identification of a small molecule inhibitor of 3 … · 3-phosphoglycerate dehydrogenase to target...
TRANSCRIPT

Correction
BIOCHEMISTRYCorrection for “Identification of a small molecule inhibitor of3-phosphoglycerate dehydrogenase to target serine biosynthesisin cancers,” by Edouard Mullarky, Natasha C. Lucki, Reza BeheshtiZavareh, Justin L. Anglin, Ana P. Gomes, Brandon N. Nicolay,Jenny C. Y. Wong, Stefan Christen, Hidenori Takahashi, Pradeep K.Singh, John Blenis, J. David Warren, Sarah-Maria Fendt, John M.Asara, Gina M. DeNicola, Costas A. Lyssiotis, Luke L. Lairson, andLewis C. Cantley, which appeared in issue 7, February 16, 2016, ofProc Natl Acad Sci USA (113:1778–1783; first published February 1,2016; 10.1073/pnas.1521548113).The editors note that the date on which this manuscript was
sent for review was originally published incorrectly as December 7,2015. The date should instead appear as November 2, 2015.
www.pnas.org/cgi/doi/10.1073/pnas.1602228113
www.pnas.org PNAS | March 15, 2016 | vol. 113 | no. 11 | E1585
CORR
ECTION

Identification of a small molecule inhibitor of3-phosphoglycerate dehydrogenase to targetserine biosynthesis in cancersEdouard Mullarkya,b,c, Natasha C. Luckid, Reza Beheshti Zavarehd, Justin L. Anglind, Ana P. Gomesa,e,Brandon N. Nicolayf, Jenny C. Y. Wonga,b, Stefan Christeng,h, Hidenori Takahashii,j,1, Pradeep K. Singhk,l, John Blenisa,e,J. David Warrenk,l, Sarah-Maria Fendtg,h, John M. Asaraj, Gina M. DeNicolaa,b, Costas A. Lyssiotism,n,2,Luke L. Lairsond,o,2, and Lewis C. Cantleya,b,2
aMeyer Cancer Center, Weill Cornell Medical College, New York, NY 10065; bDepartment of Medicine, Weill Cornell Medical College, New York, NY 10065;cBiological and Biomedical Sciences Graduate Program, Harvard Medical School, Boston, MA 02115; dThe California Institute for Biomedical Research,La Jolla, CA 92037; eDepartment of Pharmacology, Weill Cornell Medical College, New York, NY 10065; fMassachusetts General Hospital Cancer Center,Harvard Medical School, Charlestown, MA 02129; gLaboratory of Cellular Metabolism and Metabolic Regulation, Department of Oncology, KatholiekeUniversiteit Leuven, 3000 Leuven, Belgium; hLaboratory of Cellular Metabolism and Metabolic Regulation, Vesalius Research Center, Vlaams Instituut voorBiotechnologie Leuven, 3000 Leuven, Belgium; iDepartment of Systems Biology, Beth Israel Deaconess Medical Center and Harvard Medical School, Boston,MA 02115; jDivision of Signal Transduction, Department of Medicine, Beth Israel Deaconess Medical Center and Harvard Medical School, Boston, MA 02115;kDepartment of Biochemistry, Weill Cornell Medical College, New York, NY 10065; lMilstein Chemistry Core Facility, Weill Cornell Medical College, NewYork, NY 10065; mDepartment of Molecular and Integrative Physiology, University of Michigan, Ann Arbor, MI 48109; nDivision of Gastroenterology,Department of Internal Medicine, University of Michigan, Ann Arbor, MI 48109; and oDepartment of Chemistry, The Scripps Research Institute, La Jolla,CA 92037
Contributed by Lewis C. Cantley, December 23, 2015 (sent for review November 2, 2015; reviewed by Eyal Gottlieb and Brent R. Stockwell)
Cancer cells reprogram their metabolism to promote growth andproliferation. The genetic evidence pointing to the importance of theamino acid serine in tumorigenesis is striking. The gene encoding theenzyme 3-phosphoglycerate dehydrogenase (PHGDH), which cata-lyzes the first committed step of serine biosynthesis, is overexpressedin tumors and cancer cell lines via focal amplification and nuclearfactor erythroid-2-related factor 2 (NRF2)-mediated up-regulation.PHGDH-overexpressing cells are exquisitely sensitive to genetic ab-lation of the pathway. Here, we report the discovery of a selectivesmall molecule inhibitor of PHGDH, CBR-5884, identified byscreening a library of 800,000 drug-like compounds. CBR-5884inhibited de novo serine synthesis in cancer cells and was selec-tively toxic to cancer cell lines with high serine biosynthetic activity.Biochemical characterization of the inhibitor revealed that it was anoncompetitive inhibitor that showed a time-dependent onset ofinhibition and disrupted the oligomerization state of PHGDH. Theidentification of a small molecule inhibitor of PHGDH not only en-ables thorough preclinical evaluation of PHGDH as a target in can-cers, but also provides a tool with which to study serine metabolism.
PHGDH | inhibitor | serine | cancer metabolism
Serine is required for a plethora of anabolic processes. Serineis an abundant component of proteins and is required for the
synthesis of lipids, including sphingolipids and phosphatidylserine,a major component of cellular membranes (1–3). Alternatively,serine hydroxymethyltransferases (SHMTs) convert serine to gly-cine, concomitantly charging the folate pool with “one-carbon”units (4, 5). Both glycine and folate one-carbon units are used tomake nucleotides. Thus, serine serves numerous critically impor-tant roles in cellular metabolism.At the cellular level, serine can be imported from the extra-
cellular space via amino acid transporters (6, 7). Alternatively,serine can be synthesized from glucose via the phosphoserinepathway (8). De novo synthesis proceeds from the glycolyticintermediate 3-phosphoglycerate (3-PG) via three sequential en-zymatic reactions (Fig. 1A), the first of which is catalyzed bythe NAD+-dependent enzyme 3-phosphoglycerate dehydrogenase(PHGDH) (9). For decades, it has been known that cancer cellshave enhanced serine synthesis, which contributes to nucleotidesynthesis (10, 11). Recently, focal amplifications of the gene encodingPHGDH have been reported, particularly in breast cancers andmelanomas (12–14). Additionally, KEAP1 and nuclear factorerythroid-2-related factor 2 (NRF2) mutant non-small cell lung
cancers (NSCLCs) overexpress PHGDH (15). Proliferation ofPHGDH-amplified cancer cell lines, and other lines that over-express PHGDH without amplification, is inhibited by PHGDHknockdown. In contrast, lines that express little PHGDH are re-sistant to shRNA-mediated ablation of the pathway, presumablybecause serine import suffices (13, 14). A detailed mechanisticunderstanding of why some cancer cells are addicted to serinesynthesis despite the availability of extracellular serine for importremains unclear. Interestingly, in triple negative breast cancer(TNBC) and NSCLC, PHGDH amplification and overexpressionare associated with more aggressive disease (13–16). Thus, PHGDH
Significance
Serine supports a number of anabolic processes, including pro-tein, lipid, and nucleic acid synthesis. Cells can either importserine or synthesize it de novo. Recently, overexpression of3-phosphoglycerate dehydrogenase (PHGDH), the gene encod-ing the first committed step of serine synthesis, via focal am-plification and other mechanisms, has been identified in humancancers. Cancer cell lines that overexpress PHGDH are uniquelysensitive to PHGDH knockdownwhereas lines that express littlePHGDH are insensitive, suggesting that PHGDH may be a clini-cally interesting target. Here, we report the discovery of aspecific small molecule inhibitor of PHGDH, which enables pre-clinical evaluation of PHGDH as a target in cancer and provides atool to study the biology of de novo serine synthesis.
