pre-assessment of the speciation of 60co, 125sb, 137cs and 241am in a contaminated aquifer
TRANSCRIPT

� Corresponding
E-mail address:
0265-931X/$ - see
doi:10.1016/j.jenvr
author. Tel.: +1-705-675-1151x2400; fax: +1-705-675-4844.
[email protected] (F. Caron).
front matter # 2004 Elsevier Ltd. All rights reserved.
ad.2004.02.002
Journal of Environmental Radioactivity 77 (2004) 29–46
www.elsevier.com/locate/jenvrad
Pre-assessment of the speciation of 60Co, 125Sb,137Cs and 241Am in a contaminated aquifer
Francois Caron �, George MankariosChemistry and Biochemistry Department, Laurentian University, Sudbury, Ont., Canada P3E 2C6
Received 1 July 2003; received in revised form 1 January 2004; accepted 28 January 2004
Abstract
A sample of contaminated groundwater was analyzed using a combination of wet techni-ques to obtain geochemical information on the mobile species of 60Co, 125Sb, 137Cs and241Am. The techniques were combined in a scheme to determine the predominant characterof the radionuclides in negative or positive fractions, size separation by ultrafiltration, andtheir association with natural organic matter (NOM). The analyses indicated that the radio-nuclides of interest were predominantly in the negative fraction. Most of the 60Co and 125Sbwere in the small size fraction (<5000 Da), and 137Cs and 241Am were found with the larger,colloidal-sized material. Antimony-125 and 60Co were predominantly in the hydrophilic frac-tion, while 137Cs and 241Am were found in hydrophobic fractions. Our analysis indicatedthat 137Cs is found in the same fraction as the large-sized colloidal (hydrophobic) material,suggesting an association with NOM. The results suggested that 60Co and 241Am were asso-ciated with NOM, in different size fractions, suggesting that these two nuclides are bound todifferent sites. Finally, the 125Sb results were inconclusive, whether this nuclide is associatedwith NOM, or it is inorganic.# 2004 Elsevier Ltd. All rights reserved.
Keywords: Cobalt-60; Antimony-125; Cesium-137; Americium-241; Speciation; Contaminated ground-
water; Separation; Solid-phase extraction; SPE; Ultrafiltration
1. Introduction
Migration of radiocontaminants depends on, among others, the radioelement
itself, the aqueous speciation and the nature of the soil as an exchanging medium.

F. Caron, G. Mankarios / J. Environ. Radioactivity 77 (2004) 29–4630
In the context of the migration of low-levels of radioelements in shallow subsurfacesoils, several geochemical considerations have to be taken into account. Forinstance, the radioelement is most likely a minor component of the total load ofions, and hence the rest of the geochemical conditions will control the aqueousgeochemistry of the elements. This is further complicated that some radioisotopes(e.g., 60Co) are potentially outnumbered on a mole or atom basis compared to thestable element, such that mixing between the active and non-active isotopes takesplace. Other radiocontaminants (e.g., 241Am) are not subject to this, but they arenevertheless minor constituents of groundwaters, and not necessarily well mixed insoils because they have recently been introduced by human activities.
In previous work at the Chalk River Laboratories (CRL) on contaminatedgroundwaters, the general character of radionuclides has been determined, parti-cularly with respect to associations between radioactive contaminants and organics(Killey et al., 1984; Champ et al., 1984; Champ and Robertson, 1986; Cooper et al.,1995; Robertson et al., 2000; Caron et al., 2002). Particularly, it has been suggestedthat aqueous speciation of some radionuclides (60Co, 137Cs, 241Am, 239Pu, 244Cm,plus a few others) plays an important role in sorption, hence retardation of con-taminants in aquifers (Robertson et al., 2000; Caron et al., 2002). The latter worksdemonstrated the importance of using radionuclides present in a contaminatedaquifer to obtain geochemical information. ‘‘Native’’ radionuclides (i.e., resultingfrom long-term mixing of radioisotopes intentionally discharged, or added acciden-tally to the natural system), were equilibrated with soil material for sorptionexperiments, along with the same radionuclides from artificial tracer solutions. Thetracer solutions differ from the ‘‘native’’ solutions because the artificial species havenot necessarily had time to equilibrate, and hence, potentially, would not reflect thefield speciation. In all the cases in the two studies above, the radionuclides in thecontaminated groundwater (which reflect the ‘‘native’’ speciation), all exhibitedlower sorption to the soil material (as per their Kd values1), compared to the arti-ficial tracers. For example, the Kd of Cs with groundwater extracts (reflecting the‘‘native’’ speciation) was less than 10 ml/g, whereas it was 580–3280 ml/g with tra-cers. Order-of-magnitude differences were observed with other radionuclides as well.
The authors of these studies concluded that the aqueous speciation of the ‘‘tra-cers’’ was different from the speciation of the ‘‘native’’ radionuclides of the sameelements, resulting in a different degree of sorption. Natural organic matter (NOM)was suspected as the main factor influencing the speciation of the elements (activeand non-active elements alike). There was no specific information on the speciesother than the negatively or positively charged character, and this is needed tounderstand the migration processes and interactions with soils, and to better quan-tify the migration behaviour of radionuclides in soils.
In our work, a sample preparation scheme was modified to get specific infor-mation on the aqueous species of the radionuclides present in the groundwater at
1 Kd is defined as (concentration of a contaminant on the solid phase)/(concentration of contaminant
in aqueous phase). The units are in volume/mass (in ml/g here).

31F. Caron, G. Mankarios / J. Environ. Radioactivity 77 (2004) 29–46
the same contaminated site. Our objective was to determine the major character ofthe aqueous radionuclides in contaminated groundwater, with respect to simple,but useful bulk properties: positive or negative character, size fractionation, hydro-philicity/hydrophobicity. The operations define the fractions of radionuclidesfound in the contaminated groundwater sample: (i) the predominance of negativelyversus positively charged species; (ii) ultrafiltration (5000 Da molecular weight cut-off—MWCO); and (iii) solid-phase extraction (SPE). The target radionuclides inthis work are those easily measurable by gamma spectroscopy, 60Co, 125Sb, 137Csand 241Am. Results on other radionuclides (beta and alpha emitters) will be repor-ted subsequently.
