 1 phút, sau đó mới xử lý với dung dịch diệt nấm khuẩn. Đồng thời người ta có thể thêm các chất làm tăng sức căng bề mặt như Tween 80 vào dung dịch diệt nấm khuẩn.

184
Bảng 4.2. Các dung dịch diệt nấm khuẩn, nồng độ và thời gian xử lý
Tác nhân vô trùng Nồng độ Thời gian xử lý Hiệu quả
Calcium hypochlorid 9-10% 5 - 30' Rất tốt Sodium hypochlorid 2% 5 - 30' Rất tốt H2O2 10-12% 5 - 15' Tốt Nước Brôm 1-2% 2 - 10' Rất tốt HgCl2 0,1-1% 2 - 10' Khá tốt
Vô trùng mẫu cấy là một thao tác quan trọng nhưng ít khi thành công ngay
lần đầu tiên. Tuy nhiên nếu kiên trì tìm nồng độ và thời gian thích hợp để khử trùng thì sau vài lần thử chắc chắn sẽ đạt kết quả. II. THỤ PHẤN VÀ NUÔI CẤY PHÔI (HỮU TÍNH) IN VITRO
Ở thực vật có hoa, phôi là những thực thể có hình thái rõ rệt và đảm nhiệm chức năng trong giai đoạn trung gian của thời kỳ chuyển tiếp giữa pha giao tử thể và bào tử thể. Ở thực vật bậc cao, các phôi trong hạt phát triển từ sản phẩm kết hợp giữa các loại giao tử khác nhau và được gọi là phôi hữu tính. Bên cạnh các phôi hữu tính, trong quá trình nuôi cấy in vitro thực vật còn có khả năng phát sinh phôi từ những tế bào sinh dưỡng được gọi là phôi vô tính.
Nuôi cấy phôi hữu tính là một hướng nghiên cứu in vitro được sử dụng trong nhân giống và chọn giống thực vật. Hanning (1904) là người đầu tiên đã thí nghiệm nuôi cấy phôi trưởng thành của cây Cochleria và Raphanus. Sau đó là nhiều công trình của các nhà nghiên cứu khác nuôi phôi tách rời từ các hạt chính. Dieterich (1942) cho rằng môi trường khoáng Knop bán lỏng có chứa 2,5-5,0% saccharose có thể duy trì phôi phát triển bình thường.
Laibach (1925, 1929) đã thí nghiệm với tổ hợp lai khác loài giữa Linim perenne và L.austriacum và nhận thấy hạt của chúng không có khả năng nảy mầm. Bằng cách tách phôi từ hạt và nuôi cấy in vitro, ông đã thu nhận được cây lai. Kể từ đó nuôi cấy phôi hữu tính tách rời đã được sử dụng rộng rãi trong việc tạo các cá thể lai mà phương pháp lai truyền thống khó thực hiện được.
1. Tính bất hợp của giao tử trước khi thụ tinh - thụ phấn in vitro
Trong tự nhiên quá trình thụ phấn của thực vật xảy ra theo trình tự hạt phấn chín rơi trên nuốm nhụy nảy mầm tạo thành ống phấn, ống phấn xuyên dọc vòi nhụy tiếp cận túi phôi. Hai tinh tử đơn bội của hạt phấn tiến hành thụ tinh kép. Nếu một hạt phấn lạ của loài khác rơi trên núm nhụy, nhụy sẽ tạo ra một số chất ức chế sự phát triển của ống phấn, làm biến dạng ống phấn ngăn cản sự thụ tinh đó chính là tính bất hợp của giao tử trước khi thụ tinh.
Để khắc phục trở ngại đó, người ta tiến hành nghiên cứu quá trình thụ phấn trong ống nghiệm.
Quá trình thụ phấn - tạo hợp tử in vitro gồm các bước: - Kích thích hạt phấn nảy mầm

185
- Kích thích sinh trưởng ống phấn - Nuôi noãn và thụ tinh noãn - Nuôi hợp tử thành hạt
Một thời gian dài phương pháp này chỉ thành công ở một số thực vật thuộc họ Papaveraceae, họ Caryophyllaceae, họ Solanaceae do ở các họ này bầu chứa nhiều noãn nên dễ nuôi cấy.
Ở họ hòa thảo Gramineae bầu chỉ chứa một noãn nên rất khó nuôi cấy đồng thời hạt phấn cũng rất khó kích thích nảy mầm.
2. Tính bất hợp của giao tử sau khi thụ tinh - Nuôi cấy phôi in vitro
Trong một số trường hợp khác khi hạt phấn lạ rơi lên nuốm nhụy, ống phấn vẫn phát triển bình thường và quá trình thụ tinh xảy ra nhưng hạt không phát triển được. Nguyên nhân là giữa nội nhũ và phôi đã hình thành cơ chế ức chế sự phát triển của phôi, đây là trường hợp hay gặp khi tiến hành lai xa.
3. Kỹ thuật nuôi cấy phôi hữu tính Có 2 vấn đề quan trọng trong nuôi cấy phôi hữu tính là kỹ thuật tách phôi và
thành phần của môi trường nuôi cấy. - Tách phôi
Nuôi cấy phôi hữu tính được khởi đầu bằng quá trình tách phôi từ vật liệu thực vật. Ở những loài cây họ đậu, phôi thường có kích thước lớn, rất thuận tiện cho quá trình thu nhận phôi.
Các phôi hữu tính được hình thành trong môi trường vô trùng của mô noãn và mô bầu hoa. Do đó, trong nhiều trường hợp có thể tách phôi ngay và đưa vào môi trường nuôi cấy ở điều kiện vô trùng.
Trong quá trình thu nhận phôi, cần hạn chế đến mức thấp nhất sự tổn thương của dây treo phôi. Dây treo phôi sẽ phát triển đến kích cỡ lớn nhất khi phôi hoàn thành giai đoạn phát triển hình cầu và bắt đầu chuyển sang giai đoạn hình tim. Do có kích thước nhỏ và cấu trúc mong manh cho nên khó tách được dây treo phôi còn nguyên vẹn cùng với phôi để đưa vào nuôi cấy. Một nghiên cứu gần đây cho thấy dây treo phôi có vai trò quan trọng đối với sự sinh trưởng và phát triển của phôi non. Tỷ lệ hình thành cây con từ phôi non nuôi cấy đã giảm đáng kể khi loại bỏ dây treo. Điều này đã được xác nhận trong nuôi cấy phôi non của các loài Capesella bursa-pastoris (Monnier, 1978), Phaseolus coccineus (Yeung và Sussex, 1979). - Thành phần môi trường
Nhiều tổ hợp muối khoáng khác nhau đã được sử dụng trong nuôi cấy phôi, trong đó môi trường MS có biểu hiện kích thích sinh trưởng phôi mạnh nhất đối với nhiều loại thực vật, tuy nhiên tỷ lệ sống của phôi thấp. Trên cơ sở môi trường MS, Monnier đã đưa ra môi trường mới đảm bảo tỷ lệ sống cao của phôi và khả năng thúc đẩy sinh trưởng tốt đối với phôi của một số loài thực vật. Thành phần môi trường này có hàm lượng cao các ion K+, Ca2+ nhưng hàm lượng NH4
+ thấp. Trong môi trường nuôi cấy phôi hữu tính, đường đóng vai trò rất quan trọng.
Trong nhiều trường hợp đường saccharose cho kết quả tốt hơn nhiều các đường

186
khác. Nồng độ saccharose có thể dùng 0,5-18%. Ngoài ra một số chất tự nhiên như nước dừa, dịch chiết malt, casein thủy phân là những chất cần trong nuôi cấy phôi.
Các chất kích thích sinh trưởng như GA3, auxin, cytokinin thường được dùng trong nhiều loại nuôi cấy phôi. Auxin thường được dùng với nồng độ thấp. Cytokinin thường dùng trong nuôi cấy phôi là kinetin do có vai trò đặc biệt trong sự phát triển của phôi. GA3 có vai trò thúc đẩy sự nảy mầm sớm của phôi như quá trình nuôi cấy phôi lúa mỳ.
Muốn thành công trong lai xa không có con đường nào khác là tìm ra kỹ thuật vượt qua sự bất tuơng hợp giữa phôi và nội nhũ sau khi lai.
Kỹ thuật nuôi phôi phân lập có khả năng giải quyết tốt vấn đề này. LaiBach lần đầu tiên nuôi được phôi non của cây lai bất dục thành cây non khoẻ mạnh.
4. Một số khó khăn trong nuôi cấy phôi hữu tính 4.1. Môi trường dinh dưỡng
Các môi trường dinh dưỡng hiện nay đang sử dụng đang có áp suất thẩm thấu nhỏ hơn dịch noãn được nuôi cấy trên môi trường đó. Rất có thể môi trường không thích hợp, vì vậy người ta đang tìm cách sử dụng dịch chiết của nội nhũ. Năm 1974, Kruse đưa ra công thức "vú em" gồm có môi trường dinh dưỡng, nội nhũ khoẻ và phôi lai. Vai trò của nội nhũ khỏe cho phôi lai những chất cần thiết, cây cho nội nhũ có thể là cây khác loài của cùng một chi.
4.2. Phát sinh mô sẹo
Khi nuôi phôi non của tổ hợp lai: Hordeum vulgare × Secale cereale các tác giả Clauss và Kunert thấy phôi phát triển thành khối mô sẹo điều này chỉ có thể giải thích là do mối tương tác giữa phôi và môi trường dinh dưỡng không bình thường như giữa phôi và nội nhũ, dù sau đó có thể tái sinh cây hoàn chỉnh từ khối mô sẹo thì cây tái sinh cũng sẽ mang những thay đổi vì mô sẹo thường không ổn định về mặt di truyền.
III. NHÂN GIỐNG IN VITRO 1. Ý nghĩa của việc nhân giống cây trồng bằng phương pháp nuôi cấy mô tế bào
Phương pháp nhân giống in vitro thực chất là một tiến bộ vượt bậc của các phương pháp nhân giống vô tính cổ điển như giâm cành, giâm chồi, chiết, ghép... Một trong những ưu việt của phương pháp nhân giống cây trồng bằng phương pháp nuôi cấy mô tế bào thực vật là việc sử dụng mô nuôi cấy có kích thước nhỏ. Với kích thước nhỏ như vậy, sự tương tác giữa các tế bào trong mô cũng như sự tiếp xúc với môi trường dinh dưỡng dễ dàng và đơn giản hơn nhiều. Do đó mô nuôi cấy dễ dàng phân hóa và tái sinh.
Ngoài ra, vi nhân giống còn có những ưu việt mà các phương pháp khác không có như: có thể nhân giống cây trồng ở quy mô công nghiệp, phương pháp này có hệ số nhân rất cao và cho các cá thể hoàn toàn đồng nhất về mặt di truyền...
Có thể kể đến một vài ứng dụng cụ thể của vi nhân giống như sau: - Duy trì và nhân nhanh các kiểu gene hiếm làm vật liệu cho công tác chọn
tạo giống.

187
- Nhân nhanh với hiệu quả kinh tế cao các loài hoa và cây cảnh gặp nhiều hạn chế khi trồng bằng hạt.
- Nhân nhanh và duy trì các cá thể đầu dòng tốt để cung cấp hạt giống các loài rau, cây cảnh và các cây trồng khác.
- Nhân nhanh và kinh tế các kiểu gene quý của cây lấy gỗ trong lâm nghiệp và gốc ghép trong nghề trồng cây ăn trái, cây cảnh. 2. Các phương thức nhân giống in vitro
2.1. Tái sinh cây mới từ cấu trúc sinh dưỡng 2.1.1. Nuôi cấy mô phân sinh (đỉnh sinh trưởng)
Khái niệm mô phân sinh chỉ đúng khi mẫu vật nuôi cấy được tách từ đỉnh sinh trưởng có kích thước trong vòng 0,1mm tính từ chóp của tháp sinh trưởng. Trong thực tế thường gặp khó khăn lớn trong việc nuôi thành công các đỉnh sinh trưởng riêng rẽ như vậy nên người ta chỉ áp dụng hình thức nuôi cấy mô phân sinh với mục đích làm sạch virus cho cây trồng.
Độ lớn của chồi (mô) nuôi cấy có mối tương quan chặt chẽ với tỷ lệ sống, khả năng cảm ứng và mức độ ổn định về mặt di truyền. Nếu độ lớn chồi tăng thì tỷ lệ sống và tính ổn định tăng, nhưng có thể làm giảm khả năng cảm ứng của chồi/mô nuôi cấy và ngược lại. Tuy nhiên, nếu xét về hiệu quả kinh tế (thể tích bình nuôi, lượng dung dịch môi trường dinh dưỡng ...): nếu độ lớn tăng thì hiệu quả kinh tế giảm, nếu độ lớn giảm thì hiệu quả kinh tế tăng. Do đó, phải có sự kết hợp hài hòa giữa các yếu tố trên để tìm ra kích thước mẫu tối ưu.
Ở các đối tượng như phong lan, dứa, mía, chuối, với mục đích nhân giống in vitro đỉnh sinh trưởng được tách với kích thước 5-10 mm nghĩa là bao gồm toàn bộ mô phân sinh và một phần mô xung quanh.
Một đỉnh sinh trưởng nuôi cấy ở điều kiện thích hợp sẽ tạo ra một hay nhiều chồi và mỗi chồi sẽ phát triển thành một cây hoàn chỉnh. Xét về nguồn gốc của các cây đó có 3 khả năng:
- Cây phát triển từ chồi đỉnh (chồi ngọn)
- Cây phát triển từ chồi nách phá ngủ
- Cây phát triển từ chồi mới phát sinh
Có hai phương thức phát triển cây hoàn chỉnh từ nuôi cấy đỉnh sinh trưởng:
- Phát triển cây trực tiếp Hình thức này thường gặp chủ yếu ở các đối tượng cây hai lá mầm
(dicotyledone) như: hoa cúc, cẩm chướng, khoai tây, thuốc lá ...
Quá trình tái sinh: Mầm (đỉnh sinh trưởng) Chồi nách Cây
Hình 4.5. Mô phân sinh đỉnh

