mutations in hemophilia b, occur at the argl”-val activation site or
TRANSCRIPT

THE JOURNAL OF BIOLOGICAL CHEMISTRY 0 1990 by The American Society for Biochemistry and Molecular Biology, Inc.
Vol. 265, No. 19, Issue of July 5, pp. 108’76-10663,199O Printed in U. S. A.
Mutations in Hemophilia B, Occur at the Argl”-Val Activation Site or in the Catalytic Domain of Factor IX*
(Received for publication, September 18, 1989)
Rogier M. Bertina$, Irma K. van der Linden, Pierro M. Mannuccit, Hanneke H. Reinalda-Poot, Rosemiek Cupers, Swibertus R. Poor& and Pieter H. Reitsma From the Haemostasis and Thrombosis Research Unit, University Hospital, 2300 RC L.&den, The Netherlands and $A. Bianchi Bonomi Hemophilia and Thrombosis Centre, University of Milan, l-20122 Milan, Italy
Hemophilia B, is characterized by a strikingly pro- longed plasma ox brain prothrombin time. In an at- tempt to find an explanation for this phenomenon we have analyzed various aspects of the B, variants factor IX Deventer, factor IX Milano, factor IX Novara, and factor IX Bergamo.
Proteolytic cleavage by factor XIa was normal in two B, variants, but absent at the ArglBo-Val bond in the other two. In the latter variants Arg”’ was re- placed by either Trp or Gln, whereas Val”’ + Phe and Pro368 + Thr replacements have occurred in the var- iants that were normally cleaved by factor XIa.
In all four variants the B, effect could be neutralized with a single monoclonal antibody against factor IX. Also, after treatment with factor XIa, none of the B, variants reacted with antithrombin III (in contrast to normal factor IXa). Purified factor IX Deventer (one of the variants with a replacement of Arg”‘), either with or without pretreatment with factor XIa, was found to be a more effective competitive inhibitor of the factor VIIa-tissue factor-induced factor X activa- tion than similarly treated normal factor IX. In addi- tion, this inhibitory effect was much more pronounced when bovine tissue factor was used instead of human tissue factor.
We propose that the normal activation of factor IX not only produces a conformational change around the active site serine that allows efficient substrate binding and catalysis, but that the same conformational change is instrumental in effectively dissociating factor IXa from the activating factor VIIa-tissue factor complex. Amino acid replacements that disrupt this conforma- tional transition directly (e.g. Pro”” + Thr near the catalytic center) or indirectly (mutations at the Arg”‘- Val activation site) therefore lead to a combination of 1) the loss of coagulant activity and 2) an inhibitory effect in the ox brain prothrombin time assay.
Factor IX, a liver-derived zymogen of a serine protease of M, 55,000, is one of the key components of the extrinsic and intrinsic coagulation pathways (for review see Ref. 1). Factor IX circulates in plasma as a zymogen and is activated by limited proteolysis by either factor XIa (intrinsic pathway) or
* The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
$ ?‘o whom correspondence should be addressed: Haemostasis & Thrombosis Research Unit, University Hospital, Bldg. 1 C2-R, P. 0. Box 9600, 2300 RC Leiden, The Netherlands.
the factor VIIa-tissue factor complex (extrinsic pathway) (2, 3).
During activation the Arg’45-Ala and Arglso-Val bonds are cleaved, with release of an activation peptide of 35 amino acids, leaving a disulfide-linked two-chain molecule with an NHz-terminal light chain and a COOH-terminal heavy chain. The latter chain harbors the catalytic center that requires factor X as a substrate.
A defect or deficiency of the factor IX protein results in the recessive X-linked bleeding disorder hemophilia B (Christmas disease). In a global coagulation test that measures the intrin- sic pathway (activated partial thromboplastin time) the defect shows up as a prolongation of the clotting time. When one measures the extrinsic pathway using the prothrombin time, however, no abnormality is observed since factor VIIa-tissue factor can bypass factor IX by directly activating factor X.
An exception is formed by a small subgroup of patients with severe hemophilia B, called hemophilia B, after the index family, that in addition to a prolongation of the acti- vated partial thromboplastin time also shows a strikingly prolonged prothrombin time when ox brain is used as a source of tissue factor (4-7). In some patients the B, phenomenon may be explained by a mild factor VII deficiency (8, 9), but in others an aberrant factor IX molecule was held responsible (4, 10, 11). This was demonstrated by the observation that in plasma from the latter patients the B, effect could be neu- tralized by antibodies to factor IX. Furthermore, the most thoroughly studied B, variant to date, factor IX Lake Elsi- nore, was shown to act as a much more potent competitive inhibitor of factor VIIa-tissue factor-catalyzed factor Xa for- mation than normal factor IX (12).
The molecular defect in the Lake Elsinore variant is an Ala + Val change at position 390 near the substrate binding pocket of the catalytic domain (13). Also factor IX Angers, another factor IX-B,,, variant, has a defect in the catalytic domain: Gly3g6 + Arg (14). Indirect evidence indicates, how- ever, that some B, variants possess mutations that block cleavage of the Arglso-Val bond resulting in incomplete acti- vation of factor IX molecules (10, 15, 16). Recently, this has been confirmed for factor IX Hilo, a factor IX-B, variant in which Arg’% has been replaced by Gln (17,18). This suggests that there are at least two subgroups of B, variants, one with mutations in the catalytic domain and one with mutations at the ArglBO-Val activation site.
Here we report the molecular defects in four hemophilia B, variants, uiz. factor IX Deventer, factor IX Novaro, factor IX Milano, and factor IX Bergamo. The observed defects confirm the notion that B, mutations either occur at the Arg”‘-Val bond or near the binding pocket of the catalytic domain. Furthermore, kinetic data obtained with one of the variants (factor IX Deventer) indicate that a mutation in the Arg’*‘-
10876
by guest on March 22, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Mutations in Hemophilia 3, 10877
Val bond, like a mutation in the catalytic domain, as described for factor IX Lake Elsinore, results in a variant factor IX molecule that acts as a potent competitive inhibitor of extrin- sic factor X activation.
EXPERIMENTAL PROCEDURES
Patients--In this study we used plasma and genomic DNA from four unrelated patients with the hemophilia B, phenotype. All four patients had a severe bleeding tendency. The Dutch patient (factor IX Deventer) has been described previously (10). Larger volumes of this patient’s plasma were obtained by plasmapheresis after informed consent of the patient. Laboratory data on the three Italian patients have been published previously by Parekh et al. (19). Small aliquots of plasma of these patients were transported deep-frozen to Leiden. It was not possible to obtain sufficient plasma from these patients for the purification of their abnormal factor IX molecules.
Blood Collection-Blood was collected in 0.1 volume of 0.11 M trisodium citrate, and platelet-poor plasma was prepared by centrif- ugation of the blood at 3,000 rpm for 10 min. Platelet-free plasma was obtained by subsequent centrifugation at 16,000 X g for 20 min at 4 “C. Plasma samples were stored at -70 ‘C. Pooled normal plasma was prepared from the platelet-free plasma of 66 healthy volunteers (46 males, 20 females) and stored at -70 “C.
