structure-reactivity probes for active site shapes of cholesterol esterase by carbamate inhibitors
TRANSCRIPT

Structure-reactivity probes for active site shapes of cholesterol esterase bycarbamate inhibitors
Gialih Lin *, Chuen-Tzwu Shieh, Yi-Chin Tsai, Ching-Ing Hwang, Chun-Ping Lu,Gan-Hong Chen
Department of Chemistry and Institute of Biochemistry, National Chung-Hsing University, Taichung 402, Taiwan
Received 8 January 1999; received in revised form 8 March 1999; accepted 19 March 1999
Abstract
4,4P-Biphenyl-di-N-butylcarbamate (1), (S)-1,1P-bi-2-naphthyl-2,2P-di-N-butylcarbamate (S-2), (S)-1,1P-bi-2-naphthyl-2-N-butylcarbamate-2P-butyrate (S-3), 2,2P-biphenyl-di-N-butylcarbamate (4), 2,2P-biphenyl-2-N-octadecylcarbamate-2P-N-oc-tylcarbamate (5), 2,2P-biphenyl-2-N-octadecylcarbamate-2P-N-phenylcarbamate (6), 2,2P-biphenyl-2-N-butylcarbamate-2P-butyrate (7), 2,2P-biphenyl-2-N-butylcarbamate-2P-ol (8), 2,2P-biphenyl-2-N-octylcarbamate-2P-ol (9), (R)-1,1P-bi-2-N-naphthyl-2-butylcarbamate-2P-ol (R-10), and glyceryl-1,2,3-tri-N-butylcarbamate (11) are prepared and evaluated for theirinhibition effects on porcine pancreatic cholesterol esterase. All inhibitors are irreversible inhibitors of the enzyme.Carbamates 1^3 and 7^10 are the first alkyl chain and esteratic binding site-directed irreversible inhibitors due to the fact thatthe reactivity of the enzyme is protected by the irreversible inhibitor, trifluoroacetophenone in the presence of thesecarbamates. Carbamate 1 is the least potent inhibitor for the enzyme probably due to the fact that the inhibitor moleculeadopts a linear conformation and one of the carbamyl groups of the inhibitor molecule covalently interacts with the firstalkyl chain binding site of the enzyme while the other carbamyl group of the inhibitor molecule exposes outside the activesite. With near orthogonal conformations at the pivot bond of biaryl groups, one carbamyl group of carbamates S-2, S-3,and R-10 covalently binds to the first alkyl chain binding site of the enzyme while the other carbamyl, butyryl, or hydroxygroup can not bind covalently to the second alkyl chain binding site probably due to the orthogonal conformations.Carbamates 4^9 and 11 are very potent inhibitors for the enzyme probably due to the fact that all these molecules freelyrotate at the pivot bond of the biphenyl or glyceryl group and therefore can fit well into the bent-shaped first and secondalkyl chains binding sites of the enzyme. Although, carbamates 4^6 and 11 are irreversible inhibitors of cholesterol esterase,the enzyme is not protected but further inhibited by trifluoroacetophenone in the presence of these carbamates. Therefore,carbamates 4^6 and 11 covalently bind to the first alkyl chain binding site of the enzyme by one of the carbamyl groups andmay also bind to the second alkyl chain binding site of the enzyme by the second carbamyl group. Besides the bent-shapedconformation, the inhibition by carbamate 6 is probably assisted by a favorable Z-Z interaction between Phe 324 at thesecond alkyl chain binding site of the enzyme and the phenyl group of the inhibitor molecule. For cholesterol esterase,carbamates 8^10 are more potent than carbamates S-2, 4, and 5 probably due to the fact that the inhibitor molecules interact
0167-4838 / 99 / $ ^ see front matter ß 1999 Published by Elsevier Science B.V. All rights reserved.PII: S 0 1 6 7 - 4 8 3 8 ( 9 9 ) 0 0 0 7 3 - 4
Abbreviations: ACS, alkyl chain binding site; CEase, pancreatic cholesterol esterase; CRL, Candida rugosa lipase; EA, elementalanalysis ; ES, esteratic site; HRMS, high resolution mass spectra; kapp, the ¢rst-order rate constants; ki, bimolecular rate constant; LHIS,leaving group hydrophilic binding site; LHOS, leaving group hydrophobic binding site; MPLC, medium pressure liquid chromatography;MS, mass spectra; OH, oxyanion hole; PNPB, p-nitrophenyl butyrate; PPL, porcine pancreatic lipase; PSL, Pseudomona species lipase;SACS, the second alkyl chain or group binding site; TFA, tri£uoroacetophenone; TLC, thin layer chromatography
* Corresponding author. Fax: 886-4-286-2547.
BBAPRO 35905 6-5-99
Biochimica et Biophysica Acta 1431 (1999) 500^511
www.elsevier.com/locate/bba

with the second alkyl chain binding site of the enzyme through a hydrogen bond between the phenol hydroxy group of theinhibitor molecules and the His 435 residue in that site. ß 1999 Published by Elsevier Science B.V. All rights reserved.
Keywords: Cholesterol esterase; Carbamate inhibitor
1. Introduction
Pancreatic cholesterol esterase (CEase, EC3.1.1.13) has been intensely studied recently due tocorrelation between absorption of dietary cholesteroland the enzyme activity [1,2]. Cholesteryl esters, ret-inyl esters, acylglycerols, vitamin esters, and phos-pholipids are natural substrates of CEase [3^6].Although its role in cholesterol absorption in partic-ular is controversial, CEase plays a major role indigestive lipid absorption in the upper intestinal tract[1,2,7]. It has been demonstrated that CEase is in-volved directly in lipoprotein metabolism, in that theenzyme catalyzes the conversion of large LDL tosmaller, denser, more cholesteryl ester-rich lipopro-teins, and that the enzyme may also regulate serumcholesterol levels [8]. CEase has also been reported tofunction in these roles by acting as a cholesteroltransfer protein [9]. Serine proteases, serine lipases,and CEase share the same catalytic mechanism asserine proteases [10,11] in that they have a Ser-His-Asp (Glu) catalytic triad. The conservation of thiscatalytic triad suggests that serine proteases, serinelipase, and CEase share a common mechanism forsubstrate hydrolysis, that is, formation of a discreteacyl enzyme intermediate via the active site serinehydroxy group. Serine protease, serine lipases, andCEase may also be expected to be inhibited by thesame classes of mechanism-based inhibitors. It hasbeen demonstrated that diethyl-p-nitrophenol phos-phate [12], hexadecylsulfonyl £uoride [13], £uoroke-tones [14,15], borinic acid [16], chloroisocoumarin[17], L-lactones [18,19], L-lactams [20], and carba-mates [21^26] are mechanism-based inhibitors ofthese enzymes. The CEase-catalyzed hydrolysis oflipid substrates via a serine protease mechanism hasbeen proposed by Quinn [27,28]. Meanwhile, CEasehas also been used in resolution of binaphthols, spi-robiindanols [29,30], and inositol derivatives [31].
