purkinje cell dysfunction and alteration of long-term
TRANSCRIPT

Purkinje cell dysfunction and alteration of long-termsynaptic plasticity in fetal alcohol syndromeLaurent Servais*†‡, Raphael Hourez*, Bertrand Bearzatto*, David Gall*, Serge N. Schiffmann*, and Guy Cheron†§
Laboratories of *Neurophysiology and §Neurophysiology and Movement Biomechanics, Universite Libre de Bruxelles (ULB), B-1070 Brussels, Belgium;†Laboratory of Electrophysiology, Universite Mons-Hainaut (UMH), 7000 Mons, Belgium
Edited by Hans Thoenen, Max Planck Institute of Neurobiology, Martinsried, Germany, and approved April 13, 2007 (received for review August 18, 2006)
In cerebellum and other brain regions, neuronal cell death becauseof ethanol consumption by the mother is thought to be the leadingcause of neurological deficits in the offspring. However, little isknown about how surviving cells function. We studied cerebellarPurkinje cells in vivo and in vitro to determine whether function ofthese cells was altered after prenatal ethanol exposure. We ob-served that Purkinje cells that were prenatally exposed to ethanolpresented decreased voltage-gated calcium currents because of adecreased expression of the �-isoform of protein kinase C. Long-term depression at the parallel fiber–Purkinje cell synapse in thecerebellum was converted into long-term potentiation. This likelyexplains the dramatic increase in Purkinje cell firing and the rapidoscillations of local field potential observed in alert fetal alcoholsyndrome mice. Our data strongly suggest that reversal of long-term synaptic plasticity and increased firing rates of Purkinje cellsin vivo are major contributors to the ataxia and motor learningdeficits observed in fetal alcohol syndrome. Our results show thatcalcium-related neuronal dysfunction is central to the pathogen-esis of the neurological manifestations of fetal alcohol syndromeand suggest new methods for treatment of this disorder.
calcium � cerebellum � protein kinase � long-term depression �motor learning
Fetal alcohol syndrome (FAS) is the leading cause of intel-lectual disability in the Western world with a prevalence of
1 to 1.5 cases per 1,000 live births (1) and a lifetime cost of careof approximately $1.4 million per case. This cost is mainlybecause of ethanol toxicity in the developing central nervoussystem, causing intellectual disability, deficits in learning, andfine-motor dysfunction (2).
The cerebellum is one of the main targets of in utero ethanoltoxicity (3). Within the cerebellum, Purkinje cells (PCs) arehighly sensitive to ethanol. PCs constitute the sole output of thecerebellar cortex and thus have a central functional role inintegration. All animal models of FAS display a reduction in thenumber of PCs by �20% (4), and various authors have proposedthat neuronal loss is the only cause of cerebellar deficits in FAS.One study conducted on adult anesthetized rats with FAS led tothe conclusion that PCs that survive ethanol administrationfunction normally (5). Thomas et al. (6) found a correlationbetween total PCs number and motor performance. Theseexperimental data led to the assumption that surviving PCsfunction normally and that motor coordination impairment inFAS results only from a quantitative defect of PCs. This iscrucially important from a therapeutic point of view becausevery few options exist to replace dead neurons. Many studieshave therefore focused on different ways to decrease PC loss inFAS (7, 8). However, different models of ataxia that result fromPC death per se (pcd mice, SV4 mice, T147 transgenic mice) havedemonstrated that considerable neuropathology can occur with-out the manifestation of a neurological phenotype and thatataxia occurs in mice only after there is loss of 50–75% of thePCs (9–11). Because FAS models are characterized by a non-progressive loss of only 20% of PCs, the presence of an associ-ated neuronal dysfunction can contribute to the observed ataxic
phenotype. Because the understanding of the relative contribu-tion of neuronal dysfunction and cell death to neurologicaldeficits occurring in FAS is central for the evaluation of potentialprevention or therapeutics, we looked for a neuronal dysfunctionof surviving PCs by using combined in vivo and in vitro recordingsin a mouse model of this condition.
ResultsHistological and Behavioral Alterations in the Mouse Model of FAS. A20% loss of PCs was observed in 3- or 7-week-old FAS mice whencompared with corresponding control groups (P � 0.001, Fig. 1A and B). This reduction was similar at 3 and 7 weeks (P � 0.05,Fig. 1B). FAS mice demonstrated significant impairment inmotor coordination in both rotarod and runway assays (P �0.0001, two-way ANOVA for repeated measures (Fig. 1 C and D,days 1–3). Eyelid conditioning, a much more cerebellar specifictask, was studied in 10 FAS and 10 control mice. Comparison ofthe daily percentage of conditioned response during eyelidconditioning demonstrated a significant difference in motorlearning between FAS and control mice (P � 0.013, two-wayANOVA for repeated measures) (Fig. 1 E and F). Mortality andethanol levels in FAS mice are available in supporting informa-tion (SI) Materials and Methods.
FAS PCs Had Increased Simple Spike Firing, Supporting Fast Oscilla-tions in Vivo. PC firing regulation is crucial in motor coordination.Thus, we studied the spontaneous PC firing in seven FAS andfive control alert mice. A total of 66 PCs were recorded (42 inFAS and 24 in control mice). PCs from FAS mice had anincreased simple spike firing rate (Fig. 2 A and B). In contrast,the complex spike firing rate was normal in FAS mice (Fig. 2C),demonstrating that decreased firing of inferior olivary neuronswas not responsible for the increased PC simple spike firing rate.During �30% of the recording time in FAS mice, a fast localfield potential oscillation (LFPO) was noticed (LFPO index �5.8 � 2.4; frequency � 196 � 37 Hz; Fig. 2 D and E, lower traces).In contrast, no LFPO was recorded in controls (Fig. 2 D and E,upper traces). As observed in other mouse models presentingfast LFPO, 11 FAS PCs recorded during these fast oscillationepisodes demonstrated a tight phase-locking with the oscillation,as illustrated by the wave trigger averaged traces of simple spikes
Author contributions: L.S. and R.H. contributed equally to this work; S.N.S. and G.C.contributed equally to this work; L.S., R.H., D.G., S.N.S., and G.C. designed research; L.S.,R.H., and B.B. performed research; L.S., R.H., and B.B. analyzed data; and L.S., R.H., S.N.S.,and G.C. wrote the paper.