Proteins-The coagulation proteins factor VII (1750 units/mg), factor IX (250 units/mg) and factor X (120-160 units/mg) were purified from human plasma essentially as described previously (20- 22). Factor IXaP was prepared from isolated factor IX by activation with factor XIa/Ca*+ (IO). Factor XIa was isolated from human plasma by CeIite adsorption, elution of the Celite with I M NaCI (23), and affinity chromatography on heparin-Sepharose, Factor Xao! was prepared from purified factor X by activation with the isolated factor k activator from Russell’s viper venom coupled to Sepharose in the Dresence of CaCl? (24). The factor X activator was isolated from the ‘crude venom of k&sell’s viper venom as described by Kisiel et al. (25). Factor VIIa was prepared from isolated factor VII by activation by @-XIIa (26) as described previously (22). Antithrombin III was isolated from human plasma by affinity chromatography on heparin- Sepharose (27). Tissue factor apoprotein (TFAP)’ was purified from Triton X-100 extracts of washed human brain by affinity chromatag- raphy on factor VII-Sepharose (21). Isolated TFAP was stored in small aliquots in TEA buffer containing 0.2 mg of ovalbumin/ml and 0.01% Triton X-100 at -20 “C.
Factor IX Deventer was isolated from the plasma of a Dutch patient with hemophilia B, by affinity chromatography on B5-IgG- Sepharose. B5 is a murine monoclonal antibody against human factor IX (28). 490 ml of titrated plasma supplemented with 10 mM benz- amidine was passed through 17 ml of B5-IgG Sepharose (1 mg of IgG/ml Sepharose); the column was washed-with i200 ml of 20 mM sodium citrate. 0.9% NaCl. 10 mM benzamidine (uH 7.5) and eluted with a linear gradient (0-i M KCNS) in the wash buffer (2 X 150 ml). Fractions containing factor IX antigen were pooled and dialyzed against 20 mM sodium citrate, 100 mM NaCl, 10 mM benzamidine (pH 7.5). The factor IX Deventer pool then was applied to a DEAE- Sephadex column (-5 ml) equilibrated in the dialysis buffer. After washing. the factor IX Deventer was eluted by increasing the salt concen&ation to 0.5 M NaCl. Protein containingfractions were pooled and dialvzed against 10 mM Tris-HCl. 0.9% NaCl (DH 8.0). BV this proceduie 800 ;g of factor IX Deventer was isolate; Essentially the same procedure was used to isolate factor IX normal.
All isolated proteins were found to be more than 95% pure after analysis by SDS-polyacrylamide gel electrophoresis (29) and staining with Coomassie Brilliant Blue R-250 or, in the case of TFAP, silver staining (30). Molar concentrations of the isolated coagulation pro- teins w&e calculated from the protein content (31) by using reported molecular weights and absomtion coefficients (A&% of 11.6, 13.3, and 13.9 for fact; X, factor Ik, and factor VII, respectively. Molar concentrations of factor Xa and factor IXa were determined by active site titration as described previously (20). All isolated proteins were stored in small aliquots at -20 “C.
Crude bovine tissue factor was kindly provided by Dr. T. H. Janson (Nycomed AS, Oslo, Norway) as a lyophilized powder. This was reconstituted with 10 ml of TEA buffer, containing 100 mM NaCl,
’ The abbreviations used are: TFAP, tissue factor apoprotein; TEA, triethanolamine; SDS-PAGE, sodium dodecyl sulfate-polyacrylamide gel electrophoresis; bp, base pair(s).
0.2 mg of ovalbumin/ml, and 0.05% Triton X-100, and dialyzed extensively against the same buffer (without the ovalbumin). The reconstituted bovine tissue factor preparation contained 0.21 mM phospholipid. Small aliquots of the preparation were stored at -20 “C.
Non-Ca(II)-dependent antibodies against human factor IX were isolated from crude rabbit antiserum by immunoaffinity chromatog- raDhv on factor IX-Sepharose as described previously (32). The &m&opurified IgG was coupled to horseradish peroxidase using the reagent N-succinimidyl-3-(2-piridylthio)propionate (33).
Phospholipids-Sonicated phospholipid vesicles were prepared from isolated human brain phospholipids (“cephalin”) (34) as de- scribed previously (20). Phospholipid concentrations were determined by phosphate analysis after HClO, combustion (35).
Analytical Methods-Factor IX procoagulant activity was meas- ured by kaolin-activated partial thromboplastin times, using severe hemophilia B plasma as a substrate plasma (36). Factor IX antigen was measured by electro-immunoassay (37). Factor IXa was either measured by coagulation assay (10) or by measuring the concentration of antithrombin III binding sites with an immunoradiometric assay (38). Both assays were calibrated with a preparation of isolated, active site-titrated factor IXa& The ox brain prothrombin time was meas- ured using the Thrombotest’” reagent (Nycomed, Oslo, Norway) as prescribed by the manufacturer.
SDS-polyacrylamide gel electrophoresis was performed according to Laemli (39). Protein bands were stained with Coomassie Brillant Blue R-250.
Factor Xa activity was measured using a sensitive and specific spectrophotometric &say: 100 pl of sample was added to 800 ~1 of a buffer containing 10 mM EDTA, 50 mM triethanolamine (pH 8.3). 100 mM NaCl, 0.05 mg of ovalbumin/ml, and 5.4 mM S-2337. The reaction mixture was incubated at 37 “C for 10 min after which the reaction was stonped bv the addition of 100 ~1 of 0.5 M benzamidine. From the abso&nce & 405 nm, the concentration of factor Xa in the sample was calculated using isolated active site-titrated factor Xa as a reference.
Transfer to nitrocellulose sheets (Schleicher & Schuell, Dassel, West Germany) was performed according to the method of Towbin (40). Subsequently factor IX antigens were identified using either a murine monoclonal anti-factor IX antibody (B5) and goat anti-mouse IgG-horseradish peroxidase conjugate, or immunopurified non- Ca(II)-dependent anti-factor IX IgG coupled to horseradish peroxi- dase. .Perbxidase activity was visu&zed with 3,3’-diaminobenzidine.
Activation of Factor X bv Factor Vlla-TFAP-The influence of normal factor IX and factor IX Deventer on the extrinsic activation of factor X was measured in a system of purified coagulation factors: 75 ~1 of cephalin (150 PM) was added to 150 ~1 of TEA buffer (pH 7.5) containing 30 mM CaCl* and incubated at 37 “C for 15 min. After the subsequent addition of 30 ~1 of TFAP (0.67 nM, freshly prepared from a 23 nM stock solution), 15 pl of factor VIIa (300 PM), and 30 ~1 of TEA buffer containing 0.2 mg of ovalbumin/ml (TEA-OVA), the mixture was incubated for another I5 min to allow equilibrium formation of the phospholipid-TFAP-factor VIIa complex. Then, 75 pl of factor IX ndrmai-fact& IX Deventer or TEA-OVA were added before the reaction was started bv the addition of 75 ul of factor X. At 0, 4, and 8 min loo-~1 samples were withdrawn and directly analyzed for factor Xa with a spe&rophotometric assay. During this ueriod maximallv 5-18% of factor X was converted into factor Xa. Factor VIIa acti;ity was calculated from the linear increase in factor Xa concentration and expressed as moles of Xa/mol VIIa/min.
