semihydrogenation of acetylene on the (010) surface of ... · pdf filesemihydrogenation of...
TRANSCRIPT

Semihydrogenation of Acetylene on the (010) Surface of GaPd2: GaEnrichment Improves SelectivityM. Krajcí*,† and J. Hafner‡
†Institute of Physics, Slovak Academy of Sciences, Bratislava, SK-84511, Slovakia‡Faculty for Physics, Center for Computational Materials Science, Vienna University, Vienna, A-1090, Austria
ABSTRACT: Ab-initio density-functional calculations have been used to investigate thestructure and stability of the (010) surfaces of the intermetallic compound GaPd2 andtheir activity and selectivity for the catalytic semihydrogenation of acetylene to ethylene.The calculations of the surface energies show that Ga-enrichment of the surface isenergetically favored under Ga-rich preparation conditions. The bulk-terminatedstoichiometric GaPd2(010) surface is catalytically active but does not exhibit the desiredselectivity. The high Pd concentration in the surface leads to the formation of Pd3 tripletsfavoring a strong binding of ethylene and its further hydrogenation to ethyl. In contrastGa-enriched GaPd2(010) surfaces provide an excellent selectivity for the formation ofethylene. Selectivity increases with increasing number of Ga atoms in the vicinity of theactive Pd atoms. The atomistic scenarios for the dissociative adsorption of hydrogen, thediffusion of atomic hydrogen, and the hydrogenation reactions of acetylene to vinyl andfurther to ethylene demonstrate that on the selective surfaces the catalytically activecenters are triangular configurations of one Pd and two Ga atoms binding acetylenethrough di-σ bonds in Ga−Ga bridge sites. Ethylene, on the other hand is only weakly π-bonded on top of the Pd atom such thatdesorption is easier than further hydrogenation to ethyl. It is remarkable that the atomic structure of one of the Ga-enrichedGaPd2(010) surfaces is very similar to that of the (001) surface of the Ga3Pd5 compound and to the structure of the (210)surface of GaPd. The structural similarity of the surfaces results in comparable catalytic properties. The comparison with theresults for the semihydrogenation of acetylene to ethylene on the (210) surfaces of GaPd and AlPd and on the (100) surface ofAl13Co4 suggests that the concept of catalytically active centers consisting of transition-metal−sp-bonded-metal complexes hasgeneral validity.
1. INTRODUCTION
Selectivity is a ubiquitous challenge in the development ofimproved catalysts.1,2 During recent years the productselectivity in the partial hydrogenation of alkynes to alkeneshas received particular attention.3−6 The reasons behind thisinterest are the industrial importance of the process and themodel character of this at least in principle rather simplereaction. The semihydrogenation of acetylene to ethylene is animportant step in the industrial polymerization of ethylene topolyethylene because it is necessary to clean the feed-stockfrom acetylene which could poison the polymerization catalyst.The requirements on the hydrogenation catalyst are an efficientand very selective conversion of acetylene to ethylene, whileavoiding a full hydrogenation to ethane and the formation ofother side-products, e.g., by oligomerization.Palladium is known to be very active in various hydro-
genation and reforming reactions, but its properties must bemodified such that under certain conditions the selectivehydrogenation of carbon−carbon triple bonds is promoted,while the reaction stops at the stage of double bonds.Semihydrogenation would be favored if the adsorption energyof the alkene is much lower than that of the alkyne such thatdesorption of the partially hydrogenated product would bemore favorable than further hydrogenation. A reduced
adsorption strength can be achieved by mixing Pd with asecond metal (often called a cocatalyst or promoter).4 Thepromoters which are integrated in the Pd lattice can be (i)noble metals (Cu, Ag, or Au) or (ii) simple or sp metals such asGa, Al, Zn, Sn, or Bi.It turns out, however, that this is not sufficient. The reaction
between coadsorbed unsaturated hydrocarbon molecules canlead to undesired side products by oligomerization. Suppressionof oligomerization can be achieved by isolation of the activesites.7,8 Isolation of the surface Pd atoms reduces theconcentration of di-σ adsorbed ethylene at neighboring siteswhich could react to form ethylidyne or vinylidene species asprecursors to oligomerization or to the formation of carbona-ceous deposits, leading to a deactivation of the catalyst.Furthermore, at high hydrogen to hydrocarbon ratios the
high reactivity of Pd-based catalysts toward hydrogen can leadto the adsorption of hydrogen in subsurface sites or even theformation of bulk β-Pd-hydride.9 The hydrogen dissolved in thebulk can diffuse to the surface and hydrogenate adsorbates. Atlow partial pressures of hydrogen dissociation of hydrocarbons
Received: March 12, 2014Revised: May 16, 2014Published: May 16, 2014
Article
pubs.acs.org/JPCC
© 2014 American Chemical Society 12285 dx.doi.org/10.1021/jp5025075 | J. Phys. Chem. C 2014, 118, 12285−12301

can lead to the formation of carbonaceous deposits orincorporation of C atoms in subsurface sites (surfacecarbides).10,11 The formation of a surface carbide hinders thesupply of hydrogen from subsurface sites that could lead to anundesired continued hydrogenation. Carbonaceous depositsseparate the active sites but, at high coverages, also lead todeactivation. Surface carbides can block the diffusion ofhydrogen from the bulk to the surface, thus changing selectivity.In summary, selectivity depends strongly on the phases of thecatalyst formed during the reaction: hydrides favor theformation of alkanes, while carbides are selective to alkenes.12,13
Density-functional calculations have contributed to elucidatethe conditions for achieving a high selectivity to ethylene in thehydrogenation of acetylene over Pd-based alloy catalysts.Neurock et al.14−16 and Norskov et al.17,18 have calculatedthe potential-energy profile for the hydrogenation of acetyleneover Pd(111) and Pd−Ag(111) surfaces. It was shown that themain effect of the cocatalyst is to reduce the adsorptionenergies of acetylene and ethylene, while the influence on theactivation energies is rather modest. Selectivity is achieved if thedesorption energy of ethylene is lower than the activationenergy for further hydrogenation. It was found that thebeneficial effect of Ag is very sensitive to the local surfacecomposition and coverage, which is in turn influenced byadsorbates and hence subject to change under reactionconditions. Gonzalez et al.19 have shown that while undervacuum conditions Ag segregates to the surface, even a lowcoverage of one-quarter of a monolayer of hydrogen issufficient to reverse the trend and to drive all Ag atoms tosubsurface positions.Hence the problem with bimetallic catalysts is that the
composition and/or structure of their active surface is subjectto change under reaction conditions, affecting both selectivityand reactivity. This has motivated the search for novel materialswith intrinsic catalytic properties matching those of Pd−Csubsurface alloys at optimal reaction conditions, but which canbe reproducibly prepared in a controlled manner and exhibitbetter stability.6 A class of compounds meeting theserequirements are intermetallic compounds. These are single-phase materials with an ordered crystal structure which isdifferent from that of the constituent elements. The oftenrather complex crystal structure is stabilized by an at leastpartially covalent interaction between the atoms of differentspecies, resulting also in a strong alteration of the electronicstructure and hence of the chemical reactivity and catalyticproperties compared to the elements forming the compound.The recent work of the group around Schlogl has identifiedintermetallic compounds of Pd with Ga or Zn as highly efficientand selective catalysts for a number of reactions: semi-hydrogenation of acetylene,6,20−23 methanol steam reforming,and methanol synthesis.24−27 Also compounds free of preciousmetals such as Al13Co4 and Al13Fe4
28,29 have been identified ashighly selective stable catalysts for the semihydrogenation ofacetylene.For GaPd and Ga7Pd3 compounds selectivities of 75% and
excellent stability with time on stream have been reported forthe semihydrogenation of acetylene.6,20−22 These effects havebeen attributed to the smallness of the active sites and to themodification of the electronic structure, which also preventside-reactions such as oligomerization and a coke formationwhich can take place on large surface ensembles. On the otherside it was observed that the improvement of the activity of thecatalysts obtained by increasing the active surface by milling of
larger grains or by etching the surfaces lead to a decrease ofselectivity.6,22 This was explained by the introduction of defectsto the pristine structure of the GaPd crystal. The improvedselectivity was tentatively attributed to a smaller percentage ofdi-σ bonded hydrocarbon species that are prone to furtherhydrogenation or to oligomerization.6,29
Recently we have used ab initio density-functional-theory(DFT) methods to investigate the surface structure andstability of several catalytically active intermetallic compoundsand to develop atomistic models for the hydrogenationreactions on their surfaces, including the (100) surface oforthorhombic Al13Co4,
30−32 the pseudo-5-fold (210) surface ofB20-type AlPd,33,34 and the (210) and the polar {111} surfacesof the isostructural GaPd compound.35−37 The important resultof these studies is that the catalytic activity of these surfaces ispromoted not by the transition metal (TM) atoms (Co, Pd)alone, but that the active centers consists of small, in most casestriangular clusters of two simple metal (Al, Ga) atoms and oneTM atom. Acetylene is preferably adsorbed in a di-σ bridgingconfiguration between two simple metal atoms and vinyl (theproduct of the first hydrogenation step) is in most cases di-σadsorbed in an asymmetric configuration between one of thesimple metal atoms and the neighboring TM atom, whileethylene is π-adsorbed only rather weakly on-top of the TMatom. The strong binding of acetylene and vinyl to the activecenter facilitates the attack of hydrogen on the hydrocarbonmolecule and promotes the activity of the catalyst. Theselectivity of the catalyst results from a desorption energy ofweakly π-bound ethylene that is lower than the activationenergy for further hydrogenation. The distance between theactive centers is large enough to suppress undesired side-reactions such as oligomerization which depend on thecoupling between carbon atoms in different reactants.The main difficulty in developing an atomistic scenario of the
reactions catalyzed by complex intermetallic compounds is theinsufficient knowledge of their surface properties. While thelow-index surfaces of elemental close-packed metals have beenstudied for decades and are today well understood,38 thesurfaces of intermetallic compounds are largely unexplored.The complexity of the problem is particularly evident in thecase of B20-type GaPd whose surfaces have recently beenstudied both experimentally39,40 and theoretically.33,35−37
Because of the lack of inversion symmetry of the structure(space group P213) the crystals exist in two enantiomorphicforms and the {111} surfaces have polar character. While the 2-fold {100} and the pseudo-5-fold {210} surfaces consist ofperiodically repeated layers with the same stoichiometry as thebulk, for the polar (111) and (1 11 ) surfaces normal to the 3-fold direction up to ten possible surface terminations differingin structure and composition have to be considered.37 Whilethe structure of the {210} surfaces can be described by atriangle-rectangle tiling and the active centers consist ofperiodically distributed triangular arrangements of two Gaand one Pd atom promoting a high activity and selectivity forthe semihydrogenation of acetylene, the possible terminationsof the 3-fold surface show very different catalytic activities andselectivities.37 Ga-rich surfaces have been found to becatalytically inactive because they cannot dissociate hydrogenand initiate the hydrogenation reaction. Highly corrugated Pd-rich surfaces bind hydrogen very strongly such that anydiffusion is impossible under normal reaction conditions.However, such surfaces are energetically rather unfavorableand can exist only under extreme values of the chemical
The Journal of Physical Chemistry C Article
dx.doi.org/10.1021/jp5025075 | J. Phys. Chem. C 2014, 118, 12285−1230112286