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Abbreviations: EPSC, excitatory postsynaptic current; FAS, fetal alcohol syndrome; LFPO,local field potential oscillation; LTD, long-term depression; LTP, long-term potentiation; PC,Purkinje cell; VGCC, voltage-gated calcium channels.
‡To whom correspondence should be addressed at present address: Service de NeurologiePediatrique, Hopital Robert Debre, 48 Boulevard Serurier, 759535 Paris France. E-mail:[email protected].
This article contains supporting information online at www.pnas.org/cgi/content/full/0607037104/DC1.
© 2007 by The National Academy of Sciences of the USA
9858–9863 � PNAS � June 5, 2007 � vol. 104 � no. 23 www.pnas.org�cgi�doi�10.1073�pnas.0607037104
Dow
nloa
ded
by g
uest
on
Nov
embe
r 6,
202
1
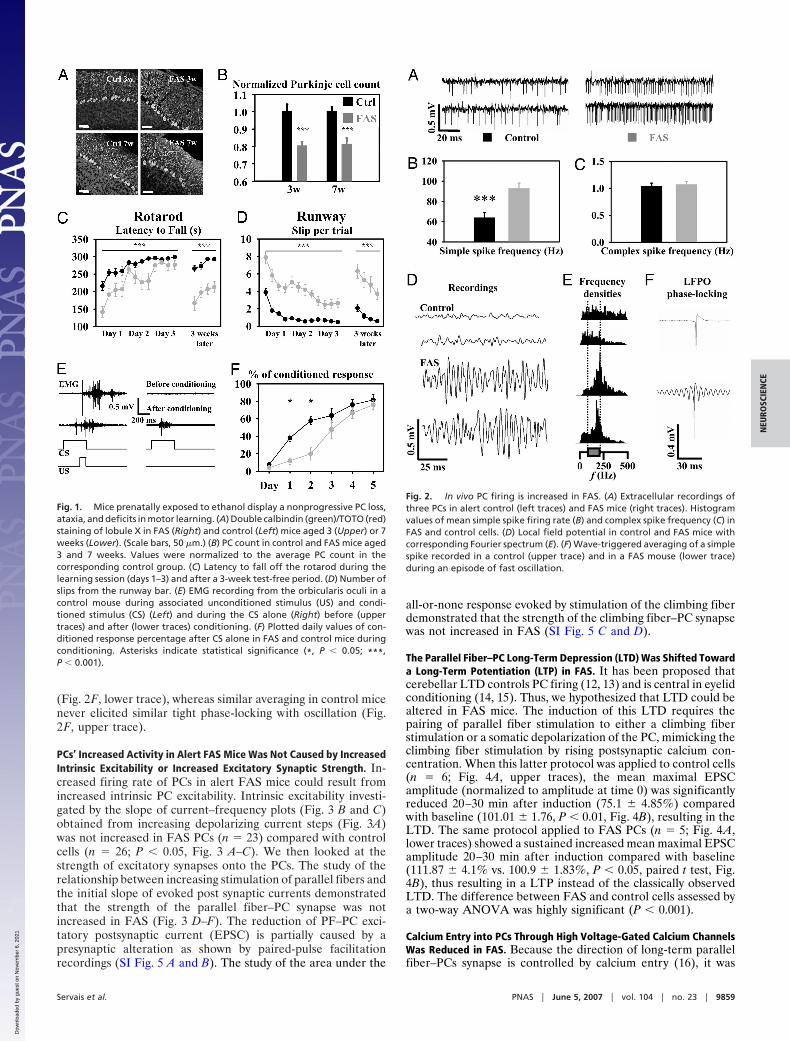
(Fig. 2F, lower trace), whereas similar averaging in control micenever elicited similar tight phase-locking with oscillation (Fig.2F, upper trace).