Activation of Factor IX in Barium Citrate Eluates by Factor Xla- 1 ml of titrated plasma was mixed with 80 ~1 of 1 M BaC12 and 20 ~1 of 1 M benzamidine for 30 min at room temperature. After centrifu- gation the pellet was washed once with 0.5 ml of 0.5 M BaCl*, 20 mM benzamidine and centrifuged. The pellet was then dissolved in 0.5 ml of 0.1 M Tris-HCl, 0.1 M EDTA, 20 mM benzamidine (pH 7.5) and dialyzed extensively against 50 mM Tris-HCI, 100 mM NaCl (pH 7.5). 90 ~1 of the dialyzed barium citrate eluate was incubated for 30 min at 37 “C with 9 ~1 of CaC& (50 mM) and 18 rl of factor XIa (0.25 fig/ ml). At time 0 and 30 min samples were taken for SDS-PAGE/ immunoblotting (20 pl plus 15 ~1 of EDTA-containing sample buffer) and analysis of factor IXa by immunoradiometric assay (10 ~1 plus 990 ~1 of EDTA containing assay buffer).
Identification of Point Mutations-Since our hypothesis is that in the haemophilia B, variants mutations have either occurred at the Arg’80-Val activation site or in the catalytic domain of the factor IX protein, we limited the analysis of the factor IX nucleotide sequence to exons 6 and 8 which code for the regions of interest (41). For the DNA analysis whole blood was collected from the patients and DNA
by guest on March 22, 2018
http://ww
w.jbc.org/
Dow
nloaded from

10878 Mutations in Hemophilia B,
was extracted from the white cell fraction using standard procedures. For the amplification of exon 6 and its flanking regions, two
oligonucleotides, “““” GTTCCATTTGCCAATGAGAA ‘“‘J” and ““““CCTGTGTCTTGCCAGCTGAGC?“’ (numbering as in Ref. 41), each linked with an EcoRI site at the h/-end, were synthesized. These primers were then used in a polymerase chain reaction as described before (42, 43). The resulting DNA segment was purified in ultra-low melting temperature agarose and cloned into Ml3 phage using estab- lished cloning procedures. The resulting recombinant Ml3 phages were subsequently sequenced using a commercial kit (Boehringer, Mannheim, Federal Republic of Germany). Since the Taq polymerase occasionally introduces errors during the amplification reaction, care was taken to sequence an adequate number of individual clones to verify the authenticity of the mutations reported in this paper. Furthermore, each of the mutations was confirmed by checking for altered restriction enzyme recognition sites (see “Results”). The amplification of the coding region of exon 8 proceeded similarly but with the two primers ‘““““TCTGTGTATGTGAAATACTG:‘D’XX and ‘“““GTTAGTGAGAGGCCCTGTTA’l”“‘. The resulting DNA frag- ment was again purified on agarose but this time directly sequenced using each of the amplification primers, as described before (43).
Materials-Ovalbumin (five times recrystallized) and tetrameth- ylenediamine were obtained from Koch-Light Laboratorium (Colnbrook, Slough, Berks, United Kingdom) and benzamidine hy- drochloride from Aldrich Europe (Beerse, Belgium). Heparin-Sepha- rose, protein A-Sepharose, CNBr-activated Sepharose, and DEAE- Sephadex A-50 were from Pharmacia Fine Chemicals (Uppsala, Swe- den); TEA from Fluka (Bucks, Austria). Trizma (Tris base) and 3,3’- diaminobenzidine were from Sigma. Goat anti-mouse horseradish peroxidase conjugate, SDS, acrylamide, and dithiothreitol were pur- chased from Rio-Rad. N,N’-Methylenehisacrylamide was from Merck-Schuckart (Hohenbrunn, West Germany). Coomassie Bril- liant Blue R-250 was from Serva (Heidelberg, West Germany). The chromogenic substrate N-benzoyl-I,-isoleucyl-L-glutamyl(piperidyl)- I,-glycyl-L-arginine-p-nitroanialide (S-2337) was a product of Kabi- Vitrum (Stockholm, Sweden). All other chemicals were analytical grade products from Merck (Darmstadt, West Germany).
RESULTS
B,, Effect and Neutralization by Anti-factor IX-Table I shows the B, effect (prolongation of the ox brain prothrombin time) for the four variants under investigation. It is clear from this table that plasmas from all four variants show a markedly prolonged clotting time (78-89 s) when compared to a pooled normal plasma (35 s). Furthermore, this prolongation of the clotting time is almost completely neutralized by the addition of a monoclonal antibody to factor IX (B5) which indicates that in all four patients an abnormal factor IX molecule is involved in the B,, effect. The concentration of monoclonal antibody used in these experiments (0.1 volume of ascites liquid) was found to be sufficient to give maximal shortening of the ox brain prothrombin time. The variation in prothrom- bin times in the presence of the B5 antibody is due to variation in the factor VII, X, and II content of the patient plasmas.
The Dignificance of the observation that the prothrombin time of pooled normal plasma was also shortened by the monoclonal antibody (see Table I) was established in a sepa- rate experiment. In the presence of buffer the prothrombin
TABLE I Laboratory findirzgs in four unrelated patients with hemophilia R,
FIX, factor IX; FIX Ag, factor IX antigen; FIX C, factor IX procoagulant activity.
Ox brain prothrombin time FIX Ag FIX C
-Anti FIX +Anti FIX (Br,)
units/ml s
FIX Deventer 1.30 co.01 89 41 FIX Novara 1.12 <O.Ol 80 33 FIX Milan0 1.30 co.01 78 41 FIX Bergamo 1.56 co.01 84 5” Normal pool 1.00 1.00 35 ;1
time was found to be 36.9 + 0.4 s (n = 8), while this was 32.0 & 0.7 s (n = 8) in the presence of monoclonal antibody. This observation is in agreement with earlier reports (44) and indicates that normal factor IX itself is also an inhibitor, albeit weak, of the prothrombin time.
Proteolysis of Factor IX by Factor XZa-One of the aims of this study is to (re)investigate the heterogeneity in the various B,,, variants with regard to proteolytic sensitivity to factor XIa. Fig. 1 shows this heterogeneity for the four variants. Factor IX in barium citrate eluates was activated with factor XIa, run on a nonreduced SDS-PAGE gel, blotted to nitro- cellulose, and probed with the B5-monoclonal antibody. In this system the release of the activation peptide as a result of the XIa-catalyzed Arg”‘j-Ala and Arg”“-Val cleavages shows as a decrease in apparent size from 66 to 46 kDa. All variants show a similar mobility (66 kDa) before the addition of factor XIa. Normal factor IX, factor IX Milano, and factor IX Bergamo appear as 46 kDa proteins after activation with factor XIa. Factor IX Deventer and factor IX Novara, how- ever, do not show this reduction in molecular weight, indicat- ing that the activation peptide cannot be released by treat- ment with factor XIa. For factor IX Deventer this finding confirms the earlier observation that purified factor IX De- venter is normally cleaved at the Arg’“‘-Ala position but not at the Arg’““-Val position (10).
