infection à répétition
TRANSCRIPT

Infections à répétition et « immunodéficiences »

Infections à répétition
• Plaintes fréquentes en consultation– Infections ORL à répétition– Infections bronchopulmonaires à répétition
– Abcès à répétition– Herpès récidivant– Cellulites et érysipèles des membres inférieurs– Cystites – Fatigue chronique et fausses infections à répétition
(mauvaises interprétations sérologiques)

Infections à répétition
• Immunodéficiences vraies sont rares
• Le plus souvent facteurs anatomiques ou environnementaux favorisants
• Anomalies de l’immunité loco-régionale (difficile à explorer et à traiter)

Infections à répétition
• Plaintes fréquentes en consultation– Infections ORL et bronchopneumonies à répétition
• Tabac, allergie, anatomie sinusale, exposition à de jeunes enfants…; bronchectasies, BPCO
– Abcès à répétition• Portage de Staph. Aureus au niveau des fosses nasales
– Herpès récidivant• stress
– Cellulites et érysipèles des membres inférieurs• Altération irréversible du réseau lymphatique
– Cystites• Activité sexuelle
– Fatigue chronique et fausses infections à répétition (mauvaises interprétations sérologiques)

Infections à répétition
• La « rentabilité » des bilans immunologiques est souvent faible

Tableaux cliniques en fonction du compartiment immunitaire
déficient

Déficits de l’immunité humorale
• Surtout infections bactériennes (notamment ORL et bronchopulmonaires)– Germes encapsulés
• Streptococcus pneumoniae• Hemophilus influenzae• Streptococoques du groupe B• Neisseria meningitidis
– Staphylococcus aureus– Bacilles entériques gram-

Déficits de l’immunité humorale
• Infections virales– Souvent primo-infections normales en
fréquence et en sévérité
– Déficit de la mémoire à long terme• Rougeoles et varicelles à répétition
– Infections sévères à entérovirus (atteintes neurologiques)
– Infections parasitaires : giardiases

Immunité à médiation cellulaire
• Germes intracellulaires – parce que seul le système d’échantillonnage des
protéines de l’intérieur de la cellule (présentation par des molécules de classe I ou II) permet au système immunitaire de percevoir ces agents qui se cachent dans les cellules (les anticorps ne peuvent les atteindre)
– les systèmes de microbicidie oxydative des macrophages ont absolument besoin des cytokines de type Th1 (IFN-γ, IL-12) pour être activés

Immunité à médiation cellulaire
– infections mycotiques (candidose mucocutanée,…)– infections virales (svt virus latents de la famille herpès)– pneumocystoses
– bactéries intracellulaires (mycobactéries)
– svt déficits qualitatifs de l’immunité humorale• taux d’anticorps normal mais anticorps de mauvaise qualité (rôle
des lymphocytes T dans l’immunopoïèse B)
– Infections qui peuvent toucher le nouveau-né (pas de transfert des lymphocytes T maternels)

Déficits du complément

Donc deux activités antimicrobiennes du complément :
• founir des opsonines (C3b et iC3b)
• lyser directement les bactéries (Gram-)


Donc déficit en
• C1, C2, C4 : peu d’infections
• C3 : infections à germes encapsulés (défaut d’opsonisation)
• C5 à C9 : infections à Neisseria– Neisseria gonorrhoeae– Neisseria meningitidis
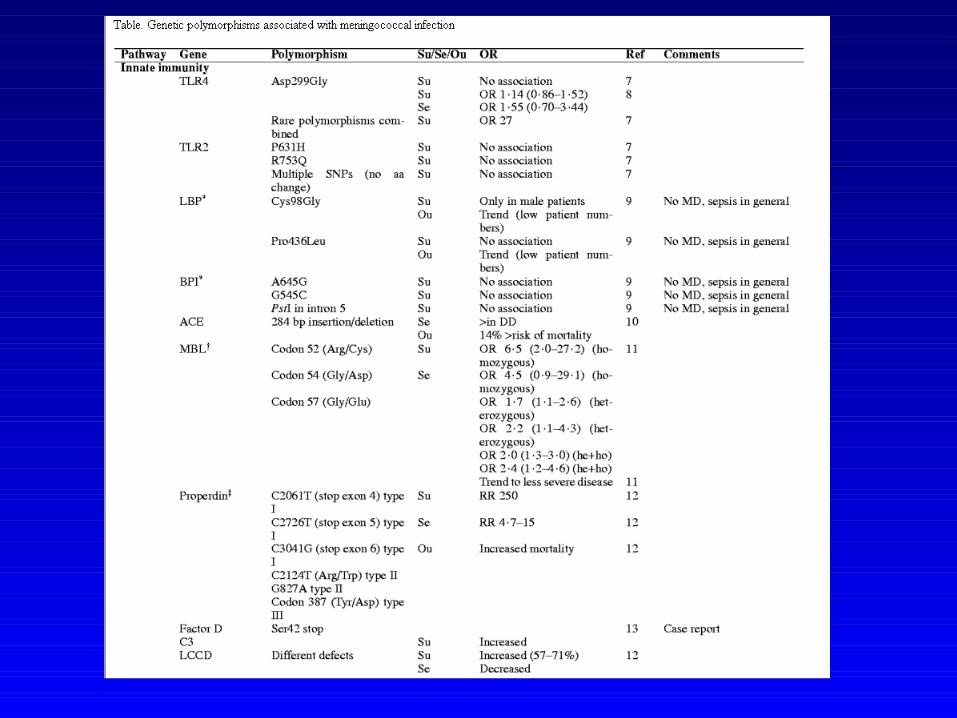

