myelodysplastic syndromes: molecular pathogenesis and genomic changes
TRANSCRIPT

REVIEW ARTICLE
Myelodysplastic syndromes: molecular pathogenesisand genomic changes
Florian Nolte & Wolf-K. Hofmann
Received: 11 October 2007 /Accepted: 15 April 2008 /Published online: 31 May 2008# Springer-Verlag 2008
Abstract Myelodysplastic syndromes (MDS) are charac-terized by ineffective hematopoiesis presenting with pe-ripheral cytopenias in combination with a hyperplastic bonemarrow and an increased risk of evolution to acute myeloidleukemia. The classification systems such as the WHOclassification mainly rely on morphological criteria and aresupplemented by the International Prognostic ScoringSystem which takes cytogenetical changes into consider-ation when determining the prognosis of MDS but wideintra-subtype variations do exist. The pathomechanismscausing primary MDS require further work. Developmentand progression of MDS is suggested to be a multistepalteration to hematopoietic stem cells. Different molecularalterations have been described, affecting genes involved incell-cycle control, mitotic checkpoints, and growth factorreceptors. Secondary signal proteins and transcriptionfactors, which gives the cell a growth advantage over itsnormal counterpart, may be affected as well. The accumu-lation of such defects may finally cause the leukemictransformation of MDS.
Keywords Angiogenesis . Bone marrow stroma cells .
Cell cycle control . Deletion 20q . DNA-methylation .
Gene expression profiling . JAK2-mutations .
Mitochondrial DNA .Moleculargenetic changes .
Myelodysplastic syndrome . SNP-analysis . 5q-syndrome
Introduction
Myelodysplastic syndromes (MDS) are heterogeneousdiseases characterized by ineffective hematopoiesis and anincreased risk of evolution to acute myeloid leukemia(AML). MDS can occur in younger people, but it is mainlya disease of the elderly with a dramatic increase of theincidence in the decades above 60 years [1].
The underlying causes in the pathogenesis of MDSremain elusive. Analysis of cytogenetic abnormalities,G6PD isoenzymes, restriction-linked polymorphisms, andX-linked DNA polymorphisms of the androgen receptor(HUMARA) have shown that MDS is a clonal abnormalityof the hematopoietic stem cell characterized by defectivematuration and in advanced stages uncontrolled prolifera-tion. However, the defects of the hematopoietic stem cellsin MDS are not well characterized. Moreover, it has not yetbeen elucidated whether—beside myeloid cells—bonemarrow stromal cells (BMSC) and lymphoid cells arederived from the stem cell clone.
Knudson’s model of the ‘two hits’ provides the basis ofthe concept of a multistep pathogenesis in the developmentof MDS (Fig. 1). Lessons learned from hereditary cancersled to the perception that loss or inactivation of one allelerarely is sufficient to result in the development of tumors orexpansion of a malignant clone. More likely, loss of thecorresponding allele or additional alterations are necessaryfor the penetrance of clonal cells [2]. These alterations caninfluence expression of cell-cycle-related genes, transcrip-tion factors, and tumor suppressor genes. Particularly MDSin early stages with its relatively slow (but increasedtendency) to AML progression might be a prototype ofthe multistep concept in leukemogenesis with accumulationof cellular and molecular defects during the initiation anddisease progression.
Ann Hematol (2008) 87:777–795DOI 10.1007/s00277-008-0502-z
F. Nolte (*) :W.-K. HofmannDepartment of Hematology and Oncology,University Hospital Benjamin Franklin,Charité, Hindenburgdamm 30,12203 Berlin, Germanye-mail: [email protected]

A wide range of different aberrations have beendescribed in MDS, such as loss or multiplication of geneticmaterial and structural alterations. However, in only 50% ofMDS cases conventional cytogenetics can detect suchaberrations [3]. Epigenetic changes such as DNA hyper-methylation and histone deacetylation have been implicatedas potential mechanisms in the development and progres-sion of MDS, which already resulted in promisingtherapeutic approaches in a subset of patients.
The recently available microarray techniques are majoradvances in the molecular research of diseases. Applicationof single-nucleotide polymorphism (SNP) arrays offershigh-density screening of the whole genome providing theopportunity to identify cryptic lesions so far not detectableby conventional cytogenetics. Data obtained from SNParrays can be analyzed for deletions and amplifications andthe identification of loss of heterozygosity (LOH) is basedon a far higher resolution compared to conventional cyto-genetics. In addition, gene expression profiling by micro-arrays is a powerful new tool to assess the expression of alarge number of genes in a single experiment. In the future,array-based techniques will be useful tools in both furtherelucidation of the underlying defects in MDS and develop-
ment of more specific classifications of the diseases con-cerning the risk assessment of affected patients resulting—hopefully—in more targeted therapeutic approaches forthese patients.
Distinct chromosomal aberrations in MDS
Inactivation of tumor suppressor genes is a crucial event inoncogenesis. A proposed mechanism for inactivation ofgenes, either associated with normal differentiation and/orgenes involved in regulation of the cell cycle, is that oneallele of the gene sustains a mutation, and then, the normalremaining allele is lost through a variety of possible mech-anisms, such as deletion or recombination [4]. MDS isoften associated with cytogenetic deletions such as −5/5q-,−7/7q-, suggesting that genes located at these sites behaveas tumor suppressor genes and their alteration is acontributing cause of MDS [5, 6]. As illustrated below,several specific syndromes are characterized by significantgenetic defects resulting in characteristic phenotypicalchanges. We anticipate that these list will continue to growrapidly.
Fig. 1 Multistep pathogenesis in MDS. The initial genetic insult uponthe hematopoietic stem cell can be caused by chemicals, radiation,cytotoxic drugs, or random endogenous mutations. The accumulationof several alterations, which may affect cell-cycle control, transcriptionof tumor suppressors, results in the expansion of the MDS clone withaffected cells showing an increased apoptosis at early stages. Theprogression to leukemia is probably not dependent on the order of
occurrence of genetic alterations but is dependent on the genes that arealtered. The final step, leukemic transformation, may be enhanced byalteration of additional genes including tumor suppressor genes/protooncogenes and/or by hypermethylation of critical targets. Thelater stage of leukemogenesis is associated with a decreased apoptosisand enhanced proliferation
778 Ann Hematol (2008) 87:777–795

Deletion 5q
Deletions on the long arm of chromosome 5 are the mostfrequent genetic abnormalities in MDS recorded so far.They can either occur as a sole aberration or combined withone or more defects. The major commonly deleted region(CDR) has been delineated at band 5q31.1.
A sole deletion on the long arm of chromosome 5 be-tween the bands 31 and 33 associated with macrocyticanemia, normal or elevated peripheral thrombocytes, andless than 5% blasts in the bone marrow characterizes the5q- syndrome, which has a favorable prognosis with only25% evolution to AML after 15 years. Moreover, patientswith 5q- syndrome show excellent response rates whentreated with the immunomodulatory drug lenalidomide [7].However, the exact mechanism and genes being affectedare still under investigation.
Recently, Pellagatti et al. [8] could demonstrate thatlenalidomide led to upregulation and increased proteinexpression of the SPARC gene in erythroblasts in vitro. TheSPARC gene is a tumor suppressor gene with antiprolifer-ative, antiadhesive, and antiangiogenic functions, and it islocated at 5q31–32, which makes it an interesting target forfurther investigation, as deletions on chromosome 5normally affect only one of the chromosomes and upregu-lation of SPARC on the remaining not deleted chromosomecould compensate for the loss of the other gene. In a murinemodel, SPARC−/− mice showed a marked thrombocytope-nia and a reduced erythroid colony formation, suggestingthat this gene might contribute to the myelodysplasticphenotype [9]. The same group found several other genesbeing significantly downregulated in CD34+ cells of 5q-patients.
The early-growth response gene 1 (EGR1) is a memberof the WT 1 transcription factor family, and it is located inthe CDR on chromosome 5. It is involved in cellularresponse to growth factors, mitogens, and stress stimuli.Beside animal models, there is evidence that low EGR1expression is pathogenetically involved in different cancertypes in humans partially by regulation of p53 [10–12].Joslin et al. [13] found that EGR−/− and EGR+/− miceyielded a higher susceptibility to develop myeloid malig-nancies and suggested haploinsufficiency of EGR1 as aninitiating event in the development of MDS and AML.
Inactivation of tumor suppressor genes is widelyaccepted as an important event in leukemogenesis. Recent-ly, Liu et al. [14] could demonstrate that patients with MDSor AML harboring a del(5q) show an increased methylationof CpG islands of the promoter region of α-catenin.Moreover, they also showed that in del(5q) carrying HL-60 cells modification of histone tails is frequent andreversible when treated with the histone deacetylaseinhibitor trichostatin A.
The ribosomal protein S14 is a component of the 40Ssubunit of ribosomes, which are responsible for proteinsynthesis. The RPS14 gene is located within the CDR onchromosome 5.
Groundbreaking data published by Ebert et al. [15]demonstrated that haploinsufficiency of RPS14 leads to thecharacteristic phenotype of the 5q- syndrome with impairederythroid differentiation and a preservation of megakaryo-poiesis. In an exhausting functional genomic approach, theysystematically knocked down all 41 genes lying within theCDR on chromosome 5 in CD34+ hematopoietic progen-itors by using lentiviral short hairpin RNA and found thatknock down of RPS14 lead to the characteristic phenotypeof the 5q- syndrome. Moreover, induction of a forcedexpression of RPS14 in primary bone marrow cells frompatients with 5q- syndrome lead to the reconstitution oferythropoiesis and megakaryopoiesis. In addition, over-expression of RPS14 in primary bone marrow cells frompatients without a del(5) had no effect on the phenotype. Itwas shown that haploinsufficiency of RPS14 led to a blockin processing pre-ribosomal RNA and formation of the 40Ssubunit of the ribosomes. Since research on biallelicinactivation of genes within the CDR has been unsuccessfulso far, these data underscore the importance of translationalaberrations in hematological malignancies. However, sincealteration of subunits of the ribosomes would affect theexpression of a wide range of genes, it is unlikely that thesole aberration of RPS14 leads to the 5q- syndrome andother genes must be involved. In Table 1, genes are listedthat are mapped to the CDR and for which data were foundon the NCBI database concerning a possible involvement inthe pathogenesis of hematopoietic malignancies or othertypes of cancer, respectively.
Aberrations of chromosome 7
In a large survey on 2,124 MDS patients, Haase et al. [3]analyzed numeric and structural cytogenetic abnormalitiesin 2,072 patients in whom cytogenetic analysis wassuccessfully performed. Of the patients, 52.3% showedclonal abnormalities with aberrations involving 5q beingthe most frequently detected in 30% of cases followed bykaryotypic abnormalities on chromosome 7 either −7 or del(7q) (21%). Moreover, these alterations on chromosome 7were associated with a worse prognosis and a mediansurvival of 14 months. However, genes on chromosome 7responsible for this course of disease have not yet beenidentified.
The oxysterol-binding protein-like 3 gene (OSBPL3)encodes for a member of intracelluar lipid receptors, whichare highly expressed in hematopoietic progenitor cells [16].Oxysterols—products of the cholesterol oxidation—areknown inhibitors of cell proliferation including hematopoietic
Ann Hematol (2008) 87:777–795 779

