myelodysplastic syndromes, aplastic anemia, & other bone marrow
TRANSCRIPT

Myelodysplastic Syndromes,
Aplastic Anemia, & Other Bone
Marrow Failure States
ASCP 10-22-11
Friederike Kreisel, MD
Washington University

Faculty/Author/Speaker Disclosure
The faculty/speakers for this live session do
not have relevant relationships with
commercial interests to disclose
Credit type: HEME, SURGCredit type: HEME, SURG

Myelodysplastic Syndromes (MDS)
• Clonal proliferation combined with
increased apoptosis
– Cytopenia(s)– Cytopenia(s)
– Dysplasia in one or more myeloid lines
– Ineffective hematopoiesis
– Increased risk of transformation to acute
myeloid leukemia (~30% of cases)

Myelodysplastic Syndromes (MDS)
• Incidence has increased from 3.3 per 100,000 in
2001 to 3.8 per 100,000 in 2006
– ~10% of MDS patients are <50 years old
– Exceedingly rare in children (0.01 per 100,000)– Exceedingly rare in children (0.01 per 100,000)
• The increase has been attributed to
– Enhanced awareness
– Aging population
– Availability of more effective therapies, making
hematologists more likely to pursue the diagnosis

Pathophysiology of MDS• Genetic alterations
– del(5q) is best understood MDS-associated molecular abnormality: RPS14 (codes for a ribosomal subunit) has been shown to induce a MDS phenotype when experimentally inhibited and to correct the MDS phenotype when resupplied to cells with del(5q) phenotype when resupplied to cells with del(5q)
• Epigenetic alterations– DNA hypermethylation – silences gene transcription by hypermethylation of CpG islands in promotor regions – silencing of tumor suppressor genes – may to some degree be reversed by cytidine analogues (i.e. decitabine) that irreversibly bind DNA methyltransferases – decreased methylation throughout the genome

Case #1
• 69-year old female with pancytopenia:
– WBC: 2.2 (4.5-11 K/cumm)
– Hemoglobin: 10.6 (13.0-17.0 g/dl)– Hemoglobin: 10.6 (13.0-17.0 g/dl)
– Platelets: 103 (140-400 K/cumm)
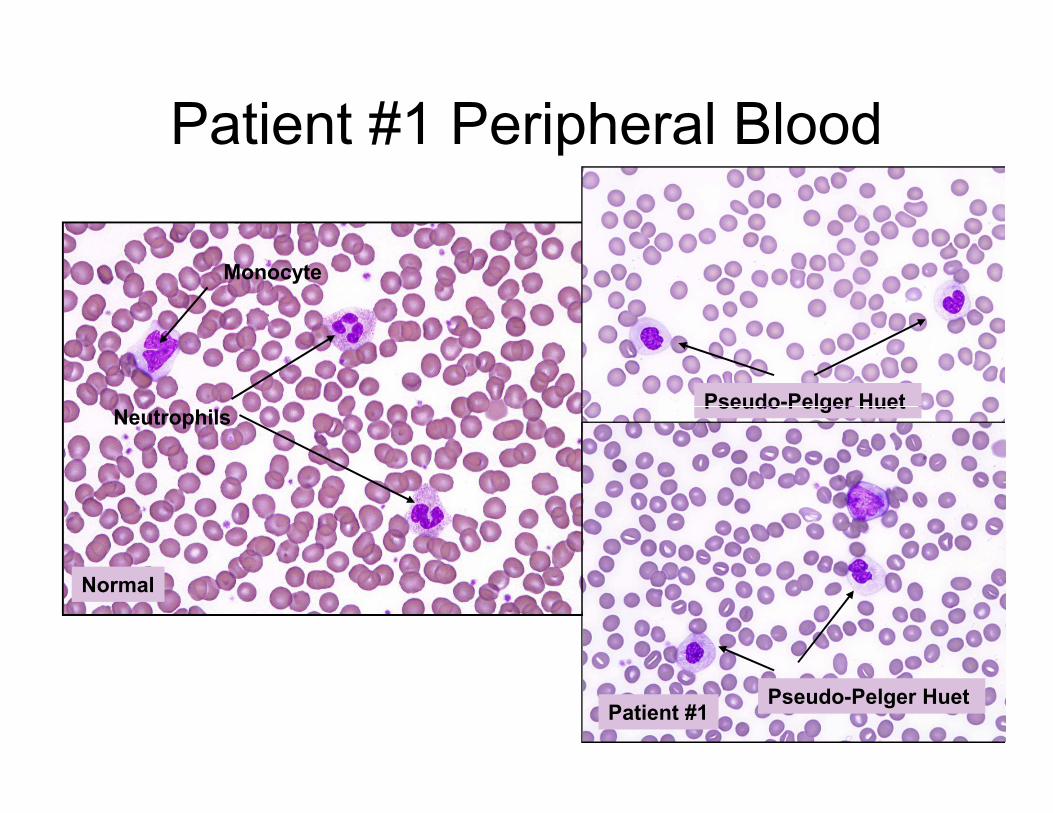
Patient #1 Peripheral Blood
Neutrophils
Monocyte
Pseudo-Pelger Huet
Normal
Neutrophils
Patient #1
Pseudo-Pelger Huet
Pseudo-Pelger Huet
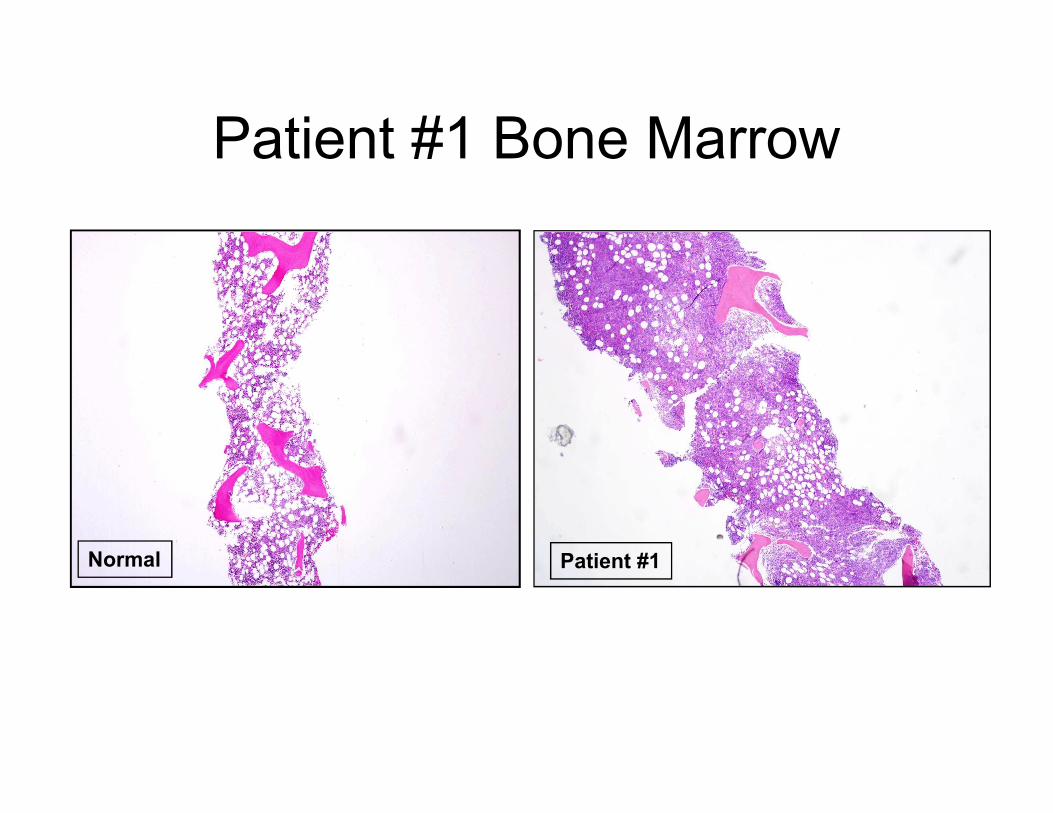
Patient #1 Bone Marrow
Normal Patient #1
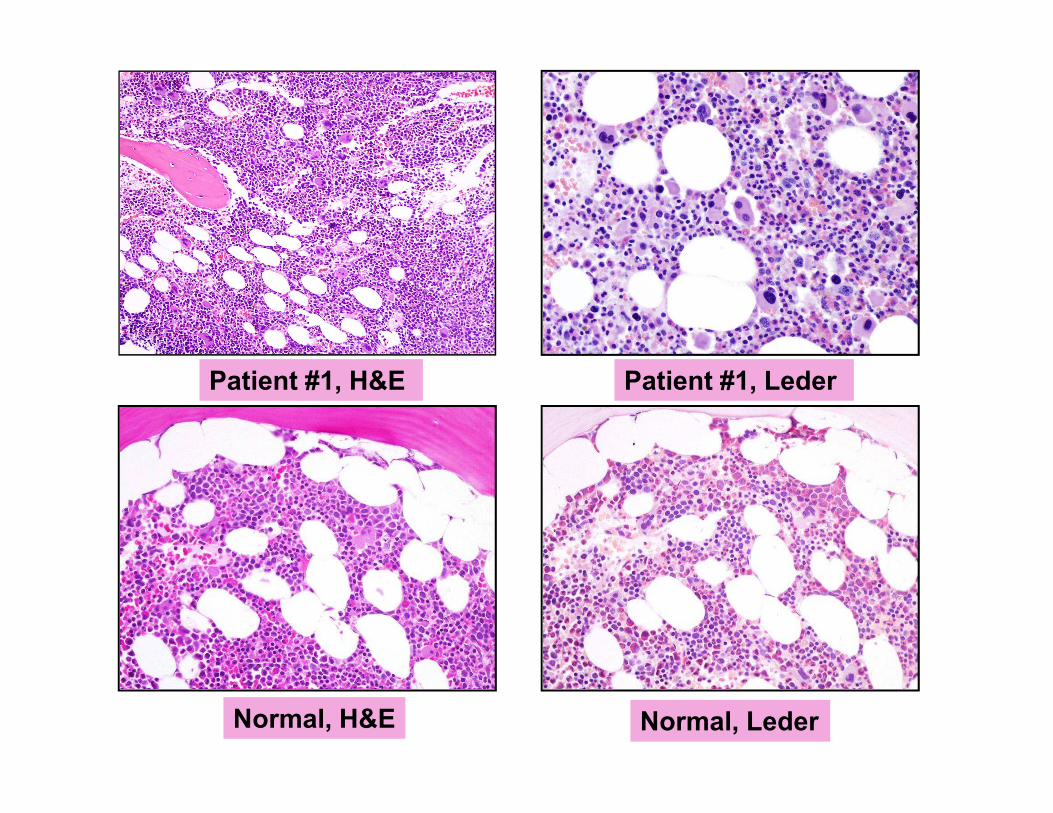
Patient #1, LederPatient #1, H&E
Normal, H&E Normal, Leder

Spicules
Normal
Patient #1
Spicules
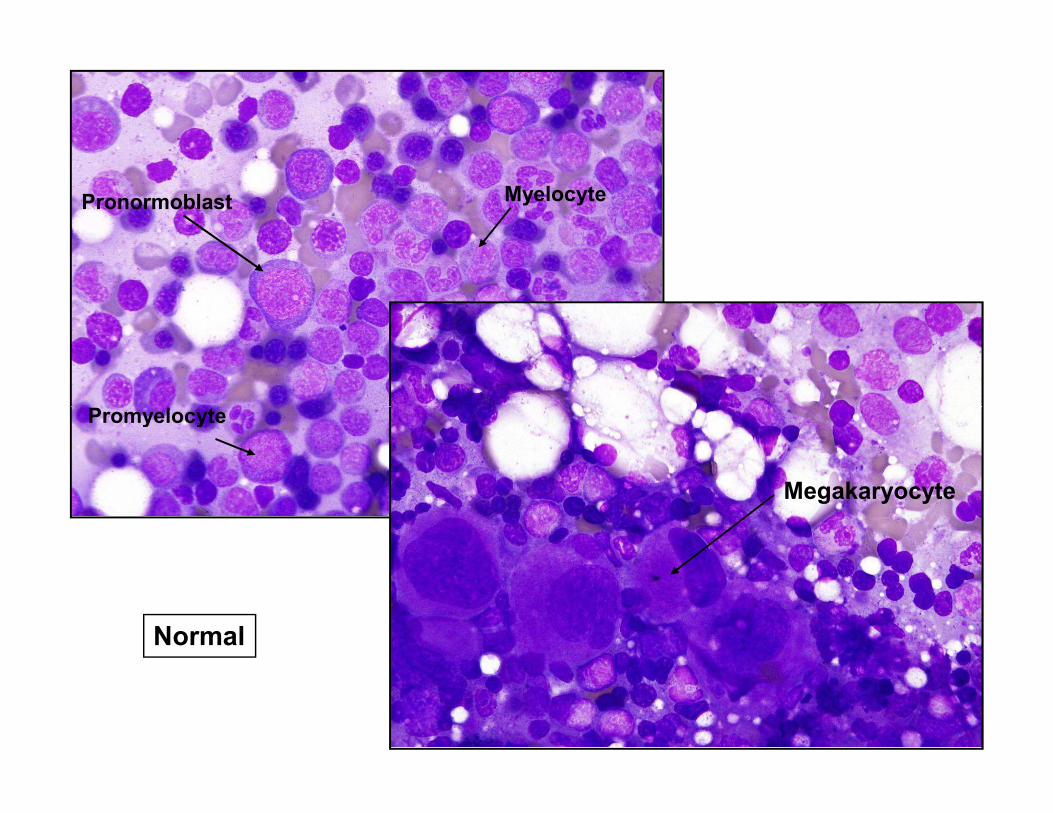
Promyelocyte
Pronormoblast Myelocyte
Normal
Megakaryocyte
Promyelocyte
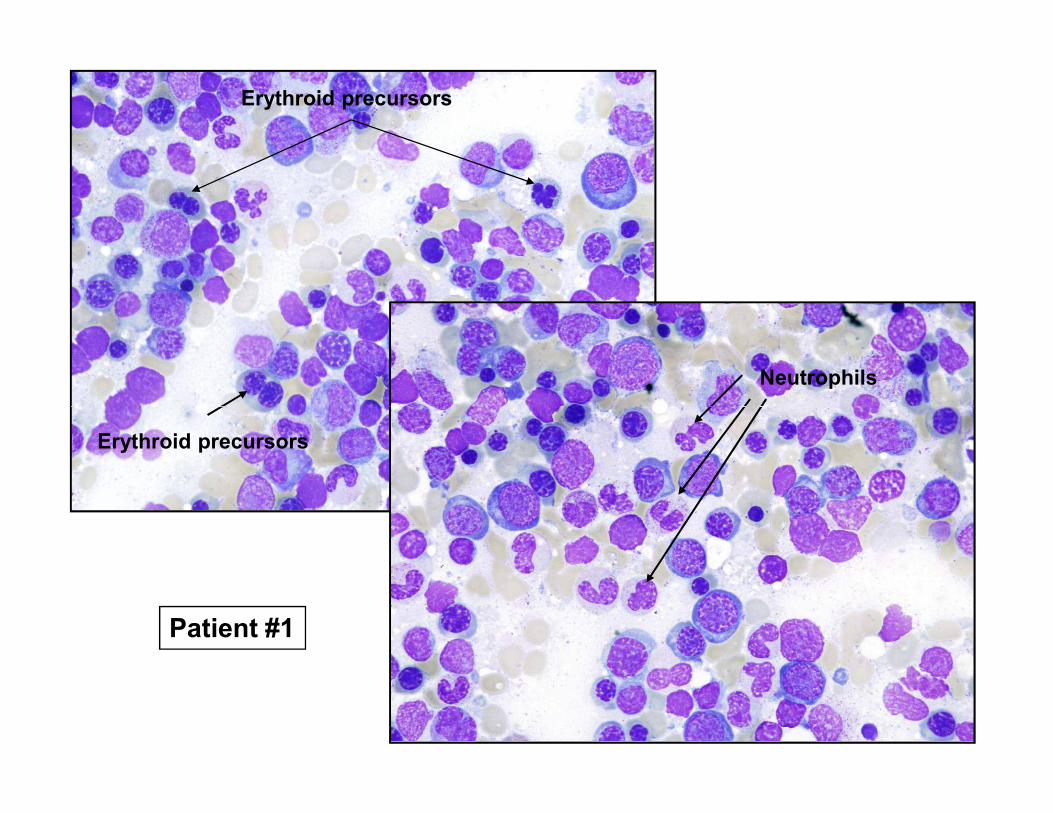
Erythroid precursors
Neutrophils
Erythroid precursors
Patient #1
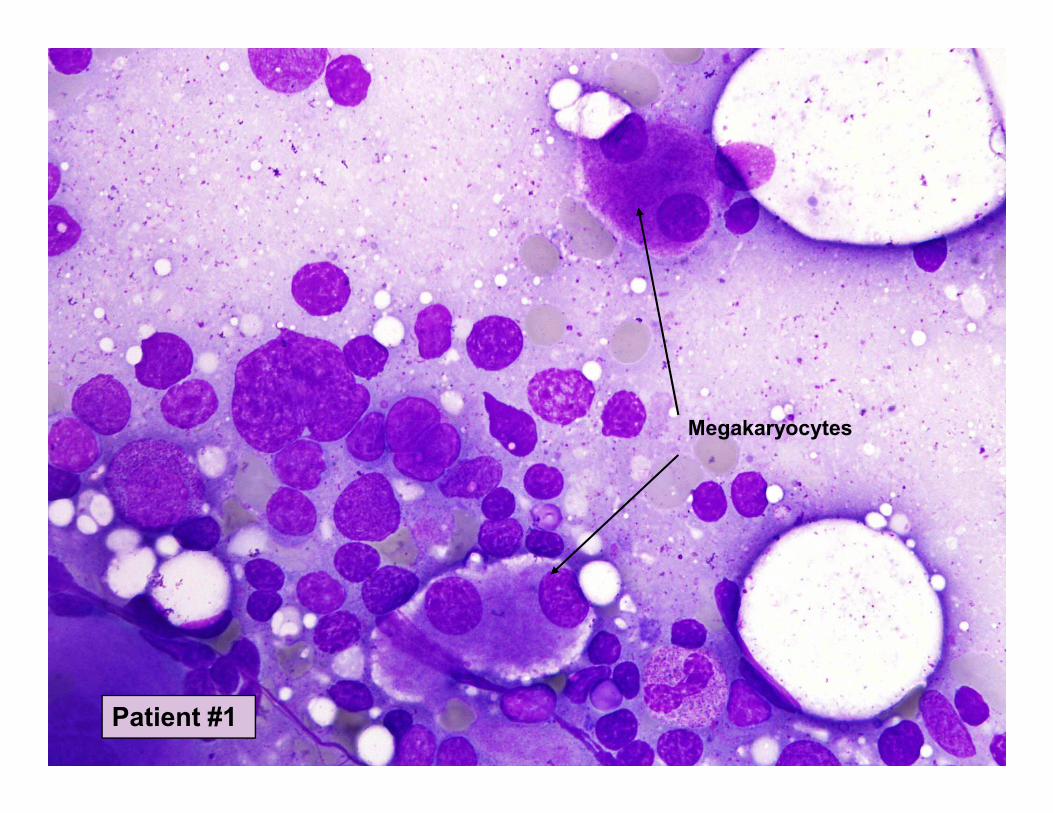
Megakaryocytes
Patient #1
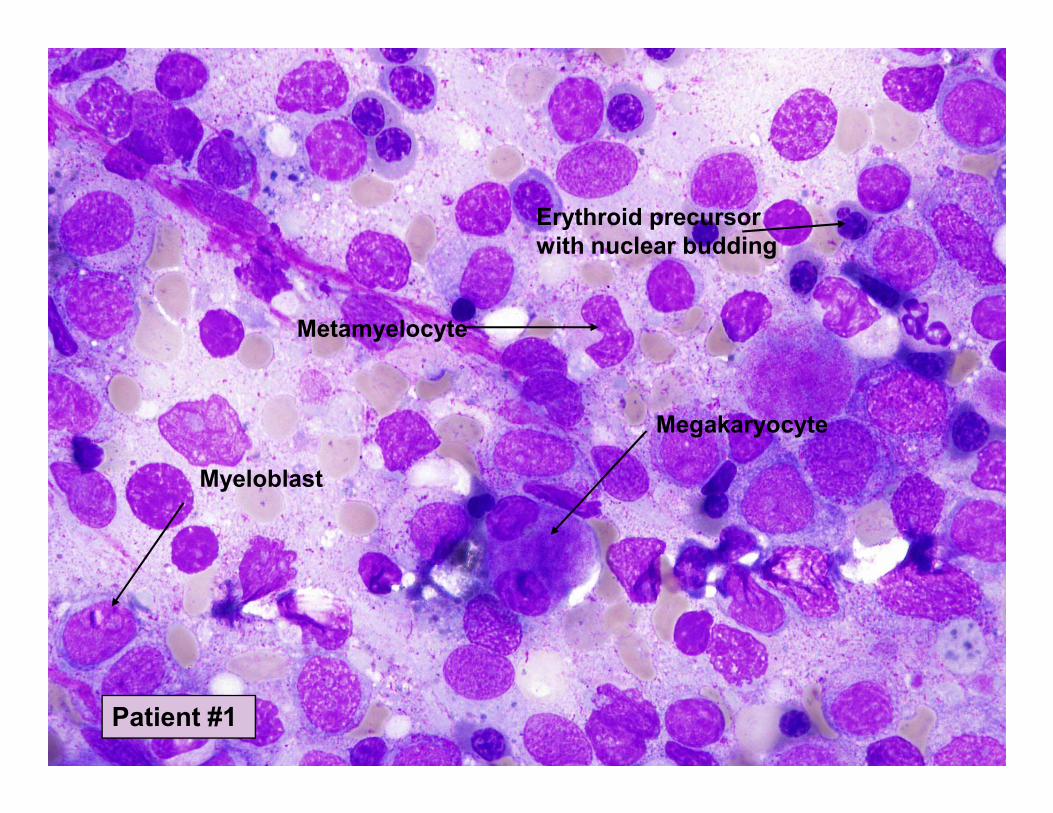
Erythroid precursor
with nuclear budding
Metamyelocyte
Myeloblast
Megakaryocyte
Patient #1
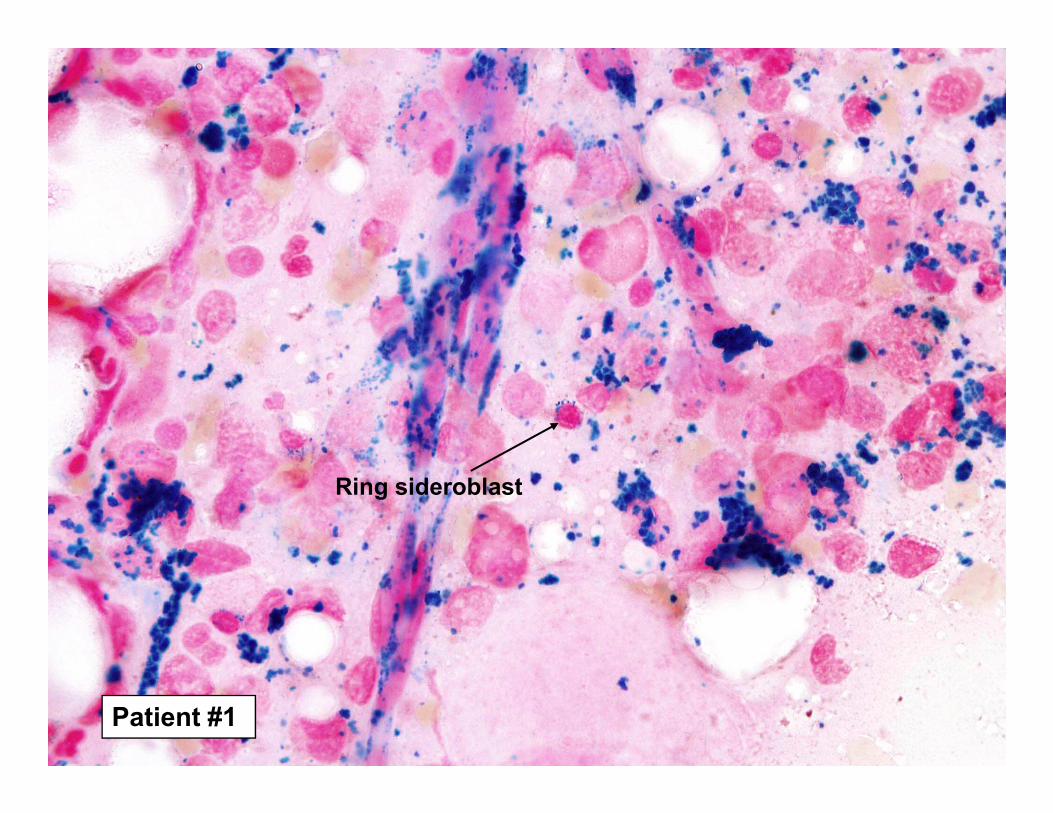
Ring sideroblast
Patient #1
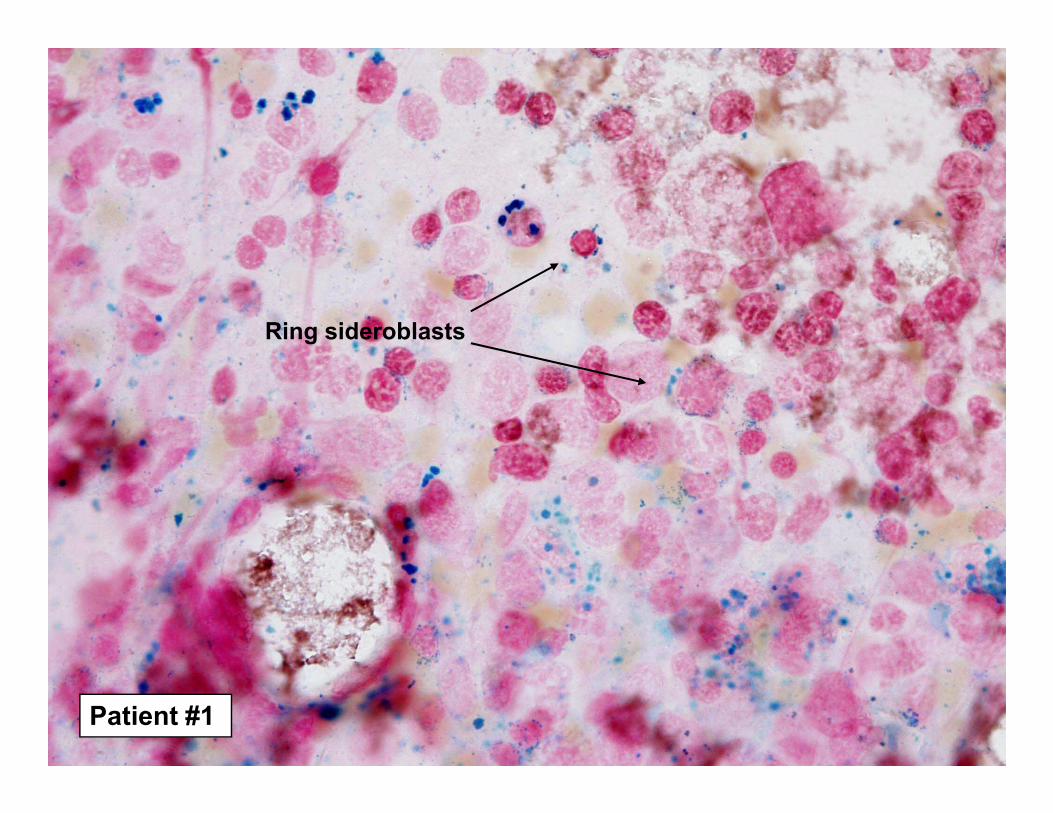
Ring sideroblasts
Patient #1