potential in the reactive atmosphere. Only surfaces with amixed Pd/Ga occupancy are catalytically active. Surfaces withprotruding isolated Pd atoms can hydrogenate acetylene andvinyl rather easily, but reactions on these surfaces are notselective, because ethylene molecules adsorbed on-top of aprotruding atom can easily be attacked by coadsorbed hydrogenatoms. On the other surfaces with mixed composition Pd atomsform isolated triplets. Because the migration of hydrogen atomsbetween different triplets is a strongly activated process, asufficient activity of the catalyst can be achieved only if firsthydrogen is adsorbed dissociatively, followed by the activatedcoadsorption of acetylene on the same Pd3 triplet. A veryimportant feature determining the selectivity for semihydroge-nation is that acetylene is strong di-σ-bound in a bridgingconfiguration between two Pd atoms, while ethylene is onlyweakly π-bonded on-top of a Pd atom.37 A similar change fromstrong di-σ bonding of acetylene to weak π-bonding of ethylenehas also been found on the {210} surfaces of GaPd andAlPd.33,35 However, because of the complex sequentialcoadsorption of hydrogen and acetylene requires a higheractivation energy, the catalytic activity of the A(1 11 ) surfacewith isolated Pd3 triplets is lower than that of the (210) surface.An activity and selectivity equal to that of the (210) surface canbe achieved only if additional Ga atoms are located in the largehole between the isolated Pd3 triplets.37 However, such asurface is energetically competitive with other surfaceterminations only in a narrow interval of the chemical potential.The important lesson from this analysis is that catalyticperformance of the 3-fold {111} surfaces depends crucially onthe preparation conditions and thermal history of the GaPdcatalyst, while the pseudo-5-fold {210} surfaces provide highactivity and selectivity under any conditions.Recently it has been demonstrated that the Pd-rich
compound GaPd2 is an efficient and selective catalyst formethanol steam-reforming25−27 and for the semihydrogenationof acetylene.24,41 The selectivity achieved with nanostructuredGaPd2 catalysts is comparable to that provided by other Pd−Gacompounds.41 Nanostructured particles of Pd−Ga intermetalliccompounds were formed by the reaction of Pd nanoparticleswith the Ga2O3 support in a reducing atmosphere26,27 or byreducing a Ga3+ complex by Pd-activated hydrogen.23,42 Thestoichiometry of the Pd−Ga compound formed upon reductionin hydrogen depends on the temperature and on the galliapolymorph chosen as supporting material. At low reductiontemperatures GaPd2 is formed, while higher temperatures favorthe formation of Ga-enriched compounds (Ga3Pd5, GaPd). Liet al.42 reported a higher selectivity for ethylene formation fromacetylene for Ga3Pd5 than for GaPd2, and Armbruster et al.41
found that on nanostructured catalysts the reaction on GaPdshows better selectivity than on GaPd2, suggesting that Ga-enrichment favors an increased selectivity.A common property of all compounds formed by simple and
transition metals is that the surface energies of the simplemetals are always much lower than those of the correspondingtransition metal surfaces. For example, the surface energy ofZn(0001) is only 22% of that of Pd(111). As the surface energyof an alloy or intermetallic compound depends mainly on thechemical composition of the surface layer, for PdZn a strongsurface segregation was expected and experimentally confirmed.For the PdZn(111) surface a Zn concentration of up to 75 at. %was measured by XPS.44 Hence Ga enrichment must also beconsidered for the surface of GaPd2. The investigationspresented below demonstrate that Ga-enrichment of the
surface layer is favored at least under the Ga-rich conditionsused in the preparation of the nanostructured GaPd2 catalysts.In the present work we have investigated the influence of the
surface stoichiometry on the catalytic performance of GaPd2 forthe semihydrogenation of acetylene using ab initio DFTmethods. GaPd2 has a layered structure consisting of a singletype of atomic planes stacked in the [010] direction.43 Thelayered structure suggests that the (010) planes are also thenatural cleavage planes, hence our investigations focus on the(010) surfaces.There exists an interesting relation between the Ga-enriched
GaPd2(010) surface and one of the possible surfaces of theGa3Pd5 compound. Ga3Pd5 has also a layered structureconsisting of a two types of atomic planes stacked in the[001] direction. In a Ga rich environment the energeticallypreferred Ga3Pd5 surface is the (001) surface containing anequal number of Ga and Pd atoms. The atomic structure of thissurface is essentially the same as one of the studied Ga-enrichedGaPd2(010) surfaces.We demonstrate that the observed high selectivity of
reactions on the GaPd2(010) surface originates from activesites in Ga-enriched local environments. We shall show that thesame active sites can be found also on the Ga3Pd5(001) surfaceas well as on the previously studied GaPd(210) surface.35
In section 2 we briefly review the methods used in ourcalculations. In section 3 we present our results for thestructure of the GaPd2(010) surface, discuss the interatomicbonding, and introduce surface models representing the Ga-enriched surfaces. The adsorption of molecular and atomichydrogen and the diffusion of H atoms on the surfaces isdiscussed in section 4, the adsorption of acetylene and ethylenemolecules in section 5. Atomistic reaction paths for thehydrogenation of acetylene are presented in section 6. Theconditions for achieving a high selectivity are analyzed in detailin section 7. In section 8 we present our conclusions, includingalso a detailed comparison with the semihydrogenation ofacetylene on the surfaces of the GaPd, AlPd, and Al13Co4compounds.
2. COMPUTATIONAL METHODSTotal-energy and electronic structure calculations have beenperformed using the Vienna ab initio simulation package(VASP).45,46 VASP performs an iterative solution of the Kohn−Sham equations of density functional theory (DFT) within aplane−wave basis. The electron−ion interaction is describedwithin the projector-augmented wave (PAW) method.47 Weused the semilocal PBE exchange-correlation functional in thegeneralized gradient approximation proposed by Perdew etal.48,49 The basis set contains plane waves with a kinetic energyup to Ecut−off = 700 eV. The self-consistency iterations werestopped when total energies are converged to within 10−6 eV.The atomic structure of the surfaces has been optimized bystatic relaxations using a quasi-Newton method and theHellmann−Feynman forces acting on the atoms. Transition-states were determined using the climbing-image nudged-elasticband method.50 Transition states have been further optimizedby the dimer method.51,52 For the structural optimizations andtransition-state searches convergence criteria of 10−4 eV fortotal energies and 0.05 eV/Å for forces acting on the atomshave been applied.Surface calculations have been performed for slab models cut
from the bulk structure of GaPd2. For the calculations ofadsorption energies and reaction paths we have used a slab 8.4
The Journal of Physical Chemistry C Article
dx.doi.org/10.1021/jp5025075 | J. Phys. Chem. C 2014, 118, 12285−1230112287

Å thick consisting of 4 atomic layers with fixed coordinates ofthe atoms in the bottom layer, altogether 48 atoms. Periodicallyrepeated slabs are separated by a vacuum layer of 14 Å. Thesurface area contains two surface cells and lateral dimensions ofthe computational cell are 7.88 Å × 11.17 Å. Total energies ofadsorption complexes and of states along the reaction pathwere calculated using a 4 × 3 × 1 k-point mesh for Brillouin-zone integration. No zero-point-energy corrections wereincluded. We note that the calculated binding and activationenergies depend on coverage. To compare activities andselectivities of different surfaces it is thus important to performthe calculations at the same or at very similar coverages andwith the same computational setting.
3. STRUCTURE OF GAPD2(010) SURFACE
GaPd2 crystallizes in the Co2Si structure (Pearson symboloP12, space group Pnma, No. 62). The orthorhombic unit cellcontains four Ga and eight Pd atoms. The lattice parametersdetermined by our calculations and X-ray diffraction43 (inbrackets) are a = 5.5858(5.4829) Å, b = 4.0999(4.0560) Å, andc = 7.8789(7.7863) Å. The structure can be described by thestacking of one type of flat layers along the [010] direction atinterlayer distances of b/2 = 2.05 Å. The unit cell contains twolayers mutually related by the inversion. Hence any surfaceperpendicular to the [010] direction exposes the same surfacelayer with the same stoichiometry as the bulk. In each atomicplane there is only one crystallographic type of Ga and twotypes of Pd atoms, Pd1 and Pd2, see Figure 1a.The interatomic bonding in GaPd2 is characterized by a
strong, at least partially covalent bonding between Pd−Ganearest neighbor pairs, weak Pd−Pd bonding, and mutualrepulsion between Ga atoms. The covalent character of thebonding is demonstrated by the differential charge density
distribution, but it is also obvious from the short nearest-neighbor distances. In the bulk structure each Pd1 atom has adistorted tetrahedral coordination by four Ga atoms atdistances of 2.57 and 2.58 Å. Each Pd2 atom has two Ganearest neighbors at a distance of 2.59 Å and one at 2.65 Å. Theshortest Pd1−Pd1, Pd1−Pd2, and Pd2−Pd2 distances are of2.94, 2.86, and 2.87 Å, respectively. The large distance of 3.04 Åbetween nearest Ga−Ga neighbors indicates the repulsionbetween the nearest Ga atoms.The (010) surface does not reconstruct; the modest
structural relaxation (reduction of the d12 interlayer distancefrom 2.05 to 2.024 Å and a very modest puckering of thesurface layer with an amplitude of 0.078 Å) preserves the bulk-terminated surface structure. Each Pd1 atom is bound in thesurface layer to two Ga1 neighbors at short distances 2.56 and2.65 Å with enhanced covalent bonding and to one Ga1 atomat a larger distance of 2.93 Å, as well as to one subsurface Ga1atom at 2.59 Å. The Pd2 atom has only one surface Ga1neighbor at a distance of 2.54 Å, and two subsurface Ga1neighbors at the distances 2.50 and 2.79 Å. The Ga1 atoms inthe surface are rather strongly bound to Pd1 and Pd2 neighborslocated on both sides approximately in the [001] direction(horizontal in Figure 1a) and show a weaker bonding to twoPd1 atoms located on both sides in the [100] direction (verticalin Figure 1a).Figure 1a shows that on the GaPd2(010) surface Pd3 triplets
consisting of one Pd1 atom and two Pd2 atoms exist. SimilarPd3 triplets exist on the Pd(111) surface and also on Pd-rich{111} surface terminations of GaPd. We shall see, as on theseGaPd 3-fold surfaces, the strong adsorption of H atoms in the3-fold hollows of the Pd3 triplets significantly complicates thediffusion of atomic hydrogen and is responsible for the lowerselectivity of a hydrogenation reaction on this surface.
Figure 1. Model surfaces: (a) clean GaPd2(010) surface, model G0 consists of two surface cells repeated in the [001] direction (vertical), (b)GaPd2(010) with one Pd2 atom per surface cell replaced by Ga, model G1, (c) GaPd2(010) with two Pd2 atoms per surface cell replaced by Ga,model G2, (d) clean Ga3Pd5(001) surface, model E0 consists of 2 surface cells repeated in the [001] direction (horizontal). The atomic structure ofthe top layer of the Ga enriched GaPd2(010) G1 surface is essentially the same as that of the Ga3Pd5(001) E0 surface.
The Journal of Physical Chemistry C Article
dx.doi.org/10.1021/jp5025075 | J. Phys. Chem. C 2014, 118, 12285−1230112288