188
- Phát triển cây thông qua giai đoạn protocorm Hình thức này thường gặp chủ yếu ở các đối tượng cây một lá mầm
(monocotyledone) như phong lan, dứa, huệ...từ một đỉnh sinh trưởng ban đầu cùng lúc tạo ra hàng loạt protocorm (proembryo) và các protocorm này có thể tiếp tục phân chia thành các protocorm mới hoặc phát triển thành cây hoàn chỉnh. Bằng phương thức này chỉ trong một thời gian ngắn người ta có thể thu được hàng triệu cá thể. Quá trình tái sinh:
Mầm (đỉnh sinh trưởng) Protocorm Cây
Hình thức nhân giống này đã được áp dụng thành công trên nhiều đối tượng cây trồng, đặc biệt là các đối tượng hoa lan, và mang lại hiệu quả kinh tế đặc biệt cao.
2.1.2. Tái sinh cây hoàn chỉnh từ các bộ phận khác của cây Ngoài mô phân sinh và đỉnh sinh trưởng là bộ phận nuôi cấy dễ thành công
thì các bộ phận còn lại của cơ thể thực vật (ngoại trừ các mô đã hóa gỗ) cũng đều có thể sử dụng cho việc nhân giống in vitro được, chẳng hạn như:
- Đoạn thân: thuốc lá, cà chua, cam, chanh, cúc...
- Mảnh lá: thuốc lá, cà phê, ca cao, lan...
- Các bộ phận của hoa: cúc, đồng tiền, súp lơ, thuốc lá...
- Vảy củ: hành, lyli, huệ...
- Đoạn mầm: măng tây, dâu tây...
Quá trình tái sinh:
Mô Chồi Cây Sự phát sinh hình thái trực tiếp từ mẫu nuôi cấy bắt đầu từ các tế bào nhu mô
nằm ở trong biểu bì hoặc ngay phía dưới bề mặt của thân. Một số tế bào này trở thành mô phân sinh và các túi nhỏ gọi là thể phân sinh (meristemoids) phát triển. Khả năng cảm ứng của mô thực vật cũng tùy thuộc vào nồng độ của các chất điều hòa sinh trưởng thực vật (phytohormone), chủ yếu phụ thuộc vào tỷ lệ nồng độ giữa auxin và cytokinin. Tỷ lệ này lớn hơn 1 thì thường có khuynh hướng tạo rễ; tỷ lệ này nhỏ hơn 1 thường có khuynh hướng tạo chồi; nếu tỷ lệ này bằng 1 thì mô nuôi cấy thường phát sinh callus.
2.2. Nhân giống in vitro thông qua giai đoạn mô sẹo (callus)
Nếu tái sinh cây hoàn chỉnh trực tiếp từ mẫu vật cấy ban đầu thì thu được nhanh chóng các cây khá đồng nhất về mặt di truyền, tuy nhiên trong nhiều trường hợp mô nuôi cấy không tái sinh cây ngay mà phát triển thành khối mô sẹo. Tế bào mô sẹo khi cấy chuyền nhiều lần sẽ không ổn định về mặt di truyền, để tránh tình trạng đó cần sử dụng mô sẹo vừa phát sinh để thu được cây tái sinh đồng nhất về di truyền.
Hình 4.5. chồi bất định

189
Quá trình tái sinh: Mô mô sẹo chồi, phôi vô tính cây
mảnh lá mô sẹo hình thành trên bề mặt lá khối mô sẹo
không phân hóa
Hình 4.6. mô sẹo phát sinh từ nuôi cấy mảnh lá
2.3. Nhân giống thông qua phát sinh phôi vô tính - công nghệ hạt nhân tạo 2.3.1. Phôi vô tính
Street và Reinert (1958) là hai tác giả đầu tiên mô tả sự hình thành phôi vô tính từ các tế bào đơn của cà rốt (Daucus carota). Ở một số loài, sự phát sinh phôi vô tính hình thành trực tiếp từ phôi bất định (adventitious embryos) nằm trong phôi tâm (nucellar embryos). Đến nay, công nghệ phôi vô tính được coi là công nghệ rất có triển vọng cho nông nghiệp trong thế kỷ 21.
Phôi vô tính là các cá thể nhân giống (propagules) có cực tính bắt nguồn từ các tế bào soma. Chúng rất giống phôi hữu tính (zygotic embryo) ở hình thái, quá trình phát triển và sinh lý, nhưng do không phải là sản phẩm của quá trình thụ tinh giữa giao tử đực và giao tử cái, vì vậy không có quá trình tái tổ hợp di truyền (geneetic recombination) do đó các phôi vô tính có nội dung di truyền giống hệt với các tế bào soma đã sinh ra chúng.
Ở trường hợp phôi hữu tính, sự kết hợp giữa giao tử đực và cái cho ra hợp tử (zygote). Hợp tử phân chia nhiều lần tạo nên phôi hữu tính có cấu trúc hai cực: rễ và ngọn. Khi hợp tử phát triển, miền sinh trưởng rễ và miền sinh trưởng ngọn cùng phát triển và cuối cùng tạo nên cây hoàn chỉnh, qua các giai đoạn phôi học như sau: - Trường hợp cây hai lá mầm: Dạng cầu dạng thủy lôi dạng có lá mầm - Trường hợp cây một lá mầm: Dạng cầu dạng scutellar dạng diệp tiêu

190
Hình 4.7. Giai đoạn phôi hình cầu, phôi hình thủy lôi và cây tái sinh từ phôi vô tính của cây cam
Ở rất nhiều cây, người ta nhận thấy các tế bào đang phân chia vô tổ chức đã
tạo nên callus khi nuôi cấy. Có thể thay đổi hướng phát triển của chúng để tạo ra các phôi vô tính với các bước phát sinh hình thái rất giống với trường hợp phôi hữu tính. Điểm khác nhau cơ bản giữa phôi hữu tính và phôi vô tính là phôi hữu tính luôn luôn đi kèm với nội nhũ là cơ quan dự trữ năng lượng và chất dinh dưỡng phục vụ cho quá trình nảy mầm, còn ở phôi vô tính hoàn toàn không có nội nhũ.
Khả năng tạo phôi vô tính trong nuôi cấy mô thực vật, ngoài các điều kiện môi trường, điều kiện nuôi cấy, còn phụ thuộc rất lớn vào loài, vào các giống (cultivars), dòng (strains) trong cùng một loài. Khả năng này đã được chứng minh là do một hoặc vài gene phụ trách. Vì vậy, bằng biện pháp lai tạo có thể chuyển khả năng tạo phôi vô tính cao từ cây này qua cây khác.
2.3.2. Công nghệ hạt nhân tạo
Hạt nhân tạo là phôi vô tính bọc trong một lớp vỏ polymer như agar, agarose, alginate... Trong cấu trúc lưới của lớp vỏ đó, nước, chất dinh dưỡng được cung cấp thay cho nội nhũ, giúp cho phôi vô tính có thể nảy mầm trở thành cây hoàn chỉnh.
Hạt nhân tạo gồm có ba phần: - Phôi vô tính - Vỏ bọc polymer - Màng ngoài
Có nhiều loại polymer tự nhiên đã được thử nghiệm dùng cho công nghệ phôi vô tính, trong đó alginate đựoc coi là tốt nhất. Alginate la một polymer sinh học, được chiết từ rong biển, chủ yếu là các loài thuộc chi Sargassum. Alginate do các phân tử manuronic acid gắn với nhau tạo thành, giống như các phân tử glucose tạo nên cellulose. Đặc điểm quan trọng nhất của alginate là chúng ở dạng hòa tan trong nước khi kết hợp với các ion hóa trị một như: Na+, K+, NH4
+... và lập tức chuyển sang dạng không tan trong nước khi kết hợp với các ion hóa trị hai hoặc đa hóa trị như: Ca2+, Mg2+, Al3+... Nếu nhỏ một giọt dung dịch sodium alginate vào dung dịch CaCl2 thì sodium alginate ở phần diện tích ngoài của giọt sẽ chuyển hóa ngay thành calcium alginate và tạo nên một màng không thấm nước hình thành các viên alginate.
2.4. Nhân giống trong các nồi phản ứng sinh học
Trước đây, các nồi phản ứng sinh học (bioreactor) hay còn gọi các bình lên men (fermenter) chủ yếu được dùng cho công nghệ vi sinh. Ngày nay, trên cơ sở các thiết bị đó, với một số cải tiến, nhiều tác giả đã nhân giống thành công phôi vô tính, các thể chồi, cụm chồi hoặc củ nhỏ trên nhiều đối tượng thực vật như cà phê, dứa, khoai tây...
Phôi vô tính cà phê được sản xuất thành công ở Brasil trong các nồi phản ứng sinh học có dung tích 2-4 lít. Nồi vận hành theo các nguyên tắc của một bình lên men, có thể có cánh khuấy hoặc dùng bọt khí thổi từ dưới lên. Mỗi mẻ có thể thu được 4-5 triệu phôi vô tính cà phê. Có một phương pháp nữa là thay vì bơm khí vào bình phản ứng, dịch lỏng nuôi cấy (môi trường mới) được bơm vào nồi và hút

191
ra (môi trường cũ) theo chu kỳ ngắn, nhờ vậy mô và tế bào thực vật có đủ oxy và chất dinh dưỡng để phát triển mạnh. Phương pháp này được ứng dụng thành công khi nuôi cấy cụm chồi dứa.
Ngoài ra, người ta có thể sản xuất củ giống trong các nồi lên men chẳng hạn như sản xuất giống khoai tây củ bi.
Tóm lại có các phuơng thức phổ biến để tạo cây hoàn chỉnh trong nhân giống in vitro
3. Các giai đoạn trong quy trình nhân giống vô tính in vitro Quy trình nhân giống vô tính in vitro được thực hiện thông qua các bước sau
3.1. Giai đoạn I - cấy khởi đầu
- Lựa chọn mẫu vật Mẫu dùng trong nuôi cấy mô tế bào thực vật có thể là hầu hết các cơ quan
hay bộ phận của cây như chồi ngọn, chồi bên, phiến lá, cuống lá, vảy củ, đoạn thân, cuống hoa... Tùy theo điều kiện tiếp xúc với môi trường mà các mẫu thực vật có chứa ít hay nhiều mầm bệnh. Vì vậy trước khi tiến hành nhân giống in vitro cần phải chọn lọc cẩn thận các cây mẹ (cây cho nguồn mẫu nuôi cấy). Các cây này cần phải khỏe mạnh, không bị bệnh. Việc lựa chọn mẫu cũng cần phải căn cứ vào trạng thái sinh lý và tuổi của mẫu. Mẫu non có mức độ nhiễm mầm bệnh ít hơn, phản ứng với môi trường nhanh, dễ tái sinh, đặc biệt là trong nuôi cấy mô sẹo, phôi.
- Nuôi cấy khởi động Đưa mẫu vật từ ngoài vào nuôi cấy vô trùng phải đảm bảo yêu cầu sau:
- Tỷ lệ nhiễm thấp - Tỷ lệ sống cao - Tốc độ sinh trưởng nhanh
Kết quả bước này phụ thuộc nhiều vào cách lấy mẫu như đã trình bày ở trên. Chọn đúng phương pháp khử trùng sẽ đưa lại tỷ lệ sống cao, và môi trường dinh dưỡng thích hợp sẽ đạt được tốc độ sinh trưởng nhanh
Cây trưởng thành
Nuôi cấy cơ quan
Nuôi cấy callus
Nuôi cấy phôi
Nuôi cấy tế bào
chồi cây
callus chồi cây
callus tế bào đơn
phôi cây
chồi, cụm chồi, phôi, củ nhỏ
cây

192
Môi trưòng dinh dưỡng phổ biến thường được dùng nhiều nhất hiện nay là môi trường khoáng MS.
Tùy theo từng loài, từng bộ phận nuôi cấy và từng mục đích nuôi cấy mà bổ sung các hàm lượng và thành phần phytohormone khác nhau.
Một số môi trường dinh dưỡng thích hợp: - Muối khoáng: môi trường White (1943), Heller (1953), Murashige-
Skoog (1962), Vacin-Went. - Chất hữu cơ:
Saccharose: 1-6% Vitamin: B1, B6, inositol, acid nicotinic Amino acid: Arg, Asg, Tyr, Glu, …
- Phytohormone: Auxin: IAA, IBA, NAA, 2,4-D Cytokinin: BAP Gibberellin: GA3
3.2. Giai đoạn II - nhân nhanh Ở giai đoạn này bao gồm nhiều lần cấy chuyền mô lên các môi trường nhân
nhanh nhằm kích thích tạo cơ quan phụ hoặc cấu trúc khác mà từ đó cây hoàn chỉnh có thể phát sinh. Những khả năng tạo cây đó là:
- Phát triển chồi nách - Tạo phôi vô tính - Tạo đỉnh sinh trưởng mới
Trong giai đoạn này cần nghiên cứu các tác nhân kích thích phân hóa cơ
quan, đặc biệt là chồi: - Bổ sung tổ hợp hormone sinh trưởng mới: tăng cytokinin giảm auxin. Tăng
tỷ lệ auxin/cytokinin sẽ kích thích mô nuôi cấy tạo rễ và ngược lại sẽ kích thích phát sinh chồi.
- Tăng cường ánh sáng 16 h/ngày với cường độ tối thiểu 1000 lux. Ánh sáng tím là thành phần quan trọng kích thích phân hóa mạnh. Ánh sáng đỏ có ảnh hưởng giống cytokinin, nó tạo nên sự tích lũy cytokinin trong mô của một số loài, chính lượng cytokinin này đã góp phần kích thích quá trình phát sinh cơ quan và tạo chồi từ những mô nuôi cấy in vitro.
- Bảo đảm chế độ nhiệt 20-27°C

193
Hình 4.8. Ảnh hưởng của tỷ lệ auxin/cytokinin lên khả năng phát sinh cơ quan của mô nuôi cấy
Tuy nhiên, cần lưu ý ảnh hưởng của số lần cấy chuyền đến sự sinh trưởng
của mô nuôi cấy. Nếu số lần cấy chuyền quá lớn thì sẽ dẫn đến hiện tượng cây bị thoái hóa và sinh trưởng kém, có thể mất đi những đặc tính ban đầu của cây mẹ. Do đó, ngoài mục đích là xác định phương thức nhân nhanh bằng môi trường dinh dưỡng và điều kiện khí hậu tối thích, ở giai đoạn này chúng ta cần phải xác định số lần cấy chuyển hợp lý.
3.3. Giai đoạn III - tạo rễ
Giai đoạn này bao gồm quá trình kéo dài chồi để đạt kích thước thích hợp và tạo rễ, tức là quá trình tạo cây hoàn chỉnh. Môi trường lúc này không cần bổ sung cytokinin mà có thể chỉ bổ sung auxin. Đối với một số loài, mô/chồi khi đạt kích thước thích hợp thì rễ tự phát sinh không cần phải bổ sung auxin ví dụ như các loài hoa lan, cúc, cẩm chướng, lyli… Tuy nhiên đối với một số loài đặc biệt là các loài thân gỗ thì việc bổ sung auxin là rất cần thiết. Nồng độ auxin tối ưu được xác định dựa trên tỷ lệ tạo rễ, số lượng rễ và chiều dài rễ. Độ dài của rễ không được quá lớn để tránh đứt, gãy, hư hỏng khi chuyển cây in vitro ra ngoài môi trường.
Ví dụ như khi nghiên cứu ảnh hưởng của IBA lên khả năng tạo rễ của chồi Aronia arbutifolia, người ta xác định được nồng độ 1,0 mg/l là tối ưu vì ở nồng độ này tỷ lệ tạo rễ và số lượng rễ/chồi cao nhưng chiều dài rễ thấp.
Bảng 4.3. Ảnh hưởng của IBA lên khả năng tạo rễ của chồi Aronia
arbutifolia sau 4 tuần nuôi cấy
IBA (mg/l) Tỷ lệ tạo rễ Số lượng rễ Chiều dài rễ (mm)
0,0 37 2,4 23,2 0,05 43 3,5 18,1 0,1 55 4,1 14,2 0,5 71 5,7 6,5 1,0 84 7,1 4,3

194
(nguồn: Trigiano, R.N. và Gray, D.J., Plant Tissue Culture Concept and
Laboratory Exercises, First Edition, CRC Press LLC, Boca Raton, FL, 1996.)
3.4. Giai đoạn IV - đưa ra ngoài đất Đây là giai đoạn quan trọng bao gồm công việc huấn luyện cây in vitro thích
nghi với điều kiện khí hậu thay đổi: nhiệt độ, ánh sáng, độ ẩm, sự mất nước, sâu bệnh và chuyển từ trạng thái dị dưỡng sang tự dưỡng hoàn toàn. Các nghiên cứu về cấu trúc của lá khoai tây nuôi cấy in vitro so với lá khoai tây trồng bên ngoài cho thấy chúng rất khác nhau, điều đó chứng tỏ phải tiến hành thích nghi dần cây nhân giống in vitro với điều kiện tự nhiên. Quá trình thích nghi ở đây phải được hiểu như là quá trình thay đổi những đặc điểm sinh lý, giải phẫu của bản thân cây non đó. Thời gian tối thiểu cho sự thích nghi là 2-3 tuần, trong thời gian này cây phải được bảo vệ trước những yếu tố bất lợi :
- Mất nước nhanh làm cho cây bị héo khô - Nhiễm vi khuẩn và nấm làm cho cây bị thối mục - Cháy lá do nắng
Hình 4.9. Các giai đoạn nuôi cấy mô
3.5. Một số lưu ý - Việc sản sinh các hợp chất độc từ mô nuôi cấy
Trong nuôi cấy mô thường quan sát thấy hiện tượng hóa nâu hay đen mẫu. Hiện tượng này là do mẫu nuôi cấy có chứa nhiếu chất tanin hoặc hydroxyphenol (thường có nhiều ở mô già).
Các phương pháp loại trừ hóa nâu: + Bổ sung than hoạt tính vào môi trường nuôi cấy (0,1 - 0,3%). Phương
pháp này đặc biệt có hiệu quả trên các loài phong lan như Phalenopsis, Cattleya. Tuy nhiên, than hoạt tính có thể làm chậm quá trình sinh trưởng của cây do than hoạt tính có khả năng hấp phụ một số chất điều tiết sinh trưởng và dinh dưỡng.