Cytogenetics
• Fluorescence in situ hybridization for MDS
panel:
– -7/del7q (D7S486): negative
– -5/del5q (EGR1): negative
– del20q (D20s108): negative
– 12p13 (ETV6): negative
– +8 (CEP8): negative
– del13q (D13S319): negative
• Normal conventional karyotype

DiagnosisHypercellular marrow for age with multilineage
dysplasia; no excess blasts seen
Comment: Dyspoiesis is a morphologic change that
can be due to myelotoxic / myelosuppressive
drugs, nutritional deficiency, chronic viral infection, drugs, nutritional deficiency, chronic viral infection,
severe or sustained inflammatory conditions, or
primary myelodysplastic syndrome (MDS).
Diagnosis of MDS, specifically, refractory
cytopenia with multilineage dysplasia may be
considered if all secondary causes are excluded.
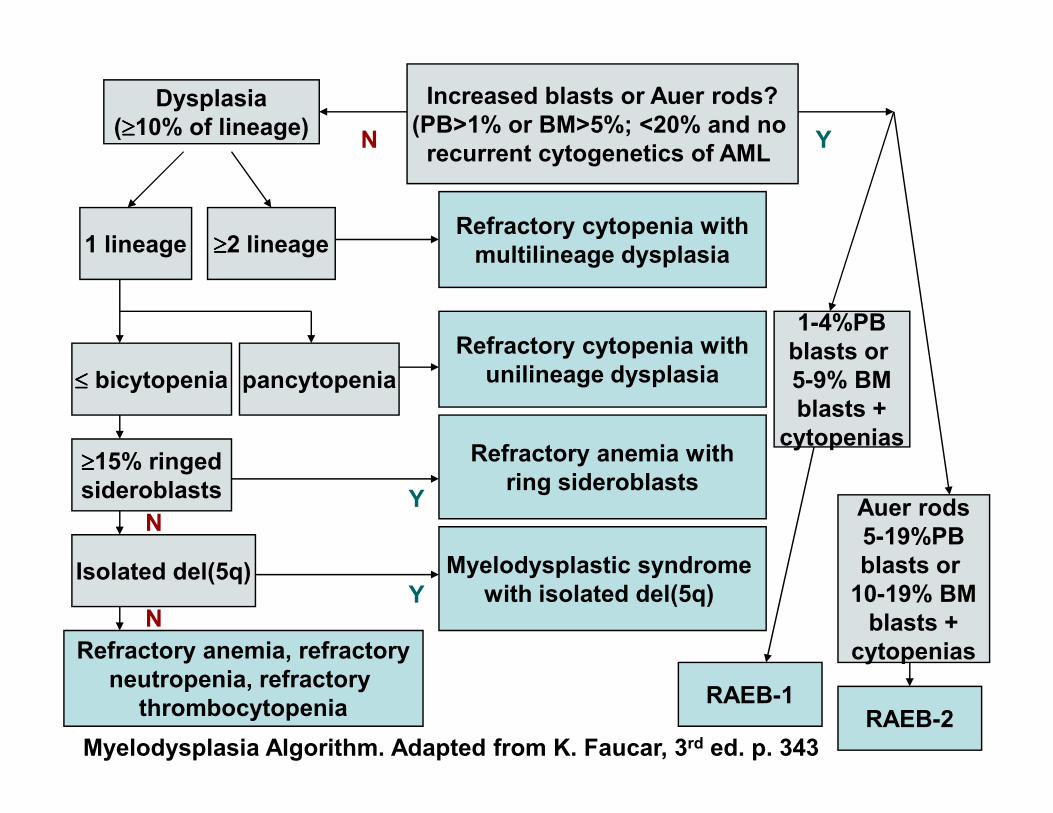
Dysplasia
(≥≥≥≥10% of lineage)
Increased blasts or Auer rods?
(PB>1% or BM>5%; <20% and no
recurrent cytogenetics of AMLN
1 lineage ≥≥≥≥2 lineageRefractory cytopenia with
multilineage dysplasia
≤≤≤≤ bicytopenia pancytopenia
Refractory cytopenia with
unilineage dysplasia
Y
1-4%PB
blasts or
5-9% BM
blasts +
≥≥≥≥15% ringed
sideroblasts
Refractory anemia with
ring sideroblastsY
Isolated del(5q) Myelodysplastic syndrome
with isolated del(5q)
Refractory anemia, refractory
neutropenia, refractory
thrombocytopenia
YN
N
blasts +
cytopenias
Auer rods
5-19%PB
blasts or
10-19% BM
blasts +
cytopenias
RAEB-2RAEB-1
Myelodysplasia Algorithm. Adapted from K. Faucar, 3rd ed. p. 343

Approach to the Diagnosis of MDS
• Correlate with CBC (cytopenia or cytosis?)
• Perform an iron stain on aspirate, core or clot section– Ringed sideroblasts can be found in
• Myelodysplastic syndrome• Myelodysplastic syndrome
• Sideroblastic anemia
• Alcoholism
• Hyposplenism
• Lead poisoning
• Cooper deficiency
• Zinc intoxication
• Drugs (isoniazid, chloramphenicol)
From Color Atlas
of Hematology,
E. Glassy

Approach to the Diagnosis of MDS
• Check laboratory values for
– Reticulocytes
– Cooper
– Vit. B12 and folate
• Correlate with clinical presentation • Correlate with clinical presentation
(splenomegaly?, HIV status?, age, sudden or
insidious onset of symptoms, medications:
cotrimoxazole, zinc containing cold remedies)
• Perform flow cytometry to assess for clonal B-cell
population (hairy cell leukemia?) or abnormal T-
cells (T-cell LGL leukemia?)

Approach to the Diagnosis of MDS
• Conventional cytogenetics: Essential diagnostic test; requires adequate BM aspirate specimen
• Fluorescence in situ hybridization for MDS panel (fast turn-around-time 24-48hrs):– -7/del7q (D7S486); 5/del5q (EGR1); del20q
(D20s108); 12p13 (ETV6); +8 (CEP8); del13q (D20s108); 12p13 (ETV6); +8 (CEP8); del13q (D13S319)
• Flow cytometry: Not required for either blast enumeration or assessment of dysplasia
• Diagnostic interpretation: Requires integration of clinical, hematologic, morphologic, and genetic findings– Provide WHO 2008 subtype
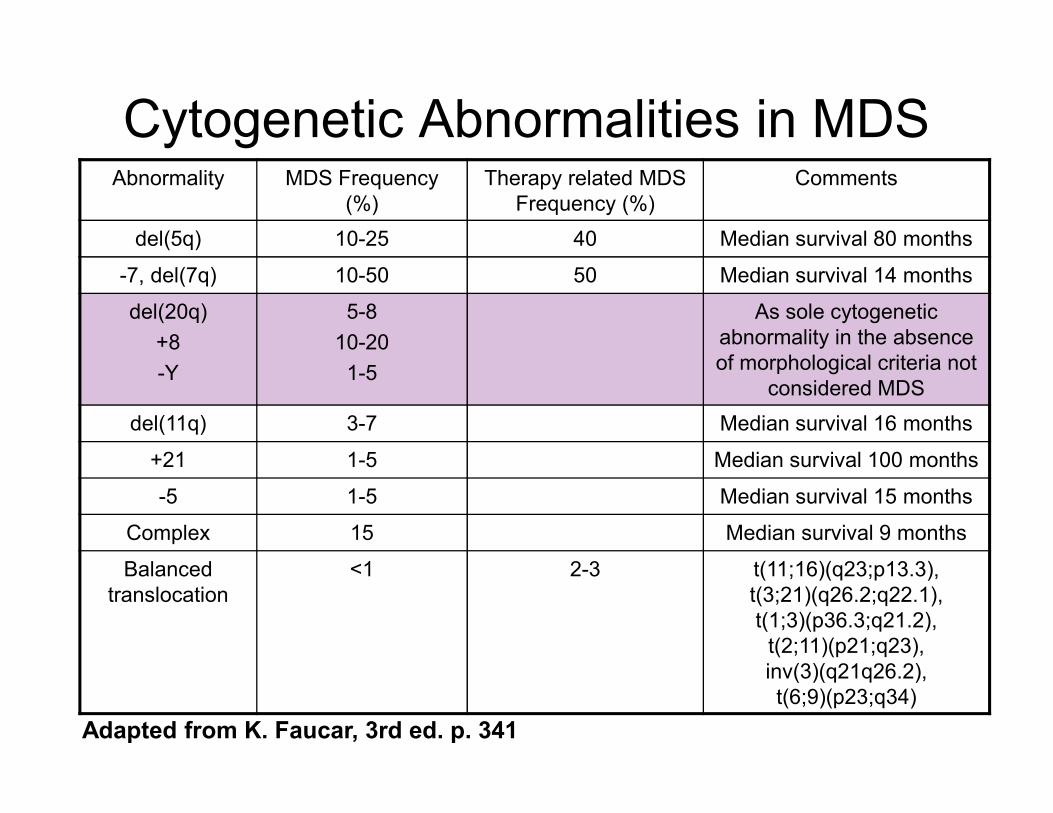
Cytogenetic Abnormalities in MDS Abnormality MDS Frequency
(%)
Therapy related MDS
Frequency (%)
Comments
del(5q) 10-25 40 Median survival 80 months
-7, del(7q) 10-50 50 Median survival 14 months
del(20q)
+8
-Y
5-8
10-20
1-5
As sole cytogenetic
abnormality in the absence
of morphological criteria not
considered MDS
del(11q) 3-7 Median survival 16 months
+21 1-5 Median survival 100 months
-5 1-5 Median survival 15 months
Complex 15 Median survival 9 months
Balanced
translocation
<1 2-3 t(11;16)(q23;p13.3),
t(3;21)(q26.2;q22.1),
t(1;3)(p36.3;q21.2),
t(2;11)(p21;q23),
inv(3)(q21q26.2),
t(6;9)(p23;q34)
Adapted from K. Faucar, 3rd ed. p. 341

Case #2
• 31-year old male who was transferred to
the hematology/oncology service with
chief complaint of pancytopenia, fatigue,
and dyspnea on exertion:and dyspnea on exertion:
• Complete blood count:
– WBC: 2.9 (4.5-11 K/cumm)
– Hemoglobin: 7.7 (13.0-17.0 g/dl)
– Platelets: 62 (140-400 K/cumm)
Patient #2
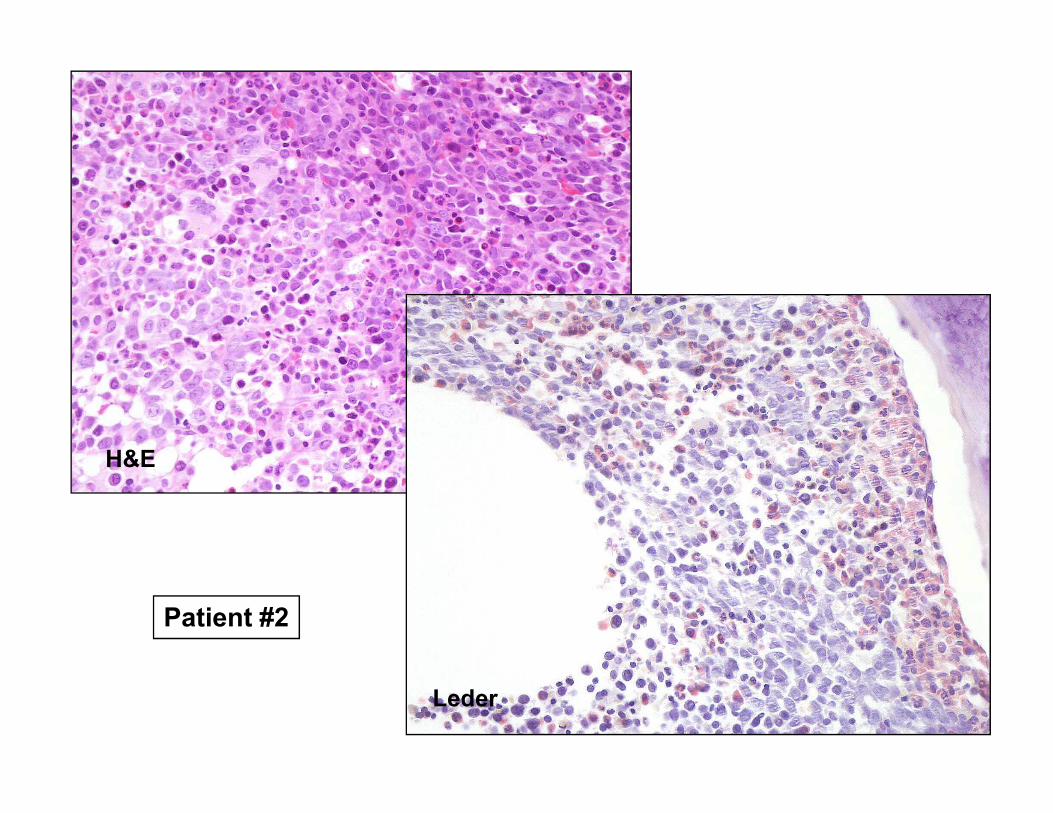
Patient #2
Leder
H&E

Patient #2
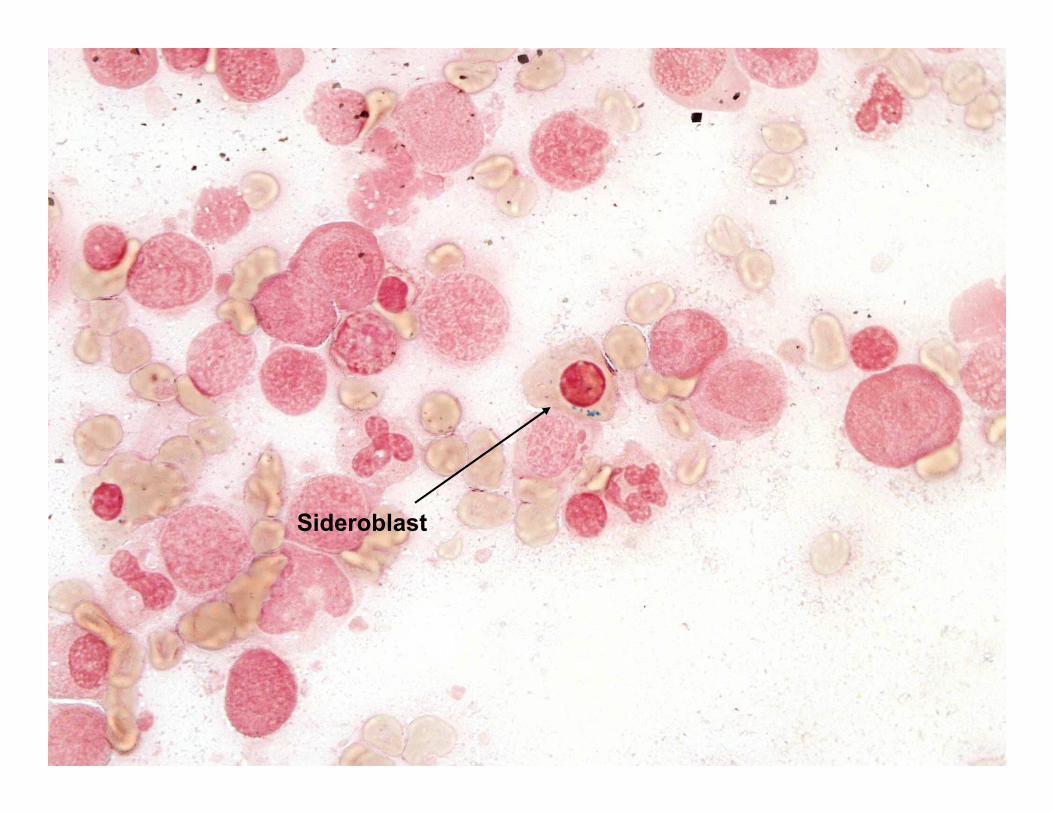
Sideroblast

Diagnosis
Hypercellular marrow with erythroid
hyperplasia and megaloblastic changes;
no excess blasts seen
Review of laboratory data revealed a B12
deficiency of 77 (211-911 pg/ml).

Diagnosis (without clinical or
laboratory information)
Hypercellular marrow with erythroid hyperplasia and megaloblastic changes; no excess blasts seen
Comment: Dyspoiesis is a morphologic change that can be due to myelotoxic / that can be due to myelotoxic / myelosuppressive drugs, nutritional deficiency, chronic viral infection, severe or sustained inflammatory conditions, or primary myelodysplastic syndrome (MDS). Diagnosis of MDS may be considered if all secondary causes are excluded, and established if a MDS associated genetic abnormality is present .

