sci. usa vol. cell protein required kinase cdc28 cyclins … division cycle in g1 is controlled...
TRANSCRIPT

Proc. Natl. Acad. Sci. USAVol. 92, pp. 4651-4655, May 1995Cell Biology
Cdc37 is required for association of the protein kinase Cdc28with G1 and mitotic cyclins
(cell cycle/cyclin-dependent kinase)
MONICA R. GERBER*t, ALISON FARRELL*t, RAYMOND J. DESHAIEStI, IRA HERSKOWITZt, AND DAVID 0. MORGAN*tDepartments of *Physiology and tBiochemistry and Biophysics, University of California, San Francisco, CA 94143-0444
Contributed by Ira Herskowitz, January 30, 1995
ABSTRACT Studies of the temperature-sensitive cdc37-1mutant of Saccharomyces cerevisiae suggest that Cdc37 isrequired for passage through the G1 phase of the cell cycle, butits precise function is not known. We have investigated the roleof Cdc37 in the regulation of the cyclin-dependent proteinkinase Cdc28. We find that G1 arrest in the cdc37-1 mutant isaccompanied by a decrease in the Cdc28 activity associatedwith the G1 cyclin Cln2. This defect appears to be caused bya decrease in the binding of Cdc28 and Cln2. cdc37-1 mutantsalso exhibit a defect in the binding and activation of Cdc28 bythe mitotic cyclin Clb2. Thus Cdc37 may be a regulator thatis required for the association of Cdc28 with multiple cyclins.
In the budding yeast Saccharomyces cerevisiae, commitment toa new cell division cycle in G1 is dependent on nutritionalconditions and other extracellular signals. Cells arrest in G1when starved of nutrients or when treated with mating pher-omone. During nutrient arrest, cells stop growing and areunable to mate, whereas in pheromone arrest the cell divisioncycle is blocked but cell growth continues and mating can occur(1, 2).Our understanding of cell cycle control has been greatly
enhanced by the analysis of temperature-sensitive mutants thatexhibit a G1 arrest like that seen during pheromone treatment(3). Screens for such mutants have led to the identification ofseveral genes, including CDC28, CDC36, CDC37, and CDC39(3, 4). Mutations in CDC36 and CDC39 result in constitutiveactivation of the mating pheromone pathway (5), whereasCDC28 is more directly involved in cell cycle control (1, 2, 6).The function of CDC37 is unknown.The product of the CDC28 gene is a member of the highly
conserved family of cyclin-dependent kinases (CDKs), whoseactivation at specific cell cycle stages requires association withcyclin regulatory subunits (2, 7-9). The commitment to a newcell division cycle in G1 is controlled by complexes of Cdc28and the G1 cyclins Clnl, -2, and -3. G1 arrest by matingpheromone involves inhibition of specific Cdc28-Cln com-plexes and decreased synthesis of Clnl and Cln2. Cdc28function is also required later in the cell cycle: progressthrough S phase and mitosis requires activation of Cdc28 byS-phase cyclins (Clb5 and -6) and mitotic cyclins (Clbl, -2, -3,and -4), respectively (see ref. 2 for review).
Unlike CDC28, CDC37 is poorly understood. Cell cycledefects in cdc37 mutants have not been extensively character-ized, and little is known about the Cdc37 protein. Its predictedamino acid sequence does not display any significant homol-ogies that might suggest a function (4, 10). To explore the roleof Cdc37, we have begun a biochemical analysis of the Cdc37protein in wild-type cells and in the temperature-sensitivecdc37-1 mutant. Because Cdc28 plays a key role at or near theG1 arrest point of cdc37-1 cells, we have also analyzed theeffect of the cdc37-1 mutation on Cdc28 activity. Our findings
The publication costs of this article were defrayed in part by page chargepayment. This article must therefore be hereby marked "advertisement" inaccordance with 18 U.S.C. §1734 solely to indicate this fact.
4651
demonstrate that mutation of CDC37 results in reduced acti-vation of Cdc28, apparently due to a defect in Cdc28-cyclinbinding. These studies demonstrate that in wild-type cellsCdc37 positively regulates Cdc28-cyclin binding.
