nedd8-targeting drug mln4924 elicits dna rereplication by
TRANSCRIPT

Therapeutics, Targets, and Chemical Biology
NEDD8-Targeting Drug MLN4924 Elicits DNA Rereplicationby Stabilizing Cdt1 in S Phase, Triggering CheckpointActivation, Apoptosis, and Senescence in Cancer Cells
Jie Jessie Lin1, Michael A. Milhollen2, Peter G. Smith2, Usha Narayanan2, and Anindya Dutta1
AbstractMLN4924 is a first-in-class experimental cancer drug that inhibits the NEDD8-activating enzyme, thereby
inhibiting cullin-RING E3 ubiquitin ligases and stabilizing many cullin substrates. The mechanism by whichMLN4924 inhibits cancer cell proliferation has not been defined, although it is accompanied by DNArereplication and attendant DNA damage. Here we show that stabilization of the DNA replication factorCdt1, a substrate of cullins 1 and 4, is critical for MLN4924 to trigger DNA rereplication and inhibit cellproliferation. Even only 1 hour of exposure to MLN4924, which was sufficient to elevate Cdt1 for 4–5 hours, wasfound to be sufficient to induce DNA rereplication and to activate apoptosis and senescence pathways. Cells in Sphase were most susceptible, suggesting that MLN4924 will be most toxic on highly proliferating cancers.Although MLN4924-induced cell senescence seems to be dependent on induction of p53 and its downstreameffector p21Waf1, we found that p53�/� and p21�/� cells were even more susceptible than wild-type cells toMLN4924. Our results suggested that apoptosis, not senescence, might be more important for the antiproli-ferative effect of MLN4924. Furthermore, our findings show that transient exposure to this new investigationaldrug should be useful for controlling p53-negative cancer cells, which often pose significant clinical challenge.Cancer Res; 70(24); 10310–20. �2010 AACR.
Introduction
Duplication of the genetic material is a key event in the cellcycle. In eukaryotes, replication origins are recognized andbound by a 6-subunit complex called ORC (Origin RecognizingComplex; refs. 1–3). Cdc6 and Cdt1 are subsequently recruitedindependently to those sites in late M or early G1 phase (1, 3,4), followed by the recruitment of MCM2–7 complex to initiateDNA replication (5, 6). It is vitally important that the initiationof replication at replication origins is tightly controlled suchthat it occurs only once during the cell cycle. Mammalian cellshave developed different mechanisms to prevent reinitiationand subsequent rereplication of DNA within the same cellcycle. One suchmechanism is the inactivation of Cdt1 during Sand G2 phases (7, 8). After replication initiation, Cdt1 is eitherinhibited by a small protein called Geminin (9, 10) or degradedby 2 distinct E3 ligases, cdk-dependent SCFskp2 and Cul4-
DDB1cdt2 in S or G2/M phase (8, 11). Deregulation of thosepathways by depletion of Geminin, Cul4, or Cdt2 activates (orstabilizes) Cdt1 and consequently induces DNA rereplicationin different systems (7, 12–14).
Studies have shown that cullin-RING ligases (CRL), a sub-class of E3 ligases that includes both SCFskp2 and CRL4Cdt2, aremodified by an ubiquitin-like protein, NEDD8, which subse-quently facilitates their ligase activities (15–18). Thus, throughthe modulation of this activity, the NEDD8 pathway regulatesthe abundance of CRL substrates. MLN4924, a potentialcancer drug currently in phase I clinical trials, is a smallmolecule inhibitor of NEDD8-activating enzyme (NAE; refs. 19,20). MLN4924 treatment in HCT116 human colon cancer–derived cell line inhibits NAE, and therefore the NEDD8conjugation pathway, resulting in an increase in proteinabundance of CRL substrates such as Cdt1 (21). This isaccompanied by an increase in the percentage of cells contain-ing more than 4N DNA, indicating DNA rereplication wasoccurring. Cells treated with MLN4924 also undergo signifi-cant apoptosis contributing to the drug's antiproliferativeactivity. Various CRL substrates play critical functions incellular growth and survival pathways and the questionremained as to which substrates are critical for MLN4924-induced rereplication and apoptosis.
In this article, we examine whether Cdt1 is the key factor forthe induction of DNA rereplication in HCT116 cells treatedwith MLN4924. Among the different approaches for stimulat-ing Cdt1 activation, MLN4924 shares a similarity with thatof Cdt2 depletion in inactivating the CRL4cdt2 E3 ligase, as
Authors' Affiliations: 1Department of Biochemistry and Molecular Genet-ics, University of Virginia, Charlottesville, Virginia; and 2Discovery,Millennium Pharmaceuticals, Inc., Cambridge, Massachusetts
Note: Supplementary data for this article are available at Cancer ResearchOnline (http://cancerres.aacrjournals.org/).
Corresponding Author: Anindya Dutta, University of Virginia School ofMedicine, Box 800733, Jordan 1240, 1300 Jefferson Park Ave, Charlot-tesville, VA 22908. Phone: 434-924-1227; Fax: 434-924-5069; E-mail:[email protected]
doi: 10.1158/0008-5472.CAN-10-2062
�2010 American Association for Cancer Research.
CancerResearch
Cancer Res; 70(24) December 15, 201010310
Research. on April 9, 2019. © 2010 American Association for Cancercancerres.aacrjournals.org Downloaded from

opposed to Geminin depletion, which activates Cdt1 by adifferent pathway. We verified this hypothesis and detected asynergistic effect between MLN4924 treatment and Geminindepletion. Transient exposure of cells to MLN4924 led to DNArereplication, as well as activation of the apoptosis andsenescence pathways. This allowed us to test whether aspecific part of the cell cycle was particularly susceptible orresistant to MLN4924. Finally, we compared the sensitivity ofwild-type (WT) HCT116 cells and isogenic p53�/� or p21�/�
HCT116 cells to MLN4924 and discovered that WT HCT116cells were less susceptible toMLN4924-induced cell death. Theresults indicate that p53-deficient cancer cells may be moresensitive to MLN4924, emphasizing the therapeutic opportu-nity with this class of investigational drugs.
Materials and Methods
Cell lines and chemicalsHuman colorectal cancer cell lines HCT116 (WT, p53�,
p21�) were cultured in McCoy's 5A-modified medium(HyClone) supplemented with 10% fetal bovine serum and1% penicillin-streptomycin. Isogenic p21�/� and p53�/�
HCT116 cell lines were described earlier (22). MillenniumPharmaceuticals Inc. provided MLN4924, which was thendissolved in DMSO (Sigma). The concentration of Z-Vad-FMK (Calbiochem) used was 50 mmol/L. The concentrationof nocodazole (Sigma) used was 40 ng/mL.
siRNAShort interfering (siRNA) oligonucleotides (Invitrogen)
were made to the following target sequences (sense): GL2(control), AACGUACGCGGAAUACUUCGA; Cdt1, GCAAU-GUUGGCCAGAUCAA; Cdc6, GAUCGACUUAAUCAGGUAU;Mcm7, GAUGUCCUGGACGUUUACA; Geminin (Gem), UGC-CAACUCUGGAAUCAAA (12); Cdt2, GAAUUAUACUG-CUUAUCGA. Transfections were conducted with 20 nmol/LsiRNA oligonucleotide duplexes with Lipofectamine RNAi-MAX (Invitrogen) according to the instructions of manufac-turer.
Antibodies and immunoblottingRabbit anti-Cdt1, rabbit anti-Geminin, and rabbit anti-Cdt2
were raised as described (9, 23). The purchased antibodieswere mouse anti-p21 (Lab Vision/Neomarkers); mouse anti-b-actin, mouse anti-Chk1, mouse anti-Chk2 (Sigma); rabbitanti-Chk1-P-S317, rabbit anti-Chk2-P-T68; rabbit anti-PARP,rabbit anti-H3-P-S10 (Upstate). Cells were lysed as described(24), and Western blot analysis was conducted according tostandard procedures.
Flow cytometric analysis (FACS)Cells were harvested by trypsinization and fixed with 70%
ethanol overnight at �80�C. Cells were then stained andanalyzed as described before (12). For FACS analysis withboth propidium iodide (PI) and bromodeoxyuridine (BrdU)double staining, cells were labeled with 10 mmol/L BrdU(Sigma) and then harvested as described earlier (12).
Time-lapse movie analysisHCT116 cells were plated at 15,000 cells per well in 6-well
culture plates (Becton Dickinson). For continuous treatment,cells were treated with 1 mmol/L MLN4924 for 72 hours. Forwashout treatment, HCT-116 cells were treated with 1 mmol/LMLN4924 for 8 hours, washed with fresh media to removecompound, and then maintained in fresh compound-freemedia for 8 days. Time-lapse movie images were taken attimes indicated using an automated TE2000U microscope(Nikon Instruments) with Hoffman-modulation optics, 20�objective, with environmental control, and an Orca-ER CCDcamera (Hamamatsu) controlled with MetaMorph imagingsoftware (Molecular Devices).