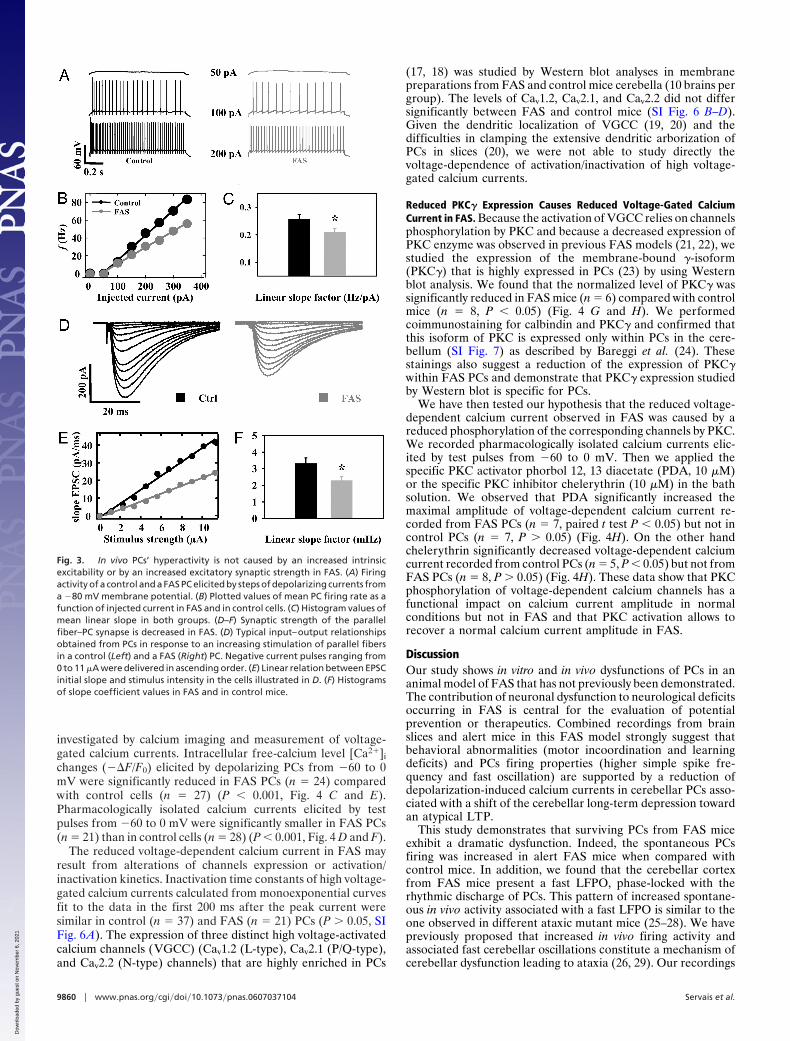
PCs’ Increased Activity in Alert FAS Mice Was Not Caused by IncreasedIntrinsic Excitability or Increased Excitatory Synaptic Strength. In-creased firing rate of PCs in alert FAS mice could result fromincreased intrinsic PC excitability. Intrinsic excitability investi-gated by the slope of current–frequency plots (Fig. 3 B and C)obtained from increasing depolarizing current steps (Fig. 3A)was not increased in FAS PCs (n � 23) compared with controlcells (n � 26; P � 0.05, Fig. 3 A–C). We then looked at thestrength of excitatory synapses onto the PCs. The study of therelationship between increasing stimulation of parallel fibers andthe initial slope of evoked post synaptic currents demonstratedthat the strength of the parallel fiber–PC synapse was notincreased in FAS (Fig. 3 D–F). The reduction of PF–PC exci-tatory postsynaptic current (EPSC) is partially caused by apresynaptic alteration as shown by paired-pulse facilitationrecordings (SI Fig. 5 A and B). The study of the area under the
all-or-none response evoked by stimulation of the climbing fiberdemonstrated that the strength of the climbing fiber–PC synapsewas not increased in FAS (SI Fig. 5 C and D).
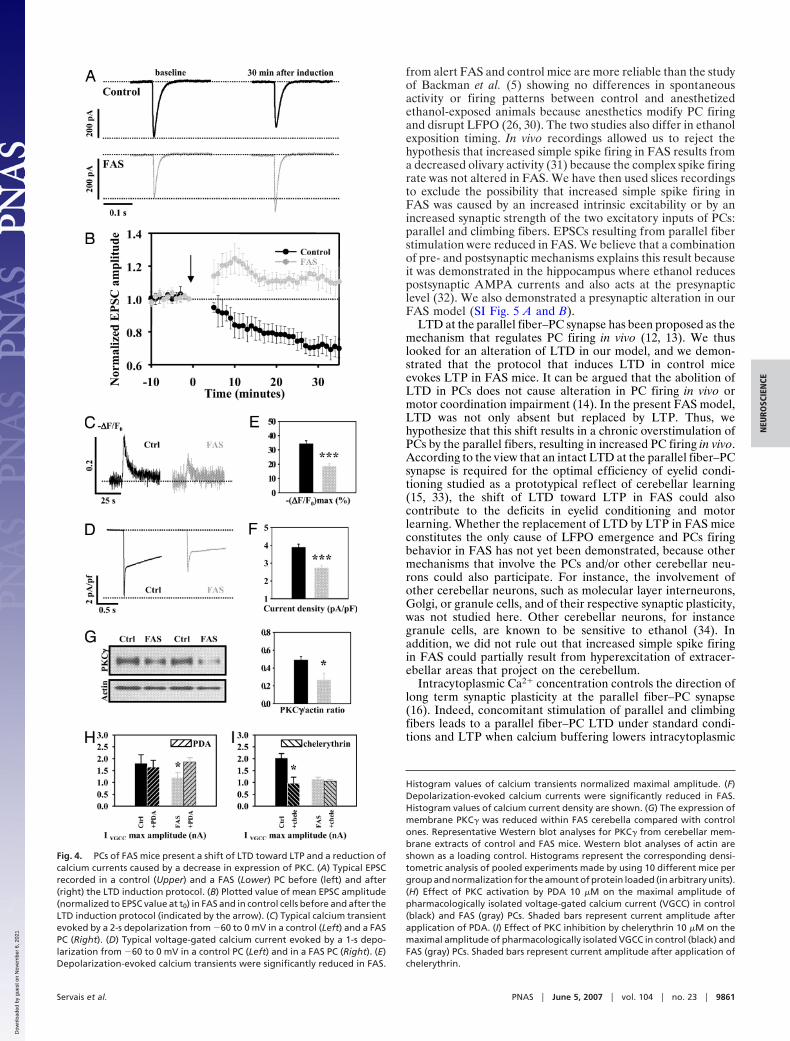
The Parallel Fiber–PC Long-Term Depression (LTD) Was Shifted Towarda Long-Term Potentiation (LTP) in FAS. It has been proposed thatcerebellar LTD controls PC firing (12, 13) and is central in eyelidconditioning (14, 15). Thus, we hypothesized that LTD could bealtered in FAS mice. The induction of this LTD requires thepairing of parallel fiber stimulation to either a climbing fiberstimulation or a somatic depolarization of the PC, mimicking theclimbing fiber stimulation by rising postsynaptic calcium con-centration. When this latter protocol was applied to control cells(n � 6; Fig. 4A, upper traces), the mean maximal EPSCamplitude (normalized to amplitude at time 0) was significantlyreduced 20–30 min after induction (75.1 � 4.85%) comparedwith baseline (101.01 � 1.76, P � 0.01, Fig. 4B), resulting in theLTD. The same protocol applied to FAS PCs (n � 5; Fig. 4A,lower traces) showed a sustained increased mean maximal EPSCamplitude 20–30 min after induction compared with baseline(111.87 � 4.1% vs. 100.9 � 1.83%, P � 0.05, paired t test, Fig.4B), thus resulting in a LTP instead of the classically observedLTD. The difference between FAS and control cells assessed bya two-way ANOVA was highly significant (P � 0.001).