MATERIALS AND METHODSYeast Strains and Manipulations. All experiments were
performed with strains of the A364a background. Mutantstrains were extensively backcrossed into this background.Characterization of Cdc37 was performed in wild-type(RD204-4C) and cdc37-1 (RD249-2D) strains. Experiments inFigs. 2-4 were performed with wild-type (RD219-2C), cdc37-1(RD249-4B), and cdc28-4 (RD705) strains in which the en-dogenous CLN2 gene was replaced with a version in whichCln2 is fused to a C-terminal triple epitope tag from influenzahemagglutinin (11, 12). The experiment in Fig. 4A was per-formed with strains carrying an expression vector (pDK11, agift of D. Kellogg, University of California, San Francisco)encoding the entire Clb2 sequence fused to glutathione S-transferase (GST) and driven by the GALl promoter.For a-factor arrest, 120-ml cultures were grown at 24°C to
an OD600 of 0.5. a-Factor (1.0 tLg/ml) was added for 2 hr. Formitotic arrest, 25-ml cultures were grown at 24°C to an OD600of 0.3. Cells were transferred to medium containing benomyl(60 ,ug/ml) and nocodazole (20 ,g/ml) for 2 hr at 24°C.A 5.8-kb genomic DNA fragment containing the CDC37
gene was a gift of S. Reed (Scripps Institute, La Jolla, CA). Toconstruct the cdc37A allele, a fragment of CDC37 (coding foraa 48-506) was replaced with LEU2 in plasmid pMG1. Wild-type diploid cells were transformed with linearized pMG1, andgene replacement was checked by Southern blot analysis.
Antibodies. Rabbit antiserum was raised against a GST-Cdc37 fusion protein (containing aa 48-506 of Cdc37) andaffinity purified on antigen columns (13). Polyclonal antibod-ies were raised against a C-terminal Cdc28 peptide (R.J.D.,unpublished work). Monoclonal antibody (12CA5) against theinfluenza hemagglutinin epitope was obtained from BabCO.Affinity-purified anti-Clb2 antiserum was a gift from D.Kellogg.
Lysate Preparation. In Figs. 1-3, logarithmic-phase cellswere resuspended in 700 ptl of ice-cold 20 mM Tris-HCl, pH7.4/0.1% Triton X-100/100 mM NaCl/5 mM EDTA/50 mMP-glycerophosphate/50 mM NaF/1 mM phenylmethylsulfonylfluoride/1 mM dithiothreitol with aprotinin (2 ,tg/ml) andleupeptin (1 tLg/ml). One milliliter of glass beads was added,and cells were lysed at 4°C by two pulses (60 s) in a mini-BeadBeater (Biospec Products, Bartlesville, OK). Lysates wereclarified by centrifugation at 14,000 x g for 10 min at 4°C. InFig. 4, lysates were prepared by lysis with glass beads in 50mMHepes, pH 7.6/1 M NaCl/1 mM EGTA/0.2% Tween 20/1
Abbreviations: CDK, cyclin-dependent kinase; GST, glutathione S-transferase.tPresent address: Division of Biology 156-29, California Institute ofTechnology, Pasadena, CA 91125.

Proc. Natl. Acad Sci. USA 92 (1995)
mM phenylmethylsulfonyl fluoride with aprotinin (2 ,tg/ml)and leupeptin (1 Lag/ml).
Immunoblotting. Immunoblots of cell lysates were probed asdescribed (14) with anti-Cdc37 antibodies (1:1000), anti-Cdc28antibodies (1:1000), monoclonal antibody 12CA5 (14 uag/ml),or anti-Clb2 antibodies (1:2000). In some experiments, blotswere incubated with 125I-labeled secondary antibodies andquantified with a PhosphorImager (Molecular Dynamics).
Metabolic Labeling. Cells growing at 30°C (wild type) or24°C (cdc37-1) were transferred to minimal medium lackingmethionine (SD-Met) at an OD600 of 0.15, grown to an OD600of 0.6, harvested by centrifugation, and resuspended in 3 ml ofSD-Met containing 50taCi of [35S]methionine per OD unit ofcells (1 ,tCi = 37 kBq). Cells were cultured for 1 hr at 30°C(wild type) or 24°C (cdc37-1). Lysates (50 t,g of protein) wereincubated for 2 hr at 4°C with 1 ,jg of anti-Cdc37 antibodiesand protein A-Sepharose. Immune complexes were washedthree times with lysis buffer and analyzed by SDS/PAGE andautoradiography.
