regulation of mdm2 and the p53 family by the nedd8 pathway · pathway and identify nedp1 as a...
TRANSCRIPT

Regulation of MDM2 and the p53 family by the NEDD8
pathway
by
Ian Robert Watson
A thesis submitted in conformity with the requirements for the degree of Doctor of Philosophy
Laboratory Medicine and Pathobiology University of Toronto
© Copyright by Ian Robert Watson 2010

ii
Regulation of MDM2 and the p53 family by the NEDD8 pathway
Ian Robert Watson
Doctor of Philosophy
Laboratory Medicine and Pathobiology
University of Toronto
2010
Abstract
NEDD8 is an ubiquitin-like protein sharing approximately 60% amino acid identity with
ubiquitin and has biological roles in cell cycle progression, viability and development. Recently,
a number of oncoproteins and tumor suppressors have been identified as NEDD8 substrates,
including MDM2 and p53. MDM2 is an oncogenic E3 ligase that promotes NEDD8
modification and ubiquitin-mediated degradation of the tumor suppressor transcription factor,
p53. Cellular stresses such as DNA damage lead to p53 activation due, in part, to MDM2
destabilization by mechanisms that are not completely understood. Studies in mice demonstrate
the biological role of MDM2 is to negatively regulate p53 function, however, when
overexpressed or amplified, MDM2 has p53-independent oncogenic functions presumably due to
the regulation of additional substrates. One such substrate may be the p53 family member, p73.
p73 exists as multiple isoforms and accumulating evidence suggests that the N-terminal isoforms
dictate its role in tumorigenesis. The full-length pro-apoptotic TAp73 isoforms are induced by
chemotherapies and are able to transactivate p53-target genes to initiate cell cycle arrest and
apoptosis. Conversely, the N-terminally truncated ΔNp73 isoforms lack the transactivation

iii
domain (TAD) and consequently act as dominant-negative inhibitors for all TA isoforms of the
p53 family, and are overexpressed in human tumors. Here, we report that TAp73, but not
ΔNp73, is covalently modified by NEDD8 in an MDM2-dependent manner, attenuating its
transactivation function and promoting cytoplasmic localization of neddylated TAp73. These
results provide the first evidence of a covalent post-translational modification exclusively
targeting the TA isoforms of p73, and identify the MDM2-TAp73 interaction as a promising
therapeutic target. We also demonstrate that the stability of MDM2 is regulated by the NEDD8
pathway and identify NEDP1 as a chemotherapy-induced isopeptidase that deneddylates MDM2,
resulting in MDM2 destabilization, concomitant with p53 activation. This study identifies a
novel p53 activating mechanism in response to chemotherapy. In conclusion, the work presented
herein has helped characterize the function of NEDD8 modification of MDM2 and the p53
family, and identify mechanisms by which MDM2 and the NEDD8 pathway may be targeted in
the development of anti-cancer therapeutics.

iv
Acknowledgments
First, I would like to thank my supervisors Dr. Michael Ohh and Dr. Meredith Irwin. Throughout
the duration of my PhD they have never wavered in their support, fostered my research potential
and given me confidence in my research abilities. I could not have asked for a better supervisory
team. I would also like to thank my advisory committee members, Dr. Dwayne Barber and Dr.
Samuel Benchimol, who have provided exceptional guidance and have always offered their time
graciously. I owe a great deal of gratitude to my undergraduate supervisor, Dr. Sandy Der, for
first providing me the opportunity to study in the department of Laboratory Medicine and
Pathobiology and Dr. Irene Hwang, the PhD student whose training has stayed with me to this
day. Throughout the duration of my study, I have had the great opportunity to work alongside a
number of talented trainees in the labs of Dr. Ohh and Dr. Irwin who have provided at one time
or another, assistance, direction and advice. In particular, I would like to thank: Dr. Alvaro
Blanch, who was a key contributor in Chapter 2, and has always taken the time to provide advice
in matters related to the lab, career, and life in general—I will sadly miss his guidance when I am
gone; Dr. Loretta Lau, who was always there for me to provide help and direction during her
time as the senior lab member; Bryan Li, who was a key contributor to Chapter 3—I appreciate
all his hard work and efforts; our lab manager, Lynn Cheng, for always being there for lab
matters and anything beyond; Dr. Fiona Robinson, for her careful reading of this thesis; and
Joanne Lau, my fellow PhD student, who shared all the same experiences with me as we started
our graduate studies together.
Personally, I would like to thank my parents for their support throughout my education
and research studies. Most importantly, I would like to thank my wife, Christine DeSantis, for
her patience, encouragement and understanding. She never ceases to amaze me with her
thoughts, consideration, and awareness. I am truly appreciative of her support; without her I
would not be where I am today.

v
Table of Contents
Abstract ii
Acknowledgments iv
Table of Contents v
List of Figures viii
Abbreviations x
Chapter 1:
Introduction to ubiquitin-like proteins, the p53 family and MDM2 1
1 INTRODUCTION 1
1.1 Ubiquitin-proteasome pathway 1
1.1.2 NEDD8: Ubiquitin-like protein (UBL) 4
1.1.3 The role of NEDD8 in cancer 6
1.1.4 p53: The guardian of the genome 7
1.1.5 Structure and function of the p53 family 10
1.1.6 p63 and p73: Dual tumor suppressor and oncogenic functions 13
1.1.7 p63 and p73: Roles in development 14
1.1.8 p63 and p73: Mouse models of cancer 15
1.1.9 p63 and p73: Alterations in human cancer and role in chemotherapy response 16
1.1.10 Mechanisms of p53 activation in response to DNA damage 17
1.1.11 MDM2 regulation of p53 18
1.1.12 MDM2 studies in mice 21
1.1.13 MDM2: p53-independent oncogenic functions 22
1.1.14 MDM2 destabilization: Mechanism of p53 activation in response to DNA damage 23
1.1.15 Regulation of the p53 family by ubiquitin and ubiquitin-like modifications 24
1.2 SIGNIFICANCE 26
CHAPTER 2:
MDM2-mediated NEDD8 modification of TAp73 regulates its transactivation function 28
2.1 HYPOTHESIS AND RATIONALE 28

vi
2.2 RESULTS AND DISCUSSION 30
2.2.1 TAp73 is modified by NEDD8 via MDM2 30
2.2.2 NEDP1 deneddylates modified TAp73β 34
2.2.3 Np73 does not undergo MDM2-mediated neddylation 36
2.2.4 Neddylation of p73 occurs under physiological conditions 38
2.2.5 The neddylation pathway attenuates TAp73 transcriptional activity 40
2.2.6 NEDD8 modification of TAp73 promotes cytoplasmic localization 44
2.3 DISCUSSION 48
2.4 FUTURE DIRECTIONS 50
2.5 MATERIALS AND METHODS 52
2.5.1 Cells 52
2.5.2 Antibodies 52
2.5.3 Plasmids 53
2.5.4 Immunoprecipitation and immunoblotting 53
2.5.5 Subcellular fractionation 54
2.5.6 Dual-luciferase reporter assay 55
CHAPTER 3:
Chemotherapy induces NEDP1-mediated destabilization of MDM2 56
3.1 HYPOTHESIS AND RATIONALE 56
3.2 RESULTS AND DISCUSSION 58
3.2.1 Neddylation stabilizes MDM2 58
3.2.2 NEDP1-mediated deneddylation promotes MDM2 destabilization 62
3.2.3 Chemotherapy increases NEDP1 levels 64
3.2.4 NEDP1 modulates p53-apoptotic response to chemotherapy 68
3.3 DISCUSSION 71
3.4 FUTURE DIRECTIONS 73
3.5 MATERIAL AND METHODS 75
3.5.1 Cells 75

vii
3.5.2 Antibodies and reagents 75
3.5.3 Plasmids 76
3.5.4 Immunoprecipitation and immunoblotting 77
3.5.5 Protein turnover assays 77
3.5.6 RNAi 78
3.5.7 TUNEL assays 78
Chapter 4: CONCLUSIONS AND FUTURE DIRECTIONS 80
APPENDIX 87
REFERENCES 98

viii
List of Figures
Page
Chapter 1
Figure 1.1.1 General overview of the ubiquitin and ubiquitin-like protein 4
conjugation pathways
Figure 1.1.2 Structure of the p73 isoforms 11
Figure 1.1.3 Schematic representation of the gene structure of the p53 family 12-13
Figure 1.1.4 Structure of MDM2 protein 20
Figure 1.1.5 MDM2 promotion of p53 ubiquitylation 21
Chapter 2
Figure 2.1 TAp73 is modified by NEDD8 32-33
Figure 2.2 NEDP1, a NEDD8 specific cysteine protease, deneddylates modified 35
TAp73
Figure 2.3 The Np73 isoform lacking the MDM2-binding site is not conjugated 37
by NEDD8
Figure 2.4 Endogenous p73 is modified by NEDD8 39
Figure 2.5 NEDD8 pathway inhibits TAp73-mediated transactivation 42-43
Figure 2.6 Neddylated TAp73 species are found preferentially in the cytoplasm 46-47
Chapter 3
Figure 3.1 Neddylation of MDM2 increases its protein stability 60-61
Figure 3.2 NEDP1-mediated deneddylation decreases MDM2 stability 63
Figure 3.3 Chemotherapy increases NEDP1-mediated p53 activation 66

ix
Figure 3.4 NEDP1 levels increase in response to chemotherapy independent of 67
ATM and p53 status
Figure 3.5 siRNA-mediated downregulation of NEDP1 enhances chemoresistance 69-70
Figure 3.6 A model of NEDP1-mediated activation of p53 apoptotic response 70
Chapter 4 Figure 4.1 MLN4924 inhibits NEDD8 conjugation by targeting the NEDD8- 84
activating enzyme (NAE)
Appendix
Figure A.1 TAp73 is modified by NEDD8, but not ubiquitin, in the presence of 87
overexpressed NEDD8
Figure A.2 Endogenous p73 is modified by NEDD8 88
Figure A.3 TAp73 1-344 truncation mutant is not modified by NEDD8 89-90
Figure A.4 MDM2(C464A) RING finger mutant does not promote cytoplasmic 91
neddylated TAp73 species localization
Figure A.5 Ectopic NEDD8 expression prolongs MDM2 half-life 92
Figure A.6 Chemotherapy induces the expression of NEDP1 93
Figure A.7 Ectopic expression of p53 has negligible effect on NEDP1 expression 94
Figure A.8 siRNA-mediated NEDP1 knockdown decreases p53 activation in 95-96
response to doxorubicin
Figure A.9 DNA-PK status determines induction of NEDP1 in glioma cell lines 97

x
Abbreviations
AMP: adenosine 5’-monophosphate
APP-BP1: amyloid precursor protein binding protein 1
ARF-BP1: alternate reading frame binding protein 1
Asp: asparagine
ATM: ataxia telangiectasia mutated
ATP: adenosine-5’-triphosphate
ATR: ataxia telangiectasia and Rad3 related
BAX: BCL2-associated X
BCA3: breast cancer-associated gene 3
Bcl2: B-cell leukemia/lymphoma 2
Bcl-xL: B-cell leukemia/lymphoma xL
c-Cbl: cellular Casitas B-lineage lymphoma
Chk1/2: checkpoint kinase 1/2
CHX: cycloheximide
CNS: central nervous system
CBP: CREB binding protein
CHO: Chinese hamster ovary
COP: constitutive photomorphogenesis 1
cPARP: cleaved poly ADP-ribose polymerase
CPT: camptothecin
CRL: Cullin-RING finger E3 ubiquitin ligases
CSN5: COP9 signalosome subunit 5
Cul: Cullin
DAXX: death-domain associated protein
DBD: DNA-binding domain
DCN1: defective in Cul neddylation 1
DMs: double minutes
DNA-PK: DNA-dependent protein kinase
DOX: doxorubicin
DR4/5: death receptor 4/5
DUB: deubiquitylating enzyme
E1: activating enzyme
E2: conjugating enzyme
E3: ligase enzyme
E6-AP: E6-associated protein
EGFR: epidermal growth factor receptor
ENU: N-ethyl-iV-nitrosourea
FBXO11: F-box protein 11
GADD45: growth arrest and DNA damage 45
Gln: glutamine
Gly: glycine
GSK-3: glycogen synthase kinase-3

xi
HA: hemagglutinin
HAUSP: herpes-associated ubiquitin-specific protease
HECT: homologous to E6-AP carboxyl terminus
HEK293: human embryonic kidney 293
HIPK2: homeodomain-interacting protein kinase 2
His: histidine
HNSCC: head and neck squamous cell carcinomas
IHC: immunohistochemistry
JNK: c-Jun N-terminal kinases
Leu: leucine
LIF: leukemia inhibitory factor
Lys: lysine
mdm2: murine double minute (mouse)
MDM2: murine double minutes 2 (human)
NAE: NEDD8-acitivating enzyme
NCS: neocarzinostatin
NEDP1: NEDD8-specific protease 1
NEDD: neural precursor cell expressed developmentally regulated
NES: nuclear export signal
NLS: nuclear localization signal
OD: oligomerization domain
p38 MAPK: p38 mitogen-activated protein kinase
PARC: parkin-like cytoplasmic
P/CAF: p300/CBP-associated factor
PERP: p53 apoptosis effector related to PMP-22
Phe: phenylalanine
PIAS: protein inhibitor of activated STAT
Pirh2: p53-induced RING-H2
PKC: protein kinase C
Pro: proline
PUMA: p53 up-regulated modulator of apoptosis
RASSF1A: ras association domain-containing protein 1 A
Rbx1: ring-box 1
RCE1: Rub1 conjugating enzyme 1
RING: really interesting novel gene
RT-PCR: reverse transcription polymerase chain reaction
SAM: sterile alpha motif
SAOS2: sarcoma osteogenic 2 (cell line)
SCF: Skp1/Cdc53/F-box
Ser: serine
SDS-PAGE: sodium dodecyl sulfate polyacrylamide gel electrophoresis
SSP3: SUMO-1-specific protease 3
STS: soft tissue sarcoma
SUMO: small ubiquitin-like modifier
SUSP4: SUMO-specific protease 4

xii
SV40: Simian virus 40
TAD: transactivation domain
Thr: threonine
Trp: tryptophan
TUNEL: terminal deoxynucleotidyl transferase dUTP nick end labeling
Tyr: tyrosine
Uba3: ubiquitin-like modifier activating enzyme 3
Ubc12: ubiquitin-conjugating enzyme 12
Ube2F: ubiquitin-conjugating enzyme E2F
UBL: ubiquitin-like protein
UV: ultraviolet
VHL: von Hippel-Lindau
Zn: Zinc

1
Chapter 1:
Introduction to ubiquitin-like proteins, the p53 family and MDM2
Excerpts from this work have been published in the review article:
Ubiquitin and ubiquitin-like modifications of the p53 family. Watson IR, Irwin MS. Neoplasia.
2006 Aug;8(8):655-66. Review.
1 INTRODUCTION
1.1 Ubiquitin-proteasome pathway
Nearly 30 years ago, ubiquitin was discovered as the heat-stable active component responsible
for protein degradation in a cell-free ATP-dependent proteolysis assay [1, 2]. Ubiquitin is a 76
amino acid polypeptide whose evolutionary conservation is demonstrated by the fact that only
three amino acids differ between human and yeast ubiquitin homologs, and which received its
name due to its ubiquitous expression in eukaryotes [3]. The best characterized function of
ubiquitin is that of targeting substrate proteins for degradation via the 26S proteasome, work
which earned Aaron Ciechanover, Avram Hershko and Irwin Rose the Nobel Prize in Chemistry
in 2004 (reviewed in [4]). The process of covalent conjugation of ubiquitin to substrates, known
as ubiquitylation, occurs through sequential steps catalyzed by ubiquitin activating (E1),
conjugating (E2), and ligase (E3) enzymes, and can be reversed by the action of deubiquitylating

2
enzymes (DUBs) (illustrated for ubiquitin and ubiquitin-like proteins in Figure 1.1.1.).
Ubiquitin is first synthesized as a precursor, and is processed at a conserved C-terminal glycine
residue by the hydrolase activity of DUBs exposing a glycine-glycine motif that serves as the
attachment site for target substrates. The exposed C-terminal glycine of ubiquitin is adenylated
by an E1 activating enzyme in an ATP-dependent manner and transferred to an E1 cysteine side
chain via a thiol ester linkage. Activated ubiquitin is subsequently transferred to an E2
conjugating enzyme forming another thiol ester linkage. Lastly, an E3 ligase transfers ubiquitin
to the –amino group of a substrate lysyl residue of a target substrate resulting in the formation
of an isopeptide bond [5]. E4 enzymes catalyze the extension of multiubiquitin chains [6], and
degradation is mediated by the 26S proteasome, which is composed of a cylindrical core 20S
catalytic component flanked by 19S regulatory complexes made up of multiple ATPases [7].
Polyubiquitylated chains made up of at least four ubiquitins linked at Lys48 are recognized by
the 19S component, which unfolds tagged protein substrates that are rapidly hydrolysed by the
trypsin, chymotrypsin and peptidylglutamyl peptidase-like activities of the 20S subunit,
producing short peptides. The ubiquitin conjugation cascade is hierarchical: eukaryotic genomes
encode a single or at most a few E1s, a moderate number of E2s (approximately 60 in mammals),
and an even greater number of E3s [5, 8].
E3 ubiquitin ligases determine the substrate specificity of the ubiquitylation process, and
are generally categorized into two broad classes: either homologous to E6-associated protein
carboxyl terminus (HECT) domain, or really interesting novel gene (RING) finger domain
containing E3s [5, 8, 9]. The HECT E3 ligases generally have an extensive N-terminal region
that contains the substrate recognition binding domain and an approximately 350 amino acid C-
terminal HECT domain originally identified in the cellular protein, E6-associated protein [10].

3
HECT E3 ligases promote ubiquitylation of substrates by forming a thiol ester intermediate with
ubiquitin, following transfer from an E2 conjugating enzyme, at a conserved cysteine
approximately 35 amino acids from the C-terminus of the HECT domain [5, 11]. In contrast,
RING finger E3 ligases are adaptor proteins possessing the consensus sequence Cys-X2- Cys-X(9-
39)-Cys-X(1-3)-His-X(2-3)-Cys/His-X2-Cys-X(4-48)-Cys-X2-Cys in which the cysteines and
histidines facilitate zinc binding [9]. The RING domain is thought to serve two important
functions to promote ubiquitylation: to recruit E2 conjugating enzymes to the substrate and to act
as cofactors that enhance substrate modification by the E2 [8].
In addition to mediating protein degradation, ubiquitylation regulates multiple cellular
functions, including kinase activation, DNA repair, transcriptional modulation, protein
localization and trafficking [5, 11, 12]. That such diverse functions may be mediated by
ubiquitin conjugation is made possible, in part, by the wide variety of potential ubiquitin
modifications. Substrates can be modified by monoubiquitin, multiple monoubiquitins, and
polyubiquitin chains that can be linked to any of the seven ubiquitin lysyl residues (Lys6-,
Lys11-, Lys27-, Lys29-, Lys33-, Lys48-, Lys63-linked chains), whereby both the linkage type
and chain length can act as functionally distinct signals [12, 13]. For example, it is well
established that Lys48-linked chains promote proteasomal degradation, whereas Lys63-linked
chains and monoubiquitin have non-proteolytic functions.

4
Figure 1.1.1. General overview of the ubiquitin and ubiquitin-like protein conjugation
pathways. (1) There are approximately 10 ubiquitin-like proteins (UBL), of which ubiquitin, the
SUMO family, and NEDD8 are the most commonly studied. All three are synthesized as precursors
that are processed at a conserved C-terminal glycine residue by the hydrolase activity of
deubiquitinating, desumoylating, and deneddylating enzymes exposing a glycine-glycine motif that
serves as the attachment site for target substrates. (2) The exposed C-terminal glycine of
ubiquitin/SUMO/NEDD8 is adenylated by an activating (E1) enzyme in an ATP-dependent manner
and transferred to a cysteine side chain of an E1 via a thiol ester linkage. (3) Activated
ubiquitin/SUMO/NEDD8 is subsequently transferred to a conjugating (E2) enzyme forming another
thiol ester linkage. (4) Lastly, a ligase (E3) transfers ubiquitin/SUMO/NEDD8 to the –amino group
of a substrate lysyl residue resulting in the formation of an isopeptide bond. Covalent modification
of substrates can be reversed by the action of deconjugating enzymes for ubiquitin, SUMO, and
NEDD8.
1.1.2 NEDD8: Ubiquitin-like protein (UBL)
In 1992, Kumar et al. carried out a subtractive cloning screen between a cDNA library from
mouse neural precursor cells and mouse adult mRNA to identify genes involved in development
and differentiation in the central nervous system (CNS) [14]. Ten neural precursor cell-
expressed, developmentally down-regulated (NEDD) genes were identified, including the eighth
gene discovered, NEDD8. Studies subsequently determined NEDD8 to be an ubiquitin-like
protein (UBL) sharing approximately 60% amino acid identity with ubiquitin [15]. Analogous to

5
ubiquitylation, the neddylation enzymatic cascade is composed of an E1 activating enzyme (the
APP-BP1/Uba3 heterodimer), E2 conjugating enzymes (Ubc12 or UBE2F), E3 ligases (RBX1,
MDM2, c-CBL, SCFFBX011
, and DCN1), and deneddylating enzymes (COP9 signalosome and
NEDP1) (see Figure 1.1.1.). To date, all reported NEDD8 E3 ligases are RING finger proteins,
with the exception of DCN-1 [16]. Like ubiquitylation, neddylation results in the formation of
an isopeptide bond linking the terminal carboxyl group of Gly76 of NEDD8 with the -amino
group of a lysyl residue of a receptive substrate [17].
Studies from a number of model organisms have demonstrated essential roles for NEDD8
in cell cycle control, embryogenesis, and viability (reviewed in [18]). In mice, deletion of one of
the components of the NEDD8-activating enzyme (NAE), Uba3, results in lethality at embryonic
day 5.5 due to increased apoptosis in the inner cell mass causing blastocyst death in the
preimplantation stage [19]. Similarly, Nedd8-null D. melanogaster mutants die at the first-instar
larval stage [20]. Interestingly, an intact NEDD8 pathway is required for viability in S. pombe
stains but not in S. cerevisiae [21, 22]. In the plant A. thaliana, NEDD8 conjugating enzyme
homolog, RCE1, mutants have a reduced growth phenotype [23]. RNAi-mediated knockdown of
E1 and E2 NEDD8 homologs in C. elegans caused hypersensitivity to ENU-induced apoptosis in
germ cells [24], and other developmental defects [25, 26]. In summary, loss of NEDD8 function
results in cell cycle defects, increased cell death and development defects in a variety of model
organisms.
The most commonly studied substrates of NEDD8 are the Cullin family of proteins,
which are involved in the assembly of a class of Cullin-RING finger E3 ubiquitin ligase
complexes (CRLs). NEDD8 conjugation to Cullins increases the ubiquitin ligase activity of
CRLs by promoting their binding to ubiquitin E2 conjugating enzymes [27]. Additionally, since

6
2004, a number of studies have demonstrated that NEDD8, like ubiquitin, modifies a variety of
substrates having an array of biological effects.
1.1.3 The role of NEDD8 in cancer
The initial screen that discovered NEDD8 also identified the proto-oncogenes, NEDD4 and
NEDD9 [28, 29]. Interestingly, a role for NEDD8 in cancer is supported by a number of
findings. First, as detailed above, a number of model organisms have demonstrated biological
roles for NEDD8 in processes relevant to tumorgenesis including cell proliferation and viability.
Second, the Cullin family, which are the best characterized substrates of NEDD8, generally have
roles regulating cell cycle transitions. Third, the overexpression and/or amplification of Cullins
Cul4A, Cul7 and PARC (a Cul7 structurally-related protein) have been observed in human
cancers [30, 31]. Fourth, general elevation in the level of NEDD8 conjugation in oral squamous
cell carcinoma tumor cell lines has been observed, while inhibition of the NEDD8 pathway
decreased proliferation in these cell lines [32]. Fifth, the general inhibitor of the NEDD8
pathway, MLN4924 (Millenium Pharmaceuticals Inc.), targeting the E1 NEDD8-activating
enzyme (NAE) possesses tumor growth inhibiting properties [33]. Finally, many of the newly
discovered substrates of NEDD8 are established tumor suppressor or oncoproteins, including
pVHL, p53, MDM2, BCA3 and EGFR (reviewed in [34]).

7
1.1.4 p53: The guardian of the genome
p53 was first identified as a SV40 T-antigen binding protein over 25 years ago, and was
originally thought to be an oncogene [35, 36]. However, in the late 1980s it became apparent
that p53 oncogenic functions were attributable to the study of mutant-derived clones [37]. Now
with over 50,000 published p53 studies to date, our understanding of the role of p53 as a tumor
suppressor derived from in vitro studies, model organisms, and human patient data, has greatly
improved. p53 is a sequence-specific DNA-binding transcription factor that plays a central role
in the response to oncogenic stimuli (such as oncogene activation) and cytotoxic stress (such as
DNA damage, chemotherapy treatment, hypoxia, growth factor and nucleotide depletion) by
initiating cell-cycle arrest, senescence or apoptosis. p53 is able to carry out these functions by
inducing transcription of genes that regulate cell cycle arrest (p21, 14-3-3ζ, GADD45), the
intrinsic apoptotic pathway (BAX, PUMA, Noxa), and the extrinsic apoptotic pathway (Fas,
PERP, DR4/5, PIDD) (reviewed in [38]). Furthermore, p53 is able to regulate apoptosis via
transcription-independent mechanisms. Studies have shown cytosolic p53 forms a complex with
the anti-apoptotic Bcl-2 family protein, Bcl-xL [39]. In response to cytotoxic stress, p53-induced
PUMA displaces p53 from Bcl-xL allowing p53 to directly activate BAX and Bak to promote
mitochondrial permeabilization and cytochome c release [40]. Since p53 has a higher affinity for
Bcl-xL compared to Bak [41], cytoplasmic p53 has been suggested to mediate apoptosis via
sensitizing mechanisms due to engagement of the anti-apoptotic Bcl2 proteins, in addition to
directly activating the pro-apoptotic Bcl2 family members by processes that are not completely
understood (reviewed in [42]). As mentioned above, p53 can induce a number of responses to
cytotoxic stress, such as cell cycle arrest, senescence and apoptosis. The factors that determine

8
the ultimate p53-induced cell fate are complex and remain an important area of p53 research.
Studies to date have shown that p53 interacting proteins, chromatin binding factors, the
respective cell-type and tissue, and the intensity and type of stimuli all play roles in mediating
the outcome of the p53 response (reviewed in [43]).
In his 1992 review paper in Nature, Dr. David Lane proposed a model that has been well
supported to date, stating p53 acts as the ―guardian of the genome‖ whereby p53 accumulates
when DNA is damaged, arresting cells to allow for repair, or inducing apoptosis in the case that
the damage is irreversible [44]. p53 plays a vital role in tumor suppression, due to the fact loss
of its function allows for the cancer enabling characteristic of genomic instability required to
transform the physiology of a normal cell to that of a cancer cell [45]. p53 is thought to be
inactivated in virtually all cancers either by direct mutation, occurring in over 50% of human
cancers, or indirectly, though alterations in pathways that impinge upon p53 function [46]. 74%
of p53 mutations are point mutations creating missense codons, which are present predominantly
in the DNA-binding domain (95%) [47]. In the early 1990s, Malkin et al. demonstrated that
germline mutations in the p53 gene constitutes the genetic defect underlying Li-Fraumeni
syndrome; a rare autosomal-dominant cancer predisposition syndrome proposed by Drs. Li and
Fraumeni in 1969 [48]. Given the importance of p53 in the DNA damage pathway, one would
expect p53 status would serve as a good indicator of patient response to chemotherapy treatment.
Surprisingly, studies revealed inconsistent results regarding p53 mutation status and
chemotherapy responsiveness and patient survival. This is due, in part, to the use of
immunohistochemistry (IHC) to detect p53 mutations, chosen because mutations generally
increase p53 protein expression; however, this is not always the case. Recent studies employing
more rigorous gene sequencing methods to determine p53 status have, for the most part, reported

9
an association between p53 mutation and poor drug response as well as poor overall and disease-
free survival (reviewed in [47]).
Studies with mice have been important in elucidating the role of p53 in tumorigenesis and
development. In 1992, Donehower et al. were the first to report that nearly 75% of p53-/- mice
developed tumors before 6 months of age. The majority of these mice developed malignant
lymphomas; however, sarcomas were also observed [49]. No overt developmental defects were
originally reported, however, there was some evidence of immunological defects in these p53-/-
mice. Lowe et al. also demonstrated that p53-/- thymocytes were resistant to lethal effects of
ionizing radiation, thereby confirming a role for p53 in DNA damage-induced cell death [50].
Subsequent studies reported that approximately 25% of p53-/- embryos developed exencephaly,
exhibiting an inappropriate neural outgrowth and a failure of neural tube closure in the midbrain
[51]. More recent studies reported that p53-/- female mice have defects in reproduction due to
implantation failure attributed to the lack of expression of a p53-target gene, LIF [52]. More
sophisticated p53 mouse models of cancer, such as mice with p53 point mutations commonly
observed in Li-Fraumeni patients, develop a wider spectrum of tumors with a higher frequency
of metastasis, more closely resembling the human condition than p53-null mice [53, 54].
Importantly, a number of studies demonstrated that the restoration of p53 function in p53-null
mice, using a number of sophisticated approaches, can lead to tumor regression by inducing
apoptosis, differentiation and senescence thus confirming the therapeutic benefit of reactivating
p53 as a possible cancer treatment [55-57].

