genome-wide dna methylation assays nadia khan, rick smith, and anna kuperman epigenetics 2012
TRANSCRIPT

Genome-Wide DNA Methylation Assays
Nadia Khan, Rick Smith, and Anna KupermanEpigenetics 2012

Introduction• Most Genome Wide Approaches
were adapted from technologies originally developed for detecting methylation at the level of a single gene
• Advantages of a Genome Wide Approach– Scale of information
• Whole chromosome• Whole genome
– Wider regulatory networks– Facilitates comparative and
population level analysis
Tollefsbol 2009
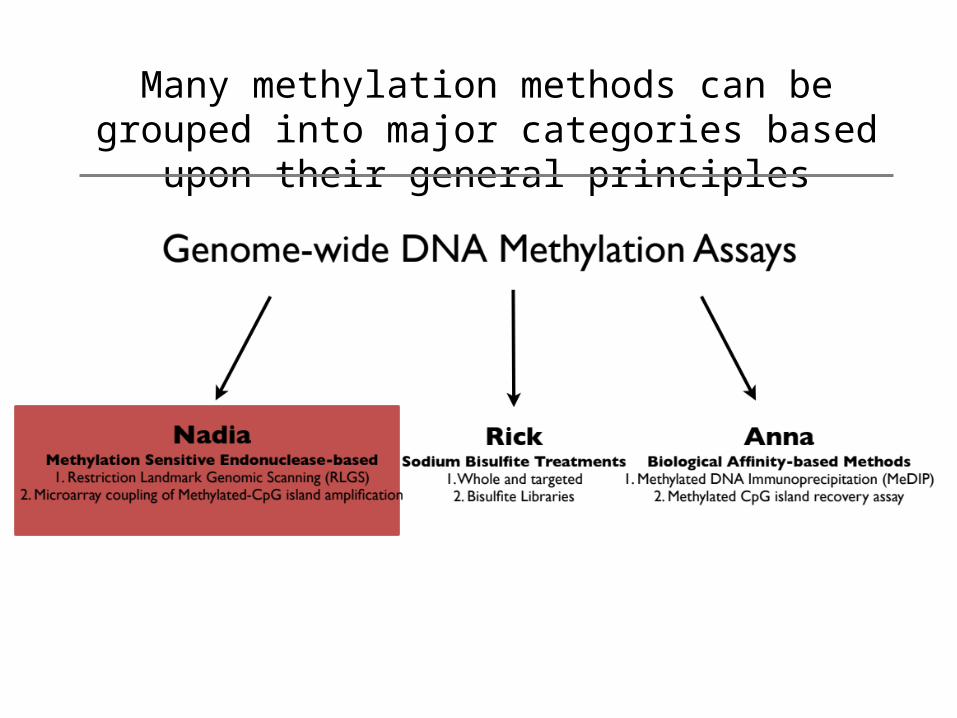
Many methylation methods can be grouped into major categories based upon their general principles

Concept of Methylation Sensitive Endonuclease Assays
• Use a restriction enzyme(s) that is methylation specific and separate the unmethylated from the methylated
• Unmethylated regions enzyme-sensitive Methylated regions enzyme-resistant
• Identify multiple de novo methylated areas across genomes vs. one specific area– 2D gel and scintillation counting– Array
Laird 2010

Restriction Landmark Genomic Scanning (RLGS)
• Cleave genome into pieces based upon restriction landmarks (sites)
• Radioactively label cleaved ends• Separate using 1D and 2D gel electrophoresis• Quantitate signal by amount of fluoresence in
gel– Intensity = copy number of the restriction site
Tollefsbol 2009

• NotI:• Radioactively
labels ends• LEAST specific
• EcoRV: • MORE specific
• HinfI:• MOST specific
Eng et al. 2000

