interaction of anti-phospholipid antibodies with late...
TRANSCRIPT

Interaction of Anti-Phospholipid Antibodies With LateEndosomes of Human Endothelial Cells
Beatrix Galve-de Rochemonteix, Toshihide Kobayashi, Corinne Rosnoblet, Margaret Lindsay,Robert G. Parton, Guido Reber, Emmanuel de Maistre, Denis Wahl, Egbert K.O. Kruithof,
Jean Gruenberg, Philippe de Moerloose
Abstract—Anti-phospholipid antibodies (APLAs) are associated with thrombosis and/or recurrent pregnancy loss. APLAsbind to anionic phospholipids directly or indirectly via a cofactor such asb2-glycoprotein 1 (b2GPI). The lipid target ofAPLA is not yet established. Recently, we observed that APLAs in vitro can bind lysobisphosphatidic acid (LBPA). Theinternal membranes of late endosomes are enriched in this phospholipid. The current study was undertaken to determineto what extent binding of APLA to LBPA is correlated with binding to cardiolipin and tob2GPI and to determinewhether patient antibodies interact with late endosomes of human umbilical vein endothelial cells (HUVECs) and thusmodify the intracellular trafficking of proteins. Binding of patient immunoglobulin G (n537) to LBPA was correlatedsignificantly with binding to cardiolipin. Although LBPA binding was correlated to a lesser extent withb2GPI binding,we observed thatb2GPI binds with high affinity to LBPA. Immunofluorescence studies showed that late endosomes ofHUVECs contain LBPA. Patient but not control antibodies recognized late endosomes, but not cardiolipin-richmitochondria, even when we used antibodies that were immunopurified on cardiolipin. Incubation of HUVECs withpatient plasma samples immunoreactive toward LBPA resulted in an accumulation of the antibodies in late endosomesand led to a redistribution of the insulinlike growth factor 2/mannose-6-phosphate receptor from the Golgi apparatus tolate endosomes. Our results suggest that LBPA is an important lipid target of APLA in HUVECs. These antibodies areinternalized by the cells and accumulate in late endosomes. By modifying the intracellular trafficking of proteins, APLAcould contribute to several of the proposed pathogenic mechanisms leading to the antiphospholipid syndrome.(Arterioscler Thromb Vasc Biol. 2000;20:563-574.)
Key Words: anti-phospholipid antibodiesn late endosomesn lysobisphosphatidic acidn b2-glycoprotein In human endothelial cells
The anti-phospholipid antibody (APLA) syndrome (APS)is a condition characterized by various clinical manifes-
tations in combination with the presence of APLAs. It hasbeen suggested that many of the APLAs bind indirectly toanionic phospholipids via phospholipid-binding proteins. Ofthese, b2-glycoprotein I (b2GPI) is the most intensivelystudied, but other phospholipid-binding proteins such asprothrombin, annexin V, protein C, thrombomodulin, proteo-glycans, or other antigens on endothelial cells (ECs) orplatelets have also been mentioned.1–4
The mechanisms of the prothrombotic state associated withthe APS are poorly understood but may involve modificationsof platelet functions and monocyte tissue factor expression.5–7
Moreover, several pathogenic mechanisms for APLA havebeen proposed that affect ECs.1,4,8–13 Taken together, thesestudies suggest that APLAs may, at least in part, exert theirthrombogenic effect by inhibiting the activated protein
C–thrombomodulin anticoagulant pathway on ECs, by acti-vating ECs to become prothrombotic or by inducing apopto-sis of ECs that leads to deendothelialization and exposure ofthe thrombogenic subendothelium. It remains to be estab-lished whether these proposed mechanisms are independentor reflect common underlying mechanisms.
Late endosomes reside on the pathway leading to deg-radation in the lysosomes and function as an importantprotein-sorting station between secretory and degradationpathways. A unique feature of late endosomes is that theycontain a complex system of poorly characterized internalmembranes in their lumen. Using baby hamster kidney(BHK) cells, we recently observed that these internalmembranes contain large amounts of a unique anionicphospholipid, lysobisphosphatidic acid (LBPA), that isrecognized by APLAs.14 Incubation of BHK cells with amonoclonal anti-LBPA antibody and with APLA from a
Received March 17, 1999; revision accepted June 9, 1999.From the Division of Angiology and Hemostasis, University Hospital Geneva (B.G.R., C.R., G.R., E.K.O.K., P.M.), Switzerland; the Department of
Biochemistry, University of Geneva (T.K., J.G.), Geneva, Switzerland; the Centre for Microscopy and Microanalysis, Department of Physiology andPharmacology (M.L.) and the Centre for Molecular and Cellular Biology (R.G.P.), University of Queensland, Australia; and the University Hospital ofNancy, Nancy, France (E.M., D.W.).
Correspondence to Dr Philippe de Moerloose, Haemostasis Unit, University Hospital Geneva, 1211 Geneva 14, Switzerland. [email protected]
© 2000 American Heart Association, Inc.
Arterioscler Thromb Vasc Biol.is available at http://www.atvbaha.org
563
by guest on May 26, 2018
http://atvb.ahajournals.org/D
ownloaded from

few patients resulted in accumulation of the antibodies inlate endosomes.
The current study was undertaken to investigate in moredetail to what extent (direct or indirect) the binding of APLAsto intracellular LBPA may constitute a plausible mechanismfor the thrombogenic effects of APLA. To this end, westudied (1) to what extent, in an ELISA assay, binding ofAPLA to LBPA is correlated with immunoreactivities towardcardiolipin and towardb2GPI; (2) whether LBPA is detect-able in late endosomes of human ECs; and (3) whetherantibodies from patients with APLA are internalized, retainedin the late endosomes of EC, and interfere with the normalprotein-sorting functions of endosomes in these cells. Theresults show that late endosomes of human ECs containLBPA and that APLAs accumulate in late endosomes andmodify intracellular protein trafficking.
MethodsSubjectsPlasma was obtained from 37 patients with APLAs who werereferred to the Hemostasis units of the University Hospitals ofNancy, France, and Geneva, Switzerland. The presence of APLAswas defined by the presence of a lupus anticoagulant and/or apositive result in an anti-cardiolipin antibody assay. Patients werediagnosed as having primary APS (n516); APS secondary tosystemic lupus erythematosus (SLE, n59); SLE or overlap syn-drome without manifestations of APS (n55); neoplasia (n52); orpsychosis treated with neuroleptics (n51). Moreover, in 4 patientslupus anticoagulant was discovered preoperatively; in these patientslupus anticoagulant activity remained strongly positive.1 year afterthe preoperative work-up. None of these 4 patients had clinicalmanifestations of APS. As a control group, blood was drawn from 15healthy persons.