Measure cell growth and clonogenicityThe number of viable cells was estimated with a cell
proliferation assay (MTT) kit (Promega) according to themanufacturer's instructions. Cells were seeded into 96-wellplates at 500 cells per well, treated with DMSO or MLN4924,and incubated for 7 days before MTT assay. Cell clonogenicityassay was conducted as described (25). Cells were seeded into6-well plates at 3 � 103 cells per well. DMSO or 1 mmol/LMLN4924 was added for 8 hours. Cells were washed twice withPBS and incubated in fresh medium after the washout.Medium was changed every 2–3 days and the colonies werestained with crystal violet to show cell clonogenicity. OD595was measured to quantify cell colony numbers and normal-ized to DMSO-treated control sample to obtain cell survivalrate.
SA-b-gal staining assay for senescenceSenescence b-galactosidase staining assay was conducted
in a 6-well plate with staining kit (Cell Signaling Technology;no. 9860). Cells were washed with PBS, fixed, and stainedfollowing manufacturer's instruction. Stained plates werechecked under a microscope for development of blue color.For each sample, SA-b-gal–positive and total cell numberswere counted from 5 different microscopic fields (roughly>200 cells per field).
Results
Stabilization of Cdt1 protein is critical for MLN4924-induced rereplication in HCT116 cells
Consistent with previous results (21), we observed rereplica-tion after 20 hours of treatment of HCT116 cells withMLN4924(Fig. 1A). To investigate whether the regulation of Cdt1 proteinlevel plays a role in MLN4924-induced rereplication, HCT116cells were treated with siRNA oligonucleotides targeting Cdt1for 48 hours prior to the addition ofMLN4924. After 20 hours ofMLN4924 treatment, Cdt1 protein level increased significantlyas expected (Fig. 1B, lanes 1 and 2) and more than 40% of cellswere determined to have rereplicated, containing >4N DNAcontent. However, in the cells depleted of Cdt1 by siRNA, thepercentage of rereplicating cells reduced to 15% (Fig. 1A). Inthese cells, Cdt1 protein expression was effectively repressed(Fig. 1B, lanes 1, 3, and 4), although at a higher exposure, Cdt1protein level was observed to be modestly induced in
Effect of Transient Exposure of HCT116 to MLN4924
www.aacrjournals.org Cancer Res; 70(24) December 15, 2010 10311
Research. on April 9, 2019. © 2010 American Association for Cancercancerres.aacrjournals.org Downloaded from
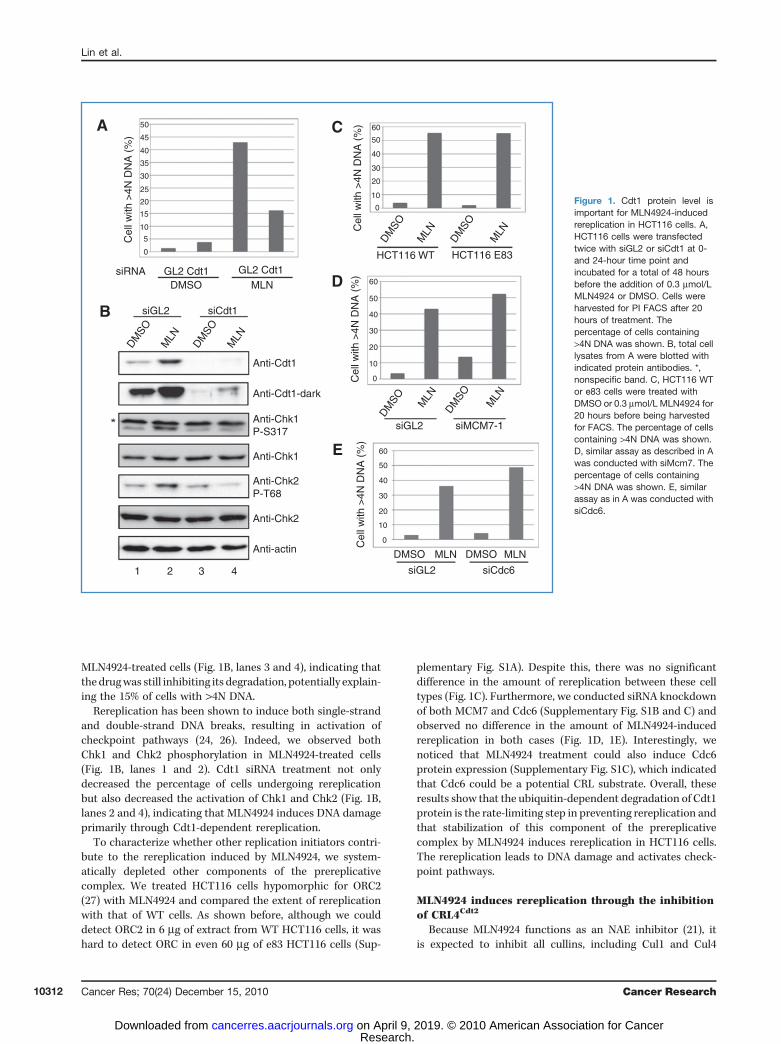
MLN4924-treated cells (Fig. 1B, lanes 3 and 4), indicating thatthe drugwas still inhibiting its degradation, potentially explain-ing the 15% of cells with >4N DNA.
Rereplication has been shown to induce both single-strandand double-strand DNA breaks, resulting in activation ofcheckpoint pathways (24, 26). Indeed, we observed bothChk1 and Chk2 phosphorylation in MLN4924-treated cells(Fig. 1B, lanes 1 and 2). Cdt1 siRNA treatment not onlydecreased the percentage of cells undergoing rereplicationbut also decreased the activation of Chk1 and Chk2 (Fig. 1B,lanes 2 and 4), indicating that MLN4924 induces DNA damageprimarily through Cdt1-dependent rereplication.
To characterize whether other replication initiators contri-bute to the rereplication induced by MLN4924, we system-atically depleted other components of the prereplicativecomplex. We treated HCT116 cells hypomorphic for ORC2(27) with MLN4924 and compared the extent of rereplicationwith that of WT cells. As shown before, although we coulddetect ORC2 in 6 mg of extract from WT HCT116 cells, it washard to detect ORC in even 60 mg of e83 HCT116 cells (Sup-
plementary Fig. S1A). Despite this, there was no significantdifference in the amount of rereplication between these celltypes (Fig. 1C). Furthermore, we conducted siRNA knockdownof both MCM7 and Cdc6 (Supplementary Fig. S1B and C) andobserved no difference in the amount of MLN4924-inducedrereplication in both cases (Fig. 1D, 1E). Interestingly, wenoticed that MLN4924 treatment could also induce Cdc6protein expression (Supplementary Fig. S1C), which indicatedthat Cdc6 could be a potential CRL substrate. Overall, theseresults show that the ubiquitin-dependent degradation of Cdt1protein is the rate-limiting step in preventing rereplication andthat stabilization of this component of the prereplicativecomplex by MLN4924 induces rereplication in HCT116 cells.The rereplication leads to DNA damage and activates check-point pathways.
MLN4924 induces rereplication through the inhibitionof CRL4Cdt2
Because MLN4924 functions as an NAE inhibitor (21), itis expected to inhibit all cullins, including Cul1 and Cul4
Anti-Cdt1
DMSO MLN
siGL2 siCdt1
GL2 Cdt1GL2 Cdt1
0
5
10
15
20
25
30
35
40
45
50
0
HCT116 WT
siGL2 siMCM7-1
siGL2 siCdc6
HCT116 E83
10
20
30
40
50
60C
D
E
A
B
0
10
20
30
40
50
60
0
10
20
30
40
50
60
siRNA
Cel
l with
>4N
DN
A (
%)
Cel
l with
>4N
DN
A (
%)
Cel
l with
>4N
DN
A (
%)
Cel
l with
>4N
DN
A (
%)
DMSO
MLN
DMSO
MLN
DMSO
MLN
DMSO
MLN
DMSO MLN
DMSO
MLNAnti-Cdt1-dark
Anti-Chk1P-S317
Anti-Chk2P-T68
Anti-Chk2
Anti-actin
1 2 3 4
Anti-Chk1
MLNDMSO MLNDMSO
Figure 1. Cdt1 protein level isimportant for MLN4924-inducedrereplication in HCT116 cells. A,HCT116 cells were transfectedtwice with siGL2 or siCdt1 at 0-and 24-hour time point andincubated for a total of 48 hoursbefore the addition of 0.3 mmol/LMLN4924 or DMSO. Cells wereharvested for PI FACS after 20hours of treatment. Thepercentage of cells containing>4N DNA was shown. B, total celllysates from A were blotted withindicated protein antibodies. *,nonspecific band. C, HCT116 WTor e83 cells were treated withDMSO or 0.3 mmol/L MLN4924 for20 hours before being harvestedfor FACS. The percentage of cellscontaining >4N DNA was shown.D, similar assay as described in Awas conducted with siMcm7. Thepercentage of cells containing>4N DNA was shown. E, similarassay as in A was conducted withsiCdc6.
Lin et al.