RLGS in-use
Takamiya, et al. 2009

Microarray Coupling of Methylated CpG Island Amplification (MCAM)
• Cleave genomic DNA with SmaI (methylation-sensitive)
• Cut received pieces again with XmaI; create sticky ends
• Amplify pieces using PCR• Hybridize onto a microarray• Analyze fluorescence reads and identify
corresponding genomic address
Tollefsbol 2009

Tollefsbol 2009

MCA
M in
-use
Estecio 2007


Sodium Bisulfite Methods
• Bisulfite Sequencing• Targeted and Whole Genome approaches• Bisulfite Libraries
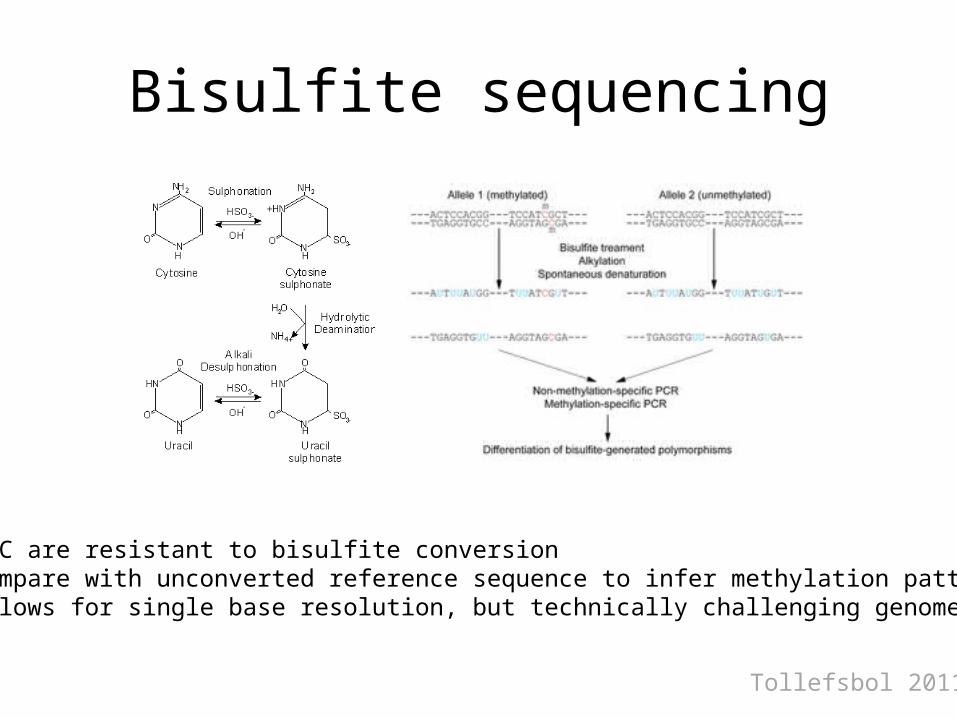
Bisulfite sequencing
• m-C are resistant to bisulfite conversion• Compare with unconverted reference sequence to infer methylation pattern• Allows for single base resolution, but technically challenging genome wide
Tollefsbol 2011

Whole Genome Approach
• Pair Bisulfite Conversion with Next Generation Sequencing (NGS)– Massively parallel sequencing• Roche 454, Illumina, SOLiD platforms• Quick, relatively cheap, large scale analysis
Tollefsbol 2011

Pyrosequencing

Whole Genome Approach
• Pair Bisulfite Conversion with Next Generation Sequencing (NGS)– Massively parallel sequencing
• Roche 454, Illumina, SOLiD platforms• Quick, relatively cheap, large scale analysis
• Tenable for relatively small genomes– Arabidopsis thaliana
• Significant challenges for mammalian genomes– Reduced complexity of the genome– Short sequence reads– Solutions
• Longer sequence reads• Targeted approaches
Cokus et al. 2008; Tollefsbol 2011

Targeted Bisulfite Sequencing
• Reduced Representation Bisulfite Sequencing (RRBS)
• Molecular Inversion Probes (MIP)– Padlock Probes