Calcium Entry into PCs Through High Voltage-Gated Calcium ChannelsWas Reduced in FAS. Because the direction of long-term parallelfiber–PCs synapse is controlled by calcium entry (16), it was
Fig. 1. Mice prenatally exposed to ethanol display a nonprogressive PC loss,ataxia, and deficits in motor learning. (A) Double calbindin (green)/TOTO (red)staining of lobule X in FAS (Right) and control (Left) mice aged 3 (Upper) or 7weeks (Lower). (Scale bars, 50 �m.) (B) PC count in control and FAS mice aged3 and 7 weeks. Values were normalized to the average PC count in thecorresponding control group. (C) Latency to fall off the rotarod during thelearning session (days 1–3) and after a 3-week test-free period. (D) Number ofslips from the runway bar. (E) EMG recording from the orbicularis oculi in acontrol mouse during associated unconditioned stimulus (US) and condi-tioned stimulus (CS) (Left) and during the CS alone (Right) before (uppertraces) and after (lower traces) conditioning. (F) Plotted daily values of con-ditioned response percentage after CS alone in FAS and control mice duringconditioning. Asterisks indicate statistical significance (*, P � 0.05; ***,P � 0.001).
Fig. 2. In vivo PC firing is increased in FAS. (A) Extracellular recordings ofthree PCs in alert control (left traces) and FAS mice (right traces). Histogramvalues of mean simple spike firing rate (B) and complex spike frequency (C) inFAS and control cells. (D) Local field potential in control and FAS mice withcorresponding Fourier spectrum (E). (F) Wave-triggered averaging of a simplespike recorded in a control (upper trace) and in a FAS mouse (lower trace)during an episode of fast oscillation.
Servais et al. PNAS � June 5, 2007 � vol. 104 � no. 23 � 9859
NEU
ROSC
IEN
CE
Dow
nloa
ded
by g
uest
on
Nov
embe
r 6,
202
1
investigated by calcium imaging and measurement of voltage-gated calcium currents. Intracellular free-calcium level [Ca2�]i
changes (��F/F0) elicited by depolarizing PCs from �60 to 0mV were significantly reduced in FAS PCs (n � 24) comparedwith control cells (n � 27) (P � 0.001, Fig. 4 C and E).Pharmacologically isolated calcium currents elicited by testpulses from �60 to 0 mV were significantly smaller in FAS PCs(n � 21) than in control cells (n � 28) (P � 0.001, Fig. 4 D and F).
The reduced voltage-dependent calcium current in FAS mayresult from alterations of channels expression or activation/inactivation kinetics. Inactivation time constants of high voltage-gated calcium currents calculated from monoexponential curvesfit to the data in the first 200 ms after the peak current weresimilar in control (n � 37) and FAS (n � 21) PCs (P � 0.05, SIFig. 6A). The expression of three distinct high voltage-activatedcalcium channels (VGCC) (Cav1.2 (L-type), Cav2.1 (P/Q-type),and Cav2.2 (N-type) channels) that are highly enriched in PCs
(17, 18) was studied by Western blot analyses in membranepreparations from FAS and control mice cerebella (10 brains pergroup). The levels of Cav1.2, Cav2.1, and Cav2.2 did not differsignificantly between FAS and control mice (SI Fig. 6 B–D).Given the dendritic localization of VGCC (19, 20) and thedifficulties in clamping the extensive dendritic arborization ofPCs in slices (20), we were not able to study directly thevoltage-dependence of activation/inactivation of high voltage-gated calcium currents.
Reduced PKC� Expression Causes Reduced Voltage-Gated CalciumCurrent in FAS. Because the activation of VGCC relies on channelsphosphorylation by PKC and because a decreased expression ofPKC enzyme was observed in previous FAS models (21, 22), westudied the expression of the membrane-bound �-isoform(PKC�) that is highly expressed in PCs (23) by using Westernblot analysis. We found that the normalized level of PKC� wassignificantly reduced in FAS mice (n � 6) compared with controlmice (n � 8, P � 0.05) (Fig. 4 G and H). We performedcoimmunostaining for calbindin and PKC� and confirmed thatthis isoform of PKC is expressed only within PCs in the cere-bellum (SI Fig. 7) as described by Bareggi et al. (24). Thesestainings also suggest a reduction of the expression of PKC�within FAS PCs and demonstrate that PKC� expression studiedby Western blot is specific for PCs.
