spring viremia of carp virus n protein suppresses fish ... various plasmids by using x-tremegene hp...
TRANSCRIPT

of July 10, 2018.This information is current as
Signaling ProteinTargeting the Mitochondrial Antiviral
1 Production byφSuppresses Fish IFNSpring Viremia of Carp Virus N Protein
ZhangYong-AnZhang, Xu-Jie Zhang, Dan-Dan Chen, Pin Nie and
Long-Feng Lu, Shun Li, Xiao-Bing Lu, Scott E. LaPatra, Nu
http://www.jimmunol.org/content/196/9/3744doi: 10.4049/jimmunol.1502038March 2016;
2016; 196:3744-3753; Prepublished online 18J Immunol
MaterialSupplementary
8.DCSupplementalhttp://www.jimmunol.org/content/suppl/2016/03/17/jimmunol.150203
Referenceshttp://www.jimmunol.org/content/196/9/3744.full#ref-list-1
, 25 of which you can access for free at: cites 58 articlesThis article
average*
4 weeks from acceptance to publicationFast Publication! •
Every submission reviewed by practicing scientistsNo Triage! •
from submission to initial decisionRapid Reviews! 30 days* •
Submit online. ?The JIWhy
Subscriptionhttp://jimmunol.org/subscription
is online at: The Journal of ImmunologyInformation about subscribing to
Permissionshttp://www.aai.org/About/Publications/JI/copyright.htmlSubmit copyright permission requests at:
Email Alertshttp://jimmunol.org/alertsReceive free email-alerts when new articles cite this article. Sign up at:
Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists, Inc. All rights reserved.Copyright © 2016 by The American Association of1451 Rockville Pike, Suite 650, Rockville, MD 20852The American Association of Immunologists, Inc.,
is published twice each month byThe Journal of Immunology
by guest on July 10, 2018http://w
ww
.jimm
unol.org/D
ownloaded from
by guest on July 10, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

The Journal of Immunology
Spring Viremia of Carp Virus N Protein Suppresses FishIFNw1 Production by Targeting the Mitochondrial AntiviralSignaling Protein
Long-Feng Lu,*,†,1 Shun Li,*,1 Xiao-Bing Lu,*,† Scott E. LaPatra,‡ Nu Zhang,*,†
Xu-Jie Zhang,*,x Dan-Dan Chen,* Pin Nie,* and Yong-An Zhang*
For a virus to replicate efficiently, it must try and inhibit host IFN expression because IFN is an important host defense at early stages
after viral infection. For aquatic viruses, the mechanisms used to escape the hosts IFN system are still unclear. In this study, we show
that the N protein of spring viremia of carp virus (SVCV) inhibits zebrafish IFNw1 production by degrading the mitochondrial
antiviral signaling protein (MAVS). First, the upregulation of IFNw1 promoter activity stimulated by polyinosinic:polycytidylic
acid, retinoic acid–inducible gene I (RIG-I) or MAVS was suppressed by the SVCV infection. However, the upregulation by the
downstream factor of the RIG-I–like receptor signaling pathway, TANK-binding kinase 1, was not affected. Notably, at the protein
level, MAVS decreased remarkably when cells were infected with SVCV. Second, consistent with the result of the SVCV infection,
overexpression of the N protein of SVCV blocked the IFNw1 transcription activated by MAVS and downregulated MAVS
expression at the protein level but not at the mRNA level. Further analysis demonstrated that the N protein targeted MAVS
for K48-linked ubiquitination, which promoted the degradation of MAVS. These data indicated that fish MAVS could be degraded
by the N protein of SVCV through the ubiquitin-proteasome pathway. To our knowledge, this is the first article of a fish RIG-I–like
receptor pathway interfered by an aquatic virus in an ubiquitin-proteasome manner, suggesting that immune evasion of a virus
also exists in lower vertebrates. The Journal of Immunology, 2016, 196: 3744–3753.
The IFN system is the first line of defense of the hostagainst viral invasion. Once host cells are infected byvirus, the retinoic acid–inducible gene I (RIG-I)–like re-
ceptors (RLRs) sense the viral RNAs and activate IFN expression(1–4). In this process, a member of the RLR family recognizesviral RNAs and activates a downstream adaptor molecule, themitochondrial antiviral signaling protein (MAVS, also known asVISA/IPS-1/Cardif), which initiates TANK-binding kinase 1(TBK1) and canonical IKK complex (IKKa/b/g) to activate IFNregulatory factor 3/7 (IRF3/7) and NF-kB, which translocate into
the nucleus and initiate the expression of IFN (5–10). This sets upthe antiviral status of the cells.However, viruses have evolved a number of elaborate strategies
to avoid being eliminated. One of those mechanisms is to blockthe production of IFN (11). In mammals, MAVS seems to be animportant target for virus. Generally, two mechanisms are usedto inhibit the expression of IFN by targeting MAVS, includingcleavage and degradation. As examples, MAVS can be cleaved bythe NS3/4A serine protease of hepatitis C virus and the cysteineprotease 3Cpro of coxsackievirus B3 (12, 13), or degraded by thehepatitis B virus X and open reading frame (ORF)-9b of coro-naviruses in an ubiquitin (Ub)–proteasome manner (14, 15).In aquaculture, a number of viruses have been identified as the
etiological agents of aquatic animal diseases (16), however, themechanism(s) used by aquatic viruses to escape the host IFNsystem are still unclear. Spring viremia of carp virus (SVCV) isthe causative agent of SVC and causes significant mortality incommon carp (Cyprinus carpio). It is a negative-sense ssRNAvirus that belongs to the genus Vesiculovirus of the familyRhabdoviridae (17). The genome of SVCV is ∼11 kb and encodesfive proteins including the nucleoprotein (N), phosphoprotein (P),matrix protein (M), glycoprotein (G), and viral RNA–dependentRNA polymerase (L) (18). The availability of sequence informa-tion for the entire SVCV genome and homology analysis withmammalian rhabdoviruses allowed more accurate deduction of thefunctions of the proteins of SVCV. The N protein interacts withthe viral RNA to form the nucleocapsid. The P and L proteinsassociate with the nucleocapsid, which are required for the tran-scription and replication of SVCV. The M protein takes part in theassembly and budding of SVCV, and the G protein binds to cel-lular receptors and mediates viral endocytosis (17, 19).There is some information available concerning the host response
to an SVCVinfection (20, 21). For example, by using high-throughput
*Institute of Hydrobiology, Chinese Academy of Sciences, Wuhan 430072, China;†University of Chinese Academy of Sciences, Beijing 100049, China; ‡ResearchDivision, Clear Spring Foods, Inc., Buhl, ID 83316; and xCollege of Fisheries andLife Science, Shanghai Ocean University, Shanghai 201306, China
1L.-F.L. and S.L. contributed equally to this work.
ORCIDs: 0000-0002-9492-2940 (P.N.); 0000-0002-9956-3879 (Y.-A.Z.).
Received for publication September 15, 2015. Accepted for publication February 18,2016.
This work was supported by National Key Basic Research Program of China Grant2014CB138601 and National Natural Science Foundation of China Grant 31172431.
Address correspondence and reprint requests to Dr. Yong-An Zhang, Institute ofHydrobiology, Chinese Academy of Sciences, 7 Donghu South Road, Wuhan430072, China. E-mail address: [email protected]
The online version of this article contains supplemental material.
Abbreviations used in this article: CPE, cytopathic effect; EPC, epithelioma papulo-sum cyprini (cell); HA, hemagglutinin; IRF, IFN regulatory factor; ISRE, IFN-stimulated response element; MAVS, mitochondrial antiviral signaling protein; MOI,multiplicity of infection; ORF, open reading frame; PFA, paraformaldehyde; poly(I:C),polyinosinic:polycytidylic acid; qPCR, quantitative real-time PCR; RIG-I, retinoicacid–inducible gene I; RLR, RIG-I–like receptor; SVCV, spring viremia of carpvirus; TBK1, TANK-binding kinase 1; TM, transmembrane; Ub, ubiquitin; ZF4,zebrafish embryo fibroblast-like (cell); ZFL, zebrafish liver (cell).
Copyright� 2016 by TheAmericanAssociation of Immunologists, Inc. 0022-1767/16/$30.00
www.jimmunol.org/cgi/doi/10.4049/jimmunol.1502038
by guest on July 10, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

methods such as pathway-targeted microarrays and transcriptomicand proteomic analyses, many immune- and autophagy-related geneshave been identified to be involved in an SVCV infection (22–25).The expression of IFN could be elicited by an SVCV infection, andoverexpression of IFN could significantly increase the survival ofzebrafish embryos infected with SVCV (26–28). Similar to mam-mals, it has been shown that zebrafish MAVS plays an important rolein IFN activation, which can significantly decrease the probabilityof infection with SVCV (29). In the RLR pathway, the truncatedsplicing variant of MAVS blocked the IFN induction by RIG-I, in-dicating the position of MAVS in the signaling pathway is conserved(30, 31). These results demonstrate that the defense mechanisms offish against SVCV infection are similar to that of mammals, espe-cially regarding IFN production being activated by MAVS.Until now, there have been few studies regarding the mechanisms
used by aquatic viruses to interfere with fish IFN production.In study reported herein we show that the N protein of SVCV couldinhibit host IFN expression by degrading MAVS in a Ub–pro-teasome manner. These results suggest that the immune evasion byan aquatic virus by interfering with the host IFN response alsoexists in lower vertebrates.