10
1.1.5 Structure and function of the p53 family
In the late 1990s, two additional p53 family members were discovered. In 1997, p73 was
serendipitously found in a hybridization screen for IRS-1 binding domain containing proteins,
and a year later, p63 was identified [58, 59]. Both p63 and p73 share approximately 25%, 60%
and 35% amino acid identity with p53 in the N-terminal transactivation domain (TAD), central
DNA-binding domain (DBD), and C-terminal oligomerization domain (OD), respectively. In
addition, certain p63 and p73 isoforms have additional domains not found in p53 (see Figure
1.1.2). For example, both p63 and p73 α isoforms have a sterile alpha motif (SAM), which
generally functions to mediate protein-protein interactions. Interestingly, p63 and p73 genes
give rise to multiple mRNAs that, upon translation, yield many different protein isoforms (see
Figure 1.1.3). These variant proteins contain different domains as a result of alternative
splicing, alternative promoter usage, and alternative initiation of translation (reviewed in [60]).
Seven C-terminal p73 variants have been described that are generated by alternative splicing (α,
β, γ, , ε, δ, and ε) [61-63] (Figure 1.1.3B). In addition, the p73 gene encodes isoforms with five
distinct N-termini including the full-length TAp73, and four N-terminally truncated variants,
collectively termed ΔTAp73 or ΔNp73, that lack the TAD. The N-terminally truncated isoforms
are generated as a result of transcription from an alternative promoter within intron 3 (ΔNp73),
translation from an alternative initiation site (ΔN’p73), and by alternative pre-mRNA splicing of
exons 2 and or 3 (ΔEx2p73 and ΔEx2/3p73) [63-65]. In theory, p73 pre-mRNA can be
processed to yield more than 30 different mature mRNAs encoding over 30 proteins. Similar to
p73, p63 encodes distinct N-terminal isoforms that include the full-length TAp63 and the N-
terminally truncated ΔNp63 isoforms, and three C-terminal variants (α, β, and γ) due to

11
alternative pre-mRNA processing (Figure 1.1.3C) [60]. It was long believed that, in contrast to
p63 and p73, the p53 gene encoded just one predominant mRNA resulting in a single protein.
However, studies clearly demonstrate the existence of multiple p53 N-terminal and C-terminal
isoforms and their function and role in tumorigenesis represents an interesting new area of p53
research (reviewed in [66]) (see Figure 1.3A for specific details).
Figure 1.1.2. Structure of the p73 isoforms. The C-terminal splicing patterns generating p73 , ,
, , , and isoforms and the N-terminal TA and N isoforms are shown. Full-length TAp73
isoforms have pro-apoptotic properties, whereas the N-terminally truncated Np73 isoforms, which
lack the TAD due to the use of a promoter within intron 3 and alternative N-terminal mRNA splicing,
have anti-apoptotic properties. TAD, transactivation domain; DBD, DNA-binding domain; OD,
oligomerization domain; PY motifs, proline-rich motif; SAM, sterile alpha motif.
12
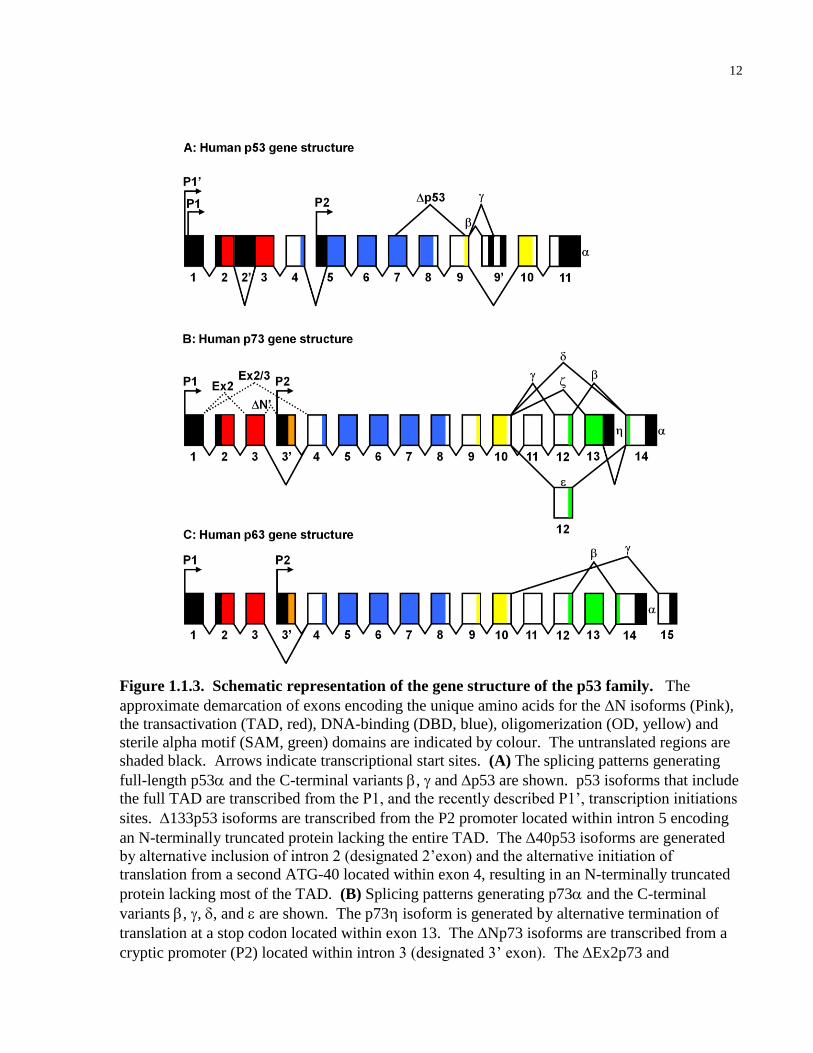
Figure 1.1.3. Schematic representation of the gene structure of the p53 family. The
approximate demarcation of exons encoding the unique amino acids for the N isoforms (Pink),
the transactivation (TAD, red), DNA-binding (DBD, blue), oligomerization (OD, yellow) and
sterile alpha motif (SAM, green) domains are indicated by colour. The untranslated regions are
shaded black. Arrows indicate transcriptional start sites. (A) The splicing patterns generating
full-length p53 and the C-terminal variants , and p53 are shown. p53 isoforms that include
the full TAD are transcribed from the P1, and the recently described P1’, transcription initiations
sites. 133p53 isoforms are transcribed from the P2 promoter located within intron 5 encoding
an N-terminally truncated protein lacking the entire TAD. The 40p53 isoforms are generated
by alternative inclusion of intron 2 (designated 2’exon) and the alternative initiation of
translation from a second ATG-40 located within exon 4, resulting in an N-terminally truncated
protein lacking most of the TAD. (B) Splicing patterns generating p73 and the C-terminal
variants , , , and are shown. The p73 isoform is generated by alternative termination of
translation at a stop codon located within exon 13. The Np73 isoforms are transcribed from a
cryptic promoter (P2) located within intron 3 (designated 3’ exon). The Ex2p73 and

13
Ex2/3p73 isoforms are generated by the indicated alternative splicing. The N’p73 isoforms
are generated from the P1 promoter, however, alternative inclusion of intron 3 (designated 3’
exon) allows for initiation of translation within the 3’ exon producing a protein indistinguishable
from Np73. (C) The splicing patterns generating p63, , and are shown. The Np63
isoforms are transcribed from a cryptic promoter (P2) located within intron 3 (designated exon
3’). Exon size and approximate contribution of exons to the indicated functional domains are not
to scale.
1.1.6 p63 and p73: Dual tumor suppressor and oncogenic
functions
p73 was originally thought to be an important tumor suppressor in neuroblastoma due to its
chromosomal location on 1p36, a region frequently deleted in neuroblastoma and other cancers
[58]. However, it has become apparent that p73, as well p63, do not behave as prototypical
tumor suppressors in human cancers. The majority of studies have generally reported opposing
functions for TA and ΔN isoforms of p63 and p73. While both the TA and ΔN p63 and p73
isoforms are able to bind p53 DNA-binding sites [59, 67-69], generally, only TA isoforms
possessing the TAD are able to transactivate promoters of p53-target genes to induce cell cycle
arrest and apoptosis [59, 65, 68, 70, 71]. The ΔN isoforms appear to act as dominant-negative
inhibitors of TA isoforms of the p53 family, by forming hetero-oligomers with them that
generate abortive transcriptional complexes [59, 63, 65, 72, 73] and by competing directly for
p53 DNA-binding sites [59, 67-69]. As illustrated below, human and mouse studies generally
support a paradigm in which the balance between the various ―tumor suppressor‖ pro-apoptotic
TA, and ―oncogenic‖ anti-apoptotic N, p53 family isoforms determine whether the predominant
signalling pathway leads to apoptosis or survival in both development and tumorigenesis.

14
1.1.7 p63 and p73: Roles in development
In contrast to p53-null mice, which had an obvious cancer phenotype and less profound
developmental defects, the original p63 and p73 knockout mice had striking ectodermal and
neuronal developmental abnormalities, respectively, and a less discernible cancer phenotype [64,
74]. p73-/- mice displayed neurological abnormalities due to hippocampal dysgenesis, olfactory
neuron defects [64], sympathetic neuron loss and cortical thinning [73, 75]. This result initially
appeared counterintuitive in the context of previous reports describing a pro-apoptotic role for
p53 in the developmental programmed cell death of sympathetic neurons downstream of TrkA
and p75NTR signalling (reviewed in [76]). However, detailed analysis determined ΔNp73 is the
predominant isoform expressed in the fetal murine nervous system, and thus, loss of this ―anti-
apoptotic‖ p73 isoform led to enhanced apoptosis in cortical, as well as sympathetic, neurons
[73]. The mechanism whereby ΔNp73 promotes survival is likely a combination of inactivation
of the full–length pro-apoptotic p53 family proteins (TAp63, TAp73 and p53) as well as
regulation of mitochondrial pathways (reviewed in [77]). It should be noted that p73-/- mice also
displayed pheromonal and inflammatory defects, however, the role of p73 in these processes is
less well understood. In contrast, the full-length TA, and not ΔN, isoforms of p63 are
predominantly expressed in the developing murine nervous system. TAp63γ was shown to be an
essential pro-apoptotic protein in sympathetic neurons functioning alone, and in collaboration
with p53, further highlighting the importance of isoform-specific expression during development
[78]. In addition, p63-/- mice have significant limb and craniofacial malformations, as well as
failure of skin and other epithelial tissue development. Interestingly, germline mutations in p63
have been reported in patients with ectodermal dysplasia syndromes that present with symptoms

15
including cleft palate (reviewed in [79]). It is important to note that the first p63 and p73
knockout mice were designed to lack all TA and N isoforms.
1.1.8 p63 and p73: Mouse models of cancer
The p53 family has distinct roles in tumorigenesis. Unlike p53, which is mutated in over 50% of
all human cancers and thought to be inactivated in the remainder, p63 and p73 mutations are
rarely observed in cancers (reviewed in [80]). In addition, unlike p53-/- mice and mice
engineered to express tumor-derived p53 mutant proteins, p63-/- and p73-/- mice are not tumor
prone [64, 74]. However, mounting evidence suggests that the relative expression and stability
of the different N-terminal isoforms of p63 and p73 play a role in tumorigenesis, which the
germline knockout mice described above could not specifically address. To examine this
possibility, a number of groups have generated TAp63- and TAp73-specific knockout mice, in
addition to carefully studying aged heterozygous p63+/- and p73+/- mice as well as compound
heterozygous p53/p63/p73 knockout mice. Studies of aged heterozygous mice reported three
important findings that support tumor suppressor roles for p63 and p73 [81]. First, aged p63+/-
and p73+/- mice develop spontaneous tumors and pre-malignant lesions, and loss of the second
allele of p63 and p73 respectively, was demonstrated in several of these tumors. Second, loss of
p63 or p73 cooperates with loss of p53 in tumor development since compound p63+/-; p53+/-
and p73+/-; p53+/- mice develop a different spectrum of tumors than p53+/- mice. Finally, in
comparison to p53+/- mice, compound heterozygous mice for both p53 and p63 or p73 exhibit
both larger tumor burdens and a higher incidence of metastatic lesions. Notably, another p63+/-

16
mouse generated on a different genetic background using an alternative gene targeting strategy
did not develop tumors, but rather demonstrated features of premature aging [82]. Studies of
TAp63-specific knockout mouse revealed a role for TAp63 in DNA damage-induced oocyte
death suggesting that p63 acts as the ―guardian of the female germline‖, while TAp63-specific
deletion in skin-derived progenitor cells induced hyperproliferation and increased genomic
instability, culminating in senescence [83, 84]. Further support for a tumor suppressor role of
TAp63 isoforms has recently been reported as reintroduction of TAp63, β and γ into p63-null
MEFs induced senescence in vitro via a p21-dependent mechanism [85]. TAp63 also inhibited
Ras-driven transformation and tumour formation of p53–/– MEFs in vivo following
subcutaneous injection in nude mice. TAp73-deficient mice provided the most convincing
evidence that p73 functions as a tumor suppressor. 73% of TAp73-/- mice developed
spontaneous tumors, with lung adenocarcinomas being the most frequent cancer, and displayed
hippocampal dysgenesis, infertility, and aging defects [86]. Importantly, the mouse data
described above support numerous studies from human patient data reporting an association
between the relative imbalance of the TA and N isoforms of p63 and p73 and tumor
development and/or progression and poor responsiveness to chemotherapy.
1.1.9 p63 and p73: Alterations in human cancer and role in
chemotherapy response
TAp73 levels are induced by a wide variety of chemotherapeutic agents [87-89], while blocking
TAp73 function promotes cell survival and leads to enhanced chemoresistance [90-92].

17
Conversely, the ―oncogenic‖ N isoforms of p53 family proteins are overexpressed in certain
tumors, and are preferentially degraded in response to chemotherapy [93, 94]. Np73 expression
has been reported to be elevated in a number of human cancers, including breast, ovarian,
hepatocellular, prostate, colon and neuroblastoma [95-98]. In several of the tumors mentioned
above, increased Np73 expression is associated with poor patient prognosis, and this is thought
to be due to the ability of Np73 to inhibit p53 and TAp73, resulting in decreased apoptotic
response and chemoresistance [65, 99-101]. Furthermore, elevated Np63 expression has been
found in primary head and neck squamous cell carcinomas (HNSCC), and other squamous
epithelial malignancies such as cervical, lung and esophageal cancers [102-104]. Np63
overexpression in HNSCC cells promotes survival of these tumor cells via inhibition of TAp73-
dependent apoptosis by both competition for promoter binding and physical interaction with
TAp73 [92]. Conversely, loss of expression of the tumor suppressor–like TAp63 and TAp73
isoforms has been observed in many tumors including leukemias, bladder cancers, mammary
tumors and squamous cell carcinomas (reviewed in [105]). Therefore, understanding the
regulatory mechanisms that differentially modulate TA and N isoform activity and stability are
of particular interest given their therapeutic relevance in human cancers.
1.1.10 Mechanisms of p53 activation in response to DNA
damage
In the early 1990s, a variety of DNA-damaging agents, including X-rays, UV radiation and
chemotherapeutic agents, were found to rapidly increase p53 protein levels. This rapid induction

18
occurred in the absence of significant up-regulation of p53 mRNA, and it appeared that post-
translational stabilization was the primary mechanism mediating this increase in p53 protein
expression [106]. Since this initial observation, numerous studies have reported a number of
mechanisms regulating p53 stability in response to a variety of stimuli, most often via post-
translational modifications involving stress-induced kinases. Kinases reported to mediate p53
phosphorylation events include: ATM, ATR, Chk1, Chk2, CK2, DNA-PK, GSK3β, HIPK2,
JNK, p38 MAPK, and PKC (reviewed in [107]). Reported stress-regulated p53 phosphorylation
sites include: Ser6, Ser9, Ser15, Thr18, Ser20, Ser33, Ser37, Ser46, Thr81, Ser366, Ser376,
Thr377, Ser378, Thr387 and Ser392 (reviewed in [108]). N-terminal phosphorylation is thought
to activate p53 by disrupting binding with the E3 ligase, MDM2 (described in detail below).
Furthermore, in response to DNA damage, p53 is acetylated on numerous lysines by
acetyltransferases p300/CBP and P/CAF, which increases its DNA-binding and transactivation of
target genes [109]. It should be noted that p73 is similarly phosphorylated and acetylated in
response to DNA-damaging agents by a subset of the mediators listed above including Chk1,
HIPK2 and p300/CBP, resulting in activation of p73 [110-112]. Furthermore, the DNA damage-
induced tyrosine kinase c-abl regulates p73, but not p53, by promoting phosphorylation of Tyr99
to induce apoptosis [87].
1.1.11 MDM2 regulation of p53
MDM2 was first identified from a spontaneously transformed murine Balb/c cell line, 3T3DM,
that possesses an average of 25-30 double minutes (DMs), which are small, acentromeric,

19
extrachromosomal nuclear bodies [113]. In the 1980s, Dr. Donna George reasoned that these
DMs contained cellular oncogenes, and initiated a series of studies that identified murine double
minute 2 (mdm2) as the gene conferring the tumorigenic potential in 3T3DM cells [114]. A year
later mdm2 was identified as a p53 interacting protein providing insight into a potential
mechanism of MDM2-mediated tumorigenesis [115]. In 1993, it was demonstrated that human
MDM2, which is located on chromosome 12q13-14, is amplified in over a third of sarcomas,
highlighting an oncogenic role in human cancers [116]. MDM2 amplification has been reported
in approximately 10% of all human cancers examined to date, with the highest incidence
occurring in sarcomas (soft tissue tumors (30%) and osteosarcomas (20%)), and tumors of the
lung (15%), central nervous system (10%), and breast (6%) (reviewed in [46]). MDM2 is also
overexpressed in human tumors by amplification-independent mechanisms, such as increased
transcription and enhanced translation [117-119]. It should be noted that the majority of reported
human tumors with amplified MDM2 possess wild-type p53 suggesting MDM2 amplification
constitutes an alternative mechanism to inactivate p53 during tumorigenesis. Furthermore, small
molecule inhibitors such as Nutlins and RITA that target the interaction between MDM2 and
p53, appear to have clinical promise in the treatment of these patients [120, 121].
A number of in vitro and mouse studies have been essential in elucidating both the
molecular mechanisms and the importance of MDM2 regulation of p53 in tumorigenesis and
development. MDM2 is a RING finger E3 ligase that interacts with the hydrophobic stretch
within the p53 N-terminus, and negatively regulates p53 stability by promoting ubiquitylation of
multiple lysines located in the C-terminal and DNA-binding domains (see Fig 1.1.4, and 1.1.5
and for illustrations) [122-126]. Studies have shown that MDM2 mediates both p53 mono-
ubiquitylation promoting nuclear export and polyubiquitylation promoting degradation [127,

20
128]. The factors that determine the ubiquitin-linkage type promoted by MDM2 include the
relative expression of MDM2 (low levels of MDM2 are thought to mediate mono-ubiquitylation
while high levels mediate polyubiquitylation), and the presence of E4 enzymes (p300 and Y11)
that aid in the polyubiquitylation process [128-130]. Recent studies have also demonstrated that
MDM2 can promote conjugation of NEDD8 to p53, inhibiting its transactivation potential [131].
Other modes of MDM2 inhibition of p53 include MDM2 interaction with the N-terminal TAD of
p53, disrupting its association with co-activators such as p300, and ubiquitin-mediated inhibition
of p53 DNA binding activity [115, 132-134]. Interestingly, MDM2 itself is a p53-target gene,
and as a result MDM2 regulation of p53 activity creates an autoregulatory feedback loop that
allows for evasion of apoptosis in the event that the cell is able to repair damaged DNA induced
by cytotoxic stress [135].
Figure 1.1.4. Structure of MDM2 protein. The domains of the human MDM2 protein are
indicated. Amino acid demarcation indicated in brackets below. p53/p73 BD, p53/p73 binding
domain (18-101); NLS, nuclear localization signal (181-185); NES, nuclear export signal (191–
205); ACIDIC, Acidic domain (237-288); Zn FINGER, Zinc finger (289-331); RING finger
(438-478).

21
Figure 1.1.5. MDM2 promotion of p53 ubiquitylation. See text for details.
1.1.12 MDM2 studies in mice
Deletion of mdm2 in mice results in lethality at embryonic day 5.5, and interestingly, this
phenotype is rescued by concomitant deletion of p53 [136, 137]. mdm2-/-;p53-/- mice appear to
develop normally, suggesting that the biological role of mdm2, in mice, is confined to the
negative regulation of the apoptotic activity of p53. Additional studies employing tissue-specific
deletion of mdm2 have confirmed this notion. First, specific deletion of mdm2 from the CNS of
mice elicited hydranencephaly due to increased apoptosis in neuronal progenitor cells, and this
phenotype was rescued by simultaneous loss of p53 [138, 139]. Second, conditional deletion of
mdm2 from smooth muscle cells of the intestine resulted in severe cell loss and lesions in the
intestinal walls eventually leading to death, which was also completely rescued on a p53-null
background [140]. Lastly, a study employing an inducible p53 model on an mdm2-null
background demonstrated that expressing p53 at near ―endogenous levels‖ in the absence of

22
mdm2, resulted in severe apoptosis in classical radiation-sensitive tissues (bone marrow, thymus,
spleen, small intestine and colon) and cell cycle arrest in radiation-insensitive tissues (brain,
heart, lung, liver, kidney) [141]. These studies are consistent with the idea that the sole
biological role of MDM2 is to negatively regulate p53 function during development and in
terminally differentiated cells, since each of the phenotypes associated with mdm2 loss described
above are rescued by concomitant loss of p53. Importantly, the finding that an mdm2 point
mutant defective for ligase activity was unable to rescue the embryonic lethality of mdm2-null
mice, demonstrated the E3 ligase activity of MDM2 is biologically necessary to inhibit p53
function [142]. Therefore, studying the E3 ligase function of MDM2 is critical to understanding
the MDM2-p53 axis.
1.1.13 MDM2: p53-independent oncogenic functions
Although the data described above demonstrate that the physiological role of MDM2 in mice is
to negatively regulate p53 function, a number of in vitro, mouse and human patient studies
suggest that, when amplified or overexpressed, MDM2 gains oncogenic functions independent of
p53. First, MDM2 has been shown to transform cells in culture, independent of its ability to
inhibit p53 [143]. Second, transgenic mice overexpressing mdm2 in a p53-null background have
a higher incidence of sarcomas than p53-null mice, suggesting that mdm2 can regulate the
function of additional players in sarcoma tumorigenesis [144]. This finding was also supported
by a second study that reported the incidence of sarcomas to be higher in p53-/-;mdm2+/- than in
p53-/-;mdm2-/- mice [145]. Third, a small, but consistently reported subset of soft tissue
sarcomas and bladder cancer patients present with both p53 mutation and MDM2 amplification,

23
and individuals with both aberrations have a worse prognosis than patients with a singular defect
in MDM2 or p53 [146-148]. Nevertheless, the fact that the majority of human tumors with
amplified MDM2 possess wild-type p53 supports the idea that MDM2 negatively regulates p53
to induce tumorigenesis. In summary, the data described above are consistent with a model in
which the physiological role of MDM2 is to negatively regulate p53; however, when amplified
or overexpressed, MDM2 promotes tumorigenesis through p53-dependent and p53-independent
pathways.
1.1.14 MDM2 destabilization: Mechanism of p53 activation in
response to DNA damage
The widely accepted model for p53 activation in response to DNA damage involves the
activation of stress-induced kinases that phosphorylate the p53 N-terminus disrupting MDM2
interaction, enabling p53 transactivation of genes involved in DNA repair, cell cycle arrest or
apoptosis. However, recent biochemical and in vivo studies have suggested MDM2 protein
destabilization also plays a vital role in the activation of p53 in response to DNA damage, and
mechanisms that mediate this process are increasingly being described. Early evidence that
MDM2 may be directly involved in the response to DNA damage came from the observation that
ATM-mediated phosphorylation of MDM2 preceded p53 induction [149]. Dr. Geoffrey Wahl’s
group subsequently demonstrated this phosphorylation event destabilized MDM2 in response to
radiomimetic drug treatment [150]. Interestingly, this study observed that treating cells with a
proteasome inhibitor preventing degradation of MDM2 but permitting N-terminal

24
phosphorylation of p53 in response to DNA damage abrogated the activation of p53. While
genetic loss of mdm2 promotes a robust p53 apoptotic response in mice, transgenic mice
expressing p53 point mutations in N-terminal phosphorylation sites targeted by stress-induced
kinases have mild cell cycle arrest and apoptotic defects and low incidence of tumorigenesis
[151-155]. Considering the above findings, it appears that modulation of MDM2 levels is a
biologically important component of the DNA damage response [46]. Since these initial
findings, studies have emerged elucidating molecular mechanisms regulating MDM2 stability in
response to chemotherapy and UV exposure. For example, studies have shown that ATM-
mediated phosphorylation of MDM2 abrogates MDM2 binding to the deubiquitylating enzyme,
HAUSP, resulting in increased ubiquitylation and degradation of MDM2 [156]. MDM2
interacting proteins, such as DAXX and RASSF1A, have been reported to modulate the HAUSP-
MDM2 interaction in response to DNA damage [157, 158]. In response to UV exposure, the
SUMO-specific protease, SUSP4, desumoylates MDM2 promoting MDM2 autoubiquitylation
and subsequent degradation [159]. As illustrated above, understanding the pathways that
modulate MDM2 stability may reveal important novel regulators of p53 in response to DNA
damage, in tumorigenesis and development.
1.1.15 Regulation of the p53 family by ubiquitin and ubiquitin-
like modifications
In addition to MDM2, a number of other E3 ubiquitin ligases target p53 for degradation, which
include but are not limited to, the RING finger E3s, Pirh2 and COP1, and the HECT E3, ARF-

25
BP1 (reviewed in [160]). Interestingly, both Pirh2 and COP1 are themselves p53-target genes,
and as a result participate in a negative autoregulatory feedback loop analogous to that of
MDM2. Studies have also observed COP1 and Pirh2 overexpression in breast and ovarian
adenocarcinoma tissues [161] and lung tumor samples, respectively [162]. However, the role of
these more recently described E3 ligases in the regulation of p53 in development has not yet
been reported. In contrast to p53, the regulation of p63 and p73 stability via the ubiquitin-
proteasomal pathway is less well characterized. One important finding is that the stability of the
pro-apoptotic TA, and anti-apoptotic ΔN, p63 and p73 isoforms are differentially regulated by
ubiquitylation in response to chemotherapy. Maisse et al. demonstrated that ΔNp73, but not
TAp73, is rapidly degraded in response to DNA-damaging agents in a proteasomal-dependent
manner [93]. Westfall et al. also observed increased ubiquitylation, and decreased total ΔNp63α
protein levels, in a response to ultraviolet radiation (UV) and paclitaxel treatment [94].
However, elucidation of the molecular mechanisms that mediate this important therapeutically
relevant process have yet to be described. Initial studies examining p73 stability naturally
investigated the best characterized p53 E3 ligase, MDM2. The three residues Phe19, Trp23, and
Leu26 in the p53 N-terminus that directly contact MDM2 are conserved in p63 and p73 [58,
122, 163, 164]. Surprisingly, in contrast to the well characterized relationship with p53, MDM2
does not promote degradation of p73. Instead MDM2 overexpression results in p73 stabilization
[165-167].
Studies have also demonstrated that the p53 family is modified by the UBL, SUMO-1.
Two studies independently confirmed that p53 is covalently modified by SUMO-1 at Lys386 in
the C-terminus, resulting in increased transactivation function [168, 169]. Consistent with these
observations, Muller et al. reported that the p53 K386R mutant, which is defective for SUMO-1

26
conjugation, had slightly impaired apoptotic activity [170]. Three members of the PIAS family
of E3 SUMO ligases, PIAS1, PIASx, and PIASy were later found to interact with p53, and both
PIAS1 and PIASx were reported to promote sumoylation of p53 [171-174]. However, there
have been conflicting reports as to the functional consequence of p53 sumoylation and the role of
the PIAS family in the regulation of p53 (reviewed in [175]). Like p53, both p63 and p73 are
sumoylated. PIAS1 promotes sumoylation of the p73 isoform resulting in its targeting to the
nuclear matrix [176, 177]. PIAS1 was also shown to stabilize p73, but this effect was
independent of its sumoylation function. Interestingly, PIAS1 has been shown to inhibit p73
transactivation of p21 in a sumoylation-dependent manner, thus regulating the G1/S phase
transition of the cell cycle [177]. Sumoylation of p63 has been shown to destabilize p63 [178]
and appears to inhibit its transactivation activity, including target genes involved in cell
differentiation and limb morphogenesis [179, 180].
1.2 SIGNIFICANCE
Chapter 2: MDM2-mediated NEDD8 modification of TAp73 regulates its
transactivation function.
MDM2 has p53-independent oncogenic functions, which may include the regulation of
additional p53 family members. MDM2 is known to regulate p73 function, but in contrast to
p53, does not promote its polyubiquitylation and degradation. In addition to promoting
ubiquitylation, MDM2 was shown to promote p53 neddylation. In chapter 2, we characterize the

27
novel process of MDM2-mediated neddylation of TAp73, and identify the MDM2-TAp73
interaction as a therapeutically relevant target for cancer treatment.
Chapter 3: Chemotherapy induces NEDP1-mediated destabilization of
MDM2
MDM2 destabilization plays an important role in activating p53 in response to DNA damage;
however, the molecular mechanisms that mediate these processes are not completely understood.
MDM2 is a known substrate of NEDD8, however, the pathways that control MDM2 neddylation
and the biological stimuli that regulate these processes, are unknown. In chapter 3, we
investigate the components that regulate MDM2 neddylation, and identify a novel mechanism of
p53 activation involving modulation of MDM2 stability by a NEDD8-specific protease, NEDP1,
in response to chemotherapy treatment.