The three-dimensional structure of Candida rugosalipase (CRL), a homologue of CEase, has been re-ported [32]. The structures of human and rat pancre-
atic CEase have been generated by molecular model-ing [33]. Two di¡erent crystal structures of bovinepancreatic CEase have been reported recently[34,35]. Although di¡erent activation mechanismsfor CEase have been proposed, the shape of the ac-tive site is similar to that of CRL. CEase, like otherserine lipases, possesses a Ser 194 (209)1-His 435(449)-Asp 320/Glu (341) active site triad that is in-volved in nucleophilic and general acid-base cataly-sis, and a neighboring oxyanion hole, the H-bondingpeptide NH functions of Gly 107 (123), Ala 108/Gly(124), and Ala 195 (210), that stabilizes the incipientcarbonyl C-03 of the ester function during turnover.Therefore, the active site of CEase like CRL mayconsist of at least ¢ve major binding sites (Fig. 1):(a) an alkyl chain binding site (ACS) that binds tothe acyl chain of the substrate, (b) an oxyanion hole(OH) that stabilizes the tetrahedral intermediate, (c)an esteratic site (ES), comprised of the active siteserine which would attack the re face of the estercarbonyl, (d) a leaving group hydrophobic bindingsite (LHOS) or/and the second alkyl chain or groupbinding site (SACS) that binds to the cholesterol partof cholesterol ester or the second fatty acid chain oftriacylglyceride and is in a crevice above the catalyticsite and only the ¢rst ¢ve carbon atoms of fatty acidchain are well ordered, and (e) a leaving group hy-drophilic binding site (LHIS) that binds to the hy-drophilic part of the leaving group and is located atthe opposite direction of ACS. Inhibition of CRLwith sulfonyl chloride:enzyme ratios of 10:1 andgreater results in a second site of covalent modi¢ca-tion at His (449) [32]. In addition to sulfonyl chlorideinhibitor bound to Ser (209) of the ES-ACS of CRL,a second inhibitor molecule has covalently modi¢edHis (449) of the SACS of CRL and has also induceda conformational change for the enzyme at SACS(the modi¢ed His side chain is rotated away from
1 The numbering system is the residue number for CEase andthat between the parentheses is for CRL.
BBAPRO 35905 6-5-99
G. Lin et al. / Biochimica et Biophysica Acta 1431 (1999) 500^511 501

its position in the intact triad) [32]. The two inhibitormolecules bind in a head to head arrangement andare bridged by two water molecules which hydrogenbond to one oxygen atom from each inhibitor. Onlythe ¢rst ¢ve carbons of this second alkyl chain arewell ordered. These carbon atoms fold between andmake extensive van der Waals contacts with thephenyl rings of Phe (344) and Phe (448). Therefore,the overall shape of three alkyl chains binding siteslooks like the Y letter (Fig. 1) and the bending anglebetween ACS and SACS is somewhere between 90³and 180³. That the blocking ACS or SACS of PPL
by a carbamate inhibitor causes the enhancement ofenantioselectivity for the resolution of 1-indanolstrongly supports two binding sites for the alkylgroups in PPL [36]. With di¡erent conformationsand bending angles at the pivot bond of biarylcarba-mates, carbamates 1^11 are therefore suitable for thisstudy (Fig. 2). Atropisomeric carbamate inhibitorssuch as compounds S-2, S-3, and R-10 with a bend-ing angle around 90³ at the pivot bond of binaphthylgroup (Fig. 3A) but without the ability to freely ro-tate the pivot bond to racemize at room temperaturehave been reported to show di¡erent stereoselectiv-
Fig. 1. Interactions between substrates and CEase. A: Cholesteryl ester and CEase. B: Triacylglyceride and CEase.
BBAPRO 35905 6-5-99
G. Lin et al. / Biochimica et Biophysica Acta 1431 (1999) 500^511502

ities for CEase [24,26]. In this paper, we thereforedesign a similar type of inhibitors 2,2P-biphenylcar-bamates (4^9) which can freely rotate at the pivotbond to racemize at room temperature (Fig. 3B).In other words, all these compounds can adopt anyV-shaped conformations with the bending angle be-tween 90³ and 180³, which are similar to manualdocking of cholesteryl linoleate into the active siteof CEase [35]. Meanwhile, we proposed that an `ex-cess' of carbamate inhibitor such as carbamates 4^6may covalently bind to Ser 194 of the ES-ACS ofCEase by one of the carbamyl group and may alsocovalently interact with His 435 of the SACS of theenzyme by the second carbamyl group. Unlike car-bamates 2^10, carbamate 1 adopts a linear confor-mation for two carbamyl groups (Fig. 3C) and cannot dock into the active site of CEase well. Carba-mate 11 is an analogue for the natural substrate tri-acylglyceride and therefore may also bind to the en-
zyme at both ACS and SACS by any two of thethree carbamyl groups.
In the presence of substrate, the mechanism forinactivation of active site-directed irreversible carba-mate inhibitors of CEase has been proposed inScheme 1 [21,22].
Since the inhibition follows ¢rst-order kineticsover the observed time period, the rate of hydrolysisof EIP must be signi¢cantly slower than the rate of
Scheme 1. Kinetic scheme for inactivation of CEase in the pres-ence of substrate.
Fig. 2. Structures of carbamates 1^11.