Conditions Mimicking MDS
Clinically and Morphologically
– Viral infections (HIV, Parvovirus B19)
– Vit. B12 and folate deficiency
– Toxic agents (lead, chemotherapy, arsenic,
zinc)
– Cooper deficiency
– T-cell large granular lymphocytic leukemia
– Hairy cell leukemia
– Medication (e.g. Cotrimoxazole)
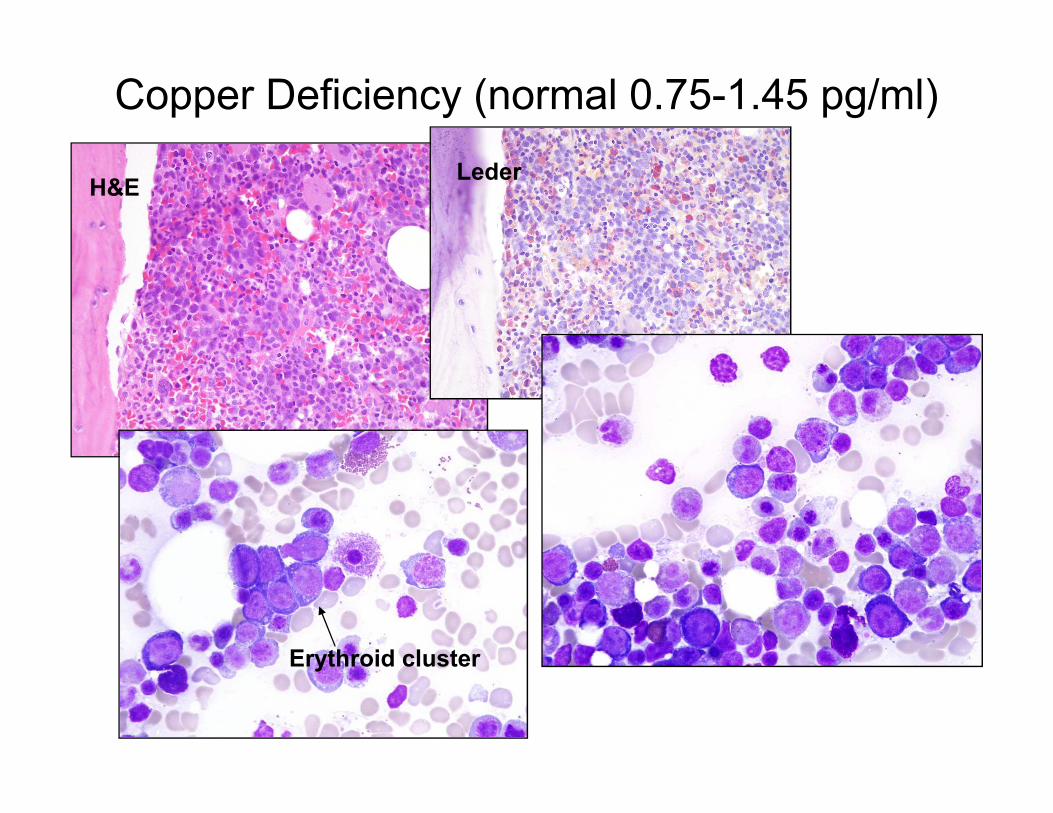
Copper Deficiency (normal 0.75-1.45 pg/ml)
H&ELeder
Erythroid cluster
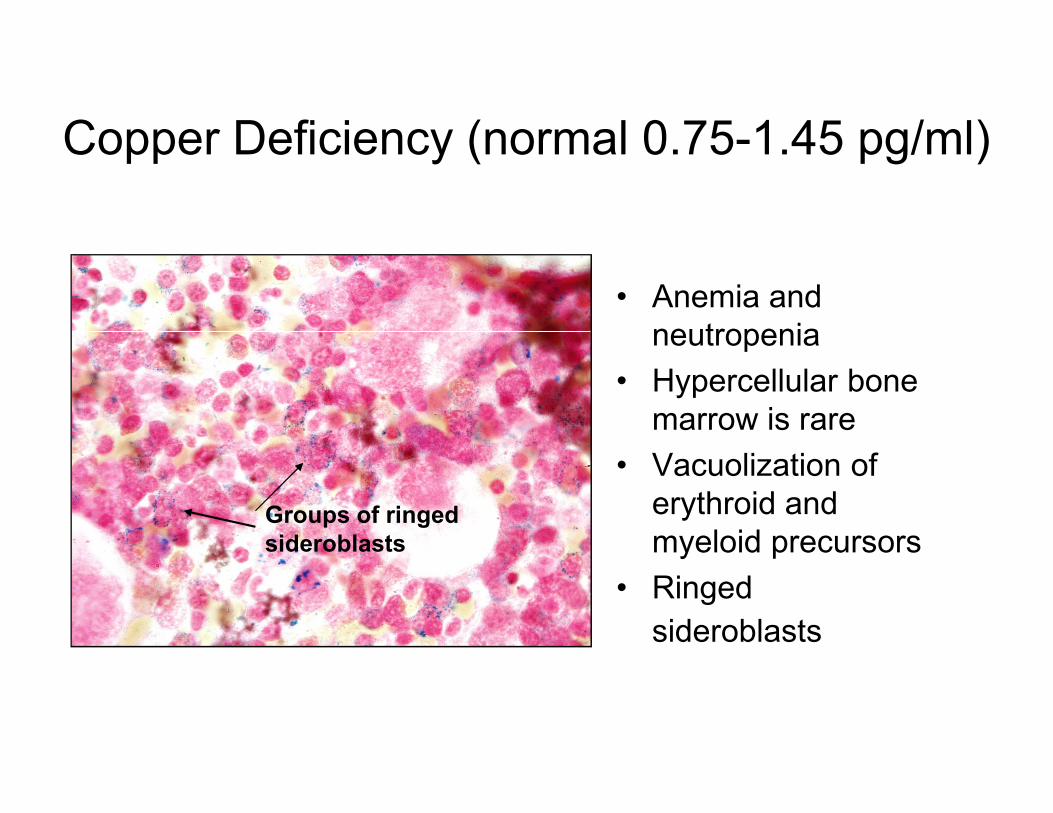
Copper Deficiency (normal 0.75-1.45 pg/ml)
• Anemia and
neutropenia
• Hypercellular bone
marrow is raremarrow is rare
• Vacuolization of
erythroid and
myeloid precursors
• Ringed
sideroblasts
Groups of ringed
sideroblasts

HIV-Infection Related BM Changes
Leder
MegakaryocytesH&E
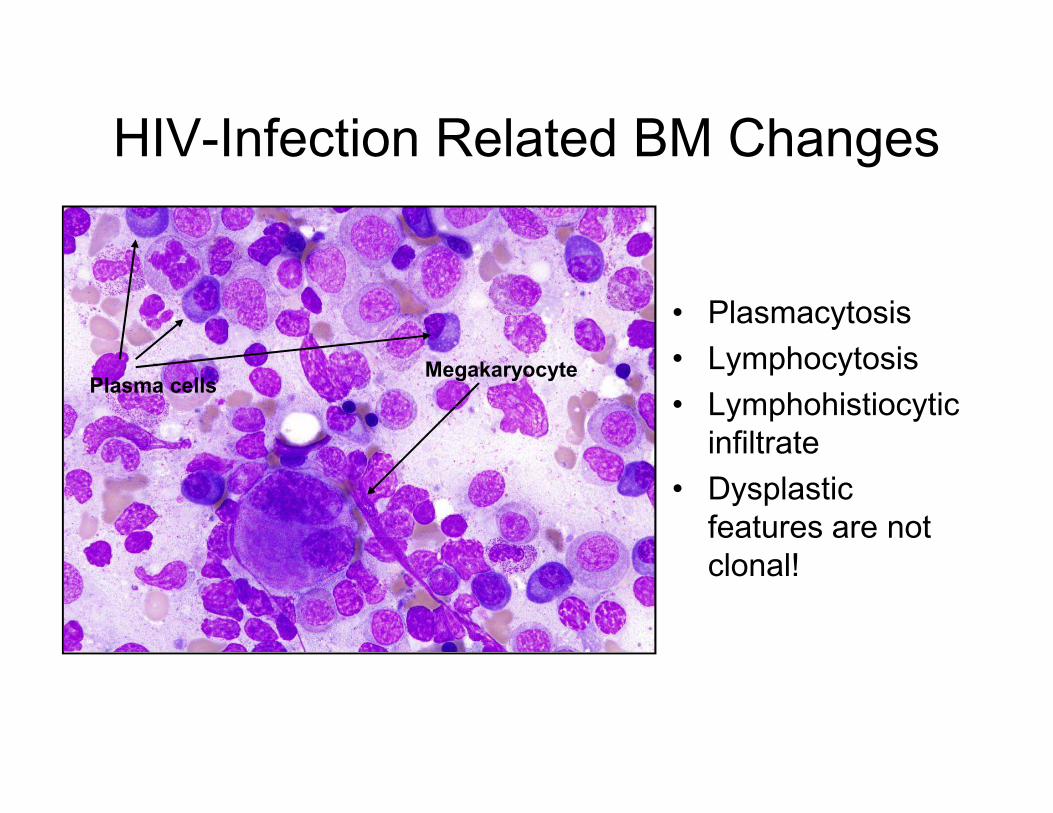
HIV-Infection Related BM Changes
• Plasmacytosis
• Lymphocytosis
• Lymphohistiocytic Plasma cells
Megakaryocyte
• Lymphohistiocytic
infiltrate
• Dysplastic
features are not
clonal!
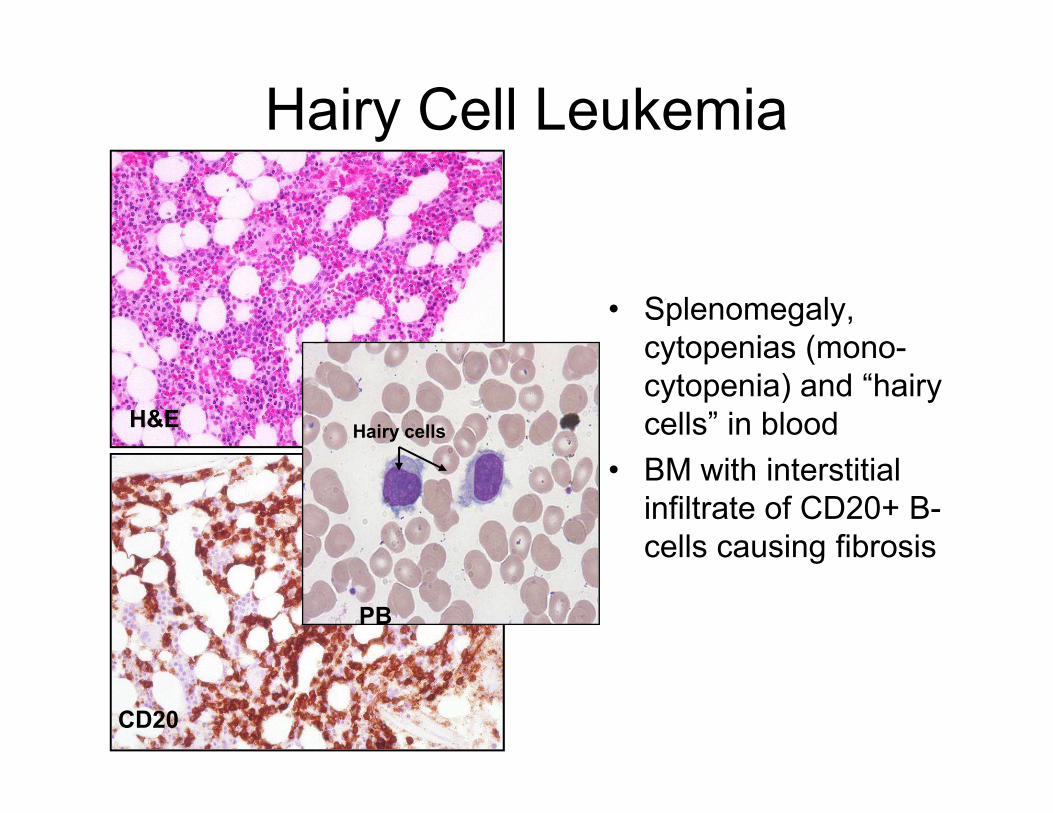
Hairy Cell Leukemia
• Splenomegaly,
cytopenias (mono-
cytopenia) and “hairy
cells” in bloodH&E cells” in blood
• BM with interstitial
infiltrate of CD20+ B-
cells causing fibrosis
CD20
PB
Hairy cellsH&E
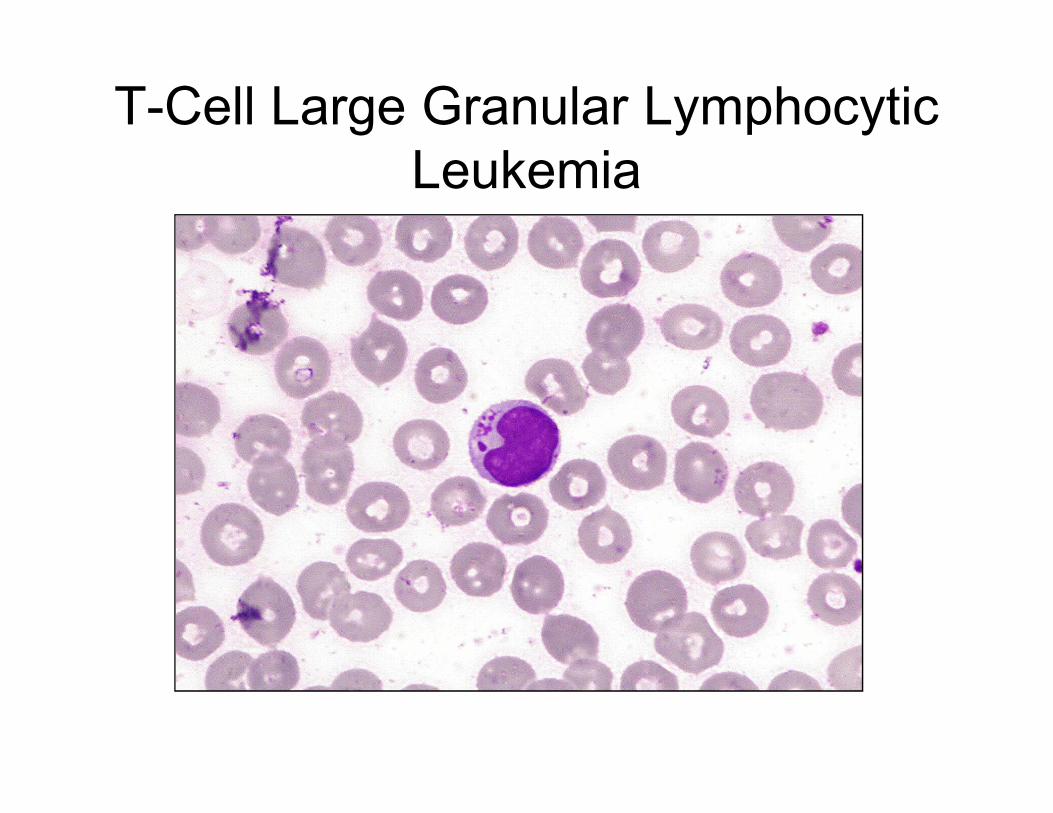
T-Cell Large Granular Lymphocytic
Leukemia
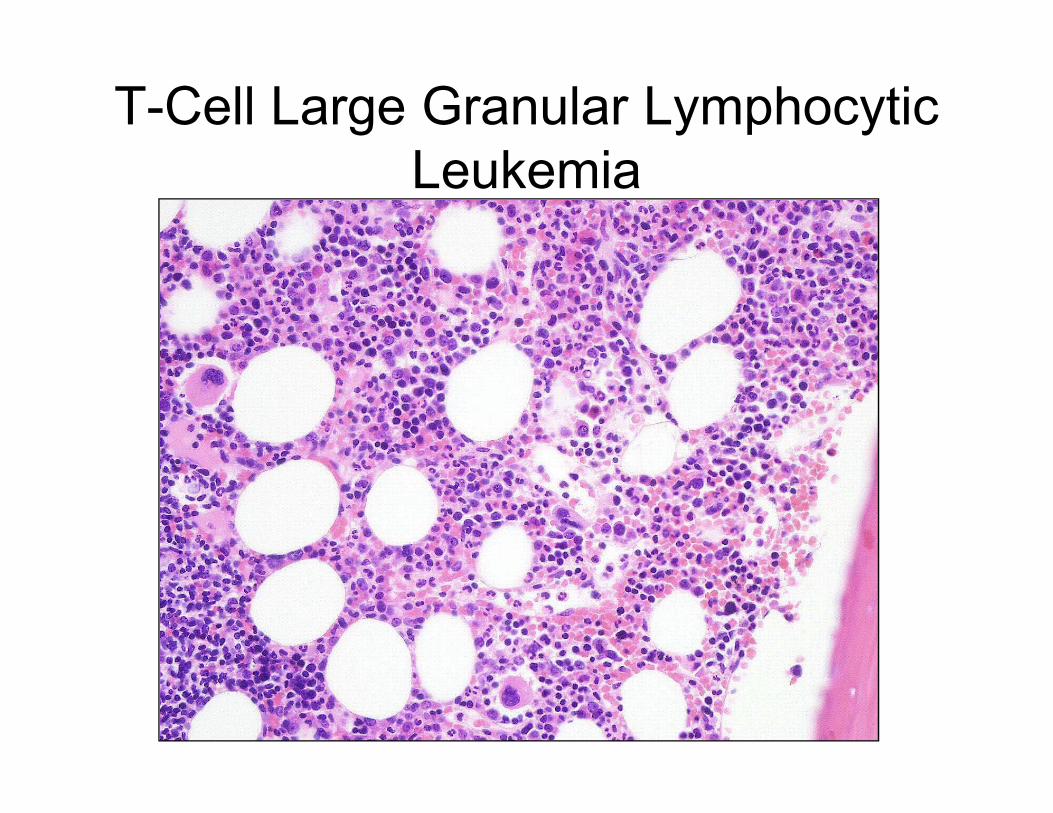
T-Cell Large Granular Lymphocytic
Leukemia
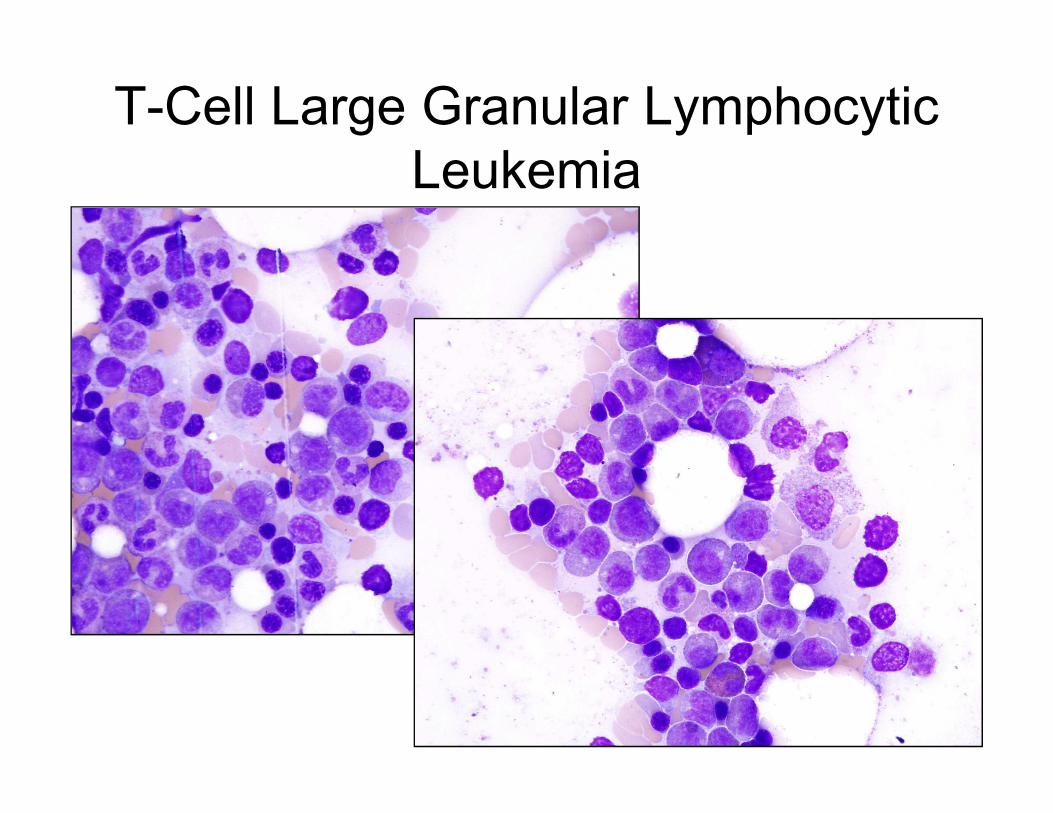
T-Cell Large Granular Lymphocytic
Leukemia
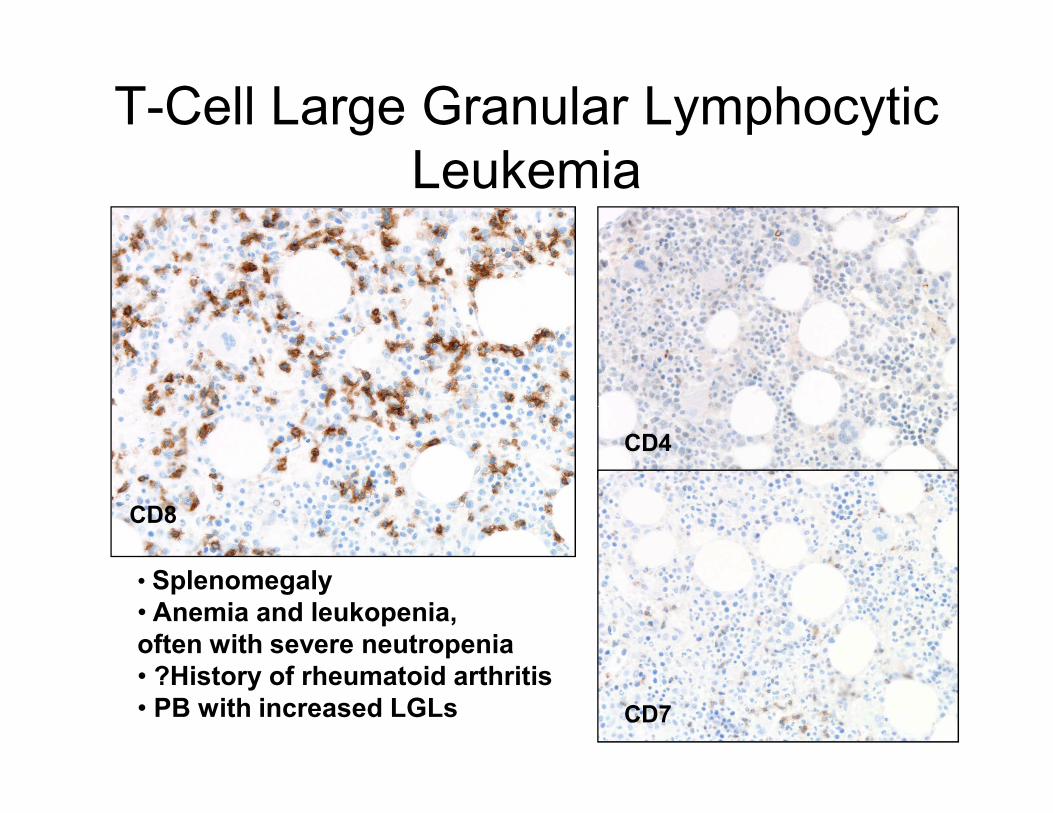
T-Cell Large Granular Lymphocytic
Leukemia
CD8
CD4
CD7
• Splenomegaly
• Anemia and leukopenia,
often with severe neutropenia
• ?History of rheumatoid arthritis
• PB with increased LGLs
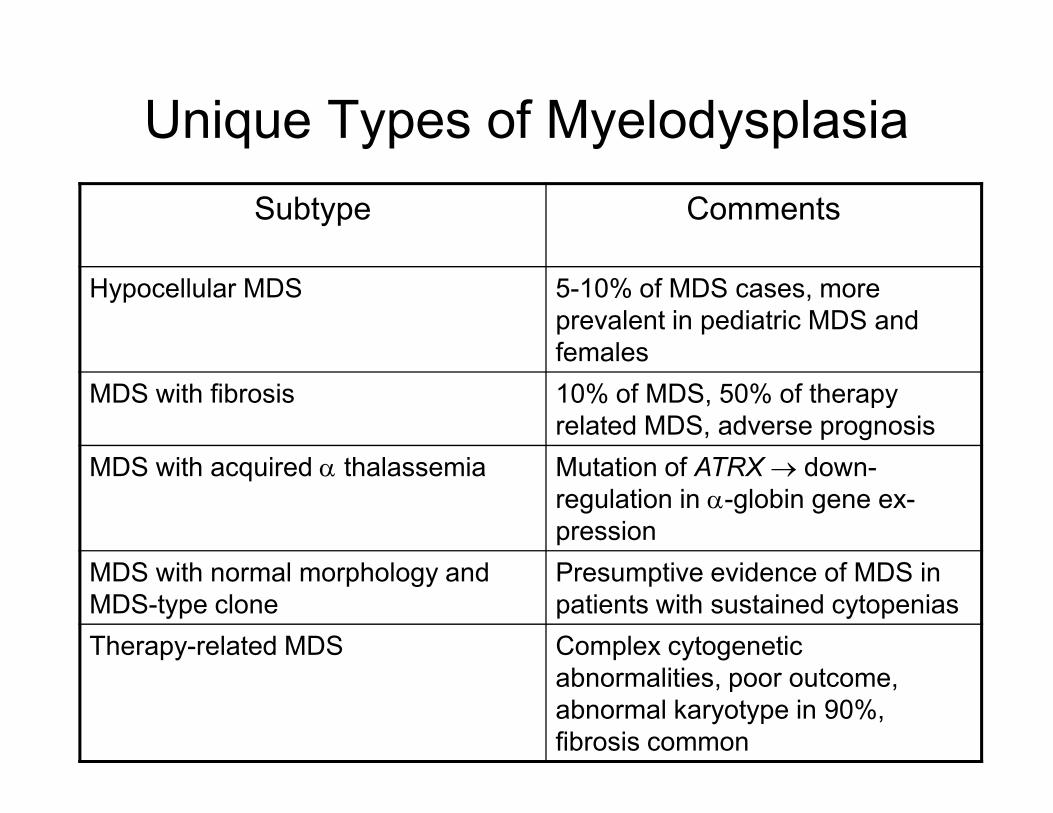
Unique Types of Myelodysplasia
Subtype Comments
Hypocellular MDS 5-10% of MDS cases, more
prevalent in pediatric MDS and
females
MDS with fibrosis 10% of MDS, 50% of therapy MDS with fibrosis 10% of MDS, 50% of therapy
related MDS, adverse prognosis
MDS with acquired α thalassemia Mutation of ATRX → down-
regulation in α-globin gene ex-
pression
MDS with normal morphology and
MDS-type clone
Presumptive evidence of MDS in
patients with sustained cytopenias
Therapy-related MDS Complex cytogenetic
abnormalities, poor outcome,
abnormal karyotype in 90%,
fibrosis common

Helpful Clues in the Diagnosis of MDS
• Poor specimen quality causes diagnostic errors
• The diagnosis of MDS requires integration of clinical, hematologic, morphologic, and cytogenetic features
• Exclude non-neoplastic causes of cytopenia →megaloblastic anemia, cooper deficiency, and chronic viral infections show the most overlap megaloblastic anemia, cooper deficiency, and chronic viral infections show the most overlap with MDS
• Megakaryoblasts, monoblasts, and promonocytes should be included in the blast percentage
• Conventional cytogenetics is the most valuable specialized test in MDS, perform routinely!

