a multianalyte electrochemical immunosensor based on patterned carbon nanotubes modified substrates...
TRANSCRIPT

A multianalyte electrochemical immunosensor based on patternedcarbon nanotubes modified substrates for detection of pesticides
Guozhen Liu n, Wenqi Guo, Dandan SongKey Laboratory of Pesticide and Chemical Biology of Ministry of Education, College of Chemistry, Central China Normal University, Wuhan 430079, PR China
a r t i c l e i n f o
Article history:Received 28 June 2013Received in revised form5 September 2013Accepted 5 September 2013Available online 16 September 2013
Keywords:ImmunosensorMicrocontact printingAryl diazonium saltsSingle walled carbon nanotubes
a b s t r a c t
A novel multianalyte electrochemical immunosensor based on the assembly of patterned SWNTs onglassy carbon (GC) substrates was developed for simultaneous detection of endosulfan and paraoxon.Based on aryldiazonium salt chemistry, forest of SWNTs can be patterned on GC substrates by C–Cbonding using micro contact printing (MCP), which provides an interface showing efficient electrontransfer between biomolecules and electrodes. Then redox molecules FDMA and PQQ can be attached tothe SWNTs, respectively followed by the attachment of specific epitopes and antibodies. The modifiedsensing surfaces were characterized by XPS, SEM, AFM and electrochemistry. Based on the currentchange of specific redox probes, the fabricated immunosensor array can be used for simultaneousdetection of endosulfan and paraoxon by a displacement assay. In phosphate buffer solution (50 mM,pH 7.0), there is a linear relationship between electrochemical signal of FDMA and the concentration ofendosulfan over the range of 0.05–100 ppb with a detection limit of 0.05 ppb; the linear range betweenelectrochemical signal of PQQ and the concentration of paraoxon is 2–2500 ppb with a detection limit of2 ppb. The immunosensor array demonstrates high repeatability, reproducibility, stability and selectivityfor the detection of endosulfan and paraoxon.
& 2013 Elsevier B.V. All rights reserved.
1. Introduction
Immunochemical techniques such as immunoassay based on theantibody-hapten reaction have lately gained great attention for theanalysis of agrochemicals because of their specificity, cost-effective-ness, and high sample throughput (Gabaldon et al., 2003). Sensorsthat can be used for detecting many species simultaneously are ofparticular importance for routine analysis in the field. The ability toperform multiple analyses on a single sensing surface has a numberof advantages (Rowe et al., 1999) over performing multiple parallelanalyses on different substrates, such as a single set of positive andnegative controls can be used for all the assays, use of a singlesubstrate provides more effective (and valid) comparisons of theexperimental data and controls, and performing multiple assayssimultaneously decreases the assay times compared with sequentialanalyses et al. Studies on optical biosensors capable of simultaneousdetection of multiple analytes are very active (Anderson et al.,2000; Blawas et al., 1998; Ekins and Chu, 1993; Venkatanarayananet al., 2012). However, demonstration of the ability to use a singlesensor substrate for simultaneous, multi-analyte detection based onelectrochemical biosensors is limited (Plowman et al., 1999; Rowe
et al., 1999; Taitt et al., 2002). Maquieira's group has successfullydeveloped a multianalyte immunosensor for on-line determinationof organic compounds (Gonzalez-Martinez et al., 2001). However,the reproducibility is low, and the effect of saturation could reducethe number of analytes that can be monitored.
Single walled carbon nanotubes (SWNTs) have attracted increas-ing attention since their discovery two decades ago (Iijima, 1991),due to their unique structural, mechanical and electric properties.Especially SWNTs have good biocompatibility characteristics, whichis beneficial to enhance electron transfer and to maintain thebiological activity of enzyme (Gooding et al., 2003). With thedeepening of research on properties of SWNTs, more and morepeople use SWNTs in surface modification for biosensors. The mostapplied method to anchor SWNTs is based on the formation ofamide bonds from the reaction between the amines located on themodified electrode and the carboxylate groups at the ends of side-wall defects of nanotubes (Garrett et al., 2010). Carbon nanotubescan also be fabricated to substrates based on H-bonding interactionsbetween the carboxylate groups of on nanotubes and the –OHgroups at the ITO surface (Diakowski et al., 2010). Pinson andcoworkers firstly reported the covalent attachment of nanotubesto substrates by C–C bonds using diazonium salt reactions (Joyeuxet al., 2009), and they claimed that nanotubes were tethered to thesurface in an approximately horizontal orientation with respect tothe surface. Recent studies by Ferri's group show vertically alignedSWNTs are covalently modified on surfaces through C–C bondings
Contents lists available at ScienceDirect
journal homepage: www.elsevier.com/locate/bios
Biosensors and Bioelectronics
0956-5663/$ - see front matter & 2013 Elsevier B.V. All rights reserved.http://dx.doi.org/10.1016/j.bios.2013.09.009
n Corresponding author. Tel.: þ86 27 6786 7535.E-mail address: [email protected] (G. Liu).