BBAPRO 35905 6-5-99
G. Lin et al. / Biochimica et Biophysica Acta 1431 (1999) 500^511 503

formation of EIP(k2Ek3) [37]. Therefore, values ofKi and k2 can be calculated from Eq. 1 [21,38]:
kapp � k2�I�Ki 1� �S�
Km
� �� �I�
: �1�
In Eq. 1, kapp values are the ¢rst-order rate constantswhich can be obtained according to Hosie's method[21]. Bimolecular rate constant, ki = k2/Ki, is relatedto overall inhibitory potency.
2. Materials and methods
2.1. Materials
CEase from porcine pancreas and PNPB were ob-tained from Sigma; other chemicals were obtainedfrom Aldrich. Silica gel used in liquid chromatogra-phy (Licorpre Silica 60, 200^400 mesh), MPLC col-
umn (LiChroprep Si 60) and TLC plates (Kieselgel60 F254) were obtained from Merck. Elutants werehexane-ethyl acetate gradient and detection was withan UV lamp as well as an UV detector (Linear UV-160 or ISCO UA-6). Other chemicals and biochem-icals were of the highest quality available commer-cially.
2.2. Instrumental methods
1H and 13C NMR spectra were recorded at 300and 75.4 MHz (Varian-VXR 300 spectrometer), re-spectively. The 1H chemical shift was referred to in-ternal Me4Si. UV spectra were recorded on a spec-trophotometer (Hewlett Packard 8452A or BeckmanDU-650) UV with a cell holder circulated with awater bath (HOTECH 631-D). MS and HRMSwere recorded at 70 eV on a Joel JMS-SX/SX-102Amass spectrophotometer. EA (elemental analysis)was performed on a Heraeus instrument. IR spectra
Fig. 3. Three possible conformations of carbamates 1^10. A: The rigidly V-shaped (orthogonal) conformation of carbamates 2, 3, and10. The pivot bond angles 93^106³ are measured from the stable conformation after MM-2 energy minimization by CSC Chem 3Dplus1. B: The £exibly V-shaped conformation of carbamates 4^9. The free rotation at the pivot bond makes these compounds adoptthe bent V-shaped conformation easily. C: The linear conformation of carbamate 1.
BBAPRO 35905 6-5-99
G. Lin et al. / Biochimica et Biophysica Acta 1431 (1999) 500^511504

were recorded on a Hitachi 270-30 spectrophotome-ter. The MM-2 calculation for the bending angle ofthe pivot bond of biaryl group was performed on apersonal computer by the software CSC Chem 3Dplus1.
2.3. Synthesis of carbamates 1^11
Dicarbamates 1, S-2, 4, and 11 were preparedfrom the condensation of the corresponding alcoholwith 2.4 or 3.6 equivalents of butyl isocyanate in thepresence of a catalytic amount of pyridine in di-chloromethane at 25³C for 48 h (86^90% yield).Monocarbamates 8, 9, and R-10 were preparedfrom the condensation of the corresponding alcoholwith 1.2 equivalent of the corresponding isocyanatein the presence of a catalytic amount of pyridine indichloromethane at 25³C for 48 h (84^92% yield).Carbamates 5 and 6 were prepared from the conden-sation of the corresponding monocarbamate with 1.2equivalent of the corresponding isocyanate in thepresence of a catalytic amount of pyridine in di-chloromethane at 25³C for 48 h (62^79% yield). Car-bamates S-3 and 7 were prepared from the conden-sation of the corresponding monocarbamate with 1.2equivalent of butyryl chloride in the presence of acatalytic amount of pyridine in dichloromethane at25³C for 48 h (65^80% yield). Carbamates 1^11 werepuri¢ed (up to 99.9%) and characterized by 1H and13C NMR in IR spectra, MS, HRMS, and EA.
S-1,1P-bi-2-naphthyl-2,2P-di-N-butylcarbamate (S-2), S-1,1P-bi-2-naphthyl-2-N-butylcarbamate-2P-buty-rate (S-3), and R-1,1P-bi-2-naphthyl-2-N-butylcarba-mate-2P-ol (R-10) were characterized previously [26].
4,4P-Biphenyl-di-N-butylcarbamate (1) : IR (KBr)3460 (NH) and 1724 (C = O) cm31 ; 1H NMR(CDCl3, 300 MHz) N/ppm 0.96 (t, J = 7 Hz, 6H, g-CH3), 1.41 (sextet, J = 7 Hz, 4H, Q-CH2), 1.58 (m,4H, L-CH2), 3.26 and 3.32 (ABq, J = 7 Hz, 4H, K-CH2), 5.02 (t, J = 4 Hz, 2H, NH), 7.17^7.54 (m, 8H,biphenyl-H); 13C NMR (CDCl3, 75.4 MHz) N/ppm13.66 (g-C), 19.84 (Q-C), 31.84 (L-C), 40.94 (K-C),121.88 (biphenyl C-3), 127.70 (biphenyl C-2),137.70 (biphenyl C-1), 150.52 (biphenyl C-4),154.62 (C = O); MS (EI): m/z (%) 384 (0.67), 285(10), 186 (100); EA: Anal. Calcd. for C22H28O4N2 :C, 68.71; H, 7.34; N, 7.29. Found C, 68.58; H, 7.31,N, 7.31.
2,2P-Biphenyl-di-N-butylcarbamate (4) : IR (KBr)3356 (NH) and 1714 (C = O) cm31 ; 1H NMR(CDCl3, 300 MHz) N/ppm 0.88 (t, J = 7 Hz, 6H, g-CH3), 1.24 (sextet, J = 7 Hz, 4H, Q-CH2), 1.37 (m,4H, L-CH2), 3.06 and 3.12 (ABq, J = 7 Hz, 4H, K-CH2), 5.39 (br, 2H, NH), 7.22^7.40 (m, 8H, biphen-yl-H) ; 13C NMR (CDCl3, 75.4 MHz) N/ppm 13.61(g-C), 19.55 (Q-C), 31.58 (L-C), 40.61 (K-C), 122.91,125.44, 128.65, 130.68, and 130.99 (biphenyl C-1 andC-3^C-6), 148.52 (biphenyl C-2), 156.71 (C = O); MS(EI): m/z (%) 384 (0.61), 285 (42), 186 (100); EA:Anal. Calcd. for C22H28O4N2 : C, 68.71; H, 7.34; N,7.29. Found C, 68.85; H, 7.32, N, 7.31.