Histone H1 Kinase Assays. For analysis of Cln2-associatedkinase activity, cell lysates (200 jig) were incubated with 1 ,tgof monoclonal antibody 12CA5 and protein A-Sepharose for3 hr at 4°C. Immune complexes were washed three times andincubated for 10 min at 24°C in a 30-,ul kinase reaction mixturecontaining 10 mM Hepes (pH 7.6), 1 mM dithiothreitol, 10mM MgCl2, 5 ,tg of histone HI, 50 ,tM ATP, and 1.0 tCi of[y-32P]ATP. Reaction products were analyzed by SDS/PAGEand autoradiography. To measure GST-Clb2-associated ki-nase activity, 50 Ag of lysate was incubated for 2 hr at 4°C with30 ,tl of glutathione-agarose beads. The beads were washedfour times and incubated for 30 min at 24°C in a 20-tul kinasereaction mixture containing 50 mM Hepes (pH 7.6), 5 ,Ag ofhistone H., 2 mM MgC12, 1 mM EGTA, 0.3 mM ATP, 2.5 ,ACiof [y-32P]ATP, and 5 mM reduced glutathione. Clb2-associated kinase activity was measured by the same methodin anti-Clb2 immunoprecipitates.
Binding of Cdc28 and Cyclins. To assess Cdc28-Cln2 bind-ing, lysates were subjected to immunoaffinity chromatographyon a column of monoclonal antibody 12CA5 (15). To assessCdc28-Clb2 binding, lysates (500 ,tg) were loaded on columnscontaining 50 Atg of anti-Clb2 antibodies covalently coupled to100 ,tl of protein A-Sepharose. Columns were washed with 2.5ml of 20 mM Hepes, pH 7.6/250 mM NaCl/5 mM EDTA/0.1% Triton X-100 and 1.5 ml of 10mM sodium phosphate, pH7.4/75 mM NaCl. Bound proteins were eluted with 100 mMtriethylamine at pH 11.
RESULTSCDC37 Encodes an Essential 58-kDa Protein. To begin our
analysis of Cdc37 function, we characterized the Cdc37 proteinby immunoblot analysis of yeast lysates with an anti-Cdc37antibody. Lysates of wild-type cells contained an immunore-active protein with an apparent mobility of -68 kDa (Fig. 1A).Lysates from the cdc37-1 mutant contained a less abundantprotein of -44 kDa. The level of Cdc37 protein was similar inexponentially growing and stationary-phase cells (data notshown) and did not change significantly during the cell cycle(see below, Fig. 3A). We also examined the Cdc37 protein byimmunoprecipitation from cells metabolically labeled with[35S]methionine (Fig. 1B). No labeled proteins consistentlyassociated with wild-type or mutant Cdc37 under these con-ditions.During the course of this work, we have found that the
previously reported CDC37 sequence (4) is incomplete. Weconstructed a low-copy expression vector in which the pub-lished CDC37 open reading frame was placed under thecontrol of the galactose-inducible GALl promoter. This plas-mid failed to complement the cdc37-1 mutant, and immuno-blotting of cell lysates revealed a galactose-induced Cdc37
A 24°C 37°C
Ki" K
kDa
143-97-
50-
35-
21-
14-
1 2 3 4
B WT cdc37-1I-+I_ + _ +
kDa
97-
66-
4'5 -' "..'45- I
.0
31-
1 2 3 4
FIG. 1. Characterization of Cdc37 protein in wild-type and cdc37-1mutant cells. (A) Anti-Cdc37 immunoblots of total protein isolatedfrom wild-type (WT, lanes 1 and 3) and cdc37-1 (lanes 2 and 4) cellsgrown at 24°C (lanes 1 and 2) or 37°C (lanes 3 and 4). (B) Immuno-precipitation of extracts from wild-type (lanes 1 and 2) and cdc37-1(lanes 3 and 4) cells metabolically labeled with [35S]methionine, withanti-Cdc37 (+, lanes 2 and 4) or no antibody (-, lanes 1 and 3).
