glutathione and ischemia-reperfusion injury in the perfused rat liver
TRANSCRIPT

Free Radical Biology & Medicine, Vol. 12, pp. 271-279, 1992 0891-5849/92 $5.00 + .00 Printed in the USA. All fights reserved. Copyright © 1992 Pergamon Press Ltd.
Original Contribution
G L U T A T H I O N E A N D I S C H E M I A - R E P E R F U S I O N I N J U R Y
I N T H E P E R F U S E D R A T L I V E R
MASAHIRO OKUDA, HSIEN-CHANG LEE, BRITTON CHANCE, and CHELLAPPA KUMAR
Department of Biochemistry and Biophysics, University of Pennsylvania, School of Medicine, Philadelphia, PA 19104°6089, U.S.A.
(Received 17 September 1991 ; Accepted 4 November 1991 )
Abstract--Using the isolated perfused rat liver, we investigated the relationship of glutathione (GSH) with reactive oxygen species (ROS) generation and liver cell damage during ischemia/reperfusion in normal and GSH-depleted conditions. Luci- genin-enhanced chemiluminescence was used as a sensitive index of tissue ROS generation. After 30 minutes of equilibration, livers were subjected to global ischemia for various times (60 or 90 minutes) and then reperfused for another 120 minutes. Intracellular ROS levels increased sharply at the onset of reperfusion and then declined slowly. After 30 to 60 minutes of reperfusion, ROS levels started to increase progressively in a linear fashion. However, sinusoidal glutathione disulfide release did not increase during reperfusion in the same livers, suggesting that intracellular ROS generation is too low to cause a significant increase in GSH oxidation. Pretreatment with phorone (300 mg/kg intrapentoneally [ip]), which reduced hepatic GSH by 90%, did not cause any difference in intracellular ROS generation compared with the control livers. There were also no significant differences in lactate dehydrogenase and thiobarbituric acid reactive substances (TBARS) release between the control and phorone-treated livers during reperfusion after various times ofischemia. These data indicate that ROS generation in the normal isolated perfused liver during ischemia/reperfusion is extremely low and intracellular GSH does not serve as a major intracellu- lar defense system against such a low oxidative stress.
Keywords--Oxygen radicals, Ischemia reperfusion, Chemiluminescence, Lucigenin, Glutathione, Liver, Lipid peroxidation, Phorone, Free radicals
INTRODUCTION
Reactive oxygen species (ROS) have been implicated in the pathogenesis of ischemia-reperfusion injury in a variety of organs) Usually, cells have a variety of defense systems against such oxidative stress. When these defense systems are weakened, organs or cells are more susceptible to oxidative stress. The glutathi- one (GSH) system is an important endogenous an- tioxidant that is found in particularly high concentra- tion in the liver. 2 The importance of GSH for protec- tion against oxidative stress has been shown in the perfused heart 3 and in in situ liver: '5 Under condi- tions of intracellular oxidative stress, GSH is oxidized to glutathione disulfide (GSSG) by ROS. Recently, the GSSG production and eitlux from the liver has
Address correspondence to M. Okuda, MD, PhD, Department of Biochemistry and Biophysics, University of Pennsylvania, School of Medicine, D-501 Richards Building, Philadelphia, PA 19104- 6089, U.S.A.
been widely used as an index for oxidative stress. 6-9
Using this method, Jaeschke et al) ° reported that there was no evidence for large formation of ROS during ischemia reperfusion in the isolated perfused liver, in which very little, if any, increase in the secre- tion of GSSG into bile or perfusate occurred and no changes in the GSSH content were seen during reper- fusion following hepatic ischemia. On the other hand, a dramatic increase of the plasma concentration of GSSG (but not in bile) has been observed in in vivo postischemic liver.~,~2 These results suggest that sig- nificant oxidative stress occurs in the extracellular space but not in the intracellular space. However, the question still remains as to whether the lack of evi- dence for intracellular oxidative stress evaluated by GSSG formation is due to the extremely low level of ROS generation in postischemic liver or due to other endogenous antioxidants (superoxide dismutase [SOD], catalase, vitamin E, etc.) scavenging the ROS before significant interaction occurs with the GSH system. The aim of this study was to establish the role
271

272 M. OKUDA el al.
of hepatic endogenous GSH in intracellular antioxi- dant defense during hepatic ischemia reperfusion by evaluating ROS generation and liver cell damage under normal and GSH-depleted conditions.
In the present study, we used lucigenin-enhanced chemiluminescence to evaluate the intracellular ROS generation. The usefulness of the enhanced chemilu- minescence technique has been described else- where/3-L7 Lucigenin-enhanced chemiluminescence is able to detect mainly intracellular ROS noninva- sively and continuously. ~6-~9 Although lucigenin has been demonstrated to be sensitive to superoxide, it can also react with hydrogen peroxide and emit light. 16 The GSH system removes hydrogen peroxide and also acts as a natural scavenger for superox- ide. 2°'2~ Therefore, one would expect that ifintracellu- lar GSH is a primary cellular defense system against ROS, the chemiluminescence response would be sig- nificantly enhanced during reperfusion after ischemia in GSH-depleted liver. This forms the basis of the pres- ent study.
