quantitative real-time pcr for equine cytokine mrna in nondecalcified bone tissue embedded in methyl...
TRANSCRIPT

Quantitative Real-Time PCR for Equine Cytokine mRNA inNondecalcified Bone Tissue Embedded in Methyl Methacrylate
C. M. Leutenegger,1,2 B. von Rechenberg,1 J. B. Huder,2 K. Zlinsky, 1 C. Mislin,2 M. K. Akens,1 J. Auer,1 H. Lutz 2
1Musculosceletal Research Unit, Department of Veterinary Surgery, University of Zurich, Switzerland2Clinical Laboratory, Department of Internal Veterinary Medicine, University of Zurich, Winterthurerstrasse 260, CH-8057 Zurich, Switzerland
Received: 5 December 1998 / Accepted: 10 June 1999
Abstract. Specific amplification and quantitation ofnucleic acid sequences by the polymerase chain reaction(PCR) has been extensively used for the detection of viralinfection and gene expression. Although successful ampli-fication of DNA and RNA sequences extracted from paraf-fin embedded tissue have been described, there are presentlyno reports available regarding RNA analysis from bone andcalcified tissues embedded in hydrophobic acrylic resin.Here we describe a general method for quantitation of spe-cific mRNA sequences extracted from undecalcified bonesections, fixed in paraformaldehyde, and embedded in ahydrophobic acrylic resin. Total RNA was extracted fromdefined regions of single 50mm sawed sections. TheseRNA preparations are suitable for quantitative PCR analysisof mRNA of different cytokines. In addition, the universallyexpressed housekeeping GAPDH mRNA proved to be use-ful as an amplification control and to correct for the degreeof RNA degradation, which may vary considerably amongsamples. Reverse transcribed mRNA was amplified andquantitated in Real-Time PCR using a fluorescein labeledinternal TaqMan® probe.
Key words: Real-Time PCR — Bone — Quantitation—Cytokines — Equine.
Since the introduction of the polymerase chain reaction(PCR), the number of reports using fromalin-fixed and par-affin-embedded (FFPE) tissues for nucleic acid analysis hasincreased substantially. While DNA extracted from FFPEtissue has been successfully used for many applications,reports describing the analysis of RNA extracted from FFPEtissues have been rare [1–4]. However, most reports explor-ing transcription of certain mRNA usein situ hybridization,which may be difficult to establish for the detection of raremRNA species. In addition, this technique is also morecomplicated than hybridization analysis or Real-Time PCR.
Paraffin embedding of bone and other calcified tissuesrequires decalcification. Depending on the size of thesamples, this decalcification process takes several weeks tomonths and increases the possibility of losing RNA due todegradation. As an alternative for histological sections,bone and calcified tissue may be embedded in hydrophobicacrylic resin based on methyl methacrylate (MMA) [5]. The
embedding process may be completed within 3 weeks.These sections demonstrate excellent morphological pres-ervation and are ideal for computerized image analysis. Fur-thermore, one report showing successfulin situ hybridiza-tion has demonstrated that cellular RNA is still accessiblefor hybridization [6].
Obviously, RNA detection by PCR in FFPE or MMA-embedded (MMAE) tissue is hampered by technical prob-lems restricting general application of this approach. Prob-lems may be related to (1) the higher level of RNA degra-dation prior to fixation caused by the short half-life ofmRNA and the high content of RNases in some tissues; (2)the degradation and modification of RNA in the fixativeused, such as formaldehyde/paraformaldehyde; (3) the long-lasting preparation process during MMA embedding; (4)steriometrical hindrance of probe binding to the targetmRNA due to the presence of protein and/or acrylic resin;and (5) the sensitivity of the reverse transcriptase reactiondue to inhibitors present in crude preparations or inad-equately purified RNA. Therefore, employing referencegenes in parallel reactions is needed to avoid false-negativeresults and to normalize for the degree of RNA degradation.For this purpose, some universally expressed mRNAs, cod-ing for housekeeping genes, have been used [7].