Materials and MethodsCells and viruses
Epithelioma papulosum cyprini (EPC) cells were maintained in medium199 (Invitrogen) supplemented with 10% FBS (Invitrogen). Zebrafish liver(ZFL) cells were cultured in 50% L-15 (Invitrogen), 35% DMEM-HG(Invitrogen), and 15% Ham’s F12 medium (Invitrogen) supplementedwith 0.15 g/l sodium bicarbonate (Sigma-Aldrich), 15 mM HEPES(Sigma-Aldrich), and 10% FBS. Zebrafish embryo fibroblast-like (ZF4)cells were grown in a 1:1 mixture of DMEM and Ham’s F12 mediumsupplemented with 10% FBS. All cells were maintained at 28˚C and 5%CO2. SVCV was propagated in EPC cells until cytopathic effect (CPE) wascomplete, and the culture medium was harvested and stored at280˚C untilneeded.
Plasmid construction and reagents
The plasmids encoding RIG-I-Nter, TBK1, and IRF7 in the pcDNA3.1(+)vector (Invitrogen) and the plasmids containing IFNw1pro-Luc, IFNw2pro-Luc, IFNw3pro-Luc, IFNw4pro-Luc, and NF-kB-Luc in the pGL3-Basicluciferase reporter vector (Promega) were constructed as described pre-viously (31–33). The IFN-stimulated response element (ISRE) luciferasereporter construct (ISRE-Luc) containing five ISRE motifs in series waspurchased from Stratagene. The ORF of MAVS was amplified by PCRfrom ZF4 cells and cloned into the pcDNA3.1(+) and pHAGE-CMV-MCS-PGK (BD Clontech) vectors. The pcDNA3.1-HA-IRF7 plasmid was con-structed by subcloning the DNA fragment encoding HA-IRF7 into thepcDNA3.1(+) vector. To generate the MAVS-mCherry, TBK1-mCherry,and IRF7-mCherry expression plasmids, the cDNA fragments encodingMAVS, TBK1, and IRF7 were cloned into the pCS2-mCherry vector(BD Clontech). The MAVS_tv2-Flag plasmid was generated by insertingMAVS_tv2 cDNA into the pHAGE-CMV-MCS-PGK vector. The cDNAfragments of N, P, and G genes were amplified by RT-PCR from the RNAof SVCV infected cells and then cloned into the pcDNA3.1(+) vector.C-terminally GFP-tagged N (N-GFP) was generated by inserting the ORFof the N protein of SVCV into the pEGFP-N3 vector (BD Clontech). ThepcDNA3.1-HA-N, pcDNA3.1-N-Flag and pcDNA3.1-HA-P plasmids wereconstructed by inserting the DNA fragments encoding HA-N, N-Flag andHA-P (P protein of SVCV) into the pcDNA3.1(+) vector. The cDNAfragment of N was also subcloned into the pCMV-Myc vector (BDClontech). All constructs were confirmed by DNA sequencing. The pri-mers including the restriction enzyme cutting sites used for plasmid con-struction are listed in Supplemental Table I. MG132 and polyinosinic:polycytidylic acid [poly(I:C)] were purchased from Sigma-Aldrich andused at a final concentration of 20 mM/ml and 1 mg/ml, respectively.
Transient transfection and virus infection
EPC cells were seeded in 10-cm2 dishes or 24-well plates and transfectedwith various plasmids by using X-tremeGENE HP DNA TransfectionReagent (Roche), according to the manufacturer’s instructions. For theantiviral assay, EPC cells seeded in 24-well plates were transfected with
0.5 mg pcDNA3.1-N or the empty vector. At 24 h posttransfection, cellswere infected with SVCVat a multiplicity of infection (MOI) of 1, 10, 100,or 1000. After 2 or 3 d, aliquots of the supernatant were harvested fordetermination of virus titers, and cell monolayers were stained with 1%crystal violet for visualizing CPE. For virus titration, 200 ml culture me-dium was collected at 48 h postinfection and used for plaque assay. Thesupernatants were subjected to 3-fold serial dilutions and then added(100 ml) onto a monolayer of EPC cells cultured in a 96-well plate. After48 or 72 h, the medium was removed, and the monolayers were washedwith PBS, fixed by 4% paraformaldehyde (PFA), and stained with 1%crystal violet. The virus titer was expressed as 50% tissue culture infec-tious dose (TCID50/ml).
Luciferase activity assay
EPC cells were seeded in 24-well plates and 24 h later cotransfectedwith 250 ng of the luciferase reporter plasmid (IFNw1pro-Luc, ISRE-Luc or NF-kB-Luc) and 25 ng of the pRL-TK vector (Promega). TheRenilla luciferase internal control was used to normalize the expressionlevels of the transefected plasmids. The empty vector pcDNA3.1(+)was used to maintain equivalent amounts of DNA in each well. Thetransfection of poly(I:C) was performed at 24 h before the cells wereharvested. At 48 h posttransfection, the cells were washed in PBS andlysed for measuring luciferase activity by Dual-Luciferase Reporter
FIGURE 1. SVCV inhibits poly(I:C)-triggered IFNw1 activation. (A)
Induction of IFNw1, IFNw2, IFNw3, and IFNw4 promoters by poly(I:C).
EPC cells seeded in 24-well plates overnight were transfected with 0.5 mg
IFNw1pro-Luc, IFNw2pro-Luc, IFNw3pro-Luc, or IFNw4pro-Luc, and
25 ng pRL-TK was used as an internal control. At 24 h posttransfection,
cells were treated with poly(I:C) (1 mg/ml) or left untreated (null). The
luciferase assay was performed 24 h after stimulation. (B) SVCV inhibited
poly(I:C)-mediated activation of IFNw1 promoter in a dose-dependent
manner. EPCs seeded in 24-well plates overnight were transfected with
0.25 mg IFNw1pro-Luc and 25 ng pRL-TK (control). At 24 h post-
transfection, cells were transfected with poly(I:C) for 4 h and then infected
with SVCV (MOI = 1, 10, 100, or 1000). The luciferase assays were
measured 48 h after stimulation, and fold activation was determined by
comparing the luciferase activity in control cells. Error bars are the SDs
obtained by measuring each sample in triplicate. Asterisks indicate sig-
nificant differences from control (*p , 0.05).
The Journal of Immunology 3745
by guest on July 10, 2018http://w
ww
.jimm
unol.org/D
ownloaded from
Assay System according to the manufacturer’s instructions (Promega).The results were obtained from more than three independent experi-ments, each performed in triplicate.
Quantitative real-time PCR
Total RNAs were extracted using the TRIzol reagent (Invitrogen). cDNAwas synthesized using a GoScript reverse transcription system (Promega),according to the manufacturer’s instructions. Quantitative real-time PCR(qPCR) was performed with Fast SYBR Green PCR Master Mix (Bio-Rad)on the CFX96 Real-Time System (Bio-Rad). PCR conditions were asfollows: 95˚C for 5 min and then 40 cycles of 95˚C for 20 s, 60˚C for 20 s,and 72˚C for 20 s. All primers used for qPCRs are shown in SupplementalTable I, and b-actin was used as an internal control. The relative foldchanges were calculated by comparison with the corresponding controlsusing the 22DDCt method. Three independent experiments were conductedfor statistical analysis purposes.
Immunoblot analysis
Whole-cell extracts were separated by 10% SDS-PAGE and transferred topolyvinylidene difluoride membranes (Bio-Rad). The membranes wereblocked for 1 h at room temperature in TBST buffer (25 mM Tris-HCl,150 mM NaCl, and 0.1% Tween 20 [pH 7.5]) containing 5% nonfat drymilk and probed with various primary Abs at an appropriate dilutionovernight at 4˚C, washed three times with TBST, and then incubated withsecondary Abs for 1 h at room temperature. After three additional washeswith TBST, the membranes were stained with Immobilon TM WesternChemiluminescent HRP Substrate (Millipore) and detected using anImageQuant LAS 4000 system (GE Healthcare). Rabbit polyclonal anti-MAVS and anti-IRF7 antisera were made by immunization of rabbits withprokaryotic expressed zebrafish MAVS_tv2 and IRF7-DBD as described ina previous report (31). The antiserum against MAVS_tv2 or IRF7 wasdiluted 1:2000. The other Abs were also diluted including anti–b-actin(Cell Signaling Technology) at 1:1000, anti-Flag/HA (Sigma-Aldrich) at1:2000, anti-myc (Santa Cruz Biotechnology) at 1:2000, and HRP-conjugated anti-rabbit IgG or anti-mouse IgG (Thermo Scientific) at1:5000. The results were the representative of three independent experi-ments. ImageJ was used for quantifying the protein levels based on theband density obtained from Western blot analysis.