MATERIALS AND METHODS
Male Sprague-Dawley rats (200-250 g body wt) were purchased from Charles River Co. (Wilmington, MA). Each rat, fasted for 24 h before use, was heparin- ized (sodium heparin, 1000 units/kg, ip) and anesthe- tized (sodium pentobarbital, 50 mg/kg, ip) before liver isolation. Some rats were pretreated with phor- one (300 mg/kg body wt) intraperitoneally I h prior to liver isolation. The lowest level of hepatic GSH was obtained after 1 h to 4 h ofintraperitoneal injection of phorone in vivo. 22 The livers from rats without phor- one treatment served as controls. The liver isolation and perfusion was carded out according to Miller. 23 After surgery, livers were excised from the abdominal cavity and transferred to a light-tight perfusion box for chemiluminescence measurements. The perfusate was maintained at 36.0 _ 0.5°C throughout the study using a water jacket. Livers were perfused with blood- and albumin-free Krebs-Henseleit (K-H) bicarbonate buffer, pH 7.4, bubbled continuously with a gas mix- ture containing 95% 02/5% CO/, and filtered through a 0.45-ttm millipore filter. The inflow was through the portal vein, and the outflow was through the inferior vena cava. The hepatic artery was ligated. Both the inflow and outflow vessels were cannulated, and the perfusate was pumped through the liver with a peris- taltic pump (Master flex, Cole Parmar Instrument Co., Chicago, IL) at a constant flow rate of 3-3.5 ml/ min per gram wet weight of the liver in a nonrecircu- lating system.
Liver chemiluminescence
The lucigenin-enhanced chemiluminescence was monitored continuously as a measure of tissue ROS generation. 15-~8 The chemiluminescence apparatus and light-tight perfusion box have been described in detail elsewhere. L3-17 Chemiluminescence was mea- sured using a home-built high-performance single photon counting system with an EMI 9658 photo- multiplier tube, responsive in the range of 300-900 nm. The photomultiplier tube was cooled to -20°C by a FACT 50 MK III cooler (Thorn EMI Gencon Inc., Fairfield, N J) to decrease the dark count. Chemi- luminescence data are expressed as counts per second (cps). To compare the chemiluminescence intensities from different livers, photoemission intensities were normalized to unit surface area of the liver (cps/cm2).
Experimental protocol
At the beginning of each experiment, a stable dark count was recorded and then the shutter was opened. All livers were perfused for 10 minutes with the stan- dard K-H buffer, and the spontaneous chemilumines- cence was recorded. Subsequently, lucigenin (N,N'- dimethyl-9,9'-biacridinium dinitrate) was infused con- tinuously into the perfusate to a final concentration in perfusate of 0.01 uM for the remaining perfusion pe- riod, except during global ischemia. After 20 minutes of lucigenin perfusion, the livers underwent 60 min- utes or 90 minutes of zero-flow global ischemia and were reperfused for 120 minutes. Ischemia and reper- fusion were performed in both the control and phor- one-treated livers. Each experimental group con- tained five livers. During global ischemia, the perfu- sion chamber was maintained at 26 _+ 0.5°C using a thermocontroller.
Release of lactate dehydrogenase and thiobarbituric acid reactive substances
Effusate was collected at specific time intervals to determine lactate dehydrogenase (LDH) activity and thiobarbituric acid reactive substances (TBARS) con- centrations. LDH activity in effusate was determined using Sigma diagnostic kit DG 1340-KI (Sigma Chem- ical Co., St. Louis, MO). TBARS were measured us- ing a modified thiobarbituric acid fluorometric methodZ4:1 ml ofeffusate was mixed thoroughly with 2 ml of a reagent containing 15% trichloroacetic acid, 0.375% thiobarbituric acid, and 0.25 N hydrochloric acid and was heated for 20 minutes at 95°C in the test tube heater; after cooling, samples were centrifuged at 1000 g for 10 minutes, and the fluorescence of the

Glutathione and liver ischemia 273
supernatant was determined with 515 nm excitation and 553 nm emission in a Luminescence Spectrome- ter (Model LS-30, Perkin Elmer, Buckinghamshire, England). Concentration of TBARS in the effusate was obtained by comparison with malondialdehyde (1,1,3,3-tetraethoxypropane) standards, and the TBARS release rate was determined using the flow rate and the liver wet weight.