Cas clinique
• Mme LRG 38 ans• ATCD RAA• Méningites à répétition
– 10 ans– 37 ans– 2 épisodes l’année dernière
• Nature méningococcique démontrée sur les deux dernières poussées
• Evolution bénigne dans les quatre cas, aucune séquelle

Cas clinique
• Bilan CHR– CH50 effondré, C3 et C4 normal
• Exploration complémentaire labo Erasme– C6 indosable– MBL effondrée
• Déficit double MBL et C6

Déficit des phagocytes
• Germes : E. Coli, Klebsiella pneumoniae, Pseudomonas aeruginosa, enterobacter, serratia, Staph. Epidermidis, Candida, Aspergillus
• Déficits quantitatifs– Neutropénies et agranulocytoses
• Voir cours d’immunologie
• Déficits qualitatifs– Maladies granulomateuses chroniques
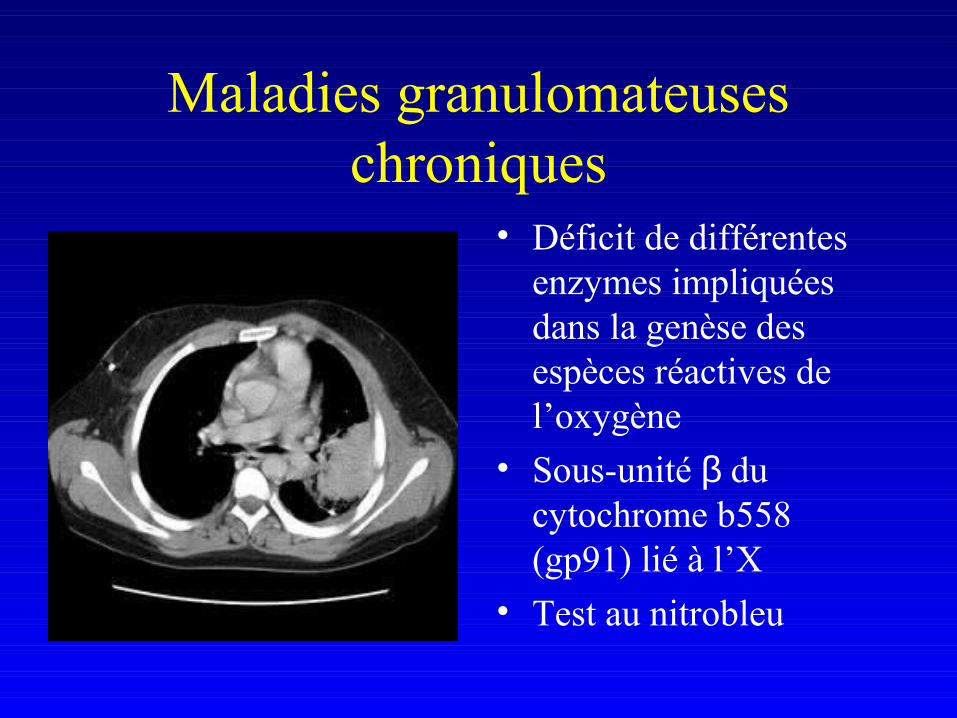
Maladies granulomateuses chroniques
• Déficit de différentes enzymes impliquées dans la genèse des espèces réactives de l’oxygène
• Sous-unité β du cytochrome b558 (gp91) lié à l’X
• Test au nitrobleu

Quelques déficits immunitaires à composant génétique
prédominante
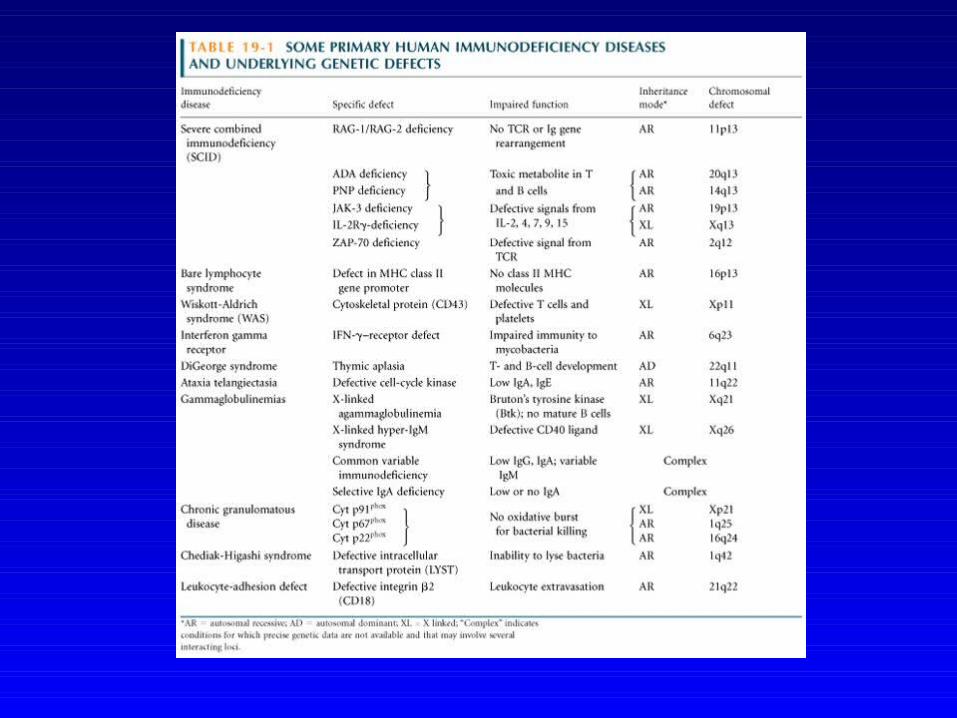

