grand rounds - nyu langone health · impaired cyclic amp generation and phos ... hypomagnesemia...
TRANSCRIPT

GRAND ROUNDS 4.16.13

Case 61 y.o F referred to renal clinic for evaluation of hypomagnesemia. Patient known to have hypomagnesemia for over 10 years.
Found to have low Mg on routine labs Has been on magnesium supplements for several years Denies h/o tetany, muscle cramps/spasms, muscle weakness,
arrhythmias, palpitations, seizures On one of the follow up visits reported severe muscle
weakness on discontinuation of SR magnesium supplements
ROS: positive for joint pain-knees, right shoulder, right hip pain Negative for nausea, vomiting, chronic diarrhea, poor appetite, chest pain, shortness of breath

PMHx: Chondrocalcinosis with superimposed OA of knees HLD Osteoporosis fracture of left wrist Family history: Mother – Hypertension Father- Arthritis, Diabetes Mellitus No family history of kidney disease or hypomagnesemia

Meds: Mg Oxide 800 mg 3 tabs bid SR Magnesium 84 mg 3 tabs bid Calcium carbonate 500 mg + vitamin D Naprosyn/Aleve bid PRN Fosamax 70 mg q weekly( took for 5-6 years, not taking currently)

Physical Exam: VS: afeb BP 110/60 HR 74 Gen: NAD Lungs: clear to ausc b/l CVS: RRR, normal S1S2 Abd: soft, non tender, non distended, bs +ve LE: no edema

Na K Cl bicarb
BUN
Cr Glu Ca Phos
141 4.3 103 32 29 0.6 103 9.4 3.5
Mg mg/dl
12/2002 0.7
1/2003 0.6
2/2003 0.8
5/2004 0.9
6/2005 0.7
11/2006 <1.3
10/2007 <1.3
1/2008 0.8
12/2009 1.2
10/2012 1.1
1/2013 1.0
24 urinary magnesium
24h urinary Calcium
2002 48mg/24hour
81 mg
2003 42 mg/24 hour
PTH
2/2003 22.5
10/2007 12.7
10/2012 33.7

Na K Mg P Ca Nh Cl Sul
11/5/12 247 71 237 0.542 195 40 226 54
11/3/12 184 82 191 0.771 134 27 169 38
Normal range
50-150 20-100 30-120 0.6-1.2 M<250 F<200
15-60 70-250 20-80
Vol 24 SSCaOx
Ox Cit SSCaP pH SSUA UA
11/5/12 1.56 6.17 43 1361 1.76 7.170 0.07 0.662
11/3/12 1.38 7.65 56 <21 1.97 6.877 0.14 0.620
Normal Range
0.5-4L 6-10 20-46 M>450 F>550
0.5-2 5.8-6.2 0-1 M<0.80 F<0.75

Renal Ultrasound: Normal sized kidneys, right 10.7 cm Left 10.5cm No hydronephrosis No calculus No solid renal lesion

Diagnosis: Renal Magnesium wasting disorder Etiology: ?

Magnesium Magnesium ion being the second most abundant
intracellular cation has a crucial role in fundamental metabolic processes such as DNA and protein synthesis, oxidative phosphorylation, enzyme function, ion channel regulation and neuromuscular excitability.
Magnesium is found in a wide variety of foods, and at particularly high levels in unrefined whole grain cereals, green leafy vegetables, nuts, seeds, peas and beans.
Balanced western diet contains approximately 360 mg
of magnesium per day; only about 120 mg of this is absorbed in the intestine.

Absorption Gastrointestinal magnesium absorption is mediated by a
saturable transcellular active pathway, as well as by non-saturable paracellular passive transport
Intestine secretes about 40 mg of magnesium per day and about 20 mg is absorbed in the large bowel.
Magnesium homeostasis is maintained by urinary excretion of approximately 100 mg/day
Regulation of renal magnesium excretion maintains physiologic serum concentrations at between 0.75 and 0.95 mmol/l (1.8-2.3 mg/dl) in healthy humans.

Renal Magnesium Handling Kidney is the major regulator of total
magnesium homeostasis In the setting of hypomagnesemia, the kidney
decreases magnesium excretion to as little as 0.5% of the filtered load.
Conversely in the setting of hypermagnesemia, up to 80% of the filtered load can be excreted.
A proportion of Mg is protein bound, such that only 70% of total plasma magnesium is ultrafilterable.