The combination of techniques is unique, aiming to lead to new information onthe behaviour of the target radionuclides. A staged approach is necessary to verifywhether analysis reflects the conditions in the sample, and what we can learn aboutthe geochemistry at every step of the way. It is understood that the fractions areoperationally defined and the term ‘‘speciation’’ cannot be used rigorously, rather,we are reporting that the radionuclides are associated with a fraction. Direct analy-sis is needed in each fraction to ascertain the actual species, but for the intent ofour work, the usefulness lies in the presence of a radionuclide in a given fraction.This work will be useful for remediation purposes and other health and safetyaspects.
2. Methodology
2.1. Field site and sampling
The sample comes from a contaminated plume at the CRL, approximately 175km west of Ottawa, Ontario (Canada). The location within the CRL site is agroundwater flow system, less than 50 m downstream from a former infiltrationpit, which has been used in the past to discard low-level aqueous radioactive wastes(Fig. 1). This pit was in use from 1956 to 1992, followed by episodic dischargesuntil its permanent closure in 1995. The pit was excavated in a sand dune, and thenfilled with coarse aggregate. It has two parts: one is circular, of diameter approxi-mately 24 m, while the other one is trapezoidal, approximately 30 m � 20 m indimensions, and a depth of ~3–5 m. The pit is open to the air, and its bottom is1–2 m above an aquifer that discharges to a perennial wetland feeding a smallstream, ~200 m downstream. The pit is behind a fenced area, and several boreholesare available for sampling, outside this perimeter.
The residence time of the contaminated groundwater in the aquifer is ~180–670d, from the pit to the wetland/stream. An estimated total volume of water of
3:3 � 105 m3 was discarded during the operation of the pit, which contained a totalof 230 TBq of beta emitters, and 0.31 TBq of alpha emitters (Killey and Munch,1984).
Our sample was taken from borehole LDA 22 by inserting a submersible pumpin the borehole. After several litres of groundwater were passed, the sample was

F. Caron, G. Mankarios / J. Environ. Radioactivity 77 (2004) 29–4632
taken directly into two collapsible carboys. The samples were kept in the dark and
on ice until processing at a different location (done within 48 h at our Sudbury
Laboratory, ~350 km away). Dark plastic sheets were used at all times to protect
the sample from the sunlight.
2.2. Sample processing
Upon arrival at the processing location, the sample was filtered with a 0.45 lm
cartridge filter (A/G technologies) and stored in the dark for the other processing
ig. 1. Map showing the sampling location. Inset: location of the Chalk River Laboratorie
F s.
33F. Caron, G. Mankarios / J. Environ. Radioactivity 77 (2004) 29–46
operations. At that stage, the sample was allowed to warm to room temperature,but it was always kept in the dark. Separate aliquots of the filtrate were used forthese operations (see Fig. 2): extraction with a cation exchange resin (Purolite C-100, separated from NRW-37 LC mixed resin), ultrafiltration (5000 Da MWCOcartridge, A/G Technologies), and SPE with a C18 sorbent cartridge (Millipore,Sep-Pak Plus, Milford, MA). All fractions were measured before and after proces-sing for mass balance (see Appendix A); only one value for 137Cs was ambiguous.Our interpretation of the individual fractions is based on the total of the fractions.
2.2.1. Cation exchange resin extractionThis step, based on past experiments (Champ et al., 1984; Robertson et al., 2000;
Caron et al., 2002), consists of passing the sample through a strong acid cationexchange resin, to remove the cationic species present in solution. A 4-l aliquot waspassed through 90 ml of resin, which had previously been converted to its Na+
form. This quantity of resin was calculated to contain approximately 10 times thenominal cationic content of the sample (see Rowat et al., 1999 for the compo-sition). After the sample had passed, the aqueous extract and the dried resin wereanalyzed by gamma spectroscopy.
The resin extractions define the major character of the radionuclide species: the‘‘cationic fraction’’ is based on the gamma emitters left on the ion exchange resin,while the fraction passing through the resin (solution extract) is called ‘‘anionicfraction’’ henceforth. This is an operational definition, and it is recognized that thenegative or positive species would need to be confirmed with other techniques. Theterminology used in our work assumes that the resins are a homogeneous support
Fig. 2. Flowchart of sample processing.

F. Caron, G. Mankarios / J. Environ. Radioactivity 77 (2004) 29–4634
with functional groups that would outcompete the NOM functional sites by elec-trostatic attraction. It is possible that labile complexes are destroyed or dissociatedby competition with exchange sites, and this would overpredict the material asso-ciated with the cationic fraction. In addition, humic/fulvic materials contain aminegroups (Thurman, 1985) which would be positively charged at the pH of interest.It is conceivable that a moiety containing amines is attracted by the strong func-tional groups of the resin. If this was a significant mechanism, and if radionuclidesare associated with this NOM, it would also overpredict the cationic fraction.Finally, it is understood that neutral species could be present in the sample, andthese would be reported in the anionic fraction (i.e., unretained by the cationexchange resin). These factors were minimal when similar samples were extractedwith both anionic and cationic resins (Robertson et al., 2000).
2.2.2. Ultrafiltration (5000 Da MWCO)Ultrafiltration was used to concentrate the material that could be classified as
colloidal (passing through a 0.45 lm filter, but retained by a 5000 Da filter), whileisolating the ‘‘dissolved’’ and other material (less than 5000 Da in size). A 3-l ali-quot was processed to concentrate the 5000 Da retentate down to ~0.5 l, to keep ashort processing time, and to save some of the sample for other operations.Although a leak in the filtration apparatus was discovered in the sample tray nearthe end of the filtration run, all fractions (including the recovered leaked material)were analyzed by gamma spectroscopy and corrected for the loss. Our radioisotoperesults show a good mass balance (except for 137Cs—as pointed out earlier), andthe leak did not invalidate our conclusions.
2.2.3. Solid phase extractionThe extraction with C18 cartridges defines the radionuclides associated with
organics. The SPE operation was performed in triplicate. Each cartridge was firstconditioned by ‘‘wetting’’ it with 20 ml of methanol (as per the manufacturer’sinstructions; HPLC grade), followed by 500 ml Milli-Q water; both these fractionswere discarded. The subsamples (0.35–0.5 l) were passed through separate car-tridges, each followed by a 20 ml rinse with Milli-Q water. The extracted aqueoussolutions and the rinse were spared. Following this, 25 ml of methanol were usedto elute off the SPE cartridge content. Each extraction generated three fractions(see Fig. 2): the aqueous extract, the methanol extract, and the spent cartridge. Allthe fractions were analyzed by gamma spectroscopy, which also enabled us toobtain a mass balance.