With a surface area of 7.88 Å2 per atom the GaPd2(010)surface is rather sparsely occupied. For comparison on Pd(111)the surface area is 6.77 Å2/atom, on the GaPd(210) surface35 itis 6.87 Å2/atom, and on a relatively dense GaPd(111) surfaceterminated by planes of Pd3 and Ga3 triplets forming a doublelayer (the PG termination in the terminology of our previouswork37) it is 7.11 Å2/atom. Although the relaxation of the cleanGaPd2(010) surface is rather modest, larger lateral displace-ments of surface atoms induced by the adsorption of reactantsand their hydrogenation can be expected.We have denoted the pristine model of the GaPd2(010)
surface with the same stoichiometry as the bulk as G0. Thecalculated surface energy is γ[GaPd2(010)] = 1.31 J/m2, almostthe same as that of the Pd(111) surface, γ[Pd(111)] = 1.33 J/m2 and only somewhat lower than the surface energy of 1.51 J/m2 of the Pd(100) surface. The surface energy of GaPd2(010)is 12% higher than the concentration-weighted average of theenergies of the (100) surfaces of face-centered cubic Pd and oforthorhombic α-Ga(100) (γ[Ga(100)] = 0.48 J/m2). Weassume that the higher value of the surface energy than theaverage value originates mainly from the more open characterof the surface, see above. We note that in the case of theGaPd(210) surface the concentration-weighted average of thesurface energies of the components is 8% higher than thecalculated surface energy, and in the case of the (100) surface ofGaPd the surface energy is almost exactly (within 1%) equal tothe average of the surface energies of the components.As already noted in the Introduction it can be expected that
Ga-enrichment of the surface will substantially reduce thesurface energy. To investigate this effect we have prepared twoadditional surface models. In the model G1, see Figure 1b, oneof the Pd2 atoms per surface cell is replaced by a Ga atom, inthe model G2, see Figure 1c, both Pd2 atoms are replaced byGa. We have found that Ga substitutions of the Pd1 atoms areenergetically unfavorable. The surface energies of the Ga-enriched, nonstoichiometric surfaces in equilibrium with thebulk have been calculated in the grand-canonical formal-ism36,53−55 as a function of the chemical potential ΔμGa = μGa −μGa0 , where μGa
0 = −2.67 eV is the chemical potential of bulk Gain its stable phase oP8; the results are presented in Figure 2.
The allowed values of the chemical potential ΔμGa aredetermined by the limits where the pure metals begin tocondense on the surface. Equilibrium with the bulk requiresthat the composition-weighted sum of the chemical potentialsof Ga and Pd is equal to the cohesive energy per formula unit.The calculated surface energies show that the stoichiometricsurface G0 is a stable surface for values of ΔμGa from −2.01 to−0.84 eV, whereas the Ga enriched surfaces G1 and G2 areenergetically favored for ΔμGa from −0.84 to −0.42 eV andfrom −0.42 to 0 eV, respectively.In addition we have prepared two stoichiometric slab models,
denoted as S1 and S2, where one or two Pd2 atoms in thesurface layer are exchanged with Ga atoms from the subsurfacelayer. The surface energies of these Ga-segregated surfaces are γ= 1.66 and 2.08 J/m2 for one and two Pd/Ga exchanges,respectively. The surface energies of the S1 and S2 surfaces aresignificantly higher than that of the bulk-terminated surface G0,see Figure 2a. The increased energy is due to the formation ofantisite defects in the subsurface layer. The exchange of onepair of Ga−Pd atoms per surface cell costs 0.95 eV.These results show that, due to the strong binding between
unlike atoms, segregation of Ga to the surface is energeticallyunfavorable. However, if the nanostructured GaPd2 catalyst isprepared by the coreduction of ionic metal precursors andannealing the chemical potential μGa is close to the Ga-rich endof allowed allowed interval, as confirmed by the observation ofthe formation of elemental Ga at the surface of Pd.23 Hence theGaPd2(010) surface will be Ga enriched and very likely containlocal configurations corresponding to our models G1 or G2.On the Ga-enriched surfaces the environment of the Pd
atoms is changed. On the G0 surface a Pd1 atom has three Ganeighbors, on G1 four, and on G2 five. Below we willdemonstrate that the Ga atoms in the environment of a Pd1atom substantially change the catalytic properties, particularlythe selectivity of the catalyst. We show that the selectivityincreases with an increasing number of Ga neighbors of thecatalytically active Pd atom. The arrangement of atoms on theG0 and G2 surfaces has 21 symmetry. For each site withcoordinates (x, y), there exists an equivalent site withcoordinates (0.5 − x,0.5 + y). On the G1 surface this symmetry
Figure 2. Dependence of the surface energies on the chemical potential ΔμGa: (a) G0, stoichiometric GaPd2(010) surface; G1 and G2, GaPd2(010)surface with one or two Pd2 atoms per surface cell replaced by Ga; S1 and S2, GaPd2(010) surfaces with one or two Pd2 surface atoms exchangedwith Ga atoms from the subsurface layer, cf. text. (b) E0, the Ga3Pd5(001) surface termination with the equal concentration of Ga and Pd atoms; P,the Pd-rich Ga3Pd5(001) surface termination; E1, the Ga-enriched Ga3Pd5(001) E0 surface.
The Journal of Physical Chemistry C Article
dx.doi.org/10.1021/jp5025075 | J. Phys. Chem. C 2014, 118, 12285−1230112289

is broken, therefore, e.g., for Pd1 two structurally inequivalentsites Pd1 and Pd1′ have to be distinguished. The Pd1 and Pd1′atoms differ in their bonding to neighboring Ga atoms. EachPd1 atom is coordinated in the surface layer by four Ga atoms:three at short distances of 2.58, 2.61, and 2.69 Å indicating astrong covalent bonding and one at a larger distance of 2.93 Å.Pd1 is bound also to one subsurface Ga atom at 2.60 Å. In theenvironment of the Pd1′ atom there are also five Ga atoms.Strong covalent bonding is observed with one subsurface Gaatom at a distance of 2.55 Å and with two Ga neighbors in thesurface at distances of 2.56 and 2.64 Å. The remaining Ganeighbors are located at distances of 2.93 Åand 3.31 Å. Hencein the surface layer each Pd1′ atom has also four Ga neighbors,but one of them is at a larger nonbonding distance.3.1. Comparison with the Ga3Pd5(001) and GaPd(210)
Surfaces. As noted in the Introduction, Ga3Pd5 and GaPd havealso been identified as active and selective catalysys foracetylene semihydrogenation.21,22,42 It is remarkable that thesurfaces that are expected to contribute to the catalytic activityof these compounds have an atomic structure very similar tothe GaPd2(010) G1 surface.The Ga3Pd5 compound crystallizes in the Rh5Ge3 structure
(Pearson symbol oP16, space group Pbam, No. 55). Theorthorhombic unit cell contains six Ga and ten Pd atoms. Thelayered structure consists of two alternating planes stacked inthe [001] direction. One of them, denoted as P, is a Pd richplane with 6 Pd and 2 Ga atoms per surface cell. The secondcontains 4 Ga and 4 Pd atoms per surface cell, it is denoted asE0. The surface energies calculated as a function of thechemical potential ΔμGa, see Figure 2b, shows that the E0termination is energetically preferred to a P termination forΔμGa > −1.02 eV. Figure 1d demonstrates that the atomicstructure of the Ga3Pd5(001) E0 surface is similar to that of theGaPd2(010) G1 surface, but the arrangement of the atoms inthe Ga3Pd5(001) E0 surface plane has 21 symmetry, while thissymmetry is broken on the G1 surface. It is also remarkable thatin the range of the chemical potentials ΔμGa where the G1 andE0 surfaces are stable their surface energies have similar valuesof ∼1.20 J/m2, see Figure 2.One can consider also a Ga enrichment of the E0 surface
consisting of a replacement of one of the Pd atoms per surfacecell by a Ga atom. The structure of this surface denoted as E1 issimilar to that of the G2 surface of GaPd2, see Figure 1c. This
surface is stabilized under preparation conditions correspond-ing to the highest values of the chemical potential ΔμGa, seeFigure 2.GaPd crystallizes in the FeSi (B20) structure (Pearson
symbol cP8, space group P213, no. 198). The cubic unit cellcontains four Ga and four Pd atoms. The catalytic activity ofthe (210) and (111) surfaces of this compound have beenstudied in detail in our previous works.35,37 For the polar 3-foldsurfaces it was found that preparation by crystal growth leads toa termination by Ga-terminated surfaces without Pd atoms inthe top atomic plane, which are catalytically inactive.37 Catalyticactivity is highest on the (210) surface. The atomic structure ofthe GaPd(210) surface is also very similar to that of theGaPd2(010) G1 surface, see Figure 2 in our previous work.35
The surface energy of the GaPd(210) surface of 1.08 J/m2 isalso comparable with that of the G1 surface. The somewhatlower value can be explained by the denser packing of thesurface atoms discussed above.From the similarity of the atomic structures of the Ga
enriched GaPd2(010) and the Ga3Pd5(001) and GaPd(210)surfaces one can expect a similar catalytic properties of thecorresponding surfaces. Moreover, we shall demonstrate thatthe reaction centers promoting the selective acetylene hydro-genation at all three compounds are the same.In the following sections we shall focus on the catalytic
properties of the GaPd2(010) surfaces only. The results for theGa3Pd5(001) surfaces will be published elsewhere. For theGaPd2(010) surfaces we present results for the adsorption ofmolecular and atomic hydrogen, for the diffusion of hydrogenatoms, the adsorption of acetylene and its hydrogenation tovinyl and ethylene on the G0, G1, and G2 surfaces. Thecoverage is one adsorbed molecule (or atom) per 12 surfaceatoms, the results can directly be compared with those foradsorption on the Pd(111) and GaPd(111) surfaces at thesame coverage37 and for the GaPd(210) surface35 at a slightlylower coverage of 1/16.
4. ADSORPTION AND DIFFUSION OF HYDROGEN
In this section we summarize our results for the adsorption ofmolecular and atomic hydrogen, the dissociation of hydrogenmolecules, and the diffusion of hydrogen atoms on the threeGaPd2 surfaces differing in their chemical composition.Adsorption energies are determined as the energy difference
Figure 3. Adsorption sites of H2 molecules (larger circles) and H atoms (smaller circles) on the stoichiometric GaPd2(010) surface, model G0 (a),and on the Ga-enriched surfaces, model G1 (b), and model G2 (c). Because of the 21 symmetry of G0 and G2 surfaces to each site with thecoordinates (x, y), there exists an equivalent site with coordinates (0.5 − x,0.5 + y) coordinates. On the G1 surface this symmetry is broken.
The Journal of Physical Chemistry C Article
dx.doi.org/10.1021/jp5025075 | J. Phys. Chem. C 2014, 118, 12285−1230112290