We have then tested our hypothesis that the reduced voltage-dependent calcium current observed in FAS was caused by areduced phosphorylation of the corresponding channels by PKC.We recorded pharmacologically isolated calcium currents elic-ited by test pulses from �60 to 0 mV. Then we applied thespecific PKC activator phorbol 12, 13 diacetate (PDA, 10 �M)or the specific PKC inhibitor chelerythrin (10 �M) in the bathsolution. We observed that PDA significantly increased themaximal amplitude of voltage-dependent calcium current re-corded from FAS PCs (n � 7, paired t test P � 0.05) but not incontrol PCs (n � 7, P � 0.05) (Fig. 4H). On the other handchelerythrin significantly decreased voltage-dependent calciumcurrent recorded from control PCs (n � 5, P � 0.05) but not fromFAS PCs (n � 8, P � 0.05) (Fig. 4H). These data show that PKCphosphorylation of voltage-dependent calcium channels has afunctional impact on calcium current amplitude in normalconditions but not in FAS and that PKC activation allows torecover a normal calcium current amplitude in FAS.
DiscussionOur study shows in vitro and in vivo dysfunctions of PCs in ananimal model of FAS that has not previously been demonstrated.The contribution of neuronal dysfunction to neurological deficitsoccurring in FAS is central for the evaluation of potentialprevention or therapeutics. Combined recordings from brainslices and alert mice in this FAS model strongly suggest thatbehavioral abnormalities (motor incoordination and learningdeficits) and PCs firing properties (higher simple spike fre-quency and fast oscillation) are supported by a reduction ofdepolarization-induced calcium currents in cerebellar PCs asso-ciated with a shift of the cerebellar long-term depression towardan atypical LTP.
This study demonstrates that surviving PCs from FAS miceexhibit a dramatic dysfunction. Indeed, the spontaneous PCsfiring was increased in alert FAS mice when compared withcontrol mice. In addition, we found that the cerebellar cortexfrom FAS mice present a fast LFPO, phase-locked with therhythmic discharge of PCs. This pattern of increased spontane-ous in vivo activity associated with a fast LFPO is similar to theone observed in different ataxic mutant mice (25–28). We havepreviously proposed that increased in vivo firing activity andassociated fast cerebellar oscillations constitute a mechanism ofcerebellar dysfunction leading to ataxia (26, 29). Our recordings
Fig. 3. In vivo PCs’ hyperactivity is not caused by an increased intrinsicexcitability or by an increased excitatory synaptic strength in FAS. (A) Firingactivity of a control and a FAS PC elicited by steps of depolarizing currents froma �80 mV membrane potential. (B) Plotted values of mean PC firing rate as afunction of injected current in FAS and in control cells. (C) Histogram values ofmean linear slope in both groups. (D–F) Synaptic strength of the parallelfiber–PC synapse is decreased in FAS. (D) Typical input–output relationshipsobtained from PCs in response to an increasing stimulation of parallel fibersin a control (Left) and a FAS (Right) PC. Negative current pulses ranging from0 to 11 �A were delivered in ascending order. (E) Linear relation between EPSCinitial slope and stimulus intensity in the cells illustrated in D. (F) Histogramsof slope coefficient values in FAS and in control mice.
9860 � www.pnas.org�cgi�doi�10.1073�pnas.0607037104 Servais et al.
Dow
nloa
ded
by g
uest
on
Nov
embe
r 6,
202
1
from alert FAS and control mice are more reliable than the studyof Backman et al. (5) showing no differences in spontaneousactivity or firing patterns between control and anesthetizedethanol-exposed animals because anesthetics modify PC firingand disrupt LFPO (26, 30). The two studies also differ in ethanolexposition timing. In vivo recordings allowed us to reject thehypothesis that increased simple spike firing in FAS results froma decreased olivary activity (31) because the complex spike firingrate was not altered in FAS. We have then used slices recordingsto exclude the possibility that increased simple spike firing inFAS was caused by an increased intrinsic excitability or by anincreased synaptic strength of the two excitatory inputs of PCs:parallel and climbing fibers. EPSCs resulting from parallel fiberstimulation were reduced in FAS. We believe that a combinationof pre- and postsynaptic mechanisms explains this result becauseit was demonstrated in the hippocampus where ethanol reducespostsynaptic AMPA currents and also acts at the presynapticlevel (32). We also demonstrated a presynaptic alteration in ourFAS model (SI Fig. 5 A and B).
LTD at the parallel fiber–PC synapse has been proposed as themechanism that regulates PC firing in vivo (12, 13). We thuslooked for an alteration of LTD in our model, and we demon-strated that the protocol that induces LTD in control miceevokes LTP in FAS mice. It can be argued that the abolition ofLTD in PCs does not cause alteration in PC firing in vivo ormotor coordination impairment (14). In the present FAS model,LTD was not only absent but replaced by LTP. Thus, wehypothesize that this shift results in a chronic overstimulation ofPCs by the parallel fibers, resulting in increased PC firing in vivo.According to the view that an intact LTD at the parallel fiber–PCsynapse is required for the optimal efficiency of eyelid condi-tioning studied as a prototypical reflect of cerebellar learning(15, 33), the shift of LTD toward LTP in FAS could alsocontribute to the deficits in eyelid conditioning and motorlearning. Whether the replacement of LTD by LTP in FAS miceconstitutes the only cause of LFPO emergence and PCs firingbehavior in FAS has not yet been demonstrated, because othermechanisms that involve the PCs and/or other cerebellar neu-rons could also participate. For instance, the involvement ofother cerebellar neurons, such as molecular layer interneurons,Golgi, or granule cells, and of their respective synaptic plasticity,was not studied here. Other cerebellar neurons, for instancegranule cells, are known to be sensitive to ethanol (34). Inaddition, we did not rule out that increased simple spike firingin FAS could partially result from hyperexcitation of extracer-ebellar areas that project on the cerebellum.