protein -5 kDa smaller than the wild-type protein. Wesequenced the CDC37 gene 5' of the published sequence andfound an upstream start codon that added 57 aa to thepredicted sequence. This start codon is preceded by a stopcodon 138 bp further upstream. The new start codon is foundin the context 5'-AAG-TCA-AAA-ATG-GCC-ATT-GAT-TAC-TCT-AAG-TGG-GAT-AAA-ATT-3', where the begin-ning of the published sequence is underlined. The revised openreading frame encodes a protein of 506 aa, with a predictedmolecular mass of 58.4 kDa. Galactose-induced overexpres-sion of the revised open reading frame rescued the growthdefect of the cdc37-1 mutant at 37°C and resulted in theoverproduction of a Cdc37 protein that migrated in polyacryl-amide gels at the same anomalous position (-68 kDa) as thewild-type protein (data not shown). Thus, the amino-terminal57 residues are required for Cdc37 function.We also analyzed the Cdc37 coding sequence in the cdc37-1
mutant. The mutant cdc37 gene from cdc37-1 cells was isolatedby standard gap-repair methods (16), and sequencing of thecoding region revealed that codon 360 (CAA in wild-typeCDC37) was changed to a stop codon (TAA; data not shown).Premature termination at this codon would be consistent withthe smaller size of the mutant protein (Fig. 1). Interestingly,overexpression of the mutant gene on a low-copy plasmid(resulting in expression of the mutant protein at levels 2- to3-fold higher than normal mutant levels) allowed slow butsignificant growth of cdc37-1 cells at 37°C (data not shown).Thus, the carboxyl-terminal region is not strictly required forCdc37 function, but truncation of this region appears to causea decrease in the steady-state concentration of Cdc37 protein(Fig. 1A). Low Cdc37 levels may be the primary cause of thegrowth defect in cdc37-1 cells.To confirm that CDC37 is an essential gene, we constructed
a diploid strain in which one chromosomal copy of the CDC37gene was replaced with the LEU2 gene. Analysis of sporesproduced by this diploid indicated that CDC37 is essential at30°C (data not shown). Spores bearing the CDC37 deletionformed microcolonies containing between 4 and =60 cells.The terminal phenotype of these cells was heterogeneous:many cells were enlarged and elongated with multiple projec-tions, suggesting that Cdc37 may be required at multiple cellcycle stages.
4652 Cell Biology: Gerber et al,
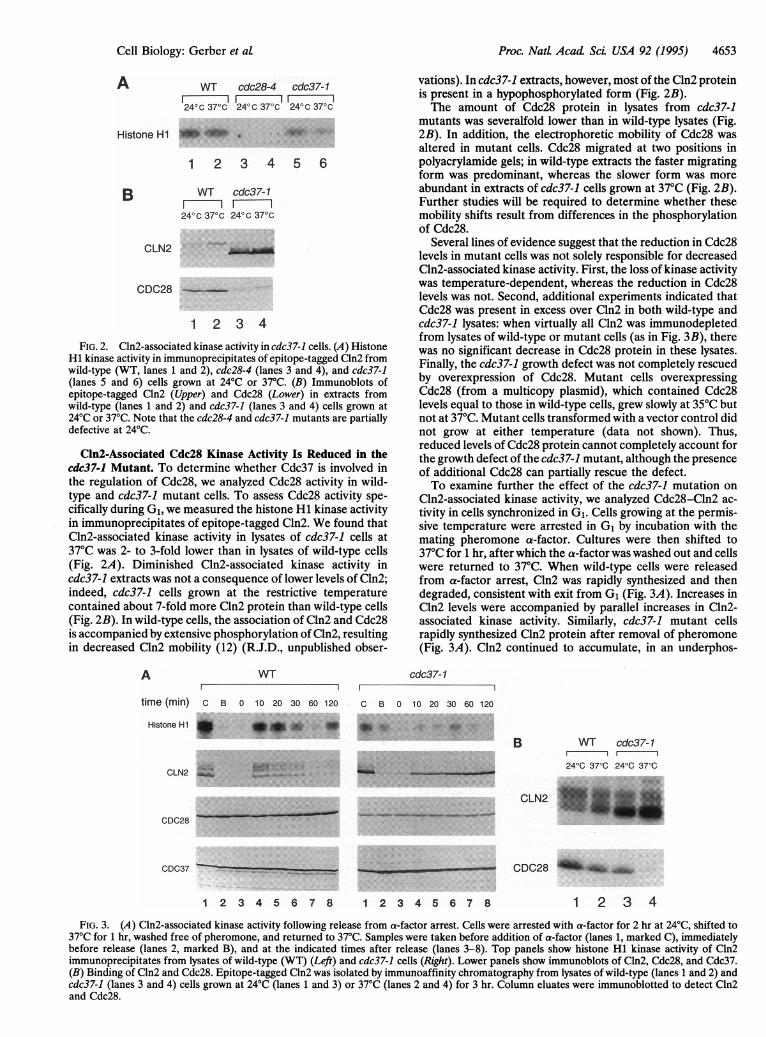
Proc. NatL Acad Sci. USA 92 (1995) 4653
A WT cdc28-4 cdc37-1
2437 241 37 24cI
3724"C 370C 24"C 37"C 24"c 37"c
Histone H1 VI AMi *
1 2 3 4 5 6
B WT cdc37-1I I I 1240C 370C 240C 370C
CLN2 ..
