fast and cost-effective genetic mapping in apple … · 1 fast and cost-effective genetic mapping...
TRANSCRIPT

1
1
Fast and cost-effective genetic mapping in apple using next-generation sequencing 1 Kyle M. Gardner*, Patrick Brown§, Thomas F Cooke†, Scott Cann*, Fabrizio Costa‡, Carlos 2 Bustamante†, Riccardo Velasco‡, Michela Troggio‡ and Sean Myles* 3 4 * Department of Plant and Animal Sciences, Faculty of Agriculture, Dalhousie University, Nova 5 Scotia, Canada 6 § Department of Crop Sciences, University of Illinois, Urbana, Illinois, USA 7 † Department of Genetics, Stanford School of Medicine, Stanford University, Stanford, 8 California, USA 9 ‡ Genetics and Molecular Biology Department, IASMA Research Center, San Michele all'Adige, 10 Italy 11 12 Data available via the Dryad Digital Repository: http://doi.org/10.5061/dryad.55t54 13 14 15 Running title: Mapping apple skin color using GBS 16 Keywords: Next-generation DNA sequencing, Genotyping-By-Sequencing, apple, Malus, QTL, 17 SNP 18 19 20 *Corresponding author: 21 Sean Myles 22 Department of Plant and Animal Sciences 23 Faculty of Agriculture 24 Dalhousie University 25 Kentville, Nova Scotia 26 B2N 5E3 27 Canada 28 Phone: +1-902-365-8460 29 Fax: +1-902-679-2311 30 Email: [email protected] 31
G3: Genes|Genomes|Genetics Early Online, published on July 16, 2014 as doi:10.1534/g3.114.011023
© The Author(s) 2013. Published by the Genetics Society of America.

2
2
ABSTRACT 1 Next-generation DNA sequencing (NGS) produces vast amounts of DNA sequence data, 2 but it is not specifically designed to generate data suitable for genetic mapping. Recently 3 developed DNA library preparation methods for NGS have helped solve this problem, 4 however, by combining the use of reduced representation libraries with DNA sample 5 barcoding to generate genome-wide genotype data from a common set of genetic markers 6 across a large number of samples. Here we use such a method, called genotyping-by-7 sequencing (GBS), to produce a data set for genetic mapping in an F1 population of apples 8 (Malus x domestica) segregating for skin color. We show that GBS produces a relatively 9 large, but extremely sparse, genotype matrix: over 270,000 SNPs were discovered, but 10 most SNPs have too much missing data across samples to be useful for genetic mapping. 11 After filtering for genotype quality and missing data, only 6% of the 85 million DNA 12 sequence reads contributed to useful genotype calls. Despite this limitation, using existing 13 software and a set of simple heuristics, we generated a final genotype matrix containing 14 3967 SNPs from 89 DNA samples from a single lane of Illumina HiSeq and used it to 15 create a saturated genetic linkage map and to identify a known QTL underlying apple skin 16 color. We therefore demonstrate that GBS is a cost effective method for generating 17 genome-wide SNP data suitable for genetic mapping in a highly diverse and heterozygous 18 agricultural species. We anticipate future improvements to the GBS analysis pipeline 19 presented here that will enhance the utility of next-generation DNA sequence data for the 20 purposes of genetic mapping across diverse species. 21
22 23 24 25 26 27 28 29 30 31

3
3
INTRODUCTION 1 The introduction of new high-throughput DNA sequencing technologies has dramatically 2 reduced sequencing costs and increased the pace of genomics research. One of the primary goals 3 of genomics research is to establish relationships between genotypes and phenotypes. In 4 agriculture, such genotype-phenotype associations form the basis of genomics-assisted breeding 5 programs that aim to accelerate the breeding of improved varieties. Long-lived woody perennials 6 are expensive to breed using traditional methods and therefore stand to benefit more from the 7 genomics revolution than most other agricultural species. Offspring from breeding programs can 8 be genetically screened and discarded at the seedling stage without incurring the enormous 9 expense of growing them for years to fruit-bearing maturity for evaluation (BUS et al. 2000; 10 KELLERHALS et al. 2000; DIRLEWANGER et al. 2004; DI GASPERO AND CATTONARO 2010). 11 Most forms of genetic mapping require the collection of genome-wide polymorphism data, 12 where genotypes from a set of common loci are obtained from a set of samples. While next-13 generation DNA sequencing technologies produce vast amounts of DNA sequence data, they 14 were not designed to generate these types of genotype data. Recently, however, next generation 15 sequencing technologies have been coupled with reduced representation libraries and DNA 16 barcoding to simultaneously identify and genotype a common set of polymorphic loci across a 17 set of samples in a single experiment. The two most common of these methods include 18 Genotyping-By-Sequencing (GBS; ELSHIRE et al. 2011) and RADseq (BAIRD et al. 2008) and 19 the present study focuses on GBS. In addition to the low per sample cost, there are several 20 benefits to using sequence-based genotyping methods over microarray-based technologies 21 (MYLES 2013). For example, polymorphism discovery and genotyping is completed in a single 22 step, which not only saves time, but also reduces the ascertainment bias inherent in the process of 23 developing genotyping microarrays. Moreover, as reference genomes, alignment methods and 24 genotype calling algorithms improve, raw sequence data collected today will become more 25 valuable in the future as improved methods will enable more information to be extracted from 26 the original raw files. 27 Despite difficulties in experimental design, due to self incompatibility and high heterozygosity, 28 there are a wide variety of apple genetic maps constructed from bi-parental crosses. Most of 29 these linkage maps have been built with low throughput genetic markers such as microsatellites 30 (CELTON et al. 2009; FERNÁNDEZ-FERNÁNDEZ et al. 2012) and AFLPs (LIEBHARD et al. 2003; 31

