special feature the 2010 nephrology quiz and questionnaire...
TRANSCRIPT

The 2010 Nephrology Quiz and Questionnaire: Part 2Richard J. Glassock (Co-Moderator),* Anthony J. Bleyer (Co-Moderator),† Joanne M. Bargman (Discussant),‡
and Fernando C. Fervenza (Discussant)§
Presentation of the Nephrology Quiz and Questionnaire (NQQ) has become an annual “tradition” at the meetings ofthe American Society of Nephrology. It is a very popular session judged by consistently large attendance. Members ofthe audience test their knowledge and judgment on a series of case-oriented questions prepared and discussed byexperts. They can also compare their answers in real time, using audience response devices, to those of programdirectors of nephrology training programs in the United States, acquired through an Internet-based questionnaire.As in the past, the topics covered were transplantation, fluid and electrolyte disorders, end-stage renal disease anddialysis, and glomerular disorders. Two challenging cases representing each of these categories along with single bestanswer questions were prepared by a panel of experts (Drs. Hricik, Palmer, Bargman, and Fervenza, respectively). The“correct” and “incorrect” answers then were briefly discussed, after the audience responses and the results of thequestionnaire were displayed. The 2010 version of the NQQ was exceptionally challenging, and the audience, for thefirst time, gained a better overall correct answer score than the program directors, but the margin was small. Lastmonth we presented the transplantation and fluid and the electrolyte cases; in this issue we present the remainingend-stage renal disease and dialysis and the glomerular disorder cases. These articles try to recapitulate the sessionand reproduce its educational value for a larger audience—that of the readers of the Clinical Journal of the AmericanSociety of Nephrology. Have fun.
Clin J Am Soc Nephrol 6: 2534–2547, 2011. doi: 10.2215/CJN.06500711
ESRD and Dialysis Case 1: Joanne M. Bargman(Discussant)
The patient is a 47-year-old woman of African-Caribbean descent who has been on conventionalthrice-weekly hemodialysis since 1997. The cause ofher kidney disease was hypertensive nephrosclerosis.She was refused listing for a deceased donor kidneytransplant because of problems with compliance. Thepatient refused attempts at permanent vascular accessuntil 2005, when she had a left brachio-cephalic arte-rio-venous (AV) fistula constructed. However, shedeveloped stenoses of the left cephalic, subclavian,and innominate veins, and a right-sided tunneled in-ternal jugular catheter was placed through which shehas dialyzed for the last 4 years. Other past medicalhistory included a ruptured appendix with surgicalperitonitis in 2000 and a recent breast lump for whicha biopsy was pending.
During a routine hemodialysis session, the patient com-plained of the sudden onset of severe headache and diz-ziness. This was followed by an acute deterioration in herlevel of consciousness (Glasgow Coma Scale score 11/15)without any lateralizing signs or focal deficits. The patientwas afebrile, and the vital signs were normal.
ESRD and Dialysis Question 1AWhich ONE of the following is the MOST likely cause
of the sudden change in her sensorium (Figure 1)?A. Internal jugular catheter-related sepsis.B. Acute intravascular hemolysis.C. Acute intracranial hypertension.D. Pulmonary leukostasis syndrome.E. Dialysis disequilibrium syndrome.
ESRD and Dialysis Question 1BWhich ONE of the following would be the MOST
appropriate immediate action (after protecting air-way) (Figure 2)?
A. Blood cultures.B. Hemodialysis line change over a guide wire.C. Lengthen the hemodialysis session by 1 hour to
optimize solute clearance.D. Neurology opinion.E. Computed tomography (CT) of the brain.
Discussion of Case 1 (Questions 1A and 1B)The correct answer for Question 1A is C. The sud-
den occurrence during the hemodialysis sessionstrongly suggests that the neurologic event is relatedto the dialysis process itself. However, as can be seenfrom the choices in Question 1A, there are multipleadverse events that can occur during hemodialysis.The best choice would be an event that leads to ageneralized decline in sensorium without focal neu-rologic deficit or other symptoms and signs, includingshortness of breath or fever. The patient complainedof severe headache soon after the initiation of hemo-dialysis, which was followed by decline in level ofconsciousness to precoma. Although the differentialdiagnosis is broad, the symptoms and signs suggestraised intracranial pressure.
What is it about the hemodialysis session that couldhave increased the intracranial pressure? With the atten-dant anticoagulation during hemodialysis, the patient isat risk for an intracranial bleed or subarachnoid hemor-rhage. The patient under discussion had a newly diag-nosed breast mass awaiting biopsy. If this mass werecancerous, it is conceivable that she could have had an
*David Geffen Schoolof Medicine at UCLA,Los Angeles, California;†Wake Forest UniversitySchool of Medicine,Winston-Salem, NorthCarolina; ‡Division ofNephrology, UniversityHealth Network,Toronto, Canada; and§Division ofNephrology andHypertension, MayoClinic, Rochester,Minnesota
Correspondence: Dr.Richard J. Glassock, 8Bethany, LagunaNiguel, CA 92677.Phone: 949-388-8885;Fax: 949-388-8882;E-mail: [email protected]
2534 Copyright © 2011 by the American Society of Nephrology www.cjasn.org Vol 6 October, 2011
Special Feature

intracranial bleed from a metastasis. Another explanation forintracranial hypertension, however, could be related to im-paired venous drainage from the brain. It was already knownfrom previous venous mapping studies that this patient hadstenoses of the left cephalic, subclavian, and innominateveins. That meant that the major drainage pathway couldoccur only through the right internal jugular vein to thesuperior vena cava, and this is where the dialysis catheterwas situated. If the right-sided venous drainage were com-promised, venous congestion and consequent intracranial hy-pertension could ensue.
Other hemodialysis-related emergencies have to be consid-ered in a patient with a sudden decline during the treatment.Options B, acute intravascular hemolysis, and D, pulmonaryleukostasis syndrome, are rare but important events that canoccur during dialysis. The former is characterized by circula-tory collapse accompanied by pinkish discoloration of thedialysate from erythrocyte destruction, and the latter byshortness of breath and hypoxemia (1). The patient, on theother hand, maintained good BP and had no breathing diffi-culties, and the organ dysfunction was clearly confined to thecentral nervous system. Therefore, options B and D are muchless likely. Dialysis disequilibrium can lead to altered level ofconsciousness but would be extremely unlikely in this patienton routine dialysis for years.
Line sepsis (Option A) unfortunately remains a seriouscomplication of hemodialysis. Well functioning arterio-venous fistulas cannot be successfully developed in manypatients on hemodialysis (2), and a high percentage of pa-tients, such as the one under discussion, continue to dialyze
via a tunneled internal jugular catheter. Any patient with thiskind of catheter presenting with fever on hemodialysis has agreater than 75% chance of having catheter-related bactere-mia (3), even in the absence of obvious exit site infection.Signs of infection can present either off or on dialysis, butcertainly the sudden onset of fever during a hemodialysisrun, especially if accompanied by chills or rigors, stronglysuggests line sepsis. The patient described in this case com-plained of headache, which could be a prodrome of sepsis,but proceeded to neurologic deterioration without fever orcirculatory collapse. Despite the very high prevalence of linesepsis as a complication of hemodialysis, the absence of fever,rigors, and chills argue against this diagnosis. However,given how common line sepsis is, our first consideration (andthat of many of the respondents in the quiz) was that thispresentation was a particularly severe form of catheter-re-lated sepsis.
The best answer for Question 1B is E. As discussedabove, the presentation was principally neurologic, and aCT would be important to rule out intracranial bleed,metastasis, cerebral edema, and other central nervous sys-tem catastrophes.
The CT scan showed generalized edema without any focallesions. The patient continued to have a decline in level ofconsciousness and needed intubation for airway protection.She was transferred to the Neuro Intensive Care Unit.
ESRD and Dialysis Question 1CWhich ONE of the following statements is MOST correct
(Figure 3)?A. She should be started on broad-spectrum antibiotics
for a presumed central nervous system bacterial infection.B. Anti-neutrophil cytoplasmic auto-antibodies should
be urgently measured to rule out vasculitis and pulsecorticosteroid given empirically.
C. She should be put on the priority list for deceaseddonor renal transplant.
D. She has superior vena cava syndrome and shouldhave dilation of the stenotic veins.
E. She is intolerant of hemodialysis and should be tran-sitioned to peritoneal dialysis.
Discussion of Case 1 (Question 1C)The most correct answer is D. If the reader thought that
the deterioration was a result of line sepsis, then broadspectrum antibiotics would be the obvious choice. Indeed,
Figure 3. | Answers for Case 1, Question 1C, ESRD and Dialysis.
Figure 1. | Answers for Case 1, Question 1A, ESRD and Dialysis.
Figure 2. | Answers for Case 1, Question 1B, ESRD and Dialysis.
Clin J Am Soc Nephrol 6: 2534–2547, October, 2011 Nephrology Quiz and Questionnaire, Glassock et al. 2535

in the face of such a catastrophic event, if the workingdiagnosis were line sepsis, it would be tempting to removeor exchange the line as more definitive therapy (reviewedin 4) in addition to antibiotics. Although vasculitis hasprotean manifestations, including those referable to thecentral nervous system, the presentation is not usually sodramatic unless there were an accompanying major bleedor thrombotic event, which was not seen on the CT scan.Furthermore, empiric high-dose corticosteroid would berisky in this patient without a working diagnosis. AnswersC and E suggest a change in modality to either deceaseddonor kidney transplant or peritoneal dialysis. Indeed, as a“long-term” issue, the patient appears to be running out ofvenous access for hemodialysis, so consideration should begiven to proactively transition to another form of renalreplacement therapy. However, given the history of com-pliance problems she may not be a candidate for transplantor a home-based dialysis modality. Furthermore, the pastepisode of surgical peritonitis (ruptured appendix) maymean that the patient does not have a usable peritonealcavity. In any case, modality change will not address thecurrent problem.