In vivo ubiquitination assay
EPC cells were transiently transfected with 5 mg MAVS-Flag, 4 mg Myc-N,and 1 mg of the hemagglutinin (HA)-Ub, HA-Ub-K48O, or HA-Ub-K63Oexpression plasmids. At 18 h posttransfection, the cells were treated with20 mM MG132. Samples were harvested at 24 h posttransfection, lysedusing a radioimmunoprecipitation assay lysis buffer (1% Nonidet P-40,50 mM Tris-HCl [pH 7.5], 150 mM NaCl, 1 mM EDTA, 1 mM NaF,1 mM Na3VO4, 1 mM PMSF, and 0.25% sodium deoxycholate) thatcontained the protease inhibitor mixture (Sigma-Aldrich). The sampleswere denatured by heating for 10 min in 1% SDS and diluted with lysisbuffer until the concentration of SDS was decreased to 0.1%. The dilutedsupernatants were then immunoprecipitated overnight at 4˚C with constantagitation with 30 ml anti-Flag agarose conjugate (Sigma-Aldrich).Immunoprecipitated protein was washed three times with lysis buffer andresuspended in 23 SDS sample buffer. The immunoprecipitates andwhole-cell lysates were analyzed by immunblotting with various Abs thatare indicated.
Fluorescent microscopy
EPC cells were seeded in a 6-well plate and transfected with variousplasmids for 24 h. Then, the cells were washed twice with PBS and fixedwith 4% PFA for 1 h. Images were obtained using a fluorescent microscope(Nikon) through a 310 objective lens.
Statistics analysis
Data are expressed as mean6 SD of at least three independent experiments(n $ 3). The p values were calculated by one-way ANOVAwith Dunnett’sposthoc test (version 19 SPSS Statistics; IBM). A p value , 0.05 wasconsidered statistically significant.
ResultsSVCV blocks the expression of IFNw1
Previous studies have shown that many strategies are used byviruses to counteract host IFN production. Whether the expressionof fish IFN could be inhibited by SVCV was explored by the lu-ciferase reporter gene assay. Because poly(I:C) mimics viral RNA,
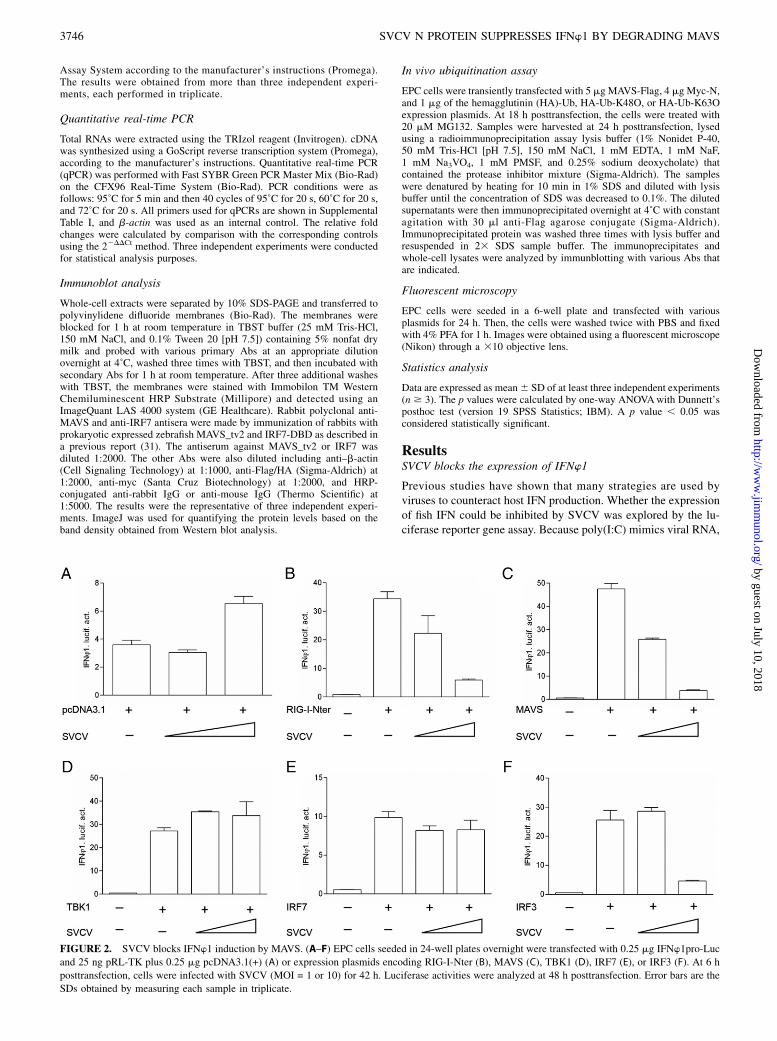
FIGURE 2. SVCV blocks IFNw1 induction by MAVS. (A–F) EPC cells seeded in 24-well plates overnight were transfected with 0.25 mg IFNw1pro-Luc
and 25 ng pRL-TK plus 0.25 mg pcDNA3.1(+) (A) or expression plasmids encoding RIG-I-Nter (B), MAVS (C), TBK1 (D), IRF7 (E), or IRF3 (F). At 6 h
posttransfection, cells were infected with SVCV (MOI = 1 or 10) for 42 h. Luciferase activities were analyzed at 48 h posttransfection. Error bars are the
SDs obtained by measuring each sample in triplicate.
3746 SVCV N PROTEIN SUPPRESSES IFNw1 BY DEGRADING MAVS
by guest on July 10, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

it was chosen for inducing the expression of the four type I IFNs(IFNw1 to IFNw4) in zebrafish. As shown in Fig. 1A, the activa-tion of IFNw1 and IFNw3 promoters were significantly upregu-lated by transfection with poly(I:C); however, the IFNw2 andIFNw4 promoters were unaffected. IFNw1 and IFNw3 had similarregulation patterns and IFNw1 but not IFNw3 could be upregu-lated by IRF3 (32, 33). In agreement with the results above, theIFNw1 promoter (IFNw1pro) activity was again induced by poly(I:C);however, the induction was remarkably suppressed by infectionwith SVCV in a dose-dependent manner (Fig. 1B). These resultsdemonstrated that the upregulation of IFNw1 by poly(I:C) couldbe blocked by SVCV.
SVCV inhibits IFNw1 induction by targeting MAVS
Similar to mammals, fish RLR signaling cascades have also beenreported to activate IFN expression (29). To determine whether thezebrafish RLR pathway could be disrupted by an SVCV infection,the N-terminal domain of RIG-I (RIG-I-Nter), MAVS, TBK1,IRF3, and IRF7 were cotransfected with IFNw1pro following in-fection with different titers of SVCV. As shown in Fig. 2A, alow level of expression of IFNw1 could be induced with a highconcentration of SVCV. In addition, overexpression of the RLR
cascades led to a higher induction of IFNw1pro activity. However,the activation of IFNw1 driven by RIG-I-Nter and MAVS wereblocked by SVCV in a dose-dependent manner (Fig. 2B, 2C),whereas the induction of IFNw1 by TBK1 and IRF7 were notaffected (Fig. 2D, 2E). Interestingly, the induction of IFNw1 byIRF3 was significantly blocked by a high concentration of SVCV,which might be due to SVCV developing multiple strategies toevade host immune surveillance (Fig. 2F). Collectively, becauseMAVS is a unique adaptor protein downstream of RIG-I and up-stream of TBK1, these data indicated that SVCV disrupts theactivation of IFNw1 by targeting MAVS.
MAVS is degraded during SVCV infection
To understand how SVCV blocks MAVS signal transduction, theexpression pattern of MAVS was analyzed in SVCV-infected cells.Remarkably, immunoblotting revealed that the abundance ofMAVS was substantially decreased postinfection with SVCV for24 h. However, the IRF7 concentration did not appear to change(Fig. 3A). Similarly, a reduction in MAVS-Flag and no effect onHA-IRF7 were observed in cells overexpressing exogenousMAVS-Flag or HA-IRF7 postinfection with SVCV (Fig. 3B, 3C).MG132, a potent inhibitor of the proteasome, was used to treat
FIGURE 3. MAVS is degraded in SVCV-infected cells. (A) SVCV infection degraded endogenous MAVS protein. ZFL cells were infected with SVCV
(MOI = 10) for 24 h, and the cell lysates were fractionated by SDS-PAGE and immunoblotted (IB) using MAVS antiserum. Membranes were reprobed with
IRF7 antiserum and b-actin Ab (internal control). (B and C) SVCV infection degraded exogenous MAVS protein. EPC cells were seeded in 6-well plates
and transfected with 3 mg MAVS-Flag (B) or HA-IRF7 (C), and after 24 h, cells were infected with SVCV (MOI = 10) for 24 h prior to being harvested for
IB analysis of whole-cell extracts with Flag or HA and b-actin Abs. (D) IB analysis for MAVS in ZFL cells infected with SVCV for 24 h and treated with
DMSO or MG132 (10, 20, or 40 mM) for 8 h. Values represent percentages of MAVS proteins normalized against b-actin and compared with the control
signal. (E) SVCV infection had no effect on the transcription of mavs. ZFL cells were infected with SVCV (MOI = 10) at various time points. RNA was
extracted, and the mavs and ifnw1 transcripts were analyzed by qPCR. b-actin was used as an internal control for normalization. The relative expression
represents the fold induction relative to the expression level in control cells (set to 1). Error bars represent SDs obtained by measuring each sample in
triplicate. Asterisks indicate significant differences from control (*p , 0.05).