MaterialsUnless stated otherwise, all biochemical reagents and chemicals usedin this study were from Sigma Chemical Co or Fluka and of thehighest grade available. The composition of the PBS used was 0.27mol/L NaCl, 5.4 mmol/L KCl, 10 mmol/L Na2HPO4, and1.76 mmol/L KH2PO4 at pH 7.4.
The following antibodies were used: monoclonal antibody(MoAb) 6C4 that binds LBPA14; affinity-purified rabbit polyclonalanti–C-terminal p23 antibody15; MoAb anti–human-b2GPI (9G1, agift from Dr J. Arvieux, Grenoble, France)16; rabbit antibodiesagainst the insulinlike growth factor 2/mannose-6-phosphate recep-tor (IGF2/M6PR, a gift from Dr B. Hoflack, Institut Pasteur, Lille,France); rabbit anti-human von Willebrand factor (vWF) antibodies(Dako); rhodamine-conjugated goat anti-mouse antibodies (Jackson;these antibodies were passed over immobilized rabbit antibodies toremove cross-reactivity); FITC-conjugated goat anti-rabbit antibod-ies (Organon Teknika), DTAF-conjugated, affinity-purified F(ab9)2
fragment goat anti-human immunoglobulins; and TRITC-conjugated, affinity-purified pure donkey anti-human IgG (H1L; thelatter 2 were from Jackson).
Patient IgGs were isolated by passage of 200mL of plasma overa 200-mL column of protein A–Sepharose CL-4B (AmershamPharmacia Biotech). After extensive washing (5 mL),bound materialwas eluted with 0.1 mol/L glycine-HCl, pH 2.2, and immediatelyneutralized with 0.1 volume of 2 mol/L Tris, pH 8.0. The proteincontent of the purified IgG preparations was assayed by the bicin-choninic acid protein assay (Pierce Chemical Co).
LBPA was purified from BHK lipid extracts by preparativethin-layer chromatography after silica gel and DEAE column chro-
Figure 1. A, Molecular structures of LBPA and cardiolipin. Eachglycerol residue of LBPA contains 1 fatty acid molecule. Theposition of fatty acid in glycerol is not determined. B, Compari-son of purified LBPA with cardiolipin. LBPA, 2.3 mg, and 2 mg ofcardiolipin were applied to a silica gel 60 high-performance thin-layer chromatography plate (Merck), and components migratedin a chloroform/methanol/32% ammonia (65:25:5, vol/vol/vol)solvent. Lipids were visualized after being charred with cupricacetate.
Figure 2. Correlation between binding of IgG from patients withAPLA or from controls to cardiolipin, LBPA, and b2GPI. IgGsamples were purified from the plasma of healthy controls(n515) or patients with APLA (n5 37) and analyzed by ELISA onmicrotiter plates coated with LBPA, cardiolipin, or b2GPI. A,Comparison between control IgG and patient IgG in the 3assays. The horizontal lines represent the upper limit of the nor-mal range, which is defined as the mean12SD of the normalpopulation. B, Correlation between LBPA and cardiolipin bindingfor patient IgG. C, Correlation between LBPA and b2GPI bindingfor patient IgG.
564 Arterioscler Thromb Vasc Biol. February 2000
by guest on May 26, 2018
http://atvb.ahajournals.org/D
ownloaded from

matography. The purity of LBPA was assessed by thin-layer chro-matography and mass spectrometry.14 Bovine heart cardiolipin wasfrom Fluka.
To determine the phospholipid composition of human umbilicalvein ECs (HUVECs), cells were metabolically labeled for 20 hourswith 32P[Pi]. After extraction, phospholipids were separated by 2Dchromatography. The first dimension was run with chloroform/methanol/32% ammonia (65:25:5, vol/vol/vol) and the second di-mension with chloroform/acetone/methanol/acetic acid/water(50:20:10:12.5:5, vol/vol/vol/vol/vol). Radioactive lipids were de-tected by autoradiography and then quantified by using the Molec-ular Imager System (Bio-Rad GS-363).
Assays for APLAs and Anti-b2GPI AntibodiesThe binding of APLAs to cardiolipin or to LBPA was assayed onELISA plates coated with the respective phospholipids.17 Microwellplates (Nunc) were coated overnight with a solution (30mL/well) of50 mg/mL of either cardiolipin (Fluka) or LBPA in 98% ethanol and2% chloroform. As controls for nonspecific binding of patient IgG tothe ELISA plates, we used wells treated with ethanol alone.Thereafter the plates were blocked with 10% FCS (Life Technolo-gies) in PBS for 2 hours at room temperature and washed 3 timeswith PBS. In some experiments, we used LBPA- or cardiolipin-coated plates that were blocked with 1% BSA (Fluka) by a 2-hourincubation. Plasma samples (100mL, diluted 1:100) or IgG (100mLat the final concentration of 20mg/mL) were added in duplicate. Theplates were then incubated for 2 hours at room temperature andwashed 3 times. Alkaline phosphatase–conjugated goat anti-humanIgG was added to each well and incubated for 90 minutes at roomtemperature. After 3 washes, the wells were incubated at 37°C withp-nitrophenyl phosphate (Sigma), and alkaline phosphatase activitywas measured at 405 nm in a microplate reader (Molecular Devices).For each plasma or IgG,the binding on uncoated wells wassubtracted from that on phospholipid-coated wells.
Maxisorb g-irradiated ELISA plates (Nunc) were coated for 2hours at room temperature with humanb2GPI (50 mg/mL), leftovernight at 4°C, and washed before use. The coated plates wereincubated for 1 hour at room temperature with plasma (diluted 1/50)or with IgG (20mg/mL). After 3 washes in PBS–Tween 20 (0.01%),the plates were exposed to alkaline phosphatase–conjugated goatanti-human IgG and incubated for 90 minutes at room temperature.After 3 washes, the wells were incubated at 37°C withp-nitrophenylphosphate (Sigma), and alkaline phosphatase activity was measuredat 405 nm in a microplate reader. On each plate, dilutions of anAPLA-positive control plasma were included. Sample blanks (un-coated wells) were included. All results are expressed in arbitraryunits with respect to a standard curve made with the positive control.
For the binding ofb2GPI to cardiolipin or LBPA, plates werecoated with cardiolipin or LBPA as described above. Then the plateswere treated with 3% BSA for 2 hours at room temperature andincubated for 2 hours with 5mg/mL purified humanb2GPI (a giftfrom Dr J. Arvieux). The plates were washed 3 times with PBS andthen incubated for 90 minutes with dilutions of an MoAb (9G1) tohumanb2GPI. The plates were washed 3 times and incubated for 1hour with horseradish peroxidase–conjugated goat anti-mouse IgG(Bio-Rad); after being washed,o-phenylenediamine dihydrochloridesubstrate (0.4 mg/mL) from Sigma was added for 30 minutes. Thecolor development was blocked by addition of 3 mol/L HCl and theoptical density read at 490 nm.