The C18 support is a hydrophobic, non-polar stationary phase, which retainshydrophobic and neutral NOM (Garnier et al., 1997; Caron et al., 1996). Thematerial of the aqueous solution passing through the cartridge (‘‘aqueous extract’’)is defined as hydrophilic, whereas the portion retained on the cartridge is definedas hydrophobic. The NOM displaced by methanol is weakly bound to the C18 sup-port, and it could contain hydrophilic and neutral NOM. Similarly, the hydro-phobic material remaining on the cartridge after methanol extraction likelyrepresents highly hydrophobic NOM (Caron et al., 1996). Hence, in line with this

35F. Caron, G. Mankarios / J. Environ. Radioactivity 77 (2004) 29–46
description, the radionuclides associated with these fractions are called ‘‘hydrophilic’’(aqueous extract), ‘‘hydrophobic-weak’’ (methanol extract), and ‘‘hydrophobic-strong’’ (residual on the C18 support after methanol extraction). It is also under-stood that the radionuclides detected in the hydrophilic fraction are not necessarilyassociated with NOM, as it could be uncomplexed or complexed with inorganicligands. The radionuclides are likely associated with NOM in the other two frac-tions.
2.2.4. Sample preparation and gamma spectroscopyThe gamma spectrometer was a Canberra model GC1020 hyperpure Ge detector
(Meriden, CT), controlled by a DSA-1000 digital processor and the Genie 2000software. The detector was housed in a lead castle (Canberra). The reproducibilityof the readings was usually better than 2% between replicates, except for 241Am,which was ~5–10%; this was acceptable since the levels were close to the detectionlimits for this radioelement.
The method used to analyze the aqueous samples was modified from Grummitand Lahaie (1978). The samples (0.3–1 l each) were poured in a polyethylene (PE)liner, previously placed in a flat glass dish, and placed on a hot plate and under IRlamps to gently evaporate the water overnight. The dry PE film was carefully fol-ded and pressed (25 t) to make a 2 cm � 0:5 cm disk. The disk was placed in aplastic jar (Qorpak), and on the detector for reading. Similarly, the ion exchangeresin was air-dried in a PE-lined tray, and poured into a Qorpak jar. The methanolextract of the SPE runs was analyzed directly, while the empty SPE cartridge wasplaced in an empty Qorpak jar. All the samples were counted overnight (20 h).
The spectrometer was calibrated using a NIST-traceable multi-isotope Gammaspectrometer calibration standard (QCY-48, AEA Technology, Burlington, MA).The efficiencies were calculated by interpolation using the Genie 2000 software.Standards of appropriate geometries were prepared (folded PE sheet, cartridge,methanol solution), and the samples were corrected using standards of the corre-sponding geometry.
3. Results
The results of the total radionuclides and the ion exchange extractions are shownin Table 1. The totals were lower than for recent monitoring results (Rowat et al.,1999), but they were close to those of Robertson et al., (2000). The four targetradionuclides (60Co, 125Sb, 137Cs, and 241Am) were in the fractions of predominantanionic character (negative complexes or otherwise), from ~90% to 9%. This is alsoconsistent with previous work done on this plume (Killey et al., 1984; Champ et al.,1984; Robertson et al., 2000).
The ultrafiltration results (Table 2) indicate that most of the 60Co and 125Sb ispresent in the small molecular weight fraction, whereas 137Cs and 241Am are asso-ciated with the larger fraction. The table also shows the values (corrected for thelosses), and the amount of small material below the weight cut-off present in the

F. Caron, G. Mankarios / J. Environ. Radioactivity 77 (2004) 29–4636
retentate. For each fraction, the correction was done according to this formula:
CRetentate � VRetentate ¼ CHigh MW � VHigh MW þ CSmall MW � VSmall MW ð1Þ
where C is the concentration of radionuclide in the sample; V is the volume of thesample; subscripts ‘‘Retentate’’ is self-explanatory; ‘‘High MW’’ represents the truematerial of size higher than the nominal size cut-off; ‘‘Small MW’’ refers to thetrue filtered material.
This correction is based upon the composition of the retentate fraction. Theretentate contains large material retained by the membrane by size, plus otherresidual material of small size that cannot be pushed across the membrane sincethe full volume of solution is not filtered. The portion of small material in theretentate is corrected using the filtered material (‘‘Small MW’’ of Eq. (1)). Thisrelationship was also used to correct for the losses.
The corrected values using Eq. (1) show that almost the totality of the 137Cs and241Am are associated with high molecular weight material (Table 2) while substan-tial amounts of 60Co and 125Sb were with the small material. This trend is consist-ent with previous work for 241Am and 60Co (Champ et al., 1984; Killey et al., 1984;Caron et al., 2002), while no corresponding values are known and 137Cs, and just afew are available for Sb (see Fig. 1 of Filella et al., 2002).
Table 2
Summary of the radionuclide distribution in the filtration step, corrected for the loss (5000 Da nominal
weight cut-off; see Appendix A for raw results)
6
0Co (Bq/l) 1 25Sb (Bq/l) 137Cs (Bq/l) 241Am (Bq/l)Original solution
(0.45 lm filtrate)
3
4.5 1.6 25.5 1.4R
elative distribution (corrected—see text and Eq. (1)), %> 5000 Da 1
7.1 6.7 98.2 95< 5000 Da 8
2.9 9 3.3 1.8 5aa This value is an estimate based on the reported count for the filtrate. This value may or may not be
real, and it is placed here as best estimate (this value is below the MDA).
Table 1
Summary of the radionuclide concentrations of radionuclides in the sample, and their distribution after
the ion exchange extraction step (see details in Appendix A)
6
0Co (Bq/l) 1 25Sb (Bq/l) 137Cs (Bq/l) 241Am (Bq/l)Original solution
(0.45 lm filtrate)
3
4.5 1 .6 25.5 1.4R
elative distribution% Cationic
9.7 6 a 3.7 10a% Anionic 9
0.3 9 4 96.3 90a This value is an estimate based on the reported count for the cationic fraction (unstripped resin).
This value may or may not be real, and it is placed here as best estimate (this value is below the mini-
mum detectable activity—MDA).