between the relaxed adsorbate/substrate complex and theenergies of the clean substrate plus the energy of the adsorbedspecies in the gas-phase. The stable and metastable adsorptionsites for molecular and atomic hydrogen on the three surfacesare shown in Figure 3, the calculated adsorption energies formolecular and atomic hydrogen are summarized in Table 1. Onthe G0 and G2 surfaces the local 21 symmetry simplifies theanalysis of the adsorption sites. On the G1 surface thissymmetry is broken. This means that there are now twoinequivalent adsorption sites on top of a Pd1 atom labeled PT1and PT3.4.1. Adsorption of Molecular Hydrogen. On all three
surfaces the only stable adsorption site for a hydrogen moleculeis on top of a Pd atom. On the G0 surface H2 adsorbs moststrongly in site PT1 at a height of 1.81 Å above a Pd1 atom,with an adsorption energy of Eb = −22 kJ/mol. The adsorptionenergy decreases to −15 kJ/mol for the strongest binding sitePT3 on the G1 surface (see Table 1). On the G2 surface theadsorption is even slightly endothermic, but the position of themolecule in site PT1 at a distance of 2.04 Å above the Pd atomis a well-defined local minimum. Adsorption energy and heightof the H2 molecule can be compared with a binding energy ofEb = −24 kJ/mol and a height of 1.82 Å on a Pd(111) surface.The decrease of the adsorption energy is correlated with anincreasing coordination of the Pd atom by Ga: the PT1 siteshave three, four, and five Ga neighbors on the G0, G1, and G2surfaces.4.2. Adsorption of Atomic Hydrogen. The adsorption
energies for hydrogen atoms compiled in Table 1 are definedrelative to molecular hydrogen, i.e., half of the binding energyof a hydrogen molecule has been subtracted. This means thatfor Eb ≈ 0, the binding of the hydrogen atom to the surface is asstrong as the binding in the hydrogen molecule.On the G0 surface the energetically most favorable position
of a hydrogen atom is in the 3-fold hollow PH1 within a Pd3triplet with Eb = −40 kJ/mol. Binding within the 4-fold hollowPH2 between two Ga and two Pd atoms and in a bridgeposition GP1 between a Ga and a Pd atom is considerablyweaker, even endothermic with respect to molecular hydrogen.On the G1 surface an exothermic (negative) adsorption energyis found only for the PP1 and PP2 bridge position between twoPd atoms, adsorption in Ga−Pd bridge sites is significantlyweaker. On the G2 surface the strongest adsorption sites foratomic hydrogen are in Ga−Pd bridge sites (GP1 and GP2 inFigure 3). Note that the Pd−Pd, Ga−Pd, and Ga−Ga bridgesites occupied by H atoms are different from the bridge sitesbinding acetylene in a di-σ configuration (see below). If it isnecessary to distinguish the bridge sites for H-adsorption from
the bridge sites occupied by other molecular species, thehydrogen bridge sites are labeled with a prefix H, e.g. H-PPx, H-GPx, and H-GGx (x = 1, ...). For simplicity the bridge sitesoccupied by acetylene are labeled without any prefix by PPx,GPx, and GGx only. The strong binding of a H atom in thecenter of the Pd3 triplet of the G0 surface is comparable to theadsorption on Pd(111) were adsorption energies of −58 and−54 kJ/mol are calculated for the fcc and hcp hollows at thesame coverage, and to the strong binding in a Pd3 triplet on theGaPd(111) surface.37 As already noted the trapping of H atomsin the hollow site in the center of a Pd3 triplet (as on theGaPd(111) PG surface37) significantly complicates catalyticproperties. A similar observation holds for the Pd3 tripletssurrounded by Ga atoms on the GaPd2 G0 surface. Because theadsorption of hydrogen atoms inside the hollows is muchhigher than their binding energies in the surrounding GPxbridge sites, the Pd3 triplets act as traps for hydrogen atoms.
4.3. Dissociation of Hydrogen. For a dissociativeadsorption of a hydrogen molecule it is necessary that thebinding of the dissociated atoms to the surface is strong enough(relative to the adsorption energy of the molecule) to providethe energy required to split the molecular bond. On the G0surface dissociation of a H2 molecule adsorbed in site PT1 canlead to H atoms bound in PH1 and GP1 sites. The activationenergy for this process is Ea = 34 kJ/mol, the adsorption energyof the coadsorbed atoms is −6 kJ/mol, the difference withrespect to the sum of the individual adsorption energies of 17kJ/mol arises from the repulsion between H atoms coadsorbedat close distance. On the G1 surface H2 adsorbed in a PT1 sitecan dissociate into atoms bound at the PP1 and GP1 sites, withan activation energy of Ea = 52 kJ/mol and a coadsorptionenergy of +12 kJ/mol of the atoms, increased by 24 kJ/molrelative to the individual adsorption energies. On the G2surface for the process PT1 → GP2 + GP4 the activationenergy is Ea = 46 kJ/mol and the final coadsorption energy is+13 kJ/mol, even reduced by 4 kJ/mol relative to the sum ofthe individual adsorption energies. On all three surfacesdissociation is slightly endothermic with a heat of reactionvarying between 16 kJ/mol (G0) and 10 kJ/mol (G2). For arather high coverage of 2/3 on Pd(111) the activation energyfor dissociation across an on-top site into atoms in the fcc andhcp hollows is 20 kJ/mol.56 At a coverage of 1/12 the activationenergy for H2 dissociation into atoms in two fcc hollows is 11kJ/mol. A typical activation energy for H2 dissociation on theGaPd(210) surface is 49 kJ/mol. The general conclusion is thatwith increasing Ga content, the activation energy for thedissociation of hydrogen increases.
Table 1. Adsorption Energies Eb of Molecular and Atomic Hydrogen (in kJ/mol, Relative to the Molecular Species in the GasPhase and the Clean Surface) in Different Sites on the (010) Surface of GaPd2
a
G0 surface G1 surface G2 surface
adsorbate site description Eb [kJ/mol] site description Eb [kJ/mol] site description Eb [kJ/mol]
H2 PT1 top −22 PT1,PT3 top −3, −15 PT1 top 3H2 PT2 top −16 PT2 top −5H GP1 bridge 17 GP1, GP5 bridge 15,16 GP1 bridge 10H PH1 hollow −40 GP2, GP6 bridge 23, 8 GP2 bridge 3H PH2 hollow 14 PP1, PP2 bridge −3, −21 GP3 bridge 19H GP4 bridge 14H PH1 hollow 34H GG1 bridge 17
aSee Figure 3 for the definition of the nomenclature and text.
The Journal of Physical Chemistry C Article
dx.doi.org/10.1021/jp5025075 | J. Phys. Chem. C 2014, 118, 12285−1230112291

4.4. Diffusion of Atomic Hydrogen. A sufficient mobilityof the hydrogen atoms on the surface of the catalyst, such thatthey can approach the coadsorbed reactant, is a necessarycondition for the catalytic activity of a surface. Activationenergies for typical elementary jumps between local metastableadsorption sites have been compiled in Table 2. On the G0
surface diffusion is highly anisotropic. The activation energy fordiffusion along the [100] direction, along a chain of GP1 andPH2 sites is only 17 kJ/mol. For diffusion in the [001]direction jumps from PH1 to PH2 sites require an activationenergy which is nearly four times higher. As already noted thePH1 sites in the center of the Pd3 triplets act as traps for Hatoms, similar as were observed also for some terminations ofthe GaPd(111) surface. On the G1 surface activation energiesfor elementary diffusion jumps range between 11 kJ/mol (GP1to GP2) and 33 kJ/mol (GP2 to GP5). The activation energiesfor diffusive jumps on the G2 surface are of the same order ofmagnitude.On the GaPd(210) surface the highest calculated activation
energy35 for a diffusive jump is 48 kJ/mol, on a Pd(111) surfacethe activation energy for jumps between fcc and hcp 3-foldhollows across a bridge site is about 15 kJ/mol. In the formercase the higher activation energy is caused by the strongercorrugation of the (210) surface of the compound, therelatively low value for diffusion on the pure Pd surface iscorrelated to the strong binding of a H atom in both the 3-foldhollows and in bridge sites.57
5. ADSORPTION OF ACETYLENE AND ETHYLENE
Figure 4 shows the locally (meta)stable adsorption sites ofacetylene and ethylene molecules on the stoichiometric G0 and
the Ga-enriched G1 and G2 GaPd2(010). The adsorptionenergies calculated for these sites are compiled in Table 3. Onthe G0 surface C2H2 preferably binds in a di-σ configuration toa Pd−Pd bridge position PP1 near the center of the Pd3 tripletwith an adsorption energy of Eb = −125 kJ/mol. Because of theapproximate 3-fold symmetry of the Pd3 triplet sites PP2 andPP3, rotated by about 60°, have similar adsorption energies of−127 and −114 kJ/mol, respectively. In Figure 4 the PP2 andPP3 sites are marked by ellipses below the PP1 site. Verysimilar binding configurations of acetylene in the Pd3 triplet hasbeen reported for the Pd(111) surface and confirmed byscanning tunneling microscopy.58 In this case 3-fold symmetryof the triplet is perfect, the three configurations are energeti-cally degenerate and separated only by a modest rotationalbarrier. Our calculations for a lower surface coverage equal tothat on the GaPd2 surface confirm the geometric features andyield an adsorption energy of Eb = −194 kJ/mol.On the Ga-enriched G1 surface the energetically preferable
adsorption sites for C2H2 are Ga−Ga bridge (GGx) and Ga−Pd (GPx) sites, while on G2 only Ga−Ga bridge forms stableadsorption complexes with acetylene. Typical di-σ bondedadsorption complexes on the three surfaces are shown in Figure5. Ga−Pd bridge configurations around the isolated Pd1 arepossible, but are found to be unstable against a transformationinto a Ga−Pd bridge configuration by a simple rotation [seeFigure 4c]. The strong binding of the acetylene molecule to Gaatoms is a remarkable and somewhat surprising observation.Generally it is assumed that on alloys of transition-metals withnoble or simple metals the transition-metal atoms provide thestrongest adsorption sites and are essential for any catalyticactivity. The function of the simple or noble metal atoms wouldbe merely to separate the active centers and to moderate theirchemical reactivity. Our findings demonstrate that simple metalatoms such as Ga not only modify the reactivity of the Pdatoms, but also participate in the catalytic reactions. Strong di-σbonding configurations of acetylene in Ga−Ga (or Al−Al)bridge sites have also been found on other catalytically activesurfaces of intermetallic compounds: on GaPd(210),35 on theisostructural AlPd(210) surface,33 but also on the Al13Co4(100)surface.31
It is interesting to analyze the variation of the adsorptionenergy of C2H2 on two selected sites, on-top of the Pd1 atom(site PT1) site and in a Ga−Ga bridge position (GG1 site),
Table 2. Typical Activation Energies for Diffusive Jumps onthe G0, G1, and G2 (010) Surfaces of GaPd2 (in kJ/mol)a
G0 surface G1 surface G2 surface
diff. jump Ea diff. jump Ea diff. jump Ea
GP1 → PH2 17 PP1 → PP2 28 GG1 → PH1 24PH1 → PH1 57 GP1 → GP2 11 GP3 → GG1 30PH1 → PH2 62 GP2 → GP5 33
aFor the nomenclature of the start- and end-points of diffusive jumps,see Figure 3.
Figure 4. Adsorption sites of C2H2 molecules (ellipses) on the GaPd2(010) surface, model G0 (a), model G1 (b), and model G2 (c). On the G0 andG2 surfaces the sites have 21 symmetry, and at G1 this symmetry is broken.
The Journal of Physical Chemistry C Article
dx.doi.org/10.1021/jp5025075 | J. Phys. Chem. C 2014, 118, 12285−1230112292

