thomas a. edison - core.ac.uk · (ii) !predicted mirnas do not functionally target the 3!utr of...
TRANSCRIPT

Catholic University of Leuven Group Biomedical Sciences Faculty of Medicine Department of Cardiovascular Sciences Division of Cardiology
CCGGMMPP SSIIGGNNAALL TTRRAANNSSDDUUCCTTIIOONN IINN HHYYPPEERRTTRROOPPHHIICC AANNDD TTOOXXIICC CCAARRDDIIOOMMYYOOPPAATTHHYY
Sara Vandenwijngaert
Jury: Promoter: Prof. Dr. Stefan Janssens Chair: Prof. Dr. Paul Herijgers Secretary: Prof. Dr. Johan Van Cleemput Jurymembers: Prof. Dr. Michaela Kuhn (University of Würzburg)
Prof. Dr. Guido De Meyer (University of Antwerp) Prof. Dr. Johan Van Cleemput (KU Leuven)
Prof. Dr. Yicheng Ni (KU Leuven) Leuven, 25.03.2013 Doctoral thesis in Biomedical Sciences


OOUURR GGRREEAATTEESSTT WWEEAAKKNNEESSSS LLIIEESS IINN GGIIVVIINNGG UUPP
TTHHEE MMOOSSTT CCEERRTTAAIINN WWAAYY TTOO SSUUCCCCEEEEDD IISS AALLWWAAYYSS TTOO TTRRYY JJUUSSTT OONNEE MMOORREE TTIIMMEE
Thomas A. Edison


i
TABLE OF CONTENTS
DANKWOORD – PREFACE LIST OF ABBREVIATIONS !
CHAPTER 1: INTRODUCTION .................................................................................................... 1!
1.1! Tackling heart failure .............................................................................................................. 1!
1.1.1 Determinants of contractile function ...................................................................................... 1!
1.1.2 Aetiology of heart failure ........................................................................................................ 2!
1.1.3 Chronic pressure overload-induced heart failure .................................................................. 4!
(i)! Aetiology ............................................................................................................................... 4!
(ii) ! Pathophysiology ................................................................................................................... 5!
1.1.4 Anthracycline-induced heart failure ..................................................................................... 25!
(i) ! Clinical application of anthracyclines .................................................................................. 25!
(ii) ! Acute and chronic cardiotoxicity and risk factors ................................................................ 26!
(iii) !Pathophysiology ................................................................................................................. 27!
1.2! Cyclic GMP signalling in the cardiovascular system ........................................................ 36!
1.2.1 Synthesis by guanylate cyclases ........................................................................................ 36!
(i) ! Nitric oxide-mediated biosynthesis of cGMP ...................................................................... 36!
(ii) ! Natriuretic peptide-mediated biosynthesis of cGMP ........................................................... 38!
1.2.2 Activation of effector molecules ........................................................................................... 39!
(i) ! Cyclic GMP-dependent protein kinases .............................................................................. 39!
(ii) ! Phosphodiesterases ........................................................................................................... 39!
1.2.3 Breakdown by PDEs ........................................................................................................... 41!
1.2.4 Compartmentalisation of cGMP signalling .......................................................................... 41!
1.2.5 Cyclic GMP regulation of the cardiovascular system .......................................................... 45!
(i) ! Vascular smooth muscle cells ............................................................................................ 45!
(ii) ! Vascular endothelial cells ................................................................................................... 46!
(iii) !Cardiac myocytes ............................................................................................................... 47!
1.3! Cyclic GMP signalling in heart failure ................................................................................. 50!
1.3.1 The role of cGMP signalling in cardiac hypertrophy and failure .......................................... 50!
1.3.2 The role of cGMP signalling in anthracycline-induced cardiac injury and failure ................ 54! CHAPTER 2: RATIONALE AND OBJECTIVES ............................................................................ 59! CHAPTER 3: MATERIALS AND METHODS ................................................................................ 63!

ii
3.1! Patients .................................................................................................................................. 63!
3.2! Experimental animals ........................................................................................................... 63!
3.2.1 Transgenic mouse models .................................................................................................. 63!
(i) ! Constitutive cardiac myocyte-specific overexpression of PDE5 (PDE5-TG) ...................... 63!
(ii) ! Conditional cardiac myocyte-specific dominant negative mutation of the !1-subunit of sGC
(DNsGC!1) ......................................................................................................................... 64!
3.2.2 Mouse models of heart failure ............................................................................................. 65!
(i) ! Mouse model for pressure overload-induced cardiac hypertrophy ..................................... 65!
(ii)! Mouse model for anthracycline-induced cardiotoxicity ....................................................... 66!
3.2.3 Assessment of cardiac function .......................................................................................... 67!
(i) ! Transthoracic echocardiography (TTE) .............................................................................. 67!
(ii) ! Haemodynamic measurements .......................................................................................... 67!
3.3! In vitro experiments .............................................................................................................. 68!
3.3.1 Murine neonatal cardiac myocytes ...................................................................................... 68!
(i) ! Isolation method ................................................................................................................. 68!
(ii) ! Induction of mechanical stretch .......................................................................................... 68!
3.3.2 Murine adult cardiac myocytes ............................................................................................ 69!
(i)! Isolation method .................................................................................................................. 69!
(ii) ! Induction of cardiac myocyte hypertrophy and hypoxia ...................................................... 70!
3.3.3 Murine cardiac endothelial cells .......................................................................................... 70!
(i) ! Isolation method ................................................................................................................. 70!
3.3.4 Human cardiac endothelial cells ......................................................................................... 71!
(i) ! Isolation method ................................................................................................................. 71!
(ii) ! Induction of mechanical stretch .......................................................................................... 71!
3.3.5 Force measurements in murine cardiac myocytes .............................................................. 71!
3.4! Determination of cardiac cyclic nucleotide levels ............................................................. 73!
3.4.1 After chronic pressure overload or doxorubicin administration ........................................... 73!
3.4.2 At baseline following in vivo stimulation of sGC activity ...................................................... 73!
3.4.3 At baseline following ex vivo stimulation of sGC activity ..................................................... 73!
3.5! Transcriptional and translational analysis ......................................................................... 74!
3.5.1 Quantitative real-time PCR (RT-qPCR) ............................................................................... 74!
3.5.2 Immunoblot analysis ........................................................................................................... 76!
3.5.3 Immunohistochemistry ........................................................................................................ 76!
(i) ! Histological stainings .......................................................................................................... 76!
(ii) ! Microscopic analysis ........................................................................................................... 78!

iii
3.6! Profiling of cardiac miRNA expression ............................................................................... 79!
3.6.1 Affymetrix miRNA Microarray .............................................................................................. 79!
3.6.2 nCounter miRNA expression assay .................................................................................... 79!
3.6.3 Experimental validation of miRNA targets ........................................................................... 80!
(i) ! MicroRNA target site identification ...................................................................................... 80
(ii) MicroRNA target site validation…………………………………………………………………. 82
3.7! Statistical analysis ................................................................................................................ 82! CHAPTER 4: ROLE OF CGMP SIGNALLING IN THE ADVERSE CARDIAC RESPONSE TO CHRONIC
PRESSURE OVERLOAD ......................................................................................................... 85!
4.1! Increased PDE5 expression in LV tissue of patients with severe aortic stenosis ......... 85!
4.1.1 Elevated PDE5 expression is localised in scattered cardiac myocytes and endothelial
cells.................................................................................................................................... 85!
4.1.2 Mechanical stretch induces PDE5 expression in cardiac endothelial cells in vitro ............ 88!
4.2! Increased PDE5 expression in cardiac tissue of mice subjected to chronic LV pressure
overload ................................................................................................................................. 89!
4.3! Increased PDE5 expression in cardiac myocytes contributes to cardiac dysfunction
and dilatation in mice with chronic pressure overload ..................................................... 92!
4.3.1 Cardiac myocyte-specific PDE5 overexpression aggravates LV dysfunction and dilatation
after sustained pressure overload ...................................................................................... 92!
4.3.2 Enhanced cardiac myocyte PDE5 expression does not affect cardiac hypertrophy and
extracellular matrix remodelling after chronic pressure overload ........................................ 96!
4.3.3 Elevated cardiac myocyte PDE5 expression limits the increase in myocardial cGMP levels
in response to chronic pressure overload ......................................................................... 100!
4.3.4 Increased PDE5 expression in cardiac myocytes is associated with reduced SERCA2
expression and greater cardiac myocyte passive force after chronic pressure overload.. 101!
4.4! In search of underlying mechanisms of increased cardiac myocyte PDE5 expression
after sustained LV pressure overload ............................................................................... 104!
4.4.1 PDE5 mRNA levels are not elevated in adult murine cardiac myocytes 4 weeks after
TAC…………………………………………………………..…………………………………………….. 104!
4.4.2 PDE5 mRNA levels are not induced in hypoxic adult murine cardiac myocytes .............. 105!
4.4.3 PDE5 protein expression is not increased in mechanically stretched neonatal murine
cardiac myocytes .............................................................................................................. 106!
4.4.4 Altered miRNA profiles in the pressure overloaded heart do not appear to regulate PDE5
expression…………………………………………………….…………………………………………...108!

iv
(i) ! Profiling of differentially expressed miRNAs after chronic pressure overload using the
Affymetrix and NanoString platform .................................................................................. 108!
(ii) ! Predicted miRNAs do not functionally target the 3!UTR of PDE5 ..................................... 112! CHAPTER 5: ROLE OF CGMP SIGNALLING IN DOXORUBICIN-INDUCED CARDIOTOXICITY .......... 119!
5.1! Baseline phenotype of mice with a dominant negative mutation of sGC!1 in cardiac
myocytes…………………………………………………………………………………………… 119!
5.1.1 Cardiac myocyte-specific dominant negative mutation of sGC"1 decreases NO-stimulated
cGMP levels ...................................................................................................................... 119!
5.1.2 Dominant negative mutated sGC"1 in cardiac myocytes does not affect basal cardiac
function ............................................................................................................................. 121!
5.2! Decreased sGC activity in cardiac myocytes aggravates cardiac dysfunction and
dilatation in mice after chronic doxorubicin administration ........................................... 123!
5.2.1 Cardiac myocyte-specific dominant negative mutation of sGC"1 does not affect survival
and cachexia in doxorubicin-treated mice ........................................................................ 123!
5.2.2 Decreased sGC activity in cardiac myocytes amplifies LV dysfunction and dilatation after
chronic doxorubicin treatment ........................................................................................... 124!
5.2.3 Cardiac structure after chronic doxorubicin treatment is not affected by dominant negative
mutation of sGC"1 in cardiac myocytes ........................................................................... 127!
5.2.4 Dominant negative mutation of sGC"1 in cardiac myocytes increases expression of Fas
and Fas ligand after chronic doxorubicin treatment .......................................................... 128!
5.2.5 Cardiac myocyte-specific decreased sGC activity increases lipid peroxidation in hearts of
doxorubicin-treated mice .................................................................................................. 129!
5.3! Reversal of the dominant negative mutation of sGC!1 abrogates increased LV
dysfunction and dilatation after chronic doxorubicin treatment .................................... 132! CHAPTER 6: DISCUSSION .................................................................................................... 137!
6.1! Increased cardiac myocyte PDE5 expression in human and murine pressure overload
hypertrophy contributes to adverse LV remodelling ...................................................... 137!
6.2! Decreased sGC activity in cardiac myocytes aggravates LV dysfunction and dilatation
in mice chronically treated with doxorubicin ................................................................... 141!
6.3! Decreased cGMP levels in cardiac myocytes: a molecular hallmark heralding adverse
LV remodelling and dysfunction ................................................................................................. 145! SUMMARY .......................................................................................................................... 147!

v
SAMENVATTING .................................................................................................................. 149! REFERENCES CURRICULUM VITAE SUPPLEMENTS!

vi

vii
DANKWOORD – PREFACE
De weg tot dit proefschrift was zoals het beklimmen van de Gasthuisberg; de ene dag
trotseerde ik moeizaam al fietsend regen en wind, de andere dag duwde ik vlot het
gaspedaal van de auto in. In deze laatste meters wil ik graag enkele mensen bedanken.
Allereerst wil ik mijn oprechte dank betuigen aan mijn promotor Prof. Dr. Stefan Janssens.
Beste Stefan, ik bewonder je onuitputtelijke gedrevenheid om patiënten te helpen, zowel
in het ziekenhuis, als door het leggen van de wetenschappelijke grondslag voor nieuwe
therapieën. Jouw bezieling en enthousiasme werken zeer aanstekelijk en vormden dan
ook de aanzet tot dit doctoraat. Tijdens deze periode was je ondanks talrijke andere
verantwoordelijkheden steeds bereid om mee na te denken over de koers van dit
onderzoek - en waar nodig bij te sturen - en om mijn schrijfsels kritisch na te lezen.
Verder had ik ook het geluk te kunnen rekenen op Prof. Dr. Jozef Bartunek.
Jozef, bedankt voor je toewijding en de constructieve suggesties. I!m also greatly indebted
to Prof. Dr. Kenneth Bloch. Dear Ken, your renowned scientific expertise has immensely
contributed to my research and writings.
I further want to thank the members of my jury: Prof. Dr. Yicheng Ni, Prof. Dr. Johan Van
Cleemput, Prof. Dr. Guido De Meyer, and Prof. Dr. Michaela Kuhn, for providing a
welcome diversity of perspectives, and the efforts made to further improve this manuscript.
A special word of thanks goes to Prof. De Meyer and Prof. Kuhn who travelled to Leuven
to participate in this discussion. My gratitude also goes to Prof. Dr. Paul Herijgers, the
chair of my doctoral committee, for ensuring my defence runs smoothly.
In het labo kon ik rekenen op onze core: Hilde, Ellen en Nina. Als ik zeg dat zonder hen
het labo vierkant zou draaien, is dat zacht uitgedrukt. Hilde, weinig technieken zijn jou
vreemd, zonder jouw ervaring en efficiëntie was dit doctoraat niet gekomen tot wat het nu
is. Ellen, jouw nauwgezette manier van werken is het in vivo deel van deze thesis zeker
ten goede gekomen! Nina, bedankt voor de hulp met de histologie en de vele uurtjes aan
de microscoop. Ladies, naast de praktische ondersteuning zorgen jullie ook voor een zeer
fijne werksfeer, bedankt hiervoor.

viii
I wish to thank my other colleagues Ann Sophie, Ming, Melissa, Dieter, and Peter, for the
good times in the lab. Ann Sophie, een toffere mede-PhD!er had ik me niet kunnen
wensen, ik hoop dat je doctoraat een pareltje wordt! Peter, my PDE5 partner-in-science,
thank you for your input and help with experiments. Voor cardio-expertise kon ik ook
steeds afzakken naar het 7de. Ilse, Patricia, Kristel, Ronald, Christel, Eef, Diogo,…
bedankt voor de vele protocollen, de aangename samenwerkingen en toffe babbels!
Ook een welgemeende merci aan Marijke, helaas geen collega meer, maar des te meer
een vriendin. Ik heb van jou veel geleerd op de werkvloer, maar ook daarbuiten.
Glenn, jouw scherpe analytische geest en wetenschappelijke drive zijn een voorbeeld
geweest. Bedankt ook voor de fijne avonden op de AHA congressen!
Hierbij waren eveneens de Boston guys vaak van de partij: Manu en Patrick, merci!
Laurens… de appel valt niet ver van de boom. Het was steeds gezellig als je in het labo
was, hopelijk hebben we je iets bijgebracht.
Furthermore, I could always count on my colleagues of the Luttun lab. Special thanks
goes to Tom, who answered my many questions with a smile. Also, a warm thank you to
Petra and Boukje for the sorting, and Giulia, it was fun working with you!
Ook aan onze overburen van de Lijnen groep: bedankt voor het fijne contact!
Zelfs voorbij de landsgrenzen kon ik rekenen op hulp, hierbij denk ik graag terug aan de
prettige samenwerking met Dr. Noortje Bax en Prof. Carlijn Bouten van de TU Eindhoven.
I!m also grateful to our Hungarian collaborators, Dr. Agnes Balogh and Prof. Zoltan Papp,
for the fruitful joint effort to unravel the underlying mechanisms of our observations.
Wie ik natuurlijk ook niet kan vergeten hier: de party crew! Merci Fred, Ester, Ellen, Tokke,
Wouter, Aernout, Kevin, Ine, Ilse, Bieke, Hanne, Domi, en Stefan voor de fijne
laboweekends, kerstmarkten (of zal ik zeggen jenevermarkten) en nachtelijke escapades!
Fred, op jou kan ik altijd rekenen, een maatje uit de duizend! Dat we nog vaak samen de
eindstreep van de 20 km mogen halen in Brussel! Blondie & blondie, jullie zijn fantastisch,
bedankt om er steeds te zijn voor mij! En natuurlijk ook een extra merci aan hét danstalent
van Thrombogenics, altijd feest! Verder ook een dikke kus voor Wout en onze Nutty
Professor, die de feestjes & weekends altijd extra wisten op te vrolijken!

ix
Ik wil ook graag Melissa bedanken; eerst klasgenoot, daarna ook vriendin en kotgenootje
(ik vergeet nooit onze legendarische gangcantus!) … nu hebben we beiden een
doctoraatsdiploma op zak. Ik ben blij dat je erbij was al die jaren! Eveneens een
welgemeende merci aan de andere Leuvense chicas: Valerie, Sanne, Daniëlle en
Stephanie. Jullie hebben het leven buiten het labo een stuk aangenamer gemaakt, merci
voor de vele fijne avonden en reisjes! Val & San, de volgende Aperol in de bergen is op
mijn kosten! Mijn Kempische achterban is ook van ontelbaar belang geweest.
Hanne, in mijn hart sinds het 6de leerjaar, merci voor alles (teveel om hier op te sommen!).
Carine, bedankt om er altijd te zijn, voor de vele onvergetelijke avonturen, en om geduldig
te zijn als ik niet meteen tijd had om af te spreken.
Daarenboven wil ik graag mijn familie bedanken voor de onvoorwaardelijke steun
gedurende al die jaren. Tantes en nonkels, jullie wisten niet altijd even goed waar ik mee
bezig was in het labo, maar waren steeds oprecht geïnteresseerd en bezorgd.
Mijn oma!s… altijd zo blij dat ik het zo goed deed op “t school”, ik vind het jammer dat ze
er niet meer bij kunnen zijn vandaag. Laura, ondanks al ons gekibbel vroeger (daar zijn
zussen voor!), ben ik trots dat jij mijn zus bent! Veel succes nog hier in Leuven en ook
daarbuiten! Mama en papa, woorden schieten tekort… Bedankt om me de kans te geven
om te studeren wat ik wilde en steeds klaar te staan. Nooit was iets teveel gevraagd,
merci! Ten slotte wil ik ook Manu (aka “den Beerens”) bedanken. Omgaan met mij tijdens
mijn ochtendhumeuren en PhD stress-momenten was ongetwijfeld niet altijd even
gemakkelijk, je hebt het glansrijk doorstaan! Hierna is het aan jou, ik zal op de eerste rij
staan om te supporteren!
Sara

x

xi
LIST OF ABBREVIATIONS 3!UTR 3! untranslated region
ABC-PO avidin-biotin-peroxidase complex
AC adenylate cyclase
Ang II angiotensin II
ANOVA analysis of variance
ANP atrial natriuretic peptide
Apaf apoptosis protease activator protein
APC allophycocyanin
ARK adrenoceptor kinase
AS aortic stenosis
ATP adenosine 5!-triphosphate
AVR aortic valve replacement
Bad Bcl-2 associated death promoter
Bak Bcl-2 antagonist/killer
Bax Bcl-2 associated X protein
BCA bicinchoninic acid
Bcl-2 B-cell leukaemia/lymphoma-2
BDM 2,3-butanedione monoxime
BH Bcl-2 homology
BH4 tetrahydrobiopterin
BNP brain natriuretic peptide
BrdU 5-bromo-2!-deoxyuridine
CaMK Ca2+/calmodulin-dependent protein kinase
cAMP cyclic 3!, 5!-adenosine monophosphate
Cdk cyclin-dependent protein kinase
cGMP cyclic guanosine 3',5'-monophosphate
CN calcineurin
CNG cyclic nucleotide-gated
CNP C-type natriuretic peptide
CO cardiac output
CT cardiotrophin

xii
CTGF connective tissue growth factor
CVD cardiovascular disease
DAB 3,3!-diaminobenzidine
DAG diacylglycerol
DAPI 4',6-diamidino-2-phenylindole
DETA/NO diethylenetriamine/NO
DMEM Dulbecco!s modified eagle medium
DMSO dimethyl sulfoxide
DNsGC!1 dominant negative mutant of sGC!1
ECE endothelin-converting enzyme
ECG electrocardiogram
ECL enhanced chemiluminescence
ECM extracellular matrix
EDPVR end-diastolic pressure-volume relationship
EDTA ethylene diamine tetraacetic acid
EDV end-diastolic volume
EF ejection fraction
EGM endothelial cell growth medium
Egr early growth response protein
ERK extracellularly responsive kinase
ESP end-systolic pressure
ESV end-systolic volume
ET-1 endothelin-1
FACS fluorescence-activated cell sorting
FAM 6-carboxy-fluorescein
FBS fetal bovine serum
FN fibronectin
FS fractional shortening
GAPDH glyceraldehyde-3!-phosphate-dehydrogenase
GC guanylate cyclase
GFP green fluorescent protein
GLUT glucose transporter
GPCR G protein-coupled receptor

xiii
GPX glutathione peroxidase
GSK glycogen synthase kinase
GSNO S-nitrosoglutathione
GTP guanosine 5!-triphosphate
HAT histone acetyltransferase
HBSS Hank!s balanced salt solution
HDAC histone deacetylase
HF heart failure
HR heart rate
HRP horseradish peroxidase
HW/BW heart weight to body weight ratio
HW/TL heart weight to tibia length ratio
I-1 protein phosphatase inhibitor-1
IP intraperitoneal
IBMX 3-isobutyl-1-methyl-xanthine
IGF insulin growth factor
IHC immunohistochemistry
IL interleukin
IP3 inositol 1,4,5-trisphosphate
IRAG IP3 receptor-associated PKG substrate
JAK janus kinase
JNK c-Jun N-terminal kinase
KHD kinase like homology domain
L-NAME N #-nitro-L-arginine methyl ester
LIF leukaemia inhibitory factor
LIMMA linear models for microarray analysis
LV left ventricle
LVID left ventricular internal diameter
MAPK mitogen-activated protein kinase
MDA malondialdehyde
MEF myocyte enhancer factor
MHC myosin heavy chain
miRNA micro ribonucleic acid

xiv
mitoKATP channel mitochondrial ATP-sensitive K+-channel
MKP MAPK phosphatase
MLC(K) myosin light chain (kinase)
MMP matrix metalloproteinase
MMP ($%m) mitochondrial membrane potential
MPTP mitochondrial permeability transition pore
MVEC microvascular endothelial cells
NADPH nicotinamide adenine dinucleotide phosphate
NCX Na+/Ca2+-exchanger
NFAT nuclear factor of activated T cells
NHE Na+/H+-exchanger
NO(S) nitric oxide (synthase)
NP(R) natriuretic peptide (receptor)
PBS phosphate buffered saline
PCR polymerase chain reaction
PDE phosphodiesterase
PDE5-TG (cardiac myocyte-specific) overexpression of PDE5
PDK phosphoinositide-dependent kinase
PE phenylephrine
pGC particulate guanylate cyclase
PGC PPAR-& coactivator
PI3K phosphoinositide 3-kinase
PIP2 phosphatidyl inositol 4,5-bisphosphate
PKA protein kinase A
PKB protein kinase B (also known as Akt)
PKC protein kinase C
PKG protein kinase G
PLC phospholipase C
PLN phospholamban
PP protein phosphatase
PPAR peroxisome proliferator-activated receptor
PV pressure-volume
Rb retinoblastoma protein

xv
RGS regulator of G protein-coupled signalling
RIPA radio-immunoprecipitation assay
RISC RNA-induced silencing complex
RMA robust multichip average
ROCK Rho-associated protein kinase
ROS reactive oxygen species
RT-qPCR quantitative real-time PCR
RV right ventricle
RyR ryanodine receptor
SDS sodium dodecyl sulphate
SDS-PAGE SDS polyacrylamide gel electrophoresis
SERCA sarcoplasmic reticulum Ca2+-ATPase
sGC soluble guanylate cyclase
SR sarcoplasmic reticulum
STAT signal transducer and activator of transcription
SV simian virus
SV stroke volume
TAC transverse aortic constriction
TAMRA 6-carboxy-tetramethyl-rhodamine
TGF transforming growth factor
Tie tyrosine kinase with immunoglobulin-like and EGF-
like domains
TIMP tissue-inhibitor of metalloproteinase
TRPC channel canonical transient receptor potential channel
TSA tyramide signal amplification
TTE transthoracic echocardiography
VEGF vascular endothelial growth factor
VSMC vascular smooth muscle cell
WT wild-type

xvi

CChhaapptteerr 11
INTRODUCTION


Introduction 1
CHAPTER 1: INTRODUCTION
1.1 Tackling heart failure Despite major therapeutic advances during the past decades, cardiovascular disease
(CVD) continues to be the leading cause of death worldwide, accounting for 30% of
deaths anually (17.3 million). By 2030, it is predicted that 23.6 million people will die from
CVD (WHO, 2011). Cardiovascular disease comprises a variety of disorders affecting the
heart and blood vessels, including coronary heart disease, cerebrovascular disease,
hypertension, peripheral artery disease, rheumatic heart disease, congenital heart
disease, and heart failure (HF). Heart failure is a complex syndrome with characteristic
clinical signs and symptoms, and a compromised cardiac function. The failing heart is
unable to generate sufficient cardiac output (CO) to meet the metabolic requirements of
the body and accommodate venous return. Cardiac dysfunction precipitates changes in
vascular function, blood volume, and neurohumoral status to help maintain cardiac output
(primarily by the Frank-Starling mechanism) and arterial blood pressure. Although these
compensatory changes can initially offset reduced cardiac performance, they become key
co-conspirators in the disease process, ultimately increasing the likelihood of organ failure
and worsening clinical prognosis.1
Despite improved medical management of HF, this condition remains a major cause of
morbidity and mortality (5-year mortality rates about 50% according to the Framingham
Heart Study2). Thus, a better understanding of the molecular mechanisms underlying this
debilitating condition is needed in the hope of devising novel clinical interventions.
1.1.1 Determinants of contractile function
The amount of blood pumped out by the heart over a given time period is known as
cardiac output (CO), which in turn is the product of heart rate and stroke volume (SV), and
ranges between 4-8 l/min under basal resting conditions. In addition, other factors such as
synergistic ventricular contraction, ventricular wall and pericardial integrity, and valvular
competence all affect CO.
Stroke volume is the amount of blood ejected by the ventricle per heartbeat, and is
affected by three main factors: preload, afterload, and intrinsic contractility. Preload is
defined as the ventricular wall tension at the end of diastole. In clinical terms, it is the
stretch on the ventricular fibres just before contraction, often approximated by the end-

Chapter 1 2
diastolic volume or pressure. Afterload denotes the ventricular wall tension during
contraction; the resistance that must be overcome for the ventricle to eject its content, and
is often approximated by the systolic ventricular (or arterial) pressure. Finally, contractility
is the inotropic state of the heart independent of preload or afterload, and reflects chemical
or hormonal influences on the force of contraction.3
1.1.2 Aetiology of heart failure
Chronic heart failure may result from a wide variety of cardiovascular insults. From a
pathophysiological point of view, the underlying causes can be grouped into those that
(1) impair ventricular contractility, (2) increase afterload, or (3) impair ventricular relaxation
and filling (Figure 1). Heart failure that results from an abnormal ventricular emptying (due
to impaired contractility or excessive afterload) is termed systolic dysfunction, whereas
heart failure caused by abnormal diastolic relaxation or ventricular filling is termed diastolic
dysfunction. However, systolic and diastolic abnormalities are not mutually exclusive,
since many patients demonstrate both. As a result, it is now common to categorise heart
failure patients into two general categories, based on left ventricular (LV) ejection fraction
(EF), a measure of cardiac performance:
(1) Heart failure with reduced EF (i.e. primarily systolic dysfunction)
(2) Heart failure with preserved EF (i.e. primarily diastolic dysfunction)
Whereas these physiological principles may be applied to both right-sided and left-sided
heart failure, the two ventricles have distinct functional characteristics. Compared with the
LV, the right ventricle (RV) is a thin-walled, highly compliant chamber that accepts its
blood volume at low pressures and ejects against a low pulmonary vascular resistance.
As a result of its high compliance, the RV easily copes with a wide range of filling volumes
without significant changes in its filling pressures. Conversely, the RV is quite susceptible
to failure in situations that present a sudden increase in afterload, such as acute
pulmonary embolism. Of note, the most common cause of right-sided heart failure is the
presence of left-sided heart disease. Under these conditions, the RV is confronted with
excessive afterload due to elevated pulmonary vascular pressures resulting from LV
dysfunction.3

Introduction 3
Figure 1. Conditions that cause left-sided heart failure through impairment of ventricular systolic or diastolic function *Importantly, in chronic stable stages the conditions in this box may instead result in heart failure with preserved ejection fraction, due to compensatory ventricular hypertrophy and increased diastolic stiffness (diastolic dysfunction).

Chapter 1 4
1.1.3 Chronic pressure overload-induced heart failure
(i) Aetiology
Advanced aortic stenosis
Aortic stenosis (AS, Figure 2) is often caused by age-related degenerative calcific
changes of the valve. Calcific changes that progress to AS may also develop in patients
with congenitally deformed aortic valves (1-2% of population have bicuspid native aortic
valves). In addition, aortic stenosis can also result from chronic rheumatic valve disease,
although the prevalence of this condition has decreased dramatically.
In age-related degenerative AS, cumulative “wear and tear” of valve motion over many
years leads to endothelial and fibrous damage, causing calcification of the trileaflet valve.
However, there is also evidence of a common aetiology with atherosclerotic vascular
disease. Studies have shown that, as in atherosclerosis, valve tissue of patients with this
form of AS displays cellular proliferation, inflammation, lipid accumulation, and increased
margination of macrophages and T lymphocytes.4
Figure 2. Aortic stenosis Aortic valve stenosis - or aortic stenosis - occurs when the aortic orifice (normally 3 cm2) is narrowed. This narrowing prevents the valve from fully opening, causing obstruction of the blood flow into the aorta. There is a long latent period of increasing obstruction and myocardial overload, during which the asymptomatic patient has a normal life span. However, once angina, syncope, or heart failure develops, survival is greatly reduced.
Uncontrolled arterial hypertension
In approximately 90% of affected patients, the cause of blood pressure elevation is
unknown, a condition termed primary or essential hypertension. High blood pressure
attributed to a definable cause is termed secondary hypertension. Although generally

Introduction 5
asymptomatic, high blood pressure can result in devastating effects on many organs,
especially the blood vessels, heart, kidney, brain, and retina.
(ii) Pathophysiology
The heart is capable of remodelling in response to environmental demands. Whereas
exercise, pregnancy, and postnatal growth induce physiological growth, a variety of stimuli
can also cause pathological growth. When LV afterload is increased, due to AS or
hypertension, significant elevation of LV pressure is necessary to eject blood into the
aorta. The ventricle responds to this increased systolic pressure by increasing muscle
mass through the initiation of a hypertrophic response. At early stages, this compensatory
cardiac hypertrophy results in reduced ventricular wall stress and improved cardiac
contraction. The defining features of hypertrophy are an increase in cardiac myocyte size,
enhanced protein synthesis, and a higher organisation of the sarcomere. These changes
in cellular phenotype are preceded and accompanied by the re-induction of the “fetal gene
program” (induction of natriuretic peptides, c-myc, c-fos, and ' myosin heavy chain).
Unfortunately, cardiac hypertrophy is a double-edged sword, beneficial in some respects
and deleterious in others. Although it helps to preserve ventricular performance,
hypertrophy also impairs coronary blood flow reserve and reduces diastolic function.
With prolonged stress, the heart undergoes irreversible decompensation, resulting in
dilatation of the failing heart. However, the mechanisms that determine progression of
long-standing hypertrophy to heart failure remain incompletely understood.
Untangling the molecular web of cardiac hypertrophy
Hypertrophic stimuli activate a variety of membrane-bound receptors coupled to multiple
intracellular signalling cascades. There are several points of convergence and divergence
in the transduction of these biomechanical stress cascades to the nucleus, which
ultimately alter transcriptional regulation of gene expression and induce long-term
phenotypic change. Here, recent insights into molecular signalling pathways involved in
cardiac hypertrophy and failure are summarised (Figure 3), with various in vitro and in vivo
studies demonstrating the importance of these pathways (Table 1).

Chapter 1 6

Introduction 7
Figure 3. Cardiac myocyte signalling pathways involved in the pathophysiology of cardiac hypertrophy and failure Stress stimuli are transduced by many intracellular signalling pathways and ultimately result in changes in cardiac myocyte function and growth. For simplicity, only some of the known interactions and feedback loops are shown. Stress stimuli include neurohormones (natriuretic peptides, angiotensin II, endothelin-1), neurotransmitters (catecholamines), cytokines, and growth factors. After these ligands bind to cell-surface receptors, the signal is transmitted to protein kinases, which in turn activate signalling nodes. These pathways are involved in physiological responses, however, in the failing heart, stress stimuli are more abundant, thereby amplifying these pathways and generating imbalances among them. AC indicates adenylate cyclase; AR, adrenergic receptor; CaM, calmodulin; cAMP, cyclic adenosine 3!, 5!-monophosphate; CN, calcineurin; DAG, diacylglycerol; ERK, extracellularly responsive kinase; HDAC, histone deacetylase; GSK, glycogen synthase kinase; JNK, c-Jun N-terminal kinase; MAPK, mitogen-activated protein kinase; MEF, myocyte enhancer factor; MLCK, myosin light chain kinase; mRNA, messenger RNA; miRNA, microRNA; NFAT, nuclear factor of activated T cells; PI3K, phosphoinositide 3-kinase; PKA, cAMP-dependent protein kinase; PKC, protein kinase C; PLC, phospholipase C; ROCK, Rho-associated protein kinase; and STAT, signal transducer and activator of transcription.
Hypertrophic signalling via G protein-coupled receptors
G protein-coupled receptors (GPCR) play an important role in the regulation of cardiac
function and adaptation to changes in haemodynamic burden. The most important
myocardial GPCRs include adrenergic (comprised of several subtypes of "- and #-
adrenergic receptors) and muscarinic receptors. These receptors are coupled to three
principal classes of heterotrimeric guanosine 5!-triphosphate (GTP)-binding proteins - Gs,
Gq/G11, and Gi - which transduce the agonist- or antagonist-induced signal to intracellular
effectors such as enzymes and ion channels. All heterotrimeric G proteins consist of the
subunits G" and G#$, which upon receptor activation dissociate and independently activate
intracellular signalling pathways.
Gs Signalling
The most abundant adrenergic receptor in cardiac tissue is the #1-receptor, coupled to Gs,
which in turn activates adenylate cyclase (AC). Adenylate cyclase catalyses synthesis of
cyclic 3!, 5!-adenosine monophosphate (cAMP), acting as a second messenger by
interacting with protein kinase A (PKA), eventually resulting in positive chronotropic,
inotropic, and lusitropic effects on the heart. Heart failure is accompanied by impaired #-
receptor function through both a decreased number of receptors and functional

Chapter 1 8
uncoupling.5 The latter is believed to be mediated by #-adrenoceptor kinase (#ARK) 1,
which phosphorylates the receptor and thereby rapidly decreases its sensitivity to further
agonist stimulation.
Gq Signalling
Angiotensin II (Ang II), endothelin-1 (ET-1), and "-adrenergic receptors are coupled to
Gq/11 which in turn activates phospholipase C (PLC), and have all been shown to be
sufficient to mediate cardiac myocyte hypertrophy upon agonist stimulation.6
PLC hydrolyses phosphatidyl inositol 4,5-bisphosphate (PIP2), resulting in the formation of
two second messengers, inositol 1,4,5-trisphosphate (IP3) and diacylglycerol (DAG).
Subsequently, IP3 stimulates intracellular Ca2+-release from the sarcoplasmic reticulum
(SR) and DAG activates protein kinase C (PKC).
Gi Signalling
Both cardiac muscarinic and #2-adrenergic receptors couple through Gi, thereby inhibiting
AC and directly opposing Gs-dependent signalling. Of note, Gi is upregulated in human
heart failure and basal AC activity decreased, suggesting that this mechanism may
contribute to the cardiomyopathic phenotype.7, 8 Moreover, Gi is upregulated in
hypertension-induced hypertrophy prior to onset of overt failure, indicating that Gi
upregulation may precede decompensation.9
Calcineurin-NFAT signalling The serine-threonine phosphatase calcineurin is expressed in multiple tissues and
consists of a catalytic A subunit and a regulatory B subunit. Elevations in cytoplasmic
Ca2+-concentrations promote the association of calmodulin with calcineurin and
subsequent activation of the enzyme. Calcineurin dephosphorylates transcription factors
of the nuclear factor of activated T cells (NFAT) family, thereby unmasking nuclear
localisation signals, which in turn results in translocation of NFAT proteins to the nucleus.
MAPK pathways
Mitogen-activated protein kinase (MAPK) pathways provide an important link between
external stimuli and the nucleus via phosphorylation and regulation of multiple
transcription factors. MAPKs can be divided into three major subfamilies: extracellularly

Introduction 9
responsive kinases (ERKs), c-Jun N-terminal kinases (JNKs), and p38 MAPKs, all
inhibited by MAPK phosphatase 1 (MKP-1) and playing a significant role in hypertrophic
signalling. p38 phosphorylates several transcription factors involved in hypertrophic gene
expression, including myocyte enhancer factor (MEF) 2 and NFAT3.10, 11 ERK also
phosphorylates GATA4, a zinc finger transcription factor that is highly expressed in
cardiac myocytes and regulates the expression of several important hypertrophic signals,
such as atrial and brain natriuretic peptide (ANP, BNP), " myosin heavy chain ("MHC),
#MHC, and ET-1. Pressure overload leads to elevated GATA4 activity, and
overexpression of GATA4 both in vitro and in vivo is sufficient to drive hypertrophy.12
Moreover, myocardial deletion of GATA4 revealed its absolute requirement in
compensating for increased wall stress during pressure overload.13
PI3K/Akt/GSK-3!-dependent signalling
Phosphoinositide 3-kinases (PI3Ks) constitute a family of enzymes that exhibit both
protein and lipid kinase activity and have been linked to signalling in many cellular
functions, particularly during cell growth, survival, and proliferation. These enzymes can
be activated by several kinases, such as GPCRs, including "- and #2-adrenergic
receptors. It was shown that PI3K is activated in pressure overload hypertrophy in a G#$-
dependent manner.14
One of the principal targets of PI3K signalling is the serine/threonine kinase Akt, also
known as protein kinase B (PKB). This kinase is activated via binding of PI3K-
phosphorylated phosphoinositides, which in turn results in its translocation to the
membrane. Full activation requires additional phosphorylation events, including
phosphorylation by phosphoinositide-dependent kinase 1 (PDK1).
Two well-defined direct downstream targets of Akt are likely candidates to be the
mediators of PI3K/Akt-induced hypertrophy: glycogen synthase kinase (GSK) 3# and the
mammalian target of rapamycin (mTor). Rapamycin, an immunosuppressive drug, binds
to its intracellular receptor, and this complex subsequently associates with mTor,
a serine/threonine kinase implicated in the regulation of protein translation. Binding of
rapamycin inhibits the activity of mTor, thus resulting in impaired protein synthesis and a
decreased cell size.
In addition to mTor, Akt also phosphorylates GSK-3#, rendering it inactive. GSK-3#
negatively modulates hypertrophy by phosphorylating NFAT proteins, thereby masking

Chapter 1 10
their nuclear import sequences and promoting translocation to the cytoplasm and
transcriptional inactivation.15 Cyclic guanosine 3', 5'-monophosphate (cGMP) and cGMP-
dependent protein kinase (PKG) are also endogenous negative modulators of stress-
response signalling and will be discussed further in this introduction (cfr. 1.2).
Gp130/STAT3 signalling Gp130 is a receptor for several cytokines, including interleukin (IL) 6/11, leukaemia
inhibitory factor (LIF), and cardiotrophin (CT) 1. Although gp130 and CT-1 are expressed
in multiple tissues, CT-1 induces cardiac myocyte hypertrophy in vitro.16 Upregulation of
LIF, CT-1, and IL-6 is induced by Ang II.17 Induction of gp130-dependent signalling leads
to activation of both MAPK and janus kinase (JAK) / signal transducer and activator of
transcription (STAT) pathways. Specifically, STAT3 is translocated to the nucleus in
response to gp130 activation, which results in induction of genes involved in hypertrophy
and survival pathways.
Small GTP-binding proteins
Small G proteins provide a critical link between cell membrane receptors and various
signalling pathways. The small G protein family consists of multiple members, regulating
diverse cellular processes such as cell growth, division and survival, organisation of the
cytoskeleton, membrane trafficking, and cellular motility. These proteins are activated by
binding of GTP, resulting in hydrolysis of GTP to GDP through their GTPase activity,
thereby returning the molecules to their inactive state. Five families of small G proteins
have been described (Rho, Ras, ARFs, Rab, and Ran), each consisting of several
members.
Signalling of Ras, the first small G protein implicated in cardiac hypertrophy, is coupled to
multiple downstream effectors participating in the hypertrophic response, including Raf
and the MAPK pathways. Activated Ras was shown to promote nuclear localisation of
NFAT3, whereas a dominant negative Ras-mutant was able to abrogate the phenylephrine
(PE)-induced increase in NFAT activity.18
The Rho family of small G proteins, consisting of Rho, Rac, and Cdc42 subfamilies,
regulates cytoskeletal organisation of non-muscle cells, as well as cardiac myocytes.
In addition, several hypertrophic signalling cascades can be influenced by Rho-dependent
signalling in muscle cells. RhoA activates a variety of protein kinases, including Rho-

Introduction 11
associated protein kinase (ROCK), which in turn promotes activation of myosin light chain
kinase (MLCK). Myosin light chain kinase, which can also be activated by
Ca2+/calmodulin, is sufficient to increase sarcomeric organisation in vitro, one of the
hallmarks of the hypertrophic phenotype.19
Transcriptional control of cardiac hypertrophy by MEF2/HDAC Many Ca2+-dependent signalling molecules, including calcineurin, Ca2+/calmodulin-
dependent protein kinase (CaMK) and MAPKs, are sufficient to evoke a hypertrophic
phenotype in cardiac myocytes and to induce reprogramming of cardiac gene expression.
Given that multiple pathways can elicit a similar molecular response, it appears likely that
hypertrophic pathways ultimately converge on common endpoints and downstream
targets. A major candidate in this regard is the transcription factor MEF2, which integrates
multiple Ca2+/calmodulin-dependent signalling pathways in muscle cells, as well as
neurons and T lymphocytes.20 Despite high expression levels, MEF2 proteins display only
basal transcriptional activity in the adult myocardium and only become active upon
stimulation, thus fulfilling the criteria for a potential integrator of pathological growth
signals.21, 22 The activity of MEF2 is controlled by direct association with histone
deacetylases (HDACs), which deacetylate nucleosomal histones, thereby promoting
chromatin condensation and transcriptional repression when recruited to target genes via
binding of specific transcription factors such as MEF2.20 This activity is opposed by
histone acetyltransferases (HATs), which relax chromatin and thereby activate target
genes. HDACs can be categorised into three classes, of which class II HDACs are
preferentially expressed at high levels in striated muscle and neurons.
Experimental research supports the notion that many, if not all, hypertrophic stimuli
converge in the nucleus and that class II HDACs in concert with MEF2 constitute the key
integrators of these signals.
MicroRNAs
MicroRNAs (miRNAs) are endogenous ~22 nt RNAs acting as negative regulators of gene
expression by inhibiting mRNA translation or promoting mRNA degradation. Growing
evidence shows involvement of miRNAs in many physiological and pathological
processes. In mammals, miRNAs originate from a primary transcript (pri-miR), which is
transcribed by RNA polymerase II and regulated by transcription factors in a similar way

Chapter 1 12
as conventional mRNAs (Figure 4). The pri-miR undergoes nuclear cleavage by a
ribonuclease III called Drosha and the double-stranded DNA binding protein Pasha
(DGCR8) to generate a hairpin-shaped pre-miRNA. These intermediates are transported
to the cytoplasm by the nuclear export factor exportin 5. Within the cytoplasm, the
ribonuclease III Dicer and its cofactors process the precursors into 19- to 25-nucleotide
miRNA-miRNA* duplexes. The double-stranded RNA molecule dissociates, and one
strand is incorporated into the RNA-induced silencing complex (RISC). The miRNA-loaded
RISC is capable of binding to target mRNAs, and the 5! proximal “seed” region
(nucleotides 2 to 8) appears to be the primary determinant of the pairing specificity of the
miRNA to the 3! untranslated region (3!UTR) of a target mRNA.23 In addition to Watson-
Crick base pairing, the efficiency of transcriptional repression also depends on the number
and configuration of mismatches between the miRNA and the target mRNA, the
secondary structure of the surrounding region, and the number of target sequences on the
mRNA.24
Among the many miRNAs, it has been reported that miR-1, miR-29, miR-30, miR-133, and
miR-150 have often been found to be downregulated, and miR-21, miR-23a, miR-125,
miR-195, miR-199, and miR-214 are upregulated with hypertrophy.25-30 Three miRNAs
have been described as muscle specific; miR-1, miR-133, and miR-208. MicroRNA-1
appears to be the most abundant miRNA in the heart (40% of cardiac miRNAs) and has
been shown to target key components of the Ca2+-mediated hypertrophic signalling
cascade, including calmodulin and MEF2.31 MicroRNA-133 is transcribed together with
miR-1, and may control myocardial remodelling through inhibition of connective tissue
growth factor (CTGF), RhoA, and Cdc42.32, 33 MicroRNA-208a is encoded by an intron in
the "MHC gene and is only expressed in the heart. Although the expression level of miR-
208 remains stable during cardiac stress, it appears to fulfil a dominant function in
regulating cardiac hypertrophy and remodelling.34 Of note, a subset of the miRNAs
involved in cardiac pathology is enriched in cardiac fibroblasts compared to cardiac
myocytes, including miR-21 and members of the miR-29 family.
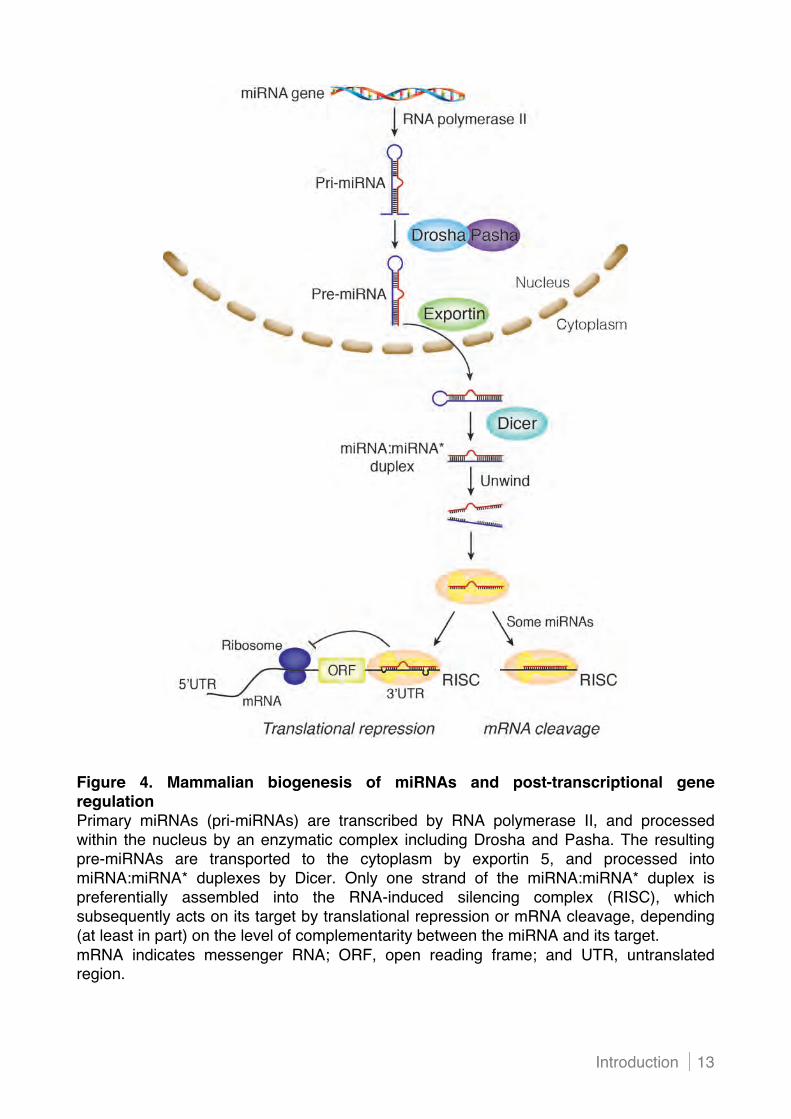
Introduction 13
Figure 4. Mammalian biogenesis of miRNAs and post-transcriptional gene regulation Primary miRNAs (pri-miRNAs) are transcribed by RNA polymerase II, and processed within the nucleus by an enzymatic complex including Drosha and Pasha. The resulting pre-miRNAs are transported to the cytoplasm by exportin 5, and processed into miRNA:miRNA* duplexes by Dicer. Only one strand of the miRNA:miRNA* duplex is preferentially assembled into the RNA-induced silencing complex (RISC), which subsequently acts on its target by translational repression or mRNA cleavage, depending (at least in part) on the level of complementarity between the miRNA and its target. mRNA indicates messenger RNA; ORF, open reading frame; and UTR, untranslated region.