37F. Caron, G. Mankarios / J. Environ. Radioactivity 77 (2004) 29–46
The SPE results (Table 3) indicated a contrasting behaviour for the radio-nuclides. Cobalt-60 and 125Sb were mostly in the hydrophilic fraction, whereas137Cs and 241Am were in nearly equal proportions between the hydrophilic and thestrong hydrophobic fractions. Only 60Co was in measurable quantity in the weakhydrophobic fraction (~29% of the total). The presence of 137Cs in the hydrophilicfraction was not surprising, but it was surprising to find approximately the sameamount of it retained on the cartridge (strong hydrophobic fraction). The behav-iour of 241Am was similar, but this is not surprising because the association of241Am with NOM is well documented (see e.g., Cooper et al., 1995; Artinger et al.,1998).
4. Discussion
4.1. Cobalt-60
The 60Co in our sample was predominant in the anionic fraction, it was associa-ted with the <5000 Da fraction, it was mostly in the hydrophilic fraction, with asignificant portion in the weak hydrophobic fraction. The majority of the 60Cofound in the small molecular fraction (~83%) is consistent with previous samplingnear this site. Cooper et al. (1995) have found that 91% of the 60Co passed througha 3000 Da membrane, while Killey et al. (1984) reported that 60.9% passedthrough a 1000 Da filter, but only 13.9% through a 500 Da membrane. The differ-ences between our studies may be due to a change in the nature of the organics,especially that, at the time our sample was taken, the pit had not been used foryears, while this was not the case in the other studies.
The specific association of 60Co association with organics is undetermined. Itsassociation with NOM is well accepted, and this association is strong and it does
Table 3
Summary of the distribution of the radionuclides after the solid-phase extraction step (see raw data in
Appendix A)
60Co (Bq/l)
125Sb (Bq/l) 1 37Cs (Bq/l) 241Am (Bq/l)Original solution—
re-analysis (0.45 lm
filtrate)
33.8
1.9 3 1.4 1.5Relative distribution (%)
Average � stdevb
Average � stdev A verage � stdev Average � stdevOrganic
‘‘hydrophobic’’
28.8
0.8 9a – ~ 0a – 8a –Not retained
‘‘hydrophilic’’
66.7
1.0 82 1 .9 4 6 3.2 57 2.9Other (?)
4.5 0.3 9a – 5 4 3.0 35 4.1a These values are estimated based on the reported count for the fractions specified. The numbers
value may or may not be real; they are reported here as best estimate (the values are below the MDA).b stdev: absolute standard deviation among replicates (see data in Appendix A). An ‘‘stdev’’ is mean-
ingless for the fractions whose values are below MDA, and is hence not reported.

F. Caron, G. Mankarios / J. Environ. Radioactivity 77 (2004) 29–4638
not appear easily reversible (Cooper and McHugh, 1983; Killey et al., 1984;Robertson et al., 2000). There does seem to be a distribution of binding sites andorganics: the most abundant site would be weak, equilibrate fast and in a reversiblemanner, while a less abundant but stronger binding site would take up Co in aslow and nearly irreversible manner. For example, in waste leachates, Caron et al.(1996) have separated organics by liquid chromatography, and they have reportedthat a majority of 60Co co-eluted with hydrophilic organics, while some 60Co wasfound with hydrophobic organics. Fortin and Caron (2000) have reported the com-plexing capacity of these leachates, which was probably dominated by hydrophilicorganics; however, the calculation of a (conditional) stability constant, logK, withthe leachate organics led to ambiguous results. This was potentially caused by aslow exchange of Co with organics and incomplete mixing of the 60Co and inactiveCo in the leachate. Mandal et al. (1999a, b) have reported the binding of Co withorganics, along with kinetic rate constants and the relative abundance of weak andstrong sites. Their experiments spanned over ~2 h, which was in the same order oftime as in Fortin and Caron (2000), but there are indications that binding withstrong sites on organics could be in the order of months (Caron et al., 1996). Gar-nier et al. (1997) have also suggested binding of 60Co with two kinetically distinctsites.
In the current study, finding a majority of 60Co in the hydrophilic fraction and aweak hydrophobic fraction extracted by SPE, plus the dominance of this radio-nuclide in the smaller-sized fraction are consistent with previous findings.
4.2. Antimony-125
Antimony is redox-sensitive, with two prominent states, Sb(III) and Sb(V). Itwas likely as Sb(III) at the time of sampling, under the mildly reducing conditionsof LDA-22 (based on Rowat et al., 1999; not measured by us). Its presence asSb(V) is possible under mildly oxidizing conditions and mid-pH. The conversionfrom Sb(III) to Sb(V) could be slow and kinetically limited (Champ et al., 1984),but conversion reaction rates could be fast in the presence of Fe and Mn oxyhydr-oxides, in the order of a day (Belzile et al., 2000). This ambiguity was pointed outin a recent survey (Filella et al., 2002), however the same work mentioned thatorganics could stabilize Sb in its (III) oxidation state, hence adding to the uncer-tainty of defining the dominant oxidation state. In our situation, however, oxy-hydroxides are not abundant, and this is not likely a factor. Bacterial oxidation tothe (V) oxidation state is negligible (Belzile et al., 2000).
Inorganic Sb(III) is expected to hydrolyze in water to form the dominant species
SbðOHÞ03 at near-neutral pH—as aqueous or hydroxide precipitate (Brookins,
1988; Filella et al., 2002). On the other hand, in its (V) oxidation state, the pre-dominant species at mid-pH would be SbðOHÞ�6 (Filella et al., 2002). Its Sb(V)
form could also be as SbO�3 , similarly to other oxyanions of the same group N
(nitrate), P (phosphate), and As (arsenate). The presence of methylated antimonyunder anoxic conditions is a minor component (<10% of the total Sb; see Filellaet al., 2002).

39F. Caron, G. Mankarios / J. Environ. Radioactivity 77 (2004) 29–46
In our study, we are reporting that 125Sb is of a predominant negative character,with a small amount in the cationic fraction. It is not significantly retained by theSPE or ultrafiltration. Inorganic Sb(V) could explain this pattern, but the associ-ation of 125Sb with hydrophilic organics cannot be discounted. The element isreported to associate with organics (Filella et al., 2002 and references therein; Chenet al., 2003), although there is no certainty about the dominant oxidation state. Wehave observed some, but limited exchange with resins, and the association of Sbwith the small-sized fraction (<5000 Da) is consistent with the data compiled byFilella et al. (2002).
