phospholipase c 2 activation redirects vesicle trafficking by
TRANSCRIPT

Phospholipase C�2 Activation Redirects Vesicle Traffickingby Regulating F-actin*
Received for publication, April 11, 2015, and in revised form, September 30, 2015 Published, JBC Papers in Press, October 2, 2015, DOI 10.1074/jbc.M115.658328
Masaki Yamaga, D. Michelle Kielar-Grevstad, and Thomas F. J. Martin1
From the Department of Biochemistry, University of Wisconsin, Madison, Wisconsin 53706
Background: Ca2� and PI(4,5)P2 regulate F-actin and vesicle exocytosis in neuroendocrine cells.Results: Phospholipase C�2 knockdown inhibits Ca2�-stimulated PI(4,5)P2 hydrolysis, F-actin disassembly, and vesicle recruit-ment in PC12 cells.Conclusion: Phospholipase C�2, which localizes with actin, links Ca2� rises to F-actin disassembly and vesicle trafficking.Significance: The results reveal a new role for phospholipase C�2 as a Ca2�-dependent regulator of actin cytoskeletal dynamicsand vesicle trafficking.
PI(4,5)P2 localizes to sites of dense core vesicle exocytosis inneuroendocrine cells and is required for Ca2�-triggered vesicleexocytosis, but the impact of local PI(4,5)P2 hydrolysis on exo-cytosis is poorly understood. Previously, we reported that Ca2�-dependent activation of phospholipase C�2 (PLC�2) catalyzesPI(4,5)P2 hydrolysis, which affected vesicle exocytosis by regu-lating the activities of the lipid-dependent priming factorsCAPS (also known as CADPS) and ubiquitous Munc13-2 inPC12 cells. Here we describe an additional role for PLC�2 invesicle exocytosis as a Ca2�-dependent regulator of the actincytoskeleton. Depolarization of neuroendocrine PC12 cells with56 or 95 mM KCl buffers increased peak Ca2� levels to �400 or�800 nM, respectively, but elicited similar numbers of vesicleexocytic events. However, 56 mM K� preferentially elicited theexocytosis of plasma membrane-resident vesicles, whereas 95mM K� preferentially elicited the exocytosis of cytoplasmic ves-icles arriving during stimulation. Depolarization with 95 mM K�
but not with 56 mM K� activated PLC�2 to catalyze PI(4,5)P2
hydrolysis. The decrease in PI(4,5)P2 promoted F-actin disas-sembly, which increased exocytosis of newly arriving vesicles.Consistent with its role as a Ca2�-dependent regulator of thecortical actin cytoskeleton, PLC�2 localized with F-actin fila-ments. The results highlight the importance of PI(4,5)P2 forcoordinating cytoskeletal dynamics with vesicle exocytosis andreveal a new role for PLC�2 as a Ca2�-dependent regulator ofF-actin dynamics and vesicle trafficking.
Peptide secretion from neural and endocrine cells occurs byCa2�-dependent dense core vesicle exocytosis. Vesicles pro-ceed through sequential steps involving transport to the cellperiphery followed by plasma membrane tethering, docking,priming, and Ca2�-dependent fusion. Essential proteins andlipids function at each step in the regulated secretory pathway
(1, 2). PI(4,5)P22 is required at a reversible ATP-dependent ves-
icle priming step (3–5) but regulates other late steps as well.The binding of PI(4,5)P2 to its protein effectors is mediatedthrough interactions with pleckstrin homology (PH) and C2domains or with highly basic motifs in proteins (6 –9).PI(4,5)P2-binding proteins that function at distinct steps of theexocytic pathway include PH domain-containing CAPS (Ca2�-dependent activator protein in secretion, also known asCADPS) (6 –9), C2 domain-containing Munc13-1/2 (mamma-lian homologue of Unc-13-1/2) (9, 10), rabphilin (11), and syn-aptotagmin-1 (12, 13) and basic motif-containing syntaxin-1 (8,14).
PI(4,5)P2 hydrolysis through receptor-regulated PLC� orPLC� is an upstream initiating signal for vesicle exocytosis inneuroendocrine cells by generating inositol 1,4,5-trisphosphatefor Ca2� mobilization (15) and DAG to potentiate exocytosis(16, 17). However, there is also a major role for PI(4,5)P2 down-stream of Ca2� signaling involving distinct pools of PI(4,5)P2 (3,9). High concentration plasma membrane domains of PI(4,5)P2localize near docked vesicles (8, 18, 19) and locally regulateCAPS and Munc13 protein function (9). The Ca2�-dependentgeneration of DAG from PI(4,5)P2 potentiates vesicle exocyto-sis, but Ca2�-activated PLCs that are directly linked to vesicleexocytosis had not been identified (20). Several PLCs (e.g. PLC�or PLC�) could function as downstream effectors in Ca2� sig-naling because of their strong Ca2�-dependent activation (21).PLC�2 was recently found to be a Ca2�-dependent modulatorfor CAPS and Munc13 function in neuroendocrine PC12 cells(9).
PI(4,5)P2 plays a key role in F-actin assembly mechanisms(22–24). The actin cytoskeleton undergoes dynamic reorgani-zation associated with the Ca2�-dependent activation of vesicleexocytosis in secretory cells. F-actin functions in part as a phys-ical barrier in secretory cells to limit vesicle access to the plasmamembrane for fusion (25, 26). This actin barrier is locally dis-assembled during Ca2� rises in stimulated chromaffin cells
* This work was supported by National Institutes of Health Grants DK025861and DK040428 (to T. F. J. M.). The authors declare that they have no con-flicts of interest with the contents of this article.Author’s Choice—Final version free via Creative Commons CC-BY license.
1 To whom correspondence should be addressed: Dept. of Biochemistry, Uni-versity of Wisconsin, 433 Babcock Dr., Madison, WI 53706. Tel.: 608-263-2427; Fax: 608-265-4693; E-mail: [email protected].
2 The abbreviations used are: PI(4,5)P2, phosphatidylinositol 4,5-bisphos-phate; PH, pleckstrin homology; PLC, phospholipase C; DAG, diacylglyc-erol; TIRF, total internal reflection fluorescence; BDNF, brain-derived neu-rotrophic factor; EGFP, enhanced green fluorescent protein; C1, proteinkinase C�-C1 domain; MS, mild stimulation; SS, strong stimulation.
THE JOURNAL OF BIOLOGICAL CHEMISTRY VOL. 290, NO. 48, pp. 29010 –29021, November 27, 2015Author’s Choice © 2015 by The American Society for Biochemistry and Molecular Biology, Inc. Published in the U.S.A.
crossmark
29010 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 290 • NUMBER 48 • NOVEMBER 27, 2015
by guest on February 18, 2018http://w
ww
.jbc.org/D
ownloaded from

involving actin-severing proteins such as scinderin (27, 28). Inother cell types, PI(4,5)P2 hydrolysis catalyzed by PLC�, PLC�,or 5-phosphatase has been shown to promote F-actin disassem-bly (29 –32). However, a Ca2�-dependent PLC pathway forPI(4,5)P2 hydrolysis that reorganizes the actin cytoskeleton inneuroendocrine cells has not been identified.
Neuroendocrine cells possess a plasma membrane-residentpool of vesicles that undergo exocytosis in response to Ca2�
rises. Cytoplasmic vesicles are also recruited to the plasmamembrane for exocytosis during stimulation (33, 34). We foundthat varying Ca2� influx in PC12 cells markedly affectedwhether resident or recruited vesicles undergo exocytosis.Stronger depolarization stimulated more Ca2� entry thatuniquely promoted PI(4,5)P2 hydrolysis and F-actin disassem-bly, which in turn enhanced exocytosis of cytoplasmic vesiclesarriving during stimulation. PLC�2 was the critical linkbetween increased Ca2� and PI(4,5)P2 hydrolysis, F-actin dis-assembly, and redirected vesicle exocytosis. These studiesreveal a functional role for PLC�2 as a Ca2�-dependent regu-lator of the actin cytoskeleton and the secretory pathway inneuroendocrine cells.
Experimental Procedures
DNA Constructs—The plasmid encoding a green fluores-cence protein-tagged BDNF (BDNF-EGFP) was provided by V.Lessmann (Johannes Gutenberg Universität, Mainz, Germany).PKC�-C1-EGFP (C1-EGFP) was provided by S. Grinstein (Hos-pital for Sick Children, Toronto, Canada). EGFP-mouse PLC�1(EGFP-PLC�1) and EGFP-mouse PLC�2 (EGFP-PLC�2) wereprovided by K. Fukami (Tokyo University of Pharmacy and LifeScience). To generate PKC�-C1-mKate2 (C1-mKate2), thePKC�-C1 domain was amplified from PKC�-C1-EGFP by PCRusing the forward primer 5�-GGACTCAGATCTACCATGG-GGG-3� and reverse primer 5�-ATGTCGACTGGTACCTTG-CGCCGGC-3�. The PCR product was digested with BglII andSalI and inserted into BglII and SalI sites of mKate2-N vector.To generate EGFP-PLC�2-PH, the PLC�2-PH domain wasamplified from EGFP-PLC�2 by PCR using the forward primer5�-CTCAGATCTATGCCTGGTCCCCAGCC-3� and the re-verse primer 5�-GCGGTCGACGATGCCAGCCATGAGG-3�.The PCR product was digested with BglII and SalI and insertedinto BglII and SalI sites of EGFP-C1 vector. EGFP-PLC�2 3M rescue plasmid was generated by inducing threenonsense mutations in the shRNA targeting sequence by usingthe forward primer 5�-CGAGCCCTCTCCGATCTCGTGAA-ATATACC-3� and the reverse primer 5�-GGTATATTTCA-CGAGATCGGAGAGGGCTCG-3�.