The superior vena cava (SVC) syndrome results fromobstruction of venous drainage from the head and upperlimbs. It typically presents with edema of the face andarms, plethora, or hyperpigmentation of the face. In severecases, there can be venous hypertension of the brain, lead-ing to cerebral edema. The magnetic resonance imagingstudy in this patient showed diffuse cerebral edema andearly tonsillar herniation. The cause of SVC syndrome haschanged over the last decades. It was originally describedwhen venous drainage was compressed by tumors or otherdiseases such as fibrosing mediastinitis from histoplasmo-sis or pressure from an aneurysm in syphilitic aortitis. Inthe modern era the cause is principally from the use ofintravascular devices leading to secondary venous steno-sis. Risk factors include longer duration of the intravascu-lar devices, multiple catheter insertions, subclavian veincatheters, and left (versus right) internal jugular catheters.The latter risk can be explained by the tortuous route thatthe catheter placed on the left has to traverse crossingmidline via the innominate vein to reach the SVC. Themechanism of stenosis is not clear, but may be related toabrasion of the device against the venous wall, leading toinflammation and consequent hyperplasia of the endothe-lial and vascular smooth muscle cells. Interestingly, how-ever, venous stenosis can occur distant from the catheteritself. The patient under discussion had stenoses of the leftcephalic, subclavian, and innominate veins, without everhaving indwelling catheters in those locations. These ve-nous stenoses may be a complication of distal fistulaecausing high flow and turbulence in the proximal stenoticvein.
The patient underwent CT angiography, which showedcomplete occlusion of the right internal jugular vein andsevere stenosis of the innominate vein. Pressure in theinnominate vein was 75/36 mmHg (normal: 5 to 10mmHg) proximal to the stenosis. The stenosis was crossedusing a guide wire. Venoplasty was performed using a14-mm followed by a 16-mm Atlas balloon (www.bardpv.com) (5). Repeat measurement of pressure in the innomi-
nate vein after dilation was 18/11 mmHg. After the pro-cedure the patient’s level of consciousness rapidly im-proved and a follow-up CT scan showed resolution of thecerebral edema.
ESRD and Dialysis Case 2: Joanne M. Bargman(Discussant)
The patient is a 74-year-old woman with CKD (stage 5)on the basis of diabetic nephropathy. She has been oncontinuous ambulatory peritoneal dialysis (PD) for the last4 years. In her third year she had an episode of peritonitisthat resolved quickly with antibiotics. Routine clinic visitsin her fourth year had been unremarkable. However, oneday the patient’s daughter called the PD unit to report aproblem with both inflow and outflow of the dialysate. Theyhad noticed the flow to be slower for the past 3 days, and onthe day of the phone call there was very little flow observed.The patient came to the unit and had a supine x-ray of theabdomen, which showed that the PD catheter was in goodposition in the deep pelvis. There was a moderate amount offeces in the colon. The patient was prescribed cathartics onthe assumption that constipation was the cause of the cathetermalfunction. However, even with several bowel movementsand follow-up radiography showing clearing of the colon,there was still very poor inflow and outflow.
ESRD and Dialysis Question 2AWhich ONE of the following is LEAST likely to be the
cause of the catheter dysfunction (Figure 4)?A. Constipation.B. Intraluminal clot of the PD catheter.C. Encapsulating peritoneal sclerosis (EPS).D. Kink in the PD catheter.E. Omental wrap of the catheter.
Discussion of Case 2 (Question 2A)The “correct” answer is C. EPS is a rare but devastating
complication of PD characterized by accelerated fibrosis andsclerosis of the peritoneal membrane and submesothelial tis-sue (7,8). In advanced cases “cocooning” of the bowel ensues,leading to recurrent bowel obstruction and malnutrition.Therapy with anti-inflammatory and antifibrotic agents hasbeen used, and in the worst cases lysis of surgical adhesionsand capsule release has been undertaken.
Poor catheter flow should be approached as “one-way” or“two-way” obstruction. The patient described has two-way
Figure 4. | Answers for Case 2, Question 2A, ESRD and Dialysis.
2536 Clinical Journal of the American Society of Nephrology

obstruction (no inflow or outflow). This kind of obstruction istypically caused by narrowing or obliteration of the catheterlumen. Examples include a blood clot in the lumen or a kinkin the catheter itself. The more common problem is one-wayobstruction, where there is satisfactory inflow of dialysisfluid, but sluggish or no outflow. The commonest causes ofone-way obstruction are constipation or wrapping of theomentum around the catheter. In both instances the flow ofdialysis fluid out of the catheter into the peritoneal cavity issufficient to push the blockage (colon filled with feces, omen-tum) away. However, when the fluid is drained out of theperitoneal cavity, the suction draws the impediment upagainst the catheter, resulting in compromise to dialysateoutflow. Other causes of one-way obstruction include thefirst-ever infusion of dialysis fluid, which may not drainbecause of its movement into the para-colonic gutters, fillingup the peritoneal “dead-space,” and leaks of dialysate out ofthe peritoneal cavity (for example, into the pleural spacethorough subdiaphragmatic stomata). Because the patienthad two-way obstruction, the most likely diagnosis was ei-ther a kink in the catheter or intraluminal obstruction.
Case 2 additional findings: The patient, who is nowanuric, has now been 4 days without dialysis. She feelswell, but her BP is 150/105 mmHg, and her potassium isnow 6.0 mEq/L.
ESRD and Dialysis Question 2BALL of the following steps could be taken EXCEPT
which ONE (Figure 5)?A. Catheter study with radiocontrast media.B. Immediate catheter placement for hemodialysis.C. Oral potassium exchange resin for the hyperkalemia.D. Peritoneal dialysis catheter removal and replacement.E. Addition of an angiotensin converting enzyme inhib-
itor for the hypertension.Note: This question was not included in the Program
Director’s Questionnaire.The “correct” but wrong answer is E. Although the BP is
higher than ideal, it is temporary and will resolve withresumption of dialysis. Furthermore, there is a risk thatrenin-angiotensin system inhibition may worsen the hy-perkalemia (9,10), although this is not consistently re-ported (11,12). All of the other choices are reasonable forthis patient. She was very opposed to temporary hemodi-alysis, so attempts were made to try to repair and returnfunction to the PD catheter. The patient underwent a cath-
eter radiocontrast study which, surprisingly, showed apatent PD catheter. Radiocontrast material injected throughthe catheter traveled freely into the peritoneal cavity withno evidence of intraluminal obstruction. Because we wereunable to elucidate the cause of the catheter dysfunction,the patient was scheduled for an urgent PD catheter re-moval and replacement the next day.
ESRD and Dialysis Question 2CIn the meantime, one of the PD nurses suggests which
ONE of the following (Figure 6)?A. Instill tissue plasminogen activator (tPA) into the
catheter lumen.B. Change the catheter transfer set and connector.C. Use a stronger cathartic.D. Change from a coiled to a straight intraperitoneal
portion of the catheter when it is replaced.E. Use icodextrin instead of conventional peritoneal di-
alysis solution.
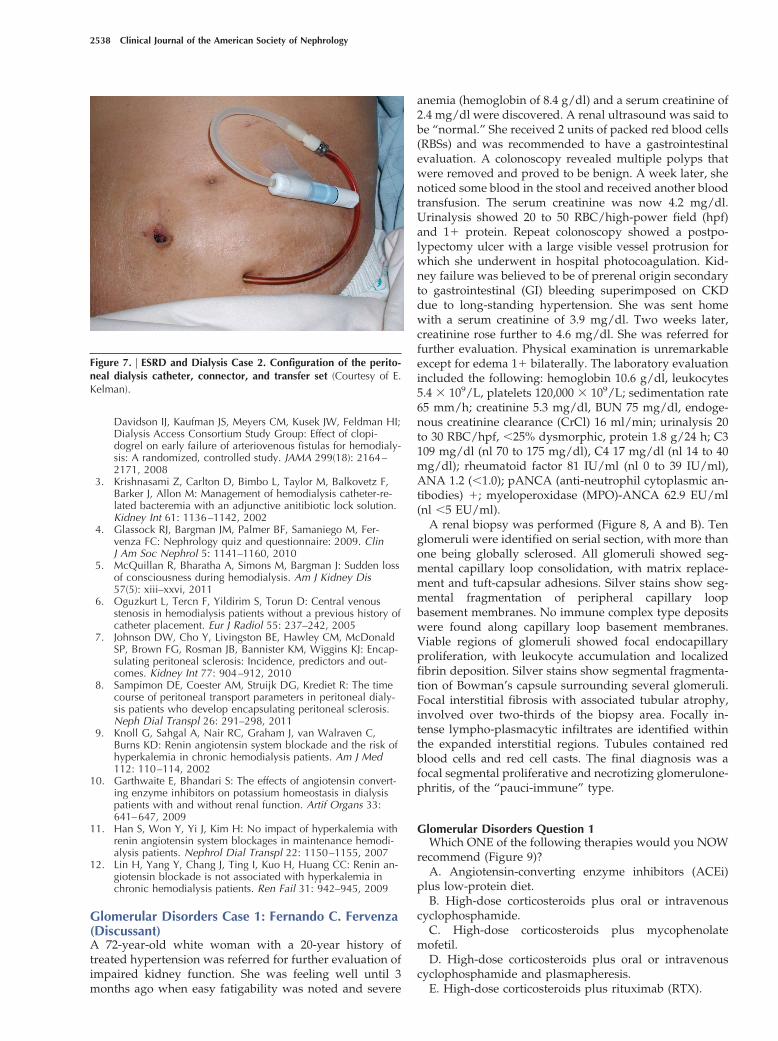
Discussion of Case 2 (Question 2C)The correct answer is B (see Figure 7). The transfer set
was removed and a flush of dialysate through the cathetershowed normal inflow and outflow. The transfer set wasreplaced with a new one, and inflow and outflow re-mained unimpeded. The problem that caused the obstruc-tion in the first place was an accumulation of fibrin in thetransfer set. Interestingly, for the radiocontrast study, theradiologist removed the transfer set to inject the dye, andthat is why the study showed normal inflow. However, theold transfer set was put back onto the catheter, and theproblem recurred. Note that the strong majority of trainingprogram directors chose instillation of tPA, whereas theradiocontrast study showed that there was no intraluminalobstruction. The patient resumed her PD and continues todo well on this therapy.
DisclosuresNone.