CDC28
1 2 3 4FIG. 2. Cln2-associated kinase activity in cdc37-1 cells. (A) Histone
H1 kinase activity in immunoprecipitates of epitope-tagged Cln2 fromwild-type (WT, lanes 1 and 2), cdc28-4 (lanes 3 and 4), and cdc37-1(lanes 5 and 6) cells grown at 24°C or 37°C. (B) Immunoblots ofepitope-tagged Cln2 (Upper) and Cdc28 (Lower) in extracts fromwild-type (lanes 1 and 2) and cdc37-1 (lanes 3 and 4) cells grown at24°C or 37°C. Note that the cdc28-4 and cdc37-1 mutants are partiallydefective at 24°C.
Cln2-Associated Cdc28 Kinase Activity Is Reduced in thecdc37-1 Mutant. To determine whether Cdc37 is involved inthe regulation of Cdc28, we analyzed Cdc28 activity in wild-type and cdc37-1 mutant cells. To assess Cdc28 activity spe-cifically during G1, we measured the histone HI kinase activityin immunoprecipitates of epitope-tagged Cln2. We found thatCln2-associated kinase activity in lysates of cdc37-1 cells at37°C was 2- to 3-fold lower than in lysates of wild-type cells(Fig. 2A). Diminished Cln2-associated kinase activity incdc37-1 extracts was not a consequence of lower levels of Cln2;indeed, cdc37-1 cells grown at the restrictive temperaturecontained about 7-fold more Cln2 protein than wild-type cells(Fig. 2B). In wild-type cells, the association of Cln2 and Cdc28is accompanied by extensive phosphorylation of Cln2, resultingin decreased Cln2 mobility (12) (R.J.D., unpublished obser-
A WT
vations). In cdc37-1 extracts, however, most of the Cln2 proteinis present in a hypophosphorylated form (Fig. 2B).The amount of Cdc28 protein in lysates from cdc37-1
mutants was severalfold lower than in wild-type lysates (Fig.2B). In addition, the electrophoretic mobility of Cdc28 wasaltered in mutant cells. Cdc28 migrated at two positions inpolyacrylamide gels; in wild-type extracts the faster migratingform was predominant, whereas the slower form was moreabundant in extracts of cdc37-1 cells grown at 37°C (Fig. 2B).Further studies will be required to determine whether thesemobility shifts result from differences in the phosphorylationof Cdc28.
Several lines of evidence suggest that the reduction in Cdc28levels in mutant cells was not solely responsible for decreasedCln2-associated kinase activity. First, the loss of kinase activitywas temperature-dependent, whereas the reduction in Cdc28levels was not. Second, additional experiments indicated thatCdc28 was present in excess over Cln2 in both wild-type andcdc37-1 lysates: when virtually all Cln2 was immunodepletedfrom lysates of wild-type or mutant cells (as in Fig. 3B), therewas no significant decrease in Cdc28 protein in these lysates.Finally, the cdc37-1 growth defect was not completely rescuedby overexpression of Cdc28. Mutant cells overexpressingCdc28 (from a multicopy plasmid), which contained Cdc28levels equal to those in wild-type cells, grew slowly at 35°C butnot at 37°C. Mutant cells transformed with a vector control didnot grow at either temperature (data not shown). Thus,reduced levels of Cdc28 protein cannot completely account forthe growth defect of the cdc37-1 mutant, although the presenceof additional Cdc28 can partially rescue the defect.To examine further the effect of the cdc37-1 mutation on
Cln2-associated kinase activity, we analyzed Cdc28-Cln2 ac-tivity in cells synchronized in G1. Cells growing at the permis-sive temperature were arrested in G1 by incubation with themating pheromone a-factor. Cultures were then shifted to37°C for 1 hr, after which the a-factor was washed out and cellswere returned to 37°C. When wild-type cells were releasedfrom a-factor arrest, Cln2 was rapidly synthesized and thendegraded, consistent with exit from G1 (Fig. 3A). Increases inCln2 levels were accompanied by parallel increases in Cln2-associated kinase activity. Similarly, cdc37-1 mutant cellsrapidly synthesized Cln2 protein after removal of pheromone(Fig. 3A). Cln2 continued to accumulate, in an underphos-
cdc37-1
time (min) c B 0 10 20 30 60 120 C B 0 10 20 30 60 120
Histone H1 I *_ _._ _*__4 _0 'IO i.. ..::
B
CDC28 .
CDC37 _.