Reactivity of Factor ZXa with Antithrombin III-To assess whether the B, effect is associated with abnormalities in the binding pocket of the catalytic domain of factor IX(a) it would be best to measure binding of the physiological substrate factor X to purified factor IXa. This was not feasible, since for three of the four hemophilia B, patients insufficient plasma was available for purification of factor IX. Therefore we resorted to measuring the formation of complexes between factor IXa and its pseudosubstrate antithrombin III as an assessment of the integrity of the substrate binding pocket. This test is commonly used in our laboratory to measure the amount of factor IXa in a sample (38). The results of such an experiment are shown in Table II. After treatment of the factor IX from the barium citrate eluate with factor XIa (see also Fig. l), samples were taken and analyzed for the presence of antithrombin III binding sites. In contrast to normal factor IX, none of the variant B,,, molecules showed detectable interaction with antithrombin III after treatment with factor XIa, which would be consistent with an abnormality in the
46 kd - 9.’
FIG. 1. Proteolytic cleavage of normal factor IX (FIX) and factor IX-B, by factor XIa. The factor IX in barium citrate eluates of normal plasma and plasma of four patients with hemophlha R,, was incubated with factor XIa as described under Experimental Procedures. Samples for SDS-PAGE and lmmunoblotting with the B5-monoclonal antibody agamst factor IX were taken before (-) and after (+) incubation with factor XIa MW, molecular weight; kd, kllodaltons.
by guest on March 22, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Mutations in Hemophilia B,
TABLE II
Generation of antithrombin III binding sites during activation of normal factor IX (FIX) and factor IX B,, with factor XIa
The factor IX in the barium citrate eluates of normal plasma and plasma of the four patients with hemophilia B,, was activated with factor XIa as described under “Experimental Procedures.” After 30 min a sample was analyzed for the formation of factor IXa (measured as antithrombin III binding sites).
Factor IXa
Normal FIX FIX Deventer FIX Novara FIX Milan0 FIX Bergamo
n&l
58 <0.15 CO.15 <0.15 co.15
substrate binding domain of factor IX(a). Point Mutations in the Factor IX Gene-The previous work
on factor IX Deventer (lo), factor IX Lake Elsinore (13), factor IX Niigata (45), factor IX Hilo (17), and factor IX Angers (14), together with the results presented this far shows that the mutations in hemophilia B,, should conceivably reside either at the cleavage sites of the activation peptide or at the carboxyl terminus of the protein. To test this hypothesis exons 6 and 8 of the factor IX gene that contain the coding information for these respective regions were sequenced for all four patients. Both exons were amplified directly from genomic DNA using the polymerase chain reaction and the nucleotide sequence was determined on either cloned frag- ments (exon 6) or directly on the amplified DNA (exon 8). The results of this analysis are shown in Figs. 2 and 3, and confirm the hypothesis.
Factor IX Deventer shows a C -+ T mutation that leads to replacement of Arg”’ by Trp. The CGG codon for Arg’*” is also mutated in factor IX Novara but in this instance a G has been mutated to A leading to a replacement by Gln. Factor IX Milan0 contains a G + T mutation in the codon for Val”’ leading to replacement by Phe. Factor IX Bergamo is the only variant that possesses a point mutation in the carboxyl- terminal domain (C + A) that leads to Pro”6R --$ Thr.
Each of the nucleotide changes also affects recognition sites for restriction enzymes. As shown in Figs. 2 and 3 (B), the mutations in factor IX Deventer, Novara, and Milan0 result in the loss of an AuaI restriction site (CNCGNG), whereas the mutation in factor IX Bergamo results in the loss of an AuaII site (CCNGG). In addition, factor IX Novara results in the creation of a new DdeI site (CTNAC). Such changes in the restriction pattern of amplified DNA fragments are very helpful as confirmation of the alterations found after sequenc- ing of fragments subcloned in M13.
Figs. 2C and 3C illustrate the conservation of the replaced amino acids in a few representative serine proteases. ArglR” is invariably found at corresponding activation sites in the se- quences shown but also in the vast majority of other serine proteases. Val ‘“I is less well conserved but in the more than 15 serine proteases sequenced to date amino acids at the corresponding position (i.e., the amino terminus of the cata- lytic domain) are always Val, Leu, or Ile. Pro”“R is also a conserved amino acid and, in addition to the sequences shown, is invariably present 3 residues carboxyl-terminal to the active site Ser in serine proteases.
Activation of Factor IX Deventer by Chymotrypsin-In fac- tor IX Deventer the replacement of Arg’*” by the bulky Trp may in theory have created a new cleavage site for the serine protease chymotrypsin. In principle, one should then be able to activate factor IX Deventer by a combination treatment with factor XIa and chymotrypsin. We have verified this
10879
FIG. 2. A, part of the nucleotide sequence of the opposite strand of exon 6 of the factor IX (FIX) gene in a normal patient and in three patients with hemophilia B,,,. Arrows in the sequence lanes of the patients indicate the various mutations in their factor IX genes. At the bottom of the panel the predicted amino acid changes for each variant are given. B, restriction enzyme digestion of the 311-bp exon 6 fragment. In normal patients a single AuaI site (CNCGNG) is present giving a 209. and 102.bp fragment. As predicted by the mutations in A, all of the three variants have lost the AvaI restriction site and no digestion is observed. In addition, the mutation in factor IX Novara predicts the introduction of a novel DdeI site which is corroborated by the digestions shown. Normally the exon 6 fragment contains two UdeI (CTNAG) sites resulting in 200., 95-, and 14bp fragments. In factor IX Novara the newly introduced DdeI site results in the digestion of the 200.bp fragment into 102. and 98-bp fragments. MW, molecular weight. C, comparison of corresponding activation sites in a few representative serine proteases from the blood coagu- lation system to illustrate the conservation of individual amino acid residues. The asterisk9 denote the two amino acids that are replaced in the B, variants and the arrow the activation cleavage site. PA, tissue plasminogen activator.
unique property of factor IX Deventer by performing the experiment shown in Fig. 4. It shows that, although to a limited extent, factor IX Deventer can become an active serine protease (measured both as coagulant activity and antithrom- bin III binding sites) concomittant with the appearance of a 46.kDa protein on SDS-PAGE. The limited extent of the effect and its transient nature on prolonged incubation with chymotrypsin (as also observed with normal factor IX) is probably due to additional chymotrypsin cleavages that ren- der the molecule inactive again (46).
The effects of factor XIa and chymotrypsin on factor IX normal and factor IX Deventer were also monitored by deter- mining at different time intervals the ox brain prothrombin times of samples diluted in factor IX-deficient plasma. From these experiments we learned that the inhibitory effect of factor IX Deventer on the prothrombin time is not influenced by incubation with factor XIa (cleavage of Arg14’-Ala bond only) and that the subsequent incubation with chymotrypsin resulted in a gradual shortening of the prothrombin time (data not shown). However, it was not possible to establish whether the shortening of the prothrombin time was due to the loss of the B, phenotype or to the introduction of factor IXa in the prothrombin time test. That the latter can con- tribute significantly to the shortening of the prothrombin time via thromboplastin-independent thrombin generation was demonstrated by prothrombin times of dilutions of factor IX normal (30 nM) and factor IXa normal (30 nM) in factor IX deficient plasma (41 and 25 s, respectively).