Release of sinusoidal GSH and GSSG and tissue GSH content
Total soluble glutathione (GSH and GSSG) in ef- fluent perfusate was measured as an index of sinusoi- dal efflux. Total GSH content of the tissue was mea- sured in acidic homogenate from freeze-clamped liver tissue. Total GSH was determined spectrophotometi- cally by measuring the formation of 5-thio-2-nitro- benzoate at 412 nm. 7 Briefly, 500 #1 of effusate was added to 1.9 ml of 100 mM potassium phosphate buffer I (pH 7.5) containing I0 mM 5,5'-dithio-bis-2- nitrobenzoic acid (DTNB), 5 mM disodium ethylene- diaminetetraacetic acid (EDTA), and 0.5 unit of GSSG reductase (Sigma Type III). Reduced nicotin- amide adenine dinucleotide phosphate (NADPH; 100 #1 of 2.2 mM) was added to the solution to initiate the reaction. After 1 minute of equilibration, the rate of absorbance change at 412 nm was measured using a U-2000 spectrophotometer (Hitachi Ltd., Tokyo, Japan). Buffer containing equal concentrations of DTNB, EDTA, GSSG reductase, and NADPH was used as the reference. For the determination of GSSG, 1 ml ofeffusate was mixed immediately with 250 #1 of 10 mM N-ethylmaleimide (NEM) in 100 mM potas- sium phosphate buffer (pH 6.5) containing 17.5 mM disodium EDTA. To separate GSSG from NEM and NEM-GSH adducts, 1 ml of the mixture was passed through a C 18 Sep-Pak cartridge (Waters Associates, Framingham, MA) followed by 1 ml of the buffer I. A 500-ul volume from a total 2 ml of combined eluate was assayed following the procedure for the total GSH measurement.
Chemicals
Lucigenin, malondialdehyde, EDTA, NEM, GSH, GSSG reductase (Type III), and NADPH were pur- chased from Sigma Chemical Co. (St. Louis, MO). DTNB and phorone were purchased from Ardrich Chemical Co. (Milwaukee, WI). All other chemicals were of analytical grade. Phorone (300 mg/kg) was diluted in 250 #1 dimethyl sulfoxide (DMSO) and then administered intraperitoneally I h before the ini- tiation of perfusion.
Table 1. Total GSH Content (#mol/g liver wt) in the Liver
Preischemia A B
Control 8.94 + 0.93 4.28 + 1.02 4.38 + 1.14 Phorone-treated 1.06 _+ 0.10 0.14 + 0.05 0.15 + 0.04
Data are mean + SD. Phorone (300 mg/kg) was administered intraperitoneally 1 hour before perfusion. The GSH content after 30 minutes of perfusion served as the preischemic value (four livers). A: GSH content at the end of experiment of the 60-minute ischemia group. B: GSH content at the end of experiment of the 90-minute ischemia group.
Statistics
All data are expressed as mean _+ SD. Intra and intergroup comparison was performed using analysis of variance followed by the Fisher protected least sig- nificant difference test. A probability o fp < .05 was considered statistically significant.
RESULTS
Table 1 shows the effect ofphorone (300 mg/kg ip) on hepatic total GSH (GSHeq = GSH + 2GSSG) con- tent. Pretreatment with phorone reduced hepatic to- tal tissue GSH by 90%, assayed after 30 minutes of initial perfusion. At the end of the experiment, both the control and phorone-treated livers showed a signif- icant decrease in total tissue GSH content compared with the preischemic level.
The time courses of changes in lucigenin-enhanced chemiluminescence in the two ischemic groups are shown in Figure 1. The spontaneous liver chemilumi- nescence was 12 _+ 1.2 c p s / c m 2 ( 2 0 livers). The addi- tion of lucigenin to the perfusate caused a small in- crease in chemiluminescence by 3-5 cps/cm 2. No dif- ferences were present between the control and phorone-treated livers in spontaneous and baseline (preischemic) lucigenin-enhanced chemilumines- cence. At the onset of ischemia, there was a prompt decrease in lucigenin-enhanced chemiluminescence, and it reached a plateau within 5 minutes. On reper- fusion, there was an immediate increase in lucigenin- enhanced chemiluminescence, which peaked in 17- 20 minutes and was followed by a slow decline. After 30-60 minutes of reperfusion, lucigenin-enhanced chemiluminescence started to increase progressively in all groups of livers. There were no statistically signif- icant differences in chemiluminescence intensifies be- tween the control and phorone-treated livers through- out the study. Also, there were no statistically signifi- cant differences in chemiluminescence intensities between 60- and 90-minute ischemia groups in the

274 M. OKUDA et al,
e~
E
d:Z
60
40
20
(A)
q ischemia ,, . y ~'
30 60 90 120 150 180 210 Perfusion time (rain)
ea
K
r =
(B) 60
40
2O
ischemia 11
,~~/ ---- Phoro~e
30 60 90 120 150 180 210 240
Perfusion time (min)
Fig. 1. Time course of changes in lucigenin-enhanced chemiluminescence of the control (O) and phorone-treated livers (O). A: 60-minute ischemia groups. B: 90-minute ischemia groups. After 10 minutes of plain Krebs-Henseleit (K-H) buffer perfusion, lucigenin was infused continuously to a final perfusate concentration of 0.01 ~M throughout the study except during ischemic period. Data are mean +_ SD.
control and phorone-treated livers throughout the study.