Biosensors and Bioelectronics 52 (2014) 360–366

(De Fuentes et al., 2011). Although the precise orientation of SWNTstethered on substrates by C–C bonds using diazonium salt reactionsis argumentative, it is widely agreed that the formed SWNTs inter-face is very stable which is in consistency with the known stabilityof tether layers grafted from diazonium salts (Liu et al., 2007).Meanwhile, the assembly of SWNTs on the tethered aryldiazoniumsalt layer has greatly increased the electronic coupling between theelectrode and outside biological environment. Our previous studieshave demonstrated that immunosensors based on SWNTs modifiedsubstrates have the ability for rapid, sensitive and quantitativeanalysis of antibodies and small molecules (Liu et al., 2011a, 2013,2012b). Venkatanarayanan et al. (2012) reported an electrochemilu-minescence sensor array based on vertically aligned SWNT , whichshows a wide linear dynamic range, a remarkably low detectionlimit of 1.1 pM for IgG and is promise for future development ofmultiplexed assays. An additional feature of this kind of immuno-sensor becomes more and more important: the immunosensorshould be able to discriminate between multiple analytes in a singlepot of samples. Fabrication of the sensing interface with differentredox reporters might be able to make this additional featurepossible. Small redox molecules (Harper et al., 2007; Liu et al.,2008; Pheeney and Barton, 2012) ferrocene derivatives (Beheshtiet al., 2012; Diakowski et al., 2010), and redox enzymes (Guo et al.,2013; Jin et al., 2012) are popular redox reporters. However, toour knowledge no report shows multiple redox reporters aresimultaneously fabricated on the sensing interfaces for multianalyteimmunosensors.
Modification of carbon surfaces with aryldiazonium salts togive covalent attached layers has attracted wide attention since itis firstly reported it 1992 (Delamar et al., 1992). Recent studies,such as in situ aryldiazonium salt fabrication (Baranton andBelanger, 2005), microcontact printing (MCP) using spontaneousreduction of aryldiazonium salts (Garrett et al., 2007), and formingmixed layers of aryldiazonium salts with something close tomolecule level control of sensing interfaces (Liu et al., 2010),demonstrate that applications of aryldiazonium salt chemistryon sensing interface have been progressively attractive. As asimple and relatively fast patterning method, MCP using adheringa poly(dimethylsiloxane) (PDMS) stamps and aryldizonium saltinks becomes an attractive route to pattern and covalently attachreactive tether layers. A broad range of substrates can be patternedby adhering a PDMS mold to the surface to form microchannels(Downard et al., 2006; Lehr et al., 2010). However, little work isreported on using patterned surfaces for multianalyte immuno-sensors for detection of pesticides.
In this contribution, as illustrated in Scheme 1, we report anovel multianalyte electrochemical immunosensor based on theassembly of SWNTs formed by MCP on glassy carbon (GC)substrates. The flow chamber modules can be made on GCsubstrates by placing a 4-channel PDMS patterning template incontact with the surface, applying pressure to create a fluid-tightand airtight seal. Two redox probes ferrocenedimethylamine(FDMA) and pyrroloquinoline quinone (PQQ) were loaded todifferent channels, respectively. Binding of the antibody to thesurface-bound epitope immerses the redox molecules in a proteinmedium. A consequence of this change in environment is theattenuation of electron transfer to the redox molecules due tothe inaccessibility of a counterion. Association or dissociation ofthe antibody with the sensing interface causes a modulation of theelectrochemistry from the redox molecules. Based on differentelectrochemistry signals from two redox reporters FDMA and PQQ,this fabricated immunosensor array can be used to simultaneouslydetect two analytes in one sample. Hence the approach weestablished here is a prototype research that is promising fordesigning portable device for on-site detection of pesticides inenvironmental monitoring.
2. Experimental section
2.1. Reagents and materials
Sodium nitrite, potassium ferricyanide, hydrochloric acid,4-phenylenediamine, aniline, 2-[4-(2-Hydroxyethyl)-1-pipera-zine]ethanesulfonic acid (HEPES), 1-ethyl-3-[3-dimethylaminopro-pyl] carbodiimide hydrochloride (EDC), N-hydroxysuccinimide(NHS), and pyrroloquinoline quinone (PQQ) were purchased fromSigma-Aldrich (Shenshi Huagong, Wuhan). SWNTs prepared bythe HiPco process were purchased from Carbon NanotechnologiesIncorporated. Cut SWNTs were prepared as reported previously(Liu et al., 1998). The aryldiazonium cations for 2-(2-(2-(4-amino-phenoxy)-ethoxy) -ethoxy)-ethanol (PEG) was custom synthesized(Liu and Gooding, 2006). Ferrocenedimethylamine (FDMA) wassynthesized by the literature method (Ossola et al., 2003). Endo-sulfan and paraoxon were purchased from Fluka (Sanyi Chemicals,Wuhan). The endosulfan hapten, anti-endosulfan IgG, paraxonhapten, and anti-paraoxon IgG were prepared as reported pre-viously (Heldman et al., 1985; Liu et al., 2013, 2012b). All otherreagents were used as received. Aqueous solutions were preparedusing Mill-Q water (418 MΩ cm). Phosphate buffered saline (PBS)solutions were 0.15 M NaCl and 0.1 M phosphate buffer, pH 7.3.Phosphate buffer solution for electrochemistry was prepared using0.1 M buffer with added 0.05 M KCl (pH 7.0).