Affinity Purification ofAnti-Cardiolipin AntibodiesA Nunc plate was coated with cardiolipin as described above. Aftera 2-hour incubation with 10% FCS, the plate was washed 3 timeswith PBS. Diluted plasmas (1:10) in PBS from 5 patients and 1control or respective IgG (60mg/mL) were adsorbed for 45 minuteson a cardiolipin-coated well. The nonadsorbed material was recov-ered and reapplied to a second cardiolipin-coated well. PBS wasadded to prevent the wells from drying out. This manipulation wasrepeated 5 times. The wells were eluted with 100mL of 0.1 mol/L
Figure 3. Binding of b2GPI to LBPA and to cardiolipin. Humanb2GPI was incubated on LBPA- (F) or cardiolipin (E)-coatedplates. Binding of b2GPI was quantified by using MoAbs tob2GPI and horseradish peroxidase–labeled secondary antibod-ies. Results are expressed as the mean of the absorbance readat 490 nm6SEM. Half-maximal binding was observed at anMoAb concentration of 2.1 nmol/L for LBPA binding and of 0.5nmol/L for cardiolipin binding.
Figure 4. Presence of LBPA in late endosomes of HUVECs. A,Lipid analysis. HUVECs were incubated for 20 hours with [32P]Pi.Lipids were extracted and separated by 2D thin-layer chroma-tography. Lipids were detected by autoradiography and quanti-fied by using the Molecular Imager System (Bio-Rad). PC indi-cates phosphatidylcholine; PE, phosphatidylethanolamine; PI,phosphatidylinositol; PS, phosphatidylserine; SM, sphingomye-lin; and CL, cardiolipin. B and C, Localization of LBPA inHUVECs by immunofluorescence analysis. Cells were processedfor double labeling by using an MoAb (6C4) specific for LBPA(B) or rabbit anti-human vWF antibodies that specifically labelWeibel-Palade bodies (C). Scale bar510 mm.
Galve-de Rochemonteix et al Anti-Phospholipid Antibodies and Endothelial Cells 565
by guest on May 26, 2018
http://atvb.ahajournals.org/D
ownloaded from

glycine-HCl, pH 2.2, and immediately neutralized with 10mL of 1mol/L Tris, pH 11.0. The immunopurified material was analyzed forbinding activity toward cardiolipin and LBPA and for binding tofixed and permeabilized HUVECs. As a control, we performed thesebinding assays with the material adsorbed to and eluted from platescoated with 10% FCS only.
Cell CultureHUVECs were isolated from umbilical cords18 and grown at 37°C ina humidified atmosphere containing 5% CO2. In brief, the umbilicalvein was washed with Krebs-Ringer bicarbonate buffer (120 mmol/LNaCl, 4.75 mmol/L KCl, 1.2 mmol/L KH2PO4, 0.6 mmol/L MgSO4,1.2 mmol/L CaCl2, 25 mmol/L NaHCO3, and 25 mmol/L HEPES, pH
Figure 5. Analysis by immunoelectron microscopy of the intracellular localization of the 6C4 antigen in ultrathin frozen sections ofHUVECs. A, Low-magnification view showing the specific labeling on the internal membranes of late endosomes (L). All gold particlesin the field are indicated by arrows. In the inset and in B, higher-magnification views of the late endosomes are shown to demonstratethe ultrastructure of the labeled elements. Note the lack of labeling associated with other membranes such as the Golgi (g) and mem-brane surrounding the nucleus (n). Scale bar5200 nm.
566 Arterioscler Thromb Vasc Biol. February 2000
by guest on May 26, 2018
http://atvb.ahajournals.org/D
ownloaded from

7.4) and incubated for 10 minutes with 1 mg/mL collagenase (CLStype 1, Worthington Biochemical). Cells were collected by flushingthe vein with 50 mL of RPMI 1640 supplemented with 10% FCS(Life Technologies). The cells were then grown in RPMI 1640containing 90mg/mL heparin (Boehringer Ingelheim), 15mg/mL ECgrowth supplement (Upstate Biotechnology), 10 mmol/L HEPES,100 U/mL penicillin, and 100mg/mL streptomycin, supplementedwith 10% FCS. Cells were passaged by trypsin-EDTA (BiochromKG) treatment at a split ratio of 1:3 and used during passage 1 or 2.Tissue-culture dishes (Falcon Becton Dickinson Labware), 24-wellplates (Costar), and glass coverslips were coated with 0.1% gelatin.Cells were identified as endothelial by indirect immunofluorescenceanalysis for the presence of Weibel-Palade bodies by the use of antivWF antibodies.
Indirect Immunofluorescence StainingHUVECs grown on glass coverslips were fixed with either methanol(220°C, 4 minutes) or 4% freshly depolymerizedp-formaldehyde inPBS, pH 7.4 (24°C, 20 minutes).p-Formaldehyde–fixed cells werewashed with PBS, treated for 20 minutes with 0.27% NH4Cl–0.38%glycine in PBS, and permeabilized for 30 minutes with 0.05%saponin and 10% FCS in PBS in the presence of primary antibody.For single-label analyses, cells were incubated sequentially with theprimary antibody and the fluorescent secondary antibody. Fordouble-label analyses, cells were incubated sequentially with amixture of primary antibodies and a mixture of fluorescent secondaryantibodies. Antibodies were diluted in 10% FCS in PBS. Allincubations were performed for 1 hour at room temperature. Afterbeing washed, coverslips were mounted in polyvinyl alcohol. Sam-ples were analyzed with a confocal laser scanning microscope(LSM410 invert, Carl Zeiss Inc) equipped with argon and helium/neon laser fluorescence at 488 and 543 nm, respectively. Fluoresceinand rhodamine signal were recorded sequentially (emission filtersBP510-525 and LP590) by using363 or 3100 plan Apochromatoil-immersion objectives.
Negative control experiments were performed by omitting theprimary antibodies or by using an irrelevant primary antibody of thesame species and IgG subclass (for MoAbs). For double-labelanalyses, we verified that FITC fluorescence gave no signal in therhodamine channel and conversely. We also confirmed that thefluorescent secondary antibodies did not cross-react with immuno-globulins from animal species other than the target species.
To determine whether APLAs accumulate in HUVECs, the cellswere cultured for 20 hours in the presence of patient plasma (1:10)in PBS or purified IgG (20mg/mL) obtained from 10 patients whowere strongly positive in all tests for APLA. Thereafter the cellswere fixed and permeabilized. Immunofluorescence analysis wasperformed as described above.
Immunogold Labeling and Electron MicroscopyTo determine the ultrastructural localization in HUVECs of theantigen that binds to the 6C4 MoAb, cryosections of fixed HUVECswere incubated with 6C4 or with plasma from a patient with APLAand prepared for immunogold labeling and electron microscopy aspreviously described.19 To determine the site of accumulation of 6C4in HUVECs, the cells were incubated for 20 hours with 6C4 (20mg/mL) and then fixed and prepared for immunoelectronmicroscopy.