Cancer Res; 70(24) December 15, 2010 Cancer Research10312
Research. on April 9, 2019. © 2010 American Association for Cancercancerres.aacrjournals.org Downloaded from
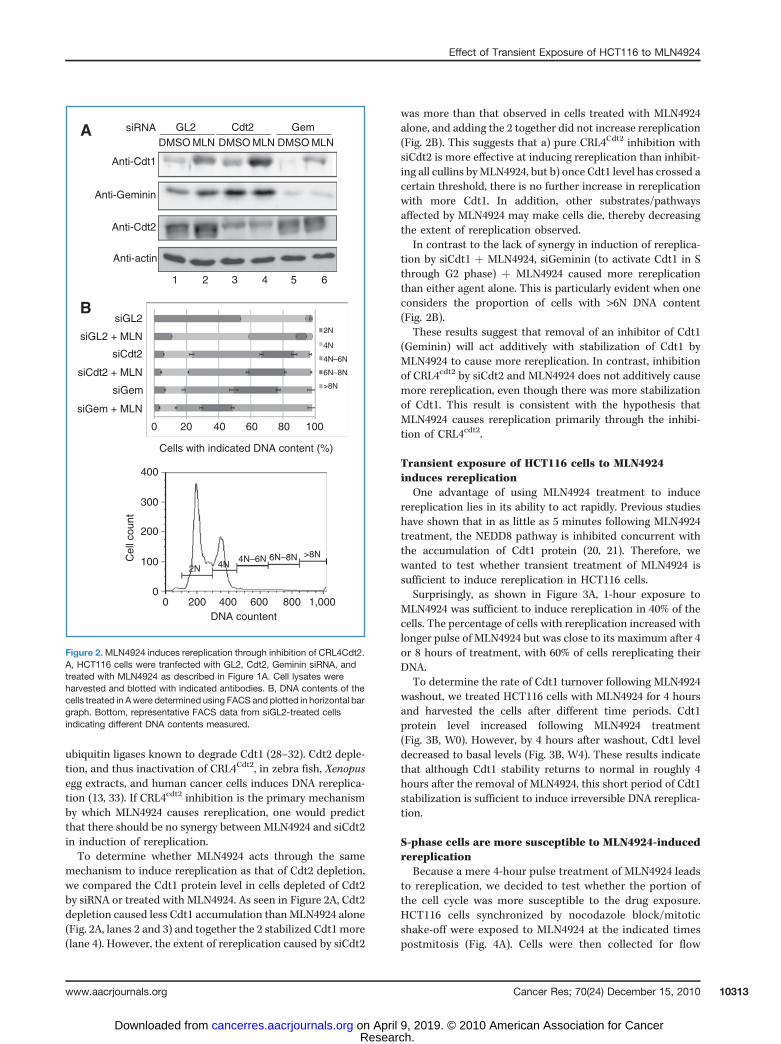
ubiquitin ligases known to degrade Cdt1 (28–32). Cdt2 deple-tion, and thus inactivation of CRL4Cdt2, in zebra fish, Xenopusegg extracts, and human cancer cells induces DNA rereplica-tion (13, 33). If CRL4cdt2 inhibition is the primary mechanismby which MLN4924 causes rereplication, one would predictthat there should be no synergy between MLN4924 and siCdt2in induction of rereplication.To determine whether MLN4924 acts through the same
mechanism to induce rereplication as that of Cdt2 depletion,we compared the Cdt1 protein level in cells depleted of Cdt2by siRNA or treated with MLN4924. As seen in Figure 2A, Cdt2depletion caused less Cdt1 accumulation thanMLN4924 alone(Fig. 2A, lanes 2 and 3) and together the 2 stabilized Cdt1 more(lane 4). However, the extent of rereplication caused by siCdt2
was more than that observed in cells treated with MLN4924alone, and adding the 2 together did not increase rereplication(Fig. 2B). This suggests that a) pure CRL4Cdt2 inhibition withsiCdt2 is more effective at inducing rereplication than inhibit-ing all cullins byMLN4924, but b) once Cdt1 level has crossed acertain threshold, there is no further increase in rereplicationwith more Cdt1. In addition, other substrates/pathwaysaffected by MLN4924 may make cells die, thereby decreasingthe extent of rereplication observed.
In contrast to the lack of synergy in induction of rereplica-tion by siCdt1 þ MLN4924, siGeminin (to activate Cdt1 in Sthrough G2 phase) þ MLN4924 caused more rereplicationthan either agent alone. This is particularly evident when oneconsiders the proportion of cells with >6N DNA content(Fig. 2B).
These results suggest that removal of an inhibitor of Cdt1(Geminin) will act additively with stabilization of Cdt1 byMLN4924 to cause more rereplication. In contrast, inhibitionof CRL4cdt2 by siCdt2 and MLN4924 does not additively causemore rereplication, even though there was more stabilizationof Cdt1. This result is consistent with the hypothesis thatMLN4924 causes rereplication primarily through the inhibi-tion of CRL4cdt2.
Transient exposure of HCT116 cells to MLN4924induces rereplication
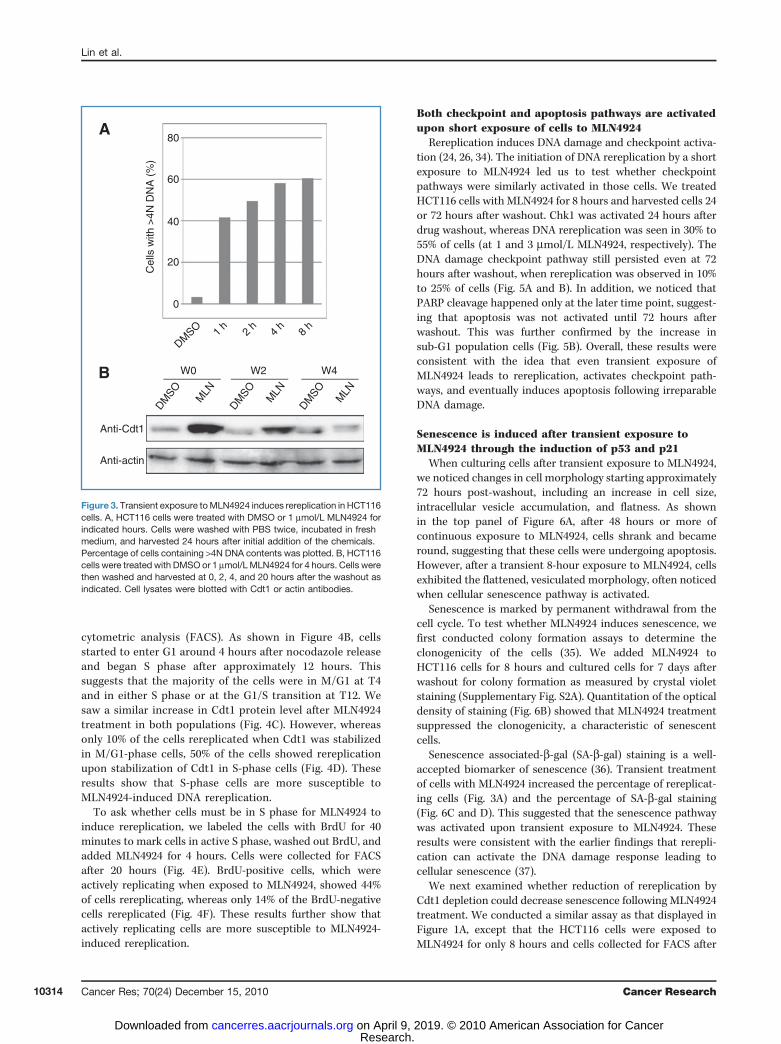
One advantage of using MLN4924 treatment to inducerereplication lies in its ability to act rapidly. Previous studieshave shown that in as little as 5 minutes following MLN4924treatment, the NEDD8 pathway is inhibited concurrent withthe accumulation of Cdt1 protein (20, 21). Therefore, wewanted to test whether transient treatment of MLN4924 issufficient to induce rereplication in HCT116 cells.
Surprisingly, as shown in Figure 3A, 1-hour exposure toMLN4924 was sufficient to induce rereplication in 40% of thecells. The percentage of cells with rereplication increased withlonger pulse of MLN4924 but was close to its maximum after 4or 8 hours of treatment, with 60% of cells rereplicating theirDNA.
To determine the rate of Cdt1 turnover following MLN4924washout, we treated HCT116 cells with MLN4924 for 4 hoursand harvested the cells after different time periods. Cdt1protein level increased following MLN4924 treatment(Fig. 3B, W0). However, by 4 hours after washout, Cdt1 leveldecreased to basal levels (Fig. 3B, W4). These results indicatethat although Cdt1 stability returns to normal in roughly 4hours after the removal of MLN4924, this short period of Cdt1stabilization is sufficient to induce irreversible DNA rereplica-tion.