J Am Soc Nephrol 19: 1451–1458, 2008

CaSR The Ca/Mg sensing receptor (CaSR), a member of the G coupled receptor
family is an important regulator of calcium and magnesium homeostasis
The CaSR consists of an extracellular domain of 612 amino acids, a 250 amino acid transmembrane domain of 7 transmembrane helices, and a 200 amino acid carboxy terminal C-tail
Mg, Sr, La, Gd, and highly positive charged organic acids such as polyamines,
aminoglycoside antibiotics, protamine, polyarginine are also able to activate the receptor, even in the absence of extracellular Ca. These polycationic agonists are termed as type I agonist
Type 2 agonists, in contrast require the presence of calcium to activate CaSR and include L-amino acids and other organic acids that act as allosteric activators of the receptor
Upon agonist binding, conformational changes in the transmembrane domain and intracellular domains trigger a number of signaling pathways. A characteristic signature of CaSR activation is the mobilization of intracellular Ca via activation of phospholipase C
CaSR is expressed widely along the nephron and exhibits segment specific polarization

Ca and magnesium homeostasis
Kidney International (2012) 82, 1157–1166

Renal Magnesium Handling In the hypomagensemic or hypocalcemic states,
the rates of calcium and Mg reabsorption in the loop of henle are increased via CASR mediated stimulation of the Na-K-2CL cotransporter and the apical ROMK channel
By contrast hypermagnesemia and hypercalcemia inhibit Na-K-2CL cotransport and activity of ROMK channel

Clinical Manifestations of Hypomagnesemia
Defined as serum Mg concentration less than 0.74mmol/l(<1.8mg/dl)
Early symptoms are non specific-lethargy and weakness
More pronounced hypomagnesemia presents with symptoms of
increased neuromuscular excitability such as tremors, carpopedal spasm, muscle cramps, tetany, generalized seizures
Hypomagnesemia can cause cardiac arrhythmias including atrial and ventricular tachycardia, prolong QT interval and torsades de pointes
Hypomagnesemia is frequently associated with other electrolyte abnormalities such as hypokalemia and hypocalcemia

Clinical Manifestations of Hypomagnesemia
Hypocalcemia is attributable to several pathophysiologic processes ◦ Concentration of PTH is inappropriately low in
many individuals ◦ Bone resistance to the effects of PT ◦ Kidney is also refractory to PTH as manifested by
impaired cyclic AMP generation and phos reabsorption ◦ Abnormalities of vitamin D metabolism has also
been described Chondrocalcinosis has been described as a complication
of chronic hypomagnesemia especially in patients with Gitelman syndrome

Etiology and diagnosis of hypomagnesemia
Hypomagnesemia results from negative magnesium balance that develops in the setting of decreased oral intake, increased gastrointestinal or renal losses
Most frequently hypomagnesemia is an acquired disorder; only in rare instances does hypomagnesemia have an underlying hereditary etiology
Measurement of urinary magnesium excretion is helpful in the
differentiation of renal from extrarenal causes of magnesium wasting
24 hour magnesium excretion in the urine is expected to be less than 1 mmol (<24 mg) per day.
It is more convenient is clinical practice to measure fractional urinary magnesium excretion

… FEMg = Umg x PCr/(0.7 x PMg) x UCr x100
In states of extra-renal magnesium wasting, the
fractional excretion is less than 2%, whereas with renal magnesium wasting the fractional excretion exceeds 4%

Nature feb 2008 Vol 4

Familial hypomagnesemia with hypercalciuria and nephrocalcinosis (FHHNC)
FHNNC is an autosomal recessive disorder
characterized by excessive renal calcium and magnesium wasting
Caused by mutations of the claudin genes-
16/paracellin-1 gene (located on chromosome 3d) and claudin 19

FHHNC phenotype Usually become evident in the first few months of life Affected individuals present with symptoms including
polyuria, polydipsia, hypomagnesemia, inappropriately high rates of urinary magnesium excretion, hypercalciuria, nephrocalcinosis, renal insufficiency and in rare instances generalized seizures
Mean age at diagnosis is 15 years with a range of 5-25 years

FHHNC Patients with claudin 19 mutations also had ocular
manifestations that included severe visual impairment, macular colobomata, horizontal nystagmus and marked myopia in addition to renal phenotype as FHHNC
Therapeutic approaches: oral magnesium
supplementation as well as thiazide diuretics to reduce calcium excretion and progression of nephrocalcinosis
Renal transplantation is curative as the primary defect
resides in the kidney