Statistical AnalysisCorrelation analysis was carried out by the Spearman test. Allcalculations were made with the computer program Stat-View II(Abacus Concepts).
ResultsComparison of Purified LBPA With CardiolipinLBPA is a structural isomer of phosphatidylglycerol, and itsstructure is related to that of cardiolipin (Figure 1A). Tocompare the immunoreactivity of purified LBPA and cardi-olipin, it was important to establish that preparations were notcross-contaminated. With the use of a basic solvent system,the 2 phospholipids were clearly separated by thin-layerchromatography. As shown in Figure 1B, we did not detect acardiolipin contaminant in our LBPA preparation. Commer-cially obtained cardiolipin also did not contain LBPA.
Correlation of the Binding of Patient IgG to LBPAWith the Binding to Cardiolipin and b2GPIWe measured binding activity in plasma and in purified IgGfrom 37 patients and 15 controls on ELISA plates coated witheither cardiolipin or LBPA. No immunoreactivity againstcardiolipin or LBPA was detected with the purified IgG ofcontrols (Figure 2A), whereas the IgG of 30 patients waspositive in both anticardiolipin and anti-LBPA ELISAs. In 7patients, no anti-LBPA and anti-cardiolipin activity wasdetected. Three of these negative patients were asymptomaticand 4 had SLE. A highly significant correlation was observedbetween anticardiolipin and anti-LBPA activities (R250.869,P,0.001; Figure 2B). Similar results were obtained whenplasmas instead of purified IgGs were used (R250.761,P,0.001; results not shown). We also analyzed and com-pared the binding of the patient IgGs tob2GPI and LBPA
Figure 6. Accumulation of an anti-LBPA MoAb (6C4) in HUVECs. Cells were incubated for 20 hours with the 6C4 antibody (A) or withcontrol antibody (C). After incubation, the cells were fixed and permeabilized. Internalized anti-LBPA antibody was revealed withrhodamine-conjugated anti-mouse antibodies. The cells incubated with the anti-LBPA antibody were doubly labeled with antibodies tovWF (B). The 6C4 antibody was detected in perinuclear vesicular structures (A), but not in Weibel Palade bodies. No internalization ofcontrol antibodies was observed (C). Scale bar510 mm.
Galve-de Rochemonteix et al Anti-Phospholipid Antibodies and Endothelial Cells 567
by guest on May 26, 2018
http://atvb.ahajournals.org/D
ownloaded from

(Figure 2C). IgGs from 27 patients exhibitedb2GPI bindingabove the normal range. A significant correlation (R250.469,P,0.001) was obtained between the binding to LBPA andb2GPI. This finding raised the question whetherb2GPI bindsto LBPA. In a direct comparative binding study, we observedthatb2GPI indeed bound to LBPA with an affinity only 4-foldlower than that obtained for the binding ofb2GPI to cardio-lipin (Figure 3).
To determine whether there were patients with IgGs thatbound directly to LBPA or cardiolipin, we also performedexperiments on LBPA- or cardiolipin-coated plates that wereblocked with albumin, ie, without serum-derived cofactor for
binding. Four patients with primary APS and 3 with APS andSLE had a high level of direct binding to LBPA. Two of thesepatients (1 primary APS and 1 APS/SLE) also had a highlevel of direct binding to cardiolipin. Furthermore, 3 patients(1 with psychosis, 1 with SLE, and 1 with neoplasia) had ahigh level of binding to cardiolipin alone.
To determine whether antibodies that bind (directly or viaa cofactor) to cardiolipin also bind to LBPA, from 5 patientswe immunopurified antibodies on cardiolipin-coated plates.Before adsorption to cardiolipin, the activity on cardiolipin-coated plates was, on average, 2-fold higher than the activityon LBPA-coated plates. The binding of these cardiolipin
Figure 7. Analysis by immunoelectron microscopy of the localization of 6C4 accumulation in HUVECs. Cells were incubated for 20hours with the 6C4 antibody and cryosectioned. Sections of 6C4-treated cells were labeled with rabbit anti-mouse IgG, followed by10-nm protein A–gold particles to detect the internalized 6C4 antibody. The gold particles are associated with late endosomes (L). Insome areas of the cell, parallel membrane arrays labeled with gold particles were detected (small arrows). g indicates Golgi; n, nucleus.Scale bar5200 nm.
568 Arterioscler Thromb Vasc Biol. February 2000
by guest on May 26, 2018
http://atvb.ahajournals.org/D
ownloaded from
affinity–purified antibodies was tested on both cardiolipin-and LBPA-coated plates. All antibodies bound to LBPA. Theratio of binding on the 2 antigens was similar to that observedbefore purification (1.8-fold).
Presence of LBPA in Late Endosomes ofHuman ECsTo determine whether LBPA was present in HUVECs, cellswere metabolically labeled with [32P]Pi. Then the lipids wereextracted and analyzed by thin-layer chromatography. Asshown in Figure 4A, a lipid was revealed that comigratedwith authentic LBPA. This lipid represented 0.7% of totalphospholipids of HUVECs. The cellular localization ofLBPA in HUVECs was visualized by indirect immunofluo-rescence and confocal microscopy with the LBPA-specificMoAb 6C4. The 6C4 antibody stained perinuclear vesicularstructures with a morphology and cellular distribution that arecharacteristic of late endosomes (Figure 4B) and are distinctfrom the elongated, rod-shaped structures of Weibel-Paladebodies (Figure 4C). Analysis by immunoelectron microscopyon cryosections of HUVECs showed that the 6C4 antibodieslabeled the internal membranes of late endosomes (Figure5A), as shown at higher magnification (Figure 5B). Nolabeling was observed on other membranes such as Golgimembranes or nuclear membranes. Taken together, the re-sults show that HUVECs contain LBPA and that the lipid islocalized within the internal membrane system of lateendosomes.
Accumulation of an Anti-LBPA MoAb in LateEndosomes of ECsBecause the late endosomes of HUVECs contain LBPA andlate endosomes are accessible to antibody endocytosed fromthe medium, we tested whether the anti-LBPA antibody 6C4could reach and then bind to its antigen on endocytosis. Cellswere incubated for 20 hours with the 6C4 MoAb, fixed, andprocessed for double immunofluorescent labeling for theMoAb and vWF. We observed in all HUVECs an accumu-lation of 6C4 within structures that closely resembled thoseobserved after direct labeling of fixed cells with 6C4 (seeFigure 4) and were distinct from Weibel-Palade bodies(Figures 6A and 6B). In contrast, control antibodies did notaccumulate intracellularly (Figure 6C). To better visualize thedistribution of the internalized 6C4 antibody, cells wereanalyzed by electron microscopy. Immunogold labeling ofcryosections showed that the antibody accumulated withinthe internal membranes of late endosomes and that thisaccumulation caused internal membranes to become morecompact (Figure 7).