Chapter 1 14
Many of the signalling pathways discussed above trigger the generation of reactive
oxygen species (ROS), a process that is increasingly recognised as an important
contributor to depressed cardiac function and maladaptive remodelling.35 There are
several sources of ROS, including the nicotinamide adenine dinucleotide phosphate
(NADPH)-oxidase system (which can be activated by Ang II and other stimuli), xanthine
oxidase, monoamine oxidases (which are important for catecholamine and serotonin
catabolism), mitochondrial electron leak, and nitric oxide synthase 3 (NOS3). The role of
NOS3 in generating ROS and contributing to cardiac dysfunction is worth highlighting,
since NOS3 is usually considered to protect against oxidative cytotoxicity, abnormal
growth, and fibrosis. In an oxidative environment and in the absence of necessary
cofactors including tetrahydrobiopterin (BH4), the normal electron transfer from the
reductase domain to the oxygenase domain of NOS3 can be impaired (%uncoupled!),
resulting in decreased synthesis of NO and increased synthesis of superoxide. It was
shown that uncoupled NOS3 activity contributes to the pathology of the hypertrophied and
failing heart.36

Introduction 15
EXPERIMENTAL MODEL CARDIAC OUTCOME Hypertrophy signalling via GPCRs
Transgenic overexpression of Ang II, ET-1, and "-adrenergic receptors in mice
Cardiac hypertrophy and subsequent cardiomyopathy with depressed contractile function37-40
Combined genetic ablation of the Gq and G11 genes in mice
Embryonic lethality due to myocardial hypoplasia41
Cardiac myocyte-specific conditional inactivation of G"q/G"11 in mice
Lack of cardiac hypertrophy or activation of the fetal gene program after TAC42
Overexpression of a dominant negative mutant of Gq in transgenic mouse hearts
Attenuated hypertrophic response to TAC43
Transgenic overexpression of PKC-# in hearts of transgenic mice
Cardiac hypertrophy and sudden death44
Transgenic overexpression of constitutively active PKC-&
Compensated cardiac hypertrophy45
Expression of a dominant negative PKC-" mutant in cardiac myocytes
Hypertrophic growth46
Overexpression of #1-AR in hearts of transgenic mice
Initially increased contractile function and responsiveness to isoproterenol, but eventually progressive deterioration of cardiac performance, cardiac myocyte hypertrophy, and fibrosis47, 48
Cardiac-directed overexpression of AC type VI in transgenic mice
Attenuated cardiomyopathic changes, including cardiac hypertrophy in intercrossed Gq transgenic mice49, 50
Transgenic overexpression of PKA in mouse hearts
Dilated cardiomyopathy associated with cardiac myocyte hypertrophy and fibrosis51
Conditional overexpression of a Gi-coupled GPCR in mice
Cardiomyopathy and lethal arrhythmias52
Calcineurin-NFAT signalling Constitutive activation of calcineurin in transgenic mouse hearts
Massive cardiac enlargement and heart failure53
Constitutive active NFAT3 mutant in transgenic mouse hearts
Similar, but less dramatic response53
Cardiac overexpression of a mutated, catalytically inactive calcineurin
Protection against hypertrophy and subsequent development of fibrosis after abdominal aortic constriction54
Calcineurin A#-deficient mice Reduction in basal heart size and resistance to diverse hypertrophic stimuli55
MAPK pathways Overexpression of MKP-1 Blocked agonist-induced hypertrophy in
cardiac myocytes and pressure overload-associated hypertrophy56
PI3K/Akt/GSK-3!-dependent signalling Overexpression of a constitutively active PI3K mutant in transgenic mice
Cardiac hypertrophy57
Dominant negative form of PI3K in transgenic mice
Reduced heart weight/body weight ratios57
Chapter 1 16
Transgenic overexpression of Akt in mice Cardiac hypertrophy without affecting systolic function58, 59
Transgenic overexpression of Akt in mice treated with rapamycin
Attenuated cardiac hypertrophy59
Neonatal cardiac myocytes treated with rapamycin
Blocked increase in cardiac myocyte size induced by PE, Ang II, or fetal calf serum60-62
Overexpression of mutant GSK-3# (resistant to phosphorylation) in cardiac myocytes
Inhibition of ET-1-mediated cardiac myocyte hypertrophy63
Overexpression of this GSK-3# mutant in hearts of transgenic animals
Blunted hypertrophic response to chronic isoproterenol administration and pressure overload64
Gp130/STAT3 signalling Double transgenic expression of IL-6 and IL-6 receptors in mice
Marked cardiac hypertrophy65
Expression of dominant negative STAT3 in cardiac myocytes
Attenuated LIF-induced hypertrophic response66
Overexpression of STAT3 in transgenic mice
Cardiac myocyte hypertrophy66, 67
Transgenic overexpression of a dominant negative mutant of the gp130 receptor in mice
Attenuated TAC-induced cardiac hypertrophy68
Gp130-deficient mice Dilated cardiomyopathy, associated with massive cardiac myocyte apoptosis after TAC69
Small GTP-binding proteins Overexpression of a constitutively activated Ras mutant in cardiac myocytes
Hypertrophic gene expression70
Overexpression of a constitutively activated Ras mutant in transgenic mouse hearts
Significant increase in cardiac mass71
Expression of dominant-negative Ras mutants in cardiac myocytes
Prevention of PE-mediated increases in cell size and protein synthesis72, 73
Expression of dominant negative RhoA mutants and inhibitors of ROCK in cardiac myocytes
Prevention of PE-, ET-1, or Gq-stimulated cardiac myocyte hypertrophy 74-76
Overexpression of RhoA in transgenic mouse hearts
Cardiac conduction abnormalities with bradycardia and, ultimately, a dilated phenotype associated with heart failure77
Constitutive activation of Rac in cardiac myocytes in vitro and in vivo
Hypertrophy78, 79
Dominant negative Rac mutant in cardiac myocytes
Prevention of PE-induced increases in protein synthesis as well as cardiac myocyte size78
Transcriptional control of cardiac hypertrophy by MEF2/HDAC Expression of mutant forms of HDAC5 in cardiac myocytes
Resistance to cardiac myocyte hypertrophy80

Introduction 17
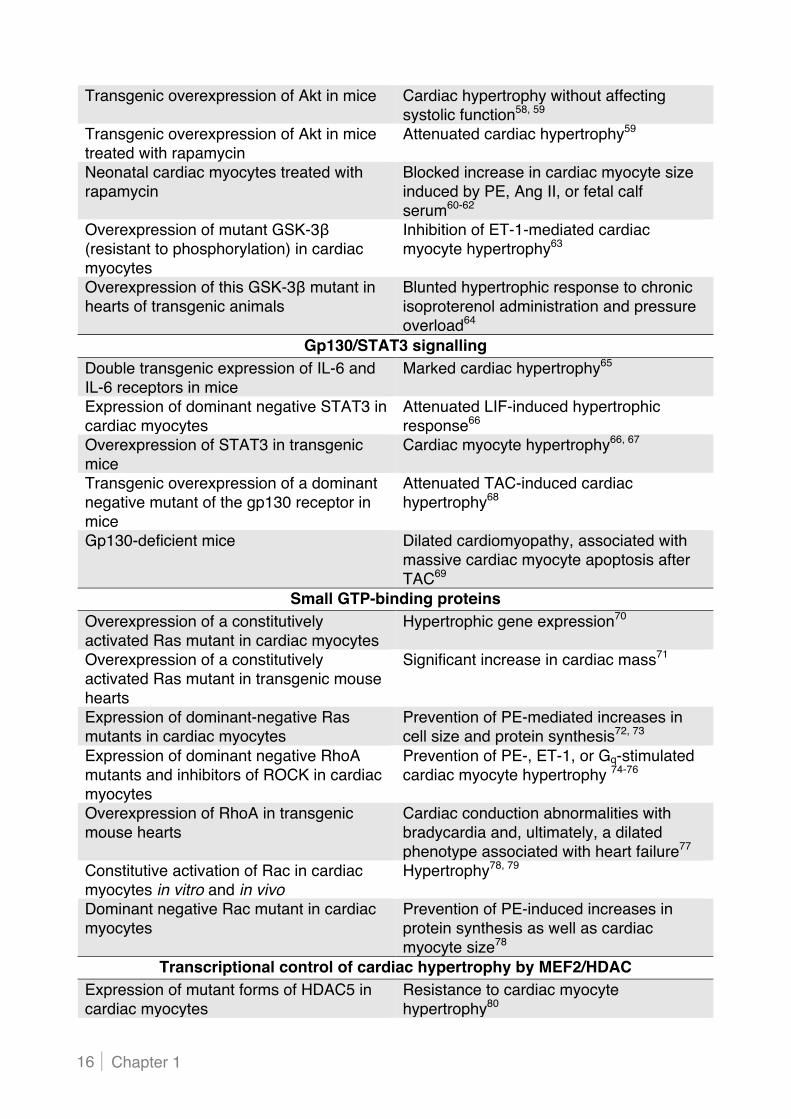
Table 1. In vivo and in vitro studies providing evidence for a role of identified signalling pathways in cardiac hypertrophy and subsequent heart failure AC indicates adenylate cyclase; Ang II, angiotensin II; AR, adrenergic receptor; ET-1, endothelin-1; GPCR, G protein-coupled receptor; GSK, glycogen synthase kinase; HDAC, histone deacetylase; IL, interleukin; LIF, leukaemia inhibitory factor; MAPK, mitogen-activated protein kinase; MEF, myocyte enhancer factor; MKP, MAPK phosphatase; NFAT, nuclear factor of activated T cell; PE, phenylephrine; PI3K, phosphoinositide 3-kinase; PKA, protein kinase A; PKC, protein kinase C; STAT, signal transducer and activator of transcription; and TAC, transverse aortic constriction.
Altered Ca2+-signalling
Calcium is the central regulator of excitation-contraction coupling, which drives muscle
contraction. Excitation-contraction coupling involves modulation of the cytosolic Ca2+-
concentration: the entry of Ca2+ in a cardiac myocyte through L-type Ca2+-channels
stimulates the release of Ca2+ from the SR through the ryanodine channel (also known as
the ryanodine receptor, RyR2), leading to activation of myofilaments (Figure 5). In addition
to Ca2+-entry through L-type Ca2+-channels, Ca2+-release through RyR2 is also modulated
by the interaction of RyR2 with calstabin 2. Resting Ca2+-concentrations are restored
mainly by re-uptake of Ca2+ into the SR by a Ca2+-ATPase (SERCA2), and removal of
Ca2+ from the cell by the Na+/Ca2+-exchanger (NCX). The activity of SERCA2 is regulated
by phospholamban (PLN). In its unphosphorylated state, phospholamban inhibits
SERCA2 activity, but upon phosphorylation (typically by PKA), this inhibition is released,
Mice lacking HDAC9 Normal cardiac size and function at early age, but spontaneous cardiac hypertrophy at advanced age Severely exaggerated response to TAC and calcineurin activation81
Mice lacking HDAC5 Profoundly enlarged hearts in response to TAC or constitutive cardiac activation of calcineurin82
MicroRNAs Forced expression of miR-195 in cardiac myocytes or in the hearts of transgenic mice
Hypertrophic growth and myocyte disarray, resulting in dilated cardiomyopathy and heart failure30
Knockdown of miR-133 in mice Hypertrophic growth of the heart with induction of fetal gene expression32
Overexpression of miR-133 in cardiac myocytes
Inhibition of agonist-induced hypertrophy32
miR-208-null mice Virtually no cardiac hypertrophy or fibrosis in response to TAC or calcineurin signalling34

Chapter 1 18
thereby increasing Ca2+-uptake. Phospholamban is potently inhibited by the
serine/threonine phosphatase PP1, which in turn is inhibited by the phosphatase inhibitor
I-1, after I-1 has been phosphorylated by PKA.
The binding of agonists to #-AR results in the activation of PKA, which leads to the
phosphorylation of L-type Ca2+-channels, RyR2, phospholamban and sarcomere proteins.
This process increases both cellular contraction and relaxation, through delivery of more
Ca2+ to the myofilaments, improved re-uptake of Ca2+ by the SR, and modulation of
myofilament Ca2+-sensitivity. The phosphorylation of RyR2 by PKA leads to dissociation of
calstabin 2, and this dissociation is proposed to increase leakage of Ca2+ from the SR and
thus arrhythmogenicity.83 Other research groups have proposed that phosphorylation of
RyR2 by CaMKII, rather than by PKA, is important for Ca2+-leakage through RyR2.84
In the failing heart, there is depressed PKA activity, reduced Ca2+-re-uptake by the SR,
increased RyR2 phosphorylation and calstabin-2 dissociation, and increased Ca2+-
extrusion through the NCX. Impaired Ca2+-re-uptake by the SR has been ascribed to a
decline in SERCA2 expression, reduced levels of phospholamban phosphorylation, and
depletion of SR Ca2+ through leaky RyR2 channels.85
An important pool of Ca2+ is also generated by Ca2+-influx into the cell through transient
receptor potential (TRP) channels. These channels are referred to as store-operated
because the opening of these channels is coupled to a decline in IP3-regulated
intracellular Ca2+-stores. Activation of these channels might have an important role in the
signalling pathways that lead to hypertrophy, with recent studies finding that the canonical
TRP channels TRPC1, TRPC3, and TRPC6 are involved in this process.86-88
In addition, intracellular Ca2+-levels rise in response to elevated Na+-concentrations via the
NCX, in turn caused by induced activity of the cardiac Na+/H+-exchanger (NHE). Its activity
is increased in several animal models of cardiac hypertrophy, including pressure
overload.89 The resulting elevation of intracellular Ca2+-levels leads to stimulation of
several signalling cascades promoting cardiac growth, including calcineurin-, CaMK-,
PKC- and MAPK-dependent pathways, providing a potential mechanism whereby NHE
might promote hypertrophy.
In addition, there is increased activation of Gq/11 protein-coupled receptor signalling in the
hypertrophied heart, which in turn increases PKC activity, thereby blocking activity of I-1
and increasing PP1 activation. This further reduces PLN phosphorylation and depresses
both cellular contraction and relaxation, by preventing re-uptake of Ca2+ by the SR.

Introduction 19
Activation of Gq/11 protein-coupled receptors also increases the amount of IP3 generated,
which interacts with receptors (IP3R) in the SR membrane to stimulate Ca2+-release.
Pools of intracellular Ca2+ activate cytosolic calmodulin-CaMKII, resulting in the activation
of NFAT, which then translocates to the nucleus, where it is involved in transcriptional regulation.

Chapter 1 20

Introduction 21
Figure 5. Calcium-handling abnormalities in myocytes of the failing heart In the failing heart, normal Ca2+-cycling becomes dysregulated by depressed PKA activity, reduced Ca2+-re-uptake by the SR, increased Ca2+-extrusion through the Na+/Ca2+-exchanger, and increased RyR2 phosphorylation and calstabin-2 dissociation. In addition, increased activation of Gq/11-protein-coupled receptor signalling represses both cellular contraction and relaxation, by preventing re-uptake of Ca2+ by the SR. Activation of Gq/11-protein-coupled receptors also increases the amount of IP3 generated, which interacts with IP3R in the SR membrane to stimulate Ca2+-release. AC indicates adenylate cyclase; AR, adrenergic receptor; CaM, calmodulin; CaMK, CaM kinase; cAMP, cyclic adenosine 3!, 5!-monophosphate; CN, calcineurin; DAG, diacylglycerol; I-1, inhibitor 1; IP3, inositol 1,4,5-trisphosphate; IP3R, IP3 receptor; NCX, Na+/ Ca2+-exchanger; NFAT, nuclear factor of activated T cells; PKA, cAMP-dependent protein kinase; PKC, protein kinase C; PLC, phospholipase C; PLN, phospholamban; PP1, protein phosphatase 1; RyR, ryanodine receptor; SERCA, sarcoplasmic reticulum Ca2+-ATPase; SR, sarcoplasmic reticulum; and TRPC, canonical transient receptor potential channels.
Dysregulated energy metabolism
The heart has a high and constant workload, and cardiac energy supply and metabolism
are tightly regulated. This regulation becomes compromised in the failing heart, which can
lead to a state of inefficiency and energy starvation. Unlike other organs, the heart has a
limited capacity for storing fuel, so substrates need to be produced efficiently and quickly,
mainly from circulating free fatty acids and, to a lesser degree, from glucose. In the failing
heart, the synthesis of adenosine 5!-triphosphate (ATP) is compromised, partially as a
result of mitochondrial dysfunction and, probably, altered substrate utilisation (i.e.
increased catabolism of glucose).
The peroxisome proliferator-activated receptor-" (PPAR-") coactivator 1 (PGC1) family of
transcriptional coactivators comprises important regulators of mitochondrial function, with
PGC1# being the best-studied family member in the heart. This coactivator functions to
increase the level of oxidative phosphorylation to meet the energy demands of cardiac
growth in response to physiological stimuli, for which the heart uses mainly free fatty
acids. The expression of the gene encoding PGC1# and of its downstream targets is
reduced in mouse models of pathological cardiac hypertrophy and failure, which is
consistent with the mitochondrial dysfunction seen in these models. Furthermore, PGC1#-
deficient mice have an exacerbated heart failure phenotype in response to pressure
overload.90
Abnormalities in ATP storage are another aspect of dysregulated energy metabolism in
failing hearts. Creatine kinase reversibly converts phosphocreatine and ADP to creatine

Chapter 1 22
and ATP when energy is needed rapidly. The ratio of phosphocreatine and ATP
concentrations has been used as a measure of this energy balance, and failing human
hearts display abnormalities in this ratio and in ATP flux.91
The density of capillaries in the heart muscle is another important parameter that affects
energy availability. In mice with hypertrophy as a result of cardiac-specific overproduction
of Akt, pathological cardiac remodelling is associated with an inability of angiogenesis to
keep up with muscle growth.92
Sarcomeric alterations The beating of the heart depends ultimately on the force generated by the sarcomere,
through the interaction of thick filaments, which are composed of myosin, with actin-
containing thin filaments (Figure 6). In rodent heart failure models, the expression ratio of
the #MHC and $MHC isoforms shifts, which can alter the contractile phenotype.
Adult rodent hearts normally contain #MHC, which has faster cross-bridge kinetics but
generates less tension than the fetal $MHC protein. However, in the setting of cardiac
hypertrophy or failure, the gene encoding $MHC is re-expressed, contributing up to half of
the total amount of MHC. Human hearts contain nearly exclusively $MHC, but changes
can still occur in failing hearts, with further reductions in the little residual expression of
#MHC.93
Besides actin and myosin, the sarcomere contains regulatory thin filaments (which consist
of troponins and #-tropomyosin), interlinking proteins (myosin-binding protein C and titin),
and a protein complex (in the Z-disk) that couples mechanical forces to signalling by
protein kinases and phosphatases. The titin-cap (T-cap or telethonin) and muscle LIM
protein (MLP or CLP) also interact with titin at the Z-disk, and are thought to form a
mechanical stretch receptor that affects sarcomere contraction. Titin has garnered
attention as an important regulator of myocardial mechanosignalling and structural
stiffness. It is the largest protein known and spans the half sarcomeric distance from the
Z-disk to the M-line. In the I-band region of the sarcomere, titin is extensible and functions
as a molecular spring that develops force in stretched sarcomeres. This force largely
determines the passive force of the cardiac myocyte and, together with collagen,
determines myocardial passive stiffness.94 The myocardium expresses two titin isoforms:
the more rigid N2B isoform and the more compliant N2BA isoform. The relative expression
of titin isoforms affects passive stiffness as indicated by the correlation between the

Introduction 23
N2BA:N2B titin isoform ratio and the ratio of end-diastolic volume to end-diastolic
pressure. In patients with end-stage HF secondary to non-ischaemic dilated
cardiomyopathy, the N2BA to N2B expression ratio was significantly increased compared
to controls, without affecting total titin levels.95, 96 These results suggest that human hearts
may adjust their passive stiffness by titin-isoform switching.
Phosphorylation-dependent control of passive force is also important in pathophysiology.
The N2B segment of cardiac titin can be phosphorylated by PKA, which initiates a
decrease in passive tension.97 More recently, it has also been demonstrated that PKG and
PKC# equally promote titin phosphorylation.98, 99 In the case of PKG, the phosphorylation
leads to a decrease in the cardiac myocyte passive tension. In the case of
phosphorylation by PKC, the global effect is an increase in passive tension.
Several studies suggest that hypophosphorylation of the PKA/PKG phosphorylation sites
on titin contributes to reduced compliance in various forms of HF with preserved ejection
fraction, including aortic stenosis and diabetes.99-101 Thus, an abnormal titin-
phosphorylation state may increase titin-based stiffness and contribute to altered diastolic
stiffness in HF.
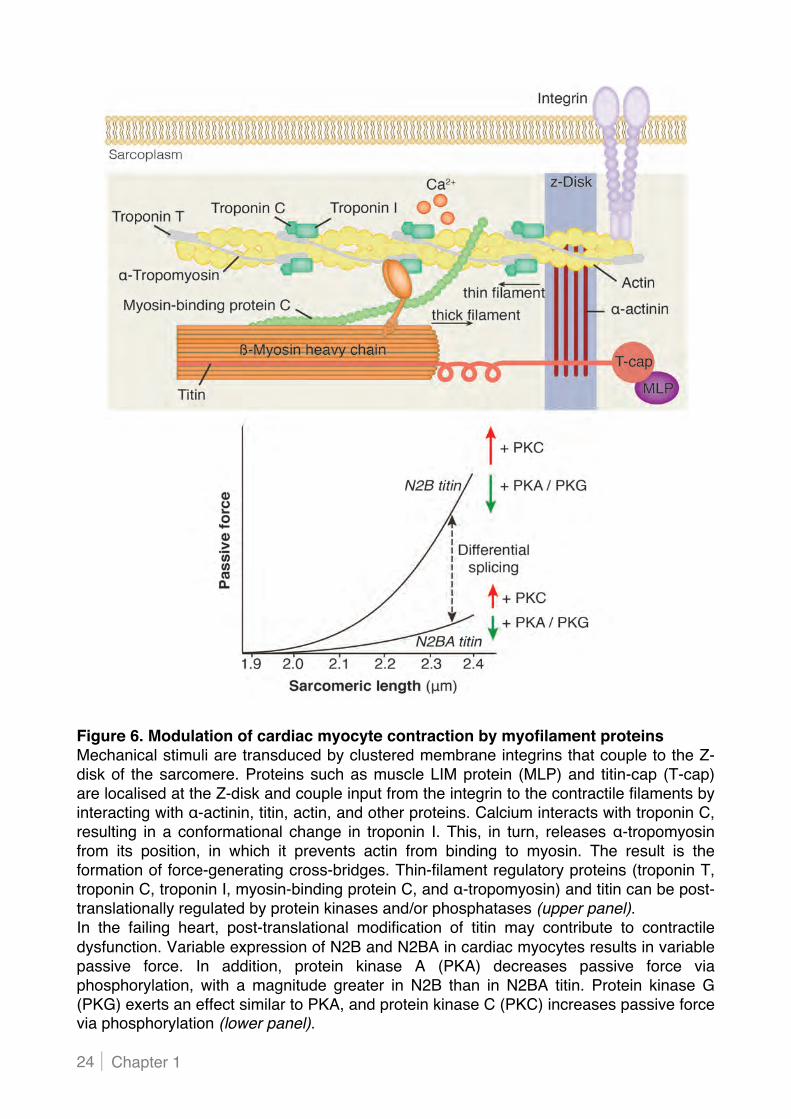
Chapter 1 24
Figure 6. Modulation of cardiac myocyte contraction by myofilament proteins Mechanical stimuli are transduced by clustered membrane integrins that couple to the Z-disk of the sarcomere. Proteins such as muscle LIM protein (MLP) and titin-cap (T-cap) are localised at the Z-disk and couple input from the integrin to the contractile filaments by interacting with #-actinin, titin, actin, and other proteins. Calcium interacts with troponin C, resulting in a conformational change in troponin I. This, in turn, releases #-tropomyosin from its position, in which it prevents actin from binding to myosin. The result is the formation of force-generating cross-bridges. Thin-filament regulatory proteins (troponin T, troponin C, troponin I, myosin-binding protein C, and #-tropomyosin) and titin can be post-translationally regulated by protein kinases and/or phosphatases (upper panel). In the failing heart, post-translational modification of titin may contribute to contractile dysfunction. Variable expression of N2B and N2BA in cardiac myocytes results in variable passive force. In addition, protein kinase A (PKA) decreases passive force via phosphorylation, with a magnitude greater in N2B than in N2BA titin. Protein kinase G (PKG) exerts an effect similar to PKA, and protein kinase C (PKC) increases passive force via phosphorylation (lower panel).

Introduction 25
1.1.4 Anthracycline-induced heart failure
Cardiotoxicity occurs during therapy with several cytotoxic drugs and may be the dose-
limiting factor in cancer treatment and hence tumour response. Furthermore, cardiotoxicity
can also be responsible for long-term side effects and may cause severe morbidity in
surviving cancer patients.
Cytotoxic drugs with potential myocardial toxicity include:
• Antibiotics: anthracyclines, bleomycin
• Alkylating agents: cyclophosphamide, ifosfamide, cisplatin, mitomycin, busulfan
• Antimetabolites: 5-fluorouracil, capecitabine, methotrexate, fludarabine, cytarabine
• Antimicrotubule agents: paclitaxel, docetaxel, etoposide, teniposide, vinca alkaloids
• Monoclonal antibodies: trastuzumab, rituximab
• Tyrosine kinase inhibitors: imatinib mesylate, sunitinib
• Miscellaneous drugs: tretinoin, pentostatin, interferon, IL-2
Of all these chemotherapeutic drugs, anthracyclines are the most notorious offenders.
(i) Clinical application of anthracyclines
Anthracyclines are among the most active and broad-spectrum antineoplastic agents used
in the treatment of several malignancies. This class of drugs comprises the naturally
occurring doxorubicin and daunorubicin, as well as the synthetic derivatives epirubicin and
idarubicin. Additional anthracyclines have attained clinical approval, including pirarubicin,
aclacinomycin A (aclarubicin), and mitoxantrone. Anthracyclines have a broad range of
clinical applications for adult and paediatric malignancies, with demonstrated efficacy for
the treatment of haematological cancers (leukaemias and lymphomas) as well as a variety
of solid malignancies (carcinomas and sarcomas).
Although there is a dose-response relationship for anthracyclines in the treatment of
cancer, there is also a dose-related cardiac toxicity that occurs with all drugs of this class.
In order to improve the efficacy and/or cardiac safety of currently approved anthracyclines,
the search for new analogues or tumour-targeted formulations continues unabated.
For example, liposomal formulations of anthracyclines, favouring accumulation in tumours,
display increased efficacy and cardiac tolerability.

Chapter 1 26
(ii) Acute and chronic cardiotoxicity and risk factors
Anthracyclines have been reported to cause both early- and late-onset cardiac effects. Although acute cardiotoxicity, manifested by electrocardiographic (ECG) changes and
depressed myocardial contractility, is commonly seen in patients during the first 24 hours
after drug infusion, the effects are generally transient and resolve spontaneously. Of far greater concern is the chronic cardiotoxic effect of anthracyclines, persisting after
discontinuation of the chemotherapeutic treatment. Clinical symptoms may include all
signs of cardiomyopathy, such as electrophysiological changes, decreased LV function,
changes in exercise-stress capacity, and overt signs of congestive HF.
The cumulative dose seems to be the most important risk factor for anthracycline-induced
cardiotoxicity. Doxorubicin, the most widely used anthracycline, exhibits greater
cardiotoxicity than the other drugs of this class. Although the total cumulative dose of
doxorubicin that is associated with the development of cardiomyopathy varies widely (from
75 to 1095 mg/m2), the median dose at which this toxicity is observed was reported to be
390 mg/m2 in 88 cases in a series of 4018 patients (overall incidence of 2.2%).102, 103
The dose-related risk was 3% with a dose of 400 mg/m2, 7% with 550 mg/m2, 18% with
700 mg/m2, and 50% with 950 mg/m2. The slope of the dose-toxicity curve increases at
about 550 mg/m2. These observations have led to 550 mg/m2 doxorubicin being
considered to be the maximum safe cumulative dose. Cardiomyopathy is reported to
develop between 0 and 231 days (median 23 days) following the final dose of doxorubicin,
however, delayed development of cardiotoxicity of up to 20 years following therapy has
been reported.103, 104 The maximum cumulative dosage needed to obtain minimal
cardiotoxicity varies among the different anthracyclines. Also, a wide variation exists in the
individual cardiac sensitivity to anthracyclines. This variation in individual sensitivity
implies that an arbitrary dose limitation of 550 mg/m2 for doxorubicin in order to avoid
cardiotoxicity may deprive some patients, who could safely receive a much higher dose of
doxorubicin, of the maximum therapeutic benefit of the drug.
Besides the cumulative dose of the anthracycline, several other factors can contribute to
the development of chronic cardiotoxicity. These include high serum peak levels of
anthracyclines, mediastinal radiation, a young or advanced age at the time of treatment,
pre-existing heart disease, hypertension, and female sex. Also combination treatment with
other anti-cancer drugs such as cyclophosphamide and mitomycin-C may possibly
increase the risk of anthracycline cardiotoxicity.

Introduction 27
(iii) Pathophysiology
Anthracyclines cause myocardial damage and induce apoptosis and necrosis in cardiac
myocytes, thereby decreasing the number of myocardial cells. The wall of the left ventricle
thins, and stress on the heart increases as a result of increased afterload.
Furthermore, the anthracycline-induced changes in the myocardium decrease contractility.
In time, elevated LV afterload and depressed contractility cause depression of the overall
LV function, leading to the development of the clinical syndrome of heart failure.
Despite a remarkably extensive literature on many aspects of cardiotoxicity,
a comprehensive unifying theory for the deleterious effects of anthracyclines on the heart
is still lacking.
Increased oxidative stress
A prominent hypothesis regarding the aetiology of anthracycline-induced cardiotoxicity is
that cardiac damage is caused by oxidative stress, i.e. the generation of ROS including
free radicals such as superoxide and hydroxyl radicals, and non-radical oxygen species
such as hydrogen peroxide. Administering doxorubicin to laboratory animals105-115 and
humans116-118 resulted in an elevation of plasma and tissue ROS, and decreased plasma
and tissue anti-oxidant levels. The high level of oxidative stress generated by
anthracyclines is accounted for by structural characteristics that allow the drugs to
participate in electron-accepting and -donating reactions.
Anthracyclines possess a unique hexose sugar, daunosamine, attached to a tetracycline
structure containing adjacent quinone and hydroquinone moieties permitting these drugs
to accept an electron and be reduced to a semiquinone radical (Figure 7).
The semiquinone radical readily donates its electron to molecular oxygen to form the
superoxide radical. Many intracellular enzymes can reduce anthracyclines, including
cytosolic xanthine oxidase and microsomal NADPH-cytochrome P450 reductase.119
Several findings also implicate a crucial role for NOS3, the NOS isoform with the highest
affinity for doxorubicin. Doxorubicin can undergo a direct reduction at the reductase
domain of NOS3, leading to enhanced superoxide formation.120
If free iron is present, the semiquinone radical can also form a complex with iron, resulting
in an anthracycline-iron free radical complex.121 This complex reduces oxygen, thereby
creating superoxide and regenerating the anthracycline.

Chapter 1 28
Superoxide is converted by superoxide dismutase to hydrogen peroxide, which is
eliminated from the body by an enzymatic anti-oxidant defence system (catalase and
glutathione peroxidase). However, in the presence of reduced iron or copper, formation of
highly toxic hydroxyl radicals occurs via a Fenton or Haber-Weiss reaction.122
Unlike hydrogen peroxide and the superoxide anion radical, hydroxyl radicals are
extremely reactive and cannot be neutralised by anti-oxidant enzymes. Instead, hydroxyl
radicals react with polyunsaturated fatty acids forming lipid peroxides, conjugated dienes,
and malondialdehyde (MDA). As a consequence, the structure of the lipid bilayer is
modified, resulting in cell membrane damage followed by cell dysfunction.
Cardiac cells are, in comparison to cells of other organs, more prone to damage by free
radicals because of the high oxidative metabolism and lower concentrations of anti-oxidant
enzymes in the heart. There is 150 times less catalase and four times less superoxide
dismutase in the heart, and rapid inactivation of glutathione peroxidase-1.123, 124
In addition, the unique structure of cardiac myocytes, in which 50% of the cell organelles
are mitochondria, serving both as a source and target for ROS, may explain why
anthracycline antibiotics are selectively toxic to the heart. Moreover, cardiac cells generate
very high levels of free radicals in the presence of doxorubicin, because of their unique
mitochondrial NADH dehydrogenase.125 Reduction of anthracyclines to the corresponding
semiquinone by this enzyme produces an extremely high level of oxidative stress.
The cationic anthracyclines are also retained in the mitochondrial inner membrane by
forming a nearly irreversible complex with cardiolipin, a phospholipid of the inner
mitochondrial membrane.126 The proteins of the electron transport chain require cardiolipin
binding to function properly, and it has been argued that disruption of the cardiolipin-
protein interface causes more superoxide formation.127 Other membrane proteins, such as
those responsible for the transfer of carnitine, can also be adversely affected by
anthracyclines, contributing to the decrease in mitochondrial function.128 This functional
disruption leads to ultrastructural pathological changes such as mitochondrial swelling and
myelin figures within the mitochondria. Anthracycline treatment also affects mitochondrial
gene expression, indicating that doxorubicin interferes with both nuclear and mitochondrial
transcriptional regulation.129, 130
Interestingly, the effect of oxidative stress in clinical cardiotoxicity is increasingly
questioned. One reason for the uncertainty is the apparent lack of protection provided by
anti-oxidants, including vitamin E and N-acetylcysteine, in long-term experimental studies

Introduction 29
and clinical trials.131, 132 Although the protective effects of carvedilol, an adrenoceptor
blocker, were tentatively attributed to its anti-oxidant properties, comparative studies of
carvedilol with other adrenolytic agents without anti-oxidant properties are missing.133
The only compound consistently found to be cardioprotective in experimental and clinical
studies is the iron chelator dexrazoxane.134-136 Dexrazoxane does not directly inactivate
free radicals but, instead, attenuates their formation through intracellular iron chelation.
Although considerable data show that anthracyclines can promote ROS formation in
cardiac tissue, the evidence that oxidative stress is the sole or the main cause of chronic
anthracycline cardiotoxicity in patients remains inconclusive.

Chapter 1 30
Figure 7. Anthracycline-induced formation of free radicals in cardiac myocytes Anthracyclines can be enzymatically reduced to a semiquinone radical, which donates its electron to oxygen to form superoxide radicals. If free iron is present, the semiquinone radical can also form a complex with iron, resulting in superoxide generation and regeneration of the anthracycline. Superoxide is converted by superoxide dismutase to hydrogen peroxide, which is eliminated by anti-oxidant enzymes, or in the presence of reduced iron or copper, is transformed to highly toxic hydroxyl radicals. Moreover, cardiac cells possess a unique mitochondrial NADH dehydrogenase, increasing oxidative stress levels by reducing anthracyclines. Anthracyclines also interfere with cardiolipin, essential for the electron transport chain. A indicates anthracycline; NAD(P)H, nicotinamide adenine dinucleotide (phosphate); NOS, nitric oxide synthase; and SOD, superoxide dismutase.

Introduction 31
Cardiac myocyte apoptosis
Apoptosis is a highly conserved, tightly regulated, and energy-dependent active form of
cell death. Apoptosis starts from two canonical signalling pathways, the extrinsic and
intrinsic pathway.
In the extrinsic pathway, binding of death ligands (FasL, TNF-#, TRAIL) with their
receptors induces recruitment and activation of caspase 8, which subsequently activates
downstream effector caspases such as caspase 3.
The intrinsic pathway is mediated by mitochondrial cytochrome c release. This process is
regulated by the members of the B-cell leukaemia/lymphoma-2 (Bcl-2) family, which
includes three groups: the anti-apoptotic members (Bcl-2, Bcl-XL, and Mcl-1), the pro-
apoptotic members (Bax, Bak), and the Bcl-2 homology 3 (BH3)-only proteins (Bad, Bid,
BNip3, and BNip3L) that enhance apoptosis via inhibition of anti-apoptotic Bcl-2 proteins
or activation of pro-apoptotic Bcl-2 associated X protein (Bax) and Bcl-2 antagonist/killer
(Bak). Activation of BH3-only proteins by stress stimuli promotes Bax/Bak translocation
from the cytosol to the outer membrane of mitochondria, resulting in increased
mitochondrial outer membrane permeabilisation. This, in turn, leads to protein release
from the intermembrane space to the cytoplasm, including the apoptogenic molecule
cytochrome c. In the cytosol, cytochrome c forms a complex with the adaptor protein
apoptosis protease activator protein-1 (Apaf-1), dATP, and caspase 9. The result is the
formation of a structure known as the apoptosome, which in turn activates caspase 9.
Both the extrinsic and intrinsic apoptotic pathways converge on the activation of the
downstream executioner caspases 3, 6, and 7, and are involved in anthracycline-induced
cardiotoxicity (Figure 8).
Extrinsic apoptotic pathway
Although cardiac myocytes are generally resistant to Fas-induced apoptosis, studies
indicate that cardiac myocyte apoptosis in doxorubicin-induced cardiomyopathy can be
executed through a Fas-mediated pathway. Increased Fas and FasL levels were shown
after doxorubicin treatment and both in vitro and in vivo studies demonstrated that
blocking of the Fas/FasL interaction with a FasL-neutralising antibody inhibited
doxorubicin-induced toxicity in cardiac myocytes.137-140 Moreover, cardiac-targeted
expression of soluble Fas, a competitive inhibitor of FasL, attenuated doxorubicin-induced
cardiotoxicity.141 Other studies showed that doxorubicin treatment of rat cardiac myocytes

Chapter 1 32
increased mitochondrial ROS production, activated the Ca2+/calcineurin signalling
pathway, and further activated NFAT4, leading to upregulation of Fas/FasL.140
The transcription factor NF-%B was also activated by ROS in doxorubicin-treated neonatal
rat cardiac myocytes and myocardium, and exerted a pro-apoptotic effect via direct
activation of apoptotic genes, including FasL, Fas, c-Myc, and p53.142-144
Intrinsic apoptotic pathway
In cardiac myocytes, the mitochondria are located near SR Ca2+-release sites, and can
capture a large quantity of the released Ca2+. Due to the significantly raised oxidative
stress, mitochondrial Ca2+-levels increase beyond a threshold. This mitochondrial Ca2+-
overload triggers mitochondrial permeability transition pore (MPTP) opening, resulting in a
loss of mitochondrial membrane potential (MMP, &'m), mitochondrial swelling, outer
membrane rupture, and consequent release of cytochrome c and apoptosis inducing
factor from mitochondria.145, 146 Numerous studies have also shown that doxorubicin-
induced cardiac myocyte apoptosis is associated with increased expression and activation
of the tumour suppressor protein p53.147-149 DNA lesions, induced by anthracycline
treatment, result in increased phosphorylation of p53, which in turn upregulates
downstream genes such as Bax, leading to activation of the intrinsic apoptotic pathway.
Moreover, anthracycline cardiotoxicity was associated with GATA4 depletion, which
sequentially caused cardiac myocyte apoptosis.150 The transcriptional factor GATA4,
strongly expressed in cardiac myocytes, has been shown to be a pivotal survival factor in
the postnatal period, and transcriptionally regulates the apoptotic pathway via activating
the gene encoding the anti-apoptotic Bcl-XL.
Introduction 33
Figure 8. Activation of apoptotic pathways by anthracyclines in cardiac myocytes Anthracyclines induce the intrinsic apoptotic pathway by inducing cytochrome c release from the mitochondria. Increased expression, activation, and phosphorylation of p53, and GATA4 depletion are as well associated with anthracycline treatment, further regulating members of the Bcl-2 family. Cardiac myocyte apoptosis in anthracycline-induced cardiomyopathy can also be executed through a Fas-mediated pathway, inducing the extrinsic apoptotic pathway. Both pathways converge on activation of downstream caspases, cleaving a variety of substrates. A indicates anthracycline; Apaf, apoptosis activating factor; Bcl-2, B-cell leukaemia/ lymphoma-2; Bcl-XL, Bcl-extra large; BH3-only, subfamily of Bcl-2 protein family with Bcl2 homology (BH) 3 domain; Bad, Bcl-2 associated death promoter; Bak, Bcl-2 antagonist/killer; Bax, Bcl-2 associated X protein; Bid, BH3 interacting-domain death agonist; casp, caspase; DISC, death-inducing signalling complex; FasL, Fas ligand; p53, (tumour) protein 53; ROS, reactive oxygen species; and &'m, mitochondrial membrane potential.
Disturbed Ca2+-homeostasis
Reactive oxygen species, produced during redox cycling of anthracyclines, target several
ion channels in cardiac myocytes, including the RyR2. In an animal model, doxorubicin-

Chapter 1 34
induced ROS production induced channel opening, SR Ca2+-release, and subsequent SR
Ca2+-store depletion.151 Opening of L-type Ca2+-channels was also triggered by ROS-
induced phosphorylation. Moreover, doxorubicin reduced SERCA2 transcription, which
could be prevented by the anti-oxidant N-acetylcysteine and blocking of ERK.152
Doxorubicin also has been reported to prolong action potential duration through ROS-
mediated inhibition of the delayed rectifier K+-current.151
Oxidative stress also induces MPTP opening with alterations in mitochondrial Ca2+-
transport. In vitro experiments showed that doxorubicin treatment caused an irreversible
decrease in the mitochondrial Ca2+-loading capacity.153
Sarcomeric alterations
Anthracyclines induce PKC-mediated phosphorylation of troponin I and C, resulting in
damage and loss of cardiac myofibrils. Anthracycline treatment also leads to decreased
expression of #-actin, troponin I, MLC2, and creatine kinase.154 This inhibition of muscle
gene expression by doxorubicin is limited to cardiac myocytes. Doses 100 times the
concentration used in cardiac cultures caused no alteration of either muscle or non-muscle
gene mRNAs in rat and human skeletal muscle cells.154
Doxorubicin also supresses the expression of GATA4, an important transcriptional
regulator of several cardiac genes, including #MHC, $MHC, and troponin I. Doxorubicin-
induced depletion of GATA4 could be responsible for the observed decrease in cardiac
troponin I expression.13
It was also shown that anthracyclines induce proteolysis of titin, possibly predisposing
cardiac myocytes to diastolic dysfunction, myofilament instability, and cell necrosis.155
Effects on non-myocyte cardiac cells
Myocytes comprise approximately 80% of the cardiac mass but constitute less than 20%
of the total cell count. Other cell types, including fibroblasts, endothelial cells, and smooth
muscle cells, provide structural and trophic support to the myocytes. Cardiac endothelial
cells and fibroblasts may be more sensitive to the toxic effects of doxorubicin than cardiac
myocytes, suggesting that cardiac myocyte deterioration may be preceded by alterations
in matrix composition and in paracrine signals or intercellular cross talk. Studies of
endothelial cells support this concept, but more studies are required to obtain a
comprehensive view and better understanding.156, 157

Introduction 35
Furthermore, it was shown that doxorubicin enhances cardiac expression and activity of
matrix metalloproteinases (MMP)-2 and -9, which is believed to contribute to
cardiomyopathy by weakening the collagenous matrix.158, 159
Finally, cardiac myocytes are more resistant to ROS formation than cardiac progenitor
cells, a pool of resident primitive cells that are self-renewing, clonogenic, and multipotent
in vitro and that regenerate myocytes and coronary vessels in vivo. Recently, it was tested
whether anthracycline-induced cardiomyopathy could be viewed as a stem cell disease.
Exposure of cardiac progenitor cells to doxorubicin resulted in an increase in ROS
production, DNA damage, p53 expression, telomere attrition, apoptosis, and decreased
cardiac progenitor cell growth. Injection of syngeneic cardiac progenitor cells in the failing
myocardium promoted regeneration of cardiac myocytes and vascular structures after
doxorubicin treatment.160 The role of cardiac progenitor cells in anthracycline-induced
cardiotoxicity is an active area of investigation, as well as other cardiotoxic mechanisms,
including neuregulin signalling, ceramide accumulation, and cannabinoid signalling.

Chapter 1 36
1.2 Cyclic GMP signalling in the cardiovascular system Over the past 15 years, the cyclic nucleotide cGMP has emerged as an important
intracellular second messenger involved in the regulation of cardiovascular homeostasis.
cGMP is synthesised from GTP by the enzyme guanylate cyclase (GC) in response to
nitric oxide (NO) or natriuretic peptides (NPs), and acts through activation of cGMP
dependent protein kinase (PKG), or modification of phosphodiesterase (PDE) enzyme
activities; the latter of which are also responsible for cGMP catabolism. As is the case with
cAMP signalling, specificity of cGMP signals is achieved through compartmentalisation of
both GCs and downstream effectors.
1.2.1 Synthesis by guanylate cyclases
Biosynthesis of cGMP from GTP is catalysed by two different GC isoforms, one which
functions as the biosensor for NO, and the other as the plasma membrane receptor for
NPs.
(i) Nitric oxide-mediated biosynthesis of cGMP
Nitric oxide is synthesised by NOS, which exists as three isoforms: neuronal NOS (NOS1
or nNOS), inducible NOS (NOS2 or iNOS), and endothelial NOS (NOS3 or eNOS).
All three isoforms have been detected in cardiac myocytes, vascular smooth muscle cells
(VSMC), and vascular endothelial cells. NOS is active as a homodimer with a central
prosthetic group, requiring BH4, oxygen, calmodulin, NADPH, flavin mononucleotide, and
flavin adenine dinucleotide, to effectively generate NO from L-arginine. Inducible NOS
synthesises NO in much larger amounts than nNOS and eNOS3.161
The biosensor of NO, soluble GC (sGC), is a heterodimer with an #-subunit and a $-
subunit, and exists in various isoforms with two different #-subunits (#1 and #2) and two
different $-subunits ($1 and $2).162 The #1$1 heterodimer is the most prevalent sGC
isoform, the #2$1 isoform is a less active cyclase, and neither the #1$2 nor #2$2
isoforms have any reported GC activity.163, 164
Activation of sGC by NO is dependent on a haem group, which is orientated between the
two subunits, principally through coordination with His-105 in the N-terminal part of the $1-
subunit. Mutation of this residue prevents haem binding and results in an sGC that retains
basal activity, but is unresponsive to NO (Figure 9).165 Early in vitro studies suggested that
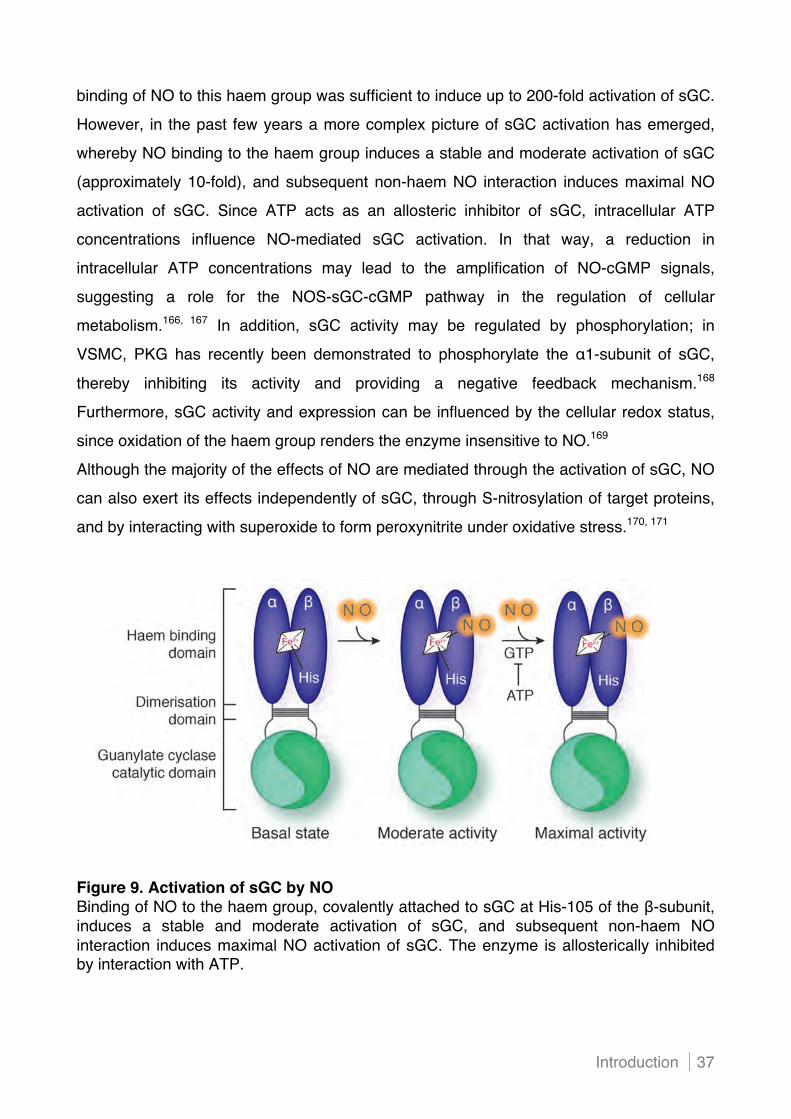
Introduction 37
binding of NO to this haem group was sufficient to induce up to 200-fold activation of sGC.
However, in the past few years a more complex picture of sGC activation has emerged,
whereby NO binding to the haem group induces a stable and moderate activation of sGC
(approximately 10-fold), and subsequent non-haem NO interaction induces maximal NO
activation of sGC. Since ATP acts as an allosteric inhibitor of sGC, intracellular ATP
concentrations influence NO-mediated sGC activation. In that way, a reduction in
intracellular ATP concentrations may lead to the amplification of NO-cGMP signals,
suggesting a role for the NOS-sGC-cGMP pathway in the regulation of cellular
metabolism.166, 167 In addition, sGC activity may be regulated by phosphorylation; in
VSMC, PKG has recently been demonstrated to phosphorylate the #1-subunit of sGC,
thereby inhibiting its activity and providing a negative feedback mechanism.168
Furthermore, sGC activity and expression can be influenced by the cellular redox status,
since oxidation of the haem group renders the enzyme insensitive to NO.169
Although the majority of the effects of NO are mediated through the activation of sGC, NO
can also exert its effects independently of sGC, through S-nitrosylation of target proteins,
and by interacting with superoxide to form peroxynitrite under oxidative stress.170, 171
Figure 9. Activation of sGC by NO Binding of NO to the haem group, covalently attached to sGC at His-105 of the $-subunit, induces a stable and moderate activation of sGC, and subsequent non-haem NO interaction induces maximal NO activation of sGC. The enzyme is allosterically inhibited by interaction with ATP.

