estudo das propriedades Óptica e elÉtrica da …‡Ão... · em função do momento dipolar do...
TRANSCRIPT

NATHÁLIA AKEMI YOSHIOKA
ESTUDO DAS PROPRIEDADES ÓPTICA E ELÉTRICA DA
CLOROFILINA CÚPRICA DE SÓDIO PARA APLICAÇÃO EM
DISPOSITIVOS CONVERSORES DE ENERGIA
Universidade Federal de Ouro Preto
Departamento de Física
Ouro Preto, Fevereiro de 2014.

NATHÁLIA AKEMI YOSHIOKA
ESTUDO DAS PROPRIEDADES ÓPTICAS E ELETRÔNICAS DA
CLOROFILINA CÚPRICA DE SÓDIO (SCC) PARA APLICAÇÃO EM
DISPOSITIVOS CONVERSORES DE ENERGIA
Universidade Federal de Ouro Preto
Departamento de Física
Ouro Preto, Fevereiro de 2014.
Dissertação de Mestrado apresentada ao
Programa de Pós-Graduação em Ciências
de Materiais do Departamento de Física
da Universidade Federal de Ouro Preto
como requisito para obtenção do título de
Mestre em Ciência de Materiais.
Orientador: Prof. Dr. Thiago Cazati

Catalogação: [email protected]
Y657e Yoshioka, Nathália Akemi.
Estudo das propriedades ótica e elétrica da clorofilina cúprica de sódio para aplicação em dispositivos conversores de energia [manuscrito]. / Nathália Akemi Yoshioka. – 2014.
91 f.: il. color., grafs., tabs. Orientador: Prof. Dr. Thiago Cazati. Dissertação (Mestrado) - Universidade Federal de Ouro Preto. Instituto de Ciências Exatas e Biológicas. Departamento de Física. Área de concentração: Física de materiais
1.Células solares - Teses. 2. Semicondutores orgânicos - Teses. I. Cazati, Thiago. II. Universidade Federal de Ouro Preto. III. Título. CDU: 621.383.51


DEDICATÓRIA
Aos meus pais, Shigeru Yoshioka e
Maria Elena do Carmo Yoshioka

AGRADECIMENTOS
À FAPEMIG, pela bolsa de estudos concedida;
Ao professor Thiago Cazati, pela orientação, seriedade e dedicação a este
trabalho;
À professora Bruna Postacchini, pelo apoio e sugestões durante a realização
deste trabalho;
Aos alunos Tomás Nogueira Ribeiro, Marcus Henrique de Araújo e Luiza de
Lazari Ferreira, pelo auxílio prestado e pela colaboração;
Ao Laboratório de Materiais Optoeletrônicos (LAMOe) do Departamento de
Física da Universidade Federal de Ouro Preto, onde a maior parte deste trabalho foi
desenvolvida;
Ao Grupo de Polímeros Bernhard Gross do Instituto de Física de São Carlos,
pela concessão do espaço para realização de partes deste trabalho;
Ao Programa de Pós-Graduação em Ciências de Materias da Universidade
Federal de Ouro Preto e ao coordenador prof. Dr. Genivaldo Júlio Perpétuo por toda a
assistência prestada;
À secretária Mariana Cristina Moreira Souza, pela atenção e solicitude;
Aos amigos e colegas de turma, em especial à Larissa, ao Márcio e ao João
Paulo pela amizade e pelo convívio;
À minha família, pela motivação, pelo incentivo e, principalmente, pelo amor
em todos os momentos.

RESUMO
Este trabalho tem como objetivo principal estudar as propriedades
optoeletrônicas da clorofilina cúprica de sódio (SCC) comercial para aplicação em
dispositivos fotovoltaicos com estruturas ITO/(PAH/SCC)n/Al e
ITO/(PAH/SCC)n/Ca/Al. A motivação para a realização deste estudo é o
desenvolvimento de células solares orgânicas de baixo custo à base de materiais com
solubilidade em água que possam ser utilizados como fontes alternativas de energia
renovável. Filmes depositados pela técnica de automontagem (LBL, do inglês layer-by-
layer) e soluções de SCC em diferentes solventes foram preparadas e suas propriedades
foram investigadas por meio da espectroscopia de absorção e fluorescência. Utilizou-se
como camada fotoativa os filmes automontados de SCC nos dispositivos fotovoltaicos
de monocamada. Os dispositivos foram, então, caracterizados eletricamente através das
medidas de condutividade dc (curvas tensão-corrente) e da espectroscopia de
impedância (ac) e medida de fotocondutividade persistente. A partir dos resultados e dos
ajustes teóricos, foi possível analisar os processos optoeletrônicos envolvidos na
conversão de energia pelo dispositivo, caracterizá-los quando a sua eficiência de
conversão e obter parâmetros importantes que influenciam o desempenho dos
dispositivos fotovoltaicos.