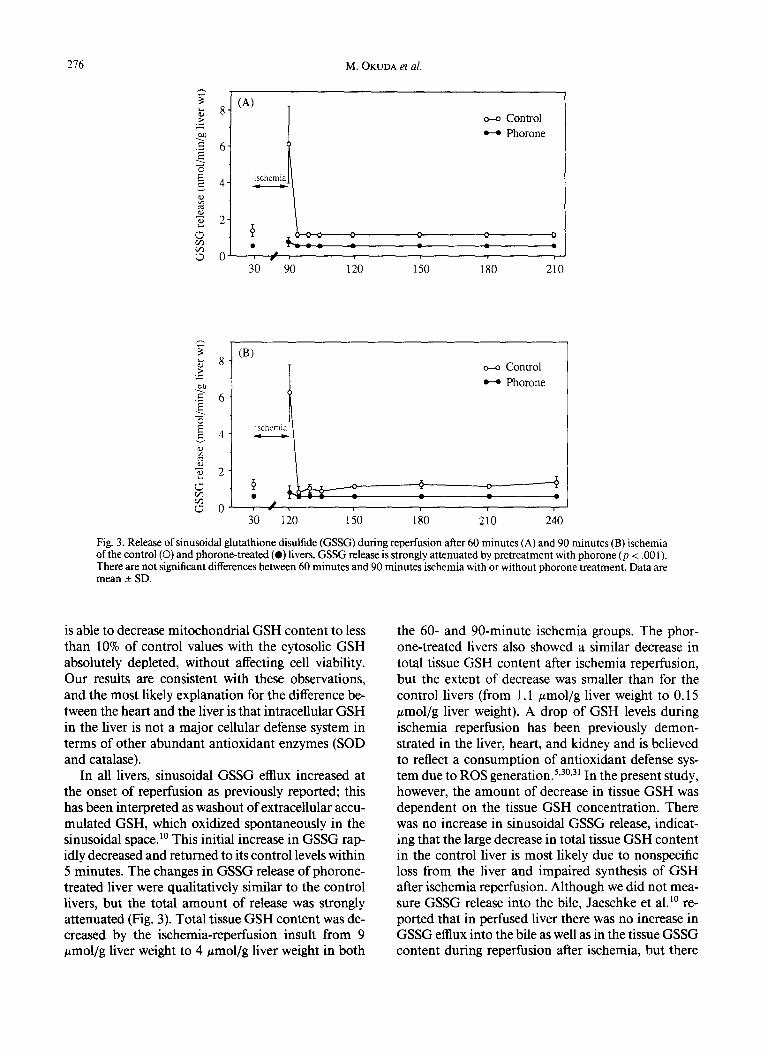
Changes in sinusoidal GSH and GSSG release are shown in Figures 2 and 3, respectively. Sinusoidal GSH and GSSG release increased only during the short washout period (30 seconds) at the onset of re- perfusion and returned to baseline levels soon thereaf- ter in both the 60- and 90-minute ischemia groups. In both 60- and 90-minute ischemia groups, GSH and GSSG release did not show any significant differences during the reperfusion period. GSH and GSSG re- lease by phorone-treated livers showed changes that were qualitatively similar to the livers, but its extent was significantly attenuated (p < .001).
Changes in LDH and TBARS release are shown in Figures 4 and 5, respectively. Both LDH and TBARS release showed a large increase only during the short washout period (30 seconds) and decreased rapidly thereafter during the first 15 minutes of reperfusion. After 15 minutes of reperfusion, LDH and TBARS release increased gradually. During the reperfusion pe-
riod, LDH and TBARS release were significantly higher than the preischemic level in all four groups (p < .05). When compared with the control and phor- one-treated livers, no significant differences were pres- ent throughout the study in both the 60- and 90-min- ute ischemia groups.