2,2P-Biphenyl-2-N-octadecylcarbamate-2P-N-octyl-carbamate (5) : 1H NMR (CDCl3, 300 MHz) N/ppm0.88 and 0.89 (t, J = 7 Hz, 6H, g-CH3), 1.25^1.80(m, 44H, L to g-1-CH2), 3.10 (q, 3JHÿCÿCÿH =3JHÿCÿNÿH = 7 Hz, 4H, K-CH2), 5.38 (q, J = 6 Hz,2H, NH), 7.25^7.29 (m, 8H, biphenyl-H) ; 13CNMR (CDCl3, 75.4 MHz) N/ppm 13.69 and 14.07(g-C) ; 19.61, 22.62, 26.51, 29.18, 29.59, 31.64, and31.78 (L to g-1-C) ; 40.64 and 40.98 (K-C) ; 122.88,125.38, 128.59, 130.60, 130.67, and 130.94 (biphenylC-1 and C-3^C-6), 148.45 (biphenyl C-2), 154.99(C = O); HRMS: Calcd. for C40H64O4N2 636.4866.Found 636.4885; EA: Anal. Calcd. for C44H64O4N2 :C, 75.41; H, 10.13; N, 4.40. Found C, 75.51; H,10.01, N, 4.49.
2,2P-Biphenyl-2-N-octadecylcarbamate-2P-N-phen-ylcarbamate (6): 1H NMR (CDCl3, 300 MHz) N/ppm0.88 (t, J = 7 Hz, 3H, g-CH3), 1.26 (m, 30H,Q to g-1-CH2), 1.42 (m, 2H, L-CH2), 3.17 (q,3JHÿCÿCÿH = 3JHÿCÿNÿH = 6 Hz, 2H, K-CH2), 5.23(m, 2H, NH), 7.04^7.29 (m, 13H, phenyl and biphen-yl-H) ; 13C NMR (CDCl3, 75.4 MHz) N/ppm 14.10(g-C) ; 22.66, 26.53, 29.23, 29.34, 29.68, and 31.90(L to g-1-C) ; 41.13 (K-C) ; 118.55, 122.60, 123.00,123.41, 125.59, 125.79, 128.79, 128.88, 128.98,131.03, 131.15, 136.12, 137.57, 147.92, 148.33,149.55 (biphenyl and phenyl), 155.14 (C = O);HRMS: Calcd. for C38H52O4N2 600.3927. Found600.3939; EA: Anal. Calcd. for C38H52O4N2 : C,75.95; H, 8.73; N, 4.66. Found C, 76.07; H, 8.62,N, 4.79.
2,2P-Biphenyl-2-N-butylcarbamate-2P-butyrate (7) :1H NMR (CDCl3, 300 MHz) N/ppm 0.86 (m, 6H,g-CH3), 1.22 (m, 4H, Q-CH2 of N-butylcarbamateand L-CH2 of butyryl), 1.58 (m, 2H, L-CH2 of N-
BBAPRO 35905 6-5-99
G. Lin et al. / Biochimica et Biophysica Acta 1431 (1999) 500^511 505

butylcarbamate), 2.34 (t, 7.2 Hz, 2H, K-CH2 of bu-tyryl), 3.02 (m, 2H, K-CH2 of N-butylcarbamate),5.05 (m, 1H, NH), 7.11^7.40 (m, 8H, biphenyl-H) ;13C NMR (CDCl3, 75.4 MHz) N/ppm 13.37 and13.56 (g-C), 17.99 and 19.45 (Q-C of N-butylcarba-mate and L-C of butyryl), 31.52 (L-C of N-butylcar-bamate), 36.00 (K-C of butyryl), 40.52 (K-C of N-butylcarbamate), 122.54, 122.73, 125.42, 125.77,128.56, 128.84, 130.48, 130.71, 130.80, and 131.46(biphenyl C-3^C-6), 130.48 and 130.71 (biphenyl C-1), 148.13 and 148.54 (biphenyl C-1), 154.27 (C = Oof N-butylcarbamate), 173.04 (C = O of butyryl);HRMS: Calcd. for C21H25O4N 355.1784. Found355.1765; EA: Anal. Calcd. for C21H25O4N: C,70.95; H, 7.09; N, 3.94. Found C, 71.08; H, 6.98,N, 4.01.
2,2P-Biphenyl-2-N-butylcarbamate-2P-ol (8) : 1HNMR (CDCl3, 300 MHz) N/ppm 0.85 (t, J = 7 Hz,3H, g-CH3), 1.20 (m, 2H, Q-CH2), 1.25 (m, 2H, L-CH2), 3.16 (m, 2H, K-CH2), 4.78 (br, 1H, NH), 5.17(br, 1H, OH), 6.92^7.48 (m, 8H, biphenyl-H) ; 13CNMR (CDCl3, 75.4 MHz) N/ppm 13.62 (g-C),19.53 (Q-C), 31.55 (L-C), 40.54 (K-C), 122.74,125.25, 128.43, 130.44, and 130.76 (biphenyl C-1and C-3^C-6), 148.46 (biphenyl C-2), 156.63(C = O); HRMS: Calcd. for C17H19O3N 285.1365.Found 285.1376; EA: Anal. Calcd. for C17H19O3N:C, 71.54; H, 6.72; N, 4.91. Found C, 71.72; H, 6.59,N, 5.02.
2,2P-Biphenyl-2-N-octylcarbamate-2P-ol (9) : 1HNMR (CD3SOCD3, 300 MHz) N/ppm 0.87 (t, J = 7Hz, 3H, g-CH3), 1.25 (m, 12H, L to g-1-CH2), 2.86(br, 1H, NH), 2.94 (m, 2H, K-CH2), 6.74^7.43 (m,8H, biphenyl-H) ; 13C NMR (CD3SOCD3, 75.4MHz) N/ppm 13.95 (g-C), 22.10, 26.07, 28.66,29.15, and 31.25 (L to g-1-C), 40.33 (K-C), 115.60,118.40, 123.24, 124.17, 124.42, 127.65, 128.50,130.72, 131.62, and 131.75 (biphenyl C-1 and C-3^C-6), 148.52 (biphenyl C-2), 154.55 (C = O); HRMS:Calcd. for C17H19O3N 341.1990. Found 341.2001;EA: Anal. Calcd. for C21H27O3N: C, 73.86; H,7.98; N, 4.10. Found C, 73.97; H, 7.85, N, 4.20.