ABSTRACT
In this work, sodium copper chlorophyllin (SCC) optoelectronic properties were
analyzed and employed as active layer of organic photovoltaic device with
ITO/(PAH/SCC)n/Al and ITO/(PAH/SCC)n/Ca/Al structures. Organic photovoltaic
devices (OPVs) based on water-soluble materials could be low-cost production,
chlorinated solvents free, widely practical, foldable, and flexible large area devices. The
SCC film was deposited onto ITO electrode by layer-by-layer technique (LBL) which
involves the sequential deposition of multilayers by immersing the substrates alternately
into aqueous cationic solution of polyallylamine hydrochloride (PAH) and anionic
solutions of sodium copper chlorophyllin (SCC). The absorption spectra of films and
solutions were optically characterized with UV-Vis and fluorescence spectroscopies.
Photovoltaic devices were characterized by the current density–voltage, impedance
spectroscopy and persistent photocondutivity measurement. Experimental data were
fitted by theoretical models and important parameters of devices performance were
found.

LISTA DE FIGURAS
Figura 1: Exemplos de aplicação de materiais orgânicos semicondutores em OLEDs e em OPVs ......................................................................................................................... 15 Figura 2: Estrutura molecular da porfirina de base livre ............................................. 17 Figura 3: Espectro de absorção na região visível típico de uma porfirina .................. 18 Figura 4: Orbitais HOMO (abaixo) e LUMO (acima) das porfirinas .......................... 19 Figura 5: a) Diagrama de orbitais mostrando as transições possível para as porfirinas; b) Diagrama de estados mostrando os possíveis estados excitados para as porfirinas. 19 Figura 6: Espectro de absorção das porfirinas contendo os principais níveis de energia, sendo que a banda Soret corresponde a uma transição π-π* e a banda Q corresponde a uma transição n-π*. ................................................................................ 20 Figura 7: Exemplos de metaloporfirinas: (a) clorofila, (b) heme, (c) ftalocianina de cobre e (d) ftalocianina de manganês. ........................................................................... 22 Figura 8: Estrutura molecular da clorofila (a) e da clorofilina cúprica de sódio (SCC) (b) ................................................................................................................................... 23 Figura 9: Diagrama de Jablonski adaptado [29] ......................................................... 24 Figura 10: Dispositivo fotovoltaico orgânico com estrutura tipo sanduíche. Nessa arquitetura exemplificada, o dispositivo é composto por uma camada fotoativa (material orgânico) depositada entre o eletrodo Cátodo (Al) e o Ânodo (ITO) semitransparente ............................................................................................................ 27 Figura 11: Estrutura química da clorofilina cúprica de sódio (SCC) e do hidrocloreto de polialilamina (PAH) .................................................................................................. 29 Figura 12: Ilustração do ciclo de deposição de bicamadas de clorofilina cúprica de sódio pela técnica de automontagem ............................................................................. 31 Figura 13: Curva de crescimento dos filmes de (PAH/SCC) obtidos em soluções com diferentes pHs em função do tempo de imersão ............................................................. 32 Figura 14: Filmes de (PAH/SCC)80 fabricados utilizando soluções de PAH com pH ácido (à esquerda) e pH básico (à direita)..................................................................... 33 Figura 15: Solução de SCC (a) a direita, em pH 7,05; (b) a esquerda, em pH 3,48 [74]. ........................................................................................................................................ 33 Figura 16: Etapas do procedimento adotado para a remoção da camada de ITO da superfície do substrato: (a) lâmina de ITO,(b) proteção (fita adesiva) sobre a região da lâmina da qual não se deseja remover o ITO, (c) remoção do ITO por meio do ataque químico e (d) obtenção do eletrodo de ITO na configuração desejada. ........................ 34 Figura 17: Medidas (em mm) da máscara utilizada na metalização dos eletrodos dos dispositivos fotovoltaicos. ............................................................................................... 35 Figura 18: Geometria dos eletrodos de ITO sobre substrato de vidro e dos eletrodos metálicos utilizados na confecção do dispositivo. .......................................................... 35 Figura 19: Diagrama de bandas simplificado para um dispositivo onde os eletrodos são o ITO e o alumínio e a camada fotoativa composta por um material orgânico semicondutor. (a) materiais antes do contato; (b) materiais após o contato; (c) em tensão reversa aplicada, diodo retificador; (d) tensão direta aplicada igual ao potencial de built-in; (e) com o aumento da polarização, surge a corrente elétrica em