Helpful Clues in the Diagnosis of MDS
• MDS with associated monocytosis of ≥ 1X109/L are included in MDS/MPN category
• MDS in children is rare may be linked to underlying constitutional disorders or prior chemotherapy
• ~10% of MDS in adults are either hypocellular or fibroticfibrotic
• Exclude low blast count AML from RAEB-2 by cytogenetic assessment for AML-defining translocations
• CD34 immunohistochemistry is useful in assessing overall number and localization of blasts in the core

References
1. David Bowen et al. Guidelines for the diagnosis and therapy of adult
myelodysplastic syndromes. British Journal of Haematology, 2003, 120:187-200.
2. Roos J Leguit et al. The pathology of bone marrow failure. Histopathology, 2010,
DOI:10.1111/j.1365-2559.2010.03612.x
3. Afsaneh Barzi et al. Myelodysplastic syndromes: A practical approach to diagnosis
and treatment. Cleveland Clinic Journal of Medicine, 2010, 77:37-44.
4. Mikkael A Sekeres. The epidemiology of myeodysplastic syndromes.
Hematology/Oncology Clinics of North America, 2010, 24:287-294.
5. Rami S. Komrokji et al. Myelodysplastic syndromes: classification and Risk 5. Rami S. Komrokji et al. Myelodysplastic syndromes: classification and Risk
Stratification. Hematology/Oncology Clinics of North America, 2010, 24:443-457.
6. John R. Feussner et al. Arsenic-induced bone marrow toxicity: ultrastructural and
electron-probe analysis. Blood, 1979, 53:820-827.
7. Monte S. Willis et al. Zinc-induced copper deficiency. American Journal for Clinical
Pathology, 2005, 123:125-131.
8. Steven H. Swerdlow et al. WHO classification of tumours of haematopoietic and
lymphoid tissues. 4th Edition IARC, Lyon, 2008
9. Kathryn Foucar et al. Bone Marrow Pathology. 3rd Edition ASCP Press, Chicago,
2010

Bone Marrow Failure Syndromes
Michele Paessler, DO
Children’s Hospital of Philadelphia
University of Pennsylvania School of Medicine

Bone Marrow Failure • Definition
• Criteria
• Classification
• Work up
• Differential Diagnosis including
important diagnoses to exclude
• Inherited bone marrow failure
syndromes that should be considered
• Other Important tests to consider

Bone Marrow Failure
• Definition:
– Inability of the marrow to produce circulating
mature cells.
– This may result from:
• Reduction in number of progenitors and • Reduction in number of progenitors and
subsequently paucity of differentiated precursors
in the periphery:
– Aplastic anemia
• Increased number of differentiated precursors
but reduction of mature products in the periphery
– Ineffective hematopoiesis (ie myelodysplastic
syndrome)
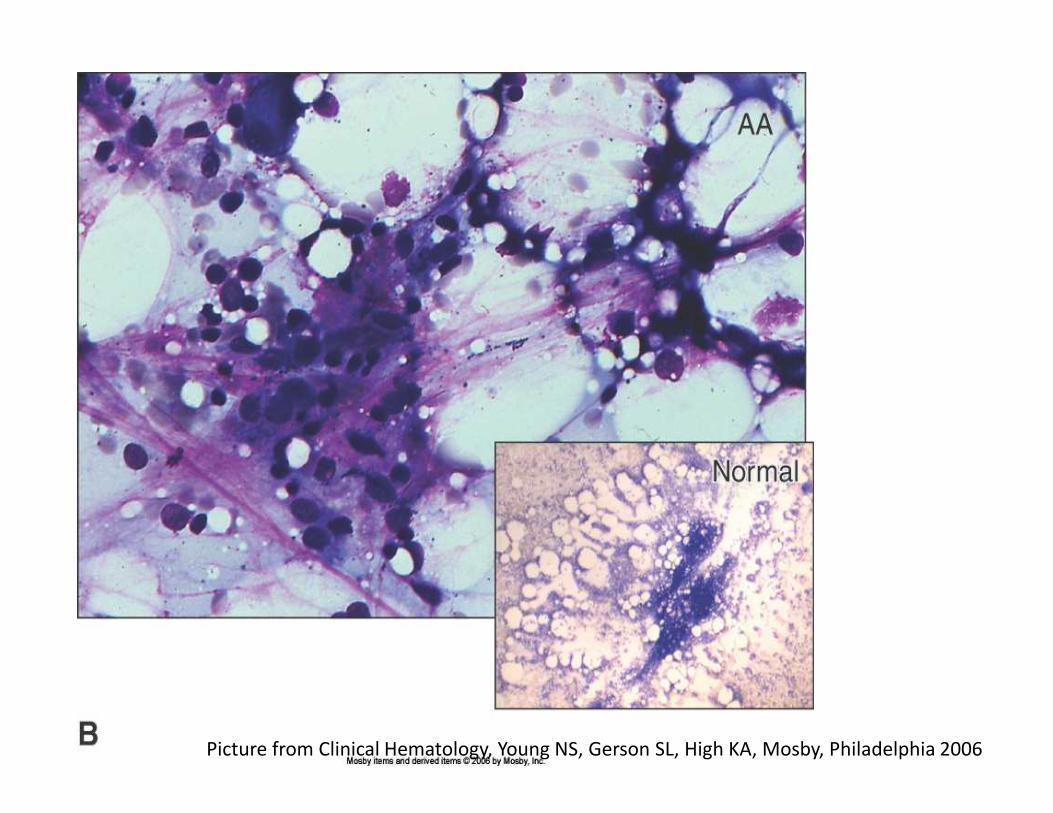
Picture from Clinical Hematology, Young NS, Gerson SL, High KA, Mosby, Philadelphia 2006
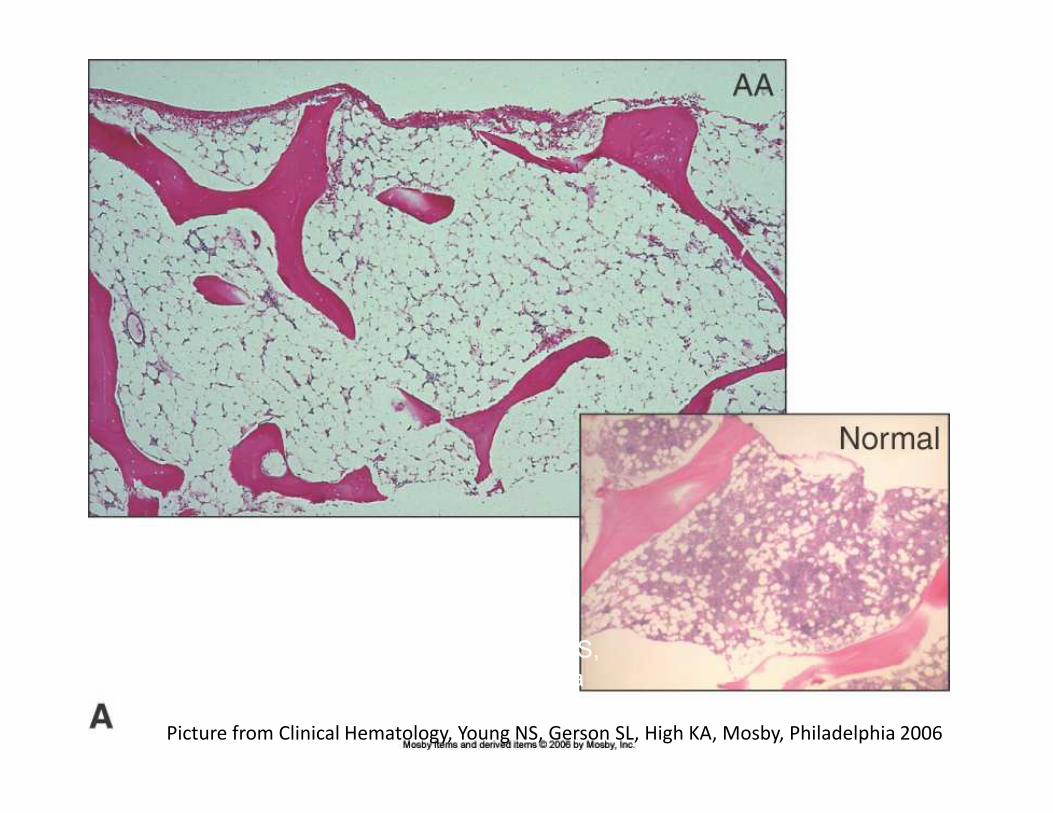
Picture from Clinical Hematology, Young NS, Gerson SL, High KA, Mosby, Philadelphia 2006
Picture from Clinical Hematology, Young NS,
Gerson SL, High KA, Mosby, Philadelphia
2006

Diagnostic Algorithm for
Aplastic Anemia• Step 1: Establish criteria
• Step 2: Exclude Malignancy
• Step 3: Etiology
• Step 4: Prognostic Data• Step 4: Prognostic Data

Criteria for Diagnosis of Aplastic
AnemiaMust have two of the following in the peripheral blood:
• ANC <0.5 x 10 9/L
• Platelet count <20 x10 9/L
• Reticulocyte count< 20 x 10 9/L
Bone marrow must show:• Biopsy with <25% of normal cellularity for age OR
• 25-50% of normal cellularity for age, with <30% of the cells being hematopoietic
Bone Marrow Pathology, 3rd Edition, Volume 1. Foucar 2010
pg 138

What defines a hypocellular
marrow?• Pancytopenia and a hypocellular/aplasticmarrow– Hypocellular is considered <25% of the NORMAL cellularity for AGE.• Newborn 80-100% cellularity
• 1-3 month 80-100% cellularity• 1-3 month 80-100% cellularity
• Child (>1 year) 60-80% cellularity
• Adult (30-70 years) 40-70% cellularity
• Adult (>70 years) ~25% cellularity
• Requires bone marrow biopsy and aspirate– Core biopsy is recommended > 2 cm for accurate determination of cellularity
• Cellularity can be variable
• Subcortical marrow is hypocellularAdapted from Foucar, Bone Marrow Pathology,3 rd Edition, ASCP Press 2010 pg 32

Classification of Aplastic Anemia
ACQUIRED (80-90%)
Idiopathic (~70%)
Secondary (~10-20%)Drugs/toxins
Viruses (HIV, Hepatitis,
EBV)
Autoimmune (SLE)
INHERITED (10-20%)
Fanconi anemia
Dyskeratosis congenita
Shwachman-Diamond Autoimmune (SLE)
Hepatits (non viral)
Nutritional deficiencies
(Vit B 12/Folate)
PNH
Malignant infiltration (
tumors)
Nonmalignant
infiltration (storage
disorders)
Other (anorexia
nervosa, pregnancy)
Shwachman-Diamond
syndrome
Amegakaryocytic
thrombocytopenia
Familial aplastic
anemias
Pearson syndrome
Adapted from Clinical
Hematology, Young NS,
Gerson SL, High KA,
Mosby, Philadelphia
2006
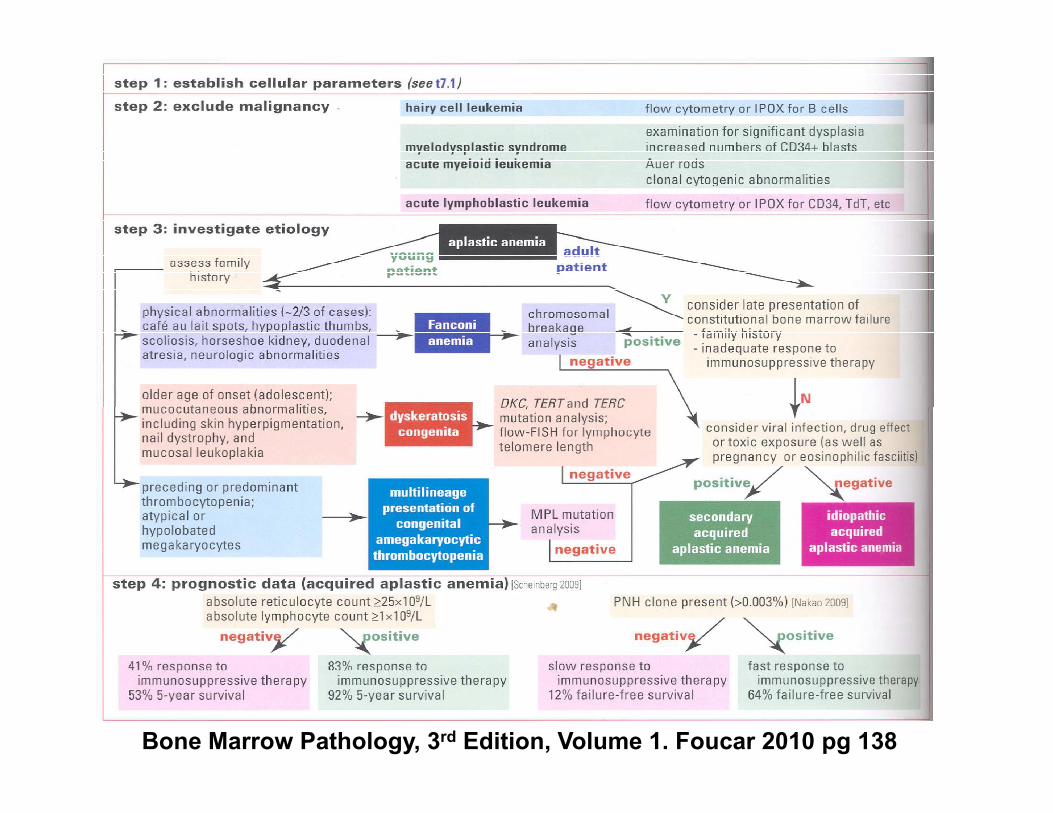
Bone Marrow Pathology, 3rd Edition, Volume 1. Foucar 2010 pg 138

Diagnostic Algorithm for
Aplastic Anemia• Step 1: Establish criteria
• Step 2: Exclude Malignancy
• Step 3: Etiology
• Step 4: Prognostic Data• Step 4: Prognostic Data

Hematopoietic Malignancies that
Masquerade as Aplastic Anemia
• Acute Leukemia CD34
– Acute lymphoblastic leukemia (TdT)
– Acute myeloid leukemia (MPO)
• Myelodysplastic syndrome CD34
Immunohistochemical Panel
– Dysplasia, Auer rods
• Hairy cell leukemia CD20
• T-cell LGL CD3
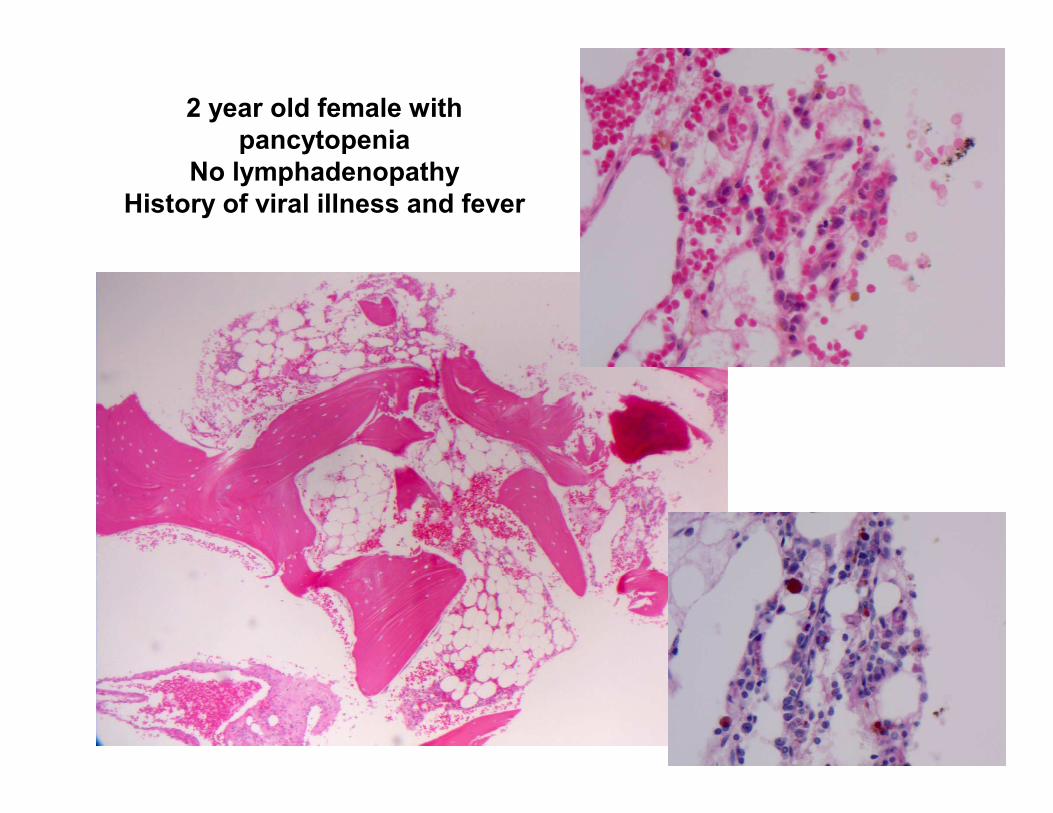
2 year old female with
pancytopenia
No lymphadenopathy
History of viral illness and fever
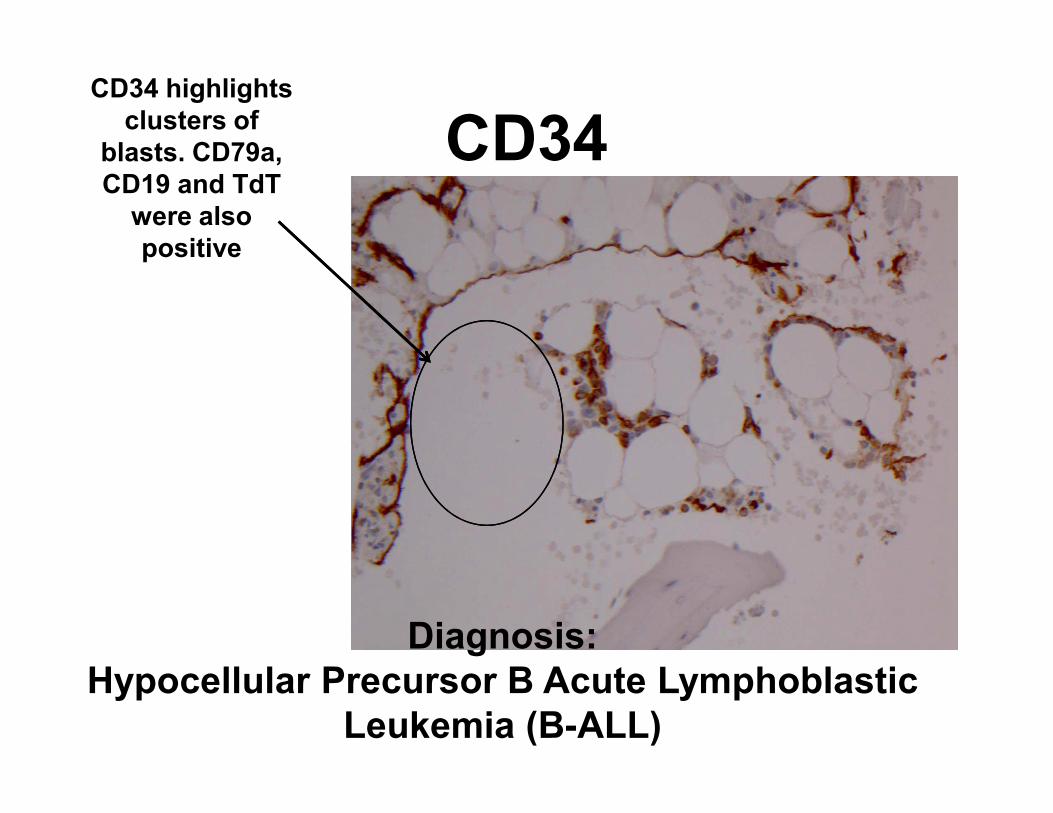
CD34CD34 highlights
clusters of
blasts. CD79a,
CD19 and TdT
were also
positive
Diagnosis:
Hypocellular Precursor B Acute Lymphoblastic
Leukemia (B-ALL)
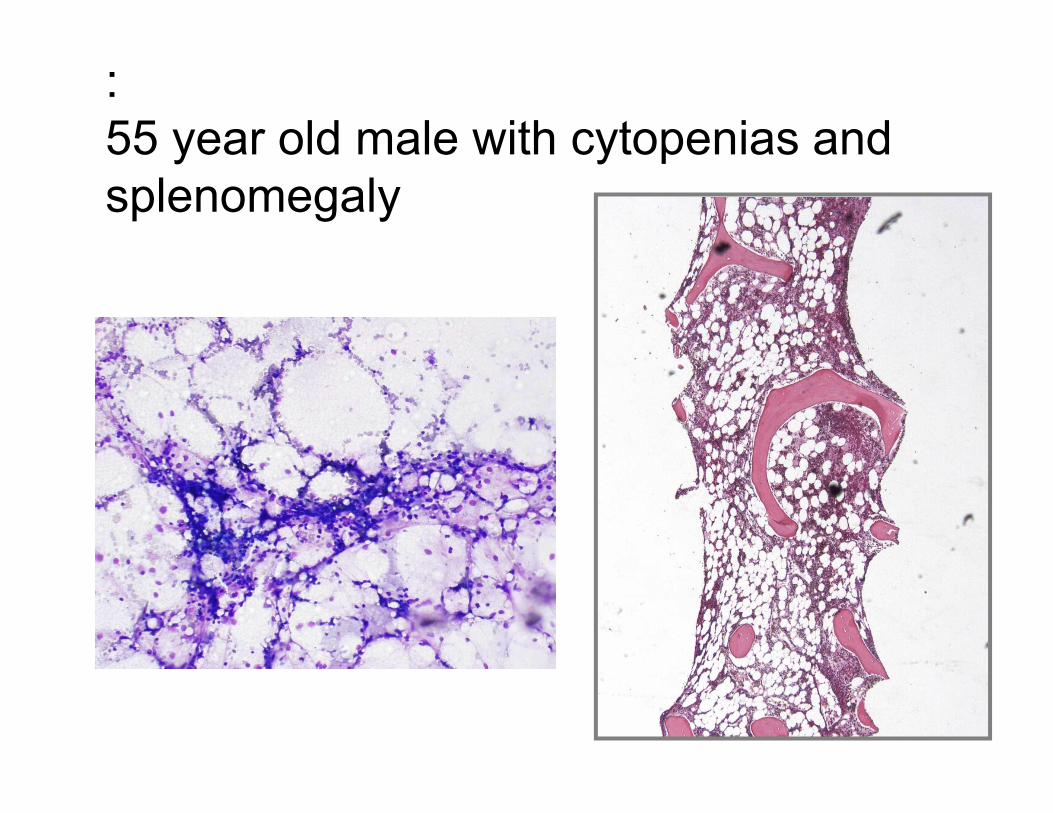
:
55 year old male with cytopenias and
splenomegaly
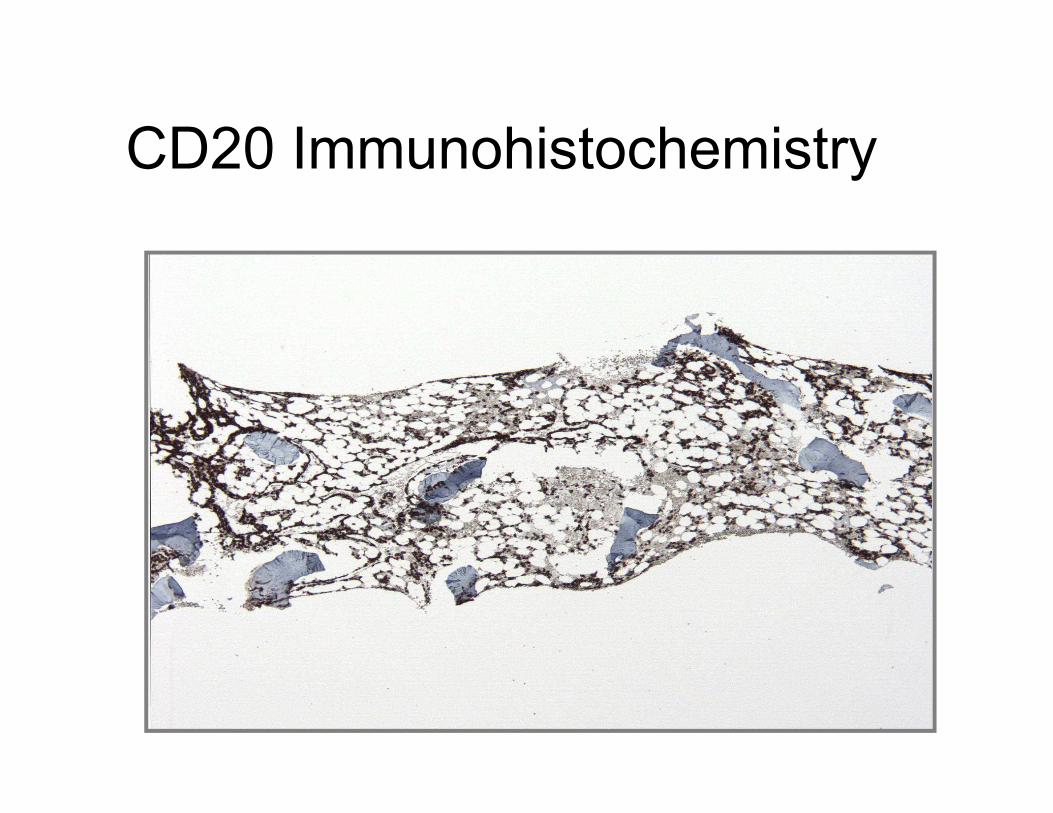
CD20 Immunohistochemistry
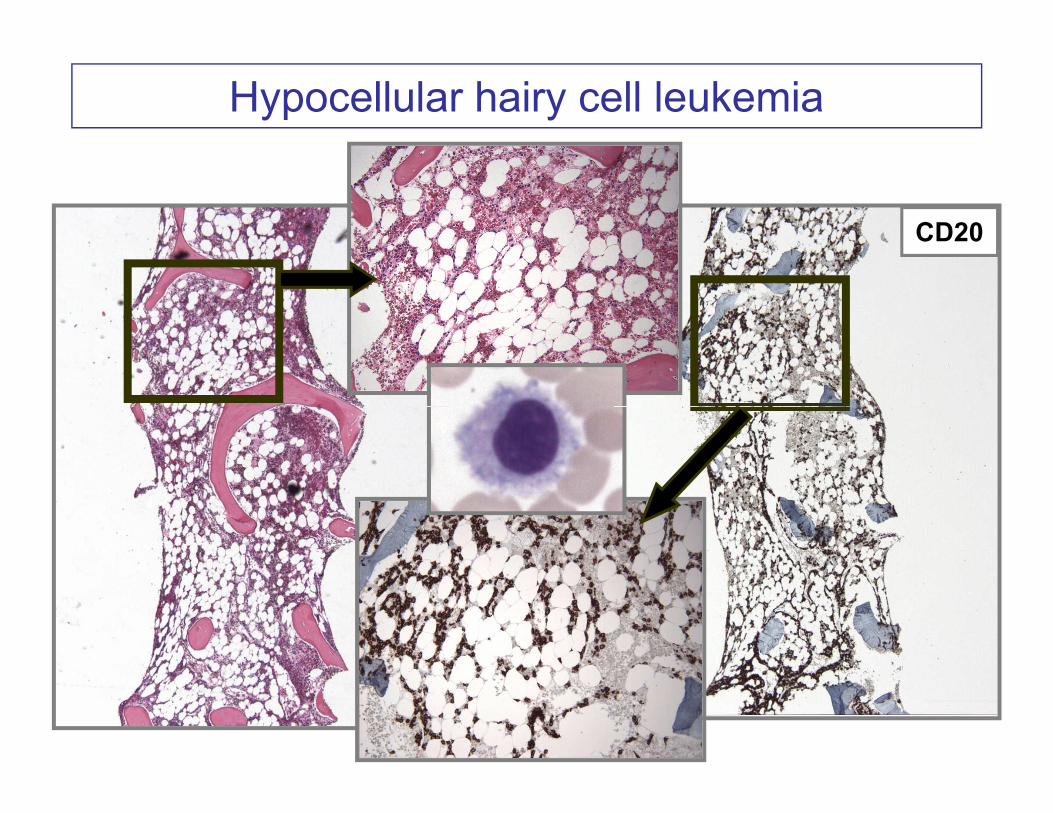
Hypocellular hairy cell leukemia
CD20
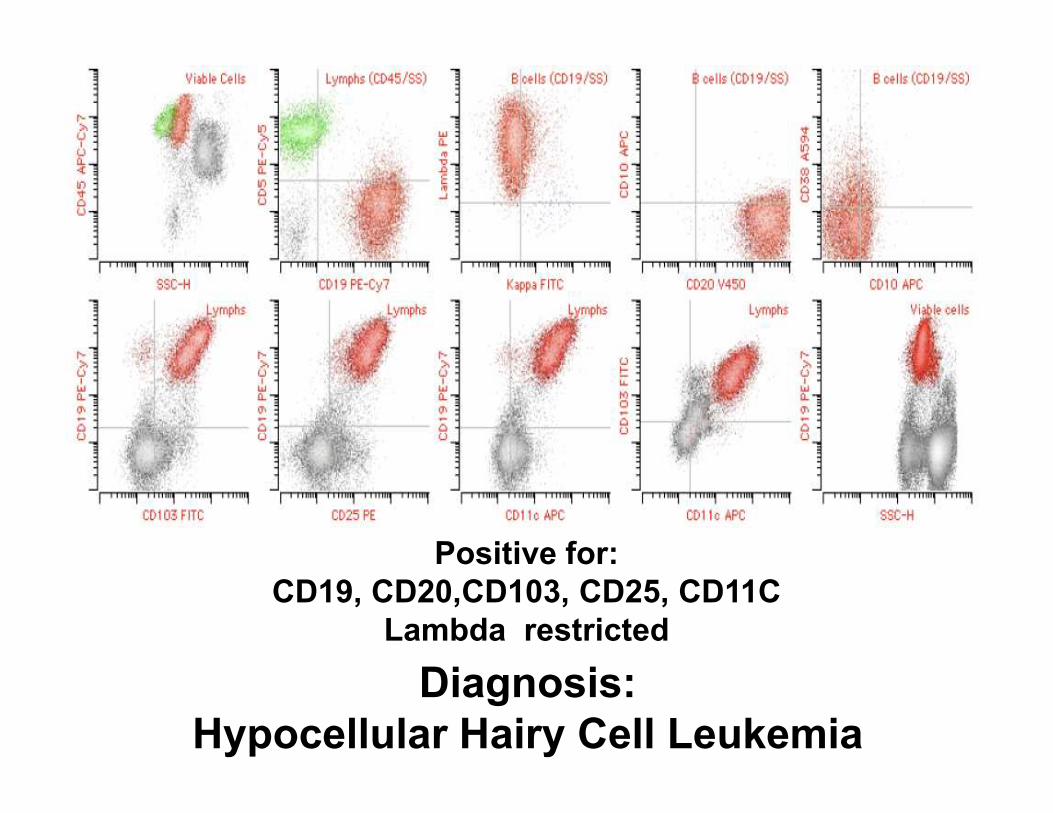
Diagnosis:
Hypocellular Hairy Cell Leukemia
Positive for:
CD19, CD20,CD103, CD25, CD11C
Lambda restricted