References1. Salem M, Invanovich PT, Ing TS, Daugirdas JT: Adverse
events of dialyzers manifesting during the dialysis session.Neph Dial Transpl 9 [Suppl 2]: 127–137, 1994
2. Dember LM, Beck GJ, Allon M, Delmez JA, Dixon BS,Greenberg A, Himmelfarb J, Vazquez MA, Gassman JJ,Greene T, Radeva MK, Braden GL, Ikizler TA, Rocco MV,
Figure 5. | Answers for Case 2, Question 2B, ESRD and Dialysis. Figure 6. | Answers for Case 2, Question 2C, ESRD and Dialysis.
Clin J Am Soc Nephrol 6: 2534–2547, October, 2011 Nephrology Quiz and Questionnaire, Glassock et al. 2537
Davidson IJ, Kaufman JS, Meyers CM, Kusek JW, Feldman HI;Dialysis Access Consortium Study Group: Effect of clopi-dogrel on early failure of arteriovenous fistulas for hemodialy-sis: A randomized, controlled study. JAMA 299(18): 2164–2171, 2008
3. Krishnasami Z, Carlton D, Bimbo L, Taylor M, Balkovetz F,Barker J, Allon M: Management of hemodialysis catheter-re-lated bacteremia with an adjunctive anitibiotic lock solution.Kidney Int 61: 1136–1142, 2002
4. Glassock RJ, Bargman JM, Palmer BF, Samaniego M, Fer-venza FC: Nephrology quiz and questionnaire: 2009. ClinJ Am Soc Nephrol 5: 1141–1160, 2010
5. McQuillan R, Bharatha A, Simons M, Bargman J: Sudden lossof consciousness during hemodialysis. Am J Kidney Dis57(5): xiii–xxvi, 2011
6. Oguzkurt L, Tercn F, Yildirim S, Torun D: Central venousstenosis in hemodialysis patients without a previous history ofcatheter placement. Eur J Radiol 55: 237–242, 2005
7. Johnson DW, Cho Y, Livingston BE, Hawley CM, McDonaldSP, Brown FG, Rosman JB, Bannister KM, Wiggins KJ: Encap-sulating peritoneal sclerosis: Incidence, predictors and out-comes. Kidney Int 77: 904–912, 2010
8. Sampimon DE, Coester AM, Struijk DG, Krediet R: The timecourse of peritoneal transport parameters in peritoneal dialy-sis patients who develop encapsulating peritoneal sclerosis.Neph Dial Transpl 26: 291–298, 2011
9. Knoll G, Sahgal A, Nair RC, Graham J, van Walraven C,Burns KD: Renin angiotensin system blockade and the risk ofhyperkalemia in chronic hemodialysis patients. Am J Med112: 110–114, 2002
10. Garthwaite E, Bhandari S: The effects of angiotensin convert-ing enzyme inhibitors on potassium homeostasis in dialysispatients with and without renal function. Artif Organs 33:641–647, 2009
11. Han S, Won Y, Yi J, Kim H: No impact of hyperkalemia withrenin angiotensin system blockages in maintenance hemodi-alysis patients. Nephrol Dial Transpl 22: 1150–1155, 2007
12. Lin H, Yang Y, Chang J, Ting I, Kuo H, Huang CC: Renin an-giotensin blockade is not associated with hyperkalemia inchronic hemodialysis patients. Ren Fail 31: 942–945, 2009
Glomerular Disorders Case 1: Fernando C. Fervenza(Discussant)A 72-year-old white woman with a 20-year history oftreated hypertension was referred for further evaluation ofimpaired kidney function. She was feeling well until 3months ago when easy fatigability was noted and severe
anemia (hemoglobin of 8.4 g/dl) and a serum creatinine of2.4 mg/dl were discovered. A renal ultrasound was said tobe “normal.” She received 2 units of packed red blood cells(RBSs) and was recommended to have a gastrointestinalevaluation. A colonoscopy revealed multiple polyps thatwere removed and proved to be benign. A week later, shenoticed some blood in the stool and received another bloodtransfusion. The serum creatinine was now 4.2 mg/dl.Urinalysis showed 20 to 50 RBC/high-power field (hpf)and 1� protein. Repeat colonoscopy showed a postpo-lypectomy ulcer with a large visible vessel protrusion forwhich she underwent in hospital photocoagulation. Kid-ney failure was believed to be of prerenal origin secondaryto gastrointestinal (GI) bleeding superimposed on CKDdue to long-standing hypertension. She was sent homewith a serum creatinine of 3.9 mg/dl. Two weeks later,creatinine rose further to 4.6 mg/dl. She was referred forfurther evaluation. Physical examination is unremarkableexcept for edema 1� bilaterally. The laboratory evaluationincluded the following: hemoglobin 10.6 g/dl, leukocytes5.4 � 109/L, platelets 120,000 � 109/L; sedimentation rate65 mm/h; creatinine 5.3 mg/dl, BUN 75 mg/dl, endoge-nous creatinine clearance (CrCl) 16 ml/min; urinalysis 20to 30 RBC/hpf, �25% dysmorphic, protein 1.8 g/24 h; C3109 mg/dl (nl 70 to 175 mg/dl), C4 17 mg/dl (nl 14 to 40mg/dl); rheumatoid factor 81 IU/ml (nl 0 to 39 IU/ml),ANA 1.2 (�1.0); pANCA (anti-neutrophil cytoplasmic an-tibodies) �; myeloperoxidase (MPO)-ANCA 62.9 EU/ml(nl �5 EU/ml).
A renal biopsy was performed (Figure 8, A and B). Tenglomeruli were identified on serial section, with more thanone being globally sclerosed. All glomeruli showed seg-mental capillary loop consolidation, with matrix replace-ment and tuft-capsular adhesions. Silver stains show seg-mental fragmentation of peripheral capillary loopbasement membranes. No immune complex type depositswere found along capillary loop basement membranes.Viable regions of glomeruli showed focal endocapillaryproliferation, with leukocyte accumulation and localizedfibrin deposition. Silver stains show segmental fragmenta-tion of Bowman’s capsule surrounding several glomeruli.Focal interstitial fibrosis with associated tubular atrophy,involved over two-thirds of the biopsy area. Focally in-tense lympho-plasmacytic infiltrates are identified withinthe expanded interstitial regions. Tubules contained redblood cells and red cell casts. The final diagnosis was afocal segmental proliferative and necrotizing glomerulone-phritis, of the “pauci-immune” type.
Glomerular Disorders Question 1Which ONE of the following therapies would you NOW
recommend (Figure 9)?A. Angiotensin-converting enzyme inhibitors (ACEi)
plus low-protein diet.B. High-dose corticosteroids plus oral or intravenous
cyclophosphamide.C. High-dose corticosteroids plus mycophenolate
mofetil.D. High-dose corticosteroids plus oral or intravenous
cyclophosphamide and plasmapheresis.E. High-dose corticosteroids plus rituximab (RTX).
Figure 7. | ESRD and Dialysis Case 2. Configuration of the perito-neal dialysis catheter, connector, and transfer set (Courtesy of E.Kelman).
2538 Clinical Journal of the American Society of Nephrology

Discussion of Case 1 (Question 1)If one restricts the answer strictly to evidence-based
medical practice, then the correct answer is B. However, aswill be discussed below, answer D can also be accepted ascorrect, under certain circumstances.
ANCA-associated vasculitis (AAV) comprises three het-erogeneous syndromes: Wegener granulomatosis (WG),now known as granulomatous polyangiitis (GPA), micro-scopic polyangiitis (MPA), and the Churg-Strauss syn-drome (CSS) (1). These three multisystem disorders arecharacterized by necrotizing small vessel vasculitis withpredilection for kidney, lungs, and peripheral nervous sys-tem involvement that share the occurrence of ANCAs inmost patients at the time of initial presentation (2–5).
Untreated, systemic AAV follows a progressive coursewith a fatal outcome due to vital organ failure. Glucocor-ticosteroids (GCS) alone do not induce disease remission inthe great majority of patients. This fact was initially dem-onstrated by Hollander and Manning who reported on 26patients with GPA treated with GCS alone, and althoughmean patient survival was extended from 5 to 12 months,eventually every patient died despite high-dose GCS (6). Asimilar experience was describe by Fauci and Wolff who in1973 wrote: “Although corticosteroids may decrease in-
flammatory reactions and improve symptoms early in thedisease, they ceased to be of any benefit when the diseaseprogressed to significant pulmonary and renal involve-ment. ” (7). The introduction by Fauci and coworkers ofcombined therapy with the use of high-dose GCS andcyclophosphamide (CYC) revolutionized the treatment ofAAV (8–10). The use of this combined therapy was capableof inducing remission in 65% to 90% of cases and quicklybecame the standard of care for patients with severe AAV(4,9–13).
However, it was soon recognized that GCS combinedwith CYC do not regularly cure the disease but rather turnit into a chronically relapsing disease process, with up to50% of the patients who respond to initial therapy relaps-ing within the first 3 to 5 years (11). In addition, not allpatients respond to CYC, and CYC is associated withserious acute and long-term adverse effects, e.g., bone mar-row suppression, infection, infertility, secondary malig-nancies, hemorrhagic cystitis, which result in treatment-related morbidity and mortality rivaling that caused by theunderlying disease (8,9).
Therefore, approaches that could minimize the toxicityof CYC have been vigorously sought. One of these ap-proaches has been to use intravenous pulse rather thanoral CYC. A recent randomized, controlled trial foundintravenous pulse CYC combined with GCS equally effec-tive as daily oral CYC combined with GCS in inducingremission in patients with newly diagnosed severe AAVwith renal involvement (88.1% versus 87.7% at 9 months)(14). The absolute cumulative CYC dose in the daily oralgroup was approximately double that in the pulse group(15.9 versus 8.2 g; P � 0.001), and not surprisingly, leuko-penia was more frequent in the oral CYC group. There wasno difference in overall mortality rate, with five deaths inthe pulse CYC group versus nine deaths in the daily oralgroup (P � 0.79). Thirteen patients in the pulse CYC groupand six in the daily oral group relapsed after achievingremission. Unfortunately, the limited follow-up of thestudy precludes an evaluation of the long-term rate ofremission in patients treated with pulse CYC. This is animportant issue based on previous publication by the samegroup, suggesting that lower cumulative doses with theuse of pulse CYC may come at the expenses of higherrelapse rates (15).