195
+ Bổ sung polyvinyl pyrolindone (PVP) có tác dụng khử nâu hóa tốt ở mẫu một số cây ăn quả (táo, hồng).
+ Sử dụng mô non, gây vết thương nhỏ nhất khi khử trùng. + Ngâm mẫu vào dung dịch acid ascorbic hoặc citric vài giờ trước khi cấy. + Nuôi cấy mẫu trong môi trường lỏng, oxy thấp, không có ánh sáng (1 - 2
tuần). + Cấy chuyển mẫu liên tục trong 1-2 tuần
- Hiện tượng thủy tinh hóa Trong quá trình nhân nhanh in vitro thường xuất hiện hiện tượng cây bị
“thủy tinh hóa” làm cho thân lá mọng nước, trong suốt, cây rất khó sống khi đưa ra ngoài môi trường do bị mất nước rất mạnh. Hiện tượng này thường xảy ra khi nuôi cấy trong môi trường lỏng hay môi trường ít agar (sự trao đổi khí thấp).
Để tránh hiện tượng thủy tinh hóa có thể tiến hành một số giải pháp như: + Giảm sự hút nước bằng cách tăng nồng độ đường hoặc các chất gây thẩm
thấu cao. + Giảm nồng độ các chất chứa nitơ trong môi trường. + Giảm sự sản sinh ethylen trong bình nuôi cấy + Xử lý abscicic acidhoặc một số chất ức chế sinh trưởng. + Tăng cường độ ánh sáng và giảm nhiệt độ phòng nuôi cây.
4. Nhân giống in vitro và các đặc điểm không di truyền Bên cạnh những đặc điểm di truyền ở thực vật người ta còn phát hiện được
hàng loạt đặc điềm (hình thái và sinh lý) không di truyền, đó là các đặc điểm epigeneetic. Quá trình nhân giống in vitro có ảnh hưởng tới các đặc điểm không di truyền này.
4.1. Hiện tượng các đặc điểm epigeneetic được lưu lại
Nghiên cứu nhân giống vô tính về cây Phyllanthus amarys cho thấy: đây là loài cây có thân thẳng đứng và cành ngang giống lá kép nhưng lại ở nách lá và có hoa nên tạm coi như là cành ngang. Lá ở thân đứng xếp theo kiểu xoắn ốc, nhưng ở cành ngang là tương đối so le. Ở nách lá có cành ngang, chồi nách phát triển mạnh thành cành thẳng đứng như thân. Nếu cắt cành ngang về ươm thì sẽ thu được cành ngang dài hàng mét. Cành ngang phát triển vô hạn theo một chương trình “ngang” định trước. Nếu ươm cành thẳng thì sẽ thu được cây thẳng đứng. Như vậy, mô phân sinh của cành khác mô phân sinh của thân và khi nhân giống vô tính các bộ phận khác nhau sẽ thu được các dạng cây khác nhau. Ở đây, mô phân sinh đỉnh điều khiển mô phân sinh cành phát triển theo hướng ngang. Nếu cắt bỏ mô phân sinh đỉnh sớm thì cành ngang sẽ phát triển thành cành đứng.
Tế bào sinh trưởng đỉnh điều khiển sự hoạt động của các tế bào khác (tương tự như ở động vật). Hiện tượng điều khiển hướng phát triển này có một ý nghĩa thực tiễn sâu sắc.

196
Các cây cà phê và ca cao cũng có đặc điểm xếp cành tương tự như vậy, do đó khi tiến hành nhân giống vô tính các loài cây này phải chú ý đến hiện tượng điều khiển hướng phát triển như ở Phyllanthus. 4.2. Hiện tượng các đặc điểm epigeneetic không được lưu lại
Nhân giống vô tính in vitro giống dứa Cayen không gai thông qua phân chia protocorm người ta thu được một tỷ lệ đáng kể các cá thể có gai.
Bình thường trong kỹ thuật trồng dứa người ta sử dụng hom dứa có kích thước khá lớn 15-30 cm. Đặc điểm không gai đã được chương trình hóa trong các đọt có kích thước lớn như vậy. Vậy thì tại sao khi tách đỉnh sinh trưởng và nuôi cấy thành protocorm và cây dứa non thì gai lại xuất hiện? Có thể ở giai đoạn sinh lý sớm của protocorm và quá trình nuôi cấy đỉnh sinh trưởng phân lập, mô phân sinh của những cây dứa non tương lai đã được giải phóng một phần khỏi ảnh hưởng của tổ chức điều khiển và vì thế chúng phát triển tự do theo đặc điểm có gai của thế hệ xa xưa.
Hiện nay, vấn đề này đang được quan tâm vì một số đặc tính chống chịu như chịu mặn, chịu bệnh, chịu lạnh là những đặc tính epigeneetic và liệu những đặc tính này có còn được lưu giữ lại thông qua nhân giống in vitro hay không.
IV. NUÔI CẤY BAO PHẤN VÀ TẠO GIỐNG CÂY TRỒNG 1. Vấn đề đơn bội ở thực vật
Hầu hết các loại cây trồng đều có mức bội thể lớn hơn 1, phổ biến là nhị bội (2n) và tứ bội (4n). Vậy mỗi đặc điểm di truyền ở những cơ thể này đều bị 2 hay nhiều allele của một gene chi phối. Nếu đó là cơ thể dị hợp tử thì sự biểu hiện tính trạng của gene đó phụ thuộc vào tính trạng lặn hay trội của chúng. Vì vậy, mức bội thể lý tưởng để tiến hành nghiên cứu di truyền tính trạng phải là mức đơn bội hoặc là các mức đa bội khác nhưng chúng phải đồng nhất tuyệt đối. Trong cơ thể thực vật chỉ có thể giao tử (hạt phấn, noãn chưa thụ tinh) là những tế bào đơn bội. Vì vậy, bằng phương pháp nuôi cấy hạt phấn, bao phấn hay noãn chưa thụ tinh có thể tái tạo được cây trực tiếp thông qua sự phân chia và biệt hóa của tế bào. Những cây tái sinh đó chính là những cây đơn bội (n).
Người ta có thể thu được đơn bội trong tự nhiên bằng hai cách: - Chọn lọc đơn bội xuất hiện ngẫu nhiên với tỷ lệ rất thấp - Lai xa và sau khi một bộ nhiễm sắc thể thoái biến sẽ còn lại một bộ nhiễm sắc thể đơn bội.
Cả hai cách này mới chỉ thành công ở một số cây thuộc họ hòa thảo. Năm 1924, Blakeslee và cộng sự đã chứng minh rằng có thể thu được các
dòng nhị bội thuần đồng hợp bằng cách nhị bội hóa các thể đơn bội. Guka và Maheshwari (1964) là hai nhà khoa học Ấn Độ lần đầu tiên đã tạo thành công cây đơn bội qua nuôi cấy bao phấn cà độc dược. Nhờ những đóng góp đáng kể của Nitsch (1973-1976) và Noieel (1973), phương pháp này đã trở thành phương pháp rất hiệu quả để tạo thành hàng loạt cây đơn bội. Đây cũng là nguyên liệu lý tưởng để nghiên cứu di truyền các tính trạng và cũng là nguyên liệu quý cho công tác tạo giống. Hiện nay, hơn 170 loài cây đã có thể tạo được từ con đường đơn bội.
Gần đây, người ta cũng đã nuôi cấy các tế bào trứng (noãn chưa thụ tinh) phát triển thành cây đơn bội, đặc biệt trên đối tượng ngô, cho phép nhanh chóng tạo

197
ra hàng loạt cây đơn bội. Đây chính là một giải pháp hữu hiệu trong ứng dụng đơn bội vào công tác giống cây trồng. Vì rằng, thông thường bằng con đường tự phối, muốn đạt được dòng đồng tử tuyệt đối phải mất từ 5-7 thế hệ, trong khi bằng con đường đơn bội và đa bội hóa sau đó, chỉ cần một thế hệ là đủ.
Téoulé và Demarly (1988) cho rằng bình quân con người tiết kiệm được 5 năm nhờ sử dụng kỹ thuật đơn bội vào việc tạo giống mới. Việc đưa cây đơn bội vào chương trình tạo giống lúa mì, lúa mạch, lúa nước, ớt, thuốc lá đã trở thành phổ biến. Pháp, Trung Quốc, Nhật Bản đang là những nước đi đầu trong lĩnh vực này, cho đến nay Trung Quốc đã tạo ra hàng trăm giống lúa bắt đầu từ nuôi cấy bao phấn.
Kỹ thuật tạo cây đơn bội in vitro thông qua kích thích tiểu bào tử phát triển thành cây trong nuôi cấy bao phấn và hạt phấn, noãn chưa thụ tinh cho phép nhanh chóng tạo ra hàng loạt cây đơn bội là một lối thoát rất tốt đối với lĩnh vực ứng dụng đơn bội vào nghiên cứu di truyền và tạo giống cây trồng.
Cây đơn bội có thể sử dụng vào các mục đích sau: - Nghiên cứu di truyền về mối tương tác của các gene - Tạo đột biến ở mức đơn bội - Tạo dòng đồng hợp tử tuyệt đối phục vụ công tác giống cây trồng
2. Phương pháp tạo cây đơn bội in vitro
2.1. Tạo cây đơn bội bằng nuôi cấy bao phấn, hạt phấn 2.1.1. Nguyên lý
Nuôi cấy hạt phấn đơn nhân (tiểu bào tử) tách rời hay các bao phấn có chứa các hạt phấn đơn nhân trên môi trường dinh dưỡng nhân tạo phù hợp sẽ kích thích các hạt phấn này phát triển thành cây đơn bội. Sự phát sinh cây đơn bội từ hạt phấn được gọi là sự sinh sản đơn tính đực (androgenesis).
Có 2 phương thức cơ bản trong nuôi cấy bao phấn, hạt phấn: - Các bao phấn được nuôi trên môi trường có agar và sự phát sinh phôi hoặc
mô sẹo xảy ra trong bao phấn. - Hạt phấn được tách rời khỏi bao phấn hoặc bằng phương pháp cơ học hoặc
do sự nứt nẻ tự nhiên của bao phấn và được nuôi cấy trong môi trưởng đặc.
2.1.2. Kỹ thuật nuôi cấy bao phấn, hạt phấn Kỹ thuật tạo cây đơn bội là một kỹ thuật phức tạp, có thể tham khảo dưới
đây những bước chính trong quy trình tạo cây đơn bội từ nuôi cấy bao phấn, hạt phấn.
- Chọn bao phấn và hạt phấn Giai đoạn phát triển đặc thù của bao phấn, hạt phấn tại thời điểm nuôi cấy là
nhân tố quan trọng nhất đối với sự thành công của phát sinh phôi. Ở thực vật hạt

198
kín, mỗi chồi hoa có thể chứa các bao phấn ở các giai đoạn phát triển khác nhau. Vì vậy, các chồi hoa phải được kiểm tra để xác định tất cả các giai đoạn phát triển giúp lựa chọn những bao phấn, hạt phấn có độ tuổi phù hợp cho nuôi cấy.
Nitsch (1967) nhận thấy cây đơn bội chỉ thu được khi cấy bao phấn chứa hạt phấn ở giai đoạn thích hợp, bắt đầu từ thể 4 nhân đến ngay sau khi nguyên phân đầu tiên. Ở thuốc lá (Nicotiana tobacum), cây kỳ nham đen (Hyscyamus niger), lúa (Oryza sativa) hạt phấn ở giai đoạn đơn nhân muộn cho hiệu quả tạo phôi cao nhất. Ngược lại, ở một số loài như đại mạch, giai đoạn tốt nhất cho nuôi cấy là hạt phấn đơn nhân sớm.
Ngoài ra, kết quả tạo cây đơn bội còn phụ thuộc nhiều vào trạng thái sinh lý của cây bố mẹ. Tuy nhiên trạng thái sinh lý của cây lại liên quan đến điều kiện môi trường như quang chu kỳ, cường độ ánh sáng, nhiệt độ và dinh dưỡng khoáng. Khả năng thành công của nuôi cấy là cao nhất với những bao phấn thu trong lần trổ hoa đầu tiên và giảm dần trong những lần trổ hoa tiếp theo. Sử dụng bao phấn từ những cây sinh trưởng dưới cường độ ánh sáng cao, ngày ngắn sẽ cho hiệu quả tạo phôi cao hơn (Dunwell, 1976).
Hình 4.10. Quy trình nuôi cấy bao phấn
- Tiền xử lý bao phấn và hạt phấn Để làm tăng kết quả của quá trình nuôi cấy bao phấn và hạt phấn, nhiều nhà
nghiên cứu đã tiến hành xử lý mẫu trước khi cấy. Theo nhiều tác giả, xử lý nhiệt độ thấp cho các hoa sau khi cắt khỏi cây và trước khi tách bao phấn để cấy sẽ kích thích sự phân chia của các tiểu bào tử để tạo cây đơn bội. Chế độ xử lý lạnh phụ thuộc vào loại cây. Ví dụ lúa Japonica xử lý ở 100C trong 2-3 tuần; lúa Indica xử lý ở 70C trong 1 tuần, thuốc lá xử lý ở 2-50C trong 2-3 ngày.
- Chọn môi trường tái sinh phù hợp Những môi trường thường được sử dụng trong nuôi cấy bao phấn và hạt
phấn là môi trường của Murashige và Skoog (MS - 1962), Linsmayer và Skoog (LS