Glyceryl-1,2,3-tri-N-butylcarbamate (11) : 1HNMR (CDCl3, 300 MHz) N/ppm 0.91 (t, J = 7 Hz,9H, g-CH3), 1.35 (m, 6H, Q-CH2), 1.47 (m, 6H, L-CH2), 3.16 (m, 6H, K-CH2), 4.20 (m, 4H, glyceryl-CH2), 4.76 (br, 3H, NH), 5.12 (m, 1H, glyceryl-CH) ;13C NMR (CDCl3, 75.4 MHz) N/ppm 13.62 (g-C),
19.77 (Q-C), 31.86 (L-C), 40.75 (K-C), 63.04 (glyceryl-CH2), 70.25 (glyceryl-CH), 156.02 (C = O); HRMS:Calcd. for C18H35O6N3 389.2526. Found 389.2535;EA: Anal. Calcd. for C18H35O6N3 : C, 55.49; H,9.06; N, 10.79. Found C, 55.53; H, 9.17; N, 10.92.
2.4. Enzyme kinetics and data reduction (Fig. 4)
All kinetic data were obtained by using an UV-visible spectrophotometer (Hewlett Packard 8452Aor Beckman DU-650) that was interfaced to a per-sonal computer. Kaleida Graph1 (version 2.0) andOrigin (version 4.0) were used for all least squarescurve ¢ttings. The CEase inhibition was assayed bythe Hosie method [21]. The temperature was main-tained at 25.0 þ 0.1³C by a refrigerated circulatingwater bath. All reactions were performed in sodiumphosphate bu¡er (pH 7.01) containing NaCl (0.1 M),acetonitrile (2% by volume), substrate (PNPB, 50WM), and varying concentrations of inhibitors. TheKm value for CEase-catalyzed hydrolysis of PNPBwas calculated to be 140 þ 10 WM by ¢tting initialvelocities versus substrate concentration to the Mi-chaelis^Menten equation. Requisite volumes of stocksolution of substrate and inhibitors in acetonitrilewere injected into reaction bu¡ers via Gilson Pipet-men. CEase was dissolved in sodium phosphatebu¡er (0.1 M, pH 7.01). Reactions were initiatedby injecting enzyme and monitored at 410 nm onthe UV-visible spectrometer. First-order rate con-stants (kapp values) for inhibition of CEase were de-
Fig. 4. The plot of kapp of CEase-catalyzed hydrolysis of PNPBversus the concentration of carbamate 6. [PNPB] = 50 WM. TheKm value used in calculation was 0.14 þ 0.01 mM.
BBAPRO 35905 6-5-99
G. Lin et al. / Biochimica et Biophysica Acta 1431 (1999) 500^511506
termined as described by Hosie et al. Values of Ki
and k2 can be obtained by ¢tting the data of kapp and[I] to Eq. 1 by non-linear least squares. Duplicate setsof data were collected for each inhibitor concentra-tion.
2.5. Return of activity and protection by TFA (Figs. 5and 6)
For the return of activity study, CEase was incu-bated with a carbamate (the concentration is twice Ki
value of the carbamate) in the absence and presenceof TFA (4 WM), a known competitive inhibitor of theenzyme (Ki = 0.34 þ 0.03 WM) [15] before the inhibi-tion reaction. After a period of 9 min to allow for90% inhibition by the carbamate, the enzyme solu-tion was diluted into bu¡er. Aliquots of the dilutedenzyme/inhibitor solutions were taken at time inter-vals and tested for activity (initial rate) with PNPB(0.2 mM). Corresponding controls were performed inthe absence of inhibitor to correct for spontaneousloss in enzyme activity. The initial rates were calcu-lated using a linear least squares program and the¢rst-order rate constants for the return of activitywere calculated by linear least squares analysis ofplots of log((A3Ar)/(AO3Ar)) against time. The
temperature was maintained at 25.0 þ 0.1³C by a re-frigerated circulating water bath. All the other pro-cedures followed those of Hosie et al. [21].
3. Results
Carbamates 1^11 were synthesized by general pro-cedures (62^92% yield) and puri¢ed (up to 99.9%)and characterized by 1H and 13C NMR, HRMS,and IR spectra and elemental analysis.
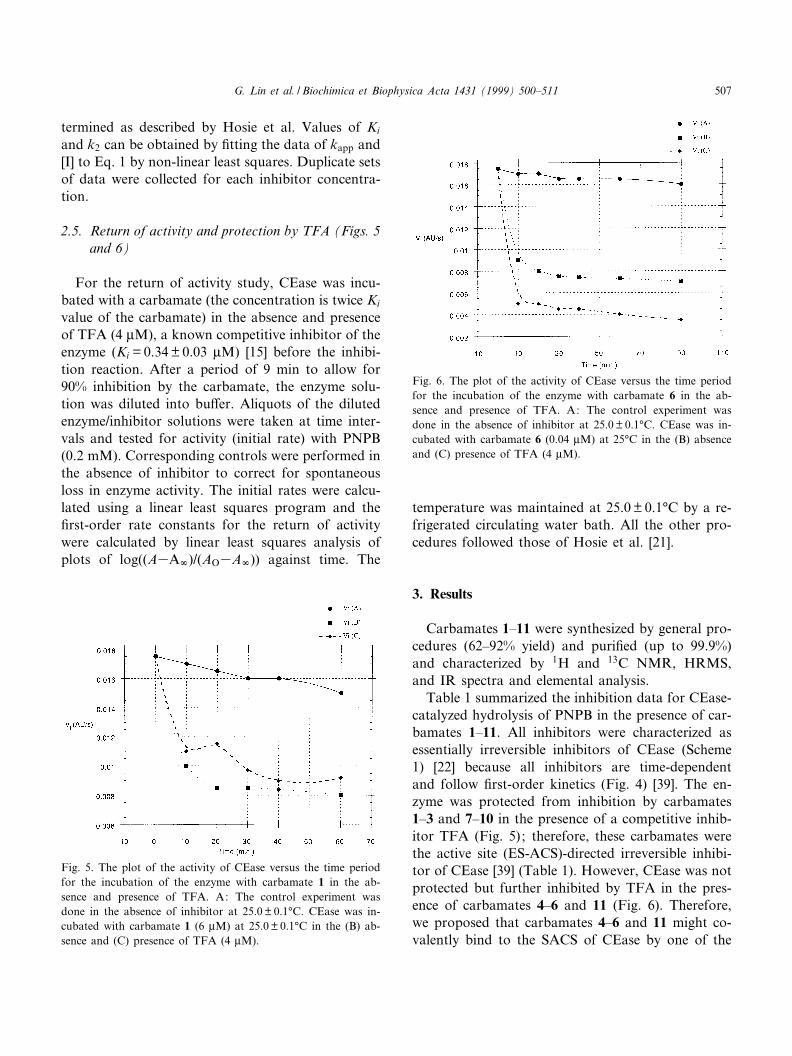
Table 1 summarized the inhibition data for CEase-catalyzed hydrolysis of PNPB in the presence of car-bamates 1^11. All inhibitors were characterized asessentially irreversible inhibitors of CEase (Scheme1) [22] because all inhibitors are time-dependentand follow ¢rst-order kinetics (Fig. 4) [39]. The en-zyme was protected from inhibition by carbamates1^3 and 7^10 in the presence of a competitive inhib-itor TFA (Fig. 5); therefore, these carbamates werethe active site (ES-ACS)-directed irreversible inhibi-tor of CEase [39] (Table 1). However, CEase was notprotected but further inhibited by TFA in the pres-ence of carbamates 4^6 and 11 (Fig. 6). Therefore,we proposed that carbamates 4^6 and 11 might co-valently bind to the SACS of CEase by one of the