Intracytoplasmic Ca2� concentration controls the direction oflong term synaptic plasticity at the parallel fiber–PC synapse(16). Indeed, concomitant stimulation of parallel and climbingfibers leads to a parallel fiber–PC LTD under standard condi-tions and LTP when calcium buffering lowers intracytoplasmic
Fig. 4. PCs of FAS mice present a shift of LTD toward LTP and a reduction ofcalcium currents caused by a decrease in expression of PKC. (A) Typical EPSCrecorded in a control (Upper) and a FAS (Lower) PC before (left) and after(right) the LTD induction protocol. (B) Plotted value of mean EPSC amplitude(normalized to EPSC value at t0) in FAS and in control cells before and after theLTD induction protocol (indicated by the arrow). (C) Typical calcium transientevoked by a 2-s depolarization from �60 to 0 mV in a control (Left) and a FASPC (Right). (D) Typical voltage-gated calcium current evoked by a 1-s depo-larization from �60 to 0 mV in a control PC (Left) and in a FAS PC (Right). (E)Depolarization-evoked calcium transients were significantly reduced in FAS.
Histogram values of calcium transients normalized maximal amplitude. (F)Depolarization-evoked calcium currents were significantly reduced in FAS.Histogram values of calcium current density are shown. (G) The expression ofmembrane PKC� was reduced within FAS cerebella compared with controlones. Representative Western blot analyses for PKC� from cerebellar mem-brane extracts of control and FAS mice. Western blot analyses of actin areshown as a loading control. Histograms represent the corresponding densi-tometric analysis of pooled experiments made by using 10 different mice pergroup and normalization for the amount of protein loaded (in arbitrary units).(H) Effect of PKC activation by PDA 10 �M on the maximal amplitude ofpharmacologically isolated voltage-gated calcium current (VGCC) in control(black) and FAS (gray) PCs. Shaded bars represent current amplitude afterapplication of PDA. (I) Effect of PKC inhibition by chelerythrin 10 �M on themaximal amplitude of pharmacologically isolated VGCC in control (black) andFAS (gray) PCs. Shaded bars represent current amplitude after application ofchelerythrin.
Servais et al. PNAS � June 5, 2007 � vol. 104 � no. 23 � 9861
NEU
ROSC
IEN
CE
Dow
nloa
ded
by g
uest
on
Nov
embe
r 6,
202
1

Ca2� concentration. Thus, we reasoned that LTD could bealtered in the present FAS model if calcium entry were reduced.Indeed, we demonstrated a reduction of voltage-gated calciumcurrents and of calcium entry after depolarization, as observedin whole-brain dissociated neurons prenatally exposed to ethanol(35). Ethanol-induced reduction of calcium entry in developingPCs depolarized by quisqualate application had already beennoted (36).
In contrast to observations at the cerebellar level by Coesmanset al. (16), a high calcium threshold for LTP and a low calciumthreshold for LTD induction have been demonstrated at thehippocampal level (37, 38). A LTP induction protocol applied tohippocampal pyramidal cells would thus lead to a LTD if calciumentry were reduced. Interestingly, the appearance of such ahippocampal LTD after a LTP induction protocol has beenobserved in vivo in guinea pigs prenatally exposed to ethanol(39). We believe that, analogous to our demonstration in cere-bellar PCs, a reduction of calcium entry through voltage-gatedcalcium channels in hippocampal pyramidal cells prenatallyexposed to ethanol is the best explanation for this observation.The mechanism leading from calcium-mediated cell dysfunctionto learning deficits that we suggest at the PC level could also berelevant for noncerebellar deficits in FAS. IntracytoplasmicCa2� concentration is also crucial for other aspects of PCelectrical properties (40). This might explain the lower intrinsicexcitability of PCs in FAS mice presenting reduced voltage-gatedcalcium currents.
The proposed sequence of events leading from decreasedcalcium currents into PCs to learning deficits and ataxia mayresult from reduced current activation, increased current inac-tivation, or reduced calcium channel expression. We demon-strated that the expression of voltage-gated calcium channelsinvolved in the high-voltage calcium current (L, P/Q, and Ntypes) was not reduced in FAS (SI Fig. 6 B–D). We alsodemonstrated that the inactivation time constant is similar inFAS and control PCs (SI Fig. 6A). This suggests that a reducedactivation of calcium channels is responsible for the reducedcurrent.
The activation/inactivation of calcium channels is known to bemodulated by different intracellular signaling cascades involvingG proteins, cAMP-dependent protein kinase (PKA), proteinkinase C, and calcium/calmodulin (41). Because the calcium/cal-modulin modulation alters the inactivation kinetics of VGCC(42–44) and because the inactivation time constant was notaltered in our FAS model (SI Fig. 6A), this mechanism can beruled out. Some studies have found increased cAMP levels orPKA activity in FAS (45, 46); this would lead to an increasedcalcium current (41), so this mechanism can also be ruled out inour model because a reduction of the calcium current wasobserved. The activity of G proteins appears unchanged in FAS(47). Finally, PKC directly increases the activity of Cav1.2 andCav2 channels and reverses G protein inhibition of these chan-nels (41). PKC expression and/or activity have been found to bereduced in FAS models (21, 22). Accordingly, we found that theexpression of membrane PKC� [the main isoform expressed inPCs and not in other cerebellar neurons as demonstrated byimmunohistochemistry (21) and in situ hybridization (23)] issignificantly reduced in the cerebellum of FAS mice as demon-strated by Western blotting. We have then tested the effect of aPKC activator and a PKC inhibitor on the maximal amplitude ofvoltage-dependent calcium current. Our data show that PKCinhibition has a significant impact on calcium current amplitudein control mice but not in FAS mice, whereas PKC activation hasno significant effect in control mice but is able to increasecalcium current amplitude in FAS mice. These data suggest thatbasal phosphorylation of voltage-dependent calcium current islacking in FAS and can be recovered by PKC activation. Weconclude that the reduced expression of PKC causes the reduc-
tion in amplitude of calcium current in FAS PCs, compared withcontrols, through an alteration of the phosphorylation state andof the activation of these channels.