Purified Factor IX Deventer as a Competitive Inhibitor of Factor X Formation-Previous work with the Lake Elsinore
by guest on March 22, 2018
http://ww
w.jbc.org/
Dow
nloaded from
Mutations in Hemophilia B,,
C . Factor IX Q t D s G ‘ P H Factor II E G D s t c P F F?otcin c E G D s G G P M IPA 0 G D 5 G G P L Plarmi” Q G D s G t P L
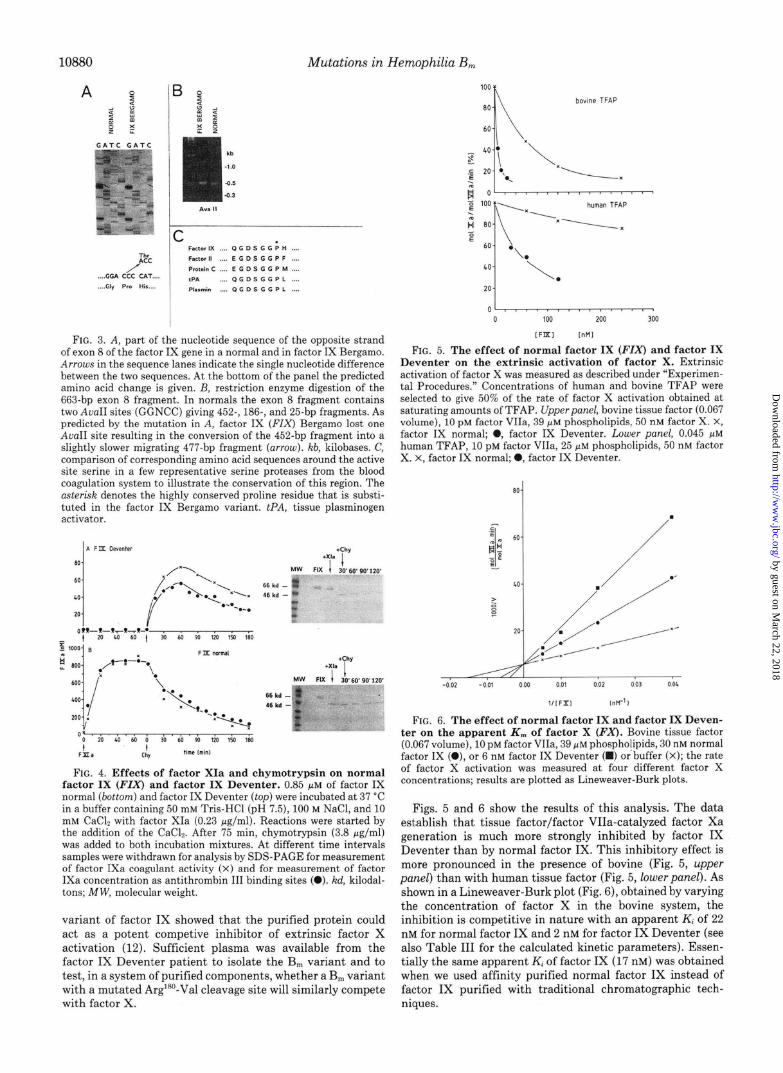
FIG. 3. A, part of the nucleotide sequence of the opposite strand of exon 8 of the factor IX gene in a normal and in factor IX Bergamo. Arrotu.s in the sequence lanes indicate the smgle nucleotide difference between the two sequences. At the bottom of the panel the predicted ammo acid change is given. B, restriction enzyme digestion of the 663-bp exon 8 fragment. In normals the exon 8 fragment contains two AvaII sites (GGNCC) givmg 452., 186-, and 25-bp fragments. As predicted by the mutation m A, factor IX (FIX) Bergamo lost one AvnII site resulting m the conversion of the 452-bp fragment into a slightly slower migrating 477-bp fragment (arrow). kb, kilobases. C, comparison of corresponding amino acid sequences around the active site serine m a few representative serine proteases from the blood coagulation system to illustrate the conservation of this region. The nstersk denotes the highly conserved proline residue that is substi- tuted in the factor IX Bergamo variant. PA, tissue plasminogen activator.
FIG. 4. Effects of factor XIa and chymotrypsin on normal factor IX (FIX) and factor IX Deventer. 0.85 ELM of factor IX normal (bottom) and factor IX Deventer (top) were incubated at 37 “C in a buffer containing 50 mM Tris-HCl (pH 7.5), 100 M NaCl, and 10 mM CaCb with factor XIa (0.23 &ml). Reactions were started by the addition of the CaCl,. After 75 min, chymotrypsin (3.8 fig/ml) was added to both incubation mixtures. At different time intervals samples were withdrawn for analysis by SDS-PAGE for measurement of factor IXa coagulant activity (x) and for measurement of factor IXa concentration as antithrombin III binding sites (0). kd, kilodal- tons; MW, molecular weight.
variant of factor IX showed that the purified protein could act as a potent competive inhibitor of extrinsic factor X activation (12). Sufficient plasma was available from the factor IX Deventer patient to isolate the B, variant and to test, in a system of purified components, whether a B,, variant with a mutated Arg”‘I-Val cleavage site will similarly compete with factor X.
0 >I...I.III,.(I~ 0 100 200 300
IFIX lllM1
FIG. 5. The effect of normal factor IX (FIX) and factor IX Deventer on the extrinsic activation of factor X. Extrinsic activation of factor X was measured as described under “Experimen- tal Procedures.” Concentrations of human and bovme TFAP were selected to give 50% of the rate of factor X activation obtained at saturating amounts of TFAP. Upperpanel, bovine tissue factor (0.067 volume), 10 pM factor VIIa, 39 j.~cM phosphohpids, 50 nM factor X. X, factor IX normal; 0, factor IX Deventer. Lower pane/, 0.045 pM human TFAP, 10 pM factor VIIa, 25 ELM phospholipids, 50 nM factor X. X, factor IX normal; a, factor IX Deventer.
so-
-002 0 01 000 0 01 002 0 03 004
l/IFXI 1°C /
FIG. 6. The effect of normal factor IX and factor IX Deven- ter on the apparent K, of factor X (FX). Bovine tissue factor (0.067 volume), 10 PM factor VIIa, 39 pM phospholipids, 30 nM normal factor IX (O), or 6 nM factor IX Deventer (W) or buffer (X); the rate of factor X activation was measured at four different factor X concentrations; results are plotted as Lmeweaver-Burk plots.