Fig. 6. The plot of the activity of CEase versus the time periodfor the incubation of the enzyme with carbamate 6 in the ab-sence and presence of TFA. A: The control experiment wasdone in the absence of inhibitor at 25.0 þ 0.1³C. CEase was in-cubated with carbamate 6 (0.04 WM) at 25³C in the (B) absenceand (C) presence of TFA (4 WM).
Fig. 5. The plot of the activity of CEase versus the time periodfor the incubation of the enzyme with carbamate 1 in the ab-sence and presence of TFA. A: The control experiment wasdone in the absence of inhibitor at 25.0 þ 0.1³C. CEase was in-cubated with carbamate 1 (6 WM) at 25.0 þ 0.1³C in the (B) ab-sence and (C) presence of TFA (4 WM).
BBAPRO 35905 6-5-99
G. Lin et al. / Biochimica et Biophysica Acta 1431 (1999) 500^511 507

carbamyl group and to the ES-ACS of the enzyme bythe second carbamyl group. Carbamate 1 adopted alinear conformation for two carbamyl groups andshowed the least potency of inhibition toward theenzyme (Table 1). With a hydroxy or phenyl group,carbamates 8^10 and 6 were very potent inhibitors ofCEase (Table 1).
4. Discussion
Carbamate 1 is essentially irreversible inhibitor ofCEase and adopts a linear conformation for twocarbamyl groups (Fig. 3C). Therefore, we ¢nd thatcarbamate 1 is di¤cult to ¢t into the V-shaped activesite of CEase [34,35] (Fig. 1). Thus, carbamate 1binds to the ACS of CEase with its one carbamylgroup while the other carbamyl group is outsidethe active site. Therefore, this inhibitor shows theleast potency of inhibition toward the enzyme (Table1).
We have pointed out previously that carbamatesS-2, S-3 and R-10 are better inhibitors than theirenantiomers for CEase [24,26]. All these three carba-mates adopt an orthogonal conformation (Fig. 3A)at the pivot bond of binaphthyl group with the bend-ing angle between 93^106³ from MM-2 calculationand are the ES-ACS-directed irreversible inhibitorsof CEase due to the enzyme protected by TFA(Fig. 5). Therefore, one carbamyl group of carba-mates S-2, S-3, and R-10 covalently binds to theACS of the enzyme while the other carbamyl, butyr-yl, or hydroxy group can not bind covalently to the
SACS probably due to the orthogonal conformation(Fig. 7).
Carbamates 4^9 and 11 are very potent inhibitorsof CEase probably due to the fact that all these mol-ecules freely rotate at the pivot bond of the biphenylor glycerol group and therefore can ¢t well into thebent-shaped ACS-ES-SACS of the enzyme. There-fore, the overall shape of ACS-ES-SACS is bentlike a V letter with the bending angle somewherebetween 120^160³ and is probably identical to thesite for manual docking cholesteryl linoleate intoCEase [35]. Carbamates 4^6 and 11 are irreversibleinhibitors of CEase but the enzyme activity is notprotected by these carbamates in the presence ofTFA (Fig. 6). Therefore, carbamates 4^6 and 11may covalently bind to the SACS of CEase by oneof the carbamyl groups and may also covalently re-act with the ES-ACS of the enzyme by the secondcarbamyl group (Fig. 8). Moreover, carbamate 6 isthe most potent inhibitor in this study. Besides theoptimum conformation for carbamate 6, the inhibi-tion by the carbamate is also assisted by a favorableZ-Z interaction between phenylalanine residue (Phe324) at the SACS of CEase and the phenyl groupof the inhibitor molecule [32] (Fig. 8). The interac-tion with the n-octyl group of carbamate 5 is notstrong as that of the phenyl group due to the lackof the Z-Z interaction. The assistance of the phenylcarbamate group in this reaction strongly supportsthat CEase like CRL exists of more than one aro-matic residue in the SACS of CEase.
Carbamates 8^10 are more potent inhibitors ofCEase than carbamates S-2, 4, and 5 probably due
Table 1Kinetic data for the irreversible inhibition of CEase by carbamates 1^11
Inhibitors Ki (WM) kc (1033 s31) ki (M31 s31) Protection by TFAa
1 3.0 þ 0.5 0.20 þ 0.02 70 þ 10 YesS-2 2.0 þ 0.2 1.6 þ 0.2 800 þ 100 YesS-3 9 þ 3 0.6 þ 0.2 80 þ 40 Yes4 0.16 þ 0.03 0.22 þ 0.01 1,400 þ 300 No5 0.11 þ 0.01 0.87 þ 0.05 7,900 þ 800 No6 0.018 þ 0.002 0.40 þ 0.06 20,000 þ 3,000 No7 1.3 þ 0.3 1.2 þ 0.1 900 þ 200 Yes8 0.13 þ 0.02 8 þ 1 60,000 þ 10,000 Yes9 0.12 þ 0.02 9.6 þ 0.9 80,000 þ 10,000 YesR-10 0.8 þ 0.1 10 þ 1 13,000 þ 2,000 Yes11 0.39 þ 0.03 0.14 þ 0.01 360 þ 40 NoaThe protection of the enzyme by TFA in the presence of the carbamate.