The Journal of Immunology 3747
by guest on July 10, 2018http://w
ww
.jimm
unol.org/D
ownloaded from
cells for 8 h to further identify the degradation pattern of MAVS.Interestingly, the expression of MAVS was rescued gradually with theincreasing concentration of MG132, indicating that MAVS was de-graded in an Ub-proteasome manner (Fig. 3D). The expression ofmavs at the mRNA level was also analyzed. As shown in Fig. 3E,compared with ifnw1, there was no significant change in the tran-scription of mavs after challenge with SVCV from 0 to 24 h. Theseresults suggested that SVCV reduces the expression of MAVS by anUb-proteasome system at the protein level but not at the mRNA level.
N protein blocks the activation of IFNw1pro
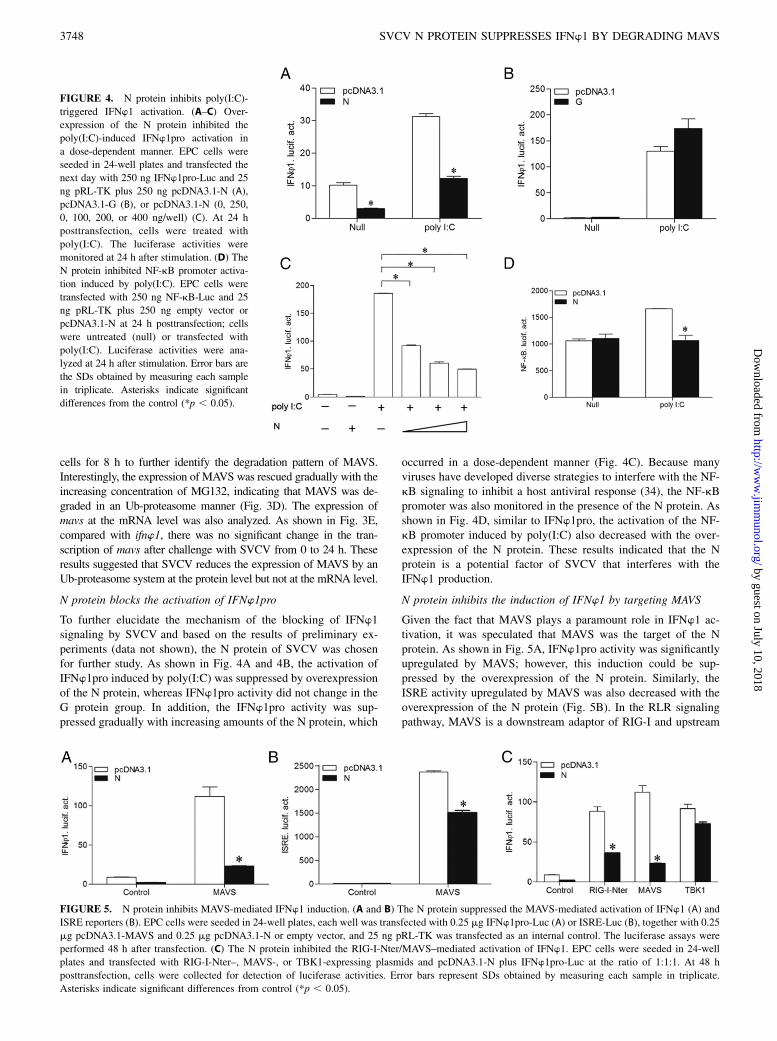
To further elucidate the mechanism of the blocking of IFNw1signaling by SVCV and based on the results of preliminary ex-periments (data not shown), the N protein of SVCV was chosenfor further study. As shown in Fig. 4A and 4B, the activation ofIFNw1pro induced by poly(I:C) was suppressed by overexpressionof the N protein, whereas IFNw1pro activity did not change in theG protein group. In addition, the IFNw1pro activity was sup-pressed gradually with increasing amounts of the N protein, which
occurred in a dose-dependent manner (Fig. 4C). Because manyviruses have developed diverse strategies to interfere with the NF-kB signaling to inhibit a host antiviral response (34), the NF-kBpromoter was also monitored in the presence of the N protein. Asshown in Fig. 4D, similar to IFNw1pro, the activation of the NF-kB promoter induced by poly(I:C) also decreased with the over-expression of the N protein. These results indicated that the Nprotein is a potential factor of SVCV that interferes with theIFNw1 production.
N protein inhibits the induction of IFNw1 by targeting MAVS
Given the fact that MAVS plays a paramount role in IFNw1 ac-tivation, it was speculated that MAVS was the target of the Nprotein. As shown in Fig. 5A, IFNw1pro activity was significantlyupregulated by MAVS; however, this induction could be sup-pressed by the overexpression of the N protein. Similarly, theISRE activity upregulated by MAVS was also decreased with theoverexpression of the N protein (Fig. 5B). In the RLR signalingpathway, MAVS is a downstream adaptor of RIG-I and upstream
FIGURE 4. N protein inhibits poly(I:C)-
triggered IFNw1 activation. (A–C) Over-
expression of the N protein inhibited the
poly(I:C)-induced IFNw1pro activation in
a dose-dependent manner. EPC cells were
seeded in 24-well plates and transfected the
next day with 250 ng IFNw1pro-Luc and 25
ng pRL-TK plus 250 ng pcDNA3.1-N (A),
pcDNA3.1-G (B), or pcDNA3.1-N (0, 250,
0, 100, 200, or 400 ng/well) (C). At 24 h
posttransfection, cells were treated with
poly(I:C). The luciferase activities were
monitored at 24 h after stimulation. (D) The
N protein inhibited NF-kB promoter activa-
tion induced by poly(I:C). EPC cells were
transfected with 250 ng NF-kB-Luc and 25
ng pRL-TK plus 250 ng empty vector or
pcDNA3.1-N at 24 h posttransfection; cells
were untreated (null) or transfected with
poly(I:C). Luciferase activities were ana-
lyzed at 24 h after stimulation. Error bars are
the SDs obtained by measuring each sample
in triplicate. Asterisks indicate significant
differences from the control (*p , 0.05).
FIGURE 5. N protein inhibits MAVS-mediated IFNw1 induction. (A and B) The N protein suppressed the MAVS-mediated activation of IFNw1 (A) and
ISRE reporters (B). EPC cells were seeded in 24-well plates, each well was transfected with 0.25 mg IFNw1pro-Luc (A) or ISRE-Luc (B), together with 0.25
mg pcDNA3.1-MAVS and 0.25 mg pcDNA3.1-N or empty vector, and 25 ng pRL-TK was transfected as an internal control. The luciferase assays were
performed 48 h after transfection. (C) The N protein inhibited the RIG-I-Nter/MAVS–mediated activation of IFNw1. EPC cells were seeded in 24-well
plates and transfected with RIG-I-Nter–, MAVS-, or TBK1-expressing plasmids and pcDNA3.1-N plus IFNw1pro-Luc at the ratio of 1:1:1. At 48 h
posttransfection, cells were collected for detection of luciferase activities. Error bars represent SDs obtained by measuring each sample in triplicate.
Asterisks indicate significant differences from control (*p , 0.05).
3748 SVCV N PROTEIN SUPPRESSES IFNw1 BY DEGRADING MAVS
by guest on July 10, 2018http://w
ww
.jimm
unol.org/D
ownloaded from
activator of TBK1. When the N protein was overexpressed, theinduction of IFNw1 by RIG-I-Nter and MAVS was remarkablyinhibited; however, no apparent effects were observed on IFNw1activation induced by TBK1 (Fig. 5C). These results suggestedthat the N protein might inhibit the transcription of IFNw1 inducedby the RLR axis through the negative regulation of MAVS.