with the increasing number of Ga nearest neighbors on the G0,G1, and G2 surfaces. On the PT1 site the adsorption energy ofC2H2 is Eb = −48, ∼−37, and −22 kJ/mol on the G0, G1, andG2 surfaces, respectively. Due to the broken symmetry of theG1 surface, the Pd1 and Pd1′ surface atoms are not equivalentand we have taken the average binding energies in the PT1 andPT3 sites. With increasing number of Ga neighbors thestrength of the π-binding in Pd on-top sites decreases. On theother hand, the adsorption energy of acetylene in the GG1bridge sites increases: Eb = −84, ∼−115, and −132 kJ/mol onthe G0, G1, and G2 surfaces, reflecting a stronger di-σ bindingto the Ga atoms. Mixed Ga−Pd bridges form locally stableadsorption sites only on the G1 surface.Locally stable adsorption sites for ethylene exist only on-top
of a Pd atom (PTx sites, see Figure 4). The average adsorptionenergy decreases from Eb = −57 kJ/mol on the G0 to Eb = −29kJ/mol on the G2 surface (see Table 3) with an increasingnumber of Ga neighbors. As will be demonstrated below (seeSection 7) the weaker binding of ethylene on Ga-enrichedsurfaces is essential for achieving an improved selectivity.
6. SEMIHYDROGENATION OF ACETYLENE
6.1. Hydrogenation on the Stoichiometric G0 Surface.On the bulk-terminated GaPd2(010) G0 surface acetylenehydrogenation proceeds entirely within the Pd3 triplet, seeFigure 5a. In a possible starting configuration C2H2 is adsorbedin the PP2 site with an adsorption energy of Eb = −127 kJ/mol
[see Figure 4a] and a hydrogen atom is coadsorbed in one ofthe H-PH1 sites at a large distance of 5.6 Å from the acetylenemolecule [see Figure 3a]. The energy of this coadsorbedconfiguration of Eb = −168 kJ/mol is essentially equal to thesum of the individual adsorption energies, because thescreening of the H atom in the triplet hollow site H-PH1minimizes any repulsion between the coadsorbates. By diffusionjumps the H atom from the distant H-PH1 site can move toanother H-PH1 hollow close to the adsorbed C2H2, leading to aslight reduction of the adsorption strength, Eb = −162 kJ/mol.This configuration of C2H2 and coadsorbed H is shown inFigure 5a. Hydrogenation of C2H2 to C2H3 (vinyl) proceedsover the Pd2 atom and requires an activation energy of Ea = 65kJ/mol. The energy of the transition state with respect to theenergies of the reactants in the gas phase is ETS1 = −97 kJ/mol.The reaction is slightly exothermic, ΔEb = −11 kJ/mol. Theenergy profile of the reaction is presented in Figure 6a, thediffusion paths of the hydrogen atoms are indicated only by thedashed lines (for the activation energies of diffusive jumps, seeSection 4.4). The energy level marked by dashed linescorresponds to a configuration of the reactant with anadditional hydrogen atom coadsorbed in a distant site withthe lowest total energy.Further hydrogenation of vinyl to ethylene can proceed by
attacking C2H3 by another H atom over the Pd1 atom or thePd2 atom. The later reaction requires a lower activation energy.If the additional H atom is first coadsorbed in a distant H-PH1
Table 3. Adsorption Energies Eb (in kJ/mol) of Acetylene and Ethylene on the Stoichiometric G0 and the Ga-Enriched G1 andG2 (010) Surfaces of GaPd2, Relative to the Molecular Species in the Gas Phase and the Clean Surfacea
G0 surface G1 surface G2 surface
adsorbate site description Eb site description Eb site description Eb
C2H2 PT1 top −48 PT1,PT3 top −26, −47 PT1 top −22C2H2 PT2 top −52 PT2 top −26C2H2 GG1 bridge −87 GG1,GG2 bridge −117, −114 GG1 bridge −132C2H2 PP1 bridge −125 GP1 bridge −103 GG2 bridge −143C2H2 PP2 bridge −127 GP2 bridge −102 GG3 bridge −120C2H2 PP3 bridge −114 GP3 bridge −123 GG4 bridge −135C2H2 GP4 bridge −151C2H4 PT1 top −57 PT1,PT3 top −36, −55 PT1 top −29C2H4 PT2 top −54 PT2 top −35
aFor the nomenclature of the adsorption sites see Figure 4. On the G0 surface [Figure 4a] the PP2 and PP3 sites are located below PP1, rotated byabout 60°. On the G1 surface [Figure 4b] the GP4 sites is drawn below GP3, again rotated by about 60°.
Figure 5. (a−c) Di-σ bonded adsorption complexes of C2H2 molecules in the PP2, GG1, and GG4 bridge sites of the G0, G1, and G2 surfaces,respectively. The coadsorbed H atoms initiate the hydrogenation reactions. The red triangles mark the atoms forming the reaction centers.
The Journal of Physical Chemistry C Article
dx.doi.org/10.1021/jp5025075 | J. Phys. Chem. C 2014, 118, 12285−1230112293
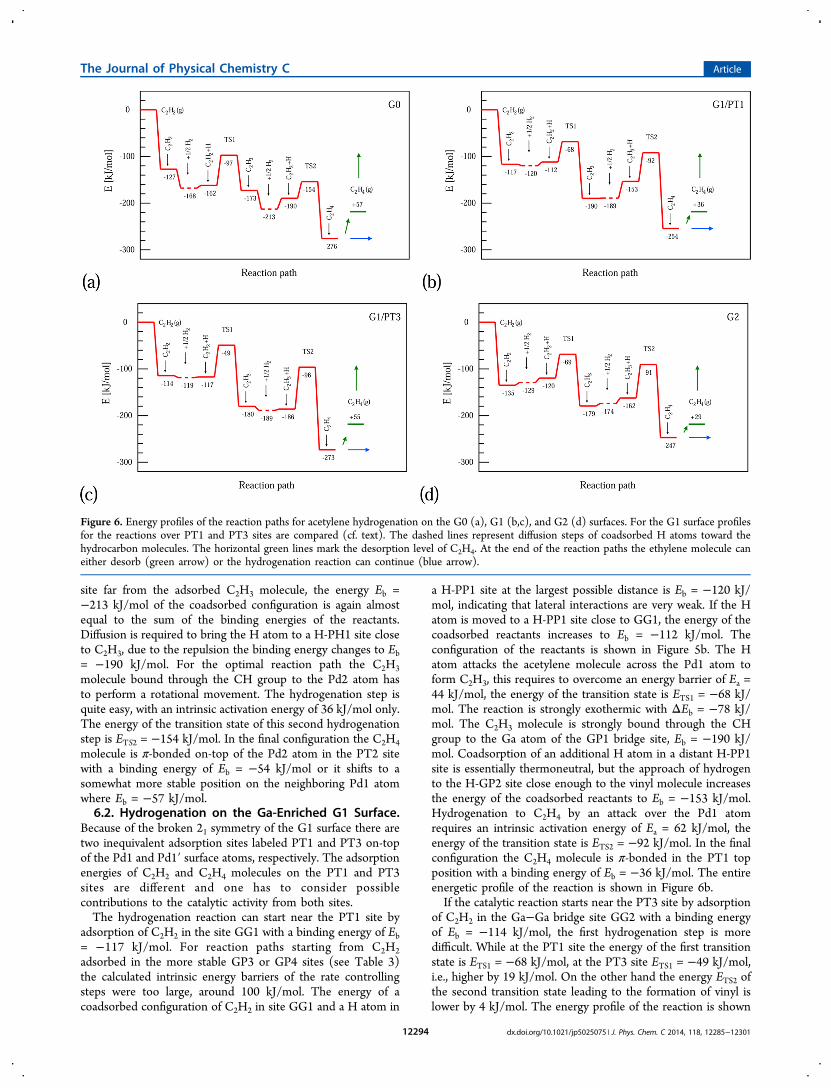
site far from the adsorbed C2H3 molecule, the energy Eb =−213 kJ/mol of the coadsorbed configuration is again almostequal to the sum of the binding energies of the reactants.Diffusion is required to bring the H atom to a H-PH1 site closeto C2H3, due to the repulsion the binding energy changes to Eb= −190 kJ/mol. For the optimal reaction path the C2H3molecule bound through the CH group to the Pd2 atom hasto perform a rotational movement. The hydrogenation step isquite easy, with an intrinsic activation energy of 36 kJ/mol only.The energy of the transition state of this second hydrogenationstep is ETS2 = −154 kJ/mol. In the final configuration the C2H4molecule is π-bonded on-top of the Pd2 atom in the PT2 sitewith a binding energy of Eb = −54 kJ/mol or it shifts to asomewhat more stable position on the neighboring Pd1 atomwhere Eb = −57 kJ/mol.6.2. Hydrogenation on the Ga-Enriched G1 Surface.
Because of the broken 21 symmetry of the G1 surface there aretwo inequivalent adsorption sites labeled PT1 and PT3 on-topof the Pd1 and Pd1′ surface atoms, respectively. The adsorptionenergies of C2H2 and C2H4 molecules on the PT1 and PT3sites are different and one has to consider possiblecontributions to the catalytic activity from both sites.The hydrogenation reaction can start near the PT1 site by
adsorption of C2H2 in the site GG1 with a binding energy of Eb= −117 kJ/mol. For reaction paths starting from C2H2adsorbed in the more stable GP3 or GP4 sites (see Table 3)the calculated intrinsic energy barriers of the rate controllingsteps were too large, around 100 kJ/mol. The energy of acoadsorbed configuration of C2H2 in site GG1 and a H atom in
a H-PP1 site at the largest possible distance is Eb = −120 kJ/mol, indicating that lateral interactions are very weak. If the Hatom is moved to a H-PP1 site close to GG1, the energy of thecoadsorbed reactants increases to Eb = −112 kJ/mol. Theconfiguration of the reactants is shown in Figure 5b. The Hatom attacks the acetylene molecule across the Pd1 atom toform C2H3, this requires to overcome an energy barrier of Ea =44 kJ/mol, the energy of the transition state is ETS1 = −68 kJ/mol. The reaction is strongly exothermic with ΔEb = −78 kJ/mol. The C2H3 molecule is strongly bound through the CHgroup to the Ga atom of the GP1 bridge site, Eb = −190 kJ/mol. Coadsorption of an additional H atom in a distant H-PP1site is essentially thermoneutral, but the approach of hydrogento the H-GP2 site close enough to the vinyl molecule increasesthe energy of the coadsorbed reactants to Eb = −153 kJ/mol.Hydrogenation to C2H4 by an attack over the Pd1 atomrequires an intrinsic activation energy of Ea = 62 kJ/mol, theenergy of the transition state is ETS2 = −92 kJ/mol. In the finalconfiguration the C2H4 molecule is π-bonded in the PT1 topposition with a binding energy of Eb = −36 kJ/mol. The entireenergetic profile of the reaction is shown in Figure 6b.If the catalytic reaction starts near the PT3 site by adsorption
of C2H2 in the Ga−Ga bridge site GG2 with a binding energyof Eb = −114 kJ/mol, the first hydrogenation step is moredifficult. While at the PT1 site the energy of the first transitionstate is ETS1 = −68 kJ/mol, at the PT3 site ETS1 = −49 kJ/mol,i.e., higher by 19 kJ/mol. On the other hand the energy ETS2 ofthe second transition state leading to the formation of vinyl islower by 4 kJ/mol. The energy profile of the reaction is shown
Figure 6. Energy profiles of the reaction paths for acetylene hydrogenation on the G0 (a), G1 (b,c), and G2 (d) surfaces. For the G1 surface profilesfor the reactions over PT1 and PT3 sites are compared (cf. text). The dashed lines represent diffusion steps of coadsorbed H atoms toward thehydrocarbon molecules. The horizontal green lines mark the desorption level of C2H4. At the end of the reaction paths the ethylene molecule caneither desorb (green arrow) or the hydrogenation reaction can continue (blue arrow).
The Journal of Physical Chemistry C Article
dx.doi.org/10.1021/jp5025075 | J. Phys. Chem. C 2014, 118, 12285−1230112294

in Figure 6c. As ethylene formation is the rate-controlling step,a reaction over the PT3 site can contribute to the catalytic
activity of the G1 surface as well as that over the PT1 site. Theselectivities of both reaction paths will be discussed in section 8.
Figure 7. Reaction paths for the hydrogenation of adsorbed ethylene to ethyl on the Pd(111) surface (a), the stoichiometric GaPd2(010) surface(model G0) (b), and the Ga-enriched GaPd2(010) surfaces with one Ga atom per surface cell replacing a Pd2 atom (model G1) (c, d), and with twoGa atoms per surface cell replacing Pd2 atoms (model G2) (e). Green arrows indicate the desorption energy of a single C2H4 molecule, red arrowsindicate the energy difference ΔEselect between the energy of the transition state ETS for the formation of ethyl and the energy level ED for ethylenedesorption. A positive value indicates selectivity for semihydrogenation.
The Journal of Physical Chemistry C Article
dx.doi.org/10.1021/jp5025075 | J. Phys. Chem. C 2014, 118, 12285−1230112295