Antibodies and Reagents—Anti-mouse PLC�2 polyclonalantibody was kindly provided by K. Fukami, anti-PLC�1 (D-7)mouse monoclonal antibody was purchased from Santa CruzBiotechnology, Inc. (Dallas, TX), and anti-GAPDH monoclonalantibody was purchased from Ambion (Austin, TX). Fluo-4AM and Alexa Fluor 568 phalloidin were purchased fromMolecular Probes, Inc. (Eugene, OR). Other materials andchemicals were obtained from commercial sources.
Cell Culture and Transfection—PC12 cells were cultured inDulbecco’s modified Eagle’s medium (Sigma) supplementedwith 5% horse serum and 5% calf serum at 37 °C in an air plus
10% CO2 atmosphere at constant humidity. Transfections forplasmid DNAs were performed by electroporation using anECM 830 system (BTX, Holliston, MA) set at 230 V, 8 ms, and1 pulse. PC12 cells (grown to �80% confluence in a 10-cm dish)suspended in 0.5 ml of cytomix buffer (25 mM HEPES, 120 mM
KCl, 10 mM KH2PO4, 0.15 mM CaCl2, 5 mM MgCl2, 2 mM
EGTA, pH 7.6) were transfected with 10 –50 �g of plasmidDNA(s) using a 4-mm gap size cuvette. Transfections forsiRNAs were performed by electroporation using an ECM830set at 90 V, 8 ms, and 1 pulse. PC12 cells were transfected with1.33 �M siRNA and 2.5 �g of plasmid DNA using a 1-mm gapsize cuvette.
Monitoring of DAG Generation on the Plasma Membrane—PC12 cells were transfected with 40 �g of C1-mKate2 plasmidDNA or co-transfected with 25 �g of C1-mKate2 and 25 �g ofEGFP, EGFP-PLC�1, or EGFP-PLC�2 plasmid DNAs andplated on poly-D-lysine-coated (Sigma) and type I collagen-coated (BD Biosciences) 35-mm glass bottom dishes (MatTekCorp., Ashland, MA). After a 48-h incubation, the culturemedium was replaced with basal buffer (15 mM HEPES, pH 7.4,145 mM NaCl, 5.6 mM KCl, 2.2 mM CaCl2, 0.5 mM MgCl2, 5.6mM glucose, 0.5 mM ascorbic acid, 0.1% BSA), and then cellswere stimulated with 56 (moderate stimulation; MS) and 95 mM
K� (strong stimulation; SS) depolarization buffer (basal bufferadjusted to 95 mM NaCl and 56 mM KCl or 56 mM NaCl and 95mM KCl). Cells were imaged on a Nikon total internal reflectionfluorescence (TIRF) microscope evanescent wave imaging sys-tem used with a TE2000-U inverted microscope (Nikon) and anApo TIRF �100, numerical aperture 1.45 (Nikon) objectivelens. EGFP and mKate2 fluorescence were excited with the488-nm laser line and the 514-nm laser line, respectively.Images were acquired at 250-ms intervals with a CoolSNAP-ESdigital monochrome CCD camera system (Photometrics, Tuc-son, AZ) controlled by Metamorph software (Universal Imag-ing Corp., Downingtown, PA). All data analysis was conductedwith ImageJ software.
TIRF Analysis of BDNF-EGFP Secretion—PC12 cells weretransfected with 30 �g of BDNF-EGFP plasmid DNA andplated on poly-D-lysine- and collagen-coated 35-mm glass bot-tom dishes. After a 48-h incubation, the culture medium wasreplaced with basal buffer, and cells were stimulated with MS orSS buffer. Cells were imaged on the Nikon TIRF microscope at250-ms intervals with a CoolSNAP-ES digital monochromeCCD camera system (Photometrics) controlled by Metamorphsoftware (Universal Imaging Corp.). The penetration depth(1/e) of the evanescent field was estimated to be 160 nm basedon a calibration with fluorescent beads. At this penetrationdepth, resident vesicles (estimated d � 100 nm) at the plasmamembrane are evident, whereas vesicles enmeshed deeper inthe 400-nm actin cortex (34), termed non-resident cytoplasmicvesicles, are dim or not evident. Exocytic events were manuallycounted and scored for whether exocytosis occurred from ves-icles resident in the evanescent field for �0.5 s (resident) or�0.5 s (non-resident) prior to fusion. All data analysis usedMetamorph software.
Ca2� Imaging—PC12 cells were plated on poly-D-lysine- andcollagen-coated 35-mm glass bottom dishes. After a 24-h incu-bation, cells were washed, and the culture medium was replaced
Phospholipase C�2 Activation in Ca2�-regulated Exocytosis
NOVEMBER 27, 2015 • VOLUME 290 • NUMBER 48 JOURNAL OF BIOLOGICAL CHEMISTRY 29011
by guest on February 18, 2018http://w
ww
.jbc.org/D
ownloaded from

with basal buffer. Cells were loaded with fluo-4 by incubationwith 2 �M fluo-4, AM and 0.02% Pluronic� F-127 (MolecularProbes) mixture at room temperature for 30 min in the dark.Cells were then washed with basal buffer and incubated at 37 °Cfor 20 min to allow de-esterification of loaded dye in basalbuffer. Cells were stimulated with MS or SS buffer, and imageswere acquired at 250-ms intervals on an epifluorescence micro-scope (Nikon). Cells were treated with 5 �M ionomycin and 5mM EGTA to obtain maximum (fmax) and minimum (fmin) fluo-rescence values, respectively. Average fluorescence intensity ateach time point (ft) was measured using Metamorph software.Relative fluorescence intensity of fluo-4-Ca2� (F) and concen-tration of intracellular Ca2� ([Ca2�]i) were determined as F �(ft � fmin)/(fmax � fmin) and [Ca2�]i � KD F/(1 � F), where KDfor fluo-4 is 345 nM.
Knockdown of PLC�2 by shRNA and siRNA—PC12 cells wereco-transfected with 30 �g of pSM2-PLC�2 shRNA vector;V2MM_89060 (shRNA 1) and V2MM_197066 (shRNA 2)targeting mouse PLC�2 mRNA (accession number NM_001113360) sequence corresponding to nucleotides 2117–2135(CCCTCTCGGACCTAGTGAA) and 2119 –2137 (CTCTCG-GACCTAGTGAAAT), respectively (Open Biosystems, Hunts-ville, AL); or pSM2 empty vector (Open Biosystems) and 10 �gof C1-mKate2 plasmid DNAs and plated on poly-D-lysine- andcollagen-coated 35-mm glass bottom dishes for TIRF analysisand 6-well dishes for Western blotting. After a 72-h incubation,cells were lysed and subjected to blotting analysis with anti-PLC�2 polyclonal antibody and anti-GAPDH monoclonal anti-body. For rescue experiments, PC12 cells were triple-transfectedwith 30 �g of pSM2-PLC�2 shRNA or pSM2 empty vector, 10�g of C1-mKate2 plasmid DNA, and 5 �g of EGFP-PLC�2 3Mplasmid DNA. Monitoring of DAG generation was performedas described above. PC12 cells were co-transfected with 35 �gof pSM2-PLC�2 shRNA or pSM2 empty vector and 15 �g ofBDNF-EGFP plasmid DNAs and plated on poly-D-lysine- and coll-agen-coated 35-mm glass bottom dishes for TIRF analysis and a6-well dish for blotting. After a 72-h incubation, blotting analysisand TIRF analysis were performed as described above. Knock-down of PLC�2 by endoribonuclease-prepared siRNA(target sequence corresponds to nucleotides 1406 –1942 ofmRNA isolated from PC12 cells) was performed as describedpreviously (9).
Co-localization Analysis—Co-localization analysis of signalscorresponding to PLC�2 and F-actin was performed using apixel by pixel analysis algorithm with Fiji-win32 software. Thecalculated percentage of random co-localization was sub-tracted from co-localization values.
Quantification of Cortical F-actin—EGFP-, EGFP-PLC�1-,and EGFP-PLC�2-overexpressing and PLC�2 knockdown(EGFP co-transfection marker indicated that transfection effi-ciency was 82%) PC12 cells were plated on poly-D-lysine- andcollagen-coated 35-mm glass bottom dishes. After a 48-h (or72-h for PLC�2 knockdown) incubation, cells were treated withMS or SS buffers at room temperature for 0.5, 1, 2, and 3 min.After treatment, the cells were immediately fixed with 3.7%formaldehyde in PBS at room temperature for 8 min and per-meabilized by incubation with 0.1% Triton X-100 in PBS con-taining 1% BSA in PBS at room temperature for 10 min. F-actin
was visualized with Alexa Fluor 568-phalloidin at room tem-perature for 20 min followed by washing. Cells were imaged ona TIRF microscope with a CoolSNAP-ES digital monochromeCCD camera system controlled by Metamorph software, anddata analysis utilized Metamorph software.