199
- 1965), Bourgin và Nitsch (N - 1967), Gamborg (B5 - 1968), Chu Chin Ching (N6 - 1976). Tùy theo đối tượng nuôi cấy khác nhau mà sử dụng môi trường tương ứng. Tuy nhiên, vẫn có một số quy luật chung.
Cây hòa thảo cần nhiều auxin, đặc biệt là 2,4-D ở nồng độ cao để khởi động sự phân chia đầu tiên (đối với lúa 10-5 mol 2,4-D trong 12 ngày), cây họ cà cần ít hơn 10-6 mol.
Hàm lượng đường cao: đối với ngô 60 - 120g saccharose/l, lúa 50 - 60g saccharose/l, cây họ cà 20 - 40g/l
Các hỗn hợp chất tự nhiên như: dịch chiết khoai tây, nước dừa... tỏ ra có tác dụng tốt trong nhiều môi trường nuôi cấy bao phấn. Nguyên tố đa lượng và vi lượng ít có ảnh hưởng trong nuôi cấy bao phấn.
- Chọn lọc cây đơn bội Không phải tất cả các cây tái sinh khi nuôi cấy bao phấn là cây đơn bội, do
đó cần phải xác định cây đơn bội. Có thể xác định cây đơn bội bằng các phương pháp: đếm số lượng nhiễm sắc thể, đo gián tiếp hàm lượng DNA của tế bào, trồng cây tái sinh và so sánh với cây mẹ về hình thái, kích thước, khả năng sinh trưởng...
Việc nuôi cấy hạt phấn tách rời nhìn chung cũng tuân theo các quy trình nêu trên. Tuy nhiên, cần phải tách các hạt phấn khỏi bao phấn khi nuôi cấy. Các hạt phấn này thường được nuôi cấy trong môi trường bán lỏng.
2.2. Tạo cây đơn bội bằng nuôi cấy noãn chưa thụ tinh Sự hình thành cây đơn bội từ noãn thụ tinh được gọi là sinh sản đơn tính cái
hay trinh nữ sinh (gynogenesis). Nghiên cứu tạo cây đơn bội bằng noãn chưa thụ tinh đã được White (1932)
và Maheshwari (1958) tiến hành trên cây Antirhinum và Cooperia pendunculata nhưng không thu được kết quả. Thành công đầu tiên trong lĩnh vực này là việc thu nhận được mô sẹo đơn bội từ nuôi cấy noãn cây Ginkgo biola (Tulekee, 1964). Tuy nhiên, trong thời gian này người ta tập trung vào nhiều nghiên cứu tạo cây đơn bội bằng con đường nuôi cấy bao phấn ở nhiều đối tượng thực vật khác nhau hơn. Nhưng ở những loại cây như hành, củ cải đường, hoa hướng dương ... việc tạo cây đơn bội bằng con đường sinh sản đơn tính đực không đạt kết quả. Hơn nữa việc tạo cây đơn bội theo con đường này thường có tỷ lệ cây bạch tạng cao do sự mất cân bằng giữa di truyền nhân và di truyền tế bào chất. Chính vì vậy, đến khoảng những năm 70 của thế kỷ 20 các nhà nghiên cứu đã tập trung giải quyết vấn đề tạo cây đơn bội bằng nuôi cấy noãn chưa thụ tinh và thu được nhiều thành tựu. Đến cuối thập kỷ này đã có hơn 100 báo cáo về tạo phôi từ nuôi cấy noãn chưa thụ tinh.
Tỷ lệ tạo cây đơn bội bằng con đường nuôi cấy noãn chưa thụ tinh biến động ở các loại cây khác nhau. Đối với hành, củ cải đường, tỷ lệ này là 5-20%, ở lúa 1,5-12%, ở dâu tằm 3-6% ... Tuy nhiên kỹ thuật nuôi cấy noãn chưa thụ tinh còn nhiều khó khăn và phức tạp do việc tách tế bào trứng rất khó và dễ gây tổn thương. Nhằm tăng hiệu quả của quá trình này, người ta đang tập trung nghiên cứu các yếu tố như kiểu gene cây mẹ, giai đoạn phát triển của túi phôi, chế độ xử lý nhiệt độ, môi trường nuôi cấy ...

200
Trong các cây ngũ cốc, ở ngô việc nuôi cấy noãn chưa thụ tinh tương đối đơn giản và thu được nhiều thành công hơn cả. Phương pháp nuôi cấy cùng một lúc nhiều noãn chưa thụ tinh trên phần lõi bắp ngô đã được thực hiện thành công ở Trung Quốc và Việt Nam: bằng phương pháp này đã tạo được trực tiếp các cây ngô đơn bội in vitro với tỷ lệ 4-5%.
Hướng nghiên cứu tạo cây đơn bội và tạo dòng thuần đồng hợp tử đang phát triển khá mạnh ở Việt Nam trong việc chọn tạo các giống lúa đặc sản, lúa ưu thế lai, ngô, ... (Viện Di truyền Nông nghiệp, Viện Công nghệ sinh học, Viện Sinh học nhiệt đới, Viện nghiên cứu ngô, Viện cây lương thực).
3. Ứng dụng của đơn bội
- Nghiên cứu về phôi học thực nghiệm: chủ yếu trên các đối tượng mà phôi phát triển từ tiểu bào tử trong nuôi cấy bao phấn thông qua giai đoạn phát sinh phôi đơn tính.
- Nghiên cứu về tế bào học: cây đơn bội có thể sinh trưởng và phát triển tới giai đoạn ra hoa nhưng bất dục. Cây đơn bội có thể phát hiện nhiều điều lý thú về quan hệ tương tác giữa các nhiễm sắc thể vì bộ nhiễm sắc thể đơn bội không thể phân chia giảm nhiễm bình thường được.
- Nghiên cứu đột biến và di truyền: trong bộ gene đơn bội không có mối quan hệ tính trội mà chỉ có mối quan hệ bổ sung giữa các gene.
- Cải thiện giống cây trồng: tạo dòng đồng hợp tử thông thường thông qua con đường tự phối nếu muốn đạt được dòng đồng hợp tử của bộ gene 2x thì phải qua 10 thế hệ và bộ gene 4x thì phải qua 30 thế hệ, bằng nuôi cấy đơn bội hoặc đa bội thì chỉ qua 1 thế hệ.
4. Một số tồn tại trong nuôi cấy đơn bội Hiện tượng bạch tạng trong nuôi cấy đơn bội
Ở các đối tượng cây hai lá mầm: Datura, Atropa, Nicotiana… khi nuôi cấy bao phấn cây đơn bội thường phát triển trực tiếp từ tiểu bào tử và ít xuất hiện cây bạch tạng nhưng ở những đối tượng cây một lá mầm: lúa nước, lúa mì … cây hoàn chỉnh phát sinh thông qua giai đoạn mô sẹo thì tỷ lệ cây bị bạch tạng chiếm khá cao 20-30%. Tỷ lệ cây bạch tạng phụ thuộc vào các yếu tố:
- Tuổi của mô sẹo cấy chuyển từ môi trường tạo mô sẹo sang môi trường tái sinh cây.
- Nhiệt độ nuôi cấy: nhiệt độ cao thường làm tăng số lượng cây bạch tạng. Nghiên cứu về siêu cấu trúc tế bào lá cây bạch tạng cho thấy trong tiền lạp thể của cây bạch tạng không có ribosome. Như vậy quá trình sinh tổng hợp protein của các lạp thể này không hoàn chỉnh dẫn đến việc lạp vô sắc không phát triển thành lục lạp được. Có giả thuyết cho rằng đó là do kết quả của hiệu ứng mẹ, hiệu ứng mẹ biểu hiện rõ ở một số đặc điểm di truyền tế bào chất, khi thụ phấn chỉ có nhân của tế bào sinh dục đực được chuyển sang tế bào noãn vì vậy các tính trạng di truyền tế bào chất chỉ di truyền theo dòng mẹ. Hạt phấn là tế bào chứa rất ít nguyên sinh chất tức là số lượng ty thể và tiền lục lạp cũng rất ít, khi nuôi những tế bào này thành những cơ thể thực vật hoàn chỉnh có thể xảy ra hiện tượng mất cân đối trong tương tác di truyền giữa nhân và cơ quan tử, dẫn đến sai lệch trong quá trình phát sinh cơ quan tử đặc biệt là lục lạp.

201
Sức sống của các cá thể lưỡng dị bội Các cá thể đơn bội có sức sống kém hơn các cá thể dị hợp. Tính dị hợp bảo
lưu tăng dần vì những thay đổi đứt khúc của nhiễm sắc thể không sửa chữa được. Trong trường hợp này, cây đơn bội phải tiến hành tự thụ phấn để tích lũy trị số dị hợp bảo lưu nhằm tăng sức sống. V. NUÔI CẤY TẾ BÀO TRẦN & TRIỂN VỌNG ỨNG DỤNG
Các tế bào thực vật có tính toàn năng, có thể nuôi cấy, điều khiển sự phát sinh hình thái của chúng. Có thể nói dòng thông tin di truyền chứa trong nhân tế bào đã quyết định mọi đường hướng của quá trình thực hiện tính toàn năng của tế bào. Vì vậy, hoàn toàn có thể nuôi cấy tạo cây hoàn chỉnh từ khối nguyên sinh chất chứa nhân tế bào. Từ đây đưa ra khái niệm nuôi cấy tế bào trần thực vật (protoplast). Tế bào trần là những tế bào thực vật bị loại bỏ thành tế bào.
Nuôi cấy tế bào trần thành công sẽ cho phép mở ra khả năng biến nạp các gene vào tế bào (trước kia thường bị vỏ tế bào ngăn cản) cũng như tạo ra khả năng dung hợp tế bào trần-gắn hai tế bào trần lại thành một tế bào với hai bộ thông tin di truyền tạo nên một thể lai vô tính.
Con người đã biết đến tế bào trần từ lâu qua hiện tượng co nguyên sinh chất và rất nhiều nhà khoa học cũng muốn tách lớp vỏ tế bào để tiến hành các thí nghiệm trên các khối nội dung này của tế bào (Kuster 1910). Tuy nhiên phải đến năm 1960, Cocking mới tìm được phương pháp tách tế bào bằng các enzymee phân giải lớp vỏ bên ngoài tế bào và thu được tế bào trần. Tình trạng tế bào trần chỉ có tính chất tạm thời sau đó tế bào tự hình thành lớp vỏ khá nhanh. Sự ra đời của tế bào trần đã mở ra cho con người hàng loạt triển vọng mới trong lĩnh vực giống cây trồng.
Cho đến nay việc hoàn thiện các phương pháp nuôi cấy mô tế bào trần từ khâu tách đến nuôi cấy, tái sinh cây hoàn chỉnh đã thành công trên hàng loạt đối tượng cây trồng quan trọng: cà chua (Zapata, 1977), cải dầu (Kartha, 1974), khoai tây (Shepand và Tohen 1977), lúa (Fujimuna 1985), lúa mì (Lorz, 1991)... Đây cũng là nền tảng quan trọng cho các biện pháp lai soma và chuyển nạp gene nhằm cải tạo các giống cây trồng.
1. Kỹ thuật nuôi cấy tế bào trần 1.1. Tách tế bào trần
Đặc trưng của tế bào thực vật là có thành cellulose bao quanh, sau đó là đến màng nguyên sinh chất. Thành cellulose giữ cho tế bào có hình dáng nhất định, còn các hợp chất pectin nằm trong thành có nhiệm vụ liên kết gắn các tế bào với nhau thành mô. Khi tách thành cellulose khỏi tế bào, màng nguyên sinh chất cho phép protoplast có thể hấp thu vào tế bào các dạng phân tử (nucleic acid, protein), các cơ quan tử (lục lạp, ty thể, nhân tế bào) theo cơ chế amip.
Các phương pháp được sử dụng trong tách và nuôi tế bào trần có mức độ thành công phụ thuộc vào bản chất của nguyên liệu ban đầu. Những quy trình cơ bản đều dựa chủ yếu vào thực nghiệm và thay đổi cho phù hợp với mỗi loại nguyên liệu. Ngoài ra, phương pháp tách cũng ảnh hưởng đến sức sống của tế bào trần và do đó liên quan đến quá trình nuôi cấy sau này.

202
Trong thực nghiệm người ta thường dùng mô thịt lá để tách tế bào trần. Những lá được tạo ra trong nuôi cấy mô in vitro là những vật liệu ban đầu lý tưởng cung cấp một lượng lớn tế bào trần khoẻ mạnh. Ngoài ra, các mẫu dùng trong tách tế bào trần phải thu từ những cây có trạng thái sinh lý tốt. Nếu mẫu lấy trực tiếp từ ngoài tự nhiên thì phải từ các cây không xử lý với thuốc trừ sâu và thuốc diệt cỏ.
- Phương pháp tách tế bào trần Chức năng chính của thành tế bào là duy trì áp suất vỏ lên khối nguyên sinh
chất và bởi vậy ngăn cản sự hấp thụ một lượng nước lớn làm vỡ tế bào. Do đó, sức trương của tế bào sống luôn cân bằng với áp lực cơ học của thành tế bào. Khi tách vỏ, tế bào bị vỡ khi không có lực nén của vỏ. Để khắc phục hiện tượng này cần phải sử dụng một dung dịch làm co nguyên sinh khi tách vỏ tế bào. Các dung dịch thường dùng là dung dịch đường mannitol, sorbitol, nồng độ sử dụng khoảng 0,3-0,7M tùy theo đối tượng thực vật.
Có hai phương pháp: cơ học và hóa học trong đó phương pháp hóa học có hiệu quả hơn rất nhiều so vơí phương pháp cơ học.
Với phương pháp cơ học người ta cho tế bào vào máy ly tâm siêu tốc vì vậy dễ gây tổn thương tế bào.
Đối với phương pháp hóa học người ta sử dụng hỗn hợp enzymee: Cellulase Hemicellulase Pectinase Sorbitol Mannitol CaCl2.2H2O CaH4(PO4)2.H2O
Sorbitol va malnitol làm tăng áp suất thẩm thấu để tránh vỡ màng tế bào.Tùy theo từng đối tượng từng loại mô mà thay đổi nồng độ cho thích hợp. Các hỗn hợp enzymee thường được sử dụng ở pH 5,5-5,8 trong 3-8 h.
Có thể tóm tắt các bước cơ bản để tách tế bào trần như sau: Vô trùng bề mặt lá Lá được ngâm trong dung dịch co nguyên sinh Loại bỏ biểu bì mặt dưới hoặc cắt mẫu lá thành các lát mỏng thuận lợi
cho sự xâm nhập của enzymee Xử lý với hỗn hợp enzymee Tinh sạch các tế bào trần Chuyển các tế bào trần vào các môi trường nuôi cấy thích hợp.
Ngoài ra, người ta cũng tách protoplast từ callus, tế bào thu được từ nuôi cấy dịch huyền phù. Protoplast tách từ nuôi cấy dịch huyền phù tế bào có khả năng tái sinh cây lớn hơn hẳn so với các protoplast tách từ tế bào thịt lá.
1.2. Các yếu tố ảnh hưởng đến sinh trưởng và phát triển của tế bào trần
- Môi trường nuôi cấy

