who drug information · depleted levels of endogen-ous antioxi-dants, reduced expression of trophic...
TRANSCRIPT

265
WHO Drug Information Vol 21, No. 4, 2007
WHO Drug Information
Contents
World Health Organization
Access to Essential MedicinesWHO celebrates thirty years of essential
medicines 267
Pharmacovigilance FocusEstrogen therapy and symptoms of
parkinsonism: a review usingWHO’s ADR data base 270
Safety and Efficacy IssuesPerflutren injectable: serious cardio-
pulmonary reactions 281Gefitinib: failure of overall survival
vs docetaxel 281Rituximab and progressive multifocal
leukoencephalopathy 282Implanon®: interactions and contra-
ceptive failure 283Atypical antipsychotic agents and
extrapyramidal effects 283Thiazolidinediones and reduced bone
density 284Renal impairment with zoledronic acid 285Epoetins: unexplained excess mortality 285Contraindicated use of sibutramine and
cardiovascular reactions 287Gadolinium-containing contrast agents:
nephrogenic systemic fibrosis 287Swedish Adjustable Gastric Band:
incidents leading to explantation 288
Regulatory Action and NewsInfluenza virus vaccines: Southern
hemisphere 2008 290Withdrawal of cough medicines
containing clobutinol? 290Rimonabant warning: psychiatric
conditions 290
Lumiracoxib registration cancelled 291Lumiracoxib: withdrawal of higher doses 291Ixabepilone approved for advanced
breast cancer 292Second-generation smallpox vaccine
approved 292Regulatory updates 293
Generic MedicinesGeneric substitution in Jamaica:
challenges to improving effectiveness 294Frequently asked questions about generic
medicines 300
Quality Assurance UpdateLatest developments in ICH quality 304
Consultation DocumentsInternational PharmacopoeiaOxytocin 309Oxytocin injection 313
Recent Publications,Information and EventsGMP for herbal medicines 315Quality of antiretrovirals in African
countries 315The Health Journey 315Human rights guidelines for pharma-
ceutical companies 316ARV price report from WHO 316
Proposed InternationalNonproprietary Names: List 98 317

266
WHO Drug Information Vol 21, No. 4, 2007World Health Organization
Announcement
The 13th International Conferenceof Drug Regulatory Authorities (ICDRA)will be hosted by the Swiss Agency forTherapeutic Products (Swissmedic) incollaboration with the World Health
Organization
The ICDRA will take placein Berne, Switzerland
from 16 to 19 September 2008
Updated information will be provided regularly at:http://www.icdra.ch
or
http://www.who.int/medicines/icdra/en/index/html

267
WHO Drug Information Vol 21, No. 4, 2007
Access to Essential Medicines
WHO celebrates thirty years of essential medicines
Essential Medicines List:A primary health care tool2007 marks the 30th anniversary of theWorld Health Organization’s Model List ofEssential Medicines (EML). The EML wascreated in 1977 with the aim of providing amodel for governments to select medicineswhich address local public health needs andto serve as a powerful tool for the promo-tion of primary health care by rationalizingthe selection and use of medicines as wellas their cost.
The EML is revised by a committee of in-dependent experts every two years to re-flect developments and new health chal-lenges. The March 2007 version addressesmany global priority conditions, such asmalaria, HIV/AIDS, tuberculosis, reproduc-tive health and chronic diseases.
Currently, 156 of the 193 WHO MemberStates have adopted essential medicineslists. Some countries have provincial orstate lists as well.
Essential medicines are those medicines that address the priority health care needsof a given population. Selection is based on health relevance, quality, safety, effi-cacy and comparative cost-effectiveness. Essential medicines should be availablein suitable amounts and dosage forms and provided through a functioning healthsystem. The selection of essential medicines is a cornerstone of national medicinepolicies.

268
WHO Drug Information Vol 21, No. 4, 2007
Access to medicines in developing coun-tries is difficult for several reasons. Theseinclude poor supply and distribution sys-tems, insufficient health facilities and staff,low investment in health and the high costof medicines. The EML is a tool that canhelp manage the purchasing and distribu-tion of medicines and the selection of qual-ity assured and cost-effective products.
• Pharmaceuticals represent 15 to 30% ofhealth spending in transitional econo-mies and 25 to 66% in developingcountries.
• In developing countries such medicinesare the largest health expense for poorhouseholds.
• In 2004, a survey carried out in Ugandashowed that among 28 medicines onthe country’s national essential medi-cines list only 55% could be found inhealth facilities where treatment isoffered for free. If the inhabitants had tobuy the same medicines ‘out-of-pocket’,prices were found to be 13.6 timeshigher for branded products and 2.6times higher for generics than theinternational pricing reference.
• The Report of the Commission onMacroeconomics and Health (2001)estimates that by 2015 over 10 milliondeaths per year could be averted byscaling up interventions for communica-ble and noncommunicable diseases,and maternal and perinatal conditions.The majority of these interventionsdepend on essential medicines.
Pioneer countriesMozambiqueIn 1977, a few months before WHOpublished the first EML, Mozambique hadalready created its national pharmacopeiaand a list consisting of 430 essentialmedicines. The country has increasedlocal access to medicines from 10% ofthe population in 1975 to 80% in 2007.
A WHO survey carried out in 2006,reports that patients interviewed at thedispensing area of public facilities werepaying a median of 2 800 Metical for theirmedicines plus fees — equivalent to ahalf hour’s wage for the lowest paidunskilled government worker.
Access to Essential Medicines

269
WHO Drug Information Vol 21, No. 4, 2007
According to the same survey, among465 medicine samples tested for regula-tory purposes only 34 (7.4%) failedidentity or assay.
PeruIn 1960, Peru created a list of basicmedicines in an attempt to address atleast the most pressing pharmaceuticalneeds of the population.
In 1971, the country promoted the BasicMedicines Programme, stimulating thecreation and use of the first national list ofessential medicines.
Sri LankaSri Lanka (formerly Ceylon) created amedicines list for purchase by the statehealth care system in 1959. In addition,the Ceylon Hospitals Formulary waspublished providing information for theuse of these medicines. They also set upan international procurement systemwhich decreased costs and at the sametime increased the availability of thesemedicines.
In spite of industry opposition, Sri Lankaintroduced a state controlled monopoly in1972, to procure medicines for the entirecountry through the creation of the StatePharmaceutical Corporation (SPC) thusextending the initiative to the privatesector. After 1977, due to pressure fromthe private sector, the Sri Lankan govern-ment granted permission to companies toimport multiple brands of medicines.
In 1987, Sri Lanka created the StatePharmaceutical Manufacturing Corpora-tion (SPMC) with the aim of importing rawmaterials and manufacturing genericessential medicines. Ever since, govern-ment resistance to privatization and
monopoly on procurement has keptquality as well as prices under controleven in the private sector.
A critical factor of Sri Lanka’s success isuniversal education, which has resulted ingreater awareness of the importance ofhealth and a strong demand for healthservices in general. Health professionaleducation and training are tailored to thenational medicine supply policy andsystem, including the essential medicinesconcept.
Widely recognized and adoptedMany international organizations, includ-ing UNICEF and UNHCR, as well asnongovernmental organizations andinternational non-profit supply agencies,have adopted the essential medicinesconcept. Essential Medicines Lists guidethe international procurement and supplyof medicines, schemes that reimbursemedicine costs, donations, and localproduction.
The International Federation of the RedCross and Red Crescent Societies andMédecins Sans Frontières (MSF) — aswell as professional bodies such as theBritish Medical Association and theInternational Pharmaceutical Federation(FIP) — have also adopted the essentialmedicines approach and base theirmedicine supply system mainly on theEML. The EML has been extensivelyused to develop international lists forspecial indications, such as TheInteragency New Emergency Health Kit(1998); the UN List of Emergency ReliefItems; or Essential Medicines for Repro-ductive Health (2006).
Source: World Health Organization. http://www.who.int/medicines
Access to Essential Medicines

270
WHO Drug Information Vol 21, No. 4, 2007
Pharmacovigilance Focus
Parkinson disease (PD) is a chronic,progressive degenerative disorder that ischaracterized by the selective degenera-tion of mesencephalic dopaminergicneurons in the Substantia nigra parscompacta (SNpc) (1). The loss ofdopamine afferents results in reduceddopamine being released into the targetfield of these neurons, the corpus stria-tum (2) and the manifestation of anextrapyramidal motor dysfunction, char-acterized by tremor, rigidity, and bradyki-nesia (3). In recent years, several geneshave been linked to autosomally reces-sive forms of PD. However, the vast
majority of PD cases appear to arisesporadically (4) and the exact etiology ofthe dopaminergic degeneration in thesecases remains unknown. A number ofmechanisms have been proposed,including the role played by oxidativestress, glutamate-induced excitotoxicity,depleted levels of endogen-ous antioxi-dants, reduced expression of trophicfactors, inflammatory processes and adysfunctional protein degradation (1, 5).In addition, it has been suggested thatexposure to environmental toxins, suchas pesticides and herbicides, either aloneor in synergy with endotoxic and/or
It has long been known that fewer women are diagnosed with Parkinson diseasethan men. The seemingly protective effect offered to females against degenera-tive disease of the central nervous system may relate to estrogen, although themechanism of action on the dopaminergic system is poorly defined. With an es-timated 10–15 million women using oral contraceptives in the United States alone,this review sets out to examine the evidence for a possible relationship betweenParkinson disease and estrogens in women.
A review of the current literature available on the topic was performed by consult-ing Medline and by performing a search of case-reports contained in the WorldHealth Organization’s International Drug Monitoring Programme database for pos-sible Parkinson disease-related symptoms that may arise from estrogen therapy.The results, whilst conflicting, seem to establish that estrogen protects womenfrom obtaining the disease, or some features of it. Intensive research is nowneeded to establish a relationship between estrogen therapy and development ofparkinsonism.
Estrogen therapy and symptoms of parkinsonism:a review using WHO’s ADR data base
“Use of WHO’s global database to compare hormone replacement therapy and parkinsoniansymptoms in women”, a study by Ilse S. Pienaar and William M.U. Daniels, Department ofMedical Physiology, University of Stellenbosch, South Africa; I. Ralph Edwards and KristinaStar, Signal Detection and Analysis Department, The Uppsala Monitoring Centre, WHO Collabo-rating Centre for International Drug Monitoring, Sweden; Karl J. Morten. Nuffield Department ofObstetrics & Gynaecology, University of Oxford, The Womens’ Centre, John Radcliffe Hospital,United Kingdom. Reprints of the review can be obtained from: [email protected]

271
WHO Drug Information Vol 21, No. 4, 2007 Pharmacovigilance Focus
genetic predisposing factors, may contri-bute towards damage sustained by DAneurons (5, 7). This may especially betrue when exposure takes place duringthe developmental period, when neuronsare particularly vulnerable.
Several epidemiological studies havereported a 1.5 to 2 times higher incidenceand prevalence of PD in men comparedto women (7–12) such as US and Euro-pean studies reporting a male preponder-ance for PD, (7, 13–15). However, afemale preponderance is reported to existfor Parkinson disease in Japan, (16) anddiscrepancies are regularly reported interms of incidence, symptoms, medica-tion effects and treatment response (17).Examples of these inconsistencies are ahigher prevalence of very late-onset PDfound in females (18) and lifetime fluctua-tions in the progressive course of PD infemale patients (19).
The marked sex difference in the rate ofthe disease suggest that the brain’shormonal environment during adulthoodmay be an important point of departure inthe search for an explanation. It has beensuggested that the reproductive femalehormones estrogen, progesterone andluteinizing hormone, as is experienced bywomen during the post-menopausal life-phase, may exert a protective effectwithin a neuronal environment, withrespect to neuronal degeneration, growth,and susceptibility to toxins.
Evidence for this was shown in animalmodels where an apparent neuroprotect-ive effect was exerted by estrogen whenthe hormone was administered prior to orcoinciding with the toxic insult necessaryfor simulating symptoms of PD (20–22).Estrogen exerts its effects on the centralnervous system (CNS) via both genomicmechanisms (to modulate the synthesis,release and metabolism of neurotrans-mitters, neuropeptides and neuroster-oids), as well as through non-genomic
mechanisms, by influencing electricalexcitability, synaptic function and morpho-logical features. Such influences may notonly exert an influence on the malepreponderance observed in a diseaseassociated with the nigrostriatal dopamin-ergic system, such as PD (23) but mayalso be pivotal in the pathogenesis ofother age-related neurodegenerativediseases, such as Alzheimer disease(24).
Animal studies indicate that estrogenaffects the synthesis, release and meta-bolism of dopamine via its effects on DAuptake (25) sites and may also modulatedopamine receptor expression andfunction (26), thereby influencing behav-iours mediated by the basal ganglia (27).In addition, a neuroprotective effect forestrogen has been demonstrated inanimal models of PD that employ neuro-toxic insults to selective neuronal popula-tions. For example, this has successfullybeen demonstrated (28–29) for 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine(MPTP) toxicity in primates, while similarresults have been revealed using the 6-hydroxydopamine rat model (20). Further-more, a loss of dopaminergic neurons inthe Substantia nigra of non-humanprimates exceeding 30% as a result ofestrogen deprivation has been reported(30), while chronic estrogen replacementwas found to restore dopaminergicfunction in ovariectomized rats (31).
Although there are a number of reportson the beneficial effects of estrogen onthe CNS of animals, human data arescarce and often contradictory (32, 33).Evidence on whether estrogens stimulateor inhibit the human dopaminergic systemis inconsistent and ranges from reportsthat the use of estrogen alone (in theabsence of progesterone) may increasethe risk of developing PD among womenwho have undergone a hysterectomy(34), to studies in which no modifyingeffect had been observed (35).

272
WHO Drug Information Vol 21, No. 4, 2007Pharmacovigilance Focus
Such inconsistencies are difficult toexplain, especially in light of evidencesuggesting that neither dopamine-sensi-tive cells, located in the originatingmesencephalic region nor those in theterminating basal ganglia show the abilityto concentrate estrogen (32). Instead,these findings evoke confusion andprovide an inadequate foundation forpractice guidelines.
Menopause is a normal milestone experi-enced annually by at least 2 millionwomen in the USA alone (36) and in-volves the cessation of ovarian secre-tions, such as estrogen. Due to improve-ments made in medical care delivery overthe past few decades, average lifeexpectancy has increased, accounting foraging of western world populations at aprogressive rate (37). However, theaverage age at which menopause com-mences and terminates remains constant,so that women can currently expect tolive up to one half of their adult lives afterthe menopausal period (38).
Many women are concerned about therelationship between menopause andhealth due to reports that post-menopau-sal estrogen deficiency leaves womenmore exposed to neurotoxic assault.Furthermore, due to an aging population,an increasing number of people canexpect to experience age-related disor-ders associated with abnormalities of thedopaminergic system, including PD (13).Evidence to encourage such concernsinclude reports of less severe symptomsduring the early phase of PD in womenwho receive estrogen replacementtherapy, a reduced anti-parkinsonianthreshold dose for levodopa, (39) andgender differences in terms of symptomsand response to levodopa treatment. Onestudy (40) made use of a ‘neuroendocrinechallenge’, a technique widely used forexploring neurotransmitter tone in hu-mans for detecting the effects of estrogenon the dopaminergic response. Theinvestigators used apomorphine-induced
growth hormone (GH) secretion via post-synaptic dopamine (D
2) receptors, located
in the median eminence of the hypo-thalamus (41), to investigate centraldopaminergic responsivity in femalepatients. Their results indicate that post-menopausal women who undergo long-term estrogen therapy, have enhancedGH responses, compared to women whoare naive to estrogen therapy, therebysuggesting increased dopaminergic tone.Interestingly, a study has reported acomparatively worse decline in the D2dopamine receptor concentration forwomen over men (42). These receptorsgenerally decline to a significant extent inhumans as a result of advancing age,particularly in frontal and basal gangliabrain regions (42).
Realization as to the importance estrogenplays in the development and mainte-nance of not only the female reproductivesystem, but its modulatory effect on theCNS as well, has resulted in the pharma-ceutical development of a vast range ofsteroidal and nonsteroidal compoundsthat interact with the estrogen receptors.Globally, more than 60 million women useoral contraceptive therapy, (43) whichentails utilizing either a combinationproduct containing both estrogen andprogestin, or single entity productscontaining progestin only. With circulatingestrogen levels diminishing in post-menopausal women, HRT is the standardtreatment for restoring decreasing hor-mone levels as a preventative stepagainst discomfort and possible healthproblems.
Due to reports emerging during the mid-1970’s that postmenopausal women whomake use of estrogen alone stand asignificantly increased risk of developingendometrial- and breast cancer, (44)progestin, a synthetic version of proges-terone, was added to the standard HRTregimen in order to counterbalance theeffects of estrogen. In addition, there isevidence linking HRT with a marginally