28
CHAPTER 2:
MDM2-mediated NEDD8 modification of TAp73 regulates its transactivation function
This work has been published:
Watson IR, Blanch A, Lin DC, Ohh M, Irwin MS. Mdm2-mediated NEDD8 modification of
TAp73 regulates its transactivation function. J Biol Chem. 2006 Nov 10;281(45):34096-103.
Epub 2006 Sep 14.
2.1 HYPOTHESIS AND RATIONALE
The p73 gene encodes for two functionally distinct N-terminal isoforms: full-length pro-
apoptotic TAp73 that can transactivate known p53-target genes and the N-terminally truncated
anti-apoptotic ΔNp73 proteins that lack the transactivation domain (TAD) and act as dominant-
negative inhibitors for all full-length TA isoforms of the p53 family [105]. Although p73 is not
commonly mutated in human cancer, accumulating evidence suggests the relative expression of
TA and ΔN p73 isoforms play a role in tumorigenesis. Np73 expression is elevated in a
number of human cancers and poor prognosis has been observed in tumors with high
Np73:TAp73 ratios [65, 99-101]. p53 is an important determinant of chemotherapy response in
human tumors, and evidence of a similar role for p73 was demonstrated by the fact that many
chemotherapy drugs induce TAp73 specifically, and interference with TAp73 activity leads to
chemoresistance [90-92]. Conversely, Np73 specific anti-sense treatment leads to enhanced
sensitivity to the chemotherapeutic agents [65]. Furthermore, 73% of TAp73-specific knockout

29
mice developed spontaneous tumors implicating a tumor suppressive role for TAp73 [86]. Since
the relative balance between the different N-terminal isoforms appears to be an important
determinant of apoptosis, tumorigenesis and chemosensitivity, elucidating regulatory
mechanisms, such as protein interactions and post-translational modifications that differentially
regulate TA and N isoforms have important therapeutic implications.
MDM2 is a RING finger E3 ligase that promotes p53 ubiquitylation and degradation and
is amplified in approximately 10% of human cancers. There is evidence that amplified MDM2
has p53-independent oncogenic functions and p73 may be one such target (reviewed in [181]).
To date, the role of MDM2-mediated regulation of p73 is complex, but remains poorly
characterized. MDM2 interacts with TAp73, but does not promote polyubiquitylation or
degradation, and instead stabilizes TAp73 [166, 167]. Despite this stabilization, MDM2 was
shown to inhibit TAp73-mediated transcription of target genes and apoptosis [167]. The
molecular mechanisms governing MDM2-mediated stabilization and transcriptional activity
remain for the most part unclear. Furthermore, regulation of p53 via post-translation
modifications that modulate activity, stability, and localization have been studied considerably,
which has led to the development of targeted therapy in cancer treatment, such as the small
molecule inhibitors Nutlins and RITA, which inhibit the MDM2-p53 interaction [120, 121] . In
contrast, post-translational modifications regulating p73 activity are less well characterized.
Studies have shown that in addition to promoting p53 ubiquitylation, MDM2 also promotes
conjugation of NEDD8 to p53 [131]. The NEDD8 pathway plays an essential biological role in
cell cycle regulation, viability and embryogenesis demonstrated in a number of model organisms
(reviewed in [18]). In light of the fact that MDM2 has been shown to bind p73 but does not
promote its polyubiquitylation and degradation, we hypothesized that MDM2 may promote

30
NEDD8 modification of p73 to regulate its function. Furthermore, since MDM2 interacts with
the p73 N-terminus, MDM2 may differentially regulate the pro-apoptotic TA and anti-apoptotic
∆N isoforms.
2.2 RESULTS AND DISCUSSION
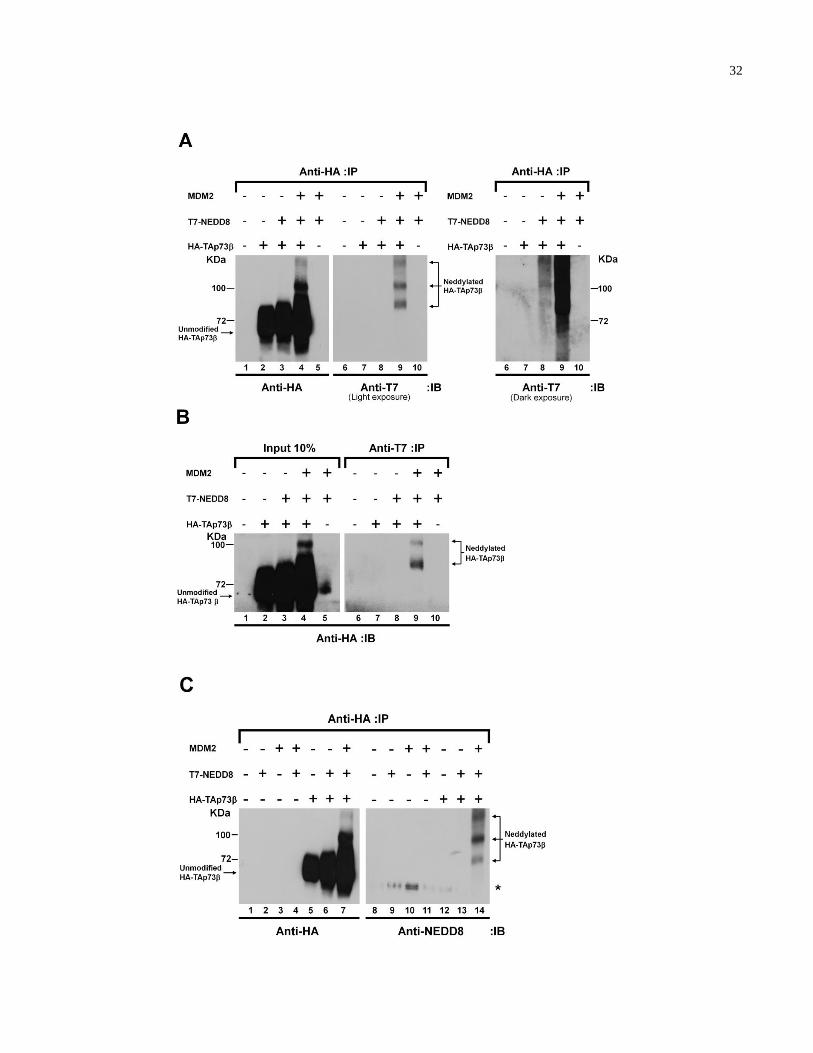
2.2.1 TAp73 is modified by NEDD8 via MDM2
MDM2 promotes NEDD8 conjugation of p53 to negatively regulate its transcriptional activity
[131]. We asked whether p73 is subjected to a similar NEDD8-dependent regulation mediated
by MDM2. HEK293 cells were transiently transfected with plasmids encoding HA-TAp73 in
combination with plasmids encoding T7-NEDD8 and human MDM2. Cells were lysed,
immunoprecipitated with anti-HA antibody, bound proteins resolved on SDS-PAGE, and
visualized by immunoblotting with anti-HA (Fig. 2.1A, left panel) and anti-T7 (right panel)
antibodies. Slower co-migrating HA-TAp73 proteins were readily detectable in cells
transfected with plasmids encoding HA-TAp73, T7-NEDD8, and MDM2 (Fig. 2.1A, compare
lanes 4 and 9). These modified HA-TAp73 proteins were absent in cells transfected with
plasmids encoding HA-TAp73 alone (Fig. 2.1A, lanes 2 and 7). As expected, we did not
observe slower migrating bands with co-expression of MDM2 and T7-NEDD8 in the absence of
HA-TAp73 demonstrating these were not non-specific bands (Fig. 2.1A, lanes 5 and 10).
Thus, the slower migrating forms of TAp73 contain both HA and T7 epitopes, suggesting that
T7-NEDD8 covalently conjugates to HA-TAp73. Notably, in the absence of exogenous

31
MDM2, ectopic co-expression of HA-TAp73 and T7-NEDD8 generated similar slower co-
migrating HA-TAp73 (Fig. 2.1A, compare lanes 8 and 9 under dark exposure). In a
reciprocal experiment, anti-T7 immunoprecipitation was performed followed by anti-HA
Western blot analysis (Fig. 2.1B, right panel). Again, slower migrating TAp73 species
containing both HA and T7 epitopes were observed, supporting the notion that HA-TAp73 is
modified by T7-NEDD8 (Fig. 2.1B, lanes 4 and 9). Similar observations were made in the
context of other TAp73 C-terminal isoforms (see Discussion).
To further confirm that the modified TAp73 is conjugated by NEDD8, HEK293 cells
were transiently transfected with plasmids encoding HA-TAp73 in combination with T7-
NEDD8 and MDM2. Cells were then lysed, immunoprecipitated with anti-HA antibody, bound
proteins separated on SDS-PAGE, and visualized by immunoblotting with anti-HA (Fig. 2.1C,
left panel) and anti-NEDD8 (right panel) antibodies. Similar slower migrating HA-TAp73
species containing both HA and NEDD8 epitopes were observed (Fig. 2.1C, lanes 7 and 14;
compare to Fig. 2.1A). Interestingly, when performing the same experiment described above
but immunoblotting with anti-NEDD8 and anti-ubiquitin antibodies, we could observe slower
migrating HA-TAp73 species containing NEDD8 but not ubiquitin epitopes (Appendix,
Fig.A.1). Taken together, these results strongly suggest that MDM2 promotes TAp73 covalent
modification by NEDD8.
32

33
Figure 2.1. TAp73 is modified by NEDD8. (A/B/C) HEK293 cells were transfected with
plasmids encoding HA-TAp73, T7-NEDD8, and human MDM2. The cells were lysed and
immunoprecipitated with the indicated antibodies. Bound proteins were resolved by SDS-PAGE
and immunoblotted with the indicated antibodies. IP, immunoprecipitated; IB, immunoblotted; *
represents nonspecific protein.

34
2.2.2 NEDP1 deneddylates modified TAp73β
NEDP1 is a cysteine protease able to cleave NEDD8 modified substrates [182-184]. In addition,
NEDP1, but not other UBL deconjugating enzymes (e.g., SUMO-deconjugating enzyme, SSP3),
was shown to specifically deneddylate NEDD8-modified p53 [131]. In a complementary
experiment, we asked whether the physiologically relevant NEDD8-deconjugating enzyme,
NEDP1, could curtail NEDD8-modification of TAp73. HEK293 cells were transfected with
plasmids encoding HA-TAp73, MDM2, T7-NEDD8 or conjugation-defective T7-NEDD8ΔGG,
and NEDP1 or inactive NEDP1(C163A) mutant (Fig. 2.2). As previously shown in Fig. 2.1,
HA-TAp73β was modified by T7-NEDD8 in the presence of MDM2 (Fig. 2.2, lane 3).
However, co-expression of NEDP1, but not the non-functional NEDP1(C163A) mutant,
dramatically reduced the level of T7-NEDD8-modified HA-TAp73 (Fig. 2.2, lanes 4 and 5).
In addition, ectopic expression of T7-NEDD8GG, which lacks the essential C-terminal glycine-
glycine residues required for the formation of the isopeptide bond, resulted in the loss of the
modified HA-TAp73 (Fig. 2.2, lane 6). Taken together, these results suggest TAp73 is
specifically modified by NEDD8 and deconjugated by NEDP1.
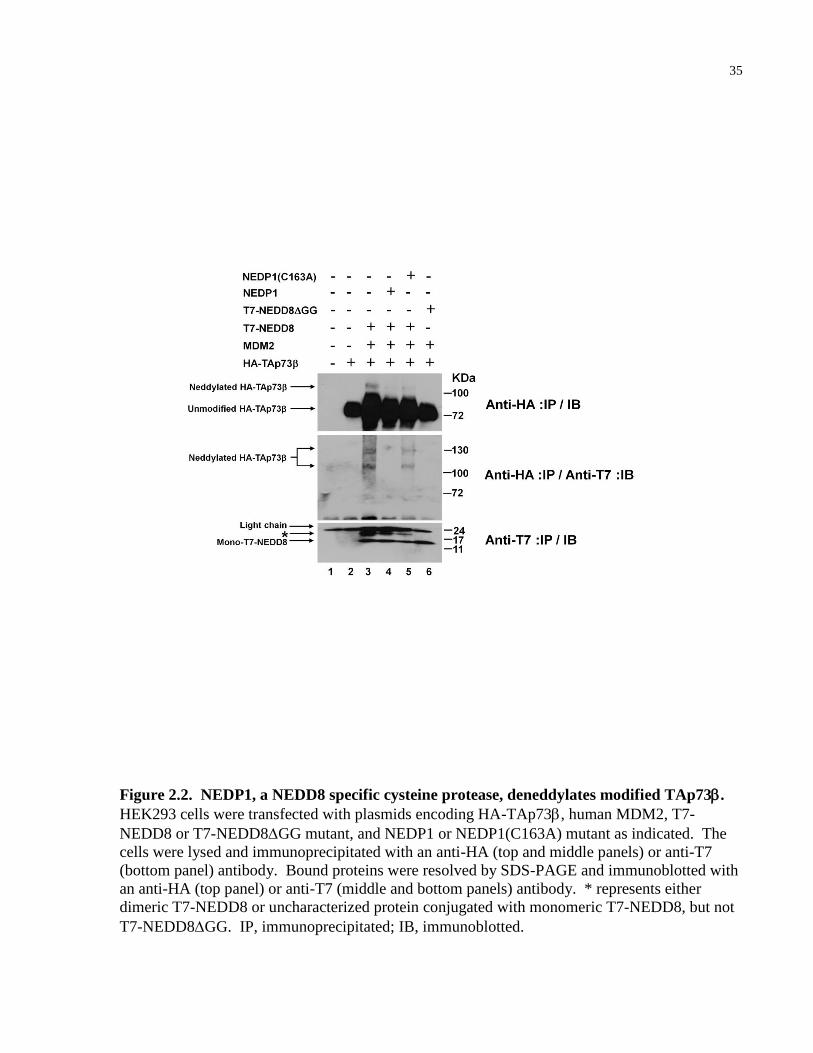
35
Figure 2.2. NEDP1, a NEDD8 specific cysteine protease, deneddylates modified TAp73.
HEK293 cells were transfected with plasmids encoding HA-TAp73, human MDM2, T7-
NEDD8 or T7-NEDD8GG mutant, and NEDP1 or NEDP1(C163A) mutant as indicated. The
cells were lysed and immunoprecipitated with an anti-HA (top and middle panels) or anti-T7
(bottom panel) antibody. Bound proteins were resolved by SDS-PAGE and immunoblotted with
an anti-HA (top panel) or anti-T7 (middle and bottom panels) antibody. * represents either
dimeric T7-NEDD8 or uncharacterized protein conjugated with monomeric T7-NEDD8, but not
T7-NEDD8GG. IP, immunoprecipitated; IB, immunoblotted.

36
2.2.3 Np73 does not undergo MDM2-mediated neddylation
MDM2 binds to the hydrophobic stretch (16-Gln-Pro-Thr-Phe-Ser-Asp-Tyr-Trp-Lys-Leu-Leu-
Pro-27) within the N-terminus of p53. In particular, Phe19, Trp23, and Leu26 residues make
direct contact with and are indispensable for binding to MDM2 [122, 163]. These critical
MDM2-binding residues are conserved in the N-terminus of TA isoforms of p73 [185].
Therefore, we asked whether the Np73 that lacks the MDM2-binding motif is capable of
being modified by NEDD8. HEK293 cells were transiently transfected with plasmids encoding
TAp73 or Np73 in combination with T7-NEDD8 and MDM2. Cells were lysed,
immunoprecipitated with an anti-p73 (ER15) antibody and immunoblotted with anti-p73
(GC15) (Fig. 2.3, left panel) or anti-NEDD8 (right panel) antibodies. While TAp73 was
neddylated (Fig. 2.3, lanes 4 and 11), there was no detectable NEDD8 conjugation to the
Np73 isoform (Fig. 2.3, lanes 7 and 14). These results suggest that MDM2-mediated
neddylation of TAp73 is dependent on its N-terminus.
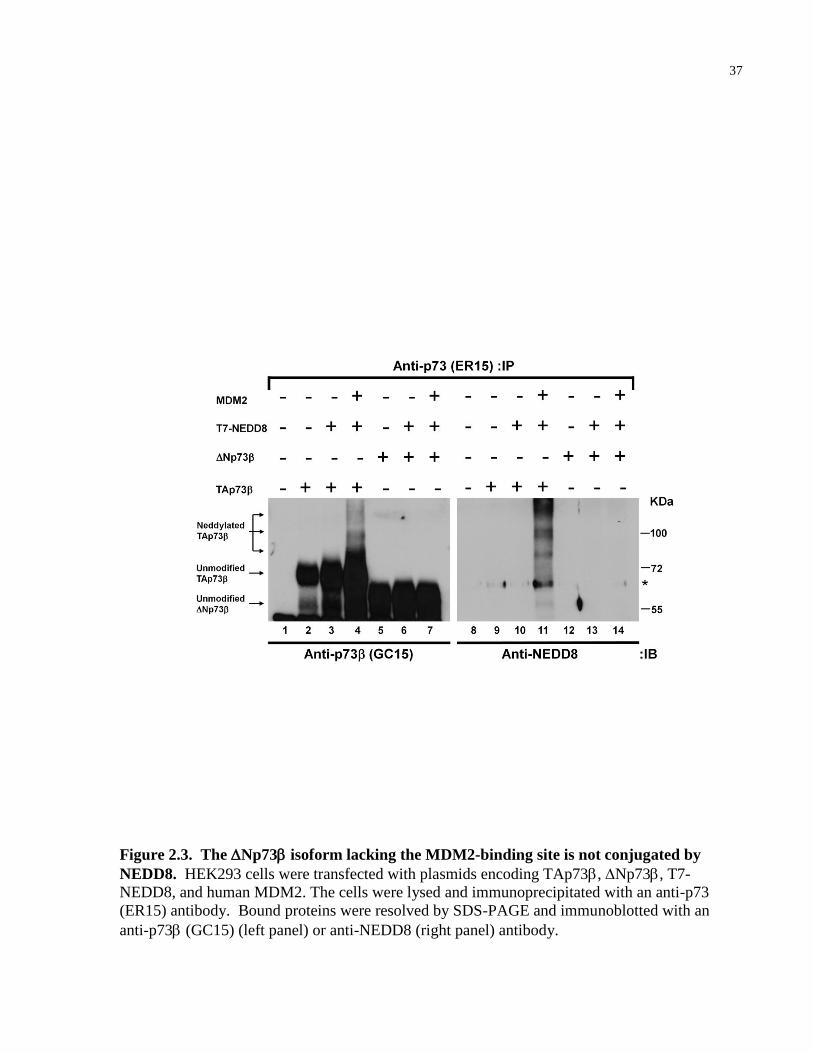
37
Figure 2.3. The Np73 isoform lacking the MDM2-binding site is not conjugated by
NEDD8. HEK293 cells were transfected with plasmids encoding TAp73, Np73, T7-
NEDD8, and human MDM2. The cells were lysed and immunoprecipitated with an anti-p73
(ER15) antibody. Bound proteins were resolved by SDS-PAGE and immunoblotted with an
anti-p73 (GC15) (left panel) or anti-NEDD8 (right panel) antibody.

38
2.2.4 Neddylation of p73 occurs under physiological
conditions
We next asked whether neddylation of p73 occurs in the absence of overexpression. HEK293
cells were lysed under denaturing conditions, immunoprecipitated with an affinity purified
polyclonal anti-p73 (2301) antibody or a control antibody, and immunoblotted with anti-p73
(Fig. 2.4, left panel) and anti-NEDD8 (right panel) antibodies. As expected, multiple
endogenous p73 proteins were observed, indicating the presence of various N- and C-terminal
p73 isoforms (Fig. 2.4, lane 1). Importantly, a co-migrating protein containing both p73- and
NEDD8-specific epitopes was observed (Fig. 2.4, compare lanes 1 and 3). Similar results were
observed using an anti-p73 antibody generated against the N-terminus of TAp73 (H-79, Santa
Cruz) (Appendix, Fig. A.2), suggesting that, in agreement with the above overexpression data,
the modified forms of p73 are most likely the full-length TA isoforms. However, unlike in the
case of overexpression, a single slower migrating p73 protein is determined to contain NEDD8
under physiologic conditions. While the reason for this discrepancy is undetermined, it is likely
due to the variations in the stoichiometry of the various components of the NEDD8 pathway,
MDM2 and p73 between experiments conducted under physiologic conditions and
overexpression.
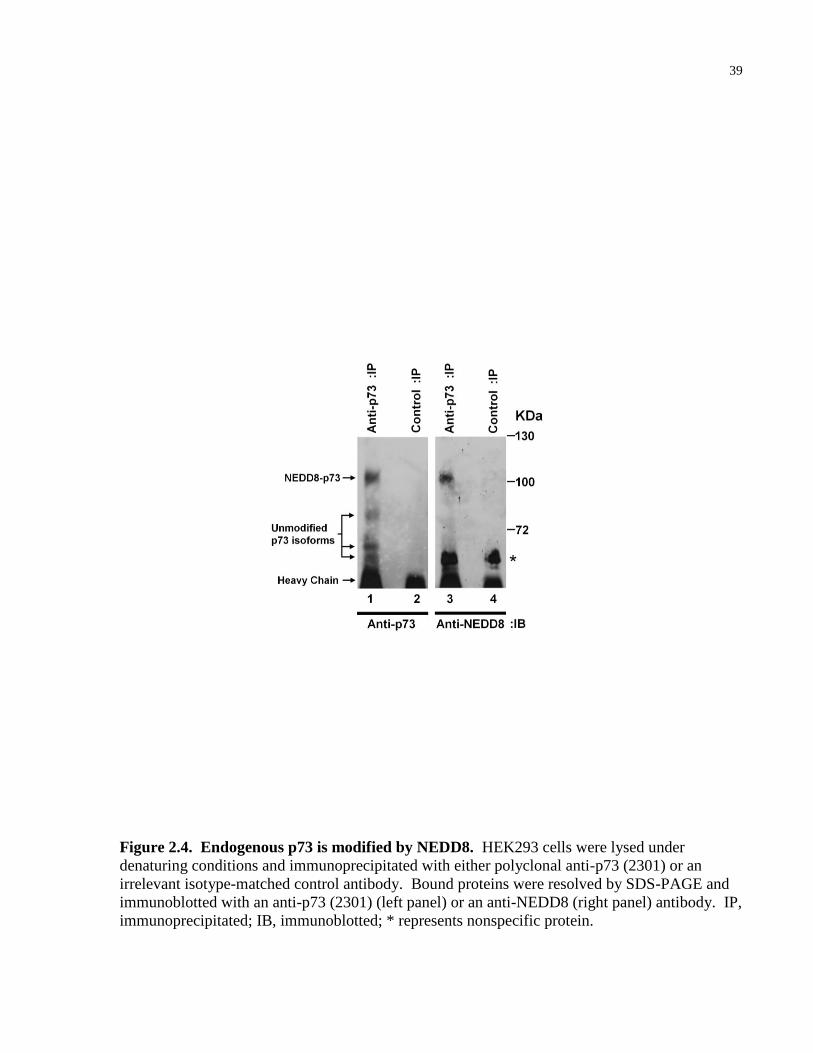
39
Figure 2.4. Endogenous p73 is modified by NEDD8. HEK293 cells were lysed under
denaturing conditions and immunoprecipitated with either polyclonal anti-p73 (2301) or an
irrelevant isotype-matched control antibody. Bound proteins were resolved by SDS-PAGE and
immunoblotted with an anti-p73 (2301) (left panel) or an anti-NEDD8 (right panel) antibody. IP,
immunoprecipitated; IB, immunoblotted; * represents nonspecific protein.

40
2.2.5 The neddylation pathway attenuates TAp73
transcriptional activity
Our results show that MDM2 promotes NEDD8 modification of TAp73. To address the
functional significance of TAp73 neddylation, we used a well-characterized ts41 CHO cell line
with a temperature-sensitive mutation in the SMC gene (the hamster homologue of human APP-
BP1, a component of the E1 NAE), to determine whether an intact NEDD8 pathway affects
TAp73 transcriptional activity [186]. The ts41 CHO cells were transfected with either p53 or
HA-TAp73 with a PG-13-luciferase reporter construct that contains multiple p53-binding sites
upstream of the luciferase gene or a p53/p73-target gene p21 promoter-driven luciferase reporter
construct. Cells were then maintained at either the permissive (33°C) or non-permissive
temperature (39°C). The shift to the non-permissive temperature significantly increased the
transcriptional activity of HA-TAp73 as demonstrated by the elevated PG-13-luciferase and
p21-luciferase reporter activity (Fig. 2.5A, B), but had a negligible effect on the MG-15-
luciferase reporter, which contains mutated p53-binding elements upstream of the luciferase gene
(data not shown). Recently, MDM2-mediated neddylation of p53 was shown to inhibit its
transcriptional activity [131] and thus, as expected, a similar increase in the PG-13-luciferase
reporter activity was observed with p53 upon inactivation of the NEDD8 pathway (Fig. 2.5A).
Furthermore, we did not observe a change in the transcriptional activity of HA-TAp73
in the wild-type CHO cells between 33°C and 39°C (Fig. 2.5B) demonstrating that the increase
in TAp73 activity in ts41 cells was not attributable to the temperature change. In
complementary experiments, a reconstitution of wild-type APP-BP1 in ts41 cells inhibited HA-

41
TAp73 transcriptional activity in a dose-dependent manner at the non-permissive temperature
(Fig. 2.5C). Furthermore, ectopic expression of human MDM2 and T7-NEDD8 in p53-/-
SAOS2 cells inhibited both HA-TAp73 and HA-TAp73 transcriptional activity as measured
by p21-luciferase reporter activity, which was alleviated with co-expression of NEDP1 (Fig.
2.5D, E). These results strongly suggest that an intact NEDD8 pathway attenuates TAp73
transcriptional activity.

42

43
Figure 2.5. NEDD8 pathway inhibits TAp73-mediated transactivation. (A/B) The ts41 or
wild-type CHO cells were transiently transfected at 33°C with plasmids encoding p53 or HA-
TAp73 and a PG-13-luciferase or p21-luciferase reporter construct with a Renilla-luciferase
construct. 24 h post-transfection, cells were either maintained overnight at the permissive
temperature of 33°C (open bars) or shifted to the non-permissive temperature of 39°C (solid
bars) where the NEDD8 pathway is inactive, for an additional 24 h. Subsequently, the dual
luciferase assay was performed to measure p53 and TAp73 transcriptional activities. (C)
Reintroduction of the wild-type APP-BP1 at the non-permissive temperature (39°C) inhibits
TAp73 transcriptional activity. The ts41 CHO cells were transiently transfected with HA-
TAp73 with increasing amounts of myc-APP-BP1 (5x, 10x and 15x). 24 h post-transfection,
cells were shifted from 33°C to 39°C for an additional 24 h prior to measurement of the TAp73
transcriptional activity. (D) Ectopic expression of MDM2 and NEDD8 inhibits HA-TAp73 and
HA-TAp73 transcriptional activity. SAOS2 cells were transiently transfected with HA-
TAp73 or HA-TAp73 alone or with MDM2 and T7-NEDD8, and transcriptional activity was
measured following an incubation period of 48 h at 37°C. (E) Ectopic expression of NEDP1
alleviates MDM2 and NEDD8 inhibition of HA-TAp73 transcriptional activity. The
experiment was performed as described above, but with co-tranfection of NEDP1. The results
shown are representative of three independent experiments performed in triplicate.

44
2.2.6 NEDD8 modification of TAp73 promotes cytoplasmic
localization
We next examined the subcellular localization of neddylated TAp73. HEK293 cells were
transiently transfected with plasmids encoding HA-TAp73 in combination with human MDM2
and T7-NEDD8 or T7-NEDD8GG. As mentioned previously, MDM2 stabilizes TAp73 via an
unknown mechanism [166, 167]. Therefore, cells overexpressing HA-TAp73 alone were
transfected with higher amounts of plasmid encoding HA-TAp73 as compared with those
transfected with plasmids encoding HA-TAp73 and MDM2 to achieve comparable levels of
HA-TAp73 protein. The isolated cytoplasmic and nuclear lysates were immunoprecipitated
and immunoblotted with an anti-HA antibody, as well as anti--Tubulin and anti-hnRNP
antibodies for validation of fractionation efficiency (Fig. 2.6A). Expression of HA-TAp73
alone or in combination with MDM2 and conjugation-defective T7-NEDD8ΔGG resulted in the
absence of neddylated HA-TAp73 (Fig. 2.6A, lanes 1, 5, 7). Importantly, in the presence of
MDM2 and T7-NEDD8, T7-NEDD8-modified HA-TAp73 was detected and preferentially
localized in the cytoplasm (Fig. 2.6A, lane 3). The level of unmodified HA-TAp73 was also
frequently higher in the cytoplasmic fraction. In addition, where the cytoplasmic ectopic
expression level of HA-TAp73 was comparable, neddylated HA-TAp73 was not detected in
cells transfected with plasmids encoding MDM2 and T7-NEDD8ΔGG (Fig. 2.6B). These results
suggest that MDM2 may exert its inhibitory effect on TAp73 transcriptional activity, at least in
part, by promoting TAp73 cytoplasmic localization via NEDD8 conjugation of TAp73 and
argue against the notion that the neddylation of HA-TAp73 is simply due to the higher

45
expression level of HA-TAp73 in the cytoplasm. Previous studies have also demonstrated
some effects of MDM2 overexpression on TAp73 subcellular localization. For example, MDM2
promoted TAp73 redistribution from the nucleus to the paranuclear regions, and upon
expression of the MDM2-related protein MDMX, TAp73 was found primarily in the cytoplasm
[187]. In addition, TAp73 redistribution to the cytosol and nuclear aggregation was observed in
the setting of ectopic MDM2 expression [188]. However, these studies were not performed in
the context of NEDD8.
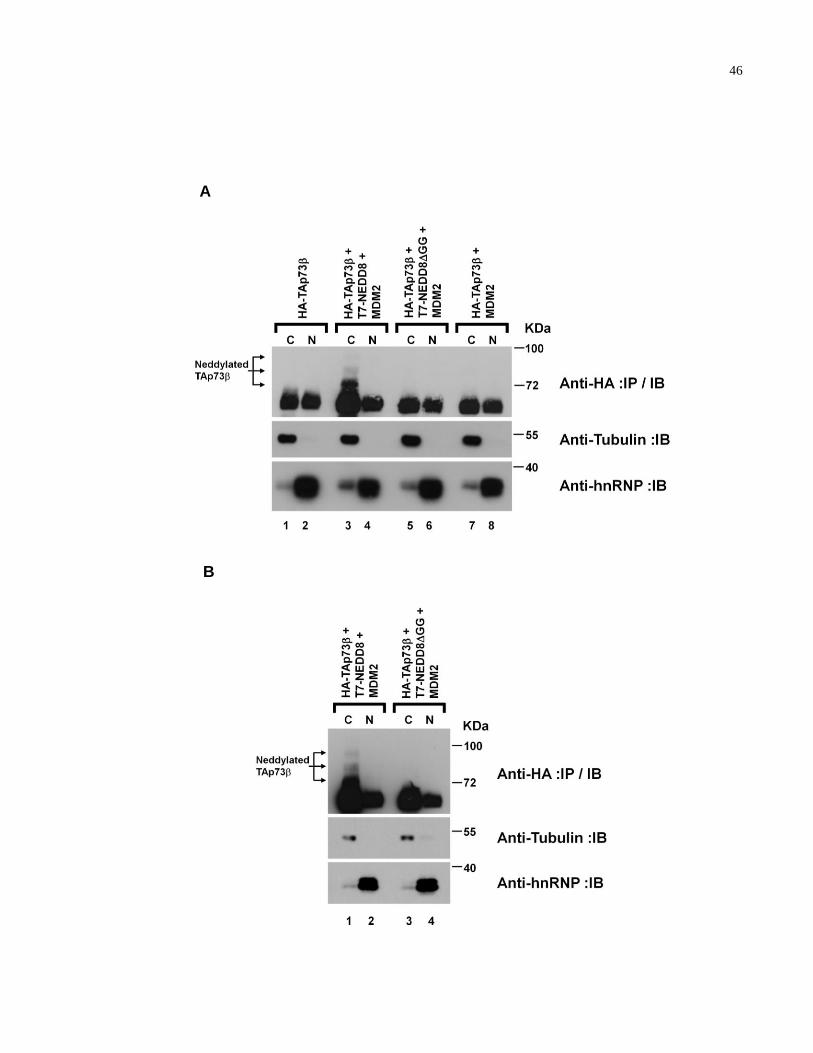
46
A
B

47
Figure 2.6. Neddylated TAp73 species are found preferentially in the cytoplasm. (A/B)
HEK293 cells were transiently transfected with the indicated plasmids. The total amount of
DNA transfected were equilibrated with empty pcDNA3 plasmids. The isolated cytoplasmic and
nuclear lysates were immunoprecipitated and immunoblotted with an anti-HA antibody (top
panel). Whole cell extracts (30 g) of subcellular fractions were analyzed by anti--Tubulin
(middle panel) and anti-hnRNP (bottom panel) immunoblots. Note that in (A), cells transfected
with plasmids encoding HA-TAp73 alone were transfected with 10x the amount of HA-
TAp73 plasmid compared to co-transfections that included MDM2.