The possibility that Sb is incorporated into organic molecules cannot be dis-counted, similarly to nitrogen and phosphorus, but it is likely to be in small quan-tities. This Sb would be eventually found into humic/fulvic material. A directconfirmation is needed.
4.3. Cesium-137
Cesium-137 was predominantly in the anionic fraction, and it was found almostexclusively in the larger-sized colloidal fraction (>5000 Da). About half of it wasextracted by the SPE cartridge, in the strong-hydrophobic fraction. These observa-tions, together, support the association of Cs with colloidal NOM. This would con-stitute a unique finding.
Most reports indicate that Cs poorly associates with organics, however ourresults are consistent with those of previous studies at Chalk River (Champ et al.,1984; Champ and Robertson, 1986; Robertson et al., 2000). These authors havereported the presence of 137Cs in the negatively charged fraction (extraction similarto ours), and they have proposed that NOM was responsible for this, but withoutverification. Our study takes this hypothesis one step further with ultrafiltrationand SPE, and our results are consistent with a 137Cs-NOM association. It is under-stood that our work does not demonstrate a direct association, and a differenttechnique (e.g., voltammetry, spectroscopy, etc.) should be used to demonstratesuch an association.
In reviews, Krupka et al. (1999), mentioned that Cs+ forms weak complexeswith humic material, while von Gunten and Benes (1995) have reported only onereference which mentioned the association of 137Cs with organics in groundwaters.
A parallel can be drawn between aqueous speciation and sediment speciation,using sequential extractions of soils and/or sediments. In their review, von Guntenand Benes (1995) have indicated that 137Cs is generally associated with clay miner-als by ion exchange. In just a few cases, association with organics appeared impor-tant (Bunzl et al., 1998)—but such reports should be interpreted cautiously,because the portion associated with organics may be overestimated (Clark et al.,1996).
There are reports that the speciation of 137Cs in sediments depends upon thesource (e.g., atmospheric deposition of particles versus aqueous discharge), themechanisms of accumulation, and the conditions in the sediments (von Gunten andBenes, 1995). Atmospheric 137Cs from human activities (testing or accidents) does

F. Caron, G. Mankarios / J. Environ. Radioactivity 77 (2004) 29–4640
not mix well with soils or sediments (which contains stable Cs and could containradioactive 134Cs or 137Cs) compared to Cs from aqueous discharge. Backgroundfallout values in the Ottawa River basin would explain only <10 mBq/l of 137Cs infreshwaters (Cornett et al., 1995), hence this source of 137Cs only plays a minorrole in groundwater speciation.
Finally, the inorganic aqueous speciation of Cs does not provide an explanationfor our observations. Cesium is the largest simple cation among the elements, itshydration structure is simple (Wulfsberg, 2000). Its hydrated radius is 228 pm(1 pm ¼ 10�12 m) (ibid), which is too small to be retained by a 5 kDa membrane(~3000–5000 pm, based on the filtration spectrum; Osmonics, 1996). Calculationsusing the Latimer relationship (Wulfsberg, 2000) suggests no hydrolysis in waterfor the full pH range, while in their review, Krupka et al. (1999) concluded thatCs does not have the tendency to form aqueous complexes in soil and aqueousenvironments.
To conclude, our findings are consistent with other work done at Chalk River.Our results are also consistent with a 137Cs-NOM association; however, the natureof this association is unknown at this moment and it needs to be investigated insubsequent experiments.
4.4. Americium-241
Americium is the only element of interest that only has radioactive isotopes. Itsinorganic speciation is expected to be dominated by hydrolysis and carbonates atneutral pH. Americium is expected in its (III) oxidation state, its first hydrolysisin water is at pH ~8 (calculated with the Latimer relationship; Wulfsberg, 2000),and only a small proportion of carbonato complexes is expected (Allard andTorstenfelt, 1985).
In our sample, 241Am was found mostly in the negatively charged fraction, andit was almost completely retained by ultrafiltration. Its relatively large portion inthe strong hydrophobic fraction (SPE cartridge) is consistent with the associationof 241Am with NOM in other studies (Artinger et al., 1998; Schuessler et al., 2000;Cooper et al., 1995, to name a few). Our sample somewhat contrasts the findingsof Cooper et al. (1995), because their chromatographic separation suggested that241Am was associated with the smaller-sized NOM. The work at the Gorleben site(Artinger et al., 1998; Schuessler et al., 2000) is particularly significant, because theauthors deal with the kinetics of association/dissociation of Am with NOM. Theirexperiments extended over several thousands of hours (months), and they obtainedboth kinetic and equilibrium constants, with more than one binding site.
This behaviour for 241Am seems parallel to that of 60Co, but these two radio-nuclides unlikely compete for the same sites, because they are associated with dif-ferent size fractions, and their predominance with weak/strong hydrophobicmaterial is different. The electronic structure of the two elements provides a poten-tial explanation: 60Co (2+ nominal charge) has a d7 electron configuration, it is aborderline acid (Pearson concept; see Wulfsberg, 2000), whereas 241Am would be aslightly harder acid, due to its charge (3+) and a lower electronegativity. The

41F. Caron, G. Mankarios / J. Environ. Radioactivity 77 (2004) 29–46
charge/radius ratios are similar, and although we calculated a pKa of ~8 for the
first hydrolysis of both cations, the partially filled orbitals of Co2+ (d7) would
enable an intimate binding with borderline bases like N atoms. NOM contains
nitrogen (~2–3%, Thurman, 1985), plus the affinity of Co for porphyrin structures
is well known. For example, plant detritus which contain chlorophyll would be an
origin of porphyrins. The electronic structure of Am (configuration f 6) does not
allow such an intimate binding, and it would associate with harder bases, such as
oxygen atoms in functional groups.
4.5. Interpretation of the geochemical information
It was not surprising to find all the radionuclides of interest in anionic extracts.
In fact, the likely scenario is that most of the radionuclides are still near the source,
or retained on the geological material (sand or coarse aggregate) by various
mechanisms. Our sample would contain the material that does not significantly
adsorb near the source. Given this assessment, however, the radionuclides in our
sample likely exhibit some weak sorption with geological material (Robertson et al.,
2000; Caron et al., 2002). The nature and the dynamics of this sorption are needed
for safety assessment and other health-related issues. Our work provides a focusing
tool to analyze the aqueous species that would interact with soils.