cells. Moreover, there is evidence that OSBPL3 is involvedin cell adhesion via an interaction with the R–Raspathway [17]. Another gene that is located on chromo-some 7 and is involved in cholesterol processing is theSTARD3NL gene.
Chromosome 7 contains the A-cluster of the so-calledhomeobox genes, namely, HOXA1, HOXA2, HOXA3,
HOXA4, HOXA5, HOXA6, HOXA7, HOXA9, andHOXA10. These genes encode for DNA-binding transcrip-tion factors, which have been implicated in differentiationand stem cell commitment in hematopoiesis.
HOXA9 is located on band 7q36. Heinrichs et al. [18]compared the HOXA9 expression in CD34+ and CD34−cells from 13 MDS patients to healthy subjects and found
Table 1 Genes in the CDR on chromosome 5
Gene Common name (NCBI) Role in carcinogenesis/leukemogenesis?
TNIP1 TNFAIP3 interacting protein 1 Downregulated in del(5q) [9]NFκB inhibition [107]EGF/ERK inhibition [108]
CSF1R Colony-stimulating factor 1 receptor Expression levels associated with different types of myeloid leukemia [109]Downregulated in del(5q) [9]
CDX1ANXA6
Caudal-type homeobox 1 Downregulated after promoter methylation in intestinal cancer [110]Annexin A6 Regulation p120/GAP and RAS [111]
Downregulated in del(5q) [9]ZNF300 Zinc finger protein 300 Transcription repressor [112]CD74 CD74 molecule, major histocompatibility
complex, class II invariant chainLeukemic cutaneous infiltrates [113]
SPARC Secreted protein, acidic, cysteine-rich Cell cycle progressionUpregulation upon lenalidomide treatment [8]Downregulated in del(5q) [9]
PPARGC1B Peroxisome proliferator-activated receptorgamma, coactivator 1 beta
Estrogen receptor co-activatorGenetic variations assciated with familial breast cancer [114]Upregulation associated with improved oxidative phosphorylation inpresence of nonsense mtDNA mutations [115]
PDGFRB Platelet-derived growth factor receptor,beta polypeptide
Proliferation, survival, developmentInhibition by Sorafenib [116]Various fusion-transcripts described [117–121]Downregulated in del(5q) [9]
IL17B Interleukin 17B TNFα release from monocytesDCTN4 Dynactin 4 (p62) TNFα/IL-1 signaling [122]CSNK1A1 Casein kinase 1, alpha 1 Promotion of survival by interfering with RXR [123]ATOX1 ATX1 antioxidant protein 1 homolog (yeast) Antioxidant against superoxide and hydrogen peroxide
Proliferation, cell cycle control [124]Downregulated in del(5q) [9]
G3BP GTPase activating protein (SH3 domain)binding protein 1
RAS pathwayDownregulated in del(5q) [9]
GPX3 Glutathione peroxidase 3 (plasma) Detoxification of hydrogen peroxideHighly methylated in different cancers [125, 126]
NDST1 N-deacetylase/N-sulfotransferase (heparanglucosaminyl) 1
AngiogenesisExtracellular matrixMutation reduced Erk expression and increased apoptosisin endothelial cells [127]
Downregulated in del(5q) [9]CAMK2A Calcium/calmodulin-dependent protein
kinase (CaM kinase) II alphaCell cycle control in osteosarcoma [128]HDAC interaction [129]IKK/ NFκB cascade [130]
SLC36A1 Solute carrier family 36 (proton/aminoacid symporter), member 1
Amino acid transportDownregulated in del(5q) [9]
RPS14 Ribosomal protein S14 Inhibition by siRNA led to a 5q- phenotype and reconstitution of RPS14expression in 5q-samples restored erythropoiesis and megakaryopoeisis [15]
Only genes for which data concerning carcinogenesis/leukemogenesis were available on the NCBI database are listed.
780 Ann Hematol (2008) 87:777–795
an increased HOXA9 expression in CD34+ cells in MDSpatients compared to CD34− cells and healthy controls.However, no data exist about decreased HOXA9 expressionin MDS, but HOXA9 seems to be required for myeloiddifferentiation [19]. Molecular aberrations involvingHOXA9 might be associated with a poor prognosis inMDS [20].
Interestingly, LOC647034, which is mapped to band7q32.3, seems to encode for a gene that shares similarity tothe ribosomal protein S14. However, no elucidatinginformation concerning LOC647034 was obtainable onthe NCBI database.
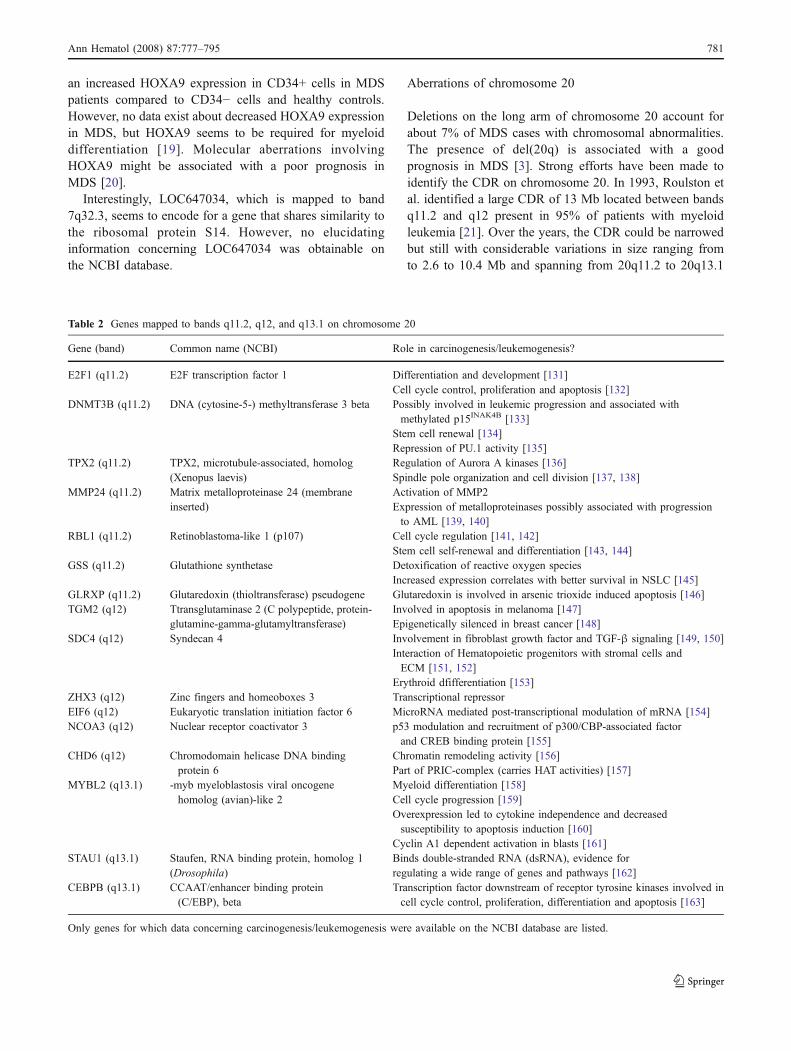
Aberrations of chromosome 20
Deletions on the long arm of chromosome 20 account forabout 7% of MDS cases with chromosomal abnormalities.The presence of del(20q) is associated with a goodprognosis in MDS [3]. Strong efforts have been made toidentify the CDR on chromosome 20. In 1993, Roulston etal. identified a large CDR of 13 Mb located between bandsq11.2 and q12 present in 95% of patients with myeloidleukemia [21]. Over the years, the CDR could be narrowedbut still with considerable variations in size ranging fromto 2.6 to 10.4 Mb and spanning from 20q11.2 to 20q13.1
Table 2 Genes mapped to bands q11.2, q12, and q13.1 on chromosome 20
Gene (band) Common name (NCBI) Role in carcinogenesis/leukemogenesis?
E2F1 (q11.2) E2F transcription factor 1 Differentiation and development [131]Cell cycle control, proliferation and apoptosis [132]
DNMT3B (q11.2) DNA (cytosine-5-) methyltransferase 3 beta Possibly involved in leukemic progression and associated withmethylated p15INAK4B [133]
Stem cell renewal [134]Repression of PU.1 activity [135]
TPX2 (q11.2) TPX2, microtubule-associated, homolog(Xenopus laevis)
Regulation of Aurora A kinases [136]Spindle pole organization and cell division [137, 138]
MMP24 (q11.2) Matrix metalloproteinase 24 (membraneinserted)
Activation of MMP2Expression of metalloproteinases possibly associated with progressionto AML [139, 140]
RBL1 (q11.2) Retinoblastoma-like 1 (p107) Cell cycle regulation [141, 142]Stem cell self-renewal and differentiation [143, 144]
GSS (q11.2) Glutathione synthetase Detoxification of reactive oxygen speciesIncreased expression correlates with better survival in NSLC [145]
GLRXP (q11.2) Glutaredoxin (thioltransferase) pseudogene Glutaredoxin is involved in arsenic trioxide induced apoptosis [146]TGM2 (q12) Ttransglutaminase 2 (C polypeptide, protein-
glutamine-gamma-glutamyltransferase)Involved in apoptosis in melanoma [147]Epigenetically silenced in breast cancer [148]
SDC4 (q12) Syndecan 4 Involvement in fibroblast growth factor and TGF-β signaling [149, 150]Interaction of Hematopoietic progenitors with stromal cells andECM [151, 152]
Erythroid dfifferentiation [153]ZHX3 (q12) Zinc fingers and homeoboxes 3 Transcriptional repressorEIF6 (q12) Eukaryotic translation initiation factor 6 MicroRNA mediated post-transcriptional modulation of mRNA [154]NCOA3 (q12) Nuclear receptor coactivator 3 p53 modulation and recruitment of p300/CBP-associated factor
and CREB binding protein [155]CHD6 (q12) Chromodomain helicase DNA binding
protein 6Chromatin remodeling activity [156]Part of PRIC-complex (carries HAT activities) [157]
MYBL2 (q13.1) -myb myeloblastosis viral oncogenehomolog (avian)-like 2
Myeloid differentiation [158]Cell cycle progression [159]Overexpression led to cytokine independence and decreasedsusceptibility to apoptosis induction [160]
Cyclin A1 dependent activation in blasts [161]STAU1 (q13.1) Staufen, RNA binding protein, homolog 1
(Drosophila)Binds double-stranded RNA (dsRNA), evidence forregulating a wide range of genes and pathways [162]
CEBPB (q13.1) CCAAT/enhancer binding protein(C/EBP), beta
Transcription factor downstream of receptor tyrosine kinases involved incell cycle control, proliferation, differentiation and apoptosis [163]
Only genes for which data concerning carcinogenesis/leukemogenesis were available on the NCBI database are listed.
Ann Hematol (2008) 87:777–795 781