Chapter 1 38
(ii) Natriuretic peptide-mediated biosynthesis of cGMP
Natriuretic peptides comprise a family of polypeptide mediators secreted by the heart and
vasculature with fundamental roles in the regulation of blood volume, systemic vascular
resistance, central venous pressure, and cardiac contractility. The principal NPs are atrial
natriuretic peptide (ANP), brain natriuretic peptide (BNP), and C-type natriuretic peptide
(CNP). ANP and BNP are secreted primarily by the atria and ventricles of the heart,
respectively, while CNP is secreted by the vascular endothelium. Cardiac production and
release of ANP and BNP are triggered mainly by increases in myocardial wall stretch
and/or pressure but may also be influenced by neurohumoral factors.172 The NPs have
vasodilatory effects and promote natriuresis and diuresis. In addition, they exert
autocrine/paracrine effects within the heart itself. Natriuretic peptides exert their biological
effects in the cardiovascular system by binding to membrane-associated GC receptors,
known as NPRs or particulate GCs. There are at least seven mammalian membrane-
associated GCs, but two subtypes, NPR-A and NPR-B, are responsible for the majority of
the physiological effects of NPs. NPR-A is activated by ANP and BNP, which both bind to
NPR-A with relatively high affinity, but ANP is more potent than BNP in receptor activation.
NPR-B mediates the paracrine action of CNP in vascular regeneration and endochondral
ossification. Whereas NPR-A is widely expressed throughout the cardiovascular system,
NPR-B is highly abundant in vascular endothelium and smooth muscle, but its presence in
the heart is thought to predominantly localise to the non-myocyte population (mostly
fibroblasts).173, 174 However, studies have also reported CNP/NPR-B signalling in
myocytes, and have shown anti-hypertrophic and pro-apoptotic effects in cardiac
myocytes.175-177
NPR-A consists of an extracellular ligand binding domain, a transmembrane spanning
region, an intracellular protein kinase like homology domain (KHD), a hinge region, and a
C-terminal GC catalytic domain. The KHD is central to regulating GC activity and
modulating receptor sensitivity.178 In the basal state, NPR-A is phosphorylated and the GC
catalytic domain of the receptor is repressed. Upon hormone ligand binding, the kinase
homology domain of NPR-A becomes dephosphorylated, the catalytic domain is activated,
and cGMP generated.

Introduction 39
1.2.2 Activation of effector molecules
Cyclic GMP exerts its physiological actions in the cardiovascular system by activating
cGMP-dependent protein kinases and phosphodiesterases.
(i) Cyclic GMP-dependent protein kinases
Three PKG isotypes have been identified, two of which are splice variants of a single
gene. PKG type I (PKG-I), consisting of an #- and $-isoform, is the prominent isotype in
the cardiovascular system. PKG-I is expressed at very high levels in VSMC and
endothelial cells and at lower levels in cardiac myocytes. PKG exists as a homodimer,
with each subunit consisting of three functional domains: an N-terminal domain,
a regulatory domain, and a kinase domain. The N-terminal domain mediates PKG
homodimerisation, suppresses kinase domain activity in the absence of cGMP, and
interacts with target substrate proteins. Upon cGMP binding to specific sites in the
regulatory domain, PKG undergoes a conformational change, resulting in the release of
the N-terminus inhibition of the kinase domain, which then catalyses the phosphorylation
of a target substrate protein.
(ii) Phosphodiesterases
The PDEs comprise a 21-gene superfamily categorised into 11 isoenzymes (PDE1-
PDE11) with a total of more than 50 isoforms. Each break the phosphodiester bond in
cGMP and/or cAMP resulting in the linear GMP or AMP. Cyclic GMP regulates the activity
of PDE 2, 3, 5, and 9. Of these, PDE 2, 3, and 5 are known to be expressed in cardiac
myocytes; PDE 3 and 5 are expressed in VSMC; and PDE 2, 3, and 5 are expressed in
vascular endothelial cells.179 While expressed in the heart, the cardiovascular role of
PDE9 remains to be elucidated.
Cyclic GMP-activated PDEs
cGMP selectively activates PDE 2 and 5 by binding to regulatory GAF domains in the N-
terminus. PDE2 is thought to be plasma membrane associated and hydrolyses both cAMP
and cGMP. Upon cGMP binding, PDE2 undergoes a conformational change and
increases its enzymatic activity for cAMP. Its dual-substrate specificity allows it to mediate

Chapter 1 40
negative cross-talk between cGMP and cAMP signalling pathways. Through PDE2
activation, cGMP is able to reduce cAMP signalling and affect cardiac function.
Whereas PDE2 can hydrolyse both cAMP and cGMP, PDE5 selectively hydrolyses cGMP.
PDE5 has a high affinity for cGMP, and its catalytic activity increases by an order of
magnitude when cGMP binds to its regulatory GAF domain. Catalytic activity is further
enhanced by phosphorylation, mostly by PKG, which stabilises cGMP binding. Of the
three N-terminal variants (PDE5A1-3), PDE5A1 and PDE5A2 are widely distributed, while
PDE5A3 has been suggested to be specifically expressed in smooth muscle cells and
cardiac myocytes. There is no known difference in activity among these isoforms.
In cardiac myocytes, PDE5 is not localised at the sarcolemma, but in the z-bands. Studies
have shown that the subcellular localisation of PDE5A to myocyte z-bands depends upon
NOS-NO-cGMP signalling. In mice genetically lacking NOS3, or those with NOS
chronically inhibited, PDE5A distribution within the myocyte became diffuse.180 PDE5 is a
well-established regulator of vascular smooth muscle contraction through regulation of
cGMP, with high levels of expression in the lung and corpus cavernosum, resulting in
therapeutic use of PDE5 inhibitors to treat pulmonary hypertension and erectile
dysfunction.181, 182
Cyclic GMP-inhibited PDEs
Although PDE3 is a dual-substrate enzyme, this enzyme largely targets cAMP, and can be
competitively inhibited by cGMP. At low cGMP levels, PDE3 may have a greater role in
controlling cGMP levels. Two isoforms, PDE3A and PDE3B, have been identified in
humans, with three N-terminal variants of PDE3A expressed in cardiac myocytes and
vascular smooth muscle, and the single isoform of PDE3B expressed in cardiac myocytes.
Both PDE3A and PDE3B are activated by PKA-mediated phosphorylation. PDE3B, and
possibly PDE3A, can also be activated by PI3K/Akt signalling triggered by insulin, insulin
growth factor (IGF)-1, and leptin.183, 184
In the cardiovascular system, PDE3 is involved in regulating cardiac myocyte and vascular
smooth muscle contractility, as well as the vascular smooth muscle phenotype switch and
stress response.185 PDE3 regulates cardiac myocyte contractility and relaxation via L-type
Ca2+-channels and SERCA2, respectively.186, 187

Introduction 41
1.2.3 Breakdown by PDEs
In addition to the above-mentioned PDE isoforms, two other isoforms also hydrolyse
cGMP and are relevant to the cardiovascular system; PDE1 and PDE9. The PDE1 family
is activated by Ca2+/calmodulin binding, whereas PKA-mediated phosphorylation of PDE1
reduces Ca2+/calmodulin binding affinity, thereby inhibiting PDE1-mediated cyclic
nucleotide hydrolysis. The three PDE1 isoforms display dual-specificity for cAMP and
cGMP catabolism: PDE1A and PDE1B target cGMP over cAMP (but with less specificity
than PDE5 or PDE9); while PDE1C hydrolyses both cyclic nucleotides with equal Km.
Both PDE1A and PDE1C have been identified in human hearts, whereas only PDE1A has
been identified in rodent hearts.188, 189 While PDE1A likely regulates vascular smooth
muscle contraction, PDE1C is involved in VSMC proliferation and apoptosis.190 PDE1C is
also expressed in cardiac myocytes, and recent studies have reported an important role in
both cAMP and cGMP hydrolysis in human myocardium in vitro.188
PDE9 is expressed in brain and heart, and has the highest affinity for cGMP of all the
cGMP-hydrolysing PDEs. Its function remains unknown and is an area of active
investigation.
1.2.4 Compartmentalisation of cGMP signalling
Compartmentalisation of cGMP signalling is accounted for by differential spatial
distribution of sGC and pGC, PKG isoforms, and PDE isoforms, as well as the distribution
of other proteins that associate with cGMP signalling (Figure 10). In addition to spatial
regulation by different components of the cGMP synthetising pathways, evidence for NO-
cGMP signal compartmentalisation also exists. Soluble GC has long been considered as
the cytosolic GC isoform, however, more recently, NO-sensitive sGC has also been noted
to associate with the plasma membrane of various cell types.191-194
PDEs also play a very important role in the compartmentalisation of cGMP signalling.
In cardiac myocytes stimulated with NO donors or NPs, a membrane localised cGMP pool
was detected predominantly with NP stimulation. Moreover, this pool was differentially
enhanced following PDE2 versus PDE5 inhibition. This model of spatially distinct cGMP
pools was tested by examining sub-sarcolemmal [cGMP] changes in isolated cardiac
myocytes using recombinant cyclic nucleotide-gated (CNG) channels as cyclic nucleotide
sensors. Administration of NO donors resulted in moderate increases of sarcolemmal
CNG current (ICNG), whereas activation of the sarcolemmal pGC by administration of ANP

Chapter 1 42
and BNP resulted in much greater increases in ICNG. In both cases, application of a non-
specific PDE inhibitor strongly potentiated ICNG, indicating that PDEs play a role in limiting
cGMP signals downstream of both pGC and sGC. Subsequently, using selective inhibitors
for PDE2 and PDE5, it was demonstrated that PDE5, and - to a lesser extent - PDE2, are
involved in the compartmentalisation of sGC-cGMP signals, whereas PDE2 alone limits
the spread of pGC-cGMP signals.195
The functional differences between the NP-cGMP and NO-cGMP pools were highlighted
by studies of $-adrenergic response modulation in intact mouse hearts and isolated
myocytes. Stimulation of cardiac contractility by isoproterenol (a non-selective $-AR
agonist) was compared under conditions of ANP stimulation (NP-cGMP) versus PDE5A
inhibition (NO-cGMP). Whereas PDE5A inhibition and selective elevation of the NO-cGMP
pool blunted the $-adrenergic cardiac response, ANP-triggered increase in the NP-cGMP
pool did not affect the $-adrenergic cardiac response.180, 196 This differential modulation of
PDE5A inhibition on the $-adrenergic cardiac response was completely abrogated in
myocytes in which NOS3 was genetically absent or pharmacologically inhibited, or in
which sGC was pharmacologically inhibited.180 More recently, the $3-AR and PKG have
been demonstrated to be important upstream and downstream mediators underlying the
effects of PDE5 inhibition on $-AR mediated increases in cardiac contractility; the former
coupling to NOS3 activation, and the latter decreasing myofilament Ca2+-sensitivity by
promoting troponin I phosphorylation.197
In addition to being involved in the hydrolysis and compartmentalisation of cGMP signals,
PDE2 acts as an integration point for cross-talk between cAMP and cGMP signals.
$-AR-mediated regulation of cardiac contractility is dependent on the
compartmentalisation of cAMP-PKA signals downstream of $-AR activation; in isolated
cardiac myocytes $-AR stimulation results in greater activation of cAMP synthesis in the
PKA Type II compartment (PKA is classified as a Type I or Type II enzyme depending
upon the associated R subunit, i.e. RI or RII), leading to the phosphorylation of
downstream targets involved in the regulation of cardiac contractility such as
phospholamban and troponin I.198 Subsequently, elevations in [cGMP]i have been
demonstrated to modify these cAMP signals downstream of $-AR activation; in the
presence of the NO donor SNAP, the rises in cAMP signals in the PKA Type I and II
compartments are inverted, with an enhanced cAMP response in the PKA Type I
compartment, and an attenuated response in the Type II compartment. On the other hand,

Introduction 43
application of ANP selectively reduces the $-AR-mediated cAMP response in the PKA
Type II compartment, due to the activation of cAMP hydrolysis by PDE2. On the other
hand, elevated [cGMP]i enhances the cAMP response in the PKA Type I compartment
through cGMP-dependent inhibition of cAMP hydrolysis by PDE3.199
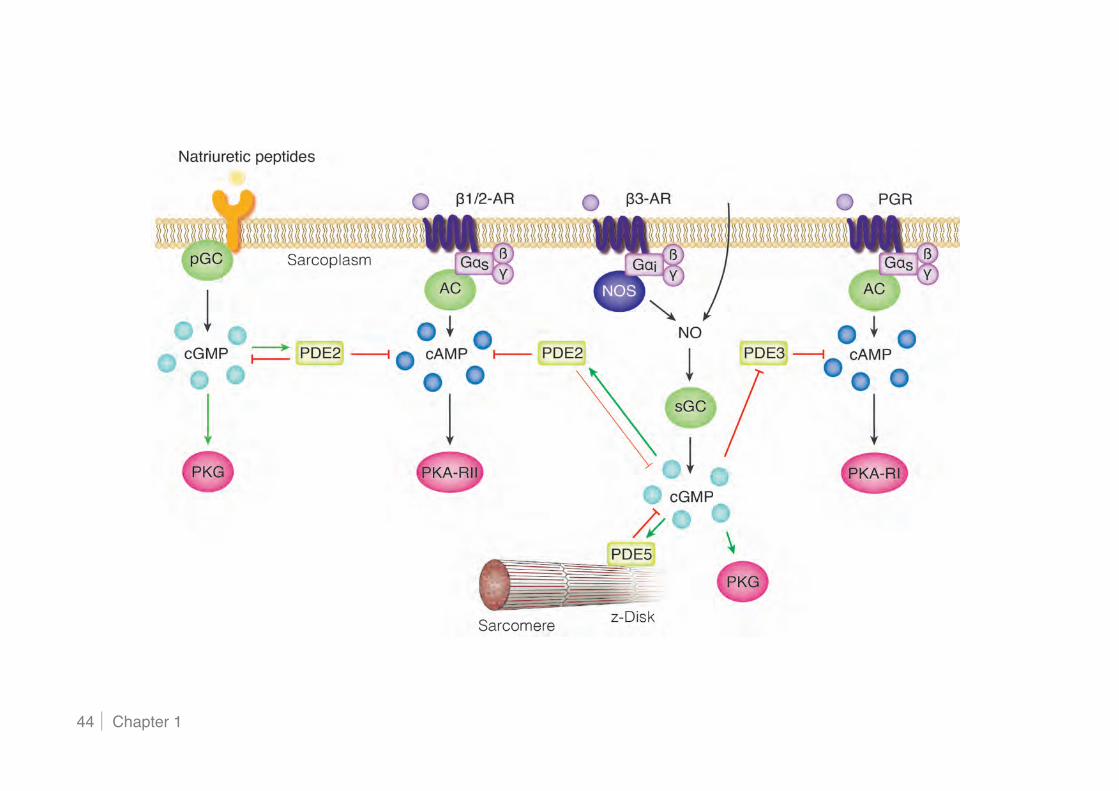
Chapter 1 44

Introduction 45
Figure 10. Compartmentalisation of cGMP signals and cross-talk between cGMP and cAMP signals The cGMP-activated PDE2 plays a key role in the compartmentalisation of cGMP signals downstream of pGC. Given its dual-substrate specificity, it also plays an important role in the compartmentalisation of cAMP signals. The compartmentalisation of sGC-derived cGMP signals is dependent on the cGMP-specific PDE5, which is involved in the regulation of !-AR stimulated contractile responses, through a signalosome with the !-AR, NOS3, sGC, and PKG. PDE2, although playing a minor role in the compartmentalisation of sGC-derived cGMP signals, acts to inhibit cAMP-dependent responses downstream of !-AR stimulation. In addition, through cGMP-mediated inhibition of cAMP hydrolysis by PDE3, sGC-derived cGMP can also potentiate cAMP-dependent signalling. Activational and inhibitory effects are indicated in green and red, respectively. AC indicates adenylate cyclase; AR, adrenergic receptor; cAMP/cGMP, cyclic adenosine/guanosine 3", 5"-monophoshate; NO(S), nitric oxide (synthase); PDE, phosphodiesterase; pGC and sGC, particulate and soluble guanylate cyclase; PGR, prostaglandin receptor; and PKA and PKG, protein kinase A and G.
1.2.5 Cyclic GMP regulation of the cardiovascular system
(i) Vascular smooth muscle cells
Cyclic GMP regulates vascular tone, and VSMC proliferation and differentiation.200, 201
Vascular tone is regulated by changes in intracellular free Ca2+-concentrations within the
smooth muscle cells. Contraction of VSMC is triggered by the receptor-mediated
generation of the second messenger IP3, which induces release of free Ca2+ from
intracellular stores, further provoking influx of extracellular Ca2+ via Ca2+-channels.
The rise in intracellular Ca2+ activates Ca2+/calmodulin-dependent MLCK, which
phosphorylates MLC to activate myosin ATPase and trigger VSMC contraction.
Reduction of the intracellular Ca2+-concentration results in vasorelaxation. Cyclic GMP
regulates the intracellular free Ca2+-level within VSMC through several PKG-mediated
mechanisms, including inhibition of IP3-mediated Ca2+-release from intracellular stores,
removal and sequestration of intracellular Ca2+ through Ca2+-pump mechanisms, and both
direct and indirect inhibition of the influx of extracellular Ca2+ through voltage-gated Ca2+-
channels. Aside from regulating intracellular Ca2+-concentrations, cGMP-PKG signalling
modulates vascular tone by altering Ca2+-sensitisation and thin filament interaction.
The importance of PKG in modulating vascular tone is emphasised by PKG-I deficient
mice, which die at a young age, and are afflicted by impaired smooth muscle
relaxation.202-205

Chapter 1 46
Another important role of cGMP-PKG signalling in VSMC is the regulation of
proliferation.206 Proliferation of VSMC involves MAPKs, cyclins, cyclin-dependent protein
kinases (Cdk), Cdk inhibitors, the retinoblastoma protein (Rb), and E2F, the transcription
factor inducing S-phase gene expression. Cyclic GMP-PKG activation directs an anti-
proliferative signal in VSMC, downregulating transcript levels of cyclins and upregulating
mRNA levels of MAPK phosphatase-1 and p16 (the gene encoding Cdk inhibitor 2A).
PKG is also involved in VSMC differentiation.206 The phenotypic switch of VSMC between
a contractile, “differentiated” state, and a synthetic or secretory, “dedifferentiated” state,
involves the differential expression of contractile proteins, extracellular matrix proteins,
signal transduction proteins, and growth factors as well as their receptors. Vascular
smooth muscle cells dedifferentiate from the “contractile” phenotype to the
“synthetic/secretory” phenotype in response to vascular injury or during in vitro culture.
During the dedifferentiation process, VSMC can proliferate, migrate, and produce
extracellular matrix proteins. This process is associated with loss of PKG expression and
transcriptional downregulation of contractile proteins, thus cGMP-dependent PKG
activation in VSMC favours the contractile phenotype.
(ii) Vascular endothelial cells
The physiological consequences of altered cGMP-PKG signalling in vascular endothelium
have not been studied as extensively as in other cardiovascular cell types. In vascular
endothelial cells, cGMP-PKG signalling regulates cell motility, migration, and proliferation,
all of which are vital to angiogenesis, and vascular permeability.
Angiogenesis involves vascular endothelial cell motility, migration, and proliferation,
physiological processes mediated by cGMP signalling. The central role of cGMP signalling
in angiogenesis has been demonstrated by studies examining NO- and ANP-induced
cGMP generation. In NOS3-/- knockout mice, vascular endothelial growth factor (VEGF)
fails to induce neovascularisation.207, 208 Furthermore, a model of hind-limb ischaemia in
NOS3-/- knockout mice demonstrated defective neovascularisation due to reduced
mobilisation of endothelial progenitor cells.209 Similarly, in vitro studies showed that VEGF-
stimulated capillary-like tube network formation in cultured endothelial cells requires NOS3
activation and NO-cGMP signalling.210-212 Small interfering RNA-mediated knockdown of
NPR-A or PKG prevents ANP-induced endothelial tube formation, demonstrating the
importance of ANP-mediated cGMP-signalling in endothelial function.213

Introduction 47
Vascular permeability and endothelial barrier function are predominantly determined by
endothelial cell contraction. Both NO- and ANP-triggered cGMP signalling have been
shown to improve endothelial barrier function, and protect against vascular injury in the
systemic and pulmonary vasculature.214-224
(iii) Cardiac myocytes
Cyclic GMP negatively modulates contractility, accelerates relaxation, and reduces the
stiffness of cardiac myocytes (Figure 11). These effects might be mediated by direct PKG
phosphorylation of proteins, including cardiac troponin I, L-type Ca2+-channels,
phospholamban, and titin (cfr. 1.1.3 - ii).
The role of PKG in the negative inotropy induced by cGMP was demonstrated by a
reduction of contractile force in electrically stimulated myocardium from wild-type mice by
cGMP, and the lack of effect on the contractile force in myocardium from cardiac-specific
PKG-I knockout mice.204 Furthermore, the difference between the wild-type and knockout
myocardium persisted even in experiments performed in the presence of forskolin, an
activator of the !-adrenergic cAMP pathway, thereby confirming the specificity of altered
cGMP signalling in reducing contractile force.
Cyclic GMP modulation of myocyte contractility can be initiated by either NO or NP.225-227
Interestingly, NO has a bimodal effect on myocardial contractility; low concentrations of
NO increase myocardial contractility, while high concentrations exert a negative inotropic
effect. Low concentrations of NO can activate AC without activating GC, inducing
production of cAMP and not cGMP.228 At low concentrations, cGMP also inhibits PDE3-
mediated cAMP hydrolysis. Accumulation of cAMP then activates PKA, leading to the
opening of sarcolemmal voltage-gated and sarcoplasmic RyR Ca2+-channels, and
ultimately improving myocyte contractility.83
The negative inotropic effect of NO has partially been attributed to a cGMP-PKG mediated
reduction of myofilament Ca2+-responsiveness, though the exact mechanism was initially
unclear. The role of PKG in the contractile response to NO was elegantly established by
isolated myocyte contractility studies in which the intracellular Ca2+-transient was
assessed when the cells were exposed to an NO donor, in the presence or absence of
sGC inhibitors. The NO donor significantly increased resting myocyte cell length, and
accelerated the relaxation time, without changing either the amplitude or kinetics of the
intracellular Ca2+-transient.229 Inhibiting either sGC or PKG abrogated the effect of the NO

Chapter 1 48
donor. Additionally, direct activation of PKG mimicked the myocyte relaxation induced by
the NO donor. Hearts treated with the NO donor also demonstrated increased
phosphorylation of troponin I.
In addition, the L-type voltage-gated Ca2+-channel has also been identified as a
phosphorylation target of PKG.23 Moreover, PKG further regulates intracellular free Ca2+-
levels by inhibition of IP3-mediated Ca2+-release from intracellular stores, and
phosphorylation of phospholamban.
Differential activation of PDEs is also believed to modulate the contractile effect of NO-
cGMP. At high NO levels, cGMP production is triggered, and activation of PDE2 by cGMP
leads to cAMP hydrolysis, and thus the cGMP signal predominates. It has also been
suggested that the variable effects of NO are not simply due to its concentration, but also
relate to the specific NOS isoform generating NO.230-234
Pro-apoptotic effects of cGMP have been reported in VSMC, endothelial cells, and cardiac
myocytes, and appear to be mediated by PKG.235-238 The downstream mechanisms in
vascular cells may involve activation of JNK and/or phosphorylation and inactivation of !-
catenin. In cardiac myocytes, cGMP decreases the mRNA levels of the anti-apoptotic Bcl-
2 homologue Mcl-1.236

Introduction 49
Figure 11. cGMP-PKG signalling negatively modulates contractility, accelerates relaxation, and reduces the stiffness of cardiac myocytes Cyclic GMP-activated PKG phosphorylates cardiac troponin I, thereby reducing myofilament Ca2+-responsiveness. The negative inotropic effect of cGMP-PKG is also mediated by an inhibitory effect on L-type Ca2+-channels. PKG further regulates the intracellular free Ca2+-level by inhibition of IP3-mediated Ca2+-release from intracellular stores, and phosphorylation of phospholamban. Finally, phosphorylation of titin by PKG leads to a decrease in the cardiac myocyte passive tension. cGMP indicates cyclic guanosine 3", 5"-monophosphate; IP3R, inositol 1,4,5-trisphosphate receptor; NO(S), nitric oxide (synthase); PKG, protein kinase G; pGC and sGC, particulate and soluble guanylate cyclase; PLN, phospholamban; RyR, ryanodine receptor; SERCA2, sarcoplasmic reticulum Ca2+-ATPase; and SR, sarcoplasmic reticulum.

Chapter 1 50
1.3 Cyclic GMP signalling in heart failure Since cGMP is a vital modulator of cardiovascular homeostasis, dysfunction at any level of
the cGMP signalling pathway is involved in many cardiovascular diseases. Endothelial cell
dysfunction contributes to hypertensive disease, both systemic and pulmonary, and
atherosclerosis. In addition, VSMC dysfunction is involved in systemic and pulmonary
hypertension, and in ischaemic heart disease. Finally, cardiac myocyte dysfunction plays
a role in ischaemic and hypertrophic heart disease, cardiomyopathy, and heart failure.
1.3.1 The role of cGMP signalling in cardiac hypertrophy and failure
cGMP-PKG signalling in cardiac myocytes serves to counter the detrimental effects of
continued hypertrophic stress.
In vitro studies have demonstrated that exogenous administration of all 3 NPs can inhibit
cardiac myocyte hypertrophy induced by a variety of G#q-agonists, including Ang II, ET-1,
and PE.176, 239, 240 Furthermore, the role of endogenous ANP in inhibiting cardiac myocyte
hypertrophy has been demonstrated in studies using a peptide antagonist, which
competitively and selectively inhibits NP binding to pGC.241 However, in vivo animal
studies provide more compelling evidence for this pathway in protecting the heart from
deleterious remodelling. The anti-hypertrophic cardiac effect of cGMP was initially
suggested by the exacerbation of cardiac hypertrophy in various knockout mice lacking
components of the cGMP signalling pathway. Global NPR-A-/- knockout mice
demonstrated salt-resistant arterial hypertension, with a disproportional degree of cardiac
hypertrophy and fibrosis.242-244 Cardiac-selective NPR-A-/- knockout mice displayed an
exaggerated hypertrophic response when chronic pressure overload was induced by
transverse aortic constriction (TAC) and Ang II infusion.245, 246 Nppa-/- knockout mice,
deficient in preproANP, developed salt-sensitive arterial hypertension and cardiac
hypertrophy.247 Similarly, mice overexpressing a dominant negative mutant of pGC
(lacking catalytic activity) showed a worsened cardiac hypertrophic response to pressure
overload.248 Mice with a constitutively active pGC in cardiac myocytes displayed an
attenuated hypertrophic response to both TAC and chronic !-AR stimulation.249
Cardiac remodelling data from genetically engineered mice lacking sGC activity is sparser.
Mice with global deletion of the !1-subunit of sGC, which leads to total loss of sGC
enzyme activity, die before reaching maturity due to gastrointestinal dismotility.250
Targeted deletion of the #1-subunit of sGC causes gender-specific hypertension, but did

Introduction 51
not affect global LV function, although cardiac myocytes isolated from these mice showed
impaired sarcomere shortening at baseline.251, 252 Currently, published reports describing
the effects of cardiac-specific knockout or overexpression of sGC are lacking.
Mice lacking NOS3 and/or NOS1 also develop cardiac hypertrophy and dysfunction when
subjected to increased afterload imposed by TAC.253, 254 In addition, NOS uncoupling is a
significant component of the pathophysiology of myocardial hypertrophy. Sustained
pressure overload activates ROS, which decrease the bioavailability of the NOS cofactor
BH4. Exogenous BH4 administration has been shown to recouple NOS and reverse
advanced hypertrophy in mice with TAC-induced pressure overload.255
Increasing evidence suggests a marked correlation between the development of heart
failure and cGMP hydrolysis as a result of increased PDE5 gene function. Although only
expressed at low levels in cardiac myocytes under normal physiological conditions,
PDE5 expression is increased in animal models of heart failure and in patients with RV
hypertrophy, or advanced HF.256-258 Recently, also cardiac myocyte PDE1 has been
shown to be upregulated in several rodent heart failure models (TAC, isoproterenol, and
Ang II).189
Multiple experimental studies have provided evidence that inhibition of cGMP hydrolysis
has a beneficial effect against adverse cardiac remodelling. In mice, the PDE5 inhibitor
sildenafil attenuated cardiac remodelling induced by pressure overload following TAC.
Also, sildenafil was able to reverse mild pre-existing cardiac remodelling and to prevent
further advancement of cardiac remodelling and deterioration of cardiac function in cases
of more advanced, pre-existing disease.259, 260 Moreover, in patients with stable systolic
heart failure, treatment with sildenafil improves LV diastolic function, cardiac geometry,
and clinical status.261 However, it is possible that some of sildenafil"s cardiac effects are
attributable to inhibition of PDE1.262 Given the wide expression of PDE1 in the
cardiovascular system, including in cardiac myocytes, possible cross-reactivity is
relevant.188 Moreover, the effects of PDE5 inhibitors on cardiac remodelling could involve
targeting of other non-myocyte cell types (including fibroblasts, smooth muscle cells, and
endothelial cells), or modulation of LV afterload via vasodilatation of systemic resistance
vessels.

Chapter 1 52
The exact role of cGMP-PKG activation in protecting against hypertrophic signalling
remains to be confirmed. The best studied downstream mechanism by which cGMP-PKG
signalling exerts its anti-hypertrophic action is the inhibition of the Ca2+-calcineurin-NFAT
pathway.53, 263-266 cGMP-PKG signalling may reduce intracellular Ca2+-concentrations,
thereby inhibiting calcineurin activation, although other mechanisms cannot be excluded.
This was shown in a study in which calcineurinA!-deficient mice developed reduced
hypertrophy in response to pressure overload, but this moderate hypertrophy was still
inhibited by PDE5 inhibition.267 Interestingly, pressure overload-induced activation of
CaMKII was inhibited by sildenafil in both WT and calcineurinA!-deficient mice.
Further evidence supporting cGMP modulation of CaMKII activation comes from pGC-/-
knockout mice, in which the observed hypertrophic phenotype is associated with the
upregulation of CaMKII expression, increased CaMKII auto-phosphorylation, and
phosphorylation of CaMKII downstream targets.262, 268-270 Given that elevations in
intracellular cGMP concentrations are ineffective in preventing increases in NFAT
transcriptional activity in isolated cardiac myocytes overexpressing a constitutively active
calcineurin mutant, and that inhibition of Ca2+-dependent signalling pathways underlies the
anti-hypertrophic effect of the cGMP-PKG pathway, the search for PKG targets accounting
for its inhibition of both calcineurin-NFAT signalling and CaMKII signalling has focused on
upstream Ca2+-handling. A target involved, however indirectly, in intracellular Ca2+-
handling that has been implicated in the anti-hypertrophic effects of cGMP is the
sarcolemmal NHE.246, 269, 271, 272 The activity of NHE is closely linked to Ca2+-handling via
the NCX. Recently, a genetic model has provided evidence that increased NHE activity is
able to generate Ca2+-signals that induce cardiac hypertrophy and failure.272 Evidence for
cGMP-dependent inhibition of NHE activity is provided by pGC-/- knockout mice and PDE5
inhibition studies. Cardiac hypertrophy in pGC-/- knockout mice has been associated with
increased NHE activity, and chronic treatment of these mice with an NHE inhibitor is
associated with regression of adverse cardiac remodelling. Interestingly, the anti-
hypertrophic effect of NHE inhibition was associated with decreased activation of the
CaMKII pathway, without attenuating the activation of the calcineurin pathway.269
Conversely, PDE5 inhibition has been associated with decreased NHE activity.273
During the last years, canonical transient receptor potential channels (TRPC) have
emerged as important regulators of pathological cardiac remodelling.274 Specifically,
TRPC3 and TRPC6 have been identified as initiators and regulators of Ca2+-signals

Introduction 53
leading to calcineurin activation in cardiac myocytes and non-cardiac cells, and both have
been demonstrated to be regulated by PKG.87, 88, 275, 276 To date, most work has focused
on TRPC6 as a PKG target in cardiac myocytes. In isolated cardiac myocytes, both ANP
and PDE5 inhibition have been demonstrated to increase the phosphorylation of TRPC6,
thereby suppressing channel current, via PKG-dependent mechanisms.277-279 Knockdown
of TRPC3 and TRPC6 expression and pharmacological inhibition of TRPC channels
abolished the inhibitory effects of ANP on elevations and [Ca2+]i-oscillations as a result of
G#q-stimulation, indicating the significance of TRPC channels as a target for the anti-
hypertrophic effects of cGMP.268, 278 In NPR-A-/- knockout mice, displaying elevated
TRPC6 expression, pharmacological inhibition of TRPC channels was associated with a
regression of cardiac myocyte hypertrophy and, conversely, overexpression of TRPC6 in
mice lacking NPR-A was associated with an exaggerated cardiac hypertrophy.278
Another target that has been implicated in the anti-hypertrophic effects of cGMP, and
could account for decreased Ca2+-signalling and reduced calcineurin activation, is the
regulator of G protein-coupled signalling (RGS) family of proteins. These proteins play an
important role in turning off GPCR signalling by acting as GTPase-activating proteins,
thereby accelerating the rate of GTP-hydrolysis by G# and subsequent reconstitution of
the GDP-bound heterotrimeric G protein complex. They also terminate PLC-mediated IP3
synthesis and subsequent IP3-mediated Ca2+-release from intracellular stores. In cardiac
myocytes, PKG-mediated phosphorylation and activation of RGS2 and RGS4 has been
shown to contribute to the anti-hypertrophic effects of cGMP. In RGS2-/- knockout mice,
PDE5 inhibition did not attenuate the development of cardiac hypertrophy or the activation
of calcineurin signalling pathways following pressure overload, despite similar increases in
PKG activity as seen in wild-type mice.280 Furthermore, in isolated myocytes from RGS2-/-
knockout mice, ANP is unable to inhibit elevations in intracellular [Ca2+] following G#q-
stimulation.268 Similarly, overexpression of a dominant negative form of RGS4 in isolated
cardiac myocytes is associated with an attenuation of the inhibitory effects of ANP on IP3
production and cardiac myocyte hypertrophy following G#q-stimulation.281 Accordingly, the
expression and phosphorylation of RGS4 were attenuated in NPR-A-/- knockout mice and,
furthermore, the adverse cardiac remodelling in these mice could be rescued by cardiac-
specific overexpression of RGS4.281

Chapter 1 54
Cardiac disease progression inevitably leads to heart failure. Also here, dysregulated
cGMP signalling is a key mediator. Natriuretic peptide signalling gains greater prominence
in heart failure as plasma NP concentrations become markedly elevated.282-284
Vasodilatory and diuretic responses to ANP are blunted in animal models and in patients
with heart failure.285-289 The blunted response has been attributed to a reduction in NPR-A
expression, as seen in the pulmonary vasculature of heart failure patients and in the
systemic vasculature of rat models of high-output heart failure.290, 291 Others have noted a
decreased responsiveness of NPR-A to ANP, without significant change in NPR-A
receptor expression levels.292 Upregulation of NPR-C, the NP clearance receptor, has also
been suggested as the mechanism by which NPR responsiveness is diminished.292, 293
Similarly, reduced NO bioavailability in heart failure is thought to be related to decreased
expression and/or activity of NOS3 in the failing myocardium of patients and experimental
animals.294-298
1.3.2 The role of cGMP signalling in anthracycline-induced cardiac injury and failure
To date, very little is known concerning the role of cGMP signalling in doxorubicin-induced
cardiotoxicity.
Mice with a heterozygous deletion of endothelin-converting enzyme-1 (ECE-1), the
enzyme producing ET-1, displayed attenuated cardiac systolic dysfunction after
doxorubicin treatment in comparison to treated wild-type mice. Cardiac cGMP content and
serum ANP levels were increased in these mice, and impairment of cardiac mitochondrial
biogenesis inhibited.299
Also, It has been shown that NOS3-dependent ROS formation plays a role in doxorubicin-
induced myocardial dysfunction, since mice with a cardiac myocyte-specific
overexpression of NOS3 displayed increased sensitivity to doxorubicin-induced
cardiotoxicity, and genetic disruption of NOS3 transcription protected against doxorubicin-
induced cardiac dysfunction, injury and mortality in mice.300
In addition, treatment of mice with sildenafil prior to doxorubicin administration inhibited
cardiac myocyte apoptosis, preserved mitochondrial membrane potential and myofibrillar
integrity, and prevented LV dysfunction as well as ST segment prolongation.301
Similarly, tadalafil also improved LV function and prevented cardiac myocyte apoptosis in
doxorubicin-induced cardiomyopathy.302 The cardioprotective effects of these
pharmacological PDE5 inhibitors were conferred to NO-cGMP-PKG-mediated

Introduction 55
phosphorylation of the inner membrane mitochondrial ATP-sensitive K+-channels
(mitoKATP). Interestingly, PDE5 inhibition has also been experimentally shown to enhance
the chemotherapeutic effect of doxorubicin. It was shown that sildenafil and vardenafil
induced apoptosis and had anti-proliferative effects in B-cell chronic lymphatic
leukaemia.303 Moreover, the combination of vardenafil and doxorubicin significantly
improved survival, and reduced the tumour size in brain tumour-bearing rats.304
Recently, a murine study showed that sildenafil is a powerful sensitiser of doxorubicin-
induced killing of prostate cancer cells, while providing concurrent cardioprotection.305
Thus, prophylactic treatment with PDE5 inhibitors might become a promising therapeutic
intervention for both enhancing the chemo-sensitivity of doxorubicin and managing the
clinical concern of doxorubicin-induced cardiotoxicity in patients.

Chapter 1 56

CChhaapptteerr 22
RATIONALE AND OBJECTIVES


Rationale and objectives 59
CHAPTER 2: RATIONALE AND OBJECTIVES
Heart failure is a syndrome with characteristic signs and symptoms, and objective
evidence of cardiac dysfunction, as a result of which the heart can no longer provide
sufficient blood flow and/or pressure to meet the body"s demands. The result is a cascade
of symptoms, including progressive fatigue and dyspnea, and the syndrome often follows
a chronic ominous course leading to severe incapacitation and compromised survival.
In the past quarter century, much progress has been made in understanding the molecular
and cellular processes that contribute to heart failure, which has led to a considerable
change in the scope of available therapies. However, chronic heart failure remains a major
cause of morbidity and mortality, with a continuously increasing worldwide prevalence.
New treatments that target pathogenesis are needed to halt and reverse the devastating
consequences of this disease.
Growing evidence suggests that the cGMP pathway may be a key element in the
pathophysiology of heart failure, and its pharmacological modulation may represent a
promising therapeutic approach. In this thesis, the role of cGMP signalling was
investigated in two important and clinically relevant pathological conditions heralding heart
failure; chronic LV pressure overload and chemotherapy-induced cardiotoxicity.
The experimental studies in this thesis are therefore presented in two major parts and
address the following main objectives:
Objective 1: To explore myocardial PDE5 expression in human and experimental hearts exposed to chronic LV pressure overload, and investigate its role in adverse cardiac remodelling (Chapter 4)
PDE5, a cGMP-catalysing enzyme, has been shown to play a key role in vascular smooth
muscle tone, and its high expression in corpus cavernosal and pulmonary vascular
smooth muscle is targeted by pharmacological PDE5 inhibitors to treat erectile dysfunction
and pulmonary hypertension. Under normal physiological conditions, PDE5 expression in
the heart is low, but, in human RV hypertrophy and advanced LV failure, PDE5 expression
is increased in cardiac myocytes, suggesting that this enzyme is involved in the adaptation
to increased stress.256-258 Moreover, pharmacological PDE5 inhibition suppressed cardiac
hypertrophy and improved LV function in mice with chronic LV pressure overload, and
reversed mild pre-established cardiac hypertrophy.260 However, it is possible that some of

Chapter 2 60
sildenafil"s beneficial cardiac effects are attributable to inhibition of PDE1, and could
involve targeting of other non-myocyte cardiac cell types, or modulation of LV afterload via
vasodilatation of systemic resistance vessels.
In chapter 4, we examined myocardial PDE5 expression in patients with severe aortic
stenosis, and in mice with TAC-induced chronic pressure overload. In addition, we studied
the role of PDE5 expression in cardiac myocytes during adverse LV remodelling,
by subjecting transgenic mice with a cardiac myocyte-specific PDE5 overexpression to
sustained pressure overload.
Objective 2: To explore the role of nitric oxide-stimulated cGMP signalling in
anthracycline-induced cardiotoxicity (Chapter 5) The cardiotoxicity of anthracyclines, a class of highly effective chemotherapeutic agents,
is dose-dependent, thereby limiting their clinical implementation at optimal anti-tumour
efficacy. Doxorubicin is the most widely used anthracycline, and considerable efforts are
made to elucidate the cause of its cardiotoxicity, and prevent or reverse the mechanisms
leading to ventricular dysfunction. An indication for the role of cGMP signalling in
doxorubicin-induced cardiotoxicity was provided by the observation that doxorubicin-
induced cardiotoxicity was attenuated in mice treated with PDE5 inhibitors. However, to
date little is known on the importance of cGMP signalling in the pathophysiology and
pathogenesis of doxorubicin-mediated cardiac injury.
In chapter 5, we further investigate the role of NO-stimulated cGMP in anthracycline-
induced cardiotoxicity, by evaluating cardiac remodelling after chronic doxorubicin
administration in mice with a cardiac myocyte-specific decrease of active sGC, the
enzyme mediating NO-stimulated cGMP synthesis.

CChhaapptteerr 33
MATERIALS AND METHODS


Materials and Methods 63
CHAPTER 3: MATERIALS AND METHODS
3.1 Patients
Cardiac tissue samples were obtained by Prof. Dr. P. Herijgers (Gasthuisberg University
Hospital, Leuven, Belgium) from the LV outflow tract of patients with isolated severe aortic
stenosis and chronic pressure overload undergoing aortic valve surgery. The ethics review
board of Gasthuisberg University Hospital approved the protocol, and all patients provided
informed consent.
Cardiac tissue samples obtained at autopsy from hearts without documented prior cardiac
disease were studied as controls, and provided by Prof. Dr. E. Verbeken (KU Leuven,
Leuven, Belgium).
3.2 Experimental animals
All animals were housed in a temperature-controlled (22°C) room with a 12-12 hour light-
dark cycle. Experiments were performed conforming the 2010/63/EU Directive, and
approved by the institution"s animal ethics committee (KU Leuven, Leuven, Belgium).
3.2.1 Transgenic mouse models
(i) Constitutive cardiac myocyte-specific overexpression of PDE5 (PDE5-TG)
The bovine PDE5 cDNA306, kindly provided by Prof. D. J. Corbin (Vanderbilt University
School of Medicine, Nashville, USA), was ligated into a plasmid containing the !MHC
promoter, driving transgene expression specific in cardiac myocytes, and a c-myc tag
(Glu-Gln-Lys-Leu-Ile-Ser-Glu-Glu-Asp-Leu). A restriction fragment that contained the full
expression cassette was microinjected into FVB mouse eggs by Dr. L Schoonjans (KU
Leuven, Leuven, Belgium). Oocytes were implanted into a pseudo-pregnant Swiss foster
mother, and offspring were screened via polymerase chain reaction (PCR) to confirm
successful transgene integration. Transgenic founders were backcrossed for 6
generations onto a C57BL/6N (Taconic) background. Wild-type (WT) littermates served as
controls in the experiments.

Chapter 3 64
(ii) Conditional cardiac myocyte-specific dominant negative mutation of the !1-subunit of
sGC (DNsGC!1)
The dominant negative sGC!1 mutant (DNsGC!1), derived from rat sGC!1 cDNA by site-
directed mutagenesis, has a point mutation in the catalytic region of sGC!1 at position
529 (D529A, Asp to Ala).307 This dominant negative mutant will compete with sGC!1 and
sGC!2 for binding to sGC!1, thereby inhibiting the formation of the two known
catalytically active sGC heterodimers. Targeting of sGC!1 was opted, since knockdown of
sGC!1 was shown to be embryonically lethal in medaka fish, and sGC!1 knockout mice
showed fatal gastrointestinal obstruction.250, 308
Inhibition of NO-stimulated cGMP production in vitro by the dominant negative sGC!1
mutant was confirmed in the laboratory of Prof. Dr. K.D. Bloch (Massachusetts General
Hospital and Harvard Medical School, Boston, USA).
The dominant negative mutant was inserted, together with a Flag tag (Asp-Tyr-Lys-Asp-
Asp-Asp-Asp-Lys), in a plasmid containing a tetracycline-responsive promoter element
(TetO) in the laboratory of Prof. Dr. K.D. Bloch. This construct was microinjected into FVB
zygotes in the G1 phase by Dr. L. Schoonjans, afterwards implanted into a pseudo-
pregnant Swiss foster mother. Offspring were genotyped using qPCR and transgenic
founders were backcrossed for 6 generations onto a C57BL/6N (Taconic) background.
The second mouse line, kindly provided by Prof. Dr. D.A. Dichek (University of
Washington Medical Center, Seattle, USA), expresses the tetracycline transactivator (tTA)
gene under the control of the !MHC promoter (!MHC-tTA) on a C57BL/6 background.309
The tTA gene encodes a transcriptional activator that binds to and activates transcription
from the tetO promoter element. Crossing the !MHC-tTA mice with the tetO-DNsGC!1
mice produced dual heterozygous mice, in which the tTA protein can bind to tetO and
induce DNsGC!1 expression in cardiac myocytes. Transcriptional activator binding to
DNA is blocked in the presence of the tetracycline antibiotic doxycycline.
Therefore, transgene induction is achieved by removing doxycycline from the diet (Tet-off
system, Figure 12). Dual heterozygotes and WT littermates were conceived and raised in
the presence of doxycycline (doxycycline containing chow, 200mg/kg, Harlan) to prevent
expression of DNsGC!1 during development. When the mice were 4-6 weeks old,
doxycycline was withdrawn

Materials and Methods 65
Figure 12. Expression of the dominant negative mutant of sGC!!1 in cardiac myocytes in dual heterozygotes by doxycycline withdrawal By halting doxycycline administration in dual heterozygous mice, repression of the tetracycline transactivator (tTA)-dependent transactivation will be abolished, and these mice will express dominant negative mutated sGC!1 in cardiac myocytes.
3.2.2 Mouse models of heart failure
(i) Mouse model for pressure overload-induced cardiac hypertrophy
Transverse aortic constriction (TAC) in mice is a common method to induce cardiac
hypertrophy and failure. Since significant gender differences in the LV adaptation to
pressure overload have been reported, chronic pressure overload was induced exclusively
in male mice (body weight 23-30g) by TAC during 10 weeks.310 Mice were anaesthetised
with sodium pentobarbital (Nembutal, Ceva Santé Animale, 40-70 mg/kg body weight,
intraperitoneally (IP)), and ventilated at 150 breaths per minute (0.25 ml tidal volume,
MiniVent, Harvard Apparatus). Body temperature was monitored and maintained at 37°C
using a rectal probe and heating pad (TC-1000, CWE Inc.). The transverse aortic arch
was ligated (7-0 silk suture) between the left and right common carotid arteries with an
overlying 27-gauge needle (Figure 13). Subsequently, the needle was removed, leaving a
region of stenosis. Following TAC, all layers of muscle and skin were closed using a 6.0
ticron suture, the wound was treated with an antiseptic, and an analgesic (buprenorphine,
Schering-Plough, 0.1 mg/kg subcutaneously) was administered during the first two days.

Chapter 3 66
1. Aortic arch
2. Right common carotid artery
3. Left common carotid artery
Constriction of aorta
Figure 13. Transverse aortic constriction in mice In mice, the transverse aortic arch was ligated between the left and right common carotid arteries during 10 weeks, thereby inducing chronic LV pressure overload.
After euthanasia of mice, the degree of cardiac hypertrophy was assessed by determining
normalised heart weight. Hearts were weighed (without auricles), and normalised to body
weight (HW/BW). To account for differences in body weight between groups, heart
weights were also normalised to tibia lengths (HW/TL), which do not alter post-maturation.
(ii) Mouse model for anthracycline-induced cardiotoxicity
Cardiotoxicity was induced by administering mice a cumulative dose of 24 mg
doxorubicin/kg body weight (Adriablastin, Pfizer) by a weekly IP injection of 2 mg/kg during
12 weeks. Control mice were treated with physiological saline (0.9%).
Furthermore, to examine the effect of doxorubicin treatment on proliferation and
differentiation of cardiac progenitor cells residing in the myocardium, 5-bromo-2"-
deoxyuridine (BrdU) was administered during the first 4 weeks of treatment. BrdU is a
thymidine analogue that incorporates into DNA upon replication, and serves as a marker
for dividing cells. Before starting the treatment, osmotic mini-pumps (Alzet) containing
BrdU (Sigma-Aldrich), dissolved in dimethyl sulfoxide (DMSO, 100 mg/ml), were implanted
subcutaneously in mice sedated with 1.5% isoflurane (Ecuphar). The infusion of 0.25 $l
per hour via the pumps (delivery rate: 25 ng BrdU per hour) was maintained for 28 days,
whereafter the pumps were removed again.