S-phase cells are more susceptible to MLN4924-inducedrereplication
Because a mere 4-hour pulse treatment of MLN4924 leadsto rereplication, we decided to test whether the portion ofthe cell cycle was more susceptible to the drug exposure.HCT116 cells synchronized by nocodazole block/mitoticshake-off were exposed to MLN4924 at the indicated timespostmitosis (Fig. 4A). Cells were then collected for flow
400
300
200
100
00 200 400
DNA countent
Cel
l cou
nt
600 800 1,000
2N 4N4N–6N 6N–8N >8N
siGL2
siGL2 + MLN
siCdt2
siCdt2 + MLN
siGem + MLN
B
0 20 40
Cells with indicated DNA content (%)
60 80 100
2N
4N
4N–6N
6N–8N
>8NsiGem
Anti-Cdt1
Anti-Geminin
Anti-Cdt2
DMSO
1 2 3 4 5 6
MLN DMSO MLN DMSO MLNA siRNA GL2 Cdt2 Gem
Anti-actin
Figure 2.MLN4924 induces rereplication through inhibition of CRL4Cdt2.A, HCT116 cells were tranfected with GL2, Cdt2, Geminin siRNA, andtreated with MLN4924 as described in Figure 1A. Cell lysates wereharvested and blotted with indicated antibodies. B, DNA contents of thecells treated in Awere determined using FACS and plotted in horizontal bargraph. Bottom, representative FACS data from siGL2-treated cellsindicating different DNA contents measured.
Effect of Transient Exposure of HCT116 to MLN4924
www.aacrjournals.org Cancer Res; 70(24) December 15, 2010 10313
Research. on April 9, 2019. © 2010 American Association for Cancercancerres.aacrjournals.org Downloaded from
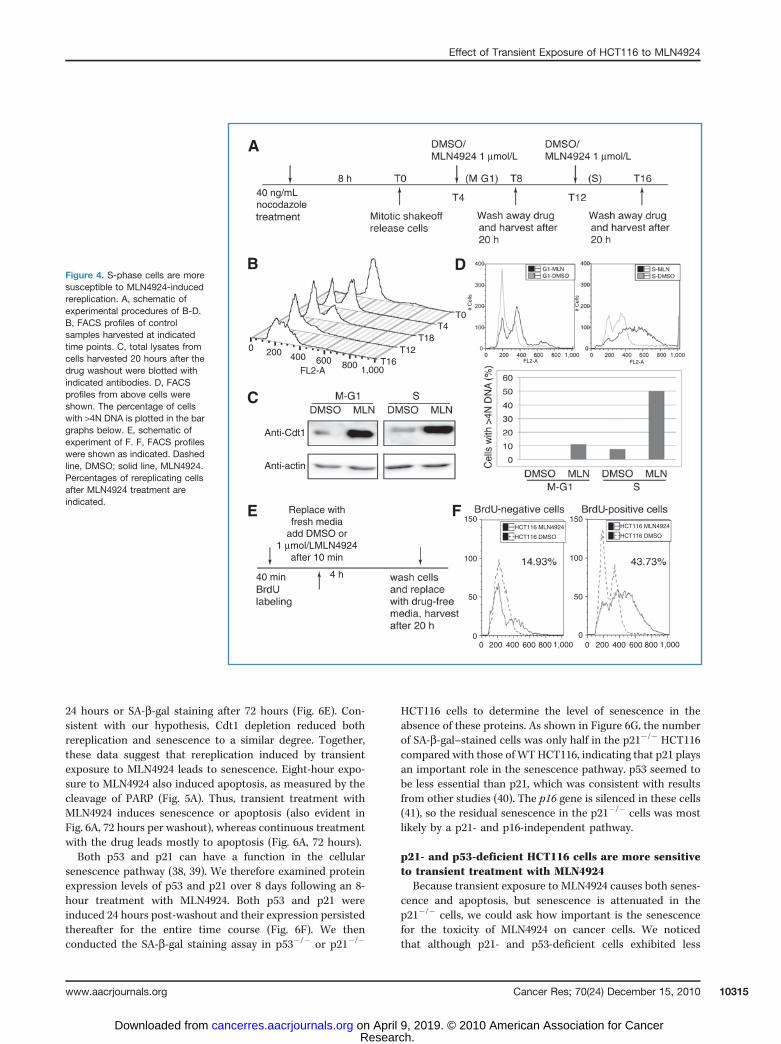
cytometric analysis (FACS). As shown in Figure 4B, cellsstarted to enter G1 around 4 hours after nocodazole releaseand began S phase after approximately 12 hours. Thissuggests that the majority of the cells were in M/G1 at T4and in either S phase or at the G1/S transition at T12. Wesaw a similar increase in Cdt1 protein level after MLN4924treatment in both populations (Fig. 4C). However, whereasonly 10% of the cells rereplicated when Cdt1 was stabilizedin M/G1-phase cells, 50% of the cells showed rereplicationupon stabilization of Cdt1 in S-phase cells (Fig. 4D). Theseresults show that S-phase cells are more susceptible toMLN4924-induced DNA rereplication.
To ask whether cells must be in S phase for MLN4924 toinduce rereplication, we labeled the cells with BrdU for 40minutes to mark cells in active S phase, washed out BrdU, andadded MLN4924 for 4 hours. Cells were collected for FACSafter 20 hours (Fig. 4E). BrdU-positive cells, which wereactively replicating when exposed to MLN4924, showed 44%of cells rereplicating, whereas only 14% of the BrdU-negativecells rereplicated (Fig. 4F). These results further show thatactively replicating cells are more susceptible to MLN4924-induced rereplication.
Both checkpoint and apoptosis pathways are activatedupon short exposure of cells to MLN4924
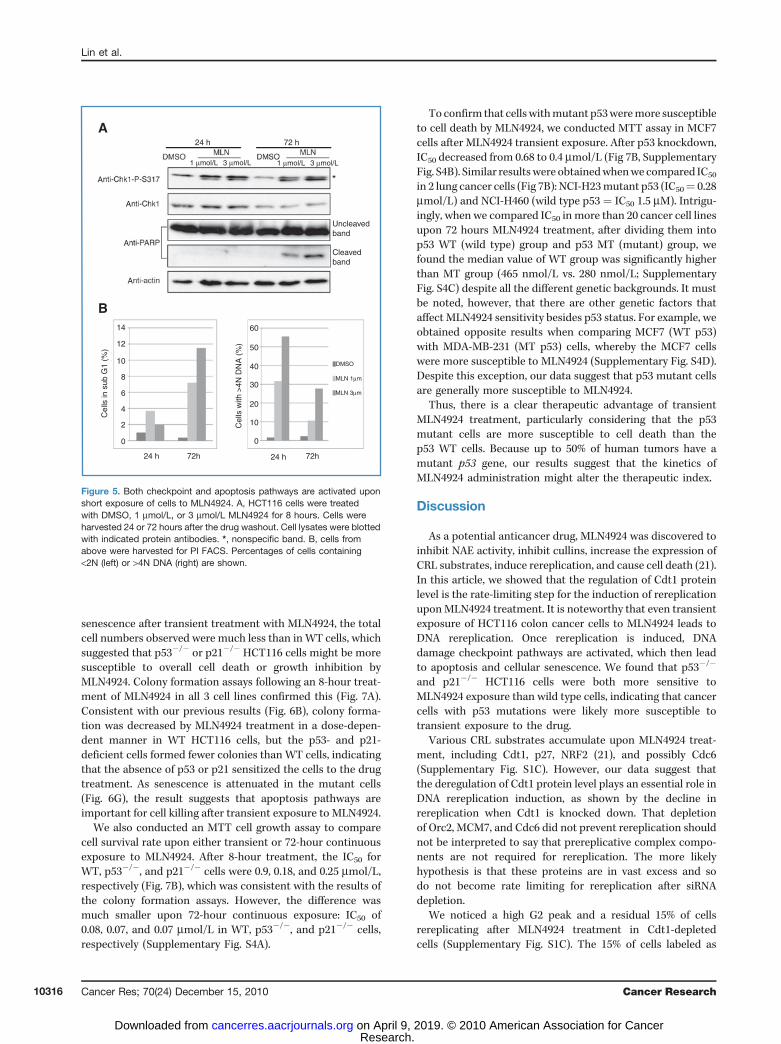
Rereplication induces DNA damage and checkpoint activa-tion (24, 26, 34). The initiation of DNA rereplication by a shortexposure to MLN4924 led us to test whether checkpointpathways were similarly activated in those cells. We treatedHCT116 cells with MLN4924 for 8 hours and harvested cells 24or 72 hours after washout. Chk1 was activated 24 hours afterdrug washout, whereas DNA rereplication was seen in 30% to55% of cells (at 1 and 3 mmol/L MLN4924, respectively). TheDNA damage checkpoint pathway still persisted even at 72hours after washout, when rereplication was observed in 10%to 25% of cells (Fig. 5A and B). In addition, we noticed thatPARP cleavage happened only at the later time point, suggest-ing that apoptosis was not activated until 72 hours afterwashout. This was further confirmed by the increase insub-G1 population cells (Fig. 5B). Overall, these results wereconsistent with the idea that even transient exposure ofMLN4924 leads to rereplication, activates checkpoint path-ways, and eventually induces apoptosis following irreparableDNA damage.