MAVS is degraded by overexpression of N protein
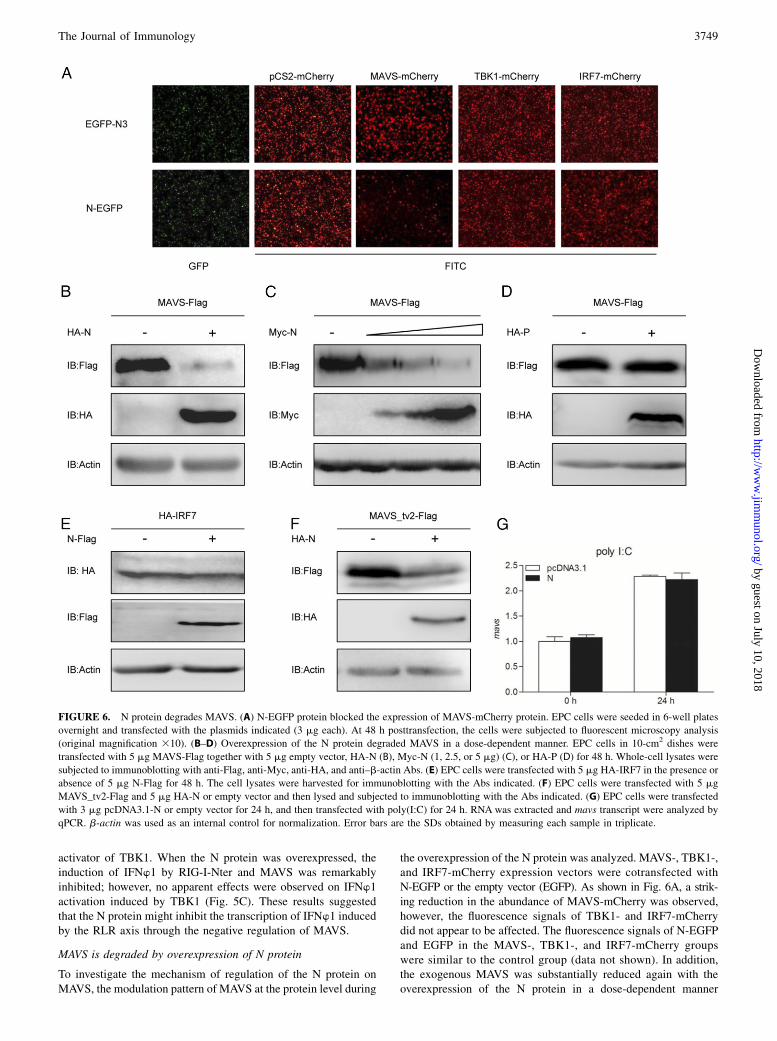
To investigate the mechanism of regulation of the N protein onMAVS, the modulation pattern of MAVS at the protein level during
the overexpression of the N protein was analyzed. MAVS-, TBK1-,and IRF7-mCherry expression vectors were cotransfected withN-EGFP or the empty vector (EGFP). As shown in Fig. 6A, a strik-ing reduction in the abundance of MAVS-mCherry was observed,however, the fluorescence signals of TBK1- and IRF7-mCherrydid not appear to be affected. The fluorescence signals of N-EGFPand EGFP in the MAVS-, TBK1-, and IRF7-mCherry groupswere similar to the control group (data not shown). In addition,the exogenous MAVS was substantially reduced again with theoverexpression of the N protein in a dose-dependent manner
FIGURE 6. N protein degrades MAVS. (A) N-EGFP protein blocked the expression of MAVS-mCherry protein. EPC cells were seeded in 6-well plates
overnight and transfected with the plasmids indicated (3 mg each). At 48 h posttransfection, the cells were subjected to fluorescent microscopy analysis
(original magnification 310). (B–D) Overexpression of the N protein degraded MAVS in a dose-dependent manner. EPC cells in 10-cm2 dishes were
transfected with 5 mg MAVS-Flag together with 5 mg empty vector, HA-N (B), Myc-N (1, 2.5, or 5 mg) (C), or HA-P (D) for 48 h. Whole-cell lysates were
subjected to immunoblotting with anti-Flag, anti-Myc, anti-HA, and anti–b-actin Abs. (E) EPC cells were transfected with 5 mg HA-IRF7 in the presence or
absence of 5 mg N-Flag for 48 h. The cell lysates were harvested for immunoblotting with the Abs indicated. (F) EPC cells were transfected with 5 mg
MAVS_tv2-Flag and 5 mg HA-N or empty vector and then lysed and subjected to immunoblotting with the Abs indicated. (G) EPC cells were transfected
with 3 mg pcDNA3.1-N or empty vector for 24 h, and then transfected with poly(I:C) for 24 h. RNA was extracted and mavs transcript were analyzed by
qPCR. b-actin was used as an internal control for normalization. Error bars are the SDs obtained by measuring each sample in triplicate.
The Journal of Immunology 3749
by guest on July 10, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

(Fig. 6B, 6C). As a control, the P protein of SVCV had little effecton MAVS (Fig. 6D). In contrast, the content of exogenous IRF7did not change with the overexpression of the N protein (Fig. 6E).Our previous study showed that a truncated MAVS (MAVS_tv2)in zebrafish, lacking the C-terminal transmembrane (TM) domain,could negatively regulate IRF7-mediated IFNw1 production (31).To further characterize the degradation region of MAVS,MAVS_tv2 was used. As shown in Fig. 6F, the abundance ofMAVS_tv2 also decreased when cotransfected with the N protein,suggesting that the TM domain of MAVS is not the target of the Nprotein. Consistent with the result that the transcription of mavswas not suppressed by SVCV, it was also not decreased by theoverexpression of the N protein (Fig. 6G). These results indicatedthat overexpression of the N protein of SVCV specifically pro-motes the degradation of MAVS at the protein level and the targetregion is beyond the TM domain.
N protein promotes ubiquitination of MAVS
The results above suggested that MAVS was degraded by SVCVin a proteasome-dependent manner. To delineate the mechanismsresponsible for the degradation of MAVS by the N protein ofSVCV, cells were cotransfected with MAVS-Flag and HA-N inthe presence of MG132. As shown in Fig.7A, the band of MAVS in
the cells coexpressing the N protein was weaker than that in thecontrol cells; however, the reduction of MAVS was significantlyrescued when treated with MG132. In addition, the repression ofMAVS was rescued with the increasing concentration of MG132,which also displayed a dose-dependent manner (Fig. 7B). Tofurther clarify whether MAVS was degraded by the Ub-proteasome pathway, MAVS-Flag, Myc-N, and HA-Ub werecotransfected in the presence or absence of MG132. Followingimmunoprecipitation of MAVS-Flag, immunoblot analysisrevealed that the N protein promoted the ubiquitination of MAVS(Fig. 7C). K48 and K63, the lysines at positions 48 and 63 ofubiquitin linked with polyubiquitin chains, are two canonicalpolyubiquitin chain linkages. A series of cases have reported thatthe target proteins were degraded by K48-linked polyubiquitinchains in a proteasome-dependent manner, whereas their functionswere stabilized and enhanced by K63-linked polyubiquitin chains(35–37). To dissect the pattern of ubiquitination of MAVS throughK48- or K63-linked ubiquitination, cells were cotransfected withMAVS-Flag, Myc-N, HA-Ub-K48O, or HA-Ub-K63O. As shownin Fig. 7D, the N protein promoted K48-linked ubiquitination ofMAVS but not K63-linked ubiquitination. These results indicatedthat the K48-linked Ub-proteasomal degradation of MAVS wastriggered by the N protein of SVCV.
FIGURE 7. N protein mediates ubiquitination of MAVS. (A) The N protein induced MAVS degradation was rescued in the presence of MG132. EPC
cells were transfected with 5 mg MAVS-Flag and 5 mg HA-N or empty vector. At 24 h posttransfection, the cells were treated with DMSO or MG132 for
6 h. Then, the cells were harvested for immunoblotting with anti-Flag, anti-HA, and anti–b-actin Abs. (B) The N protein–induced MAVS degradation was
rescued by MG132 in a dose-dependent manner. EPC cells were seeded in 6-well plates overnight and transfected with 1.5 mg MAVS-Flag and 1.5 mg
HA-N or empty vector. At 24 h posttransfection, the cells were treated with DMSO or MG132 (10, 20, or 40 mM) for 6 h. Then, the cells were harvested for
immunoblotting with the Abs indicated. (C) The N protein promoted the ubiquitination of MAVS. EPC cells were transfected with 5 mg MAVS-Flag, 4 mg
Myc-N or empty vector, and 1 mg HA-Ub. At 18 h posttransfection, the cells were treated with DMSO or MG132 for 6 h. Cell lysates were immuno-
precipitated (IP) with anti-Flag–agarose conjugate and immunoblotted with anti-HA and anti-Flag Abs. The whole-cell lysates (WCL) were immunoblotted
with the Abs indicated. (D) The N protein mediated K48-linked ubiquitination of MAVS in vivo. EPC cells were transfected with 5 mg MAVS-Flag, 4 mg
Myc-N or empty vector, and 1 mg HA-Ub-K48O or HA-Ub-K63O. At 18 h posttransfection, the cells were treated with DMSO or MG132 for 6 h. At 24 h
posttransfection, immunoprecipitation and immunoblot analyses were performed with the Abs indicated. The WCL were immunoblotted with the Abs
indicated.