[22, 23]. Beside hypothetical proteins and open readingframes, bands 20q11.2, 20q12, and 20q13.1 contain severalinteresting genes, which could be of relevance in thepathogenesis and course of MDS. Table 2 summarizes thedata supporting an involvement in carcinogenesis andleukemogenesis.
The E2F1 gene is located on band q11.2 and encodes fora transcription factor, which is involved in cell-cyclecontrol and regulation of proliferation and p53-mediatedapoptosis. Interestingly, Saberwal et al. [24] could showincreased levels of E2F1 in mononuclear bone marrow cellsof MDS patients as compared to healthy controls. More-over, elevated E2F1 levels were associated with increasedcyclin D1 expression suggesting possible role in increasedapoptosis in MDS patients [25]. Using the myeloid celllines NB4 and U937, Gery et al. [26] could demonstratethat E2F1 is required for C/EBPε-induced granulocyticdifferentiation. However, no data concerning a decreasedE2F1 expression in MDS have been published so far.
Alteration of signal transduction
RAS pathway
The RAS subfamily consists of the H-, N-, and K-RASgenes (along with the RAL and RAP genes). They play animportant role in proliferation and differentiation of cellsand have been implicated in cellular transformation.
Briefly, RAS proteins cycle between a guanosine triphos-phate (GTP)- and a guanosine diphosphate (GDP)-boundstate with the GTP-bound protein being the active formthat activates complex downstream signaling cascades in-cluding Raf and mitogen-activated protein kinase (MAPK)leading to the activation of transcription factors, such asNF-IL6, ELK-1, c-Jun, and c-Myc, which play a criticalrole in cell proliferation and differentiation. The forma-tion of the active GTP-bound RAS is promoted by theso-called guanine nucleotide exchange factors, whileGTPase-activating proteins (GAP) increase the hydrolysisof GTP rendering RAS inactive. In addition, another crucialprocess in the activation of RAS proteins is the transloca-tion from the cytoplasm to the inner plasma membrane,which requires the prenylation (farnesylation) of the RasC-terminal cysteine a process that is catalyzed by farnesyl-transferases [27].
RAS mutations occur in about 10–15% of MDS cases,with N-RAS being most frequently mutated. The ‘hotspots’ in which point mutations most frequently occur arelocated either at codon 12, 13, or 61. Mutations of this generesult in an activated protein, which stimulates its down-stream targets such as RAF and MAPK. MDS with mutantN-RAS may be associated with a worse prognosis [28].
Irrespective of RAS mutations, other alterations can leadto an uncontrolled activation of the RAS pathway. Loss offunction of GAPs can lead to increased levels of RAS-GTP[29], resulting in an uncontrolled active state of the RASpathway (Fig. 2).
Fig. 2 RAS mutations in MDS.Under normal conditions, RAScycles between the inactive(e.g., GDP bound) state and theactive (e.g., GTP bound) state.Mutations in the RAS generesult in a permanent GTPbound active state of RAS withconsecutive activation or inacti-vation of downstream effectors.Arrow-up upregulated; arrow-down downregulated
782 Ann Hematol (2008) 87:777–795

JAK2 mutations
The importance of Janus tyrosine kinases in regulatingcytokine-dependent gene expression and cellular develop-ment and survival is well established.
As cytokine receptors lack an intrinsic catalytic activity,Janus kinases play a crucial role in the “membrane-to-nucleus” signaling cascade. Briefly, activation of Jakkinases upon ligand binding leads to the creation ofdocking sites for cytoplasmic-signaling molecules knownas the “signal transducers and activators of transcription”(STAT). STATs—upon activation via tyrosine phosphory-lation by Jak kinases—dimerize and translocate to thenucleus. By binding to the DNA, they regulate theexpression of certain genes.
Among the Jak kinase family, JAK2 has recently gaineda lot of interest, as it seems to play a crucial role in thepathogenesis of myeloproliferative syndromes (MPS). Sev-eral groups [30–32] identified a valine-to-phenylalanin sub-stitution at amino acid position 617 of JAK2 (JAK2V617F).This amino acid substitution is due to a G-to-T alteration inexon 12 of JAK2, which is located on the short arm ofchromosome 9 [30]. As a result of this mutation, cells showa hypersensitivity to cytokines due to a constitutivephosphorylation activity of the kinase that promotescytokine hypersensitivity [31]. Downstream effectors ofJAK2V617F are depicted in Fig. 3.
In up to 97% of patients with polycythemia vera, 57%with essential thrombocythemia, and 50% of patientssuffering from idiopathic myelofibrosis, the JAK2V617F
mutation could be detected in several studies while notpresent in healthy controls [30–32], indicating that it playsan important role in the pathogenesis of MPS.
However, the JAK2V617F mutation is infrequent intypical MDS and is found in about 5% of cases [33, 34].Recent findings, however, revealed JAK2V617F in a certainsubgroup of MDS, called the refractory anemia with ringedsideroblasts and thrombocytosis (RARS-T). This entitycontributes to the subgroup of overlapping syndromesMDS/MPS of the WHO classification presenting with bothhallmarks of MDS and MPS. Several groups reportedJAK2V617F as a common feature in RARS-T, detectable inthe majority (>60%) of patients [33–39]. Besides a markedthrombocytosis, the patients do not present with otherclinical features of MPS. However, it still remains contro-versial whether RARS-T corresponds to two differentsimultaneous entities or to a unique disease.
FLT3 mutations
The FLT3 gene encodes for a tyrosine kinase receptor andis involved in the regulation of proliferation, differentiation,and apoptosis in hematopoietic cells. Mutations in theFLT3 gene are less common in MDS but frequent in AML
Fig. 3 JAK2 mutations.JAK2V617F mutations lead tohypersensitivity to cytokinesdue to a constitutive phosphor-ylation activity of the kinase.Arrow-up upregulated;arrow-down downregulated
Ann Hematol (2008) 87:777–795 783

(5% and 25–30%, respectively) and predict a worse out-come in AML [40, 41]. The most frequent mutations ofFLT3 include an internal tandem duplication within thejuxtamembrane domain and point mutation in the tyrosinekinase domain (TKD) resulting in a ligand-independentactivation of the FLT3 pathway. Whether FLT3 is associ-ated with transformation of MDS to AML was examined ina group of 97 MDS patients [cases with refractory anemiawith excess blasts in transformation (RAEB-t) wereexcluded] [42]. Only three patients presenting with RAEBat diagnosis showed a mutation of the FLT3 gene, whereasin patients with early stages of MDS, detection of FLT3mutations failed. This finding is in accordance with otherstudies that could not detect FLT3 alterations in early MDSstages [43]. However, of 97 patients, 42 progressed toAML: All three patients already presented with FLT3mutations at the time of diagnosis (all of them RAEB),and three patients acquired this mutations and progressed toAML within a month.
In a large survey, Bacher et al. [43] analyzed 381patients with MDS and 4,130 patients with AML. Theirresults confirmed FLT3 mutations being more frequent inAML compared to MDS, and they found an increase ofFLT3 mutations in advanced stages of MDS compared toearly stages. Noteworthy, the FLT3-TKD mutation wasmore frequently detectable in refractory anemia (RA) andRARS (3.6%) than in patients with RAEB (0.0%).However, FLT3-TKD mutations had the highest frequencyin de novo AML (5.5%).
In conclusion, it seems likely that FLT3 mutations play acritical role in the progression to AML in a subset of MDSpatients and are rather a ‘second hit’ than a ‘first hit’ in thedevelopment of MDS. Moreover, patients with mutations inthe FLT3 gene could benefit from small-molecule TKinhibitors such as PKC412.
NPM1 mutations
The nucleophosmin 1 (NPM1) gene is mapped to band q35 onchromosome 5 and encodes for a multifunctional protein thathas been implicated in tumorigenesis of various types of solidtumors and hematological malignancies. NPM1 containsfunctional domains that account for diverse biologicalfunctions. By shuttling between the nucleus and the cyto-plasm, NPM1 takes part in various cellular processes such asribosomal biogenesis, response to stress stimuli, control ofcellular ploidy and maintenance of genomic stability, DNArepair processes, and regulation of DNA transcription throughmodulation of chromatin condensation [44]. Due to the widerange of different cellular processes, NPM1 has beenattributed as a proto-oncogene and a tumor suppressor gene.
Mutations of NPM1 are found in about 35% of patientswith AML with a normal karyotype [45]. The presence of
NPM1 mutations predict a relatively good response to induc-tion chemotherapy and a good prognosis in these patients.
In a mouse model, haploinsufficiency of NPM1 has beenshown to result in a higher susceptibility to hematologicalmalignancies compared to wild-type mice [46]. In addition,haploinsufficiency resulted in a MDS-like phenotype.
In contrast to the high frequency of NPM1 mutations inAML, these mutations are relatively rare in MDS. In 38patients with MDS, Zhang et al. [47] found mutations ofNPM1 only in two patients (5.2%): one being diagnosedwith a RA and the other with a RAEB-1. These findings arein accordance to the results from other groups [48, 49].
However, recently, Boehrer et al. [50] published dataabout off-target antineoplastic effects of erlotinib, an in-hibitor of the epidermal growth factor receptor (EGFR), inpatients with AML and MDS. Erlotinib induced differ-entiation, cell-cycle arrest, and apoptosis in EGFR-negativemyeloblasts and was associated with a nucleocytoplasmictranslocation of NPM1 and p14ARF. This translocation wasnot observed in myeloblasts insensitive to apoptosis induc-tion by erlotinib.
The aforementioned data suggest that NPM1 mutationsmight play a role in a subset of MDS cases, but the exactcontribution to initiation and progression of MDS needsfurther research.
Alterations of transcription factors and cell-cyclecontrol
AML1 gene
The human AML1 gene (also known as CBFA2, PEBP2αB,or RUNX1), located in the 21q22 chromosomal band,encodes for one of the two subunits forming a heterodimerictranscription factor, the human core-binding factor (CBF).AML1 protein contains a highly evolutionary conserveddomain of 128 amino acids called runt domain, responsiblefor both heterodimerization with the beta subunit of CBF andfor DNA binding. AML1 is normally expressed in allhematopoietic lineages and acts to regulate the expressionof various genes specific to hematopoiesis playing a pivotalrole in myeloid differentiation [51]. Mutations of AML1occur in about 25% of AML cases (mainly in AML-M0)and are frequently observed in MDS with excess of blasts[52]. AML1 mutations not only result in loss of functionphenotypes but can show dominant negative effects onnormal wild-type AML1 function [6].
It is interesting to note that AML1-mediated transcrip-tional activation involves the direct binding of the tran-scriptional coactivators p300 and CBP to the transcriptionactivation domains of AML1. These coactivators have in-trinsic histone acetyltransferase (HAT) activity (see below)
784 Ann Hematol (2008) 87:777–795