4
4
KENIS AND KEULEMANS 2005) resulting relatively low marker density across assembled linkage 1 groups. Recently there has been a shift towards the high throughput identification of single 2 nucleotide polymorphisms (SNPs) in apple spurred on by decreasing DNA sequencing costs and 3 the availability of an apple (Malus x domestica, cultivar ‘Golden Delicious’) reference genome 4 (VELASCO et al. 2010). Chagne et al. (2012a) detail the creation of a SNP genotyping microarray 5 that assays 8,000 SNPs discovered from low coverage sequencing of 27 cultivars. To date, the 6 apple 8K SNP array has been used to create saturated linkage maps in bi-parental cross 7 populations (ANTANAVICIUTE et al. 2012) and to perform genomic selection (KUMAR et al. 2012) 8 and genome-wide association (KUMAR et al. 2013) in diverse breeding material. While SNP 9 arrays are widely used, the high levels of polymorphism in many agricultural species like apples 10 often results in unreliable or useless genotype calls because of highly variable probe-sequence 11 hybridization (e.g. MILLER et al. 2013). In addition, the ascertainment bias inherent in the design 12 of SNP genotyping microarrays results in only a small fraction of the queried loci being 13 polymorphic in any given bi-parental cross (MICHELETTI et al. 2011). For example, only about a 14 third of the SNPs on the apple 8K SNP array were observed to be polymorphic in a ‘Royal 15 Gala’בGranny Smith’ segregating population (CHAGNE et al. 2012a). 16 It is evident that GBS offers several advantages over competing technologies and is quickly 17 becoming the genotyping method of choice in many agricultural systems (POLAND AND RIFE 18 2012; MYLES 2013). For example, GBS has been recently used for a variety of applications 19 including saturating an existing genetic map in rice (SPINDEL et al. 2013); creating high density 20 genetic maps in wheat and barley (POLAND et al. 2012b); performing genomic selection in wheat 21 (POLAND et al. 2012a); ordering of a draft genome sequence in barley (CONSORTIUM 2012; 22 MASCHER et al. 2013a); and characterizing germplasm diversity in maize and switchgrass (LU et 23 al. 2013; ROMAY et al. 2013). Almost all GBS studies to date have focussed on inbred lines, 24 because genotype calling in highly heterozygous crops using next-generation DNA sequence 25 data requires more data and is far more complicated. The present study addresses this issue by 26 presenting a pipeline for GBS SNP calling in apples and follows recently published work on 27 GBS workflows developed for other heterozygous crops like grape (BARBA et al. 2013) and 28 raspberry (WARD et al. 2013). Using a single lane of Illumina HiSeq data, we identify a robust 29 set of SNPs and use them to generate a saturated genetic linkage map of the apple genome and 30 map a major QTL for apple skin colour in an F1 population. 31

5
5
MATERIALS AND METHODS 1 2 Population description and phenotyping 3 The ‘Golden Delicious x Scarlet Spur’ population investigated here is planted at the experimental 4 orchard of the Foundation Edmund Mach (FEM) in San Michele all’Adige, Italy. Each individual 5 progeny is represented by a single tree, grafted on M9 rootstock and planted in 2003. The 6 population has been grown and maintained following standard agronomical practice for fruit 7 thinning, canopy pruning, chemical fertilization and disease control. Due to the large variation in 8 ripening time among progeny, phenotyping was repeated three times during the harvesting 9 season. Skin color was scored as a binary trait: trees had apples with either yellow/green skin 10 (like Golden Delicious) or apples with red skin (like Scarlet Spur). 11 12 GBS library construction 13 GBS libraries were constructed using the two-enzyme modification of the original GBS protocol 14 (ELSHIRE et al. 2011; POLAND et al. 2012). DNA was extracted using commercial extraction kits. 15 Restriction/ligation reactions were performed in 96-well plates using 500 ng of DNA from each 16 individual, digestion with HindIII-HF and MspI (New England Biolabs, Ipswich, MA), and 17
0.1M and 10M of A1 and A2 adapters per well, respectively. Libraries were pooled, size-18
selected on a 1% agarose gel, column-cleaned using a PCR purification kit (Qiagen, Valencia, 19 CA), and amplified for 12 cycles using Phusion DNA polymerase (NEB). Average fragment size 20 was estimated on a Bioanalyzer 2100 (Agilent, Santa Clara, CA) using a DNA1000 chip 21 following a second column-cleaning, and library quantification was performed using PicoGreen 22 (Invitrogen, Carlsbad, CA). Pooled libraries were adjusted to 10 nmol and sequenced with 100 23 bp, single-end reads on the HiSeq2000 (Illumina, San Diego, CA). 24 25 SNP calling 26 We created a custom bioinformatics pipeline, employing custom python scripts and existing 27 software, to process raw GBS sequence data from a single lane of an Illumina HiSeq sequencer 28 into SNP genotype tables (Figure S1). DNA barcode deconvolution and basic sequence quality 29 filtering was carried out using a custom python program (barcode_splitter.py). This program 30 splits the raw Illumina fastq file into 96 separate fastq files based on the barcode sequences 31