273
WHO Drug Information Vol 21, No. 4, 2007 Pharmacovigilance Focus
increased risk of stroke, heart diseaseand breast cancer (45). An androgen,usually testosterone, may be added tothe drug regimen to aid in restoringlowered energy levels, libido and preventosteoporosis following menopause.
HRT has been applied as a therapeutictool beyond its original design intention.Attempts are made to include HRT as atherapeutic strategy towards a range oftarget diseases that primarily affect thereproductive system, including breastcancer and uterine dysfunctions (46),while beneficial effects for the use of HRThave also been reported for treatingosteoporosis and coronary artery disease(47). Compounds directed at inducingspecific and potent estrogen-receptoractivity in non-traditional target tissues,such as the central nervous system(CNS) may also possess therapeuticpotential, by either modulating brainneurotransmission or via neuroprotectiveactivity. HRT has been found to protectthe CNS against the development ofdiseases such as Alzheimer diseasewhen administered after menopause (48).
However, a large, randomized, double-blind, placebo-controlled clinical trial ofpostmenopausal HRT failed to replicatethese results (49). Studies performed onhuman patients suffering from extrapy-ramidal disorders, such as PD, illustrateestrogen’s behaviour-mediating effect ondopamine within the basal ganglia (50),and biochemical evidence suggests thatestrogen regulates dopaminergicneurotransmission in the basal ganglia(51, 52). In addition, various gynaecologicbenefits are associated with HRT, such asan ability to reduce the incidence ofsecondary amenorrhoea (53), benignbreast neoplasm (54), prevention of iron-deficiency anaemia (55), a reduced risk ofendometrial cancer (56–59), prevention ofectopic pregnancy (60–63) the formationof functional ovarian cysts (64), andpelvic inflammatory disease (65).
Valid concerns exist that the substantialhormonal changes associated withnatural or surgically-induced menopausemay have adverse reactions and contrib-ute towards the initiation and/or develop-ment of various diseases. Concern isespecially provoked by several studiesindicating significantly higher incidence ofcardiovascular abnormalities includingstroke, due to thrombo-embolism, sub-arachnoid haemorrhage and cerebralvenous thrombosis, in users of oralcontraceptives, particularly high estrogenformulations (66).
Similarly, a number of neurologicalalterations suggesting estrogen involve-ment have been documented. For in-stance, it has been postulated thatchorea may be due to the direct dopamin-ergic action of estrogens or due todopamine accumulating in the braincaused by its competitive binding to thedopamine-degrading enzyme catechol-o-methyltransferase (COMT) (66). Furthersupport for estrogen’s influence on brainfunction stems from experiments showingthat the expression of COMT is regulatedby ovarian steroids (51, 52, 67).
Evidence for a role for estrogen in PD isless clear with some studies suggestingthat estrogen replacement therapy maybe useful for female patients during theearly phases of the disease and beforeinitiating levodopa therapy (68). However,others report only a slight anti-parkinso-nian effect to 17ß-estradiol therapy or nobeneficial PD effects (70), thereby sup-porting an anti-dopaminergic effect forestrogens on parkinsonian symptoms(71).
In the absence of investigations to estab-lish insight into the effects that estrogenexerts on the dopaminergic system,especially in terms of long-term therapeu-tic influences, the beneficial resultsgained from HRT are seen to significantlyoutweigh the risks involved, and HRTcontinues to gain widespread prescriptive

274
WHO Drug Information Vol 21, No. 4, 2007Pharmacovigilance Focus
support. The current article outlines theapparent paradox of the effects of estro-gens on the CNS with particular referenceto whether the provision of hormonereplacement therapy will worsen orreduce the chance of PD or PD-likesymptoms, or alter specific attributes ofthe PD syndrome.
MethodThe present study is a review of currentlyavailable data to assess the relativesafety of supplementary hormone therapyto women. The rationale for this is thatthe number of women reaching theirmidlife years and beyond who seek pro-fessional advice regarding HRT is likely todouble. Yet the studies that have investi-gated the relative safety of estrogentreatment in the context of developingPD-related symptoms are limited. At bestthe studies are inconclusive with findingssuggesting both an associated risk (23,35) as well as no associated risk fordeveloping the disease (45) being re-ported. In view of the conflicting evidence,we revisited existing published data, inconjunction with the WHO Programme forInternational Drug Monitoring, through itsCollaborating Centre based in Uppsala,Sweden, to examine the increased riskthat HRT may specifically hold for devel-oping PD-related symptoms.
The WHO Programme for InternationalDrug Monitoring database containsapproximately 3.7 million case reports ofsuspected adverse drug reactions thathave been filed as reports since 1968,with a total of 81 countries currentlyparticipating in the programme (72). Suchspontaneous reports provide a first-linesurveillance method for the detection ofnew adverse drug reactions (ADRs). Thisis extremely useful because of the size-able population surveyed and the timelyreporting of unexpected adverse reac-tions (73).
A data mining search (74) was performedto extract drug-ADR combinations occur-ring more frequently than expected. A
positive drug-reaction association impliesa significant difference from the entireglobal report (75). The search was doneusing the anatomical therapeutic chemi-cal (ATC) classification to cover allestrogen-containing compounds. Reportswith drugs belonging to any of the follow-ing ATC groups were included in thesearch:
• hormonal contraceptives for systemicuse/progestogens and estrogens;
• estrogens/natural and semisyntheticestrogens; synthetic estrogen;
• estrogen combined with other drugs;
• androgens and estrogens;
• androgen alone;
• progestogen and estrogen in combina-tion; and
• progestogens and estrogens in combi-nation.
The following WHO-preferred terms wereused during the search for therapy-associated movement disorders: Bradyki-nesia, Dyskinesia, Hypokinesia, Choreo-athetosis, Hypertonia, Tremor, Extrapy-ramidal disorder and Parkinsonismaggravated. In the WHO adverse reactionterminology, there were 4 terms related to‘Parkinson disease’. Three subtermswere included under the preferred term‘extrapyramidal disorder’, namely ‘Parkin-son syndrome’, ‘Parkinsonism’ and‘Parkinsonian gait’, while the fourth termwas the preferred term ‘Parkinsonismaggravated’, that did not contain addition-ally included terms.
Results and DiscussionVery few reports of PD per se wererevealed during the search, suggestingthat PD is either unrecognized as anadverse effect of estrogen-containingcompounds or that a weak relationshipexists between the two. However, it isacknowledged that under-reporting and

275
WHO Drug Information Vol 21, No. 4, 2007 Pharmacovigilance Focus
failure to recognize adverse reactionswith a long latency are two weaknessesinherent to this particular reportingsystem.
Reports to the WHO Programme indicatea disproportionately high amount ofchorea compared to all other drugs in thedatabase, though this does not reach95% confidence level significance. Differ-ential diagnosis of chorea, in addition toSydenham chorea, include Wilson dis-ease; encephalitis; Huntington chorea;drug intoxication; benign familial chorea;pregnancy; systemic lupus erythemato-sus; Henoch-Schonlein purpura; polycyth-emia vera; hypocalcaemia; hyperthyroid-ism; carbon monoxide poisoning; cerebralinfarction, and intracranial tumour (76).
Chorea involves rhythmic motor oscilla-tions characterized by brief, irregularmuscle contractions that are not repetitiveor rhythmic in nature, but rather appear tocontinually flow from one muscle to thenext, often co-presenting with athetosis,adding twisting and writhing movementsby the hands, feet, trunk, neck and face,to the already existing disorder. Choreamay be drug-induced and this is the mostcommonly occurring type of chorea foundin practice (78, 79). More than 40% of allPD patients, depending on age anddosage used (80, 81) develop choreaafter 5–10 years of continual levodopause with increasing numbers of patientsaffected as time elapses (82). It isthought that chorea is due to the drug’sability to stimulate postsynaptic dopaminereceptors in a pulsatile fashion, evokingchanged responses amongst the neuro-nal networks that interconnect the basalganglia with the frontal cortical motorregions (80). Such excessive thalamo-cortical facilitation thereby overactivatesthe neurotransmitter dopamine (80).
However, inconsistencies between theproposed model and clinical evidencehave been observed, including theabsence of increased excitatory thalamo-cortical drive following pallidotomy, which
should effectively worsen chorea (83).Current opinion on what constituteshyperkinetic movement disorders, suchas chorea, maintain that more complexalterations in the temporal and spatialfiring pattern of the globus pallidus areresponsible for the clinical phenomenon(80, 83, 23, 11, 84). A paradox seems toexist as to the effects of estrogens on theCNS, with particular reference to whetherthe provision of hormone replacementtherapy will worsen or reduce the chanceof PD, or alter specific attributes of the PDsyndrome.
Following the first report of an associationbetween chorea and oral contraceptivesin 1966 (85), various additional case-reports and epidemiology-based studieshave revealed chorea to be a rare compli-cation of estrogen-containing oral contra-ceptive therapy, such as the use of topicalvaginal cream containing conjugatedestrogen (86). One large-scale study (87)reported that 12% of all patients thatparticipated in the study, developedchorea soon after starting estrogen-containing oral contraceptive treatment,with 6% developing chorea gravidarumand 2% developing the disorder shortlyfollowing delivery.
In contrast to certain other drugs found tocause this anomaly and which tend tofunction at a more universally choreigeniclevel, OCs prerequisitely require exist-ence of basal ganglia dysfunction in orderto induce adverse effects (88). Suchadverse drug reactions are only expectedto occur as recurring choreic episodes,such as Sydenham chorea, chorea due tosystemic lupus erythematosus, choreagravidarum (89) or when levodopa-induced (80), with the time betweeninitiation of therapy and appearance ofchoreiform movements varying from 6days to 9 months (76). This observationsupports previous reports that womenpresent more often with tremor than men(77), indicating a mild form of motordeterioration and striatal degeneration.

276
WHO Drug Information Vol 21, No. 4, 2007Pharmacovigilance Focus
During future studies it therefore remainsessential to consider controlling for suchpossible variation. The relatively welldescribed incidence of chorea associatedwith estrogens, is therefore a clearconcern to patients using levodopa whenchorea is one of the most troublesomeadverse effects relating to treatment withthat drug. There is a need to knowwhether there is a potential-stimulatinginteraction by estrogens.
SummaryThere are several ways human studiescould be performed with different com-parisons made between genders, agegroups, with either the use of HRT or not,as well as by carefully differentiating thecomponents of PD. Recent reports of acorrelation between estrogen-basedtherapy and chorea were found to belacking in recent literature, and largestudies on PD with sufficient power toexamine the differential effects of estro-gen on the PD syndrome have not beenconducted.
However, the current review highlightsthat there is indeed cause for concern inthis regard, because of the conflictingresults presented, with a definite need forfurther investigation in order to extrapo-late for the relationship between femalegonodal hormones and the nigrostriataldopaminergic system. The absence ofevidence for any signal on the adverseeffects of estrogen-containing compoundson PD in the WHO database could beinterpreted as a failure to recognize asignal of a causative effect or worsenedPD, even though the database reflectedthe known occurrence of chorea as aside-effect to estrogen-based therapy.None the less, the result does not conflictwith the possibility that estrogens mayprotect from all or aspects of the PDsyndrome (19).
The regulatory effect of estrogen on theactivity of dopamine-containing fibres
originating in the midbrain and terminat-ing in the basal ganglia and on dopa-mine-sensitive cells in the basal ganglia iscomplex and the mechanism of action isunclear since cells in neither of theseregions concentrate estrogen. Whileestrogen may act indirectly via the cat-echol-estrogens and prolactin, it has beendemonstrated that estrogen can actdirectly on the striatum also (31). Thesefindings relate to the effects of estrogenon human extrapyramidal disorders.However, estrogen appears to pose onlylimited efficacy in the treatment of dyski-netic disorders, despite its apparentantidopaminergic effect in humans.Intensified research efforts are called forbefore agreement can be reached withregards to the locus, direction, or themechanism of OC and HRT action onbasal ganglia function.
References
1. Arai H, Furuya T, Mizuno Y Mocizuki H.Inflammation and infection in Parkinson’sdisease. Histol Histopathol 2006; 21(6):673-678.
2. Ascherio A, Chen H, Swarzchild MA, ZhangSM, Colditz GA, Speizer FE. Caffeine,postmenopausal estrogen and risk of Parkin-son’s disease. Neurology 2003; 60:790-795.
3. Olanov CW, Schapira AH, Agid Y.Neuroprotection for Parkinson’s diseaseprospects and promises. Ann Neurol 2003;53(Suppl 3):S1-S2.
4. Warner TT, Schapira AHV. Genetic andenvironmental factors in the cause of Parkin-son’s disease. Ann Neurol 2003; 53:S16-S25.
5. Schapira AH. Etiology of Parkinson’sdisease. Neurology 2006; 66(10, suppl.4):S10-23.
6. Marchetti B, Serra PA, Tirolo C et al.Glucocorticoid receptor-nitric oxide crosstalkand vulnerability to experimental Parkinsonismpivotal role for glia-neuron interactions. BrainRes Rev 2005; 48:302–321.

277
WHO Drug Information Vol 21, No. 4, 2007 Pharmacovigilance Focus
7. Van den Eeden SK, Tanner CM, BernsteinAL, Fross RD, Leimpeter A., Bloch DA.,Nelson LM. Incidence of Parkinson’s disease:variation by age, gender and race/ethnicity.American J Epidemiol 2003; 157:1015-1022.
8 De Lau LM, Giesbergen PC, de Rijk MC etal. Incidence of parkinsonism and Parkinsondisease in a general population: the Rotter-dam study. Neurology 2004, 163:1240-1244.
9 Wooten GF, Currie LJ, Bovbjerg VE et al.Are men at greater risk for Parkinson’sdisease than women? J Neurol NeurosurgPsychiatry 2004; 75:637-639.
10. Schrag A, Ben-Shlomo Y, Quinn N. Crosssectional prevalence survey of idiopathicParkinson’s disease and parkinsonism inLondon. BMJ 2000; 321:21-22.
11. Claveria LE, Duarte J, Sevillano MD et al.Prevalence of Parkinson’s disease inCantelejo, Spain: a door-to-door survey. MovDisord 2002; 17:242-249.
12. Benito-Leon J, Bermejo-Pareja F,Rodriguez J et al. Prevalence of PD and othertypes of Parkinsonism in three elderly popula-tions of central Spain. Mov Disord 2003;18:267-274.
13. Tanner CM, Ashton DA. Epidemiology ofParkinson’s disease and akinetic syndromes.Curr Opin Neurol 2000; 13:427-430.
14. Baldereschi M, Di Carlo A, Rocca WA etal. Parkinson’s disease and parkinsonism in alongitudinal study: two-fold higher incidence inmen. ILSA working group. Italian longitudinalstudy on Aging. Neurology 2000; 55:1358-1363.
15. Mayeux R, Marder K, Cote LJ, Denaro J,Hemeneqildo N, Mejia H et al. The frequencyof idiopathic Parkinson’s disease by age,ethnic group, and sex in northern Manhattan,1988-1983. American J Epidemiol 1995;142:820-827.
16. Kimura H, Kurimura M, Wada M,Kawanami T, Kurita K, Suzuki Y et al. Femalepreponderance of Parkinson’s disease inJapan. NeuroEpidemiol 2002; 21(6):292-296.
17. Shulman LM, Bhat V. Gender disparitiesin Parkinson’s disease. Expert Rev Neurother2006; 6(3):407-416.
18. Bower JH, Maraganore DM, McDonnellSK, Rocca WA. Incidence and distribution ofparkinsonism in Olmsted County, Minnesota,1976-1990. Neurology 1999; 52:1214-1220.
19. Sato K, Hatano T, Yamashiro K,Kagohashi M, Nishioka K, Izawa N et al.Prognosis of Parkinson’s disease: time tostage III, IV, V, and to motor fluctuations. MovDisord 2006; 21(9):1384-1395.
20. Dluzen DE. Estrogen decreases corpusstriatal neurotoxicity in response to 6-hydroxy-dopamine. Brain Res 1997; 767:340-344.
21. Gao X, Dluzen DE. Tamoxifen abolishesestrogen’s neuroprotective effect uponmethamphetamine induced neurotoxicity ofthe nigrostriatal dopaminergic system. Neuro-science 2001; 103:385-394.
22. Gajjar TM, Anderson LI, Dluzen DE. Acuteeffects of estrogen upon methamphetamineinduced neurotoxicity of the nigrostriataldopaminergic system. J Neural Transm 2003;110:1215-1224.
23. Morale MC, Serra PA, L’episcopo F, TiroloC, Caniglia S, Testa N. Estrogen, neuro-inflammation and neuroprotection in Parkin-son’s disease: glia dictates resistance versusvulnerability to neurodegeneration. Neuro-science 2006; 138(3):869-878.
24. Benedetti MD, Maraganore DM, BowerJH, McDonnell SK, Peterson BJ, Ahlskog Jenet al. Hysterectomy, menopause, and estro-gen use preceding Parkinson’s disease: anexploratory case-control study. Mov Disord2001;16: 830-837.
25. Becker JB. Direct effect of 17â-estradiolon striatum: sex differences in dopaminerelease. Synapse 1990; 5(2):157-164.
26. Di Paolo T, Falardeau P, Morissette M.Striatal D-2 dopamine agonist binding sitesfluctuate during the rat estrous cycle. Life Sci1988; 43(8):665-627.