Diagnostic Algorithm for
Aplastic Anemia• Step 1: Establish criteria
• Step 2: Exclude Malignancy
• Step 3: Etiology
• Step 4: Prognostic Data• Step 4: Prognostic Data

Inherited Forms Associated with
Multilineage Bone Marrow Failure
• Primarily (but not always!!!!) associated with pediatric and young
adult
– Reports of patients being diagnosed in 5th-8th decades
• Can affect single lineage or multilineage
• Associated with increased risk of developing hematopoietic and
other malignanciesother malignancies
– Important to identify these patients
• Two disorders classically present with aplastic or hypoplasticbone marrow failure involving all hematopoietic lineages:
– Fanconi anemia
– Dyskeratosis congenita
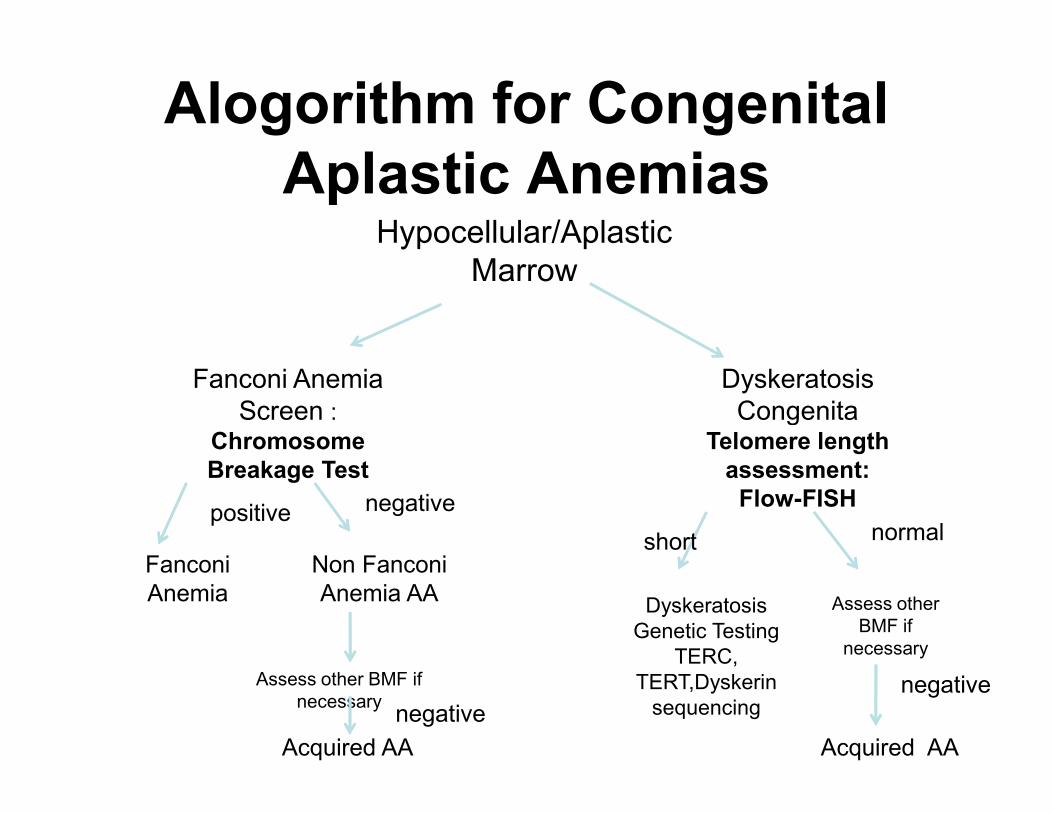
Alogorithm for Congenital
Aplastic AnemiasHypocellular/Aplastic
Marrow
Fanconi Anemia
Screen :
Dyskeratosis
CongenitaScreen : Chromosome
Breakage Test
CongenitaTelomere length
assessment:
Flow-FISH
Fanconi
Anemia
positive
Non Fanconi
Anemia AA
negative
Dyskeratosis
Genetic Testing
TERC,
TERT,Dyskerin
sequencing
Assess other
BMF if
necessary
short normal
Acquired AA
Assess other BMF if
necessary
Acquired AA
negative
negative
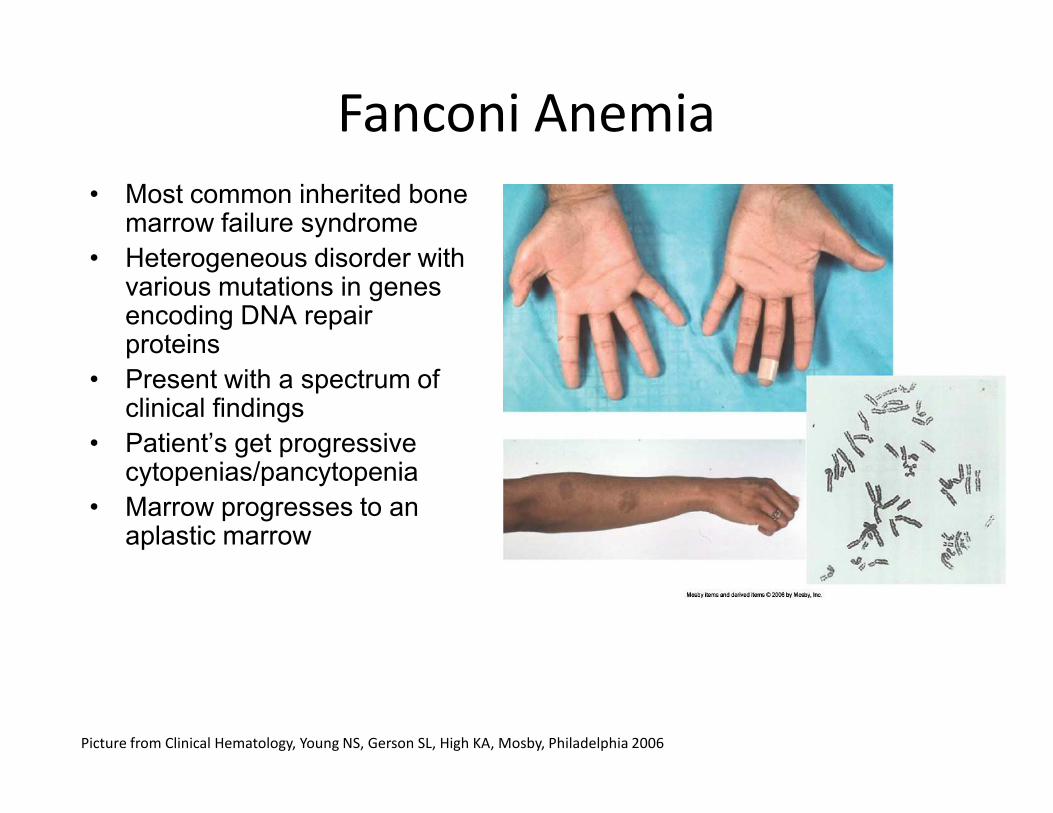
• Most common inherited bone marrow failure syndrome
• Heterogeneous disorder with various mutations in genes encoding DNA repair proteins
• Present with a spectrum of clinical findings
Fanconi Anemia
clinical findings
• Patient’s get progressive cytopenias/pancytopenia
• Marrow progresses to an aplastic marrow
Picture from Clinical Hematology, Young NS, Gerson SL, High KA, Mosby, Philadelphia 2006
Fanconi Anemia

Fanconi Anemia Test
Chromosome Breakage Test
• Best screening
method:
• Chromosome
Breakage test
• Genetic testing is • Genetic testing is
usually performed
if there is a
positive
chromosome
breakage test
Shimamura, A. Hematology; 2006:63-71

Case History
• 14 year old male
• Pancytopenia
• Mild immunodeficiency
• Small stature• Small stature
• Family history:
– Mother small stature and history of anemia
– Father small(ish)
– 3 brothers normal size
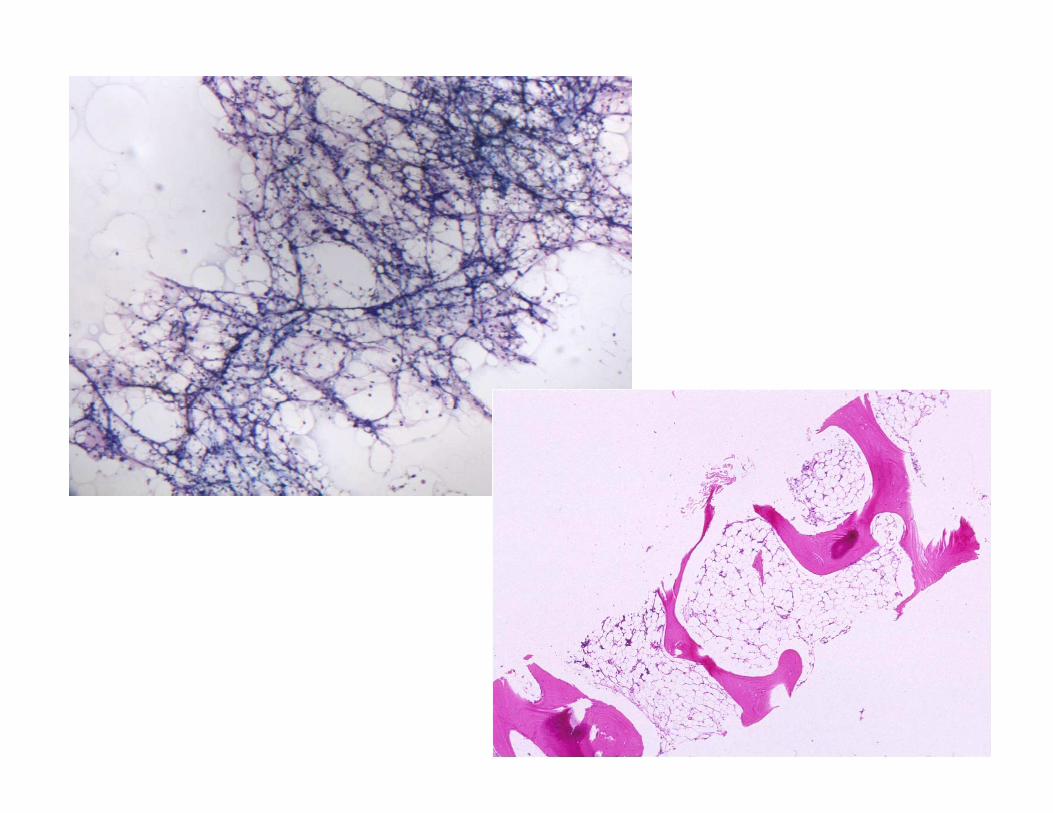