In this report we tested the feasibility of quantitativereverse transcribed (RT) Real-Time PCR [8] in MMAEcompared with fresh material. Tissue of horses affected withsubchondral cystic lesions (SCL) was harvested during ar-throscopic surgery or after slaughter. Fresh tissues weresubjected either directly to total RNA extraction or to rou-tine fixation and MMAE procedures. Total RNA extractedfrom different samples of fresh and paraformaldehyde-fixedMMAE tissue were used to quantitative RT-PCR for thedetection of mRNA coding for the housekeeping gene glyc-eraldehyde-3-phosphate dehydrogenase (GAPDH), the pro-inflammatory cytokines interleukin (IL) 1b and IL-6, andthe inducible form of NO synthase (iNOS).
Materials and Methods
Fixation and Embedding of Tissue
Fibrous tissue of horses affected with SCL was harvested duringarthroscopic surgery, immediately frozen in liquid nitrogen, andstored at −80°C or immediately fixed in 4% buffered paraformal-dehyde at 4°C for 6–8 hours. Thereafter, tissue was embedded inparaffin blocks under standard conditions. Blocks of bone withSCL in the vicinity of the joints were harvested immediately afterslaughter. Embedding of undecalcified bone blocks containing the
Correspondence to:C. M. Leutenegger DVM, Clinical Labora-tory, Department of Internal Veterinary Medicine
Calcif Tissue Int (1999) 65:378–383
© 1999 Springer-Verlag New York Inc.

cystic lesions was carried out in an acrylic resin based on MMA(Leica HistoCour, Heidelberg, Germany) according to the manu-facturers instructions. From MMAE and FFPE tissues, 5mm sec-tions were cut with a microtome (Leica RM 2155) and stained withtoluidine blue for histological evaluation. In addition, 50mm sec-tions of the MMAE and FFPE tissues were cut with a sawingmicrotome (Leica SP 1600). Special areas in the periphery andcenter of MMAE tissue sections corresponding to tissues contrib-uting to different stages of SCL were excised using a microscissor.
RNA Extraction
Excised parts of MMAE sections and the 50mm FFPE sectionswere immediately stored in a chaotropic reagent (Trizol, Gibco,Basel, Switzerland) to prevent RNA degradation. After an incu-bation step of several minutes, the soaked tissue was mechanicallycrushed in a plotter for 1 minute. Total RNA was extracted ac-cording to the manufacturers recommendations. A total RNA pel-let was disolved in 10 mM Tris-HCl, 1 mM EDTA (pH 7.4) madein diethyl pyrocarbonate (DEPC, Fluka, Buchs, Switzerland)-treated water. Contaminating genomic DNA was digested withRNase-free DNAse I (Promega, Dubendorf, Switzerland) at 37°Cfor 1 hour followed by an inactivation step at 95°C for 5 minutes.
RNA Analysis
OD260 and OD280 were used for quantitation and determination ofthe purity of the preparation. Separation of total RNA was accom-plished on 1.4% agarose gels to analyze the size and integrity ofnucleic acids as described [9]. Briefly, RNA was disolved in sterilewater and sterile loading buffer (0.25% (w/v) bromophenol blue,0.25% (w/v) xylene cyanol, 30% (w/v) glycerol, 1.2% SDS, 60mM sodium phosphate (pH 6.8). The mixture was incubated at75°C for 5 minutes followed by immediate loading of the sampleonto a 1.4% gel. The gel was electrophoresed in 10 mM sodiumphosphate buffer, pH 6.8, containing 1ml of 10 mg/ml ethidiumbromide (BioRad, Glattbrugg, Switzerland). Constant recirculationof the buffer was maintained to prevent the formation of an un-desirable pH gradient due to the low buffering capacity of elec-trophoretic buffer.
cDNA Synthesis
Complementary DNA (cDNA) was synthesized using 10 U ofAMV RT (Promega, Dubendorf, Switzerland) essentially accord-ing to the recommendations of the manufacturer as follows: Re-verse transcription was performed in 20ml final volume containing50 mM Tris-HCl, pH 8.3, 50 mM KCl, 8 mM MgCl2, 0.25 mMdNTPs, 4 U RNAsin, 5 mM dithiothreitol (DTT), and 1ml randomhexamer primer (Pharmacia, Du¨bendorf, Switzerland). The reac-tion proceeded at 42°C for 1 hour. After inactivation at 95°C for 5minutes, the reaction volume was adjusted to 100ml with DEPC-treated water. The cDNA was stored at −30°C until processing ofthe samples.