6
6
associated with each sample while filtering out reads containing any ambiguous bases in the 1 barcodes or restriction site remnants immediately following the barcode sequence. All sequences 2 successfully passing these basic filters were then scanned for the presence of an additional 3 restriction site remnant and, if present, the read was trimmed accordingly. Reads were also 4 trimmed if the sequence contained the common (A2) GBS adapter, indicating that the genomic 5 fragment sequenced was less than 90-100 bp in size. 6 The separate fastq files for each DNA sample were then aligned to version 1.0 of the apple 7 reference genome (VELASCO et al. 2010) using bwa (LI AND DURBIN 2009), allowing a maximum 8 of 4% sequence mismatch. Alignments were converted to the SAM format, then merged and 9 sorted into one master binary alignment file (BAM format) with SAMtools 0.1.18. (LI et al. 10 2009). SNP calling was carried out using the genome analysis toolkit (MCKENNA et al. 2010) on 11 the BAM file using a minimal set of filters that required a called SNP to have a locus quality 12 score of at least 30 given a prior probability of heterozygosity of 0.01. 13 14 SNP filtering and segregation analysis 15 SNP calls were filtered for quality by restricting the marker set to biallelic SNPs, requiring 16 genotype calls at each SNP to have a depth of coverage of six reads in each sample, 17 implementing a minor allele frequency threshold of ≥0.2, and limiting missing genotype data to a 18 maximum of 20% per SNP. All filtering was carried out using vcftools 0.1.10 (DANECEK et al. 19 2011) and the final SNP genotype tables were output into PLINK format (PURCELL et al. 2007). 20 The final SNP table contained 3967 SNPs and all of these SNPs were used to map apple color 21 using a simple chi-squared test (see below). The construction of the genetic map, however, 22 required further filtering based on the segregation pattern of the genotypes in parents and 23 offspring. 24 Linkage mapping in apple F1 populations is referred to as a double pseudo-testcross and only 25 three genotype combinations in the parental lines are informative for the construction of a 26 genetic map: when one parent is heterozygous and the other is homozygous (i.e. either AA x AB 27 or AB x AA) and when both parents are heterozygous (i.e. AB x AB). Only SNPs following 28 these segregation patterns in the parents were retained. Each SNP was subsequently tested for 29 the expected segregation ratios in the F1 progeny: heterozygous in Golden Delicious (1:1), 30 heterozygous in Scarlet Spur (1:1), and heterozygous in both Golden Delicious and Scarlet Spur 31

7
7
(1:2:1). SNPs deviating from these ratios according to a chi-squared test (P < 0.01) were not 1 included in map construction. Finally, progeny genotypes inconsistent with Mendelian 2 inheritance from the parental genotypes were set to missing. After implementing these filters, 3 2436 SNPs remained for linkage map construction. 4 5 Linkage map construction 6 A single composite linkage map was constructed using JoinMap 4.0 (STAM 1993) by combining 7 the backcross type (Aa x aa) markers segregating within each parental background and intercross 8 type (Aa x Aa) markers segregating within both parental backgrounds. Markers suspected of 9 being incorrectly phased by JoinMap, due to high pairwise linkage LOD and spurious 10 recombination fractions above 0.5, had their allele codes switched manually, but were dropped 11 from further analysis if phasing problems persisted. To increase mapping efficiency, pairs or 12 groups of loci with identical genotypes (i.e. complete linkage) were identified and a single 13 marker was chosen to represent the group. 14 Linkage groups were constructed, and ordered, with a linkage LOD of at least 6.0, a minimum 15 recombination fraction of 0.35, and a jump threshold of 5. SNPs exhibiting “suspect linkage” to 16 several loci within their assigned group (as determined by JoinMap), poorly fitting markers 17 within an ordered group, and markers that greatly inflated the linkage group size, were dropped 18 from the final mapping. All map distances were calculated using the Kosambi mapping function. 19 After applying these filters, 1994 SNPs remained within the genetic linkage map. 20 21 Mapping skin color 22 For association analysis based on the physical coordinates obtained from the reference genome, 23 the full set of 3967 SNPs were tested for association with skin color measurements using the 24 case/control single marker analysis in PLINK 1.04 (PURCELL et al. 2007) which employs a chi-25 squared test of independence of allele frequencies between cases and controls with one degree of 26 freedom at each marker independently. For linkage map based QTL analysis, R/qtl 1.26-14 27 (BROMAN et al. 2003) was used with the single binary QTL interval mapping model, scanning 28 the linkage map at 1 cM steps for the presence of a significant QTL. A significance threshold of 29 3.05 was determined by permutation tests on 1000 randomizations of the trait data (CHURCHILL 30 AND DOERGE 1994). The PLINK genotype files, JoinMap input files and phenotype data are 31

8
8
available from the Dryad Digital Repository: http://doi.org/10.5061/dryad.55t54. 1 2
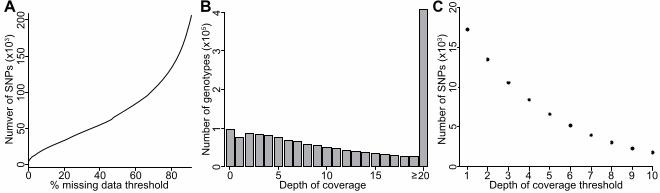
RESULTS 3 Alignment of GBS reads to the reference genome 4 GBS of a single plate of 96 DNA samples yielded 85,129,960 100bp reads using Illumina’s 5 HiSeq 2000 sequencing technology. The high proportion of reads beginning with a barcode 6 sequence (98.2%) and containing a restriction site remnant (99%) indicated that the library 7 preparation was effective and the data were of high quality. In addition, there were very low 8 occurrences of chimeric reads (~1.0%) and of reads containing downstream adapter sequences 9 (~1.2%). 10 After aligning each of the 96 samples’ reads to the ‘Golden Delicious’ v1.0 reference genome, 11 seven of the samples were found to have a relatively low numbers of reads (<150 000) that 12 uniquely aligned to the genome and these samples were subsequently dropped from further 13 analysis. In the remaining 89 samples (87 F1 progeny and 2 parents) there was an average of 14 973,896 (sd = 609,869) reads per sample and an average of 628,085 reads (sd = 393,153) 15 uniquely aligned to the reference genome (Figure 1). Despite the wide range of read counts 16 across samples, there was relative uniformity in the proportion of reads successfully mapped 17 across samples with an average of 63.5% (sd = 2.5%). 18 19 SNP calling 20 Considering only the successfully mapped reads from 89 samples, SNPs were discovered and 21 genotypes were called by analyzing the single master alignment file with GATK (MCKENNA et 22 al. 2010). After employing a minimal set of initial quality filters (see Methods), 273,835 SNPs 23 were identified. However, the resulting genotype matrix was extremely sparse: more than 75% of 24 the 273,835 SNPs contained >50% missing genotypes (Figure 2a). Restricting the analysis to 25 SNPs with <20% missing genotype data drastically reduced the number of SNPs to 30,393. It is 26 likely that many false negative genotype calls still exist in the resulting genotype table since 27 confidently calling heterozygotes in a highly heterozygous diploid species like apple requires a 28 relatively high depth of sequence coverage compared to genotype calling in inbreds. For the set 29 of 30,393 SNPs, the number of genotype calls at various sequence depth thresholds is shown in 30 Figure 2b. Despite the observation that many genotype calls are supported by >20 reads, the 31