Results
Strong Stimulation Promotes Greater Ca2� Rises andPI(4,5)P2 Hydrolysis—PI(4,5)P2 is required for regulated vesicleexocytosis and is distributed in membrane domains present atsites of exocytosis (8, 9, 19). To determine the impact ofPI(4,5)P2 hydrolysis on regulated vesicle exocytosis, we utilizeda range of stimulation conditions in the well characterizedPC12 cell model for neuroendocrine secretion. TIRF micros-copy was used to monitor the exocytosis of vesicles containingfluorescent cargo proteins (35, 36) and to detect PI(4,5)P2 hy-drolysis and DAG generation (9, 37). Depolarizing the cells byincubation in high KCl buffers promotes depolarization andCa2� influx. PC12 cells have a low density of L-type Ca2� chan-nels so that substantial depolarization in KCl buffers is requiredto elicit Ca2� increases (38, 39). We found that 56 mM KCl (MS)was optimal for promoting maximal number of dense core ves-icle fusion events of BDNF-EGFP within 3 min (Fig. 1A). Stron-ger depolarization at 95 mM KCl (SS) did not further increasethe number of fusion events (Figs. 1A and 3C). Analysis of thetime courses of averaged cumulative fusion events that fit wellto an exponential function indicated that the time course ofSS-evoked fusion events tended to be slower than MS-evokedfusion events (time constant � values for MS and SS were 19.7 2.3 s and 29.5 4.9 s, respectively, p � 0.09). MS conditionspromoted a peak rise in [Ca2�] to �400 nM, whereas strongerdepolarization with SS conditions evoked a peak [Ca2�] rise to�800 nM (Fig. 1B), similar to a previous report (39). The resultsindicate that Ca2�-dependent vesicle exocytosis is saturated bythe Ca2� concentration rises elicited by MS conditions. In sub-sequent studies, we utilized MS and SS conditions to promoteoptimal (�400 nM) or greater than optimal (�800 nM) Ca2�
rises, respectively.Under the MS and SS buffer stimulation conditions, lipid
signaling events differed markedly. A fluorescent PKC�-C1-mKate2 probe was used to detect DAG generation in theplasma membrane by TIRF microscopy (37). Under MS condi-tions, where vesicle exocytosis was maximally stimulated (Fig.1A), plasma membrane DAG levels did not differ from those inunstimulated cells (Fig. 1, C and D). By contrast, SS conditionsresulted in a rapid (�15 s), transient increase in DAG levels, asinferred from the translocation of the C1-mKate2 protein (Fig.1, C and D). The translocated C1-mKate2 was evident as brightpuncta and diffuse fluorescence (Fig. 1C), which suggested thathigh concentration domains of DAG may be generated fromhigh concentration domains of PI(4,5)P2 (9) followed by diffu-sion. Using a PLC�4-PH-mKate2 domain probe with TIRF (9),we detected a corresponding partial loss of PI(4,5)P2 from theplasma membrane under SS but not under MS buffer condi-tions (Fig. 1E). The results indicate that PI(4,5)P2 hydrolysiswith DAG generation was only promoted at Ca2� concentra-tions higher (SS conditions) than those needed to maximallystimulate vesicle exocytosis (MS conditions).
Phospholipase C�2 Activation in Ca2�-regulated Exocytosis
29012 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 290 • NUMBER 48 • NOVEMBER 27, 2015
by guest on February 18, 2018http://w
ww
.jbc.org/D
ownloaded from

PLC�2 Mediates PI(4,5)P2 Hydrolysis at Elevated Ca2�
Levels—The greater Ca2� elevation under SS conditions prob-ably stimulated DAG generation from the Ca2�-dependentactivation of a PI(4,5)P2-hydrolyzing PLC. Because PLC�1 andPLC�2 are expressed in PC12 cells (data not shown) (40) andare strongly activated by Ca2� (21), we determined which ifeither was responsible for DAG generation in response to Ca2�
elevations under SS conditions. We first determined the effectof PLC�1 and PLC�2 overexpression on DAG generation. APKC�-C1-mKate2 probe that monitored DAG was translo-cated to the plasma membrane in EGFP-expressing controlcells under SS conditions but not under MS conditions (Fig. 2, A(top) and B), whereas expression of a EGFP-PLC�2 proteinenhanced DAG generation even under MS conditions (Fig. 2, A(middle) and B). This contrasted with cells expressing EGFP-PLC�1 (Fig. 2, A (bottom panels) and B), where there was noDAG generation beyond that of control cells. These findingsindicate that PLC�2 rather than PLC�1 responds to Ca2� influxby generating DAG in PC12 cells, which was consistent with in
vitro studies indicating the greater Ca2� sensitivity for PLC�2activation compared with PLC�1 (41).
To determine whether endogenous PLC�2 was responsiblefor DAG generation in control cells under SS conditions, weutilized shRNA plasmids and siRNA that were effective inreducing PLC�2 by more than 90 and 80%, respectively (Fig. 2,C and D). Depletion of PLC�2 did not affect the expression levelof PLC�1 (Fig. 2E). In cells depleted of PLC�2 by either shRNAplasmid or siRNA, DAG generation in response to SS buffer wascompletely abolished (Fig. 2, F–H). Both shRNA plasmids,but not a control plasmid, had similar effects (data notshown). To confirm that the loss of DAG generation inPLC�2-depleted cells was due to the lack of PLC�2, we con-ducted rescue experiments with an shRNA-resistant EGFP-PLC�2 construct (EGFP-PLC�2 3M) that contained threenucleotide substitutions in the shRNA target. Expression ofEGFP-PLC�2 3M restored DAG generation (Fig. 2, F and G).These findings indicate that PLC�2 is the major PLC in PC12cells that is activated at the plasma membrane by the higher
FIGURE 1. Depolarization in 95 mM KCl elicits greater Ca2� influx and DAG generation. A, depolarization-dependent increases in the number of BDNF-EGFPfusion events. PC12 cells expressing BDNF-EGFP were stimulated with the indicated KCl-containing buffers with 2.2 mM CaCl2. Images were acquired by TIRFmicroscopy at 4 Hz, and fusion events were identified and shown as cumulative plots (mean S.E. (error bars), n � 10 cells). B, peak intracellular [Ca2�] at 5–10s in PC12 cells loaded with fluo-4 was determined for cells incubated in 56 mM KCl (MS) (n � 9 cells) and 95 mM KCl (SS) (n � 10 cells) buffers. Mean values S.E.are shown (*, p � 0.005). C, DAG generation under MS and SS conditions was determined by monitoring PKC�-C1-mKate2 translocation to the plasmamembrane by TIRF microscopy at 4 Hz. D, relative fluorescence intensities of images from C were plotted (mean S.E., n � 10 –14 cells). E, plasma membranePI(4,5)P2 levels in PC12 cells incubated in MS and SS buffers. Cells expressing PLC�4-PH-mKate2 were incubated in MS or SS buffers for the times indicated andimaged by TIRF microscopy at 4 Hz. Representative images (top) indicate a transient decrease in PI(4,5)P2 levels in cells incubated with SS but not MS buffers.The relative fluorescence of cell footprints was quantitated at 10-s intervals (mean S.E., n � 8 cells; *, p � 0.005; **, p � 0.05). The PI(4,5)P2 decrease in SSbuffers was partial.
Phospholipase C�2 Activation in Ca2�-regulated Exocytosis
NOVEMBER 27, 2015 • VOLUME 290 • NUMBER 48 JOURNAL OF BIOLOGICAL CHEMISTRY 29013
by guest on February 18, 2018http://w
ww
.jbc.org/D
ownloaded from

Ca2� concentrations promoted by strong depolarizationunder SS conditions.
Strong Stimulation Shifts Exocytosis from Docked to NewlyArrived Vesicles—PI(4,5)P2 is required for dense core vesicleexocytosis (3, 18, 42). However, the SS conditions that pro-moted PI(4,5)P2 hydrolysis by PLC�2 did not reduce the totalnumber of exocytic events in 5 min (Fig. 3C). This is accountedfor by the fact that PI(4,5)P2 hydrolysis is partial under SS stim-ulation conditions (Fig. 1E) and that the DAG generated (Fig.1D) further activates ubMunc13-2 (9). To determine more fullythe impact of PI(4,5)P2 hydrolysis on Ca2�-triggered vesicleexocytosis, we examined individual exocytic events by TIRFmicroscopy. As previously characterized for PC12 cells (34, 36,43), two types of evoked vesicle exocytic events were observed:from resident vesicles present at the plasma membrane for�0.5 s before fusion or from non-resident cytoplasmic vesiclesthat arrived �0.5 s before fusion in stimulated cells (Fig. 3A).The vesicle exocytosis assay utilized in TIRF studies employingBDNF-EGFP cargo has been extensively characterized (35).Resident and non-resident vesicles that fuse are readily distin-
guished from non-fusing, vesicles as shown by the traces offluorescent changes in Fig. 3B. Fusing vesicles exhibit hallmarkfeatures of an initial brightening upon fusion pore formation(due to vesicle pH change) followed by slow dimming as thefusion pore closes and the vesicles re-acidify in cavicapture exo-cytosis (Fig. 3B, left). In accord with this, a similar brighteningwas obtained by neutralizing vesicle pH by treatment with 50mM NH4Cl (Fig. 3B, left, arrow). The initial fluorescence of res-ident vesicles prior to fusion is greater than that for non-resi-dent vesicles, which is at background (Fig. 3, A and B (left)). Bycontrast, non-fusing vesicles that approach the plasma mem-brane and dock or approach the plasma membrane and leavelack a fusion spike (Fig. 3B, right). Such vesicles are rare instimulated cells and are readily distinguished from fusing vesi-cles. Under MS stimulation conditions, �70% of exocyticevents occurred from resident vesicles and �30% from non-resident vesicles (Fig. 3D), similar to previous studies (36, 43).By contrast, under SS stimulation conditions, the number ofexocytic events from resident vesicles decreased, and thosefrom non-resident vesicles increased (Fig. 3D), resulting in
FIGURE 2. PLC�2 generates DAG at the plasma membrane in response to high K�-induced depolarization. A, effect of PLC overexpression on DAGgeneration under resting or MS and SS conditions. EGFP, EGFP-PLC�2, and EGFP-PLC�1 were co-expressed with PKC�-C1-mKate2 in PC12 cells. After 48 h, thecells were incubated in resting conditions or in MS or SS buffers, and images were acquired at 4 Hz by TIRF microscopy. Images show the point of maximalfluorescence increase with insets showing the distribution of EGFP and EGFP fusion proteins before incubation by epifluorescence microscopy. B, averages ofrelative fluorescence intensity of PKC�-C1-mKate2 in cell footprints during incubation in MS (top) or SS buffer (bottom) from TIRF images similar to A are plotted(n � 9 cells). C and D, PLC�2 levels were determined in PC12 cells transfected with a control shRNA plasmid (C) or siRNA (D) or with either of two PLC�2 shRNAplasmids (C) or PLC�2 siRNA (D). E, expression levels of PLC�1 in PLC�2 KD PC12 cells. PLC�1 was detected by Western blotting with anti-PLC�1 antibody in cellstreated with PLC�2 shRNA 2. F, PLC�2 is required for DAG generation elicited under SS conditions. PC12 cells were co-transfected with PLC�2 shRNA 2 (n � 12cells) or with empty vector (n � 10 cells) along with an expression plasmid for PKC�-C1-mKate2. For rescue studies, PC12 cells were triple-transfected withPLC�2 shRNA 2, PKC�1-C1-mKate2, and EGFP-PLC�2 3M plasmid (n � 12 cells). Expressed EGFP-PLC�2 3M is shown in the inset. After 72 h, cells were incubatedin SS buffer for the indicated times. Images were acquired at 4 Hz by TIRF microscopy with representative frames shown. G and H, the relative fluorescenceintensity of PKC�-C1-mKate2 in cell footprints during incubation is shown (mean S.E. (error bars), n � 10 –12 cells).