48
2.3 DISCUSSION
Accumulating evidence suggests that the relative expression and stability of the N-terminal pro-
apoptotic TA and anti-apoptotic ΔN p73 isoforms contribute to a role in tumorigenesis [105].
TAp73 transactivates known p53-target genes in contrast to ΔNp73 that lacks the TAD and acts
as dominant-negative inhibitors for all full-length TA isoforms of the p53 family by forming
hetero-oligomers that generate an abortive transcriptional complex or by competing directly for
p53 DNA binding sites [59, 63, 65, 68, 70-73]. The tumor suppressor role for TAp73 is
supported by the findings that TAp73-/- mice are tumor prone, TAp73 is induced by a wide
variety of chemotherapeutic agents [86-89] and blocking TAp73 function leads to
chemoresistance and survival [91, 92]. In contrast, anti-apoptotic ΔNp73 blocks chemotherapy-
induced apoptosis in tumor cells that retain wild-type p53, and elevated expression of ΔNp73 has
been associated with tumor progression, poor prognosis, and increased cancer recurrence [65, 95,
99, 101]. Thus, the role of p73 in tumorigenesis hinges on the balance between the expression of
TA and ΔN isoforms. However, post-translational modifications differentially governing the
expression and/or activity of TA and ΔNp73 isoforms have yet to be clearly characterized.
Here, we report the first evidence of a covalent post-translational modification
exclusively targeting the TA isoform of p73. We demonstrate that TAp73, but not ΔNp73, is
covalently modified by NEDD8 in an MDM2-dependent manner. The failure of ΔNp73
neddylation is likely due to the absence of the critical MDM2-binding domain located within the
N-terminus. In addition, a NEDD8-specific cysteine protease, NEDP1, deconjugated neddylated
TAp73, and an intact NEDD8 pathway attenuated TAp73 transcriptional activity in ts41 CHO
cells. Furthermore, the co-expression of MDM2 with NEDD8, but not with a conjugation-

49
defective NEDD8ΔGG mutant, promoted accumulation of NEDD8-modified TAp73 in the
cytoplasm. These results, taken together, suggest that neddylation of TAp73β via MDM2 may
exert its inhibitory effect on TAp73 transcriptional activity, at least in part, by altering its
subcellular localization.
MDM2 is one of the major regulators of p53 stability via the ubiquitin pathway.
Interestingly, MDM2, rather than promoting the degradation of TAp73, stabilizes TAp73 (33-
35). The mechanism underlying MDM2-mediated stabilization of TAp73 is currently unknown.
We have also noted increased levels of other TAp73 C-terminal isoforms upon ectopic
expression of MDM2 and/or NEDD8 (see Fig. 2.1 and 2.3; Appendix, Fig. A.3). Therefore, it
is tempting to speculate that MDM2-mediated neddylation of TAp73 results, not only in
subcellular redistribution of TAp73, but also in its stabilization. Recently, Bernassola et al have
shown that p73α accumulated under the non-permissive temperature in ts41 cells, suggesting a
role of the NEDD8 pathway in the stabilization of p73α [189]. In the context of our study, there
are several reasons to consider that may explain why increased MDM2-mediated stabilization of
TAp73 is associated with decreased transactivation function of TAp73. The direct binding of
MDM2 to the N-terminal TAD and/or competitive disruption of p300/CBP binding to the N-
terminal TAD by MDM2 may inhibit TAp73 transcriptional activity [167]. Furthermore, it is
formally possible that MDM2 regulates NEDD8 modification of lysyl residues that are targets of
acetylation by transcriptional co-activators [110]. Thus, there are multiple modes of TAp73
regulation mediated by MDM2. The elucidation of the precise molecular mechanisms that
orchestrate the various post-translational regulatory modifications and the molecular signals that
initiate MDM2-mediated neddylation influencing the function of TAp73 are outstanding
questions that remain to be resolved.

50
2.4 FUTURE DIRECTIONS
To determine the molecular mechanism of MDM2-mediated NEDD8-dependent TAp73 cytoplasmic redistribution
We have observed all of the TAp73 C-terminal isoforms tested to date are modified by NEDD8
(Appendix, Fig. A.3A). However, MDM2 is not able to promote neddylation of the C-terminal
truncated mutant 1-344 (Appendix, Fig. A.3A, B). Interestingly, lysines common to all
NEDD8-modified TAp73 C-terminal isoforms not present in the TAp73 1-344 truncated mutant
are located in the nuclear localization (NLS) and export (NES) signals respectively (Appendix,
Fig. A.3B). We also observed the enzymatically inactive RING finger MDM2(C464A) mutant
does not promote cytoplasmic neddylated TAp73 in subcellular fractionation experiments
(Appendix, Fig. A.4). We suspect that NEDD8 modification results in nuclear export of TAp73,
but we cannot rule out the possibility that MDM2-mediated neddylation sequesters TAp73 in the
cytoplasm, or TAp73 is preferentially deneddylated in the nucleus. To address such alternatives
future studies employing a combination of MDM2 and TAp73 mutants, as well as number of
inhibitors, can be employed to address these issues as described below.
For example, to determine whether MDM2 promotes nuclear export of neddylated
TAp73, Leptomycin B, a nuclear export inhibitor that prevents the proper function of the CRM1
protein known to modulate both p53 and p73 subcellular localization would be a valuable tool
[190, 191]. In the case MDM2 promotes nuclear export of TAp73 via NEDD8 modification, we
should observe an accumulation of neddylated TAp73 in the nucleus in subcellular fractionation
and immunofluorescence experiments following LMB treatment. However, LMB also
modulates MDM2 localization. Therefore, determining whether an MDM2 mutant lacking a

51
functional NES, which sequesters MDM2 in the nucleus [127], is able to promote cytoplasmic
TAp73 will be important in determining whether MDM2-mediated neddylation of TAp73
promotes nuclear export or cytoplasmic sequestration. Future studies should investigate the
specific molecular mechanism by which MDM2 neddylation promotes nuclear export of TAp73,
if this is the determined mechanism. For example, NEDD8 modification may unmask the NES
of TAp73, or alternatively, NEDD8 may act as a functionally distinct signal promoting nuclear
export. To address the first mechanism, one could determine CRM1 binding to TAp73 in the
ts41 CHO cell lines under permissive and non-permissive temperatures. To address whether
NEDD8 acts as a functionally distinct signal for nuclear export, NEDD8-TAp73 chimeric
proteins may be a useful approach. Lastly, to determine whether TAp73 is preferentially
deneddylated in the nucleus, studies using the cysteine alkylating agent N-ethylmaleimide
(NEM), which inhibits cysteine proteases responsible for deneddylation may prove informative.
To determine the role of DNA damage in the regulation of p73 neddylation
The mechanisms involved in the induction of TAp73 by chemotherapy treatment are not
completely understood. Furthermore, DNA damage destabilizes MDM2, and siRNA targeting
MDM2 results in enhanced chemosensitivity in both wild-type and mutant p53 cells [192]. UV
radiation has already been shown to decrease p53 neddylation. Considering MDM2-mediated
neddylation of TAp73 inhibits its transactivation function and TAp73 is induced in response to
chemotherapy, we hypothesize that TAp73 neddylation is curtailed in response to DNA damage.
Future studies should determine whether chemotherapy modulates levels of neddylated TAp73
and determine potential mechanisms mediating this effect, which include: DNA damage-induced

52
decrease in MDM2 levels; decreased binding of MDM2 to TAp73; increased NEDD8
deconjugation mediated by deneddylating enzymes; or decreased neddylation activity of NEDD8
E2 enzymes.
2.5 MATERIALS AND METHODS
2.5.1 Cells
HEK293 human embryonic kidney and SAOS2 cells were obtained from American Type Culture
Collection (Rockville, MD) and maintained in Dulbecco’s modified Eagle’s medium (Gibco;
Grand Island, NY) supplemented with 10% heat-inactivated fetal bovine serum (Hyclone; Logan,
UT) at 37°C in a humidified 5% CO2 atmosphere. The ts41 and wild type Chinese hamster ovary
(CHO) cell lines were maintained essentially as previously described [186].
2.5.2 Antibodies
Monoclonal anti-hemagglutinin (HA) (12CA5) and polyclonal anti-HA (Y11) antibodies were
obtained from Boehringer (Indianapolis, IN) and Santa Cruz (Santa Cruz, CA), respectively.
Monoclonal anti-T7 antibody was obtained from Novagen (Madison, WI). Polyclonal anti-
NEDD8 antibody was obtained from Alexis Biochemicals (Lansen, Switzerland). Monoclonal
anti-p73 (ER15) antibody was previously described [67]. Monoclonal anti-p73 (GC15)
antibody was obtained from Oncogene Research (La Jolla, CA). Monoclonal anti--Tubulin
antibody was obtained from Sigma (St. Louis, MO). Monoclonal anti-hnRNP antibody was
obtained from Abcam (Cambridge, MA). Polyclonal anti-p73 (2301) antibodies were raised

53
against a GST fusion of the alpha C-terminus of murine p73. Rabbits were immunized and
boosted at six-week intervals using standard methods, and antibodies were subsequently affinity
purified.
2.5.3 Plasmids
pcDNA3-T7-NEDD8 and pcDNA3-T7-NEDD8GG were described previously [193]. The
pCMV-human MDM2 plasmid was generously provided by Dr. Samuel Benchimol. The
pcDNA3-HA-TAp73 and pcDNA3-myc-Np73 plasmids were previously described and
generously provided by Dr. Gerry Melino [61] and Dr. Freda Miller, respectively [73]. The
pcDNA3.1/V5-His-NEDP1 and pcDNA3.1/V5-His-NEDP1(C163A) plasmids were gifts from
Dr. Ronald T. Hay [183]. pcDNA3-myc-APP-BP1 was generously provided by Dr. Dimitris P.
Xirodimas [131]. pcDNA3-p53 and PG-13-luciferase reporter constructs were previously
described [194]. The PG-13-luciferase reporter plasmid that contains 13 contiguous p53 DNA-
binding sites upstream of the luciferase gene and the MG-15-luciferase reporter plasmid that
contains mutated p53 DNA-binding sites upstream of the luciferase gene were kindly provided
by Dr. Wafik El-Deiry.
2.5.4 Immunoprecipitation and immunoblotting
Cells growing in monolayers were removed from 10 cm tissue culture plates by scraping and
collected by centrifugation. The cells were washed once with cold PBS, resuspended in 10%
PBS, and lysed in 90% lysis buffer [pH 7.35] (20 mM Tris, 250 mM NaCl, 3 mM EDTA, 3 mM
EGTA, 0.5% NP-40, 2 mM DTT, 5 mM NEM, 2 mM iodoacetamide) containing 1% sodium

54
dodecyl sulfate (SDS) and supplemented with complete protease inhibitors (Roche; Indianapolis,
IN). The samples were incubated at 100°C for 20 min, followed by centrifugation at 13,200 rpm
for 10 min to remove cell debris. Protein concentrations were determined by Bradford method
and whole cell lysates were diluted 10 times with lysis buffer without SDS in preparation for the
immunoprecipitation procedure. Immunoprecipitation and immunoblotting procedures were
performed as previously described [67]. Briefly, immunoprecipitations were carried out with the
indicated antibody for 2 h at 4°C with protein A-Sepharose (Amersham Biosciences; Sweden).
Immunoprecipitates were washed five times with NETN buffer (2 M Tris [pH 8.0], 5 M NaCl,
0.5 M EDTA [pH 8.0], 0.5% NP-40) eluted by boiling in SDS-containing buffer, and separated
by SDS-polyacrylamide gel electrophoresis (PAGE). Proteins were transferred to nitrocellulose
membranes (Bio-Rad; Hercules, CA) for Western analysis.
2.5.5 Subcellular fractionation
Cells were trypsinized, washed twice with cold PBS, and pelleted by centrifugation at 1000 rpm
at 4°C for 3 min. Cells were resuspended in 250 l of lysis buffer A (10 mM HEPES [pH 7.9],
10 mM KCl, 1.5 mM MgCl2, 0.34 M sucrose, 10% glycerol, 1 mM DTT) containing 0.1% Triton
X-100 and supplemented with complete protease inhibitors (Roche; Indianapolis, IN). Following
a 7 min incubation on ice, the nuclear fraction was pelleted by centrifugation at 3600 rpm for 5
min at 4°C, and the cytoplasmic cell fraction was decanted. The nuclear pellet was washed once
with buffer A without Triton X-100. The nuclear pellet was resuspended in 100 l of lysis buffer
B (0.2 mM EGTA [pH 8], 3 mM EDTA [pH 8], 1 mM DTT) and incubated on ice for 30 min
while undergoing periodic vortexing. The nuclear fraction was collected following

55
centrifugation at 4000 rpm for 5 min at 4°C. Protein concentrations were determined by
Bradford method, and immunoprecipitations were performed as described above.
2.5.6 Dual-luciferase reporter assay
The ts41 and wild-type CHO cells were transiently transfected using Fugene 6 (Roche;
Indianapolis, IN) with 20 ng of pcDNA3-HA-TAp73 or pcDNA3-p53, 20 ng of either PG-13-
or p21-luciferase reporter construct, and 0.1 ng of Renilla-luciferase construct. In cases where
APP-BP1 and TAp73 were co-expressed in the ts41 cells, 10 ng of pcDNA3-HA-TAp73 was
co-transfected with either 50, 100 or 150 ng of pcDNA3-myc-APP-BP1. Following transfection,
cells were incubated overnight at 33°C, and were maintained either at 33°C or shifted to 39°C for
an additional 24 h. SAOS2 cells were similarly transfected with 20 ng of pcDNA3-HA-TAp73β
or HA-TAp73 in combination with 80 ng of pCMV-human MDM2 and 100 ng of pcDNA3-T7-
NEDD8 where indicated, followed by an incubation period of 48 h at 37°C. The total amount of
transfected DNA was equilibrated to 200 ng using an empty pcDNA3 plasmid. Luciferase and
Renilla activities were measured using Dual-Luciferase Reporter Assay System (Promega;
Madison, WI) according to the manufacturer’s instructions on Lumat LB 9507 luminometer
(Berthold Technologies; Germany).

56
CHAPTER 3:
Chemotherapy induces NEDP1-mediated destabilization of MDM2
This work has been published:
Watson IR, Li BK, Roche O, Blanch A, Ohh M, Irwin MS. Chemotherapy induces NEDP1-
mediated destabilization of MDM2. Oncogene. 2010 Jan 14;29(2):297-304. Epub 2009 Sep 28.
3.1 HYPOTHESIS AND RATIONALE
Deregulation of the p53 pathway is observed in most human cancers. Approximately 50% of
human tumors harbor inactivating mutations in the p53 gene, while the remaining tumors exhibit
suppressed, albeit wild-type, p53 activity owing to a variety of mechanisms including
amplification of MDM2 [46]. MDM2 is a critical regulator of p53 during development,
demonstrated by the embryonic lethality of mdm2-null mice that is rescued by concomitant loss
of p53 [136, 137]. Conversely, a study employing an inducible p53 mouse model on an mdm2-
null background demonstrated that expressing p53 at near ―endogenous levels‖ in the absence of
mdm2, resulted in severe apoptosis in classical radiation-sensitive tissues and cell cycle arrest in
radiation-insensitive tissues [141]. Finally, an E3 ligase-defective mdm2 mutant was unable to
rescue the embryonic lethality of mdm2-null mice, demonstrating the biological importance of
MDM2 E3 activity in the negative regulation of p53 [142].

57
In response to DNA damage, p53 becomes phosphorylated by several kinases within the
MDM2-binding domain, which prevents MDM2-p53 interaction (reviewed in [108]). The
stabilization of p53 then leads to DNA repair, cell cycle arrest, senescence or apoptosis.
However, a number of recent studies have demonstrated the importance of MDM2
destabilization in response to DNA damage, which promotes p53 activation [150, 195]. First,
MDM2 phosphorylation by DNA damage-induced kinases precedes p53 induction [149].
Second, pre-treatment of tumor cell lines with proteasome inhibitor preventing degradation of
MDM2 but permitting N-terminal phosphorylation of p53 in response to DNA damage,
abrogated the activation of p53 [150]. Third, transgenic mice expressing p53 point mutations in
N-terminal phosphorylation sites targeted by stress-induced kinases have mild apoptotic defects
and low incidence of tumorigenesis (reviewed in [46]). In contrast, genetic loss of mdm2
promotes a robust p53 apoptotic response in mouse studies described above.
NEDD8 is an ubiquitin-like protein that regulates protein function via covalent
modification of substrates such as Cullins, BCA3, EGFR, ribosomal L11 protein, pVHL, p73 and
p53 (reviewed in [34]). For example, NEDD8 modification of Cullins not only regulates the
enzymatic activity of CRLs [18], but also influences Cullin protein stability [196, 197].
Neddylation of p53 via MDM2 was shown to inhibit p53 transcriptional activity [131]. MDM2
itself is also subject to NEDD8 modification; however, the molecular mechanisms governing
MDM2 neddylation and its biological significance remain unclear. In our studies examining
MDM2 neddylation of TAp73, we observed that ectopic expression of NEDD8 dramatically
increased the expression of MDM2. We hypothesized that neddylation, promoted by the known
NEDD8 E2 conjugating enzyme, Ubc12, leads to MDM2 stabilization. Conversely, we
hypothesized that deneddylation mediated by NEDD8-specific proteases would promote MDM2

58
destabilization. Interestingly, recent studies have reported that the activity of the COP9
signalosome deneddylating complex increases in response to apoptotic stimuli, and protein levels
of the ubiquitin E2 enzyme, Ubc5b/c, decrease in response to DNA damage [198, 199]. In light
of the findings that MDM2 destabilization plays an important role in p53 activation, we
hypothesized that the expression and/or activity of NEDD8-specific cysteine proteases and
Ubc12 are modulated by DNA damage, and as a result, regulate MDM2 destabilization and
subsequent activation of p53.
3.2 RESULTS AND DISCUSSION
3.2.1 Neddylation stabilizes MDM2
Ectopic expression of T7-NEDD8, but not T7-NEDD8GG mutant that lacks the essential C-
terminal glycine-glycine motif required for covalent conjugation of substrates, promoted
significant MDM2 neddylation (Fig. 3.1A). While this observation is consistent with a previous
report showing MDM2 is a substrate for NEDD8 modification [131], we noted a dramatic dose-
dependent increase in MDM2 levels with increasing amounts of T7-NEDD8 (Fig. 3.1B), which
was not attributable to changes in MDM2 transcript levels as determined by semi-quantitative
RT-PCR (data not shown). A similar induction of MDMX, a MDM2 structural homologue that
lacks E3 ligase activity, was not observed with increasing amounts of T7-NEDD8 (Fig. 3.1B).
We next asked whether the known NEDD8 E2 conjugating enzyme, Ubc12, modulates MDM2
levels. HEK293A cells transfected with increasing amounts of His-Ubc12 showed a dose-
dependent increase in MDM2 (Fig. 3.1C, left panels). However, cells expressing increasing

59
amounts of the dominant-negative Ubc12(C111S), which forms a stable interaction with NEDD8
(Fig. 3.1C, bottom right panel) and therefore has limited ability to transfer NEDD8 to target
substrates, showed a striking dose-dependent decrease in MDM2 (Fig. 3.1C, right panels). We
next asked whether the observed changes in MDM2 expression levels were due to alterations in
MDM2 protein stability. Endogenous MDM2 half-life was approximately 30-40 min in cells
expressing T7-NEDD8GG mutant or an empty plasmid (Fig. 3.1D). In contrast, cells
ectopically expressing T7-NEDD8 showed a near three-fold increase in MDM2 half-life to 1.6 h
(Fig. 3.1D). Similar results were observed in U2OS osteosarcoma cell line (Appendix, Fig.
A.5). Notably, only a minor fraction of MDM2 was observed to be neddylated at a given time
(see Fig. 3.1A). However, conjugation and de-conjugation of ubiquitin and ubiquitin-like
proteins such as NEDD8 and SUMO is a dynamic process and hence, a greater proportion of
MDM2 is likely subjected to NEDD8 modification. In support of this hypothesis, recent studies
have shown that mutants lacking the NEDP1 homologues in S. pombe and D. melanogaster
significantly increased the profile of neddylated proteins that otherwise would not have been
detected [200, 201]. These results suggest that NEDD8 conjugation promotes MDM2 protein
stabilization.
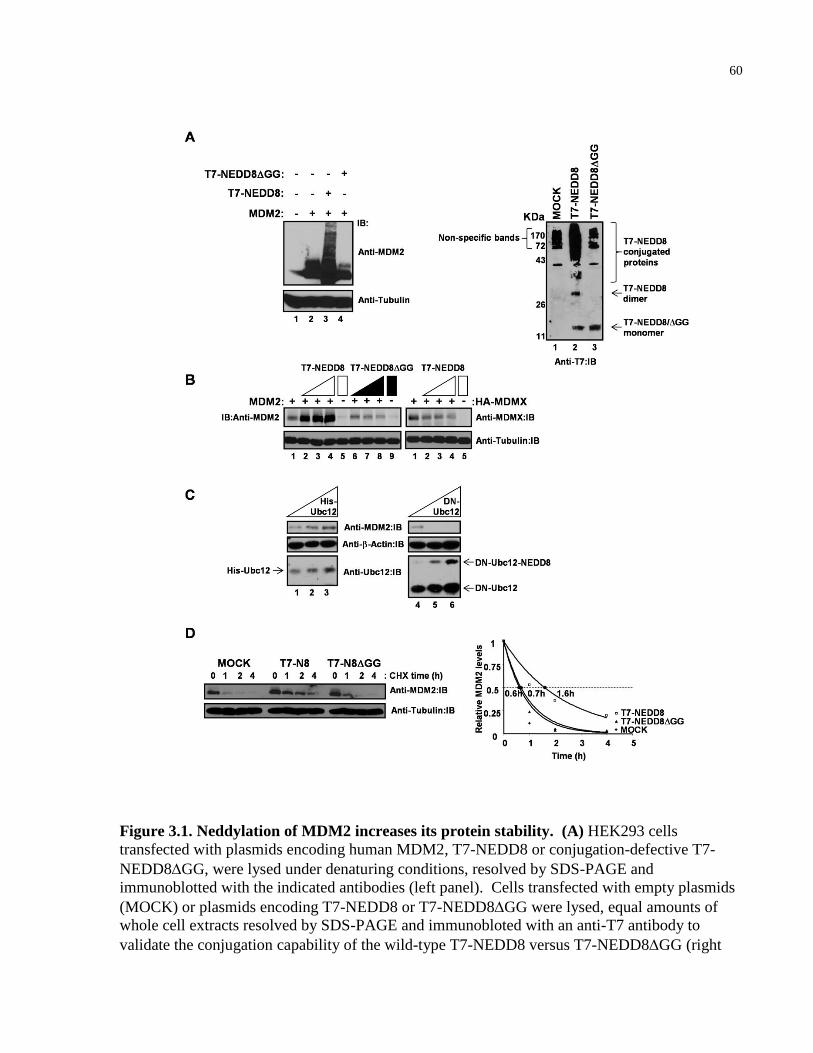
60
Figure 3.1. Neddylation of MDM2 increases its protein stability. (A) HEK293 cells
transfected with plasmids encoding human MDM2, T7-NEDD8 or conjugation-defective T7-
NEDD8GG, were lysed under denaturing conditions, resolved by SDS-PAGE and
immunoblotted with the indicated antibodies (left panel). Cells transfected with empty plasmids
(MOCK) or plasmids encoding T7-NEDD8 or T7-NEDD8GG were lysed, equal amounts of
whole cell extracts resolved by SDS-PAGE and immunobloted with an anti-T7 antibody to
validate the conjugation capability of the wild-type T7-NEDD8 versus T7-NEDD8GG (right

61
panel). (B) Cells were transfected with plasmids encoding MDM2 (left panel) or human HA-
MDMX (right panel) with increasing amounts of T7-NEDD8 or T7-NEDDGG. Cells were
lysed under denaturing conditions, equal amounts of whole cell extracts resolved by SDS-PAGE
and immunoblotted with either anti-MDM2 or anti-MDMX antibodies. (C) Cells transfected
with plasmids encoding MDM2 with increasing amounts of His-Ubc12 or dominant-negative
Ubc12(C111S) (DN-Ubc12), resolved by SDS-PAGE and immunoblotted with the indicated
antibodies. (D) Cells were transfected with empty plasmids (MOCK) or plasmids encoding T7-
NEDD8 or T7-NEDD8GG. 48 h post-transfection, cells were treated with 40 µg/mL
cycloheximide (CHX) for the indicated time periods, lysed, equal amounts of lysates resolved by
SDS-PAGE, and immunoblotted with the indicated antibodies (left panel). Protein levels of
endogenous MDM2 were quantified by densitometry using Image J (http://rsb.info.nih.gov/ij)
software and plotted as a percentage of the no treatment baseline. A trendline was derived to
determine the half-life of MDM2 (right panel). IB: immunoblot.

62
3.2.2 NEDP1-mediated deneddylation promotes MDM2
destabilization
A corollary prediction is that deneddylation would lead to MDM2 destabilization. The two most
commonly studied NEDD8-specific proteases are the COP9 signalosome complex, in which
CSN5/JAB1 possesses the protease activity, and the more recently identified NEDP1 [182-184].
Furthermore, the activity of the COP9 signalosome has been reported to be modulated by
apoptotic stimuli, such as chemotherapy treatment [198, 199]. Thus, we asked whether CSN5 or
NEDP1 played a role in MDM2 deneddylation. Interestingly, ectopic expression of myc-
NEDP1, but not HA-CSN5, markedly attenuated the levels of neddylated MDM2 in a dose-
dependent manner under conditions that favored robust MDM2 neddylation (Fig. 3.2A).
Furthermore, cysteine protease activity of NEDP1 is required for MDM2 deneddylation as the
catalytically defective myc-NEDP1(C163A) mutant failed to deneddylate MDM2 (Fig. 3.2B).
Importantly, expression of NEDP1 decreased MDM2 levels while an ubiquitin-specific
isopeptidase HAUSP increased MDM2 levels (Fig. 3.2C). Moreover, ectopic expression of
myc-NEDP1 appeared to accelerate the turnover of endogenous MDM2 in contrast to the myc-
NEDP1(C163A) mutant (Fig. 3.2D). These results demonstrate that deneddylation destabilizes
MDM2.
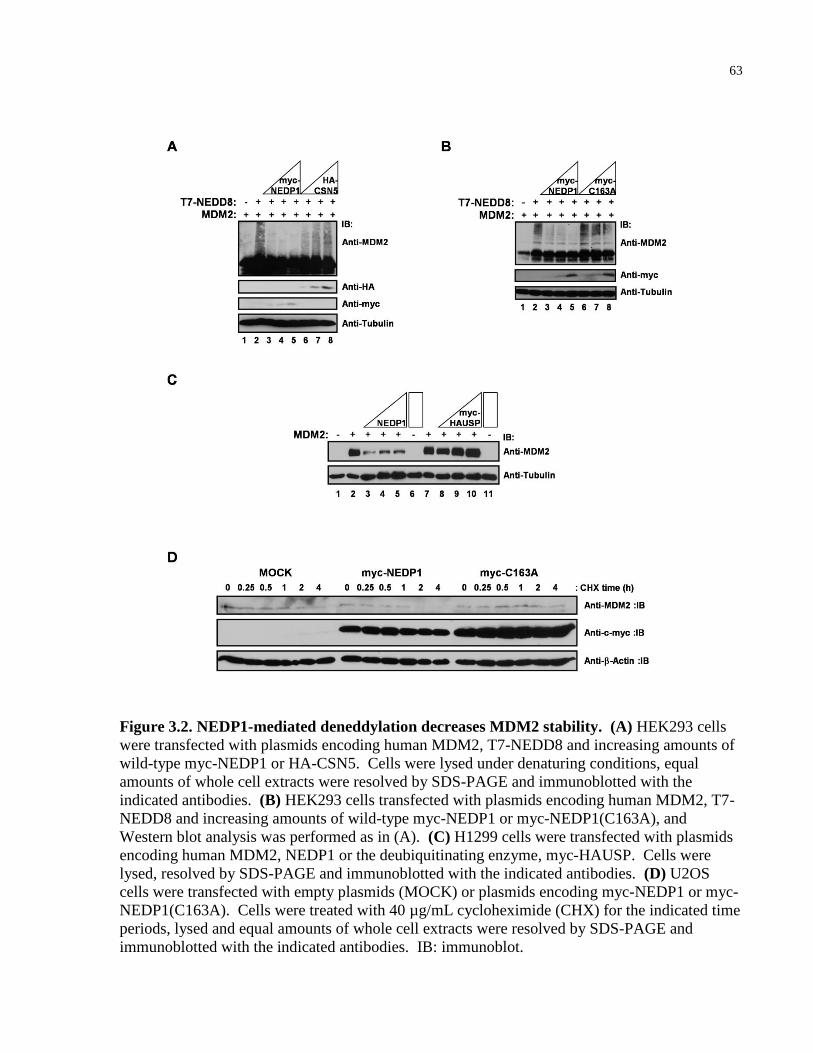
63
Figure 3.2. NEDP1-mediated deneddylation decreases MDM2 stability. (A) HEK293 cells
were transfected with plasmids encoding human MDM2, T7-NEDD8 and increasing amounts of
wild-type myc-NEDP1 or HA-CSN5. Cells were lysed under denaturing conditions, equal
amounts of whole cell extracts were resolved by SDS-PAGE and immunoblotted with the
indicated antibodies. (B) HEK293 cells transfected with plasmids encoding human MDM2, T7-
NEDD8 and increasing amounts of wild-type myc-NEDP1 or myc-NEDP1(C163A), and
Western blot analysis was performed as in (A). (C) H1299 cells were transfected with plasmids
encoding human MDM2, NEDP1 or the deubiquitinating enzyme, myc-HAUSP. Cells were
lysed, resolved by SDS-PAGE and immunoblotted with the indicated antibodies. (D) U2OS
cells were transfected with empty plasmids (MOCK) or plasmids encoding myc-NEDP1 or myc-
NEDP1(C163A). Cells were treated with 40 µg/mL cycloheximide (CHX) for the indicated time
periods, lysed and equal amounts of whole cell extracts were resolved by SDS-PAGE and
immunoblotted with the indicated antibodies. IB: immunoblot.

64
3.2.3 Chemotherapy increases NEDP1 levels
MDM2 destabilization in response to DNA-damaging agents has been shown to play a critical
role in the activation of p53. Recently, the activity of COP9 signalosome deneddylating complex
was shown to increase in response to apoptotic stimuli, whereas ubiquitin E2 conjugating
enzyme, Ubc5b/c, protein levels were shown to decrease in response to doxorubicin treatment
[198, 199]. Considering the present observation that the NEDD8 pathway modulates MDM2
stability, we asked whether DNA-damaging chemotherapeutic agents regulate the components of
the NEDD8 pathway, in particular NEDP1 and Ubc12. Although we did not observe discernible
changes in Ubc12 levels in a variety of cell lines following treatment with several chemotherapy
drugs (data not shown), NEDP1 proteins levels were consistently induced in several cancer cell
lines in response to multiple chemotherapeutic agents, including topoisomerase inhibitors
doxorubicin and camptothecin and the radiomimetic neocarzinostatin (NCS) (Fig. 3.3A,
Appendix, Fig. A.6). Notably, the increase in NEDP1 levels corresponded with elevated p53
and p53-target gene products, PUMA and p21, which were consistent with diminishing MDM2
expression (Fig. 3.3A). In addition, the rise in NEDP1 upon chemotherapy treatment paralleled
the appearance of cleaved PARP, a marker of apoptosis (Fig. 3.3A). In support of a role of
NEDP1 in activating p53, ectopic expression of myc-NEDP1 decreased MDM2 levels and
correspondingly increased p53 and p53-target PUMA levels in MCF7 cells treated with
doxorubicin (Fig. 3.3B). Furthermore, doxorubicin treatment of p53-/- SAOS2 osteosarcoma
cells increased NEDP1 levels (Fig. 3.4A, B, and C) and ectopic expression of p53 in p53-/-
H1299 lung adenocarcinoma cells had negligible effect on NEDP1 expression in contrast to a
known p53-target, p21 (Appendix, Fig A.7), suggesting that NEDP1 is not a target of p53. In

65
addition, the induction of NEDP1 was similar in AT cells (GM16666) and AT isogenic clonal
cells reconstituted with wild-type ATM (GM16667) (Fig. 3.4D, E, and F). These results suggest
that the induction of NEDP1 does not require p53 or ATM and support the notion that the
activation of p53 upon chemotherapy treatment is in part due to the induction of NEDP1-
mediated destabilization of MDM2.