DISCUSSION
The main observation arising from the present study is that GSH depletion does not increase intra- cellular ROS generation (evaluated by lucigenin-en- hanced chemiluminescence), liver cell damage (evalu- ated by LDH release), or lipid peroxidation (evalu- ated by TBARS release) during ischemia reperfusion in the isolated blood-free perfused rat liver. Concern- ing lucigenin-enhanced chemiluminescence, our ap- proach is based on the assumption that lucigenin competes with endogenous defense substances (SOD, catalase, GSH, vitamin E) to react with ROS and emit light. Therefore, chemiluminescence would be af-

Glutathione and liver ischemia 275
._>
L~
140
1201 (A)
100-
8o] 601 ischemia
40-
201, ~
0 : /
o--o Control Phorone
30 90 120 150 180 210
140
120~
-~ 100-
~ 80
~ 60
~ 40
-v 20
(B)
ischemia
o-o Control ~-4 Phorone
° . ~ - - o - - - - - - - ~ ff ~
30 120 150 180 210 240
Fig. 2. Release of sinusoidal glutathione (GSH) during reperfusion after 60 minutes (A) and 90 minutes (B) ischemia of the control (O) and phorone-treated (O) livers. GSH release is strongly attenuated by pretreatment with phorone (p < .001). There are not significant differences between 60 minutes and 90 minutes ischemia with or without phorone treatment. Data are mean _+ SD.
fected both by the amount of generation of ROS and the status of endogenous defense systems. One would expect that decreased endogenous defense systems (e.g., GSH depletion) would cause increased chemilu- minescence during oxidative stress. In the present study, GSH depletion was accomplished to 10% of control, which is consistent with previous reports . 25 If intracellular GSH constitutes the large part of the cellular defense system, the chemiluminescence re- sponse would be affected significantly by 90% deple- tion of GSH. However, the present results demon- strate that responses in lucigenin-enhanced chemilu- minescence caused by ischemia-reperfusion insults were not affected by 90% GSH depletion.
In the isolated perfused rat heart, Barsacchi et al. 3 demonstrated that the GSH depletion caused an in- crease in chemiluminescence associated with an in- crease of malondialdehyde content in the heart and an impairment of heart function. These observations may indicate that the decreased GSH levels rendered the organ susceptible to the oxidative stress. In the
report of Barsacchi et al., however, phorone was in- fused directly to the heart. Higher doses of phorone often lead to death of the animals. Therefore, the di- rect toxic effect of phorone itself may be responsible for the impaired heart functions and increased lipid peroxidation. Another possible explanation for the difference between the liver and the heart is that the content of intracellular defense substances other than GSH (SOD and catalase) in myocytes is less than in hepatocytes. 26,27 Further, Romero and R o m ~ , 2s re-
viewing the effects of GSH depletion of the liver and the heart that were previously reported, postulated that the mitochondrial GSH pool in the heart is smaller than that in the liver. They also pointed out that hepatic GSH depletion per se promotes neither lipid peroxidation nor increases in chemilumines- cence except in phenobarbital-treated animals, where cytochrome P450 induction was complete. 22 Romero a n d Sies 29 reported that a combined phorone-BSO (L- buthionine sulfoximine, an inhibitor of y-glutamyl- cystein synthetase) treatment of isolated hepatocytes
276 M. OKUDA et al.
._>
E
E
O
¢D
(A) 8
6
4
2
0
ischemia
o
,, ,
30 90 120
o-o Control
Phorone
i
150 180 210
8 .>_
• E 6
E 8 4
~ 2
r ~
~ o
(B)
ischemia
30 120 15O
o---o Control
Phorone
6 - - w - -
, i |
180 210 240
Fig. 3. Release of sinusoidal glutathione disulfide (GSSG) during reperfusion after 60 minutes (A) and 90 minutes (B) ischemia of the control (O) and phorone-treated (e) livers. GSSG release is strongly attenuated by 10retreatment with phorone (p < .001). There are not significant differences between 60 minutes and 90 minutes ischemia with or without phorone treatment. Data are mean _+ SD.
is able to decrease mitochondrial GSH content to less than 10% of control values with the cytosolic GSH absolutely depleted, without affecting cell viability. Our results are consistent with these observations, and the most likely explanation for the difference be- tween the heart and the liver is that intracellular GSH in the liver is not a major cellular defense system in terms of other abundant antioxidant enzymes (SOD and catalase).