9
9
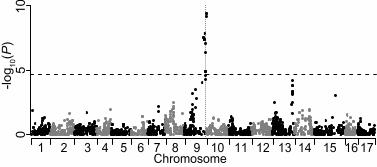
number of SNPs for downstream analysis drops rapidly as the minimum depth of coverage 1 threshold is increased from 1 to 10 while implementing a missing data threshold of 20% (Figure 2 2c). We further reduced the number of markers to 3967 SNPs by applying a minimum depth of 3 coverage threshold of six sequence reads for a genotype call (i.e. any genotype with fewer than 4 six supporting sequence reads was set to missing) and then re-filtering for SNPs with <20% 5 missing genotypes. The choice of six reads per genotype as a threshold was arbitrary: it was 6 chosen as a tradeoff between increased confidence in genotype calls and the number of SNPs 7 retained for mapping. This set of 3967 SNPs was used to map skin color using a chi-squared test. 8 To create the genetic map, a final set of 2436 SNPs were retained after filtering for Mendelian 9 inconsistencies and segregation distortion (see Methods). 10 11 Genetic map construction 12 The final set of 2436 SNPs was separated into the three segregation types: 884 heterozygous in 13 Golden Delicious, 1044 heterozygous in Scarlet Spur, and 508 heterozygous in both parental 14 varieties. Once this set of SNPs was imported into JoinMap 4.0, 442 segregated identically to at 15 least one other SNP (i.e. no recombinants observed) and were dropped from further mapping. 16 The remaining 1994 SNPs were successfully grouped into 17 linkage groups (Figure S2), as 17 expected for the apple genome, at a conservative LOD threshold of six. Only 35 loci were found 18 to be unlinked to any group, suggesting a high degree of saturation of the assembled mapping 19 groups. Once ordered, the linkage groups spanned 1272 cM, with individual linkage groups 20 ranging from 56 cM to 96 cM. Across linkage groups, the average marker density was high with 21 a marker found every 0.68 cM (±0.13 SD). 22 23 QTL mapping for apple skin color 24 Single marker analysis using the final set of 3967 SNPs identified 15 SNPs significantly 25 associated with skin color after applying a conservative Bonferroni correction for multiple 26 testing (P < 1.26x10-5; Figure 3). These SNPs cluster within a single interval on chromosome 9 27 (bounded by coordinates 29,305,493 and 33,701,563). There was also a cluster of SNPs on the 28 distal portion of chromosome 13 that showed a suggestive association with skin color, but all fell 29 short of the corrected significance threshold (Figure 3). 30 Interval mapping using 1994 SNPs detected one highly significant QTL for skin color on the 31

10
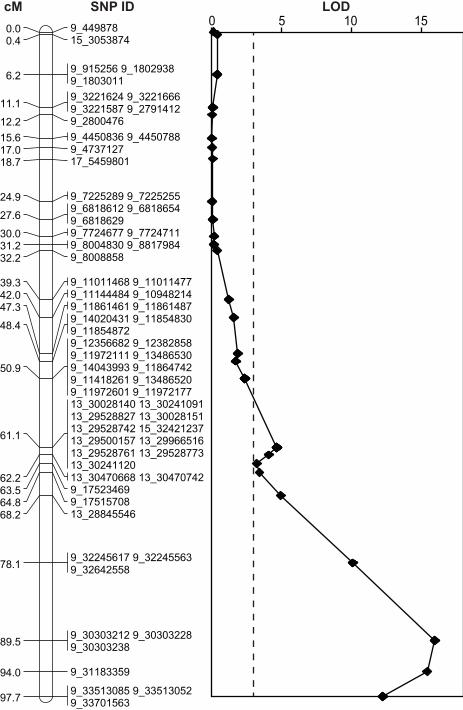
10
distal portion of chromosome nine, at position 89.5 cM, near SNP 9_30303238 (Figure 4). The 1 “2 LOD” confidence interval for this QTL extended to a 14 cM region around the point estimate 2 of the QTL position. 3 4
DISCUSSION 5 A major consideration when conducting GBS experiments is the choice of restriction enzyme 6 used to generate the reduced representation library (RRL). This choice determines the tradeoff 7 between the number of fragments in the library and the sequencing depth of the fragments. There 8 are several GBS library preparation methods currently in use, including the original single 9 enzyme ApeKI protocol described in Elshire et al. (2011); a double digest employing enzymes 10 with differing restriction site lengths (Pst I/Msp I; POLAND et al. 2012b); and a multi-step 11 procedure that combines the double restriction enzyme digest with selective PCR amplification 12 (SONAH et al. 2013). While there was ultimately no explicit test of library fragment composition 13 in the present study, using the HindIII-MspI enzyme combination followed by size selection we 14 observed a very small proportion of sequence reads that required adapter trimming (1.2%), 15 suggesting that most fragments were larger than 100bp and were thus suitable for GBS. 16 However, the number of DNA sequence reads across individual samples varied by more than an 17 order of magnitude (0.15 million to 3 million; Figure 1), and we ultimately excluded seven 18 samples’ sequence data from analyses due to low read counts. These observations highlight the 19 sensitivity of GBS to DNA sample uniformity. Recent GBS library preparation methods have 20 been shown to improve the uniformity in read counts across samples (SONAH et al. 2013) and 21 further improvements are expected. 22 23 Due to the apple’s high heterozygosity and ancestral polyploidy, there are major challenges in the 24 assembly of its genome. It is estimated that the assembled portion of the current apple reference 25 assembly represents only 71% of its ~750Mb genome (VELASCO et al. 2010). Despite this 26 constraint, we restricted our analysis to sequences aligning to this assembly in order to enable a 27 comparison of our inferred linkage groups to the physical map. Approximately 40% of the 28 sequence reads did not map to the assembled genome and were thus excluded from further 29 analysis (Figure 1). These excluded reads are a combination of DNA sequences from the ~29% 30 of the genome that is not represented in the current assembly, DNA sequences that mapped to 31