2.2. Apparatus
All electrochemical experiments were conducted using theGaossUnion EC510 potentiostat (GaossUnion, China). All experi-ments utilized a 3 mm disk GC working electrode, a Pt secondaryelectrode and a Ag/AgCl (3.0 M NaCl) reference electrode. X-rayphotoelectron spectra (XPS) were collected from GC plates on a VGmultilab 2000 spectrometer with a monochromated Al Kα source(1486.6 eV), hemispherical analyzer and multichannel detector.SEM was carried out using a Hitachi S-900 SEM (Berkshire,England). Atomic force microscopy (AFM) images were taken onGC plates using a Digital Instruments Dimension 3100 scanningprobe microscope.
2.3. Preparation of aryldiazonium cation ink and microcontactprinting
Aryldiazonium salt modified SWNTs were prepared accordingto the literature (de Fuentes et al., 2011). Specifically, cut SWNTs(1 mL, 1 mg/mL) was added to the mixture of HCl (1 mL, 0.5 M),p-nitroaniline (10 mM), and NaNO2 (10 mg) which was left to reactovernight. The functionalized SWNTs suspension was centrifuged,and the residue was washed with water and acetonitrile. Then theresidue (p-nitrobenzene modified SWNTs) was treated in acetoni-trile solution containing 5 mM PEG diazonium salts for 6 h toattach PEG molecules to the sidewall of SWNTs. The PEG functio-nalized SWNTs suspension was centrifuged, and the residue waswashed with acetonitrile. Finally p-nitrobenzene/PEG-modifiedSWNTs were suspended in a basic solution (pH 10) and treatedwith NaBH4 for 1 h to reduce –NO2 to –NH2. Subsequently themixture was centrifuged and rinsed with water. The p-aminophe-nyl/PEG-modified SWNTs were suspended in HCl (0.5 M) contain-ing NaNO2 (10 mg) to get the aryldiazonium cation ink.
PDMS stamps were immersed in the aryldiazonium cation inkfor 10 min, dried to tackiness in a stream of N2 gas, and placed onthe substrate for 25 min to obtain the GC-Ph-SWNTs modifiedinterface. All printed samples were rinsed with acetonitrile solu-tion followed by Milli-Q water and dried in a stream of N2 prior toanalysis or further treatment. Control samples were prepared in
G. Liu et al. / Biosensors and Bioelectronics 52 (2014) 360–366 361

the same way but using a solution from which NaNO2 had beenomitted (“blank” ink).
2.4. Fabrication of the sensing interface
An absolute ethanol solution containing 40 mM DCC and 5 mMFDMA was loaded into the SWNTs modified interfaces (channel1 and 3) for 6 h. Then channel 1 and 3 were rinsed with copiousamounts of water before loading 1 mg mL�1 endosulfan hapten in0.1 M PBS for 2 h at 4 1C. The endosulfan hapten modified electro-des were rinsed with copious amounts of water and PBS beforeloading a PBS solution of endosulfan monoclonal IgG for 30 min at4 1C. Simultaneously, an 10 mM HEPES buffer (pH 7.5) solutioncontaining 20 mM EDC/1 mM NHS and 1 mM PQQ was loaded intothe SWNTs modified interfaces (channel 2 and 4) for 6 h. Channel2 and 4 were treated with the same manner as channel 1 and3 except that endosulfan hapten was replaced by paraoxon haptenand endosulfan monoclonal IgG was placed by paraoxon mono-clonal IgG. Then the formed interface is ready for electrochemistrymeasurement using cyclic voltammetry (CV) and square wavevoltammetry (SWV).
3. Results and discussion
Fig. 1 shows the C1s and N1s signals of the (a and c) bare GCsurface; (b and d) after modification of 4-aminophenyl/PEG-modified SWNTs onto the GC surface. C1s scan of the bare GCplate was dominated by the graphitic carbon peak at 284.4 eVtogether with a broader peak on the high binding energy 285.2 eVdue to the presence of low levels of oxidized carbon species on theelectrode surface. After modification of 4-aminophenyl/PEG-mod-ified SWNTs, a peak at 288.8 eV, which is typical of C1s from–COOH groups, was observed (Fig. 1b), indicating the presence of–COOH groups on GC surfaces. The ether carbon species at286.3 eV was also observed, indicating the successful attachmentof PEG molecules (Liu et al., 2012b). Being different from the N1ssignals of bare GC surface, the N1s signal at 400.2 eV was detectedon GC-Ph-SWNTs surface (Fig. 1d), which is attributed to –NH2
groups (Liu et al., 2012b). The presence of –NH2 groups indicatesthat only partial of –NH2 groups on SWNTs have been converted todiazonium cations (–N2
þ) attaching to GC surfaces and some –NH2
groups still exist on SWNTs. The incompletely conversion of –NH2
groups to –N2þ groups was observed previously (Liu et al., 2011b).