278
WHO Drug Information Vol 21, No. 4, 2007Pharmacovigilance Focus
27. Roy EJ, Buyer DR, Licari VA. Estradiol inthe striatum: effects on behavior and dopa-mine receptors but no evidence for membranesteroid receptors. Brain Res Bull 1990;25(2):221-227.
28. Dluzen DE, McDermott JL, Liu B. Estro-gen alters MPTP-induced neurotoxicity infemale mice: effects on striatal dopamineconsentrations and release. J Neurochem1996; 66:688-695.
29. Miller DB, Ali SF, O’Callaghan JP, LawsSC. The impact of gender and estrogen onstriatal dopaminergic neurotoxicity. Ann NYAcad Sci 1998; 844:153-165.
30. Leranth C, Roth RH, Elsworth JD, NaftolinF, Horvath TL, Redmond DE Jr. Estrogen isessential for maintaining nigrostriataldopamine neurons in primates: implicationsfor Parkinson’s disease and memory. JNeurosci 2000; 20(23):8604-8609.
31. Ohtani H, Nomoto M, Douchi T. Chronicestrogen treatment replaces striatal dopamin-ergic function in ovariectomized rats. BrainRes 2001; 900(2):163-168.
32. Van Hartesveldt C, Joyce JN. Effects ofestrogen on the basal ganglia. NeurosciBiobehav Rev 1986; 10(1):1-14.
33. Kompoliti K. Estrogen and movementdisorders. Clin Neuropharmacol 1999;22(6):318-326.
34. Marder K, Tang MX, Alfaro B, Mejia H,Cote L, Jacobs D et al. Postmenopausalestrogen and Parkinson’s disease with andwithout dementia. Neurology 1998; 50:1141-1143.
35. Currie LJ, Harrison MB, Trugman JM,Bennett JP, Wooten GF. Postmenopausalestrogen use affects risk for Parkinsondisease. Arch Neurol 2004; 61:886-888.
36. Henderson VW. Menopause and disordersof the central nervous system. MinervaGinecol 2005; 57(6):579-592.
37. Ernst, RL, Hay JW. The US economic andsocial costs of Alzheimer’s disease revisited.Am J Public Health 1994; 84:1261-1264.
38. Vliet EL, Davis VL. New perspectives onthe relationship of hormone changes toaffective disorders in the perimenopause.NAACOGGS Clin Issu Perinat WomensHealth Nurs 1991; 2:453-471.
39. Blanchet PJ, Fang J, Hyland K, Arnold LA,Mouradian MM, Chase TN. Short-term effectsof high dose 17 [beta]-estradiol in postmeno-pausal PD patients: a crossover study.Neurology 1999; 53:91-95.
40. Lyons KE, Hubble JP, Troster AI, PahwaR, Koller WC. Gender differences in Parkin-son’s disease. Clin Neuropharmacol 1998;21:118-121.
41. Meltzer HY. Clinical evidence for multipledopamine receptors in man. CommunPsychopharmacol 1979; 3(6):457-470.
42. Wong DF, Brousolle EP, Wand G. In vivomeasurement of dopamine receptors inhuman brain by positron emission tomogra-phy. Age and sex differences. Ann NY AcadSci 1988; 515:203-214.
43. Tierney MC Luine VN. Effects of estro-gens and SERMs on the central nervoussystem. J Soc Obstet Gynaecol Can 1997;19:46-56.
44. Sismondi P, Biglia N, Giai M et al. HRT,breast and endometrial cancers: strategiesand intervention options. Maturitas 1999;32:131-139.
45. Gaspard U, van den Brule F, Pintiaux A,Foidart JM. Clinical study of the month.Benefit/risk balance of postmenopausalestrogen-progestin treatment in peril in theWomen’s Health Initiative study: practicalattitude of the clinician. Rev Med Liege 2002;57(8):556-562.
46. Cyr M, Calon F, Morissette M, Di Paolo T.Estrogenic modulation of brain activity:implications for schizophrenia and Parkinson’sdisease. J Psychiatry Neurosci 2000; 27(1):12-27.
47. Epperson CN, Wisner KL, Yamamoto B.Gonadal steroids in the treatment of mooddisorders. Psychosom Med 1999; 61:676-697.

279
WHO Drug Information Vol 21, No. 4, 2007 Pharmacovigilance Focus
58. Schlesselman JJ. Risk of endometrialcancer in relation to use of combined oralcontraceptives. A practitioner’s guide to meta-analysis. Hum Reprod 1997; 12:1851–1863.
59. Weiderpass E, Adami HO, Baron JA et al.Risk of endometrial cancer following estrogenreplacement with and without progestins. JNatl Cancer Inst 1999; 91:1131.
60. Sivin I. International experience withNorplant® and Norplant®-2 contraceptives.Stud Fam Plann 1988; 19:81-94.
61. Sivin I. International experience withNorplant® and Norplant®-2 contraceptives.Stud Fam Plann 1988; 19:81-94.
62. Franks AL, Beral V, Cates, Jr W, HogueCJR. Contraception and ectopic pregnancyrisk. Am J Obstet Gynecol 1990; 163:1120–1123.
63. Sivin I. Dose- and age-dependent ectopicpregnancy risks with intrauterine contracep-tion. Obstet Gynecol 1991; 78:291–298.
64. Peterson HB, Xia Z, Hughes JM et al. Therisk of ectopic pregnancy after tubal steriliza-tion. N Engl J Med 1997; 336:762-767.
65. Lanes SF, Birmann B, Walker AM, SingerS. Oral contraceptive type and functionalovarian cysts. Am J Obstet Gynecol 1992;166: 956–961.
66. Panser LA, Phipps WR. Type of oralcontraceptive in relation to acute, initialepisodes of pelvic inflammatory disease.Contraception 1991; 43:91–99.
67. Schipper HM. Sex hormones in stroke,chorea, and anticonvulsant therapy. SeminNeurol 1988; 8(3):181-186.
68. Xie T, Ho SL, Ramsden D. Characteriza-tion and implications of estrogen downregulation of human catechol-O-methyl-transferase gene transcription. Mol Pharmacol1999; 56:31-38.
69. Saunders-Pullman R, Gordon-Elliot J,Parides M, Fahn S, Saunders HR, BressmanS. The effect of estrogen replacement on earlyParkinson’s disease. Neurology 1999;52:1417-1421.
48. Hogervorst E, Williams J, Budge M,Riedel W, Jolles J. The nature of the effect offemale gonadal hormone replacement therapyon cognitive functions in post-menopausalwomen: a meta-analysis. Neuroscience 2000;101(3):485–512.
49. Shumaker SA, Legault C, Kuller L, RappSR, Thal L, Lane DS et al. Conjugates equineestrogen and the incidence of probabledementia and mild cognitive impairment inpostmenopausal women. JAMA 2004;291:2947–2958.
50. Hruska RE, Silbergeld EK. Increaseddopamine receptor sensitivity after estrogentreatment using the rat rotation model.Science 1980; 208(4451):1466-1468.
51. Di Paolo T. Modulation of brain dopaminetransmission by sex steroids. Rev Neurosci1994; 5:27-41.
52. Becker JB. Gender differences in dopa-minergic function in striatum and nucleusaccumbens. Pharmacol Biochem Behav 1999;64:803-812.
53. Romer T, Schmidt T Foth D. Pre- andpostoperative hormonal treatment in patientswith hysteroscopic surgery. Contrib GynecolObstet 2000; 20:1-12.
54. Kramer EA, Seeger H, Kramer B,Wallwiener D, Mueck AO. The effect ofprogesterone, testosterone and syntheticprogestogens on growth factor- and estradiol-treated human cancerous and benign breastcells. Eur J Obstet Gynecol Reprod Biol 2006;129(1):77-83.
55. Grimes DA. The safety of oral contracep-tives: epidemiologic insights from the first 30years. Am J Obstet Gynecol 1992; 166:1950-1954.
56. Pettersson B, Adami HO, Bergstrom R,Johansson ED. Menstruation span - a time-limited risk factor for endometrial carcinoma.Acta Obstet Gynecol Scand 1986; 65:247–255.
57. Kelsey JL, LiVolsi VA, Holford TR et al. Acase-control study of cancer of the endo-metrium. Am J Epidemiol 1982; 116:333–342.

280
WHO Drug Information Vol 21, No. 4, 2007
70. Tsang KL, Ho SL & Lo SK. Estrogenimproves motor disability in parkinsonianpostmenopausal women with motor fluctua-tions. Neurology 2000; 54(12):2292-2298.
71. Strijks E, Kremer JA, Horstink MW. Effectsof female sex steroids on Parkinson’s diseasein postmenopausal women. Clin Neuro-pharmacol 1999; 22:93-97.
72. Session DR, Pearlstone MM, JewelewiczR, Kelly AC. Med Hypotheses 1994; 42:280-282.
73. Olsson S. The role of the WHO pro-gramme on international drug monitoring incoordinating worldwide drug safety efforts.Drug Safety 1998; 19(1):1-10.
74. Smith CC, Bennett PM, Pearce HM,Harrison PI, Reynolds DJM, Aronson JK,Grahame-Smith D.G. Adverse drug reactionsin a hospital general medical unit meritingnotification to the committee on Safety ofMedicines. Brit J Clin Pharmacol 1996;42:423-429.
75. Bate A, Lindquist M, Edwards IR et al. ABayesian neural network method for adversedrug reaction signal generation. Eur J ClinPharmacol 1998; 54:315-321.
76. Edwards IR. Adverse drug reactions:finding the needle in the haystack. Brit Med J1997; 315:500.
77. Dove DJ. Chorea associated with oralcontraceptive therapy. Am J Obstet Gynecol1980;137(6):740-742.
78. Haaxma CA, Bloem BR, Borm GF, OyenWJ, Leenders KL, Eshuis S et al. Genderdifferences in Parkinson’s disease. J NeurolNeurosurg Psychiatry 2006; 78(8):819-824.
79. Cardoso F, Seppi K, Mair KJ, WenningGK, Poewe W. Seminar on Choreas. LancetNeurol 2006; 5(7):589-602.
80. Wenning GK, Kiechl S, Seppi K. Muller J,Hogl B, Saletu M, Rungger G, Gasperi A,Willeit J, Poewe W. Prevalence of movementdisorders in men and women aged 50–89
years (Bruneck Study Cohort): a population-based study. Lancet Neurol 2005; 4:815-820.
81. Fahn S. The spectrum of levodopa-induced dyskinesias. Ann Neurol 2000;47(4):S2-9.
82. Schrag A, Quinn N. Dyskinesias and motorfluctuations in Parkinson’s disease. A commu-nity-based study. Brain 2000; 123:2297-2305.
83. Obeso JA, Rodriguez-Oroz MC, China P,Lera G, Rodriguez M, Olanow CW. Theevolution and origin of motor complications inParkinson’s disease. Neurology 2000; 55(suppl. 4):S13-S20.
84. Liu Y, Postupna N, Falkenberg J,Anderson ME. High frequency deep brainstimulation: What are the therapeutic mecha-nisms? Neurosci Biobehav 2006 (Epub aheadof print).
85. Moro E, Lang AE, Strafella AP et al. Bi-lateral globus pallidus stimulation for Hunting-ton’s disease. Ann Neurol 2004; 56:290-294.
86. Fernando SJ. An attack of chorea compli-cating oral contraceptive therapy. Practitioner1966; 197(178):210-211.
87. Caviness JN, Muenter MD. An unusualcause of recurrent chorea. Mov Disord 1991;6(4):355-357.
88. Cervera R, Asherson RA, Font J, Tikly M,Pallares L, Chamorro A et al. Chorea in theantiphospholipid syndrome. Clinical, radiologicand immunologic charactersitics of 50 patientsfrom our clinics and the recent literature.Medicine 1997; 76(3):203-212
89. Miranda M, Cardoso F, Giovannoni G,Church A. Oral contraceptive induced chorea:another condition associated with anti-basalganglia antibodies. J Neurol NeurosurgPsychiatry 2004; 75:327-328.
90. Karageyim AY, Kars B, Dansuk R, Unal O,Turan MC. Chorea gravidarum: a case report.J Matern Fetal Neonatal Med 2002; 12:353-354.
Pharmacovigilance Focus

281
WHO Drug Information Vol 21, No. 4, 2007
Safety and Efficacy Issues
Perflutren injectable: seriouscardiopulmonary reactionsCanada — Perflutren Injectable Suspen-sion (Definity®) is an ultrasound contrastindicated for ultrasound imaging ofcardiac structures (ventricular chambersand endocardial borders) and function(regional wall motion) in adult patientswith suboptimal echocardiograms. It isalso indicated for ultrasound imaging ofthe liver and kidneys in adult patients.The manufacturer of this product hascirculated information pertaining to postmarketing reports of serious cardio-pulmonary reactions, including fatalities.
As of 30 September 2007, a total of 99cases of serious cardiopulmonary reac-tions have been reported to the company,with none reported from Canada. In post-marketing use, four patients experiencedfatal cardiac arrests either during or within30 minutes of administration. Three of the4 patients had pre-existing conditions,including severe congestive heart failureand one on mechanical ventilation forrespiratory failure.
Other uncommon but serious reactionsobserved during or shortly followingadministration included cardiac or respira-tory arrest, loss of consciousness, convul-sions, symptomatic arrhythmias (atrialfibrillation, supraventricular tachycardia,and ventricular tachycardia or fibrillation),hypotension, respiratory distress orcardiac ischemia.
Patients should be assessed for thepresence of any condition that precludesDefinity® administration and should bemonitored during and for 30 minutesfollowing administration, including vitalsign measurements and electrocardio-
graphy in all patients and cutaneousoxygen saturation in patients at risk ofhypoxaemia. Resuscitation equipmentand trained personnel should be readilyavailable.
Conditions that preclude administrationinclude:
• Worsening or clinically unstable conges-tive heart failure
• Acute myocardial infarction or acutecoronary syndromes
• Serious ventricular arrhythmias or highrisk for arrhythmias
• Respiratory failure
• Severe emphysema, pulmonary embolior other conditions that cause pulmo-nary hypertension.
Reference: Health Canada, Medeffect, 18October 2007 at http://www.hc-sc.gc.ca
Gefitinib: failure of overallsurvival vs docetaxelCanada — The manufacturer of gefitinib(Iressa®) 250 mg tablets has informedhealthcare professionals taking part in theIressa® Patient Registry (IPR) of thefailure to demonstrate non-inferiority inoverall survival versus docetaxel. Theresults are from a Japanese multicentre,Phase III study in patients with ad-vanced, metastatic, or recurrent non-small cell lung cancer, who failed one ortwo chemotherapy regimens.
The study failed the primary objective todemonstrate non-inferiority of Iressa® to

282
WHO Drug Information Vol 21, No. 4, 2007Safety and Efficacy Issues
docetaxel in overall survival. Whencompared to docetaxel, some secondaryendpoints showed benefit with Iressa®use, but other secondary endpoints didnot.
No new safety findings were noted.There were 3 treatment-related deathsdue to interstitial lung disease in theIressa® arm and none in the docetaxelarm. No patients in Canada shouldreceive Iressa® unless they are enrolledin the IPR.
Reference: Communication from AstraZeneca through Health Canada, Medeffect, athttp://www.hc-sc.gc.ca
Rituximab and progressivemultifocal leukoencephalo-pathyAustralia — The Adverse Drug Reac-tions Advisory Committee (ADRAC) hasreceived a report of the rare neurologicaldisease, progressive multifocal leuko-encephalopathy, associated with the useof rituximab (Mabthera®) (1).
Rituximab is an immunosuppressantavailable in Australia since 1998 for thetreatment of certain types of non-Hodgkinlymphoma. Its indications were extendedin December 2006 to include use withmethotrexate for the treatment of severeactive rheumatoid arthritis in patients notresponding to, or unable to take, a tumournecrosis factor antagonist.
In December 2006, the US Food andDrug Administration (FDA) noted that twopatients had died after treatment withrituximab for systemic lupus erythemato-sus (2). The cause of death was progres-sive multifocal leukoencephalopathy(PML), a viral infection of the braincaused by reactivated JC virus. JC virusis a human polyomavirus that causeswidespread infection in childhood and islatent in about 80 percent of adults (2).
PML is a rare but devastating disease,occurring in fewer than 1 per 1500patients with lymphoma treated withrituximab in clinical trials; no cases werereported in approximately 3000 patientswith rheumatoid arthritis (3). Symptoms ofPML include mental deterioration, confu-sion, vision loss, difficulty speaking, andloss of balance. PML usually results indeath or severe disability. There is noknown effective treatment other than tostop medicines that are interfering withthe immune system.
In the case reported to ADRAC, a patientwith Waldenstrom Macroglobulinaemiapresented with altered vision and a newleft homonymous hemianopia one monthafter commencing rituximab. He had acomplicated history and had receivedother immunosuppressive treatmentincluding fludarabine, corticosteroids andirradiation. He died 5 months later, withwidespread lesions of PML found atautopsy (1).
Rituximab use is increasing and it ispossible that patients will present to GPswhen new symptoms develop. Patientstreated with rituximab who present withnew neurological signs or symptomsshould be referred for evaluation. ThePrecautions/spontaneous reportingsections of the rituximab Product Informa-tion have been recently updated toinclude information relating to PML.
Extract from Australian Adverse DrugReactions Bulletin Volume 26, Number 4,August 2007.
Reference
1. Ng C, Slavin MA, Seymour JF. ProgressiveMultifocal Leukoencephalopathy ComplicatingWaldenstrom’s Macroglobulinemia. Leukemiaand Lymphoma 2003; 44: 1819-1821.http://www.fda.gov/cder/drug/infopage/rituximab/default.htm http://www.fda.gov/cder/drug/infopage/rituximab/rituximabQA.htm