Senescence is induced after transient exposure toMLN4924 through the induction of p53 and p21
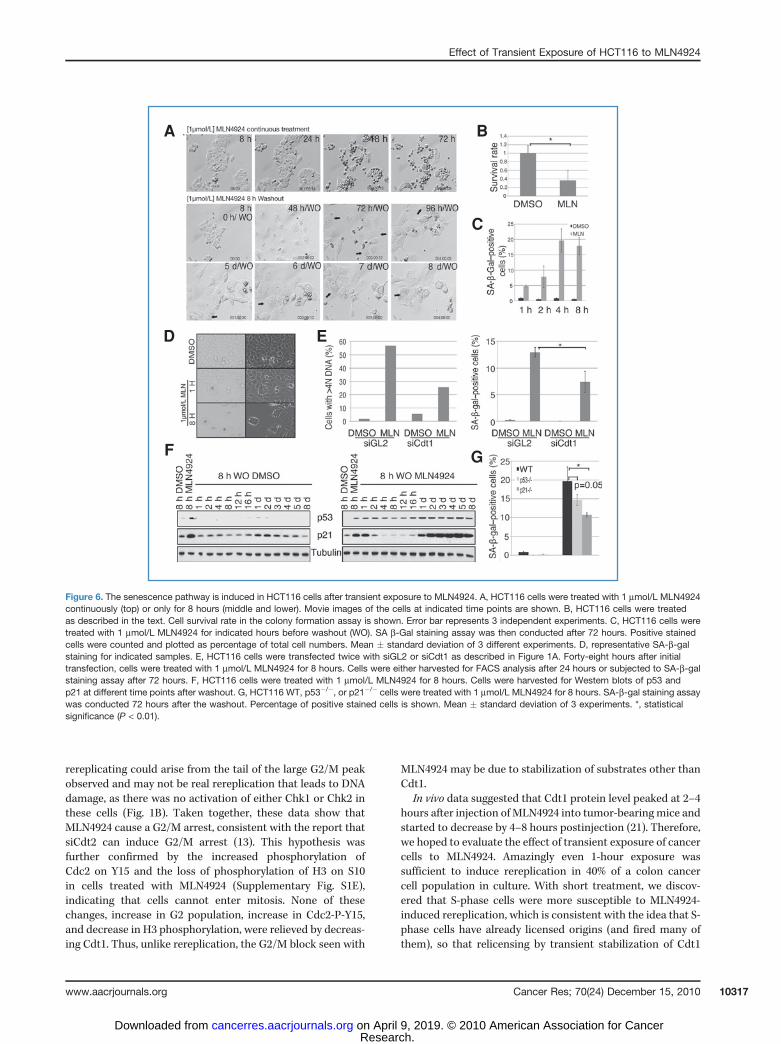
When culturing cells after transient exposure to MLN4924,we noticed changes in cell morphology starting approximately72 hours post-washout, including an increase in cell size,intracellular vesicle accumulation, and flatness. As shownin the top panel of Figure 6A, after 48 hours or more ofcontinuous exposure to MLN4924, cells shrank and becameround, suggesting that these cells were undergoing apoptosis.However, after a transient 8-hour exposure to MLN4924, cellsexhibited the flattened, vesiculated morphology, often noticedwhen cellular senescence pathway is activated.
Senescence is marked by permanent withdrawal from thecell cycle. To test whether MLN4924 induces senescence, wefirst conducted colony formation assays to determine theclonogenicity of the cells (35). We added MLN4924 toHCT116 cells for 8 hours and cultured cells for 7 days afterwashout for colony formation as measured by crystal violetstaining (Supplementary Fig. S2A). Quantitation of the opticaldensity of staining (Fig. 6B) showed that MLN4924 treatmentsuppressed the clonogenicity, a characteristic of senescentcells.
Senescence associated-b-gal (SA-b-gal) staining is a well-accepted biomarker of senescence (36). Transient treatmentof cells with MLN4924 increased the percentage of rereplicat-ing cells (Fig. 3A) and the percentage of SA-b-gal staining(Fig. 6C and D). This suggested that the senescence pathwaywas activated upon transient exposure to MLN4924. Theseresults were consistent with the earlier findings that rerepli-cation can activate the DNA damage response leading tocellular senescence (37).
We next examined whether reduction of rereplication byCdt1 depletion could decrease senescence following MLN4924treatment. We conducted a similar assay as that displayed inFigure 1A, except that the HCT116 cells were exposed toMLN4924 for only 8 hours and cells collected for FACS after
80
B
A
60
40
20
Cel
ls w
ith >
4N D
NA
(%
)
0
W0
Anti-Cdt1
Anti-actin
W2 W4
DMSO
DMSO
MLN
DMSO
MLN
DMSO
MLN
1 h
2 h
4 h
8 h
Figure 3. Transient exposure toMLN4924 induces rereplication in HCT116cells. A, HCT116 cells were treated with DMSO or 1 mmol/L MLN4924 forindicated hours. Cells were washed with PBS twice, incubated in freshmedium, and harvested 24 hours after initial addition of the chemicals.Percentage of cells containing >4N DNA contents was plotted. B, HCT116cells were treated with DMSO or 1 mmol/LMLN4924 for 4 hours. Cells werethen washed and harvested at 0, 2, 4, and 20 hours after the washout asindicated. Cell lysates were blotted with Cdt1 or actin antibodies.
Lin et al.
Cancer Res; 70(24) December 15, 2010 Cancer Research10314
Research. on April 9, 2019. © 2010 American Association for Cancercancerres.aacrjournals.org Downloaded from
24 hours or SA-b-gal staining after 72 hours (Fig. 6E). Con-sistent with our hypothesis, Cdt1 depletion reduced bothrereplication and senescence to a similar degree. Together,these data suggest that rereplication induced by transientexposure to MLN4924 leads to senescence. Eight-hour expo-sure to MLN4924 also induced apoptosis, as measured by thecleavage of PARP (Fig. 5A). Thus, transient treatment withMLN4924 induces senescence or apoptosis (also evident inFig. 6A, 72 hours per washout), whereas continuous treatmentwith the drug leads mostly to apoptosis (Fig. 6A, 72 hours).Both p53 and p21 can have a function in the cellular
senescence pathway (38, 39). We therefore examined proteinexpression levels of p53 and p21 over 8 days following an 8-hour treatment with MLN4924. Both p53 and p21 wereinduced 24 hours post-washout and their expression persistedthereafter for the entire time course (Fig. 6F). We thenconducted the SA-b-gal staining assay in p53�/� or p21�/�
HCT116 cells to determine the level of senescence in theabsence of these proteins. As shown in Figure 6G, the numberof SA-b-gal–stained cells was only half in the p21�/� HCT116compared with those of WT HCT116, indicating that p21 playsan important role in the senescence pathway. p53 seemed tobe less essential than p21, which was consistent with resultsfrom other studies (40). The p16 gene is silenced in these cells(41), so the residual senescence in the p21�/� cells was mostlikely by a p21- and p16-independent pathway.
p21- and p53-deficient HCT116 cells are more sensitiveto transient treatment with MLN4924
Because transient exposure to MLN4924 causes both senes-cence and apoptosis, but senescence is attenuated in thep21�/� cells, we could ask how important is the senescencefor the toxicity of MLN4924 on cancer cells. We noticedthat although p21- and p53-deficient cells exhibited less
Figure 4. S-phase cells are moresusceptible to MLN4924-inducedrereplication. A, schematic ofexperimental procedures of B-D.B, FACS profiles of controlsamples harvested at indicatedtime points. C, total lysates fromcells harvested 20 hours after thedrug washout were blotted withindicated antibodies. D, FACSprofiles from above cells wereshown. The percentage of cellswith >4N DNA is plotted in the bargraphs below. E, schematic ofexperiment of F. F, FACS profileswere shown as indicated. Dashedline, DMSO; solid line, MLN4924.Percentages of rereplicating cellsafter MLN4924 treatment areindicated.