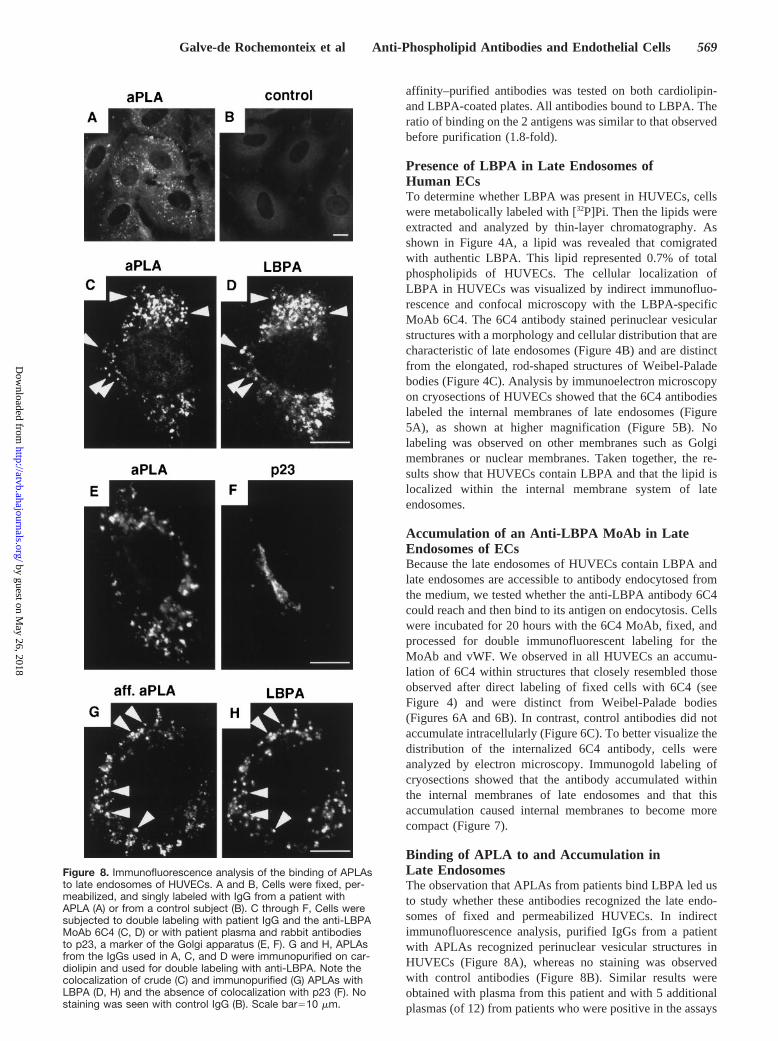
Binding of APLA to and Accumulation inLate EndosomesThe observation that APLAs from patients bind LBPA led usto study whether these antibodies recognized the late endo-somes of fixed and permeabilized HUVECs. In indirectimmunofluorescence analysis, purified IgGs from a patientwith APLAs recognized perinuclear vesicular structures inHUVECs (Figure 8A), whereas no staining was observedwith control antibodies (Figure 8B). Similar results wereobtained with plasma from this patient and with 5 additionalplasmas (of 12) from patients who were positive in the assays
Figure 8. Immunofluorescence analysis of the binding of APLAsto late endosomes of HUVECs. A and B, Cells were fixed, per-meabilized, and singly labeled with IgG from a patient withAPLA (A) or from a control subject (B). C through F, Cells weresubjected to double labeling with patient IgG and the anti-LBPAMoAb 6C4 (C, D) or with patient plasma and rabbit antibodiesto p23, a marker of the Golgi apparatus (E, F). G and H, APLAsfrom the IgGs used in A, C, and D were immunopurified on car-diolipin and used for double labeling with anti-LBPA. Note thecolocalization of crude (C) and immunopurified (G) APLAs withLBPA (D, H) and the absence of colocalization with p23 (F). Nostaining was seen with control IgG (B). Scale bar510 mm.
Galve-de Rochemonteix et al Anti-Phospholipid Antibodies and Endothelial Cells 569
by guest on May 26, 2018
http://atvb.ahajournals.org/D
ownloaded from

measuring IgG binding to LBPA, cardiolipin, andb2GPI. Theclinical characteristics of these patients were primary APS(n54) and APS combined with SLE (n52). After analyzingthe other 6 patients, we saw either no staining or highbackground fluorescence. The clinical characteristics of thesepatients were primary APS (n51), APS combined with SLE(n53), type II diabetes (n51), and neoplasia (n51).
The vesicular structures stained with APLAs colocalizedwith LBPA (Figures 8C and 8D). To exclude the possibilitythat the colocalization of patient antibodies and anti-LBPAMoAb was due to nonspecific binding of secondary antibod-ies to APLA antibodies, cells were doubly labeled withpatient plasma and antibodies against p23, a Golgi membraneprotein15 (Figures 8E and 8F). Clear segregation of fluores-cent secondary antibodies indicates that the observed colo-calization was not due to nonspecific absorption of secondaryantibodies. These results indicate that APLAs from certain
APS patients recognize late endosomes. Because IgGs frompatients are heterogeneous, we also used IgGs from 2 patientsand 1 control that were immunopurified on cardiolipin (seeabove). We also observed specific staining of late endosomesbut not of mitochondria, which contain cardiolipin (Figures8G and 8H). No staining was seen with the control IgG. OurELISA results showed that anti-LBPA activity was copurifiedwith anti-cardiolipin activity. These results confirm thatanti-cardiolipin/-LBPA antibodies, but not other antibodies inpatient IgGs, recognized late endosomes. Analysis by immu-noelectron microscopy of cryosections of HUVECs showedthat the APLA labeled the internal membranes of lateendosomes (Figure 9).
As an obvious next step, HUVECs were then incubatedfor 20 hours at 37°C in the presence of plasma1:10 frompatients with APLA in the medium and then analyzed byimmunofluorescence. APLAs from all 10 analyzed patients
Figure 9. Intracellular localization of the antigen recognized by APLA in ultrathin frozen sections of HUVECs. Gold particles in the fieldare indicated by arrows and show the specific labeling on the internal membranes of late endosomes. Scale bar5200 nm.
570 Arterioscler Thromb Vasc Biol. February 2000
by guest on May 26, 2018
http://atvb.ahajournals.org/D
ownloaded from

were endocytosed by all HUVECs and then accumulatedintracellularly, whereas we did not observe intracellularaccumulation of control antibodies (Figures 10A and 10B).Colocalization of internalized antibodies with LBPA indi-cates that the antibodies accumulate in late endosomes.