In all livers, sinusoidal GSSG efflux increased at the onset of reperfusion as previously reported; this has been interpreted as washout of extracellular accu- mulated GSH, which oxidized spontaneously in the sinusoidal space2 ° This initial increase in GSSG rap- idly decreased and returned to its control levels within 5 minutes. The changes in GSSG release of phorone- treated liver were qualitatively similar to the control livers, but the total amount of release was strongly attenuated (Fig. 3). Total tissue GSH content was de- creased by the ischemia-reperfusion insult from 9 #mol/g liver weight to 4 #mol/g liver weight in both
the 60- and 90-minute ischemia groups. The phor- one-treated livers also showed a similar decrease in total tissue GSH content after ischemia reperfusion, but the extent of decrease was smaller than for the control livers (from 1.1 ~mol/g liver weight to 0.15 gmol/g liver weight). A drop of GSH levels during ischemia reperfusion has been previously demon- strated in the liver, heart, and kidney and is believed to reflect a consumption of antioxidant defense sys- tem due to ROS generation) '3°'3~ In the present study, however, the amount of decrease in tissue GSH was dependent on the tissue GSH concentration. There was no increase in sinusoidal GSSG release, indicat- ing that the large decrease in total tissue GSH content in the control liver is most likely due to nonspecific loss from the liver and impaired synthesis of GSH after ischemia reperfusion. Although we did not mea- sure GSSG release into the bile, Jaeschke et al. ~° re- ported that in perfused liver there was no increase in GSSG effiux into the bile as well as in the tissue GSSG content during reperfusion after ischemia, but there

Glutathione and liver ischemia 277
,.d
500
400
300
200
100
0
o-o Control
H Phorone
(g)
ischemia
? , / ,
30 90 120 150 180 210
.... 500'
> 400'
~. 300
~ 200
"~ 100
0
(B)
ischemia
o---o Control H Phorone
30 120 150 180 210 240
Fig. 4. Release of hepatic lactate dehydrogenase (LDH) during reperfusion after 60 minutes (A) and 90 minutes (B) ischemia of the control (O) and phorone-treated (e) livers. Data are mean _+ SD.
was a large increase in GSH effiux in perfusate during the initial period of reperfusion after 120 minutes of ischemia. They suggested that increased GSH ettlux represents nonspecific leakage from hepatocytes and that there is little, if any, dependence on increased intracellular ROS generation.
There was no increase in sinusoidal GSSG release during reperfusion after various times ofischemia (60 or 90 minutes). Nevertheless, lucigenin-enhanced che- miluminescence detected increased ROS generation during ischemia reperfusion in the same liver. Usually, spontaneous chemiluminescence responses can only be seen under severe oxidative stress, such as the administration of hydrogen peroxide or mena- dione. 32'33 It was not possible to detect ROS genera- tion as a spontaneous chemiluminescence response during hepatic ischemia/reperfusion (data not shown), and responses could only be observed in the presence of enhancers. Therefore, lucigenin-en- hanced chemiluminescence can detect extremely low levels of ROS in tissue. On the other hand, severe oxidative stress, such as the administration of hydro- gen peroxide, causes significant GSSG f o r m a t i o n 7'1°
that is accompanied by a very large spontaneous che- miluminescence response. 32 Taken together, the pres- ent results indicate that the GSH system is not active enough to scavenge the low level of ROS produced during the ischemia-reperfusion insult.
However, in the in situ model, the selective in- crease of the GSSG concentration in plasma during reperfusion after ischemia and the extent of liver cell damage was more severe than seen in the isolated per- fused liver.ll'12 Animals pretreated with the glutathi- one reductase inhibitor 1,3-bis(2-chloroethyl)-l-ni- troso-urea (BCNU) and subjected to hepatic ischemia reperfusion did not show any significant changes of plasma, bile, and tissue concentrations of GSSG or enhanced liver cell damage compared with postisch- emic controls, but severe liver cell damage occurred in animals treated with phorone in the same series of experiments: Also, hepatic GSH depletion by diethyl malate increases ischemia-reperfusion injury in in situ rat2 The fiver is a major organ for the synthesis of GSH, which is subsequently released into both blood and bile. 2 Therefore, the administration of phorone causes GSH depletion in the liver and also in the

278 M. OKUDA et al.
-6
rv" <
300
200
100
0
(A)
ischcrnia
o-o Control Phorone
i ~ , D , , i
30 90 120 150 180 210
300
~ 2oo- 0
& c¢:
100"
< ~- 0
(B) ~ o-.o Control J
i s c h e m i ~ .
s 30 120 150 180 210 240
Fig. 5. Release of hepatic thiobarbituric acid reactive substance (TBARS) during reperfusion after 60 minutes (A) and 90 minutes (B) ischemia of the control ((3) and phorone-treated (O) livers. Data are mean _+ SD.
plasma. 4 These observations lead to the conclusion that in the in situ model, the oxidation of GSH most likely takes place extracellularly in the hepatic sinu- soids, and hepatic GSH serves to limit ischemia-re- perfusion injury as a source of extracellular GSH but not as a major factor in intracellular defense against ROS. However, Strubelt et al . 34 demonstrated that GSH depletion enhanced ethanol-induced acute hepa- totoxicity in the isolated perfused liver. This report, along with the present observation, suggests that the hepatic GSH would become important in severe oxi- dative stress.
Leukocyte and tissue fixed macrophage (e.g., Kupffer cells) are both known to generate and release reactive oxygen species, and these cells have been shown to be potential candidates for induction of ex- traceUular oxidant stress and to contribute signifi- cantly to ischemia-reperfusion injury in the l i v e r . 35'36
The present study confirms the observation that oxi- dation of GSH does not occur during ischemia/reper- fusion in the isolated perfused liver to any significant extent, and it offers additional support to assigning an important role to the leukocyte and Kupffer cells in ischemia-reperfusion injury.