and directly bind to another HAT, P/CAF. These HATsinduce the acetylation of lysine residues in chromatin-associated histones, resulting in a change in chromatinstructure that leads to enhanced transcription [53]. As thesole loss of function of AML1 due to AML1 mutations hasbeen shown not to be sufficient to induce MDS/AMLalone, second hit events such as epigenetic alterations(methylation and deacetylation) could promote the devel-opment of AML1 mutation-positive MDS/AML.
EVI1 gene
Ecotropic viral integration site 1 (EVI1) gene is located onchromosome 3q26.2 and encodes for a relatively largenuclear protein containing ten zinc finger motifs, which hasbeen shown to act as an aggressive oncoprotein. There existtwo forms of EVI1: the 145 kDa EVI1 gene and the largerMDS1/EVI1 gene, which is a result of an in-frame splicingof the myelodysplasia syndrome gene1 to the second exonof EVI1. EVI1 and MDS1/EVI1, respectively, are scarcelyexpressed in hematopoietic cells [54], and inappropriateexpression in hematopoietic cells has been implicated in thedevelopment or progress of myeloid disorders, either AMLor MDS [55].
Inappropriate activity can result from 3q26 rearrange-ments such as translocations or inversions. However,several groups reported the frequent expression of EVI1in MDS partially without structural alteration of 3q26,suggesting that promoter deregulation being possiblyinvolved in EVI1 expression [56, 57]. The expression ofEVI1 yielded no restriction to a particular subtype accord-ing to FAB. In MDS patients, EVI1 expression is a verypoor prognostic marker and is associated with severeerythropoietin (EPO)-unresponsive anemia [58]. In addi-tion, there seems to exist an association between EVI1expression and deletions on chromosome 7 [59], a featurethat also predicts poor outcome in MDS. Concerningerythropoiesis, EVI1 represses the expression of the EPOreceptor in animal models [60], which may in part explainthe unresponsiveness to EPO in these patients. Themechanisms by which EVI1 alters gene expression arenot yet fully understood. It can act as a repressor of targetgenes on the one hand and as an activator on the otherhand. EVI1 has been connected to GATA2, a gene highlyexpressed in hematopoietic stem cells and involved inerythroid and megacaryocytic differentiation [61, 62].Moreover, recent findings by Laricchia-Robbio et al. [63]showed that EVI1 represses erythroid differentiation byinteracting with the DNA-binding protein GATA1. Theydemonstrated that point mutations disrupting the geometryof two zinc fingers of EVI1 abolished the protein–proteininteraction and led to normal erythroid differentiationin vitro.
It is interesting to note that EVI1 has been connected toboth genes encoding for proteins showing histone acetyltransferase activity, such as P/CAF, and to histone deacty-lases [64]. Whether EVI1 leads to activation or repressionof target genes seem at least in part depend on itsacetylation state.
EVI1 has also been associated with genes being involvedin apoptosis regulation. EVI1 led to an inhibition of tumorgrowth factor (TGF)-β-induced Smad3 activation. Inaddition, EVI1 resulted in an activation of the PI3K/Aktpathway [65]. Genes possibly involved in the pathobiologyof EVI1 are depicted in Fig. 4.
p53 gene
Alterations of the p53 gene occur in about 10% of MDS[66]. Changes are usually missense mutations of one allelewith the second allele being lost. Patients whose abnormalclone has an isochromosome 17q often have a p53 mutationin their cells. These mutations usually prevent the proteinfrom being able to bind to DNA, thus losing its ability totransactivate target genes such as the cyclin-dependentkinase inhibitor (CDKI) p21WAF2. Mutations of p53 areassociated with progression of the disease and poorprognosis.
Epigenetic alterations
DNA methylation
DNA methylation is a known mechanism influencingchromatin structure and transcriptional activity and has astrong impact on both normal development and pathogen-esis of hematopoietic stem cells.
Hypermethylation of CpG islands of gene promoterscatalyzed by DNA methyltransferases leads to closedchromatin structure and transcriptional silencing frequentlyresulting in silencing of tumor suppressor genes [67](Fig. 5a, b). Several fundamental biological processescan be affected by this epigenetic event in human cancer,such as DNA repair, cell-cycle control, apoptosis, anddetoxification.
Hypermethylation of genes involved in cell-cycle controland apoptosis is a common feature particularly in high-riskMDS. Important genes for cell-cycle regulation are theCDKI p15INK4b and p16INK4a. These two genes are rarelymutated or deleted [68], but transcription of the p15INK4b
gene is often silenced due to abnormal methylation of itspromoter region, and several studies indicate that roughly50% of MDS patients show this alteration [69, 70]. Othergenes being frequently affected by hypermethylation inMDS are cancer 1 (HIC1), E-cadherin (CDH1), and
Ann Hematol (2008) 87:777–795 785

estrogen receptor (ER). In addition, hypermethylation ofthese genes was associated with poor outcome in early-stage MDS [71].
Hopfer et al. [72] performed lineage specific DNAmethylation profiling at defined differentiation stages inerythro-, thrombo-, and granulopoiesis from patients withMDS and healthy controls. Promoter methylation analysiswas performed for key regulator genes involved in cell-cycle control, DNA repair, apoptosis, and differentiation.For p16, survivin, CHK2, and WT1, an association of theirmethylation status with international prognostic scoringsystem (IPSS) risk types was found. Moreover, for specifichematopoietic lineages and differentiation stages, a meth-ylation-associated messenger RNA (mRNA) downregula-tion could be demonstrated, which provides evidence thatlineage-specific methylation followed by gene silencingcontributes to MDS-specific phenotypes and possibly to thedifferent courses of MDS entities.
In contrast to genetic aberrations, silencing of genes byDNA methylation is a reversible process, and the introduc-tion of demethylating agents (e.g., 5-azacytidine and 5-aza-2′-deoxycytidine), particularly in the treatment of high-riskMDS, yielded encouraging results [73, 74].
Histone deacetylation
Acetylation and deacetylation of nucleosomal histones playa critical role in the transcriptional process. Acetylation ofamino acids of the histones catalyzed by the HAT leads to adestabilization of the histone–DNA interaction, which inturn results in an opening of the nucleosome structure and
consecutively to a permission of transcription. Opponentsof the HATs are histone deacetylases (HDAC), whichremove acetyl groups from histones that stabilizes the localchromatin structure and renders affected promoter sequen-ces inaccessible leading to silencing of genes in this region[75] (Fig. 6a, b). However, valproic acid (VPA), a knownHDAC inhibitor, has been shown to induce differentiationand apoptosis of leukemic cell lines in vitro. Kuendgenet al. [76, 77] showed that VPA has some effect in MDSwith response rates up to 30% in MDS patients according tothe international working group criteria [78]. Responserates were high (50%) in patients with a normal blast countin the bone marrow. Correlation between response rate andthe IPSS showed that patients with low-risk MDS re-sponded in up to 70% of cases.
Moreover, as it has been shown that DNA methylationand histone acetylation are closely connected, there isevidence that combination of VPA with demethylatingagents have additional effects in advanced stages of MDSand AML [79, 80].
Mitochondrial DNA mutations
Through various ways, mitochondria are involved in thehematopoietic cell homeostasis, such as production ofadenosine triphosphate (ATP) by oxidative phosphoryla-tion, participation in apoptotic pathways, and differentmetabolic processes including heme synthesis. In addition,maintenance of an adequate mitochondrial membranepotential is pivotal for ATP synthesis and cell survival.
Fig. 4 EVI1. For EVI1, a widerange of interactions with othergenes have been described—some of them are depicted inthis figure. EVI1 can act as botha repressor and an activator ofcertain genes
786 Ann Hematol (2008) 87:777–795

For some diseases, mutations of genes that encode formitochondrial enzymes have been identified. The X-linkedsideroblastic anemia, which is the most common inheritedsideroblastic anemia, is caused by mutations in the specificδ-aminolevulinic acid synthase. In addition, deletions in
mitochondrial DNA (mtDNA) are a hallmark of Pearson’ssyndrome, a rare congenital disorder with sideroblasticanemia.
Gattermann et al. described abnormalities in the cyto-chrome b gene in patients with acquired idiopathic sideroblastic
Fig. 5 DNA methylation andsuppression of tumor suppres-sor genes. a Transcription ofgenes can take place since theCpG island within the promoteris not methylated. b In case ofmethylation of the CpG islandthe gene can not be transcripted:it is silenced
Ann Hematol (2008) 87:777–795 787

anemia (AISA) and RAEB. They could also demonstratethe alteration of the mitochondrial transfer RNA (tRNA)gene in patients with MDS, namely, the mitochondriallyencoded tRNA leucine 2 (CUN). The same group found
mutations of the cytochrome c oxidase (CO) gene inpatients with AISA [81–83]. In addition, Reddy et al. [84]described cytochrome c oxidase I mutations in 13 of 20MDS patients irrespective of MDS subtype. However, a
Fig. 6 Deacetylation ofhistones. a Acetylation of aminoacids of the histones is catalyzedby histone acetyltransferases(HAT ). Acetylation leads todestabilization of histone–DNAinteraction, which results in anopening of the nucleosomestructure and consecutivelyto a permission of transcription.b Opponents of the HATs arehistone deacetylases (HDAC),which remove acetyl groupsfrom histones that stabilizes thelocal chromatin structure andrenders affected promotersequences inaccessible leadingto silencing of genes in thisregion
788 Ann Hematol (2008) 87:777–795