Author contributions: E.M., N.C.L., L.L.L., and L.C.C. designed research; E.M., N.C.L., R.B.Z.,J.L.A., A.P.G., B.N.N., J.C.Y.W., S.C., H.T., P.K.S., J.D.W., S.-M.F., J.M.A., G.M.D., and C.A.L.performed research; E.M., N.C.L., R.B.Z., J.L.A., A.P.G., B.N.N., S.C., H.T., P.K.S., J.B., J.D.W.,S.-M.F., J.M.A., G.M.D., C.A.L., L.L.L., and L.C.C. analyzed data; and E.M., C.A.L., L.L.L.,and L.C.C. wrote the paper.
Reviewers: E.G., Beatson Institute for Cancer Research; and B.R.S., Columbia/HowardHughes Medical Institute.
Conflict of interest statement: L.C.C. owns equity in, receives compensation from, andserves on the Board of Directors and Scientific Advisory Board of Agios Pharmaceuticals.Agios Pharmaceuticals is identifying metabolic pathways of cancer cells and developingdrugs to inhibit such enzymes to disrupt tumor cell growth and survival.1Present address: Frontier Research Laboratories, Daiichi Sankyo Co., Shinagawa-ku, Tokyo140-8710, Japan.
2To whom correspondence may be addressed. Email: [email protected], [email protected], or [email protected].
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1521548113/-/DCSupplemental.
1778–1783 | PNAS | February 16, 2016 | vol. 113 | no. 7 www.pnas.org/cgi/doi/10.1073/pnas.1521548113

inhibitors as a targeted therapy for these tumor types represent anexciting clinical opportunity.The studies herein detail our efforts in identifying small mole-
cule inhibitors of PHGDH. We reasoned that a PHGDH inhibitorwould have the benefits of not only providing a tool compoundwith which to study the biology of serine synthesis, but also en-abling thorough preclinical evaluation of PHGDH as a target incancers. We screened a library of 800,000 small molecules using anin vitro PHGDH assay. A cell-based assay for serine synthesis wasused to identify a lead, CBR-5884, that was active in cells. CBR-5884 selectively inhibited the proliferation of melanoma and breastcancer lines that have a high propensity for serine synthesis but hadno effect on lines that rely on extracellular serine uptake. Mech-anistically, CBR-5884 was found to be a noncompetitive inhibitor,showed a time-dependent onset of inhibition, and disrupted theoligomerization state of PHGDH.
ResultsScreening for Small Molecule Inhibitors of PHGDH. An in vitro en-zymatic assay for PHGDH activity amenable to high-throughputscreening (HTS) was developed by coupling the production ofNADH, upon 3-PG oxidation, to the reduction of resazurin toresorufin using diaphorase as the coupling enzyme (Fig. 1B). Thus,resorufin fluorescence served as a proxy for PHGDH activity.The assay was miniaturized to a 1,536-well format with aZ-factor of >0.75, indicating a high quality assay (17). A libraryof 800,000 small molecules was screened in single point formatat 13 μM (Fig. 1C). Setting a threshold Z score of −3, corre-sponding to at least 50% PHGDH inhibition, gave a 0.5% hit rate,yielding 3,906 hits. Putative hits were reassayed in triplicate andcounter-screened against diaphorase to rule out false positivestargeting diaphorase. The counter screen eliminated 3,498 com-pounds, giving 408 PHGDH inhibitors (Fig. 1D).A triaging strategy based on hit potency and selectivity was
designed. We reasoned that inhibitors specific to PHGDH wouldminimize general cellular toxicity compared with compounds thathit a variety of dehydrogenases. Thus, half maximal inhibitoryconcentrations (IC50) were determined for a panel of NAD(P)+-dependent dehydrogenases that included PHGDH, isocitratedehydrogenase (IDH1), malate dehydrogenase (MDH1), and 3α-hydroxysteroid dehydrogenase (3α-HSD). Compounds at leastfourfold more selective for PHGDH were progressed for fur-ther analysis. Based on this triaging, seven of the most potentPHGDH inhibitors were selected as lead compounds for evalua-tion in cell-based assays; selected structures are shown in Fig. 1E
(Table S1). A number of these compounds are likely to targetsulfhydryl groups and may therefore react with a PHGDH cys-teine residue. For example, both CBR-5807 and CBR-6936 con-tain sulfhydryl-reactive disulfide centers. Interestingly, CBR-5807(Disulfiram) is an approved drug dosed in humans to treat alco-holism and known to inhibit aldehyde dehydrogenase by reactingwith sulfhydryl groups (18).