The electrochemistry of patterned GC-Ph-SWNTs surfaces wasstudied in Fe(CN)63�/4� solution (1 mM; 0.05 M KCl; 0.05Mphosphate buffer; pH 7.0). As expected the obtained electrochem-istry of GC-Ph-SWNTs surfaces (Fig. S1) is similar to that for bareGC electrodes. However, the electrochemistry of 4-aminophenylmodified GC electrodes shows no Faradaic peaks for ferricyanide,suggesting SWNTs assembly can restore the electron transfer rateto that observed prior to film grafting. In addition, no redox peakat 0.3 V for –NO2 groups was observed for patterned GC-Ph-SWNTs modified GC surfaces in 0.5 M HCl (Gui et al., 2010), furtherconfirming the completely reduction of –NO2 to –NH2 in thearyldiazonium cation ink. After patterning the surface withSWNTs, the water contact angles decreased from 76731 to35721 due to the presence of hydrophilic –COOH and –NH2
groups on SWNTs. However, the water contact angles (76751)remained the same after patterning the surface with blank ink.
Fig. 2 shows the SEM images of GC substrates printed witharyldiazonium cation ink and blank ink. The surface printed withp-aminophenyl/PEG-modified SWNTs/0.5 M HCl/NaNO2 ink pro-vided clearly defined patterns, whereas surfaces printed withp-aminophenyl/PEG-modified SWNTs/0.5 M HCl ink did not showany pattern feature. It indicates the presence of NaNO2 in the ink isthe key component of the printing ink, which is able to reducesome –NH2 to –N2
þ followed by the attachment with GC surfaces.The results suggest the successful printing of aryldiazonium cationinks to form GC-Ph-SWNTs modified GC surfaces.
In order to further confirm the successful patterning of surfacewith SWNTs and the presence of –NH2 groups on patternedsurfaces, the patterned surfaces were treated with Au nanoparti-cles (AuNPs) solutions for 1 h and imaged by SEM again. Due to theaffinity between NH2 and AuNPs, lots of AuNPs (Fig. 2c) wereobserved for GC substrates printed with aryldiazonium cation inkafter incubation in AuNPs. In contrast, few AuNPs due to thephysical adsorption were observed for GC substrates printed withblank ink after incubation in AuNPs. This result further confirmsthe successful attachment of SWNTs and the presence of –NH2
groups on the interface. In addition, the density of AuNPsincreased with the treatment time between aryldiazonium cationink treated PDMS stamp and GC substrates. Maximum density ofAuNPs was obtained when PDMS stamp was placed on GCsubstrates for 25 min. Thus 25 min was selected as the optimizedtime for patterning GC substrates.
The surfaces printed with aryldiazonium cation ink were alsoimaged by AFM (Fig. S2). AFM image of GC surfaces after modification
Scheme 1. Schematic representation of fabricating an immunosensor array for simultaneous detection of endosulfan and paraoxan using PDMS patterning template.Drawings are not at scale and only the chemical linkers formed at each step are shown. PEG may react with the endsites of SWNTs, although they are not represented for thesake of clarity.
G. Liu et al. / Biosensors and Bioelectronics 52 (2014) 360–366362

with SWNTs weremuch rougher than that of the GC-Ph-NH2modifiedGC substrates (Liu et al., 2012b), suggesting that SWNTs wereperpendicularly oriented with only one end anchored on the GCsurface, which is consistent with the literature (De Fuentes et al.,2011). SWNTs here had undergone two steps of functionization. In thefirst step SWNTs reacted end-on with the aryldiazonium ions to get
p-nitrophenyl-modified SWNTs. While in the next step, p-nitrophenyl-modified SWNTs were treated with PEG diazonium ions. PEG mole-cules were attached to the sidewall of SWNTs in high possibility sincealmost all reactive endsites of SWNTs were occupied in the first step.Take the advantage of PEG having the ability to resist the non-specificprotein adsorptionwhich is essential for the sensor construction in the
279281283285287289291
Binding energy /eV
C1s
Oxidised carbon285.2 eV
Graphitic carbon284.4 eV
Bare GC
279281283285287289291
Binding energy /eV
-COOH carbon288.8 eV
C1s
Graphitic carbon284.4 eV
-C-O-C-carbon286.3 eV
GC-Ph-SWNTs
395 397 399 401 403 405 407
Binding energy /eV
Bare GCN1s
395 397 399 401 403 405 407
Binding Energy /eV
GC-Ph-SWNTsN1s
-NH2 nitrogen 400.2 eV
Fig. 1. C1s core level spectra for (a) bare GC and (b) GC-Ph-SWNTs modified GC; N1s core level spectra for (c) bare GC and (d) GC-Ph-SWNTs modified GC.