Materials and Methods 67
3.2.3 Assessment of cardiac function
(i) Transthoracic echocardiography (TTE)
Echocardiographic measurements were performed in collaboration with Prof. J. D"hooge
(KU Leuven, Leuven, Belgium). Mice were sedated with 1.5% isoflurane (Ecuphar) and
standard views were obtained in two-dimensional and M-mode using TTE.
Echocardiography was performed using a 13-MHz transducer (i13L) on a GE Vivid7
scanner (GE Healthcare), or using a MS400 transducer (VisualSonics) on a Vevo 2100
scanner (VisualSonics) for all experiments outlined in Chapter 4 and 5, respectively.
Body temperature was monitored and maintained at 37°C using a rectal probe and heating
pad (TC-1000, CWE Inc.). Image analysis was performed using the manufacturer"s
software package, and end-diastolic and end-systolic volumes were calculated based on
the half ellipsoid model (with fixed long-to-short axis ratio; volume = %*LVID3/6, with LVID
indicating LV internal diameter at the level of the papillary muscles).
(ii) Haemodynamic measurements
For invasive pressure measurements, mice were anaesthetised with urethane (Sigma-
Aldrich, 1.2-1.4 g/kg, IP) and etomidate (10 mg/kg, IP), spontaneously breathing, and
temperature was monitored and controlled using a rectal probe and heating pad (TC-1000,
CWE Inc.). Right and left common carotid arteries were exposed and a 1.4-F high fidelity
pressure-conductance catheter (Millar Instruments) was first inserted into the left and then
into the right common carotid artery for arterial pressure recordings, followed by LV
pressure measurements. All haemodynamic parameters were recorded after a 3-5 min
stabilisation period (PowerLab Recorder and LabChart software, ADInstuments), and
averaged over 15 consecutive pressure cycles.
For LV pressure-volume measurements, mice were anaesthetised with urethane (1 g/kg
BW, IP), etomidate (10 mg/kg BW, IP), morphine (1 mg/kg BW, IP), and pancuronium (1
mg/kg BW, IP), and mechanically ventilated (MiniVent, Harvard Apparatus).
A polyethylene 10 catheter was inserted in the left jugular vein for fluid support (bovine
serum albumin in physiological saline). The 1.4-F high fidelity pressure-conductance
catheter (Millar Instruments) was advanced through the right common carotid artery into
the LV. After stabilisation of the haemodynamic situation, pressure-volume (PV) loops
were recorded (PowerLab Recorder and LabChart software, ADInstruments), while the

Chapter 3 68
ventilation was momentarily turned off to avoid respiratory fluctuation of cardiac signals.
Parallel volume was determined using a bolus injection of saline (15%, 10-20 $l, three
times). The inferior caval vein was compressed between liver and diaphragm with a cotton
swab without opening the abdomen, while PV loops were recorded. Occlusion was
repeated at least three times. Finally, blood was retrieved from the inferior caval vein to
measure specific conductivity in pre-calibrated cuvettes, and mice were euthanised.
3.3 In vitro experiments
3.3.1 Murine neonatal cardiac myocytes
(i) Isolation method
Hearts were collected from C57BL/6 neonatal pups and washed in Hank"s balanced salt
solution (HBSS) without Ca2+ and Mg2+ (Gibco - Life Technologies). Pre-digestion of the
tissue was performed in a trypsin solution (0.06% in HBSS without Ca2+ and Mg2+) for 5
min at 37°C, after stirring for 4 hours at 4°C for thorough diffusion of trypsin in the tissue.
The hearts were further digested by multiple rounds of collagenase type IV treatment
(Sigma-Aldrich, 0.1%) at 37°C. The cell suspension was filtered (mesh opening 30 $m)
and centrifuged at 1200 rpm for 5 min. Next, cells were suspended in Dulbecco"s modified
eagle medium (DMEM), and loaded on a Percoll gradient solution (GE Healthcare),
followed by density gradient centrifugation at 1994 rpm during 30 min. The pellet,
containing the cardiac myocytes, was washed with DMEM, and the cardiac myocyte
number was determined using an improved Neubauer haemocytometer (Marienfeld-
Superior). Following centrifugation, the cardiac myocytes were resuspended in culture
medium (DMEM, supplemented with minimal essential medium with non-essential amino
acids (2x), 0.02 $M !-mercaptoethanol, 4.8 mM L-glutamine, and 14% heat-inactivated
fetal bovine serum (FBS)), plated onto dishes previously coated with laminin (Sigma-
Aldrich), and cultured under standard conditions (95% O2 / 5% CO2, 37°C).
(ii) Induction of mechanical stretch
To mechanically stretch cardiac myocytes, 106 cells were seeded per well of 6-well flexible
silicone elastomer bottomed BioFlex plates (Flexcell International), previously coated with
laminin (Sigma-Aldrich), and subjected to cyclic uniaxial stretch using the FX-4000
Flexercell strain unit (Flexcell International) at the Technical University of Eindhoven

Materials and Methods 69
(The Netherlands) in collaboration with Dr. N.A.M. Bax and Prof. C.V.C. Bouten.
This device uses a controlled vacuum to deform the monolayer of cells grown on top of the
flexible well surface. The vacuum produced a gradual elongation up to 10% (2h 2%, 2h
4%, 2h 6%, 2h 8%, and 40h 10%) of the flexible bottom membranes at a frequency of 60
cycles/min (1 Hz). Cells that were not stretched were plated on identical plates to avoid
variations based on attachment surface. After 48 hours, all cells were harvested.
3.3.2 Murine adult cardiac myocytes
(i) Isolation method
Three to four months old C57BL/6 mice were heparinised, and an overdose of sodium
pentobarbital (Nembutal, Ceva Santé Animale) was administered. The heart was rapidly
excised and placed in ice-cold Normal Tyrode buffer (137 mM NaCl, 5.4 mM KCl, 0.5 mM
MgCl2, 1 mM CaCl2, and 11.8 mM HEPES pH 7.45), supplemented with 0.18% D-glucose
and 10 mM 2,3-butanedione monoxime (BDM, Sigma-Aldrich). BDM is a myosin ATPase
inhibitor, preventing hypercontraction of cardiac myocytes. Next, the aorta was cannulated
under a dissection microscope, and the heart was flushed with the supplemented Normal
Tyrode buffer. Hearts were retrogradely perfused (2.5-3.0 ml/min) through the aorta using
a Langendorff perfusion apparatus with Normal Tyrode buffer for 2-3 min, Ca2+-free
perfusion buffer (pH 7.2, 130 mM NaCl, 5.5 mM KCl, 1.2 mM KH2PO4, 1.2 mM MgSO4, 6
mM HEPES, 0.36% D-glucose, and 10 mM BDM) for 10 min, and digestion buffer
(perfusion buffer containing 672 U collagenase type II per ml (Worthington), and 30 $M
CaCl2) for 8-10 min. Following 2-3 min perfusion with bovine serum albumin (Sigma-
Aldrich, 0.5% in perfusion buffer with 0.09 mM CaCl2) to inactivate the collagenase, hearts
were perfused with perfusion buffer supplemented with 0.09 mM CaCl2 during 3 min.
Afterwards, hearts were removed from the perfusion apparatus, and dispersed into cell
suspension in perfusion buffer with 0.09 mM CaCl2 using plastic pasteur pipettes. The cell
suspension was swirled during 5 min at 37°C, filtered (250 $m mesh opening), and
centrifuged at 50 g for 1 min. Damaged myocytes and non-myocytes were removed by
washing with perfusion buffer containing, sequentially, 0.5 or 1.0 $M CaCl2.
Cardiac myocytes were pelleted by centrifugation at 50 g for 1 min after each wash.
Finally, cardiac myocytes were resuspended in culture medium (M-199 medium, Gibco -
Life Technologies), supplemented with 2 mM carnitine, 5 mM taurine, 5 mM creatine (all

Chapter 3 70
Sigma-Aldrich), and 10 mM BDM, plated onto dishes previously coated with laminin
(Sigma-Aldrich), and cultured under standard conditions (95% O2 / 5% CO2, 37°C).
After 4 hours attachment, further culturing was established in culture medium containing a
lower BDM concentration (1 mM).
(ii) Induction of cardiac myocyte hypertrophy and hypoxia
Induction of cardiac myocyte hypertrophy by neurohormones, including ET-1, Ang II, or
PE, in murine cardiac myocytes is very hard to establish and is poorly described in the
literature. Therefore, adult cardiac myocytes were isolated from C57BL/6 mice 4 weeks
after TAC to obtain hypertrophied cardiac myocytes. Non-hypertrophied control cardiac
myocytes were isolated from littermates without TAC.
To induce cardiac myocyte hypoxia, adult cardiac myocytes were plated and incubated for
4 hours in normoxic conditions, and then transferred to hypoxic conditions during 6h
(SANYO hypoxia incubator set at 1% oxygen).
3.3.3 Murine cardiac endothelial cells
(i) Isolation method
Endothelial cells were purified from hearts of mice expressing green fluorescent protein
(GFP) under the direction of the endothelial-specific receptor tyrosine kinase Tie2
(tyrosine kinase with immunoglobulin-like and EGF-like domains 2) promoter (Tie2-GFP,
FVB background, provided by Prof. A. Luttun, KU Leuven, Leuven, Belgium) through
fluorescence-activated cell sorting (FACS). Hearts were digested in a collagenase I
solution (Gibco - Life Technologies, 0.15% in Gey"s balanced salt solution) at 37°C during
1 hour. Following further mechanical digestion of the tissue and filtering of the cell
suspension (100 $m mesh opening), the collagenase was inactivated by FBS-
supplemented phosphate buffered saline (PBS). Thereafter, the cell suspension was
centrifuged (7 min, 600 g), and red blood cells were lysed (lysis buffer: 50 mM NH4Cl, 10
mM CO3HK, 1 mM ethylene diamine tetraacetic acid (EDTA), pH 7.4). Following
centrifugation (7 min, 600 g), the pellet was resuspended in FACS buffer (1 mM EDTA, 25
mM HEPES pH 7.0, and 1% BSA in PBS). A GFP-positive and -negative fraction was
sorted from the cell suspension using a FACSAria cell sorter (BD Biosciences).

Materials and Methods 71
3.3.4 Human cardiac endothelial cells
(i) Isolation method
Human cardiac microvascular endothelial cells were isolated by G. Coppiello (KU Leuven,
Leuven, Belgium) from auricular biopsies of patients undergoing valve replacement
surgery. The ethics review board of Gasthuisberg University Hospital approved the
protocol, and all patients provided informed consent. The human cardiac biopsies were
processed using the protocol as described for murine cardiac endothelial cells. Cells were
plated on dishes previously coated with 0.1% gelatin (MilliPore), and cultured in
endothelial cell growth medium (EGM), supplemented with the EGM2-MV bullet kit
(Lonza), 50 U/ml penicillin, and 0.005% streptomycin. When 100% confluency was nearly
reached, the cells were trypsinised, washed, and centrifuged (7 min, 600 g).
The resuspended cell pellet was stained with a fluorescein isothiocyanate (FITC)-labelled
anti-CD31 antibody (1/10, BD Pharmingen), a biotin-labelled anti-Tie2 antibody (1/40,
eBioscience) in combination with an allophycocyanin (APC)-labelled streptavidin (1/400,
eBioscience), or a phycoerythrin-labelled anti-podoplanin antibody (1/100 Angiobio), and
the appropriate isotype control antibody. After 15 min incubation in the dark at room
temperature, the cells were washed, centrifuged (7 min, 600 g), and resuspended in FACS
buffer. The CD31-positive, Tie2-positive, and podoplanin-negative fraction was sorted and
plated on gelatin-coated culture dishes to establish a highly enriched endothelial cell
culture.
(ii) Induction of mechanical stretch
Mechanical stretch was applied on human cardiac microvascular endothelial cells as
described for murine neonatal cardiac myocytes (cfr. 3.3.1 - ii).
3.3.5 Force measurements in murine cardiac myocytes
Calcium-dependent active isometric force and its Ca2+-sensitivity, and Ca2+-independent
passive force of permeabilised cardiac myocytes were determined at 15°C using a
mechanical force measuring system, in collaboration with Dr. A. Balogh and Prof. Dr. Z.
Papp (University of Debrecen, Debrecen, Hungary). Cardiac myocytes were obtained from
the LV of mechanically disrupted frozen hearts, permeabilised using Triton X-100, and
mounted between a force transducer and an electromagnetic motor. Isometric Ca2+-
contractures were evoked by transferring cardiac myocytes from a Ca2+-free relaxing

Chapter 3 72
solution (37.34 mM KCl, 10 mM BES, 6.24 mM MgCl2, 7 mM CaEGTA, 6.99 mM Na2ATP,
and 15 mM sodium creatine-phosphate, pH 7.2) to Ca2+-containing activating solutions
(of otherwise identical compositions) at a sarcomere length of 2.3 µm. Calcium-
independent passive force was determined in relaxing solution. Active and passive force
values were standardised to cardiac myocyte cross-sectional area.
To determine the Ca2+-sensitivity of isometric force production, active forces measured at
various Ca2+-concentrations were plotted as a function of pCa (pCa = -log10[Ca2+]), and a
modified Hill’s equation was then employed to determine the [Ca2+] evoking half-maximal
force production (pCa50, Figure 14).
To determine cGMP/PKG-dependent modulation of the mechanical function of
myofilaments, Ca2+-force relationships of cardiac myocytes were determined before and
after incubations in relaxing solution supplemented with the catalytic subunit of bovine
PKG (PKG-I!, 0.01 U/mL), cGMP (10 µM), and dithiothreitol (DTT, 6 mM; all Sigma-
Aldrich) for 40 min.
Figure 14. The Ca2+-force relationship of cardiac myocytes In the Ca2+-force relationship, the Ca2+-sensitivity of isometric force production represents the [Ca2+] (plotted as a function of pCa; pCa = -log10[Ca2+]) evoking half-maximal force production. If the pCa50 increases to a higher numerical value (leftward curve shift), the myofilaments are more sensitive to Ca2+. Thus, at a given submaximal [Ca2+], a higher active tension is developed. If the pCa50 decreases to a lower numerical value (rightward curve shift), the myofilaments display a lower Ca2+-sensitivity.

Materials and Methods 73
3.4 Determination of cardiac cyclic nucleotide levels
3.4.1 After chronic pressure overload or doxorubicin administration
To measure cardiac cGMP or cAMP concentrations, frozen ventricular tissues were
pulverised in liquid nitrogen, and cyclic nucleotides were extracted in 6% trichloro-acetic
acid, incubated at 4°C during 30 min, and centrifuged (15 min, 12 500 rpm, 4°C).
Supernatant was extracted three times with water-saturated ether and vacuum-dried
overnight. The lyophilised extracts were solubilised in assay buffer, and cyclic nucleotide
concentrations were measured using an enzyme immunoassay according to the
manufacturer’s instructions (GE Healthcare).
3.4.2 At baseline following in vivo stimulation of sGC activity
Cyclic GMP was extracted from cardiac tissue of mice euthanised 5 min after increasing
NO-stimulated cGMP synthesis by intravenous bolus administration of S-
nitrosoglutathione (GSNO; 1 mg/kg body weight, via a polyethylene 10 catheter in the
jugular vein), and prior bolus administration of a non-selective PDE inhibitor
(dimethylxanthine; Theophylline, 0.162 g/kg body weight, 30 min before sacrifice).
3.4.3 At baseline following ex vivo stimulation of sGC activity
Enzyme activity of sGC in myocardial tissue samples was determined by Prof. E. Buys
(Massachusetts General Hospital and Harvard Medical School, Boston, USA).
Following homogenisation of cardiac tissue in a buffer (50 mM Tris(hydroxymethyl)amino
methane HCl (pH 7.6), 1 mM EDTA, 1 mM DTT, and 2 mM phenylmethylsulfonyl fluoride),
cardiac extracts were centrifuged (20 min, 20 000 g, 4°C), and the supernatant (containing
50 $g protein) was incubated for 10 min at 37°C in a reaction mixture containing 50 mM
Tris.HCl (pH 7.5), 4 mM MgCl2, 0.5 mM 3-isobutyl-1-methyl-xanthine (IBMX), 7.5 mM
creatine phosphate, 0.2 mg/ml creatine phosphokinase, 1 mM N &-nitro-L-arginine methyl
ester (L-NAME), and 1 mM GTP. During this time period, sGC enzyme activity was
stimulated with 1.25 mM diethylenetriamine/NO (DETA/NO), 125 $M BAY 41-2272 (both
Enzo Life Sciences), or 125 $M BAY 58-2667 (BioVision, Inc.). The reaction was
terminated by adding 0.05 M HCl. Cyclic GMP levels in the reaction mixture were
measured using an enzyme immunoassay according to the manufacturer’s instructions
(Biomedical Technologies).

Chapter 3 74
3.5 Transcriptional and translational analysis
3.5.1 Quantitative real-time PCR (RT-qPCR)
Following pulverisation of frozen ventricular tissue in liquid nitrogen, mRNA extraction was
performed using the RNeasy fibrous tissue kit (Qiagen), according to the manufacturer"s
recommendations. For mRNA isolation from cells, the RNeasy mini or micro kit (Qiagen)
was used. The concentration and quality of the extracted mRNA was assessed using a
spectrophotometer (NanoDrop, Thermo Scientific).
The mRNA was reverse transcribed using the QuantiTect Reverse Transcription kit
(Qiagen), or the SuperScript III First-Strand Synthesis System (Invitrogen - Life
Technologies) when higher cDNA yields were required.
Transcript levels were determined by RT-qPCR (StepOnePlus system, Applied
Biosystems - Life Technologies) using specific primers and the DNA-intercalating SYBR
Green dye or 6-carboxy-fluorescein (FAM)-labelled probes (TaqMan probes, Applied
Biosystems - Life Technologies). These probes contain a FAM fluorescent dye at the 5"
end and a 6-carboxy-tetramethyl-rhodamine (TAMRA) quencher at the 3" end.
The 5" exonuclease activity of the Taq polymerase will release the fluorescent FAM
molecule when probes hybridise to the target gene sequence. Primers and probes were
designed using Primer Express software (Applied Biosystems - Life Technologies, Table
2) or supplied by commercially available and validated TaqMan gene expression assays
(Applied Biosystems - Life Technologies).
Expression levels were quantified by determining the cycle number at which SYBR Green
or FAM fluorescence is higher than the threshold (Ct or threshold cycle value). Transcript
levels were always normalised to mRNA levels of a housekeeping gene (18S ribosomal
RNA or glyceraldehyde-3"-phosphate-dehydrogenase (GAPDH)), and expressed relative
to the control group according to the Livak method (2-''Ct).311
Mouse gene Primer/probe sequence Nppa Forward 5"-TCCATCACCCTGGGCTTCT-3" (ANP) Reverse 5"-AGCATTTGGTCCAATATGGCC -3" Probe 5"-CCTCGTCTTGGCCTTTTGGCTTCC-3" Bax Forward 5"-CCGGCGAATTGGAGATGA-3" (Bax) Reverse 5"-CCCAGTTGAAGTTGCCATCA-3" Probe 5"- TGGACACGGACTCCCCCCGA-3" Bcl2 Forward 5"-TGGCCTTCTTTGAGTTCGGT-3"

Materials and Methods 75
(Bcl-2) Reverse 5"-GAGAAATCAAACAGAGGTCGCAT-3" Bcl2l1 Forward 5"-ACTCATCGCCTGCCTCTCTC-3" (Bcl-XL) Reverse 5-GCCACAGCAGCAGTTTGGAT-3" Myh7 Forward 5"-AACATTCGGGCCTTCATGG-3" (!MHC) Reverse 5"-CAGCGGCTTGATCTTGAAGTAGA-3" Nppb Forward 5"-GCCAGTCTCCAGAGCAATTCA-3" (BNP) Reverse 5"-GTGAGGCCTTGGTCCTTCAA-3" Probe 5"-TCTGGGCCATTTCCTCCGACTTTTCT-3" Ctgf Forward 5"-TGACCCCTGCGACCCACA-3" (CTGF) Reverse 5"-TACACCGACCCACCGAAGACACAG-3" Fas Forward 5"-TAGAACCTCCAGTCGTGAAACCATA-3" (Fas) Reverse 5"-TTTAGCTTCCTGGATTGTCATGTC-3" Fasl Forward 5"-AGTGTCTCATTGGCACCATCTTTA-3" (FasL) Reverse 5"-CAAACATCCCTCTTACTTCTCCGTTA-3" Fn Forward 5"-CCGGTGGCTGTCAGTCAGA-3" (FN) Reverse 5"-CCGTTCCCACTGCTGATTTATC-3" Gapdh (GAPDH)
Forward 5"-TGTGTCCGTCGTGGATCTGA-3" Reverse 5"-CCTGCTTCACCACCTTCTTGA-3"
TaqMan Probe 5"-CCTGGAGAAACCTGCCAAGTATGATGACA-3" Gapdh Forward 5"-CCGCATCTTCTTGTGCAGT-3"
(GAPDH) Reverse 5"-GAATTTGCCGTGAGTGGAGT-3" SYBR Green
Npr1 Forward 5"-CAGGGCTCCCAGTGAGGAA-3" (NPR-A) Reverse 5"-AGCATCGAGCAGTGCAAGTG-3" Probe 5"-CATGCCCGAGAGGTAGCCCGA-3" Npr2 Forward 5"-AGATGCAGGGACGAAGATACCA-3" (NPR-B) Reverse 5"-AAAGTCCCCAGAATCCAAGTCTC-3" Pde5 Forward 5"-CGGCCTACCTGGCATTCTG-3" (PDE5) Reverse 5"-GCAAGGTCAAGTAACACCTGATT-3" Atp2a2 Forward 5"-CATCTGCTTGTCCATGTCACTT-3" (SERCA2) Reverse 5"- CGGTGTGATCTGGAAAATGAG-3" Probe 5"-TCTTGATCCTCTACGTGGAACCTTTGC-3" Gucy1a3 Forward 5"-CTATCTGTCCGACATCCCAATTC-3" (sGC#1) Reverse 5"-TGAGCCCGTGCCTGCT-3" Probe 5"-TCCCCGCTTGCGTCTTCTGCAAG-3" Gucy1b3 Forward 5"-TCAGTGTGGCAATGCCATCTA-3" (sGC!1) Reverse 5"-AGGGCGGACCAGAGAGAAGA-3" Probe 5"-CCAGCCTGGGAACTGCAGCCTTCT-3" Tgfb1 Forward 5"-GACCCTGCCCCTATATTTGGA-3"
(TGF-!1) Reverse 5"-GCGCCCGGGTTGTGT-3"

Chapter 3 76
Table 2. Sequences of designed primers and probes used for RT-qPCR ANP and BNP indicate atrial and brain natriuretic peptide; Bcl-2 and Bcl-XL, B-cell lymphoma 2 and extra large; Bax, Bcl-2 associated X protein; !MHC, ! myosin heavy chain; CTGF, connective tissue growth factor; Fas and FasL, Fas receptor and ligand; FN, fibronectin; GAPDH, glyceraldehyde-3"-phosphate-dehydrogenase; NPR-A and NPR-B, natriuretic peptide receptor A and B; PDE, phosphodiesterase; SERCA2, sarcoplasmic reticulum Ca2+-ATPase 2; sGC#1 and sGC!1, #1- and !1-subunit of soluble guanylate cyclase; and TGF-!1, transforming growth factor-!1.
3.5.2 Immunoblot analysis
Frozen ventricular tissue was pulverised in liquid nitrogen and homogenised in lysing
matrix D tubes (MP Biomedicals) with a ribolyser (Hybaid), using the modified radio-
immunoprecipitation assay (RIPA) lysis solution containing 2 mM Tris-HCl pH 7.4, 137
mM NaCl, 10% glycerol, 1% Triton X-100, 0.1% sodium deoxycholate, 0.1% sodium
dodecyl sulphate (SDS), 2 mM EDTA pH 7.5, 1 mM Na3VO4, 2 mM NaF, and Complete
protease inhibitor (Roche). The matrix was washed and briefly centrifuged, and the
supernatant was incubated on ice for 30 min and centrifuged at 13 500 rpm for 10 min.
Protein concentrations were determined using the bicinchoninic acid (BCA) assay
(Pierce). Immunoblot analysis was performed using SDS polyacrylamide gel
electrophoresis (SDS-PAGE, 10% or 4-12%, NuPage, Invitrogen - Life Technologies), and
proteins were transferred to nitrocellulose membranes by semi-dry electroblotting.
The membranes were blocked for 1 hour in 5% non-fat milk, and incubated overnight with
an antibody directed against SERCA2 (Thermo Scientific), calcineurin (BD Biosciences),
ERK1/2, phosphorylated ERK1/2, Akt, phosphorylated Akt, PDE5 (all Cell Signaling
Technology), 3-nitrotyrosine (Chemicon International), 4-hydroxy-2-nonenal, or
malondialdehyde (both Calbiochem). Bound antibodies were detected using horseradish
peroxidase (HRP)-conjugated secondary antibodies (Dako), and visualised using
enhanced chemiluminescence (ECL) western blotting detection reagents (GE Healthcare -
Life Sciences) or SuperSignal West Femto chemiluminescent substrates (Thermo
Scientific). Actin (Millipore) or GAPDH (Cell Signaling Technology) protein levels were
used as a loading control. 3.5.3 Immunohistochemistry
(i) Histological stainings
Human and murine tissue was incubated overnight in buffered zinc formalin fixative (Z-fix,
Materials and Methods 77
Anatech LTD), transferred to 70% ethanol, and embedded in paraffin. Six $m-thick
sections were prepared from paraffin-embedded tissue on positively charged Superfrost
Plus glass slides (MenzelGläser), and dried overnight at 37°C to ensure adhesion. Tissue
sections were stained with haematoxylin and eosin (H&E), or picrosirius red, or used for
immunohistochemistry (IHC). Prior to staining, sections were deparaffinised with xylol and
decreasing concentrations of ethanol. For IHC stainings, antigen retrieval was performed
(Table 3), and endogenous peroxidase was quenched by incubation in 0.09% H2O2 in
methanol. Primary antibodies (Table 3) were applied overnight at room temperature and
non-specific binding of the secondary antibody was prevented by prior incubation with
10% serum from the secondary antibody species for 45 min. Secondary antibodies were
labelled with biotin (Dako), in which case the signal was amplified by avidin-biotin-
peroxidase complexing (ABC-PO, Vector Laboratories) or tyramide signal amplification
(TSA, Perkin Elmer, Table 3). Peroxidase activity was detected with 0.01% H2O2/3,3"-
diaminobenzidine (DAB, Fluka), which is revealed by a brown precipitate at the antigen
site. Following counterstaining with Harris haematoxylin, tissue slides were dehydrated in
increasing concentrations of ethanol and xylol, and mounted with glass cover-slides using
DPX mounting medium (Prosan). For immunofluorescent stainings, the signal was
amplified by streptavidin, labelled with an Alexa fluor dye (Invitrogen - Life Technologies)
or TSA (Perkin Elmer), and slides mounted with glass cover-slides using ProLong Gold
anti-fade reagent with 4',6-diamidino-2-phenylindole (DAPI, Invitrogen - Life
Technologies). Images were obtained using a confocal laser scanning 510 microscope or
an Axiovert 200M imaging microscope (Zeiss).
Target Antigen retrieval Primary antibody Amplification IHC STAININGS ON HUMAN TISSUE
PDE5 Citrate buffer pH 6.1 (Dako), 20 min 95°C Home-made, 10 $g/ml ABC
Desmin Citrate buffer pH 6.1 (Dako), 20 min 95°C Dako (1/100) Streptavidin-
Alexa
vWF Citrate buffer pH 6.1 (Dako), 20 min 95°C Dako (1/25) Streptavidin-
Alexa IHC STAININGS ON MURINE TISSUE
PDE5 Citrate buffer pH 6.1 (Dako), 20 min 95°C Santa Cruz (1/100)
ABC / Streptavidin-
Alexa

Chapter 3 78
Laminin Citrate buffer pH 6.1 (Dako), 20 min 95°C Sigma-Aldrich (1/100) ABC
Lectin BS1 Citrate buffer pH 6.1 (Dako), 20 min 95°C
Sigma-Aldrich, biotin labelled (1/50) Streptavidin-HRP
!-SMA Tris-EDTA buffer pH 9, 20 min 95°C Dako (1/500) -
DNA strand breaks
Proteinase K (1/500 in PBS), 10 min room
temperature
Apoptag kit (TUNEL method*,
MilliPore) -
BrdU & Actin (#-sarcomeric)
Citrate buffer pH 6, 10 min microwave
Roche (1/50, BrdU labelling and detection kit I)
& Sigma (1/50) -
Table 3. Overview of antigen retrieval methods, primary antibodies, and amplification methods used in IHC stainings Tris-EDTA buffer: 10 mM Tris, 1 mM EDTA, 0.05% Tween 20, pH 9. PDE indicates phosphodiesterase; vWF, von Willebrand factor; Lectin BS1, lectin from Bandeiraea simplicifolia I; BrdU 5-bromo-2"-deoxyuridine; FITC, fluorescein isothio-cyanate; and ABC, avidin-biotin complex. * TUNEL method: This method identifies apoptotic cells using terminal deoxynucleotidyl transferase to transfer digoxigenin-conjugated nucleotides to DNA strand breaks. DNA fragments that have been labelled with digoxigenin-nucleotides are then allowed to bind an anti-digoxigenin antibody conjugated to a peroxidase reporter molecule. The use of chromogenic substrates allows visualisation of these antibodies.
(ii) Microscopic analysis
Microscopic analysis was performed on pictures obtained using an Axiovert 200M imaging
microscope with Axiovision software (Zeiss).
To determine cardiac myocyte width on laminin-stained tissue sections, measurements
were obtained at the level of the nucleus in longitudinally-sectioned myocytes.
To assess the degree of fibrosis in the murine LV, the area of collagen deposition was
traced on picrosirius red-stained tissue sections, using circularly polarised light allowing
evaluation of thick, tightly-packed, red birefringent collagen and thin, loosely-assembled,
green birefringent collagen. The degree of fibrosis was expressed as the area of red or
green birefringent collagen relative to the area of the examined LV tissue area.
The index of apoptosis was determined by dividing the number of apoptotic cardiac
myocytes by the area of the examined LV tissue area.
Vascular density was analysed on lectin-stained tissue sections by dividing the number of
positively stained small blood vessels by the number of cardiac myocyte nuclei.
To asses the degree of inflammation in the myocardial tissue, the number of CD45-

Materials and Methods 79
immunolabelled haematopoietic cells was divided by the number of cardiac myocyte
nuclei.
Proliferation and differentiation of cardiac progenitor cells was investigated by expressing
the number of BrdU-positive cardiac myocytes relative to the total number of cardiac
myocytes, identified by co-staining for #-sarcomeric actin.
3.6 Profiling of cardiac miRNA expression
Cardiac miRNA levels were determined by the Nucleomics Core (VIB, Leuven, Belgium).
3.6.1 Affymetrix miRNA Microarray
Cardiac tissue was homogenised and subjected to miRNA isolation using the miRNeasy
kit (Qiagen). Capillary electrophoresis (Bioanalyzer 2100; Agilent) and spectrophotometry
(NanoDrop, Thermo Scientitifc) were used to assess quality and quantity of total RNA.
MicroRNA obtained from 1 $g total RNA was labelled with a Cy3 dye (Molecular Probes)
using the mirVana miRNA labelling Kit (Ambion - Life Technologies), according to the
manufacturer"s recommendations. Labelling efficacy and specificity were tested on control
samples spiked with standard miRNAs. The array covered 609 murine miRNAs
(GeneChip miRNA Array; Affymetrix). Resulting signal intensities were normalised
according to the robust multichip average (RMA) method, and linear models for microarray
analysis (LIMMA) statistics were applied to determine alterations in miRNA levels.
Differences in miRNA levels with P<0.01 were considered significant. The resulting
P-values were corrected for multiple testing with the Benjamini-Hochberg method to
control the false discovery rate (P<0.05). To validate differentially regulated miRNAs, the
miRNA was reverse transcribed with a miRNA-specific primer, followed by RT-qPCR with
specific primers and TaqMan probe (TaqMan microRNA assays, Applied Biosystems - Life
Technologies). As an internal control, miRNA-16 and snoRNA412 primers were used.
Relative miRNA expression was analysed using the Livak method (2-''Ct).311
3.6.2 nCounter miRNA expression assay
MiRNA isolation and determination of quality and quantity was carried out as described for
the Affymetrix microRNA Microarray. The nCounter miRNA expression assay (NanoString
Technologies) delivered expression profiling of 566 miRNAs and 33 viral miRNAs (this set

Chapter 3 80
was completed with 6 positive controls, 8 negative controls, and 4 housekeeping genes for
normalisation and QC purposes). The nCounter assay involves the hybridisation of
fluorescently labelled, bar-coded probes to the miRNAs of interest, which are then counted
to quantify miRNA expression. Differential expression with P<0.01 was considered
significant.
3.6.3 Experimental validation of miRNA targets
(i) MicroRNA target site identification
Commercially available HEK293 cells, stably transfected with a lentiviral vector containing
a CMV promoter driven firefly luciferase reporter gene containing the 3"UTR of human
PDE5A (NM_001083), and a puromycin resistance gene, were purchased from Applied
Biological Materials (for inserted sequence see Supplement 1). The cells were cultured in
medium (DMEM + 10% FBS + 1% penicillin/streptomycin) supplemented with puromycin
(Sigma-Aldrich, 0.00006%) to select for stably transfected cells.
To validate predicted miRNA binding sites in the 3"UTR of PDE5 (predictive databases
consulted: TargetScan, PITA, miRanda, DIANA, miRtarget2), the stably transfected
HEK293 cells were transfected with pre-miR miRNA precursors and anti-miR miRNA
inhibitors (Ambion - Life Technologies), and luciferase activity was measured.
Briefly, 24 hours before transfection, HEK293 cells were plated at a density to guarantee
70-80% confluency at the time of transfection. Both siPORT amine transfection agent
(Ambion - Life Technologies; final dilution of 1/312.5) and pre-miR miRNA precursors or
anti-miR miRNA inhibitors (Ambion - Life Technologies, final concentration of 30 nM) were
diluted in Opti-MEM I medium (Gibco - Life Technologies), incubated at room temperature
for 10 min, and mixed. Following a subsequent 10 min incubation at room temperature,
the transfection complexes were then added to the cells (2.5 ml final volume per well of a
6-well plate). Scrambled, non-targeting, Cy3-labelled pre-miR precursors and anti-miR
inhibitors (pre-miR#1 and anti-miR#1) were used as negative controls and to monitor
transfection efficiency. After 24h transfection, cells were collected on ice using lysis buffer
(100 mM KH2PO4, 0.2% Triton X-100, and 0.5 mM DTT, pH 7.8). Next, the cell lysate was
vortexed and centrifuged at 4°C for 2 min at 12 000 rpm, and the supernatant was stored
overnight at -80°C. Firefly luciferase activity was measured in 20 $l of the supernatant
using a luminometer (MicroLumat Plus, Berthold Technologies) after addition of 100 $l
luciferase assay buffer (20 mM tricine, 1.07 mM (MgCO3)4Mg(HO)2.5H20, 0.1 mM EDTA

Materials and Methods 81
and 33.3 mM DTT), supplemented with 0.5 mM coenzyme A (MP Biomedicals), 1 mM
ATP (Roche), and 0.5 mM luciferin (Sigma-Aldrich). Firefly luciferase activity of cells
transfected with pre-miR miRNA precursors or anti-miR miRNA inhibitors was normalised
to protein concentration, as determined by a BCA assay (Pierce), and expressed relative
to normalised luciferase activity of cells transfected with scrambled miR negative controls.
In addition, the 3"UTR of murine PDE5 was cloned downstream of a firefly luciferase
gene driven by a simian virus (SV) 40 promoter in a reporter plasmid (pISO, kindly
provided by Prof. Dr. K.D. Bloch). In brief, the 3"UTR of murine PDE5 (AK031275) was
PCR amplified (Phusion high-fidelity DNA polymerase, Thermo Scientific), using cDNA
obtained from murine pulmonary tissue as a template and SacI restriction site containing
primers (forward primer: 5"-AAAGAGCTCCGGTGAGAGGTGTGAGTGTG-3" and reverse
primer: 5"-AAACTCGAGAGGGTGATGTGCTCATGGTT-3"). The PCR product was purified
from an agarose gel (high pure PCR product purification kit, Roche), digested overnight
with SacI restriction enzyme (Fermentas), and ligated in the multiple cloning site of the
pISO vector using T4 ligase enzyme (New England Biolabs, for inserted sequence see
Supplement 2).
Twenty-four hours after plating mouse embryonic fibroblasts (NIH 3T3) at a density to
guarantee 70-80% confluency at the time of transfection, the cells were transfected for 24
hours with the firefly luciferase reporter plasmid, a Renilla luciferase reporter plasmid (with
a thymidine kinase promoter), and pre-miR miRNA precursors and anti-miR miRNA
inhibitors (Ambion - Life Technologies), and luciferase activity was measured.
For transfection, lipofectamine 2000 (Invitrogen - Life technologies, final dilution of 1/500)
the firefly luciferase reporter plasmid (2 $g), the Renilla luciferase reporter plasmid (0.5
$g), and the pre-miR miRNA precursors and anti-miR miRNA inhibitors (final
concentration of 30 nM) were diluted in Opti-MEM I medium, and mixed after 5 min
incubation. Following a subsequent 20 min incubation at room temperature, the
transfection complexes were dispensed onto the cells (2.5 ml final volume per well of a 6-
well plate). After 24h transfection, cells were collected on ice using lysis buffer (dual
luciferase reporter assay system, Promega), and firefly luciferase activity was measured
using a luminometer (MicroLumat Plus, Berthold Technologies) in 20 $l cell lysate after
addition of 75 $l luciferase assay reagent (dual luciferase reporter assay system,
Promega). After quantifying the firefly luminescence, this reaction was quenched, and the

Chapter 3 82
Renilla luciferase reaction was initiated and quantified. Firefly luciferase activity of cells
transfected with pre-miR miRNA precursors or anti-miR miRNA inhibitors was normalised
to Renilla luciferase activity, and expressed relative to normalised luciferase activity of
cells transfected with scrambled miR negative controls.
(ii) MicroRNA target site validation
If a given mRNA is a genuine target of a specific miRNA, then modulation of miRNA
concentrations should correspond to a predictable change in the amount of protein
encoded by the target mRNA. Therefore, PDE5 immunoblotting is used for functional
validation of miRNA target sites.
3.7 Statistical analysis
All data are expressed as mean±standard error of the mean. Differences between groups
(except for miRNA profiling) were determined with an unpaired t-test (with Welch"s
Correction), using GraphPad Prism (GraphPad Software), or a factorial analysis of
variance (ANOVA) followed by a Bonferroni post-hoc test, using the statistical software
Statistica (StatSoft). A probability value of P<0.05 was considered statistically significant.

CChhaapptteerr 44
ROLE OF CGMP SIGNALLING IN THE ADVERSE CARDIAC RESPONSE TO
CHRONIC PRESSURE OVERLOAD


cGMP in Chronic LV Pressure Overload 85
CHAPTER 4: ROLE OF CGMP SIGNALLING IN THE ADVERSE CARDIAC RESPONSE TO
CHRONIC PRESSURE OVERLOAD
4.1 Increased PDE5 expression in LV tissue of patients with severe aortic
stenosis
Left ventricular outflow tract tissue was obtained from patients undergoing aortic valve
replacement (AVR, n=20). These patients had an aortic valve area (0.7 cm2, a mean
trans-valvular gradient of 57±3 mmHg, an increased LV mass index of 86±4 g/m2, and no
signs of obstructive coronary artery disease. Quantitative PCR and immunoblot analysis
showed greater PDE5 levels in AS patients than in controls (n=6, Figure 15A and B).
Figure 15. PDE5 expression in LV outflow tract tissue of AS patients Quantitative RT-PCR showed increased PDE5 expression (relative to 18S rRNA expression) in LV tissue of AS patients (n=20) compared to controls (n=6, A). Immunoblot and densitometric analysis confirmed elevated PDE5 levels in AS patients (B). Protein levels of GAPDH were measured to control for sample variability. *P<0.05, vs controls.
4.1.1 Elevated PDE5 expression is localised in scattered cardiac myocytes and endothelial cells
To investigate spatial distribution of increased PDE5 expression in the hearts of AS
patients, we performed immunohistochemical analyses. In cardiac tissue of control
subjects, PDE5 expression was scarcely detectable (Figure 16A), and predominantly
present in vascular smooth muscle. In contrast, in AS patients, PDE5 expression was
A B

Chapter 4 86
markedly induced in scattered cardiac myocytes (Figure 16A and B). Histological
examination of PDE5 immunoreactivity in cardiac tissue of AS patients was performed on
a minimum of 10 high-power fields from 3 sections per patient, allowing a semi-
quantitative scoring of PDE5 expression on a four-point scale from 0 to 3. Grade 0
indicated no PDE5 immunoreactivity in cardiac myocytes; grade 1, up to 1% PDE5
immunoreactive cardiac myocytes; grade 2, between 1 and 10% of PDE5 immunoreactive
cardiac myocytes; and grade 3, PDE5 immunoreactivity in >10% of the cardiac myocytes.
Subsequently, AS patients were separated into patients with modestly increased PDE5
expression (grade 1), and those with markedly increased PDE5 immunoreactivity (grade
2-3). The latter group of AS patients (n=13) had higher pulmonary capillary wedge
pressure and serum NT-proBNP levels than AS patients with grade 1 PDE5
immunoreactivity (n=7) (Table 4).
Whereas the degree of PDE5 expression in cardiac myocytes was correlated with cardiac
dysfunction and failure, we detected PDE5 immunoreactivity throughout endothelium of
capillaries and arterioles in all AS patients.
Table 4. Indices of cardiac stress and filling pressures in AS patients grouped according to cardiac myocyte PDE5 expression AS patients with markedly increased PDE5 immunoreactivity in cardiac myocytes (grade 2-3*; n=13) had higher pulmonary capillary wedge pressures (PCWP) and serum NT-proBNP levels than AS patients with grade 1 PDE5 immunoreactivity (n=7), despite similar LV mass (LVM). P<0.05, vs patients with grade 1 PDE5 immunoreactivity. * Grade 1, up to 1% PDE5 immunoreactive cardiac myocytes; Grade 2, between 1 and 10% of PDE5 immunoreactive cardiac myocytes; Grade 3, PDE5 immunoreactivity in >10% of the cardiac myocytes.
AS Patients
Grade 1 PDE5 immunoreactivity (n=7)
Grade 2-3 PDE5 immunoreactivity (n=13)
NT-proBNP (ng/l) 403±221 1090±254*
PCWP (mmHg) 11±1 18±2*
LVM (g/m2) 82±8 88±4
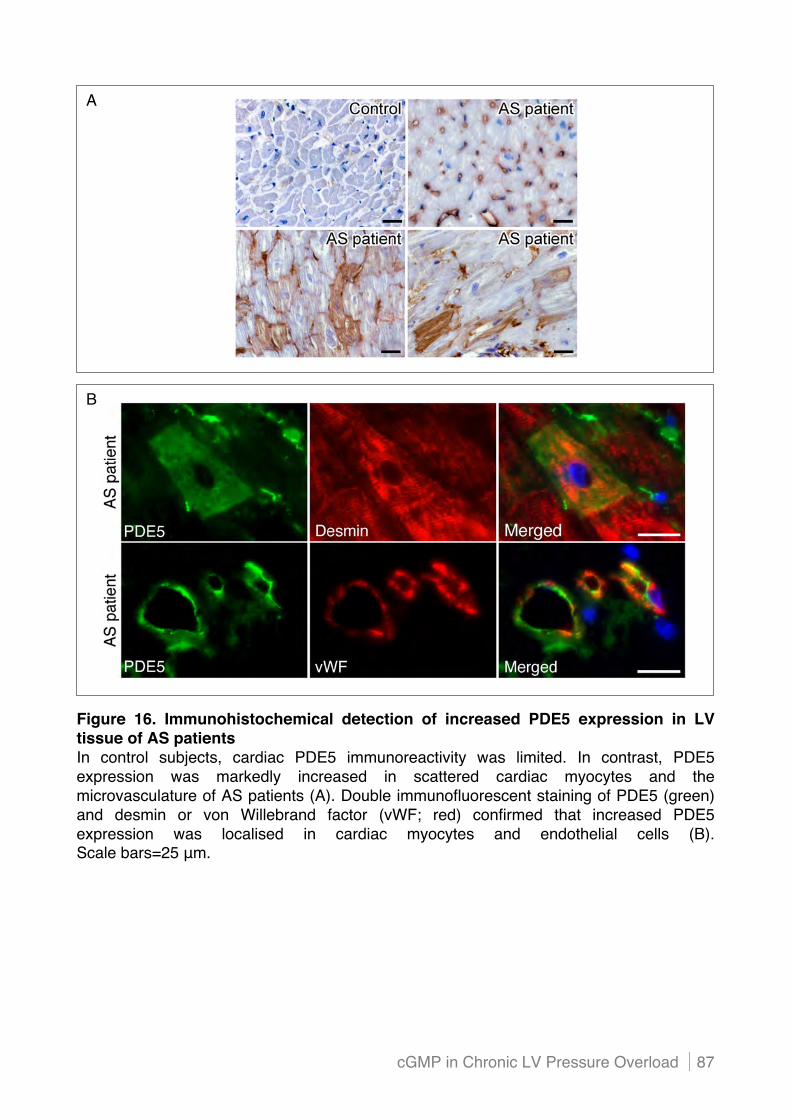
cGMP in Chronic LV Pressure Overload 87
Figure 16. Immunohistochemical detection of increased PDE5 expression in LV tissue of AS patients In control subjects, cardiac PDE5 immunoreactivity was limited. In contrast, PDE5 expression was markedly increased in scattered cardiac myocytes and the microvasculature of AS patients (A). Double immunofluorescent staining of PDE5 (green) and desmin or von Willebrand factor (vWF; red) confirmed that increased PDE5 expression was localised in cardiac myocytes and endothelial cells (B). Scale bars=25 $m.
A
B

Chapter 4 88
4.1.2 Mechanical stretch induces PDE5 expression in cardiac endothelial cells in vitro
To investigate whether mechanical load induced changes in endothelial PDE5 protein
expression, we applied cyclic stretch for 48 hours on human cardiac microvascular
endothelial cells (MVEC) in vitro. After 48 hours cyclic stretch, these cells were reoriented
and aligned perpendicularly to the stretch direction (Figure 17A), consistent with previous
reports showing alignment of endothelial cells in the direction of the minimal substrate
deformation.312-314 Perpendicular orientation of endothelial cells to uniaxial stretch is also
seen in arteries in vivo, where the endothelial cells are aligned along the longitudinal axial
direction of the arteries, which are subjected to cyclic circumferential stretch due to the
pulsatile pressure.
Immunoblot and densitometric analysis demonstrated significant upregulation of PDE5
protein expression in MVEC after 48h cyclic stretch (Figure 17B).
Figure 17. Effect of mechanical stretch on alignment and PDE5 expression in human cardiac microvascular endothelial cells After 48 hours cyclic stretch, the cardiac microvascular endothelial cells (MVEC) aligned perpendicularly to the stretch direction (A). In three independent experiments, immunoblot and densitometric analysis showed significantly increased PDE5 expression in cardiac MVEC exposed to cyclic stretch (B). Protein levels of actin were measured to control for sample variability. *P<0.05, vs unstretched MVEC.
BA

cGMP in Chronic LV Pressure Overload 89
4.2 Increased PDE5 expression in cardiac tissue of mice subjected to
chronic LV pressure overload
To investigate myocardial PDE5 expression in a mouse model of chronic pressure
overload, we performed immunoblot and immunohistochemical analyses in WT mice at
baseline and after 10 weeks TAC. Immunoblot analysis showed increased PDE5 protein
expression after chronic pressure overload (Figure 18A). Histological examination of
PDE5 immunoreactivity showed distinctly increased PDE5 expression in scattered cardiac
myocytes (Figure 18B). A semi-quantitative scoring of PDE5 expression was performed on
a four-point scale from 0 to 3 on a minimum of 20 high-power fields from 3 sections per
mouse. Grade 0 indicated no obvious PDE5 immunoreactivity in cardiac myocytes, grade
1; up to 1% of cardiac myocytes with distinctly increased PDE5 immunoreactivity, grade 2;
marked PDE5 immunoreactivity in up to 50% of cardiac myocytes, and grade 3; robust
immunoreactivity in >50% of the cardiac myocytes. In a second set of mice, PDE5
expression was quantified using PDE5 immunoblot and densitometric analysis.
In mice with a modest increase in PDE5 protein levels (>1.5 fold versus baseline) and
immunoreactivity (grade 1), we measured intermediate cardiac hypertrophy, and limited
LV dysfunction and dilatation (Table 5). In contrast, mice with markedly increased PDE5
protein levels (>10-fold versus baseline) and immunoreactivity (grade 2-3) exhibited the
most severe cardiac hypertrophy, LV dilatation, and impaired systolic function.
PDE5 immunoreactivity was also increased in vascular endothelium of arterioles and mid-
sized arteries, however, only in mice with severe adverse LV remodelling.
To examine PDE5 gene expression in cardiac endothelial cells after sustained pressure
overload, Tie2-GFP transgenic mice (expressing GFP in endothelial cells) were subjected
to TAC during 10 weeks. Unfortunately, these mice were not prone to the development of
severe adverse cardiac remodelling, probably related to the background of the mouse
strain; as HW/BW only increased from 3.9±0.1 mg/g in baseline conditions (n=9) to
5.1±0.2 mg/g (n=21) after TAC.
After 10 weeks TAC, Tie2-GFP hearts were digested, and endothelial cells were collected
by FACS. The sorted cardiac endothelial cell population displayed significantly higher
mRNA levels of the endothelial cell markers CD31, NOS3, and vascular endothelial
cadherin (VE-cadherin), compared to the non-endothelial cell population.