Extractions of gDNA
In parallel to the RNA extractions, gDNA was coextracted fromTrizol-treated samples from the phenol phase according to themanufacturers instructions. Pellets of gDNA were disolved inDEPC-treated water and quantitated by spectrophotometry.
Real-Time PCR
For each target gene, three oligonucleotides were selected usingPrimer Express software (PE Biosystems, Foster City, California):two primers and an internal oligonucleotide as a probe. The inter-
nal probe was labeled at the 58 end with the reporter dye FAM(6-carboxyfluorescein), at the 38 end with the quencher dyeTAMRA (6-carboxytetramethyl-rhodamine) and was phosphate-blocked at the 38 end to prevent extension. Reporter and quencherin proximity results in suppression of reporter fluorescence of theintact probe by Fo¨rster-type energy transfer [10]. Binding of theprobe downstream of a primer during PCR, the 58→38 exonucleaseactivity of Taq DNA polymerase releases the reporter from thevicinity of the quencher dye resulting in increased reporter fluo-rescence [11]. Fluorescence intensity is directly related to theamount of input target DNA and can be detected with an auto-mated fluorometer (ABI Prism 7700 Sequence Detection System,PE Biosystems, Foster City, California).
For all the sequences studied, the sense and the antisenseprimer were placed in two consecutive exons of the gene; theprobe spanned the junction over the two exons, between the se-quences covered by the two primers. The PCR products (ampli-cons) were held very short, between 82 and 151 basepairs, for tworeasons: short amplicons can be extended in a few seconds and donot require an extension step. Therefore, the cycling program is atwo-temperature profile and allows shortening of the time requiredfor amplification. The primer and probe sequences are described inTable 1.
The amplification was performed by addition of 12ml of thecDNA in 25 ml (50 mM KCl, 10 mM Tris-HCl, pH 8.3, 5 mMMgCl2, 100mM of each dTNP and 600 nM of each primer) with0.5 U ofTaqDNA Polymerase (Sigma, Buchs, Switzerland). Am-plification was carried out as follows: denaturation for 3 minutes at94°C, five cycles of 1 minute at 94°C and 1 minute at 64°C,followed by 40 cycles at 30 seconds at 85°C and 1 minute at 64°C.Thermal cycling and fluorescence detection was done in an ABI7700 Prism as described above.
Relative quantitation of the cytokine signals was done by nor-malizing the cytokine signals with the GAPDH signal. We there-fore cloned a stretch of 225 bp of equine GAPDH by PCR cloning(GenBank accession no. AF097178). The normalized amount oftargets was compared with each other by designating one of thesamples as a calibrator. The threshold cycle (CT) represents thePCR cycle at which an increase in reporter fluorescence above abase line signal can first be detected.
Results
Characterization of Purified RNA
We used gel electrophoresis to compare RNA purified fromfrozen, formalin-fixed, and paraffin-embedded, and forma-lin-fixed and methyl methacrylate embedded tissues fromSCL (Fig. 1). RNA prepared from fresh-frozen tissue dis-played the characteristic pattern of undegraded cellularRNA, with distinct ribosomal RNA bands. The 28S bandhad twice the intensity of the 18S band. The FFPE andMMAE fibrous tissues showed smears with barely distin-guishable bands. The former showed reduced staining ofRNA.
DNA Contamination
The RNA gel showed some stained nucleic acid at the topof the gel lanes (data not shown). Double-stranded DNAmigrates to this location [12]. From comparisons to stan-dards (not illustrated), we estimated that DNA represented0.5–5% of the total nucleic acid. After DNAse treatment, noDNA bands could be detected anymore (Fig. 1).