Claudins Claudins are integral membrane protein localized to tight
junctions Epithelial tight junction is responsible for the control of
paracellular transport between the epithelial cells and the maintenance of apical/basolateral polarity
Renal tubular epithelia in the mammalian nephron have TJs
that determine the paracellular permeability properties. Claudins appear to determine the size and charge
selectivity of the TJ
Am J Physiol Renal Physiol 290:F572-579,2006

Claudins consist of four transmembrane domains, two extracellular loops and a short COOH intracellular tail
Claudins use their extracellular domains to control paracellular transport, supposedly by formation of selective pores that are oriented parallel to the lateral memebrane.
Differences in charge selectivity are based on charged amino acid residues of the extracellular domain that line the paracellular pore and electrostatically interact with ions passing the paracellular route

Isolated recessive hypomagnesemia with normocalciuria
Isolated recessive hypomagnesemia is a rare hereditary disease that was originally described in a consanguineous family
Affected individual present with symptoms of hypomagnesemia during early infancy.
IRH is due to mutation in the EGF gene. EGF gene encodes pro-EGF, which is expressed in the basolateral membrane of the DCT.
Pro-EGF is a type I membrane protein that is sorted and subsequently inserted
into the luminal and basolateral membrane of the epithelial cells. It consists of a small intracellular C terminal tail, one trans-membrane domain, and a large extracellular amino terminal.
Pro- EGF is cleaved by extracellular proteases in the basolateral space into the active molecule EGF
EGF acts as an autocrine/paracrine magnesiotropic hormone by activating the
basolateral EGF receptor and that in turn causes increase in luminal TRPM6 activity
In IRH mutation disrupts the basolateral sorting motif and for that reason pro-
EGF can not travel to the basolateral membrane

Hypomagnesemia with secondary hypocalcemia HSH is an autosomal recessive disorder caused by
mutations of TRPM6 TRPM6 mutations cause a defect in the active
transcellular pathways of intestinal and renal magnesium absorption.
Patients with HSH present with generalized seizures due to profound hypomagnesemia
Hypocalcemia develop in the setting of hypomagnesemia induced inhibition of PTH synthesis and release from the parathyroid gland

TRPM6
Share structural homology to other TRP channels It is composed of a large intracellular amino terminus, six membrane
spanning domains that make up the channel pore and a large intracellular carboxy terminal domain
Fused to the carboxy terminus is an alpha kinase domain, which plays
role in regulating channel activity, this domain is not necessary for basal function, however autophosphorylation is a mechanism regulating channel activity
TRPM6 is a cation selective channel conduct magnesium over calcium
Intracellular Magnesium acts as a negative regulator of channel activity
J Am Soc Nephrol 19: 1451–1458, 2008

TRPM6 Recently, a new TRPM6 interacting protein, RACK1- receptor
for activated C-kinase has been described RACK-1 interacts directly with the alpha kinase domain
inhibiting channel activity
RACK-1 mediated inhibition requires autophosphorylation of residue T1851 in the kinase domain
Furthermore, the inhibition of TRPM6 activity by intracellular
Mg depends on this autophosphorylation
It is possible that TRPM6 mediated Mg influx induces phosphorylation of T1851 located in the alpha kinase domain, a process that activates the inhibitory effect of RACK1
J Am Soc Nephrol 19: 1451–1458, 2008

J Am Soc Nephrol 19: 1451–1458, 2008

Figure 1. EGF activates TRPM6 current in HEK 293 cells J Am Soc Nephrol 20:78-85,2009