APLAs Induce a Late EndosomeSorting/Trafficking DefectOne of the main functions of late endosomes is the sorting ofthe multifunctional receptor (IGF2/M6PR) for ligands bear-ing M6P, which include lysosomal enzymes and IGF2.20
IGF2/M6PR delivers newly synthesized lysosomal enzymesfrom the trans-Golgi network (TGN) to late endosomes andthen recycles back to the TGN for reuse. At steady state,IGF2/M6PR localizes predominantly to the TGN in HU-VECs. This perinuclear distribution of IGF2/M6PR was notaffected when cells were treated with control plasma (Figure11B). In contrast, incubation of HUVECs with plasma from apatient with APLA caused the IGF2/M6PR to redistribute toperinuclear vesicular structures (Figure 11A). These struc-tures were identified as late endosomes by double labelingwith the anti-LBPA MoAb (Figures 11C and 11D). Similarresults were obtained after HUVEC treatment with each ofthe plasmas from 9 other patients with APLAs. Internaliza-tion of anti-LBPA MoAb also induced a redistribution ofIGF2/M6PR from the TGN to late endosomes in HUVECs(Figures 11E and 11F).
DiscussionThere is still much debate on the underlying pathogenicmechanisms associated with APLAs. Several recent studieshave proposed that APLAs act on ECs and may compromisethe normal antithrombotic function of ECs.1,4,8–13 Thesestudies proposed, however, no obvious common mechanisms.Recently, we observed that antibodies from some patients
with APLAs also recognized LBPA, an anionic phospholipidpresent in the late endosomes of BHK cells.14
In the current study, we have demonstrated that (1) there isa strong correlation between the binding activity in plasmafrom 37 APLA patients to LBPA and to cardiolipin and, to alesser extent, tob2GPI; (2) b2GPI binds LBPA; (3) ECscontain LBPA in their late endosomes; (4) late endosomes ofpermeabilized ECs are recognized by APLAs, whereas mito-chondria, which contain cardiolipin, are not; and (5) incuba-tion of ECs with APLAs leads to accumulation of theseantibodies in late endosomes and interference with theirprotein-sorting function. Our results therefore show thatanti-LBPA antibodies are present in the plasma of patientswith APLA, may interact with LBPA directly or viab2GPI,and are able to interact with and modify the cellular physi-ology of ECs.
We observed a highly significant correlation betweencardiolipin and LBPA binding in the plasma or purifiedIgG of 37 patients with APLAs. A biochemical analysis ofthe 2 phospholipid preparations used in our study ruled outthat the strong correlation was due to contamination of theLBPA preparation with cardiolipin or vice versa. Takentogether, our results indicate that, in general, antibodiesthat bind directly or indirectly to cardiolipin also bind toLBPA. To determine whether protein cofactors mediate, atleast in part, the binding of the antibodies to LBPA, wecompared LBPA binding withb2GPI binding. We ob-served a significant correlation, albeit a much lower 1, thanthat between cardiolipin and LBPA binding. This suggeststhat only part of the interaction between the APLAs andLBPA is mediated byb2GPI and implies that other proteincofactors, such as prothrombin, annexin V, or protein C,may also contribute, or that some of the antibodies coulddirectly bind to cardiolipin and LBPA.21 Moreover, someof the patient antibodies interacted withb2GPI (or otherproteins) alone.
Figure 10. Immunofluorescence analysis of theaccumulation of APLAs in late endosomes. Aand B, Cells were incubated for 20 hours withpatient plasma (A) or with control plasma (B).After incubation, cells were fixed and permeab-ilized. Internalized antibodies were thenrevealed with DTAF-conjugated anti-humanantibodies. C and D, The cells incubated withthe APLA were doubly labeled with the anti-LBPA antibody. Note the colocalization of APLA(C) and the anti-LBPA antibody (D) in perinu-clear vesicular structures. No internalization ofcontrol antibodies was observed (B). Scalebar510 mm.
Galve-de Rochemonteix et al Anti-Phospholipid Antibodies and Endothelial Cells 571
by guest on May 26, 2018
http://atvb.ahajournals.org/D
ownloaded from

We observed that anti-cardiolipin antibodies also inter-acted with LBPA and that APLAs recognized late endosomesof HUVECs, which contain LBPA, but did not recognizemitochondria, which are rich in cardiolipin. One possibilityfor this observation is that cardiolipin is highly susceptible tooxidation. A recent study demonstrated that many APLAsbound to cardiolipin only after it had been oxidized.22 InsideECs, cardiolipin is protected from oxidation, whereas inELISA plates, cardiolipin is exposed to oxygen for a pro-longed period. Alternative explanations that mitochondrialcardiolipin in fixed, permeabilized HUVECs is inaccessibleto APLA or that cardiolipin had been washed away duringfixation cannot be excluded at present.
Our findings raise the possibility that the correlation betweenanti-cardiolipin antibodies and the clinical manifestations of the
APS reflects a pathogenic effect of the binding of APLAs toLBPA rather than to cardiolipin. The immunofluorescence andimmunoelectron microscopy results showed that ECs containLBPA in the internal membranes of late endosomes. On endo-cytosis, APLAs accumulated in late endosomes, containingLBPA, and then affected their sorting functions as shown by theredistribution of the marker protein IGF2/M6PR from the Golgiapparatus to late endosomes. The slow kinetics of internalizationof APLAs suggests that fluid-phase endocytosis is a major routefor internalization. However, we cannot exclude the possibilitythat LBPA recycles between late endosomes and plasma mem-branes and that the antibodies enter via a mechanism of receptor-mediated endocytosis.
Presently, the contribution of anionic, phospholipid-binding proteins such asb2GPI, prothrombin, annexin V, or
Figure 11. Antibody-induced redistribu-tion of IGF2/M6PR in HUVECs. A and B,Cells were incubated for 20 hours withplasma from a patient with APLA (A) orwith control plasma (B). Cells were fixedand analyzed by single labeling for IGF2/M6PR. C through F, Cells were incubatedfor 20 hours with patient plasma (C, D) orwith anti-LBPA antibody (E, F). After fixa-tion and permeabilization, cells were dou-bly labeled with rabbit anti-IGF2/M6PR(C, E) and with FITC-labeled anti-mouseantibody (D, F).
572 Arterioscler Thromb Vasc Biol. February 2000
by guest on May 26, 2018
http://atvb.ahajournals.org/D
ownloaded from

others to the accumulation of APLAs in late endosomes isunclear. Future work will be required to characterize pre-cisely the interactions that may occur within the cell betweenAPLA, LBPA, other anionic phospholipids, and anionic,phospholipid-binding proteins. However, our data alreadyindicate that at least some APLAs can bind LBPA directly,and our correlation studies show that anti-LBPA antibodiesmay be, at least in part, distinct from those reacting againstanti-b2GPI. One may thus speculate that the effects of APLAson endosomal sorting functions are caused by multiple andcomplex interactions between APLAs and LBPA,b2GPI,and/or LBPA-b2GPI complexes. Alternatively, however, it isalso possible that the observed APLA effects are caused by acompetition of (some) APLA antibodies withb2GPI for thesame LBPA binding sites within late endosome internalmembranes.