In conclusion, hepatic GSH depletion per se does not increase lucigenin-enhanced chemiluminescence during reperfusion after global ischemia. This result indicates that intracellular GSH does not constitute a large part of cellular defense systems against low levels of ROS. Both the lack of GSSG formation during isch- emia/reperfusion and the significant response in luci- genin-enhanced chemiluminescence in normal liver indicate that the GSH system is not sensitive to low- level ROS generation. However, under conditions where significantly large quantities of ROS may be produced, such as during activation of neutrophils, drug toxicities, etc., GSH oxidation may play an im- portant role in protecting the tissue against ROS-in- duced tissue damage.
Acknowledgement - - This work was supported in part by NIH grants HL 18708 and AA 01876.
REFERENCES
1. McCord, J. M. Oxygen-derived free radicals in post-ischemic tissue injury. N. Engl. J. Med. 312:159-163; 1985.
2. Kaplowitz, N.; Aw, T. Y.; Ookhtens, M. The regulation of he-

Glutathione and liver ischemia 279
patic glutathione. Annu. Rev. Pharmacol. ToxicoL 25:715- 744; 1985.
3, Barsacchi, R.; Pelosi, G.; Camici, P.; Bonaldo, L.; Maiorino, M.; Ursini, F. Glutathione depletion increases chemilumines- cence emission and lipid peroxidation in the heart. Biochim. Biophys. Acta 804:356-360; 1984.
4, Jaeschke, H. Vascular oxidant stress and hepatic ischemia/re- perfusion injury. Free. Rad. Res. Comms. 12:737-743; 1991.
5, Stein, H. J.; Oosthuizen, M. M. J.; Hinder, R. A.; Lamprechts, H. Oxygen free radicals and glutathione in hepatic ischemia/re- perfusion injury. J. Surg. Res. 50:398-402; 1991.
6. Akerboom, T. P. M.; Bilzer, M.; Sies, H. The relationship of biliary glutathione disulfide efflux and intracellular glutathione disulfide content in perfused rat liver. J. BioL Chem. 257:4248-4252; 1982.
7. Adams, J. D., Jr.; Lauterburg, B. H.; Mitchell, J. R. Plasma glutathione and glutathione disulfide in the rat: Regulation and response to oxidative stress. J. Pharmacol. Exp. Ther. 227:749- 754; 1983.
8. Jaeschke, H.; Mitchell, J. R. Use of isolated perfused organs in hypoxia and ischemia/reperfusion oxidant stress. Methods in EnzymoL 186:752-759; 1990.
9. Jaeschke, H. Glutathione disulfide as index of oxidant stress in rat liver during hypoxia. Am. J. Physiol. 258:G499-G505; 1990.
10. Jaeschke, H.; Smith, C. V.; Mitchell, J. R. Reactive oxygen species during ischemia-reflow injury in isolated perfused rat liver. J. Clin. Invest. 81:1240-1246; 1988.
11. Jaeschke, H.; Smith, C. V.; Hughes, H.; Mitchell, J. R. No evidence for oxidative hepatic damage during ischemia/reflow injury. The Pharmacologist 29:425; 1987.
12. Mitchell, J. R.; Smith, C. V.; Hughes, H.; Lenz, M. L.; Jaeschke, H.; Shappell, S. B.; Mitchael, L. H.; Entman, M. L. No evidence for reactive oxygen damage in ischemia-reflow injury. Trans. Assoc. Am. Physicians 100:54-61; 1987.
13. Okuda, M.; Ikai, I.; Chance, B.; Kumar, C. Oxygen radical production during ischemia-reperfusion in the isolated per- fused rat liver as monitored by luminol enhanced chemilumi- neseence. Biochem. Biophys. Res. Commun. 174:217-221; 1991.
14. Okuda, M.; Ikai, I.; Chance, B.; Kumar, C. Superoxide dismu- tase, but not catalase or mannitol, decreased oxygen radical formation in peffused rat heart during ischemia-reperfusion as monitored by luminol enhanced chemiluminescence. J. Appl. CardioL 6:113-121; 1991.
15. Archer, S. L.; Nelson, D. P.; Weir, E. K. Simultaneous measure- ment of O2 radicals and pulmonary vascular reactivity in rat lung. J. AppL Physiol. 67:1903-1911; 1989.
16. Archer, S. L.; Nelson, D. P.; Weir, E. K. Detection of activated 02 species in vitro and in rat lungs by chemiluminescence. J. AppL Physiol. 67:1912-1921; 1989.
17. Henry, T. D.; Archer, S. L.; Nelson, D. P.; Weir, E. K.; From, A. H. L. Enhanced chemiluminescence as a measure of oxy- gen-derived free radical generation during ischemia and reper- fusion. Circ. Res. 67:1453-1461; 1990.