Phospholipase C�2 Activation in Ca2�-regulated Exocytosis
29014 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 290 • NUMBER 48 • NOVEMBER 27, 2015
by guest on February 18, 2018http://w
ww
.jbc.org/D
ownloaded from
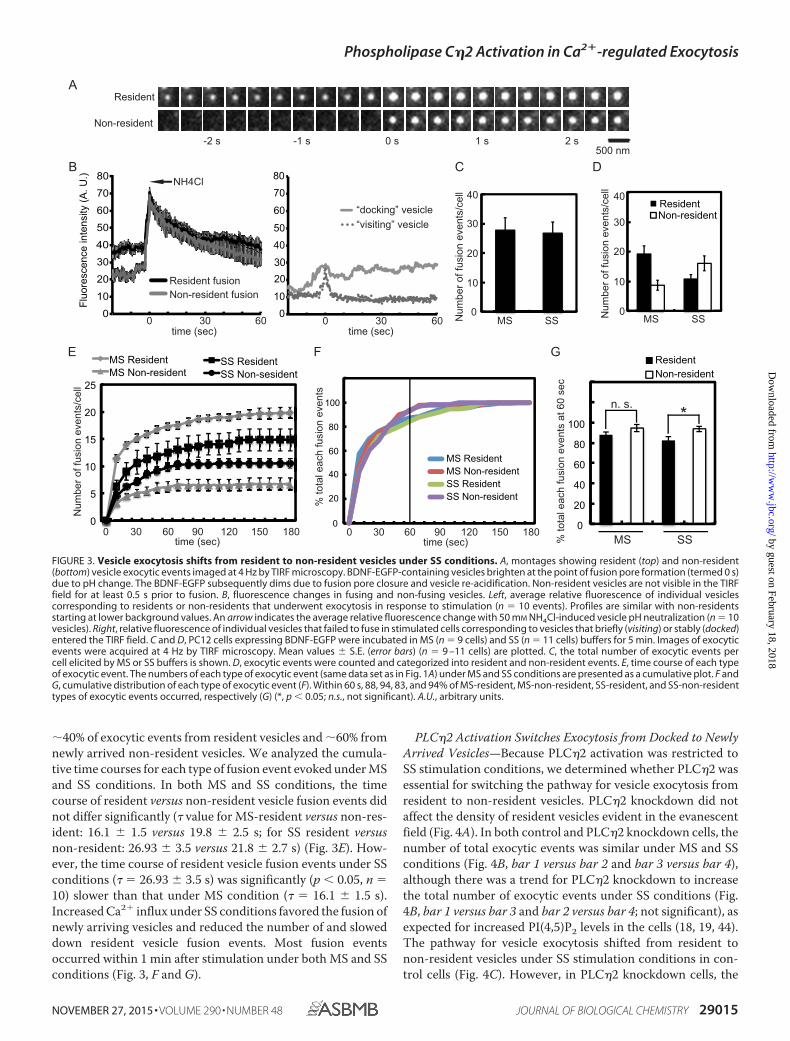
�40% of exocytic events from resident vesicles and �60% fromnewly arrived non-resident vesicles. We analyzed the cumula-tive time courses for each type of fusion event evoked under MSand SS conditions. In both MS and SS conditions, the timecourse of resident versus non-resident vesicle fusion events didnot differ significantly (� value for MS-resident versus non-res-ident: 16.1 1.5 versus 19.8 2.5 s; for SS resident versusnon-resident: 26.93 3.5 versus 21.8 2.7 s) (Fig. 3E). How-ever, the time course of resident vesicle fusion events under SSconditions (� � 26.93 3.5 s) was significantly (p � 0.05, n �10) slower than that under MS condition (� � 16.1 1.5 s).Increased Ca2� influx under SS conditions favored the fusion ofnewly arriving vesicles and reduced the number of and sloweddown resident vesicle fusion events. Most fusion eventsoccurred within 1 min after stimulation under both MS and SSconditions (Fig. 3, F and G).
PLC�2 Activation Switches Exocytosis from Docked to NewlyArrived Vesicles—Because PLC�2 activation was restricted toSS stimulation conditions, we determined whether PLC�2 wasessential for switching the pathway for vesicle exocytosis fromresident to non-resident vesicles. PLC�2 knockdown did notaffect the density of resident vesicles evident in the evanescentfield (Fig. 4A). In both control and PLC�2 knockdown cells, thenumber of total exocytic events was similar under MS and SSconditions (Fig. 4B, bar 1 versus bar 2 and bar 3 versus bar 4),although there was a trend for PLC�2 knockdown to increasethe total number of exocytic events under SS conditions (Fig.4B, bar 1 versus bar 3 and bar 2 versus bar 4; not significant), asexpected for increased PI(4,5)P2 levels in the cells (18, 19, 44).The pathway for vesicle exocytosis shifted from resident tonon-resident vesicles under SS stimulation conditions in con-trol cells (Fig. 4C). However, in PLC�2 knockdown cells, the
Resident
Non-resident
-1 s-2 1s s 2 s0 s500 nm
A
B
Fluo
resc
ence
inte
nsity
(A. U
.)
0 30 60time (sec)time (sec)
Resident fusionNon-resident fusion
“docking” vesicle“visiting” vesicle
C
Num
ber o
f fus
ion
even
ts/c
ell
MS SS0
10
20
30
40
Num
ber o
f fus
ion
even
ts/c
ell
0
10
20
30
40
MS SS
D
Resident Non-resident
0 30 600
10
20
30
40
50
60
70
80 NH4Cl
20
0
40
60
80
100
MS Resident MS Non-resident SS Resident SS Non-resident %
tota
l eac
h fu
sion
eve
nts
0 30 60 90 180120 150time (sec)
00
5
10
15
20
25
MS Resident SS Resident MS Non-resident SS Non-sesident
Num
ber o
f fus
ion
even
ts/c
ell
% to
tal e
ach
fusi
on e
vent
s at
60
sec
0 20 40 60 80
100
Resident Non-resident
MS SS
n. s.*
0 30 60 90 120 150 180time (sec)
E F G
0
10
203040
50
60
70
80
FIGURE 3. Vesicle exocytosis shifts from resident to non-resident vesicles under SS conditions. A, montages showing resident (top) and non-resident(bottom) vesicle exocytic events imaged at 4 Hz by TIRF microscopy. BDNF-EGFP-containing vesicles brighten at the point of fusion pore formation (termed 0 s)due to pH change. The BDNF-EGFP subsequently dims due to fusion pore closure and vesicle re-acidification. Non-resident vesicles are not visible in the TIRFfield for at least 0.5 s prior to fusion. B, fluorescence changes in fusing and non-fusing vesicles. Left, average relative fluorescence of individual vesiclescorresponding to residents or non-residents that underwent exocytosis in response to stimulation (n � 10 events). Profiles are similar with non-residentsstarting at lower background values. An arrow indicates the average relative fluorescence change with 50 mM NH4Cl-induced vesicle pH neutralization (n � 10vesicles). Right, relative fluorescence of individual vesicles that failed to fuse in stimulated cells corresponding to vesicles that briefly (visiting) or stably (docked)entered the TIRF field. C and D, PC12 cells expressing BDNF-EGFP were incubated in MS (n � 9 cells) and SS (n � 11 cells) buffers for 5 min. Images of exocyticevents were acquired at 4 Hz by TIRF microscopy. Mean values S.E. (error bars) (n � 9 –11 cells) are plotted. C, the total number of exocytic events percell elicited by MS or SS buffers is shown. D, exocytic events were counted and categorized into resident and non-resident events. E, time course of each typeof exocytic event. The numbers of each type of exocytic event (same data set as in Fig. 1A) under MS and SS conditions are presented as a cumulative plot. F andG, cumulative distribution of each type of exocytic event (F). Within 60 s, 88, 94, 83, and 94% of MS-resident, MS-non-resident, SS-resident, and SS-non-residenttypes of exocytic events occurred, respectively (G) (*, p � 0.05; n.s., not significant). A.U., arbitrary units.
Phospholipase C�2 Activation in Ca2�-regulated Exocytosis
NOVEMBER 27, 2015 • VOLUME 290 • NUMBER 48 JOURNAL OF BIOLOGICAL CHEMISTRY 29015
by guest on February 18, 2018http://w
ww
.jbc.org/D
ownloaded from

shift from resident to non-resident vesicle exocytosis under SSconditions failed to occur (Fig. 4C). Similar results wereobtained in PLC�2 siRNA knockdown cells (Fig. 4D). Theseresults establish that PLC�2 activation is responsible for theshift in the exocytic pathway from resident to non-resident ves-icles promoted by greater elevations in Ca2�.