Chapter 4 90
Moreover, expression levels of CD45, a haematopoietic cell marker, and podoplanin, a
marker for lymphatic endothelial cells, were significantly lower in the endothelial cell
population than in the non-endothelial cell population, indicating minor contamination with
Tie2-positive monocytes and lymphatic endothelial cells.
Quantitative RT-PCR showed comparable PDE5 gene expression in cardiac endothelial
cells from Tie2-GFP mice after 10 weeks TAC (n=8) and from mice without TAC (n=5)
(relative gene expression: 1.01±0.07 vs 1.00±0.06), confirming the absence of elevated
endothelial PDE5 expression in mice with modest adverse remodelling after sustained
pressure overload.
Figure 18. Cardiac PDE5 expression in mice subjected to sustained pressure overload
Immunoblot analysis showed increased PDE5 protein expression after chronic pressure overload (A). Protein levels of GAPDH were measured to control for sample variability. Immunohistochemical staining of PDE5 showed increased PDE5 expression in cardiac myocytes (B). Localisation of PDE5 expression in cardiac myocytes was confirmed in confocal images of double immunofluorescent staining of PDE5 (green) and desmin (red). Scale bars, 20 $m.
B
A

cGMP in Chronic LV Pressure Overload 91
Table 5. Indices of cardiac remodelling and functional parameters after 10 weeks TAC in mice grouped according to myocardial PDE5 expression Mice with prominently increased PDE5 expression (protein levels >10-fold vs baseline, and grade 2-3 PDE5 immunoreactivity*) showed the most adverse cardiac response after chronic pressure overload, evidenced by high HW/BW (post-mortem), and significant systolic dysfunction and LV dilatation (TTE). * Grade 1, up to 1% of cardiac myocytes with distinctly increased PDE5 immunoreactivity; Grade 2, marked PDE5 immunoreactivity in up to 50% of cardiac myocytes; Grade 3, robust immunoreactivity in >50% of the cardiac myocytes. HW/BW indicates heart to body weight ratio; LVIDD, LV internal diameter during diastole; EDV, end-diastolic volume; LVIDS, LV internal diameter during systole; ESV, end-systolic volume; FS, fractional shortening; EF, ejection fraction; and HR, heart rate. †P<0.05, vs baseline; *P<0.05, vs mice with moderately increased PDE5 expression.
Baseline (n=12)
10 weeks TAC
Moderately increased PDE5
expression (n=11)
Markedly increased PDE5
expression (n=6)
HW/BW (mg/g) 4.0±0.1 7.3±0.2 † 12.7±1.2 †*
LVIDD (mm) 3.8±0.1 4.3±0.1 † 5.4±0.2 †*
EDV ($l) 26±1 43±4 † 85±8 †*
LVIDS (mm) 2.7±0.1 3.7±0.1 † 5.1±0.2 †*
ESV ($l) 14±1 28±4 † 71±7 †*
FS (%) 27±2 15±2 † 6±1 †*
EF (%) 46±3 37±3 17±2 †*
HR (bpm) 485±14 495±21 547±12

Chapter 4 92
4.3 Increased PDE5 expression in cardiac myocytes contributes to cardiac
dysfunction and dilatation in mice with chronic pressure overload
To investigate whether increased PDE5 expression in cardiac myocytes contributes to
cardiac dysfunction and adverse LV remodelling in response to pressure overload, or
occurs secondary to the maladaptive cardiac response, we investigated haemodynamics
and LV remodelling in mice with a cardiac myocyte-specific overexpression of PDE5
(PDE5-TG) subjected to 10 weeks TAC.
In baseline conditions, confocal and immunoelectron microscopy showed increased PDE5
expression in cardiac myocytes of PDE5-TG mice, predominantly localised to Z-bands.
Sildenafil-inhibitable cGMP hydrolysis, a measure of PDE5 enzyme activity, was 10-fold
greater in LV tissue of PDE5-TG mice compared to WT littermates. Baseline myocardial
cGMP levels, cell shortening and Ca2+-handling in isolated cardiac myocytes, and LV
haemodynamic parameters were similar in PDE5-TG and WT mice.257
After 10 weeks TAC, post-operative survival rates did not differ in PDE5-TG (n=60) and
WT mice (n=47) (87% and 85%, respectively; P=NS).
4.3.1 Cardiac myocyte-specific PDE5 overexpression aggravates LV dysfunction and dilatation after sustained pressure overload
Pressure-volume measurements showed that during the first 10 seconds after induction of
TAC, end-systolic pressure (ESP) increased significantly and similarly in PDE5-TG and
WT mice (Figure 19A). The accompanying changes in end-systolic and end-diastolic
volumes resulted in an abrupt decrease in stroke volume (SV) and ejection fraction (EF) in
PDE5-TG and WT mice (Figure 19B), which remained similarly depressed in both
genotypes during the first 15 minutes after TAC.

cGMP in Chronic LV Pressure Overload 93
Figure 19. Haemodynamic parameters during the first fifteen minutes after TAC In PDE5-TG (n=9) and WT mice (n=7), an instantaneous and similar increase in end-systolic pressure is observed upon aortic banding, followed by a progressive decrease during the following 15 minutes (A). Simultaneously, an abrupt and equal decrease in stroke volume and ejection fraction is observed immediately after banding in both genotypes (B).
However, after 10 weeks TAC, pressure measurements demonstrated that WT mice were
able to sustain a higher pressure gradient across the fixed constriction compared to
PDE5-TG mice, consistent with better preserved LV systolic function (dP/dtmax) and
maximum developed LV pressure in WT mice (Table 6). Transgenic mice also displayed
worsened LV diastolic function (dP/dtmin), confirmed by a significantly increased Tau.
A
B

Chapter 4 94
Table 6. Haemodynamic parameters after 10 weeks TAC HW/BW indicates heart to body weight ratio; HR, heart rate; RCA-LCA gradient, gradient between right and left common carotid artery; dP/dtmax and dP/dtmin, maximum and minimum of the first derivative of LV pressure over time (i.e. maximum rate of LV pressure development and decline); and Tau, time constant for isovolumic relaxation (Weiss). *P<0.05, vs WT.
Additional pressure-volume measurements were performed in 9 WT and 15 PDE5-TG
mice, and showed a better preserved pre-load recruitable stroke work, a load-independent
measure of systolic function, in WT than in PDE5-TG mice (slope: 89.6±6.1 vs 66.8±5.2;
P<0.05). Moreover, the k1 constant of the quadratic end-diastolic pressure-volume
relationship (EDPVR, EDP=k1*exp(k2*EDV)), was significantly greater in PDE5-TG than
in WT mice (2.85±0.37 vs 0.82±0.58), demonstrating an upward shift of the EDPVR curve,
thereby emphasising the accentuated diastolic dysfunction in transgenic mice after chronic
pressure overload.
In addition, LV dimensions measured using TTE, were greater in PDE5-TG (n=49) than in
WT mice (n=35) after 10 weeks TAC (LVIDS: 4.0±0.1 vs 3.6±0.2 mm and LVIDD 4.6±0.1
vs 4.2±0.1 mm; P<0.05 for both). In PDE5-TG, the slope of the regression curves of the
linear relation between HW/BW and end-systolic and end-diastolic volumes were
significantly steeper than in WT mice, suggesting greater adverse remodelling with
increasing hypertrophic stress (Figure 20).
10 weeks TAC
WT (n=11) PDE5-TG (n=18)
HW/BW (mg/g) 7.2±0.5 6.8±0.2
HR (bpm) 580±29 519±22
RCA-LCA gradient (mmHg) 78±11 47±5*
Maximum LV pressure (mmHg) 145±7 106±5*
dP/dtmax (mmHg/s) 9811±809 5868±375*
dP/dtmin (mmHg/s) -9638±769 -5989±453*
Tau (ms) 10.9±1.1 13.6±1.0*
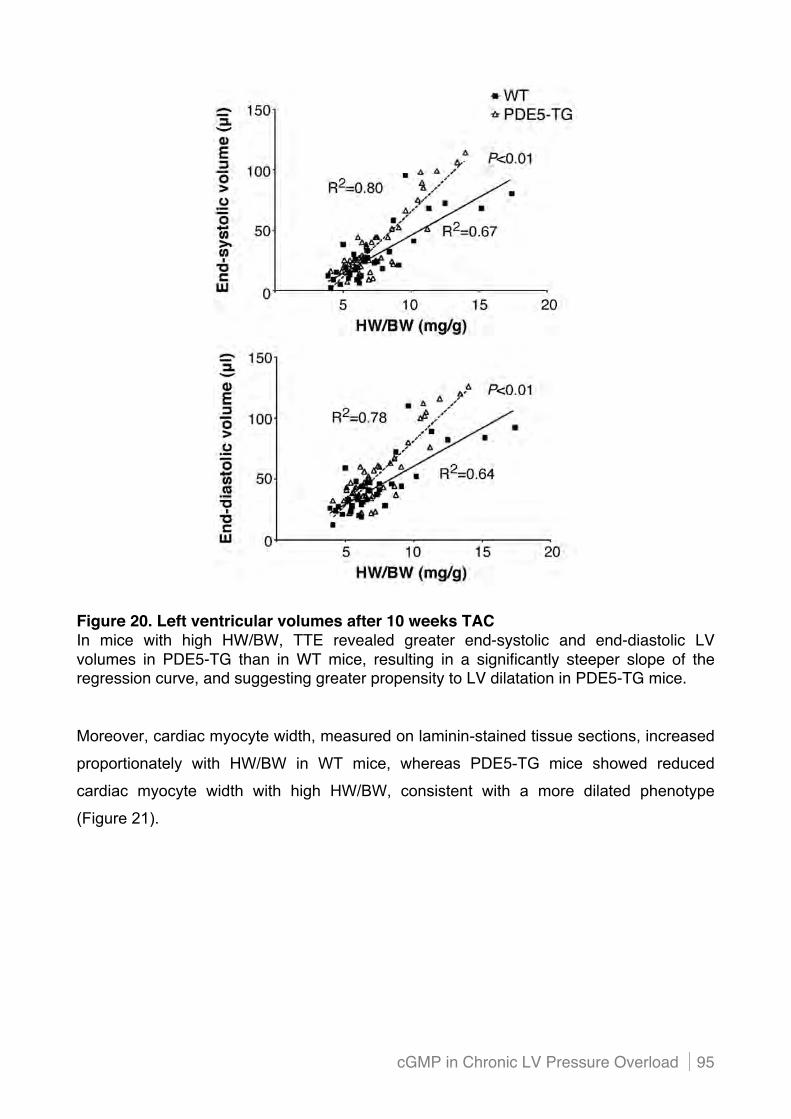
cGMP in Chronic LV Pressure Overload 95
Figure 20. Left ventricular volumes after 10 weeks TAC In mice with high HW/BW, TTE revealed greater end-systolic and end-diastolic LV volumes in PDE5-TG than in WT mice, resulting in a significantly steeper slope of the regression curve, and suggesting greater propensity to LV dilatation in PDE5-TG mice.
Moreover, cardiac myocyte width, measured on laminin-stained tissue sections, increased
proportionately with HW/BW in WT mice, whereas PDE5-TG mice showed reduced
cardiac myocyte width with high HW/BW, consistent with a more dilated phenotype
(Figure 21).

Chapter 4 96
Figure 21. Cardiac myocyte width after 10 weeks TAC Unlike in WT mice, cardiac myocyte width did not augment linearly in PDE5-TG mice with increasing HW/BW, suggesting a more dilated phenotype.
4.3.2 Enhanced cardiac myocyte PDE5 expression does not affect cardiac hypertrophy and extracellular matrix remodelling after chronic pressure overload
After 10 weeks TAC, HW/BW and HW/TL increased significantly and similarly in PDE5-TG
and WT mice compared to baseline (Table 7).
Table 7. Normalised heart weights in baseline conditions and after sustained pressure overload HW/BW indicates heart weight to body weight ratio; and HW/TL, heart weight to tibia length ratio.
Baseline
WT (n=14) PDE5-TG (n=20)
HW/BW (mg/g) 4.0±0.1
3.9±0.1
HW/TL (mg/cm) 59.2±1.3
62.8±1.4
10 weeks TAC
PDE5-TG (n=39) PDE5-TG (n=52)
HW/BW (mg/g) 7.8±0.5 7.6±0.3
HW/TL (mg/cm) 115.5±5.5
119.8±4.0

cGMP in Chronic LV Pressure Overload 97
Furthermore, upregulation of fetal gene expression was comparable in PDE5-TG (n=16)
and WT mice (n=8; ANP: 1.76±0.43 vs 1.00±0.40, BNP: 0.87±0.22 vs 1.00±0.40, !MHC:
1.11±0.32 vs 1.00±0.40; P=NS for all). Finally, levels of proteins known to be markers of
hypertrophy did not differ after 10 weeks TAC (Figure 22A). Taken together, these data
indicate a similar degree of cardiac hypertrophy in WT and PDE5-TG mice after 10 weeks
TAC.
The degree of myocardial fibrosis was also determined after 10 weeks TAC. Deposition of
thick, tightly-packed red birefringent collagen fibres, and thin, loosely-assembled green
birefringent collagen fibres did not differ between genotypes (Figure 22B), consistent with
similar mRNA levels of fibronectin (FN), transforming growth factor-!1 (TGF-!1), and
connective tissue growth factor (CTGF, Table 8).
Moreover, the degree of cardiac myocyte apoptosis did not differ in WT and PDE5-TG
mice after 10 weeks TAC, indicated by a comparable number of TUNEL-positive
myocytes/LV tissue area (Figure 22B), and consistent with comparable mRNA levels of
anti-apoptotic Bcl-2 and Bcl-XL, pro-apoptotic Bax, and Fas and its ligand FasL (Table 8).
Finally, protein levels of oxidative stress markers (3-nitrotyrosine, 4-hydroxy-2-nonenal,
and malondialdehyde) were similar in PDE5-TG and WT mice after 10 weeks TAC
(Figure 22C).

Chapter 4 98
B
C
A

cGMP in Chronic LV Pressure Overload 99
Figure 22. Cardiac hypertrophy, fibrosis, apoptosis, and oxidative stress after sustained pressure overload Protein levels of components of the cardiac hypertrophy pathway (phosphorylated ERK1/2, phosphorylated Akt, and calcineurin) were similar in both genotypes after 10 weeks TAC (A). Cardiac fibrosis and apoptosis were also comparable in pressure overloaded WT and PDE5-TG mice. To assess the degree of fibrosis in the murine LV, the area of collagen deposition was traced on Sirius red-stained tissue sections using polarised light, allowing evaluation of tightly-packed red birefringent collagen versus thin, loosely-assembled green birefringent collagen. Cardiac myocyte apoptosis was evaluated by detecting DNA strand breaks (TUNEL) (B). The levels of oxidative stress markers 3-nitrotyrosine, 4-hydroxy-2-nonenal, and malondialdehyde were similar in hearts of WT and PDE5-TG mice after chronic pressure overload (C). GAPDH protein levels were measured to control for sample variability. Scale bars=50 $m.
Table 8. Indices of cardiac fibrosis and apoptosis after chronic pressure overload FN indicates fibronectin; TGF-"1, transforming growth factor-"1; CTGF, connective tissue growth factor; Bcl-2 and Bcl-XL, B-cell lymphoma 2 and extra large; Bax, Bcl-2 associated X protein; and Fas and FasL, Fas receptor and ligand.
10 weeks TAC
WT PDE5-TG
Red birefringent collagen (% tissue area)
4.7±0.8 (n=5) 4.2±1.1 (n=5)
Green birefringent collagen (% tissue area)
1.3±0.4 (n=5) 1.5±0.7 (n=5)
FN mRNA levels 1.00±0.30 (n=13) 0.79±0.18 (n=19)
TGF-""1 mRNA levels 1.00±0.32 (n=13) 0.64±0.27 (n=19)
CTGF mRNA levels 1.00±0.25 (n=13) 1.05±0.28 (n=19)
# Apoptotic cardiac myocytes (/10 mm2)
13±4 (n=5) 18±4 (n=7)
Bcl-2 mRNA levels 1.00±0.24(n=12) 2.22±0.79 (n=19)
Bcl-XL mRNA levels 1.00±0.48 (n=8) 0.76±0.18 (n=11)
Bax mRNA levels 1.00±0.23 (n=12) 1.98±0.75 (n=18)
Fas mRNA levels 1.00±0.29 (n=13) 0.46±0.14 (n=21)
FasL mRNA levels 1.00±0.43 (n=13) 0.30±0.12 (n=21)

Chapter 4 100
4.3.3 Elevated cardiac myocyte PDE5 expression limits the increase in myocardial cGMP levels in response to chronic pressure overload
After 10 weeks TAC, the increase in myocardial cGMP levels was blunted in PDE5-TG
(0.050±0.005 pmol/mg protein, n=26) compared to WT mice (0.112±0.024 pmol/mg
protein, n=15; P<0.05). The lower myocardial cGMP levels in PDE5-TG mice were not
attributable to lower expression levels of cGMP-synthetising sGC and pGC; protein levels
of the predominant cardiac isoform of sGC, sGC#1!1 (Figure 23), and transcript levels of
NPR-A and NPR-B were not different in PDE5-TG (n=24) and WT mice (n=18; NPR-A:
0.59±0.23 vs 1.00±0.36, NPR-B: 0.92±0.13 vs 1.00±0.12; P=NS for both).
In addition, myocardial levels of cAMP, another important second messenger in the heart,
were similar in both genotypes after 10 weeks TAC (3.5±0.4 vs 4.2±0.6 pmol/mg protein in
WT and PDE5-TG, respectively; P=NS).
Figure 23. Protein levels of the !1- and #1-subunit of sGC after 10 weeks TAC Immunoblot and densitometric analysis showed similar myocardial expression of the predominant sGC isoform in WT and PDE5-TG mice after 10 weeks TAC. GAPDH protein levels were measured as a loading control.

cGMP in Chronic LV Pressure Overload 101
4.3.4 Increased PDE5 expression in cardiac myocytes is associated with reduced SERCA2 expression and greater cardiac myocyte passive force after chronic pressure overload
To investigate the molecular mechanisms of increased contractile dysfunction in PDE5-TG
mice after 10 weeks TAC, we measured cardiac levels of SERCA2. Quantitative PCR and
immunoblot analysis revealed significantly lower SERCA2 transcript and protein levels in
PDE5-TG than in WT mice after 10 weeks TAC (Figure 24), resulting in a more delayed
myocardial relaxation and thus a more deficient contractile state in PDE5-TG mice.
To examine SERCA2 ATPase activity, the phosphorylation status of phospholamban was
as well examined, but marked inter-sample variation blurred potential differential
expression in the genotypes.
Figure 24. Myocardial SERCA2 expression after 10 weeks aortic banding Transcript levels of SERCA2 were significantly lower in PDE5-TG (n=15) than in WT mice (n=12) (A). Reduced SERCA2 protein expression in PDE5-TG was demonstrated by immunoblot and densitometric analysis. Protein levels of GAPDH were measured to control for sample variability (B). *P<0.05, vs WT.
In addition, the mechanical properties of cardiac myocytes were determined after 10
weeks TAC. Active forces did not differ between WT (16 cells from 4 hearts, average
HW/BW= 8.5±2.5 mg/g) and PDE5-TG mice (20 cells from 5 hearts, average HW/BW=
8.5±0.8 mg/g) (Figure 25A). Also, Ca2+-sensitivity of isometric force production (pCa50)
A B

Chapter 4 102
was similar in permeabilised cardiac myocytes from WT and PDE5-TG mice, and was
equally decreased after pretreatment with cGMP-dependent PKG-I#.
In contrast, passive forces were significantly higher in PDE5-TG than in WT cardiac
myocytes, consistent with more pronounced diastolic dysfunction in PDE5-TG mice.
Passive forces were reduced after pretreatment with PKG-I# in both genotypes
(Figure 25B).
Figure 25. Active force and its Ca2+-sensitivity, and passive force in cardiac myocytes isolated after 10 weeks TAC Active forces were not significantly different in WT and PDE5-TG cardiac myocytes, and the Ca2+-sensitivity was comparable (A). In contrast, passive forces were significantly greater in cardiac myocytes from PDE5-TG than from WT mice (B). Pretreatment of cardiac myocytes with PKG significantly reduced the Ca2+-sensitivity of isometric force production and passive forces in both genotypes. †P<0.05, vs - PKG; *P<0.05, vs WT.
A
B

cGMP in Chronic LV Pressure Overload 103
Finally, we wanted to explore whether the aggravated cardiac dysfunction in PDE5-TG
mice after 10 weeks TAC was associated with a differential miRNA expression, thereby
interfering post-transcriptionally with genes involved in Ca2+-homeostasis or the contractile
machinery. However, miRNA microarray analysis (Affymetrix) revealed no significant
alterations in miRNA expression between WT and PDE5-TG mice after sustained
pressure overload.

Chapter 4 104
4.4 In search of underlying mechanisms of increased cardiac myocyte PDE5 expression after sustained LV pressure overload
4.4.1 PDE5 mRNA levels are not elevated in adult murine cardiac myocytes 4 weeks after TAC
On tissue sections, the width of PDE5 immunoreactive cardiac myocytes and of cardiac
myocytes without obvious PDE5 immunoreactivity was determined. Interestingly, the width
of cardiac myocytes with robustly elevated PDE5 expression was significantly greater
compared to cardiac myocytes without distinct induction of PDE5 expression (16.5±0.3 $m
in 177 cells vs 14.4±0.4 $m in 82 cells, respectively; P<0.05).
To explore whether cardiac myocyte hypertrophy induces PDE5 expression, we
determined PDE5 expression in hypertrophied cardiac myocytes in vitro. Induction of
hypertrophy in vitro is usually accomplished by incubating cells in the presence of growth-
promoting cytokines or neurohormones, including Ang II, ET-1, or PE. However, there are
virtually no published literature reports using such methodology in adult murine cardiac
myocytes (in contrast to isolated rat cardiac myocytes, which have a marked growth
response). To circumvent these limitations, we isolated adult cardiac myocytes from
C57BL/6 mice after 4 weeks TAC. In general, mice with TAC-induced pressure overload
develop cardiac hypertrophy within 1-2 weeks, and cardiac dilatation after 6-8 weeks.
By isolating cardiac myocytes after 4 weeks TAC, we expected to be able to detect
different signatures after induction of hypertrophic cellular remodelling when compared to
signatures of cardiac cells from hearts without TAC.
Transcript levels of natriuretic peptides were modestly higher in cardiac myocytes isolated
from mice 4 weeks after TAC, and PDE5 gene expression was not increased compared to
control conditions (Figure 26). The reason for this is unclear, but could be related to the
fact that for transcriptional analysis all isolated cardiac myocytes were lumped together,
whereas immunohistochemical analysis clearly showed spatial variation of the PDE
expression pattern between individual cardiac myocytes within the same pressure
overloaded heart. The underlying cause for such regional heterogeneity in PDE5
expression remains enigmatic.

cGMP in Chronic LV Pressure Overload 105
Figure 26. PDE5 mRNA levels in adult cardiac myocytes after 4 weeks TAC Transcriptional analysis did not show increased PDE5 expression levels in cardiac myocytes isolated from mice subjected to TAC during 4 weeks (n=3 experiments).
4.4.2 PDE5 mRNA levels are not induced in hypoxic adult murine cardiac myocytes
Isolated adult cardiac myocytes of C57BL/6 mice attached to culture dishes for 4 hours,
and were then incubated in hypoxic conditions (1% oxygen) during 6 hours.
Transcript levels of PDE5 did not differ between cardiac myocytes cultured in hypoxic and
normoxic conditions, despite upregulated expression of the established hypoxia-induced
glucose transporter 1 (GLUT-1) and vascular endothelial growth factor A (VEGFA) genes
(Figure 27).
Figure 27. PDE5 mRNA levels in hypoxic adult cardiac myocytes Transcriptional analysis did not show increased PDE5 expression levels in cardiac myocytes cultured in hypoxia (n=5 experiments), despite significant induction of GLUT1 and VEGFA expression. *P<0.05, vs normoxic conditions.

Chapter 4 106
Of note, neonatal murine cardiac myocytes were also cultured in hypoxic conditions.
Similarly, no hypoxia-induced PDE5 protein expression response could be observed after
48 hours, refuting the hypothesis that local hypoxic conditions in the pressure overloaded
heart would trigger PDE5 upregulation.
4.4.3 PDE5 protein expression is not increased in mechanically stretched neonatal murine cardiac myocytes
After subjecting murine neonatal cardiac myocytes to cyclic stretch during 48 hours, no
reorientation and alignment of cells could be observed (Figure 28A).
Immunoblot and densitometric analysis did not show upregulation of PDE5 protein
expression in cardiac myocytes in response to stretch, on the contrary, a trend towards
suppressed PDE5 expression in mechanically stretched cells was observed (Figure 28B).
These data suggest that increased preload is not responsible for PDE5 induction in
isolated cardiac myocytes.
A
B

cGMP in Chronic LV Pressure Overload 107
Figure 28. Effect of mechanical stretch on neonatal cardiac myocytes Cyclic stretch did not result in cell reorientation (A) or significant alterations of PDE5 expression (B) in neonatal cardiac myocytes (n=4 experiments). Protein levels of actin were measured to control for sample variability.
In a preliminary experiment, we explored a potential paracrine modulation of cardiac
myocyte PDE5 expression by endothelial cells, and exposed neonatal cardiac myocytes
during cyclic stretch to conditioned medium from stretched human cardiac microvascular
endothelial cells. However, no induction of PDE5 expression was observed in these
cardiac myocytes (Figure 29). Whether or not adult cardiac myocytes would react
differently under these experimental conditions remains to be determined.
Figure 29. PDE5 expression in neonatal cardiac myocytes exposed to conditioned medium from stretched human cardiac microvascular endothelial cells Immunoblot and densitometric analysis showed no paracrine effect of stretched human microvascular endothelial cells (conditioned EC medium) on PDE5 expression of neonatal cardiac myocytes (CM) after 48 hours cyclic stretch. Protein levels of actin were measured to control for sample variability.

Chapter 4 108
4.4.4 Altered miRNA profiles in the pressure overloaded heart do not appear to regulate PDE5 expression
We hypothesised that increased PDE5 expression in cardiac myocytes after chronic
pressure overload could be the result of mRNA regulation by miRNAs. If a given miRNA
would functionally target the 3"UTR of PDE5, thereby inhibiting its protein expression,
downregulation of this miRNA after 10 weeks TAC could abrogate this inhibition, resulting
in increased PDE5 protein expression. Therefore, we determined changes in myocardial
miRNA expression after sustained pressure overload, and further analysed downregulated
miRNAs with a predicted target in the 3"UTR of PDE5.
(i) Profiling of differentially expressed miRNAs after chronic pressure overload using the Affymetrix and NanoString platform
Affymetrix microarray analysis showed significant upregulation of 4 miRNAs and
significant downregulation of 14 miRNAs in mice after 10 weeks TAC (n=4,
HW/BW=7.0±0.4 mg/g) compared to mice at baseline (n=4, HW/BW=4.0±0.1 mg/g).
Expression of altered miRNAs was also examined by RT-qPCR, and these findings were
in good agreement with the microarray data (Figure 30).

cGMP in Chronic LV Pressure Overload 109
Figure 30. Changes in miRNA expression after chronic pressure overload After 10 weeks TAC, 3 miRNAs were ) 2-fold upregulated and 7 miRNAs were ) 2-fold downregulated compared to baseline (in bold). Additional 7 miRNAs were ) 1.5-fold downregulated, and one miRNA was ) 1.5-fold upregulated. Quantitative RT-PCR was used to confirm ) 2-fold altered miRNA expression. Expression of miRNAs was normalised to miR16 and snoRNA412 levels, and represented relative to baseline (dotted line). *P<0.05, vs baseline.
In a later stage, the miRNA expression profile of hearts subjected to increased LV
afterload was analysed using the NanoString nCounter assay, based on a novel technique
with increased sensitivity in comparison to microarrays. MiRNA levels were determined in
cardiac tissue of mice with a moderate hypertrophic response to TAC (n=4,
HW/BW=6.7±0.4 mg/g), and mice with a severe cardiac remodelling response to TAC

Chapter 4 110
(n=4, HW/BW=9.4±0.6 mg/g), and compared to miRNA profiles at baseline (n=4,
HW/BW=4.0±0.1 mg/g). In cardiac tissue of mice with an intermediate HW/BW,
37 miRNAs were significantly downregulated and 5 miRNAs were significantly upregulated
compared to baseline (Figure 31). In the hearts of mice with a high HW/BW, 16 miRNAs
were significantly downregulated and 6 miRNAs were significantly upregulated compared
to baseline. Additionally, cardiac miRNA expression in the two mice with the highest
HW/BW - showing marked PDE5 upregulation, as demonstrated by immunoblot analysis
in Figure 18A - was compared to miRNA expression in baseline conditions; 7 miRNAs
were significantly downregulated in these two mice: miR-125a-3p, miR-7a, miR-9, miR-
338-3p, miR-139-5p, miR-499, and miR-486.
It was demonstrated that nCounter expression assays are more sensitive than
microarrays, and similar in sensitivity to RT-qPCR, so quantification of differential miRNA
expression was not repeated with RT-qPCR.315

cGMP in Chronic LV Pressure Overload 111

Chapter 4 112
Figure 31. Changes in cardiac miRNA expression in mice with moderate or severe LV remodelling after sustained pressure overload After 10 weeks TAC, 37 miRNAs were downregulated and 5 miRNAs were upregulated in mice with an intermediate HW/BW. In mice with a high HW/BW after aortic banding, 16 miRNAs were downregulated and 6 miRNAs were upregulated. MiRNAs with a ) 2-fold expressional change are indicated in bold, and miRNAs that are common in both groups are underlined.
(ii) Predicted miRNAs do not functionally target the 3"UTR of PDE5
Multiple miRNA databases were scanned in search for miRNAs with downregulated
expression in the pressure overloaded heart that functionally target the 3"UTR of PDE5,
thereby modulating its expression. Three miRNAs were predicted to target the 3"UTR of
PDE5 (both in human and mouse); miR-200c, miR-181b, and miR-499. In addition, miR-
7a (both in human and mouse) was predicted to target the 3"UTR of trans-acting
transcription factor 1 (Sp1), a transcription factor binding to the upstream enhancer of the
human PDE5A promoter and to the alternative intronic human PDE5A2 promoter (the
murine PDE5 promoter has not been described to this extent).
Of these miRNAs predicted to modulate PDE5 expression, miR-499 was considered to be
the miRNA with the highest probability of functionally regulating PDE5 expression, since
this miRNA was exclusively downregulated in mice with severe adverse LV remodelling
and dysfunction after 10 weeks TAC, which display the highest PDE5 expression levels.
MicroRNA target site identification
To evaluate whether miR-499 binds to the 3"UTR of PDE5, we used HEK293 cells, stably
transfected with a reporter vector containing the 3"UTR of human PDE5 downstream of
the firefly luciferase gene. MiRNA binding to the 3"UTR of PDE5 causes interference of the
miRNA RISC complex with luciferase translation, thereby reducing luciferase activity
(Figure 32).

cGMP in Chronic LV Pressure Overload 113
Figure 32. Using 3"UTR luciferase reporter constructs for miRNA target site identification The 3"UTR sequence of human PDE5 was cloned downstream of the firefly luciferase gene. Translation of the luciferase reporter is modulated by the interaction of a miRNA with the 3"UTR of PDE5, resulting in varying luciferase activity.
Downregulation of luciferase reporter expression after transfection with miR-499 precursor
molecules (increasing the miRNA levels) would implicate binding to the 3"UTR of PDE5.
Upregulation of the luciferase reporter after transfection with miR-499 inhibitor molecules
(inhibition of endogenous miRNA-499) would be confirmatory evidence that miRNA-499
binds to the 3"UTR of PDE5. In preliminary experiments, the concentration of the
transfection agent for optimal transfection of miRNA precursors and inhibitors was
determined by visualising transfection of the scrambled miRNA negative controls (anti-
miR#1 and pre-miR#1), labelled with Cy3. In addition, the concentration of transfected pre-
miR499 and anti-miR499 molecules for optimal upregulation and downregulation of miR-
499, respectively, was determined by quantifying miR-499 levels using qPCR.
Transfection of HEK293 cells, stably transfected with a luciferase reporter vector
containing the 3"UTR of human PDE5, with pre-miR499 did not result in downregulated
luciferase expression, and transfection of the cells with anti-miR499 resulted in a modest
upregulation of luciferase expression (Figure 33).

Chapter 4 114
Figure 33. Relative luciferase activity in HEK293 cells, stably transfected with a luciferase vector containing the 3"UTR of human PDE5, after transfection with pre-miR499 and anti-miR499 Modest effects on luciferase activity were observed after transfection with pre-miR499 and anti-miR499 molecules during 24 hours. These results summarise five independent experiments, with each transfection condition in triplicate for each experiment.
MicroRNA target site validation
Pre-miR499 precursors and anti-miR499 inhibitors were also transfected into regular
HEK293 cells during 24 hours, and PDE5 protein expression was measured.
In accordance with the previous results, PDE5 protein expression was not altered after
transfection with pre-miR499 or anti-miR499 (Figure 34). These transfection experiments
were also performed with pre-miR and anti-miR molecules targeting miR-181b, 200c, and
7a expression, with similar results.
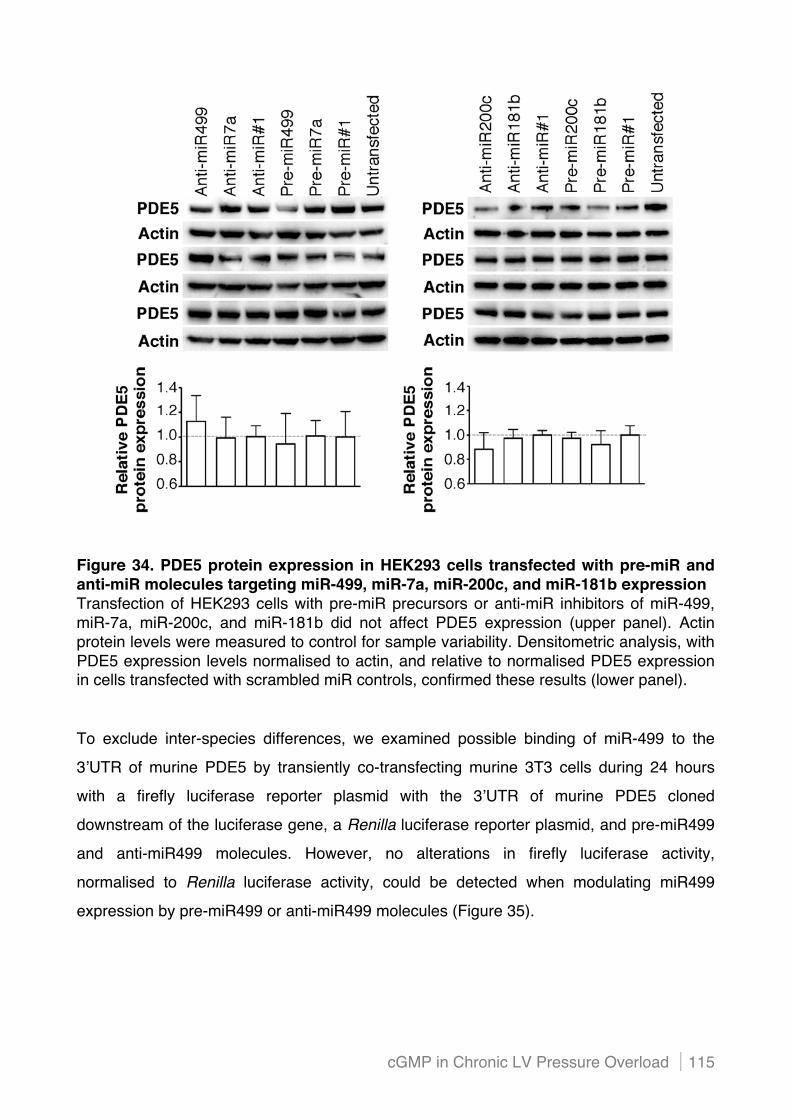
cGMP in Chronic LV Pressure Overload 115
Figure 34. PDE5 protein expression in HEK293 cells transfected with pre-miR and anti-miR molecules targeting miR-499, miR-7a, miR-200c, and miR-181b expression Transfection of HEK293 cells with pre-miR precursors or anti-miR inhibitors of miR-499, miR-7a, miR-200c, and miR-181b did not affect PDE5 expression (upper panel). Actin protein levels were measured to control for sample variability. Densitometric analysis, with PDE5 expression levels normalised to actin, and relative to normalised PDE5 expression in cells transfected with scrambled miR controls, confirmed these results (lower panel).
To exclude inter-species differences, we examined possible binding of miR-499 to the
3"UTR of murine PDE5 by transiently co-transfecting murine 3T3 cells during 24 hours
with a firefly luciferase reporter plasmid with the 3"UTR of murine PDE5 cloned
downstream of the luciferase gene, a Renilla luciferase reporter plasmid, and pre-miR499
and anti-miR499 molecules. However, no alterations in firefly luciferase activity,
normalised to Renilla luciferase activity, could be detected when modulating miR499
expression by pre-miR499 or anti-miR499 molecules (Figure 35).

Chapter 4 116
Figure 35. Relative luciferase activity in 3T3 cells transfected with a luciferase vector containing the 3"UTR of murine PDE5, and pre-miR499 and anti-miR499 Transfection with precursor and inhibitor molecules of miR-499 during 24 hours did not alter firefly luciferase activity. Firefly luciferase activity was normalised to Renilla luciferase activity, and relative to normalised luciferase activity in 3T3 cells transfected with scrambled miR controls (A). Experimental variation was assessed by transfecting 3T3 cells with a luciferase reporter plasmid without the 3"UTR of PDE5, and pre-miR499 and anti-miR499 molecules (B).
A
B

CChhaapptteerr 55
ROLE OF CGMP SIGNALLING IN DOXORUBICIN-INDUCED CARDIOTOXICITY


cGMP in Doxorubicin-induced Cardiotoxicity 119
CHAPTER 5: ROLE OF CGMP SIGNALLING IN DOXORUBICIN-INDUCED
CARDIOTOXICITY
To elucidate the role of cGMP signalling in anthracycline-induced cardiotoxicity, mice with
a cardiac myocyte-specific dominant negative mutated sGC#1 were chronically treated
with doxorubicin.
5.1 Baseline phenotype of mice with a dominant negative mutation of sGC!1 in cardiac myocytes
5.1.1 Cardiac myocyte-specific dominant negative mutation of sGC!1 decreases NO-stimulated cGMP levels
Four weeks after doxycycline withdrawal from the diet, cGMP was extracted from the
hearts of WT and DNsGC#1 mice, following GSNO-mediated sGC activation and non-
selective PDE inhibition by theophylline. Myocardial cGMP levels were significantly
induced following administration of GSNO in WT mice, but not in DNsGC#1 mice (Figure
36). Of note, after 2 weeks doxycycline withdrawal, a non-significant decrease in cardiac
cGMP levels was observed, and after 6 weeks doxycycline withdrawal, the decrease in
cGMP levels was not more pronounced than after 4 weeks.
Figure 36. Myocardial cGMP levels after 4 weeks doxycycline withdrawal Myocardial cGMP levels, normalised to protein concentrations, failed to increase after GSNO administration in DNsGC#1 (n=10) compared to WT mice (n=20). †P<0.05, vs WT without GSNO treatment; *P<0.05, vs WT treated with GSNO.

Chapter 5 120
Measurements of sGC activity in tissue homogenates from WT and DNsGC#1 mice after
4 weeks doxycycline withdrawal confirmed impaired sGC function in DNsGC#1 mice.
The quantity of cGMP, normalised to protein concentration, produced per minute was
measured after incubation with the NO donor DETA-NO or sGC activators BAY 41-2272
and BAY 58-2667, and in the presence of the non-selective PDE inhibitor IBMX.
Nitric oxide-mediated sGC activation by DETA-NO increased cGMP production to a
signifcantly lesser extent in DNsGC#1 than in WT mice (Figure 37). The same trend was
observed after incubation with the sGC activators.
Figure 37. sGC enzyme activity in hearts after 4 weeks doxycycline withdrawal Induction of sGC activity by an NO-donor (DETA NO) or sGC activators (BAY 41-2272, BAY 58-2667) was attenuated in DNsGC#1 (n=8) compared to WT mice (n=8). †P<0.05, vs baseline (-); *P<0.05, vs WT.
Immunoblot analysis showed comparable sGC#1 and sGC!1 protein levels in WT and
DNsGC#1 mice (Figure 38). Transcription levels of other components involved in cGMP
synthesis, breakdown, and signalling were similar in DNsGCa1 (n=5) compared to WT
mice (n=5) at baseline (NPR-A: 1.52±0.48 vs 1.00±0.26, NPR-B: 0.88±0.15 vs 1.00±0.15,
PDE5: 0.57±0.18 vs 1.00±0.18, and PKG-I: 1.36±0.42 vs 1.00±0.27; P=NS for all).
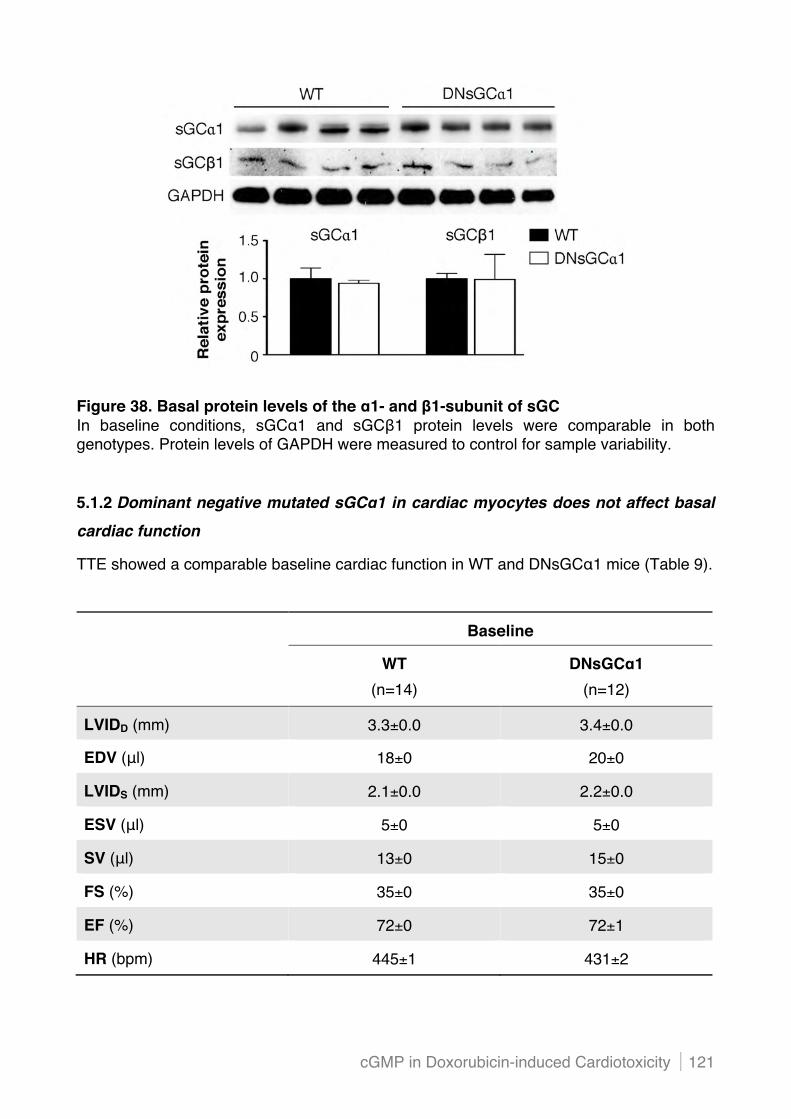
cGMP in Doxorubicin-induced Cardiotoxicity 121
Figure 38. Basal protein levels of the !1- and #1-subunit of sGC In baseline conditions, sGC#1 and sGC!1 protein levels were comparable in both genotypes. Protein levels of GAPDH were measured to control for sample variability.
5.1.2 Dominant negative mutated sGC!1 in cardiac myocytes does not affect basal cardiac function
TTE showed a comparable baseline cardiac function in WT and DNsGC#1 mice (Table 9).
Baseline
WT DNsGC!1 (n=14) (n=12)
LVIDD (mm) 3.3±0.0 3.4±0.0
EDV ($l) 18±0 20±0
LVIDS (mm) 2.1±0.0 2.2±0.0
ESV ($l) 5±0 5±0
SV ($l) 13±0 15±0
FS (%) 35±0 35±0
EF (%) 72±0 72±1
HR (bpm) 445±1 431±2

Chapter 5 122
Table 9. Cardiac imaging in baseline conditions LVIDD indicates LV internal diameter during diastole; EDV, end-diastolic volume; LVIDS, LV internal diameter during systole; ESV, end-systolic volume; SV, stroke volume; FS, fractional shortening; EF, ejection fraction; and HR, heart rate.

cGMP in Doxorubicin-induced Cardiotoxicity 123
5.2 Decreased sGC activity in cardiac myocytes aggravates cardiac dysfunction and dilatation in mice after chronic doxorubicin administration 5.2.1 Cardiac myocyte-specific dominant negative mutation of sGC!1 does not affect survival and cachexia in doxorubicin-treated mice
The clinical importance and widespread use of anthracyclines in chemotherapy has
stimulated development of experimental models to better understand mechanisms of
cardiotoxicity. One reason why pathogenesis of chronic and delayed anthracycline
cardiotoxicity remains poorly understood, is related to the selection of appropriate
experimental models of toxicity.316 Many studies on molecular or cellular pathogenic
mechanisms evaluate effects of relatively high drug concentrations within hours or days.
In contrast, the effects of chronic anthracycline cardiotoxicity require weeks to appear and
are associated with lower drug concentrations. In the present study, we therefore treated
mice with doxorubicin during 12 weeks with a weekly dose of 2 mg/kg.
After 12 weeks doxorubicin administration, mortality was not increased in DNsGC#1
(n=30, 33%) compared to WT mice (n=34, 44%; P=NS, Figure 39). All saline-treated mice
survived the treatment period.
Figure 39. Survival curve after chronic doxorubicin treatment Survival was not affected in DNsGC#1 (n=30) compared to WT mice (n=34) after chronic doxorubicin (DOX) treatment. All saline-treated mice (24 WT and 7 DNsGC#1) had survived after 12 weeks.