Figs. 5 and 6 show the results of this analysis. The data establish that tissue factor/factor VIIa-catalyzed factor Xa generation is much more strongly inhibited by factor IX Deventer than by normal factor IX. This inhibitory effect is more pronounced in the presence of bovine (Fig. 5, upper panel) than with human tissue factor (Fig. 5, lower panel). As shown in a Lineweaver-Burk plot (Fig. 6), obtained by varying the concentration of factor X in the bovine system, the inhibition is competitive in nature with an apparent K, of 22 nM for normal factor IX and 2 nM for factor IX Deventer (see also Table III for the calculated kinetic parameters). Essen- tially the same apparent K, of factor IX (17 nM) was obtained when we used affinity purified normal factor IX instead of factor IX purified with traditional chromatographic tech- niques.
by guest on March 22, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Mutations in Hemophilia B, 10881
TABLE III
Kinetic parameters for the extrinsic activation of factor X (FX) and its inhibition by normal factor IX (FIX) and factor IX Deventer
TFAP
K,,, FX (nM) K,,, (mol FXa/mol FVIIa/min)
K, FIX-normal (nM) K, FIX Deventer (nM)
Bovine HUman
67 67 180 125
22 470 2 118
1
FIG. 7. Effect of factor IXa and activated factor IX Deven- ter on the extrinsic activation of factor IX. Factor IX Deventer (0.69 PM) and normal factor IX (0.69 KM) were activated by incubation for 1 b at 37 “C with factor XIa (0.2 pg/ml) and 10 mM CaC12. Extrinsic factor X activation was measured as described under “Ex- perimental Procedures” in a system containing bovine tissue factor (0.067 volume), 10 pM factor VIIa, 39 PM phospholipids, 100 nM factor X, and various concentrations of normal factor IX (0) factor IXa/3 (0) or activated factor IX Deventer (X).
It should be realized, that factor IX normal and factor IX Deventer in fact are not competitive inhibitors but competi- tive substrates. Therefore the measured apparent K; values will be a function of the affinities of zymogen, intermediate, and product for the factor VIIa-tissue factor complex. In the experiment of Fig. 7, we compared the effects of the activation products of factor IX Deventer (factor IXcv Deventer; cleavage only of the Arg’45-Ala bond) and factor IX normal (factor IXafi; cleavage of both Arg’45-Ala and Argl”-Val bonds) on the extrinsic factor X activation. From the slopes of the lines in Fig. 7, it was calculated that the K, of factor IXa@ is 110 nM, while the K, of factor IXLV Deventer is 5 nM. The latter value is very similar to that observed for factor IX Deventer in the experiment of Fig. 6, indicating that the affinity of the zymogen and the factor IXa form of the Deventer variant for the catalytic complex is about equal. The Ki of factor IX& however is more than &fold higher than the apparent Ki observed for factor IX (see also Table III), indicating that the affinity is considerably higher for factor IX zymogen than for factor IX&. The difference between the apparent K, values of factor IX normal and factor IX Deventer (Fig. 6 and Table III) now may be explained by the observations that the conversion of the zymogen into the factor IXa form is not associated with a significant change in the affinity for the catalytic complex (Factor IX Deventer) and that the conver- sion of the zymogen form into the factor IXL$ form is asso- ciated with a considerable decrease in the affinity for the catalytic complex (factor IX normal). Together these obser- vations suggest that the conversion of the factor IXa into the factor IXa/3 form is associated with a conformational change which favours the dissociation of the final product from the catalytic complex.
DISCUSSION
The molecular heterogeneity of hemophilia B, is confirmed in the present study, where substitutions of amino acids at
three different positions (Arg”“, Val’*l, Pro36s) were identified in four unrelated patients with the B, phenotype, i.e. a severe bleeding tendency associated with the presence of an abnor- mal factor IX molecule with strongly reduced procoagulant activity, that acts as an inhibitor of the ox brain prothrombin time.
First of all, we need to establish how the observed altera- tions in the factor IX-B, molecules (see Table IV) will explain the strongly reduced procoagulant activity of these factor IX molecules. For this one would really need the three-dimen- sional structures of factor IX and factor IXa& Since these are not available, however, the extensive data on the three- dimensional structures of other serine proteases (47-49) have been used to model the serine protease part of the factor IXaP molecule (the heavy chain of factor IXafi) (50). Although these modeling studies have their limitations, e.g. with respect to the influence of the factor IXaP light chain, which is linked by a disulfide bridge to Cys*“, on the three-dimensional structure of the heavy chain, they are very useful to visualize some of the general features of serine proteases that are relevant for structure-function relationships in factor IX.
The crucial event during activation of factor IX is the cleavage of the Arg”‘-Val bond which will result in the formation of a new positively charged amino group on the amino-terminal valine. In analogy with what has been estab- lished for chymotrypsin (51, 52), this group may form an ion pair with the negatively charged carboxyl group of ASPIRE. This ion pair formation is accompanied with the movement of amino acid residues on the globular surface of the protein molecule especially in the region of the so-called substrate binding pocket (Ser384-Gly386 and Asp35g-Gln362 in factor IX) (53). It is believed that this conformational change makes the active site of a serine protease (Asp264-His221-Ser365), which is already preformed in the zymogen, accessible to its physiolog- ical substrates. At the same time the conditions are created to stabilize the P3-P,-Pl-P;-P&P; residues of the substrate in the tetrahedral configurations, which form the obligatory intermediates in the cleavage of the P,-P; bond and the subsequent deacylation of Ser365.
Because both factor XIa and factor VIIa show a preference for an arginine in the P1 position, it is not surprising that the Trp’“-Val in factor IX Deventer and the Gin”‘-Val in factor IX Novara and factor IX Hilo are not cleaved. In these variants the end product of the activation reaction is equiva- lent to factor IXLY (cleavage of the Arg14”-Ala bond), which has no factor IXa coagulant activity (10, 18). Apparently the elongation of the amino terminus of the heavy chain with the activation peptide does not lead to the formation of an ion pair between the amino-terminal Ala’46 with As~~~~, and thus prevents the formation of the substrate binding pocket. This explains why “activated” factor IX Deventer and factor IX Novara will not bind antithrombin III, and have no factor IX coagulant activity. Interestingly, cleavage of the Trp’“‘-Val
TABLE IV
Amino acid substitutions resulting in the hemophilia B, phenotype FIX, factor IX.
Variant Alteration Reference
FIX Angers” Gl ‘S + Arg FIX Bergamo ,&8
14,57 + Thr This study
FIX Deventer Arg”’ + Trp This study FIX Hilo Arg”’ + Gin 17, 18 FIX Lake Elsinore Ala3” ---f Val 12, 13 FIX Milan0 Val’R1 + Phe This study FIX Niigata Ala”” + Val 45 FIX Novara Arg’“’ + Gln This study
’ Mildly prolonged ox brain prothrombin time.
by guest on March 22, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Mutations in Hemophilia B,
bond in Factor IX Deventer with chymotrypsin resulted in the transient formation of both factor IXa activity and anti- thrombin III binding sites. Very recently this observation was confirmed by Suehiro et al. (541, who reported that factor IX- B, Nagoya, which also has the Argl*’ + Trp mutation, can be converted into active factor IXa by chymotrypsin or rat mast cell chymase.