WT cdc37-1
24°C 37°C 24°C 37°C
CLN2 *
CDC28 _,! _i,
1 2 3 4 5 6 7 8 1 2 3 4 5 6 7 8 1 2 3 4FIG. 3. (A) Cln2-associated kinase activity following release from a-factor arrest. Cells were arrested with a-factor for 2 hr at 24°C, shifted to
37°C for 1 hr, washed free of pheromone, and returned to 37°C. Samples were taken before addition of a-factor (lanes 1, marked C), immediatelybefore release (lanes 2, marked B), and at the indicated times after release (lanes 3-8). Top panels show histone H1 kinase activity of Cln2immunoprecipitates from lysates of wild-type (WT) (Left) and cdc37-1 cells (Right). Lower panels show immunoblots of Cln2, Cdc28, and Cdc37.(B) Binding of Cln2 and Cdc28. Epitope-tagged Cln2 was isolated by immunoaffinity chromatography from lysates of wild-type (lanes 1 and 2) andcdc37-1 (lanes 3 and 4) cells grown at 24°C (lanes 1 and 3) or 37°C (lanes 2 and 4) for 3 hr. Column eluates were immunoblotted to detect Cln2and Cdc28.
Cell Biology: Gerber et at.
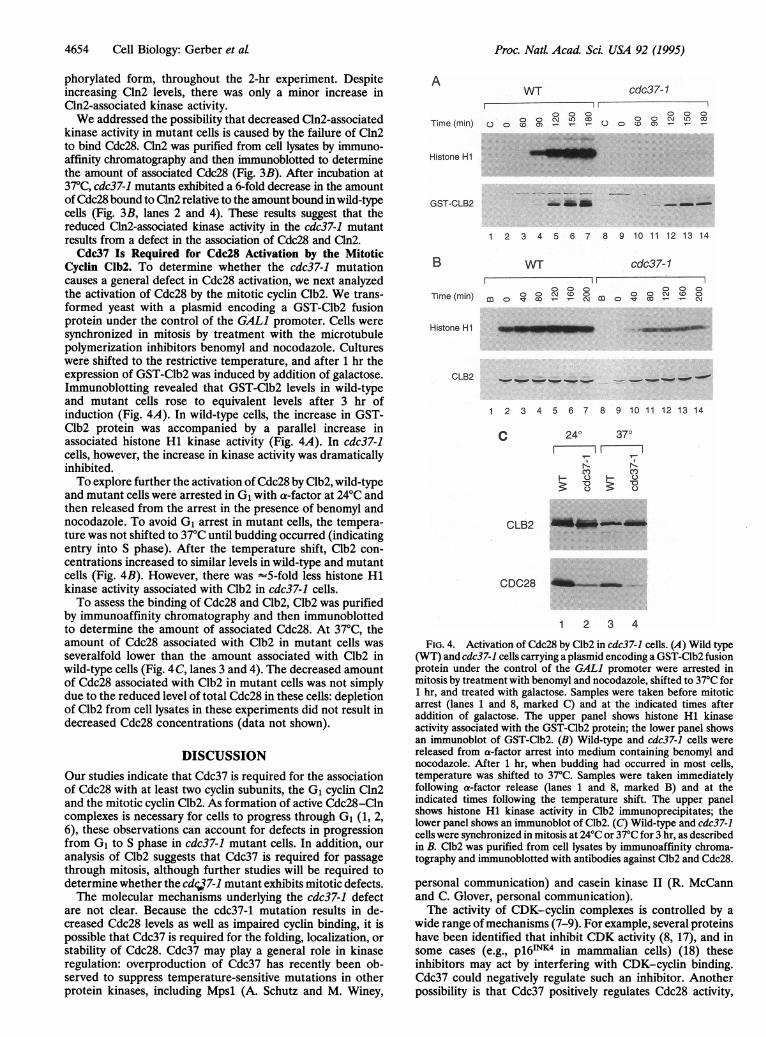
Proc. Natl. Acad Sci. USA 92 (1995)
phorylated form, throughout the 2-hr experiment. Despiteincreasing Cln2 levels, there was only a minor increase inCln2-associated kinase activity.We addressed the possibility that decreased Cln2-associated
kinase activity in mutant cells is caused by the failure of Cln2to bind Cdc28. Cln2 was purified from cell lysates by immuno-affinity chromatography and then immunoblotted to determinethe amount of associated Cdc28 (Fig. 3B). After incubation at37°C, cdc37-1 mutants exhibited a 6-fold decrease in the amountof Cdc28 bound to Cln2 relative to the amount bound in wild-typecells (Fig. 3B, lanes 2 and 4). These results suggest that thereduced Cln2-associated kinase activity in the cdc37-1 mutantresults from a defect in the association of Cdc28 and Cln2.Cdc37 Is Required for Cdc28 Activation by the Mitotic
Cyclin Clb2. To determine whether the cdc37-1 mutationcauses a general defect in Cdc28 activation, we next analyzedthe activation of Cdc28 by the mitotic cyclin Clb2. We trans-formed yeast with a plasmid encoding a GST-Clb2 fusionprotein under the control of the GALl promoter. Cells weresynchronized in mitosis by treatment with the microtubulepolymerization inhibitors benomyl and nocodazole. Cultureswere shifted to the restrictive temperature, and after 1 hr theexpression of GST-Clb2 was induced by addition of galactose.Immunoblotting revealed that GST-Clb2 levels in wild-typeand mutant cells rose to equivalent levels after 3 hr ofinduction (Fig. 4A). In wild-type cells, the increase in GST-Clb2 protein was accompanied by a parallel increase inassociated histone HI kinase activity (Fig. 4A). In cdc37-1cells, however, the increase in kinase activity was dramaticallyinhibited.To explore further the activation of Cdc28 by Clb2, wild-type
and mutant cells were arrested in G1 with a-factor at 24°C andthen released from the arrest in the presence of benomyl andnocodazole. To avoid G1 arrest in mutant cells, the tempera-ture was not shifted to 37°C until budding occurred (indicatingentry into S phase). After the temperature shift, Clb2 con-centrations increased to similar levels in wild-type and mutantcells (Fig. 4B). However, there was -5-fold less histone HIkinase activity associated with Clb2 in cdc37-1 cells.To assess the binding of Cdc28 and Clb2, Clb2 was purified