283
WHO Drug Information Vol 21, No. 4, 2007
Implanon®: interactions andcontraceptive failureAustralia — Hepatic enzyme inducingmedicines can reduce the efficacy ofcontraceptives, including implantablecontraceptives, leading to unintendedpregnancy.
Medicare Australia data indicate that370,173 etonogestrel-containing contra-ceptive implants (Implanon®) have beendispensed since 2001. The Adverse DrugReactions Advisory Committee (ADRAC)database contains 594 reports concern-ing Implanon®, 32 of which describe asuspected interaction betweenImplanon® and another medicine, result-ing in unintended pregnancy.
Medicines implicated in a possible inter-action with Implanon® leading to contra-ceptive failure include carbamazepine(26), phenytoin (4), methylphenobarbital(1) and rifampicin (1). All but 1 of theseinteractions involved medicines used totreat epilepsy. The 4 interacting medi-cines are potent inducers of CYP3A4 andother phase I and phase II enzymesystems in the liver. This enzyme induc-tion is likely to reduce plasma concentra-tions of etonogestrel which, similar toother contraceptive steroids, is catalysedby CYP3A4.
Other medicines likely to interact withetonogestrel and thus possibly reduce itscontraceptive effect or lead to break-through bleeding include primidone,oxcarbazepine, rifabutin, griseofulvin andproducts containing St John’s wort.Maximal enzyme induction is generallynot seen before two to three weeks butmay then be sustained for at least fourweeks after cessation of therapy withthese medicines.
The Product Information advises thatwomen receiving short-term treatmentwith any of the above medicines or other
hepatic enzyme inducing medicinesshould temporarily use a barrier methodin addition to Implanon®, i.e. during thetime of concomitant medicine administra-tion and for at least seven days afterdiscontinuation. For women takingrifampicin, an additional barrier methodshould be used during the time of rifam-picin administration and for 28 days afterits discontinuation.
Prescribers are reminded that in womenreceiving long-term treatment with hepaticenzyme inducing medicines, the ap-proved prescribing information recom-mends Implanon® should be removedand another, nonhormonal, contraceptivemethod should be used.
Extract from Australian Adverse DrugReactions Bulletin Volume 26, Number 4,August 2007.
Atypical antipsychotic agentsand extrapyramidal effectsAustralia — It has been suggested thatthe atypical antipsychotic agents clozap-ine, risperidone, olanzapine, aripipazoleand quetiapine may have lower propen-sity for causing extrapyramidal sideeffects (EPS) when compared with olderantipsychotic agents such as haloperidol,chlorpromazine and thioridazine (1,2)However, studies comparing the inci-dence of EPS between the older andnewer agents are often confounded,especially by previous exposure to olderagents (2).
Adverse Drug Reactions Advisory Com-mittee (ADRAC) data were searched forreports of EPS in association with thenewer antipsychotic agents, using termsrelating to nervous system disorders andmovement disorders. The number ofthese reports, as well as total number ofreports involving the medicine, is shownin the Table overleaf. Reports for ha-loperidol are included for comparison.
Safety and Efficacy Issues

284
WHO Drug Information Vol 21, No. 4, 2007
These data should be interpreted withcaution. As with most data from sponta-neous reporting, the data do not take intoaccount factors influencing reportingrates, such as level of usage and inten-sive post-market safety monitoringprograms. Reports with the newer agentsare also likely to be confounded asdescribed above, and ADRAC data forhaloperidol are likely to be substantiallymore incomplete and limited than data forthe newer agents. Therefore, the relativenumbers of EPS reports shown in theTable do not necessarily reflect relativerisk between these agents.
The most common reactions described inreports with the newer antipsychoticagents include dystonias, dyskinesias,akathisia and other non-specified EPdisorder. At the time of reporting, aboutone third of patients experiencing EPShad not recovered, with no distinctionbetween medicines in this regard.
Extract from Australian Adverse DrugReactions Bulletin Volume 26, Number 4,August 2007.
References
1. Kane JM. Tardive dyskinesia rates withatypical antipsychotics in adults: prevalenceand incidence. J Clin Psychiatry 2004; 65(Suppl. 9): 16 -20.
2. Tarsy D and Baldessarini RJ. Epidemiologyof tardive dyskinesia: Is risk declining withmodern antipsychotics? Movement Disorders2006; 21: 589-598.
Thiazolidinediones andreduced bone densityAustralia — Thiazolidinediones includerosiglitazone (Avandia® and Avanda-met®) and pioglitazone (Actos®). Thesemedicines act to increase insulin sensitiv-ity and are widely prescribed to treat typeII diabetes mellitus. Recent evidencesuggests thiazolidinediones are associ-ated with an increased risk of peripheralfractures in post-menopausal women.
The ADOPT study (1) was a randomized,double-blind, parallel group study thatfollowed the progression of 4360 recentlydiagnosed patients with diabetes mellitusfor a median of 4.0 years. The incidenceof fractures in women taking rosiglitazonewas 9.3%, compared with 5.1% in thosetaking metformin and 3.5% in thosetaking glibenclamide. The majority offractures in these patients were in thehumerus, hand, or foot. The incidence offractures of the hip or spine in womenand the incidence of fractures in maleswere similar among the 3 treatmentgroups.
A sponsor-initiated review of fracture riskin patients treated with pioglitazone for upto 3.5 years also found more fractures infemale patients taking pioglitazone thanthose taking a comparator. There was noincreased risk of fracture identified inmen.
The sponsors of rosiglitazone and piogli-tazone have updated the product infor-
Medicine Total reports EPS reports (% of total)
Aripiprazole 147 33 (22.4)Risperidone 812 159 (19.6)Quetiapine 315 42 (13.3)Olanzapine 1203 126 (10.5)Clozapine 3775 70 (1.9)
Haloperidol 753 321 (42.6)
Safety and Efficacy Issues

285
WHO Drug Information Vol 21, No. 4, 2007
mation documents for these medicinesand issued letters to healthcare profes-sionals describing the above findings.
The mechanism for increased fracturerisk was examined in a 14 week study in50 healthy postmenopausal women inNew Zealand (2). This study showedreductions in markers of bone formationin women taking rosiglitazone 8 mg/daycompared with placebo. These changeswere evident after 4 weeks and persistedfor the duration of the study. There werealso small reductions in hip and lumbarspine bone density in women takingrosiglitazone.
The full clinical significance of theserecent findings is yet to be determined.However, the risk of fracture should beconsidered for all patients, especiallywomen, taking or being considered fortreatment with thiazolidinediones. Forthese patients, as for all others with typeII diabetes mellitus, attention should begiven to assessing and maintaining bonehealth according to current standards ofcare.
Extract from Australian Adverse DrugReactions Bulletin, Volume 26, Number 5,October 2007.
Reference
1. Kahn S et al. Glycemic durability of rosigli-tazone, metformin, or glyburide monotherapy.NEJM 2006; 355: 2427-43.
2. Grey A et al. The peroxisome-proliferator-activated receptor-gamma agonist rosiglita-zone decreases bone formation and bonemineral density in healthy postmenopausalwomen: a randomized, controlled trial. J ClinEndocrin Metab 2007;92:1305-1310.
Renal impairment withzoledronic acidAustralia — There is a well-known risk ofdeterioration in renal function with intrave-nous bi-sphosphonates administered at a
rapid infusion rate. The Adverse DrugReactions Advisory Committee (ADRAC)has received few reports of renal impair-ment or failure with pamidronate and theoral bisphosphonates risedronate andalendronate, but there is a significantnumber with zoledronic acid (31 from atotal of 268 reports for this drug). Whilethe deterioration in renal function withzoledronic acid (Zometa®) was usuallyacute, in many cases it did not appear tobe related to a rapid infusion rate.
The 31 reports in association withzoledronic acid describe either renalfailure (16) or renal impairment (15). Itwas the only suspected drug in 20 of the31 reports. Interstitial nephritis wasdescribed in 3 of the reports. Zoledronicacid was being used for a variety ofindications with multiple myeloma (13cases) the most common but also breastcancer (5), prostate cancer (4), plasmacy-toma, malignant melanoma, osteoporosis,bone metastases and osteomyelitis (1case each). Only 4 reports did not specifythe reason for use.
ADRAC reminds prescribers of bisphos-phonates to pay close attention to riskfactors for renal impairment and toadhere strictly to the instructions for use.
Extract from Australian Adverse DrugReactions Bulletin, Volume 26, Number 5,October 2007.
Epoetins: unexplainedexcess mortalityEuropean Union — The EuropeanMedicines Agency (EMEA) has recentlyreviewed the safety of epoetins. Thesemedicines are used for the treatment ofanaemia in patients with chronic renalfailure and for the treatment of patientswith non-myeloid malignancies receivingchemotherapy (1).
The safety review was initiated becausedata from recent clinical trials show a
Safety and Efficacy Issues

286
WHO Drug Information Vol 21, No. 4, 2007
consistent unexplained excess mortalityin patients with anaemia associated withcancer who have been treated withepoetins.
In addition, the results of two studies anda meta-analysis have recently beenpublished suggest that treatment ofanaemia with epoetins in patients withchronic kidney disease to achieve rela-tively high target haemoglobin concentra-tions may be associated with an increasein the risk of mortality and cardiovascularmorbidity.
EMEA’s Committee for Medicinal Prod-ucts for Human Use (CHMP) and itsPharmacovigilance Working Party(PhVWP) concluded that the benefits ofthese products continue to outweigh theirrisks in the approved indications, butrecommended changes to the productinformation:
• Changes to the ‘Indication’ section(section 4.1), saying that epoetinsshould be used in the treatment ofanaemia only if associated with symp-toms.
• Changes to the ‘Posology’ section(Section 4.2), stipulating a uniformtarget haemoglobin range for allepoetins of 10 g/dL to 12 g/dL with awarning not to exceed a concentrationof 12 g/dL.
• Changes to the ‘Special Warnings andPrecautions for Use’ section (Section4.4), adding an explanation that trialshave shown a small unexplained excessmortality in association with high targethaemoglobin concentrations and theyhave not shown significant benefitsattributable to the administration ofepoetins to increase haemoglobinconcentration beyond the level neces-sary to control symptoms of anaemiaand to avoid blood transfusion.
• Changes to the ‘Pharmacodynamicproperties’ section (section 5.1) toinclude new information on results ofclinical trials which have shown signifi-cant excess mortality in patients whohave anaemia associated with variouscommon cancers who received epoetinscompared with those who did notreceive epoetins.
The changes will now be implementedthroughout the European Union. TheEMEA has requested all MarketingAuthorization Holders for centrally author-ised epoetins to submit an application fora type II variation to the marketing au-thorization. For medicinal products thatare authorized at the level of theMember States, the national competentauthorities will take further action asappropriate.
The CHMP also identified a need toincrease scientific knowledge on theeffect of epoetins. Marketing Authoriza-tion Holders have been asked to combineall available patient-level data jointly andto assess the functional activity of epoetinreceptors in different tumour types, and atdifferent stages in the life-cycle of tumourevolution.
References
1. Public Statement, Doc. Ref. EMEA/496188/2007. London, 23 October 2007. http://www.emea.europa.eu
2. Ajay K Singh et al. for the CHOIR Investiga-tors. Correction of Anaemia with Epoetin Alfain Chronic Kidney Disease. N Engl J Med2006; 355:2058-98.
3. Tilman B Drüeke et al. for the CREATEInvestigators. Normalization of HaemoglobinLevel in Patients with Chronic Kidney Diseaseand Anaemia. N Engl J Med 2006; 355: 2071-84.
4. Phrommintikul A et al.: Mortality and TargetHaemoglobin Concentrations in AnaemicPatients with Chronic Kidney Disease Treatedwith erythropoietin: a Meta-Analysis. Lancet2007; 369: 381-88.
Safety and Efficacy Issues

287
WHO Drug Information Vol 21, No. 4, 2007
Contraindicated useof sibutramine andcardiovascular reactionsCanada — Sibutramine (Meridia®), aserotonin and norepinephrine reuptakeinhibitor, is an antiobesity agent marketedin Canada since February 2001.
Sibutramine is indicated as adjunctivetherapy within a weight managementprogramme for obese patients (1). TheCanadian product monograph ofsibutramine includes several contra-indications. Noncompliance withcontraindications could result in seriousadverse reactions (ARs).
Health Canada continues to receivereports of ARs in patients usingsibutramine who have contraindications.Between 2001 and 2007, 65 reports ofcardiovascular ARs suspected of beingassociated with sibutramine were re-ceived. Thirteen of these reports involvedpatients with at least 1 contraindicatedcondition.
In 2002 and 2003, international regulatoryactions were taken, including safetynotices, concerning cardiovascular ARsassociated with sibutramine (2, 3, 4).Health Canada and other foreign regula-tory agencies reviewed the safety ofsibutramine and concluded that thebenefit-risk profile of sibutramine re-mained favourable (4).
Extract from Canadian Adverse ReactionNewsletter, Volume 17(4), 2007.
References
1. Meridia® (sibutramine hydrochloridemonohydrate) [product monograph]. Saint-Laurent (QC): Abbott Laboratories Ltd; 2005.
2. Nisoli E, Carruba MO. A benefit-risk assess-ment of sibutramine in the management ofobesity. Drug Saf 2003;26(14):1027-48. [PubMed]
3. Health Canada investigates safety ofMeridia® (sibutramine). Ottawa: HealthCanada; 2002 Mar 27. Available: www.hc-sc.gc.ca/ahc-asc/media/advisories-avis/2002/2002_21_e.html (accessed 2007 July 10).
4. Health Canada reports back to public onsafety profile of Meridia® (sibutramine).Ottawa: Health Canada; 2003 Feb 28. Avail-able: www.hc-sc.gc.ca/ahc-asc/media/advisories-avis/2003/2003_07_e.html(accessed 2007 July 10).
Gadolinium-containingcontrast agents: nephrogenicsystemic fibrosisCanada — Gadolinium (Gd)-containingmedia are used to enhance the contrastof magnetic resonance images. Thoseauthorized for sale in Canada includeMagnevist®, Omniscan®, OptiMARK®,Gadovist®, ProHance®, MultiHance®and Vasovist.
Nephrogenic systemic fibrosis (NSF),also known as nephrogenic fibrosingdermopathy (NFD), is a systemic disorderwhose most prominent and visible effectsare on the skin (1). It is associated withsignificant morbidity (2 3). The fibrosiscan extend beyond the dermis andinvolve subcutaneous tissues, musclesand internal organs (2 ,3). The diseasewas first described in the medical litera-ture in 2000,(4) but the first case wasreported in 1997 (2, 4). To date, NSF hasbeen observed only in patients withkidney disease (1).
Internationally, cases of NSF have beenobserved with 5 different Gd-containingcontrast agents (5), and a causal associa-tion has recently been suggested (6). InMarch 2007, Health Canada communi-cated this safety concern to the publicand health professionals (2, 3). As ofJune 27, 2007 (see WHO Drug Informa-tion, 21(1) 15, 2007), 5 cases of NSFsuspected of being associated with Gd-containing contrast agents have been
Safety and Efficacy Issues

288
WHO Drug Information Vol 21, No. 4, 2007
reported in Canada. This recently discov-ered disease is currently not described inthe Canadian product monographs forthese agents.
The association between NSF and theuse of Gd-containing contrast medianeeds to be characterized further. Inparticular, are other patient populations atrisk for NSF (e.g., neonates)? Does therisk vary according to the nature ofunderlying renal disorder? What is therole of dialysis in the prevention andtreatment of NSF?
Valuable information could be obtainedfrom adverse reaction (AR) reports.Spontaneous reporting can be useful toidentify the clinical spectrum of the drug-AR pair, the patient subtypes and medicalcircumstances associated with a sus-pected AR (7). It can also provide clues tothe mechanism of action whereby aproduct can lead to an AR (7). Theaddition of clinical information in reports,such as the duration of kidney diseaseand its underlying cause, laboratoryvalues (e.g. glomerular filtration rate), thetype and dose of the contrast agents, andconcomitant conditions and medications,is important to help characterize the riskfactors of NSF.
Extract from Canadian Adverse ReactionNewsletter, Volume 17(4), 2007.
References
1. Conference announcement: First annualscientific symposium on NSF and MRIcontrast. New Haven (CT): InternationalCenter for Nephrogenic Fibrosing DermopathyResearch; 2007 March 27. Available:www.icnfdr.org (accessed 2007 July 11).
2. Association of nephrogenic systemicfibrosis/nephrogenic fibrosing dermopathy(NSF/NFD) with the use of gadolinium-containing contrast agents. Ottawa: HealthCanada; 2007 March 9 & 13. Available:www.hc-sc.gc.ca/dhp-mps/medeff/advisories-avis/prof/2007/gadolinium_nth-aah_e.html
3. Updated safety information on Omniscan®and nephrogenic systemic fibrosis/nephro-genic fibrosing dermopathy (NSF/NFD).Ottawa: Health Canada; 2007 March 7 & 12.Available: www.hc-sc.gc.ca/dhp-mps/medeff/advisories-avis/prof/2007/omniscan_hpc-cps2_e.html.
4. Cowper SE, Robin HS, Steinberg SM, et al.Scleromyxoedema-like cutaneous diseasesin renal-dialysis patients. Lancet 2000;356:1000-1.
5. Gadolinium-based contrast agents formagnetic resonance imaging (marketed asMagnevist, MultiHance, Omniscan,OptiMARK, ProHance). Rockville (MD): USFood and Drug Administration; 2007 May 23.Available: www.fda.gov/cder/drug/InfoSheets/HCP/gcca_200705HCP. pdf
6. Grobner T. Gadolinium — A specific triggerfor the development of nephrogenic fibrosingdermopathy and nephrogenic systemicfibrosis? Nephrol Dial Transplant 2006;21(4):1104-8.
7. Clark JA, Klincewicz SL, Stang PE. Sponta-neous adverse event signaling methods:classification and use with health care treat-ment products. Epidemiol Rev. 2001; 23(2):191-210.
Swedish Adjustable GastricBand: incidents leading toexplantationCanada — The Swedish AdjustableGastric Band (SAGB) is an implantable,adjustable gastric band indicated for usein the treatment of morbid obesity inadults (1). It consists of a reinforcedsilicone gastric band fitted around thestomach and an injection port placedunder the skin and connected to the bandby tubing. The SAGB is designed toreduce food intake and can be inflated ordeflated as needed after implantation tomeet weight-loss requirements withoutthe need for further surgery. The SAGBwas originally licensed for sale in Canadain November 2002. A modified version of
Safety and Efficacy Issues