66
Figure 3.3. Chemotherapy increases NEDP1-mediated p53 activation. (A) MCF7 breast
adenocarcinoma cells were treated with 0.1 µM doxorubicin (DOX) (Sigma) (top panel) and
U2OS osteosarcoma cells were treated with 300 ng/mL neocarzinostatin (NCS) (Sigma) (bottom
panel). Cells were harvested at the indicated time points, lysed and equal amounts of whole cell
extract were resolved by SDS-PAGE and immunoblotted with the indicated antibodies. (B)
MCF7 cells were transfected with empty plasmid (MOCK) or plasmid encoding myc-NEDP1
and treated 48 h post-transfection with 0.1 µM DOX (Sigma). Cells were harvested at the
indicated time points, lysed and equal amounts of whole cell extracts were resolved by SDS-
PAGE and immunoblotted with the indicated antibodies. The left and right panels are derived
from identical exposure of the same immunoblot.
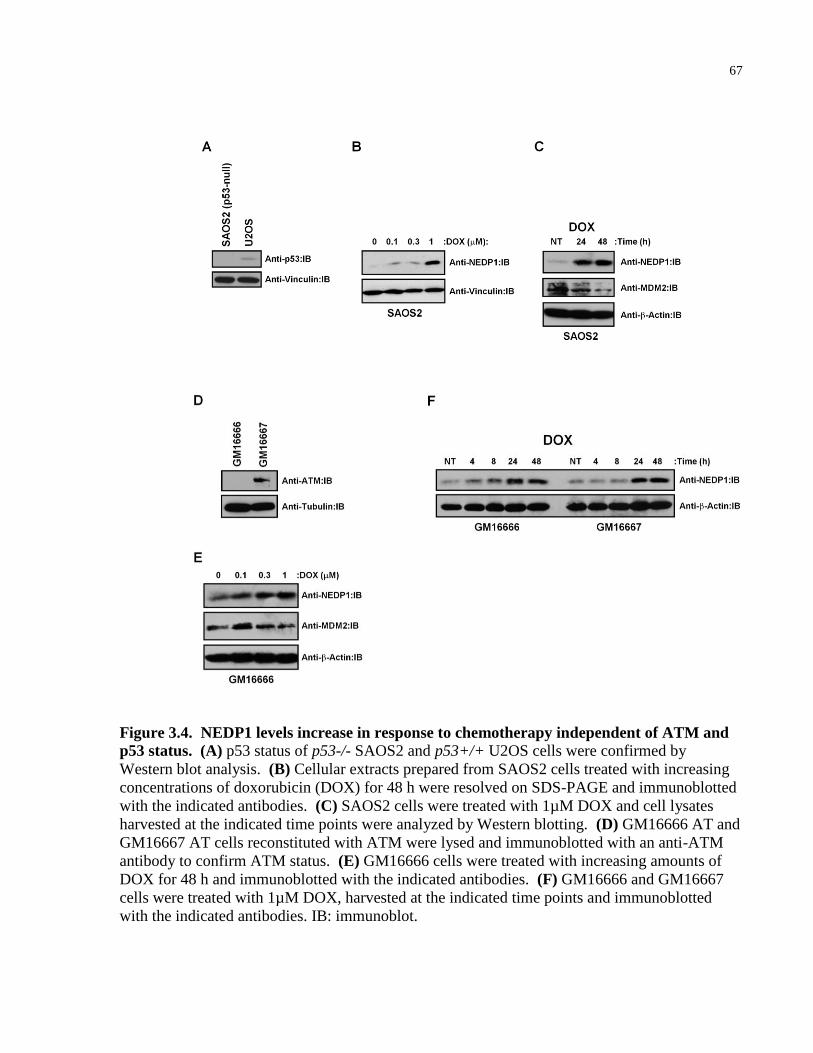
67
Figure 3.4. NEDP1 levels increase in response to chemotherapy independent of ATM and
p53 status. (A) p53 status of p53-/- SAOS2 and p53+/+ U2OS cells were confirmed by
Western blot analysis. (B) Cellular extracts prepared from SAOS2 cells treated with increasing
concentrations of doxorubicin (DOX) for 48 h were resolved on SDS-PAGE and immunoblotted
with the indicated antibodies. (C) SAOS2 cells were treated with 1µM DOX and cell lysates
harvested at the indicated time points were analyzed by Western blotting. (D) GM16666 AT and
GM16667 AT cells reconstituted with ATM were lysed and immunoblotted with an anti-ATM
antibody to confirm ATM status. (E) GM16666 cells were treated with increasing amounts of
DOX for 48 h and immunoblotted with the indicated antibodies. (F) GM16666 and GM16667
cells were treated with 1µM DOX, harvested at the indicated time points and immunoblotted
with the indicated antibodies. IB: immunoblot.

68
3.2.4 NEDP1 modulates p53-apoptotic response to
chemotherapy
The efficacy of chemotherapy is dependent on a successful execution of p53-mediated apoptosis
to override the survival signals acquired by cancer cells. For example, tumors harboring p53
mutations are associated with chemoresistance and predict a considerably worse patient
prognosis in comparison to cancers with wild-type p53 [47]. We predict that forced
downregulation of NEDP1, which we show here to be induced in response to DNA-damaging
chemotherapies, would promote chemoresistance due to enhanced MDM2-mediated suppression
of p53. In support of this assertion, siRNA-mediated knockdown of endogenous NEDP1
promoted MDM2 stabilization, decreased p53 levels and activation demonstrated by relatively
lower expression of p53-target genes (PUMA, p21 and BAX), and resulted in enhanced
chemoresistance of MCF7 cells in response to doxorubicin treatment as measured by reduced
levels of cleaved PARP and TUNEL positivity (Fig. 3.5A, B, and C, Appendix, Fig. A.8).
Similar results were observed in HeLa cervical and 786-O renal carcinoma cells possessing wild-
type p53 (Appendix, Fig. A.8). Notably, MDM2 itself is a p53-target gene, and as a result
MDM2 regulates p53 activity via an autoregulatory feedback. Thus, while the initial MDM2
stabilization upon NEDP1 knockdown leads to increased downregulation of p53, the resulting
lower p53 levels decrease p53-dependent MDM2 transactivation, ultimately attenuating MDM2
levels at later time points (Fig. 3.5A and C). These results suggest that NEDP1 plays a critical
role in activating p53 apoptotic response to DNA damage by modulating the levels of MDM2.
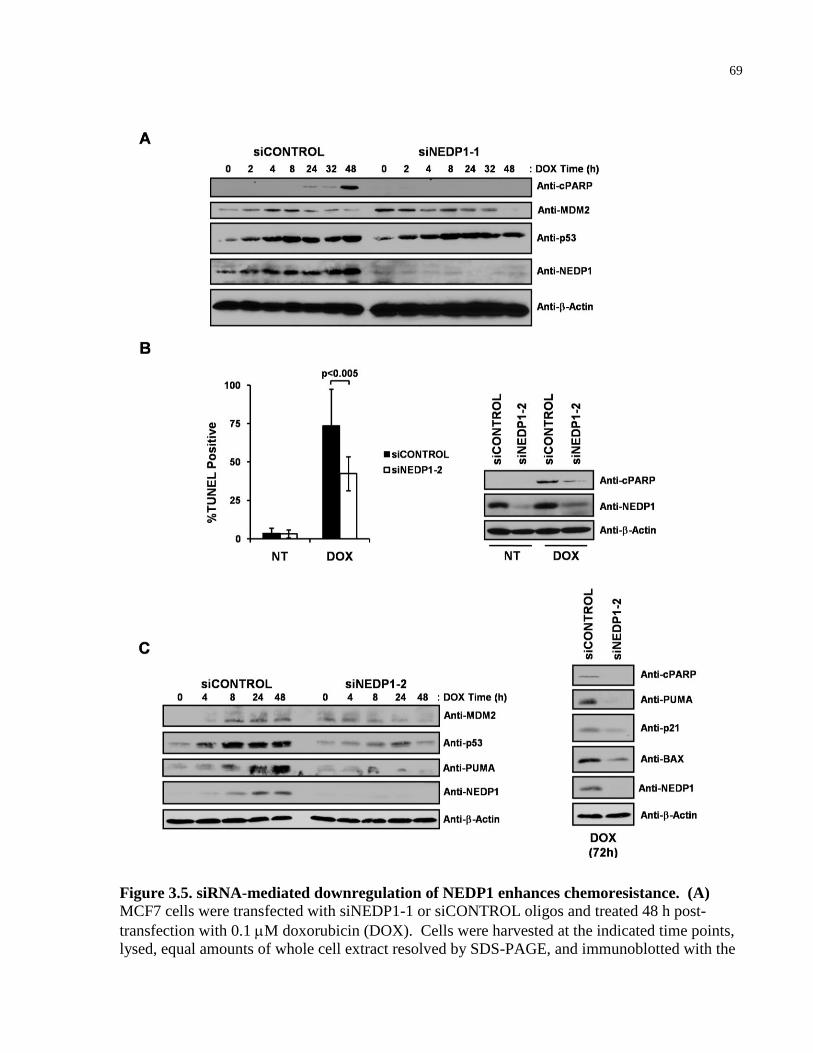
69
Figure 3.5. siRNA-mediated downregulation of NEDP1 enhances chemoresistance. (A)
MCF7 cells were transfected with siNEDP1-1 or siCONTROL oligos and treated 48 h post-
transfection with 0.1 M doxorubicin (DOX). Cells were harvested at the indicated time points,
lysed, equal amounts of whole cell extract resolved by SDS-PAGE, and immunoblotted with the

70
indicated antibodies. (B) MCF7 cells were transfected with a second siNEDP1-2 oligo and 48 h
post-transfection, cells were treated with 2 M DOX for 48 h, trypsinized and 5x104 cells were
mounted on glass slides by cytospin centrifugation and TUNEL assays were performed
according to manufacturer’s protocol (Promega). TUNEL and DAPI positive cells were counted
using Volocity software and confirmed by manual counting (left panel). Equal amounts of whole
cell extracts generated from the remaining cells were resolved by SDS-PAGE and
immunoblotted with the indicated antibodies (right panel). Similar results seen in (B) were
observed following treatment with 0.5 and 1.0 M DOX (data not shown). (C) MCF7 cells were
transfected with siNEDP1-2 or siCONTROL oligos and treated 48 h post-transfection with 0.1
M DOX. Cells were harvested at the indicated time points and Western blot analysis performed
(left panel). Cells harvested at 72 h time point were harvested and visualized by immunoblotting
with the indicated antibodies (right panel).
Figure 3.6. A model of NEDP1-mediated activation of p53 apoptotic response. Steady-state
MDM2 levels are regulated in part by the NEDD8 pathway mediated by the E2 conjugating
enzyme, Ubc12, and/or other unknown factor(s) and the NEDD8-specific isopeptidase, NEDP1.
In response to DNA damage, NEDP1 levels increase to mediate MDM2 deneddylation, resulting
in MDM2 destabilization and increased p53 activation to ultimately trigger apoptosis.

71
3.3 DISCUSSION
An essential response to DNA damage is the uncoupling of MDM2-mediated negative regulation
of p53, enabling p53-dependent DNA repair, cell cycle arrest or apoptosis. For example, in
response to radiomimetic drug, NCS, MDM2 becomes phosphorylated by ATM, which
abrogates MDM2 binding to the deubiquitylating enzyme, HAUSP, resulting in increased
ubiquitylation and degradation of MDM2 [149, 150, 195]. In response to UV, the SUMO-
specific protease, SUSP4, desumoylates MDM2 enhancing MDM2 autoubiquitylation and
destabilization [159]. Intriguingly, these mechanisms regulating MDM2 destabilization occur
early in the DNA damage response ranging from 0.5-2 h [150, 159, 195]. Here, we show that
MDM2 destabilization is mediated through increased NEDP1-mediated deneddylation of MDM2
in response to chemotherapy-induced DNA damage (see Fig. 3.6). The sustained induction of
NEDP1 ranges from 8-72 h post-treatment. Therefore, we appear to have identified a novel
mechanism of p53 activation that occurs relatively late in the DNA damage response. p53 and
TAp73 are also targets of NEDD8 and neddylation has been shown to inhibit the transactivation
function of these two transcription factors (see Chapter 2 Discussion). Similar to temporal
induction of NEDP1, Xirodimas et al. demonstrated that a decrease in p53 neddylation occurs
via unknown mechanism at later time points (post-8 h) following UV treatment [131]. However,
whether NEDP1 directly deneddylates p53 or TAp73 in response to DNA damage is not yet
known.
The two most commonly studied NEDD8-specific proteases are the COP9 signalosome
complex, in which CSN5/JAB1 possesses the protease activity, and the more recently identified
NEDP1 [182-184]. The importance of COP9 signalosome-dependent regulation of Cullin family

72
members is well established. CSN5 was also recently shown to stabilize MDM2 by inhibiting its
autoubiquitylation function [202], which is consistent with our findings that CSN5 does not
appear to curtail NEDD8-stabilized MDM2 in comparison to NEDP1 (see Fig.3.2A). NEDP1
can also deneddylate Cullins in vitro, but NEDP1 exhibits three orders of magnitude less activity
in cleaving neddylated Cul1 than COP9 signalosome [203]. Consistent with this observation,
deletion of NEDP1 homologues in S. pombe and D. melanogaster did not result in accumulation
of neddylated Cul1 or Cul3 [200, 201]. However, a significant accumulation of yet-identified
neddylated substrates was noted in both model systems upon deletion of NEDP1 homologues.
Furthermore, identification of biological stimuli that regulate components of the NEDD8
pathway is one of the outstanding questions in the field [204]. Here, we show the expression of a
NEDD8 isopetidase, NEDP1, is induced by DNA-damaging chemotherapeutic agents, and
induction in NEDP1 expression occurs in a variety of tumor cell lines upon treatment with
several chemotherapeutic drugs. Our findings suggest that the MDM2-p53 axis is a crucial target
of NEDP1 and that NEDP1-mediated activation of p53 via MDM2 destabilization is an
important event promoting chemotherapy-induced cell death. Lastly, recent studies have also
identified putative NEDD8 substrates involved in DNA repair and replication, including
mismatch repair enzymes and replicative helicases [205, 206]. Thus, it is possible that NEDP1
has important functions in the regulation of yet-defined components of the DNA damage
response.

73
3.4 FUTURE DIRECTIONS
Examining the role of NEDP1 in tumor progression and response to chemotherapy in human tumors
MDM2 is overexpressed in numerous cancers by a variety of gene amplification-independent
mechanisms (reviewed in [207]). Interestingly, NEDP1 is located on chromosome 15q23 and
genetic aberrations have been reported in this region in non-small cell lung cancers, gliomas,
oligodendrigliomas, and diffuse-type tenosynovial giant cell tumors [208-211]. Furthermore,
examining the Sanger Institute Cancer Genome database of 45 breast cancer cell lines tested for
gene copy number analysis using the Affymetrix SNP6.0 array, 14 cases of loss of
heterozygosity and a case of homozygous deletion spanning the genomic region encompassing
NEDP1 have been reported (resource described in [212]). Notably, the homozygous deletion of
NEDP1 in the HCC1187 breast carcinoma cell line is a focal deletion of less than 0.5 Mb
containing only one other gene (data not shown).
A general elevation of NEDD8 conjugation has been reported in oral squamous
carcinoma cell lines, and inhibition of the NEDD8 pathway decreased proliferation [32].
Interestingly, excessive sumoylation of MDM2 promoting its stabilization has also been reported
in oral squamous cell carcinomas [213]. However, it remains an outstanding question whether
NEDP1 loss or overexpression of the components that promote MDM2 neddylation play an
oncogenic role in mediating MDM2 overexpression in cancer. Future studies should examine
any associations between APP-BP1/Uba3, Ubc12 and NEDP1 protein expression with MDM2
expression and chemotherapy response in the candidate human tumors mentioned above. Tumor

74
profiling of NEDP1 mutation status and other genetic alterations may also provide additional
insights into its role in cancer.
Determining the mechanisms of chemotherapy-mediated NEDP1 induction and NEDP1-mediated destabilization of MDM2
We hypothesize that NEDD8-modification of MDM2 antagonizes ubiquitin-mediated
degradation of MDM2, increasing its stability. Future studies should identify the specific
NEDD8- and ubiquitin-modified residues of MDM2 to determine if there is any overlap between
the modified lysines. In vitro and in vivo ubiquitylation and neddylation assays combined with
mass spectrometry approaches will be important in addressing this question.
We consistently observed an increase in NEDP1 protein expression upon treatment with a
number of different chemotherapy agents in a variety of tumor cell lines. This effect was
observed in p53-/- cell lines (see Fig. 3.4A-C), and ectopic expression of p53 in the absence of
DNA damage did not increase NEDP1 levels, suggesting that NEDP1 is not a p53-target gene
(Appendix, Fig A.7). Concordantly, we did not observe a significant increase in NEDP1 mRNA
expression upon chemotherapy treatment (Appendix, Fig. A.8), and propose that post-
translational mechanisms mediate this process. NEDP1 levels increase regardless of ATM
status, suggesting that chemotherapy-mediated induction of NEDP1 does not require ATM (Fig.
3.4D, E, and F). Future studies should examine whether or not NEDP1 is modified by
phosphorylation or other post-translational modifications in response to DNA damage.
Differential 2-dimensional gel electrophoresis in conjunction with mass spectrometry can be
used to identify phosphorylated, or otherwise modified, NEDP1 residues following
chemotherapy treatment. Interestingly, a few predicted NEDP1 phosphosites have been

75
identified using post-translational modification predictive software, including putative DNA-PK
phospho-serine and p38 MAPK phospho-threonine sites (software described in [214]).
Interestingly, we have made the preliminary observation that NEDP1 levels increase in the
M059K human malignant glioma cell line that possess normal levels of DNA-PK, but not in the
M059J glioma cell line that was isolated from the same tumor specimen, but lack the DNA-PK
catalytic subunit (Appendix, Fig. A9). Understanding the mechanism of NEDP1-mediated
induction leading to MDM2 destabilization in response to chemotherapy may lead to novel
approaches to target the MDM2-p53 axis to sensitize tumors to chemotherapy treatment.
3.5 MATERIAL AND METHODS
3.5.1 Cells
HEK293A human embryonic kidney, H1299 lung carcinoma, MCF7 breast adenocarcinoma,
IMR5 neuroblastoma cells, 786-0 renal carcinoma, and HeLa cervical carcinoma cells were
obtained from American Type Culture Collection (Rockville). All cells were maintained in
Dulbecco’s modified Eagle’s medium (DMEM) (Gibco), supplemented with 10% heat-
inactivated fetal bovine serum (FBS) (Hyclone) at 37°C in a humidified 5% CO2 atmosphere.
3.5.2 Antibodies and reagents
Doxorubicin, camptothecin and NCS were obtained from Sigma. The following antibodies were
used in this study:

76
3.5.3 Plasmids
pcDNA3-T7-NEDD8, pcDNA3-T7-NEDD8GG, human pCMV-MDM2, pcDNA3.1/V5-His-
NEDP1 and pcDNA3.1/V5-His-NEDP1(C163A) plasmids were described previously [215].
pcDNA3-His-Ubc12 and pcDNA3-Ubc12(C111S) were kindly provided by Dr. Edward Yeh.
pcDNA3.1-HA-HDMX was kindly provided by Dr. Aart Jochemsen. pcDNA3-HA-CSN5 was
kindly provided by Dr. Xin Chen. pcDNA3.1-myc-HAUSP was kindly provided by Dr. Roger
Everett. The human non-tagged pcCDNA3-MDM2 was generated from pcDNA3-FLAG-
MDM2 provided by Dr. Carl Maki by double digestion with BamH1 and EcoR1, and ligating in-
frame into a pcDNA3 vector. The pcDNA3-myc-NEDP1 and the pcDNA3-myc-
NEDP1(C163A) plasmids were generated from pcDNA3.1/V5-His-NEDP1 and C163A mutant
plasmids provided by Dr. Ronald T. Hay. Full-length myc-NEDP1 and myc-NEDP1(C163A)
Antibody Company
p21 Cell Signaling p53 (DO-1) Calbiochem
-Tubulin Sigma
-Actin Sigma ATM Epitomics Bax Cell Signaling c-myc (9E10) Santa Cruz c-myc (A14) Santa Cruz Cleaved PARP Upstate HA Covance HDMX Bethyl MDM2 (Ab-1) Calbiochem MDM2 (SMP14) Santa Cruz NEDP1 BioMol PUMA Cell Signaling Ubc12 Boston Biochem T7 Novagen Vinculin Upstate
_______________________________
_______________________________

77
cDNAs were amplified by PCR with primers (5’-GAC GAC AAG CTT GCC ACC ATG GAG
CAG AAG CTG ATC TCC GAG GAG GAC CTG GAA TTC GAC CCC GTA GTC TTG AGT
TAC-3’ and 5’-GAC GAC CTC GAG CTA CTT TTT AGC AAG TGT GGC-3’), digested with
HindIII and Xho1, and ligated in-frame into a pcDNA3 vector.
3.5.4 Immunoprecipitation and immunoblotting
Cells lysis procedures utilized to examine MDM2 neddylation were described previously [215].
Whole cells extracts from cycloheximide experiments and DNA damage-treated cells were
prepared by standard lysis procedures using EBC buffer (50 mM Tris (pH 8.0), 120mM NaCl,
0.5% nonidet P-40) supplemented with complete protease inhibitors (Roche). Protein
concentrations were determined using the Bradford method (Bio-Rad) and equal amounts of
whole cell extracts were separated by SDS-PAGE. Proteins were transferred to nitrocellulose
membranes (Bio-Rad), and immunoblotting procedures were performed as previously described
[215].
3.5.5 Protein turnover assays
MDM2 half-life determination was performed as previously described [216]. Briefly, transfected
cells were treated with 40 µg/mL of cycloheximide (Sigma) for the indicated time periods, and
cell lysis and immunoblot procedures were performed as described above. Image J
(http://rsb.info.nih.gov/ij) software was used to perform densitometry of MDM2 signals,

78
quantifying values expressed as optical density, and graphed as a percentage against the non
cycloheximide-treated sample. A trendline was derived to determine the half-life of MDM2.
3.5.6 RNAi
siRNA transfection was performed as previously described [216] using the following siRNA
oligos (Dharmacon): NEDP1-1: 5’-CAGAGAAACUGGAGGCUUUdUdU-3’ and 5’-
AAAGCCUCCAGUUUCUCUGdUdU-3’; NEDP1-2: 5’-
GGAUGUACGUGAUAUGUAAdUdU-3’ and 5’-UUACAUAUCACGUACAUCCdUdU-3’;
and siGENOME RISC-Free Control siRNA (Dharmacon) was used as the negative control.
Briefly, cells were seeded in 1.5 cm plates at a confluency of approximately 30-50%. Oligos
were resuspended, and transfected with oligofectamine (Invitrogen), according to manufacturer’s
instructions at a final siRNA concentration of 75 nM. For chemosensitivity experiments cells
were treated with DOX 48 h post-siRNA treatment.
3.5.7 TUNEL assays
MCF7 cells were transfected with siRNAs as described above. 48 h post-transfection, cells were
treated with DOX for 48 h, trypsinized and 5x104
cells were mounted on glass slides by cytospin
centrifugation. TUNEL assays were performed according to manufacturer’s protocol (Promega)
and images were taken by an inverted epifluorescence microscope (Zeiss Axiovert 200).
TUNEL and DAPI positive cells were counted using Volocity software and confirmed by manual

79
counting. Equal amounts of whole cell extracts generated from the remaining cells were
resolved by SDS-PAGE and immunoblotted with an anti-cPARP antibody to compare with
TUNEL results.

80
Chapter 4:
Conclusions and future directions
Targeting the MDM2-TAp73 interaction as an anti-cancer strategy
Since MDM2 amplification represents an alternative mechanism to inhibit p53 function that
occurs in approximately 10% of all human tumors, a number of small molecule inhibitors have
been developed to target the MDM2-p53 interaction. The proof of concept of this approach was
established using synthetic peptides and protein aptamers in the late 1990s [217]. Subsequently,
a number of compounds have been developed, including the Nutlins, which were the first
selective MDM2 inhibitors [121]. Nutlins are a series of cis-imidazoline analogs discovered by
screening a diverse synthetic chemical library for selective small-molecule antagonists of MDM2
at the Roche Research Center in Nutley, New Jersey. Nutlins mimic the interaction with the p53
peptide, whereby the imidazoline scaffold replaces the p53 helical backbone normally occupied
by amino acid residues Phe19, Trp23, and Leu26 in the p53-binding pocket of MDM2. The
Nutlin-3a enantiomer was found to disrupt the MDM2-p53 interaction with the lowest IC50 of the
three cis-imidazoline analogs identified.
Novel chemical screens for small molecules that specifically inhibit ubiquitin E3 ligases
targeting p73 for degradation are ongoing. The caveat to this approach is that the majority of E3
ligases targeting p73 identified to date similarly regulate both TA and ΔN isoforms. However,
the widespread overexpression of oncogenic ΔNp73 in a number of human cancers limits the
clinical utility of ubiquitin E3 ligase inhibitors targeting both TA and ΔN p73. In light of the fact
the p53 N-terminal residues Phe19, Trp23, and Leu26 that directly contact MDM2 are conserved

81
in TAp73 and not Np73, and due to the fact MDM2 inhibits TAp73 transcriptional and
apoptotic activity but does not regulate ΔNp73, our lab investigated the utility of targeting the
MDM2-TAp73 interaction with Nutlin-3 [122, 163]. Studies by Lau et al. demonstrated that
Nutlin-3 could induce apoptosis in cancer lines lacking wild-type p53 in a TAp73-dependent
manner [218]. While cell viability and apoptosis assays in cancer cell lines possessing wild-type
p53 yielded IC50 values of less than 10 μM of Nutlin-3, 20-30 μM of drug were required to
achieve the same effects in cells lines with p53 deletion or mutation. Although the higher doses
required to induce TAp73-dependent cell death may argue against the clinical utility of this drug
in tumors lacking functional p53, Nutlin did disrupt MDM2-TAp73 at lower concentrations in a
dose-dependent manner, suggesting Nutlin could still be used sensitize these tumors to
chemotherapy treatment. Future studies testing combination treatment regimens with
chemotherapies and Nutlin to identify synergistic drug combinations inducing TAp73-mediated
apoptosis will be of significant clinical value. Furthermore, identifying cis-imidazoline
compounds that more effectively disrupt the MDM2-TAp73 interaction regardless of their ability
to modulate p53 may prove worthwhile since more than half of all human tumors possess mutant
p53 limiting the utility of drugs aimed at disrupting MDM2-p53 binding. In cases where p53 is
mutated, inhibition of the MDM2-p53 interaction may not be desirable due to the gain of
function activity of a subset of mutant p53 proteins, which include the ability to inhibit the pro-
apoptotic activity of TAp73 and TAp63. Therefore, compounds inhibiting the MDM2
interaction with TAp73 more effectively than p53 may have important clinical relevance.
Recently, the drug RETRA has been shown to possess anti-cancer properties attributed to the
inhibition of the interaction between mutant p53 and TAp73, and thus, could prove useful in
combination therapy with Nutlin in p53 mutant human tumors [219].

82
Since the discovery of Nutlins a number of novel compounds have been identified that
inhibit the MDM2-p53 interaction, including peptidomimetics and small molecules such as
chalcones, aryl sulphonamides, 1,4-benzodiazepine-2,4-diones, isoindolinones and spiro-
oxindoles (reviewed in [220]). Determining whether these drugs modulate the MDM2-TAp73
interaction, and the efficacy of this inhibition relative to that of p53 is a worthwhile endeavour.
Lastly, testing additional MDM2-TAp73 inhibitors, alone and in combination with other
chemotherapies in patient cohorts with human tumors possessing both MDM2 amplification and
p53 mutation such as certain soft tissue sarcomas or bladder cancers, may inform the
stratification of patient subpopulations on the basis of molecular potential to benefit from such
treatment strategies.
NEDD8-activating enzyme (NAE) inhibitor as an approach to treat cancer: Targeting the MDM2-p53/TAp73 axis?
Considering the utility of the proteasome inhibitor bortezomib (Velcade) in the treatment of
multiple myeloma and mantle cell lymphoma [221, 222], Millennium Pharmaceuticals, Inc.
tested whether inhibiting the NEDD8 pathway would have therapeutic utility in limiting cancer
progression due to the implicated oncogenic role of NEDD8 and its targeted substrates [33, 223].
Analogous to ubiquitylation, the neddylation enzymatic cascade is composed of an E1 NEDD8-
activating enzyme (NAE is composed of APP-BP1/Uba3 heterodimer), E2 conjugating enzymes
(Ubc12 or UBE2F), and E3 ligases (RBX1, MDM2, c-CBL, SCFFBX011
, and DCN1). Recently,
Millennium Pharmaceuticals, Inc. reported their discovery of MLN4924 [33], a selective
inhibitor of the NAE, that effectively inhibits NEDD8 conjugation of substrates. MLN4924 is
the final product of an iterative medicinal chemistry effort following the identification of its

83
parent compound, N6-benzyl adenosine, in a high throughput screen for NAE inhibitors.
MLN4924 is structurally related to AMP and forms a covalent adduct with NEDD8, which is
catalyzed by the NAE in a process resembling the first NEDD8 adenylation step in the
neddylation cascade reaction (See Figure 4.1 below for illustration). The resulting stable
NEDD8-MLN4924 adduct positioned within the NAE active site blocks its enzymatic activity,
shutting down further NEDD8 conjugation [223]. Treatment of a number of cancer cell lines
with MLN4924 results in S-phase defects, DNA damage and subsequent apoptosis and can
inhibit tumor growth in vitro as well as in human tumor xenograft assays [33]. Notably, the
NEDD8 pathway regulates a number of substrates, of which, the Cullin-RING finger E3
ubiquitin ligases (CRL) are the best characterized. Soucy et al. demonstrated that MLN4924
inhibits neddylation of Cullins, increasing the stability of CRL substrates including NRF2
(transcription factor), p27 (cyclin-dependent kinase inhibitor) and CDT1 (involved in replication
initiation). Considering we have demonstrated NEDD8 conjugation promotes MDM2
stabilization and inhibits TAp73 and p53 transactivation function, examining whether MLN4924
modulates MDM2 stability and p53/TAp73-mediated apoptosis may provide mechanistic insight
into MLN4924-mediated anti-cancer activity.