A wide variety of, at first sight, unrelated pathogenicmechanisms have been proposed by which APLAs affect ECsand induce a prothrombotic state.8,23,24 The incubation ofHUVECs with APLAs induces an increase in monocyteadhesion, due to an increase in E-selectin, intercellularadhesion molecule-1, and vascular cell adhesion molecule-1expression at the EC surface.10,25 APLAs may also create anacquired protein C dysfunction, because anti-b2GPI antibod-ies hampered the inactivation of factor Va by endogenousactivated protein C,26 whereas lupus anticoagulant inhibitedactivated protein C anticoagulant activity but not thrombo-modulin activity.13 Anti-prothrombin antibodies induced therecruitment of prothrombin on EC surfaces and therebyfacilitated local thrombin generation.27 Annexin V–bindingantibodies in sera from patients with lupus anticoagulantinduced apoptosis in cultured ECs.12 Interestingly, removal ofAPLAs by incubation with phospholipid liposomes did notabolish the apoptosis-inducing activities or the binding toannexin V. Incubation of ECs with APLAs increased tissuefactor expression and decreased annexin V expression at thecell surface and decreased the clotting time of recalcifiedplasma added to these cells.11,28APLAs may bind to heparin-like glycosaminoglycans on ECs and thereby inhibit localantithrombin III activity.29 It remains to be established towhat extent our finding that APLA interferes with intracel-lular protein trafficking contributes to 1 or more of thepathogenic mechanisms described above.
Our results indicate that APLAs can, as do other autoan-tibodies,30 enter into living cells. Until now, APLAs werethought to be directed to the outer membrane of ECs4 and toactivate these cells.10,23,25,31 At the EC surface,b2GPI orb2GPI-phospholipid complexes were thought to be the maintargets of APLAs, and it has been shown that both MoAbsand polyclonal anti-b2GPI antibodies can upregulate adhesionmolecule expression and interleukin-6 secretion after ECbinding24,32 and induce adherence of monocytes to ECs.10,25
However, the binding of APLAs to the EC surface has neverbeen clearly demonstrated and remains speculative in view ofscarce evidence of the loss in membrane asymmetry.25 Ourdata indicate that APLAs enter ECs and bind to late endo-somes but obviously do not exclude that APLAs first bind tothe cell surface. For example, bothb2GPI and APLA couldbind to the cell surface, enter the cell, and then accumulate inlate endosomes.
Our data indicate that LBPA is an important lipid target ofAPLA, either direct or indirect, via a protein cofactor.Furthermore, they show that incubation of ECs with APLAsleads to accumulation of these antibodies in the late endo-somes of ECs and a redistribution of the IGF2/M6PR fromthe Golgi apparatus to late endosomes.
AcknowledgmentsThis work was supported by funds granted to Ph. de M. (32-51064.97), to E.K.O.K. (31-50645.97), and to J.G. by the SwissNational Science Foundation (31-37296.93); to R.G.P. by theNHMRC of Australia; and to J.G., R.G.P., and T.K. by theInternational Human Frontier Science Program. We thank JulienChevalier for the lipid analysis and Marie-He´lene Beuchat and OanaBulla for their excellent technical assistance.
References1. Cines DB, McCrae KR. The antiphospholipid-protein syndrome.J Clin
Immunol. 1995;15:86S–100S.2. Rauch J, Janoff AS. Antibodies against phospholipids other than cardi-
olipin: potential roles for both phospholipid and protein.Lupus. 1996;5:498–502.
3. Roubey RAS. Immunology of the antiphospholipid antibody syndrome.Arthritis Rheum. 1996;39:1444–1454.
4. Kandiah DA, Sali A, Sheng Y, Victoria EJ, Marquis DM, Coutts SM,Krilis SA. Current insights in the ‘antiphospholipid’ syndrome: clinical,immunological, and molecular aspects.Adv Immunol. 1998;70:507–563.
5. Bakimer R, Shoenfeld Y. Pathogenesis of the antiphospholipid syndrome.In: Asherson RA, Cervera R, Piette JC, Shoenfeld Y, eds.The Antiphos-pholipid Syndrome. Boca Raton, Fla: CRC Press; 1996:59.
6. Arnout J. The pathogenesis of the antiphospholipid syndrome: ahypothesis based on parallelisms with heparin-induced thrombocy-topenia.Thromb Haemost. 1996;75:536–541.
7. Petri M. Pathogenesis and treatment of the antiphospholipid antibodysyndrome.Med Clin North Am. 1997;81:151–177.
8. Meroni PL, del Papa N, Gambini D, Tincani A, Balestrieri G. Antiphos-pholipid antibodies and endothelial cells: an unending story.Lupus.1995;4:169–171.
9. Roubey RAS. Mechanisms of autoantibody-mediated thrombosis.Lupus.1998;7(suppl 2):S114–S119.
10. Simantov R, La Sala JM, Lo SK, Gharavi AE, Sammaritano LR, SalmonJE, Silverstein RL. Activation of cultured vascular endothelial cells byantiphospholipid antibodies. J Clin Invest. 1995;96:2211–2219.
11. Rand JH, Wu XX, Andree HA, Ross JB, Rusinova E, Gascon-Lema MG,Calandri C, Harpel PC. Antiphospholipid antibodies accelerate plasmacoagulation by inhibiting annexin-V binding to phospholipids: a ‘lupusprocoagulant’ phenomenon.Blood. 1998;92:1652–1660.
12. Nakamura N, Ban T, Yamaji K, Yoneda Y, Wada Y. Localization of theapoptosis-inducing activity of lupus anticoagulant in an annexinV-binding antibody subset.J Clin Invest. 1998;101:1951–1959.
13. Potzsch B, Kawamura H, Preissner KT, Schmidt M, Seelig C, Muller-Berghaus G. Acquired protein C dysfunction but not decreased activity ofthrombomodulin is a possible marker of thrombophilia in patients withlupus anticoagulant.J Lab Clin Med. 1995;125:56–65.
14. Kobayashi T, Stang E, Fang KS, de Moerloose P, Parton RG, GruenbergJ. A lipid associated with the antiphospholipid syndrome regulatesendosome structure and function.Nature. 1998;392:193–197.
15. Rojo M, Pepperkok R, Emery G, Kellner R, Parton RG, GruenbergJ. Involvement of the transmembrane protein p23 in biosynthetic proteintransport.J Cell Biol. 1997;139:1119–1135.