18. Henry, T. D.; Archer, S. L.; Nelson, D. P.; Weir, E. K.; From, A. H. L. Effect of anion channel blockade on oxygen radical induced chemiluminescence in postischemic myocardium. Circulation 80(Suppl. I1):II-31; 1989.
19. Kumar, C.; Ikai, I.; Okuda, M.; Chance, B. Chemically en- hanced chemiluminescence studies as applied to perfused or- gans. FASEBJ. 5(Abstr. II):A1142; 1991.
20. Ross, D. Glutathione, free radicals and chemotherapeutic agents: Mechanism of free radical induced toxicity and glutathi- one dependent protection. Pharmacol. Ther. 37:231-249; 1988.
21. Reed, D. J. Regulation of reductive processes by glutathione. Biochem. PharmacoL 35:7-13; 1986.
22. Younes, M.; Siegers, C.-P. Mechanistic aspects of enhanced
lipid peroxidation following glutathione depletion in vivo. Chem.-Biol. Interactions 34:257-266; 1981.
23. Miller, L. L. Technique of isolated rat liver perfusion. In: Bar- tosek, I.; Guaitani, A.; Miller, L. L., eds. Isolated liver perfusion and its application. New York: Raven Press; 1973:11-52.
24. Yagi, K. A simple fluorometric assay for lipoperoxide in blood plasma. Biochem. Med. 15:212-216; 1976.
25. Plummer, J. L.; Smith, B. R.; Sies, H.; Bend, J. R. Chemical depletion of glutathione in vivo. Methods in Enzymol. 77:50- 59; 1981.
26. Halliwell, B.; Gutteridge, J. M. C., eds. Free radical in biology and medicine. 2nd ed. Oxford: Clarendon Press; 1989.
27. Marklund, S. Distribution ofCuZn superoxide dismutase and Mn superoxide dismutase in human tissues and extracellular fluids. Acta Physiol. Scand. SuppL 492:19-23; 1980.
28. Romero, F. J.; Rom~, J. Careful consideration of the effects induced by glutathione depletion in rat liver and heart. The involvement of cytosolic and mitochondrial glutathione pools. Chem.-BioL Interactions 70:29-37; 1989.
29. Romero, F. J.; Sies, H. Subcellular glutathione contents in iso- lated hepatocytes treated with L-buthionine sulfoximine. Bio- chem. Biophys. Res. Commun. 123:1116-1121; 1984.
30. Curello, S.; Ceconi, C.; Bigoli, C.; Ferrari, R.; Albertini, A.; Guarnieri, C. Changes in cardiac glutathione status after isch- emia and reperfusion. Experientia 41:42-43; 1984.
31. Linas, S. L.; Shanley, P. F.; White, C. W.; Parker, N. P.; Re- pine, J. E. 02 metabolite-mediated injury in perfused kidneys is reflected by consumption of DMTU and glutathione. Am. J. PhysioL 253:F692-FT01; 1987.
32. Cadenas, E.; Boveris, A.; Chance, B. Low-level chemilumines- cence of biological systems. In: Pryor, W. A., ed. Free radicals in biology. Vol. VI. New York: Academic Press; 1984:211-242.
33. Wefers, H.; Sies, H. Hepatic low-level chemiluminescence dur- ing redox cycling of menadione and the menadione-glutathi- one conjugate: Relation to glutathione and NAD(P)H:quinone reductase (DT-diaphorase) activity. Arch. Biochem. Biophys. 224:568-578; 1983.
34. Strubelt, O.; Younes, M.; Pentz, R. Enhancement by glutathi- one depletion of ethanol-induced acute hepatotoxicity in vitro and in vivo. Toxicology 45:213-223; 1987.
35. Jaeschke, H,; Farhood, A.; Smith, C. W. Neutrophils contrib- ute to ischemia/reperfusion injury in rat liver in vivo. FASEB J. 4:3355-3359; 1990,
36. Jaeschke, H.; Farhood, A. Neutrophil and Kupffer cell-in- duced oxidant stress and ischemia-reperfusion injury in rat liver. Am. ,L PhysioL 260:G355-G362; 1991.
ABBREVIATIONS
BCNU-- 1,3-bis(2-chloroehyl)- l-nitroso-urea cps--count per second DMSO--dimethyl sulfoxide DTNB--5,5'-dithio-bis-2-nitrobenzoic acid EDTA--ethylenediaminetetraacetic acid GSH--glutathione GSSG--glutathione disulfide ip--intraperitoneal LDH--lactate dehydrogenase NADPH--nicotinamide adenine dinucleotide phos-
phate (reduced form) NEM--N-ethylmaleimide ROS--reactive oxygen species SD--standard error SOD--superoxide dismutase TBARS--thiobarbituric acid reactive substances wt--weight