survey by Shin et al. [85] comparing mtDNA of ten patientswith MDS (four RA, three RARS, and three RAEB) andeight healthy controls could neither confirm these findingsnor support a major role for mitochondrial instability inMDS (Fig. 7).
BMSCs in MDS
BMSCs are an important component of the hematopoieticmicroenvironment. However, there is a controversy ongo-ing about the role of BMSCs in myeloid disorders such asMDS and whether BMSCs are derived from the malignantclone.
Lessons learned from allogeneic stem cell transplanta-tion (alloSCT) revealed that BMSC remain are of hostorigin, while hematopoietic cells after successful transplan-tation are of graft origin [86, 87]. As MDS patients can becured by alloSCT, this suggests that the BMSCs are intactor alterations are reversible. In addition, several reports ofin vitro and in vivo studies confirm these findings andpromote the functional normality of BMSCs [88–90]. Incontrast, Flores-Figueroa et al. characterized BMSCs fromMDS patients phenotypically and cytogenetically and couldfind cytogenetic aberrations in BMSC derived from MDSpatients in 55% of cases. Moreover, they showed alteredcytokine pattern synthesized by MDS-derived BMSCscompared to healthy controls [91, 92].
Blau et al. published data concerning cytogeneticalterations of MDS and AML-derived BMSCs as comparedto healthy controls. Cytogenetics identified in seven (44%)out of 16 MDS cases genetic aberrations in BMSCs, whilechromosomal alterations were absent in healthy controls.Among other translocations, aberrations found in BMSCsincluded t(1;7), t(4;7), and t(7;19) as well as deletions onchromosome 2 and 17. However, they were not able to findidentical chromosomal aberrations in hematopoietic cellsand BMSCs [93].
Therefore, it remains an open question if BMSCs arederived from the malignant clone in MDS and whether theyplay a role in the pathogenesis of MDS. New scientificapproaches and techniques might—hopefully—contributeto the clarification of this issue in particular and elucidatethe pathogenesis of MDS.
Angiogenesis in MDS and myeloid malignancies
Angiogenesis is recognized as an important prerequisite ingrowth and progression of solid tumors. In addition, alteredangiogenesis has been implicated as a pathobiologicalfactor in development and progression of myeloid diseases.
Aguayo et al. [94] investigated the vascularity in bonemarrow biopsies in patients with different types ofhematological malignancies. They found an increasedvascularity in MDS patients compared to healthy controls.
Fig. 7 Mitochondrial genomeand mutations that have beendescribed in MDS. CO ICytochrome C oxidase I
Ann Hematol (2008) 87:777–795 789

Moreover, plasma levels of angiogenic factors such asvascular endothelial growth factor (VEGF), basic fibroblastgrowth factor, and hepatocyte growth factor were increasedin MDS specimens. However, they failed to detect in-creased plasma levels for tumor necrosis factor-α (TNF-α)and TGF-α.
Keith et al. [95] used immunohistochemical methods forthe detection of microvessel density (MVD), and involve-ment of angiogenic factors was investigated by using real-time polymerase chain reaction–based methods. Theyconfirmed the finding of increased vascularity in bonemarrow specimens from MDS patients, but expressionof VEGF in MDS was not increased compared to con-trols. In contrast, TNF-α showed a higher expression inMDS, which is in accordance with findings from othergroups [96].
It is interesting to note that Keith et al. found thatpatients with MDS who had transformed to overt leukemiashowed a lower MVD compared to the MVD beforetransformation and to de novo AMLs. In addition, in anin vitro assay, endothelial cells from patients with RAEB-tfailed to form colony-forming unit-erythroid as comparedto endothelial cells from patients with RA, RARS, andRAEB [97].
The lower MVD in transforming MDS could at least inpart explain for the higher response rates of drugs withantiangiogenic potential (e.g., lenalidomide) in patientswith early MDS stages. Moreover, the knowledge of alteredcytokine levels provide the rationale for targeted therapieswith TNF-α inhibitors such as infliximab or VEGFinhibitors [98–100].
SNP analysis in MDS
As mentioned above, conventional cytogenetics yieldgenetic aberrations in only 50% of MDS patients. Thismay be due to the low resolution of routine metaphasecytogenetics. Moreover, metaphase karyotyping is restrict-ed to dividing cells. Undisputable, the introduction ofinterphase fluorescent in situ hybridization (FISH) im-proved cytogenetic analysis. However, FISH utilizes alimited number of probes directed to already knownlesions, and therefore, new chromosomal defects can notbe detected.
The introduction of high-density SNP microarrays offerthe opportunity to perform a global analysis of genomicDNA and provide the possibility to detect so far crypticlesions, as they cover the whole genome and the resolutionis much higher irrespective to cell division. Different in-formation can be obtained from analysis of SNP array data:beside copy number estimation (deletion/amplification),
SNP arrays allow for precise determination of LOH evenwithout loss of ploidy [uniparental disomy (UPD)].
Today, only few data exist concerning SNP analysis inMDS by application of DNA microarrays. We and othershave clearly shown that whole-genome SNP analysis is apowerful tool to identify so far hidden genetic alterations inpatients with MDS.
Nowak et al. compared purified CD34+ cells from MDSpatients to healthy controls using 500K SNP arrays. Thisapproach not only reliably confirmed a known aberrationalready found in conventional metaphase cytogenetics inMDS samples but also identified numerous new heterozy-gous deletions on chromosomes 2, 9, 13, 16, and 17ranging in size from 0.1–2.1 Mb (personal communication).In addition, numerous regions with significant UPD weredetected, which is in accordance to recently published datathat show a high prevalence of UPD in hematopoietic cellsfrom MDS patients [101].
Integration of the genomic data with gene expressionanalysis showed that genes, which were downregulated atleast 1.5-fold in regions of significant deletion, and UPDwere exclusively downregulated in those samples display-ing the aberration.
Gene expression analysis by microarrays in MDS
We and others have shown that microarray analysis canprovide sufficient data to detect genes or gene patterns,which are associated with alterations of specific cellularpathways or signal cascades in tumor cells including MDS[102–104]. The technique of gene expression profiling canalso be used for subclassification of leukemias [105] andlymphomas [106]. The approach to predict prognosis orrisk type of MDS using gene expression data is a long wayfrom practice, but the ability to predict who will do welland who will not may have a strong impact on the furtherclassification and risk definition of MDS.
Summary and future aspects
The underlying mechanisms causing primary MDS requiresfurther work. Different molecular alterations that have beendescribed suggest that it is a multistep alteration to thehematopoietic stem cells that include genes involved incell-cycle control, mitotic checkpoints, and growth factorreceptors. Secondary signal proteins and transcriptionfactors, which give the cell a growth advantage over itsnormal counterpart, may be affected as well. The accumu-lation of such defects may finally cause the leukemictransformation of MDS.
790 Ann Hematol (2008) 87:777–795

The new microarray techniques such as SNP arrays andgene expression arrays provide the opportunity to further un-derstand the pathogenesis of MDS providing more insightsinto genetic alterations and changes in cellular pathways.
Strong efforts have been made and are still undertaken toidentify specific gene expression patterns, which areassociated with different risk groups in MDS aiming tofurther minimize the different entities for a more tailoredrisk assessment or—as it has been successfully done for the5q- syndrome—identify distinct subtypes in MDS. There-fore, we believe that the prognosis of this disease may bepredicted by using gene expression analysis or SNPassociation studies. We and others have shown that micro-array analysis can be used with small amounts of DNA/RNA, which can be obtained from cells during a routinediagnostic bone marrow aspirate. These methods mayfacilitate making therapeutic decisions in cases where thediagnosis and/or risk evaluation is not possible based onmorphologic and classical cytogenetic data. The resultsfrom a number of studies using CD34+ cells and other celltypes (e.g., granulocytes) from patients with MDS arepending.
Elucidation of the underlying defects particularlyconcerning altered cellular pathways should consequentlyresult in the further design/discovery of target-specificdrugs for treatment of MDS.
References
1. Germing U, Strupp C, Kundgen A et al (2004) No increase inage-specific incidence of myelodysplastic syndromes. Haemato-logica 89:905–910
2. Knudson AG (1996) Hereditary cancer: two hits revisited. JCancer Res Clin Oncol 122:135–140
3. Haase D, Germing U, Schanz J et al (2007) New insights into theprognostic impact of the karyotype in MDS and correlation withsubtypes: evidence from a core dataset of 2124 patients. Blood110:4385–4395
4. Krug U, Ganser A, Koeffler HP (2002) Tumor suppressor genes innormal and malignant hematopoiesis. Oncogene 21:3475–3495
5. Chen Z, Sandberg AA (2002) Molecular cytogenetic aspects ofhematological malignancies: clinical implications. Am J MedGenet 115:130–141
6. Hirai H (2002) Molecular pathogenesis of MDS. Int J Hematol76(Suppl 2):213–221
7. List A, Dewald G, Bennett J et al (2006) Lenalidomide in themyelodysplastic syndrome with chromosome 5q deletion. NEngl J Med 355:1456–1465
8. Pellagatti A, Jadersten M, Forsblom AM et al (2007) Lenalido-mide inhibits the malignant clone and up-regulates the SPARCgene mapping to the commonly deleted region in 5q- syndromepatients. Proc Natl Acad Sci U S A 104:11406–11411
9. Lehmann S, O’Kelly J, Raynaud S et al (2007) Common deletedgenes in the 5q- syndrome: thrombocytopenia and reducederythroid colony formation in SPARC null mice. Leukemia21:1931–1936
10. Ferraro B, Bepler G, Sharma S, Cantor A, Haura EB (2005)EGR1 predicts PTEN and survival in patients with non-small-cell lung cancer. J Clin Oncol 23:1921–1926
11. Ronski K, Sanders M, Burleson JA et al (2005) Early growthresponse gene 1 (EGR1) is deleted in estrogen receptor-negativehuman breast carcinoma. Cancer 104:925–930
12. Krones-Herzig A, Mittal S, Yule K et al (2005) Early growthresponse 1 acts as a tumor suppressor in vivo and in vitro viaregulation of p53. Cancer Res 65:5133–5143
13. Joslin JM, Fernald AA, Tennant TR et al (2007) Haploinsuffi-ciency of EGR1, a candidate gene in the del(5q), leads to thedevelopment of myeloid disorders. Blood 110:719–726
14. Liu TX, Becker MW, Jelinek J et al (2007) Chromosome 5qdeletion and epigenetic suppression of the gene encoding alpha-catenin (CTNNA1) in myeloid cell transformation. Nat Med13:78–83
15. Ebert BL, Pretz J, Bosco J et al (2008) Identification of RPS14as a 5q- syndrome gene by RNA interference screen. Nature451:335–339
16. Gregorio-King CC, Collier GR, McMillan JS et al (2001) ORP-3,a human oxysterol-binding protein gene differentially expressedin hematopoietic cells. Blood 98:2279–2281
17. Lehto M, Mayranpaa MI, Pellinen T et al (2008) The R-Rasinteraction partner ORP3 regulates cell adhesion. J Cell Sci121:695–705
18. Heinrichs S, Berman JN, Ortiz TM et al (2005) CD34+ cellselection is required to assess HOXA9 expression levels inpatients with myelodysplastic syndrome. Br J Haematol 130:83–86
19. Bei L, Lu Y, Eklund EA (2005) HOXA9 activates transcriptionof the gene encoding gp91Phox during myeloid differentiation.J Biol Chem 280:12359–12370
20. Hatano Y, Miura I, Nakamura T et al (1999) Molecularheterogeneity of the NUP98/HOXA9 fusion transcript inmyelodysplastic syndromes associated with t(7;11)(p15;p15).Br J Haematol 107:600–604
21. Roulston D, Espinosa R III, Stoffel M, Bell GI, Le Beau MM(1993) Molecular genetics of myeloid leukemia: identification ofthe commonly deleted segment of chromosome 20. Blood 82:3424–3429
22. Bench AJ, Nacheva EP, Hood TL et al (2000) Chromosome 20deletions in myeloid malignancies: reduction of the commondeleted region, generation of a PAC/BAC contig and identifica-tion of candidate genes. UK Cancer Cytogenetics Group(UKCCG). Oncogene 19:3902–3913
23. Douet-Guilbert N, Basinko A, Morel F et al (2008) Chromosome20 deletions in myelodysplastic syndromes and Philadelphia-chromosome-negative myeloproliferative disorders: characteri-zation by molecular cytogenetics of commonly deleted andretained regions. Ann Hematol 87(7):537–544
24. Saberwal G, Lucas S, Janssen I et al (2004) Increased levels andactivity of E2F1 transcription factor in myelodysplastic bonemarrow. Int J Hematol 80:146–154
25. Saberwal G, Broderick E, Janssen I et al (2003) Involvement ofcyclin D1 and E2F1 in intramedullary apoptosis in myelodys-plastic syndromes. J Hematother Stem Cell Res 12:443–450
26. Gery S, Gombart AF, Fung YK, Koeffler HP (2004) C/EBPepsilon interacts with retinoblastoma and E2F1 duringgranulopoiesis. Blood 103:828–835
27. Beaupre DM, Kurzrock R (1999) RAS and leukemia: from basicmechanisms to gene-directed therapy. J Clin Oncol 17:1071–1079
28. Paquette RL, Landaw EM, Pierre RV et al (1993) N-rasmutations are associated with poor prognosis and increased riskof leukemia in myelodysplastic syndrome. Blood 82:590–599
Ann Hematol (2008) 87:777–795 791