Targeted Bisulfite Sequencing
• Reduced Representation Methylation Sequencing (RRMS) – Enrichment for CG-rich regions via Msp1 digestion
(5‘-CCGG-3‘)– NGS– Disadvantages: mostly un-methylated DNA
Meissner 2005; Jeddeloh 2008

Targeted Bisulfite Sequencing
Deng et al. 2009
Padlock Probes

Bisulfite Libraries• Applications and Advantages• Coverage of relevant genome regions • Facilitates large comparative study• Multiplex Sequencing• High sensitivity• Whole library amplification
• Pair with NGS or Array
Gu et al. 2011


Biological Affinity Based Methods• Basic Concept: Use antibodies that are specific
for 5meC or proteins that bind preferably to methylated genomic DNA to profile DNA methylation patterns. These patterns are detected through microarrays or parallel high through-put sequencing.
• Types– MBD affinity column (MAC)– Methylated DNA Immunoprecipitation (MeDIP)– Methylated-CpG island recovery assay
Laird 2010
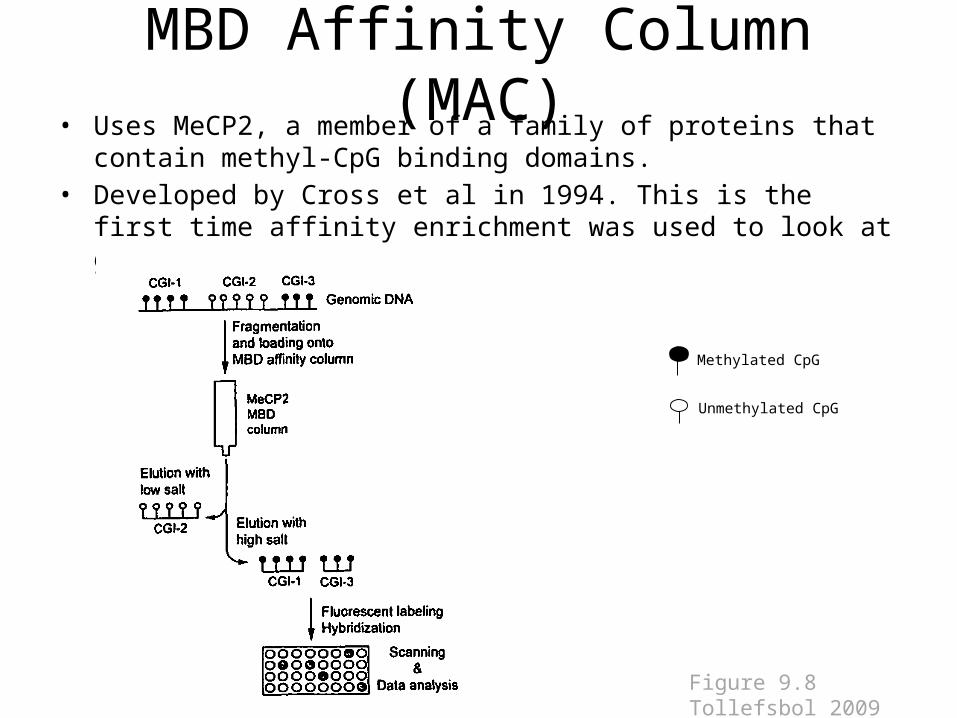
MBD Affinity Column (MAC)• Uses MeCP2, a member of a family of proteins that contain methyl-CpG
binding domains. • Developed by Cross et al in 1994. This is the first time affinity enrichment
was used to look at genome-wide methylation
Figure 9.8 Tollefsbol 2009
Methylated CpG
Unmethylated CpG

MBD Affinity Column (MAC)
• Advantages: this method is fast and efficient.• Limitations: Needs a large amount of starting
genomic DNA to pass through column purification
Figure 9.8 Tollefsbol 2009