3750 SVCV N PROTEIN SUPPRESSES IFNw1 BY DEGRADING MAVS
by guest on July 10, 2018http://w
ww
.jimm
unol.org/D
ownloaded from
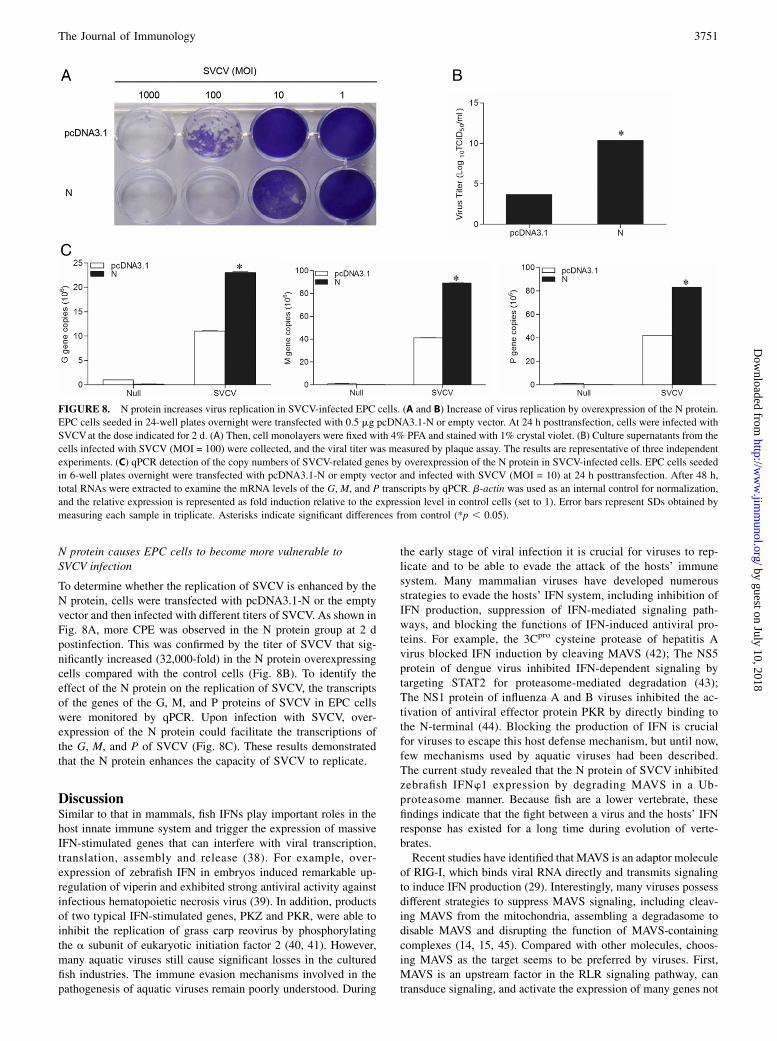
N protein causes EPC cells to become more vulnerable toSVCV infection
To determine whether the replication of SVCV is enhanced by theN protein, cells were transfected with pcDNA3.1-N or the emptyvector and then infected with different titers of SVCV. As shown inFig. 8A, more CPE was observed in the N protein group at 2 dpostinfection. This was confirmed by the titer of SVCV that sig-nificantly increased (32,000-fold) in the N protein overexpressingcells compared with the control cells (Fig. 8B). To identify theeffect of the N protein on the replication of SVCV, the transcriptsof the genes of the G, M, and P proteins of SVCV in EPC cellswere monitored by qPCR. Upon infection with SVCV, over-expression of the N protein could facilitate the transcriptions ofthe G, M, and P of SVCV (Fig. 8C). These results demonstratedthat the N protein enhances the capacity of SVCV to replicate.
DiscussionSimilar to that in mammals, fish IFNs play important roles in thehost innate immune system and trigger the expression of massiveIFN-stimulated genes that can interfere with viral transcription,translation, assembly and release (38). For example, over-expression of zebrafish IFN in embryos induced remarkable up-regulation of viperin and exhibited strong antiviral activity againstinfectious hematopoietic necrosis virus (39). In addition, productsof two typical IFN-stimulated genes, PKZ and PKR, were able toinhibit the replication of grass carp reovirus by phosphorylatingthe a subunit of eukaryotic initiation factor 2 (40, 41). However,many aquatic viruses still cause significant losses in the culturedfish industries. The immune evasion mechanisms involved in thepathogenesis of aquatic viruses remain poorly understood. During
the early stage of viral infection it is crucial for viruses to rep-licate and to be able to evade the attack of the hosts’ immunesystem. Many mammalian viruses have developed numerousstrategies to evade the hosts’ IFN system, including inhibition ofIFN production, suppression of IFN-mediated signaling path-ways, and blocking the functions of IFN-induced antiviral pro-teins. For example, the 3Cpro cysteine protease of hepatitis Avirus blocked IFN induction by cleaving MAVS (42); The NS5protein of dengue virus inhibited IFN-dependent signaling bytargeting STAT2 for proteasome-mediated degradation (43);The NS1 protein of influenza A and B viruses inhibited the ac-tivation of antiviral effector protein PKR by directly binding tothe N-terminal (44). Blocking the production of IFN is crucialfor viruses to escape this host defense mechanism, but until now,few mechanisms used by aquatic viruses had been described.The current study revealed that the N protein of SVCV inhibitedzebrafish IFNw1 expression by degrading MAVS in a Ub-proteasome manner. Because fish are a lower vertebrate, thesefindings indicate that the fight between a virus and the hosts’ IFNresponse has existed for a long time during evolution of verte-brates.Recent studies have identified that MAVS is an adaptor molecule
of RIG-I, which binds viral RNA directly and transmits signalingto induce IFN production (29). Interestingly, many viruses possessdifferent strategies to suppress MAVS signaling, including cleav-ing MAVS from the mitochondria, assembling a degradasome todisable MAVS and disrupting the function of MAVS-containingcomplexes (14, 15, 45). Compared with other molecules, choos-ing MAVS as the target seems to be preferred by viruses. First,MAVS is an upstream factor in the RLR signaling pathway, cantransduce signaling, and activate the expression of many genes not
FIGURE 8. N protein increases virus replication in SVCV-infected EPC cells. (A and B) Increase of virus replication by overexpression of the N protein.
EPC cells seeded in 24-well plates overnight were transfected with 0.5 mg pcDNA3.1-N or empty vector. At 24 h posttransfection, cells were infected with
SVCVat the dose indicated for 2 d. (A) Then, cell monolayers were fixed with 4% PFA and stained with 1% crystal violet. (B) Culture supernatants from the
cells infected with SVCV (MOI = 100) were collected, and the viral titer was measured by plaque assay. The results are representative of three independent
experiments. (C) qPCR detection of the copy numbers of SVCV-related genes by overexpression of the N protein in SVCV-infected cells. EPC cells seeded
in 6-well plates overnight were transfected with pcDNA3.1-N or empty vector and infected with SVCV (MOI = 10) at 24 h posttransfection. After 48 h,
total RNAs were extracted to examine the mRNA levels of the G, M, and P transcripts by qPCR. b-actin was used as an internal control for normalization,
and the relative expression is represented as fold induction relative to the expression level in control cells (set to 1). Error bars represent SDs obtained by
measuring each sample in triplicate. Asterisks indicate significant differences from control (*p , 0.05).
The Journal of Immunology 3751
by guest on July 10, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

only IFN, which can provide a more powerful antiviral response(46, 47). Second, the transcription, translation, and assembly ofSVCV is carried out in cytoplasm. Therefore, selecting a hostcytoplasmic protein such as MAVS as the target will avoidencoding a nuclear localization signal and transferring this intothe nucleus. Third, MAVS functions not only on the IFN systembut also on inducing host cell apoptosis. It was reported thatMAVS could be cleaved by dengue virus to retard host cellapoptosis and to maximize the use of the intracellular resource(48). Finally, MAVS is localized to the outer membrane ofmitochondria that are crucial organelles responsible for ATPproduction (49). Degradation of MAVS might hinder thefunction of mitochondria and make host cells unable to pro-vide energy for the antiviral system that could also decreasepotency.One of the host immune molecules could be the target of
several viruses or one virus could also possess a number ofdifferent strategies to elude host defense mechanisms. Previousstudies demonstrated that SVCV induced the autophagy of hostcells and upregulated the expression of related genes (23). Inthe current study, the N protein inhibited IFN expression bydegrading MAVS, which provided another strategy for SVCVto resist the host defense system. Among the other proteins ofSVCV, the M protein has been reported to interfere with theposttranscriptional machinery of the host, such as blocking thenuclear transport of spliced mRNAs and small nuclear RNAsand to slow the nuclear transport of many other molecules (50).In addition, our initial study showed that the P protein couldsuppress the activation of IFNw1 induced by poly(I:C) (data notshown). RNA viruses appear to use one protein to antagonizethe IFN response by different strategies because of their rela-tively limited genome capacity (51–54). As an RNA virus caus-ing significant mortality of fish, the functions of the differentproteins of SVCV should be studied further.In mammals, several viruses regulate the components of the
IFN signal transduction pathway through ubiquitination orphosphorylation (55, 56). Ubiquitination is a reversible cova-lent modification that regulates the stability, activity, and lo-calization of target proteins. The polyubiquitin chains linkedthrough lysine at position 48 of ubiquitin (K48) target proteinsfor proteasomal degradation. For example, AIP4 catalyzed theK48-linked ubiquitination of MAVS, in cooperation withPCBP2, leading to the degradation of MAVS (57), whereasK63-linked ubiquitination usually carries out signaling func-tions independent of proteolysis. For example, TRIM25 tar-geted RIG-I for K63-linked ubiquitination and enhanced thebinding of RIG-I to MAVS (58). In the current study, the Nprotein degraded MAVS that underwent K48-linked ubiq-uitination. However, the coimmunoprecipitation failed to showdirect interaction between the N protein and MAVS (data notshown). Thus, our working hypothesis is that the N proteinwould recruit an E3 ligase or enhance its activity and thentrigger the ubiquitination of MAVS. However, recent studiesrevealed that several E3 ligases were involved in SVCV in-fection (24); future studies should find out the exact E3 ligase(s)related with the N protein and the K48-linked ubiquitinationresidues of MAVS.In conclusion, the current study revealed a potential vital
mechanism used by the N protein of SVCV to negatively regulateMAVS through the Ub-proteasome degradation pathway (Fig. 9).These data provide new insights into the strategy of an aquaticvirus to evade host innate immunity in a lower vertebrate. Furtherstudies should focus on the functions of other proteins of SVCV inimmune evasion.