The reduced expression of PKC� might also contribute toLTD alteration because PKC� inhibition blocks LTD inductionin PCs (48, 49). Because the present FAS model is not specificfor PKC� underexpression or inhibition, it is difficult to compareit with PKC� knockout mice that present a cerebellar LTD thatis much less sensitive to PKC� inhibitors (49) than wild-typemice, probably because of the compensatory activation of otherkinases. Finally, the involvement of glutamate receptors at thelevel of excitatory synapses onto PCs in FAS could not beexcluded.
Our results demonstrate both in vivo and in vitro neuronaldysfunction of surviving FAS PCs that may be explained byaltered calcium dynamics. The relevance of this dysfunction inthe pathogenesis of learning deficits provides new opportunitiesfor therapeutic intervention in FAS.
MethodsMouse Model. FAS and control mice were generated from 3- to5-month-old NMRI mice drinking ad libitum a water solutioncontaining either ethanol 18% or sucrose 25% from one daybefore mating until delivery. At birth, ethanol was replaced bysucrose. This study was approved by local ethical committees.
Immunohistochemistry and Western blots were performed asdescribed in SI Materials and Methods.
Motor Coordination. Accelerating rotarod and runway tests wereperformed as described in SI Materials and Methods on postnataldays 24–26 and 26–28, respectively. Tests were repeated after a3-week trial-free period.
Eyelid conditioning was performed as described in SI Mate-rials and Methods. Briefly, mice were conditioned each day by theassociation (10 blocks of nine associations plus one conditionedstimulus alone) of an unconditioned stimulus (100-ms air puff onthe eyelid) and coterminated conditioned stimulus (80-db,1-kHz, 370-ms tone). Daily percentage of significant EMGresponse to conditioned stimulus alone was counted.
Patch-Clamp Recordings and Calcium Imaging on Brain Slices. Acutecerebellar slices were prepared from 15- to 20-day-old mice, andpatch-clamp recordings were performed. Calcium imaging wasperformed as described (50). Parallel fibers and climbing fiberswere stimulated as described (51, 52). LTD recordings wereobtained as described (53, 54) through parallel fiber stimulationin conjunction with a depolarizing pulse applied to PCs. Thesetechniques are detailed in SI Materials and Methods.
Single Unit Recording in Alert Mice. One- to 2-month-old FAS andcontrol mice were prepared for PCs, and local field potential andrecordings were performed in alert immobilized mice 24 h laterby using glass micropipettes as described (25) and as detailed inSI Materials and Methods.
Data Analyses and Statistics. Results are reported and illustratedas mean � SEM. Unless otherwise mentioned, means andvariance were compared by using t and F test, respectively. Allstatistical analyses were performed by using STATISTICA 6.0(Statsoft, Maisons-Alfort, France).
We thank L. Cuvelier, P. Demaret, C. Waroquier, P. Kelidis, and M. P.Dufief for expert assistance. R.H. and L.S. were supported by the FondsNational de la Recherche Scientifique (FNRS) (Belgium). This work wassupported by the Fondation Medicale Reine Elisabeth, FNRS, Ministeredes Affaires Sociales et de la Sante Publique (Belgium), the Van BuurenFoundation, Action de Recherche Concertee (2002–2007), and researchfunds of ULB and UMH.
9862 � www.pnas.org�cgi�doi�10.1073�pnas.0607037104 Servais et al.
Dow
nloa
ded
by g
uest
on
Nov
embe
r 6,
202
1

1. Burd L, Cotsonas-Hassler TM, Martsolf JT, Kerbeshian J (2003) NeurotoxicolTeratol 25:681–688.
2. Burd L, Martsolf JT (1989) Physiol Behav 46:39–43.3. Bauer-Moffett C, Altman J (1977) Brain Res 119:249–268.4. Pierce DR, Williams DK, Light KE (1999) Alcohol Clin Exp Res 23:1650–1659.5. Backman C, West JR, Mahoney JC, Palmer MR (1998) Alcohol Clin Exp Res
22:1137–1145.6. Thomas JD, Goodlett CR, West JR (1998) Brain Res Dev Brain Res 105:159–
166.7. Heaton MB, Mitchell JJ, Paiva M (2000) Alcohol Clin Exp Res 24:512–518.8. Edwards RB, Manzana EJ, Chen WJ (2002) Alcohol Clin Exp Res 26:1003–1009.9. Mullen RJ, Eicher EM, Sidman RL (1976) Proc Natl Acad Sci USA 73:208–212.