6.3. Hydrogenation on the Ga-Enriched G2 Surface.On the G2 surface we could not find reaction paths startingfrom C2H2 adsorbed in the most stable GG1 or GG2 sites withan intrinsic energy barrier for the rate controlling step lowerthan 115 kJ/mol. In the initial configuration of the optimalreaction path C2H2 is adsorbed in the site GG4, it is attacked bya H atom coadsorbed in the H-GP4 site [see Figures 3c and4c]. The binding energy of C2H2 in the GG4 site is Eb = −135kJ/mol. Co-adsorption of a H atom in a distant H-GP2 siteleads to a coadsorption energy of Eb = −129 kJ/mol, therepulsive interaction changes the binding energy by only 3 kJ/mol. Approach of the H atom to the site H-GP4 close to theadsorbed C2H2 increases the repulsive interaction only by amodest 9 kJ/mol. The configuration of C2H2 and coadsorbed His shown in Figure 5c. Hydrogenation to C2H3 requires anactivation energy of Ea = 51 kJ/mol, the reaction is exothermicwith ΔEb = −59 kJ/mol. The C2H3 molecule is bound throughthe CH group bound to the Ga atom next to the H-GP2 sitewith a binding energy of Eb = −179 kJ/mol. The nexthydrogenation step proceeds by attacking the C2H3 moleculeby an additional H atom coadsorbed in the H-GP4 site. In thefinal state the C2H4 molecule is in an top position (the PT1site), desorption requires an energy of Ed = 29 kJ/mol. Thecomplete energetic profile of the reaction is shown in Figure 6d.Its is remarkable that on the G1 and G2 surfaces the energies
of the transition states TS2 for the formation of ethylene arealmost the same: ETS2(G1) ≈ ETS2(G2). The reason is that onboth surfaces the attack of the H atom on the coadsorbedhydrocarbon molecule takes place via a Pd1 atom and thecatalytic reaction is mediated by nonbonding Pd d-states. Thismeans that the energy of the transition state of hydrogenationsteps where strong C−H bonds are formed is quiteindependent of the environment of the Pd atom. The activationenergies, however, are determined by the difference betweenthe transition-state and the coadsorption energies of thereactants and depend on the configuration of the entire activesite. On the G0 surface the reaction proceeds over the Pd2atom. However, we have found also an alternative reaction pathwhere the second hydrogenation step proceeds over the Pd1atom and the transition-state energy is also ETS2(G0) ≈ETS2(G1).The reaction mechanism we have identified for the Ga-
enriched (010) surfaces of GaPd2 is very similar to that on theGaPd(210), AlPd(210), and Al13Co4(100) surfaces.31,33,35
Hydrogenation starts from C2H2 adsorbed in a bridge positionbetween two Ga or Al atoms, which is attacked by a H atomacross a neighboring transition-metal (Pd or Co). In most casesC2H3 remains bridge-bonded to the simple-metal atoms, but ina slightly asymmetric configuration where the CH2 group is abit closer to the transition-metal (see Figure 7 of our previouswork35 and Figure 5 ref 31). The transition state for the secondhydrogenation step is shifted toward the Pd/Co atom and atthe end of the semihydrogenation process C2H4 is bonded on-top of the transition-metal atom. The active center is always atriangular configuration of two simple-metal and one transition-metal atoms. In contrast on the G0 surface the reaction occurswithin the same Pd3 triplet, as on the Pd(111) surface, seeFigure 5a.
7. ETHYLENE HYDROGENATION AND SELECTIVITYThe selectivity of the catalyst for the semihydrogenation ofacetylene to ethylene production is determined by thedifference between the activation energy for the hydrogenation
of ethylene to ethyl and the desorption energy. The positivedesorption energy is Ed = −Eb where the adsorption energy Ebof ethylene is calculated relative to C2H4(g) in the gas-phase. InFigures 6 and 7 the adsorption energy of ethylene Eb isexpressed relative to the energy of the reactants C2H2(g) andH2(g) in the gas phase. With this reference the energy ED ofethylene in the gas phase is equal to −219 kJ/mol, independentof the chosen surface; see Figure 7 where ED is marked by thegreen line.The desorption energy depends on the configuration of the
adsorbed molecule on the surface and on the coverage. At acoverage of 1/12 ethylene can be adsorbed on Pd(111) eitherin a Pd−Pd bridge position (Eb = −88 kJ/mol) or on top of aPd atom (Eb = −75 kJ/mol). On the G0 surface of GaPd2 thebinding energy of C2H4 on top of the Pd1 atom (site PT1) is Eb= −53 kJ/mol, on the PT2 site adsorption is slightly strongerby 2 kJ/mol, cf. Table 1. A Pd2−Pd2 bridge position with anadsorption energy of Eb = −43 kJ/mol is less favorable. On theGa-enriched surfaces adsorption of ethylene is stable only inPd-top sites. The PT1 states are the adsorption sites of ethyleneproduced by the semihydrogenation of acetylene, as describedin the foregoing Section. With an increasing number of Gaatoms the adsorption energy gradually decreases, Eb = −57,−36 (−55), and −29 kJ/mol on the G0, G1 (PT1 and PT3sites), and G2 surfaces, respectively. Adsorption of ethylene inon-top sites is possible in different orientations of the molecularaxis, but the energy for a rotational reorientation varies onlybetween 1 to 4 kJ/mol. Parallel to the decreasing adsorptionenergy, the height of the molecule above the Pd atom increasesfrom 1.92 Å on Pd(111) to 2.25 Å on the G2 surface.Desorption of ethylene competes with a reaction C2H4 + H
→ C2H5, further hydrogenation to ethyl. The process startswith the coadsorption of an additional H atom, first at a largedistance from the adsorbed molecule in the site with the lowesttotal energy. In this configuration the interaction between thecoadsorbates is very weak. The diffusive jumps required tobring the H atom close enough to the ethylene molecule toinitiate the hydrogenation reaction are summarized by dashedlines in Figure 7. We note that a repulsive interaction betweenthe approaching H atom and the hydrocarbon moleculeincreases the activation energies of diffusion jumps.A direct side attack of a H atom coadsorbed in a nearby site
on the C2H4 molecule is difficult because of the differentheights of the coadsorbed species. The H atom is bound tobridge or hollow sites, only a few tenths of Å above the surfaceplane, while the molecule is situated between 1.8 and 2.2 Åabove the supporting Pd atom. Hence the H atom has to breakthe bond to the surface to approach the ethylene moleculeadsorbed high above the surface. This is possible only if H isadsorbed very close, almost below the C2H4 molecule. Thehydrogenation of ethylene is essentially a two-step process. Inthe first step the H atom is inserted below the ethylenemolecule, in the second step the atom can attacks the ethylenemolecule from the bottom. The activation energy required forthe first step can be much higher than for diffusion on the cleansurface (see Section 4.4) by 7 kJ/mol [on Pd(111)] to 23 kJ/mol (on the G1 surface) more than a typical diffusion step. Theactivation energy for the following step, C−H bond formation,is much lower. The intrinsic values for the activation energiesfor the formation of ethyl are 54, 54, 47(38), and 42 kJ/mol onthe Pd, G0, G1/PT1 (G1/PT3 sites), and G2 surfaces, leadingto transition-state energies ETS of −273, −232, −201 (−220),and −170 kJ/mol, respectively. With an increasing number of
The Journal of Physical Chemistry C Article
dx.doi.org/10.1021/jp5025075 | J. Phys. Chem. C 2014, 118, 12285−1230112296

Ga atoms around the Pd atom to which ethylene is bound theenergy of the transition state gradually increases. This is incontrast to the fact, noted above, that the transition-stateenergies for the formation of ethylene are almost independentof the local environment. The reason is in the different bondingcharacter. In the previous hydrogenation steps the nonbondingPd states mediated the catalytic reactions, while here the bondbetween Pd and the C2H4 molecule has to be broken. Figure8a−d shows the atomic configurations of the correspondingtransition states.The conditions for ethylene hydrogenation found here are
very similar to those on the Al13Co4(100) surface.31 Here theactivation energy for inserting the H atom below thecoadsorbed C2H4 molecule was also found to be much higherthan that for the following hydrogenation step. In contrast, onthe {210} surfaces of GaPd the reaction is also a two-stepprocess, but the C−H bond formation is the rate-controllingstep.33,35
The selectivity of the catalyst is determined by the energydifference ΔEselect = ETS − ED between the transition-stateenergy for ethylene formation and the desorption energy levelof ethylene ED (which in our reference is −219 kJ/mol) definedabove. The catalyst is selective for the formation of ethylene ifΔEselect > 0. A similar criterion for the selective semi-hydrogenation of acetylene over NiZn has recently been usedby Studt et al.17,18
In Figure 7 ΔEselect is represented by red arrows. On Pd(111)the transition state for the hydrogenation of ethylene to ethyl islower by −55 kJ/mol lower than the desorption energy levelED, see Figure 7a. The value of ΔEselect is less negative for thestoichiometric GaPd2(010) G0 surface, ΔEselect = −13 kJ/mol,but the continued hydrogenation to ethyl remains energeticallyfavored. The pristine GaPd2(010) surface (Figure 7b) thus doesnot exhibit the sufficient selectivity. The desired selectivesemihydrogenation of acetylene to ethylene is achieved on theGa enriched G1 and G2 surfaces only (Figure 7c−e). The realGaPd2(010) surface prepared under Ga-rich conditions willcontain local atomic environments corresponding to all threesurface models G0, G1, and G2. The effective selectivity of theGaPd2(010) surface will thus come from several contributions.On the G1 surface one has to consider contributions from twosites PT1 and PT3. The hydrogenation reaction over the PT1site is selective with ΔEselect = +17 kJ/mol. The rather strongadsorption of ethylene on the PT3 site results in a contributionwith a lower selectivity, ΔEselect = −1 kJ/mol. On the other sidethe local environments typical for the G2 surface will contributewith a very high selectivity of ΔEselect = +39 kJ/mol.
We observe a correlation between the selectivity and thenumber of Ga neighbors surrounding the catalytically active Pdatoms on which the ethylene molecule is adsorbed. On theGaPd2(010) G0 surface the PT1 site is surrounded by 3 Ganeighbors, on the G1 surface by 4 Ga neighbors. Around thePT3 site on the G1 surface there are also 4 Ga neighbors, butone of them is at a rather large nonbonding distance of 3.3 Å,see section 3. This could explain the lower selectivity of thePT3 site. On the G2 surface which provides the best selectivitythe PT1 site is surrounded by 5 Ga neighbors.
8. DISCUSSION AND CONCLUSIONS
We have used ab initio DFT calculations to investigate thestability, structure, and chemical reactivity and to constructatomistic scenarios for the hydrogenation of acetylene on thestoichiometric and Ga-enriched (010) surfaces of theintermetallic compound GaPd2. The calculations of the surfaceenergies as a function of the chemical potential in the reactiveatmosphere above the surface demonstrate that Ga-enrichedsurfaces are stabilized at least at sufficiently large values of thechemical potential but that a segregation of Ga to the surface isenergetically unfavorable because the formation of Pd/Gaantisite defects in the subsurface layers costs too much energy.The calculated activation energies for hydrogen dissociation,
the diffusion of atomic hydrogen, and the rate-controlling stepof the hydrogenation reactions, as well as the energy differenceΔEselect determining the selectivity for semihydrogenation toethylene are summarized in Table 4 and compared with thecorresponding quantities calculated for the {210} surfaces ofGaPd35 and AlPd33 and on the most reactive terminations ofthe chiral {111} surfaces of GaPd (the Pd-rich A(1 11 ) and themixed A(111) surface terminations).37
On the bulk-terminated, stoichiometric surface named G0,the dissociative adsorption of hydrogen is relatively easy, butdue to the strong binding of molecular hydrogen the activationenergy for diffusive movements of a H atom can be as large asfor the rate-controlling step of the hydrogenation reactions (theformation of vinyl). The continued hydrogenation of ethyleneto ethyl is energetically preferable to the desorption of ethylene,so that the GaPd2(010) G0 surface does not show the desiredselectivity for semihydrogenation to ethylene. The G0 surface ischaracterized by the existence of very reactive Pd3 triplets whichdissociate molecular hydrogen very easily, but bind atomichydrogen and hydrocarbon molecules too strongly. Migrationof H atoms between different Pd3 triplets is hindered by a highactivation energy and the strong adsorption of ethylene enablesa further hydrogenation to ethyl.
Figure 8. Transition states for the hydrogenation of ethylene to ethyl on different surfaces: (a) Pd(111), (b) GaPd2(010), model G0, (c) model G1,and (d) model G2. The ethylene molecule adsorbed on top of a Pd atom is attacked by atomic hydrogen from the bottom.
The Journal of Physical Chemistry C Article
dx.doi.org/10.1021/jp5025075 | J. Phys. Chem. C 2014, 118, 12285−1230112297