BBAPRO 35905 6-5-99
G. Lin et al. / Biochimica et Biophysica Acta 1431 (1999) 500^511508

to the fact that the former inhibitor molecules inter-act with the ES of the enzyme through a hydrogenbond between the phenol hydroxy group of the in-hibitor molecules and the His 435 residue in that site(Fig. 7C,D,E). The free rotation at the pivot bondmakes the hydroxy group of carbamates 8 and 9more accessible to the bent-shaped active site of theenzyme for the ¢rst reversible step; therefore, carba-mates 8 and 9 are more potent inhibitors than R-10(Fig. 7C,D). The a¤nity (Ki step) for carbamate 6 toCEase is about 7-fold higher than that for carba-
mates 8 and 9 to CEase (Table 1) probably due tothe favorable Z-Z interaction for the former inhibitorand the enzyme (Fig. 8); however, the irreversible k2
step of carbamate 6 is about 20-fold slower thancarbamates 8 and 9 probably due to the fact thatthe covalent binding of carbamate 6 to the SACSof the enzyme is sterically hindered by the formationof the hydrogen bond between His 435 and the car-bonyl oxygen of the second carbamyl group. There-fore, the phenol hydroxy group assists primarily atthe k2 step (Fig. 7E) while the phenyl carbamate
Fig. 7. The proposed interactions between inhibitor molecules and active sites of CEase. A: Carbamate S-2 and the enzyme. B: Car-bamate 4 and the enzyme. C: Carbamate R-10 and the enzyme. A favorable hydrogen bonding between the phenol hydroxy group ofthe inhibitor molecule and His 435 of the enzyme is proposed. D: Carbamate 8 and the enzyme. The conformation of carbamate 8 inthis complex adopts the conformation of carbamate R-10 but is less hindered sterically for the formation of the hydrogen bond. E:The formation of the tetrahedral intermediate between carbamate 8 and the enzyme. The leaving group biphenol is intramolecularlyassisted by its phenol hydroxy proton.
BBAPRO 35905 6-5-99
G. Lin et al. / Biochimica et Biophysica Acta 1431 (1999) 500^511 509
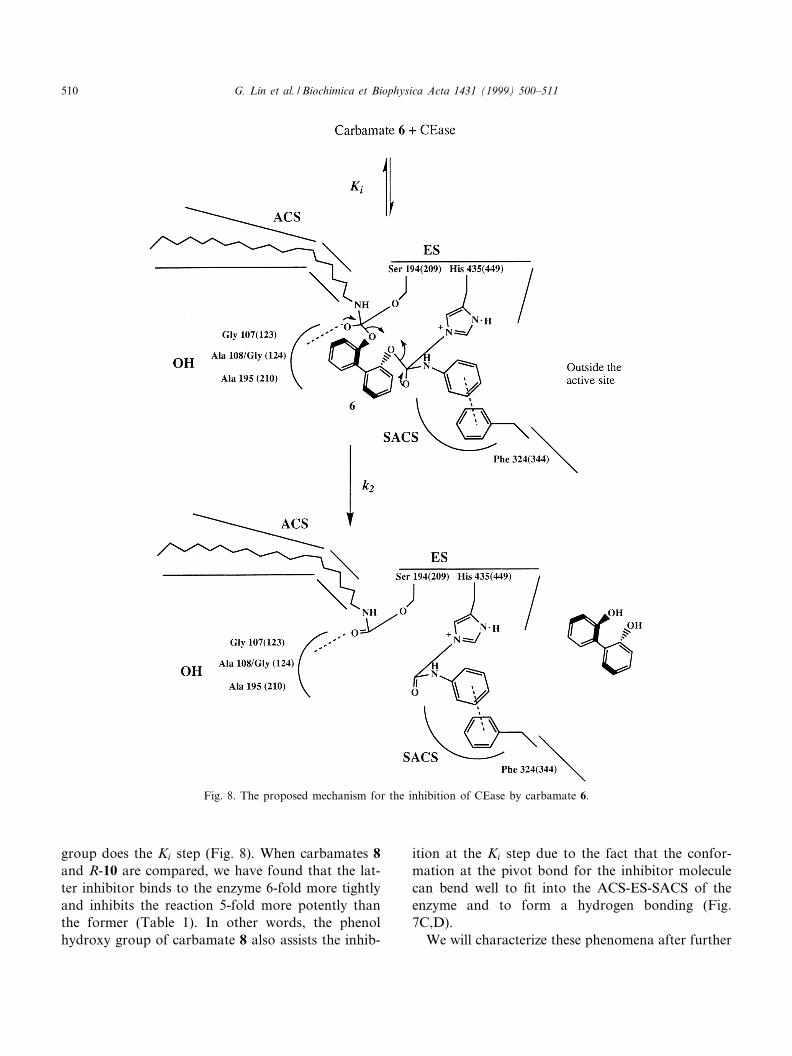
group does the Ki step (Fig. 8). When carbamates 8and R-10 are compared, we have found that the lat-ter inhibitor binds to the enzyme 6-fold more tightlyand inhibits the reaction 5-fold more potently thanthe former (Table 1). In other words, the phenolhydroxy group of carbamate 8 also assists the inhib-
ition at the Ki step due to the fact that the confor-mation at the pivot bond for the inhibitor moleculecan bend well to ¢t into the ACS-ES-SACS of theenzyme and to form a hydrogen bonding (Fig.7C,D).
We will characterize these phenomena after further
Fig. 8. The proposed mechanism for the inhibition of CEase by carbamate 6.
BBAPRO 35905 6-5-99
G. Lin et al. / Biochimica et Biophysica Acta 1431 (1999) 500^511510

study on the structure-activity correlations for moreinhibitors in future.
Acknowledgements
This work was supported by the National ScienceCouncil of Taiwan.