Methylated DNA Immunoprecipitation (MeDIP)
• Introduced in 2005 by Weber et al• Uses an antibody that specifically binds to methylated
cytosines.• Fragmented DNA is incubated with the antibodies,
immunoprecipitated, and then enrichment is quantified.• Advantages: Efficient, sensitive, large-scale analysis of
genomic methylation• Limitations: need good quality 5meC antibodies and
denatured ssDNA, which can be difficult to obtain in CpG rich genes, is required for analysis
Figure 9.8 Tollefsbol 2009
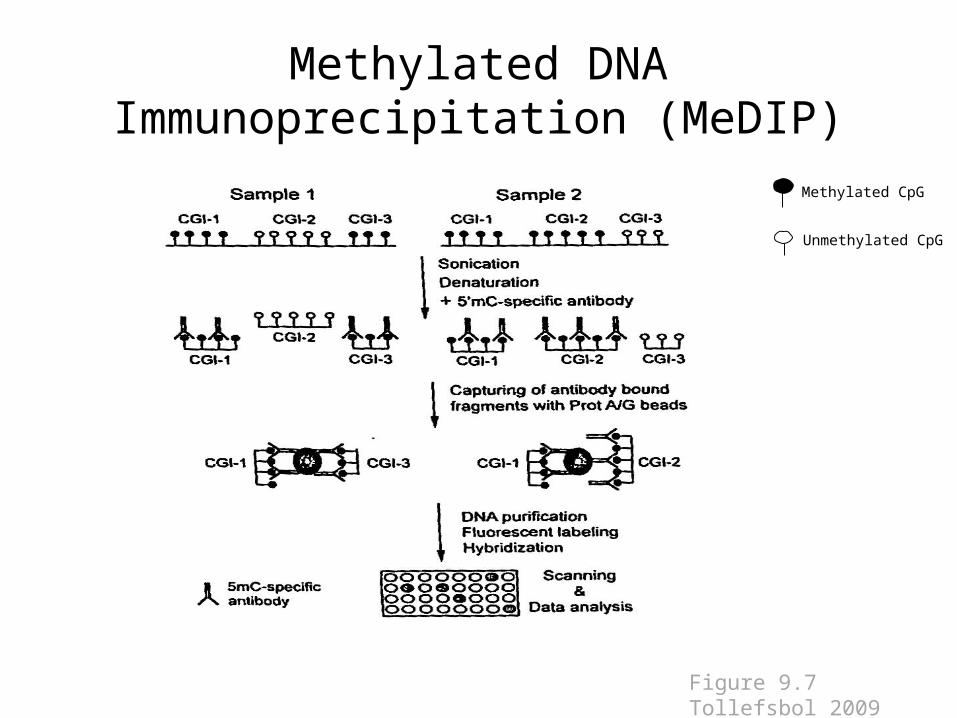
Methylated DNA Immunoprecipitation (MeDIP)
Methylated CpG
Unmethylated CpG
Figure 9.7 Tollefsbol 2009

MeDIP• How can it be used?– Identifying genes involved
in cancer development• Ex: Morris et al were able
to shortlist genes involved in renal cell carcinoma (RCC) suppression by looking at promoter regions that were frequently methylated in RCC lines, but not in normal kidney cell lines.
Morris et al. 2011

Methylated-CpG Island Recovery Assay (MIRA)
• MBD2b/MBD3L1 complex has a high affinity to methylated DNA (higher than MBD2b on it’s own; MBD3L1 has no affinity)
• MIRA developed in 2006 by Rauch et al to have a better screen for methylation patterns in lung cancer tumors so can have a better early detection
• Advantages: Can be used to examine large number of genes simultaneously, works on dsDNA, only need a few hundred nanograms of genomic DNA
Figure 9.7 Tollefsbol 2009