In factor IX Milan0 we identified a point mutation in the triplet coding for Val*‘*, which results in its substitution by a Phe. From Table I and Fig. 1, it is clear that factor IX Milan0 has no detectable procoagulant activity while it is normally converted into a factor IXa/3 form by factor XIa. The obser- vation that after cleavage of the Arg’45-Ala and Arg”*-Val bonds, the factor IXa@ Milan0 cannot bind antithrombin III strongly suggests that no stable factor IXa-antithrombin III complex can be formed, either due to a defective substrate binding pocket or due to a defect in a specific antithrombin III binding site. Recently, it has been established that, in analogy with what has been found in trypsin (55), the se- quence 294-306 in tissue plasminogen activator is involved in the formation of stable complexes between tissue plasminogen activator and its inhibitor plasminogen activator inhibitor-l (56). Because in factor IX Milan0 the alteration is not in the region 199-205, which corresponds to residues 36-41 in tryp- sin and 294-306 in tissue plasminogen activator, it is plausible that the inability of factor IXa Milan0 to form complexes with antithrombin III is caused by a defect in the formation of the substrate binding pocket itself. One explanation would be that the aromatic side chain in the amino-terminal Phe does not permit ion pair formation with ASPIRE, so that no structural rearrangements in the region of the substrate bind- ing pocket will occur. Such an explanation would be in agree- ment with the observation that none of the known serine proteases has a Phe at the amino terminus (see also Fig. 2). In fact, our observations on factor IX Milan0 may be inter- preted as the first evidence that the concept of serine protease function as delineated by the crystallographic data on chy- motrypsinogen and chymotrypsin also apply for factor IX- factor IXa.
The alteration in factor IX Bergamo is the replacement of Pro3’j8 by a Thr. Pro368 which is the third amino acid carboxyl- terminal of the active site Ser365 is conserved in all serine proteases. The observation that activated factor IX Bergamo does not bind antithrombin III suggests that no stable enzyme substrate complex can be formed. It is not clear whether this is due to a defect in the catalytic site configuration or in the substrate binding pocket itself.
The replacement of Ala3” by Val in factor IX Lake Elsinore (13) and factor IX Niigata (45) also results in the complete loss of antithrombin III binding properties. Ala3” is not absolutely conserved, but a Val in this position has never been found. The alteration in factor IX Angers, finally, is the replacement of the highly conserved GlyZg6 by an Arg (14). Using a molecular model of the active site of factor IXa based on it homology with trypsin, Vidaud et al. (57) proposed that in factor IX Angers the guanidinium group of Arg3g6 forms a salt bridge with the carboxyl group of ASPIC’, thus permitting the Arg side chain to occupy the substrate binding pocket (57). It is not known whether activated factor IX Angers still can bind antithrombin III and what is the actual prolongation of the ox brain prothrombin time of the patients plasma.
All the variants listed in Table IV have the phenotype of a prolonged ox brain prothrombin time of the patients plasma.
For two of these factor IX-B,,, molecules (factor IX Lake Elsinore and factor IX Deventer) it has now been established that the kinetic basis for the prolonged ox brain prothrombin
time is the competitive inhibition by factor IX, which itself is a substrate for the factor VIla-tissue factor complex, of ex- trinsic factor X activation (Table III, Ref. 12). Using bovine tissue factor and human coagulation factors, we observed an apparent Ki of 22 nM for normal factor IX and of 2 nM for factor IX Deventer (see Table III). Using purified human tissue factor the apparent I(, for normal factor IX was much higher (470 nM), but again the Ki for factor IX Deventer was considerably lower (118 nM). The relative insensitivity of the human extrinsic activator to normal factor IX may explain why the prolongation of the prothrombin time is only seen with bovine and not with human tissue factor.
The finding of a &IO-fold lower apparent I(, of factor IX Deventer for the inhibition of extrinsic factor X activation indicates that at the same inhibitor and substrate concentra- tions factor IX Deventer will occupy more catalytic sites in the steady state than normal factor IX. One of the possible explanations for this phenomenon is that with factor IX Deventer the Michaelis complex of enzyme and product is more stable than with normal factor IX. The experiment of Fig. 7 provides some evidence for such a hypothesis by dem- onstrating that the Ki of activated normal factor IX (factor IXafl) is 20-fold higher than the Ki of activated factor IX Deventer (“factor IX&‘). This indicates that in the case of factor IX Deventer the 20-fold decrease in the K. of the enzyme-product complex will contribute to the 5-IO-fold de- crease in the Ki for inhibition of the extrinsic factor X activation.
Mutations in the Arg’@‘-Val cleavage site, near the catalytic triad or in the substrate binding region of factor IX all result in the failure to form an intact substrate binding pocket, as evidenced by the lack of antithrombin III binding sites, and in the B, phenotype. When we assume that our findings with factor IX Deventer also apply to the other factor IX-B,,, variants, the B, phenotype may be considered a reflection of the fact that during the “activation” of factor IX (B,) by the VIIa-tissue factor complex, the enzyme-product complex is more stable than in the case of normal factor IX.
Finally we wish to propose the following model for the activation of normal factor IX and factor IX-B, variants: in normal factor IX cleavage of the ArglaO-Val bond will release the positively charged amino group on Val”’ that will form an ion pair with ASPIRE. This event is associated with a conformational change resulting both in the formation of the substrate binding pocket and in a reduced affinity of factor IXaP for the activating enzyme (dissociation enzyme-product complex). In factor IX-B,,, molecules the alteration in the molecule will prevent this conformational transition in the enzyme-product complex, thus ensuring that the product will not have the proper substrate binding pocket for catalytic activity and is not readily released from the activating com- plex.
Acknowbzdgments-we gratefully thank M. J. Mentink and R. M. Claassen-Tegelaar for secretarial help during the preparation of the manuscript.
REFERENCES
1. Thompson, A. R. (1986) Blood 67,565-572 Discinio. R. G.. Kurachi. K.. and Davie, E. W. (1978) J. Clin. 2.
3.
4.
5. 6.
Zniest: 61,1528-1538 ' ' 0sterud, B., Rapaport, S. I. (1977) Proc. Natl. Acad. Sci. U. S. A.
74,5260-5264 Fantl, P., Sawers, R. J., Marr, A. G. (1956) Au&r. Ann. Med. 5,
163-176 Hougie, C., and Twomey, J. J. (1967) Lancet i, 698-700 Denson, K. W. E., Biggs, R., and Mannucci, P. M. (1968) J. Clin.
Pathol. 21,X0-165
by guest on March 22, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Mutations in Hemophilia B,
0sterud, B., Kasper, C. K., and Prodanos, C. (1979) Thromb. Res. 15.235-243
10883
33.
34.
35.
36.
37.
38.
39. 40.
41.
42.
Bri&, E., Bertina, R. M. (1986) Thromb. Haemostasis 55, 122- 128
7.
8. 9.
10.
11.
12.
13.
14.
15.
16.
17.
18.
19.
20.
21.
22.
23.
24.
25.
26.
27.
28.
29. 30. 31.
32.
Elodi, S. (1973) Thromb. Diath. Haemorrh. 29, 247-252 Mazzucconi. M. G.. Bertina. R. M.. Romoli. D.. Orlando. M..