Diagnosis:
• Aplastic marrow, no leukemia seen.
• Recommendations:• Recommendations:
– Flow fish for telomere length
– Fanconi anemia testing
– PNH flow cytometry
Leukoplakia
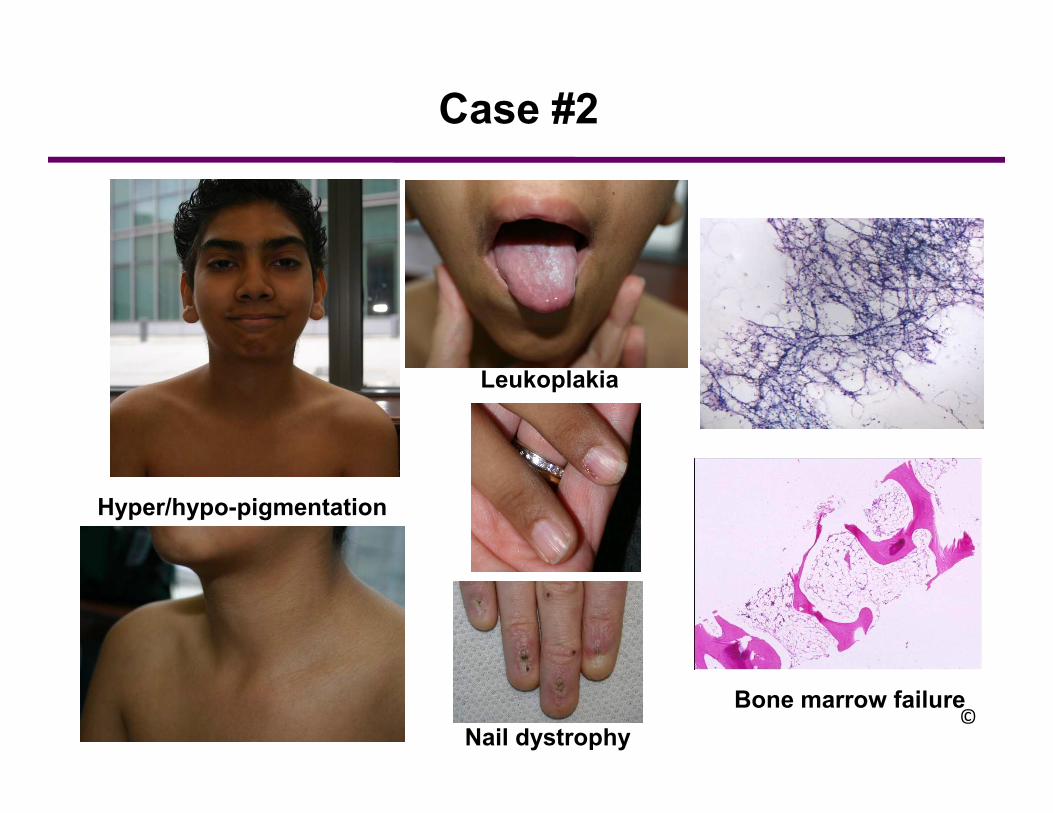
Case #2
Hyper/hypo-pigmentation
Nail dystrophy
Bone marrow failure©
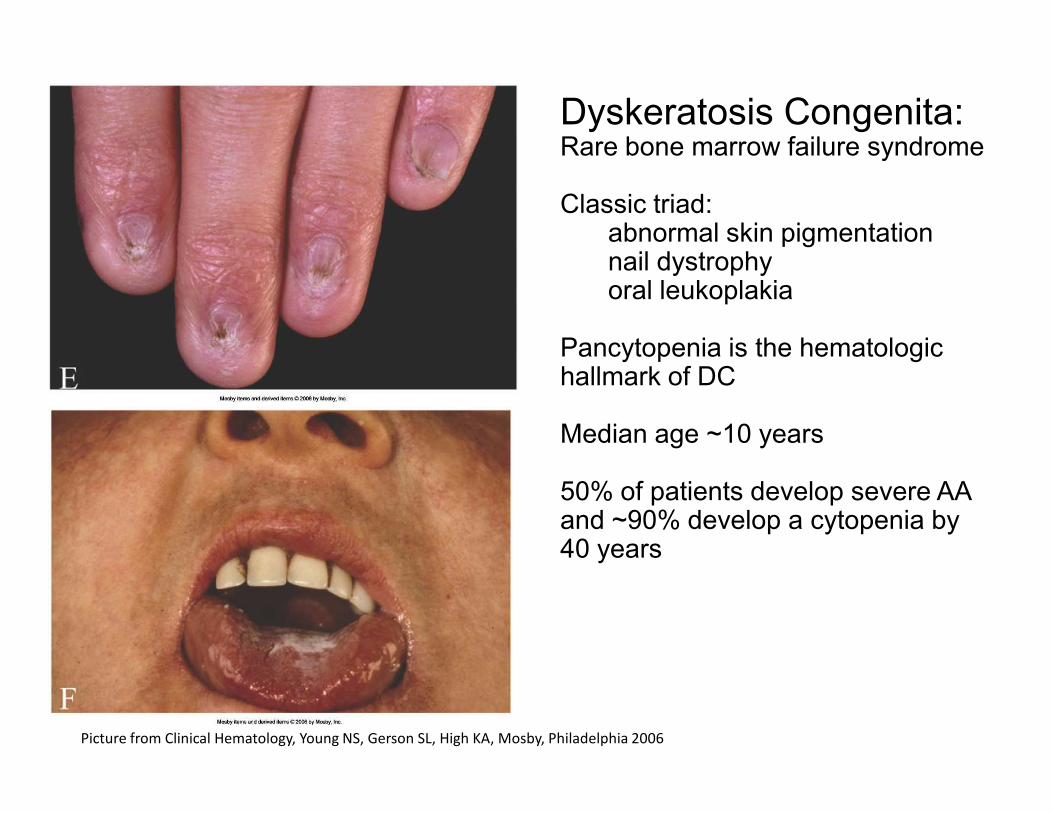
Dyskeratosis Congenita: Rare bone marrow failure syndrome
Classic triad:abnormal skin pigmentationnail dystrophyoral leukoplakia
Pancytopenia is the hematologic hallmark of DC
Picture from Clinical Hematology, Young NS, Gerson SL, High KA, Mosby, Philadelphia 2006
Median age ~10 years
50% of patients develop severe AA and ~90% develop a cytopenia by 40 years
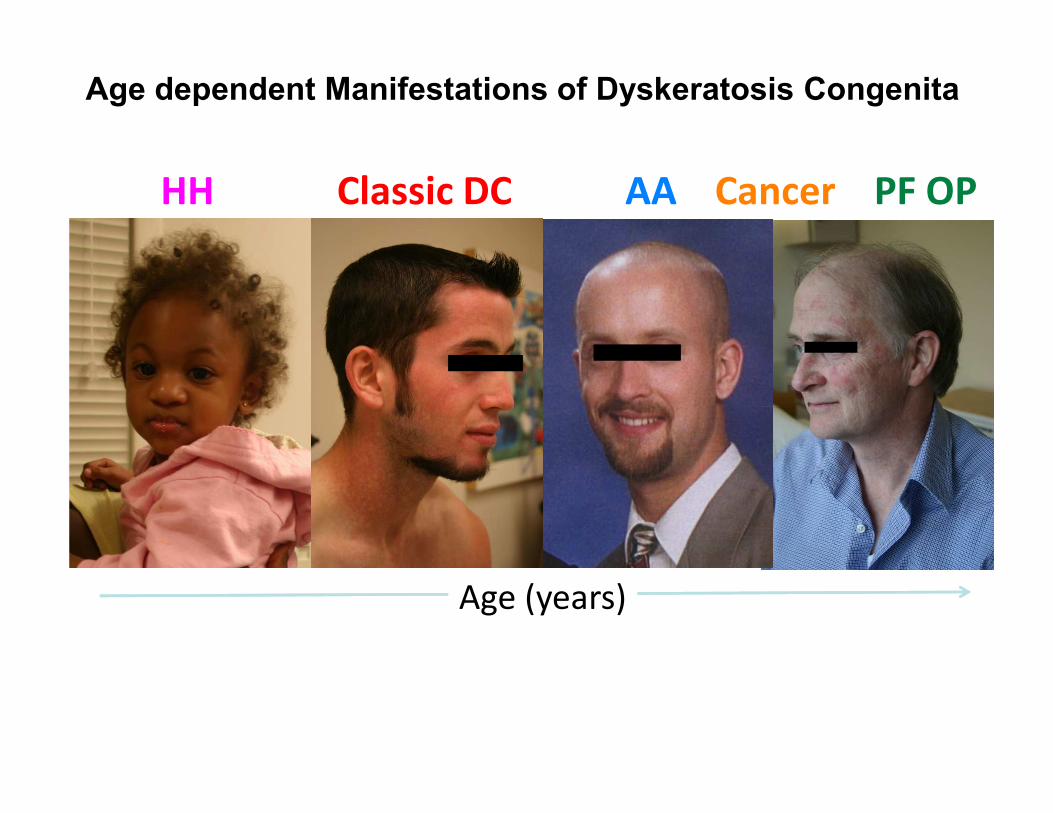
Age dependent Manifestations of Dyskeratosis Congenita
AA PF OPHH Classic DC Cancer
Age (years)
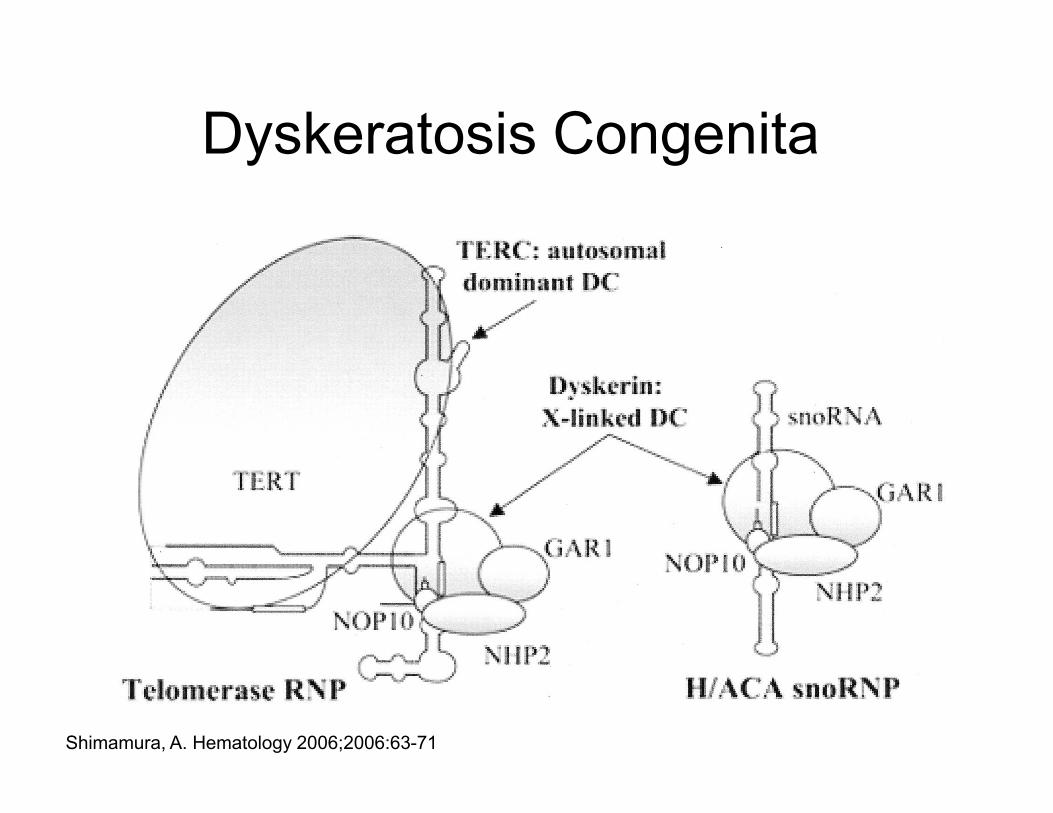
Dyskeratosis Congenita
Shimamura, A. Hematology 2006;2006:63-71
12
14
16
18
20
Re
lati
ve
Te
lom
ere
Le
ng
th (
% 4
N c
ell
lin
e)
99%
90%
75%
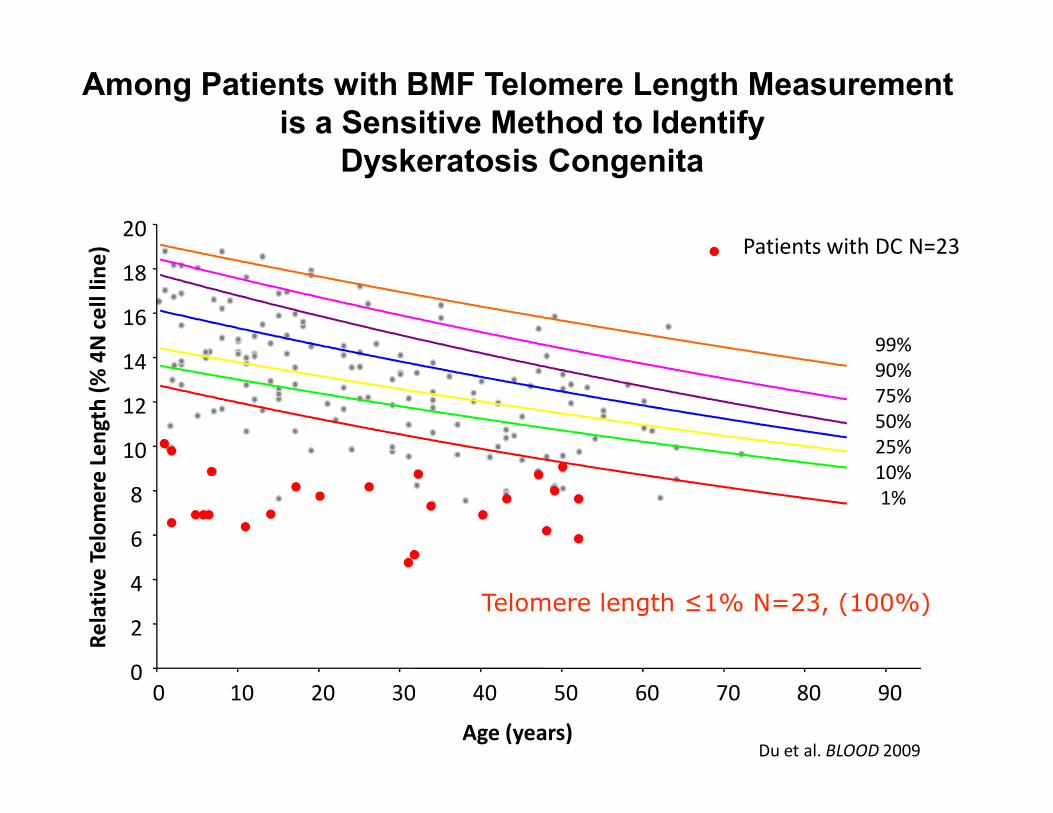
Among Patients with BMF Telomere Length Measurement
is a Sensitive Method to Identify
Dyskeratosis Congenita
Patients with DC N=23
Age (years)
0
2
4
6
8
10
12
0 10 20 30 40 50 60 70 80 90
Re
lati
ve
Te
lom
ere
Le
ng
th (
% 4
N c
ell
lin
e)
50%
25%
10%
1%
Telomere length ≤1% N=23, (100%)
Du et al. BLOOD 2009
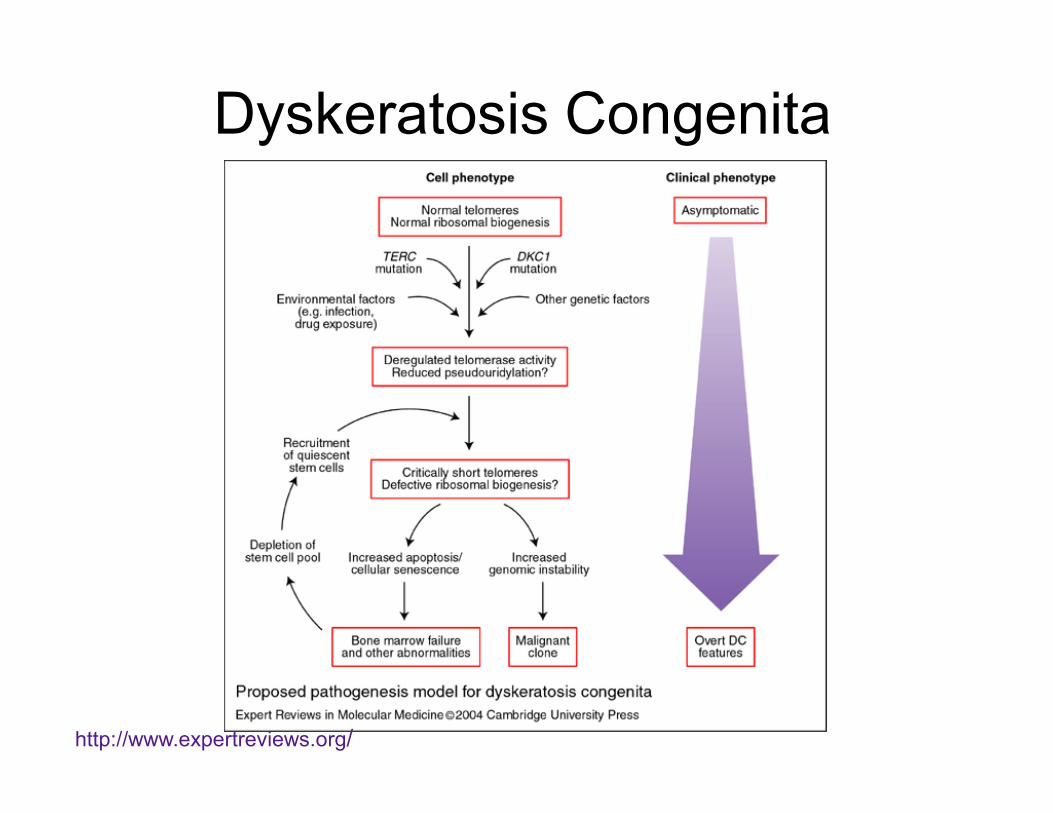
Dyskeratosis Congenita
http://www.expertreviews.org/
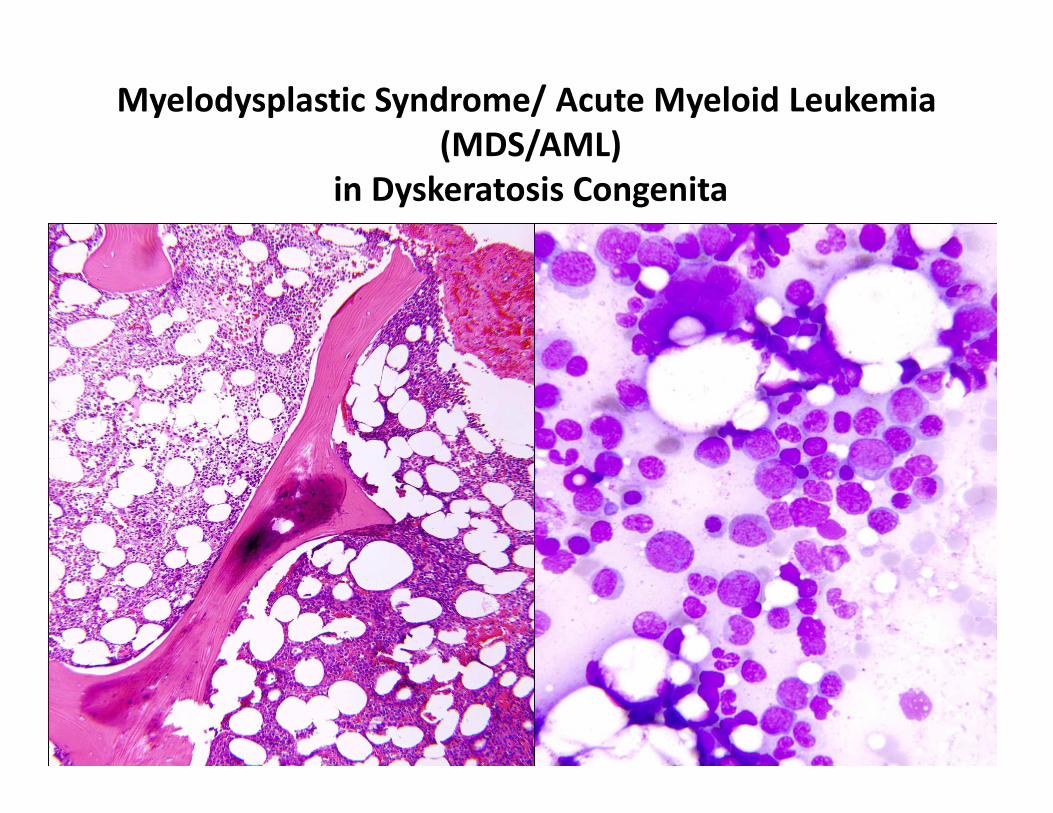
Myelodysplastic Syndrome/ Acute Myeloid Leukemia
(MDS/AML)
in Dyskeratosis Congenita

Who Should Be Screened for Inherited
Bone Marrow Failure Syndromes?
• Pediatric, adolescent or young adult
– Primarily occur in this age group including young adult
• Any congenital abnormalities
– Congenital abnormalities (including cardiac and renal)
are important clues
– 30-50% of IBMFS patients have NO associated – 30-50% of IBMFS patients have NO associated
abnormalities
• Family history of aplastic anemia, cytopenias,
pulmonary fibrosis, AML or epithelial malignancies
• Pre-transplant workup for AA
– IBMFS patients cannot undergo standard conditioning
and undergo reduced intensity conditioning therapy
pretransplant

Aplastic Anemia vs. Hypoecellular
MDS• Can be very difficult to distinguish!!!
• Aplastic anemia
– Can see compensatory cellularity
– Can be associated with macrocytic anemia
– Can show “stress” dyserythropoiesis
– Cytogenetic abnormalities can also be seen
• Monosomy 7 – associated with progression MDS/AML****
• Trisomy 8 – usually respond to immunsuppressive therapy
• Factors that favor MDS
– Increase in blasts
– Dysplasia in non-erythroid lineages (myeloid and
megakaryocytic)
- Ringed sideroblasts in erythroid dysplasia

Case Presentation:
15 year old with pancytopenia15 year old with pancytopenia
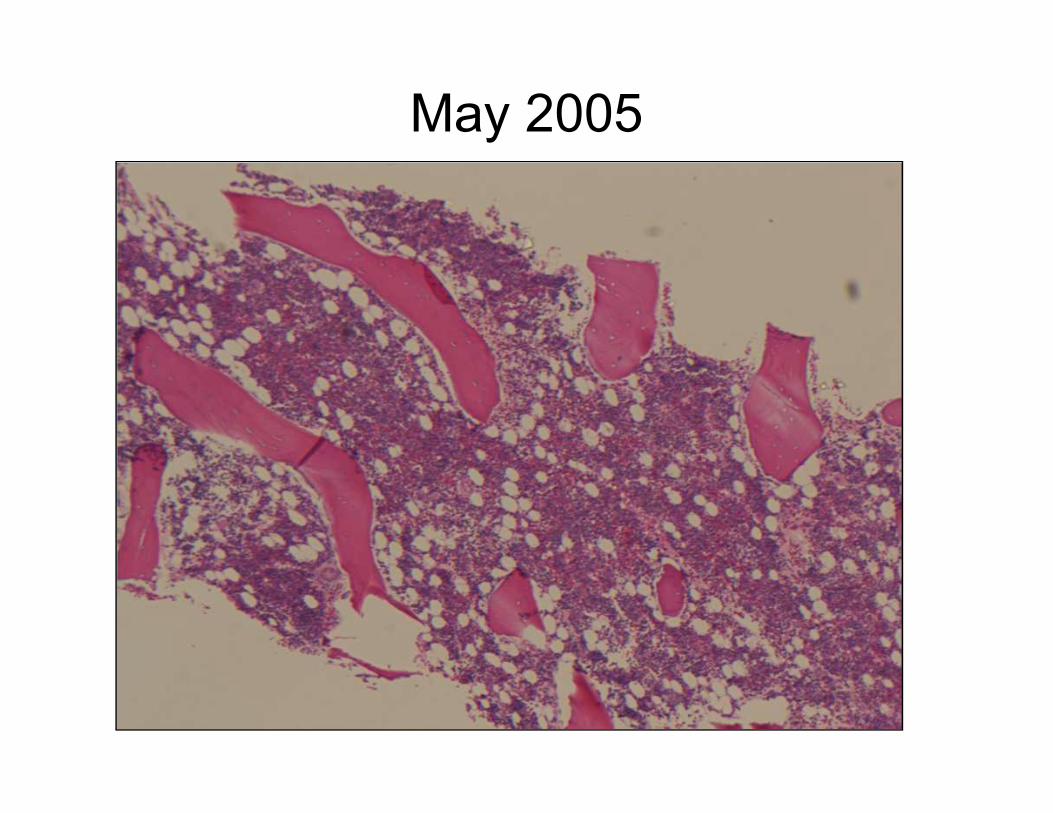
May 2005
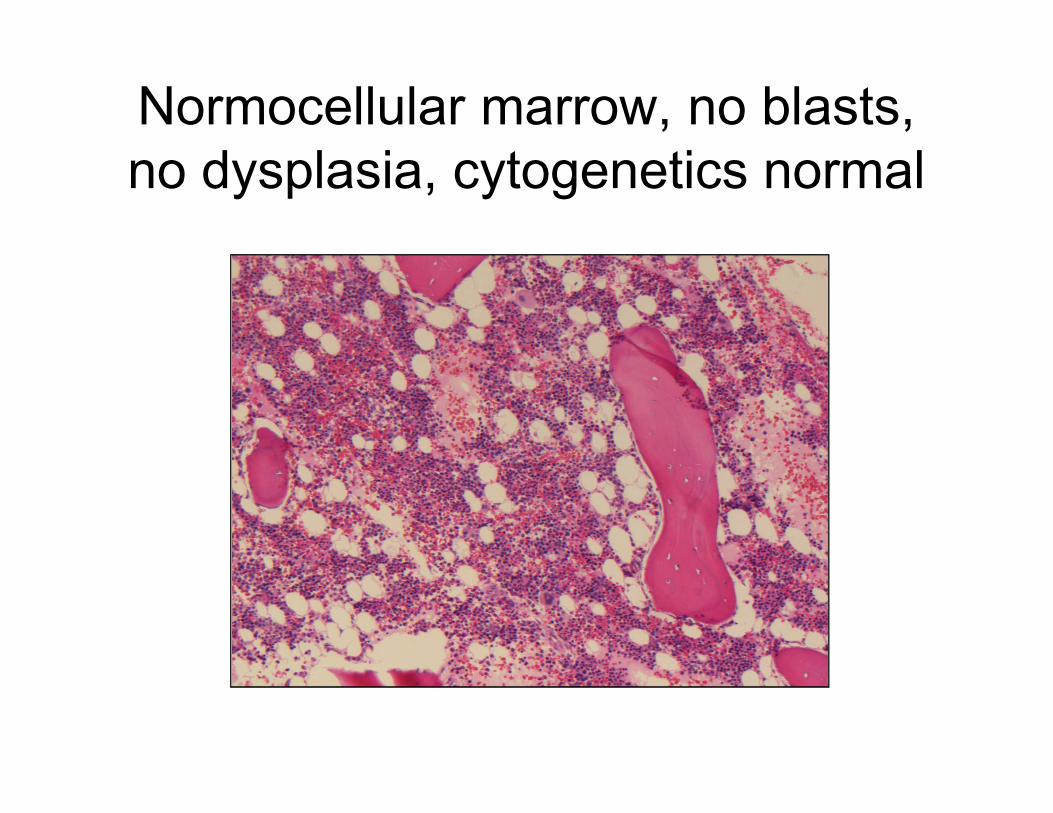
Normocellular marrow, no blasts,
no dysplasia, cytogenetics normal
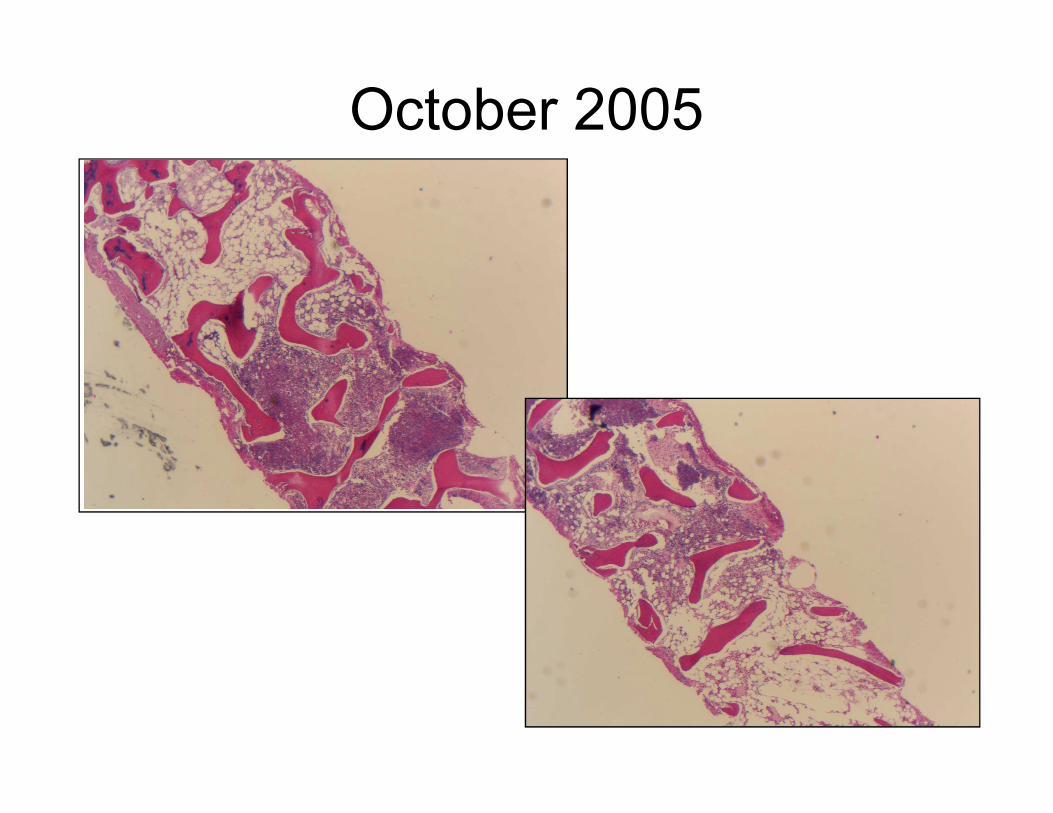
October 2005
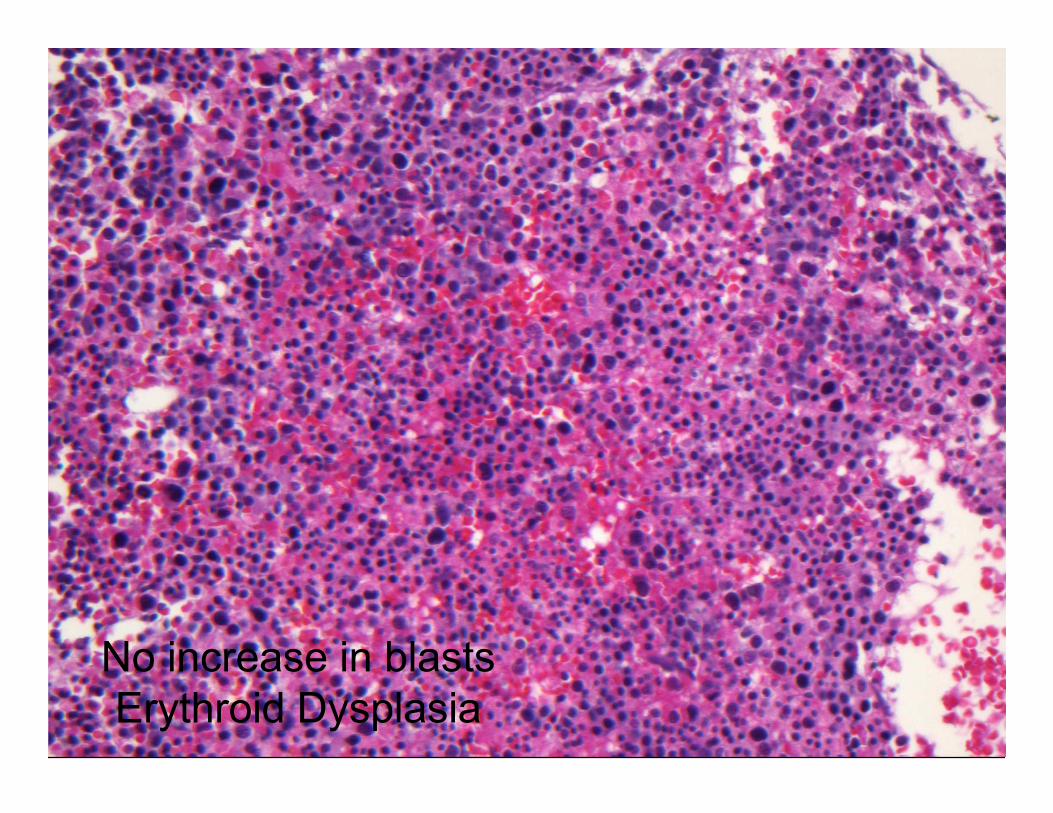
No increase in blasts
Erythroid Dysplasia