CBR-5884 Inhibits Serine Synthesis in Cells. We determined whetherany of our seven leads inhibited serine synthesis in cancer cells.To do so, we turned to gas chromatography mass-spectrometry(GCMS) with uniformly carbon-13–labeled glucose (13C6-glucose)tracing. Given the isotopic enrichment of serine, it is possible todecouple newly synthesized serine from extracellular serine orserine that was synthesized before tracer addition. Newly syn-thesized serine has a mass-shift of 3 (M+3) due to the incorpora-tion of glucose-derived 13C via 3-PG. We first investigated thekinetics of serine labeling. Serine labeling plateaued around 6 h,with ∼65% of the serine pool being 13C-labeled (Fig. S1). Theplateau phase likely reflects exchange between intra- and extra-cellular serine pools (19).With an understanding of serine-labeling kinetics, we designed
a 13C6-glucose tracing assay to acutely interrogate the effects ofcompounds on serine synthesis (Fig. 2A). Assaying serine syn-thesis with a 3-h compound treatment was preferred to longertreatments to guard against false positives that decrease serinelabeling by an indirect effect, such as generally compromisingcellular viability. Among our lead compounds, CBR-5884 was ableto decrease de novo serine synthesis by 30%; the remaining com-pounds had little effect (Fig. 2B). The dose at which CBR-5884 hadan effect on serine labeling was consistent with the in vitro biochemicalIC50 of 33 ± 12 μM for PHGDH (Fig. 2C). At such concentrations,CBR-5884 had no effect on two other NAD+-dependent de-hydrogenases, lactate dehydrogenase (LDH) and MDH1 (Fig.2C and Fig. S1). Importantly, under the acute treatment time pe-riod used in the labeling assays, CBR-5884 was not generally cytotoxicat concentrations up to 40 μM as determined by two independentcellular viability assays (Fig. S1). Therefore, decreases in serine la-beling are a direct effect of CBR-5884–mediated PHGDH inhibition.We resynthesized CBR-5884 in-house and performed a dose–
response experiment for CBR-5884 using the same acute treat-ment method as above. Serine labeling was significantly decreasedat 30 μM and trended toward a decrease at 15 μM (Fig. 2D). Im-portantly, perturbations in labeling were specific to serine in thatneither the PHGDH substrate, 3-PG, nor the end products of
A
DPrimary Screen
800K compounds
Diaphorase
3,906 Hits
(Z score < -3)
(triplicate) counter screen
Hits: 408
3PG P-Pyr P-Ser
NAD+ NADH Glu αKG
SerPSAT1 PSPHPHGDH
Glucose
Pyruvate
C
The Phosphoserine Pathway
3PG p-Pyr p-Ser
NAD+ NADH Glu αKG
PSAT1PHGDH
DiaphoraseResazurin
(Ex550/Em585)
B Screening Assay
E
SH
NH
O
CBR-9480CBR-6936
N
S
SS N
S
CBR-5807
OO
HN S
O
O
S
N
CBR-5884
SS
NH
OO
HN(dehydrogenase panel)
Z-s
core
Compounds
Fig. 1. Screening for inhibitors of PHGDH. (A) Serinesynthesis from glucose via the phosphoserine pathway:Phosphoglycerate dehydrogenase (PHGDH) oxidizesthe glycolytic intermediate 3-phosphoglycerate (3-PG) to3-phosphohydroxypyruvate (p-Pyr) using NAD+; phos-phoserine amino transferase (PSAT1) transaminatesp-Pyr to phosphoserine (p-Ser) using glutamate as a ni-trogen donor; phosphoserine phosphatase (PSPH) de-phosphorylates p-Ser to yield serine. (B) In vitro PHGDHassay. Diaphorase couples the NADH produced uponPHGDH turnover to the reduction of resazurin to fluo-rescent resorufin. Resorufin fluorescence is a proxy forPHGDH activity. PSAT1 is included to prevent productfeedback inhibition of PHGDH by p-Pyr. (C) Z-score plotfor the 800,000-compound library screened using theabove PHGDH assay. Each point represents a singlecompound. A negative score indicates inhibition.(D) Screen triaging strategy. Setting a Z-score thresholdof −3 gave 3,906 putative hits. After counter-screeningagainst diaphorase to rule out false positives and con-firming activity against PHGDH, 408 compounds re-mained. Selected compounds were profiled against apanel of metabolic NAD(P)+ dehydrogenases to ascer-tain selectivity for PHGDH. (E) Structures of represen-tative PHGDH inhibitors evaluated in cell-based assays.
Mullarky et al. PNAS | February 16, 2016 | vol. 113 | no. 7 | 1779
BIOCH
EMISTR
Y
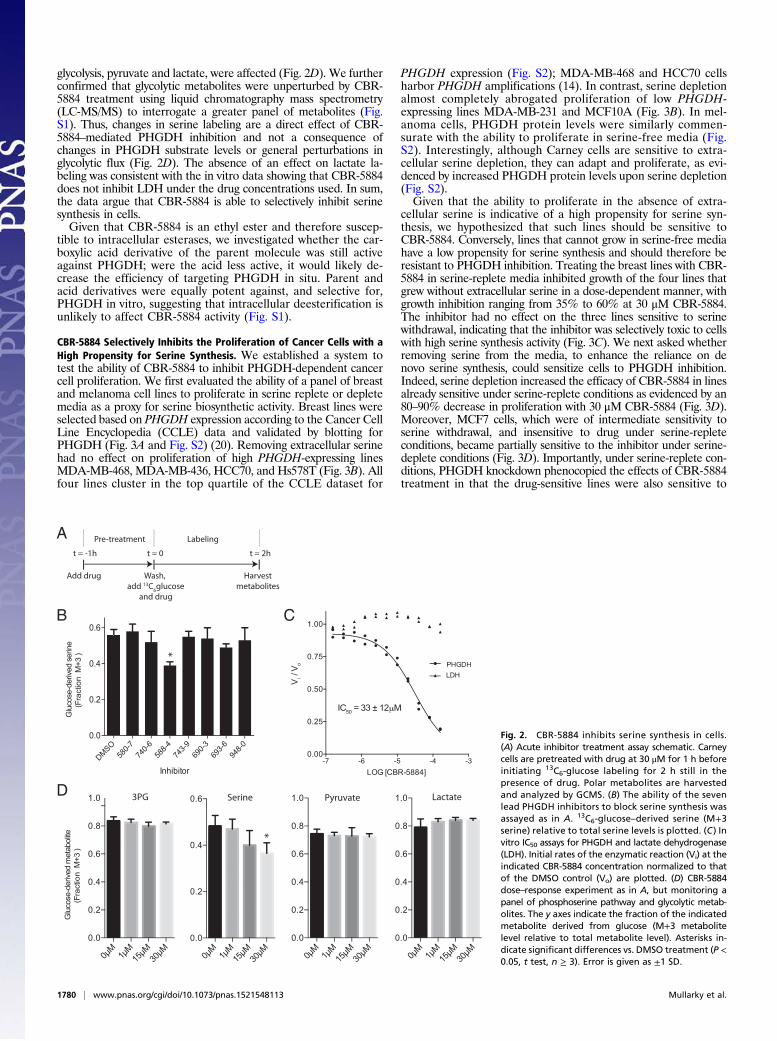
glycolysis, pyruvate and lactate, were affected (Fig. 2D). We furtherconfirmed that glycolytic metabolites were unperturbed by CBR-5884 treatment using liquid chromatography mass spectrometry(LC-MS/MS) to interrogate a greater panel of metabolites (Fig.S1). Thus, changes in serine labeling are a direct effect of CBR-5884–mediated PHGDH inhibition and not a consequence ofchanges in PHGDH substrate levels or general perturbations inglycolytic flux (Fig. 2D). The absence of an effect on lactate la-beling was consistent with the in vitro data showing that CBR-5884does not inhibit LDH under the drug concentrations used. In sum,the data argue that CBR-5884 is able to selectively inhibit serinesynthesis in cells.Given that CBR-5884 is an ethyl ester and therefore suscep-
tible to intracellular esterases, we investigated whether the car-boxylic acid derivative of the parent molecule was still activeagainst PHGDH; were the acid less active, it would likely de-crease the efficiency of targeting PHGDH in situ. Parent andacid derivatives were equally potent against, and selective for,PHGDH in vitro, suggesting that intracellular deesterification isunlikely to affect CBR-5884 activity (Fig. S1).