On the Ga-enriched GaPd2(010) G1 and G2 surfaces thedissociation of hydrogen requires a higher activation energy,but the H atoms are more mobile and the activation energy forthe rate-controlling step of the hydrogenation reactions is of thesame order of magnitude. The decisive point, however, is thatthe desorption energy of ethylene is now lower than theactivation energy for further hydrogenation to ethyl. Thereason for the different catalytic behavior of the stoichiometricG0 and the Ga-enriched G1 and G2 surfaces is thefundamentally different character of the catalytically activecenter: A Pd3 triplet on the G0, and a triangular arrangement ofone Pd and two Ga atoms on the G1 and G2 surfaces, seeFigure 5. The chemical reactivity of the Pd3 triplet leads notonly to a relatively strong binding of the ethylene molecule, italso facilitates the adsorption of a H atom at a sufficiently closedistance to enable further hydrogenation. In contrast, on the
G1 and G2 surfaces acetylene is strongly di-σ bonded to thetwo Ga atoms of the reactive center, and the adsorptionconfiguration remains essentially the same for the vinylmolecule. Ethylene in contrast is rather weakly π-bonded on-top of the Pd atom and the isolation of the Pd atom makes anattack by atomic hydrogen more difficult. On the Ga-enrichedsurfaces the distance of 5.5 Å between two active Pd1 atoms isalso large enough to avoid oligomerization by formation C−Cbonds between hydrocarbon molecules adsorbed on neighbor-ing Pd1 sites.The foregoing analysis also fully agrees with the results of our
investigations of other surfaces of catalytically active inter-metallics, see Table 4. On the {210} surfaces of GaPd35 andAlPd33 and on the (100) surface of Al13Co4 (which are selectivecatalysts for the semihydrogenation of acetylene to ethylene),the active centers are also triangular arrangements of onetransition-metal (Pd, Co) and two simple-metal (Ga, Al)atoms, and the decisive point is again the change from di-σbinding to the simple-metal atoms for acetylene, to π-bindingto the isolated transition-metal for ethylene. The comparisonwith the polar 3-fold surfaces of GaPd is also instructive.Among the possible terminations of the A(111) surfaces,36 onlythe “pG” termination with isolated Pd atoms at a height ofabout 0.6 Å above a layer composed of Ga3 triplets. On theprotruding Pd atom the binding of ethylene is strong, also asstrong as on a Pd(111) surface37 and as a consequence furtherhydrogenation is possible, the surface is not selective. On theopposite A(1 1 1) surface a ”PG” termination terminated by anouter layer of Pd3 triplets 0.5 Å above a layer of Ga3 triplet. Thissurface is catalytically active only if molecular hydrogen andacetylene are sequentially coadsorbed on the same Pd3 triplet,the coadsorption representing also the rate-limiting step. If thisis not possible, the catalytic activity is suppressed by the strongbinding of atomic hydrogen in the hollow centering the Pd3triplet.37 Both the catalytic activity and the selectivity of thissurface are improved is additional Ga atoms are deposited onthe hollows between the Pd3 (forming the “gP” termination).The screening of the Pd3 triplet by the separating Ga atomsmakes them less reactive, facilitates the mobility of H atomsand improves both activity and selectivity. However, it isnecessary to emphasize that these terminations are either stableonly under Pd-rich conditions (PG, pG) or marginally stable(gP) only in a very narrow interval of the chemicalpotential.36,37 The results compiled in Table 4 also show thatthe selectivity ΔEselect is comparable for the {210} surfaces ofGaPd and on the Ga-enriched (010) surfaces of GaPd2. Hencethe Ga-enrichment of the GaPd2(010) surface due to thepreparation conditions is essential to understand the similarcatalytic performance of both compounds observed in theexperiments.21,23,24
Figure 9 demonstrates that selectivity increases with anincreasing number of Ga neighbors around the Pd atom onwhich ethylene is adsorbed. We present here also a preliminaryresult for the selectivity of a reaction on the Ga3Pd5(001) E0surface. It is interesting that for this surface the energydifference ΔEselect = 9 kJ/mol determining selectivity is almostthe same as the average for the PT1 and PT3 sites on the G1surface. The atomic structures of GaPd2(010) G1, Ga3Pd5(001)E0, and GaPd(210) surfaces are very similar each other andthey exhibit also almost the same selectivities. The highestselectivity was found for the G2 surface, however, such surfacebecomes stable only at very high values of the chemicalpotential ΔμGa, see Figure 2a.
Table 4. Activation Energies Ea (in kJ/mol) for theDissociative Adsorption of Hydrogen, the Diffusion ofAtomic Hydrogen, the Rate-Controlling Step for theHydrogenation of Acetylene to Ethylene, and the EnergyDifference ΔEselect (also in kJ/mol) Determining theSelectivity for Semi-Hydrogenation to Ethylene for theStoichiometric (G0) and Ga-Enriched (G1, G2) (010)Surfaces of GaPd2, Compared with the CorrespondingQuantities for Catalytic Reactions on the (210) Surfaces ofGaPd35 and AlPd,33 on Selected {111} Surfaces of GaPd,37
and on the (100) Surface of Al13Co431
H2dissoc.
Hdiffusion selectivity
surface Ea Ea
rate-controlling
step coverage ΔEselect
GaPd2G0 34 <65 C2H3formation
1/12 −13
GaPd2G1/PT1 52 <35 C2H4formation
1/12 +17
GaPd2G1/PT3 42 <35 C2H4formation
1/12 −1
GaPd2G2 46 <30 C2H4formation
1/12 +39
GaPd(210) 49 <50 C2H4formation
1/16 +14
AlPd(210) 54 <45 C2H4formation
1/16 +11
GaPd(111)pGa
74 <45 H2 dissociation 1/12 −4
GaPd(1 11)PGb
59 <80 H diffusion 1/12 +6
GaPd(1 11)gPc 37 <60 H diffusion 1/12 +17Pd(111) 11 <20 C2H4
formation1/12 −54
Al13Co4(100) 17 <60 C2H3formation
1/16 +10
aThe A(111) gP surface of GaPd is terminated is terminated by adouble-layer with a single Pd atom in the top and Ga3 triplets in thesecond layer. It is stable only under Pd-rich conditions. bThe A(1 1 1)PG surface of GaPd is terminated by a double layer with Pd3 triplets inthe outer and Ga3 triplets in the lower layer. It is stable only under Pd-rich conditions. A favorable catalytic activity of the PG surface isachieved only if hydrogen and acetylene are sequentially coadsorbedon the same Pd3 triplet. No H-diffusion between the Pd3 triplets isnecessary.37 cThe A(1 1 1) gP surface of GaPd is terminated by adouble-layer with a single Ga atom in the top and Pd3 triplets in thesecond layer. It is stable only in a very narrow interval of the chemicalpotential.37
The Journal of Physical Chemistry C Article
dx.doi.org/10.1021/jp5025075 | J. Phys. Chem. C 2014, 118, 12285−1230112298

On all the surfaces of intermetallic compounds investigatedso far, the isolation of the active sites is essential for achieving ahigh selectivity for the semihydrogenation of acetylene. Whenthe concept of active site isolation was originally pro-posed7,8,20,23,40 it was assumed that only the transition-metalatoms provide the catalytic activity and that the reactants are π-bonded on-top of an isolated transition-metal atom. In ourwork we have demonstrated that the active sites are localarrangements of transition- and simple-metal atoms. Theelectronic and chemical properties of both species are stronglymodified by the covalent bonding between them, asdemonstrated by our first-principles calculations. Beforehydrogenation acetylene is adsorbed via a di-σ bond in abridging configuration between to simple-metal atoms, onlyafter hydrogenation to ethylene the adsorption configuration ischanged to a π-bond on-top of the transition-metals atom. Onthe (010) surface of GaPd2 Ga surface enrichment is necessaryto create such a configuration. The strong covalent bondingbetween the components also ensures the high stability of thecatalyst. Surface segregation, eventually under the influence ofthe adsorbed reactants, was found to be energeticallyunfavorable. The comparison of the catalytic properties ofdifferent surfaces of Ga−Pd compounds of varying composi-tions shows that to achieve a high selectivity, a Gaconcentration in the surface layer high enough to provide anefficient isolation of the Pd atoms is required; in the case ofGaPd2 surfaces this requires the Ga-enrichment favored by thepreparation process.21,23,24 The comparison of the atomisticscenarios for semihydrogenation reactions on the surfaces ofGa−Pd compounds with that for hydrogenation on theAl13Co4(100) surface suggests that these conclusions applyalso to the catalytic properties of other compound formed bytransition and simple metals.
■ AUTHOR INFORMATIONCorresponding Author*E-mail: [email protected].
NotesThe authors declare no competing financial interest.
■ ACKNOWLEDGMENTSThis work has been supported by the Austrian Ministry forScience through the Center for Computational MaterialsScience. M.K. is also thankful for support from the GrantAgency for Science of Slovakia (No. 2/0111/11), from CEXFUN-MAT, and from the Slovak Research and DevelopmentAgency (Grants No. APVV-0647-10, APVV-0076-11, APVV-0492-11). Part of the calculations were performed in theComputing Centre of the Slovak Academy of Sciences usingthe supercomputing infrastructure acquired in project ITMS26230120002 and 26210120002 (Slovak infrastructure forhigh-performance computing) supported by the Research andDevelopment Operational Programme funded by the ERDF.
■ REFERENCES(1) Somorjai, G. A.; Park, J. Y. Molecular Factors of CatalyticSelectivity. Angew. Chem., Int. Ed. 2008, 47, 9212−9228.(2) Freund, H. J. Model Systems in Heterogeneous Catalysis:Selectivity Studies at The Atomic Level. Topics Catal. 2008, 48, 137−144.(3) Borodzinski, A.; Bond, G. C. Selective Hydrogenation of Ethynein Ethene-Rich Streams on Palladium Catalysts. Part 1: Effect ofChanges to the Catalyst During Reaction. Catal. Rev. 2008, 48, 91−144.(4) Borodzinski, A.; Bond, G. C. Selective Hydrogenation of Ethynein Ethene-Rich Streams on Palladium Catalysts. Part 2: Steady StateKinetics and Effects of Palladium Particle Size, Carbon Monoxide andPromoters. Catal. Rev. 2008, 50, 379−469.(5) Lopez, N.; Vargas-Fuentes, C. Promoters in the Hydrogenationof Alkynes in Mixtures: Insights from DFT. Chem. Commun. 2012, 48,1379−1391.(6) Armbruster, M.; Behrens, M.; Cinquini, F.; Fottinger, K.; Grin,Yu.; Haghofer, A.; Klotzer, B.; Knop-Gericke, A.; Lorenz, H.; Ota, A.;et al. How to Control the Selectivity of Palladium-Based Catalysts inHydrogenation Reactions. Chem. Catal. Chem. 2012, 4, 1048−1063.(7) Shaikhutdinov, S. K.; Heemeierk, M.; Baumer, M.; Lear, T.;Lennon, D.; Oldman, R. J.; Jackson, S. D.; Freund, H.-J. Structure-Reactivity Relationships on Supported Metal Model Catalysts:Adsorption and Reaction of Ethene and Hydrogen on Pd/Al2O3/NiAl. J. Catal. 2001, 200, 330−339.(8) Shaikhutdinov, S. K.; Frank, M.; Baumer, M.; Jackson, S. D.;Oldman, R. J.; Hemminger, J. C.; Freund, H.-J. Effect of CarbonDeposits on Reactivity of Supported Pd Model Catalysts. Catal. Lett.2002, 80, 115−122.(9) Ledentu, V.; Dong, W.; Sautet, Ph. Heterogeneous Catalysisthrough Subsurface Sites. J. Am. Chem. Soc. 2000, 122, 1796−1801.(10) Rose, M. K.; Borg, A.; Mitsui, T.; Ogletree, D. F.; Salmeron, M.Subsurface Impurities in Pd(111) Studied by Scanning TunnelingMicroscopy. J. Chem. Phys. 2001, 115, 10927−10934.(11) Gracia, L. ; Catalayud, M. ; Andres, J. ; Minot, C. ; Salmeron, M.Migration of the Subsurface C Impurity in Pd(111). Phys. Rev. 2005,71, No. 033407.(12) Teschner, D.; Revay, Z.; Borsodi, J.; Havecker, M.; Knop-Gericke, A.; Schlogl, R.; Milroy, D.; Jackson, S. D.; Torres, D.; Sautet,Ph. Understanding Palladium Hydrogenation Catalysts: When theNature of the Reactive Molecule Controls the Nature of the CatalystActive Phase. Angew. Chem., Int. Ed. 2008, 47, 9274−9278.(13) García-Mota, M.; Bridier, B.; Perez-Ramírez, J.; Lopez, N.Interplay between Carbon Monoxide, Hydrides, and Carbides inSelective Alkyne Hydrogenation on Palladium. J. Catal. 2010, 273,92−102.(14) Sheth, P. A.; Neurock, M.; Smith, C. M. A First-PrinciplesAnalysis of Acetylene Hydrogenation Over Pd(111). J. Phys. Chem. B2003, 107, 2009−2017.
Figure 9. Selectivity to ethylene formation. If ΔEselect < 0 furtherhydrogenation of ethylene is preferred. The desired selectivity foracetylene semihydrogenation is obtained only for surfaces whereΔEselect > 0. The selectivity increases with the increasing number ofsurface Ga neighbors of the active Pd atoms. The selectivity of the Ga-enriched GaPd2(010) surface comes from the G0, G1, and G2contributions.
The Journal of Physical Chemistry C Article
dx.doi.org/10.1021/jp5025075 | J. Phys. Chem. C 2014, 118, 12285−1230112299