203
Môi trường nuôi cấy protoplast có một số thay đổi nhỏ so với môi trường phát sinh cơ quan từ mô nuôi cấy. Nồng độ NH4
+ cao (20 mM) kích thích sự phân chia tế bào nhưng gây độc đối với protoplast. Vì vậy, trong nuôi cấy protoplast nồng độ NH4
+ thường phải giảm từ ¼ đến ½ so với bình thường để vừa kích thích sự phân bào nhưng đồng thời cũng không gây độc đối với protoplast.
Nồng độ Ca2+ dao động trong khoảng 14-40 mM có tác dụng kích thích sự phân chia tế bào, quá trình đồng hóa, ngăn ngừa sự kết cụm, giảm sự hóa nâu của protoplast ở giai đoạn sớm của quá trình nuôi cấy.
Các hợp chất hữu cơ như inositol, nicotinic acid, vitamin B1, B6, glycin, biotin cũng được bổ sung vào môi trường nuôi cấy. Bên cạnh đó, các chất như casein, D-Ca-pantothenate, cystein, malic acid, ascorbic acid , adenin sulfate, riboflavin cũng được bổ sung vào môi trường nuôi cấy protoplast với nồng độ từ 0,01-10 mg/l để thúc đẩy quá trình tổng hợp thành tế bào và sự phân bào.
Đường sử dụng trong nuôi cấy protoplast để ổn định áp suất thẩm thấu và là nguồn cung cấp carbon. Mannitol và sorbitol với nồng độ 0,3-0,7 M để ổn định áp suất thẩm thấu, nồng độ các chất này sẽ được giảm xuống một khi protoplast đã tổng hợp đươc thành tế bào và bắt đầu phân bào. Glucose và saccharose được bổ sung vào môi trường nuôi cấy với nồng độ 0,2-0,6 M để cung cấp nguồn carbon cho tế bào.
Các chất điều hòa sinh trưởng, chủ yếu là nhóm auxin và cytokinin, được bổ sung vào môi trường nuôi cấy kích thích sự phân chia tế bào. Các chất kích thích sinh trưởng thường sử dụng là NAA và 2,4-D (0,45-10,7 µM); BA và zeatin (2-5µM).
- Điều kiện nuôi cấy Protoplast được nuôi cấy ở điều kiện nhiệt độ 20-25oC; cường độ ánh sáng 5-
30 µmol.m-2.s-1.
2. Dung hợp tế bào trần Tế bào trần là tế bào không có thành tế bào. Chính vì thế chúng có thể hòa
lẫn với nhau (dung hợp) thành một tế bào lai mang trong mình vật chất di truyền của cả hai tế bào. Khi hai hoặc nhiều tế bào trần dung hợp sẽ hợp thành một khối tế bào trần, nhân của chúng có thể kết hợp hoặc tồn tại riêng rẽ. Nếu hai nhân khác nhau cùng tồn tại sẽ tạo ra thể dị nhân, ngược lại sẽ tạo thể lai đồng nhân. Sự kết hợp nhân trong thể dị nhân hình thành tế bào trần lai thực sự gọi là thể hợp nhân. Nếu một trong hai nhân bị đào thải thì phần nguyên sinh chất của hai tế bào trần sẽ dung hợp tạo thể lai tế bào chất.
Trong điều kiện thích hợp, tế bào lai được tái sinh thành một cây lai. Quá trình lai này xảy ra ở tế bào nên gọi là lai tế bào và thông qua tế bào soma nên gọi là lai soma hay lai vô tính tế bào. Có thể nói việc dung hợp tế bào trần và tái sinh thành cây lai từ tế bào trần là một trong những thành tựu tuyệt vời của kỹ thuật cấy mô tế bào. Bằng phương pháp này giúp các nhà lai tạo giống có thể lai xa giữa các loài – điều không thể thực hiện được bằng phương pháp lai hữu tính thông thường.
Những tác giả tiên phong trong lĩnh vực này phải kể đến Carlson (1972), người đã tạo thành công cây thuốc lá dung hợp từ tế bào trần của hai loài N. glauca

204
và N. langsdorfii. Đặc biệt là Melchers, với thành tựu dung hợp thành công tế bào trần khoai tây với tế bào trần cà chua đã đánh dấu một mốc rất quan trọng trong lĩnh vực này. Ngày nay, việc sử dụng kỹ thuật dung hợp tế bào trần đã được ứng dụng rộng rãi vào các hệ thống tạo giống.
2.1. Các phương pháp dung hợp - Dung hợp tự phát
Trong quá trình phân cắt thành tế bào bằng enzyme, các tế bào trần được hình thành từ những tế bào tiếp giáp với nhau có thể dung hợp qua cầu nối sinh chất. Việc loại bỏ thành tế bào đã làm cho các cầu nối sinh chất giữa các tế bào giãn nở to lên, nguyên sinh chất và các cơ quan từ hai hoặc nhiều tế bào có thể lưu thông với nhau.
Dung hợp tự phát chỉ xảy ra trong phạm vi một loài và hiện tượng này hiếm gặp do sự tích điện âm trên bề mặt là nguyên nhân ngăn cản chúng kết hợp với nhau.
- Dung hợp cảm ứng Dung hợp cảm ứng là việc sử dụng các hóa chất, dòng điện, huyết thanh
miễn dịch... để kích thích quá trình dung hợp tế bào trần. Trong đó, phương pháp dung hợp bằng hóa chất và bằng dòng điện thường được dùng hơn cả.
Dung hợp bằng hóa chất: Có nhiều hóa chất được sử dụng để kích thích dung hợp như dung dịch các muối (NaCl, KCl, NaNO3), dextran sulfate, polyvinyl alcohol, lysolethicin, và polyethylene glycol (PEG).
Thường sử dụng nhất là polyethylene glycol (PEG 5-25%) do PEG có độc tính thấp và có tác dụng dung hợp với hầu hết các kiểu tế bào. Sự thành công của thí nghiệm dung hợp phụ thuộc vào trọng lượng phân tử của PEG. Trọng lượng phân tử của PEG thấp (<100) không thể tạo ra một sự dính kết chắc chẳn. Phân tử PEG có trọng lượng phân tử 6000 cho hiệu quả dung hợp cao nhất. Quá trình dung hợp sẽ được cải thiện hơn nếu xảy ra trong môi trường kiềm (pH 8-10) và bổ sung CaCl2 nồng độ cao (50-250 mM).
Ví dụ: Quy trình dung hợp protoplast khoai tây theo các bước sau: (theo Trigiano, R.N. và Gray, D.J. Plant Tisue culture Concept and Laboratory Exercises, First Edition, CRC Press LLC, Boca Raton, FL, 1996)
* Hòa tan protoplast (mật độ 104 tế bào/ml) trong 0,5 M glucose, 3 mM CaCl2, 0,7 mM KH2PO4.(pH 5,7).
** Bổ sung 3-200 µl dung dịch kết dính (pH 5,5) gồm 0,09 mM PEG 6000, 10 mM CaCl2, 0,7 mM KH2PO4.
*** Thêm 500 µl dung dịch (pH 10) 50 mM CaCl2, 0,5 M glucose, 50 mM đệm glycin.
**** Rửa tế bào bằng môi trường nuôi cấy sau đó nuôi cấy theo phương pháp giọt treo (hanging drops culture).
Dung hợp bằng điện AC (100 V/cm; 0,6 MHz): Đầu tiên các protoplast được dưa vào trong ngăn dung hợp nhỏ có hai dây kim loại song song đóng vai trò là các điện cực. Sử dụng điện áp thấp và trường AC dao động nhanh kích thích các protoplast sắp thành từng chuỗi tế bào giữa các điện cực (chuỗi ngọc trai). Phương

205
thức này cho phép các tế bào tiếp xúc hoàn toàn với nhau trong vài phút. Sau khi các tế bào xếp thành hàng hoàn chỉnh, quá trình dung hợp được thực hiện theo từng đợt ngắn của DC điện áp cao. Xung DC điện áp cao tạo ra sự phá vỡ thuận nghịch của màng nguyên sinh chất ở vị trí tiếp xúc giữa các tế bào tạo ra sự dung hợp và tái tổ chức lại màng một cách hợp lý. Một quá trình hoàn chỉnh bắt đầu từ lúc đưa các protoplast vào bên trong ngăn cho đến khi chuyển chúng lên môi trường nuôi cấy có thể được thực hiện trong vòng 5 phút hoặc ít hơn. 3. Ứng dụng của protoplast 3.1. Chọn dòng tế bào
Cơ thể thực vật bậc cao là cơ thể đa bào đã được phân hóa thành các tổ chức khác nhau. Nếu tiến hành xử lý đột biến cả tổng thể đó thì rất khó đạt tần số cần thiết, phải cần một số luợng rất lớn (106-108) cá thể mới có thể xuất hiện các cá thể đột biến. Sử dụng tế bào trần riêng rẽ cho phép loại bỏ mối tương tác với các tế bào bên cạnh và những thay đổi di truyền có điều kiện biểu hiện rõ ràng hơn. Ngoài ra, các tác nhân đột biến có thể gây chết hàng loạt các cá thể trồng, muốn nuôi 106-108
cá thể thực vật cần có một diện tích đất rất lớn, chi phí lao động cao, hiệu quả kinh tế kém. Bên cạnh đó, cá thể thay đổi tính trạng chưa hẳn là đột biến mà có thể là thường biến, hoặc đột biến đó không ổn định qua nhiều thế hệ. Bằng biện pháp phá vỡ thành tế bào và đưa cơ thể thực vật về trạng thái từng tế bào riêng rẽ với kích thước không lớn hơn nhiều so với cơ thể vi sinh vật đã cho phép áp dụng các kỹ thuật chọn dòng của vi sinh vật đối với thực vật. Một đĩa petri đường kính 5-7 cm cho phép nuôi 5.106 protoplast thuốc lá trong khi muốn trồng 5.106 cây thuốc lá phải cần có 100 ha đất. Vì vậy việc sử dụng tế bào trần riêng lẽ để chọn dòng đột biến là phương pháp thuận tiện và có nhiều hiệu quả nhất. 3.2. Dung hợp protoplast để lai vô tính tế bào thực vật
Triển vọng của phương pháp nuôi cấy và dung hợp tế bào trần là rất to lớn. Đáng chú ý là sự vận dụng thành công trên lĩnh vực cải thiện giống cải dầu, khoai tây BF15 với Solanum berthauliti để tạo khoai tây chống rệp. Kết quả tạo được con lai soma có đặc tính nhiều lông trên mặt lá ngăn cản sự chích hút của rệp. Viện nghiên cứu di truyền Grunbach CHLB Đức đã tổ hợp thành công đặc tính chống chịu virus ở khoai tây qua dung hợp tế bào trần. Hai hoặc nhiều tế bào trần có thể được cảm ứng dung hợp để tạo ra sản phẩm lai, mặc dù hiện tượng này vẫn chưa thành công ở nhiều thực vật. Trong một vài trường hợp, cây lai không thể được tạo ra qua phương pháp thông thường do sự bất tương hợp về sinh sản hoặc sinh lý, có thể được hình thành qua dung hợp tế bào soma. 3.3. Biến nạp thông tin di truyền
Các tế bào trần có khả năng tiếp nhận các vật liệu từ bên ngoài đưa vào trong tế bào. Do đó tế bào trần là đối tượng thích hợp cho nghiên cứu đưa nhân, lạp thể, ty thể, DNA, plasmid… vào tế bào. Quá trình biến nạp thông tin di truyền được thực hiện bằng cách nuôi cấy protoplast với Agrobacterium tumefaciens hoặc chuyển trực tiếp DNA vào tế bào bằng phương pháp xung điện, vi tiêm, siêu âm, polyethylene glycol (PEG) ...
- Chuyển gene ngoại lai vào protoplast thông qua Agrobacterium tumerfaciens: Trong phương pháp này protoplast được tách và nuôi cấy 2-3 ngày

206
trước khi đồng nuôi cấy với A. tumerfaciens mang gene cần chuyển nạp. Sau 24-30 h, protoplast được chuyển lên nuôi cấy ở môi trường có kháng sinh (kanamycin, hygromycin...) để chọn lọc những tế bào đã được biến nạp. Tỷ lệ biến nạp thành công ở phương pháp này khoảng 0,26-15,3%.
- Chuyển gene ngoại lai vào protoplast bằng PEG: PEG cùng với sự có mặt của Ca2+ là tác nhân dung hợp được sử dụng phổ biến nhất trong lai tế bào soma. Kỹ thuật này đòi hỏi phải nuôi protoplast với DNA trần có mặt PEG và CaCl2. Sau đó, các protoplast được phân lập, rửa và nuôi cấy trên môi trường chọn lọc.
- Chuyển nạp gene ngoại lai vào protoplast bằng điện xung: Thiết bị điện xung là thiết bị có khả năng tạo ra các xung điện trong thời gian rất ngắn (5-6 phần nghìn giây) và ở điện thế chính xác. Protoplast được đặt giữa hai tấm kim loại cách nhau từ 1-4 mm trong một cuvette bằng nhựa. Ở điện thế cao, xung điện tạo ra các lỗ (kích thước 30 nm) tạm thời trên màng protoplast và DNA bên ngoài có thể xâm nhập vào chất nguyên sinh và sau đó màng sẽ liền lại.
- Chuyển nạp gene ngoại lai vào protoplast bằng kỹ thuật vi tiêm: Trên hiển vi trường, DAN ngoại lai có thể được tiêm vào protoplast .
- Chuyển DNA ngoại lai vào protoplast bằng sóng siêu âm: Protoplast được xử lý nhẹ bằng siêu âm có sự hiện diện của DNA ngoại lai. Sóng siêu âm giúp DNA đi vào trong tế bào và thể hiện. VI. LÀM SẠCH BỆNH VIRUS TRONG CÔNG TÁC PHỤC TRÁNG GIỐNG CÂY TRỒNG 1. Nguyên lý làm sạch virus và duy trì tính sạch bệnh
Khi tiến hành nuôi cấy, mẫu mô thực vật phải được khử trùng, các vi sinh vật gây bệnh như nấm và vi khuẩn về cơ bản đã bị tiêu diệt. Tuy nhiên, virus vẫn có thể tồn tại ở các tế bào sống. Khác với các loại bệnh do nấm và vi khuẩn gây ra, bệnh do virus là loại bệnh khó chữa trong khi lại gây tổn thất rất lớn về năng suất, phẩm chất và lan truyền qua các thế hệ khi nhân giống vô tính, gây ra hiện tượng thoái hóa giống. Đa số các cây trồng nhân giống vô tính như khoai tây, khoai lang, các loại hoa trồng từ củ hoặc giâm cành dễ bị thoái hóa giống do nhiễm virus. Chính vì vậy việc tạo ra cây sạch virus là biện pháp bắt buộc phải tiến hành cho tất cả các cây nhân giống vô tính và cũng là biện pháp phục tráng giống cho các giống đã bị thoái hóa do virus.
Đã có rất nhiều nghiên cứu của White (1934), Limasset và Cornuet (1950), Morel và Martin (1952) cho thấy virus tồn tại ở mọi tế bào sống nhưng trong cây nhiễm bệnh, sự phân bố virus thay đổi tùy theo các cơ quan của cây: Nồng độ virus ở mô phân sinh đỉnh rễ, đỉnh chồi và lá bao thứ nhất là bằng 0 sau đó tăng dần ở các lá xa với mô phân sinh. Từ đây các ông đã đề xuất kỹ thuật nuôi cấy mô phân sinh đỉnh để tạo ra cây sạch virus từ cây đã nhiễm bệnh.
Theo Mathews (1970) và Wang (1980) thì mô phân sinh đỉnh không có virus có thể do các lý do sau đây:
- Virus vận chuyển trong cây nhờ mô dẫn, hệ thống này không có ở mô phân sinh đỉnh.
- Trong sự phân chia, các tế bào mô phân sinh đỉnh không cho phép sao chép các thông tin di truyền của virus

207
- Hệ thống vô hiệu hóa virus ở vùng mô phân sinh đỉnh mạnh hơn các vùng khác trong cây.
- Nồng độ auxin cao ở mô phân sinh đỉnh có thể ngăn cản quá trình sao chép virus.
Trong nuôi cấy tạo cây sạch bệnh, cần xác định đúng kích thước mô phân sinh đỉnh để tách chính xác, đảm bảo sạch virus. Kích thước của mô phân sinh đỉnh thay đổi tùy theo giống cây trồng và giai đoạn phát triển của chúng. Một cách tương đối, chúng ta có thể nói rằng mô phân sinh đỉnh chỉ là hình vòm ở đỉnh có kích thước khoảng 0,1-0,2 mm tính từ chóp đỉnh.
2. Các kỹ thuật làm sạch virus in vitro
- Nuôi cấy đỉnh sinh trưởng Tách chính xác đỉnh sinh trưởng có kích thước nhỏ hơn 0,3 mm, nuôi cấy
chúng trên môi trường dinh dưỡng để tái sinh thành cây hoàn chỉnh. Sau đó kiểm tra độ sạch virus ở cây tái sinh bằng phương pháp khác nhau để thu nhận được cây sạch bệnh.
Hình 4.11. Nuôi cấy mô phân sinh đỉnh để tạo cây sạch bệnh
- Nuôi cấy đỉnh sinh trưởng kết hợp xử lý nhiệt độ cao Ở nhiệt độ từ 36-370C thì một số loài virus không có khả năng nhân lên. Lợi
dụng đặc tính này có thể kết hợp phương pháp nuôi cấy đỉnh sinh trưởng với xử lý nhiệt để loại virus khỏi mẫu cấy. Biện pháp này cho phép có thể tách đỉnh sinh trưởng ở kích thước lớn hơn (0,5-1,0 mm), giúp cho việc tách và tái sinh cây thuận lợi hơn so với phương pháp tách ở kích thước 0,1-0,3 mm.