Another approach has been to avoid CYC altogether byusing mycophenolate mofetil (MMF) for remission induc-
Figure 9. | Answers for Case 1, Question 1, Glomerular Disorders.
Figure 8. | Glomerular disorders case 1. (A) A low-power photomi-crograph of the renal biopsy (hematoxylin and eosin). (B) A high-power photomicrograph of the renal biopsy shown in (A).
Clin J Am Soc Nephrol 6: 2534–2547, October, 2011 Nephrology Quiz and Questionnaire, Glassock et al. 2539

tion in patients with AAV who have failed or are intolerantof CYC. This approach has been associated with a withvariable degree of success (16–18). On the other hand, in arecent uncontrolled trial MMF used in combination withGCS was capable inducing remission in 76% of patientwith MPA who were MPO-ANCA positive and had mildto moderate renal involvement (19). The discrepancyamong study outcomes may be related to the fact that instudies where MMF performed poorly, the majority ofpatients had GPA (17,18). Furthermore, these studiesenrolled only patients who were either relapsing, resis-tant to CYC, or had contraindications for CYC use(17,18). In contrast, Silva et al. focused solely on patientswith MPO-ANCA positive MPA with active renal dis-ease, and the majority of enrolled patients were newlydiagnosed and CYC-naive (19).
In recent years, a number of single case reports andsmall case series have suggested that targeting B cells withthe use of RTX was effective in the treatment of patientswith GPA, MPA, and CSS who had been refractory or hadcontraindications to the use of CYC (20–33). These prelim-inary observations have now been confirmed by two ran-domized controlled studies that have examined the effi-cacy of RTX in inducing remission in patients with severeAAV (34,35). The major findings and differences betweenthe two studies have been recently reviewed (36). In thefirst study, RITUXVAS, Jones et al., randomized 44 patientswith newly diagnosed AAV and renal involvement in a 3:1ratio to receive standard GCS regimen plus either RTX (375mg/m2 intravenous weekly four times) with two intrave-nous CYC pulses (33 patients, the RTX group) or intrave-nous CYC for 3 to 6 months followed by azathioprine (11patients, the control group) (34). Sustained remission (de-fined as BVAS of 0 for �6 months) at 12 months wasachieved in 25 patients in the RTX group (76%) and ninepatients in the CYC group (82%) (P � 0.68). Severe adverseevents occurred in 14 patients in the RTX group (42%) andfour patients in the control group (36%) (P � 0.77). It ispossible that RTX used in combination with CYC leads toa higher frequency of adverse events than standard ther-apy with CYC, but the small number of patients involvedin this trial precludes any firm conclusion. Six of the pa-tients in the RTX group (18%) and two of 11 patients in thecontrol group (18%) died (P � 1.00).
Simultaneously published in the same journal were theresults of the RAVE trial: a multicenter randomized, dou-ble-blind, double placebo-controlled trial conducted toevaluate the efficacy and safety of RTX for remission in-duction in severe AAV in comparison to CYC (35). A totalof 197 patients with severe GPA or MPA (3:1), all positivefor PR3-ANCA or MPO-ANCA (2:1), were randomized ona 1-to-1 ratio to receive treatment with RTX (375 mg/m2
intravenous weekly four times) or CYC (2 mg/kg per day,by mouth) in combination with GCS. Initial GCS treatmentwas the same in both groups (1 to 3 g intravenous meth-ylprednisolone) and was followed by rigorously protocol-determined GCS tapering, aimed at complete discontinu-ation of GCS by month 5. Sixty-three (64%) of the patientsin the RTX arm versus 52 (53%) in the CYC arm achievedthe primary endpoint of the study, defined as diseaseremission with a BVAS of 0 in the absence of GCS therapy
at month 6, a result that met the criterion for noninferiority(P � 0.001). A larger proportion of patients in the RTXgroup achieved the primary endpoint than in the CYCgroup, and the results came close to meet the criteria forsuperiority (P � 0.09). Similarly, 71% of the patients in theRTX group versus 62% of the patients in the CYC groupreached the secondary outcome of disease remission whilereceiving a daily prednisone dose �10 mg. RTX was moreeffective than CYC in inducing remission in patients withrelapsing disease. This is an important point to considerbecause it is in this patient population that minimization ofthe cumulative CYC exposure might be beneficial. Prelim-inary analysis of the long-term follow-up of the RAVE trialshows that the benefits of RTX therapy are maintained forat least 18 months (37).
The results of these two randomized trials demonstrate,without question, that RTX, either combined with GCSalone or with GCS and CYC, is not inferior to standardtherapy for the induction of remissions in patients withnewly diagnosed severe AAV and is in fact the superior tostandard therapy in patients with relapsing disease. Majorcontraindications to the use of RTX include history ofsevere allergic reactions to humanized or chimeric mono-clonal antibodies or murine-derived protein, active sys-temic infection (including hepatitis B and HIV), severeliver disease (alanine aminotransferase or aspartate amino-transferase level �2.5 times the upper limit of normal thatcannot be attributed to underlying AAV disease), activemalignancy or history of malignancy in the last 5 years,total white blood cell count that is �4000/mm3, plateletcount that is �120,000/mm3, pregnancy, and the use of alive vaccine fewer than 4 weeks before receiving RTX.
For ethical reasons, patients with severe alveolar hem-orrhage or with serum creatinine �4.0 mg/dl at the time ofscreening were excluded from participating in the RAVEtrial, and although the RITUXVAS trial included nine pa-tients who were dialysis dependent (five of the eight pa-tients treated with RTX actually came off dialysis), furtherresearch is needed to evaluate the efficacy of RTX in thegroup of patients, as the one currently presented, whoserenal disease severity recommends the use of plasma ex-change (38). A large multicenter, international, open label,factorial design, randomized control trial to further evalu-ate the efficacy of plasma exchange in addition to immu-nosuppressive therapy and GCS in reducing death andESRD in severe AAV (PEXIVAS trial ISRCTN07757494) iscurrently under way.
Although renal biopsy findings correlate, in general,with the renal outcome in patients with AAV, the correla-tion is not great (39). In the opinion of the writer, a decisionregarding initiation of immunosuppression treatment inpatients with AAV and renal involvement depends moreon the levels of renal function before the development ofAAV, the rapidity of rise in serum creatinine, and theevidence of active vasculitis on renal biopsy, as well as theoverall clinical status of the patient. In the present case,renal function had been lost relatively quickly and thepresence of red cell casts suggested ongoing active vascu-litis. This together with the overall good clinical status ofthe patient helped in making a decision to treat the patient
2540 Clinical Journal of the American Society of Nephrology

despite the renal biopsy finding showing severe tubulo-interstitial damage.
Treating the patient only with an ACEi � low-proteindiet (option A) could be considered in patients whosedisease is clearly in remission, which is not the case here.Also, the elevated serum creatinine makes it unlikely thepatient could tolerate an ACEi.
High-dose corticosteroids plus oral or intravenous cyclo-phosphamide (option B) can be considered the most cor-rect answer in view that the patient has systemic diseaseand severe renal involvement, and if we strictly abide bythe fact that the MEPEX trial, which evaluated and sup-ported the use of plasmapheresis (plasma exchange[PLEX]) in severe AAV, only included patients with aserum creatinine �5.8 mg/dl (40). The use of high-dosecorticosteroids plus oral or intravenous cyclophosphamideplus PLEX (option D) might also be considered a correctchoice, based on current clinical practice in a number ofcenters with extensive experience in treating patients withsevere AAV, including the author’s own center. Indeed,given the severity and speed of decline in the patients renalfunction, the addition of PLEX to treatment with oral GCSand CYC could be justified by extrapolation from the re-sults of the MEPEX trial. However, the evidence for thispractice needs closer examination. Early studies of PLEX inrapidly progressive glomerulonephritis due to AAV gavemixed results (40–42). On the other hand, the MEPEX trial(38) strictly examined the benefits of the addition of PLEXversus intravenous methylprednisolone pulses to standardCYC-based therapy in 137 patients with AAV and severerenal failure (serum creatinine �5.8 mg/dl or dialysis de-pendent at presentation). The MEPEX trial demonstratedan absolute reduction in the development of irreversibleESRD of 24% (95% CI 6.5% to 41% at 12 months for patientstreated with PLEX. There were no demonstrable differ-ences in mortality at 12 months among the treatmentgroups (25% in both). Longer-term follow-up from theMEPEX trial shows identical outcomes between the treat-ment groups for ESRD or death beyond 12 months (P �0.57) (43,44). Furthermore, Walsh et al. recently published asystematic review and meta-analysis of randomized con-trolled trials comparing standard CYC-based care plusadjuvant PLEX in adult patents with either renal vasculitisor rapidly progressive glomerulonephritis (45). These au-thors concluded that there is insufficient information toreliably determine whether PLEX decreases the compositeoutcomes of ESRD or death. If adding PLEX would helppatients with lesser degrees of renal insufficiency is notclear, and this issue is currently being evaluated in a largemulticenter, international, open-label, factorial design, ran-domized controlled trial (PEXIVAS, ISRCTN07757494). Inthe present case the patient was actually treated with GCSand CYC without PLEX. Ten years later she is in completeremission of her AAV with a serum creatinine of 1.5 mg/dland a creatinine clearance of 37 ml/min. Clearly, moreresearch is needed to better define the role of PLEX inpatients with severe AAV. PLEX may be life-saving inthose patients with AAV who present with severe pulmo-nary hemorrhage (46). As discussed above, the use ofhigh-dose GCS plus mycophenolate mofetil (option C) hasbeen disappointing, and preliminary results showed suc-
cess only in patients with MPA and with serum creatininelevels �3 mg/dl. Finally, high-dose GCS plus RTX (optionE) was not considered as a correct answer in the presentcase because the RAVE trial did not include patients withserum creatinine �4 mg/dl. On the other hand, theRITUXVAS trial did include nine patients who were dial-ysis dependent, with six of the eight patients in the RTXgroup achieving sustained remission, whereas five actuallycame off dialysis. Thus, RTX appears to be effective in thegroup of patients whose renal disease severity suggests theuse of PLEX, but further research is needed to define betterevidence-based guidelines.