5. Conclusions
Radionuclides found in a contaminated aquifer (60Co, 125Sb, 137Cs and 241Am)
were analyzed for their general character in aqueous solution: the dominance in
the anionic versus the cationic fraction, the association with the size fraction,
and the presence in the hydrophobic/hydrophilic fractions. We found that most
of these radionuclides were in the anionic groundwater extract, and mostly
hydrophilic. Cesium-137 and 241Am were found in the fraction containing the
larger-sized colloidal material (>5000 Da), whereas 60Co and 125Sb were associa-
ted with smaller material. The association of the radionuclides with NOM is
likely in all cases, except perhaps for 125Sb, which could be inorganic, but we do
not have a unifying explanation for its behaviour. Cesium-137 displayed a behav-
iour consistent with a NOM association. This is consistent with previous work
done at Chalk River, but not with other findings. This radioelement is usually
associated with clays (suspended or in sediments), or as a simple cation (in aque-
ous solutions).Our work is based on operational definitions, which have known shortcomings,
but this scheme provides useful geochemical information on the major aqueous
character of the radionuclides. These steps can be expanded to isolate the radio-
nuclides in given fractions, to determine the specific nature of these interactions by
direct techniques.
F. Caron, G. Mankarios / J. Environ. Radioactivity 77 (2004) 29–4642
Acknowledgements
We thank R.W.D. Killey for ideas and field assistance at the Chalk River
Laboratories, and C.C. Davison for site access (both from AECL). This work has
benefited in large part from ideas generated several years ago by D.E. Robertson
(Battelle Pacific Northwest National Laboratories, Richland, WA), E.L. Cooper,
D.R. Champ and J. Young (AECL). The authors acknowledge the support from
the Natural Science and Engineering Research Council of Canada (NSERC). G.
Mankarios was supported by the NSERC undergraduate student research award
program (USRA) in the summer of 2002. We thank N. Belzile (Laurentian U.) for
his comments on the manuscript.
Appendix A
Tables A.1–A.3. Raw data.
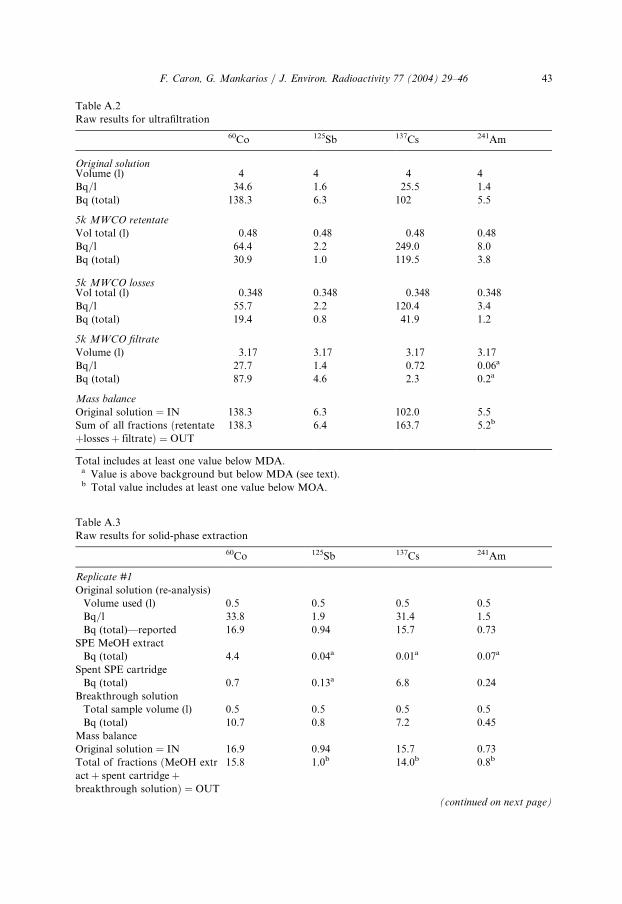
Table A.1
Raw results for the cation exchange resin extractions
60Co 1
25Sb 137Cs 241AmOriginal solution
Volume (l)
1 1 1 1Bq/l
34.6 1.6 25.5 1.1Bq (total)
34.6 1.6 25.5 1.1Operation: cation exchange resin extraction
Vol resin—analyzed (ml)
25 2 5 25 25Vol resin—total (ml)
50 5 0 50 50Bq (per sample)
1.8 0.05a 0.44 0.06aBq (total resin)
3.5 0.1a 0.9 0.1aBreakthrough solution
Volume (l)
0.965 0.965 0.965 0.965Bq/l
33.9 1.6 23.9 1.2Bq (total)
32.7i 1.6 23.0 1.1Mass balance
Original solution ¼ IN
34.6 1.6 25.5 1.4Total of fractions
ðresin þ solutionÞ ¼ OUT
36.2
1.7b 23.9 1.2b% Cationic
9.7 6b 3.7 10b% Anionic
90.3 9 4 96.3 90a Value is above background but below MDA (see text).b Total includes at least one value below MDA.
43F. Caron, G. Mankarios / J. Environ. Radioactivity 77 (2004) 29–46
Table A.2
Raw results for ultrafiltration
60Co
125Sb 137Cs 2 41AmOriginal solution
Volume (l) 4 4 4 4Bq/l
34.6 1.6 25.5 1 .4Bq (total)
138.3 6.3 102 5 .55k MWCO retentate
Vol total (l)
0.48 0.48 0.48 0 .48Bq/l
64.4 2.2 249.0 8 .0Bq (total)
30.9 1.0 119.5 3 .85k MWCO losses
Vol total (l) 0.348 0.348 0.348 0 .348Bq/l
55.7 2.2 120.4 3 .4Bq (total)
19.4 0.8 41.9 1 .25k MWCO filtrate
Volume (l)
3.17 3.17 3.17 3 .17Bq/l
27.7 1.4 0.72 0 .06aBq (total)
87.9 4.6 2.3 0 .2aMass balance
Original solution ¼ IN
138.3 6.3 102.0 5 .5Sum of all fractions ðretentate
þlosses þ filtrateÞ ¼ OUT
138.3
6.4 163.7 5 .2bTotal includes at least one value below MDA.a Value is above background but below MDA (see text).b Total value includes at least one value below MOA.