CBR-5884 Selectively Inhibits the Proliferation of Cancer Cells with aHigh Propensity for Serine Synthesis. We established a system totest the ability of CBR-5884 to inhibit PHGDH-dependent cancercell proliferation. We first evaluated the ability of a panel of breastand melanoma cell lines to proliferate in serine replete or depletemedia as a proxy for serine biosynthetic activity. Breast lines wereselected based on PHGDH expression according to the Cancer CellLine Encyclopedia (CCLE) data and validated by blotting forPHGDH (Fig. 3A and Fig. S2) (20). Removing extracellular serinehad no effect on proliferation of high PHGDH-expressing linesMDA-MB-468, MDA-MB-436, HCC70, and Hs578T (Fig. 3B). Allfour lines cluster in the top quartile of the CCLE dataset for
PHGDH expression (Fig. S2); MDA-MB-468 and HCC70 cellsharbor PHGDH amplifications (14). In contrast, serine depletionalmost completely abrogated proliferation of low PHGDH-expressing lines MDA-MB-231 and MCF10A (Fig. 3B). In mel-anoma cells, PHGDH protein levels were similarly commen-surate with the ability to proliferate in serine-free media (Fig.S2). Interestingly, although Carney cells are sensitive to extra-cellular serine depletion, they can adapt and proliferate, as evi-denced by increased PHGDH protein levels upon serine depletion(Fig. S2).Given that the ability to proliferate in the absence of extra-
cellular serine is indicative of a high propensity for serine syn-thesis, we hypothesized that such lines should be sensitive toCBR-5884. Conversely, lines that cannot grow in serine-free mediahave a low propensity for serine synthesis and should therefore beresistant to PHGDH inhibition. Treating the breast lines with CBR-5884 in serine-replete media inhibited growth of the four lines thatgrew without extracellular serine in a dose-dependent manner, withgrowth inhibition ranging from 35% to 60% at 30 μM CBR-5884.The inhibitor had no effect on the three lines sensitive to serinewithdrawal, indicating that the inhibitor was selectively toxic to cellswith high serine synthesis activity (Fig. 3C). We next asked whetherremoving serine from the media, to enhance the reliance on denovo serine synthesis, could sensitize cells to PHGDH inhibition.Indeed, serine depletion increased the efficacy of CBR-5884 in linesalready sensitive under serine-replete conditions as evidenced by an80–90% decrease in proliferation with 30 μM CBR-5884 (Fig. 3D).Moreover, MCF7 cells, which were of intermediate sensitivity toserine withdrawal, and insensitive to drug under serine-repleteconditions, became partially sensitive to the inhibitor under serine-deplete conditions (Fig. 3D). Importantly, under serine-replete con-ditions, PHGDH knockdown phenocopied the effects of CBR-5884treatment in that the drug-sensitive lines were also sensitive to
B C
D PyruvateSerine
0.0
0.2
0.4
0.6
Inhibitor
Glu
cose
-der
ived
serin
e(F
ract
ion
M+3
)
-7 -6 -5 -4 -30.00
0.25
0.50
0.75
1.00
LOG [CBR-5884]
V i/ V
o PHGDHLDH
3PG
IC50 = 33 ± 12 M
0μM
1μM
15μM
30μM
0.0
0.2
0.4
0.6
0.8
1.0
0.0
0.2
0.4
0.6
0μM
1μM
15μM
30μM
0.0
0.2
0.4
0.6
0.8
1.0
0μM
1μM
15μM
30μM
0.0
0.2
0.4
0.6
0.8
1.0 Lactate
*
*
Glu
cose
-der
ived
met
abol
ite(F
ract
ion
M+3
)
Pre-treatment Labeling
t = -1h
Add drug
t = 0
Wash, add 13C6glucose
and drug
Harvestmetabolites
t = 2h
A
DMSO58
0-74
0-58
8-74
3-69
0-69
3-94
8-7 6 4 9 3 6 0
0μM
1μM
15μM
30μM
Fig. 2. CBR-5884 inhibits serine synthesis in cells.(A) Acute inhibitor treatment assay schematic. Carneycells are pretreated with drug at 30 μM for 1 h beforeinitiating 13C6-glucose labeling for 2 h still in thepresence of drug. Polar metabolites are harvestedand analyzed by GCMS. (B) The ability of the sevenlead PHGDH inhibitors to block serine synthesis wasassayed as in A. 13C6-glucose–derived serine (M+3serine) relative to total serine levels is plotted. (C ) Invitro IC50 assays for PHGDH and lactate dehydrogenase(LDH). Initial rates of the enzymatic reaction (Vi) at theindicated CBR-5884 concentration normalized to thatof the DMSO control (Vo) are plotted. (D) CBR-5884dose–response experiment as in A, but monitoring apanel of phosphoserine pathway and glycolytic metab-olites. The y axes indicate the fraction of the indicatedmetabolite derived from glucose (M+3 metabolitelevel relative to total metabolite level). Asterisks in-dicate significant differences vs. DMSO treatment (P <0.05, t test, n ≥ 3). Error is given as ±1 SD.
1780 | www.pnas.org/cgi/doi/10.1073/pnas.1521548113 Mullarky et al.

PHGDH knockdown (Fig. 3E and Fig. S2). Furthermore, aswith the drug treatments, growing cells in serine-free mediaenhanced the growth inhibitory effect of PHGDH knockdown(Fig. 3F). Similar trends were observed for the melanoma panelin terms of both the selectivity of CBR-5884 for cells with a highpropensity for serine synthesis and the increased efficacy underserine-deplete conditions (Fig. S2). Finally, the acid derivativeof compound 5884 was not effective on MDA-MB-468 cells,likely owing to poor membrane permeability, and is thereforenot a viable alternative to the parent compound (Fig. S2).