289
WHO Drug Information Vol 21, No. 4, 2007
the device, the SAGB Quick Close(SAGB-QC), was added to the licence aspart of a device licence amendment inAugust 2004 (2).
Although band erosion is listed amongthe possible adverse events (2), thedevice labelling for patients states thatthe overall rate of re-operation is low andthat extensive use of the SAGB has led toa method where failure is uncommon (3).By definition, band erosion is “a situationwhere a part of the band has erodedthrough the full-thickness gastric wall andmigrated into the lumen (4). This repre-sents a total failure of the gastric bandingprocedure (5).
Health Canada has received 19 reports ofincidents suspected of being associatedwith the SAGB and 17 with the SAGB-QC. Thirteen of the 36 reports necessi-tated removal of the band. Other reportsdescribed incidents such as band slip-page, band leakage, abscess, dysphagiaand regurgitation. In 35 of the 36 reports,band explantation was reported as anoutcome.
Since band erosion is often asymptomaticor only mildly symptomatic initially andsince the condition is best diagnosed bygastroscopy, which may not be includedin the follow-up of asymptomatic patients,the true incidence of band erosion isunderestimated in the literature and itsdiagnosis can be markedly delayed (4, 5).Moreover, band erosion is associatedwith dense scarring and distortion oftissues, which can complicate revisionprocedures (5).
The complication rates and outcomesassociated with SAGB and reported in theliterature are variable. Although theauthors of some studies have concluded
that use of the SAGB demonstratesacceptable levels of safety and effective-ness (6, 7), others have reported highlong-term complication and failure ratesand poor long-term outcomes (4, 5). Themedical literature suggests that alterna-tive treatment options should be consid-ered and gastric banding should beperformed only in carefully selected andfully informed patients (5).
Extract from Canadian Adverse ReactionNewsletter, Volume 17(4), 2007.
References
1. Swedish Adjustable Gastric Band [Canadianinstructions for use]. Baar (SWI): ObtechMedical AG; 2000.
2. Swedish Adjustable Gastric Band QuickClose. Zug (SWI): Ethicon Endo-Surgery incooperation with Obtech Medical AG; 2003.
3. Swedish Adjustable Gastric Band QuickClose. Zug (SWI): Ethicon Endo-Surgery incooperation with Obtech Medical AG; 2003.
4. Gustavsson S, Westling A. Laparoscopicadjustable gastric banding: complications andside effects responsible for the poor long-termoutcome. Semin Laparosc Surg 2002;9(2):115-24.
5. Suter M, Calmes JM, Paroz A, et al. A 10-year experience with laparoscopic gastricbanding for morbid obesity: high long-termcomplication and failure rates. Obes Surg2006;16(7):829-35.
6. Steffen R, Biertho L, Ricklin T, et al.Laparoscopic Swedish adjustable gastricbanding: a five-year prospective study. ObesSurg 2003;13(3):404-11.
7. Zehetner J, Holzinger F, Tiraca H, et al. A 6-year experience with the Swedish adjustablegastric band. Prospective long-term audit oflaparoscopic gastric banding. Surg Endosc2005;19(1):21-8.
Spontaneous monitoring systems are useful in detecting signals of relatively rare, serious and unexpected adversedrug reactions. A signal is defined as "reported information on a possible causal relationship between an adverse eventand a drug, the relationship being unknown or incompletely documented previously. Usually, more than a single reportis required to generate a signal, depending upon the seriousness of the event and the quality of the information". Allsignals must be validated before any regulatory decision can be made.
Safety and Efficacy Issues

290
WHO Drug Information Vol 21, No. 4, 2007
Regulatory Action and News
Influenza virus vaccines:Southern hemisphere 2008World Health Organization — It isrecommended that vaccines for use in the2008 influenza season (Southern hemi-sphere winter) contain the following:
• an A/Solomon Islands/3/2006 (H1N1)-like virus (A/Solomon Islands/3/2006 isa current vaccine virus):
• an A/Brisbane/10/2007 (H3N2)-likevirus;
• a B/Florida/4/2006-like virus.
As in previous years, national controlauthorities should approve the specificvaccine viruses used in each country.National public health authorities areresponsible for making recommendationsregarding the use of the vaccine.
Reference: World Health Organization.Weekly Epidemiological Record, 2007;82: 351
Withdrawal of coughmedicines containingclobutinol?European Union — Following a reviewof the safety of clobutinol-containingcough medicines (Silomat®), the Euro-pean Medicines Agency (EMEA) hasconcluded that the risks of these medi-cines outweigh the benefits and hasrecommended that marketing authoriza-tions be withdrawn.
Silomat® is available without a prescrip-tion in a number of EU Member States forthe short-term treatment of irritable, non-productive cough..
Having considered all available evidence,it was concluded that the use of clobutinolis associated with a risk of prolongation ofthe QT interval which is known to belinked to fainting and disruption of theheart rhythm, especially when taken inhigher doses. The opinion will now besent to the European Commission for theadoption of a decision applicable in all EUMember States.
Reference: Press release, Ref. EMEA/480863/2007. London, 18 October 2007.http://www.emea.europa.eu
Rimonabant warning:psychiatric conditionsEuropean Union — The EuropeanMedicines Agency (EMEA) has recom-mended contraindicating rimonabant(Acomplia®) in patients with ongoingmajor depression or who are beingtreated with antidepressants, because ofthe risk of psychiatric side effects.
Doctors in the EU have already beenwarned about this since June 2006 butthe Agency’s Committee for MedicinalProducts for Human Use (CHMP) hasnow recommended upgrading this warn-ing. Acomplia® has been authorized inthe EU since June 2006 as an adjunct todiet and exercise for the treatment ofobese or overweight adult patients.Psychiatric side effects, in particulardepression, were identified as the mainsafety issue at the time of approval. Theywere reflected in the medicine’sproduct information as a warning thatdoctors should not prescribe Acomplia®in patients with uncontrolled seriouspsychiatric conditions such as majordepression.

291
WHO Drug Information Vol 21, No. 4, 2007 Regulatory Action and News
At the 16-19 July 2007 meeting, theCHMP concluded that the benefits ofAcomplia® continue to outweigh its risks,except in patients with ongoing majordepression or those taking antidepres-sants.
Reference: Press Release, Doc. Ref. EMEA/329826/2007, London, 19 July 2007 availableat www.emea.europa.eu
United States of America —Rimonabantis a first in class cannabinoid receptor 1(CB1) antagonist/inverse agonist forweight management. The Food and DrugAdministration’s Endocrinologic andMetabolic Drugs Advisory Committee meton 13 June 2007 to discuss an ongoingdata review. The Committee decided thatmore detailed long-term safety informa-tion with larger patient groups wasneeded with regard to neurological andpsychiatric side effects associated withthe drug including seizure, depression,anxiety, insomnia, aggressiveness andsuicidal thoughts.
Reference: http://www.fda.gov
Lumiracoxib registrationcancelledAustralia — The Therapeutic GoodsAdministration (TGA) has cancelled theregistration of the osteoarthritis drug,lumiracoxib (Prexige®) because ofserious liver side effects associated withthe use of the drug.
Lumiracoxib is a cox-2 inhibitor indicatedfor symptomatic relief in the treatment ofosteoarthritis, relief of acute pain, includ-ing post-operative pain and pain relatedto dental procedures and relief of paindue to primary dysmenorrhoea. It wasfirst approved in Australia in July 2004and listed on the Pharmaceutical BenefitsScheme (PBS) in 2006. As of 10th August2007, the TGA has received 8 reports ofserious liver adverse reactions to thedrug, including two deaths and two livertransplants.
The Adverse Drug Reactions AdvisoryCommittee (ADRAC) has recommendedthe cancellation of registration due to theseverity of the reported side effects.
Reference: Media statement, 11 August 2007at http://www.tga.gov.au
Lumiracoxib: withdrawalof higher dosesNew Zealand — The Medicines andMedical Devices Safety Authority,Medsafe, has withdrawn supply of 200mg and 400 mg tablets of the cox-2 anti-inflammatory medicine lumiracoxib(Prexige®).
The decision has been reached afterMedsafe reviewed local and internationalsafety data relating to reports of severeliver damage in patients using this medi-cation at doses of 200 mg and above. Inmaking the decision, Medsafe discussedthe overall risks and benefits of the use ofPrexige® with medicines regulators inAustralia, Singapore and the UnitedKingdom.
Medsafe also reviewed the safety ofPrexige® 100 mg indicated for use inosteoarthritis. The data available fromclinical trials and reported side effects inthe United Kingdom, Europe, Canada orSouth America indicate that severe liverdamage with Prexige® 100 mg/day israre. The frequency with which liverdamage is reported for Prexige® 100 mgdoes not appear to be significantlydifferent from that seen for other anti-inflammatory medicines.
Medsafe has accepted the interim adviceof the Medicines Adverse ReactionsCommittee (MARC) that Prexige® 100mg should remain on the market and itssafety be closely monitored.
Reference: Communication from Medsafe, 21August 2007, at http://www.medsafe.govt.nz/hot/alerts.asp

292
WHO Drug Information Vol 21, No. 4, 2007Regulatory Action and News
Ixabepilone approved foradvanced breast cancerUnites States of America — The Foodand Drug Administration (FDA) hasapproved ixabepilone (Ixempra®), a newanti-cancer treatment, for use in patientswith metastatic or locally advanced breastcancer who have not responded tocertain other cancer drugs. Ixabepilone isadministered by intravenous infusion.
Ixabepilone was approved for use in com-bination with another cancer drug, cape-citabine, in patients who no longer benefitfrom two other chemotherapy treatments.These prior treatments included ananthracycline (such as doxorubicin orepirubicin) and a taxane (such as paclit-axel or docetaxel). Ixabepilone was alsoapproved for use alone in patients whono longer benefit from an anthracycline, ataxane and capecitabine.
Ixempra has been shown to bind tocancer cell microtubules, which arestructures within cells that help to supportand shape them. Microtubules also play arole in cell division. The safety andefficacy of ixabepilone in combinationwith capecitabine were evaluated in 752patients in a randomized clinical trialcomparing the combination to capecitab-ine alone. This combination therapydemonstrated improvements in delayingcancer progression or death compared tocapecitabine alone.
Ixabepilone’s significant side effectsincluded peripheral neuropathy (numb-ness, tingling or burning in the hands orfeet) and bone marrow suppression.Other commonly observed toxicitiesincluded constipation, nausea, vomiting,muscle paint, joint pain, fatigue andgeneral weakness.
Women taking ixabepilone should avoidtaking drugs that are strong inhibitors ofCYP3A4.
Ixabepilone should not be taken bywomen who have had severe allergicreactions to drugs that containCremophor or its derivatives, or bywomen who have baseline bone marrowsuppression determined by low whiteblood cell or platelet count.
The combination of Ixempra® andcapecitabine should not be given topatients with moderate or severe liverimpairment due to the increased risk oftoxicity and death.
Reference: FDA News, 22 October 2007 athttp://www.fda.gov
Second-generation smallpoxvaccine approvedUnited States of America — The Foodand Drug Administration (FDA) haslicensed a new vaccine to protect againstsmallpox, a highly contagious diseasewith the potential to be used as a deadlybioterror weapon.
A worldwide vaccination programmeeradicated smallpox in the population.The last case of naturally occurringsmallpox in the USA was in 1949 and thelast case in the world was reported inSomalia in 1977. Known stockpiles of thevirus are kept only in two approved labsin the United States and Russia.
Smallpox is caused by the variola virus, avirus that emerged in human populationsthousands of years ago. It spreadsthrough close contact with infectedindividuals or contaminated objects, suchas bedding or clothing. There is no FDA-approved treatment for smallpox and theonly prevention is vaccination.
Because ACAM2000 contains live vac-cinia virus, care must be taken to preventthe virus from spreading from the inocula-tion site to other parts of the body, and toother individuals.

293
WHO Drug Information Vol 21, No. 4, 2007
Reference: FDA News, 1 September 2007 athttp://www.fda.gov
Regulatory updatesEuropean Union — A summary of recentregulatory developments is available fromthe European Commission.
B.1.1.2 Notice to applicants for medici-nal products for human useEudraLex Vol 2A, Chapter 3 - CommunityReferral (for human medicinal products)(revised version reflects the current legalframework for referral procedures)
EudraLex Vol 2A, Chapter 7 - GeneralInformation (Revision from July 2007)(revised verions includes updated infor-mation from different EU Member Statesand contains a major update of section3.2 “National, Mutual Recognition andDecentralised Procedures: “additionaldata” requested)
EudraLex Vol 2C - Guideline on thepackaging information of medicinalproducts for human use authorized by theCommunity (Revision 12, August 2007)(Remark: Revision 12 was publishedshortly after Revision 11 in order toinclude corrections related to the require-ments by Italy; revision 11 has beenamended to include updates requestedby Italy and Sweden)
EudraLex Vol 6A, Chapter 3 - CommunityReferral (for veterinary medicinal prod-ucts). (revised version reflects currentlegal framework for referral procedures)
A.4.6.1 Compassionate UseAccording to Art. 83 of Reg. 726/2004 –Definition, Requirements and Procedure.
A.4.6.1.1 Principles of Application of theCompassionate Use Provision.
A.4.6.1.2 Compassionate Use Procedure
CHMP Guideline on Compassionate Use.
In July 2007 the “Guideline on Compas-sionate Use of Medicinal Products,Pursuant to Article 83 of Regulation (EC)No. 726/2004” was finally adopted byCHMP and implemented on July 19,2007. It aims at providing guidance andexplanations as well as definitions inrelation to the criteria and the procedureforeseen in Art. 83 of Regulation (EC) No.726/2004 and to give information regard-ing the documentation which has to besubmitted. Chapter 4.6.1 has accordinglybeen supplemented and the presentationof the principle of compassionate use hasbeen extended by Chapters 4.6.1.1 and4.6.1.2.
A.2.4.3 Inspections Unit of the EMEAA.2.4.3.1 Responsibilities of the Inspec-tion UnitA.2.4.3.2 Inspectors Working Groups
EMEA Inspections Unit - GMPD and GCPInspectors Working Groups - Mandateand Objectives. Shortly after the enteringinto force of the new European MarketingAuthorization System Ad hoc InspectionServices Groups were set up for thesectors of GMP and GCP. At the end of2006, the Ad Hoc Inspection ServicesGroups changed names and became“Inspectors Working Groups”. Mid 2007,the two Inspector Working Groups pub-lished documents on their mandate andobjectives. These documents give reasonto extend the description of the EMEAInspections Unit, updating Chapter A2.4.3 and extending it by Chapters A2.4.3.1 and 2.4.3.2.
Reference: Informatio available at http://www.drugregulatoryaffairs.eu

294
WHO Drug Information Vol 21, No. 4, 2007
Generic Medicines
Offering patients generic substitutes toinnovator brands of drugs is an encour-aged practice of many countries becauseof the cost saving benefits that can begained to patient management [1–5]. In1993, out of a need to reduce the cost ofdrugs to patients, the Ministry of Health inJamaica amended the Pharmacy Act, tomandate pharmacists to offer all patientssubstitution of innovator brands of pre-scription drugs with less expensivegeneric brands, thus allowing the patientto choose [6]. Along with the authoritygiven to pharmacists, the amendmentadditionally allowed physicians to request“no substitution” of innovator brand bygeneric, or a generic brand by anotherbrand.
In Jamaica, the pharmaceutical marketcan be quite complex to assess, as manygroups are involved and the success ofmandates to encourage generics involves
much more than a reliance on costbenefit. In a number of cases, genericshave been assessed as therapeuticallyinequivalent [7] and this has had anegative influence on perceptions amonghealth professionals. In 2003, a newspa-per article in the Jamaica Gleaner re-ported that both physicians and pharma-cists expressed dissatisfaction withgeneric substitution as mandated by thePharmacy Act because generics wereplagued with incidents of therapeuticinequivalence [8].
In 2006, The Fair Trading Commission, incollaboration with The Consumer AffairsCommission and The University ofTechnology, examined some of the issuesthat influenced the sale of innovator andgeneric products by conducting surveysamong patients, physicians and pharma-cists. These surveys involved the use ofvalidated questionnaires, comprising
In 1993, an Amendment to the Pharmacy Act introduced generic substitution of inno-vator brands to Jamaica. Although the Amendment gives prescribers the opportunityto indicate “no substitution” on prescriptions, implementation of the generic medi-cines policy has been largely hindered by doubts concerning therapeutic equiva-lence. In addition, reservations among many physicians and pharmacists have beenobserved concerning implementation of the policy.
This review summarizes information collected among health professionals and pa-tients in Jamaica on the knowledge and perceptions surrounding generic substitutionof medicines. It concludes that only through evidence-based awareness programmesand responsive pharmacovigilance systems will physicians, pharmacists and the publicgain confidence in the concept of generic substitution.
Generic substitution in Jamaica:challenges to improving effectiveness
Article proposed by Gossell-Williams, M., Department of Basic Medical Sciences, PharmacologySection, University of the West Indies, Jamaica; Harriott, K., Jamaica Fair Trading Commission,Kingston 5. Jamaica. Reprints of this article can be obtained from Maxine Gossell-Williams, Tel:876-927-2216, Fax: 876-9773823. Email: [email protected]

295
WHO Drug Information Vol 21, No. 4, 2007
adequately represented sample sizes andproportionately stratified island-widegroups (i.e. Kingston Metropolitan area,other towns and rural areas) [9]. Theresults suggest that, in 2006, the percep-tion that generics are therapeuticallyinequivalent is still of concern amongphysicians and pharmacists. This willpresumably affect acceptance of substi-tutability and success of the Amendment.This review discusses some of thefindings related to knowledge of genericdrugs by patients and the prevailing lackof confidence that physicians and phar-macists have in substitutability andproposes some action points.
Patient understanding of thegeneric conceptThis component of the survey wasconducted among patients, eighteenyears and older (N=1030). The majoritydid not fully appreciate the term “genericmedicine”, as 63.6% (N=1,020; non-responsive=10) had either never heardthe term or were familiar with it, but notsure what it meant. Therefore, mostpatients were not adequately informedabout the difference or similarities be-tween innovator and generic medicines.Interestingly, of those who were familiarwith and understood the term (N=371), alittle less than half stated only that gener-
Figure 1. Defining generic medicines
Che
aper
, equ
ally
effe
ctiv
e
Equ
ally
effe
ctiv
e
Che
aper
Che
aper
, les
s ef
fect
ive
less
effe
ctiv
e
Mad
e fr
om n
atur
al in
gred
ient
s
Mad
e fr
om m
an m
ade
ingr
edie
nts
Sub
stitu
te fo
r br
ande
d m
edic
atio
n
Imita
tion/
copy
, nor
orig
inal
Alte
rnat
ive
to b
rand
Can
’t ex
plai
n
Oth
er
Generic Medicines
% o
f p
ati
en
ts