16. Arvieux J, Pouzol P, Roussel B, Jacob MC, Colomb MG. Lupus-likeanticoagulant properties of murine monoclonal antibodies tob2-glyco-protein I.Br J Haematol. 1992;81:568–573.
17. Reber G, Tremblet C, Bernard C, Mermillod B, de Moerloose P. Anti-cardiolipin antibodies and thrombosis: buffer’s influence on the detectionand quantitation of anticardiolipin measured by ELISA.Thromb Res.1990;57:215–226.
18. Jaffe EA, Nachman RI, Becker CG, Minick CR. Culture of humanendothelial cells derived from umbilical veins: identification by mor-phologic and immunologic criteria.J Clin Invest. 1973;52:2745–2756.
19. Griffiths G, McDowell A, Back R, Dubochet J. On the preparation ofcryosections for immunocytochemistry.J Ultrastruct Res. 1984;89:65–78.
Galve-de Rochemonteix et al Anti-Phospholipid Antibodies and Endothelial Cells 573
by guest on May 26, 2018
http://atvb.ahajournals.org/D
ownloaded from

20. Kornfeld S. Structure and function of the mannose-6-phosphate/insulin-like growth factor II receptors.Annu Rev Biochem. 1992;62:307–330.
21. Ikematsu W, Luan F-L, La Rosa L, Beltrami B, Nicoletti F, Buyon JP,Meroni PL, Balestrieri G, Casali P. Human anticardiolipin monoclonalautoantibodies cause placental necrosis and fetal loss in Balb/c mice.Arthritis Rheum. 1998;41:1026–1039.
22. Horkko S, Miller E, Dudl E, Reaven P, Curtiss LK, Zvaifler NJ,Terkeltaub R, Pierangeli SS, Branch DW, Palinski W, Witztum JL.Antiphospholipid antibodies are directed against epitopes of oxidizedphospholipids: recognition of cardiolipin by monoclonal antibodies toepitopes of oxidized low density lipoprotein.J Clin Invest. 1996;98:815–825.
23. Del Papa N, Guidali L, Spatola L, Bonra P, Borghi MO, Tincani A,Balestrieri G, Meroni PL. Relationship between anti-phospholipid andanti-endothelial antibodies, III:b2-glycoprotein I mediates the antibodybinding to endothelial membranes and induce the expression of adhesionmolecules.Clin Exp Rheumatol. 1995;13:179–185.
24. Del Papa N, Guidali L, Sala A, Buccellati C, Khamashta MA, IchikawaK, Koike T, Balestrieri G, Tincani A, Hughes GRV, Meroni PL. Endo-thelial cell as target of antiphospholipid antibodies: human polyclonal andmonoclonal anti-b2-glycoprotein I antibodies react in vitro with endothe-lial cells through adherentb2-glycoprotein I and induce endothelial acti-vation.Arthritis Rheum. 1997;40:551–561.
25. George J, Blank M, Levy Y, Meroni P, Damianovich M, Tincani A,Shoenfeld Y. Differential effects of anti-b2-glycoprotein 1 antibodies on
endothelial cells and on the manifestations of experimental antiphos-pholipid syndrome.Circulation. 1998;97:900–906.
26. Galli M, Ruggeri L, Barbui T. Differential effects of anti-b2-glycoproteinI and antiprothrombin antibodies on the anticoagulant activity of activatedprotein C.Blood. 1998;91:1999–2004.
27. Rao LV, Hoang AD, Rapaport SI. Mechanism and effects of the bindingof lupus anticoagulant IgG and prothrombin to surface phospholipid.Blood. 1996;88:4173–4182.
28. Branch DW, Rodgers GM. Induction of endothelial cell tissue factoractivity by sera from patients with antiphospholipid syndrome: a possiblemechanism of thrombosis.Am J Obstet Gynecol. 1993;168:206–210.
29. Shibata S, Harpel PC, Gharavi A, Rand JH, Fillit H. Autoantibodies toheparin from patients with antiphospholipid antibody syndrome inhibitformation of antithrombin III-thrombin complexes.Blood. 1994;83:2532–2540.
30. Alarcon-Segovia D, Ruiz-Arguelles A, Llorente L. Broken dogma: pen-etration of autoantibodies into living cells.Immunol Today. 1996;17:163–164.
31. Le Tonqueze M, Dueymes M, Piette J-C. Role ofb2-glycoprotein 1 in theantiphospholipid binding to endothelial cells.Lupus. 1995;4:179–186.
32. Del Papa N, Sheng YH, Raschi E, Kandiah DA, Tincani A, KhamashtaMA, Atsumi T, Hughes GRV, Ichikawa K, Koike T, Balestrieri G, KrillisSA, Meroni PL. Humanb2-glycoprotein I binds to endothelial cellsthrough a cluster of lysine residues that are critical for anionic phos-pholipid binding and offers epitopes for anti-b2-glycoprotein I antibodies.J Immunol. 1998;160:5572–5578.
574 Arterioscler Thromb Vasc Biol. February 2000
by guest on May 26, 2018
http://atvb.ahajournals.org/D
ownloaded from

Gruenberg and Philippe de MoerlooseJeanRobert G. Parton, Guido Reber, Emmanuel de Maistre, Denis Wahl, Egbert K. O. Kruithof,
Béatrix Galve-de Rochemonteix, Toshihide Kobayashi, Corinne Rosnoblet, Margaret Lindsay,Cells
Interaction of Anti-Phospholipid Antibodies With Late Endosomes of Human Endothelial
Print ISSN: 1079-5642. Online ISSN: 1524-4636 Copyright © 2000 American Heart Association, Inc. All rights reserved.
Greenville Avenue, Dallas, TX 75231is published by the American Heart Association, 7272Arteriosclerosis, Thrombosis, and Vascular Biology
doi: 10.1161/01.ATV.20.2.5632000;20:563-574Arterioscler Thromb Vasc Biol.
http://atvb.ahajournals.org/content/20/2/563World Wide Web at:
The online version of this article, along with updated information and services, is located on the
http://atvb.ahajournals.org//subscriptions/
at: is onlineArteriosclerosis, Thrombosis, and Vascular Biology Information about subscribing to Subscriptions:
http://www.lww.com/reprints
Information about reprints can be found online at: Reprints:
document. Question and AnswerPermissions and Rightspage under Services. Further information about this process is available in the
which permission is being requested is located, click Request Permissions in the middle column of the WebCopyright Clearance Center, not the Editorial Office. Once the online version of the published article for
can be obtained via RightsLink, a service of theArteriosclerosis, Thrombosis, and Vascular Biologyin Requests for permissions to reproduce figures, tables, or portions of articles originally publishedPermissions:
by guest on May 26, 2018
http://atvb.ahajournals.org/D
ownloaded from