F-actin Disassembly Is Necessary for the Shift in the ExocyticPathway—Cortical F-actin acts as a barrier or cage to restrictvesicle access to the plasma membrane for fusion (25, 26).Because PI(4,5)P2 regulates the actin cytoskeleton during vesi-cle exocytosis (45, 46), we determined whether PLC�2-cata-lyzed PI(4,5)P2 hydrolysis caused disassembly of F-actin, whichwould increase the access of non-resident vesicles to the plasma
membrane. The assembled state of cortical F-actin was assessedby fluorescent phalloidin staining of cells viewed by TIRFmicroscopy. The assembled state of cortical F-actin did notsignificantly change after 0.5, 1, 2, or 3 min after application ofMS buffer. By contrast, in SS buffer, cortical F-actin stronglydecreased by 0.5 min (�42%) and 1 min (�62%) (Fig. 5, A and B)but reassembled by 2–3 min (Fig. 5B). The F-actin dynamicscorrelated with the dynamics of PI(4,5)P2 hydrolysis/DAG gen-eration (Fig. 1, C–E). Moreover, non-resident vesicle fusionevents were completed within this period (Fig. 3, E–G). Thedata suggest that PI(4,5)P2 hydrolysis-dependent cortical F-ac-tin disassembly might enable non-resident vesicle fusion eventsunder SS condition.
Pharmacological treatments were used to determinewhether F-actin disassembly was necessary or sufficient to shiftthe exocytic pathway from resident to non-resident vesicles.Treatment with jasplakinolide, an F-actin-stabilizing drug,inhibited fusion events under both MS and SS conditions (Fig.5C, bar 1 versus bar 3 and bar 2 versus bar 4). In control cells(�Jasp), the number of exocytic events from resident vesicleswas reduced, and that from non-resident vesicles was increasedunder SS conditions (Fig. 5D, bars 1– 4). Treatment with jas-plakinolide (�Jasp) mainly suppressed exocytic events fromnon-resident vesicles, especially under SS conditions (Fig. 5D,bars 5– 8). These data suggested that stabilization of F-actin byjasplakinolide preferentially affected the exocytosis of non-res-ident vesicles, thereby preventing an increase under SS condi-tions. Conversely, we promoted F-actin disassembly by treat-ment with halichondramide (Hali), an actin filament-severingand capping drug (47). Under MS conditions, halichondramidetreatment increased the number of non-resident vesicle exo-cytic events without affecting the number of resident events(Fig. 5E). Halichondramide treatment mimicked the effect of SSconditions in enhancing non-resident exocytic events underMS condition. These data indicated that F-actin disassemblywas sufficient to enhance the exocytosis of non-resident vesi-cles. Overall, the opposite effects of jasplakinolide andhalichondramide treatment were consistent with a role forF-actin disassembly in enabling the fusion of non-residentvesicles.
PLC�2 Regulates F-actin Disassembly—The previous resultsindicated that PLC�2 activation shifts exocytosis from residentto non-resident vesicles and that F-actin disassembly was inpart responsible for the shift. To determine the relationship ofPLC�2 to cortical F-actin, co-localization studies were con-ducted. TIRF microscopy revealed that EGFP-PLC�2 wasdistributed in punctate and in filamentous structures, withthe latter co-localizing with F-actin (Fig. 6, A and B). BecausePLC�2 was reported to localize to the plasma membrane bybinding to PI(4,5)P2 via its PH domain (41, 48), the observedfilamentous distribution of EGFP-PLC�2 (Fig. 6A) couldresult from binding to PI(4,5)P2-rich membrane domainsthat co-localize along F-actin filaments. Alternatively, thefilamentous distribution might correspond to direct bindingof EGFP-PLC�2 to F-actin or to actin-binding proteins. Todistinguish these alternatives, we determined the localiza-tion of EGFP-PLC�2 following treatment with latrunculin Ato disassemble F-actin. Latrunculin A treatment effectively
0
10
20
30
40
50
60
MS SS MS SSCtrl
shRNAPLCη2 shRNA
# of
fusi
on e
vent
s / c
ell
n. s.
n. s.
B
ResidentNon-resident
0
10
20
30
40
50
60
MS SS MS SSCtrl shRNA PLCη2 shRNA
# of
fusi
on e
vent
s / c
ell
n. s.
n. s.
***
C
A
Ctrl sh
RNA
PLCη2
shRNA
n. s.
Vesi
cles
/ m
m2
0
1
2
D
ResidentNon-resident
# of
fusi
on e
vent
s / c
ell
# of
fusi
on e
vent
s / c
ell
MS SS MS SSCtrl siRNA PLCη2 siRNA
*n. s.
n. s. n. s.
0
5
10
15
20
25
30
FIGURE 4. Knockdown of PLC�2 affects the shift in exocytosis from resi-dent to non-resident vesicles. Cells were co-transfected with BDNF-EGFPand shRNA plasmid targeting PLC�2 (n � 12 cells) or empty vector (n � 10cells). A, bar graphs show the average density of vesicles in the TIRF field forcontrol and PLC�2 knockdown cells (mean S.E. (error bars)). B and D, PLC�2knockdown or control cells were incubated in MS (n � 7– 8 cells) and SS (n �7– 8 cells) buffers. The total exocytic events for 5 min were counted (B), cate-gorized into resident and non-resident events, and expressed as number/cell(C). D, PLC�2 knockdown by siRNA or control cells were incubated in MS (n �8 control cells, n � 13 KD cells) and SS (n � 8 control cells, n � 9 KD cells)buffers. The total exocytic events for 5 min were counted and categorizedinto resident and non-resident events and expressed as number/cell (mean S.E.; *, p � 0.05; **, p � 0.001; n.s., not significant).
Phospholipase C�2 Activation in Ca2�-regulated Exocytosis
29016 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 290 • NUMBER 48 • NOVEMBER 27, 2015
by guest on February 18, 2018http://w
ww
.jbc.org/D
ownloaded from

disassembled F-actin (Fig. 6C) and eliminated the filamen-tous distribution of EGFP-PLC�2 (Fig. 6D) detected by TIRFmicroscopy. Latrunculin treatment produced small struc-tures and puncta of EGFP-PLC�2 with some nearly residualactin-containing structures (Fig. 6D, arrows). By contrast, thePH domain of PLC�2 (EGFP-PLC�2-PH), which localized tothe plasma membrane similarly to full-length EGFP-PLC�2 byepifluorescence (Fig. 6E), localized rather differently intobroadly distributed small puncta rather than filaments by TIRFmicroscopy, and this distribution was not altered by latrunculinA treatment (Fig. 6F). The results suggest that the filamentousdistribution of EGFP-PLC�2 near the plasma membrane ismediated by a direct interaction with F-actin or actin-bindingproteins utilizing a domain of PLC�2 other than its PH domain.
To further link the activation of PLC�2 to the state ofF-actin assembly, we determined the effect of PLC overex-pression on F-actin disassembly. In control EGFP-express-ing cells, cortical F-actin visualized by fluorescent phalloidinwas disassembled when cells were incubated under SS butnot MS conditions (Fig. 7, A and D). Cells expressing EGFP-PLC�1 exhibited changes very similar to control cells (Fig. 7,B and D). By contrast, overexpression of EGFP-PLC�2resulted in the disassembly of cortical F-actin even under MSconditions (Fig. 7, C and D), which was similar to the resultsfor DAG generation (Fig. 2, A and B). Last, to confirm thatPLC�2 activation affects F-actin disassembly, we deter-mined the impact of PLC�2 knockdown. In control cells,cortical F-actin was disassembled under SS but not MS con-ditions (Fig. 7, E and F). By contrast, neither MS nor SSconditions induced cortical F-actin disassembly in PLC�2
knockdown cells (Fig. 7, E and F). The results indicate thatPLC�2 activation promotes F-actin disassembly.
Discussion
Studies in many cell types have described the important roleof PI(4,5)P2 in enabling F-actin assembly (24). In addition, mul-tiple roles for F-actin in vesicle trafficking and exocytosis havebeen characterized for neuroendocrine cells (49). It has beenshown that increases in cytoplasmic Ca2� trigger F-actin disas-sembly (25, 28, 49 –52), which increases the access of cytoplas-mic recruitment vesicles to the plasma membrane for fusion(33, 34). The current study reveals that PLC�2 is a critical linkbetween Ca2� rises and the disassembly of the F-actin cytoskel-eton for regulating vesicle trafficking to the plasma membranefor fusion.
PLC�2 is mainly expressed in neural and endocrine secretorycells, but a functional cellular role for the enzyme has not pre-viously been characterized (41, 53, 54). The strong Ca2� depen-dence of the activation of PLC�2 suggested that it was a Ca2�-dependent effector for unidentified neural/endocrine pro-cesses (41). G�� subunits also activate PLC�2, which couldindicate receptor-regulated roles for this enzyme as well (55).However, PLC�2 knock-out mice did not exhibit obvious phe-notypes that would suggest a functional role (56). By contrast,the loss of PLC�2 in PC12 cells attenuated the evoked fusion ofvesicles recruited to the plasma membrane under enhancedCa2� influx conditions. PLC�2 was activated at cytoplasmicCa2� levels (�800 nM) greater than those required to elicitmaximal vesicle exocytosis (�400 nM) in PC12 cells, which cor-responds closely to the observed in vitro Ca2�-dependent acti-
A B
Rel
ativ
e flu
ores
cenc
e (F
t / F
0)
DCResidentNon-resident
# of
fusi
on e
vent
s / c
ell
0
10
20
30
-Jasp +JaspMS SS MS SSMS SS MS SSSMS SS MS SS
40
MS SS MS SS
# of
fusi
on e
vent
s / c
ell
0
10
20
30
40
-Jasp +JaspCtrl Hali
n. s.
n. s.