296
WHO Drug Information Vol 21, No. 4, 2007
ics were cheaper, suggesting that therewas a lack of appreciation that genericsare expected to provide similar efficacy tothat of the innovator (See figure 1).
Patient acceptance of substitution wasnot significantly influenced by income,health insurance coverage or whetherthey were compliant to drug therapy. Mostof the patients who understood the termgeneric indicated that physicians andpharmacists were their most crediblesource of information (Table 1). Addition-ally, the majority indicated that they wouldnot request substitution of prescribedmedicines, whether it be innovator orgeneric (Figure 2). Therefore a patient’s
choice to accept substitution with genericmedicines will depend on the confidencetheir physician has in generic productsand particularly of therapeutic equiva-lence. This is supported by pharmacists.
Physician views ontherapeutic equivalenceSurveyed physicians (N=242) repre-sented 10% of registered physicians as at2005. The majority were males (66.8%),practising for over ten years (52.8%) andwriting between 10 to 20 prescriptions perweekday (33.1%) of which 50% would bewritten for generic medicines. Whenphysicians were asked about their per-ception of therapeutic equivalence
Table 1. Patients: credibility of information sources
Figure 2. “My doctor knows best”Patients who understood the term “generic medicine” were asked if they would request substitu-tion of a medicine prescribed by their physician.
A. Patients who would request generic medi-cine at the pharmacy although physician pre-scribed innovator (N=355; total non-respond-ents=16)
Yes = 19.7%No = 65.4%Why no:
Reason No. patientsMy doctor knows best 181It would not be safe to do so 18Other reasons 32Non-respondents 4
B. Patients who would request innovatormedicine at the pharmacy although physi-cian prescribed generic (N=361; total non-respondents=10)
Yes = 14.4 %No = 73.4%Why no:
Reason No. patientsMy doctor knows best 215It would not be safe to do so 11Other reasons 34Non-respondents 2
Generic Medicines
Information source Number of patients(Total = 371)*
Physician 288Pharmacist 35Family/Friend 16Drug manufacturer/importer 12Ministry of Health 13Internet 5Testimonials (word of mouth by other users of medication) 3
* Values in columns do not balance because of non-responders

297
WHO Drug Information Vol 21, No. 4, 2007
(N=238; non-respondents=4), 45%indicated that they believed genericswere therapeutically equivalent (Figure3). However, more than 25% indicateddoubt, based mainly on the reputation ofthe manufacturer or quality of the genericdrug. This demonstrates an uncertaintyregarding the ability of generics to substi-tute for innovators.
Pharmacist views on efficacyPharmacists located in thirty-six pharma-cies, representing 10% of pharmaciesregistered as at 2005, were surveyed.The majority of respondents were fe-males (75%), practising for more than 10years (47.25%) and filling 30–40 prescrip-tions on a weekday (22.2%). Pharmacistswere specifically asked to give views onthe efficacy of generics when comparedwith innovators. The majority indicated alack of confidence in generics, as only44.4% thought they were of the sameefficacy as innovators, and this percep-tion was based mainly on feedback frompatients (Figure 4). Fourteen of the 36pharmacists indicated doubt in efficacydue to factors such as the individualsituation of a patient, type of illness,
manufacturer’s reputation and quality ofthe generic product/excipient.
Challenging awarenessMost physicians indicated a need toincrease awareness of the advantages ofgeneric medicines in Jamaica (61.6%,N=242). Pharmacists expressed similarsentiments and indicated that the greatestneed was among patients. They sug-gested that strategies to increase aware-ness should involve better provision ofinformation to patients by physicians andpharmacists. Because of the trust pa-tients place in health professional advice,any doubt in the value of generics amongphysicians and pharmacists should beaddressed, while there is an urgent needfor increased confidence in the therapeu-tic equivalence of generics.
For such an effort to succeed, knowledgeof where these groups obtain credibleinformation is a key. Both pharmacistsand physicians were asked to list their topsource of credible information. Pharma-cists indicated that they relied mainly onmedical journals, as well as drug manu-facturers, through seminars and their
Dependent factors (N=71)
Top reasons No. physicians
Quality of generics/ excipients 29Manufacturer’s reputation 21
Figure 3. Physician perceptions and therapeutic equivalence:
Yes
No
Depends
Generic Medicines

298
WHO Drug Information Vol 21, No. 4, 2007
representatives for educational material.While these two sources were importantto physicians as sources of credibleinformation, consultation with otherphysicians was also a major factor (Table2). Since a manufacturer’s reputationinfluences the confidence of profession-als, manufacturers of generic productsshould consider providing more evidencedemonstrating therapeutic equivalence oftheir products.
A positive role for pharmacovigilancePharmacovigilance can also have asignificant impact on increasing confi-dence in the therapeutic equivalence ingenerics. Through active pharmacovigi-lance, medicines (both generic andinnovator) associated with therapeuticfailure or adverse effects could be bettermonitored for potential problems. Cur-rently, the Ministry of Health relies on thespontaneous reporting method, Pharm-
Figure 4. Pharmacist perceptions of generic efficacy
Table 2. Physicians and pharmacists: credibility of information sources
Information source No. physicians No. pharmacists(Total = 242) * (Total = 36)*
Medical Journals 66 12Drug manufacturers/drug representatives 66 12Physicians 64 0Pharmacists 13 2Ministry of Health 11 4Internet 8 2
* Values in columns do not balance due to non-responders.
Greater efficacy
Same efficacy
Less efficacy
Depends
Dependent factors
Top reasons no. pharmacists
Patient factor/ type of illness 6Quality of generics/ excipients 5Manufacturer’s reputation 2
Generic Medicines

299
WHO Drug Information Vol 21, No. 4, 2007
watch, but the success of this methodrequires continuous oversight [10–12].Programmes to encourage physicians,pharmacists, other health professionalsand patients to submit reports of prob-lems with drug therapy may need to beconsidered. Confidence in the pro-gramme can be further reinforced byensuring that appropriate action is takenin response to all reports.
It is important to recognize that doubts ingeneric substitutability reflected in thissurvey are similar to those reported overten years ago [8] and therefore consid-eration should be given to more pro-active methods of pharmacovigilance.Action must be taken to ensure thatoffering patients cheaper drugs does nottranslate into inferior therapy, thus com-promising patient healthcare.
The authors wish to thank the Jamaica FairTrading Commission and The ConsumersAffairs Commission for their collaboration inthe surveys.
References
1. Sagardui-Villamor J, Lacalle Rodriguez-Labajo M and Casado-Buendia S. Substitutionof generic for brand medicines in primary care.Factors associated to refuse the change. AtenPrimaria 2005; 36(9):489-93.[Abstract only]
2. Tilson L McGowan B, Ryan M and Barry M.Generic drug utilisation on the GeneralMedical Services (GMS) scheme in 2001. IrMed J 2003; 96(6):176-9.
3. Kjonniksen I, Lindbaek M and Granas AG.Patients’ experiences with and attitudes togeneric substitution. Tidsskr Nor Laegeforen2005; 125(12):1682-4.[Abstract only]
4. Tilson L, Bennett K and Barry M. Thepotential impact of implementing a system ofgeneric substitution on the community drugschemes in Ireland. Eur J Health Econ. 2005;6(3):267-73.
5. Andersson K, Sonesson C, Petzold M,Carlsten A and Lonnroth K. What are theobstacles to generic substitution? An assess-ment of the behaviour of prescribers, patientsand pharmacies during the first year of genericsubstitution in Sweden. PharmacoepidemiolDrug Saf 2005; 14(5):341-8.
6. The Pharmacy act: The Pharmacy (Amend-ment) Regulation. The Jamaica GazetteSupplement, Proclamations, rules andregulations. 1993; Vol CXVI (29A).
7. Gleiter CH, Gundert-Remy U. Bio-inequivalence and drug toxicity. How great isthe problem and what can be done? Drug Saf1994; 11:1-6.
8. Glenda Anderson. Doctors reject ‘bad’generic drugs. Jamaica Gleaner, 22 June2003.
9. Fair Trading Commission. An Assessmentof Impediments to Competition in the Pharma-ceutical Sector in Jamaica. 2007.
10. Thiessard F, Roux E, Miremont-Salamé G,Fourrier-Réglat A, Haramburu F, Tubert-BitterP, Bégaud B. Trends in spontaneous adversedrug reaction reports to the French pharma-covigilance system (1986-2001). Drug Saf2005; 28(8):731-40.
11. Bracchi RC, Houghton J, Woods FJ,Thomas S, Smail SA, Routledge PA. Adistance-learning programme in pharmacovigi-lance linked to educational credits is associ-ated with improved reporting of suspectedadverse drug reactions via the UK yellow cardscheme. Br J Clin Pharmacol. 2005;60(2):221-3
12. van Grootheest K, Olsson S, Couper M,de Jong-van den Berg L. Pharmacists’ role inreporting adverse drug reactions in an interna-tional perspective. Pharmacoepidemiol DrugSaf. 2004; 13(7):457-64.
.
Generic Medicines

300
WHO Drug Information Vol 21, No. 4, 2007
When the patent of an innovator drug ex-pires, other manufacturers can make ge-neric versions. A generic drug contains thesame active ingredient as another product,but is marketed under a different name. InAustralia, the Phar-maceutical BenefitsAdvisory Commit-tee (PBAC) recog-nises the inter-changeability of dif-ferent brands con-taining the sameactive ingredient,providing thesebrands are provento be bioequivalent(1–4).
What isbioequivalence?Two products arebioequivalent whenthey produce suchsimilar plasma con-centrations of theactive ingredientthat their clinical ef-fects can be ex-pected to be thesame. In a standard bioequivalence testboth products are administered on sepa-rate occasions to healthy volunteers.Bioequivalence is then determined by com-
paring the peak plasma concentration(Cmax), time to achieve a maximal concen-tration (Tmax) and the extent of absorption(area under the concentration-time curve,AUC) of the products (Fig. 1).
These studies arewell suited to iden-tifying potentiallysignificant differ-ences in the deliv-ery characteristicsof the active sub-stance of differentproducts. Thesame bioequiva-lence principlesapply to new drugswhen different for-mulations of anactive ingredientare compared.B i o e q u i v a l e n tproducts aremarked with a su-perscript a or b inthe Schedule ofPharmaceuticalBenefits (5).
Is bioequivalence clinically important?Yes, only those products that have beenproven to be bioequivalent should be usedinterchangeably. On scientific grounds there
Frequently asked questions about generic medicines
In Australia, generic products must be bioequivalent to the innovator brand nameproduct, or the market leader, before they are approved. Australia has rigorous sci-entifically based evaluation procedures for generic medicines based on the interna-tionally accepted principle of bioequivalence. Under the Pharmaceutical BenefitsScheme, generic substitution is only permitted if two products are bioequivalent.Consumers should be encouraged to know and record the name of the active ingre-dient (International Nonproprietary Name) in the medicines they are receiving toavoid confusion between different brands of medicines. Healthcare professionalshave a key role in helping consumers understand any real or perceived differences(or lack thereof) between different brands of medicines. Prescribing generics helpsto contain health costs.
Figure 1. Bioequivalence analysis –a hypothetical bioequivalence study
Mean concentration-time curves for two brands of a drugafter single oral doses.
Time after dose (hours)
The original brand : generic medicine ratio for AUC is 0.99(90% CI 0.91 to 1.04) and for Cmax is 0.99% CI 0.92 to 1.07)
Cmax peak plasma concentrationAUC area under the concentration-time curveCI confidence interval
Reprinted from NPS News, 2006;44-3.
Generic medicineCmax = 662 ng/mLAUC = 3030 ng.h/mL
Original brandCmax = 660 ng/mLAUC = 3000 ng.h/mL
drug
con
cent
ratio
n (n
g/m
L)
Generic Medicines

301
WHO Drug Information Vol 21, No. 4, 2007
is no reason to be concerned about substi-tuting a generic product for a branded prod-uct that is flagged as being bioequivalent(5). Switching inequivalent products maylead to lower or higher blood concentrationsof a drug in a patient. This may increasethe risk of therapeutic failure or drug-related toxicity.
The precise extent to which inequivalencebetween two formulations will affect theclinical response depends on their phar-macological and/or therapeutic proper-ties. It depends specifically on which partof the drug concentration-effect curve isaffected by any concentration difference(4). For example, if the drug is usuallydosed close to the upper flat part of thedose response curve, then large changesin plasma concentration will result in onlysmall changes in therapeutic response oradverse effects. Theoretically, this is agreater concern for drugs with a narrowtherapeutic index, such as carbama-zepine, digoxin and sodium valproate.However, this is not as problematic asmay be predicted because patients takingthese drugs are generally closely moni-tored (either by measuring concentrationsor effects). For drugs with wider safetymargins, there should be no concernsabout a change in response when switch-ing from one bioequivalent brand toanother.
Which medicines should notbe substituted?Products that are not bioequivalentshould not be substituted for each other.For example, metoprolol is available asboth an intermediate release and amodified release tablet. These doseforms are not bioequivalent and shouldnot be substituted. There are two innova-tor brands of warfarin available in Aus-tralia. These have not been proven to bebioequivalent and so it is recommendedthat warfarin products should not besubstituted.
There has been considerable debateregarding the bioequivalence of drugswith a narrow therapeutic index, thatis, drugs for which a small change inblood drug concentration leads to signifi-cant change in therapeutic response ortoxicity (6).
These drugs generally display relativelyminor variability within a patient from dayto day but often display considerablevariability between patients (4,6). Takentogether this implies that the dose re-quired to achieve the same concentrationin the body, and therefore the samepharmacological effect, might be quitedifferent between different patients.However, within a patient the doserequirements are unlikely to vary greatlyover time and between doses while thepatient is clinically stable. Bioequivalenceprinciples and criteria equally apply tomedicines with a narrow safety margin(6,7).
Can people have a reaction to theexcipients in different products?Yes, although adverse reactions toexcipients are rare. Pharmaceuticalproducts contain the active pharmacologi-cal ingredient and a range of excipientsthat are designed to deliver the activedrug optimally in a reliable and reproduc-ible manner. These excipients can bediluents, binders, fillers, surfactants,lubricants, coatings and dyes. Excipientsare generally considered ‘inactive’, butthere is some evidence to suggest thatexcipients can have an impact on patienttolerability (8). The main risk is allergy orintolerance to a specific ingredient suchas lactose. The range of excipients usedpharmaceutically is small, and the typeused in individual products must becarefully chosen so that bioequivalence isachieved. The quality and safety of allexcipients are carefully reviewed by theTherapeutic Goods Administration (TGA)and excipients can only be used if theyare safe and non-toxic. It may not be
Generic Medicines

302
WHO Drug Information Vol 21, No. 4, 2007
possible to determine which ingredients ineither generic or branded products maycause an allergic reaction given thatformulations are likely to be similar.Patients who are aware of their allergiescan refer to the ingredients listed in theConsumer Medicines Information thataccompanies the product.
How can patients avoid being con-fused by the brand name of genericproducts?Patients should be encouraged to knowand record the name of the active ingredi-ent in the medicine they are taking ratherthan the product brand name. In this waya patient will understand that the samemedicine may be available in differentbrands. This has implications for the waymedicines are labelled. Ideally, the activeingredient in the product should bedisplayed with greater or equal promi-nence to the brand name on the packag-ing as recommended by the TGA in the‘Best practice guideline on prescriptionmedicine labelling’ (9).
Public hospitals are likely to only haveone or two brands of a medicine andthese are often generic products. Aspatients move in and out of hospital it islikely that generic substitution will occur toa greater extent. This reinforces the needfor patients to be aware of and carry a listof the name of the active ingredient orgeneric name of their medicines tomaintain effective management of theircondition (10).
When deciding whether to substitute ageneric product for a branded product,one must always consider the patient’sunderstanding of their medicines and therisk of medication misadventure. Discussthis with the patient and provide appropri-ate information (3). If there is potential forconfusion on the part of the patient andthere is a risk of dose duplication, thengeneric substitution may need to beavoided (independent of the drug in-volved) unless the patient or carer fully
understands the difference between thevarious brands of the same medicine.Clearly elderly patients, those withcognitive impairment and patients takingmultiple medicines for serious chronicillness are at greatest risk of misadven-ture from their drugs.
Do community pharmacists makea bigger profit if they substitute ageneric drug?Not necessarily. Under the Brand Pre-mium Policy of the PharmaceuticalBenefits Scheme (PBS), pharmacists areallowed to substitute a generic productwhen a branded product is prescribed,unless the prescriber directs otherwise.The PBS provides a subsidy up to theprice of the cheapest brand of a drug in aparticular therapeutic area. This oftencreates a price difference betweengeneric and branded products.
The pharmacist’s profit margin variesfrom drug to drug and product to product.In the past, cost savings for communitypharmacists arose when they purchasedbulk orders of generic drugs directly frommanufacturers. This issue was not uniqueto generic products because somemanufacturers of branded medicines alsosold their products directly to communitypharmacies under price-volume agree-ments. This is one of the many economicissues that community pharmacists haveto deal with in the efficient running of theirbusinesses. Recent PBS reforms havecreated different remuneration schedulesfor generic and branded medicinesresulting in these cost savings now beingretained within the PBS.
Can the bioavailability of bioequivalentproducts differ?For two drugs to be bioequivalent, the90% confidence intervals (90% CI) for theratio of each pharmacokinetic parameter,C
max and AUC, must lie within the range
0.8–1.25 (sometimes also expressed as80–125%).
Generic Medicines