208
Có thể xử lý nhiệt độ cao 35-370C một thời gian dài cho cây mẹ trước khi tách đỉnh sinh trưởng để nuôi cấy (5-10 tuần). Hoặc có thể xử lý mẫu vật sau khi đã đưa vào nuôi cấy in vitro ở nhiệt độ 39-400C trong 1-2 tuần lễ. Ở nhiệt độ này, thường các RNA thông tin của virus sẽ bị phân giải và cây tái sinh sẽ có độ sạch virus cao.
- Nuôi cấy đỉnh sinh trưởng kết hợp với xử lý hóa chất Bên cạnh xử lý nhiệt độ cao còn có thể nuôi cấy đỉnh sinh trưởng có kích
thước lớn (0,5-1,0 mm) kết hợp với việc bổ sung vào môi trường nuôi cấy các chất kháng virus để tạo cây sạch bệnh. Các chất kháng virus như: 2-thiouracil, ribavirin, vidarabin làm tăng khả năng kháng của tế bào, mô thực vật và ức chế sự nhân bản của virus.
- Vi ghép Kỹ thuật nuôi cấy đỉnh sinh trưởng cho phép tạo cây con sạch bệnh từ cây
mẹ nhiễm bệnh. Tuy nhiên vấn đề đặt ra ở đây là các cây tái sinh sau khi đưa ra vườn ươm có sức sống kém, hơn thế nữa đối với một vài loại cây thân gỗ, việc cho ra rễ của các cây in vitro gặp rất nhiều khó khăn. Chính vì vậy người ta đã tiến hành kỹ thuật vi ghép để tận dụng khả năng sạch bệnh của đỉnh sinh trưởng đồng thời giải quyết những khó khăn trên.
Vi ghép là kỹ thuật ghép đỉnh sinh trưởng lên gốc cây sạch và kháng bệnh trong điều kiện in vitro để sản xuất cây sạch virus. Đỉnh sinh trưởng làm mắt ghép có kích thước khoảng từ 0,2-0,5 mm được tách từ chồi non đang sinh trưởng mạnh của cây mẹ trưởng thành, gốc ghép là mầm giá mới nảy mầm từ hạt của giống hoang dại hay giống địa phương. Kỹ thuật này thường được sử dụng với các cây thân gỗ đặc biệt là họ cam, chanh.
Những cây ghép thu được bằng phương pháp này hoàn toàn sạch bệnh và mang đặc điểm di truyền của cây mẹ cho mắt ghép; đồng thời tận dụng các đặc tính của gốc ghép hoang dại như tính kháng bệnh và khả năng thích ứng với môi trường địa phương.
Kỹ thuật vi ghép sẽ góp phần tạo ra những tập đoàn cây giống sạch bệnh dùng làm nguyên liệu cho sản xuất đại trà. Ngoài ra, còn dùng cho nghiên cứu sự ghép không tương hợp giữa chồi ghép và gốc ghép, và các khía cạnh về mô học và sinh lý học của sự ghép.
Hình 4.12. Kỹ thuật vi ghép

209
3. Kiểm tra, nhân nhanh và duy trì tình trạng sạch bệnh Sau khi đã có cây tái sinh, cần phải kiểm tra xem mẫu đã sạch bệnh virus hay
chưa. Các phương pháp chẩn đoán virus thường được dùng là: - Phương pháp chẩn đoán bằng mắt (bằng triệu chứng bệnh) Dựa trên những mô tả có sẵn về các dạng bệnh virus để phân biệt bệnh virus
với các triệu chứng do viroide, mycoplasma, tuyến trùng hay các bệnh không truyền nhiễm (bệnh do môi trường không thuận lợi) gây ra.
Sự nhận biết các biểu hiện virus bằng triệu chứng có thể nhìn thấy là phương pháp đơn giản nhất. Tuy vậy, chẩn đoán bằng mắt đòi hỏi phải có kinh nghiệm quan sát, so sánh vì dễ có sự nhầm lẫn giữa cây bị virus với cây bị bệnh khác như microplasma, tuyến trùng và một số bệnh sinh lý. Việc đánh giá triệu chứng cũng gặp khó khăn khi cây bị một hỗn hợp virus xâm nhiễm. Mặt khác thời điểm quan sát, điều kiện ngoại cảnh, tuổi cây, đặc điểm giống cũng là những yếu tố quan trọng để đánh giá sự nhiễm bệnh virus, hơn nữa, một điểm cần xem xét là bệnh virus thường có hiện tượng mất triệu chứng (latent period) gây ra sự nhầm lần giữa cây bị bệnh và cây khoẻ.
- Phương pháp cây chỉ thị (plant indicator) được sử dụng rộng rãi trong nghiên cứu và sản xuất.
Cây chỉ thị thực chất là cây ký chủ của virus, chúng đặc biệt mẫn cảm với virus và thường tạo các triệu chứng điển hình và biểu hiện bệnh nhanh.
Cây chỉ thị được chia làm 2 nhóm: các cây nhiễm bệnh cục bộ và các cây nhiễm bệnh hệ thống. Cây nhiễm bệnh cục bộ thường tạo ra các vết chết trên lá, thân, ... còn cây nhiễm bệnh hệ thống thường tạo triệu chứng toàn thân như khảm lá, biến vàng, lùn cây. Với mục đích chẩn đoán bệnh hại người ta thường dùng cây có triệu chứng nhiễm bộ phận, còn cây nhiễm hệ thống thường được dùng để giữ nguồn virus phục vụ cho các thí nghiệm trong phòng.
Chẩn đoán bằng cây chỉ thị là một phương pháp khá chính xác trong chẩn đoán. Nhược điểm cơ bản của phương pháp này là cần có thời gian lây nhiễm và phải chờ cây phát bệnh sau một thời kỳ ủ bệnh.
Cách thực hiện: nghiền lá cây cần kiểm tra xem có nhiễm bệnh hay không trong nước cất vô trùng. Sau đó thu lấy dịch chiết và cấy lên lá cây chỉ thị (đã biết là mẫn cảm với virus đang nghiên cứu). Nếu lá đổi màu thì mẫu thử bị nhiễm. Hoặc có thể ghép cây cần kiểm tra trên cây chỉ thị, nếu xảy ra những biến đổi như có vết hoại tử hoặc thay đổi hình dạng thì chứng tỏ cây có bị nhiễm.
- Phương pháp huyết thanh Cơ sở khoa học của phương pháp này chủ yếu dựa vào phát minh của nhà
khoa học Pasteur - hệ miễn dịch ở động vật. Phản ứng huyết thanh là kết quả của kháng nguyên khi gặp kháng thể tạo
thành kết tủa. Kết tủa này có thể quan sát được dưới tụ quang đen ở kính hiển vi quang học. Dưới màn đen của tụ quang, phản ứng kết hợp kháng nguyên – kháng thể sẽ được xác định giống như những đám mây bông. Phương pháp huyết thanh là phương pháp được sử dụng rộng rãi, đặc hiệu, nhanh chóng trong chẩn đoán virus thực vật.

210
ELISA (Enzymee Linked Immuno Sorbent Assay) là phương pháp huyết thanh hiện đại nhất hiện nay.
- Phương pháp sinh học phân tử Từ những năm 1980, các phương pháp sinh học phân tử đã phát triển mạnh
mẽ mang lại nhiều thành tựu to lớn trong việc chẩn đoán, phát hiện và nghiên cứu bản chất virus gây bệnh, đặc biệt là kỹ thuật PCR.
Nhờ các phương pháp này, chỉ cần một lượng nhỏ DNA hay RNA thu được từ tế bào chủ là có thể nhân lên thành số lượng lớn rồi đưa vào bản điện di để phát hiện vạch đặc trưng của chúng. Việc nghiên cứu sinh học phân tử đã giúp chúng ta dễ dàng phân chủng sinh học của virus và đặc biệt nghiên cứu để tạo giống chống bệnh.
Sau khi đã đảm bảo cây đã sạch virus thì việc nhân nhanh giống sạch bệnh và duy trì độ sạch virus của giống có ý nghĩa quan trọng quyết định.
Để duy trì độ sạch bệnh cần tiến hành chọn lọc, vệ sinh đồng ruộng, nhổ bỏ các cây bị tái nhiễm.
VII. TẠO ĐỘT BIẾN VÀ CHỌN DÒNG TẾ BÀO THỰC VẬT 1. Khái niệm
Biến dị dòng soma (somaclone variation) là khái niệm dùng để chỉ các biến dị thể hiện ở các tế bào, mô nuôi cấy và cây có nguồn gốc từ nuôi cấy mô (Larkin và Scowcropt, 1981). Biến dị soma còn được gọi là biến dị dòng vô tính.
Trong vi nhân giống, tính di truyền không ổn định của các cây tái sinh sau nuôi cấy được xem là một trong những hạn chế lớn của vi nhân giống, vốn yêu cầu tính đồng nhất và ổn định di truyền. Tuy nhiên, trong chọn tạo giống cây trồng, các biến dị vô tính cung cấp một tiềm năng lớn để làm phong phú thêm các kiểu gene mới vào quỹ gene cũng như giúp ích cho nghiên cứu di truyền tế bào. Trên thực tế, các tỷ lệ biến dị di truyền có thể là rất lớn và chính vì thế việc hiểu và xác định được cơ chế, nguyên nhân gây các biến dị này rất quan trọng. Qua đó chúng ta có thể điều khiển các biến dị một cách có hiệu quả theo hướng có lợi.
Kỹ thuật chọn dòng tế bào đã ra đời rất sớm trong nghiên cứu vi sinh vật nhưng đối với các thực vật bậc cao kỹ thuật mới chỉ được ứng dụng cách đây khoảng 15 năm. Có thể tiến hành chọn lọc và xử lý trên 4 mức độ:
- Mô sẹo - Tế bào đơn - Tế bào trần - Phôi
2. Các nguyên nhân chính gây biến dị soma Đã có rất nhiều nghiên cứu được tiến hành nhằm xác định nguyên nhân gây
ra các biến dị soma trong nuôi cấy mô. Bất kỳ một yếu tố có khả năng dẫn đến các thay đổi di truyền đều được xem như là một nguyên nhân gây ra các biến dị di truyền. Có thể gộp các yếu tố này thành 3 nhóm chính là nguyên nhân sinh lý, di truyền và sinh hóa. Các nguyên nhân chính gây biến dị soma là tính không đồng

211
nhất di truyền của các tế bào soma ở mẫu cấy ban đầu và tác động của các yếu tố trong quá trình nuôi cấy in vitro.
- Sự đa dạng di truyền tự nhiên của các tế bào nuôi cấy Các mẫu cấy có nguồn gốc từ một dòng đơn tính, từ hạt hay cây con được
xem như đồng nhất về mặt di truyền và khi lấy mẫu có thể có kiểu hình giống nhau. Tuy nhiên, các mẫu này trên thực tế lại bao gồm nhiều loại tế bào khác nhau như phloem, xylen, nhu mô, mô vỏ ... những tế bào này có thể có mức độ bội thể khác nhau. Nói cách khác, có sự đa dạng tế bào giữa các tế bào trong cùng một mẫu cấy. Sự đa dạng tế bào như vậy được gọi là "polysomatic" (đa bội vô tính). Các loài như lúa mỳ, thuốc lá đã được công bố là có các mô đa bội vô tính như thế.
Ngoài ra nhiều thực vật tồn tại ở dạng thể khảm. Chúng chứa các tế bào hoặc mô có cấu trúc di truyền khác nhau được phát triển từ đỉnh sinh trưởng có chứa lớp hay bộ phận mô bị đột biến. Hiện tượng này phổ biến ở các cây thân gỗ.
Sự sắp xếp các mô khác nhau về mặt di truyền trong mô phân sinh đỉnh của thực vật ảnh hưởng đến tính ổn định của các thể khảm. Các cây ở thể khảm thường có mức độ biến dị di truyền cao khi nuôi cấy in vitro.
- Tác động của các yếu tố trong quá trình nuôi cấy Những yếu tố chủ yếu dẫn đến các biến dị dòng soma trong quá trình nuôi
cấy mô, tế bao gồm: - Phương thức nhân giống in vitro: Các phương thức nhân giống khác nhau
sẽ cho tỷ lệ xuất hiện các biến dị vô tính khác nhau. Nhìn chung nếu chồi bất định được tái sinh từ một tế bào thì cơ hội để xuất hiện các biến dị soma thường là lớn hơn rất nhiều so với các chồi được tái sinh từ nhiều tế bào. Các quá trình nuôi cấy callus, huyền phù hoặc protoplast thường có nhiều biến dị soma.
- Loại mẫu cấy: Các loại mẫu cấy khác nhau thường thể hiện mức độ biến dị khác nhau. Các mẫu cấy có nguồn gốc từ các thể tiền chồi như chồi nách, chồi đỉnh thường có mức độ biến dị thấp hơn khi sử dụng các mẫu cấy có nguồn gốc không phải từ đỉnh sinh trưởng như là lá, rễ hay protoplast. Khả năng xảy ra các biến dị soma còn phụ thuộc vào kiểu gene cũng như tuổi cây mẹ. Các dòng già thường tiềm ẩn các biến dị sẵn có ở mức cao hơn các dòng trẻ. Các loài có độ bội thể càng cao và số lượng nhiễm sắc thể càng nhiều thì có tính biến dị càng cao.
- Loại và nồng độ chất điều hòa sinh trưởng sử dụng: Các cây tái sinh từ các mô nuôi cấy dài ngày trong môi trường chứa các auxin mạnh như 2,4-D hoặc 2,4,5-T thường có các sai khác. Thí dụ các cây dừa dầu tái sinh từ callus nuôi cấy dài ngày trên môi trường có chứa 2,4-D có tỷ lệ các biến dị lớn hơn rất nhiều khi trồng trên đồng. Ở nho, các tế bào phôi nuôi cấy kéo dài trong vài năm đã mất dần khả năng phân hóa và tái sinh thành cây.
- Thời gian nuôi cấy và số lần cây chuyển: Việc nuôi cấy dài ngày trong điều kiện in vitro cũng như số lần cấy chuyển sẽ làm tăng khả năng xuất hiện các biến soma.