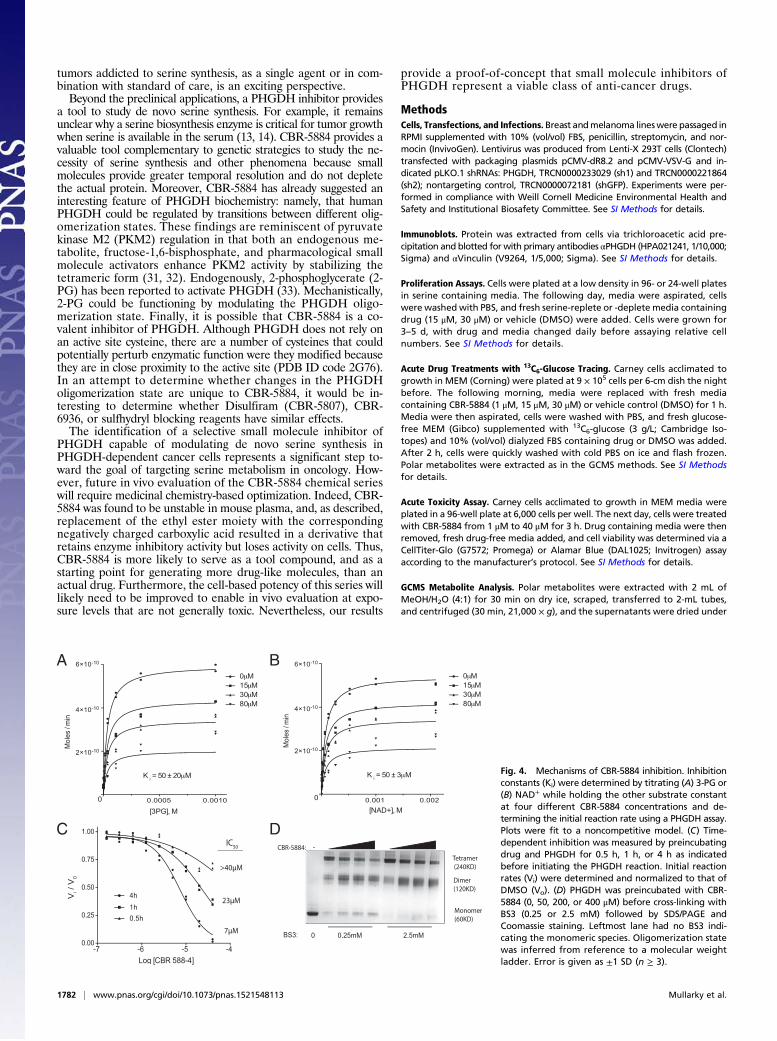
Analysis of CBR-5884 Inhibition Modality.We sought to more deeplycharacterize the mechanism by which CBR-5884 inhibits PHGDH.Inhibition constants (Ki) for CBR-5884 with respect to eachsubstrate were determined. CBR-5884 inhibited PHGDH ina noncompetitive mode with respect to both substrates, as evi-denced by a decreasing Vmax with increasing CBR-5884 con-centration. The inhibition constants were 50 ± 20 μM and 50 ±3 μM for 3-PG and NAD+, respectively (Fig. 4 A and B). Weassessed whether there was any time dependence to the onset ofinhibition by varying the time period for which drug andPHGDH were preincubated before initiating the enzymatic re-action. CBR-5884 was progressively more potent with increasingpreincubation time, culminating in an IC50 of 7 μM when drugand PHGDH were preincubated for 4 h (Fig. 4C). Intriguedby the combination of a time-dependent onset of inhibition andnoncompetitive inhibition, the latter suggesting that CBR-5884might be binding to an allosteric pocket, we speculated that CBR-5884 could be affecting the PHGDH oligomerization state, wherethe time dependency of inhibition could potentially stem fromdrug-induced conformational changes in PHGDH. To evaluatethe PHGDH oligomerization state, we incubated PHGDHwith drug and then cross-linked before SDS/PAGE. CBR-5884shifted the PHGDH equilibrium from the tetrameric to thedimeric state (Fig. 4D). No such effect was observed with LDH,
which is resistant to CBR-5884–mediated inhibition (Fig. S3).CBR-5884 still inhibited a truncated form of PHGDH, whichlacks the C-terminal domain responsible for tetramerization andis therefore a constitutive dimer (Fig. S3). Together, these resultssuggest that disruption of the tetramer might assist PHGDH in-hibition but is not necessary for inhibition.
