tropospheric ozone chemistry david plummer presented at the gcc summer school montreal, august 7-13,...
TRANSCRIPT

Tropospheric Ozone Chemistry
David Plummerpresented at the GCC Summer School
Montreal, August 7-13, 2003
Outline:
- Solar radiation and chemistry
- Tropospheric ozone production
- Methane oxidation cycle
- Nitrogen species
- A look at global tropospheric ozone
- Oxidizing capacity of the troposphere

Ozone in the atmosphere
• 90% of total column O3 is found in the stratosphere
Timeseries of ozone profiles over Edmonton for 2002. From World Ozone Data Centre (www.woudc.org)

Solar radiation and chemistry• the reaction that produces ozone in the
atmosphere:
O + O2 + M O3 + M
• difference between stratospheric and tropospheric ozone generation is in the source of atomic O
• for solar radiation with a wavelength of less than 242 nm:
O2 + hv O + O
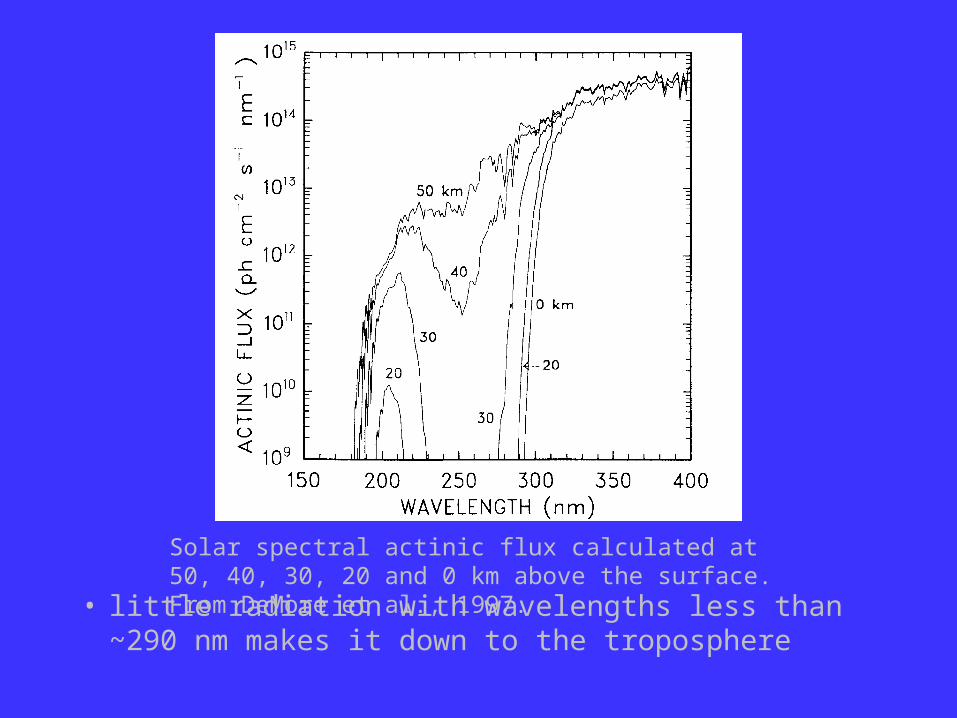
• little radiation with wavelengths less than ~290 nm makes it down to the troposphere
Solar spectral actinic flux calculated at 50, 40, 30, 20 and 0 km above the surface. From DeMore et al., 1997.

• photochemical production of O3 in troposphere tied to NOx (NO + NO2)
• for wavelengths less than 424 nm:
NO2 + hv NO + O
• but NO will react with O3
NO + O3 NO2
• cycling has no net effect on ozone

O3-NO-NO2 photochemical steady state
• consider the two reactions just seen
NO2 + hv (+O2) NO + O3 J1
NO + O3 NO2 K1
• ignoring other reactions, during daylight this forms a fast cycle in steady-state
d[NO2]/dt = Prod - Loss = 0
K1[NO][O3] = J1[NO2]
[NO]/[NO2] = J1/K1[O3]
• partioning of NOx between NO and NO2 has important implications for removal of NOx from the atmosphere

• presence of peroxy radicals, from the oxidation of hydrocarbons, disturbs O3-NO-NO2 cycle
NO + HO2· NO2 + OH·
NO + RO2· NO2 + RO·
– leads to net production of ozone