PCR Amplification of gDNA
To demonstrate that our Real-Time primer-probe systemswere able to discriminate between cytokine-specific gDNA
C. M. Leutenegger et al.: Cytokine Quantitation in Methyl Methacrylate-Embedded Bone Tissue 379

and cDNA, 100 ng of purified gDNA was subjected to Real-Time PCR. None of the reactions from three repetitions withgDNA gave positive results (Table 2).
Quantitation of mRNA Obtained from Frozen, FFPE, andMMAE Tissues
The RNA obtained from frozen, FFPE, and MMAE tissuesand corresponding cDNA preparations were subjected toPCR analysis for the detection of the universally expressedGAPDH, iNOS, and the proinflammatory cytokines IL-1b
and IL-6. Since yield varied between specimens and quan-titation by spectrophotometry was not always possible,three different concentrations of RNA were used for cDNAsynthesis and subsequent PCR. Using different amounts ofthe cDNA preparation allowed amplification under optimalconditions at least in one of the three samples. Figure 2shows representatively, the results of the GAPDH, IL-1b,iNOS, and IL-6 Real-Time PCR from defined localizationscut out from MMAE sections. All samples gave positiveresults for GAPDH; however, IL-1b, iNOS, and IL-6 ex-pression were restricted to certain areas of the SCL lesion.
RNA extracted from fresh frozen tissue of surgicallyharvested material was subjected to the same PCR proce-dure. Similar to the results obtained with the MMAE ma-terial, GAPDH and IL-6 signals were readily detected.When FFPE material from the center of SCL was used forRNA extraction, GAPDH, IL-1b, iNOS, and IL-6 expres-sion were readily quantitated.
Discussion
In this report we have shown that total RNA extracted from
Table 1. Real-Time PCR systems for the detection of equine proinflammatory cytokines andGAPDH
GAPDHGAPDH.74f : AAGTGGATATTGTCGCCATCAATGAPDH.161f : AACTTGCCATGGGTGGAATCGAPDH.108p : aTGACCTCAACTACATGGTCTACATGTTTCAb
IL-1bIL 1b.7f : GCAGTACCCGACACCAGTGAIL 1b.88r : TTTTGGGCCATCCTCCTCAIL 1b.31p : aATGACTTACTGCAGCGGCAATGAGAATGAb
IL-6IL6.155f : AGCACATTAAGTACATCCTCGGCIL6.305r : CCAGATTGGAAGCATCCGTCIL6.181p : aATCTCTGCCCTGAAAAATGAGATGTGTAACAATTTb
iNOSiNOS.279f : GGATGACTTTCGAGGACATGCiNOS.388r : GGGCCCTCTGGTCATACTTTTiNOS.310p : aAAGCGACCTCCCCATTGGCCTb
Oligonucleotides are given in 58–38 orientation. Exon junctions within the probe sequencesare underlined.a Nucleotide, to which the reporter dye FAM is coupledb Nucleotide, to which the quencher dye TMARA is coupled
Fig. 1. Electrophoretic analysis of RNA from extracted frozen,FFPE, and MMA samples. Separation of RNA on a 1% agarosegel. (A, B) total RNA from decalcified bone sections;(C) totalRNA extracted from methyl methacrylate embedded tissue;(D)total RNA extracted from fresh subchondral tissue.(M) RNA sizemarker (Promega, Dubendorf, Switzerland).