by immunoaffinity chromatography and then immunoblottedto determine the amount of associated Cdc28. At 37°C, theamount of Cdc28 associated with Clb2 in mutant cells wasseveralfold lower than the amount associated with Clb2 inwild-type cells (Fig. 4 C, lanes 3 and 4). The decreased amountof Cdc28 associated with Clb2 in mutant cells was not simplydue to the reduced level of total Cdc28 in these cells: depletionof Clb2 from cell lysates in these experiments did not result indecreased Cdc28 concentrations (data not shown).
DISCUSSIONOur studies indicate that Cdc37 is required for the associationof Cdc28 with at least two cyclin subunits, the G1 cyclin Cln2and the mitotic cyclin Clb2. As formation of active Cdc28-Clncomplexes is necessary for cells to progress through G1 (1, 2,6), these observations can account for defects in progressionfrom G1 to S phase in cdc37-1 mutant cells. In addition, ouranalysis of Clb2 suggests that Cdc37 is required for passagethrough mitosis, although further studies will be required todetermine whether the cdcqj7-1 mutant exhibits mitotic defects.The molecular mechanisms underlying the cdc37-1 defect
are not clear. Because the cdc37-1 mutation results in de-creased Cdc28 levels as well as impaired cyclin binding, it ispossible that Cdc37 is required for the folding, localization, orstability of Cdc28. Cdc37 may play a general role in kinaseregulation: overproduction of Cdc37 has recently been ob-served to suppress temperature-sensitive mutations in otherprotein kinases, including Mpsl (A. Schutz and M. Winey,
AWT cdc37-1
/1000 000
o o o~ t, 00 oo o inLO coTime (min) O Oco a) -- O o o o-N- -
Histone H1
GST-CLB2 -l-m.m
1 2 3 4 5 6 7 8 9 10 11 12 13 14
B WT cdc37-1
000 000Time (min) m c -o-oC-°- cH
Histone H1 _ _... . _---
CLB2 ,___ _-_...___, '
1 2 3 4 5 6 7 8 9 10 11 12 13 14
C 24° 37°
Cl) CY)3: 'a 'LO)
CLB2
CDC28 ~ ..
1 2 3 4
FIG. 4. Activation of Cdc28 by Clb2 in cdc37-1 cells. (A) Wild type(WT) and cdc37-1 cells carrying a plasmid encoding a GST-Clb2 fusionprotein under the control of the GALl promoter were arrested inmitosis by treatment with benomyl and nocodazole, shifted to 37°C for1 hr, and treated with galactose. Samples were taken before mitoticarrest (lanes 1 and 8, marked C) and at the indicated times afteraddition of galactose. The upper panel shows histone H1 kinaseactivity associated with the GST-Clb2 protein; the lower panel showsan immunoblot of GST-Clb2. (B) Wild-type and cdc37-1 cells werereleased from a-factor arrest into medium containing benomyl andnocodazole. After 1 hr, when budding had occurred in most cells,temperature was shifted to 37°C. Samples were taken immediatelyfollowing a-factor release (lanes 1 and 8, marked B) and at theindicated times following the temperature shift. The upper panelshows histone H1 kinase activity in Clb2 immunoprecipitates; thelower panel shows an immunoblot of Clb2. (C) Wild-type and cdc37-1cells were synchronized in mitosis at 24°C or 37°C for 3 hr, as describedin B. Clb2 was purified from cell lysates by immunoaffinity chroma-tography and immunoblotted with antibodies against Clb2 and Cdc28.