Diagnosis: Variably cellular marrow with
dyserythropoiesis, see note.
• Note: The marrow markedly hypocellular with areas of
hypercellularity. Dyserythropoiesis is present. No
megakaryocytic or myeloid dysplasia is seen. No blasts
are present. These findings may represent an evolving
aplastic anemia, but an underlying MDS cannot be aplastic anemia, but an underlying MDS cannot be
excluded. However, given that the dysplasia is limited to
the erythroid lineage and no blasts are present, an
evolving aplastic anemia is favored.
• Factors favoring Aplastic Anemia:
– No blasts
– No myeloid or megakaryocytic dysplasia
– Dysplasia limited to erythroid series with no
ringed sideroblasts (stress dyseryhropoiesis)
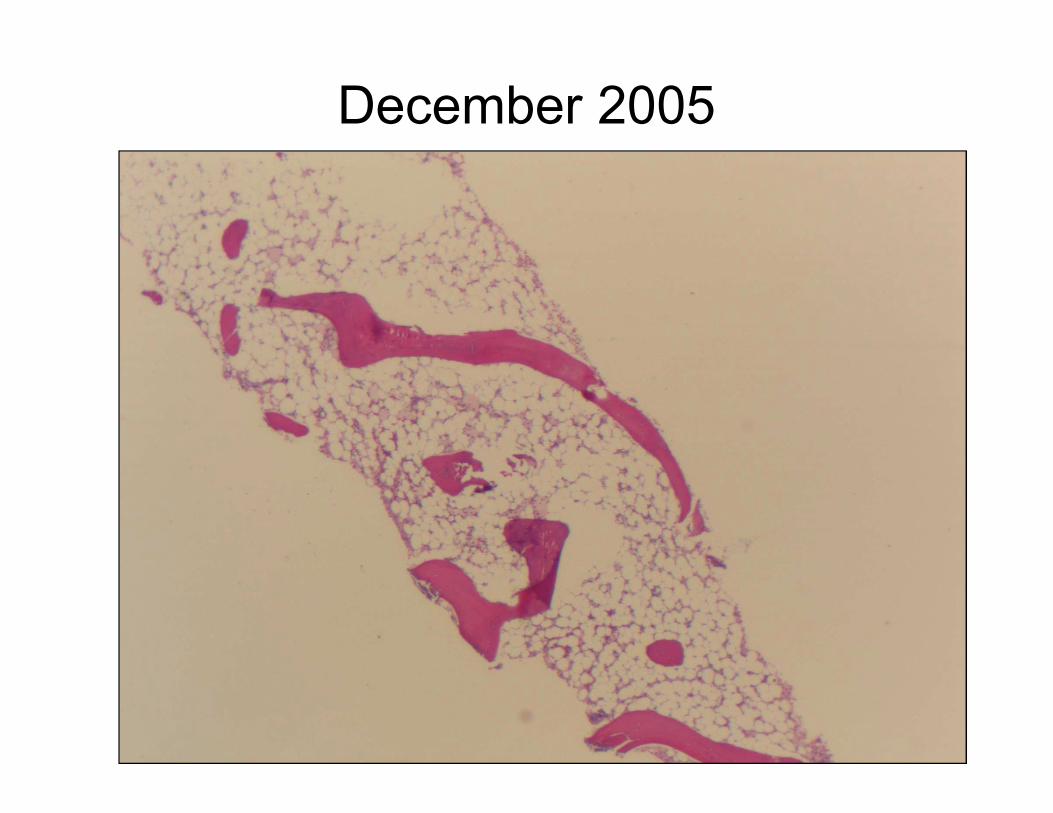
December 2005
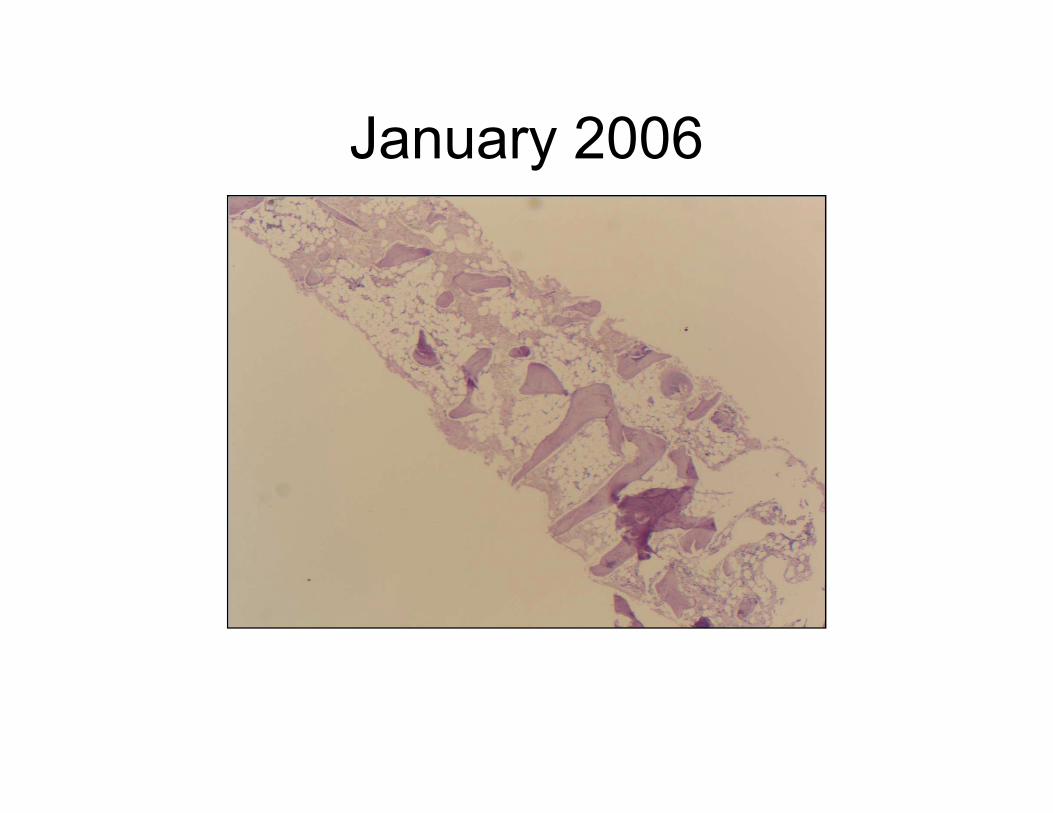
January 2006
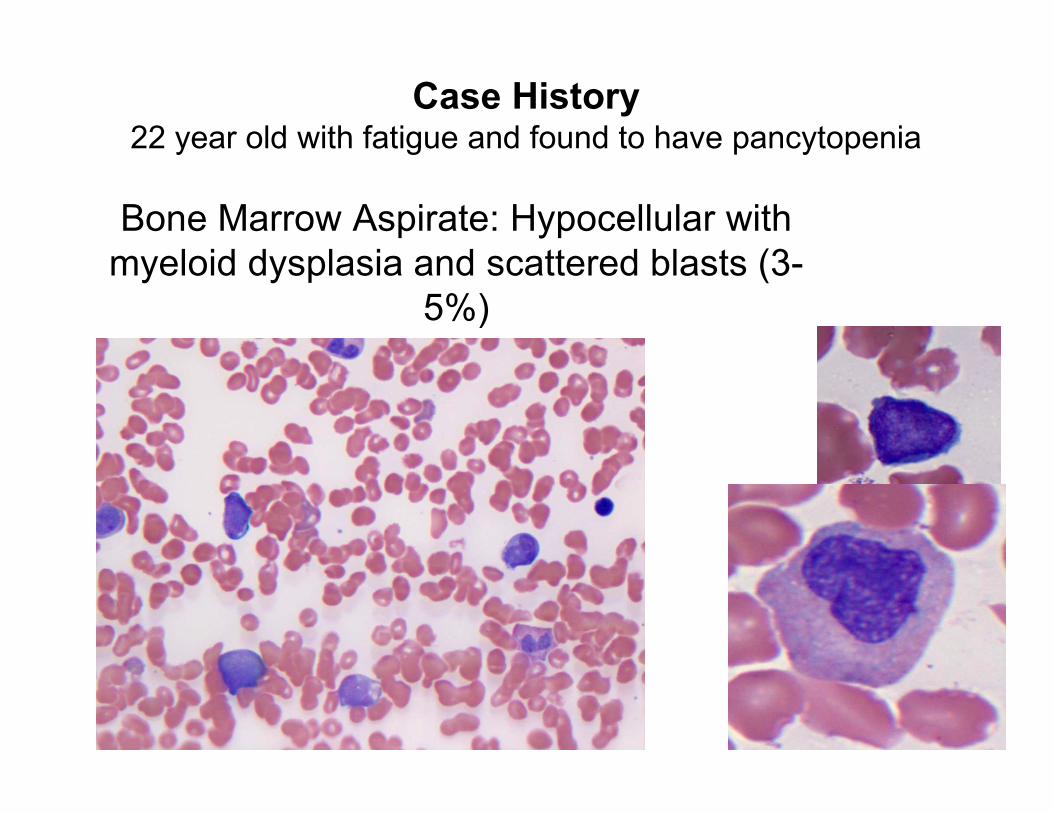
Bone Marrow Aspirate: Hypocellular with
myeloid dysplasia and scattered blasts (3-
5%)
Case History22 year old with fatigue and found to have pancytopenia
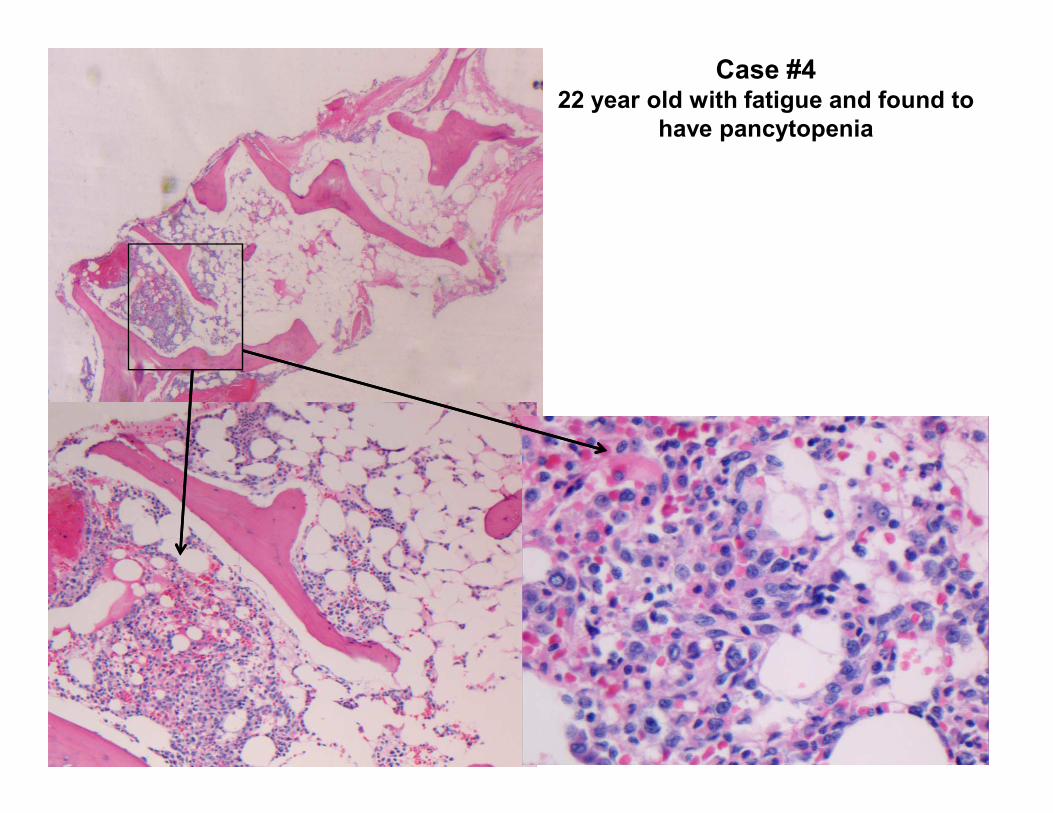
Case #4 22 year old with fatigue and found to
have pancytopenia
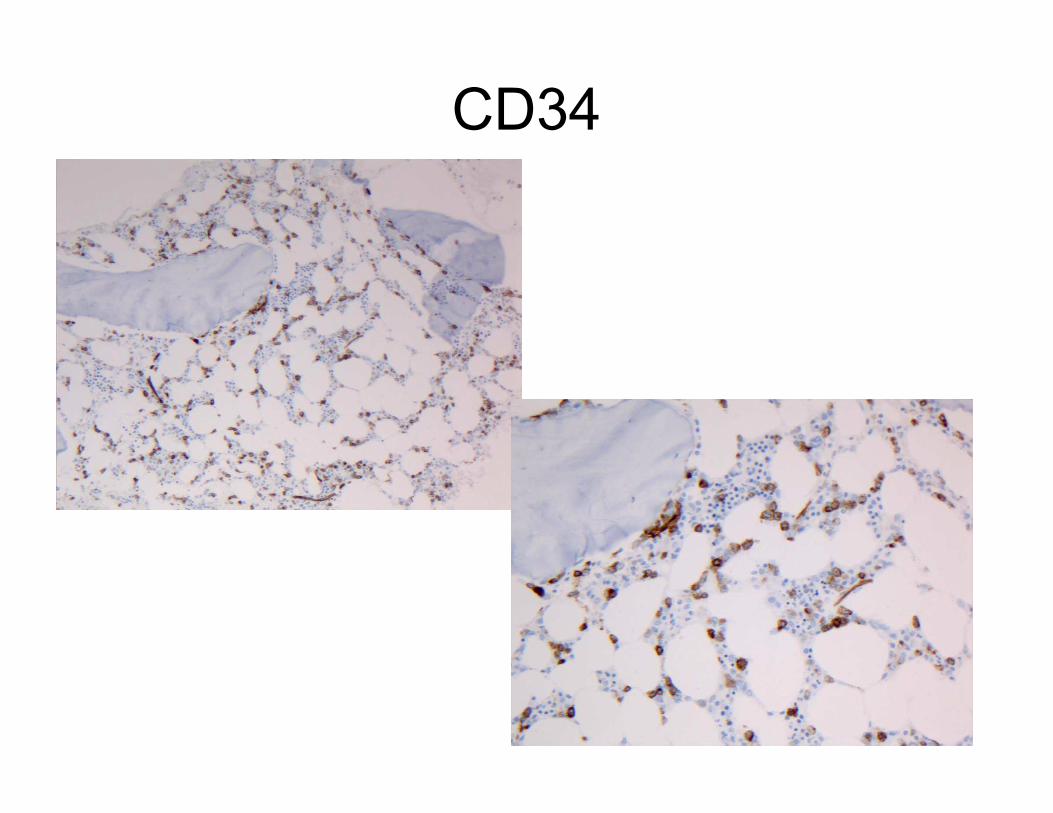
CD34
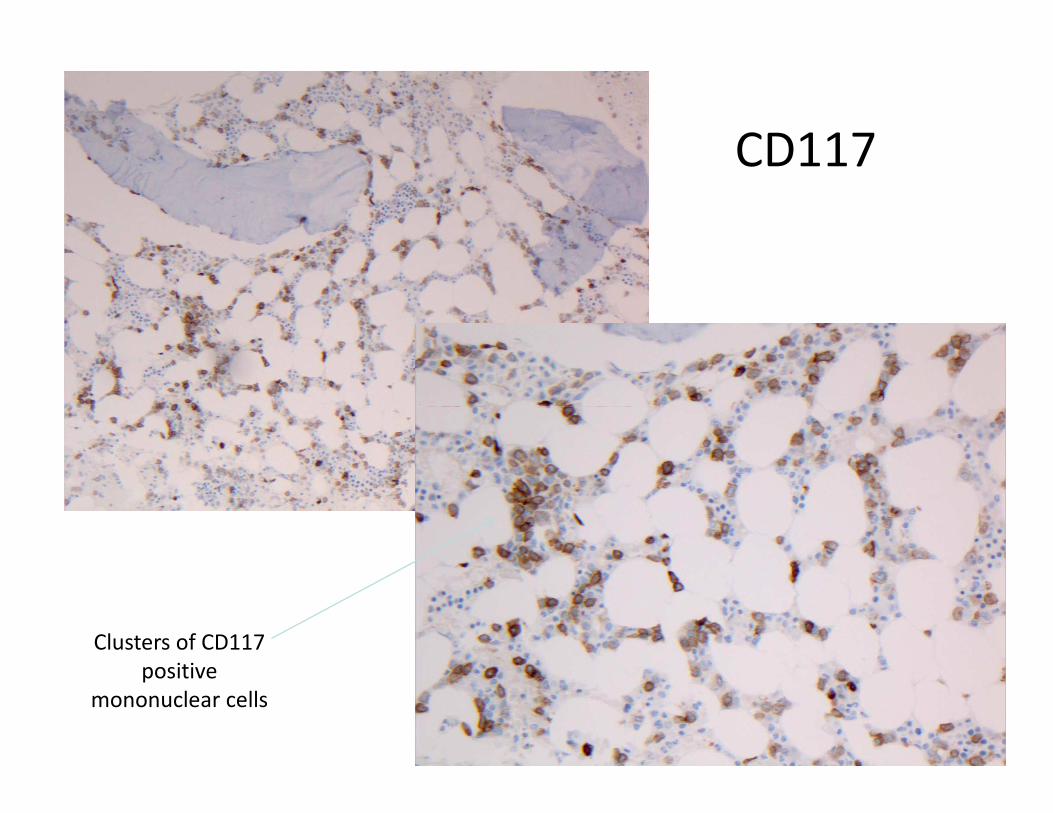
CD117
Clusters of CD117
positive
mononuclear cells

Dilemma??????
• Reason for consultation:
– Aplastic anemia OR
– Hypocellular MDS
• Factors that favor MDS• Factors that favor MDS
– Myeloid dysplasia
– Increase in blasts (~5-10%)
• Blasts seen on aspirate
• Confirmed CD34 and CD117
• Flow cytometry showed 5% myeloid blasts
• FISH showed monosomy 7

Diagnosis….
• Consistent with hypocellular MDS with
increase in blasts, best classified as
refractory anemia with excess blasts
(RAEB1)(RAEB1)
• Recommended tests based on the
patients age
– Telomere length testing
– Fanconi testing
– PNH

Diagnostic Algorithm for
Aplastic Anemia• Step 1: Establish criteria
• Step 2: Exclude Malignancy
• Step 3: Etiology
• Step 4: Prognostic Data• Step 4: Prognostic Data