• produced from ozone photolysis– for radiation with wavelengths less than 320 nm:
O3 + hv O(1D) + O2
followed by
O(1D) + M O(3P) + M (+O2O3) (~90%)
O(1D) + H2O 2 OH· (~10%)
• OH initiates the atmospheric oxidation of a wide range of compounds in the atmosphere– referred to as ‘detergent of the atmosphere’
– typical concentrations near the surface ~106 - 107cm-3
– very reactive, effectively recycled
The Hydroxyl Radical

Oxidation of CO - production of ozone
CO + OH· CO2 + H·
H· + O2 + M HO2· + M
NO + HO2· NO2 + OH·
NO2 + hv NO + O
O + O2 + M O3
CO + 2 O2 + hv CO2 + O3

What breaks the cycle?
• cycle terminated byOH· + NO2 HNO3
HO2· + HO2· H2O2
• both HNO3 and H2O2 will photolyze or react with OH to, in effect, reverse these pathways– but reactions are slow (lifetime of several days)
– both are very soluble - though H2O2 less-so• washout by precipitation
• dry deposition
– in PBL they are effectively a loss
– situation is more complicated in the upper troposphere• no dry deposition, limited wet removal

Methane Oxidation Cycle
• CH4 is simplest alkane species– features of oxidation cycle common to other organic
compounds
• long photochemical lifetime– fairly evenly distributed throughout troposphere
– concentrations ~1.8ppmv
• reactions form ‘bedrock’ of the chemistry in the background troposphere

CH4 + OH· CH3· + H2O
CH3· + O2 + M CH3O2· + M
CH3O2· + NO CH3O· + NO2
CH3O· +O2 HCHO + HO2·
HO2· + NO OH· + NO2
2{NO2 + hv (+O2) NO + O3}
CH4 + 4 O2 + 2 hv HCHO + 2O3 + H2O
• HCHO will also undergo further reactionHCHO + hv H2 + CO
H· + HCO
HCHO + OH HCO + H2O
HCO + O2 HO2· + CO
H· + O2 HO2·

Cycle limiting reactions
OH· + NO2 HNO3
HO2· + HO2· H2O2
but also
HO2· + CH3O2· CH3OOH + O2
• methyl hydroperoxide (CH3OOH) – can photolyze or react with OH with a lifetime of ~ 2
days• return radicals to system
• important source of radicals in upper tropical troposphere
– moderately soluble and can be removed from atmosphere by wet or dry deposition
• loss of radicals

Conceptually
• photolysis of ozone most significant source of OH• atmospheric oxidation of hydrocarbons initiated
by OH radical– production of peroxy radicals (HO2, RO2) which
interact with O3-NO-NO2 cycle to photo-chemically produce ozone
– produce carbonyl compounds (aldehydes and ketones) which undergo further oxidation
– recycling of OH
• termination by formation of nitric acid (OH + NO2 HNO3) or peroxides (H2O2, ROOH)

Nitrogen species
• NOx (NO + NO2) plays a critical role in the atmospheric oxidation of hydrocarbons
• short chemical lifetime – from ~ 6 hours in PBL to several days to a week in the
upper troposphere
• large variations in concentration– from 10s ppbv in urban areas to 10s pptv in remote
regions (UT and remote MBL)
• gives rise to different chemical regimes

Regional Ozone perspective - O3 production
• More accurate to talk of NOx/VOC ratio • VOC - volatile organic carbon
• High NOx/VOC environments
– OH reaction with NO2 dominates
– NO-NO2 cycling inefficient compared with NOx loss
– only found in urban areas
• Low NOx/VOC environments– high peroxy radical concentrations
– peroxy radical self-reactions become important sink for radicals
• production of H2O2 and ROOH

Global perspective
• NOx concentrations almost always low enough that ozone production is NOx limited
• globally NOx concentrations control whether local chemistry creates or destroys ozone
• for [NOx] less than ~20 pptv, chemistry results in net ozone destruction– no NOx to turn-over the NO-NO2 cycle
O3+ hv O(1D) + O2
O(1D) + H2O 2 OH·
– also
HO2· + O3 OH· + 2 O2
– particularly important in tropical marine boundary layer