11
11
multiple locations, and sequences that map to a unique position but with too many mismatches 1 (>4 mismatches per 100bp). As improvements are made to the genome assembly and to DNA 2 sequence length and quality, we anticipate significant improvements in the mapping step of the 3 GBS analysis pipeline. It is worth noting that a potential alternative is to avoid the use of a 4 reference genome altogether and to use a SNP calling pipeline that does not rely on a reference 5 genome (LU et al. 2013). 6 7 There are an enormous number of possible paths one can take from a DNA sequence file to a 8 SNP genotype table and it is known that alignment and genotype calling parameters have a 9 strong effect on the resulting quantity and quality of genome-wide SNP data (MASCHER et al. 10 2013b). Although established software does exist for SNP genotype calling from GBS data 11 (BRADBURY et al. 2007), our goal here was to demonstrate that, with simple heuristics applied 12 together with standard software packages, one can generate SNPs of sufficient quality and 13 quantity to be of utility for genetic mapping. Regardless of what tools are used, it is evident that 14 GBS generates a sparse genotype matrix due to uneven sequence coverage across samples and 15 sites: a large number of SNPs are discovered, but genotypes for these SNPs are most often 16 generated from only a small proportion of the DNA samples. For example, over 250,000 SNPs 17 were discovered in the present study, but when SNPs with >20% missing data are excluded, the 18 number of SNPs remaining for analysis is reduced to ~30,000 (Figure 2A). Moreover, the 19 number of SNPs decreases as the depth of coverage filter is increased (Figure 2B,C). We chose 20 arbitrarily to include only genotypes with ≥6 supporting reads as a trade-off between genotype 21 quality and quantity. Using these thresholds, we obtain a set of 3967 SNPs derived from only 22 6% of the sequence reads. Thus, in the end, 94% of the sequence we collected in this experiment 23 was ignored. Most GBS studies to date have focused on genotyping inbred lines, which requires 24 far less data and is statistically far simpler than genotype calling in highly heterozygous species 25 like apple or in polyploids, which are common among species of agricultural importance. GBS 26 SNP calling pipelines designed specifically for highly heterozygous and polyploid species that 27 take haplotype phasing and imputation into account promise to significantly enhance the utility 28 of GBS (e.g. LU et al. 2013; WARD et al. 2013). 29 30 Despite the high proportion of sequence reads discarded due to filtering, there remained a 31

12
12
sufficient number of markers to perform genetic mapping of apple skin color with a modest 1 sample size. By mapping skin color, we have intentionally focused on a trait with a simple 2 genetic architecture, which deviates from most QTL studies which focus on more complex traits. 3 Skin color is known to be controlled almost entirely by a single locus of large effect (i.e. 4 effectively Mendelian) and we leverage this knowledge here to verify the utility and power of 5 GBS to identify this known locus. With 3967 SNPs genotyped in 89 samples, a simple chi-6 squared test for association revealed a single significant peak from position 29.3Mb to 33.7Mb 7 on chromosome 9 (Figure 3). This peak overlaps with the R2R3 MYB transcription factor gene 8 at position 32.8Mb on chromosome 9 known to regulate apple skin color (TAKOS et al. 2006; 9 ESPLEY et al. 2007; LIN-WANG et al. 2011; ZHU et al. 2011; CHAGNE et al. 2013). In addition, a 10 set of 1994 SNPs were used to produce a well-saturated linkage map spanning 1272 cM (Figure 11 S2). This map size is consistent with the sizes of apple genetic maps from previous studies, for 12 example 1538 cM (Khan et al. 2012); 1005 cM (CHAGNE et al. 2012b); 1143 cM (Han et al. 13 2011). On average, the genetic map has a marker every 0.68cM, which is in line with the marker 14 density achieved using the apple 8K SNP array (0.5cM/marker in ANTANAVICIUTE et al. 2012; 15 0.88cM/marker in CHAGNE et al. 2012a). Using this genetic map, interval mapping revealed 16 one highly significant QTL for skin color centered at position 89.5 cM (Figure 4; Figure S3), in 17 agreement with the results from the chi-squared test for association. Thus, with a set of simple 18 heuristics for calling genotypes from GBS data, a saturated genetic map can be generated and 19 QTL mapping can robustly identify genotype-phenotype associations. 20 21 In the present study, the parents of the F1 population were included only once and had average 22 sequence coverage (Figure 1). However, since inclusion of a SNP in the genetic map relied on 23 accurate genotype calls from both parents, it may be advisable to sequence the parents of 24 mapping populations to a higher depth, i.e. include them multiple times in the plate of samples. 25 Moreover, the genetic map presented here was constructed using only SNPs that mapped to the 26 anchored portion of the reference genome in order to allow comparison between physical and 27 genetic map positions. By including the unanchored portion of the genome sequence during the 28 initial alignment stage, it is likely that additional SNPs could be identified and placed on the 29 genetic map. 30 31