Table 2. Discrimination between genomic DNA (gDNA) andcomplemetary DNA (cDNA) in Real-Time PCR with the fourprimer-probe systems for equine GAPDH, IL-1b, iNOS, and IL-6
Primer-probe system gDNA cDNA
GAPDH 40 23.83IL-1b 40 26.86a
IL-6 40 32.71a
iNOS 40 25.37a
Values are defined as the cycle when the threshold value is ex-ceeded. A CT value of 40 is indicative of a negative sample (nofluorescence)a cDNA from RNAs extracted from different localizations within aSCL lesion
C. M. Leutenegger et al.: Cytokine Quantitation in Methyl Methacrylate-Embedded Bone Tissue380
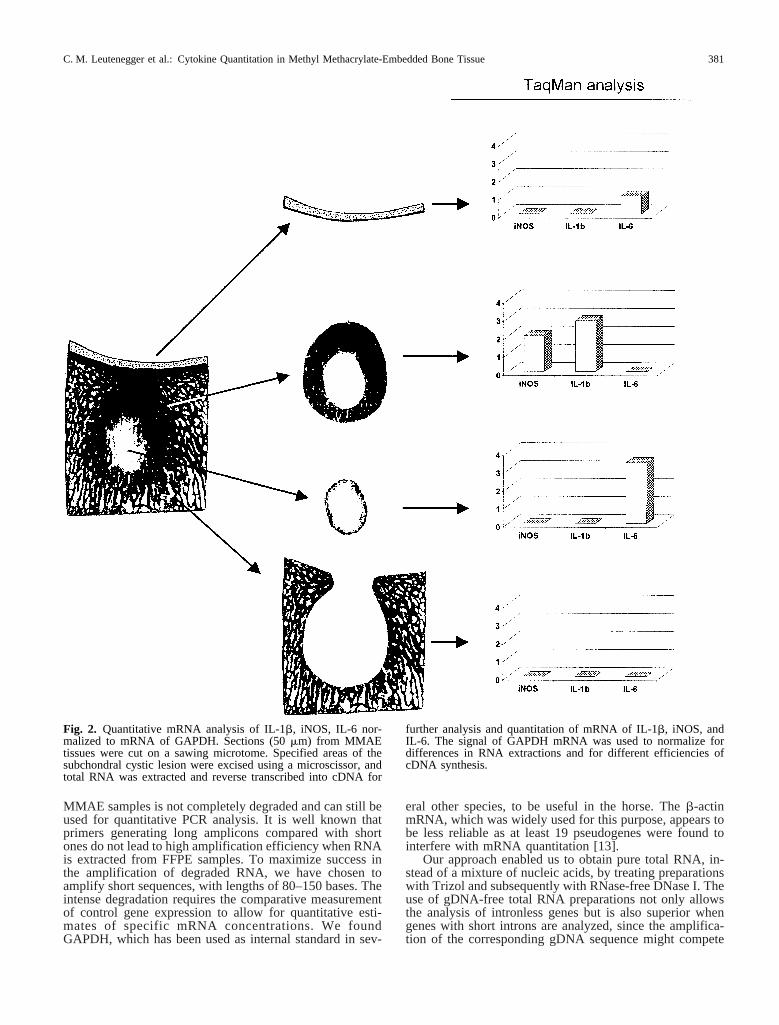
MMAE samples is not completely degraded and can still beused for quantitative PCR analysis. It is well known thatprimers generating long amplicons compared with shortones do not lead to high amplification efficiency when RNAis extracted from FFPE samples. To maximize success inthe amplification of degraded RNA, we have chosen toamplify short sequences, with lengths of 80–150 bases. Theintense degradation requires the comparative measurementof control gene expression to allow for quantitative esti-mates of specific mRNA concentrations. We foundGAPDH, which has been used as internal standard in sev-
eral other species, to be useful in the horse. Theb-actinmRNA, which was widely used for this purpose, appears tobe less reliable as at least 19 pseudogenes were found tointerfere with mRNA quantitation [13].
Our approach enabled us to obtain pure total RNA, in-stead of a mixture of nucleic acids, by treating preparationswith Trizol and subsequently with RNase-free DNase I. Theuse of gDNA-free total RNA preparations not only allowsthe analysis of intronless genes but is also superior whengenes with short introns are analyzed, since the amplifica-tion of the corresponding gDNA sequence might compete
Fig. 2. Quantitative mRNA analysis of IL-1b, iNOS, IL-6 nor-malized to mRNA of GAPDH. Sections (50mm) from MMAEtissues were cut on a sawing microtome. Specified areas of thesubchondral cystic lesion were excised using a microscissor, andtotal RNA was extracted and reverse transcribed into cDNA for
further analysis and quantitation of mRNA of IL-1b, iNOS, andIL-6. The signal of GAPDH mRNA was used to normalize fordifferences in RNA extractions and for different efficiencies ofcDNA synthesis.