DiscussionWe have reported the discovery of a PHGDH inhibitor, CBR-5884, and have shown that it inhibits serine synthesis in cells.Furthermore, CBR-5884 specifically inhibited the proliferation ofmelanoma and breast cancer lines with high levels of serine syn-thesis activity, with little effect on lines reliant on serine import.Thus, CBR-5884 is selective for lines addicted to serine synthesisand phenocopies sensitivity to PHGDH knockdown. Finally, abiochemical analysis of CBR-5884 revealed that it was a non-competitive inhibitor that showed a time-dependent onset of in-hibition and disrupted the oligomerization state of PHGDH.Recent work examining how malignant cells rewire their me-
tabolism to support growth and proliferation has revealed a numberof clinically interesting targets (21, 22). Perhaps the most promisingis the discovery of gain-of-function mutations in the isocitratedehydrogenase (IDH) enzymes that result in the production ofthe oncometabolite 2-hydroxyglutarate (23, 24). The findings trans-lated into chemical probes that yielded insights into the biologyof IDH mutations (25, 26) and led to clinical programs (e.g.,NCT02481154). The genetic evidence pointing to a role for PHGDHin cancer is similarly striking: PHGDH is one of few metabolicenzymes genetically deregulated in cancer (27, 28). Notably, el-evated PHGDH expression correlates with clinical aggressivenessand poor prognosis in TNBC (13, 16) and NSCLC (15). There is apaucity of targeted therapies for these cancers and chemother-apies are frequently used (29, 30). Thus, the clinical potential ofPHGDH inhibitors as targeted agents for TNBC and NSCLC
A
C D
E F
Vinculin
PHGDH (dark)
PHGDH (light)
MDA-M
B-468
MDA-M
B-436
Hcc70
MCF-7
MDA-M
B-231
MCF-1
0A
Hs578T
MDA-M
B-468
MDA-M
B-436
Hcc70
Hs578T
MCF-7
MDA-M
B-231
MCF-1
0A
Re
lati
ve c
ell
nu
mb
er
Re
lati
ve c
ell
nu
mb
er
Re
lati
ve c
ell
nu
mb
er
MDA-M
B-468
MDA-M
B-436
Hcc70
Hs578T
MCF-7
Re
lati
ve c
ell
nu
mb
er
B
0.00
0.25
0.50
0.75
1.00
+SER -SER
MDA-M
B-468
MDA-M
B-436
Hcc70
Hs578T
MCF-7
MDA-M
B-231
MCF-1
0A
0.00
0.25
0.50
0.75
1.00
DMSO 15μM 30μM
0.00
0.25
0.50
0.75
1.00
DMSO 15μM 30μM
Re
lati
ve c
ell
nu
mb
er
0.00
0.25
0.50
0.75
1.00
1.25shGFP sh1 sh2
0.00
0.25
0.50
0.75
1.00
shGFP sh1 sh2
MDA-M
B-468
MDA-M
B-436
Hs578T
MCF-7
MDA-M
B-231
MCF-1
0A
MDA-M
B-468
MDA-M
B-436
Hs578T
MCF-7
Fig. 3. CBR-5884 selectively inhibits the proliferationof breast cancer lines with a high propensity for ser-ine synthesis. (A) Western blot for lines grown in +SERmedia: two lanes per cell line with each lane loadedwith independent cell lysates. (B) Proliferation assayfor breast lines grown in either serine-replete (+SER)or -deplete (−SER) media. Proliferation assay for linestreated with CBR-5884 in (C ) +SER or (D) −SERmedia. Proliferation assay for lines grown in (E) +SERor (F) −SER media with PHGDH knockdown (sh1 andsh2) or a nontargeting control (shGFP). MDA-MB-468and HCC70 are PHGDH amplified. MCF-10A cells arenontransformed mammary epithelial cells; other linesare cancer cell lines. MDA-MB-231 and MCF-10A lineswere not included in −SER experiments in D and Fbecause they are sensitive to serine withdrawal. His-tograms depict mean ± SE (n ≥ 3).
Mullarky et al. PNAS | February 16, 2016 | vol. 113 | no. 7 | 1781
BIOCH
EMISTR
Y
tumors addicted to serine synthesis, as a single agent or in com-bination with standard of care, is an exciting perspective.Beyond the preclinical applications, a PHGDH inhibitor provides
a tool to study de novo serine synthesis. For example, it remainsunclear why a serine biosynthesis enzyme is critical for tumor growthwhen serine is available in the serum (13, 14). CBR-5884 provides avaluable tool complementary to genetic strategies to study the ne-cessity of serine synthesis and other phenomena because smallmolecules provide greater temporal resolution and do not depletethe actual protein. Moreover, CBR-5884 has already suggested aninteresting feature of PHGDH biochemistry: namely, that humanPHGDH could be regulated by transitions between different olig-omerization states. These findings are reminiscent of pyruvatekinase M2 (PKM2) regulation in that both an endogenous me-tabolite, fructose-1,6-bisphosphate, and pharmacological smallmolecule activators enhance PKM2 activity by stabilizing thetetrameric form (31, 32). Endogenously, 2-phosphoglycerate (2-PG) has been reported to activate PHGDH (33). Mechanistically,2-PG could be functioning by modulating the PHGDH oligo-merization state. Finally, it is possible that CBR-5884 is a co-valent inhibitor of PHGDH. Although PHGDH does not rely onan active site cysteine, there are a number of cysteines that couldpotentially perturb enzymatic function were they modified becausethey are in close proximity to the active site (PDB ID code 2G76).In an attempt to determine whether changes in the PHGDHoligomerization state are unique to CBR-5884, it would be in-teresting to determine whether Disulfiram (CBR-5807), CBR-6936, or sulfhydryl blocking reagents have similar effects.The identification of a selective small molecule inhibitor of
PHGDH capable of modulating de novo serine synthesis inPHGDH-dependent cancer cells represents a significant step to-ward the goal of targeting serine metabolism in oncology. How-ever, future in vivo evaluation of the CBR-5884 chemical serieswill require medicinal chemistry-based optimization. Indeed, CBR-5884 was found to be unstable in mouse plasma, and, as described,replacement of the ethyl ester moiety with the correspondingnegatively charged carboxylic acid resulted in a derivative thatretains enzyme inhibitory activity but loses activity on cells. Thus,CBR-5884 is more likely to serve as a tool compound, and as astarting point for generating more drug-like molecules, than anactual drug. Furthermore, the cell-based potency of this series willlikely need to be improved to enable in vivo evaluation at expo-sure levels that are not generally toxic. Nevertheless, our results
provide a proof-of-concept that small molecule inhibitors ofPHGDH represent a viable class of anti-cancer drugs.
MethodsCells, Transfections, and Infections. Breast andmelanoma lineswere passaged inRPMI supplemented with 10% (vol/vol) FBS, penicillin, streptomycin, and nor-mocin (InvivoGen). Lentivirus was produced from Lenti-X 293T cells (Clontech)transfected with packaging plasmids pCMV-dR8.2 and pCMV-VSV-G and in-dicated pLKO.1 shRNAs: PHGDH, TRCN0000233029 (sh1) and TRCN0000221864(sh2); nontargeting control, TRCN0000072181 (shGFP). Experiments were per-formed in compliance with Weill Cornell Medicine Environmental Health andSafety and Institutional Biosafety Committee. See SI Methods for details.
Immunoblots. Protein was extracted from cells via trichloroacetic acid pre-cipitation and blotted for with primary antibodies αPHGDH (HPA021241, 1/10,000;Sigma) and αVinculin (V9264, 1/5,000; Sigma). See SI Methods for details.
Proliferation Assays. Cells were plated at a low density in 96- or 24-well platesin serine containing media. The following day, media were aspirated, cellswere washed with PBS, and fresh serine-replete or -deplete media containingdrug (15 μM, 30 μM) or vehicle (DMSO) were added. Cells were grown for3–5 d, with drug and media changed daily before assaying relative cellnumbers. See SI Methods for details.