100μm 100μm
Fig. 2. SEM images of GC substrates printed with aryldiazonium cation ink (p-aminobenzene-modified SWNTs/0.5 M HCl/NaNO2) (a) before and (c) after incubation inAuNPs, and blank ink (p-aminobenzene-modified SWNTs/0.5 M HCl) (b) before and (d) after incubation in AuNPs.
G. Liu et al. / Biosensors and Bioelectronics 52 (2014) 360–366 363

following steps (Khor et al., 2011a; Liu et al., 2012b), PEG moleculeswere introduce herein.
The electrochemistry of the attached redox probes FDMA andPQQ was modulated by the binding event between sensing inter-face and analytes. Fig. 3 shows the electrochemistry of GC-Ph-SWNT modified GC surfaces after the stepwise binding of redoxprobes (FDMA and PQQ), epitopes (endosulfan antigen and para-oxon antigen), anti-epitope IgG, and after exposure antibodymodified interface to analytes (10 ppb endosulfan and 100 ppbparaoxon) in phosphate buffer. Two separate redox peaks centeredat 0.3 V and �0.2 V from FDMA and PQQ, respectively wereobserved. There was no significant change in the peak current ofredox probe after attachment of epitope, however a pronounceddecrease in peak current after incubation of GC-Ph-SWNT/redoxprobe/epitope modified surface with the specific 0.5 mg mL�1
antibody solutions was observed. Table 1 lists the current changeafter incubation of the epitope modified interfaces with proteinsolutions. Control experiments showed no significant change inpeak current when the GC-Ph-SWNT/redox probe modified GCelectrodes were incubated in antibody solution without priorattachment of epitope. In addition, SWV peak currents werealmost the same before and after treatment of the GC-Ph-SWNT/redox probe/epitope modified surfaces with other proteins (suchas anti-pig IgG or BSA) which are not specific to epitopes. Theseresults indicate that the antibody specifically associates with thespecific epitope and only a biospecific interaction leads to a changeof electrochemistry.
Both redox peaks increased with the time for incubation of thesensing interface in analyte solution containing 10 ppb endosulfanand 100 ppb paraoxon, and the current saturated when theincubation time was 5 min. In addition, the higher the concentra-tion of analyte the higher the current increased. These results areconsistent with our previous studies (Liu et al., 2008, 2013, 2012b).Decreased current density upon antibody binding reflects changes
in the interfacial microenvironment which arises from the forma-tion of an immunocomplex on the electrode surface. Formation ofthe complex blocks counterions accessing to the redox probe witha corresponding decrease in current. Due to the affinity betweenthe antibody on the sensing interface and the analyte in solution,the antibody can dissociate from epitope on the electrode surfaceand then binds with the analyte in the solution resulting in theincrease of current from redox probe. However, not all antibodiescould dissociate from the sensing interface due to the competitionbinding of antibody between epitope and analyte. The interfer-ences of other members of cyclodiene and organophosphorusinsecticides were investigated (Fig. 4). It was observed that onlythe presence of endosulfan or paraoxon in analyte solutions couldresult in the obvious current change at 0.3 V or �0.2 V, indicatingthat anti-endosulfan IgG (or anti-paraoxon IgG) were only specificfor endosulfan (or paraoxon) without significant affinity to othermembers of the cyclodiene (or organophosphorus) insecticides atthe interested concentrations. The interference study also suggeststhat there is no cross-reactivity within the neighbor sites for thisimmunosensor assay. Thus the sensing interface has high selectivityto the specific analyte, and it is possible to use the immunosensorfor the simultaneous detection of endosulfan and paraoxon by adisplacement assay.
Fig. 5 shows the calibration curve for simultaneous detection offree endosulfan and paraoxon in phosphate buffer solution underoptimal experimental conditions. The FDMA current had goodlinear relationship with the concentration of endosulfan between
-1.2
-0.8
-0.4
0
0.4
0.8
1.2
1.6
2
-0.3 -0.1 0.1 0.3 0.5
Cur
rent
/µµA
Potential /V-0.3 -0.1 0.1 0.3 0.5
Cur
rent
/µA
0.5
1
1.5
2
2.5
3
3.5
Potential /V
Fig. 3. CV and SWV of the immunosensor array after the step-wise attachment of redox probe (FDMA and PQQ), epitope (endosulfan antigen and paraoxon antigen),anti-epitope IgG and exposure to analytes (10 ppb endosulfan and 100 ppb paraoxon) in 0.05 M phosphate buffer (0.05 M KCl, pH 7.0).
Table 1Current change after incubation of epitope modified interfaces with protein solutions.