AcknowledgmentsWe thank Dr. Hong-Bing Shu (Wuhan University, Wuhan, China) for pro-
viding plasmids (HA-Ub, HA-Ub-K48O, and HA-Ub-K63O) and Dr. Xing
Liu (Institute of Hydrobiology) for providing vector (pCMV-Myc).
DisclosuresThe authors have no financial conflicts of interest.
References1. Kawai, T., and S. Akira. 2006. Innate immune recognition of viral infection. Nat.
Immunol. 7: 131–137.2. Takeuchi, O., and S. Akira. 2007. Recognition of viruses by innate immunity.
Immunol. Rev. 220: 214–224.3. Akira, S., S. Uematsu, and O. Takeuchi. 2006. Pathogen recognition and innate
immunity. Cell 124: 783–801.4. Mogensen, T. H. 2009. Pathogen recognition and inflammatory signaling in
innate immune defenses. Clin. Microbiol. Rev. 22: 240–273.5. Yoneyama, M., M. Kikuchi, T. Natsukawa, N. Shinobu, T. Imaizumi,
M. Miyagishi, K. Taira, S. Akira, and T. Fujita. 2004. The RNA helicase RIG-Ihas an essential function in double-stranded RNA‑induced innate antiviral re-sponses. Nat. Immunol. 5: 730–737.
6. Kawai, T., K. Takahashi, S. Sato, C. Coban, H. Kumar, H. Kato, K. J. Ishii,O. Takeuchi, and S. Akira. 2005. IPS-1, an adaptor triggering RIG-I‑ and Mda5-mediated type I interferon induction. Nat. Immunol. 6: 981–988.
7. Meylan, E., J. Curran, K. Hofmann, D. Moradpour, M. Binder,R. Bartenschlager, and J. Tschopp. 2005. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 437: 1167–1172.
8. Xu, L. G., Y. Y. Wang, K. J. Han, L. Y. Li, Z. Zhai, and H. B. Shu. 2005. VISA isan adapter protein required for virus-triggered IFN-b signaling. Mol. Cell 19:727–740.
9. Seth, R. B., L. Sun, C. K. Ea, and Z. J. Chen. 2005. Identification and charac-terization of MAVS, a mitochondrial antiviral signaling protein that activatesNF-kB and IRF 3. Cell 122: 669–682.
10. Honda, K., A. Takaoka, and T. Taniguchi. 2006. Type I interferon [corrected]gene induction by the interferon regulatory factor family of transcription factors.Immunity 25: 349–360.
11. Correia, S., S. Ventura, and R. M. Parkhouse. 2013. Identification and utility ofinnate immune system evasion mechanisms of ASFV. Virus Res. 173: 87–100.
12. Li, X. D., L. Sun, R. B. Seth, G. Pineda, and Z. J. Chen. 2005. Hepatitis C virusprotease NS3/4A cleaves mitochondrial antiviral signaling protein off the mi-
FIGURE 9. The degradation model of MAVS mediated by the N protein
of SVCV. Upon SVCV infection, fish RIG-I recognizes the viral RNA and
recruits MAVS and TBK1, leading to activation of IRF3/7, which induces
IFN expression. Meanwhile, SVCV-encoded N protein triggers the ubiq-
uitination and degradation of MAVS, blocking the MAVS-induced IFN
activation. Namely, the N protein of SVCV inhibits host IFN induction by
degrading MAVS through Ub-proteasome pathway.
3752 SVCV N PROTEIN SUPPRESSES IFNw1 BY DEGRADING MAVS
by guest on July 10, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

tochondria to evade innate immunity. Proc. Natl. Acad. Sci. USA 102: 17717–17722.
13. Mukherjee, A., S. A. Morosky, E. Delorme-Axford, N. Dybdahl-Sissoko,M. S. Oberste, T. Wang, and C. B. Coyne. 2011. The coxsackievirus B 3Cprotease cleaves MAVS and TRIF to attenuate host type I interferon and apo-ptotic signaling. PLoS Pathog. 7: e1001311.
14. Wei, C., C. Ni, T. Song, Y. Liu, X. Yang, Z. Zheng, Y. Jia, Y. Yuan, K. Guan,Y. Xu, et al. 2010. The hepatitis B virus X protein disrupts innate immunity bydownregulating mitochondrial antiviral signaling protein. J. Immunol. 185:1158–1168.
15. Shi, C. S., H. Y. Qi, C. Boularan, N. N. Huang, M. Abu-Asab, J. H. Shelhamer,and J. H. Kehrl. 2014. SARS-coronavirus open reading frame-9b suppressesinnate immunity by targeting mitochondria and the MAVS/TRAF3/TRAF6signalosome. J. Immunol. 193: 3080–3089.
16. Ran, X., X. Bian, Y. Ji, X. Yan, F. Yang, and F. Li. 2013. White spot syndromevirus IE1 and WSV056 modulate the G1/S transition by binding to the hostretinoblastoma protein. J. Virol. 87: 12576–12582.
17. Ahne, W., H. V. Bjorklund, S. Essbauer, N. Fijan, G. Kurath, and J. R. Winton.2002. Spring viremia of carp (SVC). Dis. Aquat. Organ. 52: 261–272.
18. Teng, Y., H. Liu, J. Q. Lv, W. H. Fan, Q. Y. Zhang, and Q. W. Qin. 2007.Characterization of complete genome sequence of the spring viremia of carpvirus isolated from common carp (Cyprinus carpio) in China. Arch. Virol. 152:1457–1465.
19. Walker, P. J., R. G. Dietzgen, D. A. Joubert, and K. R. Blasdell. 2011. Rhab-dovirus accessory genes. Virus Res. 162: 110–125.
20. Bjorklund, H. V., T. R. Johansson, and A. Rinne. 1997. Rhabdovirus-inducedapoptosis in a fish cell line is inhibited by a human endogenous acid cysteineproteinase inhibitor. J. Virol. 71: 5658–5662.
21. Gotesman, M., H. Soliman, R. Besch, and M. El-Matbouli. 2015. Inhibition ofspring viraemia of carp virus replication in an Epithelioma papulosum cyprinicell line by RNAi. J. Fish Dis. 38: 197–207.
22. Encinas, P., P. Garcia-Valtanen, B. Chinchilla, E. Gomez-Casado, A. Estepa, andJ. Coll. 2013. Identification of multipath genes differentially expressed inpathway-targeted microarrays in zebrafish infected and surviving spring viremiacarp virus (SVCV) suggest preventive drug candidates. PLoS One 8: e73553.
23. Liu, L., B. Zhu, S. Wu, L. Lin, G. Liu, Y. Zhou, W. Wang, M. Asim, J. Yuan,L. Li, et al. 2015. Spring viraemia of carp virus induces autophagy for necessaryviral replication. Cell. Microbiol. 17: 595–605.
24. Yuan, J., Y. Yang, H. Nie, L. Li, W. Gu, L. Lin, M. Zou, X. Liu, M. Wang, andZ. Gu. 2014. Transcriptome analysis of epithelioma papulosum cyprini cellsafter SVCV infection. BMC Genomics 15: 935.
25. Liu, L., Q. Li, L. Lin, M. Wang, Y. Lu, W. Wang, J. Yuan, L. Li, and X. Liu.2013. Proteomic analysis of epithelioma papulosum cyprini cells infected withspring viremia of carp virus. Fish Shellfish Immunol. 35: 26–35.
26. Adamek, M., K. L. Rakus, J. Chyb, G. Brogden, A. Huebner, I. Irnazarow, andD. Steinhagen. 2012. Interferon type I responses to virus infections in carp cells:In vitro studies on Cyprinid herpesvirus 3 and Rhabdovirus carpio infections.Fish Shellfish Immunol. 33: 482–493.