personal communication) and casein kinase II (R. McCannand C. Glover, personal communication).The activity of CDK-cyclin complexes is controlled by a
wide range of mechanisms (7-9). For example, several proteinshave been identified that inhibit CDK activity (8, 17), and insome cases (e.g., p16'NK4 in mammalian cells) (18) theseinhibitors may act by interfering with CDK-cyclin binding.Cdc37 could negatively regulate such an inhibitor. Anotherpossibility is that Cdc37 positively regulates Cdc28 activity,
4654 Cell Biology: Gerber et at

Proc. Nat. Acad Sci. USA 92 (1995) 4655
perhaps by directly catalyzing Cdc28-cyclin binding or bystimulating the activity of the CDK-activating kinase (CAK).In some cases (e.g., human CDC2 and cyclin A), the bindingof CDK and cyclin requires phosphorylation by CAK at aconserved threonine residue (Thr161 in CDC2) (19, 20). Thus,if Cdc28-cyclin binding requires phosphorylation at the equiv-alent site (Thr169), then mutation of CDC37 could inhibitCdc28-cyclin binding by reducing CAK activity. Biochemicalexplorations of these issues, both in yeast and in vertebratecells, could lead to the identification of novel mechanisms inCDK regulation.We thank Yong Gu, Doug Kellogg, Marc Kirschner, Steve Murphy,
Andrew Murray, Matthias Peter, Steve Reed, Aaron Straight, andMike Tyers for reagents and helpful advice and Rob Fisher forcomments on the manuscript. This work was supported by grants toI.H. from the National Institutes of Health and to D.O.M. from theNational Institutes of Health, the Markey Charitable Trust, the Marchof Dimes Birth Defects Foundation, and the Rita Allen Foundation.R.J.D. is a Lucille P. Markey Scholar. M.R.G. was supported byNational Institutes of Health training grant to the Medical ScientistTraining Program of the University of California, Los Angeles. A.F.is supported by a postgraduate scholarship from the Natural Sciencesand Engineering Research Council of Canada.
1. Forsburg, S. L. & Nurse, P. (1991) Annu. Rev. Cell Biol. 7,227-256.
2. Nasmyth, K. (1993) Curr. Opin. Cell Biol. 5, 166-179.3. Reed, S. I. (1980) Genetics 95, 561-577.4. Ferguson, J., Ho, J., Peterson, T. A. & Reed, S. I. (1986) Nucleic
Acids Res. 14, 6681-6697.5. Neiman, A. M., Chang, F., Komachi, K. & Herskowitz, I. (1990)
Cell Regul. 1, 391-401.6. Reed, S. I. (1992) Annu. Rev. Cell Biol. 8, 529-561.7. Draetta, G. (1993) Trends Cell Biol. 3, 287-289.8. Morgan, D. 0. (1995) Nature (London) 374, 131-134.9. Solomon, M. (1993) Curr. Opin. Cell Biol. 5, 180-186.
10. Cutforth, T. & Rubin, G. M. (1994) Cell 77, 1027-1036.11. Tyers, M., Tokiwa, G., Nash, R. & Futcher, B. (1992) EMBO J.
11, 1773-1784.12. Tyers, M., Tokiwa, G. & Futcher, B. (1993) EMBO J. 12,
1955-1968.13. Kellogg, D. R. & Alberts, B. M. (1992) Mol. Biol. Cell 3, 1-11.14. Gu, Y., Rosenblatt, J. & Morgan, D. 0. (1992) EMBO J. 11,
3995-4005.15. Gu, Y., Turck, C. W. & Morgan, D. 0. (1993) Nature (London)
366, 707-710.16. Rothstein, R. (1991) Methods Enzymol. 194, 281-301.17. Peter, M. & Herskowitz, I. (1994) Cell 79, 181-184.18. Serrano, M., Hannon, G. J. & Beach, D. (1993) Nature (London)
366, 704-707.19. Ducommun, B., Brambilla, P., Felix, M.-A., Franza, B. R.,
Karsenti, E. & Draetta, G. (1991) EMBO J. 10, 3311-3319.20. Desai, D., Wessling, H. C., Fisher, R. P. & Morgan, D. 0. (1995)
Mol. Cell. Biol. 15, 345-350.
Cell Biology: Gerber et aL