29. Basu TN, Gutmann DH, Fletcher JA et al (1992) Aberrantregulation of ras proteins in malignant tumour cells from type 1neurofibromatosis patients. Nature 356:713–715
30. Baxter EJ, Scott LM, Campbell PJ et al (2005) Acquiredmutation of the tyrosine kinase JAK2 in human myeloprolifer-ative disorders. Lancet 365:1054–1061
31. James C, Ugo V, Le Couedic JP et al (2005) A unique clonalJAK2 mutation leading to constitutive signalling causes poly-cythaemia vera. Nature 434:1144–1148
32. Jones AV, Kreil S, Zoi K et al (2005) Widespread occurrenceof the JAK2 V617F mutation in chronic myeloproliferativedisorders. Blood 106:2162–2168
33. Renneville A, Quesnel B, Charpentier A et al (2006) Highoccurrence of JAK2 V617 mutation in refractory anemia withringed sideroblasts associated with marked thrombocytosis.Leukemia 20:2067–2070
34. Szpurka H, Tiu R, Murugesan G et al (2006) Refractory anemiawith ringed sideroblasts associated with marked thrombocytosis(RARS-T), another myeloproliferative condition characterizedby JAK2 V617F mutation. Blood 108:2173–2181
35. Boissinot M, Garand R, Hamidou M, Hermouet S (2006) TheJAK2-V617F mutation and essential thrombocythemia featuresin a subset of patients with refractory anemia with ringsideroblasts (RARS). Blood 108:1781–1782
36. Ingram W, Lea NC, Cervera J et al (2006) The JAK2 V617Fmutation identifies a subgroup of MDS patients with isolateddeletion 5q and a proliferative bone marrow. Leukemia 20:1319–1321
37. Remacha AF, Nomdedeu JF, Puget G et al (2006) Occurrence ofthe JAK2 V617F mutation in the WHO provisional entity:myelodysplastic/myeloproliferative disease, unclassifiable-refractory anemia with ringed sideroblasts associated withmarked thrombocytosis. Haematologica 91:719–720
38. Wang SA, Hasserjian RP, Loew JM et al (2006) Refractoryanemia with ringed sideroblasts associated with marked throm-bocytosis harbors JAK2 mutation and shows overlappingmyeloproliferative and myelodysplastic features. Leukemia20:1641–1644
39. Zipperer E, Wulfert M, Germing U, Haas R, Gattermann N(2008) MPL 515 and JAK2 mutation analysis in MDS presentingwith a platelet count of more than 500 x 10(9)/l. Ann Hematol87:413–415
40. Kottaridis PD, Gale RE, Frew ME et al (2001) The presence of aFLT3 internal tandem duplication in patients with acute myeloidleukemia (AML) adds important prognostic information tocytogenetic risk group and response to the first cycle ofchemotherapy: analysis of 854 patients from the UnitedKingdom Medical Research Council AML 10 and 12 trials.Blood 98:1752–1759
41. Thiede C, Steudel C, Mohr B et al (2002) Analysis of FLT3-activating mutations in 979 patients with acute myelogenousleukemia: association with FAB subtypes and identification ofsubgroups with poor prognosis. Blood 99:4326–4335
42. Georgiou G, Karali V, Zouvelou C et al (2006) Serialdetermination of FLT3 mutations in myelodysplastic syndromepatients at diagnosis, follow up or acute myeloid leukaemiatransformation: incidence and their prognostic significance. Br JHaematol 134:302–306
43. Bacher U, Haferlach T, Kern W, Haferlach C, Schnittger S(2007) A comparative study of molecular mutations in 381patients with myelodysplastic syndrome and in 4130 patientswith acute myeloid leukemia. Haematologica 92:744–752
44. Grisendi S, Mecucci C, Falini B, Pandolfi PP (2006) Nucleo-phosmin and cancer. Nat Rev Cancer 6:493–505
45. Falini B, Mecucci C, Tiacci E et al (2005) Cytoplasmicnucleophosmin in acute myelogenous leukemia with a normalkaryotype. N Engl J Med 352:254–266
46. Sportoletti P, Grisendi S, Majid SM et al (2008) Npm1 is ahaploinsufficient suppressor of myeloid and lymphoid malignan-cies in the mouse. Blood 111:3859–3862
47. Zhang Y, Zhang M, Yang L, Xiao Z (2007) NPM1 mutations inmyelodysplastic syndromes and acute myeloid leukemia withnormal karyotype. Leuk Res 31:109–111
48. Thiede C, Koch S, Creutzig E et al (2006) Prevalence andprognostic impact of NPM1 mutations in 1485 adult patientswith acute myeloid leukemia (AML). Blood 107:4011–4020
49. Shiseki M, Kitagawa Y, Wang YH et al (2007) Lack ofnucleophosmin mutation in patients with myelodysplastic syn-drome and acute myeloid leukemia with chromosome 5abnormalities. Leuk Lymphoma 48:2141–2144
50. Boehrer S, Ades L, Braun T et al (2008) Erlotinib exhibitsantineoplastic off-target effects in AML and MDS: a preclinicalstudy. Blood 111:2170–2180
51. Ichikawa M, Asai T, Chiba S, Kurokawa M, Ogawa S (2004)Runx1/AML-1 ranks as a master regulator of adult hematopoi-esis. Cell Cycle 3:722–724
52. Harada H, Harada Y, Niimi H et al (2004) High incidence ofsomatic mutations in the AML1/RUNX1 gene in myelodys-plastic syndrome and low blast percentage myeloid leukemiawith myelodysplasia. Blood 103:2316–2324
53. Kitabayashi I, Yokoyama A, Shimizu K, Ohki M (1998)Interaction and functional cooperation of the leukemia-associatedfactors AML1 and p300 in myeloid cell differentiation. EMBO J17:2994–3004
54. Ohashi H, Tsushita K, Utsumi M et al (2001) Relationshipbetween methylation of the p15 gene and ectopic expression ofthe EVI-1 gene in myelodysplastic syndromes (MDS). Leukemia15:990–991
55. Nucifora G, Laricchia-Robbio L, Senyuk V (2006) EVI1and hematopoietic disorders: history and perspectives. Gene368: 1–11
56. Russell M, List A, Greenberg P et al (1994) Expression of EVI1in myelodysplastic syndromes and other hematologic malignan-cies without 3q26 translocations. Blood 84:1243–1248
57. Zoccola D, Legros L, Cassuto P et al (2003) A discriminatingscreening is necessary to ascertain EVI1 expression by RT-PCRin malignant cells from the myeloid lineage without 3q26rearrangement. Leukemia 17:643–645
58. Raza A, Buonamici S, Lisak L et al (2004) Arsenic trioxideand thalidomide combination produces multi-lineage hemato-logical responses in myelodysplastic syndromes patients,particularly in those with high pre-therapy EVI1 expression.Leuk Res 28:791–803
59. Morishita K, Parganas E, William CL et al (1992) Activation ofEVI1 gene expression in human acute myelogenous leukemiasby translocations spanning 300–400 kilobases on chromosomeband 3q26. Proc Natl Acad Sci U S A 89:3937–3941
60. Buonamici S, Li D, Chi Y et al (2004) EVI1 induces myelodys-plastic syndrome in mice. J Clin Invest 114:713–719
61. Ikonomi P, Rivera CE, Riordan M et al (2000) Overexpressionof GATA-2 inhibits erythroid and promotes megakaryocytedifferentiation. Exp Hematol 28:1423–1431
62. Sitailo S, Sood R, Barton K, Nucifora G (1999) Forcedexpression of the leukemia-associated gene EVI1 in ES cells:a model for myeloid leukemia with 3q26 rearrangements.Leukemia 13:1639–1645
63. Laricchia-Robbio L, Fazzina R, Li D et al (2006) Pointmutations in two EVI1 Zn fingers abolish EVI1-GATA1
792 Ann Hematol (2008) 87:777–795