Other nitrogen species• Peroxyacyl nitrates (PANs)
– most important being peroxyacetyl nitrate
• CH3C(O)OONO2
– formed from oxidation of acetaldehyde
CH3CHO + OH· (+ O2) CH3C(O)O2 + H2O
CH3C(O)O2 + NO2 + M CH3C(O)O2NO2 + M
– decomposition is strongly temperature dependent• from 30 minutes at 298K near the surface to several months
under upper tropospheric conditions
• NOx exported from boundary layer to remote troposphere in the form of PAN
– observations show PAN is dominant NOy compound in northern hemisphere spring troposphere
• insoluble

• N2O5
– formed byNO2 + O3 NO3 + O2
NO2 + NO3 N2O5
– most important is what happens to N2O5
N2O5 + H2O(s) 2 HNO3
– during daylight fast photolysis of NO3 limits production of N2O5:
NO3 + hv NO2 + O
Other nitrogen species

– especially important NOx sink at higher latitudes and in winter - particularly northern hemisphere
• OH concentrations much lower
The calculated reduction in NOx and O3 amounts in the MOZART model with the inclusion of N2O5 hydrolysis. From Tie et al. 2001.

NOx Sources
Estimates of annual global NOx emissions for the early 1990s. Units of Tg-N/year.
• Biomass burning includes savannah burning, tropical deforestation, temperate wildfires and agricultural waste burning
• Soil emission– enhanced by application of fertilizers– largest uncertainty is in estimates of canopy transmission
• Lightning– models use ~5.0 Tg-N/yr– scaling up from observations suggest 20 Tg-N/yr
Technological 23 - 27Aircraft 0.5Biomass burning 7.0 - 8.0Soils 5.0 - 12.0Lightning 3.0 - 20.0

An example of gridded NOx emissions

Impacts of NOx emission
• by mass, most NOx is emitted at the surface
• chemical impacts of NOx very non-linear
– limited impact in the continental PBL• high OH and high NO2/NO ratio near surface result in
a short photo-chemical lifetime
• NOx concentrations are already substantial
– per molecule, impact of NOx much greater in free troposphere
• venting to the free troposphere important• emissions that occur in free troposphere
– aircraft, lightning

Global tropospheric ozone
• Remote northern stations– spring-time maximum
• nearer to industrial emissions– broader maximum stretching through summer
Seasonal cycle of O3 concentrations at different pressure levels, derived from ozonesonde data at eight different stations in the northern hemisphere. From Logan, J. Geophys. Res., 16115-16149, 1999.

O3 at the surface
• Surface sites in industrialized regions show an even more pronounced summer-time peak
Seasonal cycle of O3 concentrations at the surface for different rural locations in the United States.
From Logan, J. Geophys. Res., 16115-16149, 1999.

Global distribution
• constructed from surface observations, ozonesondes and a bit of intuition– note very low concentrations over tropical Pacific ocean
Spatial distribution of climatological O3 concentrations at 1000hPa.
From Logan, J. Geophys. Res., 16115-16149, 1999.
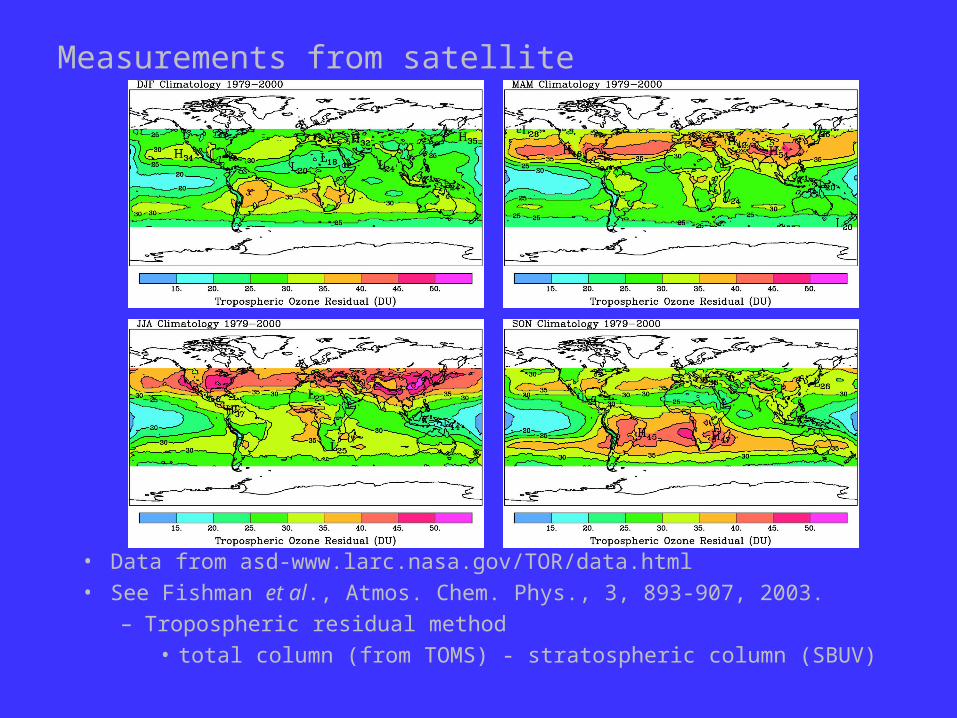
Measurements from satellite
• Data from asd-www.larc.nasa.gov/TOR/data.html• See Fishman et al., Atmos. Chem. Phys., 3, 893-907, 2003.
– Tropospheric residual method• total column (from TOMS) - stratospheric column (SBUV)