84
Figure 4.1. MLN4924 inhibits NEDD8 conjugation by targeting the NEDD8-activating
enzyme (NAE). The neddylation enzymatic cascade is composed of an E1 activating enzyme
(the NAE is composed of APP-BP1/Uba3 heterodimer), E2 conjugating enzymes, and E3 ligases.
NEDD8 is synthesized as a precursor that is processed at a conserved C-terminal glycine residue
by the hydrolase activity of deneddylating enzymes exposing a glycine-glycine motif that serves
as the attachment site for target substrates (1). The exposed C-terminal glycine of NEDD8 is
adenylated by the NAE, APP-BP1/Uba3 heterodimer, in an ATP-dependent manner and
transferred to a cysteine side chain of the NAE via a thiol ester linkage (2). MLN4924 is
structurally related to AMP and forms a covalent adduct with NEDD8, which is catalyzed by the
NAE in a process resembling the NEDD8 adenylation step. The resulting stable NEDD8-
MLN4924 adduct positioned within the NAE active site blocks its enzymatic activity, limiting
further NEDD8 conjugation [223].
Role of NEDD8-mediated regulation of MDM2 and the p53 family in the development of the nervous system
Determining the mechanisms by which NEDD8 regulates embryogenesis remains an area of
research that has yet to be fully investigated. Inactivation of the NEDD8 pathway in mice by
deletion of Uba3, a component of the NAE, results in lethality at embryonic day 5.5 due to
increased apoptosis in the inner cell mass causing blastocyst death in the preimplantation stage
[19]. Interestingly, mdm2-/- mice die at the same stage in development, which is rescued with
simultaneous deletion of p53. Our data demonstrates the NEDD8 pathway modulates MDM2
stability to negatively regulate p53 activity establishing a role for NEDD8 in the inhibition of the

85
p53 pathway in cancer cell lines. Determining whether the p53 pathway is activated in Uba3-/-
embryos, and whether concomitant loss of p53 prolongs the survival of these mice would
establish a link between these pathways in mouse development providing mechanistic insight
into the role of NEDD8 in embryogenesis.
Studies have demonstrated a role for the p53 family, MDM2 and NEDD8 in
neurodevelopment to varying degrees and whether there is a functional link between these
pathways in the nervous system has not yet been investigated. NEDD8 was originally
discovered to be highly enriched in mouse neural precursor cells and found to be largely absent
in the adult murine brain [14]. However, studies examining the role of NEDD8 in
neurodevelopment have yet to be reported. Such studies should confirm the down-regulation of
NEDD8 expression during neural precursor cell differentiation. Furthermore, tissue-specific
deletion or overexpression of NEDD8 in the nervous system would help elucidate the role of the
NEDD8 pathway in neurodevelopment.
Conversely, a role of p53 in nervous system development has been described.
Approximately 25% of p53-/- embryos die in utero because of mid-brain exencephaly due to the
overproduction of neural tissue and subsequent failure of neural tube closure [51]. This
phenotype was similar to mice with targeted mutations in the intrinsic mitochondrial apoptotic
pathway, which prompted a number of in vitro studies utilizing cultured neurons to elucidate the
role of p53-mediatied apoptosis in both developing and mature injured neurons [76]. The
ultimate survival of any given neuron is determined by the interaction of two neurotrophin
receptors: the pro-survival TrkA and the p75NTR pro-apoptotic receptors (reviewed in [224]). It
is thought that sympathetic p75NTR-mediating signalling predisposes sympathetic neurons to
cell death via p53 and TAp63-dependent mechanisms that are rescued only in the presence of

86
sufficient NGF to robustly activate TrkA signalling [78]. A link between MDM2 and
TrkA/p75NTR signalling has not been established; however, NGF/TrkA survival signalling does
require an intact PI3-K/Akt pathway, which is known to regulate MDM2 [225, 226]. In addition,
specific deletion of mdm2 from the CNS of mice elicited hydranencephaly due to increased
apoptosis of neural precursor cells, and this phenotype was rescued with simultaneous loss of
p53 [138, 139]. In light of our findings that NEDD8-modification of MDM2 results in protein
stabilization inhibiting p53 function, examining a mechanistic link between NEDD8 down-
regulation during neurodevelopment and the regulation of neural precursor cells mediated by the
MDM2-p53 axis may reveal additional layers of p53 regulation in the developing nervous
system.
The work presented here has uncovered the novel process of MDM2-mediated
neddylation of TAp73, and identified the MDM2-TAp73 interaction as a therapeutically relevant
target for cancer treatment. Furthermore, we described the components that regulate MDM2
neddylation, and identified a novel mechanism of p53 activation involving NEDP1-modulation
of MDM2 stability in response to chemotherapy treatment. Our work identifying the
components of the NEDD8 pathway that regulate MDM2 and p53/TAp73 and their functional
consequence has laid the groundwork for future translational studies. We have provided the
rationale to examine these signalling pathways in neurodevelopment, and identified molecular
anti-cancer strategies that can be exploited with drugs currently available to treat human cancer
targeting MDM2 and the NEDD8 pathway.
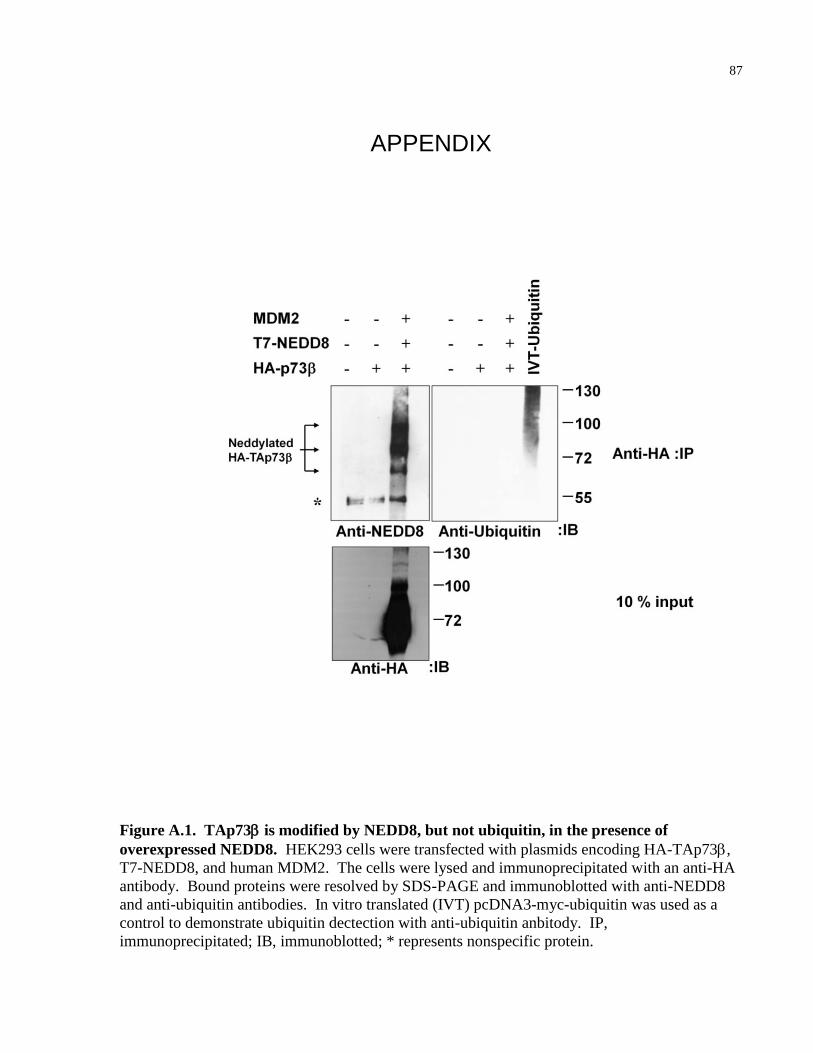
87
APPENDIX
Figure A.1. TAp73 is modified by NEDD8, but not ubiquitin, in the presence of
overexpressed NEDD8. HEK293 cells were transfected with plasmids encoding HA-TAp73,
T7-NEDD8, and human MDM2. The cells were lysed and immunoprecipitated with an anti-HA
antibody. Bound proteins were resolved by SDS-PAGE and immunoblotted with anti-NEDD8
and anti-ubiquitin antibodies. In vitro translated (IVT) pcDNA3-myc-ubiquitin was used as a
control to demonstrate ubiquitin dectection with anti-ubiquitin anbitody. IP,
immunoprecipitated; IB, immunoblotted; * represents nonspecific protein.
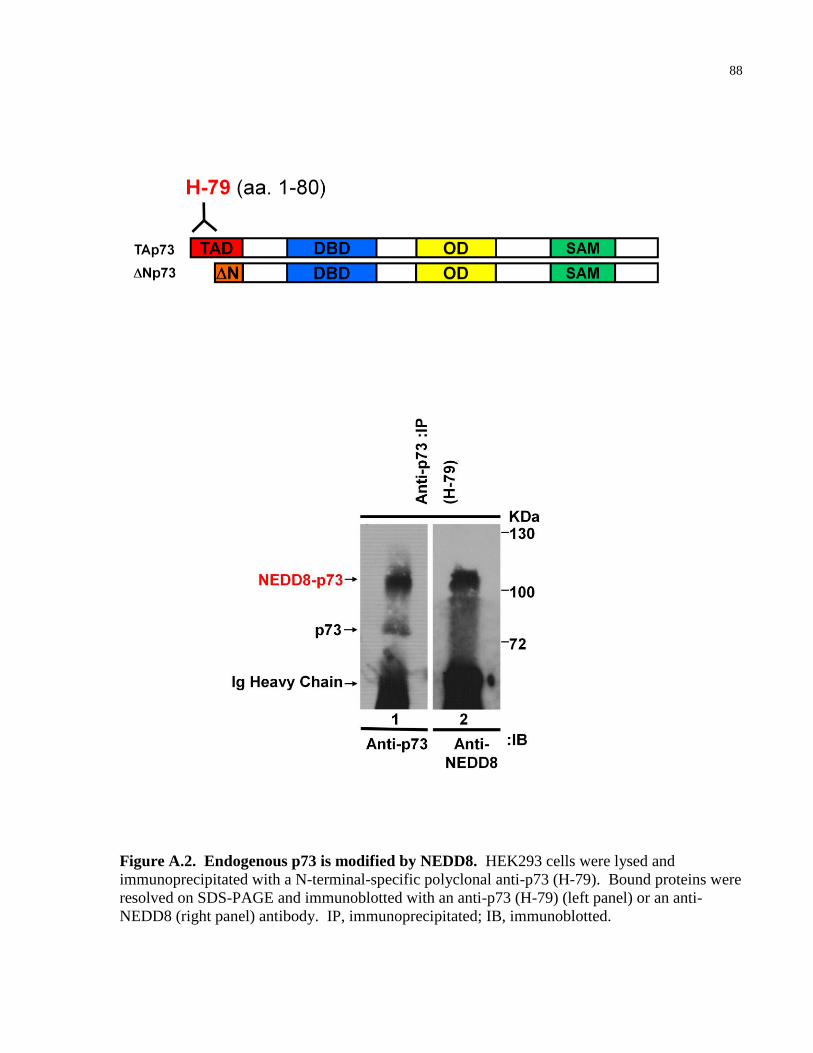
88
Figure A.2. Endogenous p73 is modified by NEDD8. HEK293 cells were lysed and
immunoprecipitated with a N-terminal-specific polyclonal anti-p73 (H-79). Bound proteins were
resolved on SDS-PAGE and immunoblotted with an anti-p73 (H-79) (left panel) or an anti-
NEDD8 (right panel) antibody. IP, immunoprecipitated; IB, immunoblotted.
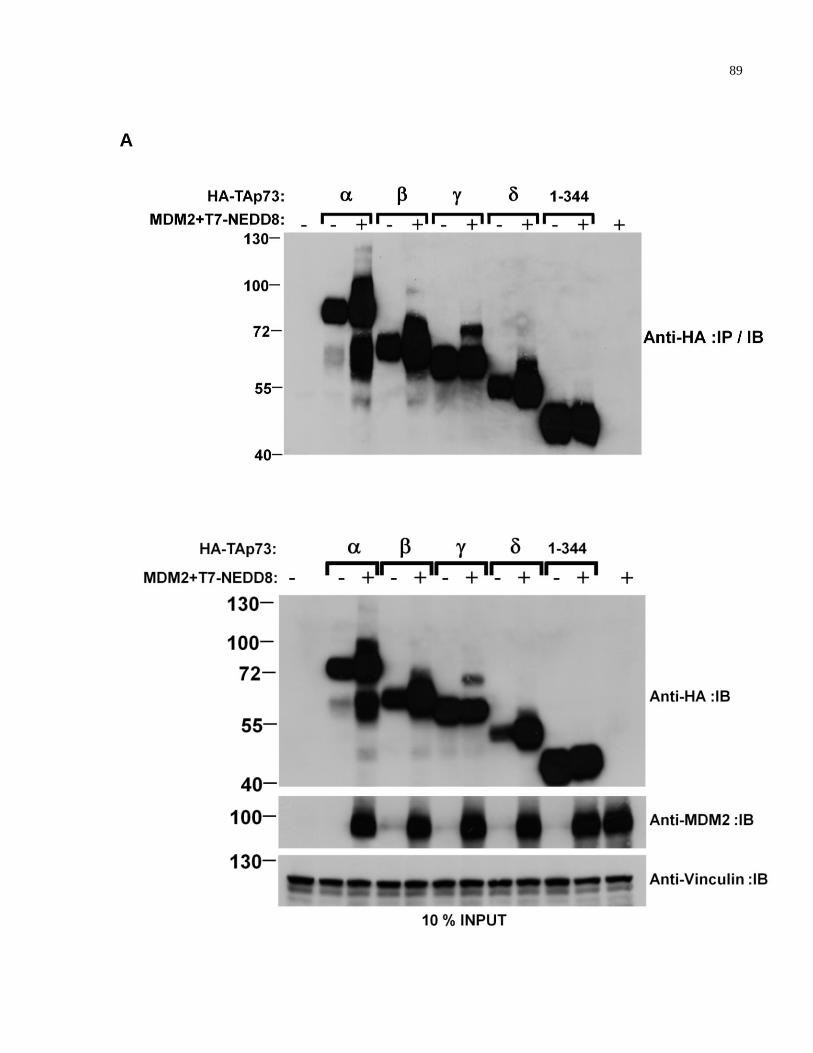
89
A
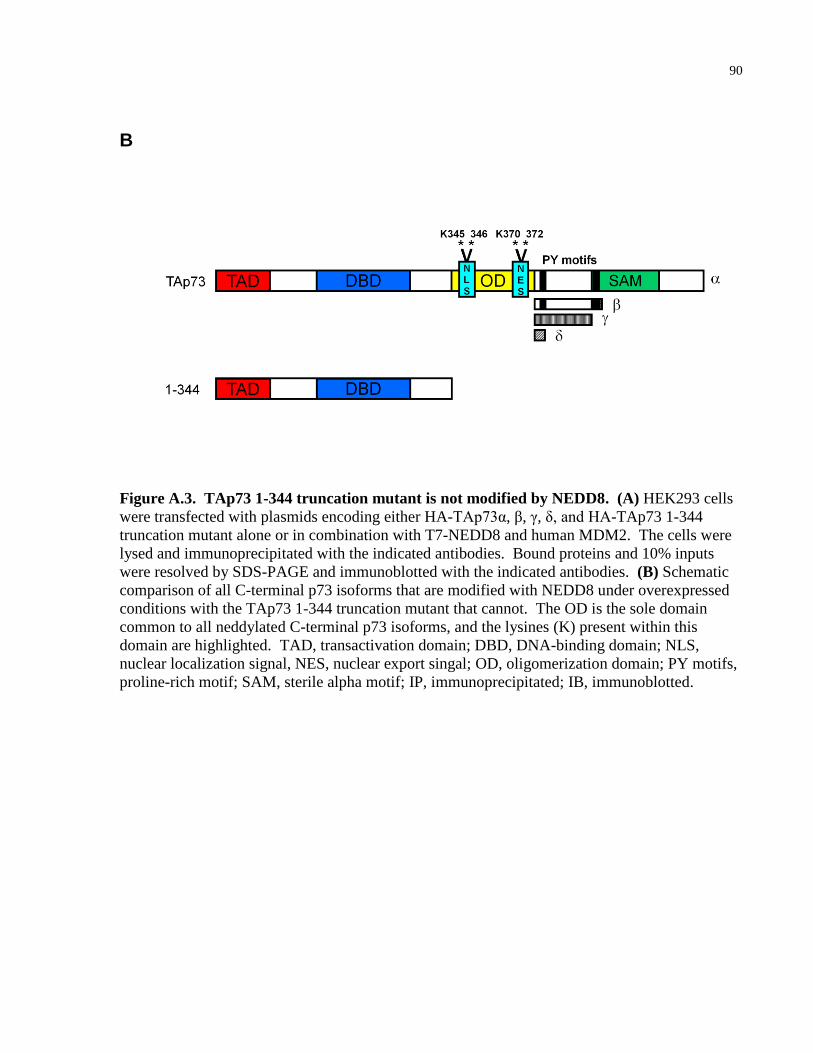
90
B
Figure A.3. TAp73 1-344 truncation mutant is not modified by NEDD8. (A) HEK293 cells
were transfected with plasmids encoding either HA-TAp73α, β, γ, δ, and HA-TAp73 1-344
truncation mutant alone or in combination with T7-NEDD8 and human MDM2. The cells were
lysed and immunoprecipitated with the indicated antibodies. Bound proteins and 10% inputs
were resolved by SDS-PAGE and immunoblotted with the indicated antibodies. (B) Schematic
comparison of all C-terminal p73 isoforms that are modified with NEDD8 under overexpressed
conditions with the TAp73 1-344 truncation mutant that cannot. The OD is the sole domain
common to all neddylated C-terminal p73 isoforms, and the lysines (K) present within this
domain are highlighted. TAD, transactivation domain; DBD, DNA-binding domain; NLS,
nuclear localization signal, NES, nuclear export singal; OD, oligomerization domain; PY motifs,
proline-rich motif; SAM, sterile alpha motif; IP, immunoprecipitated; IB, immunoblotted.
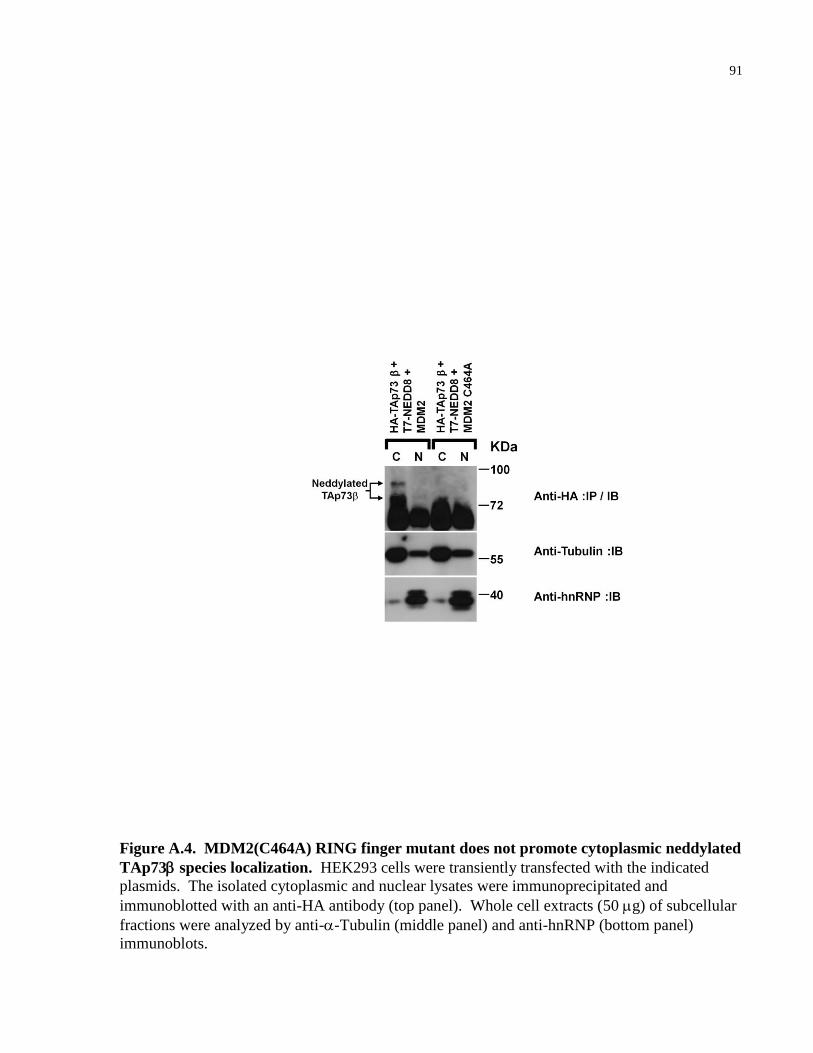
91
Figure A.4. MDM2(C464A) RING finger mutant does not promote cytoplasmic neddylated
TAp73 species localization. HEK293 cells were transiently transfected with the indicated
plasmids. The isolated cytoplasmic and nuclear lysates were immunoprecipitated and
immunoblotted with an anti-HA antibody (top panel). Whole cell extracts (50 g) of subcellular
fractions were analyzed by anti--Tubulin (middle panel) and anti-hnRNP (bottom panel)
immunoblots.

92
Figure A.5. Ectopic NEDD8 expression prolongs MDM2 half-life. U2OS cells were
transfected with plasmids encoding T7-NEDD8 or T7-NEDD8ΔGG. 48 h post-transfection,
cells were treated with 40 μg/mL cycloheximide (CHX) for the indicated time periods. Cells
were lysed, equal amounts of whole cell extracts resolved by SDS-PAGE and immunoblotted
with the indicated antibodies (left panel). Protein levels of endogenous MDM2 were quantified
by densitometry as previously described [216]. IB: immunoblot.
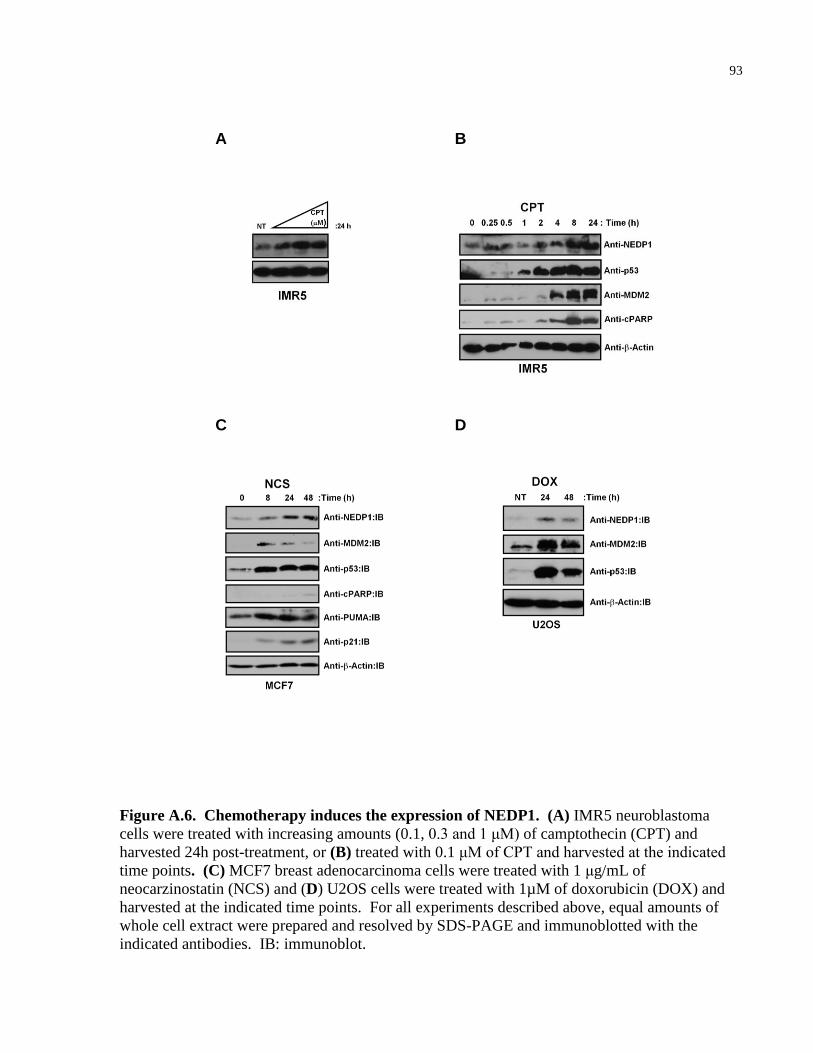
93
A B
C D
Figure A.6. Chemotherapy induces the expression of NEDP1. (A) IMR5 neuroblastoma
cells were treated with increasing amounts (0.1, 0.3 and 1 μM) of camptothecin (CPT) and
harvested 24h post-treatment, or (B) treated with 0.1 μM of CPT and harvested at the indicated
time points. (C) MCF7 breast adenocarcinoma cells were treated with 1 μg/mL of
neocarzinostatin (NCS) and (D) U2OS cells were treated with 1µM of doxorubicin (DOX) and
harvested at the indicated time points. For all experiments described above, equal amounts of
whole cell extract were prepared and resolved by SDS-PAGE and immunoblotted with the
indicated antibodies. IB: immunoblot.

94
Figure A.7. Ectopic expression of p53 has negligible effect on NEDP1 expression. p53-/-
H1299 lung adenocarcinoma cells were transfected with empty plasmid (MOCK) or plasmid
encoding HA-p53. Equal amounts of whole cell extracts were resolved by SDS-PAGE and
immunoblotted with the indicated antibodies.
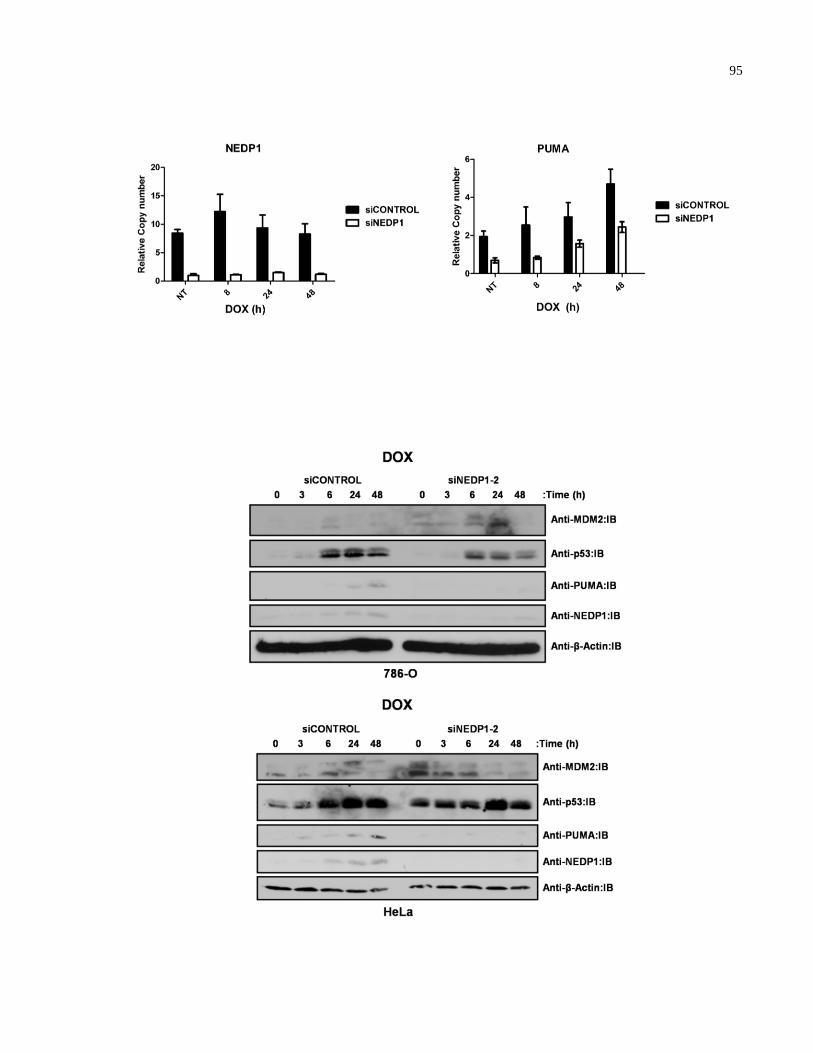
95

96
Figure A.8. siRNA-mediated NEDP1 knockdown decreases p53 activation in response to
doxorubicin. (A) MCF7 cells were transfected with siNEDP1-2 and treated 48 h post-
transfection with 0.1 μM doxorubicin and harvested at the indicated time points. mRNA was
isolated using the RNeasy kit (QIAGEN). 1 µL random hexamer (Fermentas) was incubated
with 2 µg of RNA and dH2O (to a final volume of 12 µL) for 10 min at 70°C in a thermal cycler
(MJ Research). The mixture was cooled to 4°C at which time 4 µL of 5x 1st strand reaction
buffer, 2 µL of 0.1 M DTT, 1 µL of 10 mM dNTPs, and 1 µL Superscript II reverse transcriptase
(Invitrogen) were added. cDNA synthesis was performed for 10 min at 70°C, followed by 1.5 h
at 42°C, and 15 min at 70°C in a thermal cycler. Human genomic DNA standards (Roche) or
cDNA samples were added to the qPCR reaction in a final volume of 10 µL containing 1x PCR
buffer (without MgCl2), 3 mM MgCl2, 0.25 units of Platinum Taq DNA polymerase, 0.2 mM
dNTPs, 0.3 µL SYBR Green I, 0.2 µL ROX reference dye, and 0.5 µM each primer (Invitrogen).
Amplification conditions were performed as follows: 95°C (3 min), 40 cycles of 95°C (10 s),
65°C (15 s), 72°C (20 s), followed by one cycle of 95°C (15 s), 60°C (15s) and 95°C (15 s).
qPCR was performed using the ABI Prism 7900HT Sequence Detection System (Applied
Biosystems). Gene-specific oligonucleotide primer sets for NEDP1 (5’-
GAGCAGCCCTCGTCAGTACAA -3’ and 5’-GCTTGGCGGATCCAATAGTG-3’), PUMA
(5’-GAAGGACAAAACTCACCAAACCA-3’ and 5’-AGACCCCATGCCAAATTTCA-3’) and
U1AsnRNP1 (5’-CAACGACAGCCGAGACATGTA-3’ and 5’-
AGCCTCCATCAAATACCCATT-3’) were designed using Primer Express (Applied
Biosystems). Expression levels of the various transcripts were determined by taking the average
Ct value for each cDNA sample performed in triplicate and measured against a standard plot of
Ct values from amplification of serially diluted human genomic DNA standards. Since the Ct
value is inversely proportional to the log of the initial copy number, the copy number of an
experimental mRNA can be obtained from linear regression of the standard curve. A measure of
the fold difference in copy number was determined for each mRNA. Values were normalized to
expression of U1AsnRNP1 mRNA and represented as the mean value of three independent
experiments performed in triplicate ± standard deviations. (B) 786-O renal clear cell carcinoma
(top panel) and HeLa cervical carcinoma cells (bottom panel) were transfected with siNEDP1-2
or siCONTROL and treated 48 h post-transfection with 0.1 μM DOX. Cells were harvested at
the indicated time points, lysed, equal amounts of whole cell extract resolved by SDS-PAGE and
immunoblotted with the indicated antibodies. IB: immunoblot.
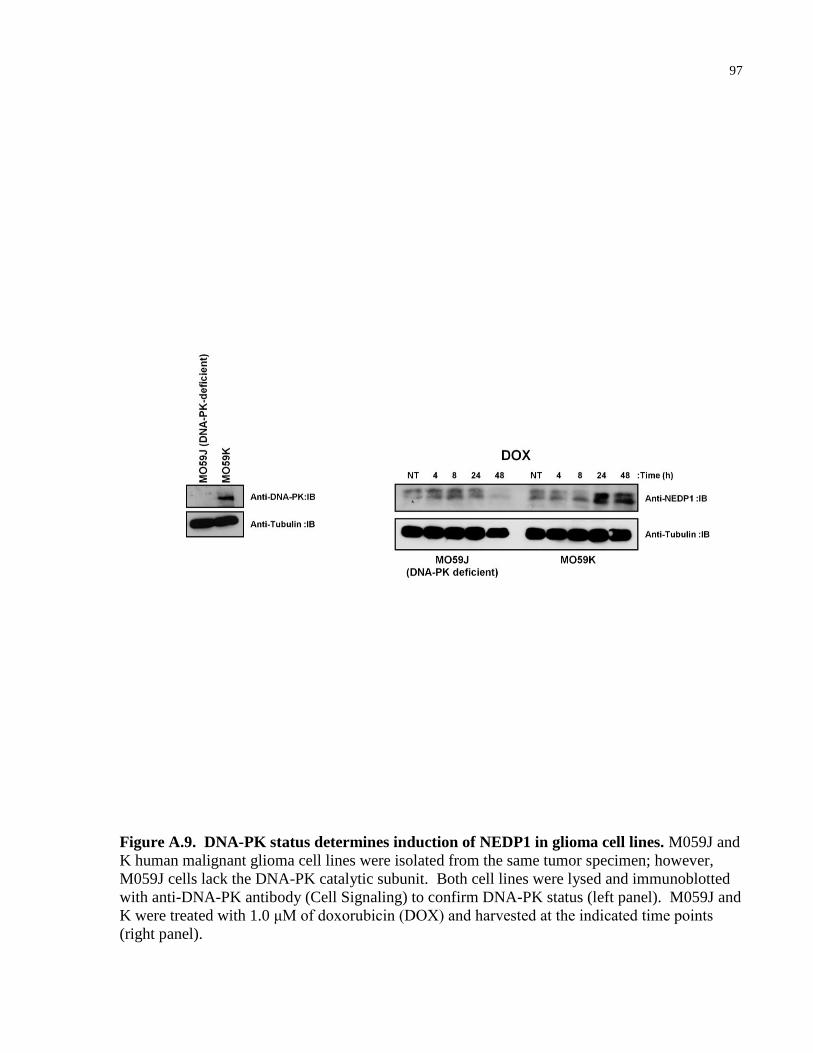
97
Figure A.9. DNA-PK status determines induction of NEDP1 in glioma cell lines. M059J and
K human malignant glioma cell lines were isolated from the same tumor specimen; however,
M059J cells lack the DNA-PK catalytic subunit. Both cell lines were lysed and immunoblotted
with anti-DNA-PK antibody (Cell Signaling) to confirm DNA-PK status (left panel). M059J and
K were treated with 1.0 μM of doxorubicin (DOX) and harvested at the indicated time points
(right panel).