DisclosuresNone.
References1. Jennette JC, Falk RJ, Andrassy K, Bacon PA, Bacon PA, Churg
J, Gross WL, Hagen EC, Hoffman GS, Hunder GG, Kallen-berg CG, McCluskey RT, Sinico RA, Rees AJ, Van Es LA,Waldherr R, Wiik A: Nomenclature of systemic vasculitides.Proposal of an international consensus conference. ArthritisRheum 37: 187–192, 1994
2. Finkelman JD, Lee AS, Hummel AM, Viss MA, Jacob GL,Homburger HA, Peikert T, Hoffman GS, Merkel PA, Spiera R,St Clair EW, Davis JC Jr., McCune WJ, Tibbs AK, YtterbergSR, Stone JH, Specks U: ANCA are detectable in nearly allpatients with active severe Wegener’s granulomatosis. Am JMed 120: 649.e9–643.e14, 2007
3. Guillevin L, Cohen P, Gayraud M, Lhote F, Jarrousse B, Cas-sassus P: Churg-Strauss syndrome. Clinical study and long-term follow-up of 96 patients. Medicine (Baltimore) 78: 26–37, 1999
4. Guillevin L, Durand-Gasselin B, Cevallos R, Gayraud M,Gayraud M, Lhote F, Callard P, Amouroux J, Casassus P, Jar-rousse B: Microscopic polyangiitis: Clinical and laboratoryfindings in eighty-five patients. Arthritis Rheum 42: 421–430,1999
5. Keogh KA, Specks U: Churg-Strauss syndrome: Clinical pre-sentation, antineutrophil cytoplasmic antibodies, and leuko-triene receptor antagonists. Am J Med 115: 284–290, 2003
6. Hollander D, Manning RT: The use of alkylating agents in thetreatment of Wegener’s granulomatosis. Ann Intern Med 67:393–398, 1967
7. Fauci AS, Wolff SM: Wegener’s granulomatosis: Studies ineighteen patients and a review of the literature. Medicine(Baltimore) 52: 535–561, 1973
8. Fauci AS: Vasculitis. J Allergy Clin Immunol 72: 211–223,1983
9. Hoffman GS, Kerr GS, Leavitt RY, Hallahan CW, HallahanCW, Lebovics RS, Travis WD, Rottem M, Fauci AS: Wegenergranulomatosis: An analysis of 158 patients. Ann Intern Med116: 488–498, 1992
10. Fauci AS, Katz P, Haynes BF, Wolff SM: Cyclophosphamidetherapy of severe systemic necrotizing vasculitis. N EnglJ Med 301: 235–238, 1979
11. Nachman PH, Hogan SL, Jennette JC, Falk RJ: Treatment re-sponse and relapse in antineutrophil cytoplasmic autoanti-body-associated microscopic polyangiitis and glomerulone-phritis. J Am Soc Nephrol 7: 33–39, 1996
12. Guillevin L, Cohen P, Mahr A, Arene JP, Mouthon L, PuechalX, Pertuiset E, Gilson B, Hamidou M, Lanoux P, Bruet A, Rui-vard M, Vanhille P, Cordier JF: Treatment of polyarteritis no-dosa and microscopic polyangiitis with poor prognosis fac-tors: A prospective trial comparing glucocorticoids and six ortwelve cyclophosphamide pulses in sixty-five patients. Arthri-tis Rheum 49: 93–100, 2003
13. Jayne D, Rasmussen N, Andrassy K, Bacon P, Tervaert JW,Dadoniene J, Ekstrand A, Gaskin G, Gregorini G, de Groot K,Gross W, Hagen EC, Mirapeix E, Pettersson E, Siegert C,Sinico A, Tesar V, Westman K, Pusey C; European Vasculitis
Clin J Am Soc Nephrol 6: 2534–2547, October, 2011 Nephrology Quiz and Questionnaire, Glassock et al. 2541

Study Group: A randomized trial of maintenance therapy forvasculitis associated with antineutrophil cytoplasmic autoan-tibodies. N Engl J Med 349: 36–44, 2003
14. de Groot K, Harper L, Jayne DR, Flores Suarez LF, GregoriniG, Gross WL, Luqmani R, Pusey CD, Rasmussen N, SinicoRA, Tesar V, Vanhille P, Westman K, Savage CO; EUVAS(European Vasculitis Study Group): Pulse versus daily oralcyclophosphamide for induction of remission in antineutro-phil cytoplasmic antibody-associated vasculitis: A random-ized trial. Ann Intern Med 150: 670–680, 2009
15. de Groot K, Adu D, Savage CO: The value of pulse cyclo-phosphamide in ANCA-associated vasculitis: meta-analysisand critical review. Nephrol Dial Transplant 16: 2018–2027,2001
16. Nowack R, Gobel U, Klooker P, Hergesell O, Andrassy K,van der Woude FJ: Mycophenolate mofetil for maintenancetherapy of Wegener’s granulomatosis and microscopic poly-angiitis: A pilot study in 11 patients with renal involvement.J Am Soc Nephrol 10: 1965–1971, 1999
17. Joy MS, Hogan SL, Jennette JC, Falk RJ, Nachman PH: A pilotstudy using mycophenolate mofetil in relapsing or resistantANCA small vessel vasculitis. Nephrol Dial Transplant 20:2725–2732, 2005
18. Stassen PM, Cohen Tervaert JW, Stegeman CA: Induction ofremission in active anti-neutrophil cytoplasmic antibody-asso-ciated vasculitis with mycophenolate mofetil in patients whocannot be treated with cyclophosphamide. Ann Rheum Dis66: 798–802, 2007
19. Silva F, Specks U, Kalra S, Hogan MC, Leung N, Sethi S, Fer-venza FC: Mycophenolate mofetil for induction and mainte-nance of remission in microscopic polyangiitis with mild tomoderate renal involvement—a prospective, open-label pilottrial. Clin J Am Soc Nephrol 5: 445–453, 2010
20. Specks U, Fervenza FC, McDonald TJ, Hogan MC: Responseof Wegener’s granulomatosis to anti-CD20 chimeric mono-clonal antibody therapy. Arthritis Rheum 44: 2836–2840,2001
21. Keogh KA, Wylam ME, Stone JH, Specks U: Induction of re-mission by B lymphocyte depletion in eleven patients withrefractory antineutrophil cytoplasmic antibody-associated vas-culitis. Arthritis Rheum 52: 262–268, 2005
22. Keogh KA, Ytterberg SR, Fervenza FC, Carlson KA, SchroederDR, Specks U: Rituximab for refractory Wegener’s granulo-matosis: Report of a prospective, open-label pilot trial. Am JRespir Crit Care Med 173: 180–187, 2006
23. Tamura N, Matsudaira R, Hirashima M, Ikeda M, Tajima M,Nawata M, Morimoto S, Kaneda K, Kobayashi S, HashimotoH, Takasaki Y: Two cases of refractory Wegener’s granuloma-tosis successfully treated with rituximab. Intern Med 46:409–414, 2007
24. Stasi R, Stipa E, Del Poeta G, Amadori S, Newland AC, Pro-lan D: Long-term observation of patients with anti-neutrophilcytoplasmic antibody-associated vasculitis treated with ritux-imab. Rheumatology (Oxford) 45: 1432–1436, 2006
25. Ferraro AJ, Day CJ, Drayson MT, Savage CO: Effective thera-peutic use of rituximab in refractory Wegener’s granulomato-sis. Nephrol Dial Transplant 20: 622–625, 2005
26. Eriksson P: Nine patients with anti-neutrophil cytoplasmicantibody-positive vasculitis successfully treated with ritux-imab. J Intern Med 257: 540–548, 2005
27. Maher LV, Wilson JG: Successful treatment of rheumatoidvasculitis-associated foot drop with rituximab. Rheumatology(Oxford) 45: 1450–1451, 2006
28. Koukoulaki M, Smith KG, Jayne DR: Rituximab in Churg-Strauss syndrome. Ann Rheum Dis 65: 557–559, 2006
29. Pepper RJ, Fabre MA, Pavesio C, Gaskin G, Jones RB, JayneD, Pusey CD, Salama AD: Rituximab is effective in the treat-ment of refractory Churg-Strauss syndrome and is associatedwith diminished T-cell interleukin-5 production. Rheumatol-ogy (Oxford) 47: 1104–1105, 2008
30. Taylor SR, Salama AD, Joshi L, Pusey CD, Lightman SL:Rituximab is effective in the treatment of refractory ophthal-mic Wegener’s granulomatosis. Arthritis Rheum 60: 1540–1547, 2009
31. Smith KG, Jones RB, Burns SM, Jayne DR: Long-term compar-
ison of rituximab treatment for refractory systemic lupus ery-thematosus and vasculitis: Remission, relapse, and re-treat-ment. Arthritis Rheum 54: 2970–2982, 2006
32. Jones RB, Ferraro AJ, Chaudhry AN, Brogan P, Salama AD,Smith KG, Savage CO, Jayne DR: A multicenter survey ofrituximab therapy for refractory antineutrophil cytoplasmicantibody-associated vasculitis. Arthritis Rheum 60: 2156–2168, 2009