T16
150
100
50
0
150HCT116 MLN4924
HCT116 DMSO
HCT116 MLN4924
HCT116 DMSO
100
50
00 200 400 600 800 1,000 0 200 400 600 800 1,000
T12T18
T4T0
# C
ells
# C
ells
0
A
B D
C
E F
200 400 600FL2-A
FL2-A FL2-A800 1,000
1,000 1,000
40 ng/mLnocodazole
40 min 4 h
8 h
Mi
Anti-Cdt1
Anti-actin
W
1 µmol/L
Replace withfresh media
add DMSO or1 µmol/LMLN4924
after 10 min
1 µmol/L
W
- -
Effect of Transient Exposure of HCT116 to MLN4924
www.aacrjournals.org Cancer Res; 70(24) December 15, 2010 10315
Research. on April 9, 2019. © 2010 American Association for Cancercancerres.aacrjournals.org Downloaded from
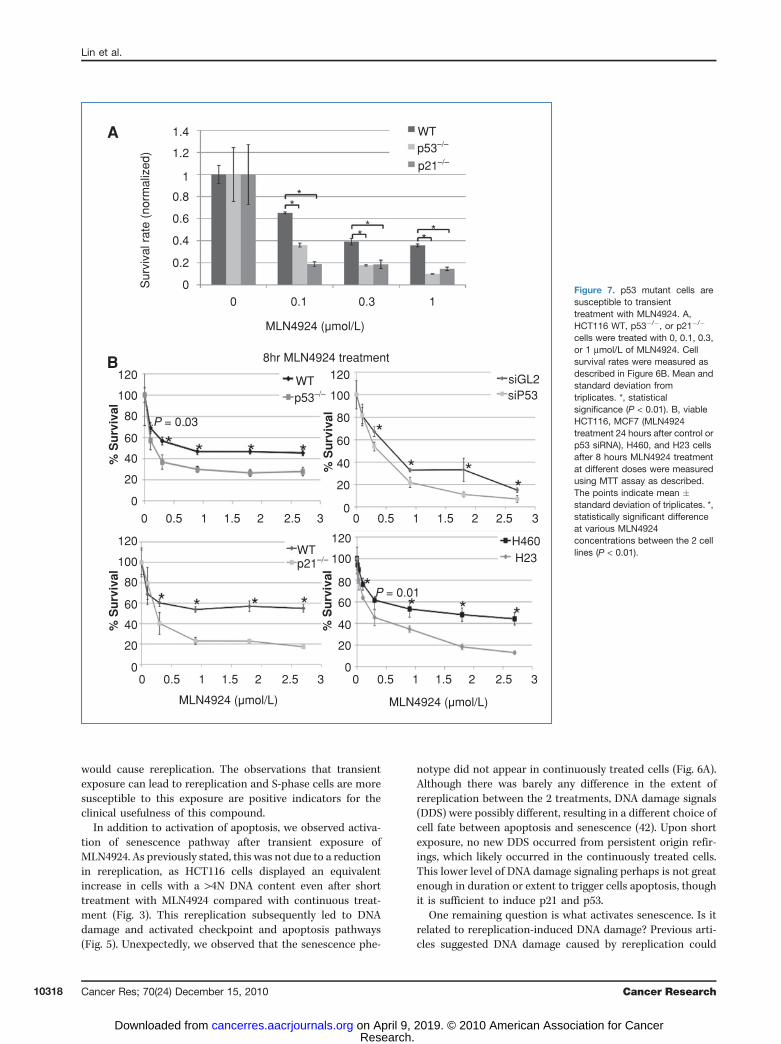
senescence after transient treatment with MLN4924, the totalcell numbers observed were much less than inWT cells, whichsuggested that p53�/� or p21�/� HCT116 cells might be moresusceptible to overall cell death or growth inhibition byMLN4924. Colony formation assays following an 8-hour treat-ment of MLN4924 in all 3 cell lines confirmed this (Fig. 7A).Consistent with our previous results (Fig. 6B), colony forma-tion was decreased by MLN4924 treatment in a dose-depen-dent manner in WT HCT116 cells, but the p53- and p21-deficient cells formed fewer colonies than WT cells, indicatingthat the absence of p53 or p21 sensitized the cells to the drugtreatment. As senescence is attenuated in the mutant cells(Fig. 6G), the result suggests that apoptosis pathways areimportant for cell killing after transient exposure to MLN4924.
We also conducted an MTT cell growth assay to comparecell survival rate upon either transient or 72-hour continuousexposure to MLN4924. After 8-hour treatment, the IC50 forWT, p53�/�, and p21�/� cells were 0.9, 0.18, and 0.25 mmol/L,respectively (Fig. 7B), which was consistent with the results ofthe colony formation assays. However, the difference wasmuch smaller upon 72-hour continuous exposure: IC50 of0.08, 0.07, and 0.07 mmol/L in WT, p53�/�, and p21�/� cells,respectively (Supplementary Fig. S4A).
To confirm that cellswithmutant p53weremore susceptibleto cell death by MLN4924, we conducted MTT assay in MCF7cells after MLN4924 transient exposure. After p53 knockdown,IC50 decreased from 0.68 to 0.4 mmol/L (Fig 7B, SupplementaryFig. S4B). Similar resultswere obtainedwhenwe compared IC50
in 2 lung cancer cells (Fig 7B): NCI-H23mutant p53 (IC50¼ 0.28mmol/L) and NCI-H460 (wild type p53 ¼ IC50 1.5 mM). Intrigu-ingly, when we compared IC50 in more than 20 cancer cell linesupon 72 hours MLN4924 treatment, after dividing them intop53 WT (wild type) group and p53 MT (mutant) group, wefound the median value of WT group was significantly higherthan MT group (465 nmol/L vs. 280 nmol/L; SupplementaryFig. S4C) despite all the different genetic backgrounds. It mustbe noted, however, that there are other genetic factors thataffect MLN4924 sensitivity besides p53 status. For example, weobtained opposite results when comparing MCF7 (WT p53)with MDA-MB-231 (MT p53) cells, whereby the MCF7 cellswere more susceptible to MLN4924 (Supplementary Fig. S4D).Despite this exception, our data suggest that p53 mutant cellsare generally more susceptible to MLN4924.
Thus, there is a clear therapeutic advantage of transientMLN4924 treatment, particularly considering that the p53mutant cells are more susceptible to cell death than thep53 WT cells. Because up to 50% of human tumors have amutant p53 gene, our results suggest that the kinetics ofMLN4924 administration might alter the therapeutic index.
Discussion
As a potential anticancer drug, MLN4924 was discovered toinhibit NAE activity, inhibit cullins, increase the expression ofCRL substrates, induce rereplication, and cause cell death (21).In this article, we showed that the regulation of Cdt1 proteinlevel is the rate-limiting step for the induction of rereplicationuponMLN4924 treatment. It is noteworthy that even transientexposure of HCT116 colon cancer cells to MLN4924 leads toDNA rereplication. Once rereplication is induced, DNAdamage checkpoint pathways are activated, which then leadto apoptosis and cellular senescence. We found that p53�/�
and p21�/� HCT116 cells were both more sensitive toMLN4924 exposure than wild type cells, indicating that cancercells with p53 mutations were likely more susceptible totransient exposure to the drug.
Various CRL substrates accumulate upon MLN4924 treat-ment, including Cdt1, p27, NRF2 (21), and possibly Cdc6(Supplementary Fig. S1C). However, our data suggest thatthe deregulation of Cdt1 protein level plays an essential role inDNA rereplication induction, as shown by the decline inrereplication when Cdt1 is knocked down. That depletionof Orc2, MCM7, and Cdc6 did not prevent rereplication shouldnot be interpreted to say that prereplicative complex compo-nents are not required for rereplication. The more likelyhypothesis is that these proteins are in vast excess and sodo not become rate limiting for rereplication after siRNAdepletion.
We noticed a high G2 peak and a residual 15% of cellsrereplicating after MLN4924 treatment in Cdt1-depletedcells (Supplementary Fig. S1C). The 15% of cells labeled as
24 h
A
A
A
14
B
A
60
50
DMSO
MLN 1µm
MLN 3µm
40
30
20
10
0
12
10
8
Cel
ls in
sub
G1
(%)
Cel
ls w
ith >
4N D
NA
(%
)
6
4
2
0
24 h 24 h 72h72h
A
1 µmol/L 3 µmol/L 1 µmol/L 3 µmol/L
72 h
Uncleavedband
Cleavedband
Figure 5. Both checkpoint and apoptosis pathways are activated uponshort exposure of cells to MLN4924. A, HCT116 cells were treatedwith DMSO, 1 mmol/L, or 3 mmol/L MLN4924 for 8 hours. Cells wereharvested 24 or 72 hours after the drug washout. Cell lysates were blottedwith indicated protein antibodies. *, nonspecific band. B, cells fromabove were harvested for PI FACS. Percentages of cells containing<2N (left) or >4N DNA (right) are shown.
Lin et al.
Cancer Res; 70(24) December 15, 2010 Cancer Research10316
Research. on April 9, 2019. © 2010 American Association for Cancercancerres.aacrjournals.org Downloaded from
rereplicating could arise from the tail of the large G2/M peakobserved and may not be real rereplication that leads to DNAdamage, as there was no activation of either Chk1 or Chk2 inthese cells (Fig. 1B). Taken together, these data show thatMLN4924 cause a G2/M arrest, consistent with the report thatsiCdt2 can induce G2/M arrest (13). This hypothesis wasfurther confirmed by the increased phosphorylation ofCdc2 on Y15 and the loss of phosphorylation of H3 on S10in cells treated with MLN4924 (Supplementary Fig. S1E),indicating that cells cannot enter mitosis. None of thesechanges, increase in G2 population, increase in Cdc2-P-Y15,and decrease in H3 phosphorylation, were relieved by decreas-ing Cdt1. Thus, unlike rereplication, the G2/M block seen with
MLN4924 may be due to stabilization of substrates other thanCdt1.