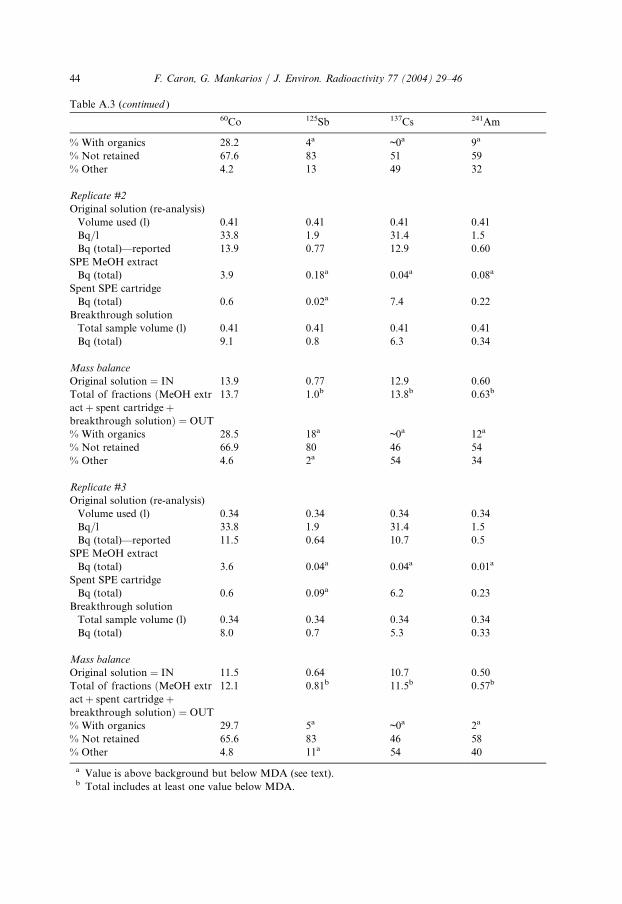
Table A.3
Raw results for solid-phase extraction
6
0Co 125Sb 1 37Cs 2 41AmReplicate #1
Original solution (re-analysis)
Volume used (l) 0
.5 0.5 0 .5 0 .5Bq/l 3
3.8 1.9 3 1.4 1 .5Bq (total)—reported 1
6.9 0.94 1 5.7 0 .73SPE MeOH extract
Bq (total) 4
.4 0.04a 0 .01a 0 .07aSpent SPE cartridge
Bq (total) 0
.7 0.13a 6 .8 0 .24Breakthrough solution
Total sample volume (l) 0
.5 0.5 0 .5 0 .5Bq (total) 1
0.7 0.8 7 .2 0 .45Mass balance
Original solution ¼ IN 1
6.9 0.94 1 5.7 0 .73Total of fractions ðMeOH extr
act þ spent cartridge þbreakthrough solutionÞ ¼ OUT
1
5.8 1.0b 1 4.0b 0 .8b(continued on next page)
F. Caron, G. Mankarios / J. Environ. Radioactivity 77 (2004) 29–4644
Table A.3 (continued )
60Co 1 25Sb 137Cs 241Am% With organics
28.2 4 a ~0a 9a% Not retained
67.6 8 3 51 59% Other
4.2 1 3 49 32Replicate #2
Original solution (re-analysis)
Volume used (l)
0.41 0 .41 0.41 0.41Bq/l
33.8 1 .9 31.4 1.5Bq (total)—reported
13.9 0 .77 12.9 0.60SPE MeOH extract
Bq (total)
3.9 0 .18a 0.04a 0.08aSpent SPE cartridge
Bq (total)
0.6 0 .02a 7.4 0.22Breakthrough solution
Total sample volume (l)
0.41 0 .41 0.41 0.41Bq (total)
9.1 0 .8 6.3 0.34Mass balance
Original solution ¼ IN
13.9 0 .77 12.9 0.60Total of fractions ðMeOH extr
act þ spent cartridge þbreakthrough solutionÞ ¼ OUT
13.7 1
.0b 13.8b 0.63b% With organics
28.5 1 8a ~0a 12a% Not retained
66.9 8 0 46 54% Other
4.6 2 a 54 34Replicate #3
Original solution (re-analysis)
Volume used (l)
0.34 0 .34 0.34 0.34Bq/l
33.8 1 .9 31.4 1.5Bq (total)—reported
11.5 0 .64 10.7 0.5SPE MeOH extract
Bq (total)
3.6 0 .04a 0.04a 0.01aSpent SPE cartridge
Bq (total)
0.6 0 .09a 6.2 0.23Breakthrough solution
Total sample volume (l)
0.34 0 .34 0.34 0.34Bq (total)
8.0 0 .7 5.3 0.33Mass balance
Original solution ¼ IN
11.5 0 .64 10.7 0.50Total of fractions ðMeOH extr
act þ spent cartridge þbreakthrough solutionÞ ¼ OUT
12.1 0
.81b 11.5b 0.57b% With organics
29.7 5 a ~0a 2a% Not retained
65.6 8 3 46 58% Other
4.8 1 1a 54 40a Value is above background but below MDA (see text).b Total includes at least one value below MDA.

45F. Caron, G. Mankarios / J. Environ. Radioactivity 77 (2004) 29–46
References
Allard, B., Torstenfelt, B., 1985. Actinide Solubilities and Speciation in a Repository Environment.
NAGRA NTB 85-18. Stockholm, Sweden.
Artinger, R., Kienzler, B., Schussler, W., Kim, J.I., 1998. Effects of humic substances on the 241Am
migration in a sandy aquifer: column experiments with Gorleben groundwater/sediment systems.
J. Contamin. Hydrol. 35, 261–275.
Belzile, N., Chen, Y.-W., Wang, Z., 2000. Oxidation of antimony (III) by amorphous iron and manga-
nese oxyhydroxides. Chem. Geol. 174, 379–387.
Brookins, D.G., 1988. Redox Diagrams. Springer-Verlag, Berlin.
Bunzl, K., Kracke, W., Schimmack, W., Zelles, L., 1998. Forms of fallout 137Cs and 239+240Pu in suc-
cessive horizons of a forest soil. J. Environ. Radioact. 39, 55–68.
Caron, F., Elchuk, S., Walker, Z.H., 1996. High-performance liquid chromatographic characterization
of dissolved organic matter from low-level radioactive waste leachates. J. Chromatogr. 739, 281–294.
Caron, F., Haas, M.K., Cooper, E.L., Robertson, D.E., 2002. Determination of liquid–solid partition
coefficients (Kd) of radionuclide anionic species from a contaminated aquifer. In: McGrail, P.J.,
Cragnolino, G.A. (Eds.), Scientific Basis for Nuclear Waste Management. Materials Research
Society Symposium Series, XXV. Materials Research Society.