303
WHO Drug Information Vol 21, No. 4, 2007
The 90% CI of 0.8–1.25 is a numericalindex and not a direct measure of thedifference in systemic concentrations ofthe active ingredient resulting fromadministration of the two products. It doesnot mean that the Cmax and AUC ratiosestimated for each formulation can varyby –20 to +25%. In reality, for a product tofit within these relatively tight confidencelimits the mean AUC and Cmax must bevery close, and any difference in bioavail-ability is certainly less than 10% (4).
ConclusionBioequivalence criteria used in Australiahave been defined and refined over manyyears and are internationally recognisedas the acceptable criteria for assessingbioequivalence (1). There is persuasiveevidence that the current internationallyaccepted limits and approaches tobioequivalence can accommodate allmedicines (6,7).
Only drugs that are marked as bioequiva-lent should be substituted for each other.Likewise, drugs that are not bioequivalentshould not be exchanged. To avoidconfusion, healthcare professionalsshould, where possible, reinforce thename of the active ingredient in themedicine, when prescribing, dispensingand administering medicines to patients.
This article appeared in The AustralianPrescriber, Number 30, 2007, pp. 41-43.Authors: Andrew J McLachlan, Professorof Pharmacy (Aged Care), Centre forEducation and Research on Ageing,Concord Repatriation General Hospitaland Faculty of Pharmacy, University ofSydney; Iqbal Ramzan, Professor ofPharmaceutics, Faculty of Pharmacy,University of Sydney; and Robert WMilne, Associate Professor, SansomInstitute, School of Pharmacy and Medi-cal Sciences, University of South Aus-tralia, Adelaide.
References
1. Birkett DJ. Generics – equal or not? AustPrescr 2003;26:85-7.
2. Hassali A, Stewart K, Kong D. Quality useof generic medicines [editorial]. Aust Prescr2004;27:80-1.
3. National Prescribing Service. Genericmedicines: same difference? NPS News2006;44. http://www.nps.org.au [cited 2007Mar 6]
4. Pearce GA, McLachlan AJ, Ramzan I.Bioequivalence: how, why, and what does itreally mean? J Pharm Pract Res 2004;34:195-200.
5. Department of Health and Ageing. Scheduleof Pharmaceutical Benefits. http://www.pbs.gov.au [cited 2007 Mar 6]
6. Benet LZ. Relevance of pharmacokineticsin narrow therapeutic index drugs. TransplantProc 1999;31:1642-4.
7. Christians U, First MR, Benet LZ. Recom-mendations for bioequivalence testing ofcyclosporine generics revisited. Ther DrugMonit 2000;22:330-45.
8. Uchegbu IF, Florence AT. Adverse drugevents related to dosage forms and deliverysystems. Drug Saf 1996;14:39-67.
9. Therapeutic Goods Administration. Bestpractice guideline on prescription medicinelabelling. 2005. http://www.tga.gov.au/pmeds/pmbestpractice.htm [cited 2007 Mar6]
10. Department of Health and Ageing. Guidingprinciples for medication management in thecommunity. 2006. http://www.health.gov.au/internet/wcms/publishing.nsf/content/apac-publications-guiding [cited 2007 Mar 6]
Generic Medicines

304
WHO Drug Information Vol 21, No. 4, 2007
Latest developmentsin ICH QualityThe International Conference on Harmo-nization of Technical Requirements forRegistration of Pharmaceuticals forHuman Use (ICH) recently convened aQuality Satellite Roundtable at Rockville,Maryland, USA. During the Satellite, theQuality Informal Implementation WorkingGroup (IWG) met from 27 to 28 Septem-ber 2007.
ICH-Q8(R1): PharmaceuticalDevelopment RevisitedThe ICH Steering Committee (SC)endorsed the development of a newguideline in October 2003 that woulddescribe the harmonized contents ofSection 3.2.P.2. “Pharmaceutical Devel-opment” within the Quality Module of theCommon Technical Document (CTD).
The final draft of the document Q8 –Pharmaceutical development (Part 1 -core guideline) was recommended foradoption to the regulatory bodies of theEuropean Union, Japan and USA inNovember 2005. The SC authorized thepreparation of Part 2, which is an adden-dum to ICH Q8 Pharmaceutical develop-ment and provides further clarification ofkey concepts outlined in the core guide-line. The SC released the document ICH-Q8(R1): Pharmaceutical Development forpublic consultation in Yokohama inNovember 2007. The structure of thedocument is outlined and arbitrarilyselected passages are excerpted, asfollows.
Approaches to pharmaceuticaldevelopmentThe theme of this section is described intable 1 overleaf, which illustrates somepotential contrasts between a minimal
Quality Assurance Update
The six members of ICH:
• European Commission of the European Union
• European Federation of Pharmaceutical Industries and Associations
• Japan Pharmaceutical Manufacturers Association
• Ministry of Health, Labor and Welfare, Japan
• Pharmaceutical research and Manufacturers of America
• US Food and Drug Administration
Non-voting observers serving as a link between ICH and non-ICH countriesand regions are:
• European Free Trade Association
• Health Canada
• International Federation of Pharmaceutical Manufacturers and Associations
• World Health Organization
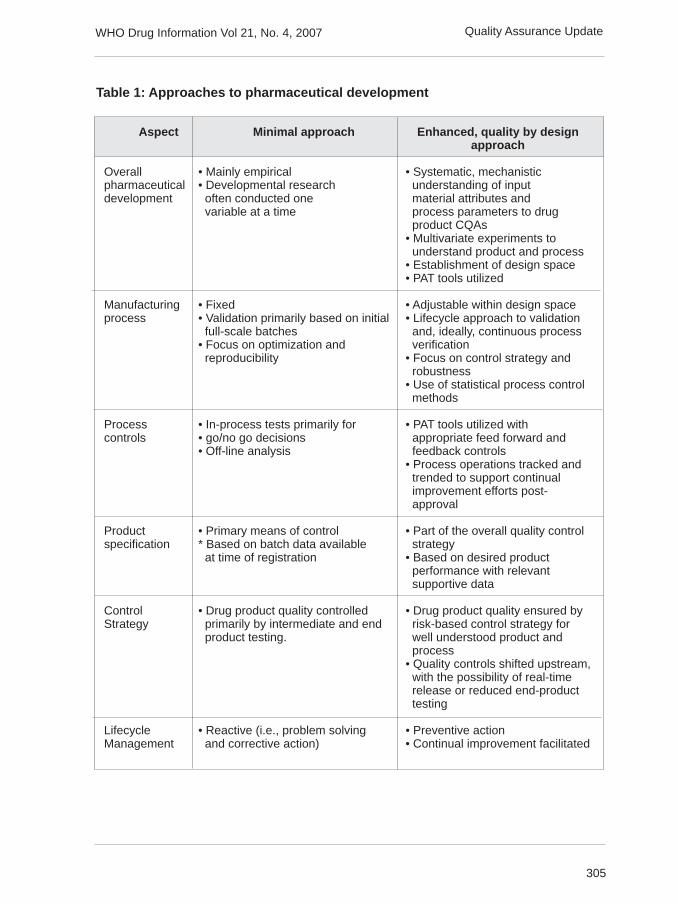
305
WHO Drug Information Vol 21, No. 4, 2007
Table 1: Approaches to pharmaceutical development
Aspect Minimal approach Enhanced, quality by designapproach
Overall • Mainly empirical • Systematic, mechanisticpharmaceutical • Developmental research understanding of inputdevelopment often conducted one material attributes and
variable at a time process parameters to drug product CQAs• Multivariate experiments to understand product and process• Establishment of design space• PAT tools utilized
Manufacturing • Fixed • Adjustable within design spaceprocess • Validation primarily based on initial • Lifecycle approach to validation
full-scale batches and, ideally, continuous process• Focus on optimization and verification reproducibility • Focus on control strategy and
robustness• Use of statistical process control methods
Process • In-process tests primarily for • PAT tools utilized withcontrols • go/no go decisions appropriate feed forward and
• Off-line analysis feedback controls• Process operations tracked and trended to support continual improvement efforts post- approval
Product • Primary means of control • Part of the overall quality controlspecification * Based on batch data available strategy
at time of registration • Based on desired product performance with relevant supportive data
Control • Drug product quality controlled • Drug product quality ensured byStrategy primarily by intermediate and end risk-based control strategy for
product testing. well understood product and process• Quality controls shifted upstream, with the possibility of real-time release or reduced end-product testing
Lifecycle • Reactive (i.e., problem solving • Preventive actionManagement and corrective action) • Continual improvement facilitated
Quality Assurance Update

306
WHO Drug Information Vol 21, No. 4, 2007
approach and an enhanced approachregarding different aspects of pharmaceu-tical development and lifecycle manage-ment. Current practices in the pharma-ceutical industry vary and typically liebetween these approaches.
The EWG concluded that pharmaceuticaldevelopment should include, at a mini-mum, the following elements:
• Defining the target product profile as itrelates to quality, safety and efficacy,considering e.g., the route of adminis-tration, dosage form, bioavailability,dosage, and stability
• Identifying critical quality attributes(CQAs) of the drug product, so thatthose product characteristics having animpact on product quality can be stud-ied and controlled
• Determining the quality attributes of thedrug substance, excipients etc., andselecting the type and amount ofexcipients to deliver drug product of thedesired quality
• Selecting an appropriate manufacturingprocess
• Identifying a control strategy.
A more systematic approach could facili-tate continual improvement and innova-tion throughout the product lifecycle (SeeICH Q10 Pharmaceutical Quality Sys-tem).
Elements of pharmaceuticaldevelopment
Target product profile. A target productprofile is a prospective summary of thequality characteristics of a new drugproduct that ideally will be achieved bythe time of submitting an application formarketing authorization.
Critical quality attributes. A criticalquality attribute (CQA) is a physical,chemical, biological, or microbiological
property or characteristic that should bewithin an appropriate limit, range, ordistribution to ensure the desired productquality. CQAs are generally associatedwith the drug substance, excipients,intermediates, and drug product.
Linking material attributes and proc-ess parameters to CQAs. Risk assess-ment tools can be used to identify andrank parameters (e.g., operational,equipment, input material) with potentialto have an impact on product qualitybased on prior knowledge and initialexperimental data.
Design space. The linkage between theprocess inputs (input variables andprocess parameters) and the criticalquality attributes can be described in thedesign space.
Selection of variables. All variables andtheir ranges within which consistentquality can be achieved should be se-lected for inclusion in the design space.
Defining and describing a design space ina submission. An example of a designspace presentation (excerpted from thedocument) is set out in Figure 1. Designspace is determined from the commonregion of successful operating ranges formultiple CQAs. The relations of twoCQAs, i.e., friability and dissolution, totwo process parameters of a granulationoperation are shown in Figures 1a and1b. Figure 1c shows the overlap of theseregions and the maximum ranges of thepotential design space.
Unit operation design space(s). Theapplicant can choose to establish inde-pendent design spaces for one or moreunit operations, or to establish a singledesign space that spans multiple opera-tions
Relationship of design space to scale andequipment. If the applicant wishes thedesign space to be applicable to multipleoperational scales, the design space
Quality Assurance Update
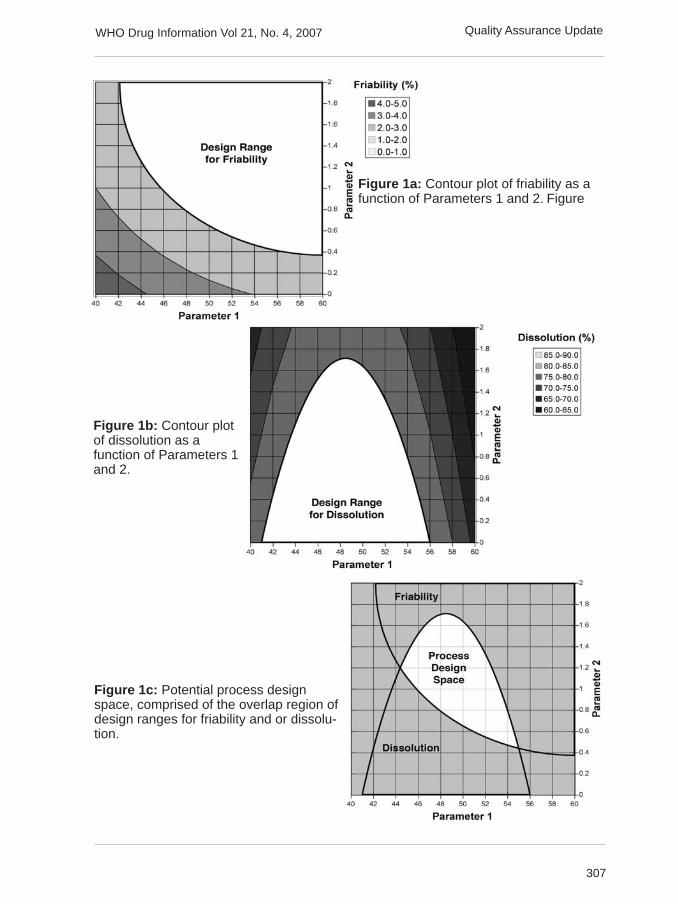
307
WHO Drug Information Vol 21, No. 4, 2007
Figure 1a: Contour plot of friability as afunction of Parameters 1 and 2. Figure
Figure 1c: Potential process designspace, comprised of the overlap region ofdesign ranges for friability and or dissolu-tion.
Figure 1b: Contour plotof dissolution as afunction of Parameters 1and 2.
Quality Assurance Update

308
WHO Drug Information Vol 21, No. 4, 2007
should be described in terms of relevantscale-independent parameters.
Design space versus proven acceptableranges. A combination of proven accept-able ranges does not constitute a designspace. (See Figures 1a and 1b.)
Design space and edge of failure. It canbe helpful to know where edges of failurecould be, or to determine potential failuremodes. However, it is not an essentialpart of establishing a design space.
Control strategy. The elements of thecontrol strategy discussed in Section P.2of the dossier should describe and justifyhow in-process controls and the controlsof input materials (drug substance andexcipients), container closure system,intermediates and end products contrib-ute to the final product quality. Thesecontrols should be based on product,formulation and process understandingand should include, at a minimum, controlof the critical parameters and attributes.
Product lifecycle management andcontinual improvement. Throughout theproduct lifecycle, companies have oppor-tunities to evaluate innovative approach-es to improve product quality (see ICHQ10).
Submission of pharmaceuticaldevelopment and related informationin Common Technical Document (CTD)formatPharmaceutical development informationis submitted in Section P.2 of the CTD.
Quality Risk Management and Productand Process Development. Risk analy-ses linking the design of the manufactur-ing process to product quality can beincluded in P.2.3.
Design Space. The product and manu-facturing process development sectionsof the application (P.2.1, P.2.2, and P.2.3)are appropriate places to summarize anddescribe product and process develop-
ment studies that provide the basis for thedesign space(s).
Control Strategy. The section of theapplication that includes the justificationof the drug product specification (P.5.6) isa good place to summarize the controlstrategy.
Drug Substance Related Information. Ifdrug substance CQAs have the potentialto affect the CQAs or manufacturingprocess of the drug product, somediscussion of drug substance CQAs canbe appropriate in the pharmaceuticaldevelopment section of the application(e.g., P.2.1).
What’s next for ICH Q8?Comments will be delivered to the ICHmeeting in fall 2008 to be considered forincorporation into the Step 2 draft. Step 2of the ICH process is reached when theICH Steering Committee agrees, basedon the report of the Expert WorkingCommittee, that there is sufficient scien-tific consensus on the technical issues forthe draft guideline or recommendation toproceed to the next stage of regulatoryconsultation or Step 3. The next step isStep 4 or adoption of the ICH guideline.Step 4 is reached when the ICH SteeringCommittee agrees, on the basis of thereport from the regulatory Rapporteur ofthe Expert Working Group, that there issufficient scientific consensus on thetechnical issues. Step 4 is followed byStep 5 or implementation.
The ICH Quality Satellite Roundtablemeeting held in Rockville, Washingtonconcluded that the principles of Q8, Q9,and Q10 are applicable to chemical andbiotech drug substances and drug prod-ucts. The Quality Informal ImplementationWorking Group (IWG) in Yokohamaproposed and the Steering Committeeagreed to the establishment of a commonformal IWG to ensure globally consistentimplementation of Q8, Q9 and Q10. Theformal IWG will start work in June 2008.
Quality Assurance Update

309
WHO Drug Information Vol 21, No. 4, 2007
International Pharmacopoeia
Consultation Documents
OXYTOCINUMOXYTOCIN
Draft proposal for the International Pharmocopoeia (September 2007).Please address any comments to Quality Assurance and Safety: Medi-cines, PSM, World Health Organization, 1211 Geneva 27, Switzerand.Fax +4122791 4730 or e-mail to [email protected]
C43
H66
N12
O12
S2
Relative molecular mass. 1007
Chemical name. L-Cysteinyl-L-tyrosyl-L-isoleucyl-L-glutamyl-L-asparaginyl-L-cysteinyl-L-prolyl-L-leucylglycinamide cyclic (1!6)-disulfide; CAS Reg. No. 50-56-6.
Other name. Alpha-hypophamine.
Description. White or almost white powder.
Solubility. Very soluble in water. It dissolves in dilute solutions of acetic acid and ofethanol.
Category. Uterine-stimulating (Oxytocic).
Storage. Oxytocin should be kept in an airtight container, protected from light, at atemperature of 2 °C to 8 °C. If the substance is sterile, store in a sterile, airtight,tamper-evident container.
Labelling. The designation on the container should state the oxytocin peptide content(C
43H
66N
12O
12S
2).
Additional information. Oxytocin is hygroscopic.
REQUIREMENTS
Definition. Oxytocin is a synthetic cyclic nonpeptide having the structure of thehormone produced by the posterior lobe of the pituitary gland that stimulates contrac-tion of the uterus and milk ejection in receptive mammals. It is available in the freeze-dried form as an acetate.