interaction and allow erythroid differentiation of murine bonemarrow cells. Mol Cell Biol 26:7658–7666
64. Chakraborty S, Senyuk V, Sitailo S, Chi Y, Nucifora G (2001)Interaction of EVI1 with cAMP-responsive element-bindingprotein-binding protein (CBP) and p300/CBP-associated factor(P/CAF) results in reversible acetylation of EVI1 and in co-localization in nuclear speckles. J Biol Chem 276:44936–44943
65. Liu Y, Chen L, Ko TC, Fields AP, Thompson EA (2006) Evi1 isa survival factor which conveys resistance to both TGFbeta- andtaxol-mediated cell death via PI3K/AKT. Oncogene 25:3565–3575
66. Lai JL, Preudhomme C, Zandecki M et al (1995) Myelodys-plastic syndromes and acute myeloid leukemia with 17pdeletion. An entity characterized by specific dysgranulopoiesisand a high incidence of P53 mutations. Leukemia 9:370–381
67. Herman JG, Baylin SB (2003) Gene silencing in cancer inassociation with promoter hypermethylation. N Engl J Med349:2042–2054
68. Nakamaki T, Bartram C, Seriu T et al (1997) Molecular analysisof the cyclin-dependent kinase inhibitor genes, p15, p16, p18and p19 in the myelodysplastic syndromes. Leuk Res 21:235–240
69. Quesnel B, Guillerm G, Vereecque R et al (1998) Methylation ofthe p15(INK4b) gene in myelodysplastic syndromes is frequentand acquired during disease progression. Blood 91:2985–2990
70. Uchida T, Kinoshita T, Nagai H et al (1997) Hypermethylation ofthe p15INK4B gene in myelodysplastic syndromes. Blood 90:1403–1409
71. Aggerholm A, Holm MS, Guldberg P, Olesen LH, Hokland P(2006) Promoter hypermethylation of p15INK4B, HIC1, CDH1,and ER is frequent in myelodysplastic syndrome and predictspoor prognosis in early-stage patients. Eur J Haematol 76:23–32
72. Hopfer O, Komor M, Koehler IS et al (2007) DNA methylationprofiling of myelodysplastic syndrome hematopoietic progenitorcells during in vitro lineage-specific differentiation. Exp Hematol35:712–723
73. Silverman LR, Demakos EP, Peterson BL et al (2002) Random-ized controlled trial of azacitidine in patients with the myelodys-plastic syndrome: a study of the cancer and leukemia group B.J Clin Oncol 20:2429–2440
74. Daskalakis M, Nguyen TT, Nguyen C et al (2002) Demethyla-tion of a hypermethylated P15/INK4B gene in patients withmyelodysplastic syndrome by 5-Aza-2′-deoxycytidine (decita-bine) treatment. Blood 100:2957–2964
75. Fenrick R, Hiebert SW (1998) Role of histone deacetylases inacute leukemia. J Cell Biochem Suppl 30–31:194–202
76. Kuendgen A, Strupp C, Aivado M et al (2004) Treatment ofmyelodysplastic syndromes with valproic acid alone or incombination with all-trans retinoic acid. Blood 104:1266–1269
77. Kuendgen A, Knipp S, Fox F et al (2005) Results of a phase 2study of valproic acid alone or in combination with all-transretinoic acid in 75 patients with myelodysplastic syndrome andrelapsed or refractory acute myeloid leukemia. Ann Hematol 84(Suppl 1):61–66
78. Cheson BD, Bennett JM, Kantarjian H et al (2000) Report of aninternational working group to standardize response criteria formyelodysplastic syndromes. Blood 96:3671–3674
79. Blum W, Klisovic RB, Hackanson B et al (2007) Phase I studyof decitabine alone or in combination with valproic acid in acutemyeloid leukemia. J Clin Oncol 25:3884–3891
80. Garcia-Manero G, Kantarjian HM, Sanchez-Gonzalez B et al(2006) Phase 1/2 study of the combination of 5-aza-2′-deoxy-cytidine with valproic acid in patients with leukemia. Blood 108:3271–3279
81. Gattermann N, Retzlaff S, Wang YL et al (1996) A hetero-plasmic point mutation of mitochondrial tRNALeu(CUN) innon-lymphoid haemopoietic cell lineages from a patient withacquired idiopathic sideroblastic anaemia. Br J Haematol93:845–855
82. Gattermann N, Retzlaff S, Wang YL et al (1997) Heteroplasmicpoint mutations of mitochondrial DNA affecting subunit I ofcytochrome c oxidase in two patients with acquired idiopathicsideroblastic anemia. Blood 90:4961–4972
83. Gattermann N, Wulfert M, Junge B et al (2004) Ineffectivehematopoiesis linked with a mitochondrial tRNA mutation(G3242A) in a patient with myelodysplastic syndrome. Blood103:1499–1502
84. Reddy PL, Shetty VT, Dutt D et al (2002) Increased incidence ofmitochondrial cytochrome c-oxidase gene mutations in patientswith myelodysplastic syndromes. Br J Haematol 116:564–575
85. Shin MG, Kajigaya S, Levin BC, Young NS (2003) MitochondrialDNA mutations in patients with myelodysplastic syndromes.Blood 101:3118–3125
86. Rieger K, Marinets O, Fietz T et al (2005) Mesenchymal stemcells remain of host origin even a long time after allogeneicperipheral blood stem cell or bone marrow transplantation. ExpHematol 33:605–611
87. Awaya N, Rupert K, Bryant E, Torok-Storb B (2002) Failure ofadult marrow-derived stem cells to generate marrow stroma aftersuccessful hematopoietic stem cell transplantation. Exp Hematol30:937–942
88. Soenen-Cornu V, Tourino C, Bonnet ML et al (2005) Mesen-chymal cells generated from patients with myelodysplasticsyndromes are devoid of chromosomal clonal markers andsupport short- and long-term hematopoiesis in vitro. Oncogene24:2441–2448
89. Alvi S, Shaher A, Shetty V et al (2001) Successful establishmentof long-term bone marrow cultures in 103 patients withmyelodysplastic syndromes. Leuk Res 25:941–954
90. Simmons PJ, Przepiorka D, Thomas ED, Torok-Storb B (1987)Host origin of marrow stromal cells following allogeneic bonemarrow transplantation. Nature 328:429–432
91. Flores-Figueroa E, Gutierrez-Espindola G, Montesinos JJ,Arana-Trejo RM, Mayani H (2002) In vitro characterizationof hematopoietic microenvironment cells from patients withmyelodysplastic syndrome. Leuk Res 26:677–686
92. Flores-Figueroa E, Arana-Trejo RM, Gutierrez-Espindola G,Perez-Cabrera A, Mayani H (2005) Mesenchymal stem cells inmyelodysplastic syndromes: phenotypic and cytogenetic charac-terization. Leuk Res 29:215–224
93. Blau O, Hofmann WK, Baldus CD et al (2007) Chromosomalaberrations in bone marrow mesenchymal stroma cells frompatients with myelodysplastic syndrome and acute myeloblasticleukemia. Exp Hematol 35:221–229
94. Aguayo A, Kantarjian H, Manshouri T et al (2000) Angiogenesisin acute and chronic leukemias and myelodysplastic syndromes.Blood 96:2240–2245
95. Keith T, Araki Y, Ohyagi M et al (2007) Regulation ofangiogenesis in the bone marrow of myelodysplastic syndromestransforming to overt leukaemia. Br J Haematol 137:206–215
96. Stifter G, Heiss S, Gastl G, Tzankov A, Stauder R (2005) Over-expression of tumor necrosis factor-alpha in bone marrowbiopsies from patients with myelodysplastic syndromes: rela-tionship to anemia and prognosis. Eur J Haematol 75:485–491
97. Campioni D, Punturieri M, Bardi A et al (2004) “In vitro”evaluation of bone marrow angiogenesis in myelodysplasticsyndromes: a morphological and functional approach. Leuk Res28:9–17
Ann Hematol (2008) 87:777–795 793