Hypogammaglobulinémies d’étiologie inconnue avec prédisposition
génétique
• Déficit en IgA
• Hypogammaglobulinémie commune variable ou immunodéficience commune variable
• Déficit en sous-classes

Hypogammaglobulinémies d’étiologie inconnue avec prédisposition
génétique
• Moins sévères que les hypogammaglobulinémies monogéniques
• Plus fréquentes
• Diagnostic occasionnel tard dans la vie
• Association à des phénomènes dysimmunitaires

Hypogammaglobulinémie commune variable
(CVH ou CVI ou CVID)
• le plus fréquent des déficits immunitaires primitifs après le déficit isolé en IgA
• syndrome probablement hétérogène caractérisé par une hypogamma-globulinémie malgré un nombre normal (ou presque) de lymphocytes B
• hypogammaglobulinémie variable selon les individus et les antigènes considérés (vs Bruton)
• Fréquemment associé à l’haplotype A1B8DR3


CVI
• Présentation– Tout âge (svt jeunes enfants ou jeunes adultes)– Infections bactériennes récurrentes
• Germes encapsulés, moraxella
– Giardiases– Hyperactivation du système immunitaire
• Splénomégalie, syndromes hémophagocytiques, hyperplasie nodulaire de l’intestin, granulomes pulmonaires ou hépatiques
– Autoimmunité (thyroïde, diabète, vitiligo, pelade)

CVI
• Complications tardives– Infections : bronchectasies, cholangites à
Campylobacter, arthrites à Ureaplasma urealyticum– Malabsorption (entéropathie de type maladie coeliaque)
– Splénomégalie, hypersplénisme
– Lymphomes B– Autoimmunité– Thymome (myasthénie grave, anémie aplastique, PTI)

Déficit sélectif en IgA
• Fréquent : 1/400 à 1/800, surtout chez les sujets atopiques
• Déficit au niveau du sérum et des sécrétions
• Etiologie hétérogène– Contexte HLA-A1B8DR3
• Une variante du CVI?
– Facteurs exogènes : chimiothérapie, phénytoïne

Déficit en IgA
• Présentation– Décours souvent bénin (sauf si déficit associé IgG2)
– Infections ORL fréquentes, plus rarement bronchectasies, diarrhée chronique (giardiases)
– Phénomènes allergiques (IgE accrues) et autoimmunitaires (LED, PR, JCA, Biermer, maladie coeliaque)
– Réactions transfusionnelles

Déficit en IgA
• Diagnostic– IgA sériques indétectables (<0.05g/l)
– Pas d’IgA salivaire, présence d’IgG et d’IgM
– IgG totales et IgM normales, IgG2 et IgG4 svt abaissées
– IgE svt accrues– Autoanticorps fréquents dont anti-IgA
– Fonction T normale

Déficit sélectif en sous-classes d’IgG
– Tout âge
– IgG2 et/ou IgG4 : la plupart des anticorps anti-polysaccharidiques sont des IgG2 donc déficit associé à infections récurrentes (pneumocoque, Haemophilus influenzae, Pseudomonas aeruginosa), bronchectasies; svt associé à déficit en IgA
– Importance clinique encore controversée

Traitement des hypogammaglobulinémies
• Immunoglobulines par voie intraveineuse (0.4 à 0.5 g/kg)
• Toutes les 3 à 4 semaines
• Viser une concentration résiduelle d’IgG supérieure à 5g/l

Prise en charge générale des patients présentant des déficits
immunitaires
• mesures générales d’hygiène
• éliminer allergènes si atopie
• kiné respiratoire
• éviter vaccins vivants (polio, fièvre jaune)

Prise en charge générale des patients présentant des déficits
immunitaires
• vaccin antigrippal et antipneumococcique
• antibiothérapies précoces
• traitements spécifiques– immunoglobulines IV pour les déficits en IgG
avec répercussion clinique– Intérêt des lysats bactériens oraux
(bronchovaxom) dans la BPCO?