212
SƠ ĐỒ NGUYÊN TẮC CHỌN DÒNG
CÂY
Mô Sẹo
Xử lý đột biến Không xử lý đột biến
Chọn lọc bằng điều kiện hoặc môi trường nuôi cấy
Tế bào sống sót
Quần lạc tế bào
Callus Kiểm tra chất
lượng Dòng mất chất
lượng
Tái sinh cây Giữ dòng ở tập đoàn
Cây
Hạt
Cây
Tế bào chết

213
3. Cách chọn dòng
- Chọn trực tiếp:Thông qua ưu thế về sinh trưởng hay sự khác nhau thấy được về màu sắc có thể chọn lọc được dòng tế bào từ quần thể tế bào. Một số dòng có khả năng chịu kháng sinh, chống chịu muối cũng có thể chọn trực tiếp từ quần thể tế bào. Hệ thống tế bào thường được sử dụng là tế bào nuôi cấy dịch lỏng (suspension cell culture) hoặc callus. Điều kiện chọn lọc ở đây là các dược tố với nồng độ khác nhau gây tác động trực tiếp lên sinh trưởng tế bào. Những tế bào có khả năng phân chia trong môi trường chứa độc tố với nồng độ tăng dần từ lần cấy chuyển này đến lần cấy chuyển khác được sàng lọc dần (chọn lọc kiểu bậc thang), hoặc có thể đưa cả quần thể tế bào vào điều kiện môi trường ức chế sinh trưởng hoàn toàn để chọn ra những tế bào sống sót. Tuy nhiên ngưỡng tối đa của tác nhân chọn lọc phải được thăm dò trước nếu không có thể ức chế sự phát triển của các tế bào đột biến trong quần thể tế bào nuôi cấy.
Thông thường người ta trộn tế bào vào môi trường thạch chứa dược tố và chờ những tế bào sống sót phân chia thành khuẩn lạc, mô sẹo, cũng có thể cấy trực tiếp lên môi trường chọn lọc chứa dược tố. Dòng chống chịu thường xuất hiện một phần khối tế bào nuôi cấy.
- Chọn dòng gián tiếp: Trong trường hợp này đặc điểm của dòng được chọn lọc là biểu hiện khuyết tật của tế bào. Ví dụ chọn dòng thiếu enzyme nitrate reductase (NR), trên môi trường chứa ClO3 những tế bào có NR sử dụng ClO3 như NO3 và khử thành ClO2. ClO2 tác dụng như một độc tố cho nên chỉ có những tế bào không có NR mới sống sót trên môi trường chọn lọc.
- Chọn tổng thể: Các tế bào dị dưỡng của thực vật thường được chọn bằng phương pháp xử lý đột biến và nuôi trên môi trường dinh dưỡng có chứa yếu tố dinh dưỡng cần thiết có khi lại chính là yếu tố gây đột biến.
4. Các hướng chính trong chọn lọc biến dị soma - Chọn dòng tế bào kháng các độc tố
Trong cuối những năm 1970 và 1980, các phytotoxin được sử dụng như các yếu tố chọn lọc thể hiện khả năng kháng bệnh. Tuy nhiên, việc tăng cường khả năng chống bệnh thường kéo theo các đặc tính không mong muốn khác như năng suất thấp. Thí dụ việc chọn lọc liên tiếp với một phytotoxin làm cho các cây khoai tây tái sinh có khả năng kháng bệnh nhưng củ lại nhỏ và chất lượng dinh dưỡng kém đi. Hiện nay, người ta thường sử dụng dịch chiết nuôi cấy của nấm, khuẩn ở các thời kỳ ủ bệnh làm yếu tố chọn lọc để phát hiện các dòng kháng bệnh. Ở đa số các trường hợp, mức độ kháng thể thể hiện ở các cây tái sinh là cao hơn bố mẹ và những thay đổi này có tính di truyền vì tính kháng bệnh được truyền đến đời con cháu thông qua sinh sản hữu tính theo quy luật Mendel.
Kiềm tra t ính di truyền
Cây không di truyền Dòng
Nghiên cứu sinh lý, sinh hóa và di truyền ứng dụng
vào sản xuất

214
- Chọn dòng tế bào chống chịu được những bất lợi của ngoại cảnh Một diện tích đất đáng kể không thích hợp cho sản xuất nông nghiệp vì đất
có hàm lượng muối, phèn hay kim loại nặng quá cao. Sự phát triển của các cây trồng thích hợp cho những vùng như vậy được đặc biệt quan tâm vì diện tích đất trồng trọt ngày càng bị thu hẹp. Việc chọn lọc biến dị dòng soma chống chịu mặn khi nuôi cấy mô đã đạt được ở một số các loài cây trồng như lúa, khoai tây, mía, cà chua. Tuy nhiên, ở hầu hết các trường hợp tính kháng là ngoại biến. Ở một số trường hợp, khả năng chống chịu muối cũng kèm theo các đặc tính không mong muốn khác như năng suất thấp hay khả năng sinh sản kém. Chỉ một số ít các nghiên cứu đã được triển khai để đánh giá sự sử dụng các biến dị dòng vô tính đối với sự chống chịu lại các kim loại nặng. Việc xác định và nhân dòng các gene có thể nâng cao tính chống chịu với muối và các kim loại nặng đang được các nhà di truyền thực vật tập trung nghiên cứu. - Chọn dòng sản xuất dư thừa các sản phẩm: chủ yếu là các amino acid và các hợp chất tự nhiên.
Hình 4.13. Quy trình chọn dòng tế bào chịu mặn của cỏ vertiver thông qua nuôi cấy mô
5. Xác định các biến dị trong nuôi cấy mô
Sau 14 ngày
MS + NaCl (2,5%)
Sau 7 ngày
MS + 1mg/ l BA MS
Sau 7 ngày
Sau 14 ngày
MS + 2 mg/l NAA
MS + 2mg/ l NAA + NaCl (0,5-2,5%)
MMôô ss ẹẹoo
MS + 1mg/ l BA +NaCl (2,5%)

215
Việc xác định các biến dị mong muốn trong nuôi cấy càng sớm càng tốt là hết sức quan trọng vì như thế không cần phải chờ đợi dài ngày trước khi các cây tái sinh có thể thể hiện ra các sai khác về kiểu hình. Các phản ứng kiểm tra sinh học in vitro rất dễ thực hiện và cho kết quả nhanh chóng. Ví dụ một cây lâu năm được dùng vào việc chọn lọc in vitro về tính kháng bệnh, có thể phải mất vài tuần hoặc vài tháng mới có được cây tái sinh để tiến hành các test cho các bệnh cụ thể. Nhưng khi nuôi cấy kép nguồn bệnh giống như môi trường nuôi cấy các tế bào đã được chọn lọc, có thể xác định nếu các tế bào chọn lọc thể hiện tính chống chịu. Khi thành công, một phân tích như vậy cho thấy sự sinh trưởng của nguồn bệnh đã giảm đi bởi các tế bào chọn lọc khi so sánh các tế bào không chọn lọc. Phân tích này đã sử dụng hết sức thành công cho các bệnh do nấm ở một số loài cây ăn quả như cam quýt, xoài và nho.
Trong thập kỷ vừa qua một số kỹ thuật phân tử có thể phân biệt được những thay đổi nhỏ ở mức acid nucleic đã được phát triển. Các kỹ thuật liên quan đến việc sử dụng các chỉ thị phân tử như RAPD, AFLP, ... được sử dụng để xác định xem có hay không một sự thay đổi di truyền bền vững ở các thế hệ tái sinh. Vì vậy, các kỹ thuật này có thể xác định các biến dị sản sinh ra trong nuôi cấy mô một cách chính xác và nhanh hơn.
6. Khả năng ứng dụng của chọn lọc biến dị soma Trong cải tiến giống cây trồng truyền thống, nhà tạo giống phải trồng một số
lượng lớn các cây trồng trong nhà kính hay trên đồng ruộng nếu như muốn chọn lọc một đặc tính cụ thể nào đó. Thí dụ nếu mục đích là phát triển dòng chống chịu mặn của một loài cụ thể, một số lượng lớn các cá thể cần được trồng và xử lý ở các nồng độ khác nhau của muối để cuối cùng xác định xem cây nào có thể chịu đựng được qua quá trình thử nghiệm. Nếu làm như thế, số nguyên liệu sử dụng sẽ bị giới hạn bởi diện tích có sẵn và thời gian. Ngoài ra các yếu tố di truyền cũng cản trở đến quá trình chọn lọc.
Các dạng biến đổi chọn được trong nuôi cấy in vitro thường xuất hiện với tần số 10-8-10-5 khi không xử lý đột biến. Nếu có xử lý đột biến tăng tần số lên 10 lần. Các tác nhân gây đột biến thường được sử dụng là: Ethyl metan sulfate (EMS), N-methyl-N-nitro-N-nitrozoguanidin, N-ethyl-nitrozoure (ENU), tia X, tia UV. Chưa có số liệu cụ thể về phổ và tần số đối với từng loại tác nhân về độ lớn của khối tế bào được xử lý, mật độ tế bào gieo trên đĩa và số nhiễm sắc thể của tế bào bị xử lý. Sử dụng protoplast của thực vật bậc cao là phương pháp tốt nhất để tiếp cận vấn đề này.
Các hệ thống nuôi cấy tế bào giúp cho các nhà chọn tạo giống có môi trường được xác định rõ ràng, nơi mà áp suất chọn lọc có thể áp dụng trên hàng ngàn các tế bào có khả năng tái sinh thành cây hoàn chỉnh. Như vậy, nhà chọn tạo giống có thể chọn lọc từ lượng rất lớn các vật liệu di truyền đồng nhất và xây dựng các thử nghiệm nhanh chóng trong một vài đĩa petri hay bình nuôi cấy. Ngoài ra một số ngành khoa học nhờ đó có thể nghiên cứu một loài nhiệt đới ở vùng ôn đới hay ngược lại vì điều kiện môi trường đặc thù có thể tạo ra ở bất kỳ đâu.
Mặc dù không được nghiên cứu nhiều nhưng các biến dị bắt nguồn từ nuôi cấy có tiềm năng nhất định trong công tác cải tiến giống cây trồng, đặc biệt là cây

216
lâu năm vốn bị cản trở bởi nền di truyền hẹp và chu kỳ tái sinh dài. Ví dụ như đối với cây chuối và cây thuốc lá bắt buộc phải nhân giống qua con đường vô tính hàng nghìn năm nay, việc cải tiến di truyền ở các loài cây này rất khó khăn vì chúng rất hiếm khi tạo thành các hạt có khả năng sinh sản. Ở trường hợp như vậy, lựa chọn duy nhất là trông mong vào các đột biến vô tính tự nhiên xảy ra với tần suất cực thấp hay các đột biến do cảm ứng.
Ở nhiều loài nhân giống vô tính, thậm chí các đột biến cảm ứng cũng rất khó thực hiện bởi số lượng cây con là rất lớn. Việc cố gắng chiếu phóng xạ cho một số lượng lớn như vậy thường dẫn đến các dạng khảm hay là bị chết. Sự phát triển hệ thống tái sinh tế bào như là sự phân hóa phôi vô tính cung cấp khả năng tiếp xúc của một số lượng lớn các tế bào tái sinh với phóng xạ gamma hay các chất đột biến hóa học như ethylmethyl sulfonate ... theo phương thức hoàn toàn được kiểm soát và như thế tăng cường được nền di truyền hiện có.
VIII. CÔNG NGHỆ TẾ BÀO TRONG BẢO QUẢN NGUỒN GENE Trong công tác giống, việc duy trì nguồn tài nguyên di truyền thực vật có vai
trò vô cùng quan trọng, làm cơ sở cho việc khai thác, sử dụng cho các công việc trước mắt cũng như trong tương lai. Vì thế, công việc bảo quản nguồn gene thực vật là một công việc không thể thiếu trong công tác giống.
Có hai phương thức bảo quản nguồn gene cây trồng: bảo quản in-situ và bảo quản ex-situ. Bảo quản in-situ là bảo quản tại chỗ, cây được duy trì tại điều kiện sinh thái tự nhiên của chúng. Bảo quản ex-situ là bảo quản các mẫu trong điều kiện nhân tạo tối ưu hay nói cách khác, các mẫu được thu gom từ nơi xuất xứ và đem bảo quản cất giữ hoặc trồng trọt tại các vườn mẫu. Bảo quản ex-situ có các phương thức sau: (1) trên vườn ươm (field geneebank); (2) bảo quản hạt trong kho lạnh (seed bank); (3) bảo quản in vitro (in vitro geneebank); (4) bảo quản DNA.
Trong bốn phương thức đó, phương thức bảo quản in vitro có vai trò quan trọng vì việc bảo quản được thực hiện trong một không gian hẹp nhưng có thể lưu giữ được khối lượng lớn cá thể, điều kiện bảo quản hoàn toàn chủ động và là biện pháp có hiệu quả đối với các cây trồng nhân giống vô tính và hạt cây dễ mất sức nảy mầm ở nhiệt độ và ẩm độ thấp. Trong vòng 20 năm gần đây đã có hơn một ngàn loài thực vật khác nhau được bảo quản theo phương thức này (Engelmann, 1997). Có hai phương pháp bảo quản in vitro: bảo quản sinh trưởng tối thiểu và bảo quản ngừng sinh trưởng tạm thời. 1. Phương pháp bảo quản sinh trưởng tối thiểu
Phương pháp này thường áp dụng cho việc bảo quản ngắn hạn các cây nhân giống vô tính in vitro (chuối, dứa, khoai tây, khoai lang...). Mẫu phải đảm bảo sạch bệnh. Mẫu đưa vào bảo quản thường ở dạng phôi, chồi, mầm, cây non. Đặc điểm của phương pháp này là kéo dài thời gian giữa hai lần cấy chuyển. Nhờ các nhân tố ức chế sự sinh trưởng mẫu cấy mà thời gian cấy chuyển dài ra giảm tới mức tối thiểu chi phí trong quá trình bảo quản. Trong quá trình bảo quản, mẫu phải được đặt trong điều kiện ánh sáng, nhiệt độ và chế độ dinh dưỡng sao cho tốc độ sinh trưởng của chúng bị giảm tới 15-20 lần so với tốc độ sinh trưởng trong điều kiện bình thường. Các yếu tố thường dùng để ức chế sự sinh trưởng của mẫu là:

217
+ Giảm nhiệt độ phòng nuôi: đây là tác nhân được dùng phổ biến nhất trong bảo quản sinh trưởng tối thiểu. Với các loài chịu lạnh nhiệt độ bảo quản từ 0-5oC. Ví dụ: chồi cây thông và cây táo giữ được 52 tuần ở 2oC, khoai tây bảo quản ở 6-12 oC có thời gian cấy chuyển là 25-52 tuần. Những loài cây nhiệt đới thường đảm bảo ở nhiệt độ cao hơn. Ví dụ: chuối giữ được 15 tháng ở nhiệt độ 15oC, cọ dầu bảo quản ở 18oC.
+ Bổ sung vào môi trường các chất ức chế sinh trưởng (abscicic acid, Chlor Cholin Cloride - CCC), các chất tăng áp suất thẩm thấu của môi trường (saccharose, mannitol...) và giảm cung cấp oxy (dầu phủ)... cũng có tác dụng ức chế sinh trưởng mạnh. Ví dụ: nuôi cấy khoai tây ở 22oC và môi trường có 5-10 mg/l ABA thì thời gian cấy chuyển kéo dài tới 12 tháng; tương tự như vậy với cây sắn khi nuôi cấy ở 2oC trong môi trường có 4% saccharose có thể kéo dài thời gian cấy chuyển tới 15 tháng.
Khi kết thúc một giai đoạn bảo quản, mẫu bảo quản cần được chuyển sang môi trường mới pha chế và đặt trong điều kiện tối thích một thời gian ngắn để kích thích sự tái sinh trưởng của mẫu trước khi bắt đầu một chu kỳ bảo quản mới.
2. Phương pháp bảo quản ngừng sinh trưởng tạm thời
Đặc điểm của phương pháp này là mẫu được bảo quản trong nitơ lỏng. Trong thời gian bảo quản tế bào, mô hoàn toàn ngừng sinh trưởng. Đây là phương pháp bảo quản dài hạn, có thể bảo quản mẫu tới 20-30 năm hoặc lâu hơn. Mẫu đem bảo quản thường ở dạng: phôi, mô sẹo, các mô nuôi cấy... Trước khi đưa vào bảo quản mẫu cần được xử lý để tránh tạo tinh thể nước trong tế bào gây chết mẫu. Quy trình bảo quản này gồm các bước sau:
- Tách mẫu nuôi cấy in vitro đang ở pha tăng trưởng mạnh để đưa vào bảo quản.
- Xử lý chống đông cho tế bào, mô của mẫu cấy: mẫu được xử lý bằng dung dịch 5-10% proline, mannitol 3-6% có bổ sung chất chống đông là dung dịch DMSO 1M + glycerol 1M + đường saccharose 2M..
- Xử lý lạnh đến nhiệt độ đóng băng ổn định của tế bào chất (-30 đến -40oC) - Bảo quản mẫu ở nhiệt độ -196oC (trong nitơ lỏng) Mẫu sau thời gian bảo quản khi lấy ra được phá băng ở nhiệt độ 37- 40oC.
Mẫu sẽ được phục hồi sinh trưởng và có thể tiếp tục nuôi cấy để tạo thành cây hoàn chỉnh.
Tài liệu tham khảo 1. Nguyễn Hoàng Lộc. 2007. Giáo trình Nhập môn Công nghệ sinh học. NXB
Đại học Huế 2. Nguyễn Văn Uyển và cs. 1993. Nuôi cấy mô thực vật phục vụ công tác giống
cây trồng. NXB Nông nghiệp, thành phố Hồ Chí Minh. 3. Nguyễn Văn Uyển. 1996. Những phương pháp công nghệ sinh học thực vật,
tập II. NXB Nông nghiệp, thành phố Hồ Chí Minh.