98. Raza A, Candoni A, Khan U et al (2004) Remicade as TNFsuppressor in patients with myelodysplastic syndromes. LeukLymphoma 45:2099–2104
99. Stasi R, Amadori S (2002) Infliximab chimaeric anti-tumournecrosis factor alpha monoclonal antibody treatment for patientswith myelodysplastic syndromes. Br J Haematol 116:334–337
100. Roboz GJ, Giles FJ, List AF et al (2006) Phase 1 study ofPTK787/ZK 222584, a small molecule tyrosine kinase receptorinhibitor, for the treatment of acute myeloid leukemia andmyelodysplastic syndrome. Leukemia 20:952–957
101. Mohamedali A, Gaken J, Twine NA et al (2007) Prevalence andprognostic significance of allelic imbalance by single nucleotidepolymorphism analysis in low risk myelodysplastic syndromes.Blood 110:3365–3373
102. Hofmann WK, de Vos S, Komor M et al (2002) Characterizationof gene expression of CD34 + cells from normal and myelodys-plastic bone marrow. Blood 100:3553–3560
103. Miyazato A, Ueno S, Ohmine K et al (2001) Identification ofmyelodysplastic syndrome-specific genes by DNA microarrayanalysis with purified hematopoietic stem cell fraction. Blood98:422–427
104. Lee YT, Miller LD, Gubin AN et al (2001) Transcriptionpatterning of uncoupled proliferation and differentiation inmyelodysplastic bone marrow with erythroid-focused arrays.Blood 98:1914–1921
105. Schoch C, Kohlmann A, Schnittger S et al (2002) Acute myeloidleukemias with reciprocal rearrangements can be distinguishedby specific gene expression profiles. Proc Natl Acad Sci U S A99:10008–10013
106. Alizadeh AA, Eisen MB, Davis RE et al (2000) Distinct types ofdiffuse large B-cell lymphoma identified by gene expressionprofiling. Nature 403:503–511
107. Mauro C, Pacifico F, Lavorgna A et al (2006) ABIN-1 bindsto NEMO/IKKgamma and co-operates with A20 in inhibitingNF-kappaB. J Biol Chem 281:18482–18488
108. Zhang S, Fukushi M, Hashimoto S et al (2002) A new ERK2binding protein, Naf1, attenuates the EGF/ERK2 nuclearsignaling. Biochem Biophys Res Commun 297:17–23
109. Casas S, Nagy B, Elonen E et al (2003) Aberrant expression ofHOXA9, DEK, CBL and CSF1R in acute myeloid leukemia.Leuk Lymphoma 44:1935–1941
110. Suh ER, Ha CS, Rankin EB, Toyota M, Traber PG (2002) DNAmethylation down-regulates CDX1 gene expression in colorectalcancer cell lines. J Biol Chem 277:35795–35800
111. Grewal T, Evans R, Rentero C et al (2005) Annexin A6stimulates the membrane recruitment of p120GAP to modulateRas and Raf-1 activity. Oncogene 24:5809–5820
112. Gou D, Wang J, Gao L et al (2004) Identification and functionalanalysis of a novel human KRAB/C2H2 zinc finger geneZNF300. Biochim Biophys Acta 1676:203–209
113. Kaddu S, Zenahlik P, Beham-Schmid C, Kerl H, Cerroni L(1999) Specific cutaneous infiltrates in patients with myeloge-nous leukemia: a clinicopathologic study of 26 patients withassessment of diagnostic criteria. J Am Acad Dermatol 40:966–978
114. Wirtenberger M, Tchatchou S, Hemminki K et al (2006)Associations of genetic variants in the estrogen receptorcoactivators PPARGC1A, PPARGC1B and EP300 with familialbreast cancer. Carcinogenesis 27:2201–2208
115. Srivastava S, Barrett JN, Moraes CT (2007) PGC-1alpha/betaupregulation is associated with improved oxidative phosphory-lation in cells harboring nonsense mtDNA mutations. Hum MolGenet 16:993–1005
116. Lierman E, Lahortiga I, Van Miegroet H et al (2007) The abilityof sorafenib to inhibit oncogenic PDGFRbeta and FLT3 mutants
and overcome resistance to other small molecule inhibitors.Haematologica 92:27–34
117. Gorello P, La Starza R, Brandimarte L et al (2008) A PDGFRB-positive acute myeloid malignancy with a new t(5;12)(q33;p13.3) involving the ERC1 gene. Leukemia 22:216–218
118. Tokita K, Maki K, Tadokoro J et al (2007) Chronic idiopathicmyelofibrosis expressing a novel type of TEL-PDGFRB chi-maera responded to imatinib mesylate therapy. Leukemia21:190–192
119. Monma F, Nishii K, Lorenzo F et al (2006) Molecular analysis ofPDGFRalpha/beta genes in core binding factor leukemia witheosinophilia. Eur J Haematol 76:18–22
120. Grand FH, Burgstaller S, Kuhr T et al (2004) p53-Bindingprotein 1 is fused to the platelet-derived growth factor receptorbeta in a patient with a t(5;15)(q33;q22) and an imatinib-responsive eosinophilic myeloproliferative disorder. Cancer Res64:7216–7219
121. Kulkarni S, Heath C, Parker S et al (2000) Fusion of H4/D10S170 to the platelet-derived growth factor receptor betain BCR-ABL-negative myeloproliferative disorders with at(5;10)(q33;q21). Cancer Res 60:3592–3598
122. Sanz L, Diaz-Meco MT, Nakano H, Moscat J (2000) Theatypical PKC-interacting protein p62 channels NF-kappaBactivation by the IL-1-TRAF6 pathway. EMBO J 19:1576–1586
123. Zhao Y, Qin S, Atangan LI et al (2004) Casein kinase 1alphainteracts with retinoid X receptor and interferes with agonist-induced apoptosis. J Biol Chem 279:30844–30849
124. Itoh S, Kim HW, Nakagawa O et al (2008) Novel role ofantioxidant-1 (atox1) as a copper dependent transcription factorinvolved in cell proliferation. J Biol Chem 283:9157–9167
125. Yu YP, Yu G, Tseng G et al (2007) Glutathione peroxidase 3,deleted or methylated in prostate cancer, suppresses prostatecancer growth and metastasis. Cancer Res 67:8043–8050
126. Lee OJ, Schneider-Stock R, McChesney PA et al (2005)Hypermethylation and loss of expression of glutathioneperoxidase-3 in Barrett's tumorigenesis. Neoplasia 7:854–861
127. Fuster MM, Wang L, Castagnola J et al (2007) Genetic alterationof endothelial heparan sulfate selectively inhibits tumor angio-genesis. J Cell Biol 177:539–549
128. Yuan K, Chung LW, Siegal GP, Zayzafoon M (2007) alpha-CaMKII controls the growth of human osteosarcoma byregulating cell cycle progression. Lab Invest 87:938–950
129. Backs J, Backs T, Bezprozvannaya S, McKinsey TA, Olson EN(2008) Histone deacetylase 5 acquires calcium/calmodulin-dependent kinase II responsiveness by oligomerization withhistone deacetylase 4. Mol Cell Biol 28(10):3437–3445
130. Liu AM, Wong YH (2005) Activation of nuclear factor {kappa}Bby somatostatin type 2 receptor in pancreatic acinar AR42J cellsinvolves G{alpha}14 and multiple signaling components: amechanism requiring protein kinase C, calmodulin-dependentkinase II, ERK, and c-Src. J Biol Chem 280:34617–34625
131. McClellan KA, Slack RS (2007) Specific in vivo roles forE2Fs in differentiation and development. Cell Cycle 6:2917–2927
132. Black AR, Azizkhan-Clifford J (1999) Regulation of E2F: afamily of transcription factors involved in proliferation control.Gene 237:281–302
133. Mizuno S, Chijiwa T, Okamura T et al (2001) Expression ofDNA methyltransferases DNMT1, 3A, and 3B in normalhematopoiesis and in acute and chronic myelogenous leukemia.Blood 97:1172–1179
134. Tadokoro Y, Ema H, Okano M, Li E, Nakauchi H (2007) Denovo DNA methyltransferase is essential for self-renewal, butnot for differentiation, in hematopoietic stem cells. J Exp Med204:715–722
794 Ann Hematol (2008) 87:777–795

135. Suzuki M, Yamada T, Kihara-Negishi F et al (2006) Site-specificDNA methylation by a complex of PU.1 and Dnmt3a/b.Oncogene 25:2477–2488
136. Evans R, Naber C, Steffler T et al (2008) Aurora A kinase RNAiand small molecule inhibition of Aurora kinases with VE-465induce apoptotic death in multiple myeloma cells. LeukLymphoma 49:559–569
137. Wittmann T, Wilm M, Karsenti E, Vernos I (2000) TPX2, Anovel xenopus MAP involved in spindle pole organization. JCell Biol 149:1405–1418
138. Eckerdt F, Eyers PA, Lewellyn AL, Prigent C, Maller JL (2008)Spindle pole regulation by a discrete Eg5-interacting domain inTPX2. Curr Biol 18:519–525
139. Ries C, Loher F, Zang C, Ismair MG, Petrides PE (1999) Matrixmetalloproteinase production by bone marrow mononuclear cellsfrom normal individuals and patients with acute and chronicmyeloid leukemia or myelodysplastic syndromes. Clin CancerRes 5:1115–1124
140. Travaglino E, Benatti C, Malcovati L et al (2008) Biological andclinical relevance of matrix metalloproteinases 2 and 9 in acutemyeloid leukaemias and myelodysplastic syndromes. Eur JHaematol 80:216–226
141. LeCouter JE, Kablar B, Hardy WR et al (1998) Strain-dependentmyeloid hyperplasia, growth deficiency, and accelerated cellcycle in mice lacking the Rb-related p107 gene. Mol Cell Biol18:7455–7465
142. Balciunaite E, Spektor A, Lents NH et al (2005) Pocket proteincomplexes are recruited to distinct targets in quiescent andproliferating cells. Mol Cell Biol 25:8166–8178
143. Clark AJ, Doyle KM, Humbert PO (2004) Cell-intrinsicrequirement for pRb in erythropoiesis. Blood 104:1324–1326
144. Walkley CR, Orkin SH (2006) Rb is dispensable for self-renewaland multilineage differentiation of adult hematopoietic stemcells. Proc Natl Acad Sci U S A 103:9057–9062
145. Allen TC, Granville LA, Cagle PT et al (2007) Expression ofglutathione S-transferase pi and glutathione synthase correlateswith survival in early stage non-small cell carcinomas of thelung. Hum Pathol 38:220–227
146. Lu J, Chew EH, Holmgren A (2007) Targeting thioredoxinreductase is a basis for cancer therapy by arsenic trioxide. ProcNatl Acad Sci U S A 104:12288–12293
147. Fok JY, Ekmekcioglu S, Mehta K (2006) Implications of tissuetransglutaminase expression in malignant melanoma. MolCancer Ther 5:1493–1503
148. Ai L, Kim WJ, Demircan B et al (2008) The transglutaminase 2gene (TGM2), a potential molecular marker for chemotherapeu-tic drug sensitivity, is epigenetically silenced in breast cancer.Carcinogenesis 29:510–518
149. Horowitz A, Tkachenko E, Simons M (2002) Fibroblast growthfactor-specific modulation of cellular response by syndecan-4.J Cell Biol 157:715–725
150. Chen Q, Sivakumar P, Barley C et al (2007) Potential role forheparan sulfate proteoglycans in regulation of transforminggrowth factor-beta (TGF-beta) by modulating assembly oflatent TGF-beta-binding protein-1. J Biol Chem 282:26418–26430
151. Gupta P, Oegema TR Jr, Brazil JJ et al (1998) Structurallyspecific heparan sulfates support primitive human hematopoiesisby formation of a multimolecular stem cell niche. Blood 92:4641–4651
152. Charnaux N, Brule S, Hamon M et al (2005) Syndecan-4 is asignaling molecule for stromal cell-derived factor-1 (SDF-1)/CXCL12. FEBS J 272:1937–1951
153. Drzeniek Z, Stocker G, Siebertz B et al (1999) Heparan sulfateproteoglycan expression is induced during early erythroiddifferentiation of multipotent hematopoietic stem cells. Blood93:2884–2897
154. Chendrimada TP, Finn KJ, Ji X et al (2007) MicroRNA silencingthrough RISC recruitment of eIF6. Nature 447:823–828
155. Lee SK, Kim HJ, Kim JW, Lee JW (1999) Steroid receptorcoactivator-1 and its family members differentially regulatetransactivation by the tumor suppressor protein p53. MolEndocrinol 13:1924–1933
156. Lutz T, Stoger R, Nieto A (2006) CHD6 is a DNA-dependentATPase and localizes at nuclear sites of mRNA synthesis. FEBSLett 580:5851–5857
157. Surapureddi S, Yu S, Bu H et al (2002) Identification of atranscriptionally active peroxisome proliferator-activated recep-tor alpha -interacting cofactor complex in rat liver andcharacterization of PRIC285 as a coactivator. Proc Natl AcadSci U S A 99:11836–11841
158. Golay J, Broccoli V, Borleri GM et al (1997) Redundantfunctions of B-Myb and c-Myb in differentiating myeloid cells.Cell Growth Differ 8:1305–1316
159. Sala A (2005) B-MYB, a transcription factor implicated inregulating cell cycle, apoptosis and cancer. Eur J Cancer 41:2479–2484
160. Grassilli E, Salomoni P, Perrotti D, Franceschi C, Calabretta B(1999) Resistance to apoptosis in CTLL-2 cells overexpressingB-Myb is associated with B-Myb-dependent bcl-2 induction.Cancer Res 59:2451–2456
161. Muller-Tidow C, Wang W, Idos GE et al (2001) Cyclin A1directly interacts with B-myb and cyclin A1/cdk2 phosphorylateB-myb at functionally important serine and threonine residues:tissue-specific regulation of B-myb function. Blood 97:2091–2097
162. Kim YK, Furic L, Parisien M et al (2007) Staufen1 regulatesdiverse classes of mammalian transcripts. EMBO J 26:2670–2681
163. Nerlov C (2007) The C/EBP family of transcription factors: aparadigm for interaction between gene expression and prolifer-ation control. Trends Cell Biol 17:318–324
Ann Hematol (2008) 87:777–795 795