C. M. Leutenegger et al.: Cytokine Quantitation in Methyl Methacrylate-Embedded Bone Tissue 381

with the cDNA sequence. To address the question of wheth-er contaminating gDNA present in the RNA samples couldinfluence the results, we subjected the nucleic acid prepa-ration to PCR without prior cDNA synthesis. None of thesamples gave a signal for GAPDH, IL-1b, iNOS, and IL-6,respectively. This control showed that treatment with excessDNase I seems to be sufficient to remove DNA contamina-tions. From conventional quantitative RT-PCR it is knownthat even in DNase-treated total RNA, residual gDNA stillis present. It is therefore important that potentially contami-nating gDNA is not amplified. This could be achieved bydesigning probes that cover an intron/exon transition.
The quality of nucleic acids extracted from MMAE tis-sue is mainly dependent on the activity of nucleases beforefixation and the embedding process. The time and quality offixation, and the duration of wet storage before embedding,are important variables. Atlhough the aldehyde groups offormalin react with the amino groups of nucleotides result-ing in cross-linking of DNA with proteins, complete cross-linking may not be present [14]. It has been described thatthe electrophoretic mobility of nucleic acids extracted fromFFPE tissue is slightly reduced, and prolonged exposure ofthe tissue to formalin leads to reduced signal intensity in dotblot and Southern blot analysis [15, 16]. This may be ex-plained in part by RNA degradation. Rapid and reproduc-ible fixation of tissue biopsies may be the reason for con-sistently successful amplification of mRNA from in ourMMAE sections.
The blurred RNA electrophoretic patterns probably re-sult from several causes, the most important of which isdegradation by tissue ribonucleases. Degradation of theRNA during preparation was of minor importance com-pared with degradation already present in the FFPE andMMA tissue since 18S and 28S rRNA fragments wereclearly visible in all preparations from frozen tissue samplesprepared with the same procedure. Some of the RNA hadreduced electrophoretic mobility, suggesting effects otherthan degradation. Formaldehyde modifies RNA by reactingwith its amino groups [14], causing irreversible denatur-ation. Such molecules unfold and have reduced mobility ongels. If the modification has only been partial, individualspecies migrate as blurs instead of discrete bands. Suchmodification does not interfere with hybridization analysis.
As total RNA extracted from different FFPE or MMAEtissues shows different levels of RNA degradation, quanti-tative PCR methods may not be used without normalizationof the degradation [17]. To obtain a reliable quantitation, thelevel of degradation was normalized using GAPDH mRNA,assuming that the level of degradation was comparable forany type of mRNA within the same sample [18, 19]. Quan-titation of mRNA expression by competitive RT-PCR re-quires several reactions with constant amounts of cDNA inthe presence of different amounts of competitor RNA at thesame time. Because only small amounts of RNA can berecovered from MMAE tissue, parallel reactions for differ-ent genes expressed would not be possible. QuantitativeReal-Time PCR therefore is a reliable alternative enablingquantitation of a certain mRNA normalized with an appro-priate housekeeping gene carried out in two PCR reactions.
The analysis of MMAE embedded tissues allows an ad-ditional methodological refinement with the analysis of cer-tain mRNA with quantitative PCR. Tissue structure is ex-cellently preserved in MMAE blocks, and sawed sectionscan be further dissected to allow localized measurements ofgene expression. Furthermore, samples from the MMAE
collection could be subjected to RNA extraction proceduresand Real-Time PCR to address new questions in certaindisease states.
In conclusion, the accurate quantitative Real-Time PCRpresented here can be performed with small amounts of totalRNA in a short time combined with GAPDH mRNA as apositive RNA and amplification control.
Acknowledgments.We thank Dr. R. Wicki, Ph.D., PE Biosystems,Switzerland, for excellent technical support with the ABI Prism7700. This paper is based on research completed as partial fulfill-ment of the requirements for a Ph.D. degree at the Universities ofBern and Zurich by CML. CML is a recipient of a Swiss NationalScience Foundation Grant (no. 823A-53569).