13
13
For 364 SNPs (18.3% of all mapped SNPs), the linkage group assignments conflicted with the 1 predicted chromosomal locations according to the reference genome. Antanaviciute et al. (2012) 2 reported a similar proportion (13.7%) using genotype data from the apple 8K SNP array. The 3 most likely reasons for these conflicts between genetic and physical maps are the high frequency 4 of paralagous genomic regions in the apple genome and the incorrect anchoring of sequences 5 during the assembly of the reference genome. Such conflicts may obscure association signals 6 and complicate the interpretation of genetic mapping results. For example, although below the 7 conservative threshold for significance, we detected a signal of association on chromosome 13 8 when SNPs were positioned according to the physical map (Figure 3). However, this same block 9 of SNPs that physically map to chromosome 13 were subsequently found to genetically map 10 close to the QTL for skin color on linkage group 9 (Figure 4). This demonstrates that caution is 11 warranted when relying on the physical map coordinates of the current reference genome 12 sequence. 13 14 It is worth noting that our modest mapping population size of 87 F1 offspring likely often 15 prevented the ordering algorithm from finding the correct order for SNPs that were closer than 16 ~2-5 cM. Over larger distances, however, the estimated map order of SNPs was generally in 17 agreement with physical coordinates from the reference genome (Fig. S2). 18 19 The present study demonstrates that a Mendelian trait (skin color) can be mapped in an apple F1 20 population using GBS data from a single lane of Illumina next-generation sequencing. 21 Considering the currents costs of acquiring SNP data from the apple 8K genotyping microarray, 22 we estimate that a similar quantity and quality of apple SNP data can be achieved using GBS 23 with a 10-100x decrease in cost. Because of the rapid improvements in DNA sequencing 24 technology, we anticipate that genotyping efforts will increasingly favor the use of GBS or 25 similar methods over the use of microarrays. The present study uses standard software tools and 26 simple heuristics to generate biological insights from GBS data. However, it is clear that 27 methodological developments required for analyzing GBS data lag far behind the technology 28 developed to generate GBS data. In order to maximize the utility of next-generation DNA 29 sequence in the future, there is a clear need for improved computational and statistical tools to 30 extract as much information as possible from the raw data, and to phase, impute, order and 31

14
14
genetically map large sets of genetic markers. 1 2
ACKNOWLEDGMENTS 3 This article was written, in part, thanks to funding from the Canada Research Chairs program 4 and the National Sciences and Engineering Research Council of Canada. 5 6
REFERENCES 7 8
Antanaviciute, L., F. Fernandez-Fernandez, J. Jansen, E. Banchi, K. Evans et al., 2012 9 Development of a dense SNP-based linkage map of an apple rootstock progeny 10 using the Malus Infinium whole genome genotyping array. BMC Genomics 13: 11 203. 12
Baird, N. A., P. D. Etter, T. S. Atwood, M. C. Currey, A. L. Shiver et al., 2008 Rapid SNP 13 Discovery and Genetic Mapping Using Sequenced RAD Markers. PLoS ONE 3: 14 e3376. 15
Barba, P., L. Cadle-Davidson, J. Harriman, J. Glaubitz, S. Brooks et al., 2013 Grapevine 16 powdery mildew resistance and susceptibility loci identified on a high-resolution 17 SNP map. Theoretical and Applied Genetics: 1-12. 18
Bradbury, P. J., Z. Zhang, D. E. Kroon, T. M. Casstevens, Y. Ramdoss et al., 2007 19 TASSEL: software for association mapping of complex traits in diverse samples. 20 Bioinformatics 23: 2633-2635. 21
Broman, K. W., H. Wu, Ś. Sen and G. A. Churchill, 2003 R/qtl: QTL mapping in 22 experimental crosses. Bioinformatics 19: 889-890. 23
Bus, V., C. Ranatunga, S. Gardiner, H. Basset, E. Rikkerink et al., 2000 Marker-24 assisted selection for pest and disease resistance in the New Zealand apple 25 breeding program. Acta Horticulturae 538: 541-547. 26
Celton, J. M., D. S. Tustin, D. Chagné and S. E. Gardiner, 2009 Construction of a dense 27 genetic linkage map for apple rootstocks using SSRs developed from Malus 28 ESTs and Pyrus genomic sequences. Tree Genetics & Genomes 5: 93-107. 29
Chagne, D., R. N. Crowhurst, M. Troggio, M. W. Davey, B. Gilmore et al., 2012a 30

15
15
Genome-Wide SNP Detection, Validation, and Development of an 8K SNP Array 1 for Apple. PLoS ONE 7: e31745. 2
Chagne, D., C. Krieger, M. Rassam, M. Sullivan, J. Fraser et al., 2012b QTL and 3 candidate gene mapping for polyphenolic composition in apple fruit. BMC Plant 4 Biology 12: 12. 5
Chagne, D., K. Lin-Wang, R. V. Espley, R. K. Volz, N. M. How et al., 2013 An Ancient 6 Duplication of Apple MYB Transcription Factors Is Responsible for Novel Red 7 Fruit-Flesh Phenotypes. Plant Physiology 161: 225-239. 8
Churchill, G. A., and R. Doerge, 1994 Empirical Threshold Values for Quantitative Trait 9 Mapping. Genetics 138: 963-971. 10
Consortium, T. I. B. G. S., 2012 A physical, genetic and functional sequence assembly 11 of the barley genome. Nature 491: 711-716. 12
Danecek, P., A. Auton, G. Abecasis, C. A. Albers, E. Banks et al., 2011 The variant call 13 format and VCFtools. Bioinformatics 27: 2156-2158. 14
di Gaspero, G., and F. Cattonaro, 2010 Application of genomics to grapevine 15 improvement. Australian Journal of Grape and Wine Research 16: 122-130. 16
Dirlewanger, E., E. Graziano, T. Joobeur, F. Garriga-Calderé, P. Cosson et al., 2004 17 Comparative mapping and marker-assisted selection in Rosaceae fruit crops. 18 Proceedings of the National Academy of Sciences of the United States of 19 America 101: 9891-9896. 20
Elshire, R. J., J. C. Glaubitz, Q. Sun, J. A. Poland, K. Kawamoto et al., 2011 A Robust, 21 Simple Genotyping-by-Sequencing (GBS) Approach for High Diversity Species. 22 PLoS ONE 6: e19379. 23
Espley, R. V., R. P. Hellens, J. Putterill, D. E. Stevenson, S. Kutty-Amma et al., 2007 24 Red colouration in apple fruit is due to the activity of the MYB transcription factor, 25 MdMYB10. The Plant Journal 49: 414-427. 26
Fernández-Fernández, F., L. Antanaviciute, M. M. Dyk, K. R. Tobutt, K. M. Evans et al., 27 2012 A genetic linkage map of an apple rootstock progeny anchored to the Malus 28 genome sequence. Tree Genetics & Genomes 8: 991-1002. 29
Han, Y., D. Zheng, S. Vimolmangkang, M. A. Khan, J. E. Beever et al., 2011 Integration 30