References
[1] S.G. Bhat, H.L. Brockman, Biochem. Biophys. Res. Com-mun. 109 (1982) 486^492.
[2] L.L. Gallo, S.B. Clark, S. Myers, G.V. Vohouny, J. LipidRes. 25 (1984) 604^612.
[3] H. Brockerho¡, R.G. Jensen, in: Lipolytic Enzymes, Aca-demic Press, New York, 1974.
[4] B. Fredrikzon, O. Hernell, L. Bla«ckberg, T. Olivecrona, Pe-diatr. Res. 12 (1978) 1048^1052.
[5] D. Kritchevsky, H.V. Kothari, Adv. Lipid Res. 16 (1978)221^226.
[6] E.A. Rudd, H.L. Brockman, in: B. Borgstro«m, H.L. Brock-man (Eds.), Lipases, Elsevier, Amsterdam, 1984.
[7] S.M. Watt, W.J. Simmonds, J. Lipid Res. 22 (1981) 157^165.[8] J. Brodt-Eppley, P. White, S. Jenkins, D.Y. Hui, Biochim.
Biophys. Acta 1272 (1995) 69^72.[9] S.C. Mayers-Payne, D.Y. Hui, H.L. Brockman, Biochemis-
try 34 (1995) 3942^3947.[10] W. Boland, C. FroMl, N. Lorenz, Synthesis 12 (1991) 1049^
1072.[11] A. Svendsen, in: P. Woolley, S.B. Petersen (Eds.), Lipases,
Their Structure Biochemistry and Application, 1994, pp. 1^21.
[12] U. Derewenda, A.M. Brzozowski, D.N. Lawson, Z.S.Derewenda, Biochemistry 31 (1992) 1532^1541.
[13] M. Leuveling Tjeenk, Y.B. Bulsink, A.J. Slotboom, H.M.Verheij, G.H. de Haas, G. Demleitner, F. Gotz, ProteinEng. 7 (1994) 579^583.
[14] F. Batoli, H.K. Lin, F. Ghomashchi, M.H. Gelb, M.K. Jain,R. Apitz-Castro, J. Biol. Chem. 269 (1994) 15625^15630.
[15] J. Sohl, L.D. Sutton, D.J. Burton, D.M. Quinn, Biochem.Biophys. Res. Commun. 151 (1988) 554^560.
[16] L.D. Sutton, J.S. Stout, L. Hosie, P.S. Spencer, D.M. Quinn,Biochem. Biophys. Res. Commun. 134 (1986) 386^392.
[17] D.G. Tew, C. Southan, S.Q. Rice, M.P. Lawrence, H. Li,H.F. Boyd, K. Moores, I.S. Gloger, C.H. Macphee,Arterioscler. Thromb. Vasc. Biol. 16 (1996) 591^599.
[18] T. Imanaka, Y. Moriyama, G.G. Ecsedi, T. Aoyagi, K.Amanuma-Muto, S. Ohkuma, Y. Takano, J. Biochem. 94(1983) 1017^1020.
[19] P. Hadvarry, H. Lengs¢eld, H. Wolfer, Biochem. J. 256(1988) 357^361.
[20] D.G. Tew, H.F. Boyd, S. Ashman, C. Theobald, C.A.Leach, Biochemistry 37 (1994) 10087^10093.
[21] L. Hosie, L.D. Sutton, D.M. Quinn, J. Biol. Chem. 262(1987) 260^264.
[22] S.R. Feaster, K. Lee, N. Baker, D.Y. Hui, D.M. Quinn,Biochemistry 35 (1996) 16723^16734.
[23] G. Lin, C.-Y. Lai, Tetrahedron Lett. 36 (1995) 6117^6120.[24] G. Lin, H.-C. Liu, Y.-C. Tsai, Bioorg. Med. Chem. Lett. 6
(1996) 43^46.[25] G. Lin, C.-Y. Lai, Tetrahedron Lett. 37 (1996) 193^196.[26] G. Lin, Y.-C. Tsai, H.-C. Liu, W.-C. Liao, C.-H. Chang,
Biochim. Biophys. Acta 1388 (1998) 161^174.[27] L.D. Sutton, J.S. Stout, D.M. Quinn, J. Am. Chem. Soc. 112
(1990) 8398^8403.[28] S.R. Feaster, D.M. Quinn, Methods Enzymol. 286 (1997)
231^252.[29] R.J. Kazlauskas, J. Am. Chem. Soc. 111 (1989) 4953^4959.[30] G. Lin, S.-H. Liu, S.-J. Chen, F.-C. Wu, H.-L. Sun, Tetra-
hedron Lett. 34 (1993) 6057^6058.[31] Y.-C. Liu, C.-S. Chen, Tetrahedron Lett. 30 (1989) 1617^
1620.[32] P. Grochulski, F. Bouthillier, R.J. Kazlauskas, A.N. Serreqi,
J.D. Schrag, E. Ziomek, M. Cygler, Biochemistry 33 (1994)3494^3500.
[33] S.R. Feaster, D.M. Quinn, B.L. Barnett, Protein Sci. 6(1997) 73^79.
[34] X. Wang, C.-S. Wang, J. Tang, F. Dyda, X.C. Zhang, Struc-ture 5 (1997) 1209^1218.
[35] J.C.-H. Chen, L.J.W. Miercke, J. Krucinski, J.R. Starr, G.Saenz, X. Wang, C.A. Spilburg, L.A. Lang, J.L. Ellsworth,R.M. Stroud, Biochemistry 37 (1998) 5107^5117.
[36] G. Lin, W.-Y. Lin, C.-T. Shieh, Tetrahedron Lett. 39 (1998)8881^8884.
[37] W.N. Aldridge, E. Reiner, in: A. Neuberger, E.L. Tatun(Eds.), Enzyme Inhibitors as Substrates, North-HollandPublishing Co., Amsterdam, 1972, pp. 123^145.
[38] G.J. Hart, R.D. O'Brien, Biochemistry 12 (1973) 2940^2945.[39] R.H. Abeles, A.L. Maycock, Acc. Chem. Res. 9 (1976) 313^
319.
BBAPRO 35905 6-5-99
G. Lin et al. / Biochimica et Biophysica Acta 1431 (1999) 500^511 511