33. Cartin-Ceba R, Keogh KA, Specks U, Sethi S, Fervenza FC:Rituximab for the treatment of Churg-Strauss syndrome withrenal involvement. Nephrol Dial Transplant, February 16,2011 [Epub ahead of print]
34. Jones RB, Cohen Tervaert JW, Hauser T, Luqmani R, MorganMD, Peh CA, Savage CO, Segelmark M, Tesar V, van Paas-sen P, Walsh D, Walsh M, Westman K, Jayne DR; EuropeanVasculitis Study Group: Rituximab versus cyclophosphamidein ANCA-associated renal vasculitis. N Engl J Med 363: 211–220, 2010
35. Stone JH, Merkel PA, Spiera R, Seo P, Langford CA, HoffmanGS, Kallenberg CG, St Clair EW, Turkiewicz A, Tchao NK,Webber L, Ding L, Sejismundo LP, Mieras K, WeitzenkampD, Ikle D, Seyfert-Margolis V, Mueller M, Brunetta P, AllenNB, Fervenza FC, Geetha D, Keogh KA, Kissin EY, MonachPA, Peikert T, Stegeman C, Ytterberg SR, Specks U, RAVE-ITN Research Group: Rituximab versus cyclophosphamide forANCA-associated vasculitis. N Engl J Med 363: 221–232,2010
36. Fervenza FC: Rituximab in ANCA-associated vasculitis: A fador a fact? Nephron Clin Pract 118: c182–c188, 2010
37. Specks U, Stone JH for the RAVE-ITN Research Group: Long-term efficacy and safety results of the RAVE trial. Clin ExpImmunol 164: 65, 2011
38. Jayne DR, Gaskin G, Rasmussen N, Abramowicz D, FerrarioF, Guillevin L, Mirapeix E, Savage CO, Sinico RA, StegemanCA, Westman KW, van der Woude FJ, de Lind van Wijn-gaarden RA, Pusey CD; European Vasculitis Study Group:Randomized trial of plasma exchange or high-dosage methyl-prednisolone as adjunctive therapy for severe renal vasculitis.J Am Soc Nephrol 18: 2180–2188, 2007
39. de Lind van Wijngaarden RA, Hauer HA, Wolterbeek R,Jayne DR, Gaskin G, Rasmussen N, Noel LH, Ferrario F,Waldherr R, Hagen EC, Bruijn JA, Bajema IM: Clinical andhistologic determinants of renal outcome in ANCA-associatedvasculitis: A prospective analysis of 100 patients with severerenal involvement. J Am Soc Nephrol 17: 2264–2274, 2006
40. Cole E, Cattran D, Magil A, Greenwood C, Churchill D, Sut-ton D, Clark W, Morrin P, Posen G, Bernstein K: A prospec-tive randomized trial of plasma exchange as additive therapyin idiopathic crescentic glomerulonephritis. The CanadianApheresis Study Group. Am J Kidney Dis 20: 261–269, 1992
41. Glockner WM, Sieberth HG, Wichmann HE, Backes E, Bam-bauer R, Boesken WH, Bohle A, Daul A, Graben N, Keller F:Plasma exchange and immunosuppression in rapidly progres-sive glomerulonephritis: A controlled, multi-center study. ClinNephrol 29: 1–8, 1988
42. Pusey CD, Rees AJ, Evans DJ, Peters DK, et al.: Plasma ex-change in focal necrotizing glomerulonephritis without anti-GBM antibodies. Kidney int 40: 757–763, 1991
43. Casian A, Jayne D: Plasma exchange in the treatment of We-gener’s granulomatosis, microscopic polyangiitis, Churg-Strauss syndrome and renal limited vasculitis. Curr OpinRheumatol 23: 12–17, 2011
44. Casian A: Plasma exchange for severe renal vasculitis: Long-term follow-up of the MEPEX trial. Clin Exp Immunol 164:50, 2011
45. Walsh M, Catapano F, Szpirt W, Thorlund K, Bruchfeld A,Guillevin L, Haubitz M, Merkel PA, Peh CA, Pusey C, JayneD: Plasma exchange for renal vasculitis and idiopathic rap-idly progressive glomerulonephritis: A meta-analysis. Am JKidney Dis 57: 566–574, 2011
46. Klemmer PJ, Chalermskulrat W, Reif MS, Hogan SL, HenkeDC, Falk RJ: Plasmapheresis tehrapy for diffsue alveolar hem-orrhage in patients with small vessel vasculitis. Am J KidneyDis 42:1149–1153, 2003
2542 Clinical Journal of the American Society of Nephrology
Glomerular Disorders Case 2: Fernando C. Fervenza(Discussant)A 73-year-old man was referred for evaluation of edemastarting in November 2009. During this time his urinebecame tea-colored and foamy. He had no prior history ofhypertension, but in October 2009 his systolic BP wasfound to be in 160 mmHg and continued to increase to 213mmHg in January 2010. During an evaluation for hyper-tension a serum creatinine of 2.1 mg/dl was noted, in-creased from a baseline of 1.0 to 1.2 mg/dl the previousyear. Urinalysis showed �50 RBC/hpf; �25% dysmorphic.Urinary protein excretion was 1.7 g/d. Fluorescence anti-nuclear antibody was 1:40, C4 was 15 mg/dl (normal), andC3 was 52 mg/dl (low). Renal ultrasound showed a rightkidney 10.9 cm and a left kidney 12.4 cm in length with nomasses, renal artery stenoses, or hydronephrosis. BP waswell controlled, and a kidney biopsy was performed. Light,immunofluorescence (IF), and electron microscopy (EM)are shown in Figure 10, A through D. The IF was positiveonly for C3.
Glomerular Disorders Question 1Based on these clinical and pathologic findings, which
ONE of the following would be the MOST useful diagnos-tic test (Figure 11)?
A. Serum free light-chain assay.B. Hepatitis B and C serology.C. Complement Factors H, I, membrane co-factor assays.D. Anti-streptolysin O titer.E. Anti-double-stranded DNA antibody.
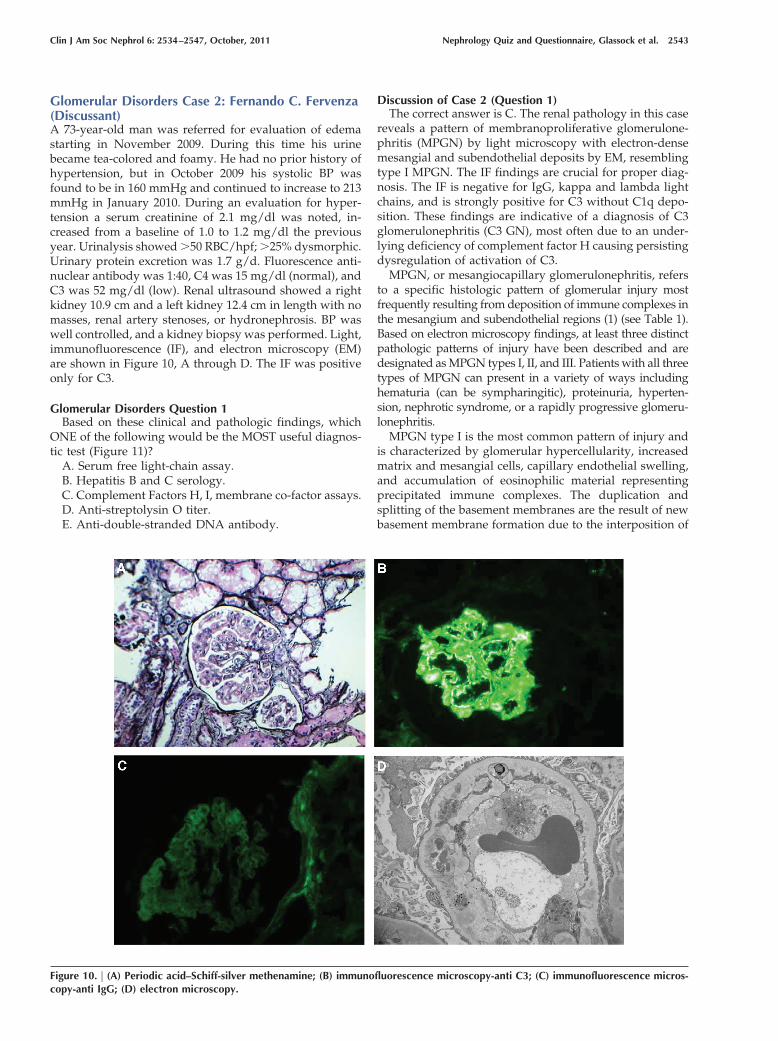
Discussion of Case 2 (Question 1)The correct answer is C. The renal pathology in this case
reveals a pattern of membranoproliferative glomerulone-phritis (MPGN) by light microscopy with electron-densemesangial and subendothelial deposits by EM, resemblingtype I MPGN. The IF findings are crucial for proper diag-nosis. The IF is negative for IgG, kappa and lambda lightchains, and is strongly positive for C3 without C1q depo-sition. These findings are indicative of a diagnosis of C3glomerulonephritis (C3 GN), most often due to an under-lying deficiency of complement factor H causing persistingdysregulation of activation of C3.
MPGN, or mesangiocapillary glomerulonephritis, refersto a specific histologic pattern of glomerular injury mostfrequently resulting from deposition of immune complexes inthe mesangium and subendothelial regions (1) (see Table 1).Based on electron microscopy findings, at least three distinctpathologic patterns of injury have been described and aredesignated as MPGN types I, II, and III. Patients with all threetypes of MPGN can present in a variety of ways includinghematuria (can be sympharingitic), proteinuria, hyperten-sion, nephrotic syndrome, or a rapidly progressive glomeru-lonephritis.
MPGN type I is the most common pattern of injury andis characterized by glomerular hypercellularity, increasedmatrix and mesangial cells, capillary endothelial swelling,and accumulation of eosinophilic material representingprecipitated immune complexes. The duplication andsplitting of the basement membranes are the result of newbasement membrane formation due to the interposition of
Figure 10. | (A) Periodic acid–Schiff-silver methenamine; (B) immunofluorescence microscopy-anti C3; (C) immunofluorescence micros-copy-anti IgG; (D) electron microscopy.
Clin J Am Soc Nephrol 6: 2534–2547, October, 2011 Nephrology Quiz and Questionnaire, Glassock et al. 2543

the mesangial cell, mesangial matrix, and endothelial celldebris along the subendothelial side of the lamina densaand give the glomeruli loops the appearance of “doublecontours.” IF usually demonstrates deposition of IgG, IgM,C3, and C4 in the mesangium and capillary walls. On EM,the deposits are primarily subendothelial (2–4).