EGF stimulated TRPM6 Current is specifically Mediated by the EGFR J Am Soc Nephrol 20:78-85,2009
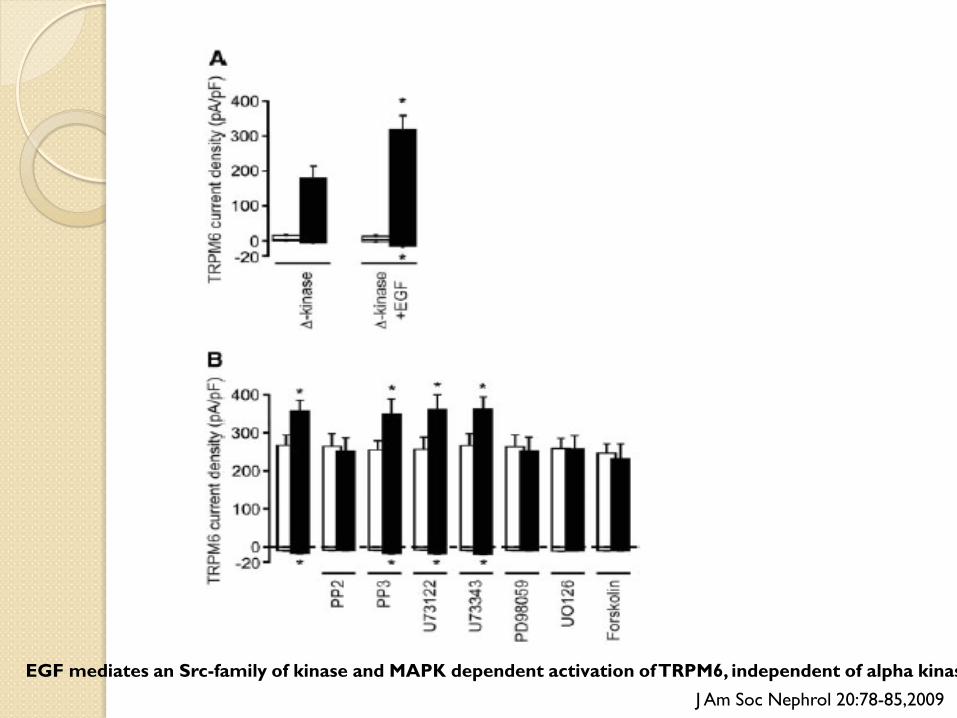
EGF mediates an Src-family of kinase and MAPK dependent activation of TRPM6, independent of alpha kinas J Am Soc Nephrol 20:78-85,2009

Stimulation of TRPM6 by EGF depends on PI3K, Rac1, and the actin cytoskeleton
J Am Soc Nephrol 20:78-85,2009

EGF increases the mobility of TRPM6
J Am Soc Nephrol 20:78-85,2009
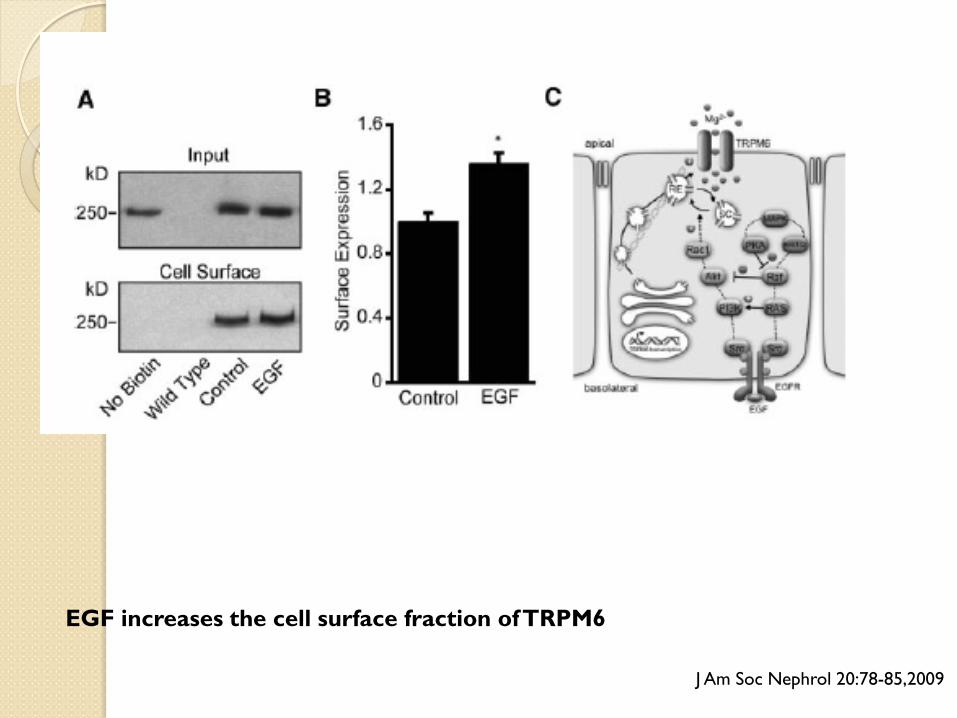
EGF increases the cell surface fraction of TRPM6
J Am Soc Nephrol 20:78-85,2009

Autosomal dominant hypomagnesemia with hypocalciuria Mutation of FXYD2 which is located on the chromosome
11q23 FXYD2 codes for the gamma subunit of Na/K ATPase which is
expressed in the basolateral memebrane of the distal convoluted tubule cells
Mutation of the FXYD2 leads to misrouting of the Na/K ATPase
gamma subunit which results in abnormal Mg reabsorption
Transcription factor HNF1B has been found to be linked to the regulation of FXYD2 gene
By use of subsequent luciferase assays, the author showed the control of FXYD2 gene expression by HNF1B