*
*
# of
fusi
on e
vent
s / c
ell
5 μm
MS
SS
0 min 0.5 min 1 min 2 min 3 min
()
0
0.5
1
1.5
0 0.5 1 2 3 0 0.5 1 2 3 (min)
MS SSn.s.
**
n.s.
ResidentNon-resident
E
0
10
20
30
FIGURE 5. Stabilization of F-actin prevents whereas drug-induced F-actin disassembly promotes a shift from resident to non-resident vesicle exocy-tosis. A, effect of incubation buffers on cortical F-actin. PC12 cells were incubated in control or MS or SS buffers for 0.5, 1, 2, and 3 min; fixed; permeabilized; andstained with Alexa Fluor 568 phalloidin for imaging by TIRF microscopy. B, the mean fluorescence intensity S.E. (error bars) of cell footprints was determined(n � 15–20 cells; *, p � 0.0001; n.s., not significant). C and D, effect of jasplakinolide (Jasp) treatment on resident and non-resident vesicle exocytic events.BDNF-EGFP-expressing cells were treated with 1 �M jasplakinolide at 37 °C for 30 min and incubated in MS or SS buffers for 5 min. The total exocytic events werecounted (C), categorized into resident and non-resident events, and plotted as number/cell (D) (mean S.E.; n � 10 –12 cells; *, p � 0.005). E, effect ofhalichondramide (Hali) treatment on the evoked exocytosis of resident and non-resident vesicles. The fusion events were counted and categorized intoresident and non-resident events. (mean S.E., n � 6 –15 cells).
Phospholipase C�2 Activation in Ca2�-regulated Exocytosis
NOVEMBER 27, 2015 • VOLUME 290 • NUMBER 48 JOURNAL OF BIOLOGICAL CHEMISTRY 29017
by guest on February 18, 2018http://w
ww
.jbc.org/D
ownloaded from

vation of PLC�2 but not PLC�1 (41). The hydrolysis ofPI(4,5)P2 with DAG generation, F-actin disassembly, and theincreased exocytosis of non-resident vesicles was also only evi-dent at the higher Ca2� concentrations. Extrapolation of theseresults into the nervous system suggests that loss of PLC�2could slow neuropeptide secretion under conditions of highdemand (e.g. sustained or high frequency stimulation), wherehigher Ca2� levels are attained. A phenotype in the PLC�2knock-out mouse (56) may only be evident under such condi-tions of stress. The current study indicates a functional role forPLC�2 as a Ca2�-dependent effector that regulates vesicle traf-ficking through its hydrolysis of PI(4,5)P2 and consequentremodeling of the actin cytoskeleton. The unanticipated local-ization of PLC�2 to F-actin supports such a role.
Compared with MS conditions, SS conditions uniquely acti-vated PLC�2, PI(4,5)P2 hydrolysis, and F-actin disassembly andpromoted a shift in the exocytic pathway toward new arrivingvesicles without markedly altering the total number of vesicle
exocytic events. PLC�2 activation both inhibited the exocytosisof resident vesicles and facilitated the exocytosis of vesicles traf-ficking to the plasma membrane during stimulation, as shownby PLC�2 knockdown. PLC�2 activation was also responsiblefor the F-actin disassembly promoted under SS conditions.However, unlike the effects of F-actin disassembly promoted byPLC�2 activation, halichondramide treatment mainly affectedthe fusion of non-resident vesicles without affecting the fusionof resident vesicles. The partial inhibition of resident vesiclefusion from PI(4,5)P2 hydrolysis might result from the inhibi-tion of the local F-actin assembly, mediated by actin-associatedproteins such as N-WASP and Arp2/3, that enhances vesicleexocytosis (57, 58). Because halichondramide causes F-actindisassembly without PI(4,5)P2 hydrolysis, it is possible thatPI(4,5)P2-dependent F-actin assembly still occurs to supportresident vesicle fusion.
The facilitation of the exocytosis of newly recruited vesiclesby PI(4,5)P2 hydrolysis is readily understood as a consequence
0 10 20 30 40 50 60 70 80
Co-
loca
lizat
ion
(%)
F-actin Merge
Act
in
DMSO Lat A
DMSOLat A
DMSO Lat A
C
D
F
A B
F-actin
with
PLCη2
/ tota
l F-ac
tin
PLCη2
with
F-actin
/ total
PLCη2
EGFP-PLCη2 EGFP-PLCη2-PHE
EGFP-PLCη2
EGFP-PLCη2E
GFP
-PLC
η2-P
H
5 μm
5 μm5 μm
5 μm
5 μm
0.5 μm
EGFP-PLCη M2 ergeActin
FIGURE 6. Co-localization of EGFP-PLC�2 and F-actin in PC12 cells. A, EGFP-PLC�2-expressing cells were fixed, permeabilized, and incubated with AlexaFluor 568 phalloidin for F-actin imaging by TIRF microscopy. Insets show portions of the image (boxed) at higher magnification. B, the co-localization ofEGFP-PLC�2 and phalloidin-stained F-actin was quantitated as described under “Experimental Procedures” (mean S.E. (error bars), n � 12 cells). C, effect oflatrunculin A (Lat A) on cortical F-actin. Cells were incubated with DMSO or with 1 �M latrunculin A at 37 °C for 5 min and fixed, permeabilized, and stained withAlexa Fluor 568 phalloidin for imaging by TIRF microscopy. D, effect of latrunculin A treatment on localization of EGFP-PLC�2. EGFP-PLC�2-expressing cellswere incubated with DMSO or with 1 �M latrunculin A at 37 °C for 5 min and imaged by TIRF microscopy. In latrunculin A-treated cells, EGFP-PLC�2 distributedto puncta and small structures that were near residual F-actin-containing structures stained with Alexa Fluor 568 phalloidin (arrows). E, the localization ofEGFP-PLC�2 and EGFP-PLC�2-PH by epifluorescence microscopy. F, effect of latrunculin A on localization of EGFP-PLC�2-PH domain. EGFP-PLC�2-PH-expressing cells were incubated with DMSO or with 1 �M latrunculin A at 37 °C for 5 min and imaged by TIRF microscopy.
Phospholipase C�2 Activation in Ca2�-regulated Exocytosis
29018 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 290 • NUMBER 48 • NOVEMBER 27, 2015
by guest on February 18, 2018http://w
ww
.jbc.org/D
ownloaded from

of the disassembly of a cortical F-actin meshwork that hindersvesicle access to the plasma membrane. This is consistent withthe inhibition and stimulation of non-resident vesicle exocy-tosis by jasplakinolide and halichondramide, respectively.PI(4,5)P2 regulates numerous actin-binding proteins, such asscinderin (also known as adseverin), gelsolin, profilin, villin,and cofilin (24). Cofilin and scinderin are sequestered byPI(4,5)P2 and released upon PI(4,5)P2 hydrolysis to sever F-ac-tin filaments (31, 45). Scinderin in chromaffin cells has beenimplicated in the Ca2�-induced disassembly of F-actin (27, 52).The Ca2�-dependent activation of PLC�2 and PI(4,5)P2 hydro-lysis would release bound scinderin and related proteins to pro-mote their actin-severing activity. Although PKC activationwas also reported to promote cortical F-actin disassembly inchromaffin cells (59), we found that overexpression of a phos-phoinositide 5-phosphatase that hydrolyzes PI(4,5)P2 withoutDAG generation closely mimicked the impact of PLC�2 over-expression on vesicle exocytosis (data not shown), which sug-gests that it is the loss of PI(4,5)P2, rather than an increase inDAG that is principally responsible for the cytoskeletal remod-eling and changes in vesicle trafficking in PC12 cells. It was alsorecently reported that PI(4,5)P2-dependent F-actin remodeling
was required for vesicle translocation to the plasma membranein chromaffin cells (60).
PI(4,5)P2 is required for multiple steps in vesicle exocytosis,which suggests additional roles for PLC�2 as a Ca2�-dependentmodulator. The priming factors CAPS and ubMunc13-2 arerequired for the regulated fusion of both resident and newlyarrived vesicles (36). Studies in PC12 cells showed that the par-tial hydrolysis of PI(4,5)P2 by PLC�2 at elevated Ca2� levels wassufficient to reduce the PI(4,5)P2-dependent activity of CAPSwhile maintaining the PI(4,5)P2-dependent membrane recruit-ment of Munc13 (9). Membrane-recruited Munc13 was furtheractivated by DAG to compensate for the loss of CAPS activity(9). DAG production at elevated Ca2� has been inferred to pro-mote augmentation of synaptic neurotransmitter release medi-ated by Munc13-2 (20), but the PLC responsible for DAG gen-eration had not been identified. Our results suggest that PLC�2may mediate synaptic augmentation promoted by elevatedCa2�. The extensive role of PI(4,5)P2 in regulating plasmamembrane-associated processes indicates that PLC�2 mayhave other functions in response to sustained Ca2� elevationsthat are linked to the actin cytoskeleton, such as endocytosis,cytokinesis, and neurite outgrowth. The current work charac-
Act
inA
ctin
A
Ctrl
KD
MS SSE
F
Ctrl KD
Rel
ativ
e flu
ores
cenc
e
MS SS
Act
inE
GFP
B
C
0
0.5
1.0
1.5
2.0
n. s. n. s.