310
WHO Drug Information Vol 21, No. 4, 2007Consultation Document
Oxytocin contains not less than 93.0% and not more than 102.0% of C43H66N12O12S2,calculated with reference to the anhydrous and acetic acid-free substance.
By convention, for the purpose of labelling oxytocin preparations, 1 mg of oxytocinpeptide (C
43H
66N
12O
12S
2) is equivalent to 600 IU of biological activity.
Identity tests
Either tests A and B, or tests C and D may be applied.
A. Examine the chromatograms obtained in the assay. The principal peak in the chro-matogram obtained with the test solution is similar in retention time to the principalpeak in the chromatogram obtained with the reference solution.
B. Carry out the examination as described under 1.7 Spectrophotometry in the infraredregion. The infrared absorption spectrum is concordant with the spectrum obtainedfrom oxytocin RS or with the reference spectrum of oxytocin.
C. Carry out test C.1. or, where UV detection is not available, test C.2.
C.1 Carry out the test as described under 1.14.1 Thin-layer chromatography, usingsilica gel R6 as the coating substance and a mixture of 70 volumes of dichloromethaneR, 30 volumes of methanol R, 6 volumes of purified water and 1 volume of glacialacetic acid R as the mobile phase. Apply separately to the plate 10 ìl of each of 2solutions in methanol containing (A) 5 mg of the test substance per ml and (B) 5 mg ofoxytocin RS per ml. After removing the plate from the chromatographic chamber, allowit to dry exhaustively in a current of cool air. Examine the chromatogram in ultravioletlight (254 nm).
The principal spot obtained with solution A corresponds in position, appearance, andintensity with that obtained with solution B.
C.2 Carry out the test as described under 1.14.1 Thin-layer chromatography, usingsilica gel R5
as the coating substance and a mixture of 70 volumes of dichloromethane
R, 30 volumes of methanol R, 6 volumes of purified water and 1 volume of glacialacetic acid R as the mobile phase. Apply separately to the plate 10 ìl of each of 2solutions in methanol containing (A) 5 mg of the test substance per ml and (B) 5 mg ofoxytocin RS per ml. After removing the plate from the chromatographic chamber, allowit to dry exhaustively in a current of cool air. Spray with ninhydrin. Heat the plate for afew minutes at 120 ˚C. Examine the chromatogram in daylight.
The principal spot obtained with solution A corresponds in position, appearance, andintensity with that obtained with solution B.
D. The absorption spectrum of a 0.30 mg/ml solution, when observed between 240nm and 330 nm, exhibits a maximum at about 275 nm; the specific absorbance(A
1cm 1%) is 14 to 16, calculated with reference to the anhydrous and acetic acid-free
substance.
Specific optical rotation. Use a 5.0 mg/ml solution and calculate with reference tothe anhydrous and acetic acid-free substance; [á]
D 20 ˚C = -24.0˚ to -28.0˚.
pH value. pH of a 20 mg/ml solution in carbon-dioxide-free water R, 3.0-6.0.

311
WHO Drug Information Vol 21, No. 4, 2007
Water. Determine as described under 2.8 Determination of water by the Karl Fischermethod, Method A, using about 0.10 g of the substance; the water content is not morethan 50 mg/g.
Acetic acid content. Determine by 1.14.4 High performance liquid chromatography,using a stainless steel column (25 cm x 4.6 mm) packed with octadecylsilyl silica gelfor chromatography R (5 ìm).
Use the following conditions for gradient elution:
Mobile phase A: Dilute 0.7 ml of phosphoric acid (~1440 g/l) TS with 900 ml purifiedwater; adjust the pH to 3.0 with sodium hydroxide (~200 g/l) TS and dilute to 1000 mlwith purified water.
Mobile phase B: Methanol R.
Time Mobile phase A Mobile phase B Comments(min) (% v/v) (% v/v)
0 – 5 95 5 Isocratic 5 – 10 95 to 50 5 to 50 Linear gradient10 – 20 50 50 Isocratic20 – 22 50 to 95 50 to 5 Linear gradient22 – 30 95 5 Isocratic re-equilibration
Prepare the following solutions:
For solution (1), dissolve 15.0 mg of the substance to be examined in a mixture of 5volumes of mobile phase B and 95 volumes of mobile phase A and dilute to 10.0 mlwith the same mixture of mobile phases.
For solution (2), prepare a 0.10 g/l solution of glacial acetic acid R in a mixture of 5volumes of mobile phase B and 95 volumes of mobile phase A.
Operate with a flow rate of 1.2 ml per minute. As a detector use an ultraviolet spectro-photometer set at a wavelength of 210 nm.
Inject alternatively 10 µl each of solutions (1) and (2). In the chromatograms obtained,the peak corresponding to acetic acid has a retention time of 3-4 min. The baselinepresents a steep rise after the start of the linear gradient, which corresponds to theelution of oxytocin from the column. Calculate the acetic acid content; not less than 60mg/g and not more than 100 mg/g.
Related substances. Carry out the test as described under 1.14.4 High performanceliquid chromatography, using a stainless steel column (25 cm x 4.6 mm) packed withoctadecylsilyl silica gel for chromatography R (5 ìm).
Use the following conditions for gradient elution:
Mobile phase A: 15 volumes of acetonitrile R, 15 volumes of phosphate buffer and 70volumes of purified water.
Consultation Document

312
WHO Drug Information Vol 21, No. 4, 2007
Mobile phase B: 70 volumes of acetonitrile R, 15 volumes of phosphate buffer and 15volumes of purified water.
Prepare the phosphate buffer by dissolving 31.2 g of sodium dihydrogen phosphatedihydrate R in 1000 ml of purified water.
Time Mobile phase A Mobile phase B Comments(min) (% v/v) (% v/v)
0 – 5 100 0 Isocratic5 – 20 100 to 94 0 to 6 Linear gradient20 – 50 94 to 60 6 to 40 Linear gradient50 – 51 60 to 100 40 to 0 Linear gradient51-65 100 0 Isocratic re-equilibration
Prepare the following solutions using mobile phase A as diluent. For solution (1) use0.50 mg of the test substance per ml. For solution (2) dilute a suitable volume ofsolution (1) to obtain a concentration equivalent to 5.0 ìg of oxytocin per ml.
For the system suitability test: prepare solution (3) using 3 ml of solution (1) and 2 mlof sulfuric acid (10 g/l), heat carefully in a boiling water-bath for 20 minutes.
Operate with a flow rate of 1.0 ml per minute. As a detector use an ultraviolet spectro-photometer set at a wavelength of 220 nm.
Maintain the column temperature at 40 ˚C.
Inject 50 ìl of solution (3). The test is not valid unless the resolution between the peakdue to oxytocin (retention time about 25 minutes) and the peak with a relative retentionof about 0.9 is not less than 1.4.
Inject alternatively 50 µl each of solutions (1) and (2).
In the chromatogram obtained with solution (1), the area of any peak, other than theprincipal peak, is not greater than 1.5 times the area of the principal peak obtainedwith solution (2) (1.5%). The sum of the areas of all peaks, other than the principalpeak, is not greater than 5.0 times the area of the principal peak obtained with solution(2) (5%). Disregard any peak with an area less than 0.1 times the area of the principalpeak in the chromatogram obtained with solution (2) (0.1%).
Assay
Either method A or method B may be applied.
A. Determine by High performance liquid chromatography as described in the test forrelated substances with the following modifications.
Prepare solution (2) as follows. Dissolve the contents of a vial of oxytocin RS in mobilephase A to obtain a concentration of 0.50 mg/ml.
Inject alternatively 50 µl each of solutions (1) and (2).
Calculate the content of oxytocin (C43
H66
N12
O12
S2) from the declared content of
C43
H66
N12
O12
S2 in oxytocin RS.
Consultation Document

313
WHO Drug Information Vol 21, No. 4, 2007
B. Dissolve about 30 mg, accurately weighed, in sufficient purified water to produce100 ml. Measure the absorbance of this solution in a 1-cm layer at the maximum atabout 275 nm, and calculate the content of C
43H
66N
12O
12S
2, using the absorptivity
value of 1.49 (A1cm
1% = 14.9).
Additional requirement for Oxytocin for parenteral useComplies with the monograph “Parenteral preparations”.
Bacterial endotoxins. Carry out the test as described under 3.4 Test for bacterialendotoxins; contains not more than 300 IU of endotoxin RS per mg.
OXYTOCINUM INJECTIOOXYTOCIN INJECTION
Draft proposal for the International Pharmocopoeia (October 2007).Please address any comments to Quality Assurance and Safety: Medi-cines, Medicines Policy and Standards, World Health Organization, 1211Geneva 27, Switzerand. Fax +4122791 4730 or e-mail [email protected]
Description. A clear, colourless liquid.
Category. Uterine-stimulating (Oxytocic).
Storage. Oxytocin injection should be kept protected from light and stored at a tem-perature between 2 °C and 8 °C.
Labelling. The designation on the container should state the content in IU per ml andthe oxytocin peptide (C43H66N12O12S2) content in mg per ml. It should also state animalsource if naturally derived, or state that it is synthetic.
Additional information. Strength in the current WHO Model list of essential medi-cines: 10 IU per ml in 1-ml ampoule.
Oxytocin injection is normally intended for intravenous or intramuscular administration.
REQUIREMENTS
Oxytocin injection complies with the monograph for “Parenteral preparations”.
Definition. Oxytocin injection is a sterile solution of Oxytocin in a suitable diluent.Oxytocin injection contains not less than 90.0% and not more than 110.0% of theamount of the peptide C43H66N12O12S2 stated on the label.
Identity tests
Either test A or test B may be applied.
A. Examine the chromatograms obtained in the assay. The principal peak in thechromatogram obtained with the test solution is similar in retention time to the principalpeak in the chromatogram obtained with the reference solution.
B. Carry out the test as described under 1.14.1 Thin-layer chromatography, usingsilica gel R5 as the coating substance and a mixture of 70 volumes of dichloromethane
Consultation Document

314
WHO Drug Information Vol 21, No. 4, 2007
R, 30 volumes of methanol R, 6 volumes of purified water and 1 volume of glacialacetic acid R as the mobile phase. Apply separately to the plate 20 ìl of each of thefollowing two solutions. For solution (A) evaporate 10.0 ml of oxytocin injection todryness at 30 ˚C under reduced pressure (not exceeding 0.6 kPa or 5 mm of mercury)and dissolve the residue in 1.0 ml of methanol R. Prepare solution (B) in methanol Rcontaining 165.0 µg/ml of oxytocin RS. After removing the plate from the chromato-graphic chamber, allow it to dry exhaustively in a current of cool air. Stain the platewith iodine vapour and examine in daylight.
The principal spot obtained with solution A corresponds in position, appearance, andintensity with that obtained with solution B.
pH value. pH of the injection, 3.0–5.0.
Assay
Determine by 1.14.4 High performance liquid chromatography, using a stainless steelcolumn (25 cm x 4.6 mm) packed with octadecylsilyl silica gel for chromatography R (5ìm).
Use the following conditions for gradient elution:
Mobile phase A: 15 volumes of acetonitrile R, 15 volumes of phosphate buffer and 70volumes of purified water.
Mobile phase B: 70 volumes of acetonitrile R, 15 volumes of phosphate buffer and 15volumes of purified water.
Prepare the phosphate buffer by dissolving 31.2 g of sodium dihydrogen phosphatedihydrate R in 1000 ml of purified water.
Time Mobile phase A Mobile phase B Comments(min) (% v/v) (% v/v)
0 – 5 100 0 Isocratic5 – 20 100 to 94 0 to 6 Linear gradient20 – 50 94 to 60 6 to 40 Linear gradient50 – 51 60 to 100 40 to 0 Linear gradient51 - 65 100 0 Isocratic re-equilibration
Use the following solutions.
For solution (1) use undiluted injection. For solution (2) dissolve the contents of vial ofoxytocin RS in mobile phase A to obtain a concentration of 16.7 µg/ml.
Operate with a flow rate of 1.0 ml per minute. As a detector use an ultraviolet spectro-photometer set at a wavelength of 220 nm.
Maintain the column temperature at 40 ˚C.
Inject alternatively 50 µl each of solutions (1) and (2).
Calculate the content of oxytocin (C43
H66
N12
O12
S2) from the declared content of
C43
H66
N12
O12
S2 in oxytocin RS.
Consultation Document

315
WHO Drug Information Vol 21, No. 4, 2007
Recent Publications,Information and EventsGMP for herbal medicinesThe safety and quality of herbal materialsand finished herbal products have be-come a major concern for health authori-ties. Requirements and methods forquality control of finished herbal productsare far more complex than for chemicaldrugs. The quality of the finished herbalproduct is also influenced by the quality ofthe raw materials used. The manufactur-ing process is one of the key steps wherequality control is measured and goodmanufacturing process (GMP) is the mostefficient tool.
Since GMP control for herbal medicinesneeds to meet technical requirements forboth herbal and chemical drugs, theWHO Guideline on good manufacturingpractices (GMP) for herbal medicinescombines the two sets of guidelines inone publication.
Reference: World Health Organization. WHOGuideline on good manufacturing practices(GMP) for herbal medicines. Available from:[email protected] or http://www.who.int/medicines
Quality of antiretroviralsin African countriesIn 2006, almost two-thirds of personsinfected with HIV were living in Sub-Saharan Africa. Provision of antiretroviraltreatment by both public and privatesector facilities has increased tenfoldbetween December 2003 and June 2006.
The first requirement of any treatmentprogramme is the availability of antiretro-virals of acceptable quality, safety andefficacy. Antiretrovirals manufactured
below established standards of qualitycan lead to therapeutic failure, develop-ment of drug resistance and toxic oradverse reactions.
A survey of the quality of antiretroviralmedicines circulating in selected Africancountries was carried out by WHO incollaboration with the national drugregulatory authorities of Cameroon, theDemocratic Republic of Congo, Kenya,Nigeria, United Republic of Tanzania,Uganda and Zambia.
Reference: World health Organization. Asurvey of the quality of antiretroviral medicinescirculating in selected African countries.Available from [email protected] orhttp:www.who.int/medicines
The Health JourneyThe Health Journey is a simple anduseful participatory methodology forunderstanding the experiences of peopleliving with HIV in trying to access and usehealth and other support services. Itprovides a starting point for planning andmonitoring community engagement andprovision of community-centred healthand support services. It is intended forindividuals, groups and organizationsworking in HIV care and support, but itcan easily be adapted to other issuesboth within and beyond the HIV field.
Outcomes from the methodology havealready contributed to improved coordina-tion of community support and healthcare for people with HIV and to reductionof stigma and discrimination in a varietyof settings, within the Caribbean, Zambia,Uganda, Myanmar and China.

316
WHO Drug Information Vol 21, No. 4, 2007Recent Publications, Information and Events
Part 1 of the health journey explains whata health journey is and who can makeuse of the methodology;
Part 2 explains: how to set up and usethe methodology; and
Part 3 provides: five different examples ofhealth journey workshops, what impactsresulted from them and a list of usefulresources.
Reference: International HIV/AIDS Alliance.The Health Journey - Understanding thedimensions of care and treatment for peoplewith HIV: a community-centred methodology.Printed copies are available from [email protected] and http://www.aidsalliance.org
Human rights guidelines forpharmaceutical companiesThe UN Special Rapporteur on the rightof everyone to the enjoyment of thehighest attainable standard of physicaland mental health, has launched forpublic consultation draft Human RightsGuidelines for Pharmaceutical Compa-nies in relation to Access to Medicines.
Access to medicines is a central featureof the right to the highest attainablestandard of health. States have primaryresponsibility for enhancing access tomedicines, as set out in the expert’sreport to the UN General Assembly lastyear (13 September 2006, A/61/338). TheSpecial Rapporteur routinely questionsGovernments about their national medi-cines policies and implementation plans.
Pharmaceutical companies have aprofound impact - both positive and
negative - on Governments’ ability torealise the right to the highest attainablestandard of health. It is time to identifywhat pharmaceutical companies shoulddo to help realize the human right tomedicine.
Reference: Draft Guidelines, and otherinitiatives of the Special Rapporteur atwww2.essex.ac.uk/human_rights_ centre/rth/ and http://www.ohchr.org/english/issues/health/right/index.htm
ARV price report from WHOThe new summary report by the WHO/AMDS Global Price Reporting Mecha-nism (GPRM) confirms that median pricesof the most commonly prescribed antiret-roviral medicines (ARVs) in fixed dosecombination (stavudine 30 mg + lamivu-dine 150 mg + nevirapine 200 mg)continued to decline during the periodfrom January to June 2007. They are nowavailable for less than US$ 100 perpatient per year (pppy) with a medianprice of US$ 77 in low-income countriesand US$ 99 in middle-income countries.
The same trend is being observed withARVs for children. However, the costs oforal solutions for infants/young childrenstill remain high. For instance, the mediancost of the most widely prescribed regi-men as oral solution (zidovudine 10 mg/ml + lamivudine 10 mg/ml + nevirapine 10mg/ml) for a five kg infant is now US$ 174(pppy) in low-income countries and US$235 (pppy) in middle-income countries.
Reference: World Health Organization. AMDSsummary Report at http://www.who.int/hiv/amds/GPRMsummaryJuly2007.pdf