Activating mutations of the CaSR Activating mutations were first described in families affected with autosomal dominant
hypocalcemia (ADH) Affected individuals present with hypocalcemia, hypercalciuria, polyuria and 50% of these
patients have hypomagnesemia
Underlying pathophysiology is a decrease CaSR set point which leads to decrease PTH release, as well as diminished renal calcium and magnesium reabsorption in the setting of low serum concentrations of divalent cations
Clinically this may be mistaken for primary hypoparathyroidism as there is decreased PTH secretion
in the setting of mild to moderate hypocalcemia
Affected individuals are at increased risk of hypercalciuria, nephrocalcinosis and even irreversible reduction of renal function in individuals who are treated with vitamin D
Only patients with symptomatic ADH should be treated with calcium and vitamin D

New Molecular players facilitating Mg reabsorption in the distal convoluted tubule
Kv1.1 Recently a mutation in the KCNA1 gene, coding for
the voltage gated K channel Kv.1 was shown to be the cause for autosomal dominant hypomagnesemia in a large Brazilian family.
Phenotype detectable from infancy consisted of recurrent muscle cramps, tetany, tremor, muscle weakness, cerebellar atrophy and myokymia
Kv1.1 colocalizes with TRPM6 along the luminal membrane
Kidney International (2010) 77, 17–22

New Molecular players facilitating Mg reabsorption in the distal convoluted tubule
Kir4.1 Mutation within KCNJ10 gene was found to be the
underlying cause of recently defined EAST syndrome- Epilepsy, ataxia, sensorineural deafness and tubulopathy (closely resembling the GS )
The KCNJ10 gene encodes the K channel Kir4.1 that is
expressed in brain, ear and in kidney
Renal phenotype is similar to GS phenotype and consists of polyuria, hypokalemic metabolic alkalosis, hypomagnesemia and hypocalciuria
Kir4.1 is expressed at the basolateral membrane of the distal
tubular epithelia
Kidney International (2010) 77, 17–22

Salt Losing tubular disorders
Gitelman syndrome and Bartter syndrome are two renal salt wasting disorders characterized by hypokalemia, metabolic alkalosis, elevated plasma renin and aldosterone levels
Hypomagnesemia and renal magnesium wasting
are distinctive features of Gitelman syndrome

Gitelman Syndrome Autosomal recessive disorder caused by mutation in the SLC12A3 gene that
encodes the sodium-chloride cotransporter ( NCCl), which is expressed in the DCT
A minority of the patients with GS have been diagnosed with mutations of
CLCNKB, the gene that encodes the basolaterally located renal chloride channel CLCKB that is expressed along the ascending limb and distal convoluted tubule
Usually present during childhood or adolescence
Cardinal symptoms include muscle weakness or tetanic episodes that are related
to profound hypomagnesemia. Hypokalemia, metabolic alkalosis, hypomagnesemia and hypocalciuria
Hypcalciuria in GS is explained by reduced entry of NaCl into the DCT cells
leading to hyperpolarization. Cellular hyperpolarization increases calcium reabsorption mediated by an apical entry via an epithelial channel and basolateral extrusion through the Na/Ca exchange
The reason for often pronunced hypomagnesemia in GS is still unknown

Bartter Syndrome
Antenatal Barter syndrome or hyperprostaglandin E syndrome Mutation in NKCC2 or ROMK Characterized by massive polyuria that manifests in
utero with the development of polyhydramnios that results in premature birth in almost all cases. Postnatally affected individuals rapidly develop salt wasting and hypokalemic metabolic alkalosis.
In addition hypercalciuria and pronunced nephrocalcinosis occur in all affected individuals
Magnesium wasting in not a common finding

Classic Bartter Syndrome: Mutation in CLCNKB encoding the basolaterally
located renal chloride channel ClC-Kb which mediates chloride efflux from the tubular epithelial cell to the interstitium along the TAL and DCT
Hypomagnesemia is detected in up to 50% of the
cases and calcium excretion is variable

Antenatal Bartter Syndrome with Sensorineural Deafness: Mutation in Barttin gene(BSND) Functional expression studies revealed, that Barttin is
a beta subunit of the renal chloride channels(ClC-Ka and –Kb)
Barttin is expressed along the kidney tubule and in the stria vascularis of inner ear
Renal phenotypic presentation-massive salt and fluid loss from birth
Hypomagnesemia has not been reported in BSND patients
Hypercalciuria and nephrocalcinosis are uncommon. Many patients develop renal failure from unknown etiology