(15) Sheth, P. A.; Neurock, M.; Smith, C. M. First-Principles Analysisof the Effects of Alloying Pd with Ag for the Catalytic Hydrogenationof Acetylene-Ethylene Mixtures. J. Phys. Chem. B 2005, 109, 12449−12466.(16) Mei, D.; Sheth, P. A.; Neurock, M.; Smith, C. M. First-PrinciplesBased Kinetic Monte Carlo Simulation of the Selective Hydrogenationof Acetylene over Pd(111). J. Catal. 2006, 242, 1−15.(17) Studt, F.; Abild-Petersen, F.; Bligaard, T.; Sørensen, R. Z.;Christensen, C. H.; Nø rskov, J. K. On the Role of SurfaceModifications of Palladium Catalysts in the Selective Hydrogenationof Acetylene. Angew. Chem., Int. Ed. 2008, 47, 9299−9302.(18) Studt, F.; Abild-Petersen, F.; Bligaard, T.; Sørensen, R. Z.;Christensen, C. H.; Nørskov, J. K. Identification of Non-PreciousMetal Alloy Catalysts for Selective Hydrogenation of Acetylene. Science2008, 320, 1320−1322.(19) Gonzalez, S.; Nyeman, K. M.; Shaikhutdinov, S.; Freund, H. J.;Illas, F. On the Promoting Role of Ag in Selective HydrogenationReactions over Pd-Ag Bimetallic Catalysts: A Theoretical Study. J.Phys. Chem. C 2007, 111, 6852−6856.(20) Kovnir, K.; Armbruster, M.; Teschner, D.; Venkov, T. V.;Jentoft, F. C.; Knop-Gericke, A.; Grin, Yu.; Schlogl, R. A NewApproach to Well-Defined, Stable and Site-Isolated Catalysts. Sci.Technol. Adv. Mater. 2007, 8, 420−427.(21) Osswald, J.; Giedigkeit, R.; Jentoft, R. E.; Armbruster, M.;Girgsdies, F.; Kovnir, K.; Ressler, T.; Grin, Yu.; Schlogl, R. Palladium-Gallium Intermetallic Compounds for the Selective Hydrogenation ofAcetylene. Part I: Preparation and Structural Investigation underReaction Conditions. J. Catal. 2008, 258, 210−218. Osswald, J.;Kovnir, K.; Armbruster, M.; Giedigkeit, R.; Jentoft, R. E.; Wild, U.;Grin, Yu.; Schlogl, R. Part II: Surface Characterization and CatalyticPerformance. J. Catal. 2008, 258, 219−227.(22) Kovnir, K.; Osswald, J.; Armbruster, M.; Teschner, D.;Weinberg, G.; Wild, U.; Knop-Gericke, A.; Ressler, T.; Grin, Yu.;Schlogl, R. Etching of the Intermetallic Compounds PdGa andPd3Ga7: An Effective Way to Increase Catalytic Activity. J. Catal. 2009,264, 93−103.(23) Armbruster, M.; Wowsnick, G.; Friedrich, M.; Heggen, M.;Cardoso-Gil, R. Synthesis and Catalytic Properties of NanoparticulateGa-Pd Compound. J. Am. Chem. Soc. 2011, 133, 9112−9118.(24) Ota, A.; Armbruster, M.; Behrens, M.; Rosenthal, D.; Friedrich,M.; Kasatkin, I.; Girgsdies, F.; Zhang, W.; Wagner, R.; Schlogl, R.Intermetallic Compound Pd2Ga as a Selective Catalyst for the Semi-Hydrogenation of Acetylene. J. Phys. Chem. C 2011, 115, 1368−1374.(25) Ota, A.; Kunkes, E. L.; Kasatkon, I.; Groppo, E.; Ferri, D.;Poceiro, B.; Navaroo Yerga, R. M.; Behrens, M. Comparative Study ofHydrotalcite-Derived Supported Pd2Ga and PdZn IntermetallicNanoparticles as Methanol Synthesis and Methanol Steam ReformingCatalysts. J. Catal. 2012, 293, 27−38.(26) Haghofer, A.; Fottinger, K.; Girgsdies, F.; Teschner, D.; Knop-Gericke, A.; Schlogl, R.; Rupprechter, G. In Situ Study of theFormation and Stability of Supported Pd2Ga MSR Catalyst. J. Catal.2012, 286, 13−21.(27) Haghofer, A.; Ferri, D.; Fottinger, K.; Rupprechtrer, G. Who isDoing the Job? Unravelling the Role of Ga2O3 in Methanol SteamReforming on Pd2Ga/Ga2O3. ACS Catal. 2012, 2, 2305−2315.(28) Armbruster, M.; Kovnir, K.; Grin, Yu.; Schlogl, R.; Gille, P.;Heggen, M.; Feuerbacher, M. Ordered Cobalt-Aluminum and Iron-aluminum Intermetallic Compounds as Hydrogenation Catalysts.European Patent Application EP09157875.7, 2009.(29) Armbruster, M.; Kovnir, K.; Friedrich, M.; Teschner, D.;Wowsnick, G.; Hahne, M.; Gille, P.; Szentmiklosi, L.; Feuerbacher, M.;Heggen, M.; et al. Al13Fe4 as a Low-Cost Alternative for Palladium inHeterogeneous Hydrogenation. Nat. Mater. 2012, 11, 690−693.(30) Krajcí, M.; Hafner, J. Catalytic Properties of Al13Co4 Studied byAb-Initio Methods. Philos. Mag. 2011, 91, 2904−2912.(31) Krajcí, M.; Hafner, J. Complex Intermetallic Compounds asSelective Hydrogenation Catalysts - A Case Study for the (100)Surface of Al13Co4. J. Catal. 2011, 278, 200−207.
(32) Krajc í, M.; Hafner, J. Surface Structures of ComplexIntermetallic Compounds: An Ab-Initio DFT Study for the (100)Surface of o-Al13Co4. Phys. Rev. B 2011, 84, No. 115410.(33) Krajcí, M.; Hafner, J. Intermetallic Compound AlPd as aSelective Hydrogenation Catalyst. J. Phys. Chem. C 2012, 116, 6307−6319.(34) Krajcí, M.; Hafner, J. Surfaces of Intermetallic Compounds: ADFT Study for B20-Type AlPd. Phys. Rev. B 2013, 84, No. 035436.(35) Krajcí, M.; Hafner, J. The (210) Surface of Intermetallic B20Compound GaPd as Selective Hydrogenation Catalyst. J. Catal. 2012,295, 70−80.(36) Krajcí, M.; Hafner, J. Structure and Chemical Reactivity of thePolar Three-Fold Surfaces of GaPd: A Density-Functional Study. J.Chem. Phys. 2013, 138, No. 124703.(37) Krajcí, M.; Hafner, J. Selective Semi-Hydrogenation ofAcetylene: Atomistic Scenario for Reactions on the Polar Three-Fold Surfaces of GaPd. J. Catal. 2014, 312, 232−248.(38) Diehl, R. D.; McGrath, R. Structural Studies of Alkali MetalAdsorption and Coadsorption on Metal Surfaces. Surf. Sci. Rep. 1996,23, 43−171.(39) Rosenthal, D.; Widmer, R.; Wagner, R.; Gille, P.; Armbruster,M.; Grin, Yu.; Schlogl, R.; Groning, O. Surface Investigation ofIntermetallic PdGa(−1−1-1). Langmuir 2012, 28, 6848−6856.(40) Prinz, J.; Gaspari, R.; Pignedoli, C. A.; Vogt, J.; Gille, P.;Armbruster, M.; Brune, H.; Groning, O.; Passerone, D.; Widmer, R.Isolated Pd Sites on Intermetallic PdGa(111) and PdGa(−1−1-1)Model Catalyst Surfaces. Angew. Chem. Int. Ed. 2012, 51, 9339−9343.(41) Armbruster, M.; Kovnir, K.; Behrens, M.; Teschner, D.; Grin,Yu.; Schlogl, R. Pd-Ga Intermetallic Compounds as Highly SelectiveSemihydrogenation Catalysts. J. Am. Chem. Soc. 2010, 132, 14745−14747.(42) Li, L.; Zhang, B.; Kunkes, E.; Fottinger, K.; Armbruster, M.; Su,D. S.; Wei, W.; Schlogl, R.; Behrens, M. Ga-Pd/Ga2O3 Catalysts: TheRole of Gallia Polymorhs, Intermetallic Compounds, and PretreatmentConditions of Selectivity and Stability in Different Reactions. Chem.Catal. Chem. 2012, 4, 1764−1775.(43) Kovnir, K.; Schmidt, M.; Waurisch, Ch.; Armbruster, M.; Prots,Yu.; Grin, Yu. Refinement of the Crystal Structure of DipalladiumGallium, Pd2Ga. Z. Kristallogr. NCS 2008, 223, 7−8.(44) Friedrich, M.; Teschner, D.; Knop-Gericke, A.; Armbruster, M.Infueance of Bulk Composition of the Intermetallic Compound ZnPdon Surface Composition. J. Catal. 2012, 285, 41−47.(45) Kresse, G.; Furthmuller, J. Efficient Iterative Schemes for Abinitio Total-Energy Calculations Using a Plane-Wave Basis Set. Phys.Rev. B 1996, 54, 11169−11186.(46) Kresse, G.; Joubert, D. From Ultrasoft Pseudopotentials to theProjector-Augumented Wave Method. Phys. Rev. B 1999, 59, 1758−1775.(47) Blochl, P. E. Projector Augmented-Wave Method. Phys. Rev. B1994, 50, 17953−17969.(48) Perdew, J. P.; Burke, K.; Ernzerhof, M. Generalized GradientApproximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865−3869.(49) Perdew, J. P.; Burke, K.; Ernzerhof, M. Emission in SymmetricHeavy Ion Reactions at Subthreshold Energies. Phys. Rev. Lett. 1997,78, 1396−1399.(50) Sheppard, D.; Terrel, R.; Henkelman, G. Optimization Methodsfor Finding Minimum Energy Paths. J. Chem. Phys. 2008, 128, No.134106.(51) Henkelman, G.; Jonsson, H. A Dimer Method for FindingSaddle Points on High Dimensional Potential Surfaces Using OnlyFirst Derivatives. J. Chem. Phys. 1999, 111, 7010−7022.(52) Heyden, A.; Bell, A. T.; Keil, F. J. Efficient Method for FindingTransition States in Chemical Reactions. J. Chem. Phys. 2005, 123, No.224101.(53) Finnis, M. Accessing the Excess: An Atomistic Approach toExcesses at Planar Defects and Dislocations in Ordered Compounds.Phys. Status Solidi A 1998, 166, 397−416.
The Journal of Physical Chemistry C Article
dx.doi.org/10.1021/jp5025075 | J. Phys. Chem. C 2014, 118, 12285−1230112300

(54) Reuter, K.; Scheffler, M. Composition, Structure, and Stability ofRuO2(110) as a Function of Oxygen Pressure. Phys. Rev. B 2001, 65,No. 035406.(55) Hafner, J.; Spisak, D. Structure and Stability of the Low-IndexSurfaces of Fe3Si: Ab-Initio DFT Investigations. Phys. Rev. B 2007, 75,No. 195411.(56) Dong, W.; Hafner, J. H2 Dissociative Adsorption on Pd(111).Phys. Rev. B 1997, 56, 15396−15403.(57) Dong, W.; Kresse, G.; Furthmuller, J.; Hafner, J. Chemisorptionof H on Pd(111): An Ab-Initio Approach with UltrasoftPseudopotentials. Phys. Rev. B 1996, 54, 2157−2166.(58) Dunphy, J. C.; Rose, M.; Behler, S.; Ogletree, D. F.; Salmeron,M.; Sautet, P. Acetylene Structure and Dynamics on Pd(111). Phys.Rev. B 1998, 57, R12705−12708.
The Journal of Physical Chemistry C Article
dx.doi.org/10.1021/jp5025075 | J. Phys. Chem. C 2014, 118, 12285−1230112301