References
1. Jackson DP, Lewis FA, Taylor GR, Boylston AW, Quirke P(1990) Tissue extraction of DNA and RNA and analysis bythe polymerase chain reaction. J Clin Pathol 43(6):499–504
2. Saito K (1998) Morphology and molecular pathology: detec-tion of hepatitis C virus RNA sequences in stained sections bymicroscopy-directed selective extraction. Rinsho Byori. Jap JClin Pathol 46(1):49–55
3. Scholte GH, Van Doorn LJ, Quint WG, Lindeman J (1997)Polymerase chain reaction for the detection of helicobacterpylori in formaldehyde sublimate-fixed, paraffin-embeddedgastric biopsies. Diagn Mol Pathol 6(4):238–243
4. Del Pozo V, De Andres B, Gallardo S, Cardaba B, De Arruda-Chaves E, Cortegano M, Jurado A, Palomino P, Oliva H,Aguilera B, Posada M, Lahoz C (1997) Cytokine mRNA ex-pression in lung tissue from toxic oil syndrome patients: a Th2immunological mechanism. Toxicol 118(1):61–70
5. Bernhards J, Weitzel B, Werner M, Rimpler M, Georgii A(1992) A new histological embedding method by low-temperature polymerisation of methyl methacrylate allowingimmuno- and enzyme histochemical studies on semi-thin sec-tions of undecalcified bone marrow biopsies. Histochemistry98(3):145–54
6. Church RJ, Hand NM, Rex M, Scotting PJ (1997) Non-isotopic in situ hybridization to detect chick sox gene mRNAin plastic-embedded tissue. Histochem J 29(8):625–629
7. Finke J, Fritzen R, Ternes P, Lange W, Dolken G (1993) Animproved strategy and a useful housekeeping gene for RNAanalysis from formalin-fixed, paraffin-embedded tissues byPCR. Biotechniques 14(3):448–453
8. Holland PM, Abramson RD, Watson R, Gelfand DH (1991)Detection of specific polymerase chain reaction product byutilizing the 58-38 exonuclease activity of Thermus aquaticusDNA polymerase. PNAS 88(16):7276–7281
9. PelleR, Murphy NB (1994) Northern hybridization: rapid andsimple electrophoretic conditions. Nucl Acid Res 27:2783–2784
10. Forster V (1948) Zwischenmolekulare Energiewanderung undFluoreszenz. Ann Phy 2:55–57
11. Heid CA, Stevens J, Livak KJ, Williams PM (1996) Real timequantitative PCR. Genome Res 6(10):986–994
12. Locker J (1979) Analytical and preparative electrophoresis ofRNA in agarose-urea. Anal Biochem 98(2):358–367
13. Leavitt J, Gunning P, Porreca P, Ng SY, Lin CS, Kedes L
C. M. Leutenegger et al.: Cytokine Quantitation in Methyl Methacrylate-Embedded Bone Tissue382

(1984) Molecular cloning and characterization of mutant andwild-type human beta-actin genes. Mol Cell Biol 4(10):1961–1969
14. Rupp GM, Locker J (1988) Purification and analysis of RNAfrom paraffin-embedded tissues. Biotechniques 6(1):56–60
15. Goelz SE, Hamilton SR, Vogelstein B (1985) Purification ofDNA from formaldehyde-fixed and paraffin-embedded hu-man tissue. Biochem Biophys Res Commun 130(1):118–126
16. Dubeau L, Chandler LA, Gralow JR, Nichols PW, Jones PA(1986) Southern blot analysis of DNA extracted from forma-lin-fixed pathology specimens. Cancer Res 46(6):2964–2969
17. Gilliland G, Perrin S, Blanchard K, Bunn HF (1990) Analysisof cytokine mRNA and DNA: detection and quantitation bycompetitive polymerase chain reaction. Proc Natl Acad SciUSA 87(7)2725–2729
18. Stanta G, Schneider C (1991) RNA extracted from paraffin-embedded human tissues is amenable to analysis by PCR am-plification. Biotechniques 11(3):304, 306, 308
19. Stanta G, Bonin S (1998) RNA quantitative analysis fromfixed and paraffin-embedded tissues: membrane hybridizationand capillary electrophoresis. Biotechniques 24(2):271–276
C. M. Leutenegger et al.: Cytokine Quantitation in Methyl Methacrylate-Embedded Bone Tissue 383