MPGN type I can be idiopathic or secondary to a variety ofdiseases (4) Traditionally, MPGN has been associated with thepresence of chronic infections including hepatitis C (5,6), shuntnephritis (7), and abscesses and endocarditis (8–11). Of these,hepatitis C virus (HCV) is by far the commonest with kidneyinvolvement due to type II cryoglobulinemia in 50% to 60% ofcases (6,12–15). In addition to chronic infections, autoimmunediseases such as systemic lupus erythematosus, Sjogren syn-drome, and rheumatoid arthritis are also associated with persis-tent circulating immune complexes and the development ofMPGN (16–18). More recently, monoclonal gammopathies havealso been associated with development of a MPGN (19–21). Infact, Sethi et al., found that MPGN type I were as or morecommonly associated with a monoclonal gammopathy thanwith HCV (21). As it stands, idiopathic MPGN type I appears tobe a vanishing disease because a possible underlying etiology islikely to be found in the majority of cases (4).
In MPGN type II or dense deposit disease (DDD), find-ings on light microscopic examination can vary widelyfrom mild mesangial hypercellularity through a mem-branoproliferative pattern to crescentic glomerulonephri-tis. C3 is present in an interrupted band pattern along theglomerular and tubular basement membranes and thebasement membranes of Bowman’s capsule. C3 in the mes-angial areas can result in a prominent spherule or ring-likepattern. However, is the presence of confluent and ex-tremely electron-dense intramembranous deposits withinglomerular, tubular, and vascular basement membranesthat are the pathognomonic feature of MPGN II or DDD. Infact, DDD is a more accurate description than MPGN IIbecause dense deposits are diagnostic, but capillary wallthickening or hypercellularity are not always present onthe biopsy (22). DDD is associated with partial lipodystro-phy (Dunnigan-Koeberling disease) and the presence ofextensive retinal degenerative changes (drusen) on indirectretinoscopy (22). These retinal changes are commonly seenin elderly patients and rarely cause visual impairment, butit is their presence in young patients that is noticeable incases of DDD. In type III MPGN, the deposits are both
subepithelial and subendothelial, and lucent areas arepresent within the glomerular basement membranes.
More recently, cases with an MPGN pattern of injury andwith extensive C3 deposition along the capillary walls andmesangium along with the absence of immunoglobulins de-position on IF microscopy have been described: the so-calledglomerulonephritis with isolated C3 deposits or C3 glomer-ulonephritis (C3 GN) (23–25). On light microscopy and on IFevaluation, these cases resemble DDD but EM examination inC3 GN does not show the typical electron-dense intramem-branous deposits characteristic of patients with DDD. Insteadthe deposits appear similar to the immune deposits noted inimmune complex-mediated MPGN.
In cases of MPGN that are due to deposition of circulatingimmune complexes, complement is activated via the classicalpathway, leading to the generation of chemotactic factors(C5a), opsonins (C3b), and the membrane attack complex(C5b-9; MAC). In these patients, classical pathway activation
Figure 11. | Answers for Case 2, Question 2, Glomerular Disorders.
Table 1. Conditions associated with a membranoproliferativepattern of injury
Chronic infectionsViral: hepatitis C, hepatitis B (rarely)Bacterial: endocarditis, infected ventriculo-atrial
shunt, visceral abscesses, leprosy, meningococcalmeningitis
Protozoa/other infections: malaria, schistosomiasis,mycoplasma, leishmaniasis
Autoimmune diseasesSLESjögren syndromeRheumatoid arthritisAntibodies against complement cascade proteins,
e.g., C3 nephritic factor, antibody against factor H(see C3 GN)
Genetic/hereditary causesInherited complement deficiencies/dysregulation:
e.g., mutations on factors H, I (see C3 GN)Monoclonal gammopathiesC3 glomerulopathy (C3 GN)
C3 glomerulonephritisDense Deposit Disease (DDD)
Chronic and healed thrombotic angiopathiesHealing phase of HUS/TTPAnti-phospholipid (anti-cardiolipin) antibodies
syndromePOEMS syndromeRadiation nephritisNephropathy associated with bone marrow
transplantationDrug-associated thrombotic angiopathiesSickle cell anemia and polycythemiaDysfibrinogenemia and other prothrombotic statesTransplant glomerulopathy
Idiopathic forms of MPGNNeed to prove that none of the conditions above
are present
HUS, hemolytic uremic syndrome; TTP, thromboticthrombocytopenic purpura; POEMS, polyneuropathy,organomegaly, endocrinopathy, monoclonal gammopathy,and skin changes; MPGN, membranoproliferativeglomerulonephritis.
2544 Clinical Journal of the American Society of Nephrology

is reflected by the presence of a normal or low C3, low C4,and low CH50.
On the other hand, MPGN type II/DDD is not due toimmune complex deposition but results from inherited oracquired dysregulation of proteins (e.g., factor H, I, MCP)involved in regulation of the alternative pathway (AP)complement cascade (Figure 12) (22, 26–28). Once acti-vated, the AP generates effector compounds that are de-livered indiscriminately to all membrane surfaces. Multi-ple complement regulators and inhibitors operate at everylevel controlling progression of the cascade and preventingself-induced damage (29). Mutations in factors H and I, orthe presence of antibodies against C3 convertase enzyme(C3Bb), also known as C3 nephritic factor (C3Nef), result inpersistent complement activation and development ofMPGN. In this situation, patients may present with low C3,normal C4, and low CH50.
Similarly, the majority of the cases of C3 glomerulone-phritis are related to the dysregulation of the alternativecomplement pathway due to loss of regulatory mecha-nisms at different steps in the alternative pathway. In thestudy by Servais et al., 19 patients were identified as havingC3 GN and were divided into two groups based on renal
pathology: group I (n � 13) was classified as showingMPGN Type I (C3GN � MPGN), and group II (n � 6) wasclassified as having mesangial and intramembranous C3deposits in the absence of mesangial proliferation (C3GNwithout MPGN) (24). Mutations in complement regulatorygenes were detected in four of six patients identified as C3GN without MPGN (heterozygous mutations in Factor H[two patients] and in factor I gene [two patients]) and intwo of 13 patients diagnosed as C3GN �MPGN (heterozy-gous mutations in factor H gene [one patient] and doubleheterozygous mutations in CD46 gene [one patient]) (24).In contrast, C3NeF was present in five of 13 patients withC3GN with MPGN and in two of six patients with C3GNwithout MPGN, one of whom had a factor H mutation.However, the presence of autoantibodies, e.g., against fac-tor H or I, was not assessed and functional assays of APactivity were not performed. We have recently evaluatedfive cases of C3 GN (30). In all these cases, AP complementcascade evaluation showed loss of regulatory control of theAP complement cascade. Thus, AP dysfunction results innot only DDD, but in C3 glomerulonephritis, a type ofMPGN that lacks the classic electron microscopic hall-marks of DDD. It is important to recognize that review ofthe histology of these cases suggests that dysregulation ofthe AP of complement produces a spectrum of morpho-logic patterns that range form a mesangial proliferativeGN (early stages) to the classic pattern of MPGN withdouble contours or even sclerosing GN (late stages). Thus,the pattern of injury likely depends on the timing of therenal biopsy as the disease progresses from a proliferativeto a sclerosing lesion. Failure to recognize this phenome-non, led, in the view of this author, to Servais et al. (24) todivide their patients into two groups while they likelyrepresent a continuum of the same pathologic process.
Evaluating the AP of complement in C3GN is importantnot only to dissect the pathologic process, but also becauseit has implications for treatment. A proposed evaluationsequence for complement-mediated MPGN includeschecking C3 and C4 serum complement levels, solublemembrane attack complex levels, alternative pathwayfunctional assays, and hemolytic assays (or CH50). If ab-normalities are found, then patients should be evaluatedfor genetic mutations and the presence of autoantibodies tocomplement cascade proteins (such as C3NeF or anti-CfH)A patient with positive autoantibodies may benefit fromimmunosuppressive therapy, whereas those cases due togenetic mutations in the complement cascade may benefitfrom treatment with drugs that inhibit formation of mem-brane attack complex, e.g., eculizumab. Eculizumab couldalso be used in patients with autoantibody-mediated dis-ease. In the present case, evaluation of the AP cascadeshowed the presence of an antibody against factor H.Treatment with high-dose corticosteroids resulted in bothimprovement of serum creatinine and proteinuria 6months after the start of therapy (serum creatinine 1.6mg/dl; proteinuria 728 mg/dl).
The differential diagnosis of MPGN includes cases dueto monoclonal gammopathy (option A), hepatitis B and C(option B), postinfectious glomerulonephritis (due to thepresence of subepithelial humps; option D), and autoim-mune disease, e.g., lupus nephritis (due to subepithelial,
Figure 12. | In vivo, the alternative pathway (AP) is constantly beingactivated at low levels. The AP is tightly regulated by a number offactors including factors B, H, and I. Amplification of soluble C3bBboccurs with low efficiency because free C3b is rapidly inactivated byfactors H and I. Antibodies to C3bBb, also known as C3 nephriticfactor (C3NeF), protects the C3Bb from inactivation resulting inconstant amplification of the AP. Similarly, mutations or autoanti-bodies to factors H (or I) can result in failure to control levels of C3bin the fluid phase, leading to constant amplification of AP comple-ment proteins. (From Smith et al. [27])
Clin J Am Soc Nephrol 6: 2534–2547, October, 2011 Nephrology Quiz and Questionnaire, Glassock et al. 2545

subendothelial, and mesangial deposits); option E). How-ever, in cases of MPGN due to a monoclonal protein, IFstudies will show Ig and light-chain restrictions on renalbiopsies (e.g., IgG and kappa light chains) (21). Similarly,chronic viral infections such as hepatitis C and B, with orwithout circulating cryoglobulins, are an important andcommon cause of MPGN (31,32). In these cases, however,the classic pathway is preferentially activated (normal orlow C3, low C4), IF demonstrates deposition of IgM, IgG,and C3, kappa and lambda in the mesangium and capillarywalls, and on EM, subendothelial immune complexes areusually seen and may have a fibrillar or immunotactoidpattern suggestive of cryoglobulin deposits. In regards todifferentiating between C3 glomerulonephritis and apostinfectious glomerulonephritis (option D) or autoim-mune proliferative glomerulonephritis (option E) are thehistologic features showing the lack of immunoglobulinson kidney biopsy IF studies in the former (the diagnosis ofSLE is suggested by a “full-house” pattern on IF). In thepresent case, the histologic pattern consisted of C3 depo-sition in the absence of Ig deposition, and the absence ofhighly electron-dense intramembranous deposits thatthickened and transformed the lamina densa on EM (34)makes C3 GN the correct diagnosis. As discussed above,these cases of MPGN with C3 deposition, just like DDD,result from dysregulation of the alternate pathway of com-plement due to mutations or antibodies to the comple-ment-regulating proteins, making evaluation of the com-plement cascade (option C) the correct answer (33).