218
4. Nguyễn Văn Uyển và Nguyễn Tiến Thắng. 1999. Những kiến thức cơ bản về công nghệ sinh học. NXB Giáo dục, Hà Nội.
5. Caponetti J. D., Gray D. J., Trigiano R. N. 1999. Plant Tissue culture concepts and laboratory exercises. CRC Press, USA.
6. Narayanaswamy S. 1994. Plant cell and tissue culture. Tata McGraw-Hill Publishing company limited, New Delhi.

LỜI NÓI ĐẦU
Công nghệ sinh học hiện đại được ra đời và phát triển như vũ bão, mang lại
những thành tựu tuyệt vời cho nhân loại. Hiện nay, công nghệ sinh học không chỉ là
công cụ nghiên cứu sắc bén cho phép nghiên cứu cơ chế của các hiện tượng sống ở
mức độ phân tử mà còn nhanh chóng trở thành một công nghệ sản xuất với những
sản phẩm hoàn toàn mới mẽ trên mọi lĩnh vực, trước hết là y tế và nông nghiệp.
Để đáp ứng cho nhu cầu học tập của sinh viên khoa Nông học, tập thể tác giả
chúng tôi được giao nhiệm vụ biên soạn giáo trình “Công nghệ sinh học thực vật”.
Nội dung giáo trình gồm 4 chương, với sự phân công biên soạn như sau:
Chương 1: Công nghệ DNA tái tổ hợp (TS. Trần Thị Lệ)
Chương 2: Các kỹ thuật chính sử dụng trong phân tích acid nucleic (TS. Trần
Thị Lệ)
Chương 3: Công nghệ chuyển gen vào tế bào thực vật (TS. Trương Thị Bích
Phượng)
Chương 4: Công nghệ nuôi cấy mô tế bào thực vật (ThS. Trần Thị Triêu Hà,
CN. Dương Thanh Thủy)
Do mới được xuất bản lần đầu nên giáo trình khó tránh khỏi thiếu sót hoặc
chưa đáp ứng được yêu cầu bạn đọc. Vì thế, chúng tôi mong nhận được nhiều ý
kiến đóng góp để lần xuất bản sau được hoàn thiện hơn.
Chúng tôi trân trọng cảm ơn PGS.TS. Nguyễn Hoàng Lộc đã đọc bản thảo,
đóng góp nhiều ý kiến quý báu và Trường Đại học Nông Lâm Huế đã hỗ trợ chúng
tôi biên soạn và xuất bản giáo trình này.
Các tác giả

211
MỤC LỤC Bài 1 CÔNG NGHỆ DNA TÁI TỔ HỢP ..................................................................................... 5
I. Khái niệm .................................................................................................................................. 5 1. Ý nghĩa của tạo dòng DNA.............................................................................................. 5 2. Tạo dòng DNA là gì? ....................................................................................................... 5 3. Tại sao sự tạo dòng DNA quan trọng như vậy? Ý nghĩa của tạo dòng đối với nghiên cứu và công nghệ sinh học ........................................................................................................... 5
II. Các enzyme tác động lên DNA ............................................................................................ 11 1.Nuclease ......................................................................................................................... 12 2. Ligase ............................................................................................................................. 13 3. Polymerase ...................................................................................................................... 13 4. Các enzyme biến đổi DNA ............................................................................................. 16 5. Topoisomerase ........ .......................................................................................................17
III. Enzym hạn chế: Restriction endonuclease ......................... ..............................................17 1. Phát hiện và kiểu tác động của enzyme hạn chế ............................................................ 18 2. Enzyme hạn chế loại II cắt DNA ở những trình tự hoàn toàn xác định ......................... 19 3. Đầu bằng và đầu dính ................................................................................................... 21 4. Số vị trí nhận biết của enzyme hạn chế trong phân tử DNA ......................................... 22 5. Quá trình cắt bằng enzyme hạn chế trong phòng thí nghiệm ......................................... 23 6. Phân tích kết quả của phản ứng cắt bằng enzyme hạn chế ............................................ 24
6.1. Sự tách các phân tử bằng điện di ...................................................................... 24 6.2. Sự hiển thị của phân tử DNA trong gel .............................................................. 26 6.3. Đánh giá độ lớn của các đoạn DNA ................................................................... 28 6.4. Lập bản đồ giới hạn phân tử DNA.......................................................................29
IV. Gắn các phân tử DNA (ligation) ....... .................................................................................31 1. Tác dụng của DNA- ligase. ...................................... ........................................................31 2. Các đầu dính tăng hiệu quả phản ứng gắn ......................................................................31 3. Tạo đầu dính từ phân tử DNA đầu bằng .........................................................................32
V. Vector tạo dòng.............. .......................................................................................................39 1. Vector plasmid ...... .......................................................................................................39
1.1. Vector tạo dòng pB322 . ......................................................................................42 1.2. pUC8: Plasmid có gene chọn lọc LacZ . .............................................................44 1.3. pGEM3Z: Sự phiên mã DNA tạo dòng in vitro ................................................45
2. Vector tạo dòng trên cơ sở thực khuẩn thể M13 ................................................... ......... 46 2.1. Phát triển vector tạo dòng M13mp2 ................................................................. ..46 2.2. M13mp7 - Những vị trí tạo dòng đối xứng ....................................................... 47
3. Vector tạo dòng là phage................................................................................................ 49 3.1. Vector thay thế và vector xen đoạn................................................... ..................53 3.2.Thí nghiệm tạo dòng với vector xen đoạn hoặc thay thế ..................................... 55
4. Các vector lai plasmid/phage ......................................................................................... 56 4.1. Cosmid ............................................................................................................... 56 4.2. Phagemid ............................. ...............................................................................58
VI. Tạo dòng gene................................................... ...................................................................58 1. Tạo dòng gene đã biết ....................................................................................................58 2. Tạo dòng gene chưa biết .. ...............................................................................................68
Bài 2 CÁC KỸ THUẬT CHÍNH SỬ DỤNG TRONG PHÂN TÍCH NUCLEIC ACID .......... 71 I. Phương pháp tách chiết DNA... ...........................................................................................71
1. Phương pháp tách DNA tổng số...................................................................................... 71 2. Tách chiết DNA plasmid ................................................................................................. 78 3. Tinh sạch DNA của phage ............................................................................................. 84
II. Kỹ thuật lai nucleic acid .............. .........................................................................................90

212
1. Kỹ thuật Southern blot .................................................................................................... 90 2. Kỹ thuật Northern blot .................................................................................................. 106 3. Kỹ thuật Western blot ................................................................................................... 106
III. Kỹ thuật xác định trình tự DNA ..................................................................................... 111 1. Phương pháp Sanger-Coulson (phương pháp enzyme hay dideoxy) ............................ 111 2. Phương pháp Maxam-Gilbert (Phương pháp hoá học) ................................................. 113 3. Phân tích trình tự DNA dài .................................................................................. ........115 4. Các khả năng xác định trình tự DNA ....................... .....................................................117 5. Giải trình tự gene bằng phương pháp tự động .............................................................. 117
IV. Kỹ thuật PCR ..................................................................................................................... 120 1. Nguyên tắc.............. .......................................................................................................120 2. Kỹ thuật PCR ......... .......................................................................................................121 3. Phân tích sản phẩm PCR ............................................................................................. 126 4. Những khó khăn do sự khiếm khuyết của Taq-polymerase ......................................... 129 5. Ứng dụng PCR ...... .......................................................................................................130
V. Các kỹ thuật xác định tính đa hình DNA dựa trên PCR................................................ 133 1. Kỹ thuật RAPD: Đa hình các đoạn DNA khuếch đại ngẫu nhiên. ............................... 133 2. Kỹ thuật AFLP (amplified fragment length polymorphism) – Sự đa hình chiều dài các phân đoạn DNA được khuếch đại ..................................................................................... 134 3. Kỹ thuật SSR (simple sequence reppeats): Đa hình đoạn trình tự lặp lại đơn giản ...... 135
VI. Thư viện hệ gene (geneomic library) và thư viện cDNA. ............ ...................................136 1. Thư viện hệ gene ( geneomic library) ......................................... .................................136 2. Thư viện cDNA ..... .......................................................................................................139
Bài 3 CÔNG NGHỆ CHUYỂN GENE VÀO TẾ BÀO THỰC VẬT ...... ................................145 I. Giới thiệu chung............ .......................................................................................................145 II. Phương pháp chuyển gene gián tiếp ..................................................................................146
1. Hệ thống Agrobacterium .............................................................................................. 146 2. Ti-plasmid là nhân tố gây biến nạp tự nhiên ở thực vật ............................ ...................148 3. T-DNA và các trật tự biên 25 bp ....................................... ...........................................149 4. Vùng vir .................. ......................................................................................................149 5. Quá trình chuyển T-DNA............................................... ..............................................150 6. Các dẫn xuất Ti-plasmid như là vector thực vật ................................... .......................151 7. Phương thức truyền gene vào thực vật nhờ Agrobacterium ................. .......................153
III. Phương pháp chuyển gene trực tiếp ................................................................................156 1. Chuyển gene vào protoplast ..... ....................................................................................156 2. Chuyển nạp DNA ngoại lai vào tế bào bằng xung điện ...................... .........................157 3. Chuyển gene bằng phương pháp bắn gene.................................................................... 157 4. Chuyển nạp DNA ngoại lai vào tế bào bằng kỹ thuật vi tiêm (microinjection) ........... 160
Bài 4 ỨNG DỤNG CỦA CÔNG NGHỆ CHUYỂN GEN VÀO TẾ BÀO THỰC VẬT
1. Chuyển gene kháng thuốc diệt cỏ ................................................................................. 160 2. Chuyển gene kháng sâu bệnh....................................................................................... 161 3. Chuyển gene gây chín chậm- DR (delayed ripening ) .................................................. 162 4. Chuyển gene tạo hoa có màu sắc và kiểu hoa mới....................................................... 164 5. Chuyển gene sản xuất protein động vật và người ........................................................167 6. Chuyển gene thay đổi protein ........................................................... ............................168 7. Chuyển gene tạo tính bất dục của cây trồng ................................................................. 169 8. Chuyển gene giúp cây trồng chống điều kiện bất lợi ................................ ...................171
Bai 5 CÔNG NGHỆ NUÔI CẤY MÔ TẾ BÀO THỰC VẬT .................................................. 176

213
I. Giới thiệu chung................................................................................................................... 176 1. Sơ lược lịch sử phát triển .............................................................................................. 176 2. Cơ sở khoa học của nuôi cấy mô tế bào thực vật ......................................................... 177 3. Một số thuật ngữ và khái niệm cơ bản ......................................................................... 177 4. Môi trường dinh dưỡng ................................................................................................ 179 5. pH của môi trường ....................................................................................................... 182 6. Vô trùng mẫu cấy ......................................................................................................... 182
II. Thụ phấn và nuôi cấy phôi (hữu tính) in vitro ................................................................ 183 1. Tính bất hợp của giao tử trước khi thụ tinh - thụ phấn in vitro..................................... 183 2. Tính bất hợp của giao tử sau khi thụ tinh - Nuôi cấy phôi in vitro ............................... 183 3. Kỹ thuật nuôi cấy phôi hữu tính ................................................................................... 184 4. Một số khó khăn trong nuôi cấy phôi hữu tính . ........................................................... 184
III. Nhân giống in vitro .......................................................................................................... 185 1. Ý nghĩa của việc nhân giống cây trồng bằng phương pháp nuôi cấy mô tế bào .......... 185 2. Các phương thức nhân giống in vitro ........................................................................... 185 3. Các giai đoạn trong quy trình nhân giống vô tính in vitro ........................................... 189 5. Nhân giống in vitro và các đặc điểm không di truyền ................................................. 192
IV. Nuôi cấy bao phấn và tạo giống cây trồng ...................................................................... 192 1. Vấn đề đơn bội ở thực vật ...................................................................... .......................192 2. Phương pháp tạo cây đơn bội in vitro ........................................................................... 193 3. Ứng dụng của đơn bội ................................................................................................... 195 4. Một số tồn tại trong nuôi cấy đơn bội ...................................................... .....................196
V. Nuôi cấy tế bào trần & triển vọng ứng dụng ................................................................... 196 1. Kỹ thuật nuôi cấy tế bào trần ........................................................................................ 197 2. Dung hợp tế bào trần..................................................................................................... 198 3. Ứng dụng của protoplast .............................................................................................. 199
VI. Làm sạch bệnh virus trong công tác phục tráng giống cây trồng ................................ 200 1. Nguyên lý làm sạch virus và duy trì tính sạch bệnh ................................... ..................200 2. Các kỹ thuật làm sạch virus in vitro ............................................................................. 201 3. Kiểm tra, nhân nhanh và duy trì tình trạng sạch bệnh .................................................. 202
VII. Tạo đột biến và chọn dòng tế bào thực vật .................................................................... 204 1. Khái niệm ..................................................................................................................... 204 2. Các nguyên nhân chính gây biến dị soma .................................................................... 204 3. Cách chọn dòng............................................................................................................. 206 4. Các hướng chính trong chọn lọc biến dị soma ............................................................. 206 5. Xác định các biến dị trong nuôi cấy mô ....................................................................... 208 6. Khả năng ứng dụng của chọn lọc biến dị soma ............................................................ 208
VIII. Công nghệ tế bào trong bảo quản nguồn gene ......................................... ...................209 1. Phương pháp bảo quản sinh trưởng tối thiểu ............................................................... 209 2. Phương pháp bảo quản ngừng sinh trưởng tạm thời ..................................................... 210