Acute Drug Treatments with 13C6-Glucose Tracing. Carney cells acclimated togrowth in MEM (Corning) were plated at 9 × 105 cells per 6-cm dish the nightbefore. The following morning, media were replaced with fresh mediacontaining CBR-5884 (1 μM, 15 μM, 30 μM) or vehicle control (DMSO) for 1 h.Media were then aspirated, cells were washed with PBS, and fresh glucose-free MEM (Gibco) supplemented with 13C6-glucose (3 g/L; Cambridge Iso-topes) and 10% (vol/vol) dialyzed FBS containing drug or DMSO was added.After 2 h, cells were quickly washed with cold PBS on ice and flash frozen.Polar metabolites were extracted as in the GCMS methods. See SI Methodsfor details.
Acute Toxicity Assay. Carney cells acclimated to growth in MEM media wereplated in a 96-well plate at 6,000 cells per well. The next day, cells were treatedwith CBR-5884 from 1 μM to 40 μM for 3 h. Drug containing media were thenremoved, fresh drug-free media added, and cell viability was determined via aCellTiter-Glo (G7572; Promega) or Alamar Blue (DAL1025; Invitrogen) assayaccording to the manufacturer’s protocol. See SI Methods for details.
GCMS Metabolite Analysis. Polar metabolites were extracted with 2 mL ofMeOH/H2O (4:1) for 30 min on dry ice, scraped, transferred to 2-mL tubes,and centrifuged (30 min, 21,000 × g), and the supernatants were dried under
A B
C D
Fig. 4. Mechanisms of CBR-5884 inhibition. Inhibitionconstants (Ki) were determined by titrating (A) 3-PG or(B) NAD+ while holding the other substrate constantat four different CBR-5884 concentrations and de-termining the initial reaction rate using a PHGDH assay.Plots were fit to a noncompetitive model. (C) Time-dependent inhibition was measured by preincubatingdrug and PHGDH for 0.5 h, 1 h, or 4 h as indicatedbefore initiating the PHGDH reaction. Initial reactionrates (Vi) were determined and normalized to that ofDMSO (Vo). (D) PHGDH was preincubated with CBR-5884 (0, 50, 200, or 400 μM) before cross-linking withBS3 (0.25 or 2.5 mM) followed by SDS/PAGE andCoomassie staining. Leftmost lane had no BS3 indi-cating the monomeric species. Oligomerization statewas inferred from reference to a molecular weightladder. Error is given as ±1 SD (n ≥ 3).
1782 | www.pnas.org/cgi/doi/10.1073/pnas.1521548113 Mullarky et al.

vacuum. Samples were derivatized as previously described (34) and analyzedon an Agilent 6890 GC instrument. Metabolite quantification was inferredfrom a standard curve, and fractional enrichment of 13C in metabolites wascorrected for the natural abundance of 13C and 15N (35, 36). See SI Methodsfor details.
LC-MS/MS Metabolite Analysis. Polar metabolites were extracted and dried asin the GCMS method. Samples were resuspended in 15 μL of HPLC gradewater. Then, 5 μL of each sample was injected and analyzed using a 5500QTRAP triple quadrupole mass spectrometer (AB/Sciex) coupled to a Prom-inence UFLC system (Shimadzu) as reported previously (37).
Protein Purification. His6-tagged pET28a PHGDH, pET28a PSAT1, and pNIC28-Bsa4 PHGDH3-314 were purified via nickel agarose (Qiagen) from BL21Escherichia coli cultures. pVB-CBD IDH1 was purified via Macroporous BeadCellulose capture, TEV protease (Sigma-Aldrich) digestion, and gel filtrationchromatography from BL21 E. coli cultures. See SI Methods for details.
PHGDH Assays. PHGDH activity was measured in 96-well plates by monitoringNADH fluorescence [excitation wavelength (Ex) 340 nm/emission wavelength(Em) 460 nm] over time. PSAT1 was included to prevent product inhibition ofPHGDH. See SI Methods for details.
LDH and MDH1 Assays. Enzyme activities were assayed using kits (for LDH,MAK06, Sigma; for MDH1, MAK196-1KT, Sigma) according to the manufac-turer’s instructions with commercially available recombinant enzyme (for LDH,59747, Sigma; for MDH1, SRP6103, Sigma). Drug, titrated as for the PHGDHIC50 assays, and enzyme were preincubated for 30 min before initiating re-action with substrate.
Cross-Linking Assays. PHGDH (1.5 μg) or LDH (2.2 μg, 59747; Sigma) was in-cubated with CBR-5884 (50 μM, 200 μM, 400 μM) or vehicle control (DMSO)in 25 mM Hepes, pH 7.3, and 1 mM NAD+ in 18 μL total volume for 30 minbefore BS3 (Pierce) cross-linking and quenching. Samples were run on SDS/PAGE and colloidal Coomassie stained (Bio-Rad). See SI Methods for details.
Primary PHGDH Screen, Diaphorase Counter Screen, and Dehydrogenase PanelSelectivity Profiling. Compounds (800,000) were screened at a single dose(13.3 μM) in 1,536-plate format against PHGDH or diaphorase quantifyingresorufin fluorescence (550/590 nm; Ex/Em) with an Envision plate reader.Results were analyzed using Genedata Screener software. Compounds witha robust Z-score of <−3 in the PHGDH screening assay and robust Z-score of>−2 in the diaphorase counter screen were selected as hits. See SI Methodsfor detailed protocols, hit selection and confirmation, and selectivity pro-filing against the dehydrogenase panel.
Chemical Syntheses. CBR-5884, ethyl 5-(furan-2-carboxamido)-3-methyl-4-thiocyanatothiophene-2-carboxylate, and the acid derivative, 5-(furan-2-carboxamido)-3-methyl-4-thiocyanatothiophene-2-carboxylic acid, synthesis wasadapted from the literature as described in SI Methods (38, 39).
ACKNOWLEDGMENTS. We thank U. Oppermann, M. G. Vander Heiden, K. R.Mattaini, and M. Yuan for technical assistance and reagents. We thankJ. Johnson, Y. Zheng, H. Shim, B. D. Ngo, and other L.C.C. laboratory membersfor helpful discussions. L.C.C. was supported by NIH Grants P01CA117969 andP01CA120964. C.A.L. was partially supported by a PanCAN-AACR Pathway toLeadership Award and a Dale F. Frey Award for Breakthrough Scientists fromthe Damon Runyon Cancer Research Foundation (Grant DFS-09-14). G.M.D.was supported by a PanCAN-AACR Pathway to Leadership Award. S.-M.F. wassupported by a Conquer Cancer Now Award from the Concern Foundation.
1. Kuge O, Hasegawa K, Saito K, Nishijima M (1998) Control of phosphatidylserinebiosynthesis through phosphatidylserine-mediated inhibition of phosphatidylserinesynthase I in Chinese hamster ovary cells. Proc Natl Acad Sci USA 95(8):4199–4203.