In vivo data suggested that Cdt1 protein level peaked at 2–4hours after injection ofMLN4924 into tumor-bearing mice andstarted to decrease by 4–8 hours postinjection (21). Therefore,we hoped to evaluate the effect of transient exposure of cancercells to MLN4924. Amazingly even 1-hour exposure wassufficient to induce rereplication in 40% of a colon cancercell population in culture. With short treatment, we discov-ered that S-phase cells were more susceptible to MLN4924-induced rereplication, which is consistent with the idea that S-phase cells have already licensed origins (and fired many ofthem), so that relicensing by transient stabilization of Cdt1
A
D
F G
E
B
C
Figure 6. The senescence pathway is induced in HCT116 cells after transient exposure to MLN4924. A, HCT116 cells were treated with 1 mmol/L MLN4924continuously (top) or only for 8 hours (middle and lower). Movie images of the cells at indicated time points are shown. B, HCT116 cells were treatedas described in the text. Cell survival rate in the colony formation assay is shown. Error bar represents 3 independent experiments. C, HCT116 cells weretreated with 1 mmol/L MLN4924 for indicated hours before washout (WO). SA b-Gal staining assay was then conducted after 72 hours. Positive stainedcells were counted and plotted as percentage of total cell numbers. Mean � standard deviation of 3 different experiments. D, representative SA-b-galstaining for indicated samples. E, HCT116 cells were transfected twice with siGL2 or siCdt1 as described in Figure 1A. Forty-eight hours after initialtransfection, cells were treated with 1 mmol/L MLN4924 for 8 hours. Cells were either harvested for FACS analysis after 24 hours or subjected to SA-b-galstaining assay after 72 hours. F, HCT116 cells were treated with 1 mmol/L MLN4924 for 8 hours. Cells were harvested for Western blots of p53 andp21 at different time points after washout. G, HCT116 WT, p53�/�, or p21�/� cells were treated with 1 mmol/L MLN4924 for 8 hours. SA-b-gal staining assaywas conducted 72 hours after the washout. Percentage of positive stained cells is shown. Mean � standard deviation of 3 experiments. *, statisticalsignificance (P < 0.01).
Effect of Transient Exposure of HCT116 to MLN4924
www.aacrjournals.org Cancer Res; 70(24) December 15, 2010 10317
Research. on April 9, 2019. © 2010 American Association for Cancercancerres.aacrjournals.org Downloaded from
would cause rereplication. The observations that transientexposure can lead to rereplication and S-phase cells are moresusceptible to this exposure are positive indicators for theclinical usefulness of this compound.
In addition to activation of apoptosis, we observed activa-tion of senescence pathway after transient exposure ofMLN4924. As previously stated, this was not due to a reductionin rereplication, as HCT116 cells displayed an equivalentincrease in cells with a >4N DNA content even after shorttreatment with MLN4924 compared with continuous treat-ment (Fig. 3). This rereplication subsequently led to DNAdamage and activated checkpoint and apoptosis pathways(Fig. 5). Unexpectedly, we observed that the senescence phe-
notype did not appear in continuously treated cells (Fig. 6A).Although there was barely any difference in the extent ofrereplication between the 2 treatments, DNA damage signals(DDS) were possibly different, resulting in a different choice ofcell fate between apoptosis and senescence (42). Upon shortexposure, no new DDS occurred from persistent origin refir-ings, which likely occurred in the continuously treated cells.This lower level of DNA damage signaling perhaps is not greatenough in duration or extent to trigger cells apoptosis, thoughit is sufficient to induce p21 and p53.
One remaining question is what activates senescence. Is itrelated to rereplication-induced DNA damage? Previous arti-cles suggested DNA damage caused by rereplication could
1.4A
B
1.2S
urvi
val r
ate
(nor
mal
ized
)
1
0.8
0.6
0.4
0.2
0
120
100
% S
urv
ival 80
60
40
20
0
120
100
% S
urv
ival 80
60
40
20
0
120
100
% S
urv
ival 80
60
40
20
0
0 0.5 1 1.5 2 2.5 3
0 0.5 1 1.5 2 2.5 3 0 0.5 1 1.5 2 2.5 3
0 0.5 1 1.5 2 2.5 3
120
100
% S
urv
ival 80
60
40
20
0
0 0.1
MLN4924 (µmol/L)
WT
WT
p53–/–
p53–/–
WTH460
siGL2siP53
H23
P = 0.01
P = 0.03
p21–/–
p21–/–
8hr MLN4924 treatment
MLN4924 (µmol/L) MLN4924 (µmol/L)
0.3 1Figure 7. p53 mutant cells aresusceptible to transienttreatment with MLN4924. A,HCT116 WT, p53�/�, or p21�/�
cells were treated with 0, 0.1, 0.3,or 1 mmol/L of MLN4924. Cellsurvival rates were measured asdescribed in Figure 6B. Mean andstandard deviation fromtriplicates. *, statisticalsignificance (P < 0.01). B, viableHCT116, MCF7 (MLN4924treatment 24 hours after control orp53 siRNA), H460, and H23 cellsafter 8 hours MLN4924 treatmentat different doses were measuredusing MTT assay as described.The points indicate mean �standard deviation of triplicates. *,statistically significant differenceat various MLN4924concentrations between the 2 celllines (P < 0.01).
Lin et al.
Cancer Res; 70(24) December 15, 2010 Cancer Research10318
Research. on April 9, 2019. © 2010 American Association for Cancercancerres.aacrjournals.org Downloaded from

activate the senescence pathway (37, 43, 44). In our hands,depletion of Geminin or Emi1 in HCT116 cells similarlyinduced senescence after 3–4 days (data not shown). Consis-tent with this idea, decrease in rereplication by depletion ofCdt1 reduced cellular senescence (Fig. 6E). Thus, the senes-cence is triggered by the rereplication-induced DNA damage.Another intriguing question is, once cell fate has been deter-mined, is it reversible? We treated cells with Z-VAD-FMKtogether with MLN4924 to inhibit cells from entering apop-tosis (Supplementary Fig. S3). However, there was no signifi-cant increase in senescence, which suggested an irreversiblecommitment to apoptotic, nonsenescent pathways.It has already been suggested that p53 and p21 levels are
increased during cellular senescence (45, 46). Multiple studieshave shown that the p53–p21 pathway is critical for senes-cence to occur in human fibroblasts and cancer cells (38, 42,47, 48). However, some researchers have observed thatalthough p53 and p21 are positive factors in senescence, theyare not necessary (49). Our results suggest that p53 and p21have important functions in initiating cellular senescenceupon MLN4924 treatment in tumor cells, but they are dis-pensable given that p53�/� or p21�/� cells showed decreasedbut not absent SA-b-gal staining (Fig. 6G).Although both p53�/� and p21�/� HCT116 cells underwent
less senescence than WT cells, both were more susceptible tocell death after transient treatment with MLN4924 (Fig. 7),suggesting a shifting of the balance toward a more apoptotic
phenotype upon intermittent treatment in those cells. Thisp53-independent susceptibility to MLN4924 is potentiallycritical for clinical applications, where nearly half of humantumors have mutated their p53 gene. Conventional che-motherapy is less effective in p53 mutant cells. Thus,MLN4924 is exceptional in its ability to target p53 mutanttumors.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We are grateful to Dr. Fred Bunz for providing us with isogenic p21�/� andp53�/� HCT116 cells. We thank members of the Dutta laboratory for readingthe manuscript and helpful discussions.
Grant Support
This work was supported by the Research Project Grant from NationalInstitutes of Health (R01-CA60499 and CA89406). J.J. Lin is supported by thePredoctoral Traineeship Award from Department of Defense Breast CancerResearch Program (W81XWH-08-1-0286).
The costs of publication of this article were defrayed in part by the paymentof page charges. This article must therefore be hereby marked advertisement inaccordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Received 06/07/2010; revised 09/04/2010; accepted 10/12/2010; publishedOnline 12/15/2010.
References1. Bell SP, Dutta A. DNA replication in eukaryotic cells. Annu Rev
Biochem 2002;71:333–74.2. Bell SP, Stillman B. ATP-dependent recognition of eukaryotic origins
of DNA replication by a multiprotein complex. Nature 1992;357:128–34.
3. Dutta A, Bell SP. Initiation of DNA replication in eukaryotic cells. AnnuRev Cell Dev Biol 1997;13:293–332.
4. Lei M, Tye BK. Initiating DNA synthesis: from recruiting to activatingthe MCM complex. J Cell Sci 2001;114:1447–54.
5. Maiorano D, Moreau J, Mechali M. XCDT1 is required for the assemblyof pre-replicative complexes in Xenopus laevis. Nature 2000;404:622–5.
6. Nishitani H, Lygerou Z, Nishimoto T, Nurse P. The Cdt1 protein isrequired to license DNA for replication in fission yeast. Nature2000;404:625–8.
7. Arias EE, Walter JC. Strength in numbers: preventing rereplication viamultiple mechanisms in eukaryotic cells. Genes Dev 2007;21:497–518.
8. Kim Y, Kipreos ET. Cdt1 degradation to prevent DNA re-replication:conserved and non-conserved pathways. Cell Div 2007;2:18.
9. Wohlschlegel JA, Dwyer BT, Dhar SK, Cvetic C, Walter JC, Dutta A.Inhibition of eukaryotic DNA replication by geminin binding to Cdt1.Science 2000;290:2309–12.
10. Tada S, Li A, Maiorano D, Mechali M, Blow JJ. Repression of originassembly in metaphase depends on inhibition of RLF-B/Cdt1 bygeminin. Nat Cell Biol 2001;3:107–13.
11. Machida YJ, Hamlin JL, Dutta A. Right place, right time, and only once:replication initiation in metazoans. Cell 2005;123:13–24.
12. Zhu W, Chen Y, Dutta A. Rereplication by depletion of geminin is seenregardless of p53 status and activates a G2/M checkpoint. Mol CellBiol 2004;24:7140–50.