*
none
MS
SS
none
none
D
0
0.5
1.0
1.5
none
MS
SS
Rel
ativ
e flu
ores
cenc
e
EGFPEGFP-PLCδ1
EGFP-PLCη2
n. s.* n. s.**
**
**
EG
FP-
PLC
δ1E
GFP
-P
LCη2
PLC
η2 K
D
PLCη2 KD5 μm
5 μm
FIGURE 7. Overexpression and knockdown of PLC�2 affect F-actin disassembly in PC12 cells. A–C, effect of PLC overexpression on cortical F-actindisassembly under MS and SS conditions. PC12 cells expressing EGFP (A), EGFP-PLC�1 (B), and EGFP-PLC�2 (C) were incubated in MS and SS buffer for 1 min,fixed, permeabilized, and incubated with Alexa Fluor 568 phalloidin. EGFP proteins were imaged by epifluorescence (top panels) and phalloidin (bottom panels)by TIRF microscopy. D, the mean fluorescence intensity S.E. (error bars) of Alexa Fluor 568 phalloidin in a footprint was determined (n � 6 –12 cells; *, p � 0.01;**, p � 0.0001). E, effect of PLC�2 knockdown on cortical F-actin disassembly. Control or PLC�2 knockdown cells were incubated in control, MS, or SS buffersfor 1 min and processed for Alexa Fluor 568 phalloidin-binding of F-actin imaged by TIRF microscopy. F, mean fluorescence intensity S.E. of phalloidin in cellfootprints (n � 8 –10 cells; *, p � 0.0005; n.s., not significant).
Phospholipase C�2 Activation in Ca2�-regulated Exocytosis
NOVEMBER 27, 2015 • VOLUME 290 • NUMBER 48 JOURNAL OF BIOLOGICAL CHEMISTRY 29019
by guest on February 18, 2018http://w
ww
.jbc.org/D
ownloaded from

terizes one of the roles of PLC�2 as a Ca2�-dependent regulatorof the actin cytoskeleton and secretory pathway in neuroendo-crine cells.
Author Contributions—M. Y. and T. F. J. M. conceived the study andwrote the manuscript. M. Y. and D. M. K.-G. performed the experi-ments, analyzed the data, and prepared the figures.
Acknowledgments—We gratefully acknowledge S. Grinstein (Hospitalfor Sick Children, Toronto, Canada) for providing the PKC�-C1-EGFP construct and K. Fukami (Tokyo University of Pharmacy andLife Science, Tokyo, Japan) for providing EGFP-PLC�2, EGFP-PLC�1constructs, and anti-PLC�2 antibody.
References1. Malsam, J., Kreye, S., Sollner, T. H., Lang, T., and Jahn, R. (2008) Mem-
brane fusion: SNAREs and regulation. Cell Mol. Life Sci. 65, 2814 –28322. Jahn, R., and Fasshauer, D. (2012) Molecular machines governing exocy-
tosis of synaptic vesicles. Nature 490, 201–2073. Hay, J. C., Fisette, P. L., Jenkins, G. H., Fukami, K., Takenawa, T., Ander-
son, R. A., and Martin, T. F. (1995) ATP-dependent inositide phosphory-lation required for Ca2�-activated secretion. Nature 374, 173–177
4. Hay, J. C., and Martin, T. F. (1993) Phosphatidylinositol transfer proteinrequired for ATP-dependent priming of Ca2�-activated secretion. Nature366, 572–575
5. Eberhard, D. A., Cooper, C. L., Low, M. G., and Holz, R. W. (1990) Evi-dence that the inositol phospholipids are necessary for exocytosis: loss ofinositol phospholipids and inhibition of secretion in permeabilized cellscaused by a bacterial phospholipase C and removal of ATP. Biochem. J.268, 15–25
6. Grishanin, R. N., Kowalchyk, J. A., Klenchin, V. A., Ann, K., Earles, C. A.,Chapman, E. R., Gerona, R. R., and Martin, T. F. (2004) CAPS acts at aprefusion step in dense-core vesicle exocytosis as a PIP2 binding protein.Neuron 43, 551–562
7. Loyet, K. M., Kowalchyk, J. A., Chaudhary, A., Chen, J., Prestwich, G. D.,and Martin, T. F. (1998) Specific binding of phosphatidylinositol 4,5-bis-phosphate to calcium-dependent activator protein for secretion (CAPS), apotential phosphoinositide effector protein for regulated exocytosis.J. Biol. Chem. 273, 8337– 8343
8. James, D. J., Khodthong, C., Kowalchyk, J. A., and Martin, T. F. (2008)Phosphatidylinositol 4,5-bisphosphate regulates SNARE-dependentmembrane fusion. J. Cell Biol. 182, 355–366
9. Kabachinski, G., Yamaga, M., Kielar-Grevstad, D. M., Bruinsma, S., andMartin, T. F. (2014) CAPS and Munc13 utilize distinct PIP2-linked mech-anisms to promote vesicle exocytosis. Mol. Biol. Cell 25, 508 –521
10. Shin, O. H., Lu, J., Rhee, J. S., Tomchick, D. R., Pang, Z. P., Wojcik, S. M.,Camacho-Perez, M., Brose, N., Machius, M., Rizo, J., Rosenmund, C., andSudhof, T. C. (2010) Munc13 C2B domain is an activity-dependent Ca2�
regulator of synaptic exocytosis. Nat. Struct. Mol. Biol. 17, 280 –28811. Chung, S. H., Song, W. J., Kim, K., Bednarski, J. J., Chen, J., Prestwich,
G. D., and Holz, R. W. (1998) The C2 domains of Rabphilin3A specificallybind phosphatidylinositol 4,5-bisphosphate containing vesicles in a Ca2�-dependent manner: in vitro characteristics and possible significance.J. Biol. Chem. 273, 10240 –10248
12. Schiavo, G., Gu, Q. M., Prestwich, G. D., Sollner, T. H., and Rothman, J. E.(1996) Calcium-dependent switching of the specificity of phosphoinosit-ide binding to synaptotagmin. Proc. Natl. Acad. Sci. U.S.A. 93,13327–13332
13. Tucker, W. C., Edwardson, J. M., Bai, J., Kim, H. J., Martin, T. F., andChapman, E. R. (2003) Identification of synaptotagmin effectors via acuteinhibition of secretion from cracked PC12 cells. J. Cell Biol. 162, 199 –209
14. van den Bogaart, G., Meyenberg, K., Risselada, H. J., Amin, H., Willig, K. I.,Hubrich, B. E., Dier, M., Hell, S. W., Grubmuller, H., Diederichsen, U., andJahn, R. (2011) Membrane protein sequestering by ionic protein-lipid in-teractions. Nature 479, 552–555
15. Tse, F. W., Tse, A., Hille, B., Horstmann, H., and Almers, W. (1997) LocalCa2� release from internal stores controls exocytosis in pituitary gonado-trophs. Neuron 18, 121–132
16. Bauer, C. S., Woolley, R. J., Teschemacher, A. G., and Seward, E. P. (2007)Potentiation of exocytosis by phospholipase C-coupled G-protein-cou-pled receptors requires the priming protein Munc13-1. J. Neurosci. 27,212–219
17. Malenka, R. C., Madison, D. V., and Nicoll, R. A. (1986) Potentiation ofsynaptic transmission in the hippocampus by phorbol esters. Nature 321,175–177
18. Milosevic, I., Sørensen, J. B., Lang, T., Krauss, M., Nagy, G., Haucke, V.,Jahn, R., and Neher, E. (2005) Plasmalemmal phosphatidylinositol-4,5-bisphosphate level regulates the releasable vesicle pool size in chromaffincells. J. Neurosci. 25, 2557–2565
19. Aoyagi, K., Sugaya, T., Umeda, M., Yamamoto, S., Terakawa, S., and Taka-hashi, M. (2005) The activation of exocytotic sites by the formation ofphosphatidylinositol 4,5-bisphosphate microdomains at syntaxin clusters.J. Biol. Chem. 280, 17346 –17352
20. Rosenmund, C., Sigler, A., Augustin, I., Reim, K., Brose, N., and Rhee, J. S.(2002) Differential control of vesicle priming and short-term plasticity byMunc13 isoforms. Neuron 33, 411– 424
21. Fukami, K., Inanobe, S., Kanemaru, K., and Nakamura, Y. (2010) Phospho-lipase C is a key enzyme regulating intracellular calcium and modulatingthe phosphoinositide balance. Prog. Lipid Res. 49, 429 – 437
22. Janmey, P. A., and Lindberg, U. (2004) Cytoskeletal regulation: rich inlipids. Nat. Rev. Mol. Cell Biol. 5, 658 – 666
23. Mao, Y. S., and Yin, H. L. (2007) Regulation of the actin cytoskeleton byphosphatidylinositol 4-phosphate 5 kinases. Pflugers Arch. 455, 5–18
24. Saarikangas, J., Zhao, H., and Lappalainen, P. (2010) Regulation of theactin cytoskeleton-plasma membrane interplay by phosphoinositides.Physiol. Rev. 90, 259 –289
25. Aunis, D., and Bader, M. F. (1988) The cytoskeleton as a barrier to exocy-tosis in secretory cells. J. Exp. Biol. 139, 253–266
26. Orci, L., Gabbay, K. H., and Malaisse, W. J. (1972) Pancreatic �-cell web:its possible role in insulin secretion. Science 175, 1128 –1130
27. Trifaró, J., Gasman, S., and Gutiérrez, L. (2008) Cytoskeletal control ofvesicle transport and exocytosis in chromaffin cells. Acta Physiol. (Oxf.)192, 165–172
28. Vitale, M. L., Rodrıguez Del Castillo, A., Tchakarov, L., and Trifaro, J. M.(1991) Cortical filamentous actin disassembly and scinderin redistribu-tion during chromaffin cell stimulation precede exocytosis, a phenome-non not exhibited by gelsolin. J. Cell Biol. 113, 1057–1067