Protein solutions Currentn (μA)
Ibefore, 0.3 V Iafter,0.3 V Ibefore,�0.2 V Iafter,�0.2 V
Anti-analyte IgG 3.42 1.17 2.12 0.811 μg mL�1 BSA 3.52 3.50 2.08 2.111 μg mL�1 anti-pig IgG 3.35 3.27 2.09 2.06
n Values correspond to the average of three separate measurements. Ibefore andIafter means the current before and after incubation of GC-Ph-SWNT/redox probe/epitope modified surfaces with the corresponding protein solutions at the con-centration of 1 μg mL�1. Anti-analyte IgG contains 0.5 μg mL�1 anti-endosulfan IgGand 0.5 μg mL�1 anti-paraoxon IgG.
0
0.2
0.4
0.6
0.8
Rel
ativ
e cu
rren
t
Pesticides
at 0.3 V
at -0.2 V
Fig. 4. Relative current at 0.3 V and �0.2 V after incubation of the sensing interfacewith the corresponding pesticide (100 ppb). The concentration of each pesticide at‘Mixture of ten pesticides’ is 10 ppb.
G. Liu et al. / Biosensors and Bioelectronics 52 (2014) 360–366364
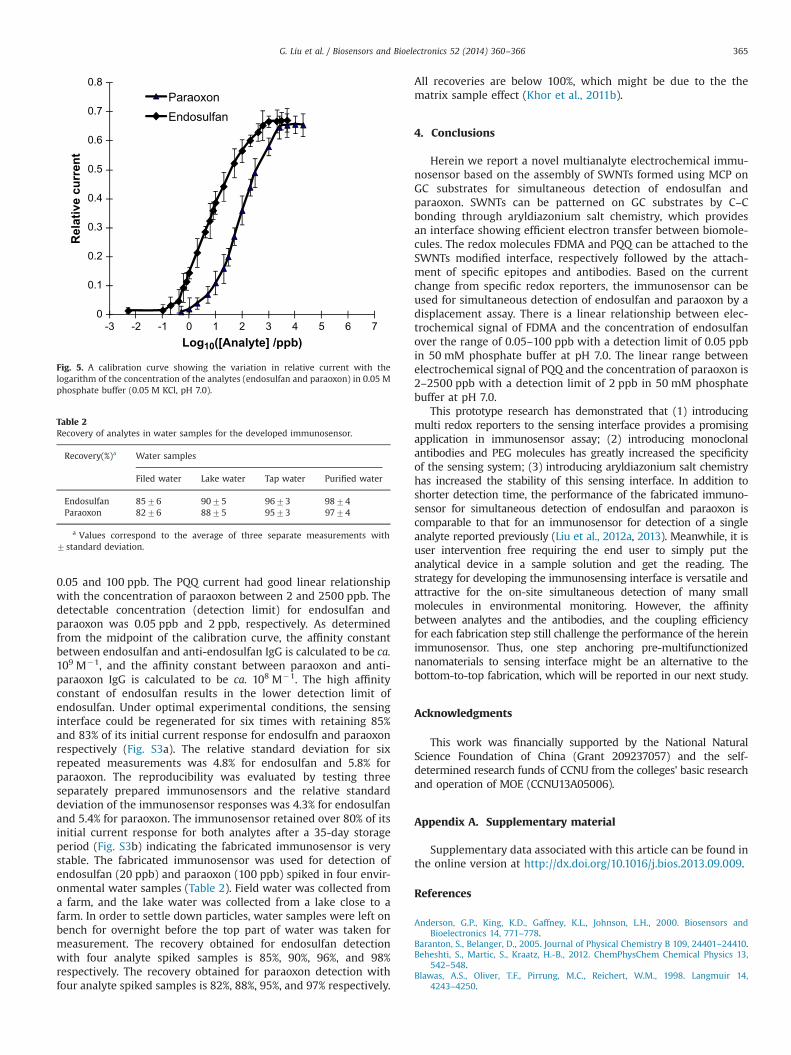
0.05 and 100 ppb. The PQQ current had good linear relationshipwith the concentration of paraoxon between 2 and 2500 ppb. Thedetectable concentration (detection limit) for endosulfan andparaoxon was 0.05 ppb and 2 ppb, respectively. As determinedfrom the midpoint of the calibration curve, the affinity constantbetween endosulfan and anti-endosulfan IgG is calculated to be ca.109 M�1, and the affinity constant between paraoxon and anti-paraoxon IgG is calculated to be ca. 108 M�1. The high affinityconstant of endosulfan results in the lower detection limit ofendosulfan. Under optimal experimental conditions, the sensinginterface could be regenerated for six times with retaining 85%and 83% of its initial current response for endosulfn and paraoxonrespectively (Fig. S3a). The relative standard deviation for sixrepeated measurements was 4.8% for endosulfan and 5.8% forparaoxon. The reproducibility was evaluated by testing threeseparately prepared immunosensors and the relative standarddeviation of the immunosensor responses was 4.3% for endosulfanand 5.4% for paraoxon. The immunosensor retained over 80% of itsinitial current response for both analytes after a 35-day storageperiod (Fig. S3b) indicating the fabricated immunosensor is verystable. The fabricated immunosensor was used for detection ofendosulfan (20 ppb) and paraoxon (100 ppb) spiked in four envir-onmental water samples (Table 2). Field water was collected froma farm, and the lake water was collected from a lake close to afarm. In order to settle down particles, water samples were left onbench for overnight before the top part of water was taken formeasurement. The recovery obtained for endosulfan detectionwith four analyte spiked samples is 85%, 90%, 96%, and 98%respectively. The recovery obtained for paraoxon detection withfour analyte spiked samples is 82%, 88%, 95%, and 97% respectively.