Recent advances in the understanding of the pathophys-iologic processes involved in the development of MPGNsuggest that the traditional EM-based classification ofMPGN as type I, II, or III may result in overlap between thetypes, often leading to poor understanding and evaluationof the patients, as well as inadequacies in treatment. La-beling a patient as having MPGN type I or III does not helpin distinguishing between an immune complex mediatedversus a complement-mediated process as cause of thedisease. A classification of MPGN that is based on thepathogenic process by analysis of IF, complemented byEM, has recently been proposed (33). Based on this newclassification, MPGN can be divided as: a) immune com-plex-mediated, or b) complement-mediated. In the opinionof the author, this new approach is more valuable for thepracticing clinician because it helps direct the correct clin-ical evaluation and design potential disease-specific treat-ments.
DisclosuresNone.
References1. Glassock RJ, Bargman JM, Palmer BF, Samaniego M, Fer-
venza FC: Nephrology quiz and questionnaire: 2009. ClinJ Am Soc Nephrol 5: 1141–1160, 2010
2. Holley K, Donadio J: Membranoproliferative glomerulone-phritis. In: Renal Pathology, with Clinical and FunctionalCorrelations, 2nd ed., edited by CC Tisher and BM Brenner,Philadelphia, PA, Lippincott Williams and Wilkins, 1994, pp294–349
3. Rennke HG: Secondary membranoproliferative glomerulone-phritis. Kidney Int 47: 643–656, 1995
4. Glassock RJ: Membranoproliferative glomerulonephritis. In:
Evidence-Based Nephrology, edited by DA Moloney and JCCraig, Hoboken, NJ, Wiley-Blackwell, 2008, pp 183–195
5. Johnson RJ, Gretch DR, Couser WG, Alpers CE, Wilson J,Chung M, Hart J, Willson R: Hepatitis C virus-associated glo-merulonephritis. Effect of alpha-interferon therapy. Kidney Int46: 1700–1704, 1994
6. Johnson RJ, Gretch DR, Yamabe H, Hart J, Carlos E. BacchiCE, Peter Hartwell P, Couser WG, Corey L, Wener MH, Alp-ers CE, Willson R: Membranoproliferative glomerulonephritisassociated with hepatitis C virus infection. N Engl J Med 328:465–470, 1993
7. Vella J, Carmody M, Campbell E, Browne O, Doyle G,Donohoe J: Glomerulonephritis after ventriculo-atrial shunt.QJM 88: 911–918, 1995
8. Boseman P, Lewin M, Dillon J, Sethi S: Marfan syndrome,MPGN, and bacterial endocarditis. Am J Kidney Dis 51: 697–701, 2008
9. Hulton SA, Risdon RA, Dillon MJ: Mesangiocapillary glomer-ulonephritis associated with meningococcal meningitis, C3nephritic factor and persistently low complement C3 and C5.Pediatr Nephrol 6: 239–243, 1992
10. West CD: Membranoproliferative hypocomplementemic glo-merulonephritis. Nephron 11: 134–146, 1973
11. West CD: Idiopathic membranoproliferative glomerulonephri-tis in childhood. Pediatr Nephrol 6: 96–103, 1992
12. Kamar N, Izopet J, Alric L, Guilbeaud-Frugier C, Rostaing L:Hepatitis C virus-related kidney disease: An overview. ClinNephrol 69: 149–160, 2008
13. D’Amico G: Renal involvement in hepatitis C infection: Cryo-globulinemic glomerulonephritis. Kidney Int 54: 650–671,1998
14. Meyers CM, Seeff LB, Stehman-Breen CO, Hoofnagle JH:Hepatitis C and renal disease: An update. Am J Kidney Dis42: 631–657, 2003
15. Roccatello D, Fornasieri A, Giachino O, Rossi D, Beltrame A,Banfi G, Confalonieri R, Tarantino A, Pasquali S, Amoroso A,Savoldi S, Colombo V, Manno C, Ponzetto A, Moriconi L,Pani A, Rustichelli R, Di Belgiojoso GB, Comotti C, Qua-renghi MI: Multicenter study on hepatitis C virus-related cryo-globulinemic glomerulonephritis. Am J Kidney Dis 49: 69–82, 2007
16. Maripuri S, Grande JP, Osborn TG, Fervenza FC, MattesonEL, Donadio JV, Hogan MC: Renal involvement in primarySjogren syndrome: A clinicopathologic study. Clin J Am SocNephrol 4: 1423–1431, 2009
17. Pereira MV, Revelo MP, Bambirra EA: [Lupic nephropathy inchildhood: morphologic analysis of 18 cases]. J Pediatr (Rio J)72: 32–34, 1996
18. Weening JJ, D’Agati VD, Schwartz MM, Seshan SV, AlpersCE, Appel GB, Balow JE, Bruijn JA, Cook T, Ferrario F, FogoAB, Ginzler EM, Hebert L, Hill G, Hill P, Jennette JC, KongNC, Lesavre P, Lockshin M, Looi LM, Makino H, Moura LA,Nagata M:. The classification of glomerulonephritis in sys-temic lupus erythematosus revisited. Kidney Int 65: 521–530,2004
19. Nasr SH, Markowitz GS, Stokes MB, Seshan SV, ValderramaE, Appel GB, Aucouturier P, D’Agati VD: Proliferative glo-merulonephritis with monoclonal IgG deposits: A distinct en-tity mimicking immune-complex glomerulonephritis. KidneyInt 65: 85–96, 2004
20. Nasr SH, Satoskar A, Markowitz GS, Valeri AM, Appel GB,Stokes MB, Nadasdy T, D’Agati VD: Proliferative glomerulo-nephritis with monoclonal IgG deposits. J Am Soc Nephrol20: 2055–2064, 2009
21. Sethi S, Zand L, Leung N, Smith RJ, Jevremonic D, HerrmannSS, Fervenza FC: Membranoproliferative glomerulonephritissecondary to monoclonal gammopathy. Clin J Am Soc Neph-rol 5: 770–782, 2010
22. Appel GB, Cook HT, Hageman G, Jennette JC, Kashgarian M,Kirschfink M, Lambris JD, Lanning L, Lutz HU, Meri S, RoseNR, Salant DJ, Sethi S, Smith RJ, Smoyer W, Tully HF, TullySP, Walker P, Welsh M, Wurzner R, Zipfel PF: Mem-branoproliferative glomerulonephritis type II (dense depositdisease): An update. J Am Soc Nephrol 16: 1392–1403, 2005
23. Habbig S, Mihatsch MJ, Heinen S, Beck B, Emmel M, Skerka
2546 Clinical Journal of the American Society of Nephrology

C, Kirschfink M, Hoppe B, Zipfel PF, Licht C: C3 depositionglomerulopathy due to a functional factor H defect. KidneyInt 75: 1230–1234, 2009
24. Servais A, Fremeaux-Bacchi V, Lequintrec M, Salomon R,Blouin J, Knebelmann B, Grunfeld JP, Lesavre P, Noel LH,Fakhouri F: Primary glomerulonephritis with isolated C3 de-posits: A new entity which shares common genetic risk fac-tors with haemolytic uraemic syndrome. J Med Genet 44:193–199, 2007
25. Fakhouri F, Fremeaux-Bacchi V, Noel LH, Cook HT, Picker-ing MC: C3 glomerulopathy: A new classification. Nat RevNephrol 6: 494–499, 2010
26. Sethi S, Gamez JD, Vrana JA, Theis JD, Bergen, HR, III, ZipfelPF, Dogan A, Smith RJH: Glomeruli of dense deposit diseasecontain components of the alternative and terminal comple-ment pathway. Kidney Int 75: 952–960, 2009
27. Smith RJ, Alexander J, Barlow PN, Botto M, Cassavant TL,Cook HT, de Cordoba SR, Hageman GS, Jokiranta TS, Kim-berling WJ, Lambris JD, Lanning LD, Levidiotis V, Licht C,Lutz HU, Meri S, Pickering MC, Quigg RJ, Rops AL, SalantDJ, Sethi S, Thurman JM, Tully HF, Tully SP, van der Vlag J,Walker PD, Wurzner R, Zipfel PF; Dense Deposit DiseaseFocus Group: New approaches to the treatment of dense de-posit disease. J Am Soc Nephrol 18: 2447–2456, 2007
28. Walker PD: Dense deposit disease: New insights. Curr OpinNephrol Hypertens 16: 204–212, 2007
29. Zipfel PF, Skerka C: Complement regulators and inhibitoryproteins. Nat Rev Immunol 9: 729–740, 2009
30. Sethi S, Fervenza FC, Zhang Y, Nasr S, Leung N, Vrana, J,Cramer C, Nestor CM, Smith RJH: Membranoproliferativeglomerulonephritis secondary to dysfunction of the alterna-tive pathway of complement. Clin J Am Soc Nephrol 6:1009–1017, 2011
31. Alpers CE, Smith KD: Cryoglobulinemia and renal disease.Curr Opin Nephrol Hypertens 17: 243–249, 2008
32. Smith KD, Alpers CE: Pathogenic mechanisms in mem-branoproliferative glomerulonephritis. Curr Opin NephrolHypertens 14: 396–403, 2005
33. Sethi S, Fervenza FC: Membranoproliferative glomerulone-phritis: Pathogenic heterogeneity and proposal for a newclassification. Semin Nephrol 31: 341–348, 2011
Presented at the Annual Meeting of the American Society of
Nephrology on November 20, 2010.
Published online ahead of print. Publication date available atwww.cjasn.org.
Clin J Am Soc Nephrol 6: 2534–2547, October, 2011 Nephrology Quiz and Questionnaire, Glassock et al. 2547