16
16
of physical and genetic maps in apple confirms whole-genome and segmental 1 duplications in the apple genome. Journal of Experimental Botany. 2
Kellerhals, M., L. Gianfranceschi, N. Seglias and C. Gessler, 2000 Marker-assisted 3 selection in apple breeding. Acta Horticulturae 521: 255-266. 4
Kenis, K., and J. Keulemans, 2005 Genetic linkage maps of two apple cultivars (Malus × 5 domestica Borkh.) based on AFLP and microsatellite markers. Molecular 6 Breeding 15: 205-219. 7
Khan, M. A., Y. Han, Y. F. Zhao, M. Troggio and S. S. Korban, 2012 A Multi-Population 8 Consensus Genetic Map Reveals Inconsistent Marker Order among Maps Likely 9 Attributed to Structural Variations in the Apple Genome. PLoS ONE 7: e47864. 10
Kumar, S., D. Chagné, M. C. A. M. Bink, R. K. Volz, C. Whitworth et al., 2012 Genomic 11 Selection for Fruit Quality Traits in Apple (Malus×domestica Borkh.). PLoS ONE 12 7: e36674. 13
Kumar, S., D. Garrick, M. Bink, C. Whitworth, D. Chagne et al., 2013 Novel genomic 14 approaches unravel genetic architecture of complex traits in apple. BMC 15 Genomics 14: 393. 16
Li, H., and R. Durbin, 2009 Fast and accurate short read alignment with Burrows–17 Wheeler transform. Bioinformatics 25: 1754-1760. 18
Li, H., B. Handsaker, A. Wysoker, T. Fennell, J. Ruan et al., 2009 The Sequence 19 Alignment/Map format and SAMtools. Bioinformatics 25: 2078-2079. 20
Liebhard, R., M. Kellerhals, W. Pfammatter, M. Jertmini and C. Gessler, 2003 Mapping 21 quantitative physiological traits in apple (Malus X domestica Borkh.). Plant 22 Molecular Biology 52: 511. 23
Lin-Wang, K. U. I., D. Micheletti, J. Palmer, R. Volz, L. Lozano et al., 2011 High 24 temperature reduces apple fruit colour via modulation of the anthocyanin 25 regulatory complex. Plant, Cell & Environment 34: 1176-1190. 26
Lu, F., A. E. Lipka, J. Glaubitz, R. Elshire, J. H. Cherney et al., 2013 Switchgrass 27 Genomic Diversity, Ploidy, and Evolution: Novel Insights from a Network-Based 28 SNP Discovery Protocol. PLoS Genet 9: e1003215. 29
Mascher, M., G. J. Muehlbauer, D. S. Rokhsar, J. Chapman, J. Schmutz et al., 2013a 30

17
17
Anchoring and ordering NGS contig assemblies by population sequencing 1 (POPSEQ). The Plant Journal 76: 718-727. 2
Mascher, M., S. Wu, P. S. Amand, N. Stein and J. Poland, 2013b Application of 3 Genotyping-by-Sequencing on Semiconductor Sequencing Platforms: A 4 Comparison of Genetic and Reference-Based Marker Ordering in Barley. PLoS 5 ONE 8: e76925. 6
McKenna, A., M. Hanna, E. Banks, A. Sivachenko, K. Cibulskis et al., 2010 The 7 Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation 8 DNA sequencing data. Genome Research 20: 1297-1303. 9
Micheletti, D., M. Troggio, A. Zharkikh, F. Costa, M. Malnoy et al., 2011 Genetic 10 diversity of the genus Malus and implications for linkage mapping with SNPs. 11 Tree Genetics & Genomes 7: 857. 12
Miller, A., N. Matasci, M. K. Aradhya, B. H. Prins, G. Y. Zhong et al., 2013 Vitis 13 phylogenomics: hybridization intensities from a SNP array outperform genotype 14 calls. PLoS ONE submitted. 15
Myles, S., 2013 Improving fruit and wine: what does genomics have to offer? Trends in 16 Genetics 29: 190-196. 17
Poland, J., J. Endelman, J. Dawson, J. Rutkoski, S. Wu et al., 2012a Genomic Selection 18 in Wheat Using Genotyping-By-Sequencing. The Plant Genome in press. 19
Poland, J. A., P. J. Brown, M. E. Sorrells and J.-L. Jannink, 2012b Development of 20 High-Density Genetic Maps for Barley and Wheat Using a Novel Two-Enzyme 21 Genotyping-by-Sequencing Approach. PLoS ONE 7: e32253. 22
Poland, J. A., and T. W. Rife, 2012 Genotyping-by-Sequencing for Plant Breeding and 23 Genetics. Plant Gen. 5: 92-102. 24
Purcell, S., B. Neale, K. Todd-Brown, L. Thomas, M. A. R. Ferreira et al., 2007 PLINK: 25 A Tool Set for Whole-Genome Association and Population-Based Linkage 26 Analyses. Am J Hum Genet 81: 559-575. 27
Romay, M., M. Millard, J. Glaubitz, J. Peiffer, K. Swarts et al., 2013 Comprehensive 28 genotyping of the USA national maize inbred seed bank. Genome Biology 14: 29 R55. 30