Tropospheric ozone budget
• derived from models– a typical budget for present-day conditions:
Strat-trop exchange 565 Tg/yr
Photochemical prod. 3314 Tg/yr
Photochemical dest. 3174 Tg/yr
Deposition 705 Tg/yr
Tropospheric burden 347 Tg
From Lelieveld and Dentener, J. Geophys. Res., 3531-3551, 105, 2000

Range of model predictions• all global models compared to available
measurements– comparisons becoming more sophisticated
– all show believable ozone
• budgets show large spread in individual termsStrat-trop exchange 300 - 1100 Tg/yr
Photochemical prod. 3000 - 5000 Tg/yr
Photochemical dest. 2500 - 4300 Tg/yr
Net chemistry -500 +600 Tg/yr
Deposition 500 - 1200 Tg/yr
Tropospheric burden 280 - 400 Tg
Adopted from von Kuhlmann et al., J. Geophys. Res., in press, 2003.

Future concerns
• How much have emissions of precursors perturbed ozone already?– Ozone is reactive
• no ice-core records
– some re-constructed records• Montsouris measurements suggested surface O3 was
~10 ppbv
– other information from model simulations• emissions, particularly biomass burning, hard to
quantify• suggest tropospheric ozone burden has increased
between 25 and 60% since pre-industrial

The more recent past
• Statistically significant negative trends of 1-2% per year found at several stations in Canada for 1980-1993 (Tarasick et al., Geophys. Res. Lett., 409-412, 22, 1995)
• trends at most other stations in NH ambiguous
Monthly averaged O3 concentration between 630 and 400 hPa from 9 ozonesonde stations located between 36 and 59N. From Logan et al. J. Geophys. Res., 104, 26373-26399, 1999.

IPCC OxComp simulations for 2100• Emissions for year 2100 were a bit of a ‘worst case’
scenario
CH4 = 4.3 ppmv; NOx = 110 Tg-N/yr (32.5)
CO = 2500 Tg/yr (1050); VOC = 350 Tg/yr (150)
• mid-latitude O3 increases by 20-30 ppbv at the surface
– puts background O3 in 60-70 ppbv range
• these models did not include impacts of global warming
– increased H2O vapour
– temperature effects on reaction rates
• increasingly coupled models– inclusion of biosphere-atmosphere interactions
– lightning

Stability of global OH
• OH originates with O3
– very reactive and very short-lived
– recycling critically important
• OH is responsible for initiating atmospheric oxidation of hydrocarbons– CH4 lifetime of ~10 years
• are changes in chemical composition of the troposphere affecting average OH?

Information from methyl chloroform
• CH3CCl3 used as solvent by industry
– atmospheric lifetime of 5-6 years• main loss by reaction with OH
• some entered stratosphere and enhanced Cl levels
– banned under Montreal protocol• use was to stop in 1996 in developed countries
– assuming one knows the sources of MCF, it is possible to calculate an average global OH by fitting to observed decay

Observed MCF concentrations at Barbados. Vertical bars represent the monthly standard deviations. Different colour symbols represent measurements made as part of different networks. See Prinn et al., J. Geophys. Res., 105, 17751-17792, 2000.

• Minor changes in the time profile of emissions can give constant OH– banking of MCF in early 1990s
– release in late 1990s
– aircraft observations of plumes of MCF in 2000 over Europe
Global average OH determined from fitting to observed MCF concentrations over 3 and 5 year periods and as a second-order polynomial. From Krol and Lelieveld, J. Geophys. Res., in press, 2002.