All recoveries are below 100%, which might be due to the thematrix sample effect (Khor et al., 2011b).
4. Conclusions
Herein we report a novel multianalyte electrochemical immu-nosensor based on the assembly of SWNTs formed using MCP onGC substrates for simultaneous detection of endosulfan andparaoxon. SWNTs can be patterned on GC substrates by C–Cbonding through aryldiazonium salt chemistry, which providesan interface showing efficient electron transfer between biomole-cules. The redox molecules FDMA and PQQ can be attached to theSWNTs modified interface, respectively followed by the attach-ment of specific epitopes and antibodies. Based on the currentchange from specific redox reporters, the immunosensor can beused for simultaneous detection of endosulfan and paraoxon by adisplacement assay. There is a linear relationship between elec-trochemical signal of FDMA and the concentration of endosulfanover the range of 0.05–100 ppb with a detection limit of 0.05 ppbin 50 mM phosphate buffer at pH 7.0. The linear range betweenelectrochemical signal of PQQ and the concentration of paraoxon is2–2500 ppb with a detection limit of 2 ppb in 50 mM phosphatebuffer at pH 7.0.
This prototype research has demonstrated that (1) introducingmulti redox reporters to the sensing interface provides a promisingapplication in immunosensor assay; (2) introducing monoclonalantibodies and PEG molecules has greatly increased the specificityof the sensing system; (3) introducing aryldiazonium salt chemistryhas increased the stability of this sensing interface. In addition toshorter detection time, the performance of the fabricated immuno-sensor for simultaneous detection of endosulfan and paraoxon iscomparable to that for an immunosensor for detection of a singleanalyte reported previously (Liu et al., 2012a, 2013). Meanwhile, it isuser intervention free requiring the end user to simply put theanalytical device in a sample solution and get the reading. Thestrategy for developing the immunosensing interface is versatile andattractive for the on-site simultaneous detection of many smallmolecules in environmental monitoring. However, the affinitybetween analytes and the antibodies, and the coupling efficiencyfor each fabrication step still challenge the performance of the hereinimmunosensor. Thus, one step anchoring pre-multifunctionizednanomaterials to sensing interface might be an alternative to thebottom-to-top fabrication, which will be reported in our next study.
Acknowledgments
This work was financially supported by the National NaturalScience Foundation of China (Grant 209237057) and the self-determined research funds of CCNU from the colleges' basic researchand operation of MOE (CCNU13A05006).
Appendix A. Supplementary material
Supplementary data associated with this article can be found inthe online version at http://dx.doi.org/10.1016/j.bios.2013.09.009.
References
Anderson, G.P., King, K.D., Gaffney, K.L., Johnson, L.H., 2000. Biosensors andBioelectronics 14, 771–778.
Baranton, S., Belanger, D., 2005. Journal of Physical Chemistry B 109, 24401–24410.Beheshti, S., Martic, S., Kraatz, H.-B., 2012. ChemPhysChem Chemical Physics 13,
542–548.Blawas, A.S., Oliver, T.F., Pirrung, M.C., Reichert, W.M., 1998. Langmuir 14,
4243–4250.
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
-3 -2 -1 0 1 2 3 4 5 6 7
Rel
ativ
e cu
rren
t
Log10([Analyte] /ppb)
ParaoxonEndosulfan
Fig. 5. A calibration curve showing the variation in relative current with thelogarithm of the concentration of the analytes (endosulfan and paraoxon) in 0.05 Mphosphate buffer (0.05 M KCl, pH 7.0).
Table 2Recovery of analytes in water samples for the developed immunosensor.
Recovery(%)a Water samples
Filed water Lake water Tap water Purified water
Endosulfan 8576 9075 9673 9874Paraoxon 8276 8875 9573 9774
a Values correspond to the average of three separate measurements with7standard deviation.
G. Liu et al. / Biosensors and Bioelectronics 52 (2014) 360–366 365

De Fuentes, O.A., Ferri, T., Frasconi, M., Paolini, V., Santucci, R., 2011. AngewandteChemie International Edition 50, 3457–3461.
Delamar, M., Hitmi, R., Pinson, J., Saveant, J.M., 1992. Journal of the AmericanChemical Society 114, 5883–5884.
Diakowski, P.M., Xiao, Y., Petryk, M.W.P., Kraatz, H.-B., 2010. Analytical Chemistry82, 3191–3197.