98
REFERENCES
1 Ciehanover, A., Hod, Y. and Hershko, A. (1978) A heat-stable polypeptide component of
an ATP-dependent proteolytic system from reticulocytes. Biochemical and biophysical
research communications. 81, 1100-1105
2 Wilkinson, K. D., Urban, M. K. and Haas, A. L. (1980) Ubiquitin is the ATP-dependent
proteolysis factor I of rabbit reticulocytes. The Journal of biological chemistry. 255,
7529-7532
3 Goldstein, G., Scheid, M., Hammerling, U., Schlesinger, D. H., Niall, H. D. and Boyse,
E. A. (1975) Isolation of a polypeptide that has lymphocyte-differentiating properties and
is probably represented universally in living cells. Proceedings of the National Academy
of Sciences of the United States of America. 72, 11-15
4 Hershko, A. (2005) The ubiquitin system for protein degradation and some of its roles in
the control of the cell division cycle. Cell death and differentiation. 12, 1191-1197
5 Pickart, C. M. (2001) Mechanisms underlying ubiquitination. Annual review of
biochemistry. 70, 503-533
6 Hoppe, T. (2005) Multiubiquitylation by E4 enzymes: 'one size' doesn't fit all. Trends in
biochemical sciences. 30, 183-187
7 Pickart, C. M. and Cohen, R. E. (2004) Proteasomes and their kin: proteases in the
machine age. Nat Rev Mol Cell Biol. 5, 177-187
8 Gao, M. and Karin, M. (2005) Regulating the regulators: control of protein ubiquitination
and ubiquitin-like modifications by extracellular stimuli. Molecular cell. 19, 581-593
9 Joazeiro, C. A. and Weissman, A. M. (2000) RING finger proteins: mediators of
ubiquitin ligase activity. Cell. 102, 549-552
10 Huibregtse, J. M., Scheffner, M. and Howley, P. M. (1993) Localization of the E6-AP
regions that direct human papillomavirus E6 binding, association with p53, and
ubiquitination of associated proteins. Molecular and cellular biology. 13, 4918-4927
11 Fang, S. and Weissman, A. M. (2004) A field guide to ubiquitylation. Cell Mol Life Sci.
61, 1546-1561
12 Kirkpatrick, D. S., Denison, C. and Gygi, S. P. (2005) Weighing in on ubiquitin: the
expanding role of mass-spectrometry-based proteomics. Nature cell biology. 7, 750-757
13 Pickart, C. M. and Fushman, D. (2004) Polyubiquitin chains: polymeric protein signals.
Curr Opin Chem Biol. 8, 610-616

99
14 Kumar, S., Tomooka, Y. and Noda, M. (1992) Identification of a set of genes with
developmentally down-regulated expression in the mouse brain. Biochemical and
biophysical research communications. 185, 1155-1161
15 Kumar, S., Yoshida, Y. and Noda, M. (1993) Cloning of a cDNA which encodes a novel
ubiquitin-like protein. Biochemical and biophysical research communications. 195, 393-
399
16 Kurz, T., Chou, Y. C., Willems, A. R., Meyer-Schaller, N., Hecht, M. L., Tyers, M.,
Peter, M. and Sicheri, F. (2008) Dcn1 functions as a scaffold-type E3 ligase for cullin
neddylation. Molecular cell. 29, 23-35
17 Kamitani, T., Kito, K., Nguyen, H. P. and Yeh, E. T. (1997) Characterization of NEDD8,
a developmentally down-regulated ubiquitin-like protein. The Journal of biological
chemistry. 272, 28557-28562
18 Pan, Z. Q., Kentsis, A., Dias, D. C., Yamoah, K. and Wu, K. (2004) Nedd8 on cullin:
building an expressway to protein destruction. Oncogene. 23, 1985-1997
19 Tateishi, K., Omata, M., Tanaka, K. and Chiba, T. (2001) The NEDD8 system is essential
for cell cycle progression and morphogenetic pathway in mice. The Journal of cell
biology. 155, 571-579
20 Ou, C. Y., Lin, Y. F., Chen, Y. J. and Chien, C. T. (2002) Distinct protein degradation
mechanisms mediated by Cul1 and Cul3 controlling Ci stability in Drosophila eye
development. Genes & development. 16, 2403-2414
21 Lammer, D., Mathias, N., Laplaza, J. M., Jiang, W., Liu, Y., Callis, J., Goebl, M. and
Estelle, M. (1998) Modification of yeast Cdc53p by the ubiquitin-related protein rub1p
affects function of the SCFCdc4 complex. Genes & development. 12, 914-926
22 Osaka, F., Saeki, M., Katayama, S., Aida, N., Toh, E. A., Kominami, K., Toda, T.,
Suzuki, T., Chiba, T., Tanaka, K. and Kato, S. (2000) Covalent modifier NEDD8 is
essential for SCF ubiquitin-ligase in fission yeast. The EMBO journal. 19, 3475-3484
23 Dharmasiri, S., Dharmasiri, N., Hellmann, H. and Estelle, M. (2003) The RUB/Nedd8
conjugation pathway is required for early development in Arabidopsis. The EMBO
journal. 22, 1762-1770
24 Gao, M. X., Liao, E. H., Yu, B., Wang, Y., Zhen, M. and Derry, W. B. (2008) The SCF
FSN-1 ubiquitin ligase controls germline apoptosis through CEP-1/p53 in C. elegans.
Cell death and differentiation. 15, 1054-1062
25 Jones, D. and Candido, E. P. (2000) The NED-8 conjugating system in Caenorhabditis
elegans is required for embryogenesis and terminal differentiation of the hypodermis.
Developmental biology. 226, 152-165

100
26 Kurz, T., Pintard, L., Willis, J. H., Hamill, D. R., Gonczy, P., Peter, M. and Bowerman,
B. (2002) Cytoskeletal regulation by the Nedd8 ubiquitin-like protein modification
pathway. Science. 295, 1294-1298
27 Kawakami, T., Chiba, T., Suzuki, T., Iwai, K., Yamanaka, K., Minato, N., Suzuki, H.,
Shimbara, N., Hidaka, Y., Osaka, F., Omata, M. and Tanaka, K. (2001) NEDD8 recruits
E2-ubiquitin to SCF E3 ligase. The EMBO journal. 20, 4003-4012
28 Kim, M., Gans, J. D., Nogueira, C., Wang, A., Paik, J. H., Feng, B., Brennan, C., Hahn,
W. C., Cordon-Cardo, C., Wagner, S. N., Flotte, T. J., Duncan, L. M., Granter, S. R. and
Chin, L. (2006) Comparative oncogenomics identifies NEDD9 as a melanoma metastasis
gene. Cell. 125, 1269-1281
29 Wang, X., Trotman, L. C., Koppie, T., Alimonti, A., Chen, Z., Gao, Z., Wang, J.,
Erdjument-Bromage, H., Tempst, P., Cordon-Cardo, C., Pandolfi, P. P. and Jiang, X.
(2007) NEDD4-1 is a proto-oncogenic ubiquitin ligase for PTEN. Cell. 128, 129-139
30 Chen, L. C., Manjeshwar, S., Lu, Y., Moore, D., Ljung, B. M., Kuo, W. L., Dairkee, S.
H., Wernick, M., Collins, C. and Smith, H. S. (1998) The human homologue for the
Caenorhabditis elegans cul-4 gene is amplified and overexpressed in primary breast
cancers. Cancer research. 58, 3677-3683
31 Kim, S. S., Shago, M., Kaustov, L., Boutros, P. C., Clendening, J. W., Sheng, Y., Trentin,
G. A., Barsyte-Lovejoy, D., Mao, D. Y., Kay, R., Jurisica, I., Arrowsmith, C. H. and
Penn, L. Z. (2007) CUL7 is a novel antiapoptotic oncogene. Cancer research. 67, 9616-
9622
32 Chairatvit, K. and Ngamkitidechakul, C. (2007) Control of cell proliferation via elevated
NEDD8 conjugation in oral squamous cell carcinoma. Molecular and cellular
biochemistry. 306, 163-169
33 Soucy, T. A., Smith, P. G., Milhollen, M. A., Berger, A. J., Gavin, J. M., Adhikari, S.,
Brownell, J. E., Burke, K. E., Cardin, D. P., Critchley, S., Cullis, C. A., Doucette, A.,
Garnsey, J. J., Gaulin, J. L., Gershman, R. E., Lublinsky, A. R., McDonald, A., Mizutani,
H., Narayanan, U., Olhava, E. J., Peluso, S., Rezaei, M., Sintchak, M. D., Talreja, T.,
Thomas, M. P., Traore, T., Vyskocil, S., Weatherhead, G. S., Yu, J., Zhang, J., Dick, L.
R., Claiborne, C. F., Rolfe, M., Bolen, J. B. and Langston, S. P. (2009) An inhibitor of
NEDD8-activating enzyme as a new approach to treat cancer. Nature. 458, 732-736
34 Xirodimas, D. P. (2008) Novel substrates and functions for the ubiquitin-like molecule
NEDD8. Biochemical Society transactions. 36, 802-806
35 Lane, D. P. and Crawford, L. V. (1979) T antigen is bound to a host protein in SV40-
transformed cells. Nature. 278, 261-263

101
36 Linzer, D. I. and Levine, A. J. (1979) Characterization of a 54K dalton cellular SV40
tumor antigen present in SV40-transformed cells and uninfected embryonal carcinoma
cells. Cell. 17, 43-52
37 Lane, D. P. and Benchimol, S. (1990) p53: oncogene or anti-oncogene? Genes &
development. 4, 1-8
38 Yu, J. and Zhang, L. (2005) The transcriptional targets of p53 in apoptosis control.
Biochemical and biophysical research communications. 331, 851-858
39 Chipuk, J. E., Kuwana, T., Bouchier-Hayes, L., Droin, N. M., Newmeyer, D. D., Schuler,
M. and Green, D. R. (2004) Direct activation of Bax by p53 mediates mitochondrial
membrane permeabilization and apoptosis. Science. 303, 1010-1014
40 Chipuk, J. E., Bouchier-Hayes, L., Kuwana, T., Newmeyer, D. D. and Green, D. R.
(2005) PUMA couples the nuclear and cytoplasmic proapoptotic function of p53.
Science. 309, 1732-1735
41 Sot, B., Freund, S. M. and Fersht, A. R. (2007) Comparative biophysical characterization
of p53 with the pro-apoptotic BAK and the anti-apoptotic BCL-xL. The Journal of
biological chemistry. 282, 29193-29200
42 Green, D. R. and Kroemer, G. (2009) Cytoplasmic functions of the tumour suppressor
p53. Nature. 458, 1127-1130
43 Aylon, Y. and Oren, M. (2007) Living with p53, dying of p53. Cell. 130, 597-600
44 Lane, D. (1992) p53, guardian of the genome. Nature. 358, 15-16
45 Hanahan, D. and Weinberg, R. A. (2000) The hallmarks of cancer. Cell. 100, 57-70
46 Toledo, F. and Wahl, G. M. (2006) Regulating the p53 pathway: in vitro hypotheses, in
vivo veritas. Nature reviews. 6, 909-923
47 Brosh, R. and Rotter, V. (2009) When mutants gain new powers: news from the mutant
p53 field. Nature reviews. 9, 701-713
48 Malkin, D., Li, F. P., Strong, L. C., Fraumeni, J. F., Nelson, C., Kim, D. H., Kaassel, J.,
Gryka, M. A., Bischoff, F. Z., Tainsky, M. A. and Friend, S. (1990) Germ line p53
mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms.
Science. 250, 1233-1238
49 Donehower, L. A., Harvey, M., Slagle, B. L., McArthur, M. J., Montgomery, C. A.,
Butel, J. S. and Bradley, A. (1992) p53-deficient mice are developmentally normal but
susceptible to spontaneous tumours. Nature. 356, 215-221

102
50 Lowe, S. W., Schmitt, E. M., Smith, S. W., Osborne, B. A. and Jacks, T. (1993) p53 is
Required for Radiation-Induced Apoptosis in Mouse Thymocytes. Nature. 362, 847-849
51 Sah, V. P., Attardi, L. D., Mulligan, G. J., Williams, B. O., Bronson, R. T. and Jacks, T.
(1995) A subset of p53-deficient embryos exhibit exencephaly. Nature genetics. 10, 175-
180
52 Hu, W., Feng, Z., Teresky, A. K. and Levine, A. J. (2007) p53 regulates maternal
reproduction through LIF. Nature. 450, 721-724
53 Lang, G. A., Iwakuma, T., Suh, Y. A., Liu, G., Rao, V. A., Parant, J. M., Valentin-Vega,
Y. A., Terzian, T., Caldwell, L. C., Strong, L. C., El-Naggar, A. K. and Lozano, G.
(2004) Gain of function of a p53 hot spot mutation in a mouse model of Li-Fraumeni
syndrome. Cell. 119, 861-872
54 Olive, K. P., Tuveson, D. A., Ruhe, Z. C., Yin, B., Willis, N. A., Bronson, R. T.,
Crowley, D. and Jacks, T. (2004) Mutant p53 gain of function in two mouse models of
Li-Fraumeni syndrome. Cell. 119, 847-860
55 Martins, C. P., Brown-Swigart, L. and Evan, G. I. (2006) Modeling the therapeutic
efficacy of p53 restoration in tumors. Cell. 127, 1323-1334
56 Ventura, A., Kirsch, D. G., McLaughlin, M. E., Tuveson, D. A., Grimm, J., Lintault, L.,
Newman, J., Reczek, E. E., Weissleder, R. and Jacks, T. (2007) Restoration of p53
function leads to tumour regression in vivo. Nature. 445, 661-665
57 Xue, W., Zender, L., Miething, C., Dickins, R. A., Hernando, E., Krizhanovsky, V.,
Cordon-Cardo, C. and Lowe, S. W. (2007) Senescence and tumour clearance is triggered
by p53 restoration in murine liver carcinomas. Nature. 445, 656-660
58 Kaghad, M., Bonnet, H., Yang, A., Creancier, L., Biscan, J. C., Valent, A., Minty, A.,
Chalon, P., Lelias, J. M., Dumont, X., Ferrara, P., McKeon, F. and Caput, D. (1997)
Monoallelically expressed gene related to p53 at 1p36, a region frequently deleted in
neuroblastoma and other human cancers. Cell. 90, 809-819
59 Yang, A., Kaghad, M., Wang, Y., Gillett, E., Fleming, M. D., Dotsch, V., Andrews, N.
C., Caput, D. and McKeon, F. (1998) p63, a p53 homolog at 3q27-29, encodes multiple
products with transactivating, death-inducing, and dominant-negative activities.
Molecular cell. 2, 305-316
60 Murray-Zmijewski, F., Lane, D. P. and Bourdon, J. C. (2006) p53/p63/p73 isoforms: an
orchestra of isoforms to harmonise cell differentiation and response to stress. Cell death
and differentiation. 13, 962-972

103
61 De Laurenzi, V., Costanzo, A., Barcaroli, D., Terrinoni, A., Falco, M., Annicchiarico-
Petruzzelli, M., Levrero, M. and Melino, G. (1998) Two new p73 splice variants, gamma
and delta, with different transcriptional activity. J Exp Med. 188, 1763-1768
62 De Laurenzi, V. D., Catani, M. V., Terrinoni, A., Corazzari, M., Melino, G., Costanzo,
A., Levrero, M. and Knight, R. A. (1999) Additional complexity in p73: induction by
mitogens in lymphoid cells and identification of two new splicing variants epsilon and
zeta [letter]. Cell death and differentiation. 6, 389-390
63 Ishimoto, O., Kawahara, C., Enjo, K., Obinata, M., Nukiwa, T. and Ikawa, S. (2002)
Possible oncogenic potential of DeltaNp73: a newly identified isoform of human p73.
Cancer research. 62, 636-641.
64 Yang, A., Walker, N., Bronson, R., Kaghad, M., Oosterwegel, M., Bonnin, J., Vagner, C.,
Bonnet, H., Dikkes, P., Sharpe, A., McKeon, F. and Caput, D. (2000) p73-deficient mice
have neurological, pheromonal and inflammatory defects but lack spontaneous tumours.
Nature. 404, 99-103
65 Zaika, A. I., Slade, N., Erster, S. H., Sansome, C., Joseph, T. W., Pearl, M., Chalas, E.
and Moll, U. M. (2002) DeltaNp73, a dominant-negative inhibitor of wild-type p53 and
TAp73, is up-regulated in human tumors. J Exp Med. 196, 765-780.
66 Bourdon, J. C. (2007) p53 and its isoforms in cancer. British journal of cancer. 97, 277-
282
67 Marin, M. C., Jost, C., Irwin, M. S., DeCaprio, J. A., Caput, D. and Kaelin, W. G. (1998)
Viral Oncoproteins Discriminate between p53 and the p53 Homolog p73. Mol. Cell. Biol.
18, 6316-6324
68 Stiewe, T., Theseling, C. C. and Putzer, B. M. (2002) Transactivation-deficient Delta TA-
p73 Inhibits p53 by Direct Competition for DNA Binding. IMPLICATIONS FOR
TUMORIGENESIS. The Journal of biological chemistry. 277, 14177-14185.
69 Stiewe, T., Zimmermann, S., Frilling, A., Esche, H. and Putzer, B. M. (2002)
Transactivation-deficient DeltaTA-p73 acts as an oncogene. Cancer research. 62, 3598-
3602.
70 Jost, C., Marin, M. and Kaelin, W. J. (1997) p73 is a human p53-related protein that can
induce apoptosis. Nature. 389, 191-194
71 Osada, M., Ohba, M., Kawahara, C., Ishioka, C., Kanamaru, R., Katoh, I., Ikawa, Y.,
Nimura, Y., Nakagawara, A., Obinata, M. and Ikawa, S. (1998) Cloning and functional
analysis of human p51, which structurally and functionally resembles p53 [see
comments] [published erratum appears in Nat Med 1998 Sep;4(9):982]. Nature medicine.
4, 839-843

104
72 Nakagawa, T., Takahashi, M., Ozaki, T., Watanabe Ki, K., Todo, S., Mizuguchi, H.,
Hayakawa, T. and Nakagawara, A. (2002) Autoinhibitory regulation of p73 by Delta
Np73 to modulate cell survival and death through a p73-specific target element within the
Delta Np73 promoter. Molecular and cellular biology. 22, 2575-2585.
73 Pozniak, C. D., Radinovic, S., Yang, A., McKeon, F., Kaplan, D. R. and Miller, F. D.
(2000) An Anti-Apoptotic Role for the p53 Family Member, p73, During Developmental
Neuron Death. Science. 289, 304-306
74 Yang, A., Schweitzer, R., Sun, D., Kaghad, M., Walker, N., Bronson, R. T., Tabin, C.,
Sharpe, A., Caput, D., Crum, C. and McKeon, F. (1999) p63 is essential for regenerative
proliferation in limb, craniofacial and epithelial development. Nature. 398, 714-718
75 Pozniak, C. D., Barnabe-Heider, F., Rymar, V. V., Lee, A. F., Sadikot, A. F. and Miller,
F. D. (2002) p73 is required for survival and maintenance of CNS neurons. J Neurosci.
22, 9800-9809
76 Jacobs, W. B., Kaplan, D. R. and Miller, F. D. (2006) The p53 family in nervous system
development and disease. Journal of neurochemistry. 97, 1571-1584
77 Irwin, M. S. and Miller, F. D. (2004) p73: regulator in cancer and neural development.
Cell death and differentiation. 11 Suppl 1, S17-22
78 Jacobs, W. B., Govoni, G., Ho, D., Atwal, J. K., Barnabe-Heider, F., Keyes, W. M.,
Mills, A. A., Miller, F. D. and Kaplan, D. R. (2005) p63 is an essential proapoptotic
protein during neural development. Neuron. 48, 743-756
79 van Bokhoven, H. and McKeon, F. (2002) Mutations in the p53 homolog p63: allele-
specific developmental syndromes in humans. Trends Mol Med. 8, 133-139.
80 Irwin, M. S. (2004) Family Feud in Chemosensitvity: p73 and Mutant p53. Cell cycle. 3,
319-323
81 Flores, E. R., Sengupta, S., Miller, J. B., Newman, J. J., Bronson, R., Crowley, D., Yang,
A., McKeon, F. and Jacks, T. (2005) Tumor predisposition in mice mutant for p63 and
p73: evidence for broader tumor suppressor functions for the p53 family. Cancer cell. 7,
363-373
82 Keyes, W. M., Wu, Y., Vogel, H., Guo, X., Lowe, S. W. and Mills, A. A. (2005) p63
deficiency activates a program of cellular senescence and leads to accelerated aging.
Genes & development. 19, 1986-1999
83 Su, X., Paris, M., Gi, Y. J., Tsai, K. Y., Cho, M. S., Lin, Y. L., Biernaskie, J. A., Sinha,
S., Prives, C., Pevny, L. H., Miller, F. D. and Flores, E. R. (2009) TAp63 prevents
premature aging by promoting adult stem cell maintenance. Cell stem cell. 5, 64-75

105
84 Suh, E. K., Yang, A., Kettenbach, A., Bamberger, C., Michaelis, A. H., Zhu, Z., Elvin, J.
A., Bronson, R. T., Crum, C. P. and McKeon, F. (2006) p63 protects the female germ line
during meiotic arrest. Nature. 444, 624-628
85 Guo, X., Keyes, W. M., Papazoglu, C., Zuber, J., Li, W., Lowe, S. W., Vogel, H. and
Mills, A. A. (2009) TAp63 induces senescence and suppresses tumorigenesis in vivo.
Nature cell biology. 11, 1451-1457
86 Tomasini, R., Tsuchihara, K., Wilhelm, M., Fujitani, M., Rufini, A., Cheung, C. C.,
Khan, F., Itie-Youten, A., Wakeham, A., Tsao, M. S., Iovanna, J. L., Squire, J., Jurisica,
I., Kaplan, D., Melino, G., Jurisicova, A. and Mak, T. W. (2008) TAp73 knockout shows
genomic instability with infertility and tumor suppressor functions. Genes &
development. 22, 2677-2691
87 Agami, R., Blandino, G., Oren, M. and Shaul, Y. (1999) Interaction of c-Abl and
p73alpha and their collaboration to induce apoptosis. Nature. 399, 809-813
88 Gong, J., Costanzo, A., yang, H., Melino, G., Kaelin, W. G., Levero, M. and Wang, J.
(1999) The tyrosine kinase c-Abl regulates p73 in apoptotic response to cisplatin-induced
DNA damage. Nature. 399, 806-808
89 Yuan, Z.-M., Shioya, H., Ishiko, T., Sun, X., Gu, J., Huang, Y., Lu, H., Kharbanda, S.,
Weichselbaum, R. and Kufe, D. (1999) p73 is regulated by tyrosine kinase c-Abl in the
apoptotic reponse to DNA damage. Nature. 399, 814-817
90 Bergamaschi, D., Gasco, M., Hiller, L., Sullivan, A., Syed, N., Trigiante, G., Yulug, I.,
Merlano, M., Numico, G., Comino, A., Attard, M., Reelfs, O., Gusterson, B., Bell, A. K.,
Heath, V., Tavassoli, M., Farrell, P. J., Smith, P., Lu, X. and Crook, T. (2003) p53
polymorphism influences response in cancer chemotherapy via modulation of p73-
dependent apoptosis. Cancer cell. 3, 387-402
91 Irwin, M. S., Kondo, K. K., Marin, M. C., Cheng, L. S., Hahn, W. C. and Kaelin, W. G.
(2003) Chemosensitivity linked to p73 function. Cancer cell. 3, 403-410
92 Rocco, J. W., Leong, C. O., Kuperwasser, N., DeYoung, M. P. and Ellisen, L. W. (2006)
p63 mediates survival in squamous cell carcinoma by suppression of p73-dependent
apoptosis. Cancer cell. 9, 45-56
93 Maisse, C., Munarriz, E., Barcaroli, D., Melino, G. and De Laurenzi, V. (2004) DNA
damage induces the rapid and selective degradation of the DeltaNp73 isoform, allowing
apoptosis to occur. Cell death and differentiation. 11, 685-687
94 Westfall, M. D., Joyner, A. S., Barbieri, C. E., Livingstone, M. and Pietenpol, J. A.
(2005) Ultraviolet radiation induces phosphorylation and ubiquitin-mediated degradation
of DeltaNp63alpha. Cell cycle. 4, 710-716

106
95 Dominguez, G., Garcia, J. M., Pena, C., Silva, J., Garcia, V., Martinez, L., Maximiano,
C., Gomez, M. E., Rivera, J. A., Garcia-Andrade, C. and Bonilla, F. (2005)
{Delta}TAp73 Upregulation Correlates With Poor Prognosis in Human Tumors: Putative
In Vivo Network Involving p73 Isoforms, p53, and E2F-1. J Clin Oncol
96 Douc-Rasy, S., Barrois, M., Echeynne, M., Kaghad, M., Blanc, E., Raguenez, G.,
Goldschneider, D., Terrier-Lacombe, M. J., Hartmann, O., Moll, U., Caput, D. and
Benard, J. (2002) DeltaN-p73alpha accumulates in human neuroblastic tumors. The
American journal of pathology. 160, 631-639.
97 Guan, M. and Chen, Y. (2005) Aberrant expression of DeltaNp73 in benign and
malignant tumours of the prostate: correlation with Gleason score. J Clin Pathol. 58,
1175-1179
98 Putzer, B. M., Tuve, S., Tannapfel, A. and Stiewe, T. (2003) Increased DeltaN-p73
expression in tumors by upregulation of the E2F1-regulated, TA-promoter-derived
DeltaN'-p73 transcript. Cell death and differentiation. 10, 612-614
99 Casciano, I., Mazzocco, K., Boni, L., Pagnan, G., Banelli, B., Allemanni, G., Ponzoni,
M., Tonini, G. P. and Romani, M. (2002) Expression of DeltaNp73 is a molecular marker
for adverse outcome in neuroblastoma patients. Cell death and differentiation. 9, 246-251.
100 Concin, N., Becker, K., Slade, N., Erster, S., Mueller-Holzner, E., Ulmer, H.,
Daxenbichler, G., Zeimet, A., Zeillinger, R., Marth, C. and Moll, U. M. (2004)
Transdominant DeltaTAp73 isoforms are frequently up-regulated in ovarian cancer.
Evidence for their role as epigenetic p53 inhibitors in vivo. Cancer research. 64, 2449-
2460
101 Concin, N., Hofstetter, G., Berger, A., Gehmacher, A., Reimer, D., Watrowski, R., Tong,
D., Schuster, E., Hefler, L., Heim, K., Mueller-Holzner, E., Marth, C., Moll, U. M.,
Zeimet, A. G. and Zeillinger, R. (2005) Clinical relevance of dominant-negative p73
isoforms for responsiveness to chemotherapy and survival in ovarian cancer: evidence for
a crucial p53-p73 cross-talk in vivo. Clin Cancer Res. 11, 8372-8383
102 Hu, H., Xia, S. H., Li, A. D., Xu, X., Cai, Y., Han, Y. L., Wei, F., Chen, B. S., Huang, X.
P., Han, Y. S., Zhang, J. W., Zhang, X., Wu, M. and Wang, M. R. (2002) Elevated
expression of p63 protein in human esophageal squamous cell carcinomas. Int J Cancer.
102, 580-583
103 Massion, P. P., Taflan, P. M., Jamshedur Rahman, S. M., Yildiz, P., Shyr, Y., Edgerton,
M. E., Westfall, M. D., Roberts, J. R., Pietenpol, J. A., Carbone, D. P. and Gonzalez, A.
L. (2003) Significance of p63 amplification and overexpression in lung cancer
development and prognosis. Cancer research. 63, 7113-7121

107
104 Sniezek, J. C., Matheny, K. E., Westfall, M. D. and Pietenpol, J. A. (2004) Dominant
negative p63 isoform expression in head and neck squamous cell carcinoma.
Laryngoscope. 114, 2063-2072
105 Moll, U. M. and Slade, N. (2004) p63 and p73: roles in development and tumor
formation. Mol Cancer Res. 2, 371-386
106 Kastan, M. B., Radin, A. I., Kuerbitz, S. J., Onyekwere, O., Wolkow, C. A., Civin, C. I.,
Stone, J. D., Woo, T., Ravinchranath, Y. and Craig, R. W. (1991) Levels of p53 protein
increase with maturation of human hematopoitic cells. Cancer research. 51, 4279-4286
107 Lavin, M. F. and Gueven, N. (2006) The complexity of p53 stabilization and activation.
Cell death and differentiation. 13, 941-950
108 Bode, A. M. and Dong, Z. (2004) Post-translational modification of p53 in tumorigenesis.
Nature reviews. 4, 793-805
109 Tang, Y., Zhao, W., Chen, Y., Zhao, Y. and Gu, W. (2008) Acetylation is indispensable
for p53 activation. Cell. 133, 612-626
110 Costanzo, A., Merlo, P., Pediconi, N., Fulco, M., Sartorelli, V., Cole, P. A., Fontemaggi,
G., Fanciulli, M., Schiltz, L., Blandino, G., Balsano, C. and Levrero, M. (2002) DNA
damage-dependent acetylation of p73 dictates the selective activation of apoptotic target
genes. Molecular cell. 9, 175-186.
111 Gonzalez, S., Prives, C. and Cordon-Cardo, C. (2003) p73alpha regulation by Chk1 in
response to DNA damage. Molecular and cellular biology. 23, 8161-8171
112 Kim, E. J., Park, J. S. and Um, S. J. (2002) Identification and characterization of HIPK2
interacting with p73 and modulating functions of the p53 family in vivo. The Journal of
biological chemistry. 29, 29
113 Cahilly-Snyder, L., Yang-Feng, T., Francke, U. and George, D. L. (1987) Molecular
analysis and chromosomal mapping of amplified genes isolated from a transformed
mouse 3T3 cell line. Somat. Cell. Mol. Genet. 13, 235-244
114 Fakharzadeh, S. S., Trusko, S. P. and George, D. L. (1991) Tumorigenic potential
associated with enhanced expression of a gene that is amplified in a mouse tumor cell
line. The EMBO journal. 10, 1565-1569
115 Momand, J., Zambetti, G. P., Olson, D. C., George, D. and Levine, A. J. (1992) The
mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-
mediated transactivation. Cell. 69, 1237-1245

108
116 Oliner, J. D., Kinzler, K. W., Meltzer, P. S., George, D. L. and Vogelstein, B. (1992)
Amplification of a gene encoding a p53-associated protein in human sarcomas. Nature.
358, 80-83
117 Bond, G. L., Hu, W., Bond, E. E., Robins, H., Lutzker, S. G., Arva, N. C., Bargonetti, J.,
Bartel, F., Taubert, H., Wuerl, P., Onel, K., Yip, L., Hwang, S. J., Strong, L. C., Lozano,
G. and Levine, A. J. (2004) A single nucleotide polymorphism in the MDM2 promoter
attenuates the p53 tumor suppressor pathway and accelerates tumor formation in humans.
Cell. 119, 591-602
118 Landers, J. E., Haines, D. S., Strauss, J. F., 3rd and George, D. L. (1994) Enhanced
translation: a novel mechanism of mdm2 oncogene overexpression identified in human
tumor cells. Oncogene. 9, 2745-2750
119 Watanabe, T., Hotta, T., Ichikawa, A., Kinoshita, T., Nagai, H., Uchida, T., Murate, T.
and Saito, H. (1994) The MDM2 oncogene overexpression in chronic lymphocytic
leukemia and low-grade lymphoma of B-cell origin. Blood. 84, 3158-3165
120 Issaeva, N., Bozko, P., Enge, M., Protopopova, M., Verhoef, L. G., Masucci, M.,
Pramanik, A. and Selivanova, G. (2004) Small molecule RITA binds to p53, blocks p53-
HDM-2 interaction and activates p53 function in tumors. Nature medicine. 10, 1321-1328
121 Vassilev, L. T., Vu, B. T., Graves, B., Carvajal, D., Podlaski, F., Filipovic, Z., Kong, N.,
Kammlott, U., Lukacs, C., Klein, C., Fotouhi, N. and Liu, E. A. (2004) In vivo activation
of the p53 pathway by small-molecule antagonists of MDM2. Science. 303, 844-848
122 Bottger, A., Bottger, V., Garcia-Echeverria, C., Chene, P., Hochkeppel, H. K., Sampson,
W., Ang, K., Howard, S. F., Picksley, S. M. and Lane, D. P. (1997) Molecular
characterization of the hdm2-p53 interaction. J Mol Biol. 269, 744-756
123 Chan, W. M., Mak, M. C., Fung, T. K., Lau, A., Siu, W. Y. and Poon, R. Y. (2006)
Ubiquitination of p53 at multiple sites in the DNA-binding domain. Mol Cancer Res. 4,
15-25
124 Haupt, Y., Maya, R., Kazaz, A. and Oren, M. (1997) Mdm2 promotes the rapid
degradation of p53. Nature. 387, 296-299
125 Honda, R., Tanaka, H. and Yasuda, H. (1997) Oncoprotein MDM2 is a ubiquitin ligase
E3 for tumor suppressor p53. FEBS Lett. 420, 25-27
126 Kubbutat, M., Jones, S. and Vousden, K. (1997) Regulation of p53 stability by Mdm2.
Nature. 387, 299-303
127 Geyer, R. K., Yu, Z. K. and Maki, C. G. (2000) The MDM2 RING-finger domain is
required to promote p53 nuclear export. Nature cell biology. 2, 569-573.