27. Lopez-Munoz, A., F. J. Roca, J. Meseguer, and V. Mulero. 2009. New insightsinto the evolution of IFNs: zebrafish group II IFNs induce a rapid and transientexpression of IFN-dependent genes and display powerful antiviral activities. J.Immunol. 182: 3440–3449.
28. Levraud, J. P., P. Boudinot, I. Colin, A. Benmansour, N. Peyrieras, P. Herbomel,and G. Lutfalla. 2007. Identification of the zebrafish IFN receptor: implicationsfor the origin of the vertebrate IFN system. J. Immunol. 178: 4385–4394.
29. Biacchesi, S., M. LeBerre, A. Lamoureux, Y. Louise, E. Lauret, P. Boudinot, andM. Bremont. 2009. Mitochondrial antiviral signaling protein plays a major rolein induction of the fish innate immune response against RNA and DNA viruses.J. Virol. 83: 7815–7827.
30. Zhang, J., Y. B. Zhang, M. Wu, B. Wang, C. Chen, and J. F. Gui. 2014. FishMAVS is involved in RLR pathway-mediated IFN response. Fish ShellfishImmunol. 41: 222–230.
31. Lu, L. F., S. Li, X. B. Lu, and Y. A. Zhang. 2015. Functions of the two zebrafishMAVS variants are opposite in the induction of IFN1 by targeting IRF7. FishShellfish Immunol. 45: 574–582.
32. Li, S., L. F. Lu, H. Feng, N. Wu, D. D. Chen, Y. B. Zhang, J. F. Gui, P. Nie, andY. A. Zhang. 2014. IFN regulatory factor 10 is a negative regulator of the IFNresponses in fish. J. Immunol. 193: 1100–1109.
33. Sun, F., Y. B. Zhang, T. K. Liu, J. Shi, B. Wang, and J. F. Gui. 2011. Fish MITAserves as a mediator for distinct fish IFN gene activation dependent on IRF3 orIRF7. J. Immunol. 187: 2531–2539.
34. Hiscott, J., T. L. Nguyen, M. Arguello, P. Nakhaei, and S. Paz. 2006. Manipu-lation of the nuclear factor-kB pathway and the innate immune response byviruses. Oncogene 25: 6844–6867.
35. Zhong, B., L. Zhang, C. Lei, Y. Li, A. P. Mao, Y. Yang, Y. Y. Wang, X. L. Zhang,and H. B. Shu. 2009. The ubiquitin ligase RNF5 regulates antiviral responses bymediating degradation of the adaptor protein MITA. Immunity 30: 397–407.
36. Tsuchida, T., J. Zou, T. Saitoh, H. Kumar, T. Abe, Y. Matsuura, T. Kawai, andS. Akira. 2010. The ubiquitin ligase TRIM56 regulates innate immune responsesto intracellular double-stranded DNA. Immunity 33: 765–776.
37. Ye, J. S., N. Kim, K. J. Lee, Y. R. Nam, U. Lee, and C. H. Joo. 2014. Lysine 63-linked TANK-binding kinase 1 ubiquitination by mindbomb E3 ubiquitin proteinligase 2 is mediated by the mitochondrial antiviral signaling protein. J. Virol. 88:12765–12776.
38. Zou, J., and C. J. Secombes. 2011. Teleost fish interferons and their role inimmunity. Dev. Comp. Immunol. 35: 1376–1387.
39. Aggad, D., M. Mazel, P. Boudinot, K. E. Mogensen, O. J. Hamming,R. Hartmann, S. Kotenko, P. Herbomel, G. Lutfalla, and J. P. Levraud. 2009. Thetwo groups of zebrafish virus-induced interferons signal via distinct receptorswith specific and shared chains. J. Immunol. 183: 3924–3931.
40. Zhu, R., Y. B. Zhang, Q. Y. Zhang, and J. F. Gui. 2008. Functional domains andthe antiviral effect of the double-stranded RNA-dependent protein kinase PKRfrom Paralichthys olivaceus. J. Virol. 82: 6889–6901.
41. Liu, T. K., Y. B. Zhang, Y. Liu, F. Sun, and J. F. Gui. 2011. Cooperative roles offish protein kinase containing Z-DNA binding domains and double-strandedRNA-dependent protein kinase in interferon-mediated antiviral response. J.Virol. 85: 12769–12780.
42. Yang, Y., Y. Liang, L. Qu, Z. Chen, M. Yi, K. Li, and S. M. Lemon. 2007.Disruption of innate immunity due to mitochondrial targeting of a picornaviralprotease precursor. Proc. Natl. Acad. Sci. USA 104: 7253–7258.
43. Ashour, J., M. Laurent-Rolle, P. Y. Shi, and A. Garcıa-Sastre. 2009. NS5 ofdengue virus mediates STAT2 binding and degradation. J. Virol. 83: 5408–5418.
44. Li, S., J. Y. Min, R. M. Krug, and G. C. Sen. 2006. Binding of the influenza Avirus NS1 protein to PKR mediates the inhibition of its activation by eitherPACT or double-stranded RNA. Virology 349: 13–21.
45. Goswami, R., T. Majumdar, J. Dhar, S. Chattopadhyay, S. K. Bandyopadhyay,V. Verbovetskaya, G. C. Sen, and S. Barik. 2013. Viral degradasome hijacksmitochondria to suppress innate immunity. Cell Res. 23: 1025–1042.
46. Lauksund, S., T. Svingerud, V. Bergan, and B. Robertsen. 2009. Atlantic salmonIPS-1 mediates induction of IFNa1 and activation of NF-kB and localizes tomitochondria. Dev. Comp. Immunol. 33: 1196–1204.
47. Sun, Q., L. Sun, H. H. Liu, X. Chen, R. B. Seth, J. Forman, and Z. J. Chen. 2006.The specific and essential role of MAVS in antiviral innate immune responses.Immunity 24: 633–642.
48. Yu, C. Y., R. L. Chiang, T. H. Chang, C. L. Liao, and Y. L. Lin. 2010. Theinterferon stimulator mitochondrial antiviral signaling protein facilitates celldeath by disrupting the mitochondrial membrane potential and by activatingcaspases. J. Virol. 84: 2421–2431.
49. Paumard, P., J. Vaillier, B. Coulary, J. Schaeffer, V. Soubannier, D. M. Mueller,D. Brethes, J. P. di Rago, and J. Velours. 2002. The ATP synthase is involved ingenerating mitochondrial cristae morphology. EMBO J. 21: 221–230.
50. Petersen, J. M., L. S. Her, and J. E. Dahlberg. 2001. Multiple vesiculoviralmatrix proteins inhibit both nuclear export and import. Proc. Natl. Acad. Sci.USA 98: 8590–8595.
51. Min, J. Y., and R. M. Krug. 2006. The primary function of RNA binding by theinfluenza A virus NS1 protein in infected cells: Inhibiting the 29-59 oligo (A)synthetase/RNase L pathway. Proc. Natl. Acad. Sci. USA 103: 7100–7105.
52. Talon, J., C. M. Horvath, R. Polley, C. F. Basler, T. Muster, P. Palese, andA. Garcıa-Sastre. 2000. Activation of interferon regulatory factor 3 is inhibitedby the influenza A virus NS1 protein. J. Virol. 74: 7989–7996.
53. Wang, X., M. Li, H. Zheng, T. Muster, P. Palese, A. A. Beg, and A. Garcıa-Sastre. 2000. Influenza A virus NS1 protein prevents activation of NF-kB andinduction of a/b interferon. J. Virol. 74: 11566–11573.
54. Mibayashi, M., L. Martınez-Sobrido, Y. M. Loo, W. B. Cardenas, M. Gale, Jr.,and A. Garcıa-Sastre. 2007. Inhibition of retinoic acid‑inducible gene I‑mediatedinduction of b interferon by the NS1 protein of influenza A virus. J. Virol. 81:514–524.
55. Hilton, L., K. Moganeradj, G. Zhang, Y. H. Chen, R. E. Randall,J. W. McCauley, and S. Goodbourn. 2006. The NPro product of bovine viraldiarrhea virus inhibits DNA binding by interferon regulatory factor 3 and targetsit for proteasomal degradation. J. Virol. 80: 11723–11732.
56. Versteeg, G. A., and A. Garcıa-Sastre. 2010. Viral tricks to grid-lock the type Iinterferon system. Curr. Opin. Microbiol. 13: 508–516.
57. You, F., H. Sun, X. Zhou, W. Sun, S. Liang, Z. Zhai, and Z. Jiang. 2009. PCBP2mediates degradation of the adaptor MAVS via the HECT ubiquitin ligase AIP4.Nat. Immunol. 10: 1300–1308.
58. Gack, M. U., Y. C. Shin, C. H. Joo, T. Urano, C. Liang, L. Sun, O. Takeuchi,S. Akira, Z. Chen, S. Inoue, and J. U. Jung. 2007. TRIM25 RING-finger E3ubiquitin ligase is essential for RIG-I‑mediated antiviral activity. Nature 446:916–920.
The Journal of Immunology 3753
by guest on July 10, 2018http://w
ww
.jimm
unol.org/D
ownloaded from