Chapter 5 124
Monitoring of body weight changes during treatment showed no difference in body weight
between doxorubicin-treated WT (9.9±0.5% weight loss compared to initial BW, n=19) and
DNsGC#1 mice (7.5±0.5% weight loss compared to initial BW, n=18; Figure 40).
This observed cachexia was in sharp contrast to the weight gain in saline-treated WT
(8.4±0.3% weight gain compared to initial BW, n=24) and DNsGC#1 mice (10.7±1.1%
weight gain compared to initial BW, n=7).
Figure 40. Body weight course during 12 weeks treatment Doxorubicin treatment of WT (n=19) and DNsGC#1 (n=18) markedly affected the body weight course compared to saline-treated WT (n=24) and DNsGC#1 mice (n=7). However, the degree of cachexia was comparable in WT and DNsGC#1 mice after chronic doxorubicin administration. †P<0.05, vs saline-treated mice.
5.2.2 Decreased sGC activity in cardiac myocytes amplifies LV dysfunction and dilatation after chronic doxorubicin treatment
After 8 weeks doxorubicin treatment, TTE demonstrated that cardiac function was not
affected in WT mice. In contrast, DNsGC#1 mice displayed LV dysfunction and dilatation
(Table 10).
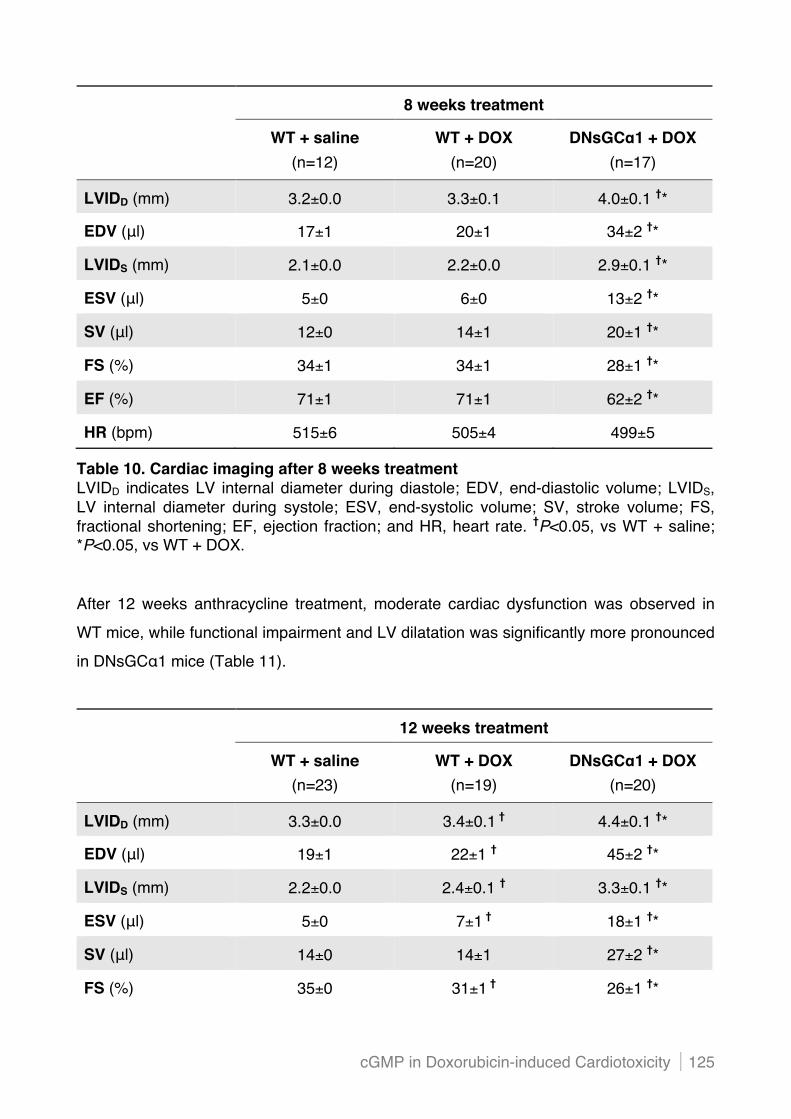
cGMP in Doxorubicin-induced Cardiotoxicity 125
8 weeks treatment
WT + saline WT + DOX DNsGC!1 + DOX
(n=12) (n=20)
(n=17)
LVIDD (mm) 3.2±0.0 3.3±0.1 4.0±0.1 †*
EDV ($l) 17±1 20±1 34±2 †*
LVIDS (mm) 2.1±0.0 2.2±0.0 2.9±0.1 †*
ESV ($l) 5±0 6±0 13±2 †*
SV ($l) 12±0 14±1 20±1 †*
FS (%) 34±1 34±1 28±1 †*
EF (%) 71±1 71±1 62±2 †*
HR (bpm) 515±6 505±4 499±5
Table 10. Cardiac imaging after 8 weeks treatment LVIDD indicates LV internal diameter during diastole; EDV, end-diastolic volume; LVIDS, LV internal diameter during systole; ESV, end-systolic volume; SV, stroke volume; FS, fractional shortening; EF, ejection fraction; and HR, heart rate. †P<0.05, vs WT + saline; *P<0.05, vs WT + DOX.
After 12 weeks anthracycline treatment, moderate cardiac dysfunction was observed in
WT mice, while functional impairment and LV dilatation was significantly more pronounced
in DNsGC#1 mice (Table 11).
12 weeks treatment
WT + saline WT + DOX DNsGC!1 + DOX
(n=23) (n=19)
(n=20)
LVIDD (mm) 3.3±0.0 3.4±0.1 † 4.4±0.1 †*
EDV ($l) 19±1 22±1 † 45±2 †*
LVIDS (mm) 2.2±0.0 2.4±0.1 † 3.3±0.1 †*
ESV ($l) 5±0 7±1 † 18±1 †*
SV ($l) 14±0 14±1 27±2 †*
FS (%) 35±0 31±1 † 26±1 †*

Chapter 5 126
EF (%) 72±1 67±2 † 59±2 †*
HR (bpm) 499±5 519±4 † 514±7
Table 11. Cardiac imaging after 12 weeks treatment LVIDD indicates LV internal diameter during diastole; EDV, end-diastolic volume; LVIDS, LV internal diameter during systole; ESV, end-systolic volume; SV, stroke volume; FS, fractional shortening; EF, ejection fraction; and HR, heart rate. †P<0.05, vs WT + saline; *P<0.05, vs WT + DOX.
Since SERCA2 plays a key role in contractile function, expression levels were quantified in
doxorubicin-treated mice, but no significant differences in SERCA2 mRNA or protein levels
were observed between DNsGC#1 and WT mice after 12 weeks doxorubicin treatment
(Figure 41).
Figure 41. Cardiac mRNA and protein levels of SERCA2 after chronic doxorubicin treatment Transcript levels of SERCA2 were similar in DNsGC#1 (n=9) and WT mice (n=9) (A). Immunoblot and densitometric analysis showed no difference in SERCA2 protein expression in both genotypes. Protein levels of GAPDH were measured to control for sample variability (B).
Also, to examine whether impaired cardiac progenitor cell proliferation and differentiation
into cardiac myocytes was involved in the enhanced functional deterioration in DNsGC#1
mice, the relative number of BrdU-positive cardiac myocytes was determined. Doxorubicin
treatment tended to reduce the percentage of BrdU-positive cardiac myocytes (1.8±0.6%
B A

cGMP in Doxorubicin-induced Cardiotoxicity 127
in 16 WT + DOX vs 3.7±1.0% in 19 WT + saline), however, to the same extent in
DNsGC#1 mice (2.2±0.7, n=17).
5.2.3 Cardiac structure after chronic doxorubicin treatment is not affected by dominant negative mutation of sGC!1 in cardiac myocytes
Light microscopic histological examination of cardiac myocytes of doxorubicin-treated WT
and DNsGC#1 mice revealed moderate pathological changes, including vacuolisations.
Potential ultrastructural differences will be explored using electron microscopy.
Doxorubicin administration induced a significant, but similar reduction in heart weight in
WT and DNsGC#1 mice (Table 12). To account for the observed cachexia due to
anthracycline treatment, heart weights were normalised to tibia length, confirming
significant reduction of heart weight after chronic doxorubicin treatment in both genotypes.
12 weeks treatment
WT + saline
DNsGC!1 + saline
WT + DOX
DNsGC!1 + DOX
(n=21)
(n=7) (n=13) (n=13)
HW (mg) 115.0±4.5 114.1±3.0 92.6±2.7 † 98.4±3.8 †
HW/TL (mg/cm) 67.1±3.4 67.2±2.3 57.0±2.1 † 55.6±2.2 †
Table 12. Heart weights after 12 weeks treatment Normalised heart weights were significantly and to the same extent reduced in WT and DNsGC#1 mice after chronic doxorubicin treatment. †P<0.05, vs saline treated mice.
In addition, vascular density was comparable in DNsGC#1 (4.6±0.2 capillaries per cardiac
myocyte nucleus; n=10) and WT hearts (4.7±0.1 capillaries per cardiac myocyte nucleus;
n=10) after 12 weeks doxorubicin treatment.
Moreover, the degree of cardiac inflammation after 12 weeks doxorubicin administration
was determined on CD45-stained tissue sections. The number of CD45-positive nuclei,
relative to the number of cardiac myocyte nuclei, was comparable in doxorubicin-treated
WT (5.5±0.5%, n=10) and DNsGC#1 mice (5.8±1.5%, n=10).

Chapter 5 128
Finally, deposition of thick, tightly-packed red birefringent collagen fibres did not differ
between WT (n=9) and DNsGC#1 mice (n=7) after chronic doxorubicin treatment
(0.89±0.29 vs 1.00±0.18% of tissue area), nor did thin, loosely-assembled green
birefringent collagen content (0.81±0.18 vs 0.44±0.14% of tissue area), consistent with
similar transcript levels of the fibrosis markers TGF-!1 (1.00±0.34 vs 1.72±0.43), and
CTGF (1.00±0.28 vs 0.47±0.13; P=NS for all comparisons).
5.2.4 Dominant negative mutation of sGC!1 in cardiac myocytes increases expression of Fas and Fas ligand after chronic doxorubicin treatment
In DNsGC#1 mice, Fas transcript levels were significantly higher than in WT mice after
chronic doxorubicin treatment. A similar trend could be observed for mRNA levels of Fas
ligand (Figure 42).
Figure 42. Cardiac mRNA levels of Fas and Fas ligand after chronic doxorubicin treatment Fas expression was significantly increased in DNsGC#1 (n=7) compared to WT mice (n=9) after 12 weeks doxorubicin treatment. †P<0.05, vs DNsGC#1 + saline (n=7); *P<0.05, vs WT + DOX.
Several experimental studies showed increased Fas and FasL levels after doxorubicin
treatment, and both in vitro and in vivo reports demonstrated that blocking Fas/FasL
interaction with a FasL-neutralising antibody inhibited doxorubicin-induced toxicity in
cardiac myocytes.137-140 Moreover, cardiac-targeted expression of soluble Fas (sFas),
a competitive inhibitor of FasL, attenuated doxorubicin-induced cardiotoxicity.141

cGMP in Doxorubicin-induced Cardiotoxicity 129
Cardioprotective effects of Fas inhibition have been attributed to apoptosis-dependent and
-independent mechanisms. One study suggesting an apoptosis-independent mechanism
showed that depletion of GATA4, a transcriptional regulator of MHC and troponin I, was
restored in doxorubicin-treated mice after sFas gene transfer, thereby preventing
sarcomeric disintegration.317 In our study, GATA4 transcript levels were similar in
doxorubicin-treated DNsGC#1 (n=7) and WT mice (n=6, 1.08±0.12 vs 1.00±0.08; P=NS).
Transcript levels of the pro-apoptotic Bax tended to be higher in DNsGC#1 (1.74±0.42,
n=7) compared to WT mice (1.00±0.19, n=8; P=0.11), with similar mRNA levels of the
anti-apoptotic Bcl-2 (0.95±0.25 vs 1.00±0.50; P=NS).
5.2.5 Cardiac myocyte-specific decreased sGC activity increases lipid peroxidation in hearts of doxorubicin-treated mice
Oxidative stress leads to the formation of MDA, a lipid peroxidation product forming stable
MDA-protein adducts, and nitrotyrosine (Figure 43).
Figure 43. Oxidative stress-induced formation of malondialdehyde-protein adducts and nitration of protein residues Superoxide reacts with nitric oxide (NO) to form peroxynitrite, which modifies proteins by nitrating tyrosine residues. Generation of reactive oxygen species also elicits lipid peroxidation, with formation of malondialdehyde as an end product. Malondialdehyde in turn reacts with lysine residues, leading to formation of stable malondialdehyde-protein adducts.
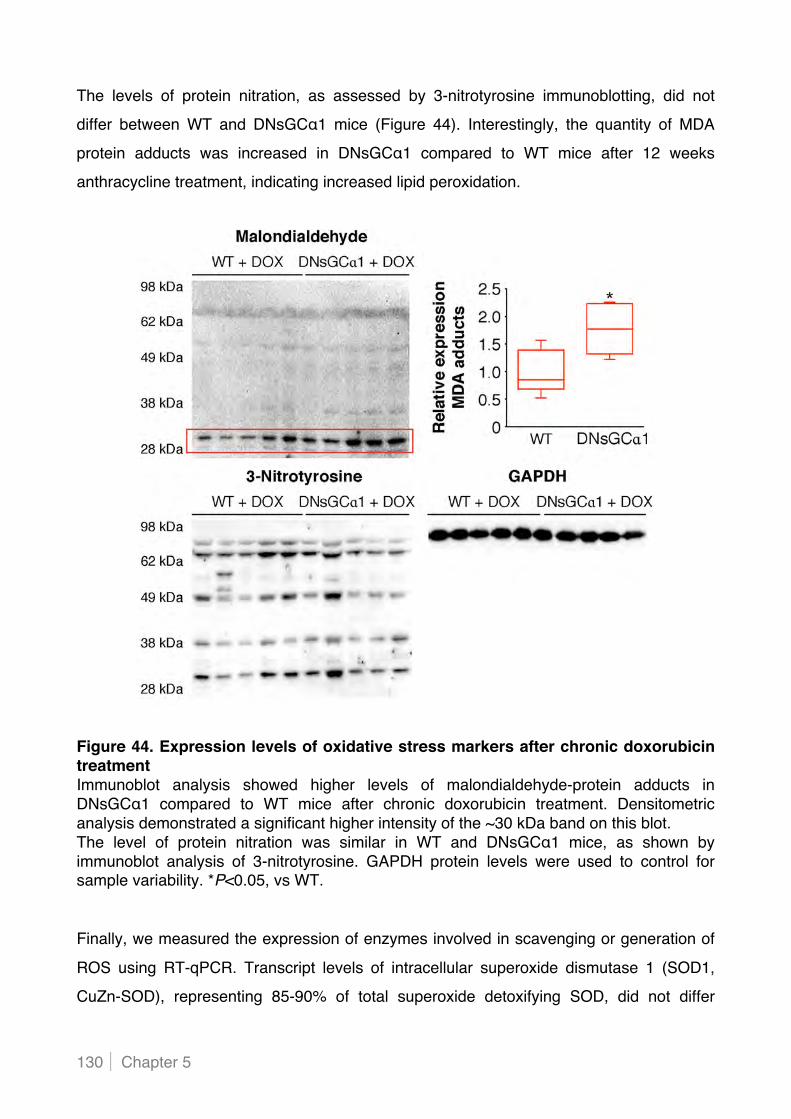
Chapter 5 130
The levels of protein nitration, as assessed by 3-nitrotyrosine immunoblotting, did not
differ between WT and DNsGC#1 mice (Figure 44). Interestingly, the quantity of MDA
protein adducts was increased in DNsGC#1 compared to WT mice after 12 weeks
anthracycline treatment, indicating increased lipid peroxidation.
Figure 44. Expression levels of oxidative stress markers after chronic doxorubicin treatment Immunoblot analysis showed higher levels of malondialdehyde-protein adducts in DNsGC#1 compared to WT mice after chronic doxorubicin treatment. Densitometric analysis demonstrated a significant higher intensity of the ~30 kDa band on this blot. The level of protein nitration was similar in WT and DNsGC#1 mice, as shown by immunoblot analysis of 3-nitrotyrosine. GAPDH protein levels were used to control for sample variability. *P<0.05, vs WT.
Finally, we measured the expression of enzymes involved in scavenging or generation of
ROS using RT-qPCR. Transcript levels of intracellular superoxide dismutase 1 (SOD1,
CuZn-SOD), representing 85-90% of total superoxide detoxifying SOD, did not differ

cGMP in Doxorubicin-induced Cardiotoxicity 131
between doxorubicin-treated WT (n=8, 1.00±0.38) and DNsGC#1 mice (n=9, 1.12±0.14),
nor did catalase mRNA levels (1.00±0.13 vs 1.39±0.19). Also, mRNA levels of the
hydrogen peroxide scavenging GPX1, the major cytoplasmic glutathione peroxidase
isoenzyme, were comparable in WT (1.00±0.14) and DNsGC#1 mice (0.89±0.12) after
chronic doxorubicin treatment. Finally, expression levels of xanthine dehydrogenase, an
enzyme generating ROS, were not different in DNsGC#1 (0.78±0.12) compared to WT
mice (1.00±0.18) (P=NS for all comparisons).

Chapter 5 132
5.3 Reversal of the dominant negative mutation of sGC!1 abrogates increased LV dysfunction and dilatation after chronic doxorubicin treatment Within 8 weeks of doxorubicin treatment, LV dysfunction and dilatation was observed in
doxorubicin-treated DNsGC#1 but not in WT mice. This provided a model to test the
impact of restoring active sGC function, and thus cGMP levels, in cardiac myocytes by
readministration of doxycycline. Both WT and DNsGC#1 mice were exposed to
doxorubicin for 12 weeks, with doxycycline added to the diet again after 8 weeks of
treatment (Figure 45). Cardiac function was assessed after 12 weeks of doxorubicin
administration and 4 weeks later.
Figure 45. Treatment protocol with reversal of the DNsGC!1 mutation After 8 weeks doxorubicin (DOX) treatment, doxycycline was added to the diet, thereby turning off expression of the mutant sGC#1. Transthoracic echocardiography (TTE) was performed after 12 weeks DOX administration and 4 weeks later.
Turning off expression of the mutant sGC#1 did not affect mortality in doxorubicin-treated
mice (32% in 22 WT vs 24% in 26 DNsGC#1; P=NS), or the degree of cachexia
(6.6±1.9% vs 7.0±2.1% BW loss, in respectively 14 WT and 20 DNsGC#1).
After 12 weeks doxorubicin treatment, cardiac dysfunction and dilatation was still more
pronounced in DNsGC#1 compared to WT mice. However, 4 weeks later, functional
deterioration and adverse remodelling had progressed further in WT mice, whereas
cardiac dysfunction and dilatation were reduced in DNsGC#1 mice, thereby eliminating
the functional disparity between the two genotypes (Table 13).

cGMP in Doxorubicin-induced Cardiotoxicity 133
12 weeks doxorubicin treatment with reversal of DNsGC!1
WT + DOX 12 weeks
WT + DOX 16 weeks
DNsGC!1 + DOX 12 weeks
DNsGC!1 + DOX 16 weeks
(n=21) (n=14)
(n=28)
(n=20)
LVIDD (mm) 3.6±0.1 3.8±0.1 †
4.2±0.1 * 3.8±0.1 †
EDV ($l) 25±2 31±2 † 40±1 * 29±1 †
LVIDS (mm) 2.4±0.1 2.7±0.1 † 3.1±0.1 * 2.5±0.0 †*
ESV ($l) 7±1 11±1 † 16±1 * 8±0 †*
SV ($l) 18±1 22±2 24±1 * 21±1
FS (%) 34±1 30±1 † 27±1 * 35±1 †*
EF (%) 71±1 65±2 † 61±1 * 73±1 †*
HR (bpm) 508±3 516±5 512±4 506±4
Table 13. Cardiac imaging after tetracycline-induced reversal of the DNsGC!1 mutation LVIDD indicates LV internal diameter during diastole; EDV, end-diastolic volume; LVIDS, LV internal diameter during systole; ESV, end-systolic volume; SV, stroke volume; FS, fractional shortening; EF, ejection fraction; and HR, heart rate. †P<0.05, vs 12 weeks time point; *P<0.05, vs WT + DOX.

Chapter 5 134
CChhaapptteerr 66
DISCUSSION


Discussion 137
CHAPTER 6: DISCUSSION
6.1 Increased cardiac myocyte PDE5 expression in human and murine pressure overload hypertrophy contributes to adverse LV remodelling In chapter 4, we report elevated myocardial PDE5 expression in patients with severe
aortic stenosis and in mice exposed to chronic pressure overload induced by TAC.
We detected a strikingly similar PDE5 expression pattern in cardiac myocytes of human
and murine hearts subjected to pronounced hypertrophic stress resulting in marked
adverse LV remodelling. To determine whether increased PDE5 levels in cardiac
myocytes contribute to the detrimental LV remodelling in response to increased load, we
took advantage of transgenic mice with cardiac myocyte-specific PDE5 overexpression.
These mice display normal haemodynamics at baseline, but develop greater LV
dysfunction and dilatation than WT littermates after 10 weeks TAC, despite a similar
degree of cardiac hypertrophy, fibrosis, apoptosis, and oxidative stress. Transgenic mice
showed a blunted myocardial cGMP response to chronic pressure overload, lower cardiac
levels of the SR Ca2+-ATPase SERCA2, and greater PKG-sensitive increases in passive
force of isolated cardiac myocytes. Taken together, these data suggest that in the context
of sustained increased afterload, PDE5 induction in cardiac myocytes increases
myocardial passive stiffness, impairs contractile function, and is associated with adverse
LV remodelling.
INCREASED PDE5 LEVELS IN CARDIAC MYOCYTES CONTRIBUTE TO ADVERSE LV
REMODELLING AFTER CHRONIC PRESSURE OVERLOAD In vivo animal studies have provided compelling evidence for a protective role of cGMP
signalling during various stress conditions leading to deleterious LV remodelling.
In human studies, enhanced myocardial PDE5 expression has been detected in patients
with right ventricular hypertrophy and advanced LV failure.256-258 In this study, we
demonstrated a patchy PDE5 cardiac myocyte expression pattern in those AS patients
with clear signs of heart failure (i.e. higher LV filling pressures and circulating BNP levels),
and in mice with marked adverse LV remodelling and dysfunction induced by chronic
pressure overload.
The role of PDE5 in hearts exposed to chronic pressure overload has been studied using
small molecule inhibitors of cGMP hydrolysis. Multiple reports have shown a beneficial

Chapter 6 138
effect of sildenafil on adverse structural and functional cardiac remodelling in mice, and
very recently also on haemodynamics in a small-scale intervention study in severe AS
patients.318 However, it is possible that some of sildenafil"s cardiac effects are attributable
to inhibition of PDE1.262 In view of widespread expression of PDE1 in the cardiovascular
system, including in cardiac myocytes, possible cross-reactivity is relevant.188 Moreover,
beneficial effects of PDE5 inhibitors on cardiac remodelling could involve targeting of other
non-myocyte cardiac cell types, or modulation of LV afterload via vasodilatation of
systemic resistance vessels. Our observations of prominent LV dysfunction and dilatation
in mice with transgenic overexpression of PDE5 in cardiac myocytes, suggest a key role
for cardiac myocyte PDE5 in adverse cardiac remodelling induced by sustained pressure
overload.
The reasons for enhanced functional impairment in PDE5-TG mice may be multiple.
First, lower SERCA2 expression is observed in transgenic than in WT mice after 10 weeks
TAC. Impaired Ca2+-handling associated with reduced SERCA2 expression and activity is
the molecular hallmark of heart failure, and a recent target for therapeutic interventions.319
Experimental research has shown that SERCA2 gene expression is regulated by peptide
growth factors, thyroid hormones, AngII, ET-1, and norepinephrine.320-324 Intracellular
signalling pathways that regulate SERCA2 gene expression are under investigation, and
compelling evidence suggests that the Raf-MAPKK-ERK cascade, which can be activated
by Ras or PKC, is both necessary and sufficient to downregulate SERCA2 gene
expression in cardiac myocytes.325 In pressure overloaded PDE5-TG hearts, reduced
cGMP levels lead, via decreased PKG activation, to decreased phosphorylation of anti-
hypertrophic regulators of G protein signalling (RGS, cfr. paragraph 1.3.1 in Introduction)
and their inhibition of G#q-coupled receptors. Increased G#q-coupled receptor activation of
PLC causes increased formation of IP3 and DAG, the latter stimulating PKC and
subsequently the Raf-MAPKK-ERK cascade. This proposed link between reduced cGMP
levels and decreased SERCA2 expression is hypothetical and requires further
investigation. Unfortunately, the phosphorylation status of phospholamban, and thus
SERCA2 activity, could not be determined in PDE5-TG and WT hearts. We hypothesise
that reduced cGMP levels in pressure overloaded PDE5-TG hearts result in decreased
PKG-mediated phosphorylation of phospholamban, leading to increased inhibition of
SERCA2 activity.

Discussion 139
Second, reduced myocardial cGMP levels in PDE5-TG mice after sustained pressure
overload may result in diminished PKG-dependent phosphorylation of the cardiac-specific
N2B element of titin, an extensible molecular spring in sarcomeres.99 PKG-mediated titin
phosphorylation causes reduction of passive forces in cardiac myocytes, thereby
representing a potential mechanism for increased passive forces in PDE5-TG cardiac
myocytes, and thus greater myocardial stiffness and diastolic dysfunction in PDE5-TG
mice. The observed in vitro cGMP/PKG-dependent reduction of passive force is consistent
with this post-translational modulation, although changes in titin isoform expression can
also modulate passive force of cardiac myocytes.99
POSSIBLE CO-CONSPIRACY OF INCREASED PDE5 EXPRESSION IN CARDIAC ENDOTHELIAL
CELLS IN PROGRESSION TO HEART FAILURE Progression from adaptive hypertrophy to heart failure has been associated with
imbalanced angiogenesis.92 Vascular endothelial cell motility, migration, and proliferation
are critical for angiogenesis, and mediated by cGMP signalling. PDE5 expression in
cardiac endothelial cells was increased in AS patients and mice with the most pronounced
adverse cardiac remodelling response to pressure overload, and may impair myocardial
perfusion and contribute to adverse cardiac remodelling and progression to heart failure.
When human cardiac microvascular endothelial cells were exposed to increased stretch in
vitro, PDE5 protein expression was significantly enhanced, suggesting a mechanosensor
trigger for endothelial cell-specific induction of PDE5 in hearts subjected to chronic
pressure overload. Endothelial cells express a variety of TRP channel isoforms, which
participate in a diverse range of vascular functions, including mechanosensing.326-330
Activation of TRP channels results in Ca2+-entry, and the subsequent elevation of [Ca2+]i stimulates NOS3 activity, leading to increased production of cGMP. Since elevated cGMP
levels activate PDE5 and increase expression through regulation of the human PDE5
intronic promoter331, this may represent a possible molecular feedback mechanism of
PDE5 upregulation in stretched endothelial cells. Involvement of TRP channel activity in
increased PDE5 expression in stretched endothelial cells can be determined using TRP
channel blockers, or indirectly, by pharmacological inhibition of NOS3 during mechanical
stretch.

Chapter 6 140
THE CONTINUED QUEST FOR MECHANISMS OF INCREASED PDE5 EXPRESSION IN CARDIAC
MYOCYTES AFTER SUSTAINED PRESSURE OVERLOAD The molecular mechanism(s) responsible for PDE5 upregulation in human and murine
cardiac myocytes after chronic pressure overload remains enigmatic. Of note, the role of
cardiac myocyte hypertrophy and of hypoxia in regulation of PDE5 expression was only
investigated at the transcriptional level, which does not necessarily reflect protein content.
In addition, we need to expand our investigation of cellular cross-talk between vascular
and cardiac cells. A possible paracrine effect of endothelial cells on cardiac myocyte
PDE5 expression during mechanical stretch may be very relevant and substantial, since
every cardiac myocyte is surrounded by multiple capillaries. The absence of an effect on
PDE5 expression in the preliminary in vitro experiment requires confirmation in additional
tests, or could reflect the possibility that distinct effects of paracrine signalling require an
adapted experimental design with e.g. an extended exposure (>48 hours) of cardiac
myocytes to stretch in combination with medium from stretched endothelial cells.
It is worthwhile to further explore this path, and even expand it since additional cell types,
such as fibroblasts, may be equally involved in the suggested modulation of PDE5
expression. Fibroblasts are not only involved in extracellular matrix (ECM) deposition and
remodelling, but have been proposed to play important roles in the myocardial response to
chemical and mechanical signals (“sentinel cells”).332
Finally, we explored the possibility of miRNA-mediated regulation of PDE5 expression.
Despite the lack of miRNA-modulated alterations in PDE5 expression in our study,
it cannot be excluded that an interplay of multiple miRNAs or other miRNAs with thus far
unrecognised targets may be involved in regulation of PDE5 expression.

Discussion 141
6.2 Decreased sGC activity in cardiac myocytes aggravates LV dysfunction and dilatation in mice chronically treated with doxorubicin In chapter 5, we report decreased myocardial sGC activity in response to NO in mice with
a cardiac myocyte-specific dominant negative mutation of the #1-subunit of sGC.
The cyclase enzyme activity was reduced by approximately 50% in a doxycycline-
regulatable manner, and resulted in greater LV dysfunction and dilatation after chronic
anthracycline treatment. Importantly, this incremental dysfunction and dilatation could be
reversed by turning off expression of the mutated sGC#1 and restoring enzymatic activity.
DIMINISHED CARDIAC MYOCYTE SGC ACTIVITY WORSENS LV DYSFUNCTION AND DILATATION
INDUCED BY CHRONIC DOXORUBICIN TREATMENT To investigate the effect of reduced cGMP bioavailability in chronic anthracycline-induced
cardiotoxicity, we administered a low dose of doxorubicin to DNsGC#1 mice for 12 weeks.
This experimental design more closely resembles the clinical scenario, in contrast to
studies in which acute effects of a single high dose of anthracyclines are examined.
Cachexia and mortality after chronic doxorubicin administration were not affected by
decreased cardiac sGC activity. In contrast, despite normal cardiac function at baseline,
doxorubicin-induced LV dysfunction and dilatation were significantly accentuated in
DNsGC#1 mice, as evidenced by echocardiographic analysis.
Elevated Fas and FasL expression levels in DNsGC#1 mice hinted towards increased
cardiac myocyte apoptosis via the extrinsic apoptotic pathway. Possible dissipation of the
mitochondrial membrane potential (*+m) and activation of the intrinsic (mitochondrial)
apoptotic pathway is yet to be investigated. An experimental study showed that sildenafil
attenuated doxorubicin-induced LV dysfunction by PKG-mediated opening of mitoKATP
channels, thereby restoring *+m.301 Conversely, a possible mechanism for worsened
cardiac dysfunction in DNsGC#1 mice could be reduced PKG-mediated opening of these
protective mitochondrial channels.
Involvement of GATA4, an important transcriptional regulator of several cardiac genes,
in augmented cardiac dysfunction in doxorubicin-treated DNsGC#1 mice could be
excluded, since GATA4 transcript levels were not altered in these mice.

Chapter 6 142
SERCA2 expression was also similar in doxorubicin-treated DNsGC#1 and WT mice.
The earlier proposed link between cGMP levels and SERCA2 gene expression via RGS
and G#q protein-coupled receptors is not applicable in the setting of doxorubicin-induced
cardiotoxicity, probably due to insufficient mechanical stress-induced G#q protein-coupled
receptor activation as observed after sustained pressure overload. Reported
downregulation of SERCA2 gene expression after doxorubicin treatment, which was
partially abolished by the anti-oxidant N-acetylcysteine, has been linked to increased
expression of the transcriptional inhibitor early growth response protein 1 (Egr-1).152
After 12 weeks doxorubicin treatment, the degree of cardiac inflammation and fibrosis was
not affected in DNsGC#1 mice, however, increased lipid peroxidation - and thus oxidative
stress - is suggested by elevated malondialdehyde-protein adduct formation. It has been
shown that ROS can activate the transcription factors NFAT4 and NF-,B in cardiac
myocytes, which in turn upregulate Fas and FasL, thereby providing a possible molecular
mechanism for Fas and FasL upregulation in DNsGC#1 mice.140, 142-144
Of note, doxorubicin-treated mice showed a tendency towards reduced proliferation of
cardiac progenitor cells. This toxic effect is especially relevant in the context of paediatric
cancer treatment, since the growing juvenile heart contains more proliferating cardiac cells
than the adult heart. This was recently confirmed in an elegant study showing that juvenile
anthracycline treatment impairs progenitor cell function and vascular development,
resulting in an adult heart that is more susceptible to stress-induced myocardial injury.333
AGGRAVATED CARDIAC DYSFUNCTION AND DILATATION AFTER CHRONIC DOXORUBICIN
TREATMENT IS ABROGATED BY TURNING OFF EXPRESSION OF THE DNSGC#1 MUTANT In contrast to WT mice, DNsGC#1 mice presented LV dysfunction and dilatation after 8
weeks doxorubicin treatment. This provided a model to test the impact of increasing and
restoring active sGC function, and thus cGMP levels, in cardiac myocytes by tetracycline-
induced resuppression of the DNsGC#1 mutation after 8 weeks of doxorubicin treatment.
Four weeks after starting tetracycline administration, it was assumed that full
resuppression of the mutation had occurred, however, this was not yet translated into a
functional improvement in DNsGC#1 mice. In contrast, 4 weeks later, cardiac dysfunction

Discussion 143
and dilatation was significantly reduced in DNsGC#1 mice, whereas cardiac function and
remodelling had deteriorated further in WT mice.
OUTSTANDING QUESTIONS AND FUTURE EXPERIMENTS The high-energy requirements of cardiac contraction are met almost exclusively by
mitochondrial oxidative phosphorylation. Besides their role as “powerhouses” of the
cardiac myocytes, mitochondria are also involved in Ca2+-signalling and necrotic and
apoptotic forms of cell death. Assessment of mitochondrial function in DNsGC#1 and WT
hearts after chronic doxorubicin treatment is therefore essential to understand the
mechanisms underlying cardiac dysfunction. Therefore, the membrane potential of cardiac
mitochondria will be examined in doxorubicin-treated DNsGC#1 and WT mice (using a
fluorescent mitochondrial selective probe), as well as mitochondrial respiration
(Oxygraph). Moreover, it has been suggested that deleterious opening of the
mitochondrial permeability transition pore is prevented by cGMP/PKG signalling, and this
will be explored in DNsGC#1 and WT hearts after chronic doxorubicin administration
(assessed by evaluation of mitochondrial swelling).334 Also, involvement of protective
mitoKATP channels in the aggravated functional impairment in DNsGC#1 mice exposed to
doxorubicin will be examined in isolated cardiac myocytes exposed to doxorubicin in the
presence of a mitoKATP agonist (e.g. diazoxide).
Furthermore, thorough examination of the degree of cardiac myocyte apoptosis in
doxorubicin-treated DNsGC#1 and WT mice requires additional evaluation of apoptosis
using the TUNEL method, quantification of activated caspase 3 and DNA fragmentation,
and annexin V/propidium iodide staining of isolated cardiac myocytes treated with
doxorubicin. Similarly, possible discrepancies in oxidative stress levels between the two
genotypes need to be confirmed by measuring myocardial superoxide formation
(lucigenin-enhanced chemiluminescence, hydroethidine staining), determining 8-hydroxy-
2'-deoxyguanosine immunoreactivity in tissue sections, and quantifying transcript levels of
additional ROS scavenging enzymes, including the mitochondrial manganese-containing
superoxide dismutase.

Chapter 6 144
Finally, cell shortening and [Ca2+]i-transients in field-stimulated cardiac myocytes will be
examined to further evaluate cardiac myocyte contractility and Ca2+-handling in DNsGC#1
and WT mice after chronic doxorubicin administration.

Discussion 145
6.3 Decreased cGMP levels in cardiac myocytes: a molecular hallmark heralding adverse LV remodelling and dysfunction CONCLUSIONS In AS patients with clear signs of heart failure, and in mice with prominent adverse LV
remodelling and dysfunction induced by chronic pressure overload, PDE5 expression is
markedly increased in cardiac myocytes. Cardiac myocyte-restricted transgenic
overexpression of PDE5 aggravates pressure overload-induced contractile dysfunction
and LV dilatation, suggesting a pathogenic role for increased cGMP hydrolysing activity,
and thus reduced cGMP levels, in cardiac myocytes. This notion is enhanced by worsened
cardiac dysfunction and dilatation in mice with a cardiac myocyte-specific reduction of
sGC activity, and thus cGMP levels. Therefore, pharmacological strategies to augment
cGMP levels in cardiac disease represent a valuable and promising therapeutic approach.
THERAPEUTIC STRATEGIES Among several strategies to enhance cGMP signalling, PDE5 inhibition has gained
significant attention, with 3 inhibitors (sildenafil, vardenafil, and tadalafil) already in clinical
use to treat erectile dysfunction and pulmonary hypertension. A randomised, placebo-
controlled clinical trial demonstrated improved LV diastolic function, cardiac geometry, and
clinical status in patients with stable systolic heart failure treated with sildenafil.261
Furthermore, anti-remodelling effects and improved cardiac kinetics following sildenafil
treatment were reported in diabetic cardiomyopathy patients.335 More information on the
clinical benefit of PDE5 inhibition in human cardiac disease will be provided by a placebo-
controlled multicentre trial (RELAX) testing sildenafil efficacy in patients with diastolic heart
failure, and a single centre study evaluating the efficacy of sildenafil in cardiomyopathy
patients with Duchenne and Becker muscular dystrophies (REVERSE-DBMD).
In light of increased myocardial PDE5 expression in AS patients reported in this doctoral
thesis, it is of major significance that a very recent small intervention study showed that a
single dose of sildenafil in severe AS patients is safe, well-tolerated, and associated with
improvements in pulmonary and systemic haemodynamics.318
Clinical studies to determine the efficacy of sildenafil treatment of cancer patients
undergoing anthracycline-based chemotherapy require more preclinical evidence of
cGMP-conferred cardioprotection in the pathogenesis of anthracycline-induced

Chapter 6 146
cardiotoxicity. Noteworthy, PDE5 inhibition enhances anti-tumour efficacy of doxorubicin in
rodent models, accentuating the possible translational relevance of PDE5 inhibition for
cancer patients requiring anthracycline-based chemotherapy.303-305
Furthermore, stimulation or activation of sGC, and/or re-coupling of NOS with BH4 might
have intriguing potential as a strategy to enhance cGMP signalling beyond inhibition of
cGMP degradation. Riociguat (BAY 63-2521) is the first sGC stimulator to make the
transition into clinical research, showing promising results in patients with pulmonary
hypertension.336, 337 Therapeutic potential of sGC stimulators continues to be explored in a
wide range of animal models, and research efforts to identify and optimise new
compounds in this class (e.g. aminopyrimidines) are ongoing.

Summary 147
SUMMARY
Heart failure is a syndrome in which the heart can no longer provide sufficient blood flow
to meet the body"s demands. This condition often follows a chronic ominous course,
leading to severe incapacitation and compromised survival. In the past quarter century,
much progress has been made in understanding the molecular and cellular processes that
contribute to heart failure, which has led to a considerable change in the scope of
available therapies. However, chronic heart failure remains a major cause of morbidity and
mortality, with a continuously increasing worldwide prevalence. Thus, the development of
novel treatments targeting the underlying pathological mechanisms is warranted to halt
and reverse the devastating consequences of this disease.
Growing evidence suggests a cardioprotective role of cyclic guanosine 3', 5"-
monophosphate (cGMP) in the pathophysiology of the heart. In the experimental studies
presented in this thesis, we observed elevated myocardial expression of
phosphodiesterase type 5 (PDE5), an enzyme catalysing cGMP breakdown, both in
patients with severe aortic stenosis and in mice with thoracic aortic constriction-induced
chronic left ventricular (LV) pressure overload. Intriguingly, we detected a strikingly similar
PDE5 expression pattern in cardiac myocytes of human and murine hearts subjected to
pronounced hypertrophic stress. Aggravated LV dysfunction and dilatation after chronic
pressure overload in mice with a transgenic cardiac myocyte-specific PDE5
overexpression suggests that increased PDE5 levels in cardiac myocytes contribute to
deleterious LV remodelling. These transgenic mice displayed a blunted myocardial cGMP
response to chronic pressure overload, lower cardiac levels of the sarcoplasmic reticulum
Ca2+-ATPase SERCA2, and elevated myocardial passive stiffness.
In a second transgenic mouse model with reduced cGMP-synthetising soluble guanylate
cyclase (sGC) activity in cardiac myocytes, doxorubicin-induced cardiotoxicity was
exacerbated, as evidenced by greater LV dysfunction and dilatation. Transgenic hearts
revealed increased lipid peroxidation and activation of the extrinsic apoptotic pathway after
chronic doxorubicin administration. A hypothesised increase in doxorubicin-induced
mitochondrial dysfunction in these transgenic hearts is currently under investigation.
Taken together, these data suggest that reduced cGMP bioavailability in cardiac myocytes
represents a molecular hallmark heralding adverse LV remodelling and dysfunction during
hypertrophic and toxic stress. Therefore, pharmacological modulation of cGMP signalling,

Summary 148
using PDE5 inhibitors or sGC stimulators/activators, may represent a promising
therapeutic approach for various cardiac pathologies resulting in heart failure, including
chronic pressure overload-induced LV remodelling and anthracycline-induced
cardiotoxicity.

Samenvatting 149
SAMENVATTING
Hartfalen is een klinisch syndroom waarbij de pompfunctie van het hart tekort schiet en de
bloedvoorziening naar weefsels en organen in het gedrang komt. Deze conditie kent vaak
een grimmig chronisch verloop, resulterend in aanzienlijke fysieke beperking en
gecompromitteerde overleving. In de afgelopen decennia is het inzicht in de moleculaire
en cellulaire processen die bijdragen tot hartfalen sterk toegenomen, met een omvangrijk
arsenaal aan beschikbare therapieën tot gevolg. Desondanks blijft chronisch hartfalen een
voorname oorzaak van morbiditeit en mortaliteit, met een voortdurend toenemende
wereldwijde prevalentie. Nieuwe behandelingen gericht op de onderliggende
pathologische mechanismen van hartfalen zijn dus onontbeerlijk om de funeste gevolgen
van deze ziekte te stuiten en terug te schroeven.
Toenemend bewijs suggereert een cardioprotectieve rol voor cyclisch guanosine 3',5'-
monofosfaat (cGMP) in de pathofysiologie van het hart. De experimentele studies in deze
thesis toonden een toegenomen myocardiale expressie aan van fosfodiesterase type 5
(PDE5), een enzyme dat de afbraak van cGMP katalyseert, in patiënten met ernstige
aorta stenose en in muizen met chronische linker ventriculaire (LV) drukoverbelasting
geïnduceerd door thoracale aorta constrictie. We observeerden een opvallend gelijkaardig
PDE5 expressiepatroon in cardiomyocyten van humane en muriene harten blootgesteld
aan aanzienlijke hypertrofe stimuli. Toegenomen LV disfunctie en dilatatie na chronische
drukoverbelasting in transgene muizen met cardiomyocyt-specifieke PDE5 overexpressie
suggereert dat verhoogde PDE5 expressie in cardiomyocyten bijdraagt aan ongunstige LV
hermodelering. Deze transgene muizen vertoonden gereduceerde myocardiale cGMP
niveau"s, lagere cardiale expressie van de Ca2+-ATPase in het sarcoplasmatisch reticulum
(SERCA2), en verhoogde myocardiale passieve stijfheid.
Een tweede transgeen muismodel met gereduceerde cGMP-synthetiserende guanylaat
cyclase (GC) activiteit in cardiomyocyten, vertoonde een meer uitgesproken doxorubicine-
geïnduceerde cardiotoxiciteit, gestaafd door toegenomen LV disfunctie en dilatatie.
De transgene harten vertoonden verhoogde lipide peroxidatie en activatie van de
extrinsieke apoptotische cascade. Een hypothethische toename van doxorubicine-
geïnduceerde mitochondriale disfunctie in deze transgene harten wordt momenteel
onderzocht.

Samenvatting 150
Samengevat suggereren deze data dat gereduceerde cGMP beschikbaarheid in
cardiomyocyten een moleculaire voorbode van nadelige LV hermodelering en disfunctie
vormt in hypertrofische en toxische cardiomyopathie. Farmacologische modulatie van
cGMP signaaltransductie, door PDE5 inhibitoren of GC stimulatoren/activatoren,
representeert een veelbelovende therapeutische benadering voor diverse cardiale
patholgieën die leiden tot hartfalen, inclusief LV hermodelering ten gevolge van
chronische drukoverbelasting en anthracycline-geïnduceerde cardiotoxiciteit.

References
REFERENCES
1. Kemp CD, Conte JV. The pathophysiology of heart failure. Cardiovasc Pathol. 2012;21:365-371 2. Levy D, Kenchaiah S, Larson MG, Benjamin EJ, Kupka MJ, Ho KK, Murabito JM, Vasan RS. Long-term
trends in the incidence of and survival with heart failure. N Engl J Med. 2002;347:1397-1402 3. Lilly SL. Pathophysiology of heart disease (fifth edition). 2011. 4. Parolari A, Loardi C, Mussoni L, Cavallotti L, Camera M, Biglioli P, Tremoli E, Alamanni F.
Nonrheumatic calcific aortic stenosis: An overview from basic science to pharmacological prevention. Eur J Cardiothorac Surg. 2009;35:493-504
5. Bristow MR, Ginsburg R, Minobe W, Cubicciotti RS, Sageman WS, Lurie K, Billingham ME, Harrison DC, Stinson EB. Decreased catecholamine sensitivity and beta-adrenergic-receptor density in failing human hearts. N Engl J Med. 1982;307:205-211
6. Nicol RL, Frey N, Olson EN. From the sarcomere to the nucleus: Role of genetics and signaling in structural heart disease. Annu Rev Genomics Hum Genet. 2000;1:179-223
7. Neumann J, Schmitz W, Scholz H, von Meyerinck L, Doring V, Kalmar P. Increase in myocardial gi-proteins in heart failure. Lancet. 1988;2:936-937
8. Eschenhagen T, Mende U, Nose M, Schmitz W, Scholz H, Haverich A, Hirt S, Doring V, Kalmar P, Hoppner W, et al. Increased messenger rna level of the inhibitory g protein alpha subunit gi alpha-2 in human end-stage heart failure. Circ Res. 1992;70:688-696
9. Bohm M, Gierschik P, Knorr A, Schmidt U, Weismann K, Erdmann E. Cardiac adenylyl cyclase, beta-adrenergic receptors, and g proteins in salt-sensitive hypertension. Hypertension. 1993;22:715-727
10. Han J, Jiang Y, Li Z, Kravchenko VV, Ulevitch RJ. Activation of the transcription factor mef2c by the map kinase p38 in inflammation. Nature. 1997;386:296-299
11. Yang TT, Xiong Q, Enslen H, Davis RJ, Chow CW. Phosphorylation of nfatc4 by p38 mitogen-activated protein kinases. Mol Cell Biol. 2002;22:3892-3904
12. Liang Q, Wiese RJ, Bueno OF, Dai YS, Markham BE, Molkentin JD. The transcription factor gata4 is activated by extracellular signal-regulated kinase 1- and 2-mediated phosphorylation of serine 105 in cardiomyocytes. Mol Cell Biol. 2001;21:7460-7469
13. Oka T, Maillet M, Watt AJ, Schwartz RJ, Aronow BJ, Duncan SA, Molkentin JD. Cardiac-specific deletion of gata4 reveals its requirement for hypertrophy, compensation, and myocyte viability. Circ Res. 2006;98:837-845
14. Naga Prasad SV, Esposito G, Mao L, Koch WJ, Rockman HA. Gbetagamma-dependent phosphoinositide 3-kinase activation in hearts with in vivo pressure overload hypertrophy. J Biol Chem. 2000;275:4693-4698
15. Zhou P, Sun LJ, Dotsch V, Wagner G, Verdine GL. Solution structure of the core nfatc1/DNA complex. Cell. 1998;92:687-696
16. Pennica D, King KL, Shaw KJ, Luis E, Rullamas J, Luoh SM, Darbonne WC, Knutzon DS, Yen R, Chien KR, et al. Expression cloning of cardiotrophin 1, a cytokine that induces cardiac myocyte hypertrophy. Proc Natl Acad Sci U S A. 1995;92:1142-1146
17. Sano M, Fukuda K, Kodama H, Pan J, Saito M, Matsuzaki J, Takahashi T, Makino S, Kato T, Ogawa S. Interleukin-6 family of cytokines mediate angiotensin ii-induced cardiac hypertrophy in rodent cardiomyocytes. J Biol Chem. 2000;275:29717-29723
18. Ichida M, Finkel T. Ras regulates nfat3 activity in cardiac myocytes. J Biol Chem. 2001;276:3524-3530 19. Aoki H, Sadoshima J, Izumo S. Myosin light chain kinase mediates sarcomere organization during
cardiac hypertrophy in vitro. Nat Med. 2000;6:183-188 20. McKinsey TA, Zhang CL, Olson EN. Mef2: A calcium-dependent regulator of cell division, differentiation
and death. Trends Biochem Sci. 2002;27:40-47 21. Naya FJ, Wu C, Richardson JA, Overbeek P, Olson EN. Transcriptional activity of mef2 during mouse
embryogenesis monitored with a mef2-dependent transgene. Development. 1999;126:2045-2052 22. Passier R, Zeng H, Frey N, Naya FJ, Nicol RL, McKinsey TA, Overbeek P, Richardson JA, Grant SR,
Olson EN. Cam kinase signaling induces cardiac hypertrophy and activates the mef2 transcription factor in vivo. J Clin Invest. 2000;105:1395-1406
23. Yang L, Liu G, Zakharov SI, Bellinger AM, Mongillo M, Marx SO. Protein kinase g phosphorylates cav1.2 alpha1c and beta2 subunits. Circ Res. 2007;101:465-474
24. Bartel DP. Micrornas: Genomics, biogenesis, mechanism, and function. Cell. 2004;116:281-297 25. Cheng Y, Ji R, Yue J, Yang J, Liu X, Chen H, Dean DB, Zhang C. Micrornas are aberrantly expressed
in hypertrophic heart: Do they play a role in cardiac hypertrophy? Am J Pathol. 2007;170:1831-1840

References
26. Ikeda S, Kong SW, Lu J, Bisping E, Zhang H, Allen PD, Golub TR, Pieske B, Pu WT. Altered microrna expression in human heart disease. Physiol Genomics. 2007;31:367-373
27. Sayed D, Hong C, Chen IY, Lypowy J, Abdellatif M. Micrornas play an essential role in the development of cardiac hypertrophy. Circ Res. 2007;100:416-424
28. Tatsuguchi M, Seok HY, Callis TE, Thomson JM, Chen JF, Newman M, Rojas M, Hammond SM, Wang DZ. Expression of micrornas is dynamically regulated during cardiomyocyte hypertrophy. J Mol Cell Cardiol. 2007;42:1137-1141
29. Thum T, Galuppo P, Wolf C, Fiedler J, Kneitz S, van Laake LW, Doevendans PA, Mummery CL, Borlak J, Haverich A, Gross C, Engelhardt S, Ertl G, Bauersachs J. Micrornas in the human heart: A clue to fetal gene reprogramming in heart failure. Circulation. 2007;116:258-267
30. van Rooij E, Sutherland LB, Liu N, Williams AH, McAnally J, Gerard RD, Richardson JA, Olson EN. A signature pattern of stress-responsive micrornas that can evoke cardiac hypertrophy and heart failure. Proc Natl Acad Sci U S A. 2006;103:18255-18260
31. Ikeda S, He A, Kong SW, Lu J, Bejar R, Bodyak N, Lee KH, Ma Q, Kang PM, Golub TR, Pu WT. Microrna-1 negatively regulates expression of the hypertrophy-associated calmodulin and mef2a genes. Mol Cell Biol. 2009;29:2193-2204
32. Care A, Catalucci D, Felicetti F, Bonci D, Addario A, Gallo P, Bang ML, Segnalini P, Gu Y, Dalton ND, Elia L, Latronico MV, Hoydal M, Autore C, Russo MA, Dorn GW, 2nd, Ellingsen O, Ruiz-Lozano P, Peterson KL, Croce CM, Peschle C, Condorelli G. Microrna-133 controls cardiac hypertrophy. Nat Med. 2007;13:613-618
33. Duisters RF, Tijsen AJ, Schroen B, Leenders JJ, Lentink V, van der Made I, Herias V, van Leeuwen RE, Schellings MW, Barenbrug P, Maessen JG, Heymans S, Pinto YM, Creemers EE. Mir-133 and mir-30 regulate connective tissue growth factor: Implications for a role of micrornas in myocardial matrix remodeling. Circ Res. 2009;104:170-178, 176p following 178
34. van Rooij E, Sutherland LB, Qi X, Richardson JA, Hill J, Olson EN. Control of stress-dependent cardiac growth and gene expression by a microrna. Science. 2007;316:575-579
35. Takimoto E, Kass DA. Role of oxidative stress in cardiac hypertrophy and remodeling. Hypertension. 2007;49:241-248
36. Takimoto E, Champion HC, Li M, Ren S, Rodriguez ER, Tavazzi B, Lazzarino G, Paolocci N, Gabrielson KL, Wang Y, Kass DA. Oxidant stress from nitric oxide synthase-3 uncoupling stimulates cardiac pathologic remodeling from chronic pressure load. J Clin Invest. 2005;115:1221-1231
37. D'Angelo DD, Sakata Y, Lorenz JN, Boivin GP, Walsh RA, Liggett SB, Dorn GW, 2nd. Transgenic galphaq overexpression induces cardiac contractile failure in mice. Proc Natl Acad Sci U S A. 1997;94:8121-8126
38. Koch WJ, Lefkowitz RJ, Rockman HA. Functional consequences of altering myocardial adrenergic receptor signaling. Annu Rev Physiol. 2000;62:237-260
39. Paradis P, Dali-Youcef N, Paradis FW, Thibault G, Nemer M. Overexpression of angiotensin ii type i receptor in cardiomyocytes induces cardiac hypertrophy and remodeling. Proc Natl Acad Sci U S A. 2000;97:931-936
40. Sakata Y, Hoit BD, Liggett SB, Walsh RA, Dorn GW, 2nd. Decompensation of pressure-overload hypertrophy in g alpha q-overexpressing mice. Circulation. 1998;97:1488-1495
41. Offermanns S, Zhao LP, Gohla A, Sarosi I, Simon MI, Wilkie TM. Embryonic cardiomyocyte hypoplasia and craniofacial defects in g alpha q/g alpha 11-mutant mice. Embo J. 1998;17:4304-4312
42. Wettschureck N, Rutten H, Zywietz A, Gehring D, Wilkie TM, Chen J, Chien KR, Offermanns S. Absence of pressure overload induced myocardial hypertrophy after conditional inactivation of galphaq/galpha11 in cardiomyocytes. Nat Med. 2001;7:1236-1240
43. Akhter SA, Luttrell LM, Rockman HA, Iaccarino G, Lefkowitz RJ, Koch WJ. Targeting the receptor-gq interface to inhibit in vivo pressure overload myocardial hypertrophy. Science. 1998;280:574-577
44. Bowman JC, Steinberg SF, Jiang T, Geenen DL, Fishman GI, Buttrick PM. Expression of protein kinase c beta in the heart causes hypertrophy in adult mice and sudden death in neonates. J Clin Invest. 1997;100:2189-2195
45. Takeishi Y, Ping P, Bolli R, Kirkpatrick DL, Hoit BD, Walsh RA. Transgenic overexpression of constitutively active protein kinase c epsilon causes concentric cardiac hypertrophy. Circ Res. 2000;86:1218-1223
46. Braz JC, Bueno OF, De Windt LJ, Molkentin JD. Pkc alpha regulates the hypertrophic growth of cardiomyocytes through extracellular signal-regulated kinase1/2 (erk1/2). J Cell Biol. 2002;156:905-919
47. Engelhardt S, Hein L, Keller U, Klambt K, Lohse MJ. Inhibition of na(+)-h(+) exchange prevents hypertrophy, fibrosis, and heart failure in beta(1)-adrenergic receptor transgenic mice. Circ Res. 2002;90:814-819