Methylated-CpG Island Recovery Assay (MIRA)
Figure 9.9 Tollefsbol 2009From activemotif.com

MIRA• Rauch et al (2006)
were able to identify lung tumor suppressor genes
• Rauch et al (2009) were able to use MIRA to characterize a human B-cell methylome at 100 bp resolution
Rauch 2006

• A lot of these assays are commercially available
• MeDIP– MagMeDIP Kit TM (Diagenode), – Methylated-DNA IP Kit (Zymo Research) and
Methylamp™ – Methylated DNA Capture Kit (Epigentek)
• MIRA– Ex: MethylCollector TM Ultra

Biological Affinity Assays
• Why are they good? – Quick and efficient genome-wide assessment of
DNA methylation• Disadvantages: – Do not give information on individual CpG
dinucleotides– Require experimental or bioniformatic adjustment
for changing CpG density at different regions of genome
Laird 2010

Complications with 5-hydroxymethylcytosine
• 5-hydroxymethylcytosine has been discovered in mammalian DNA, and is produced by an enzymatic pathway involving TET1 hydroxylase.
• All 3 methods discussed are unable to distinguish between 5mC and 5hmC.
• However, one can distinguish 5-hmc by adding a glucose to the hydroxy-group (EpiMark Kit)
Tollefsbol 2009

The Future of Genome-Wide Methylation Assays
• As more data is experimentally collected about the methylome, there will be more and more need for analysis. Bioinformatics is beginning to play a big role.
• Increasing role of sequencing as opposed to arrays.
• Nanopore sequencing could directly allow sequencing of 5meC

ReferencesCross SH et al. 1994. Purification of CpG islands using a methylated DNA binding column. Nat. Genet. 6(3):236-44.Deng J et al. 2009. Targeted bisulfite sequencing reveals changes in DNA methylation associated with nuclear reprogramming. Nat
Biotechnol. 27(4):341-2.Estecio MRH et al. 2007. High-throughput Methylation Profiling by MCA Coupled to CpG Island Microarray. Genome
Research 17(10): 1529-536.Gu H et al. 2011. Preparation of reduced representation bisulfite sequencing libraries for genome-scale DNA methylation profiling.
Nature Protocols 6:468–481.Jeddeloh JA et al. 2008. Reduced-representation methylation mapping. Genome Biology 9:231.Laird PW. 2012. Principles and Challenges of Genome-wide DNA Methylation Analysis. Nature Reviews Genetics 11:191-203.Meissner A et al. 2005. Reduced representation bisulfite sequencing for comparative high-resolution DNA methylation analysis.
Nuc. Acids Res. 33(18):5868-5877.Morris MR et al. 2011. 6.Genome-wide methylation analysis identifies epigenetically inactivated candidate tumour suppressor
genes in renal cell carcinoma. Oncogene 30(12):1390-401.Rauch TA and Pfeifer GP. 2009. Chapter 9: Methods for Assessing Genome Wide DNA methylation. In: Handbook of Epigenetics :
The New Molecular and Medical Genetics. ed. Tollefsbol T. Academic Press.Rauch TA et al. 2006. MIRA-assisted microarray analysis, a new technology for the determination of DNA methylation
patterns, identifies frequent methylation of homeodomain-containing genes in lung cancer cells. Cancer Res. 66(16)7939-47.Rauch TA et al. 2009. A human B cell methylome at 100 base pair resolution. Proc. Natl. Acad. Sci. 106(3):671-8.Takamiya et al. 2009. The Application of Restriction Landmark Genome Scanning Method for Surveillance of Non-
Mendelian Inheritance in F1 Hybrids. Comparative and Functional Genomics 2009: 1-7.Tost J. 2009. Epigenetics. Caister Academic Press.Weber M et al. 2005. Chromosome-wide and promoter-specific analyses identify sites of differential DNA methylation in normal
and transformed human cells. Nat Genet. 37(8):853-62.