18
18
Sonah, H., M. Bastien, E. Iquira, A. Tardivel, G. Légaré et al., 2013 An Improved 1 Genotyping by Sequencing (GBS) Approach Offering Increased Versatility and 2 Efficiency of SNP Discovery and Genotyping. PLoS ONE 8: e54603. 3
Spindel, J., M. Wright, C. Chen, J. Cobb, J. Gage et al., 2013 Bridging the genotyping 4 gap: using genotyping by sequencing (GBS) to add high-density SNP markers 5 and new value to traditional bi-parental mapping and breeding populations. 6 Theoretical and Applied Genetics 126: 2699-2716. 7
Stam, P., 1993 Construction of integrated genetic linkage maps by means of a new 8 computer package: JoinMap. The Plant Journal 5: 739-744. 9
Takos, A. M., F. W. Jaffé, S. R. Jacob, J. Bogs, S. P. Robinson et al., 2006 Light-10 Induced Expression of a MYB Gene Regulates Anthocyanin Biosynthesis in Red 11 Apples. Plant Physiology 142: 1216-1232. 12
Velasco, R., A. Zharkikh, J. Affourtit, A. Dhingra, A. Cestaro et al., 2010 The genome of 13 the domesticated apple (Malus x domestica Borkh.). Nature Genetics 42: 833. 14
Ward, J., J. Bhangoo, F. Fernandez-Fernandez, P. Moore, J. Swanson et al., 2013 15 Saturated linkage map construction in Rubus idaeus using genotyping by 16 sequencing and genome-independent imputation. BMC Genomics 14: 2. 17
Zhu, Y., K. Evans and C. Peace, 2011 Utility testing of an apple skin color MdMYB1 18 marker in two progenies. Molecular Breeding 27: 525-532. 19
20 FIGURE LEGENDS: 21 22 Figure 1: Results of alignment of GBS reads to the apple reference genome. For each sample, 23 the number of reads mapped and unmapped to the reference genome is shown. The read counts 24 for the parents of the F1 mapping population, Golden Delicious and Scarlet Spur, are indicated. 25 26 Figure 2: SNP and genotype counts from GBS data. a) Cumulative count of SNPs identified 27 across varying missing data thresholds. Over 200,000 SNPs are called with a very liberal 28 missing data threshold of 90%, but only 30,393 SNPs remain if only SNPs with <20% missing 29 data are retained. b) The number of genotypes called at increasing levels of sequencing depth, 30 after retaining only SNPs with <20% missing data. c) The number of SNPs retained at increasing 31

19
19
minimum thresholds of sequence depth while retaining only SNPs with <20% missing data. Here 1 we choose a minimum depth of coverage of six reads. Thus only SNPs with at least six 2 supporting reads and <20% missing genotypes were retained, which results in a set of 3967 3 SNPs. 4 5 Figure 3: Manhattan plot of a single marker association analysis for apple skin color. Each of the 6 3967 SNPs is plotted according to its physical position from the ‘Golden Delicious’ reference 7 genome and the –log10P value of the single marker association test. The horizontal dotted line 8 represents the Bonferonni-corrected P value significance threshold. The vertical dotted line 9 represents the location of the MYB transcription factor gene known to be responsible for skin 10 color variation. 11 12 Figure 4: Result of QTL analysis across the linkage group corresponding to chromosome 9 of 13 the apple genome. The left panel indicates the genetic map positions in cM of each of the SNPs 14 or groups of SNPs. Each SNP’s ID indicates its physical position according to the reference 15 genome, i.e. the physical coordinate it was assigned through alignment and SNP calling (e.g. 16 SNP 9_ 449878 mapped to position 449878 on chromosome 9 of the ‘Golden Delicious’ v1.0 17 reference genome). Note that many SNPs genetically mapping to the linkage group 18 corresponding to chromosome 9 are assigned to other chromosomes according to the reference 19 genome (e.g. SNP 13_30028140). The right panel displays the LOD scores from a QTL analysis 20 for skin color for markers that segregate in the Scarlet Spur genetic background. The horizontal 21 dashed line represents the significance threshold determined by permutation. LOD scores across 22 all linkage groups are shown in Figure S3. 23 24 Figure S1: A custom GBS analysis pipeline. Each box represents a file type, indicated by its file 25 extension, within the pipeline. Starting with a single .fastq file from the Illumina HiSeq 2000 26 sequencing machine and ending with a single set of PLINK files, the software packages and 27 custom scripts used to move from one file type to another are indicated beside each arrow. 28 Details of each step of the pipeline are provided in the Materials and Methods. 29 30 Figure S2: A composite genetic linkage map of the Malus x domestica genome. The map was 31

20
20
constructed from 1994 SNPs discovered and genotyped using GBS, including SNPs from all 1 three pseudo-testcross segregation types, in a Golden Delicious x Scarlet Spur F1 population. 2 3 Figure S3: Genome-wide LOD scores for apple fruit skin color. Each SNP is represented by a 4 single vertical line at the bottom of the plot. Only a single peak associated with skin color was 5 identified on linkage group 9 and this peak overlaps with the known apple skin color locus. The 6 horizontal dashed line represents the significance threshold determined by permutation. 7

Number of reads (x106)0 0.5 1 1.5 2 2.5 3
Mapped readsUnmapped reads
Sam
ples
Golden Delicious
Scarlet Spur
0 20 40 60 80
050
100
150
200
% missing data threshold
Num
ver o
f SN
Ps
(x10
3 )A B
01
23
4N
umbe
r of g
enot
ypes
(x10
5 )
0 5 10 15 20Depth of coverage
≥
C
510
15N
umbe
r of S
NP
s (x
103 )
020
2 4 6 8 10Depth of coverage threshold
1 3 5 7 9
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17
50
10-lo
g 10(P
)
Chromosome
9_4498780.015_30538740.4
9_915256 9_18029389_18030116.2
9_3221624 9_32216669_3221587 9_279141211.19_280047612.29_4450836 9_445078815.69_473712717.017_545980118.7
9_7225289 9_722525524.99_6818612 9_68186549_681862927.69_7724677 9_772471130.09_8004830 9_881798431.29_800885832.2
9_11011468 9_1101147739.39_11144484 9_1094821442.09_11861461 9_1186148747.39_14020431 9_118548309_1185487248.49_12356682 9_123828589_11972111 9_134865309_14043993 9_118647429_11418261 9_134865209_11972601 9_11972177
50.9
13_30028140 13_3024109113_29528827 13_3002815113_29528742 15_3242123713_29500157 13_2996651613_29528761 13_2952877313_30241120
61.1
13_30470668 13_3047074262.29_1752346963.59_1751570864.813_2884554668.2
9_32245617 9_322455639_3264255878.1
9_30303212 9_303032289_3030323889.5
9_3118335994.0
9_33513085 9_335130529_3370156397.7
0 5 10 15 cM SNP ID LOD