Avvisati, &., and Mariani, k. (1986) Thromb. Hhemostasi$ 43; 16-19
Bertina, R. M., and Van der Linden, I. K. (1982) Thromb. Haemostasis 47, 136-140
Kasper, C. K., 0sterud, B., Minami, J. Y., Shonick, W., Rapaport, S. I. (1977) Blood 50, 351-366
asterud, B., Kasper, C. K., Lavine, K. K., Prodanos, C., and Rapaport, S. I. (1981) Thromb. Haemostasis 45,55-59
Spitzer, S. G., Pendurthi, U. R., Kasper, C. K., and Bajaj, S. P. (1988) J. Biol. Chem. 263. 10545-10548
Attree, O., Vidaud, D., Vidaud, M., Amselem, S., Lavergne, J.- M., Goossens, M. (1989) Genomics 4, 266-272
Yoshioka, A., Ohkubo, Y., Nishimura, T., Tanaka, I., Fukui, H., Ogata, K., Kamiya, T., Takahashi, H. (1986) Thromb. Res. 42, 595-604
Yoshioka, A., Sakai, T., Yamamoto, K., Ohkubo, Y., Fukui, H. (1987) Thromb. Haemostasis 58, 705-708
Huang, M.-N., Kasper, C. K., Roberts, H. R., Stafford, D. W., High, K. A. (1989) Blood 73, 718-721
Monroe, D. M., McCord, D. M., Huang, M.-N., High, K. A., Lundblad, R. L., Kasper, C. K., Roberts, H. R. (1989) Blood 73,1540-1544
Parekh, V. R., Mannucci, P. M., Ruggeri, Z. M. (1978) Br. J. Haematol. 40, 643-655
Mertens, K., Cupers, R., van Wijngaarden, A., and Bertina, R. M. (1984) Biochem. J. 223,599-605
Born, V. J. J., Ram, I. E., Alderkamp, G. H. J., Reinalda-Poot, H. H., Bertina, R. M. (1986) Thromb. Res. 42,635-643
Born. V. J. J.. van Tilburz N. H.. Krommenhoek-van Es. C.. Bekina, R. &I. (1986) Thyomb. Haemostasis 56, 343-348 ’ ’
Bsterud, B., and Rapaport, S. I. (1977) Proc. Natl. Acad. Sci. U. S. A. 74,5260-5264
Mertens, K., and Bertina, R. M. (1982) Thromb. Haemostasis 47, 96-100
Kisiel, W., Hermodson, M. A., Davie, E. W. (1976) Biochemistry 15,4901-4906
Tankersley, D. L., Alving, B., Finlayson, J. S. (1982) Thromb. Res. 25, 307-317
Bertina, R. M., Broekmans, A. W., Krommenhoek-van Es, C., van Wijngaarden, A. (1984) Thromb. Haemostasis 51, l-5
Bertina, R. M., van der Linden, I. K., Muller, H. P., Derks, J., Klein-Breteler, E. (1981) Thromb. Haemostasis 46, 165
Mertens, K., Bertina, R. M. (1980) Biochem. J. 185, 647-658 Morrissey, J. (1981) Anal. Biochem. 117, 307-310 Shapiro, S. S., and Waugh, D. F. (1966) Thromb. Diath. Hae-
morrh. 16,469-490 Poor& S. R., van der Linden, I. K., Krommenhoek-van Es, C.,
43.
44. 45.
46. 47.
48.
49.
50.
51.
52.
53.
54.
55.
56.
57.
Carlsson, J., Drevin, H., Ax&, R. (1978) Biochem. J. 173, 723- 737
Van den Besselaar, A. M. H. P., Ram, I. E., Alderkamp, G. H. J., Bertina, R. M. (1982) Thromb. Haemostasis 48.54-58
Battcher, ‘C. J. F., van Gent, C. M., Pries, G. (1961) Anal. Chim. Acta 24, 203-204
Veltkamp, J. J., Drion, F. F., Loeliger, E. A. (1968) Thromb. Diath. Haemorrh. 19,279-303
Bertina, R. M., Van der Linden, I. K. (1977) Clin. Chim. Acta 77,275-286
Born, V. J. J., Reinalda-Poot, H. H., Poort, S. R., Cupers, R., Bertina, R. M. (1987) Thromb. Res. 45,661-667
Laemmli, U. K. (1970) Nature 227, 680-685 Towbin, H., Staehelin, T., Gordon, J. (1979) Proc. Natl. Acad.
Sci. U. S. A. 76,4350-4354 Yoshitake, S., Schach, B. G., Foster, D. C., Davie, E. W., Kurachi,
K. (1985) Biochemistry 25, 3736-3750 Saiki, R. K., Gelfand, D. H., Stoffel, S., Schauf, S. J., Higuchi,
R., Horn, G. T., Muller, K. B., Ehrlich, H. A. (1988) Science 230,487-491
Reitsma, P. H., Bertina, R. M., Ploos van Amstel, H. K., Riemens, A., Brilt, E. (1988) Blood 72, 1074-1076
Brstavik, K. H., Laake, K. (1978) Thromb. Res. 12, 455-465 Sugimoto, M., Miyata, T., Kawabata, S., Yoshioka, A., Fukui, H.,
Takahashi, H., Iwanaga, S. (1988) J. Biochem. (Tokyo) 104, 878-880
Enfield, D. L., Thompson, A. R. (1984) Blood 64, 821-831 Marquart, M., Walter, J., Deisenhofer, J., Bode, W., Huber, R.
(1983) Acta Chrystallogr. Sect. B. Strut. Sci. 39, 480-490 Sawyer, L., Shotton, D. M., Campbell, J. W., Wendell, P. L.,
Muirhead, H., Watson, H. C., Diamond, R., and Ladner, R. C. (1978) J. Mol. Biol. 118, 137-208
Cohen, G. H., Silverton, E. W., Davies, D. R. (1981) J. Mol. Biol. 148,449-479
Furie, B., Bing, D. H., Feldmann, R. J., Robison, D. J., Burnier, J. P., Furie, B. C. (1982) J. Biol. Chem. 257,3875-3882
Matthews, B. W., Sigler, P. B., Henderson, R., Blow, D. M. (1967) Nature 2 14,652-656
Sigler, P. W., Blow, D. M., Matthews, B. W., Hendersson, R. (1968) J. Mol. Biol. 35, 143-164
Freer, S. T., Kraut, J., Robertus, J. D., Wright, H. T., Xuong, N.- H. (1970) Biochemistry 9,1997-2009
Suehiro, K., Kawabata, S., Miyata, T., Takeya, H., Takamatsu, J., Ogata, K., Kamiya, T., Saito, H., Niho, Y., Iwanaga, S. (1989) J. Biol. Chem. 264,21257-21265
Huber, R., Kukla, D., Bode, W., Schwager, P., Bartels, K., De- isenhofer, J., Steigemann, W. (1974) J. Mol. Biol. 89, 73-101
Madison, E. L., Goldsmith, E. J., Gerard, R. D., Gething, M. J. H., Sambrook, J. F. (1989) Nature 339, 721-724
Vidaud, M., Attree, O., Schaad, O., Vidaud, D., Edelstein, S., Goossens, M. (1988) Blood 72, (Suppl. I) 313 (abstr.)
by guest on March 22, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Poort and P H ReitsmaR M Bertina, I K van der Linden, P M Mannucci, H H Reinalda-Poot, R Cupers, S R
catalytic domain of factor IX.Mutations in hemophilia Bm occur at the Arg180-Val activation site or in the
1990, 265:10876-10883.J. Biol. Chem.
http://www.jbc.org/content/265/19/10876Access the most updated version of this article at
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
http://www.jbc.org/content/265/19/10876.full.html#ref-list-1
This article cites 0 references, 0 of which can be accessed free at
by guest on March 22, 2018
http://ww
w.jbc.org/
Dow
nloaded from