References
48. Bisognano JD, Weinberger HD, Bohlmeyer TJ, Pende A, Raynolds MV, Sastravaha A, Roden R, Asano K, Blaxall BC, Wu SC, Communal C, Singh K, Colucci W, Bristow MR, Port DJ. Myocardial-directed overexpression of the human beta(1)-adrenergic receptor in transgenic mice. J Mol Cell Cardiol. 2000;32:817-830
49. Roth DM, Gao MH, Lai NC, Drumm J, Dalton N, Zhou JY, Zhu J, Entrikin D, Hammond HK. Cardiac-directed adenylyl cyclase expression improves heart function in murine cardiomyopathy. Circulation. 1999;99:3099-3102
50. Roth DM, Bayat H, Drumm JD, Gao MH, Swaney JS, Ander A, Hammond HK. Adenylyl cyclase increases survival in cardiomyopathy. Circulation. 2002;105:1989-1994
51. Antos CL, Frey N, Marx SO, Reiken S, Gaburjakova M, Richardson JA, Marks AR, Olson EN. Dilated cardiomyopathy and sudden death resulting from constitutive activation of protein kinase a. Circ Res. 2001;89:997-1004
52. Redfern CH, Degtyarev MY, Kwa AT, Salomonis N, Cotte N, Nanevicz T, Fidelman N, Desai K, Vranizan K, Lee EK, Coward P, Shah N, Warrington JA, Fishman GI, Bernstein D, Baker AJ, Conklin BR. Conditional expression of a gi-coupled receptor causes ventricular conduction delay and a lethal cardiomyopathy. Proc Natl Acad Sci U S A. 2000;97:4826-4831
53. Molkentin JD, Lu JR, Antos CL, Markham B, Richardson J, Robbins J, Grant SR, Olson EN. A calcineurin-dependent transcriptional pathway for cardiac hypertrophy. Cell. 1998;93:215-228
54. Zou Y, Hiroi Y, Uozumi H, Takimoto E, Toko H, Zhu W, Kudoh S, Mizukami M, Shimoyama M, Shibasaki F, Nagai R, Yazaki Y, Komuro I. Calcineurin plays a critical role in the development of pressure overload-induced cardiac hypertrophy. Circulation. 2001;104:97-101
55. Bueno OF, Wilkins BJ, Tymitz KM, Glascock BJ, Kimball TF, Lorenz JN, Molkentin JD. Impaired cardiac hypertrophic response in calcineurin abeta -deficient mice. Proc Natl Acad Sci U S A. 2002;99:4586-4591
56. Bueno OF, De Windt LJ, Lim HW, Tymitz KM, Witt SA, Kimball TR, Molkentin JD. The dual-specificity phosphatase mkp-1 limits the cardiac hypertrophic response in vitro and in vivo. Circ Res. 2001;88:88-96
57. Shioi T, Kang PM, Douglas PS, Hampe J, Yballe CM, Lawitts J, Cantley LC, Izumo S. The conserved phosphoinositide 3-kinase pathway determines heart size in mice. Embo J. 2000;19:2537-2548
58. Matsui T, Li L, Wu JC, Cook SA, Nagoshi T, Picard MH, Liao R, Rosenzweig A. Phenotypic spectrum caused by transgenic overexpression of activated akt in the heart. J Biol Chem. 2002;277:22896-22901
59. Shioi T, McMullen JR, Kang PM, Douglas PS, Obata T, Franke TF, Cantley LC, Izumo S. Akt/protein kinase b promotes organ growth in transgenic mice. Mol Cell Biol. 2002;22:2799-2809
60. Boluyt MO, Zheng JS, Younes A, Long X, O'Neill L, Silverman H, Lakatta EG, Crow MT. Rapamycin inhibits alpha 1-adrenergic receptor-stimulated cardiac myocyte hypertrophy but not activation of hypertrophy-associated genes. Evidence for involvement of p70 s6 kinase. Circ Res. 1997;81:176-186
61. Ono Y, Ito H, Tamamori M, Nozato T, Adachi S, Abe S, Marumo F, Hiroe M. Role and relation of p70 s6 and extracellular signal-regulated kinases in the phenotypic changes of hypertrophy of cardiac myocytes. Jpn Circ J. 2000;64:695-700
62. Sadoshima J, Izumo S. Rapamycin selectively inhibits angiotensin ii-induced increase in protein synthesis in cardiac myocytes in vitro. Potential role of 70-kd s6 kinase in angiotensin ii-induced cardiac hypertrophy. Circ Res. 1995;77:1040-1052
63. Haq S, Choukroun G, Kang ZB, Ranu H, Matsui T, Rosenzweig A, Molkentin JD, Alessandrini A, Woodgett J, Hajjar R, Michael A, Force T. Glycogen synthase kinase-3beta is a negative regulator of cardiomyocyte hypertrophy. J Cell Biol. 2000;151:117-130
64. Antos CL, McKinsey TA, Frey N, Kutschke W, McAnally J, Shelton JM, Richardson JA, Hill JA, Olson EN. Activated glycogen synthase-3 beta suppresses cardiac hypertrophy in vivo. Proc Natl Acad Sci U S A. 2002;99:907-912
65. Hirota H, Yoshida K, Kishimoto T, Taga T. Continuous activation of gp130, a signal-transducing receptor component for interleukin 6-related cytokines, causes myocardial hypertrophy in mice. Proc Natl Acad Sci U S A. 1995;92:4862-4866
66. Kunisada K, Tone E, Fujio Y, Matsui H, Yamauchi-Takihara K, Kishimoto T. Activation of gp130 transduces hypertrophic signals via stat3 in cardiac myocytes. Circulation. 1998;98:346-352
67. Kunisada K, Negoro S, Tone E, Funamoto M, Osugi T, Yamada S, Okabe M, Kishimoto T, Yamauchi-Takihara K. Signal transducer and activator of transcription 3 in the heart transduces not only a hypertrophic signal but a protective signal against doxorubicin-induced cardiomyopathy. Proc Natl Acad Sci U S A. 2000;97:315-319

References
68. Uozumi H, Hiroi Y, Zou Y, Takimoto E, Toko H, Niu P, Shimoyama M, Yazaki Y, Nagai R, Komuro I. Gp130 plays a critical role in pressure overload-induced cardiac hypertrophy. J Biol Chem. 2001;276:23115-23119
69. Hirota H, Chen J, Betz UA, Rajewsky K, Gu Y, Ross J, Jr., Muller W, Chien KR. Loss of a gp130 cardiac muscle cell survival pathway is a critical event in the onset of heart failure during biomechanical stress. Cell. 1999;97:189-198
70. Fuller SJ, Gillespie-Brown J, Sugden PH. Oncogenic src, raf, and ras stimulate a hypertrophic pattern of gene expression and increase cell size in neonatal rat ventricular myocytes. J Biol Chem. 1998;273:18146-18152
71. Hunter JJ, Tanaka N, Rockman HA, Ross J, Jr., Chien KR. Ventricular expression of a mlc-2v-ras fusion gene induces cardiac hypertrophy and selective diastolic dysfunction in transgenic mice. J Biol Chem. 1995;270:23173-23178
72. Thorburn A. Ras activity is required for phenylephrine-induced activation of mitogen-activated protein kinase in cardiac muscle cells. Biochem Biophys Res Commun. 1994;205:1417-1422
73. Abdellatif M, Packer SE, Michael LH, Zhang D, Charng MJ, Schneider MD. A ras-dependent pathway regulates rna polymerase ii phosphorylation in cardiac myocytes: Implications for cardiac hypertrophy. Mol Cell Biol. 1998;18:6729-6736
74. Sah VP, Hoshijima M, Chien KR, Brown JH. Rho is required for galphaq and alpha1-adrenergic receptor signaling in cardiomyocytes. Dissociation of ras and rho pathways. J Biol Chem. 1996;271:31185-31190
75. Hines WA, Thorburn A. Ras and rho are required for galphaq-induced hypertrophic gene expression in neonatal rat cardiac myocytes. J Mol Cell Cardiol. 1998;30:485-494
76. Kuwahara K, Saito Y, Nakagawa O, Kishimoto I, Harada M, Ogawa E, Miyamoto Y, Hamanaka I, Kajiyama N, Takahashi N, Izumi T, Kawakami R, Tamura N, Ogawa Y, Nakao K. The effects of the selective rock inhibitor, y27632, on et-1-induced hypertrophic response in neonatal rat cardiac myocytes--possible involvement of rho/rock pathway in cardiac muscle cell hypertrophy. FEBS Lett. 1999;452:314-318
77. Sah VP, Minamisawa S, Tam SP, Wu TH, Dorn GW, 2nd, Ross J, Jr., Chien KR, Brown JH. Cardiac-specific overexpression of rhoa results in sinus and atrioventricular nodal dysfunction and contractile failure. J Clin Invest. 1999;103:1627-1634
78. Pracyk JB, Tanaka K, Hegland DD, Kim KS, Sethi R, Rovira, II, Blazina DR, Lee L, Bruder JT, Kovesdi I, Goldshmidt-Clermont PJ, Irani K, Finkel T. A requirement for the rac1 gtpase in the signal transduction pathway leading to cardiac myocyte hypertrophy. J Clin Invest. 1998;102:929-937
79. Sussman MA, Welch S, Walker A, Klevitsky R, Hewett TE, Price RL, Schaefer E, Yager K. Altered focal adhesion regulation correlates with cardiomyopathy in mice expressing constitutively active rac1. J Clin Invest. 2000;105:875-886
80. Vega RB, Harrison BC, Meadows E, Roberts CR, Papst PJ, Olson EN, McKinsey TA. Protein kinases c and d mediate agonist-dependent cardiac hypertrophy through nuclear export of histone deacetylase 5. Mol Cell Biol. 2004;24:8374-8385
81. Zhang CL, McKinsey TA, Chang S, Antos CL, Hill JA, Olson EN. Class ii histone deacetylases act as signal-responsive repressors of cardiac hypertrophy. Cell. 2002;110:479-488
82. Chang S, McKinsey TA, Zhang CL, Richardson JA, Hill JA, Olson EN. Histone deacetylases 5 and 9 govern responsiveness of the heart to a subset of stress signals and play redundant roles in heart development. Mol Cell Biol. 2004;24:8467-8476
83. Marx SO, Reiken S, Hisamatsu Y, Jayaraman T, Burkhoff D, Rosemblit N, Marks AR. Pka phosphorylation dissociates fkbp12.6 from the calcium release channel (ryanodine receptor): Defective regulation in failing hearts. Cell. 2000;101:365-376
84. Curran J, Hinton MJ, Rios E, Bers DM, Shannon TR. Beta-adrenergic enhancement of sarcoplasmic reticulum calcium leak in cardiac myocytes is mediated by calcium/calmodulin-dependent protein kinase. Circ Res. 2007;100:391-398
85. Bers DM. Altered cardiac myocyte ca regulation in heart failure. Physiology (Bethesda). 2006;21:380-387
86. Ohba T, Watanabe H, Murakami M, Takahashi Y, Iino K, Kuromitsu S, Mori Y, Ono K, Iijima T, Ito H. Upregulation of trpc1 in the development of cardiac hypertrophy. J Mol Cell Cardiol. 2007;42:498-507
87. Onohara N, Nishida M, Inoue R, Kobayashi H, Sumimoto H, Sato Y, Mori Y, Nagao T, Kurose H. Trpc3 and trpc6 are essential for angiotensin ii-induced cardiac hypertrophy. Embo J. 2006;25:5305-5316
88. Kuwahara K, Wang Y, McAnally J, Richardson JA, Bassel-Duby R, Hill JA, Olson EN. Trpc6 fulfills a calcineurin signaling circuit during pathologic cardiac remodeling. J Clin Invest. 2006;116:3114-3126

References
89. Takewaki S, Kuro-o M, Hiroi Y, Yamazaki T, Noguchi T, Miyagishi A, Nakahara K, Aikawa M, Manabe I, Yazaki Y, et al. Activation of na(+)-h+ antiporter (nhe-1) gene expression during growth, hypertrophy and proliferation of the rabbit cardiovascular system. J Mol Cell Cardiol. 1995;27:729-742
90. Arany Z, Novikov M, Chin S, Ma Y, Rosenzweig A, Spiegelman BM. Transverse aortic constriction leads to accelerated heart failure in mice lacking ppar-gamma coactivator 1alpha. Proc Natl Acad Sci U S A. 2006;103:10086-10091
91. Weiss RG, Gerstenblith G, Bottomley PA. Atp flux through creatine kinase in the normal, stressed, and failing human heart. Proc Natl Acad Sci U S A. 2005;102:808-813
92. Shiojima I, Sato K, Izumiya Y, Schiekofer S, Ito M, Liao R, Colucci WS, Walsh K. Disruption of coordinated cardiac hypertrophy and angiogenesis contributes to the transition to heart failure. J Clin Invest. 2005;115:2108-2118
93. Lowes BD, Minobe W, Abraham WT, Rizeq MN, Bohlmeyer TJ, Quaife RA, Roden RL, Dutcher DL, Robertson AD, Voelkel NF, Badesch DB, Groves BM, Gilbert EM, Bristow MR. Changes in gene expression in the intact human heart. Downregulation of alpha-myosin heavy chain in hypertrophied, failing ventricular myocardium. J Clin Invest. 1997;100:2315-2324
94. Wu Y, Cazorla O, Labeit D, Labeit S, Granzier H. Changes in titin and collagen underlie diastolic stiffness diversity of cardiac muscle. J Mol Cell Cardiol. 2000;32:2151-2162
95. Makarenko I, Opitz CA, Leake MC, Neagoe C, Kulke M, Gwathmey JK, del Monte F, Hajjar RJ, Linke WA. Passive stiffness changes caused by upregulation of compliant titin isoforms in human dilated cardiomyopathy hearts. Circ Res. 2004;95:708-716
96. Nagueh SF, Shah G, Wu Y, Torre-Amione G, King NM, Lahmers S, Witt CC, Becker K, Labeit S, Granzier HL. Altered titin expression, myocardial stiffness, and left ventricular function in patients with dilated cardiomyopathy. Circulation. 2004;110:155-162
97. Yamasaki R, Wu Y, McNabb M, Greaser M, Labeit S, Granzier H. Protein kinase a phosphorylates titin's cardiac-specific n2b domain and reduces passive tension in rat cardiac myocytes. Circ Res. 2002;90:1181-1188
98. Hidalgo C, Hudson B, Bogomolovas J, Zhu Y, Anderson B, Greaser M, Labeit S, Granzier H. Pkc phosphorylation of titin's pevk element: A novel and conserved pathway for modulating myocardial stiffness. Circ Res. 2009;105:631-638, 617 p following 638
99. Kruger M, Kotter S, Grutzner A, Lang P, Andresen C, Redfield MM, Butt E, dos Remedios CG, Linke WA. Protein kinase g modulates human myocardial passive stiffness by phosphorylation of the titin springs. Circ Res. 2009;104:87-94
100. Falcao-Pires I, Hamdani N, Borbely A, Gavina C, Schalkwijk CG, van der Velden J, van Heerebeek L, Stienen GJ, Niessen HW, Leite-Moreira AF, Paulus WJ. Diabetes mellitus worsens diastolic left ventricular dysfunction in aortic stenosis through altered myocardial structure and cardiomyocyte stiffness. Circulation. 2011;124:1151-1159
101. Borbely A, Falcao-Pires I, van Heerebeek L, Hamdani N, Edes I, Gavina C, Leite-Moreira AF, Bronzwaer JG, Papp Z, van der Velden J, Stienen GJ, Paulus WJ. Hypophosphorylation of the stiff n2b titin isoform raises cardiomyocyte resting tension in failing human myocardium. Circ Res. 2009;104:780-786
102. Schwartz RG, McKenzie WB, Alexander J, Sager P, D'Souza A, Manatunga A, Schwartz PE, Berger HJ, Setaro J, Surkin L, et al. Congestive heart failure and left ventricular dysfunction complicating doxorubicin therapy. Seven-year experience using serial radionuclide angiocardiography. Am J Med. 1987;82:1109-1118
103. Von Hoff DD, Layard MW, Basa P, Davis HL, Jr., Von Hoff AL, Rozencweig M, Muggia FM. Risk factors for doxorubicin-induced congestive heart failure. Ann Intern Med. 1979;91:710-717
104. Steinherz LJ, Steinherz PG, Tan CT, Heller G, Murphy ML. Cardiac toxicity 4 to 20 years after completing anthracycline therapy. Jama. 1991;266:1672-1677
105. Milei J, Boveris A, Llesuy S, Molina HA, Storino R, Ortega D, Milei SE. Amelioration of adriamycin-induced cardiotoxicity in rabbits by prenylamine and vitamins a and e. Am Heart J. 1986;111:95-102
106. Lenzhofer R, Magometschnigg D, Dudczak R, Cerni C, Bolebruch C, Moser K. Indication of reduced doxorubicin-induced cardiac toxicity by additional treatment with antioxidative substances. Experientia. 1983;39:62-64
107. Myers CE, McGuire WP, Liss RH, Ifrim I, Grotzinger K, Young RC. Adriamycin: The role of lipid peroxidation in cardiac toxicity and tumor response. Science. 1977;197:165-167
108. Fujita K, Shinpo K, Yamada K, Sato T, Niimi H, Shamoto M, Nagatsu T, Takeuchi T, Umezawa H. Reduction of adriamycin toxicity by ascorbate in mice and guinea pigs. Cancer Res. 1982;42:309-316

References
109. Luo X, Evrovsky Y, Cole D, Trines J, Benson LN, Lehotay DC. Doxorubicin-induced acute changes in cytotoxic aldehydes, antioxidant status and cardiac function in the rat. Biochim Biophys Acta. 1997;1360:45-52
110. Zhou S, Palmeira CM, Wallace KB. Doxorubicin-induced persistent oxidative stress to cardiac myocytes. Toxicol Lett. 2001;121:151-157
111. Adachi K, Fujiura Y, Mayumi F, Nozuhara A, Sugiu Y, Sakanashi T, Hidaka T, Toshima H. A deletion of mitochondrial DNA in murine doxorubicin-induced cardiotoxicity. Biochem Biophys Res Commun. 1993;195:945-951
112. Shinozawa S, Etowo K, Araki Y, Oda T. Effect of coenzyme q10 on the survival time and lipid peroxidation of adriamycin (doxorubicin) treated mice. Acta Med Okayama. 1984;38:57-63
113. Shinozawa S, Gomita Y, Araki Y. Protective effects of various drugs on adriamycin (doxorubicin)-induced toxicity and microsomal lipid peroxidation in mice and rats. Biol Pharm Bull. 1993;16:1114-1117
114. Yamanaka N, Kato T, Nishida K, Fujikawa T, Fukushima M, Ota K. Elevation of serum lipid peroxide level associated with doxorubicin toxicity and its amelioration by [dl]-alpha-tocopheryl acetate or coenzyme q10 in mouse (doxorubicin, toxicity, lipid peroxide, tocopherol, coenzyme q10). Cancer Chemother Pharmacol. 1979;3:223-227
115. Liu XL, Sato S, Dai W, Yamanaka N. The protective effect of hepatocyte growth-promoting factor (phgf) against hydrogen peroxide-induced acute lung injury in rats. Med Electron Microsc. 2001;34:92-102
116. Faber M, Coudray C, Hida H, Mousseau M, Favier A. Lipid peroxidation products, and vitamin and trace element status in patients with cancer before and after chemotherapy, including adriamycin. A preliminary study. Biol Trace Elem Res. 1995;47:117-123
117. Erhola M, Kellokumpu-Lehtinen P, Metsa-Ketela T, Alanko K, Nieminen MM. Effects of anthracyclin-based chemotherapy on total plasma antioxidant capacity in small cell lung cancer patients. Free Radic Biol Med. 1996;21:383-390
118. Sangeetha P, Das UN, Koratkar R, Suryaprabha P. Increase in free radical generation and lipid peroxidation following chemotherapy in patients with cancer. Free Radic Biol Med. 1990;8:15-19
119. Doroshow JH. Effect of anthracycline antibiotics on oxygen radical formation in rat heart. Cancer Res. 1983;43:460-472
120. Vasquez-Vivar J, Martasek P, Hogg N, Masters BS, Pritchard KA, Jr., Kalyanaraman B. Endothelial nitric oxide synthase-dependent superoxide generation from adriamycin. Biochemistry. 1997;36:11293-11297
121. Muindi J, Sinha BK, Gianni L, Myers C. Thiol-dependent DNA damage produced by anthracycline-iron complexes. The structure-activity relationships and molecular mechanisms. Mol Pharmacol. 1985;27:356-365
122. Keizer HG, Pinedo HM, Schuurhuis GJ, Joenje H. Doxorubicin (adriamycin): A critical review of free radical-dependent mechanisms of cytotoxicity. Pharmacol Ther. 1990;47:219-231
123. Doroshow JH, Davies KJ. Redox cycling of anthracyclines by cardiac mitochondria. Ii. Formation of superoxide anion, hydrogen peroxide, and hydroxyl radical. J Biol Chem. 1986;261:3068-3074
124. Doroshow JH, Locker GY, Myers CE. Enzymatic defenses of the mouse heart against reactive oxygen metabolites: Alterations produced by doxorubicin. J Clin Invest. 1980;65:128-135
125. Davies KJ, Doroshow JH. Redox cycling of anthracyclines by cardiac mitochondria. I. Anthracycline radical formation by nadh dehydrogenase. J Biol Chem. 1986;261:3060-3067
126. Goormaghtigh E, Chatelain P, Caspers J, Ruysschaert JM. Evidence of a specific complex between adriamycin and negatively-charged phospholipids. Biochim Biophys Acta. 1980;597:1-14
127. Schlame M, Rua D, Greenberg ML. The biosynthesis and functional role of cardiolipin. Prog Lipid Res. 2000;39:257-288
128. Kashfi K, Israel M, Sweatman TW, Seshadri R, Cook GA. Inhibition of mitochondrial carnitine palmitoyltransferases by adriamycin and adriamycin analogues. Biochem Pharmacol. 1990;40:1441-1448
129. Suliman HB, Carraway MS, Ali AS, Reynolds CM, Welty-Wolf KE, Piantadosi CA. The co/ho system reverses inhibition of mitochondrial biogenesis and prevents murine doxorubicin cardiomyopathy. J Clin Invest. 2007;117:3730-3741
130. Piantadosi CA, Carraway MS, Babiker A, Suliman HB. Heme oxygenase-1 regulates cardiac mitochondrial biogenesis via nrf2-mediated transcriptional control of nuclear respiratory factor-1. Circ Res. 2008;103:1232-1240
131. Herman EH, Ferrans VJ. Preclinical animal models of cardiac protection from anthracycline-induced cardiotoxicity. Semin Oncol. 1998;25:15-21

References
132. Ladas EJ, Jacobson JS, Kennedy DD, Teel K, Fleischauer A, Kelly KM. Antioxidants and cancer therapy: A systematic review. J Clin Oncol. 2004;22:517-528
133. Kalay N, Basar E, Ozdogru I, Er O, Cetinkaya Y, Dogan A, Inanc T, Oguzhan A, Eryol NK, Topsakal R, Ergin A. Protective effects of carvedilol against anthracycline-induced cardiomyopathy. J Am Coll Cardiol. 2006;48:2258-2262
134. Herman EH, Ferrans VJ, Jordan W, Ardalan B. Reduction of chronic daunorubicin cardiotoxicity by icrf-187 in rabbits. Res Commun Chem Pathol Pharmacol. 1981;31:85-97
135. Speyer JL, Green MD, Kramer E, Rey M, Sanger J, Ward C, Dubin N, Ferrans V, Stecy P, Zeleniuch-Jacquotte A, et al. Protective effect of the bispiperazinedione icrf-187 against doxorubicin-induced cardiac toxicity in women with advanced breast cancer. N Engl J Med. 1988;319:745-752
136. Marty M, Espie M, Llombart A, Monnier A, Rapoport BL, Stahalova V. Multicenter randomized phase iii study of the cardioprotective effect of dexrazoxane (cardioxane) in advanced/metastatic breast cancer patients treated with anthracycline-based chemotherapy. Ann Oncol. 2006;17:614-622
137. Nakamura T, Ueda Y, Juan Y, Katsuda S, Takahashi H, Koh E. Fas-mediated apoptosis in adriamycin-induced cardiomyopathy in rats: In vivo study. Circulation. 2000;102:572-578
138. Lien YC, Lin SM, Nithipongvanitch R, Oberley TD, Noel T, Zhao Q, Daosukho C, St Clair DK. Tumor necrosis factor receptor deficiency exacerbated adriamycin-induced cardiomyocytes apoptosis: An insight into the fas connection. Mol Cancer Ther. 2006;5:261-269
139. Wu S, Ko YS, Teng MS, Ko YL, Hsu LA, Hsueh C, Chou YY, Liew CC, Lee YS. Adriamycin-induced cardiomyocyte and endothelial cell apoptosis: In vitro and in vivo studies. J Mol Cell Cardiol. 2002;34:1595-1607
140. Kalivendi SV, Konorev EA, Cunningham S, Vanamala SK, Kaji EH, Joseph J, Kalyanaraman B. Doxorubicin activates nuclear factor of activated t-lymphocytes and fas ligand transcription: Role of mitochondrial reactive oxygen species and calcium. Biochem J. 2005;389:527-539
141. Niu J, Azfer A, Wang K, Wang X, Kolattukudy PE. Cardiac-targeted expression of soluble fas attenuates doxorubicin-induced cardiotoxicity in mice. J Pharmacol Exp Ther. 2009;328:740-748
142. Kim DS, Woo ER, Chae SW, Ha KC, Lee GH, Hong ST, Kwon DY, Kim MS, Jung YK, Kim HM, Kim HK, Kim HR, Chae HJ. Plantainoside d protects adriamycin-induced apoptosis in h9c2 cardiac muscle cells via the inhibition of ros generation and nf-kappab activation. Life Sci. 2007;80:314-323
143. Li H, Gu H, Sun B. Protective effects of pyrrolidine dithiocarbamate on myocardium apoptosis induced by adriamycin in rats. Int J Cardiol. 2007;114:159-165
144. Wang S, Kotamraju S, Konorev E, Kalivendi S, Joseph J, Kalyanaraman B. Activation of nuclear factor-kappab during doxorubicin-induced apoptosis in endothelial cells and myocytes is pro-apoptotic: The role of hydrogen peroxide. Biochem J. 2002;367:729-740
145. Deniaud A, Sharaf el dein O, Maillier E, Poncet D, Kroemer G, Lemaire C, Brenner C. Endoplasmic reticulum stress induces calcium-dependent permeability transition, mitochondrial outer membrane permeabilization and apoptosis. Oncogene. 2008;27:285-299
146. Childs AC, Phaneuf SL, Dirks AJ, Phillips T, Leeuwenburgh C. Doxorubicin treatment in vivo causes cytochrome c release and cardiomyocyte apoptosis, as well as increased mitochondrial efficiency, superoxide dismutase activity, and bcl-2:Bax ratio. Cancer Res. 2002;62:4592-4598
147. L'Ecuyer T, Sanjeev S, Thomas R, Novak R, Das L, Campbell W, Heide RV. DNA damage is an early event in doxorubicin-induced cardiac myocyte death. Am J Physiol Heart Circ Physiol. 2006;291:H1273-1280
148. Liu X, Chua CC, Gao J, Chen Z, Landy CL, Hamdy R, Chua BH. Pifithrin-alpha protects against doxorubicin-induced apoptosis and acute cardiotoxicity in mice. Am J Physiol Heart Circ Physiol. 2004;286:H933-939
149. Liu J, Mao W, Ding B, Liang CS. Erks/p53 signal transduction pathway is involved in doxorubicin-induced apoptosis in h9c2 cells and cardiomyocytes. Am J Physiol Heart Circ Physiol. 2008;295:H1956-1965
150. Aries A, Paradis P, Lefebvre C, Schwartz RJ, Nemer M. Essential role of gata-4 in cell survival and drug-induced cardiotoxicity. Proc Natl Acad Sci U S A. 2004;101:6975-6980
151. Wang YX, Korth M. Effects of doxorubicin on excitation-contraction coupling in guinea pig ventricular myocardium. Circ Res. 1995;76:645-653
152. Arai M, Yoguchi A, Takizawa T, Yokoyama T, Kanda T, Kurabayashi M, Nagai R. Mechanism of doxorubicin-induced inhibition of sarcoplasmic reticulum ca(2+)-atpase gene transcription. Circ Res. 2000;86:8-14
153. Zhou S, Starkov A, Froberg MK, Leino RL, Wallace KB. Cumulative and irreversible cardiac mitochondrial dysfunction induced by doxorubicin. Cancer Res. 2001;61:771-777

References
154. Ito H, Miller SC, Billingham ME, Akimoto H, Torti SV, Wade R, Gahlmann R, Lyons G, Kedes L, Torti FM. Doxorubicin selectively inhibits muscle gene expression in cardiac muscle cells in vivo and in vitro. Proc Natl Acad Sci U S A. 1990;87:4275-4279
155. Lim CC, Zuppinger C, Guo X, Kuster GM, Helmes M, Eppenberger HM, Suter TM, Liao R, Sawyer DB. Anthracyclines induce calpain-dependent titin proteolysis and necrosis in cardiomyocytes. J Biol Chem. 2004;279:8290-8299
156. Zsary A, Szucs S, Keltai K, Schneider T, Rosta A, Sarman P, Fenyvesi T, Karadi I. Endothelins: A possible mechanism of cytostatics-induced cardiomyopathy. Leuk Lymphoma. 2004;45:351-355
157. Lemmens K, Segers VF, Demolder M, De Keulenaer GW. Role of neuregulin-1/erbb2 signaling in endothelium-cardiomyocyte cross-talk. J Biol Chem. 2006;281:19469-19477
158. Goetzenich A, Hatam N, Zernecke A, Weber C, Czarnotta T, Autschbach R, Christiansen S. Alteration of matrix metalloproteinases in selective left ventricular adriamycin-induced cardiomyopathy in the pig. J Heart Lung Transplant. 2009;28:1087-1093
159. Spallarossa P, Altieri P, Garibaldi S, Ghigliotti G, Barisione C, Manca V, Fabbi P, Ballestrero A, Brunelli C, Barsotti A. Matrix metalloproteinase-2 and -9 are induced differently by doxorubicin in h9c2 cells: The role of map kinases and nad(p)h oxidase. Cardiovasc Res. 2006;69:736-745
160. De Angelis A, Piegari E, Cappetta D, Marino L, Filippelli A, Berrino L, Ferreira-Martins J, Zheng H, Hosoda T, Rota M, Urbanek K, Kajstura J, Leri A, Rossi F, Anversa P. Anthracycline cardiomyopathy is mediated by depletion of the cardiac stem cell pool and is rescued by restoration of progenitor cell function. Circulation. 2010;121:276-292
161. Shah AM, MacCarthy PA. Paracrine and autocrine effects of nitric oxide on myocardial function. Pharmacol Ther. 2000;86:49-86
162. Kamisaki Y, Saheki S, Nakane M, Palmieri JA, Kuno T, Chang BY, Waldman SA, Murad F. Soluble guanylate cyclase from rat lung exists as a heterodimer. J Biol Chem. 1986;261:7236-7241
163. Harteneck C, Wedel B, Koesling D, Malkewitz J, Bohme E, Schultz G. Molecular cloning and expression of a new alpha-subunit of soluble guanylyl cyclase. Interchangeability of the alpha-subunits of the enzyme. FEBS Lett. 1991;292:217-222
164. Yuen PS, Potter LR, Garbers DL. A new form of guanylyl cyclase is preferentially expressed in rat kidney. Biochemistry. 1990;29:10872-10878
165. Wedel B, Humbert P, Harteneck C, Foerster J, Malkewitz J, Bohme E, Schultz G, Koesling D. Mutation of his-105 in the beta 1 subunit yields a nitric oxide-insensitive form of soluble guanylyl cyclase. Proc Natl Acad Sci U S A. 1994;91:2592-2596
166. Cary SP, Winger JA, Marletta MA. Tonic and acute nitric oxide signaling through soluble guanylate cyclase is mediated by nonheme nitric oxide, atp, and gtp. Proc Natl Acad Sci U S A. 2005;102:13064-13069
167. Ruiz-Stewart I, Tiyyagura SR, Lin JE, Kazerounian S, Pitari GM, Schulz S, Martin E, Murad F, Waldman SA. Guanylyl cyclase is an atp sensor coupling nitric oxide signaling to cell metabolism. Proc Natl Acad Sci U S A. 2004;101:37-42
168. Zhou Z, Sayed N, Pyriochou A, Roussos C, Fulton D, Beuve A, Papapetropoulos A. Protein kinase g phosphorylates soluble guanylyl cyclase on serine 64 and inhibits its activity. Arterioscler Thromb Vasc Biol. 2008;28:1803-1810
169. Weber M, Lauer N, Mulsch A, Kojda G. The effect of peroxynitrite on the catalytic activity of soluble guanylyl cyclase. Free Radic Biol Med. 2001;31:1360-1367
170. Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol Rev. 2007;87:315-424
171. Lima B, Forrester MT, Hess DT, Stamler JS. S-nitrosylation in cardiovascular signaling. Circ Res. 2010;106:633-646
172. Nishikimi T, Maeda N, Matsuoka H. The role of natriuretic peptides in cardioprotection. Cardiovasc Res. 2006;69:318-328
173. Hutchinson HG, Trindade PT, Cunanan DB, Wu CF, Pratt RE. Mechanisms of natriuretic-peptide-induced growth inhibition of vascular smooth muscle cells. Cardiovasc Res. 1997;35:158-167
174. Doyle DD, Upshaw-Earley J, Bell EL, Palfrey HC. Natriuretic peptide receptor-b in adult rat ventricle is predominantly confined to the nonmyocyte population. Am J Physiol Heart Circ Physiol. 2002;282:H2117-2123
175. Han B, Fixler R, Beeri R, Wang Y, Bachrach U, Hasin Y. The opposing effects of endothelin-1 and c-type natriuretic peptide on apoptosis of neonatal rat cardiac myocytes. Eur J Pharmacol. 2003;474:15-20
176. Rosenkranz AC, Woods RL, Dusting GJ, Ritchie RH. Antihypertrophic actions of the natriuretic peptides in adult rat cardiomyocytes: Importance of cyclic gmp. Cardiovasc Res. 2003;57:515-522

References
177. Tokudome T, Horio T, Soeki T, Mori K, Kishimoto I, Suga S, Yoshihara F, Kawano Y, Kohno M, Kangawa K. Inhibitory effect of c-type natriuretic peptide (cnp) on cultured cardiac myocyte hypertrophy: Interference between cnp and endothelin-1 signaling pathways. Endocrinology. 2004;145:2131-2140
178. Chinkers M, Garbers DL. The protein kinase domain of the anp receptor is required for signaling. Science. 1989;245:1392-1394
179. Maurice DH, Palmer D, Tilley DG, Dunkerley HA, Netherton SJ, Raymond DR, Elbatarny HS, Jimmo SL. Cyclic nucleotide phosphodiesterase activity, expression, and targeting in cells of the cardiovascular system. Mol Pharmacol. 2003;64:533-546
180. Takimoto E, Champion HC, Belardi D, Moslehi J, Mongillo M, Mergia E, Montrose DC, Isoda T, Aufiero K, Zaccolo M, Dostmann WR, Smith CJ, Kass DA. Cgmp catabolism by phosphodiesterase 5a regulates cardiac adrenergic stimulation by nos3-dependent mechanism. Circ Res. 2005;96:100-109
181. Croom KF, Curran MP. Sildenafil: A review of its use in pulmonary arterial hypertension. Drugs. 2008;68:383-397
182. Rosen RC, Kostis JB. Overview of phosphodiesterase 5 inhibition in erectile dysfunction. Am J Cardiol. 2003;92:9M-18M
183. Shakur Y, Holst LS, Landstrom TR, Movsesian M, Degerman E, Manganiello V. Regulation and function of the cyclic nucleotide phosphodiesterase (pde3) gene family. Prog Nucleic Acid Res Mol Biol. 2001;66:241-277
184. Patrucco E, Notte A, Barberis L, Selvetella G, Maffei A, Brancaccio M, Marengo S, Russo G, Azzolino O, Rybalkin SD, Silengo L, Altruda F, Wetzker R, Wymann MP, Lembo G, Hirsch E. Pi3kgamma modulates the cardiac response to chronic pressure overload by distinct kinase-dependent and -independent effects. Cell. 2004;118:375-387
185. Dunkerley HA, Tilley DG, Palmer D, Liu H, Jimmo SL, Maurice DH. Reduced phosphodiesterase 3 activity and phosphodiesterase 3a level in synthetic vascular smooth muscle cells: Implications for use of phosphodiesterase 3 inhibitors in cardiovascular tissues. Mol Pharmacol. 2002;61:1033-1040
186. Yano M, Kohno M, Ohkusa T, Mochizuki M, Yamada J, Hisaoka T, Ono K, Tanigawa T, Kobayashi S, Matsuzaki M. Effect of milrinone on left ventricular relaxation and ca(2+) uptake function of cardiac sarcoplasmic reticulum. Am J Physiol Heart Circ Physiol. 2000;279:H1898-1905
187. Verde I, Vandecasteele G, Lezoualc'h F, Fischmeister R. Characterization of the cyclic nucleotide phosphodiesterase subtypes involved in the regulation of the l-type ca2+ current in rat ventricular myocytes. Br J Pharmacol. 1999;127:65-74
188. Vandeput F, Wolda SL, Krall J, Hambleton R, Uher L, McCaw KN, Radwanski PB, Florio V, Movsesian MA. Cyclic nucleotide phosphodiesterase pde1c1 in human cardiac myocytes. J Biol Chem. 2007;282:32749-32757
189. Miller CL, Oikawa M, Cai Y, Wojtovich AP, Nagel DJ, Xu X, Xu H, Florio V, Rybalkin SD, Beavo JA, Chen YF, Li JD, Blaxall BC, Abe J, Yan C. Role of ca2+/calmodulin-stimulated cyclic nucleotide phosphodiesterase 1 in mediating cardiomyocyte hypertrophy. Circ Res. 2009;105:956-964
190. Rybalkin SD, Yan C, Bornfeldt KE, Beavo JA. Cyclic gmp phosphodiesterases and regulation of smooth muscle function. Circ Res. 2003;93:280-291
191. Russwurm M, Wittau N, Koesling D. Guanylyl cyclase/psd-95 interaction: Targeting of the nitric oxide-sensitive alpha2beta1 guanylyl cyclase to synaptic membranes. J Biol Chem. 2001;276:44647-44652
192. Schoser BG, Behrends S. Soluble guanylyl cyclase is localized at the neuromuscular junction in human skeletal muscle. Neuroreport. 2001;12:979-981
193. Zabel U, Kleinschnitz C, Oh P, Nedvetsky P, Smolenski A, Muller H, Kronich P, Kugler P, Walter U, Schnitzer JE, Schmidt HH. Calcium-dependent membrane association sensitizes soluble guanylyl cyclase to nitric oxide. Nat Cell Biol. 2002;4:307-311
194. Linder AE, McCluskey LP, Cole KR, 3rd, Lanning KM, Webb RC. Dynamic association of nitric oxide downstream signaling molecules with endothelial caveolin-1 in rat aorta. J Pharmacol Exp Ther. 2005;314:9-15
195. Castro LR, Verde I, Cooper DM, Fischmeister R. Cyclic guanosine monophosphate compartmentation in rat cardiac myocytes. Circulation. 2006;113:2221-2228
196. Takimoto E, Belardi D, Tocchetti CG, Vahebi S, Cormaci G, Ketner EA, Moens AL, Champion HC, Kass DA. Compartmentalization of cardiac beta-adrenergic inotropy modulation by phosphodiesterase type 5. Circulation. 2007;115:2159-2167
197. Lee DI, Vahebi S, Tocchetti CG, Barouch LA, Solaro RJ, Takimoto E, Kass DA. Pde5a suppression of acute beta-adrenergic activation requires modulation of myocyte beta-3 signaling coupled to pkg-mediated troponin i phosphorylation. Basic Res Cardiol. 2010;105:337-347

References
198. Di Benedetto G, Zoccarato A, Lissandron V, Terrin A, Li X, Houslay MD, Baillie GS, Zaccolo M. Protein kinase a type i and type ii define distinct intracellular signaling compartments. Circ Res. 2008;103:836-844
199. Stangherlin A, Gesellchen F, Zoccarato A, Terrin A, Fields LA, Berrera M, Surdo NC, Craig MA, Smith G, Hamilton G, Zaccolo M. Cgmp signals modulate camp levels in a compartment-specific manner to regulate catecholamine-dependent signaling in cardiac myocytes. Circ Res. 2011;108:929-939
200. Murad F, Rapoport RM, Fiscus R. Role of cyclic-gmp in relaxations of vascular smooth muscle. J Cardiovasc Pharmacol. 1985;7 Suppl 3:S111-118
201. Munzel T, Feil R, Mulsch A, Lohmann SM, Hofmann F, Walter U. Physiology and pathophysiology of vascular signaling controlled by guanosine 3',5'-cyclic monophosphate-dependent protein kinase [corrected]. Circulation. 2003;108:2172-2183
202. Pfeifer A, Klatt P, Massberg S, Ny L, Sausbier M, Hirneiss C, Wang GX, Korth M, Aszodi A, Andersson KE, Krombach F, Mayerhofer A, Ruth P, Fassler R, Hofmann F. Defective smooth muscle regulation in cgmp kinase i-deficient mice. Embo J. 1998;17:3045-3051
203. Sausbier M, Schubert R, Voigt V, Hirneiss C, Pfeifer A, Korth M, Kleppisch T, Ruth P, Hofmann F. Mechanisms of no/cgmp-dependent vasorelaxation. Circ Res. 2000;87:825-830
204. Wegener JW, Nawrath H, Wolfsgruber W, Kuhbandner S, Werner C, Hofmann F, Feil R. Cgmp-dependent protein kinase i mediates the negative inotropic effect of cgmp in the murine myocardium. Circ Res. 2002;90:18-20
205. Koeppen M, Feil R, Siegl D, Feil S, Hofmann F, Pohl U, de Wit C. Cgmp-dependent protein kinase mediates no- but not acetylcholine-induced dilations in resistance vessels in vivo. Hypertension. 2004;44:952-955
206. Lincoln TM, Wu X, Sellak H, Dey N, Choi CS. Regulation of vascular smooth muscle cell phenotype by cyclic gmp and cyclic gmp-dependent protein kinase. Front Biosci. 2006;11:356-367
207. Murohara T, Asahara T, Silver M, Bauters C, Masuda H, Kalka C, Kearney M, Chen D, Symes JF, Fishman MC, Huang PL, Isner JM. Nitric oxide synthase modulates angiogenesis in response to tissue ischemia. J Clin Invest. 1998;101:2567-2578
208. Fukumura D, Gohongi T, Kadambi A, Izumi Y, Ang J, Yun CO, Buerk DG, Huang PL, Jain RK. Predominant role of endothelial nitric oxide synthase in vascular endothelial growth factor-induced angiogenesis and vascular permeability. Proc Natl Acad Sci U S A. 2001;98:2604-2609
209. Aicher A, Heeschen C, Mildner-Rihm C, Urbich C, Ihling C, Technau-Ihling K, Zeiher AM, Dimmeler S. Essential role of endothelial nitric oxide synthase for mobilization of stem and progenitor cells. Nat Med. 2003;9:1370-1376
210. Bussolati B, Dunk C, Grohman M, Kontos CD, Mason J, Ahmed A. Vascular endothelial growth factor receptor-1 modulates vascular endothelial growth factor-mediated angiogenesis via nitric oxide. Am J Pathol. 2001;159:993-1008
211. Papapetropoulos A, Desai KM, Rudic RD, Mayer B, Zhang R, Ruiz-Torres MP, Garcia-Cardena G, Madri JA, Sessa WC. Nitric oxide synthase inhibitors attenuate transforming-growth-factor-beta 1-stimulated capillary organization in vitro. Am J Pathol. 1997;150:1835-1844
212. Papapetropoulos A, Garcia-Cardena G, Madri JA, Sessa WC. Nitric oxide production contributes to the angiogenic properties of vascular endothelial growth factor in human endothelial cells. J Clin Invest. 1997;100:3131-3139
213. Chen H, Levine YC, Golan DE, Michel T, Lin AJ. Atrial natriuretic peptide-initiated cgmp pathways regulate vasodilator-stimulated phosphoprotein phosphorylation and angiogenesis in vascular endothelium. J Biol Chem. 2008;283:4439-4447
214. Sabrane K, Kruse MN, Fabritz L, Zetsche B, Mitko D, Skryabin BV, Zwiener M, Baba HA, Yanagisawa M, Kuhn M. Vascular endothelium is critically involved in the hypotensive and hypovolemic actions of atrial natriuretic peptide. J Clin Invest. 2005;115:1666-1674
215. Surapisitchat J, Jeon KI, Yan C, Beavo JA. Differential regulation of endothelial cell permeability by cgmp via phosphodiesterases 2 and 3. Circ Res. 2007;101:811-818
216. Tanabe M, Ueda M, Endo M, Kitajima M. The effect of atrial natriuretic peptide on pulmonary acid injury in a pig model. Am J Respir Crit Care Med. 1996;154:1351-1356
217. Mitaka C, Hirata Y, Nagura T, Tsunoda Y, Amaha K. Beneficial effect of atrial natriuretic peptide on pulmonary gas exchange in patients with acute lung injury. Chest. 1998;114:223-228
218. Klinger JR, Warburton R, Carino GP, Murray J, Murphy C, Napier M, Harrington EO. Natriuretic peptides differentially attenuate thrombin-induced barrier dysfunction in pulmonary microvascular endothelial cells. Exp Cell Res. 2006;312:401-410