29. Sakisaka, T., Itoh, T., Miura, K., and Takenawa, T. (1997) Phosphatidyli-nositol 4,5-bisphosphate phosphatase regulates the rearrangement of ac-tin filaments. Mol. Cell. Biol. 17, 3841–3849
30. Cremona, O., and De Camilli, P. (2001) Phosphoinositides in membranetraffic at the synapse. J. Cell Sci. 114, 1041–1052
31. van Rheenen, J., Song, X., van Roosmalen, W., Cammer, M., Chen, X.,Desmarais, V., Yip, S. C., Backer, J. M., Eddy, R. J., and Condeelis, J. S.(2007) EGF-induced PIP2 hydrolysis releases and activates cofilin locallyin carcinoma cells. J. Cell Biol. 179, 1247–1259
32. Raucher, D., Stauffer, T., Chen, W., Shen, K., Guo, S., York, J. D., Sheetz,M. P., and Meyer, T. (2000) Phosphatidylinositol 4,5-bisphosphate func-tions as a second messenger that regulates cytoskeleton-plasma mem-brane adhesion. Cell 100, 221–228
33. Oheim, M., and Stuhmer, W. (2000) Interaction of secretory organelleswith the membrane. J. Membr. Biol. 178, 163–173
34. Lang, T., Wacker, I., Wunderlich, I., Rohrbach, A., Giese, G., Soldati, T.,and Almers, W. (2000) Role of actin cortex in the subplasmalemmal trans-port of secretory granules in PC-12 cells. Biophys. J. 78, 2863–2877
35. Lynch, K. L., Gerona, R. R., Kielar, D. M., Martens, S., McMahon, H. T.,and Martin, T. F. (2008) Synaptotagmin-1 utilizes membrane bending andSNARE binding to drive fusion pore expansion. Mol. Biol. Cell 19,5093–5103
36. Khodthong, C., Kabachinski, G., James, D. J., and Martin, T. F. (2011)Munc13 homology domain-1 in CAPS/UNC31 mediates SNARE bindingrequired for priming vesicle exocytosis. Cell Metab. 14, 254 –263
37. Botelho, R. J., Teruel, M., Dierckman, R., Anderson, R., Wells, A., York,
Phospholipase C�2 Activation in Ca2�-regulated Exocytosis
29020 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 290 • NUMBER 48 • NOVEMBER 27, 2015
by guest on February 18, 2018http://w
ww
.jbc.org/D
ownloaded from

J. D., Meyer, T., and Grinstein, S. (2000) Localized biphasic changes inphosphatidylinositol-4,5-bisphosphate at sites of phagocytosis. J. Cell Biol.151, 1353–1368
38. Wang, C. T., Grishanin, R., Earles, C. A., Chang, P. Y., Martin, T. F.,Chapman, E. R., and Jackson, M. B. (2001) Synaptotagmin modulation offusion pore kinetics in regulated exocytosis of dense-core vesicles. Science294, 1111–1115
39. Reber, B. F., and Reuter, H. (1991) Dependence of cytosolic calcium indifferentiating rat pheochromocytoma cells on calcium channels and in-tracellular stores. J. Physiol. 435, 145–162
40. Kim, Y. H., Park, T. J., Lee, Y. H., Baek, K. J., Suh, P. G., Ryu, S. H., and Kim,K. T. (1999) Phospholipase C-�1 is activated by capacitative calcium entrythat follows phospholipase C-� activation upon bradykinin stimulation.The J. Biol. Chem. 274, 26127–26134
41. Nakahara, M., Shimozawa, M., Nakamura, Y., Irino, Y., Morita, M., Kudo,Y., and Fukami, K. (2005) A novel phospholipase C, PLC�2, is a neuron-specific isozyme. J. Biol. Chem. 280, 29128 –29134
42. Olsen, H. L., Hoy, M., Zhang, W., Bertorello, A. M., Bokvist, K., Capito, K.,Efanov, A. M., Meister, B., Thams, P., Yang, S. N., Rorsman, P., Berggren,P. O., and Gromada, J. (2003) Phosphatidylinositol 4-kinase serves as ametabolic sensor and regulates priming of secretory granules in pancreatic� cells. Proc. Natl. Acad. Sci. U.S.A. 100, 5187–5192
43. Bai, L., Zhu, D., Zhou, K., Zhou, W., Li, D., Wang, Y., Zhang, R., and Xu, T.(2006) Differential properties of GTP- and Ca2�-stimulated exocytosisfrom large dense core vesicles. Traffic 7, 416 – 428
44. Aikawa, Y., and Martin, T. F. (2003) ARF6 regulates a plasma membranepool of phosphatidylinositol(4,5)bisphosphate required for regulated ex-ocytosis. J. Cell Biol. 162, 647– 659
45. Rodrıguez Del Castillo, A., Vitale, M. L., and Trifaro, J. M. (1992) Ca2� andpH determine the interaction of chromaffin cell scinderin with phosphati-dylserine and phosphatidylinositol 4,5-biphosphate and its cellular distri-bution during nicotinic-receptor stimulation and protein kinase C activa-tion. J. Cell Biol. 119, 797– 810
46. Bittner, M. A., and Holz, R. W. (2005) Phosphatidylinositol-4,5-bisphos-phate: actin dynamics and the regulation of ATP-dependent and -inde-pendent secretion. Mol. Pharmacol. 67, 1089 –1098
47. Chung, S. C., Lee, S. H., Jang, K. H., Park, W., Jeon, J. E., Oh, H., Shin, J., andOh, K. B. (2011) Actin depolymerizing effect of trisoxazole-containingmacrolides. Bioorg. Med. Chem. Lett. 21, 3198 –3201
48. Popovics, P., Gray, A., Arastoo, M., Finelli, D. K., Tan, A. J., and Stewart,A. J. (2013) Phospholipase C-�2 is required for retinoic acid-stimulatedneurite growth. J. Neurochem. 124, 632– 644
49. Malacombe, M., Bader, M. F., and Gasman, S. (2006) Exocytosis in neu-roendocrine cells: new tasks for actin. Biochim. Biophys. Acta 1763,1175–1183
50. Bader, M. F., Doussau, F., Chasserot-Golaz, S., Vitale, N., and Gasman, S.(2004) Coupling actin and membrane dynamics during calcium-regulatedexocytosis: a role for Rho and ARF GTPases. Biochim. Biophys. Acta 1742,37– 49
51. Chowdhury, H. H., Popoff, M. R., and Zorec, R. (2000) Actin cytoskeletonand exocytosis in rat melanotrophs. Pflugers Arch. 439, R148 –R149
52. Dumitrescu Pene, T., Rose, S. D., Lejen, T., Marcu, M. G., and Trifaro, J. M.(2005) Expression of various scinderin domains in chromaffin cells indi-cates that this protein acts as a molecular switch in the control of actinfilament dynamics and exocytosis. J. Neurochem. 92, 780 –789
53. Zhou, Y., Wing, M. R., Sondek, J., and Harden, T. K. (2005) Molecularcloning and characterization of PLC-�2. Biochem. J. 391, 667– 676
54. Stewart, A. J., Morgan, K., Farquharson, C., and Millar, R. P. (2007) Phos-pholipase C-� enzymes as putative protein kinase C and Ca2� signallingcomponents in neuronal and neuroendocrine tissues. Neuroendocrinology86, 243–248
55. Zhou, Y., Sondek, J., and Harden, T. K. (2008) Activation of human phos-pholipase C-�2 by G��. Biochemistry 47, 4410 – 4417
56. Kanemaru, K., Nakahara, M., Nakamura, Y., Hashiguchi, Y., Kouchi, Z.,Yamaguchi, H., Oshima, N., Kiyonari, H., and Fukami, K. (2010) Phospho-lipase C-�2 is highly expressed in the habenula and retina. Gene Expr.Patterns 10, 119 –126
57. Rohatgi, R., Ma, L., Miki, H., Lopez, M., Kirchhausen, T., Takenawa, T.,and Kirschner, M. W. (1999) The interaction between N-WASP and theArp2/3 complex links Cdc42-dependent signals to actin assembly. Cell 97,221–231
58. Gasman, S., Chasserot-Golaz, S., Malacombe, M., Way, M., and Bader, M.(2004) Regulated exocytosis in neuroendocrine cells: a role for subplas-malemmal Cdc42/N-WASP-induced actin filaments. Mol. Biol. Cell 15,520 –531
59. Trifaro, J., Rose, S. D., Lejen, T., and Elzagallaai, A. (2000) Two pathwayscontrol chromaffin cell cortical F-actin dynamics during exocytosis.Biochimie 82, 339 –352
60. Wen, P. J., Osborne, S. L., Zanin, M., Low, P. C., Wang, H. T., Schoenwael-der, S. M., Jackson, S. P., Wedlich-Soldner, R., Vanhaesebroeck, B., Keat-ing, D. J., and Meunier, F. A. (2011) Phosphatidylinositol(4,5)bisphosphatecoordinates actin-mediated mobilization and translocation of secretoryvesicles to the plasma membrane of chromaffin cells. Nat. Commun. 2,491
Phospholipase C�2 Activation in Ca2�-regulated Exocytosis
NOVEMBER 27, 2015 • VOLUME 290 • NUMBER 48 JOURNAL OF BIOLOGICAL CHEMISTRY 29021
by guest on February 18, 2018http://w
ww
.jbc.org/D
ownloaded from

Masaki Yamaga, D. Michelle Kielar-Grevstad and Thomas F. J. Martin2 Activation Redirects Vesicle Trafficking by Regulating F-actinηPhospholipase C
doi: 10.1074/jbc.M115.658328 originally published online October 2, 20152015, 290:29010-29021.J. Biol. Chem.
10.1074/jbc.M115.658328Access the most updated version of this article at doi:
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
http://www.jbc.org/content/290/48/29010.full.html#ref-list-1
This article cites 60 references, 27 of which can be accessed free at
by guest on February 18, 2018http://w
ww
.jbc.org/D
ownloaded from