2. de Koning TJ, et al. (2003) L-serine in disease and development. Biochem J 371(Pt 3):653–661.
3. Futerman AH, Riezman H (2005) The ins and outs of sphingolipid synthesis. Trends CellBiol 15(6):312–318.
4. Stover P, Schirch V (1990) Serine hydroxymethyltransferase catalyzes the hydrolysis of5,10-methenyltetrahydrofolate to 5-formyltetrahydrofolate. J Biol Chem 265(24):14227–14233.
5. Tibbetts AS, Appling DR (2010) Compartmentalization of mammalian folate-mediatedone-carbon metabolism. Annu Rev Nutr 30(1):57–81.
6. Palacín M, Estévez R, Bertran J, Zorzano A (1998) Molecular biology of mammalianplasma membrane amino acid transporters. Physiol Rev 78(4):969–1054.
7. Barker GA, Ellory JC (1990) The identification of neutral amino acid transport systems.Exp Physiol 75(1):3–26.
8. Snell K (1986) The duality of pathways for serine biosynthesis is a fallacy. TrendsBiochem Sci 11(6):241–243.
9. Mullarky E, Mattaini KR, Vander Heiden MG, Cantley LC, Locasale JW (2011) PHGDHamplification and altered glucose metabolism in human melanoma. Pigment CellMelanoma Res 24(6):1112–1115.
10. Snell K, Natsumeda Y, Eble JN, Glover JL, Weber G (1988) Enzymic imbalance in serinemetabolism in human colon carcinoma and rat sarcoma. Br J Cancer 57(1):87–90.
11. Snell K, Natsumeda Y, Weber G (1987) The modulation of serine metabolism inhepatoma 3924A during different phases of cellular proliferation in culture. BiochemJ 245(2):609–612.
12. Beroukhim R, et al. (2010) The landscape of somatic copy-number alteration acrosshuman cancers. Nature 463(7283):899–905.
13. Locasale JW, et al. (2011) Phosphoglycerate dehydrogenase diverts glycolytic flux andcontributes to oncogenesis. Nat Genet 43(9):869–874.
14. Possemato R, et al. (2011) Functional genomics reveal that the serine synthesispathway is essential in breast cancer. Nature 476(7360):346–350.
15. DeNicola GM, et al. (2015) NRF2 regulates serine biosynthesis in non-small cell lungcancer. Nat Genet 47(12):1475–1481.
16. Pollari S, et al. (2011) Enhanced serine production by bone metastatic breast cancercells stimulates osteoclastogenesis. Breast Cancer Res Treat 125(2):421–430.
17. Zhang JH, Chung TD, Oldenburg KR (1999) A simple statistical parameter for use inevaluation and validation of high throughput screening assays. J Biomol Screen 4(2):67–73.
18. Vallari RC, Pietruszko R (1982) Human aldehyde dehydrogenase: Mechanism of in-hibition of disulfiram. Science 216(4546):637–639.
19. Buescher JM, et al. (2015) A roadmap for interpreting (13)C metabolite labelingpatterns from cells. Curr Opin Biotechnol 34:189–201.
20. Barretina J, et al. (2012) The Cancer Cell Line Encyclopedia enables predictive mod-elling of anticancer drug sensitivity. Nature 483(7391):603–607.
21. Son J, et al. (2013) Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 496(7443):101–105.
22. Tennant DA, Durán RV, Gottlieb E (2010) Targeting metabolic transformation forcancer therapy. Nat Rev Cancer 10(4):267–277.
23. Dang L, et al. (2009) Cancer-associated IDH1 mutations produce 2-hydroxyglutarate.Nature 462(7274):739–744.
24. Parsons DW, et al. (2008) An integrated genomic analysis of human glioblastomamultiforme. Science 321(5897):1807–1812.
25. Wang F, et al. (2013) Targeted inhibition of mutant IDH2 in leukemia cells inducescellular differentiation. Science 340(6132):622–626.
26. Rohle D, et al. (2013) An inhibitor of mutant IDH1 delays growth and promotes dif-ferentiation of glioma cells. Science 340(6132):626–630.
27. Mullen AR, DeBerardinis RJ (2012) Genetically-defined metabolic reprogramming incancer. Trends Endocrinol Metab 23(11):552–559.
28. Gottlieb E, Tomlinson IPM (2005) Mitochondrial tumour suppressors: A genetic andbiochemical update. Nat Rev Cancer 5(11):857–866.
29. Foulkes WD, Smith IE, Reis-Filho JS (2010) Triple-negative breast cancer. N Engl J Med363(20):1938–1948.
30. Boolell V, Alamgeer M, Watkins DN, Ganju V (2015) The evolution of therapies in non-small cell lung cancer. Cancers (Basel) 7(3):1815–1846.
31. Anastasiou D, et al. (2012) Pyruvate kinase M2 activators promote tetramer formationand suppress tumorigenesis. Nat Chem Biol 8(10):839–847.
32. Kung C, et al. (2012) Small molecule activation of PKM2 in cancer cells induces serineauxotrophy. Chem Biol 19(9):1187–1198.
33. Hitosugi T, et al. (2012) Phosphoglycerate mutase 1 coordinates glycolysis and bio-synthesis to promote tumor growth. Cancer Cell 22(5):585–600.
34. Nicolay BN, et al. (2013) Loss of RBF1 changes glutamine catabolism. Genes Dev 27(2):182–196.
35. Antoniewicz MR, Kelleher JK, Stephanopoulos G (2006) Determination of confidenceintervals of metabolic fluxes estimated from stable isotope measurements. MetabEng 8(4):324–337.
36. Antoniewicz MR, Kelleher JK, Stephanopoulos G (2007) Elementary metabolite units(EMU): A novel framework for modeling isotopic distributions. Metab Eng 9(1):68–86.
37. Ying H, et al. (2012) Oncogenic Kras maintains pancreatic tumors through regulationof anabolic glucose metabolism. Cell 149(3):656–670.
38. Guzi JT, et al. (2007) Methods for inhibiting protein kinases. US Patent Appl 11/598,188:1–346.
39. Ambrogi V, Grandolini G, Perioli L, Rossi C (1992) Convenient synthesis of 2-amino-naphthalene-1-thiol and 3-aminoquinoline-4-thiol and cyclocondensations to 1,4-thiazino and 1,4-thiazepino derivatives. Synthesis 1992(7):656–658.
Mullarky et al. PNAS | February 16, 2016 | vol. 113 | no. 7 | 1783
BIOCH
EMISTR
Y