Downard, A.J., Garrett, D.J., Tan, E.S.Q., 2006. Langmuir 22, 10739–10746.Ekins, R.P., Chu, F., 1993. Clinical Chemistry 39, 369–370.Gabaldon, J.A., Cascales, J.M., Morais, S., Maquieira, A., Puchades, R., 2003. Food
Additives and Contaminants 20, 707–715.Garrett, D.J., Flavel, B.S., Shapter, J.G., Baronian, K.H.R., Downard, A.J., 2010.
Langmuir 26, 1848–1857.Garrett, D.J., Lehr, J., Miskelly, G.M., Downard, A.J., 2007. Journal of the American
Chemical Society 129, 15456–15461.Gonzalez-Martinez, M.A., Puchades, R., Maquieira, A., 2001. Analytical Chemistry
73, 4326–4332.Gooding, J.J., Wibowo, R., Liu, J., Yang, W., Losic, D., Orbons, S., Mearns, F.J.,
Shapter, J.G., Hibbert, D.B., 2003. Journal of the American Chemical Society125, 9006–9007.
Gui, A.L., Liu, G.Z., Chockalingam, M., Le Saux, G., Luais, E., Harper, J.B., Gooding, J.J.,2010. Electroanalysis 22, 1824–1830.
Guo, W.Q., Jiang, F.F., Chu, J.M., Song, D.D., Liu, G.Z., 2013. Journal of ElectroanalyticalChemistry 703, 63–69.
Harper, J.C., Polsky, R., Dirk, S.M., Wheeler, D.R., Brozik, S.M., 2007. Electroanalysis19, 1268–1274.
Heldman, E., Balan, A., Horowits, O., Ben-Zion, S., Torten, M., 1985. FEBS Letters 180,243–248.
Iijima, S., 1991. Nature 354, 56–58.Jin, L., Zhao, Y., Liu, X., Wang, Y., Ye, B., Xie, Z., Gu, Z., 2012. Soft Matter 8, 4911–4917.Joyeux, X., Mangiagalli, P., Pinson, J., 2009. Advanced Materials 21, 4404–4408.Khor, S.M., Liu, G.Z., Fairman, C., Iyengar, S.G., Gooding, J.J., 2011a. Biosensors and
Bioelectronics 26, 2038–2044.
Khor, S.M., Liu, G.Z., Peterson, J., Iyengar, S.G., Gooding, J.J., 2011b. Electroanalysis23, 1797–1804.
Lehr, J., Garrett, D.J., Paulik, M.G., Flavel, B.S., Brooksby, P.A., Williamson, B.E.,Downard, A.J., 2010. Analytical Chemistry 82, 7027–7034.
Liu, G.Z., Bocking, T., Gooding, J.J., 2007. Journal of Electroanalytical Chemistry 600,335–344.
Liu, G.Z., Chockalingham, M., Khor, S.M., Gui, A.L., Gooding, J.J., 2010. Electroanalysis22, 918–926.
Liu, G.Z., Gooding, J.J., 2006. Langmuir 22, 7421–7430.Liu, G.Z., Khor, S.M., Iyengar, S.G., Gooding, J.J., 2012a. Analyst 137, 829–832.Liu, G.Z., Liu, J.Q., Davis, T.P., Gooding, J.J., 2011a. Biosensors and Bioelectronics 26,
3660–3665.Liu, G.Z., Luais, E., Gooding, J.J., 2011b. Langmuir 27, 4176–4183.Liu, G.Z., Paddon-Row, M.N., Gooding, J.J., 2008. Chemical Communications,
3870–3872.Liu, G.Z., Song, D.D., Chen, F.J., 2013. Talanta 104, 103–108.Liu, G.Z., Wang, S., Liu, J.Q., Song, D.D., 2012b. Analytical Chemistry 84, 3921–3928.Liu, J., Rinzler, A.G., Dai, H.J., Hafner, J.H., Bradley, R.K., Boul, P.J., Lu, A., Iverson, T.,
Shelimov, K., Huffman, C.B., Rodriguez-Macias, F., Shon, Y.S., Lee, T.R., Colbert, D.T.,Smalley, R.E., 1998. Science 280, 1253–1256.
Ossola, F., Tomasin, P., Benetollo, F., Foresti, E., Vigato, P.A., 2003. Inorganica ChimicaActa 353, 292–300.
Pheeney, C.G., Barton, J.K., 2012. Langmuir 28, 7063–7070.Plowman, T.E., Durstchi, J.D., Wang, H.K., Christensen, D.A., Herron, J.N., Reichert, W.M.,
1999. Analytical Chemistry 71, 4344–4352.Rowe, C.A., Scruggs, S.B., Feldstein, M.J., Golden, J.P., Ligler, F.S., 1999. Analytical
Chemistry 71, 433–439.Taitt, C.R., Anderson, G.P., Lingerfelt, B.M., Feldstein, M.J., Ligler, F.S., 2002. Analytical
Chemistry 74, 6114–6120.Venkatanarayanan, A., Crowley, K., Lestini, E., Keyes, T.E., Rusling, J.F., 2012.
Biosensors and Bioelectronics 31, 233–239.
G. Liu et al. / Biosensors and Bioelectronics 52 (2014) 360–366366