109
128 Li, M., Brooks, C. L., Wu-Baer, F., Chen, D., Baer, R. and Gu, W. (2003) Mono- versus
polyubiquitination: differential control of p53 fate by Mdm2. Science. 302, 1972-1975
129 Gronroos, E., Terentiev, A. A., Punga, T. and Ericsson, J. (2004) YY1 inhibits the
activation of the p53 tumor suppressor in response to genotoxic stress. Proceedings of the
National Academy of Sciences of the United States of America. 101, 12165-12170
130 Grossman, S. R., Deato, M. E., Brignone, C., Chan, H. M., Kung, A. L., Tagami, H.,
Nakatani, Y. and Livingston, D. M. (2003) Polyubiquitination of p53 by a ubiquitin
ligase activity of p300. Science. 300, 342-344
131 Xirodimas, D. P., Saville, M. K., Bourdon, J. C., Hay, R. T. and Lane, D. P. (2004)
Mdm2-mediated NEDD8 conjugation of p53 inhibits its transcriptional activity. Cell.
118, 83-97
132 Brooks, C. L., Li, M. and Gu, W. (2007) Mechanistic studies of MDM2-mediated
ubiquitination in p53 regulation. The Journal of biological chemistry. 282, 22804-22815
133 Kobet, E., Zeng, X., Zhu, Y., Keller, D. and Lu, H. (2000) MDM2 inhibits p300-
mediated p53 acetylation and activation by forming a ternary complex with the two
proteins. Proceedings of the National Academy of Sciences of the United States of
America. 97, 12547-12552.
134 Oliner, J., Pietenpol, J., Thiagalingam, S., Gyuris, J., Kinzler, K. and Vogelstein, B.
(1993) Oncoprotein MDM2 conceals the activation domain of tumour suppressor p53.
Nature. 362, 857-860
135 Barak, Y., Juven, T., Haffner, R. and Oren, M. (1993) mdm2 expression is induced by
wild type p53 activity. The EMBO journal. 12, 461-468
136 Jones, S. N., Roe, A. E., Donehower, L. A. and Bradley, A. (1995) Rescue of embryonic
lethality in Mdm2-deficient mice by absence of p53. Nature. 378, 206-208
137 Montes de Oca Luna, R., Wagner, D. and Lozano, G. (1995) Rescue of early embryonic
lethality in mdm2-deficient mice by deletion of p53. Nature. 378, 203-206
138 Francoz, S., Froment, P., Bogaerts, S., De Clercq, S., Maetens, M., Doumont, G.,
Bellefroid, E. and Marine, J. C. (2006) Mdm4 and Mdm2 cooperate to inhibit p53 activity
in proliferating and quiescent cells in vivo. Proceedings of the National Academy of
Sciences of the United States of America. 103, 3232-3237
139 Xiong, S., Van Pelt, C. S., Elizondo-Fraire, A. C., Liu, G. and Lozano, G. (2006)
Synergistic roles of Mdm2 and Mdm4 for p53 inhibition in central nervous system
development. Proceedings of the National Academy of Sciences of the United States of
America. 103, 3226-3231

110
140 Boesten, L. S., Zadelaar, S. M., De Clercq, S., Francoz, S., van Nieuwkoop, A., Biessen,
E. A., Hofmann, F., Feil, S., Feil, R., Jochemsen, A. G., Zurcher, C., Havekes, L. M., van
Vlijmen, B. J. and Marine, J. C. (2006) Mdm2, but not Mdm4, protects terminally
differentiated smooth muscle cells from p53-mediated caspase-3-independent cell death.
Cell death and differentiation. 13, 2089-2098
141 Ringshausen, I., O'Shea, C. C., Finch, A. J., Swigart, L. B. and Evan, G. I. (2006) Mdm2
is critically and continuously required to suppress lethal p53 activity in vivo. Cancer cell.
10, 501-514
142 Itahana, K., Mao, H., Jin, A., Itahana, Y., Clegg, H. V., Lindstrom, M. S., Bhat, K. P.,
Godfrey, V. L., Evan, G. I. and Zhang, Y. (2007) Targeted inactivation of Mdm2 RING
finger E3 ubiquitin ligase activity in the mouse reveals mechanistic insights into p53
regulation. Cancer cell. 12, 355-366
143 Sigalas, I., Calvert, A. H., Anderson, J. J., Neal, D. E. and Lunec, J. (1996) Alternatively
spliced mdm2 transcripts with loss of p53 binding domain sequences: transforming
ability and frequent detection in human cancer. Nature medicine. 2, 912-917
144 Jones, S. N., Hancock, A. R., Vogel, H., Donehower, L. A. and Bradley, A. (1998)
Overexpression of Mdm2 in mice reveals a p53-independent role for Mdm2 in
tumorigenesis. Proceedings of the National Academy of Sciences of the United States of
America. 95, 15608-15612
145 McDonnell, T. J., Montes de Oca Luna, R., Cho, S., Amelse, L. L., Chavez-Reyes, A. and
Lozano, G. (1999) Loss of one but not two mdm2 null alleles alters the tumour spectrum
in p53 null mice. The Journal of pathology. 188, 322-328
146 Cordon-Cardo, C., Latres, E., Drobnjak, M., Oliva, M. R., Pollack, D., Woodruff, J. M.,
Marechal, V., Chen, J., Brennan, M. F. and Levine, A. J. (1994) Molecular abnormalities
of mdm2 and p53 genes in adult soft tissue sarcomas. Cancer research. 54, 794-799
147 Wurl, P., Meye, A., Berger, D., Bache, M., Lautenschlager, C., Schmidt, H., Kalthoff, H.,
Rath, F. W. and Taubert, H. (1997) Prognostic relevance of C-terminal Mdm2 detection
is enhanced by p53 positivity in soft tissue sarcomas. Diagn Mol Pathol. 6, 249-254
148 Wurl, P., Meye, A., Schmidt, H., Lautenschlager, C., Kalthoff, H., Rath, F. W. and
Taubert, H. (1998) High prognostic significance of Mdm2/p53 co-overexpression in soft
tissue sarcomas of the extremities. Oncogene. 16, 1183-1185
149 Khosravi, R., Maya, R., Gottlieb, T., Oren, M., Shiloh, Y. and Shkedy, D. (1999) Rapid
ATM-dependent phosphorylation of MDM2 precedes p53 accumulation in response to
DNA damage. Proceedings of the National Academy of Sciences of the United States of
America. 96, 14973-14977

111
150 Stommel, J. M. and Wahl, G. M. (2004) Accelerated MDM2 auto-degradation induced by
DNA-damage kinases is required for p53 activation. The EMBO journal. 23, 1547-1556
151 Chao, C., Hergenhahn, M., Kaeser, M. D., Wu, Z., Saito, S., Iggo, R., Hollstein, M.,
Appella, E. and Xu, Y. (2003) Cell type- and promoter-specific roles of Ser18
phosphorylation in regulating p53 responses. The Journal of biological chemistry. 278,
41028-41033
152 Chao, C., Herr, D., Chun, J. and Xu, Y. (2006) Ser18 and 23 phosphorylation is required
for p53-dependent apoptosis and tumor suppression. The EMBO journal. 25, 2615-2622
153 MacPherson, D., Kim, J., Kim, T., Rhee, B. K., Van Oostrom, C. T., DiTullio, R. A.,
Venere, M., Halazonetis, T. D., Bronson, R., De Vries, A., Fleming, M. and Jacks, T.
(2004) Defective apoptosis and B-cell lymphomas in mice with p53 point mutation at Ser
23. The EMBO journal. 23, 3689-3699
154 Sluss, H. K., Armata, H., Gallant, J. and Jones, S. N. (2004) Phosphorylation of serine 18
regulates distinct p53 functions in mice. Molecular and cellular biology. 24, 976-984
155 Wu, Z., Earle, J., Saito, S., Anderson, C. W., Appella, E. and Xu, Y. (2002) Mutation of
mouse p53 Ser23 and the response to DNA damage. Molecular and cellular biology. 22,
2441-2449
156 Meulmeester, E., Pereg, Y., Shiloh, Y. and Jochemsen, A. G. (2005) ATM-mediated
phosphorylations inhibit Mdmx/Mdm2 stabilization by HAUSP in favor of p53
activation. Cell cycle. 4, 1166-1170
157 Song, M. S., Song, S. J., Kim, S. Y., Oh, H. J. and Lim, D. S. (2008) The tumour
suppressor RASSF1A promotes MDM2 self-ubiquitination by disrupting the MDM2-
DAXX-HAUSP complex. The EMBO journal. 27, 1863-1874
158 Tang, J., Qu, L. K., Zhang, J., Wang, W., Michaelson, J. S., Degenhardt, Y. Y., El-Deiry,
W. S. and Yang, X. (2006) Critical role for Daxx in regulating Mdm2. Nature cell
biology. 8, 855-862
159 Lee, M. H., Lee, S. W., Lee, E. J., Choi, S. J., Chung, S. S., Lee, J. I., Cho, J. M., Seol, J.
H., Baek, S. H., Kim, K. I., Chiba, T., Tanaka, K., Bang, O. S. and Chung, C. H. (2006)
SUMO-specific protease SUSP4 positively regulates p53 by promoting Mdm2 self-
ubiquitination. Nature cell biology. 8, 1424-1431
160 Tai, E. and Benchimol, S. (2009) TRIMming p53 for ubiquitination. Proceedings of the
National Academy of Sciences of the United States of America. 106, 11431-11432
161 Dornan, D., Bheddah, S., Newton, K., Ince, W., Frantz, G. D., Dowd, P., Koeppen, H.,
Dixit, V. M. and French, D. M. (2004) COP1, the negative regulator of p53, is
overexpressed in breast and ovarian adenocarcinomas. Cancer research. 64, 7226-7230

112
162 Duan, W., Gao, L., Druhan, L. J., Zhu, W. G., Morrison, C., Otterson, G. A. and
Villalona-Calero, M. A. (2004) Expression of Pirh2, a newly identified ubiquitin protein
ligase, in lung cancer. J Natl Cancer Inst. 96, 1718-1721
163 Kussie, P. H., Gorina, S., Marechal, V., Elenbaas, B., Moreau, J., Levine, A. J. and
Pavletich, N. P. (1996) Structure of the MDM2 oncoprotein bound to the p53 tumor
suppressor transactivation domain. Science. 274, 948-953
164 Little, N. A. and Jochemsen, A. G. (2001) Hdmx and Mdm2 can repress transcription
activation by p53 but not by p63. Oncogene. 20, 4576-4580.
165 Balint, E., Bates, S. and Vousden, K. H. (1999) Mdm-2 binds p73 alpha without targeting
degradation. Oncogene. 18, 3923-3929
166 Ongkeko, W. M., Wang, X. Q., Siu, W. Y., Lau, A. W., Yamashita, K., Harris, A. L.,
Cox, L. S. and Poon, R. Y. (1999) MDM2 and MDMX bind and stabilize the p53-related
protein p73. Curr Biol. 9, 829-832
167 Zeng, X., Chen, L., Jost, C. A., Maya, R., Keller, D., Wang, X., Kaelin, W. G., Jr., Oren,
M., Chen, J. and Lu, H. (1999) MDM2 suppresses p73 function without promoting p73
degradation. Molecular and cellular biology. 19, 3257-3266
168 Gostissa, M., Hengstermann, A., Fogal, V., Sandy, P., Schwarz, S. E., Scheffner, M. and
Del Sal, G. (1999) Activation of p53 by conjugation to the ubiquitin-like protein SUMO-
1. The EMBO journal. 18, 6462-6471.
169 Rodriguez, M. S., Dargemont, C. and Hay, R. T. (2000) SUMO-1 conjugation in vivo
requires both a consensus modification motif and nuclear targeting. The Journal of
biological chemistry. 21, 21
170 Muller, S., Berger, M., Lehembre, F., Seeler, J. S., Haupt, Y. and Dejean, A. (2000) c-Jun
and p53 activity is modulated by SUMO-1 modification. The Journal of biological
chemistry. 275, 13321-13329
171 Kahyo, T., Nishida, T. and Yasuda, H. (2001) Involvement of PIAS1 in the sumoylation
of tumor suppressor p53. Molecular cell. 8, 713-718
172 Megidish, T., Xu, J. H. and Xu, C. W. (2002) Activation of p53 by protein inhibitor of
activated Stat1 (PIAS1). The Journal of biological chemistry. 277, 8255-8259
173 Nelson, V., Davis, G. E. and Maxwell, S. A. (2001) A putative protein inhibitor of
activated STAT (PIASy) interacts with p53 and inhibits p53-mediated transactivation but
not apoptosis. Apoptosis. 6, 221-234

113
174 Schmidt, D. and Muller, S. (2002) Members of the PIAS family act as SUMO ligases for
c-Jun and p53 and repress p53 activity. Proceedings of the National Academy of Sciences
of the United States of America. 99, 2872-2877
175 Melchior, F. and Hengst, L. (2002) SUMO-1 and p53. Cell cycle. 1, 245-249
176 Minty, A., Dumont, X., Kaghad, M. and Caput, D. (2000) Covalent Modification of
p73alpha by SUMO-1. Two-Hybrid Screening with p73 Identifies Novel SUMO-1-
Interacting Proteins and a SUMO-1 Interaction Motif. The Journal of biological
chemistry. 275, 36316-36323
177 Munarriz, E., Barcaroli, D., Stephanou, A., Townsend, P. A., Maisse, C., Terrinoni, A.,
Neale, M. H., Martin, S. J., Latchman, D. S., Knight, R. A., Melino, G. and De Laurenzi,
V. (2004) PIAS-1 is a checkpoint regulator which affects exit from G1 and G2 by
sumoylation of p73. Molecular and cellular biology. 24, 10593-10610
178 Huang, Y. P., Wu, G., Guo, Z., Osada, M., Fomenkov, T., Park, H. L., Trink, B.,
Sidransky, D., Fomenkov, A. and Ratovitski, E. A. (2004) Altered sumoylation of
p63alpha contributes to the split-hand/foot malformation phenotype. Cell cycle. 3, 1587-
1596
179 Bakkers, J., Camacho-Carvajal, M., Nowak, M., Kramer, C., Danger, B. and
Hammerschmidt, M. (2005) Destabilization of DeltaNp63alpha by Nedd4-mediated
ubiquitination and Ubc9-mediated sumoylation, and its implications on dorsoventral
patterning of the zebrafish embryo. Cell cycle. 4, 790-800
180 Ghioni, P., D'Alessandra, Y., Mansueto, G., Jaffray, E., Hay, R. T., La Mantia, G. and
Guerrini, L. (2005) The protein stability and transcriptional activity of p63alpha are
regulated by SUMO-1 conjugation. Cell cycle. 4, 183-190
181 Bouska, A. and Eischen, C. M. (2009) Murine double minute 2: p53-independent roads
lead to genome instability or death. Trends in biochemical sciences. 34, 279-286
182 Gan-Erdene, T., Nagamalleswari, K., Yin, L., Wu, K., Pan, Z. Q. and Wilkinson, K. D.
(2003) Identification and characterization of DEN1, a deneddylase of the ULP family.
The Journal of biological chemistry. 278, 28892-28900
183 Mendoza, H. M., Shen, L. N., Botting, C., Lewis, A., Chen, J., Ink, B. and Hay, R. T.
(2003) NEDP1, a highly conserved cysteine protease that deNEDDylates Cullins. The
Journal of biological chemistry. 278, 25637-25643
184 Wu, K., Yamoah, K., Dolios, G., Gan-Erdene, T., Tan, P., Chen, A., Lee, C. G., Wei, N.,
Wilkinson, K. D., Wang, R. and Pan, Z. Q. (2003) DEN1 is a dual function protease
capable of processing the C terminus of Nedd8 and deconjugating hyper-neddylated
CUL1. The Journal of biological chemistry. 278, 28882-28891

114
185 Kaghad, M., Bonnet, H., Yang, A., Creancier, L., Biscan, J.-C., Valent, A., Minty, A.,
Chalon, P., Lelias, J.-M., Dumont, X., Ferrara, P., McKeon, F. and Caput, D. (1997)
Monoallelically Expressed Gene Related to p53 at 1p36, a region frequently deleted in
neuroblastoma and other human cancers. Cell. 90, 809-819
186 Hirschberg, J. and Marcus, M. (1982) Isolation by a replica-plating technique of Chinese
hamster temperature-sensitive cell cycle mutants. Journal of cellular physiology. 113,
159-166
187 Wang, X., Arooz, T., Siu, W. Y., Chiu, C. H., Lau, A., Yamashita, K. and Poon, R. Y.
(2001) MDM2 and MDMX can interact differently with ARF and members of the p53
family. FEBS Lett. 490, 202-208.
188 Gu, J., Nie, L., Kawai, H. and Yuan, Z. M. (2001) Subcellular distribution of p53 and p73
are differentially regulated by MDM2. Cancer research. 61, 6703-6707.
189 Bernassola, F., Salomoni, P., Oberst, A., Di Como, C. J., Pagano, M., Melino, G. and
Pandolfi, P. P. (2004) Ubiquitin-dependent degradation of p73 is inhibited by PML. J Exp
Med. 199, 1545-1557
190 Inoue, T., Stuart, J., Leno, R. and Maki, C. G. (2002) Nuclear Import and Export Signals
in Control of the p53-related Protein p73. The Journal of biological chemistry. 277,
15053-15060.
191 Stommel, J. M., Marchenko, N. D., Jimenez, G. S., Moll, U. M., Hope, T. J. and Wahl, G.
M. (1999) A leucine-rich nuclear export signal in the p53 tetramerization domain:
regulation of subcellular localization and p53 activity by NES masking. EMBO. 18,
1660-1672
192 Bianco, R., Ciardiello, F. and Tortora, G. (2005) Chemosensitization by antisense
oligonucleotides targeting MDM2. Current cancer drug targets. 5, 51-56
193 Stickle, N. H., Chung, J., Klco, J. M., Hill, R. P., Kaelin, W. G., Jr. and Ohh, M. (2004)
pVHL modification by NEDD8 is required for fibronectin matrix assembly and
suppression of tumor development. Molecular and cellular biology. 24, 3251-3261
194 Marin, M. C., Jost, C. A., Brooks, L. A., Irwin, M. S., O'Nions, J., Tidy, J. A., James, N.,
McGregor, J. M., Harwood, C. A., Yulug, I. G., Vousden, K. H., Allday, M. J.,
Gusterson, B., Ikawa, S., Hinds, P. W., Crook, T. and Kaelin, W. G., Jr. (2000) A
common polymorphism acts as an intragenic modifier of mutant p53 behaviour. Nature
genetics. 25, 47-54.
195 Meulmeester, E., Maurice, M. M., Boutell, C., Teunisse, A. F., Ovaa, H., Abraham, T. E.,
Dirks, R. W. and Jochemsen, A. G. (2005) Loss of HAUSP-mediated deubiquitination
contributes to DNA damage-induced destabilization of Hdmx and Hdm2. Molecular cell.
18, 565-576

115
196 Morimoto, M., Nishida, T., Nagayama, Y. and Yasuda, H. (2003) Nedd8-modification of
Cul1 is promoted by Roc1 as a Nedd8-E3 ligase and regulates its stability. Biochemical
and biophysical research communications. 301, 392-398
197 Wu, J. T., Lin, H. C., Hu, Y. C. and Chien, C. T. (2005) Neddylation and deneddylation
regulate Cul1 and Cul3 protein accumulation. Nature cell biology. 7, 1014-1020
198 Hetfeld, B. K., Peth, A., Sun, X. M., Henklein, P., Cohen, G. M. and Dubiel, W. (2008)
The COP9 signalosome-mediated deneddylation is stimulated by caspases during
apoptosis. Apoptosis. 13, 187-195
199 Saville, M. K., Sparks, A., Xirodimas, D. P., Wardrop, J., Stevenson, L. F., Bourdon, J.
C., Woods, Y. L. and Lane, D. P. (2004) Regulation of p53 by the ubiquitin-conjugating
enzymes UbcH5B/C in vivo. The Journal of biological chemistry. 279, 42169-42181
200 Chan, Y., Yoon, J., Wu, J. T., Kim, H. J., Pan, K. T., Yim, J. and Chien, C. T. (2008)
DEN1 deneddylates non-cullin proteins in vivo. Journal of cell science. 121, 3218-3223
201 Zhou, L. and Watts, F. Z. (2005) Nep1, a Schizosaccharomyces pombe deneddylating
enzyme. The Biochemical journal. 389, 307-314
202 Zhang, X. C., Chen, J., Su, C. H., Yang, H. Y. and Lee, M. H. (2008) Roles for CSN5 in
control of p53/MDM2 activities. Journal of cellular biochemistry. 103, 1219-1230
203 Yamoah, K., Wu, K. and Pan, Z. Q. (2005) In vitro cleavage of Nedd8 from cullin 1 by
COP9 signalosome and deneddylase 1. Methods in enzymology. 398, 509-522
204 Rabut, G. and Peter, M. (2008) Function and regulation of protein neddylation. 'Protein
modifications: beyond the usual suspects' review series. EMBO reports. 9, 969-976
205 Jones, J., Wu, K., Yang, Y., Guerrero, C., Nillegoda, N., Pan, Z. Q. and Huang, L. (2008)
A targeted proteomic analysis of the ubiquitin-like modifier nedd8 and associated
proteins. Journal of proteome research. 7, 1274-1287
206 Xirodimas, D. P., Sundqvist, A., Nakamura, A., Shen, L., Botting, C. and Hay, R. T.
(2008) Ribosomal proteins are targets for the NEDD8 pathway. EMBO reports. 9, 280-
286
207 Levav-Cohen, Y., Haupt, S. and Haupt, Y. (2005) Mdm2 in growth signaling and cancer.
Growth factors (Chur, Switzerland). 23, 183-192
208 Huang, H. Y., West, R. B., Tzeng, C. C., van de Rijn, M., Wang, J. W., Chou, S. C.,
Huang, W. W., Eng, H. L., Lin, C. N., Yu, S. C., Wu, J. M., Lu, C. C. and Li, C. F.
(2008) Immunohistochemical and biogenetic features of diffuse-type tenosynovial giant
cell tumors: the potential roles of cyclin A, P53, and deletion of 15q in sarcomatous
transformation. Clin Cancer Res. 14, 6023-6032

116
209 Koschny, R., Holland, H., Koschny, T. and Vitzthum, H. E. (2006) Comparative genomic
hybridization pattern of non-anaplastic and anaplastic oligodendrogliomas--a meta-
analysis. Pathology, research and practice. 202, 23-30
210 Paunu, N., Lahermo, P., Onkamo, P., Ollikainen, V., Rantala, I., Helen, P., Simola, K. O.,
Kere, J. and Haapasalo, H. (2002) A novel low-penetrance locus for familial glioma at
15q23-q26.3. Cancer research. 62, 3798-3802
211 Suzuki, H., Ouchida, M., Yamamoto, H., Yano, M., Toyooka, S., Aoe, M., Shimizu, N.,
Date, H. and Shimizu, K. (2008) Decreased expression of the SIN3A gene, a candidate
tumor suppressor located at the prevalent allelic loss region 15q23 in non-small cell lung
cancer. Lung cancer (Amsterdam, Netherlands). 59, 24-31
212 Forbes, S. A., Bhamra, G., Bamford, S., Dawson, E., Kok, C., Clements, J., Menzies, A.,
Teague, J. W., Futreal, P. A. and Stratton, M. R. (2008) The Catalogue of Somatic
Mutations in Cancer (COSMIC). Current protocols in human genetics / editorial board,
Jonathan L. Haines ... [et al. Chapter 10, Unit 10 11
213 Katayama, A., Ogino, T., Bandoh, N., Takahara, M., Kishibe, K., Nonaka, S. and
Harabuchi, Y. (2007) Overexpression of small ubiquitin-related modifier-1 and
sumoylated Mdm2 in oral squamous cell carcinoma: possible involvement in tumor
proliferation and prognosis. International journal of oncology. 31, 517-524
214 Blom, N., Sicheritz-Ponten, T., Gupta, R., Gammeltoft, S. and Brunak, S. (2004)
Prediction of post-translational glycosylation and phosphorylation of proteins from the
amino acid sequence. Proteomics. 4, 1633-1649
215 Watson, I. R., Blanch, A., Lin, D. C., Ohh, M. and Irwin, M. S. (2006) Mdm2-mediated
NEDD8 modification of TAp73 regulates its transactivation function. The Journal of
biological chemistry. 281, 34096-34103
216 Lau, L., Hansford, L. M., Cheng, L. S., Hang, M., Baruchel, S., Kaplan, D. R. and Irwin,
M. S. (2007) Cyclooxygenase inhibitors modulate the p53/HDM2 pathway and enhance
chemotherapy-induced apoptosis in neuroblastoma. Oncogene. 26, 1920-1931
217 Bottger, A., Bottger, V., Sparks, A., Liu, W. L., Howard, S. F. and Lane, D. P. (1997)
Design of a synthetic Mdm2-binding mini protein that activates the p53 response in vivo.
Curr Biol. 7, 860-869
218 Lau, L. M., Nugent, J. K., Zhao, X. and Irwin, M. S. (2008) HDM2 antagonist Nutlin-3
disrupts p73-HDM2 binding and enhances p73 function. Oncogene. 27, 997-1003
219 Kravchenko, J. E., Ilyinskaya, G. V., Komarov, P. G., Agapova, L. S., Kochetkov, D. V.,
Strom, E., Frolova, E. I., Kovriga, I., Gudkov, A. V., Feinstein, E. and Chumakov, P. M.
(2008) Small-molecule RETRA suppresses mutant p53-bearing cancer cells through a

117
p73-dependent salvage pathway. Proceedings of the National Academy of Sciences of the
United States of America. 105, 6302-6307
220 Dey, A., Verma, C. S. and Lane, D. P. (2008) Updates on p53: modulation of p53
degradation as a therapeutic approach. British journal of cancer. 98, 4-8
221 Kane, R. C., Bross, P. F., Farrell, A. T. and Pazdur, R. (2003) Velcade: U.S. FDA
approval for the treatment of multiple myeloma progressing on prior therapy. The
oncologist. 8, 508-513
222 Kane, R. C., Dagher, R., Farrell, A., Ko, C. W., Sridhara, R., Justice, R. and Pazdur, R.
(2007) Bortezomib for the treatment of mantle cell lymphoma. Clin Cancer Res. 13,
5291-5294
223 Brownell, J. E., Sintchak, M. D., Gavin, J. M., Liao, H., Bruzzese, F. J., Bump, N. J.,
Soucy, T. A., Milhollen, M. A., Yang, X., Burkhardt, A. L., Ma, J., Loke, H. K., Lingaraj,
T., Wu, D., Hamman, K. B., Spelman, J. J., Cullis, C. A., Langston, S. P., Vyskocil, S.,
Sells, T. B., Mallender, W. D., Visiers, I., Li, P., Claiborne, C. F., Rolfe, M., Bolen, J. B.
and Dick, L. R. Substrate-assisted inhibition of ubiquitin-like protein-activating enzymes:
the NEDD8 E1 inhibitor MLN4924 forms a NEDD8-AMP mimetic in situ. Molecular
cell. 37, 102-111
224 Miller, F. D. and Kaplan, D. R. (2001) Neurotrophin signalling pathways regulating
neuronal apoptosis. Cell Mol Life Sci. 58, 1045-1053
225 Bartlett, S. E., Reynolds, A. J., Weible, M., Heydon, K. and Hendry, I. A. (1997) In
sympathetic but not sensory neurones, phosphoinositide-3 kinase is important for NGF-
dependent survival and the retrograde transport of 125I-betaNGF. Brain research. 761,
257-262
226 Vaillant, A. R., Mazzoni, I., Tudan, C., Boudreau, M., Kaplan, D. R. and Miller, F. D.
(1999) Depolarization and neurotrophins converge on the phosphatidylinositol 3-kinase-
Akt pathway to synergistically regulate neuronal survival. The Journal of cell biology.
146, 955-966