10. Feddersen RM, Ehlenfeldt R, Yunis WS, Clark HB, Orr HT (1992) Neuron9:955–966.
11. Feddersen RM, Clark HB, Yunis WS, Orr HT (1995) Mol Cell Neurosci6:153–167.
12. Miall RC, Keating JG, Malkmus M, Thach WT (1998) Nat Neurosci 1:13–15.13. Kenyon GT, Medina JF, Mauk MD (1998) J Comput Neurosci 5:71–90.14. Goossens J, Daniel H, Rancillac A, van der SJ, Oberdick J, Crepel F, De Zeeuw
CI, Frens MA (2001) J Neurosci 21:5813–5823.15. Koekkoek SK, Hulscher HC, Dortland BR, Hensbroek RA, Elgersma Y,
Ruigrok TJ, De Zeeuw CI (2003) Science 301:1736–1739.16. Coesmans M, Weber JT, De Zeeuw CI, Hansel C (2004) Neuron 44:691–700.17. Chung YH, Shin CM, Kim MJ, Shin DH, Yoo YB, Cha CI (2001) Brain Res
903:247–252.18. Park SK, Hwang IK, An SJ, Won MH, Kang TC (2003) Mol Cell 16:297–301.19. Pouille F, Cavelier P, Desplantez T, Beekenkamp H, Craig PJ, Beattie RE,
Volsen SG, Bossu JL (2000) J Physiol 527 Pt 2:265–282.20. Watanabe S, Takagi H, Miyasho T, Inoue M, Kirino Y, Kudo Y, Miyakawa H
(1998) Brain Res 791:43–55.21. McIntyre TA, Souder MG, Hartl MW, Shibley IA (1999) Brain Res Dev Brain
Res 117:191–197.22. Tanner DC, Githinji AW, Young EA, Meiri K, Savage DD, Perrone-Bizzozero
NI (2004) Alcohol Clin Exp Res 28:113–122.23. Barmack NH, Qian Z, Yoshimura J (2000) J Comp Neurol 427:235–254.24. Bareggi R, Narducci P, Grill V, Lach S, Martelli AM (1996) Biol Cell 87:55–63.25. Cheron G, Gall D, Servais L, Dan B, Maex R, Schiffmann SN (2004) J Neurosci
24:434–441.26. Servais L, Cheron G (2005) Neuroscience 134:1247–1259.27. Bearzatto B, Servais L, Roussel C, Gall D, Baba-Aissa F, Schurmans S, de
Kerchove, dA, Cheron G, Schiffmann SN (2006) FASEB J 20:380–382.28. Cheron G, Servais L, Wagstaff J, Dan B (2005) Neuroscience 130:631–637.
29. Servais L, Bearzatto B, Schwaller B, Dumont M, De Saedeleer C, Dan B, BarskiJJ, Schiffmann SN, Cheron G (2005) Eur J Neurosci 22:861–870.
30. Sato Y, Miura A, Fushiki H, Kawasaki T (1993) J Neurophysiol 69:1082–1090.31. Montarolo PG, Palestini M, Strata P (1982) J Physiol 332:187–202.32. Mameli M, Zamudio PA, Carta M, Valenzuela CF (2005) J Neurosci 25:8027–
8036.33. Thompson RF, Krupa DJ (1994) Annu Rev Neurosci 17:519–549.34. Offenhauser N, Castelletti D, Mapelli L, Soppo BE, Regondi MC, Rossi P,
D’Angelo E, Frassoni C, Amadeo A, Tocchetti A, et al. (2006) Cell 127:213–226.
35. Lee YH, Spuhler-Phillips K, Randall PK, Leslle SW (1996) Alcohol Clin ExpRes 20:921–928.
36. Gruol DL, Parsons KL (1996) Brain Res 728:166–174.37. Cummings JA, Mulkey RM, Nicoll RA, Malenka RC (1996) Neuron 16:825–
833.38. Cormier RJ, Greenwood AC, Connor JA (2001) J Neurophysiol 85:399–406.39. Richardson DP, Byrnes ML, Brien JF, Reynolds JN, Dringenberg HC (2002)
Eur J Neurosci 16:1593–1598.40. Berridge MJ (1998) Neuron 21:13–26.41. Felix R (2005) J Recept Signal Transduct Res 25:57–71.42. Zuhlke RD, Pitt GS, Deisseroth K, Tsien RW, Reuter H (1999) Nature
399:159–162.43. Lee A, Wong ST, Gallagher D, Li B, Storm DR, Scheuer T, Catterall WA
(1999) Nature 399:155–159.44. Peterson BZ, DeMaria CD, Adelman JP, Yue DT (1999) Neuron 22:549–558.45. Kumada T, Lakshmana MK, Komuro H (2006) J Neurosci 26:742–756.46. Constantinescu A, Wu M, Asher O, Diamond I (2004) J Biol Chem 279:43321–
43329.47. Druse MJ, Tajuddin NF, Eshed M, Gillespie R (1994) Alcohol Clin Exp Res
18:47–52.48. Linden DJ, Connor JA (1991) Science 254:1656–1659.49. Chen C, Kano M, Abeliovich A, Chen L, Bao S, Kim JJ, Hashimoto K,
Thompson RF, Tonegawa S (1995) Cell 83:1233–1242.50. Kano M, Schneggenburger R, Verkhratsky A, Konnerth A (1995) Neurosci Res
24:87–95.51. Llano I, Marty A, Armstrong CM, Konnerth A (1991) J Physiol 434:183–213.52. Sacchetti B, Scelfo B, Tempia F, Strata P (2004) Neuron 42:973–982.53. Konnerth A, Dreessen J, Augustine GJ (1992) Proc Natl Acad Sci USA
89:7051–7055.54. Barski JJ, Hartmann J, Rose CR, Hoebeek F, Morl K, Noll-Hussong M, De
Zeeuw CI, Konnerth A, Meyer M (2003) J Neurosci 23:3469–3477.
Servais et al. PNAS � June 5, 2007 � vol. 104 � no. 23 � 9863
NEU
ROSC
IEN
CE
Dow
nloa
ded
by g
uest
on
Nov
embe
r 6,
202
1