References
219. Furst R, Bubik MF, Bihari P, Mayer BA, Khandoga AG, Hoffmann F, Rehberg M, Krombach F, Zahler S, Vollmar AM. Atrial natriuretic peptide protects against histamine-induced endothelial barrier dysfunction in vivo. Mol Pharmacol. 2008;74:1-8
220. Klinger JR, Warburton RR, Pietras L, Hill NS. Brain natriuretic peptide inhibits hypoxic pulmonary hypertension in rats. J Appl Physiol. 1998;84:1646-1652
221. Irwin DC, Rhodes J, Baker DC, Nelson SE, Tucker A. Atrial natriuretic peptide blockade exacerbates high altitude pulmonary edema in endotoxin-primed rats. High Alt Med Biol. 2001;2:349-360
222. Irwin DC, Tissot van Patot MC, Tucker A, Bowen R. Direct anp inhibition of hypoxia-induced inflammatory pathways in pulmonary microvascular and macrovascular endothelial monolayers. Am J Physiol Lung Cell Mol Physiol. 2005;288:L849-859
223. Yin J, Hoffmann J, Kaestle SM, Neye N, Wang L, Baeurle J, Liedtke W, Wu S, Kuppe H, Pries AR, Kuebler WM. Negative-feedback loop attenuates hydrostatic lung edema via a cgmp-dependent regulation of transient receptor potential vanilloid 4. Circ Res. 2008;102:966-974
224. Brovkovych V, Gao XP, Ong E, Brovkovych S, Brennan ML, Su X, Hazen SL, Malik AB, Skidgel RA. Augmented inducible nitric oxide synthase expression and increased no production reduce sepsis-induced lung injury and mortality in myeloperoxidase-null mice. Am J Physiol Lung Cell Mol Physiol. 2008;295:L96-103
225. Zhang Q, Moalem J, Tse J, Scholz PM, Weiss HR. Effects of natriuretic peptides on ventricular myocyte contraction and role of cyclic gmp signaling. Eur J Pharmacol. 2005;510:209-215
226. Mohan P, Brutsaert DL, Paulus WJ, Sys SU. Myocardial contractile response to nitric oxide and cgmp. Circulation. 1996;93:1223-1229
227. Kojda G, Kottenberg K, Nix P, Schluter KD, Piper HM, Noack E. Low increase in cgmp induced by organic nitrates and nitrovasodilators improves contractile response of rat ventricular myocytes. Circ Res. 1996;78:91-101
228. Vila-Petroff MG, Younes A, Egan J, Lakatta EG, Sollott SJ. Activation of distinct camp-dependent and cgmp-dependent pathways by nitric oxide in cardiac myocytes. Circ Res. 1999;84:1020-1031
229. Layland J, Li JM, Shah AM. Role of cyclic gmp-dependent protein kinase in the contractile response to exogenous nitric oxide in rat cardiac myocytes. J Physiol. 2002;540:457-467
230. Xu KY, Huso DL, Dawson TM, Bredt DS, Becker LC. Nitric oxide synthase in cardiac sarcoplasmic reticulum. Proc Natl Acad Sci U S A. 1999;96:657-662
231. Zhang YH, Zhang MH, Sears CE, Emanuel K, Redwood C, El-Armouche A, Kranias EG, Casadei B. Reduced phospholamban phosphorylation is associated with impaired relaxation in left ventricular myocytes from neuronal no synthase-deficient mice. Circ Res. 2008;102:242-249
232. Martin SR, Emanuel K, Sears CE, Zhang YH, Casadei B. Are myocardial enos and nnos involved in the beta-adrenergic and muscarinic regulation of inotropy? A systematic investigation. Cardiovasc Res. 2006;70:97-106
233. Sears CE, Bryant SM, Ashley EA, Lygate CA, Rakovic S, Wallis HL, Neubauer S, Terrar DA, Casadei B. Cardiac neuronal nitric oxide synthase isoform regulates myocardial contraction and calcium handling. Circ Res. 2003;92:e52-59
234. Khan SA, Skaf MW, Harrison RW, Lee K, Minhas KM, Kumar A, Fradley M, Shoukas AA, Berkowitz DE, Hare JM. Nitric oxide regulation of myocardial contractility and calcium cycling: Independent impact of neuronal and endothelial nitric oxide synthases. Circ Res. 2003;92:1322-1329
235. Pollman MJ, Yamada T, Horiuchi M, Gibbons GH. Vasoactive substances regulate vascular smooth muscle cell apoptosis. Countervailing influences of nitric oxide and angiotensin ii. Circ Res. 1996;79:748-756
236. Wu CF, Bishopric NH, Pratt RE. Atrial natriuretic peptide induces apoptosis in neonatal rat cardiac myocytes. J Biol Chem. 1997;272:14860-14866
237. Chiche JD, Schlutsmeyer SM, Bloch DB, de la Monte SM, Roberts JD, Jr., Filippov G, Janssens SP, Rosenzweig A, Bloch KD. Adenovirus-mediated gene transfer of cgmp-dependent protein kinase increases the sensitivity of cultured vascular smooth muscle cells to the antiproliferative and pro-apoptotic effects of nitric oxide/cgmp. J Biol Chem. 1998;273:34263-34271
238. Kloss S, Bouloumie A, Mulsch A. Aging and chronic hypertension decrease expression of rat aortic soluble guanylyl cyclase. Hypertension. 2000;35:43-47
239. Hayashi D, Kudoh S, Shiojima I, Zou Y, Harada K, Shimoyama M, Imai Y, Monzen K, Yamazaki T, Yazaki Y, Nagai R, Komuro I. Atrial natriuretic peptide inhibits cardiomyocyte hypertrophy through mitogen-activated protein kinase phosphatase-1. Biochem Biophys Res Commun. 2004;322:310-319
240. Laskowski A, Woodman OL, Cao AH, Drummond GR, Marshall T, Kaye DM, Ritchie RH. Antioxidant actions contribute to the antihypertrophic effects of atrial natriuretic peptide in neonatal rat cardiomyocytes. Cardiovasc Res. 2006;72:112-123

References
241. Horio T, Nishikimi T, Yoshihara F, Matsuo H, Takishita S, Kangawa K. Inhibitory regulation of hypertrophy by endogenous atrial natriuretic peptide in cultured cardiac myocytes. Hypertension. 2000;35:19-24
242. Kuhn M, Holtwick R, Baba HA, Perriard JC, Schmitz W, Ehler E. Progressive cardiac hypertrophy and dysfunction in atrial natriuretic peptide receptor (gc-a) deficient mice. Heart. 2002;87:368-374
243. Lopez MJ, Wong SK, Kishimoto I, Dubois S, Mach V, Friesen J, Garbers DL, Beuve A. Salt-resistant hypertension in mice lacking the guanylyl cyclase-a receptor for atrial natriuretic peptide. Nature. 1995;378:65-68
244. Oliver PM, Fox JE, Kim R, Rockman HA, Kim HS, Reddick RL, Pandey KN, Milgram SL, Smithies O, Maeda N. Hypertension, cardiac hypertrophy, and sudden death in mice lacking natriuretic peptide receptor a. Proc Natl Acad Sci U S A. 1997;94:14730-14735
245. Holtwick R, van Eickels M, Skryabin BV, Baba HA, Bubikat A, Begrow F, Schneider MD, Garbers DL, Kuhn M. Pressure-independent cardiac hypertrophy in mice with cardiomyocyte-restricted inactivation of the atrial natriuretic peptide receptor guanylyl cyclase-a. J Clin Invest. 2003;111:1399-1407
246. Kilic A, Bubikat A, Gassner B, Baba HA, Kuhn M. Local actions of atrial natriuretic peptide counteract angiotensin ii stimulated cardiac remodeling. Endocrinology. 2007;148:4162-4169
247. John SW, Krege JH, Oliver PM, Hagaman JR, Hodgin JB, Pang SC, Flynn TG, Smithies O. Genetic decreases in atrial natriuretic peptide and salt-sensitive hypertension. Science. 1995;267:679-681
248. Patel JB, Valencik ML, Pritchett AM, Burnett JC, Jr., McDonald JA, Redfield MM. Cardiac-specific attenuation of natriuretic peptide a receptor activity accentuates adverse cardiac remodeling and mortality in response to pressure overload. Am J Physiol Heart Circ Physiol. 2005;289:H777-784
249. Zahabi A, Picard S, Fortin N, Reudelhuber TL, Deschepper CF. Expression of constitutively active guanylate cyclase in cardiomyocytes inhibits the hypertrophic effects of isoproterenol and aortic constriction on mouse hearts. J Biol Chem. 2003;278:47694-47699
250. Friebe A, Mergia E, Dangel O, Lange A, Koesling D. Fatal gastrointestinal obstruction and hypertension in mice lacking nitric oxide-sensitive guanylyl cyclase. Proc Natl Acad Sci U S A. 2007;104:7699-7704
251. Buys ES, Sips P, Vermeersch P, Raher MJ, Rogge E, Ichinose F, Dewerchin M, Bloch KD, Janssens S, Brouckaert P. Gender-specific hypertension and responsiveness to nitric oxide in sgcalpha1 knockout mice. Cardiovasc Res. 2008;79:179-186
252. Cawley SM, Kolodziej S, Ichinose F, Brouckaert P, Buys ES, Bloch KD. Sgc{alpha}1 mediates the negative inotropic effects of no in cardiac myocytes independent of changes in calcium handling. Am J Physiol Heart Circ Physiol. 2011;301:H157-163
253. Niu X, Watts VL, Cingolani OH, Sivakumaran V, Leyton-Mange JS, Ellis CL, Miller KL, Vandegaer K, Bedja D, Gabrielson KL, Paolocci N, Kass DA, Barouch LA. Cardioprotective effect of beta-3 adrenergic receptor agonism: Role of neuronal nitric oxide synthase. J Am Coll Cardiol. 2012;59:1979-1987
254. Ruetten H, Dimmeler S, Gehring D, Ihling C, Zeiher AM. Concentric left ventricular remodeling in endothelial nitric oxide synthase knockout mice by chronic pressure overload. Cardiovasc Res. 2005;66:444-453
255. Moens AL, Takimoto E, Tocchetti CG, Chakir K, Bedja D, Cormaci G, Ketner EA, Majmudar M, Gabrielson K, Halushka MK, Mitchell JB, Biswal S, Channon KM, Wolin MS, Alp NJ, Paolocci N, Champion HC, Kass DA. Reversal of cardiac hypertrophy and fibrosis from pressure overload by tetrahydrobiopterin: Efficacy of recoupling nitric oxide synthase as a therapeutic strategy. Circulation. 2008;117:2626-2636
256. Lu Z, Xu X, Hu X, Lee S, Traverse JH, Zhu G, Fassett J, Tao Y, Zhang P, dos Remedios C, Pritzker M, Hall JL, Garry DJ, Chen Y. Oxidative stress regulates left ventricular pde5 expression in the failing heart. Circulation. 2010;121:1474-1483
257. Pokreisz P, Vandenwijngaert S, Bito V, Van den Bergh A, Lenaerts I, Busch C, Marsboom G, Gheysens O, Vermeersch P, Biesmans L, Liu X, Gillijns H, Pellens M, Van Lommel A, Buys E, Schoonjans L, Vanhaecke J, Verbeken E, Sipido K, Herijgers P, Bloch KD, Janssens SP. Ventricular phosphodiesterase-5 expression is increased in patients with advanced heart failure and contributes to adverse ventricular remodeling after myocardial infarction in mice. Circulation. 2009;119:408-416
258. Nagendran J, Archer SL, Soliman D, Gurtu V, Moudgil R, Haromy A, St Aubin C, Webster L, Rebeyka IM, Ross DB, Light PE, Dyck JR, Michelakis ED. Phosphodiesterase type 5 is highly expressed in the hypertrophied human right ventricle, and acute inhibition of phosphodiesterase type 5 improves contractility. Circulation. 2007;116:238-248
259. Nagayama T, Hsu S, Zhang M, Koitabashi N, Bedja D, Gabrielson KL, Takimoto E, Kass DA. Sildenafil stops progressive chamber, cellular, and molecular remodeling and improves calcium handling and function in hearts with pre-existing advanced hypertrophy caused by pressure overload. J Am Coll Cardiol. 2009;53:207-215

References
260. Takimoto E, Champion HC, Li M, Belardi D, Ren S, Rodriguez ER, Bedja D, Gabrielson KL, Wang Y, Kass DA. Chronic inhibition of cyclic gmp phosphodiesterase 5a prevents and reverses cardiac hypertrophy. Nat Med. 2005;11:214-222
261. Guazzi M, Vicenzi M, Arena R, Guazzi MD. Pde5 inhibition with sildenafil improves left ventricular diastolic function, cardiac geometry, and clinical status in patients with stable systolic heart failure: Results of a 1-year, prospective, randomized, placebo-controlled study. Circ Heart Fail. 2011;4:8-17
262. Vandeput F, Krall J, Ockaili R, Salloum FN, Florio V, Corbin JD, Francis SH, Kukreja RC, Movsesian MA. Cgmp-hydrolytic activity and its inhibition by sildenafil in normal and failing human and mouse myocardium. J Pharmacol Exp Ther. 2009;330:884-891
263. Kato T, Sano M, Miyoshi S, Sato T, Hakuno D, Ishida H, Kinoshita-Nakazawa H, Fukuda K, Ogawa S. Calmodulin kinases ii and iv and calcineurin are involved in leukemia inhibitory factor-induced cardiac hypertrophy in rats. Circ Res. 2000;87:937-945
264. Taigen T, De Windt LJ, Lim HW, Molkentin JD. Targeted inhibition of calcineurin prevents agonist-induced cardiomyocyte hypertrophy. Proc Natl Acad Sci U S A. 2000;97:1196-1201
265. Zhu W, Zou Y, Shiojima I, Kudoh S, Aikawa R, Hayashi D, Mizukami M, Toko H, Shibasaki F, Yazaki Y, Nagai R, Komuro I. Ca2+/calmodulin-dependent kinase ii and calcineurin play critical roles in endothelin-1-induced cardiomyocyte hypertrophy. J Biol Chem. 2000;275:15239-15245
266. Fiedler B, Lohmann SM, Smolenski A, Linnemuller S, Pieske B, Schroder F, Molkentin JD, Drexler H, Wollert KC. Inhibition of calcineurin-nfat hypertrophy signaling by cgmp-dependent protein kinase type i in cardiac myocytes. Proc Natl Acad Sci U S A. 2002;99:11363-11368
267. Hsu S, Nagayama T, Koitabashi N, Zhang M, Zhou L, Bedja D, Gabrielson KL, Molkentin JD, Kass DA, Takimoto E. Phosphodiesterase 5 inhibition blocks pressure overload-induced cardiac hypertrophy independent of the calcineurin pathway. Cardiovasc Res. 2009;81:301-309
268. Klaiber M, Kruse M, Volker K, Schroter J, Feil R, Freichel M, Gerling A, Feil S, Dietrich A, Londono JE, Baba HA, Abramowitz J, Birnbaumer L, Penninger JM, Pongs O, Kuhn M. Novel insights into the mechanisms mediating the local antihypertrophic effects of cardiac atrial natriuretic peptide: Role of cgmp-dependent protein kinase and rgs2. Basic Res Cardiol. 2010;105:583-595
269. Kilic A, Velic A, De Windt LJ, Fabritz L, Voss M, Mitko D, Zwiener M, Baba HA, van Eickels M, Schlatter E, Kuhn M. Enhanced activity of the myocardial na+/h+ exchanger nhe-1 contributes to cardiac remodeling in atrial natriuretic peptide receptor-deficient mice. Circulation. 2005;112:2307-2317
270. Kirchhof P, Fabritz L, Kilic A, Begrow F, Breithardt G, Kuhn M. Ventricular arrhythmias, increased cardiac calmodulin kinase ii expression, and altered repolarization kinetics in anp receptor deficient mice. J Mol Cell Cardiol. 2004;36:691-700
271. Yeves AM, Garciarena CD, Nolly MB, Chiappe de Cingolani GE, Cingolani HE, Ennis IL. Decreased activity of the na+/h+ exchanger by phosphodiesterase 5a inhibition is attributed to an increase in protein phosphatase activity. Hypertension. 2010;56:690-695
272. Nakamura TY, Iwata Y, Arai Y, Komamura K, Wakabayashi S. Activation of na+/h+ exchanger 1 is sufficient to generate ca2+ signals that induce cardiac hypertrophy and heart failure. Circ Res. 2008;103:891-899
273. Perez NG, Piaggio MR, Ennis IL, Garciarena CD, Morales C, Escudero EM, Cingolani OH, Chiappe de Cingolani G, Yang XP, Cingolani HE. Phosphodiesterase 5a inhibition induces na+/h+ exchanger blockade and protection against myocardial infarction. Hypertension. 2007;49:1095-1103
274. Eder P, Molkentin JD. Trpc channels as effectors of cardiac hypertrophy. Circ Res. 2011;108:265-272 275. Bush EW, Hood DB, Papst PJ, Chapo JA, Minobe W, Bristow MR, Olson EN, McKinsey TA. Canonical
transient receptor potential channels promote cardiomyocyte hypertrophy through activation of calcineurin signaling. J Biol Chem. 2006;281:33487-33496
276. Wu X, Eder P, Chang B, Molkentin JD. Trpc channels are necessary mediators of pathologic cardiac hypertrophy. Proc Natl Acad Sci U S A. 2010;107:7000-7005
277. Koitabashi N, Aiba T, Hesketh GG, Rowell J, Zhang M, Takimoto E, Tomaselli GF, Kass DA. Cyclic gmp/pkg-dependent inhibition of trpc6 channel activity and expression negatively regulates cardiomyocyte nfat activation novel mechanism of cardiac stress modulation by pde5 inhibition. J Mol Cell Cardiol. 2010;48:713-724
278. Kinoshita H, Kuwahara K, Nishida M, Jian Z, Rong X, Kiyonaka S, Kuwabara Y, Kurose H, Inoue R, Mori Y, Li Y, Nakagawa Y, Usami S, Fujiwara M, Yamada Y, Minami T, Ueshima K, Nakao K. Inhibition of trpc6 channel activity contributes to the antihypertrophic effects of natriuretic peptides-guanylyl cyclase-a signaling in the heart. Circ Res. 2010;106:1849-1860
279. Nishida M, Watanabe K, Sato Y, Nakaya M, Kitajima N, Ide T, Inoue R, Kurose H. Phosphorylation of trpc6 channels at thr69 is required for anti-hypertrophic effects of phosphodiesterase 5 inhibition. J Biol Chem. 2010;285:13244-13253

References
280. Takimoto E, Koitabashi N, Hsu S, Ketner EA, Zhang M, Nagayama T, Bedja D, Gabrielson KL, Blanton R, Siderovski DP, Mendelsohn ME, Kass DA. Regulator of g protein signaling 2 mediates cardiac compensation to pressure overload and antihypertrophic effects of pde5 inhibition in mice. J Clin Invest. 2009;119:408-420
281. Tokudome T, Kishimoto I, Horio T, Arai Y, Schwenke DO, Hino J, Okano I, Kawano Y, Kohno M, Miyazato M, Nakao K, Kangawa K. Regulator of g-protein signaling subtype 4 mediates antihypertrophic effect of locally secreted natriuretic peptides in the heart. Circulation. 2008;117:2329-2339
282. Tikkanen I, Fyhrquist F, Metsarinne K, Leidenius R. Plasma atrial natriuretic peptide in cardiac disease and during infusion in healthy volunteers. Lancet. 1985;2:66-69
283. Burnett JC, Jr., Kao PC, Hu DC, Heser DW, Heublein D, Granger JP, Opgenorth TJ, Reeder GS. Atrial natriuretic peptide elevation in congestive heart failure in the human. Science. 1986;231:1145-1147
284. Sugawara A, Nakao K, Morii N, Yamada T, Itoh H, Shiono S, Saito Y, Mukoyama M, Arai H, Nishimura K, et al. Synthesis of atrial natriuretic polypeptide in human failing hearts. Evidence for altered processing of atrial natriuretic polypeptide precursor and augmented synthesis of beta-human anp. J Clin Invest. 1988;81:1962-1970
285. Drexler H, Finkh M, Hoing S, Toth M, Just H, Lang RE. Systemic and regional vascular effects of atrial natriuretic peptide in a rat model of chronic heart failure. Basic Res Cardiol. 1987;82:517-529
286. Riegger GA, Elsner D, Kromer EP, Daffner C, Forssmann WG, Muders F, Pascher EW, Kochsiek K. Atrial natriuretic peptide in congestive heart failure in the dog: Plasma levels, cyclic guanosine monophosphate, ultrastructure of atrial myoendocrine cells, and hemodynamic, hormonal, and renal effects. Circulation. 1988;77:398-406
287. Tsutamoto T, Kanamori T, Morigami N, Sugimoto Y, Yamaoka O, Kinoshita M. Possibility of downregulation of atrial natriuretic peptide receptor coupled to guanylate cyclase in peripheral vascular beds of patients with chronic severe heart failure. Circulation. 1993;87:70-75
288. Hirooka Y, Takeshita A, Imaizumi T, Suzuki S, Yoshida M, Ando S, Nakamura M. Attenuated forearm vasodilative response to intra-arterial atrial natriuretic peptide in patients with heart failure. Circulation. 1990;82:147-153
289. Cody RJ, Atlas SA, Laragh JH, Kubo SH, Covit AB, Ryman KS, Shaknovich A, Pondolfino K, Clark M, Camargo MJ, et al. Atrial natriuretic factor in normal subjects and heart failure patients. Plasma levels and renal, hormonal, and hemodynamic responses to peptide infusion. J Clin Invest. 1986;78:1362-1374
290. Garcia R, Bonhomme MC, Schiffrin EL. Divergent regulation of atrial natriuretic factor receptors in high-output heart failure. Am J Physiol. 1992;263:H1790-1797
291. Tsutamoto T, Kanamori T, Wada A, Kinoshita M. Uncoupling of atrial natriuretic peptide extraction and cyclic guanosine monophosphate production in the pulmonary circulation in patients with severe heart failure. J Am Coll Cardiol. 1992;20:541-546
292. Kuhn M, Voss M, Mitko D, Stypmann J, Schmid C, Kawaguchi N, Grabellus F, Baba HA. Left ventricular assist device support reverses altered cardiac expression and function of natriuretic peptides and receptors in end-stage heart failure. Cardiovasc Res. 2004;64:308-314
293. Andreassi MG, Del Ry S, Palmieri C, Clerico A, Biagini A, Giannessi D. Up-regulation of 'clearance' receptors in patients with chronic heart failure: A possible explanation for the resistance to biological effects of cardiac natriuretic hormones. Eur J Heart Fail. 2001;3:407-414
294. Bauersachs J, Bouloumie A, Mulsch A, Wiemer G, Fleming I, Busse R. Vasodilator dysfunction in aged spontaneously hypertensive rats: Changes in no synthase iii and soluble guanylyl cyclase expression, and in superoxide anion production. Cardiovasc Res. 1998;37:772-779
295. Crabos M, Coste P, Paccalin M, Tariosse L, Daret D, Besse P, Bonoron-Adele S. Reduced basal no-mediated dilation and decreased endothelial no-synthase expression in coronary vessels of spontaneously hypertensive rats. J Mol Cell Cardiol. 1997;29:55-65
296. Drexler H, Kastner S, Strobel A, Studer R, Brodde OE, Hasenfuss G. Expression, activity and functional significance of inducible nitric oxide synthase in the failing human heart. J Am Coll Cardiol. 1998;32:955-963
297. Heymes C, Vanderheyden M, Bronzwaer JG, Shah AM, Paulus WJ. Endomyocardial nitric oxide synthase and left ventricular preload reserve in dilated cardiomyopathy. Circulation. 1999;99:3009-3016
298. Wiemer G, Linz W, Hatrik S, Scholkens BA, Malinski T. Angiotensin-converting enzyme inhibition alters nitric oxide and superoxide release in normotensive and hypertensive rats. Hypertension. 1997;30:1183-1190

References
299. Miyagawa K, Emoto N, Widyantoro B, Nakayama K, Yagi K, Rikitake Y, Suzuki T, Hirata K. Attenuation of doxorubicin-induced cardiomyopathy by endothelin-converting enzyme-1 ablation through prevention of mitochondrial biogenesis impairment. Hypertension. 2010;55:738-746
300. Neilan TG, Blake SL, Ichinose F, Raher MJ, Buys ES, Jassal DS, Furutani E, Perez-Sanz TM, Graveline A, Janssens SP, Picard MH, Scherrer-Crosbie M, Bloch KD. Disruption of nitric oxide synthase 3 protects against the cardiac injury, dysfunction, and mortality induced by doxorubicin. Circulation. 2007;116:506-514
301. Fisher PW, Salloum F, Das A, Hyder H, Kukreja RC. Phosphodiesterase-5 inhibition with sildenafil attenuates cardiomyocyte apoptosis and left ventricular dysfunction in a chronic model of doxorubicin cardiotoxicity. Circulation. 2005;111:1601-1610
302. Koka S, Das A, Zhu SG, Durrant D, Xi L, Kukreja RC. Long-acting phosphodiesterase-5 inhibitor tadalafil attenuates doxorubicin-induced cardiomyopathy without interfering with chemotherapeutic effect. J Pharmacol Exp Ther. 2010;334:1023-1030
303. Zhu B, Vemavarapu L, Thompson WJ, Strada SJ. Suppression of cyclic gmp-specific phosphodiesterase 5 promotes apoptosis and inhibits growth in ht29 cells. J Cell Biochem. 2005;94:336-350
304. Black KL, Yin D, Ong JM, Hu J, Konda BM, Wang X, Ko MK, Bayan JA, Sacapano MR, Espinoza A, Irvin DK, Shu Y. Pde5 inhibitors enhance tumor permeability and efficacy of chemotherapy in a rat brain tumor model. Brain Res. 2008;1230:290-302
305. Das A, Durrant D, Mitchell C, Mayton E, Hoke NN, Salloum FN, Park MA, Qureshi I, Lee R, Dent P, Kukreja RC. Sildenafil increases chemotherapeutic efficacy of doxorubicin in prostate cancer and ameliorates cardiac dysfunction. Proc Natl Acad Sci U S A. 2010;107:18202-18207
306. McAllister-Lucas LM, Sonnenburg WK, Kadlecek A, Seger D, Trong HL, Colbran JL, Thomas MK, Walsh KA, Francis SH, Corbin JD, et al. The structure of a bovine lung cgmp-binding, cgmp-specific phosphodiesterase deduced from a cdna clone. J Biol Chem. 1993;268:22863-22873
307. Yuen PS, Doolittle LK, Garbers DL. Dominant negative mutants of nitric oxide-sensitive guanylyl cyclase. J Biol Chem. 1994;269:791-793
308. Yamamoto T, Yao Y, Harumi T, Suzuki N. Localization of the nitric oxide/cgmp signaling pathway-related genes and influences of morpholino knock-down of soluble guanylyl cyclase on medaka fish embryogenesis. Zoolog Sci. 2003;20:181-191
309. Gossen M, Bujard H. Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc Natl Acad Sci U S A. 1992;89:5547-5551
310. Fliegner D, Schubert C, Penkalla A, Witt H, Kararigas G, Dworatzek E, Staub E, Martus P, Ruiz Noppinger P, Kintscher U, Gustafsson JA, Regitz-Zagrosek V. Female sex and estrogen receptor-beta attenuate cardiac remodeling and apoptosis in pressure overload. Am J Physiol Regul Integr Comp Physiol. 2010;298:R1597-1606
311. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative pcr and the 2(-delta delta c(t)) method. Methods. 2001;25:402-408
312. Iwaki M, Ito S, Morioka M, Iwata S, Numaguchi Y, Ishii M, Kondo M, Kume H, Naruse K, Sokabe M, Hasegawa Y. Mechanical stretch enhances il-8 production in pulmonary microvascular endothelial cells. Biochem Biophys Res Commun. 2009;389:531-536
313. Chien S. Mechanotransduction and endothelial cell homeostasis: The wisdom of the cell. Am J Physiol Heart Circ Physiol. 2007;292:H1209-1224
314. Sipkema P, van der Linden PJ, Westerhof N, Yin FC. Effect of cyclic axial stretch of rat arteries on endothelial cytoskeletal morphology and vascular reactivity. J Biomech. 2003;36:653-659
315. Geiss GK, Bumgarner RE, Birditt B, Dahl T, Dowidar N, Dunaway DL, Fell HP, Ferree S, George RD, Grogan T, James JJ, Maysuria M, Mitton JD, Oliveri P, Osborn JL, Peng T, Ratcliffe AL, Webster PJ, Davidson EH, Hood L, Dimitrov K. Direct multiplexed measurement of gene expression with color-coded probe pairs. Nat Biotechnol. 2008;26:317-325
316. Lipshultz SE, Cohen H, Colan SD, Herman EH. The relevance of information generated by in vitro experimental models to clinical doxorubicin cardiotoxicity. Leuk Lymphoma. 2006;47:1454-1458
317. Miyata S, Takemura G, Kosai K, Takahashi T, Esaki M, Li L, Kanamori H, Maruyama R, Goto K, Tsujimoto A, Takeyama T, Kawaguchi T, Ohno T, Nishigaki K, Fujiwara T, Fujiwara H, Minatoguchi S. Anti-fas gene therapy prevents doxorubicin-induced acute cardiotoxicity through mechanisms independent of apoptosis. Am J Pathol. 2010;176:687-698
318. Lindman BR, Zajarias A, Madrazo JA, Shah J, Gage BF, Novak E, Johnson SN, Chakinala MM, Hohn TA, Saghir M, Mann DL. Effects of phosphodiesterase type 5 inhibition on systemic and pulmonary hemodynamics and ventricular function in patients with severe symptomatic aortic stenosis. Circulation. 2012;125:2353-2362

References
319. Jessup M, Greenberg B, Mancini D, Cappola T, Pauly DF, Jaski B, Yaroshinsky A, Zsebo KM, Dittrich H, Hajjar RJ. Calcium upregulation by percutaneous administration of gene therapy in cardiac disease (cupid): A phase 2 trial of intracoronary gene therapy of sarcoplasmic reticulum ca2+-atpase in patients with advanced heart failure. Circulation. 2011;124:304-313
320. Parker TG, Packer SE, Schneider MD. Peptide growth factors can provoke "fetal" contractile protein gene expression in rat cardiac myocytes. J Clin Invest. 1990;85:507-514
321. Hartong R, Wang N, Kurokawa R, Lazar MA, Glass CK, Apriletti JW, Dillmann WH. Delineation of three different thyroid hormone-response elements in promoter of rat sarcoplasmic reticulum ca2+atpase gene. Demonstration that retinoid x receptor binds 5' to thyroid hormone receptor in response element 1. J Biol Chem. 1994;269:13021-13029
322. Ju H, Scammel-La Fleur T, Dixon IM. Altered mrna abundance of calcium transport genes in cardiac myocytes induced by angiotensin ii. J Mol Cell Cardiol. 1996;28:1119-1128
323. Hartong R, Villarreal FJ, Giordano F, Hilal-Dandan R, McDonough PM, Dillmann WH. Phorbol myristate acetate-induced hypertrophy of neonatal rat cardiac myocytes is associated with decreased sarcoplasmic reticulum ca2+ atpase (serca2) gene expression and calcium reuptake. J Mol Cell Cardiol. 1996;28:2467-2477
324. Calderone A, Thaik CM, Takahashi N, Chang DL, Colucci WS. Nitric oxide, atrial natriuretic peptide, and cyclic gmp inhibit the growth-promoting effects of norepinephrine in cardiac myocytes and fibroblasts. J Clin Invest. 1998;101:812-818
325. Ho PD, Zechner DK, He H, Dillmann WH, Glembotski CC, McDonough PM. The raf-mek-erk cascade represents a common pathway for alteration of intracellular calcium by ras and protein kinase c in cardiac myocytes. J Biol Chem. 1998;273:21730-21735
326. Kohler R, Heyken WT, Heinau P, Schubert R, Si H, Kacik M, Busch C, Grgic I, Maier T, Hoyer J. Evidence for a functional role of endothelial transient receptor potential v4 in shear stress-induced vasodilatation. Arterioscler Thromb Vasc Biol. 2006;26:1495-1502
327. Liu CL, Huang Y, Ngai CY, Leung YK, Yao XQ. Trpc3 is involved in flow- and bradykinin-induced vasodilation in rat small mesenteric arteries. Acta Pharmacol Sin. 2006;27:981-990
328. Oancea E, Wolfe JT, Clapham DE. Functional trpm7 channels accumulate at the plasma membrane in response to fluid flow. Circ Res. 2006;98:245-253
329. Ngai CY, Yao X. Vascular responses to shear stress: The involvement of mechanosensors in endothelial cells. The Open Circulation and Vascular Journal. 2010;3:85-94
330. Kohler R, Brakemeier S, Kuhn M, Degenhardt C, Buhr H, Pries A, Hoyer J. Expression of ryanodine receptor type 3 and trp channels in endothelial cells: Comparison of in situ and cultured human endothelial cells. Cardiovasc Res. 2001;51:160-168
331. Lin CS, Chow S, Lau A, Tu R, Lue TF. Regulation of human pde5a2 intronic promoter by camp and cgmp: Identification of a critical sp1-binding site. Biochem Biophys Res Commun. 2001;280:693-699
332. Baudino TA, Carver W, Giles W, Borg TK. Cardiac fibroblasts: Friend or foe? Am J Physiol Heart Circ Physiol. 2006;291:H1015-1026
333. Huang C, Zhang X, Ramil JM, Rikka S, Kim L, Lee Y, Gude NA, Thistlethwaite PA, Sussman MA, Gottlieb RA, Gustafsson AB. Juvenile exposure to anthracyclines impairs cardiac progenitor cell function and vascularization resulting in greater susceptibility to stress-induced myocardial injury in adult mice. Circulation. 2010;121:675-683
334. Lewis GD, Lachmann J, Camuso J, Lepore JJ, Shin J, Martinovic ME, Systrom DM, Bloch KD, Semigran MJ. Sildenafil improves exercise hemodynamics and oxygen uptake in patients with systolic heart failure. Circulation. 2007;115:59-66
335. Giannetta E, Isidori AM, Galea N, Carbone I, Mandosi E, Vizza CD, Naro F, Morano S, Fedele F, Lenzi A. Chronic inhibition of cgmp phosphodiesterase 5a improves diabetic cardiomyopathy: A randomized, controlled clinical trial using magnetic resonance imaging with myocardial tagging. Circulation. 2012;125:2323-2333
336. Grimminger F, Weimann G, Frey R, Voswinckel R, Thamm M, Bolkow D, Weissmann N, Muck W, Unger S, Wensing G, Schermuly RT, Ghofrani HA. First acute haemodynamic study of soluble guanylate cyclase stimulator riociguat in pulmonary hypertension. Eur Respir J. 2009;33:785-792
337. Ghofrani HA, Hoeper MM, Halank M, Meyer FJ, Staehler G, Behr J, Ewert R, Weimann G, Grimminger F. Riociguat for chronic thromboembolic pulmonary hypertension and pulmonary arterial hypertension: A phase ii study. Eur Respir J. 2010;36:792-799

Curriculum Vitae
CURRICULUM VITAE
First name: Sara
Surname: Vandenwijngaert
Date of birth: Nov 8th, 1985
Nationality: Belgian
Education
PhD student in Biomedical Sciences September 2007 - March 2013
KU Leuven, Department of Cardiovascular Sciences, Division of Cardiology
Doctoral School programme: Emerging Concepts in Cardiovascular Medicine
Mentor: Prof. Dr. Stefan Janssens
Master in Biomedical Sciences 2003-2007
KU Leuven
Graduated magna cum laude
Master's thesis:
"Detection of cardiotoxicity after in utero exposure to doxorubicin"
KU Leuven, Department of Oncology (Division of Gynaecological Oncology) & Department
of Cardiovascular Sciences (Division of Cardiology)
Mentors: Prof. Dr. Frédéric Amant and Prof. Dr. Stefan Janssens
Secondary school 1997-2003
Latin-Sciences
PhD Fellowships
KU Leuven 2007-2008
Research Foundation Flanders (FWO) 2008-2012

Curriculum Vitae
List of Publications
Vandenwijngaert S, Pokreisz P, Hermans H, Gillijns H, Pellens M, Bax NAM, Coppiello
G, Oosterlinck W, Balogh A, Papp Z, Bouten CVC, Bartunek J, D'hooge J, Luttun A,
Verbeken E, Herregods MC, Herijgers P, Bloch KD, Janssens SP. Increased cardiac
myocyte PDE5 levels in human and murine pressure overload hypertrophy contribute to
adverse LV remodeling. Accepted for publication - PLOS ONE
Liu X, Claus P, Wu M, Reyns G, Verhamme P, Pokreisz P, Vandenwijngaert S, Dubois
C, Vanhaecke J, Verbeken E, Bogaert J, Janssens S. Placental growth factor increases
regional myocardial blood flow and contractile function in chronic myocardial ischemia.
Accepted for publication - AJP-Heart and Circulatory Physiology
Pokreisz P, Vandenwijngaert S, Bito V, Van den Bergh A, Lenaerts I, Busch C,
Marsboom G, Gheysens O, Vermeersch P, Biesmans L, Liu X, Gillijns H, Pellens M, Van
Lommel A, Buys E, Schoonjans L, Vanhaecke J, Verbeken E, Sipido K, Herijgers P, Bloch
KD, Janssens SP. Ventricular phosphodiesterase-5 expression is increased in patients
with advanced heart failure and contributes to adverse ventricular remodeling after
myocardial infarction in mice. Circulation. 2009 Jan 27;119(3):408-16
Dubois C, Liu X, Claus P, Marsboom G, Pokreisz P, Vandenwijngaert S, Dépelteau H,
Streb W, Chaothawee L, Maes F, Gheysens O, Debyser Z, Gillijns H, Pellens M,
Vandendriessche T, Chuah M, Collen D, Verbeken E, Belmans A, Van de Werf F, Bogaert
J, Janssens S. Differential effects of progenitor cell populations on left ventricular
remodeling and myocardial neovascularization after myocardial infarction. J Am Coll
Cardiol. 2010 May 18;55(20):2232-43
Presentations at International meetings
Heart Failure (European Society of Cardiology), 2009, Nice (France)
Poster presentation:
Myocardial phosphodiesterase 5 expression is increased in patients with severe aortic
stenosis and is associated with impaired functional and structural remodeling after aortic
constriction in mice

Curriculum Vitae
American Heart Association Scientific Sessions, 2009, Orlando (Florida, USA)
Poster presentation:
Myocardial phosphodiesterase 5 expression contributes to left ventricular dysfunction in
the pressure overloaded heart
Heart Failure (European Society of Cardiology), 2010, Berlin (Germany)
Oral presentation - Young Investigator Award Finalist:
Myocardial phosphodiesterase expression is increased in patients with severe aortic
stenosis and contributes to left ventricular dysfunction in pressure overloaded mice
CBCS Summer School on Cardiovascular Sciences “From Basic Mechanisms to Clinical
Application” (European Society of Cardiology), 2011, Sophia Antipolis (France)
Oral & poster presentation:
Myocardial phosphodiesterase 5 is increased in patients with severe aortic stenosis and
contributes to left ventricular dysfunction in pressure overloaded mice
American Heart Association Scientific Sessions, 2011, Orlando (Florida, USA)
Oral presentation:
Myocardial phosphodiesterase 5 is increased in patients with severe aortic stenosis and
contributes to left ventricular dysfunction in pressure overloaded mice
American Heart Association Scientific Sessions, 2012, Los Angeles (California, USA)
Poster presentation:
Cardiac soluble guanylate cyclase protects against the cardiac dysfunction induced by
chronic doxorubicin treatment in mice

Curriculum Vitae

Supplements
SUPPLEMENTS
Supplement 1. Sequence of the 3"UTR of hsa-PDE5A (NM_001083) inserted in the
lentiviral firefly luciferase vector
gcctggcgctccggccgctttgtcgaaagccggcccgactggagcaggacgaagggggagggtctcgaggccgagtcct
gttcttctgagggacggaccccagctggggtggaaaagcagtaccagagagcctccgaggcgcgcggtgccaaccatgg
agcgggccggccccagcttcgggcagcagcgacagcagcagcagccccagcagcagaagcagcagcagagggatca
ggactcggtcgaagcatggctggacgatcactgggactttaccttctcatactttgttagaaaagccaccagagaaatggtca
atgcatggtttgctgagagagttcacaccatccctgtgtgcaaggaaggtatcagaggccacaccgaatcttgctcttgtccctt
gcagcagagtcctcgtgcagataacagtgcccctggaacaccaaccaggaaaatctctgcctctgaatttgaccggcctctta
gacccattgttgtcaaggattctgagggaactgtgagcttcctctctgactcagaaaagaaggaacagatgcctctaacccctc
caaggtttgatcatgatgaaggggaccagtgctcaagactcttggaattagtgaaggatatttctagtcatttggatgtcacagc
cttatgtcacaaaattttcttgcatatccatggactgatatctgctgaccgctattccctgttccttgtctgtgaagacagctccaatg
acaagtttcttatcagccgcctctttgatgttgctgaaggttcaacactggaagaagtttcaaataactgtatccgcttagaatgga
acaaaggcattgtgggacatgtggcagcgcttggtgagcccttgaacatcaaagatgcatatgaggatcctcggttcaatgca
gaagttgaccaaattacaggctacaagacacaaagcattctttgtatgccaattaagaatcatagggaagaggttgttggtgta
gcccaggccatcaacaagaaatcaggaaacggtgggacatttactgaaaaagatgaaaaggactttgctgcttatttggcat
tttgtggtattgttcttcataatgctcagctctatgagacttcactgctggagaacaagagaaatcaggtgctgcttgaccttgctag
tttaatttttgaagaacaacaatcattagaagtaattttgaagaaaatagctgccactattatctctttcatgcaagtgcagaaatg
caccattttcatagtggatgaagattgctccgattctttttctagtgtgtttcacatggagtgtgaggaattagaaaaatcatctgata
cattaacaagggaacatgatgcaaacaaaatcaattacatgtatgctcagtatgtcaaaaatactatggaaccacttaatatcc
cagatgtcagtaaggataaaagatttccctggacaactgaaaatacaggaaatgtaaaccagcagtgcattagaagtttgctt
tgtacacctataaaaaatggaaagaagaataaagttataggggtttgccaacttgttaataagatggaggagaatactggca
aggttaagcctttcaaccgaaatgacgaacagtttctggaagcttttgtcatcttttgtggcttggggatccagaacacgcagatg
tatgaagcagtggagagagccatggccaagcaaatggtcacattggaggttctgtcgtatcatgcttcagcagcagaggaa
gaaacaagagagctacagtcgttagcggctgctgtggtgccatctgcccagacccttaaaattactgactttagcttcagtgact
ttgagctgtctgatctggaaacagcactgtgtacaattcggatgtttactgacctcaaccttgtgcagaacttccagatgaaacat
gaggttctttgcagatggattttaagtgttaagaagaattatcggaagaatgttgcctatcataattggagacatgcctttaataca
gctcagtgcatgtttgctgctctaaaagcaggcaaaattcagaacaagctgactgacctggagatacttgcattgctgattgctg
cactaagccacgatttggatcaccgtggtgtgaataactcttacatacagcgaagtgaacatccacttgcccagctttactgcc
attcaatcatggaacaccatcattttgaccagtgcctgatgattcttaatagtccaggcaatcagattctcagtggcctctccattg
aagaatataagaccacgttgaaaataatcaagcaagctattttagctacagacctagcactgtacattaagaggcgaggag
aattttttgaacttataagaaaaaatcaattcaatttggaagatcctcatcaaaaggagttgtttttggcaatgctgatgacagcttg

Supplements
tgatctttctgcaattacaaaaccctggcctattcaacaacggatagcagaacttgtagcaactgaattttttgatcaaggagac
agagagagaaaagaactcaacatagaacccactgatctaatgaacagggagaagaaaaacaaaatcccaagtatgca
agttgggttcatagatgccatctgcttgcaactgtatgaggccctgacccacgtgtcagaggactgtttccctttgctagatggctg
cagaaagaacaggcagaaatggcaggcccttgcagaacagcaggagaagatgctgattaatggggaaagcggccagg
ccaagcggaactgagtggcctatttcatgcagagttgaagtttacagagatggtgtgttctgcaatatgcctagtttcttacacact
gtctgtatagtgtctgtatttggtatatactttgccactgctgtatttttatttttgcacaacttttgagagtatagcatgaatgtttttagag
gactattacatattttttgtatatttgttttatgctactgaactgaaaggatcaacaacatccactgttagcacattgataaaagcatt
gtttgtgatatttcgtgtactgcaaagtgtatgcagtattcttgcactgaggtttttttgcttggggattattttaaataattggtttttgtgttt
tctgaattaccattttttcaagaatgtttggaatctttcctttttcaaaagtaggttaggagcaaattatcatacattctgtgacatttaa
agcctttataggatagtgaaaaatgctggctgagtggattttaagagaaataattgtatttgttaacagtgtctttttttaaaaagtta
aggcactctgaaacaaatggaaagtcctatgaaactgtattgtaaagaaaacattatttaattgatatgctgttttgtgagagaa
caggcaagacagaactttgtcacttcagtgcagtacatttttctgaaagctacccataaaatcactttcatctcacctacctgatg
caaagcaggtgaaaccttaggagatgatccagtcactgacttgattgagggataagtgtgatttagaaatggaatggccttgg
atgtctatcagtgaagaaaaatgttctgttagaagatctctctaagagtttttttccttctgagcttccttttcaaaataaaagtgaca
attgtagcattgacttgaagtgagacatggttatagataagagagtacaaaatgactctttttcctgtcaattgaaatttaaagaa
aagttttaattatataaatagcaaagggctattgccaatactagggtcaaaaatgaatttgagggaacagtgggtaagaaactt
tatgcctgaataacatttagcagtattgtgattgaaaaattgccatattttgatgtataggacaagtcaactgagatccagagaat
cctggatgtgaatgctaaacactggcccttaactcacattcaatgtattttcttcccataacatttagtatagttaatattttcttagaat
ttgagcccatttaagtggattaatattctacatgtgtgcccctaaagacacatttactcaatattggagaagtagataatgaattaa
gcaactggtctaggaaaggaaaatttgtttcaaatatgcaggaatgtttggatttggggagagtagaaggagagatttgcttgat
ttgttaacttctacctccaacccacaaaaaagatatttgatctgagtttctatcactaatttggatagaaaatttctaagggacatgg
taatccagcattctcaaggacctttcgccaaaatgtgttttccatctatgtcccgattcccctaaattttgcctaaaattcagtatgttc
cttaagtttttaaaattctgagtgtgtacaaatatcttgacataatgcagttttatttttatcattctggtaaaaaaacaaaaaataga
agcaaaacacattgtattgccattattttgtatttggtaaaggttaatctaggaagttaccaactgtttaatgctatatgtattgtatact
tgtattttcaggatattttattttttttgccatacagataaaatttgtaaggttgcccctttgtggcactggtgtgtaaaatacacagact
atcactaaaataatagttatatatacatacaggtgtatacttatgcatgcatacataaatccttagtatagaaaaattgcataaag
aatagcaatctttaataaacctttttattacattgtgatttagcagttatgctaaaatatgtacttatgctttagtagtttgtttggtcccct
ctagtatgtgtcactgagaaattttttaaagacatggtagatcgtgtttagaggctttgtatgtgtgtcattttaataagcaagaagat
atatttagattagaaatggtttggtctgcctttgaatattgtttattttactttactagttgagacattaaaggaagctgggcaatgcct
attttatttctttgttggatattttagttcatacaaagcagagtacttctttagggctggttaattggttcaaataatttttaatttcctttctag
tatcttctcaagttggaaaaatatacatacagtcctccttcaccttactctgtatttatattacccataactagcaagaagttcttgttct
agattttttgtttgtttagttataacagagtaacataccatttaattacaatttttagccagaaaagtccccactattttactaacttgtta
aaagatatctatataattgcctggccttatatttttcagtagattagaccgtgccaatcacaatcctgggtggatttgtgtaagtcact

Supplements
taacctctgtgtgcctaaacaagttgtgcttttttaaaaggagttatgtttgggcaaagcctttgtcttcaagcagaatgtcacaga
aggcagctactttataagccccaatgggccatggagaccactgtcagaaatgggatattagtctagagagaaggtgatctatt
cccacatgtcatttctaatgttgagtttccatgactgaacaaagagaatatatttattcagcttcacttgcagatcactagtgaatgt
gagatttagagctcattgagtatattgcttcaaggtacaaacccaggatgatgatgttgtcaccactgtctcttaattttgaataata
gtttcctttaataggagtattagagataagaaagtatatgaaaatatactggaaatattggattcttggagaaaactgttcagtca
cagatatattcttgcctagcagtgaagtgcctttattttcagcatagcaaataaatattagacctgttccaatttgatctacaattttttt
ctgtgtttttcaccagattgtactcctaaaacttaacaggccatcacaagcaattgtcttttgtttacaagattgatttaatatgagag
gatacaaaatgtcatcgttatcctctcttatgaacaactgtagtcaaaataaggtggcacaatttaattgttttgtatcagaaatac
actgacccaccttttattgagtcctgccacatgttaggtaccgtgctctgctgtggagacagagcagtgaccccaaggagctca
cggtccctgaaggaggtgctagagaagagacttagcttctgatactgccaatttaatgtgagaacatggggtatactgcatcat
ttccattttcatcaataacatatgttttatgcaccttctttacctgaaacttactaagaatctaccagtaaacaaacatcctgtctttttg
caagtatgaatcacttaacctgctgatagttgaagaacactttaggagttttgtattcttgtatatagtttattttttccatgtgctagcc
aggtaaagattacacagttcttctggactgttaaattgtgcatggttttggaccccttctgctctactacagagagtgaagaagaa
agtattaaagctcactttaccattccatatacttactaaaagcctgtgtaaacatgtcttaatgaatgttgttgaaagcaatgtaaat
agttgaaaatataaatttatattacagtttaagaaaaccttatgaggcatcactagccactgttaatatctatttgtattcttataccttt
tcaatatatttgaacaaatatagtttctggcactatttttatactaggaaaaggagttactatgtatattatgcttagcttttaaggcatt
ttaaataaccatgaatgttgatttcattactttcctttcctccatcacgagagtcatttcagatgactctttcatgacaaaatcacttta
aaggaacacttacctccgattcctgtataaagtcatgagatggtcaaggtggttttccattgtgcaaattcttcacctgtcagtggtt
tcctcattttgccatgctttgtaaaaataaaaagaatgatcaagtaggtatgaa

Supplements
Supplement 2. Sequence of the 3"UTR of mmu-PDE5A (AK031275) inserted in the firefly
luciferase reporter plasmid cggtgagaggtgtgagtgtgtgtgtgtgctcgtgtgcatgcatcagtaacaattaaggaaatgggggcatggaagaagttggc
gaggcaagagggtggaagggaaaggagataactacagtactcatttgtgaaattctcaacaaaatattttttgaagaggaaa
aaaaatgatgtgaatttgagaacactgtcttcagctaactatggtgttgtttgacctgagggttcttgagtgtcatcaggaaccccg
tgttctcccctcacagaaccacccggcagccttcccctgagctggtgcacaggaagggtccctctgtgttctagtgttctcattgt
actttccatacatgtcggacaaacattcatcctggaatgggaacactgacttctctaagccctgtaccctgcacttttcctgctggt
aaagcaaatctagttttatacgcttcacacaggagtgcccaggtgtaaattcaggatggagtataggaattcatcttccaagag
aacagcaaccagcgttatcttactgttcccctccaagtcttaagaaaaacattccaccttccacactggcaagattcccctcctg
ctcatccttaggtctgcctctgccttcattctccagagaccatcgagttttatatattgaatatgtattgtacatctgtcatagcacaca
gcatttccattgctccaagccgttgctggtgtgttgttcctctgaaaaatagttggtatttaattcaaacaaggcgattacttgtgttttt
gttatatagcagtattgacacctatgctagcccctatgtatgttgtaatccaaaccataggaagtgcctttgaaagtcagagtagc
atacttctatgttattaattacacctggatacttgatatcaaagcataattattagataaactttatgtccactattcttatagtccactg
gaataaccctactcttaatagatatttagaactgtatcacatcttgacaaaatatatccataggtgtggataaatcataccatttaa
agattgctttaatcactaacttaaccatgagcacatcaccct