Case History:
46 year old woman with fatigue and
bruising
• Clinical history:
• Diagnosed with acquired idiopathic
aplastic anemia on immunosuppression
• Developed hemolysis and venous
thrombosis
– Coombs negative anemia
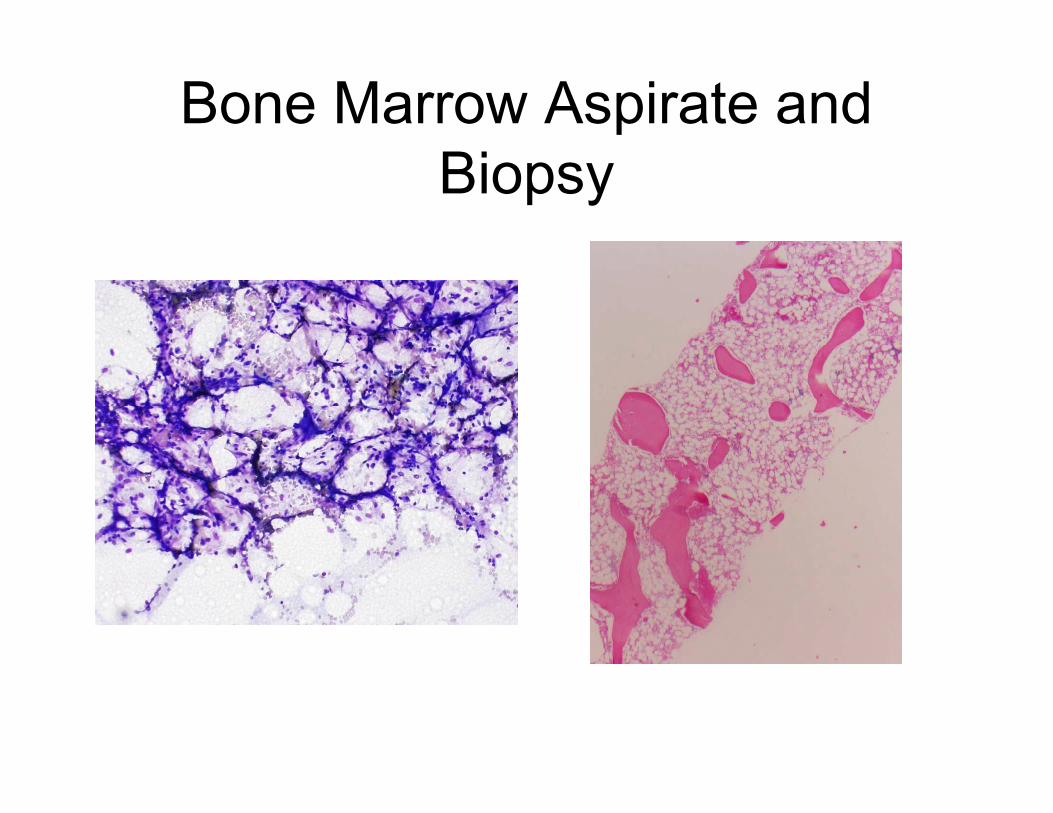
Bone Marrow Aspirate and
Biopsy
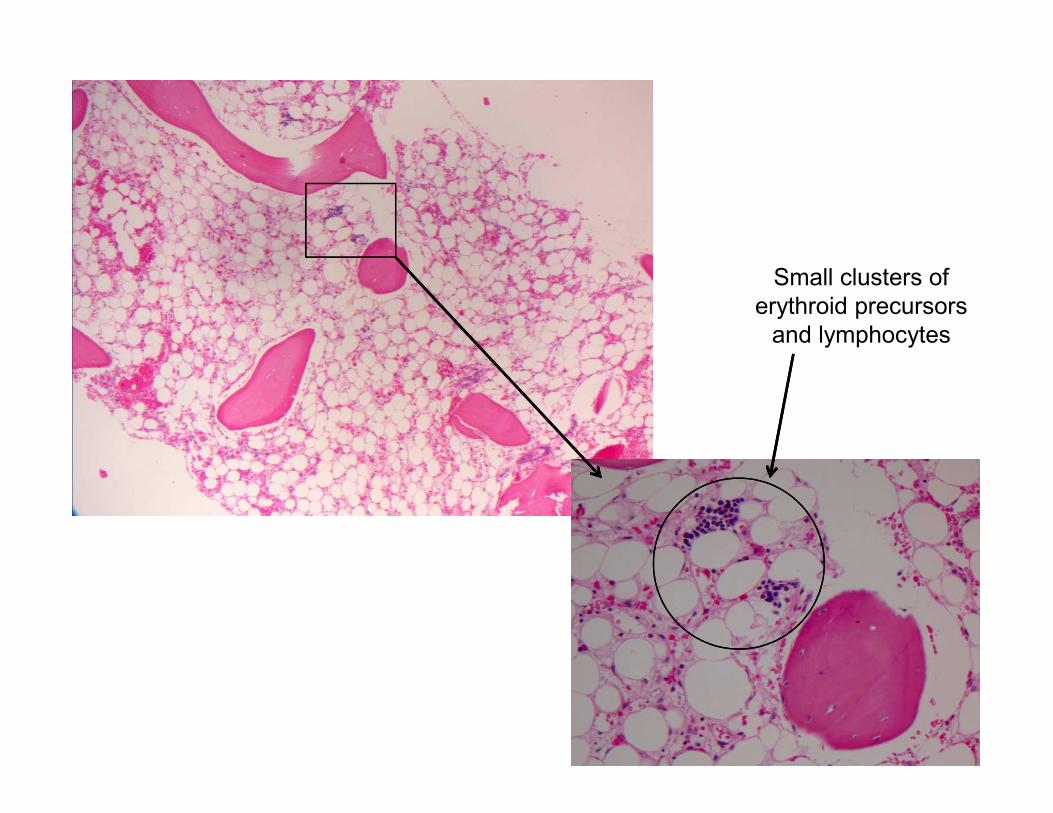
Small clusters of
erythroid precursors
and lymphocytes

Diagnosis: PNH in setting of
Aplastic Anemia• PNH is an ACQUIRED clonal disorder of
hematopoiesis
• Clone arises by somatic mutation in X-
linked phosphatidylinositol glycan class A linked phosphatidylinositol glycan class A
gene (PIG-A gene)
• Leads to deficiency in GPI anchor,
deficiency of GPI linked proteins
• Results in hemolytic anemia,
hypercoaguable state, and bone marrow
failure.
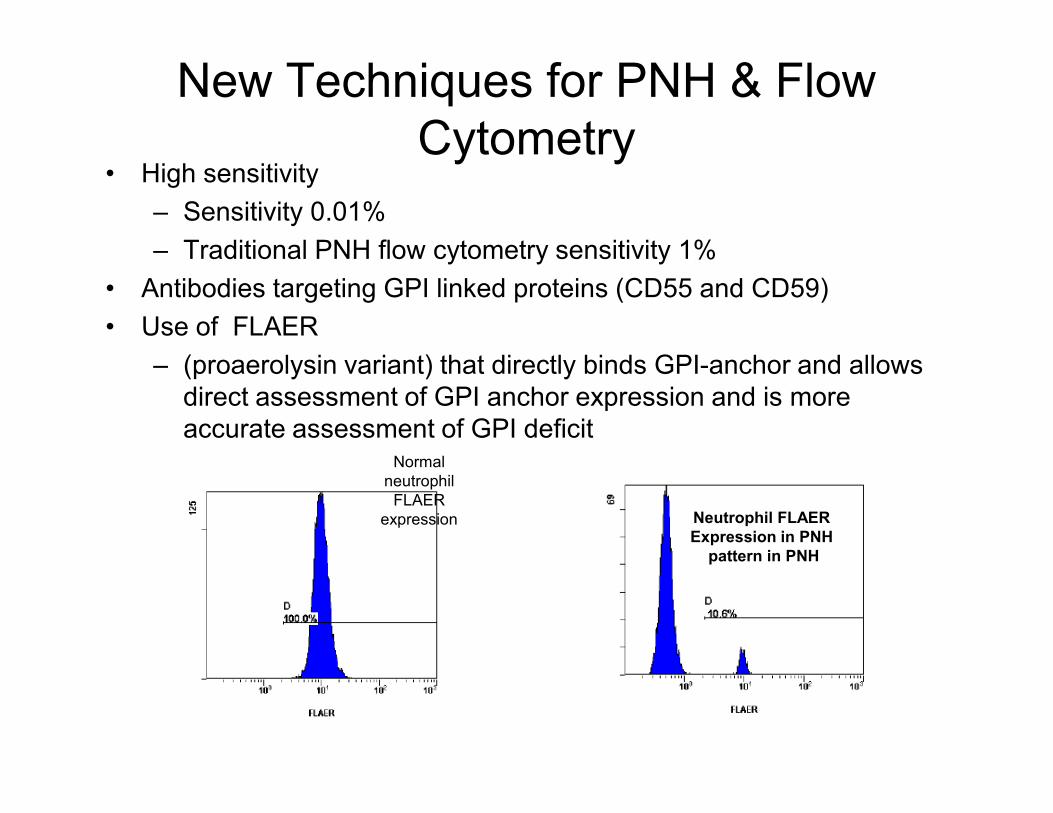
New Techniques for PNH & Flow
Cytometry• High sensitivity
– Sensitivity 0.01%
– Traditional PNH flow cytometry sensitivity 1%
• Antibodies targeting GPI linked proteins (CD55 and CD59)
• Use of FLAER
– (proaerolysin variant) that directly binds GPI-anchor and allows
direct assessment of GPI anchor expression and is more direct assessment of GPI anchor expression and is more
accurate assessment of GPI deficitNormal
neutrophil
FLAER
expression Neutrophil FLAER
Expression in PNH
pattern in PNH
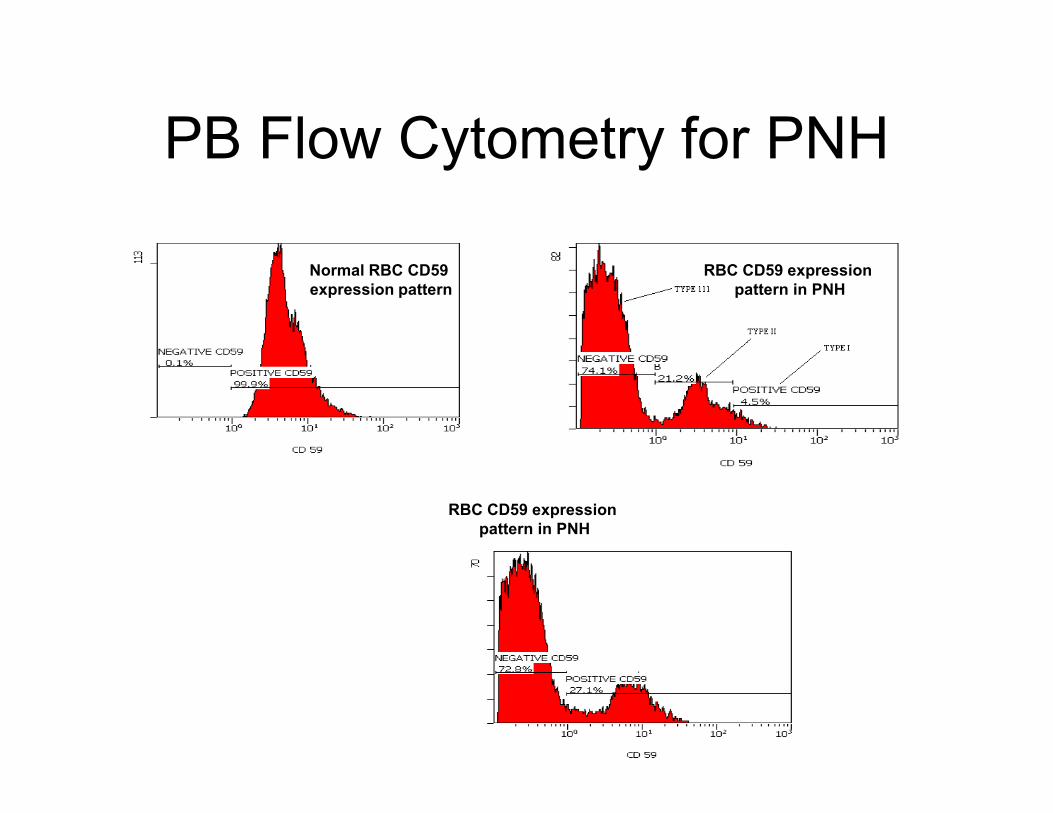
PB Flow Cytometry for PNH
Normal RBC CD59
expression pattern
RBC CD59 expression
pattern in PNH
RBC CD59 expression
pattern in PNH
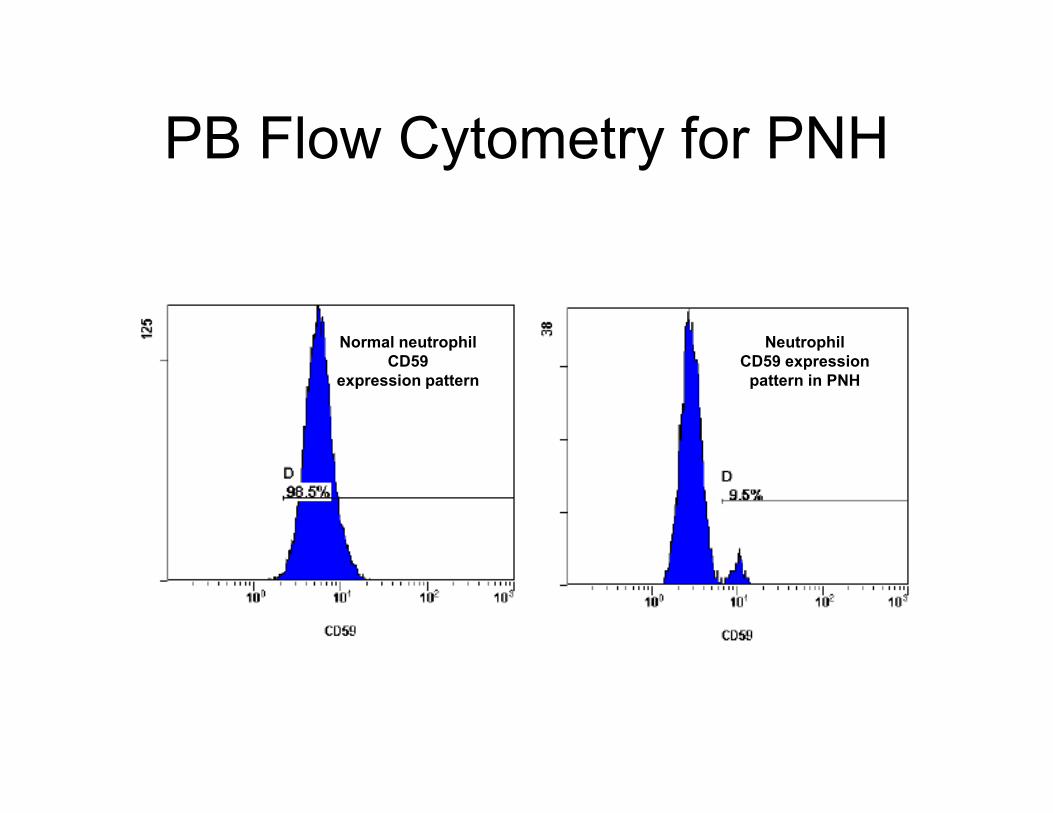
PB Flow Cytometry for PNH
Normal neutrophil
CD59
expression pattern
Neutrophil
CD59 expression
pattern in PNH

When to test for PNH• Recommended to test PNH at diagnosis of AA and then
yearly
– This is recommended for patients with inherited bone
marrow failure syndromes too not just acquired AA
– ?PNH clones may have a selective advantage in
marrow failure
• Small clones can be seen in AA in up to 40-80% of • Small clones can be seen in AA in up to 40-80% of
cases
• Clone sizes are followed and not treated any differently
than AA without a clone unless clinical symptoms
develop and there is progression to classical PNH or
clone size begins to increase
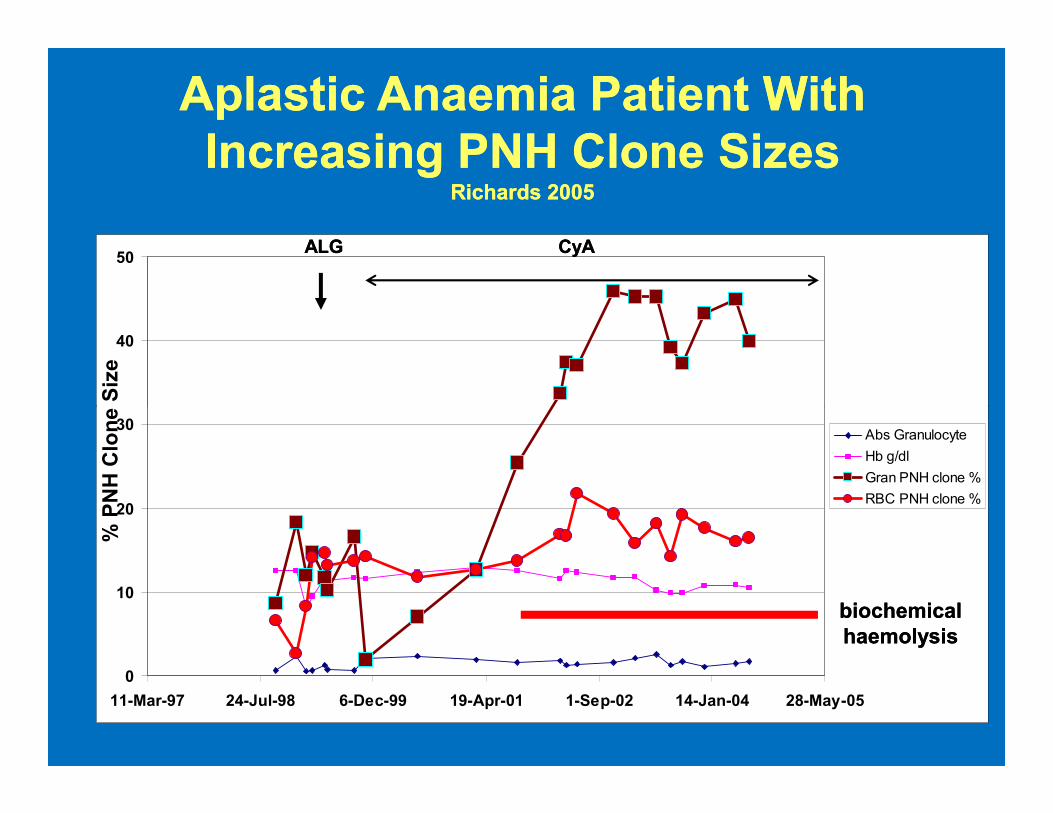
AplasticAplastic Anaemia Patient With Anaemia Patient With
Increasing PNH Clone SizesIncreasing PNH Clone SizesRichards 2005Richards 2005
40
50ALGALG CyACyA
% PNH Clone Size
0
10
20
30
11-Mar-97 24-Jul-98 6-Dec-99 19-Apr-01 1-Sep-02 14-Jan-04 28-May-05
Abs Granulocyte
Hb g/dl
Gran PNH clone %
RBC PNH clone %
biochemical biochemical
haemolysishaemolysis
% PNH Clone Size

Significance of PNH clones
• AA associated with PNH clones may have
a better prognosis and show a better
response to immunosuppression.
• MDS has been associated with PNH
clones (20% of cases)
• Prognosis of these clones in MDS is
controversial although some have shown a
better response other groups have not.

Summary• Hypocellular /aplastic marrows require a
vigorous work up
• Hypocellular presentations of
malignancies should be excluded
–ALL,AML,MDS, hairy cell leukemia, T –ALL,AML,MDS, hairy cell leukemia, T
cell LGL
– Immunohistochemical panel CD34,
CD20 and CD3
• Secondary causes of acquired AA should
be ruled out
–Clinical history, laboratory studies

Summary • Inherited bone marrow failures should be
considered
– pediatric/young patients and those with family
history and congenital anomalies
– Screening tests available – Screening tests available
• Fanconi – chromosome breakage test
• Dyskeratosis Congenita – telomere length
assessment FLOW-FISH
Prognostic data
– PNH clone
– Monitor clone in AA at diagnosis and yearly
for progression to clinical PNH

Thank You!!!
•
• Acknowledgements:
• Monica Bessler, MD, PhD• Monica Bessler, MD, PhD

Selected References• Foucar K, Reichard K, CzuchlewskiD. Bone Marrow Pathology, 3rd edition. 2010.
ASCP Press: Chicago.
• Young NS, Gerson SL, High KA, Clinical Hematology, 2006. Mosby, Philadelphia
• Shimamura A, Alter BP. Pathophysiology and management of inherited bone marrow
failure syndromes.Blood Reviews.2010;24:101-22.2010;584:3831-3838.
• Bessler M, Mason P, Link D and Wilson D. Nathan and Oski’s Hematology of Infancy
and Childhood. Pp 307-39, Saunders , Philadelphia.
• Bessler M,Wilson B, Mason P. Dyskeratosis Congenita. Federation of European
Biochemical Societies (FEBS) Letters.
• Garcia CK, Wrights WE, Shay JW. Human diseases of telomerase dysfunction: • Garcia CK, Wrights WE, Shay JW. Human diseases of telomerase dysfunction:
insights into tissue aging. Nucleic Acids Research. 2007;35(22):7406-7416.
• O’Sullivan RJ and Karlseder J. Telomeres: protecting chromosomes against genome
instability. Nat Rev Mol Cell Biol. 11, 171-181.
• De Winter JP, Joenje H. The genetic and molecular basis of Fanconi anemia. Mutat
Res. 2009;668:11-19.
• Green AM, Kupfer GM. Fanconi Anemia. Hematol Oncol Clin North AM 2009.;
23:193-214.
• Moldovan GL, D’Andrea AD. How the Fanconi anemia pathway guards the genome.
Ann Rev Genet 2009;43:223-49.
• Gluckman E, Wagner JE, Hematopoietic stem cell transplantation in childhood
inherited bone marrow failure syndrome. Bone Marrow Transplant. 2008;41:127-32.
• Gupta V, Brooker C, Tooze JA, et al. Clinical relevance of cytogenetic abnormalities
at diagnosis of acquired aplastic anemia in adults. Br J Hematol. 2006;143(1):95-9.

Selected References• Soland E. Hypocellular myelodysplasia. Hematol Oncol Clin North America.
2009;23:347-60.
• Greenberg P, CoxC, LeBeau MM, et al. International scoring system for evaluating
prognosis in myelodysplastic syndromes. Blood. 1997;89)6):2079-88.
• Kojima S, Ohara A, Tsuchida M. et al. Risk factors for evolution of acquired aplastic
anemia into myelodysplastic syndrome and acute myeloid leukemia after
immunosuppressive therapy in children. Blood.2002;100(3):P786-90.
• Wang H, Chuhjo T, Yasue S et al. Clinical significance of a minor population of
paroxysmal nocturnal hemoglobinuria-type c ells in bone marrow failure syndromes.
Blood. 2002;100:3897-3902.
• Wang SA, Pozdyakova O, Jorgensen JL et al. Detection of paroxysmal nocturnal
hemoglobinuria clones in patients with myelodysplastic syndromes and related bone
marrow diseases, with emphasis on diagnostic pitfalls and caveats. Haematologica.
2008;941:29-37.
• Young NS. Paroxysmal nocturnal hemoglobinuria and myelodysplastic syndromes:
clonal expansion of PIG-A mutant hematopoietic cells in bone marrow failure.
Haematologica. 2009;94:3-7.
• Sugimori C, Chuhjo T, Feng X, et al. Minor population of CD55 –CD59- blood cells
predicts response to immunosuppressive therapy and prognosis in patients with
aplastic anemia. Blood 2006:107:1308-14.
• Borowitz et al. Guidelines for the diagnosis and monitoring of paroxysmal nocturnal
hemoglobinuria and related disorders by flow cytometry. Cytometry B 2010; B.
• Rachidi S, Musallam K,Taher AT. A closer look at paroxysmal nocturnal
hemoglobinuria. Eur J of Int Med 2010;21:260-7.