Champ, D.R., Robertson, D.E., 1986. Chemical speciation of radionuclides in contaminant plumes at
the Chalk River Nuclear Laboratories. In: Bulman, R.A., Cooper, J.R. (Eds.), Speciation of Fission
and Activation Products in the Environment. In: Proceedings of the Speciation-85 Seminar held in
Christ Church, Oxford, UK, April 16–19. Elsevier Appl. Sci, London, UK, pp. 114–120.
Champ, D.R., Young, J.L., Robertson, D.E., Abel, K.H., 1984. Chemical speciation of long-lived radio-
nuclides in a shallow groundwater flow system. Water Poll. Res. J. Can. 19, 35–54.
Chen, Y.-W., Deng, T.-L., Filella, M., Belzile, N., 2003. Distribution and early diagenesis of antimony
species in sediments and porewaters of freshwater lakes. Environ. Sci. Technol. 37, 1163–1168.
Clark, S.B., Johnson, W.H., Malek, M.A., Serkiz, S.M., Hinton, T.G., 1996. A comparison of sequen-
tial extraction techniques to estimate geochemical controls on the mobility of fission product, acti-
nide, and heavy metal contaminants in soils. Radiochim. Acta 74, 173–179.
Cooper, E.L., McHugh, J.O., 1983. Migration of radiocontaminants in a forested wetland on the
Canadian shield: nuclide speciation and arboreal uptake. Sci. Tot. Environ. 28, 215–230.
Cooper, E.L., Haas, M.K., Mattie, J.F., 1995. Studies of the speciation of plutonium and other actinides
in natural groundwater using anion exchange resin. Appl. Radiat. Isot. 46, 1159–1173.
Cornett, R.J., Eve, T., Docherty, A.E., Cooper, E.L., 1995. Plutonium in freshwaters: sources and
behaviour in the Ottawa River Basin. Appl. Radiat. Isot. 46, 1239–1243.
Filella, M., Belzile, N., Chen, Y.-W., 2002. Antimony in the environment: a review focused on natural
waters II. Relevant solution chemistry. Earth Sci. Rev. 59, 265–285.
Fortin, C., Caron, F., 2000. Complexing capacity of low-level radioactive waste leachates for 60Co and109Cd using an ion-exchange technique. Anal. Chim. Acta 410, 107–117.
Garnier, J.-M., Pham, M.K., Ciffroy, P., Martin, J.-M., 1997. Kinetics of trace element complexation
with suspended matter and with filterable ligands in freshwater. Environ. Sci. Technol. 31,
1597–1606.
Grummit, W.E., Lahaie, G., 1978. Problems of Monitoring Radioactive Effluents. Monitoring of
Radioactive Effluents from Nuclear Facilities. International Atomic Energy Agency, Vienna, Austria,
pp. 9–18.
Killey, R.W.D., Munch, J.H., 1984. Subsurface Contaminant Transport from the Liquid Disposal Area,
CRNL: 1) Hydrogeology and Tritium Contamination near the Chemical Pit. Atomic Energy of
Canada, Ltd., Chalk River Laboratories, Chalk River, Ont., Canada, (AECL-7691).
Killey, R.W.D., McHugh, J.O., Champ, D.R., Cooper, E.L., Young, J.L., 1984. Subsurface cobalt-60
migration from a low-level waste disposal site. Environ. Sci. Technol. 18, 148–157.
Krupka, K.M., Kaplan, D.E., Whelan, G., Serne, R.J., Mattigod, S.V., 1999. Understand Variations in
the Partition Coefficient, Kd, Values. Part II: Review of Geochemistry and Available Kd Values for

F. Caron, G. Mankarios / J. Environ. Radioactivity 77 (2004) 29–4646
Cadmium, Cesium, Chromium, Lead, Plutonium, Radon, Strontium, Thorium, Tritium (3H), and
Uranium. U.S. Environmental Protection Agency, Washington, DC, (EPA-402-R-99-004B).
Mandal, R., Sekaly, A.L.R., Murimboh, J., Hassan, N.M., Chakrabarti, C.L., Back, M.H., Gregoire,
D.C., Schroeder, W.H., 1999a. Effect of the competition of copper and cobalt on the lability of
Ni(II)-organic ligand complexes. Part I. In model solutions containing Ni(II) and a well character-
ized fulvic acid. Anal. Chim. Acta 395, 309–322.
Mandal, R., Sekaly, A.L.R., Murimboh, J., Hassan, N.M., Chakrabarti, C.L., Back, M.H., Gregoire,
D.C., Schroeder, W.H., 1999b. Effect of the competition of copper and cobalt on the labiity of
Ni(II)-organic ligand complexes. Part II: in freshwaters (Rideau river surface waters). Anal. Chim.
Acta 395, 323–334.
Osmonics. 1996. The filtration spectrum. Available from: www.osmonics.com.
Robertson, D.E., Thomas, C.W., Pratt, S.L., Abel, K.H., Lepel, E.A., Schilk, A.J., Cooper, E.L.,
Caron, F., King, K.J., Killey, R.W.D., Hartwig, P.G., Mattie, J.F., Haas, M.K., Romaniszyn, E.,
Benz, M.L., Evenden, W.G., Link, S.O., Vilks, P., 2000. The Role of Organic Complexants and
Colloids in the Transport of Radionuclides by Groundwater. U.S. Nucl. Regulatory Commission,
Washington, DC, (NUREG/CR-6627, PNNL-12185).
Rowat, J.H., Killey, R.W.D., Welch, S.R., 1999. AECL-MISC-403-98. Atomic Energy of Canada Lim-
ited, Chalk River, Ontario.
Schuessler, W., Artinger, R., Kienzier, B., Kim, J.-I., 2000. Conceptual modeling of the humic colloid-
borne americium(III) migration by a kinetic approach. Environ. Sci. Technol. 34, 2608–2611.
Thurman, E.M., 1985. Organic geochemistry of natural waters. Developments in Biogeochemistry. M.
Nijhoff/Dr. W. Junk, Dordrecht, NE.
von Gunten, H.R., Benes, P., 1995. Speciation of radionuclides in the environment. Radiochim. Acta 69,
1–29.
Wulfsberg, G., 2000. Inorganic Chemistry. University Science books, Sausalito, CA.