13. Jin J, Arias EE, Chen J, Harper JW,Walter JC. A family of diverse Cul4-Ddb1-interacting proteins includes Cdt2, which is required for S
phase destruction of the replication factor Cdt1. Mol Cell 2006;23:709–21.
14. Zhong W, Feng H, Santiago FE, Kipreos ET. CUL-4 ubiquitin ligasemaintains genome stability by restraining DNA-replication licensing.Nature 2003;423:885–9.
15. Gong L, Yeh ET. Identification of the activating and conjugatingenzymes of the NEDD8 conjugation pathway. J Biol Chem 1999;274:12036–42.
16. Petroski MD, Deshaies RJ. Function and regulation of cullin-RINGubiquitin ligases. Nat Rev Mol Cell Biol 2005;6:9–20.
17. Read MA, Brownell JE, Gladysheva TB, et al. Nedd8 modification ofcul-1 activates SCF(beta(TrCP))-dependent ubiquitination of Ikappa-Balpha. Mol Cell Biol 2000;20:2326–33.
18. Chiba T, Tanaka K. Cullin-based ubiquitin ligase and its control byNEDD8-conjugating system. Curr Protein Pept Sci 2004;5:177–84.
19. Petroski MD. Mechanism-based neddylation inhibitor. Chem Biol2010;17:6–8.
20. Brownell JE, Sintchak MD, Gavin JM, et al. Substrate-assisted inhibi-tion of ubiquitin-like protein-activating enzymes: the NEDD8 E1 inhi-bitor MLN4924 forms a NEDD8-AMP mimetic in situ. Mol Cell2010;37:102–11.
21. Soucy TA, Smith PG, Milhollen MA, et al. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature 2009;458:732–6.
22. Bunz F, Dutriaux A, Lengauer C, et al. Requirement for p53 and p21 tosustain G2 arrest after DNA damage. Science 1998;282:1497–501.
23. Abbas T, Sivaprasad U, Terai K, Amador V, Pagano M, Dutta A.PCNA-dependent regulation of p21 ubiquitylation and degradationvia the CRL4Cdt2 ubiquitin ligase complex. Genes Dev 2008;22:2496–506.
24. Lin JJ, Dutta A. ATR pathway is the primary pathway for activating G2/M checkpoint induction after re-replication. J Biol Chem 2007;282:30357–62.
Effect of Transient Exposure of HCT116 to MLN4924
www.aacrjournals.org Cancer Res; 70(24) December 15, 2010 10319
Research. on April 9, 2019. © 2010 American Association for Cancercancerres.aacrjournals.org Downloaded from

25. Montes de Oca R, Andreassen PR, Margossian SP, et al. Regulatedinteraction of the Fanconi anemia protein, FANCD2, with chromatin.Blood 2005;105:1003–9.
26. ZhuWDA. An ATR- and BRCA1-mediated Fanconi anemia pathway isrequired for activating the G2/M checkpoint and DNA damage repairupon rereplication. MCB 2006;26:4601–11
27. Dhar SK, Yoshida K,Machida Y, et al. Replication from oriP of Epstein-Barr virus requires human ORC and is inhibited by geminin. Cell 2001;106:287–96.
28. Sugimoto N, Tatsumi Y, Tsurumi T, et al. Cdt1 phosphorylation bycyclin A-dependent kinases negatively regulates its function withoutaffecting geminin binding. J Biol Chem 2004;279:19691–7.
29. Takeda DY, Parvin JD, Dutta A. Degradation of Cdt1 during S phaseis Skp2-independent and is required for efficient progression ofmammalian cells through S phase. J Biol Chem 2005;280:23416–23.
30. Li X, Zhao Q, Liao R, Sun P, Wu X. The SCF(Skp2) ubiquitin ligasecomplex interacts with the human replication licensing factor Cdt1and regulates Cdt1 degradation. J Biol Chem 2003;278:30854–8.
31. Hu J, McCall CM, Ohta T, Xiong Y. Targeted ubiquitination of CDT1 bythe DDB1-CUL4A-ROC1 ligase in response to DNA damage. Nat CellBiol 2004;6:1003–9.
32. Senga T, Sivaprasad U, Zhu W, et al. PCNA is a cofactor for Cdt1degradation by CUL4/DDB1-mediated N-terminal ubiquitination. JBiol Chem 2006;281:6246–52.
33. Sansam CL, Shepard JL, Lai K, et al. DTL/CDT2 is essential for bothCDT1 regulation and the early G2/M checkpoint. Genes Dev 2006;20:3117–29.
34. McGarry TJ. Geminin deficiency causes a Chk1-dependent G2 arrestin Xenopus. Mol Biol Cell 2002;13:3662–71.
35. Stein GH, Beeson M, Gordon L. Failure to phosphorylate the retino-blastoma gene product in senescent human fibroblasts. Science1990;249:666–9.
36. Dimri GP, Lee X, Basile G, et al. A biomarker that identifies senescenthuman cells in culture and in aging skin in vivo. Proc Natl Acad Sci U SA 1995;92:9363–7.
37. Di Micco R, Fumagalli M, Cicalese A, et al. Oncogene-inducedsenescence is a DNA damage response triggered by DNA hyper-replication. Nature 2006;444:638–42.
38. Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenicras provokes premature cell senescence associated with accumula-tion of p53 and p16INK4a. Cell 1997;88:593–602.
39. Stein GH, Drullinger LF, Soulard A, Dulic V. Differential roles for cyclin-dependent kinase inhibitors p21 and p16 in the mechanisms ofsenescence and differentiation in human fibroblasts. Mol Cell Biol1999;19:2109–17.
40. Zou X, Ray D, Aziyu A, et al. Cdk4 disruption renders primary mousecells resistant to oncogenic transformation, leading to Arf/p53-inde-pendent senescence. Genes Dev 2002;16:2923–34.
41. Myohanen SK, Baylin SB, Herman JG. Hypermethylation can selec-tively silence individual p16ink4A alleles in neoplasia. Cancer Res1998;58:591–3.
42. Han Z, Wei W, Dunaway S, et al. Role of p21 in apoptosis andsenescence of human colon cancer cells treated with camptothecin.J Biol Chem 2002;277:17154–60.
43. Liontos M, Koutsami M, Sideridou M, et al. Deregulated overexpres-sion of hCdt1 and hCdc6 promotes malignant behavior. Cancer Res2007;67:10899–909.
44. Verschuren EW, Ban KH, Masek MA, Lehman NL, Jackson PK. Lossof Emi1-dependent anaphase-promoting complex/cyclosome inhibi-tion deregulates E2F target expression and elicits DNA damage-induced senescence. Mol Cell Biol 2007;27:7955–65.
45. Alcorta DA, Xiong Y, Phelps D, Hannon G, Beach D, Barrett JC.Involvement of the cyclin-dependent kinase inhibitor p16 (INK4a) inreplicative senescence of normal human fibroblasts. Proc Natl AcadSci U S A 1996;93:13742–7.
46. Atadja P, Wong H, Garkavtsev I, Veillette C, Riabowol K. Increasedactivity of p53 in senescing fibroblasts. Proc Natl Acad Sci USA1995;92:8348–52.
47. Brown JP, Wei W, Sedivy JM. Bypass of senescence after disruptionof p21CIP1/WAF1 gene in normal diploid human fibroblasts. Science1997;277:831–4.
48. Kang JY, Kim JJ, Jang SY, Bae YS. The p53-p21(Cip1/WAF1) path-way is necessary for cellular senescence induced by the inhibition ofprotein kinase CKII in human colon cancer cells. Mol Cells2009;28:489–94.
49. Roninson IB, Broude EV, Chang BD. If not apoptosis, then what?Treatment-induced senescence and mitotic catastrophe in tumorcells. Drug Resist Updat 2001;4:303–13.
Lin et al.
Cancer Res; 70(24) December 15, 2010 Cancer Research10320
Research. on April 9, 2019. © 2010 American Association for Cancercancerres.aacrjournals.org Downloaded from

2010;70:10310-10320. Cancer Res Jie Jessie Lin, Michael A. Milhollen, Peter G. Smith, et al. Activation, Apoptosis, and Senescence in Cancer Cellsby Stabilizing Cdt1 in S Phase, Triggering Checkpoint NEDD8-Targeting Drug MLN4924 Elicits DNA Rereplication
Updated version
http://cancerres.aacrjournals.org/content/70/24/10310
Access the most recent version of this article at:
Material
Supplementary
http://cancerres.aacrjournals.org/content/suppl/2010/12/13/70.24.10310.DC1
Access the most recent supplemental material at:
Cited articles
http://cancerres.aacrjournals.org/content/70/24/10310.full#ref-list-1
This article cites 49 articles, 27 of which you can access for free at:
Citing articles
http://cancerres.aacrjournals.org/content/70/24/10310.full#related-urls
This article has been cited by 32 HighWire-hosted articles. Access the articles at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
SubscriptionsReprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications
Permissions
Rightslink site. (CCC)Click on "Request Permissions" which will take you to the Copyright Clearance Center's
.http://cancerres.aacrjournals.org/content/70/24/10310To request permission to re-use all or part of this article, use this link
Research. on April 9, 2019. © 2010 American Association for Cancercancerres.aacrjournals.org Downloaded from