tensão direta. ΦITO / ΦAl: função trabalho dos eletrodos, Ip: potencial de ionização e χ: afinidade eletrônica da camada ativa. ........................................................................... 38 Figura 20: Curva característica de um dispositivo fotovoltaico, tanto no escuro quanto iluminado. A Figura mostra a tensão de circuito aberto (VOC) e a corrente de curto circuito para o dispositivo iluminado. O ponto A representa a corrente do dispositivo durante a tensão reversa (negativa), o ponto B é conhecido como corrente de curto-circuito, o ponto C é a tensão de circuito aberto e o ponto D a corrente no modo direto para V>VOC. ................................................................................................................... 40 Figura 21: Representação de um dispositivo fotovoltaico de monocamada com estrutura tipo sanduíche durante a caracterização de condutividade DC, com incidência de luz através do eletrodo de ITO ................................................................. 42 Figura 22: Espectro de impedância de um RC paralelo [] ........................................... 46 Figura 23: Espectro de impedância pelo diagrama de Argand [79] ............................ 47 Figura 24: Espectro de absorção da solução de SCC 0,05 mg/mL em água e de filmes de PAH/SCC com diferentes números de bicamadas ..................................................... 49 Figura 25: Curvas de crescimento baseada nos máximos de absorção da banda Soret (406 nm) e da banda Q (638 nm) dos filmes de (PAH/SCC)n automontados em função do número de bicamadas. A linha tracejada é guia para os olhos. ............................... 50 Figura 26: Espectros de absorção das soluções de SCC em diferentes solventes com concentração de 0,05 mg/mL (em detalhe o gráfico do coeficiente de absorção molar em função do momento dipolar do solvente) .................................................................. 51 Figura 27: Espectros de absorção das soluções de SCC em diferentes solventes a 0,05 mg/mL, normalizados em relação às principais bandas (a) Soret e (b) Q. .................... 53 Figura 28: Espectros de fluorescência de soluções de SCC em diferentes solventes à concentração de 0,05 mg/mL excitadas no comprimento de onda de suas respectivas bandas Soret ................................................................................................................... 54 Figura 29: Diagrama de energias evidenciando o efeito da relaxação do solvente. As esferas centrais representam as moléculas do soluto e as periféricas representam as moléculas do solvente, demonstrando a mudança do momento dipolar do estado excitado µe com relação ao do estado fundamental µf [29] ........................................... 55 Figura 30: Espectros de fluorescência de soluções de SCC em diferentes solventes à concentração de 0,05 mg/mL excitadas no comprimento de onda de suas respectivas banda Q .......................................................................................................................... 56 Figura 31: Espectro de absorção da solução de SCC em tolueno com concentração de 0,05 mg/mL (em preto) e seus espectros de fluorescência quando excitada em 410 nm (azul escuro) e em 645 nm (azul claro) .......................................................................... 57 Figura 32: Espectro de absorção da solução de SCC em clorofórmio com concentração de 0,05 mg/mL (em preto) e seus espectros de fluorescência quando excitada em 407 nm (azul escuro) e em 638 nm (azul claro) .................................................................... 58 Figura 33: Espectro de absorção da solução de SCC em isopropanol com concentração de 0,05 mg/mL (em preto) e seus espectros de fluorescência quando excitada em 407 nm (azul escuro) e em 630 nm (azul claro) ......................................... 59

Figura 34: Espectro de absorção da solução de SCC em etanol com concentração de 0,05 mg/mL (em preto) e seus espectros de fluorescência quando excitada em 405 nm (azul escuro) e em 625 nm (azul claro) .......................................................................... 60 Figura 35: Espectro de absorção da solução de SCC em água com concentração de 0,05 mg/mL (em preto) e seus espectros de fluorescência quando excitada em 404 nm (azul escuro) e em 627 nm (azul claro) .......................................................................... 60 Figura 36: Espectros de fluorescência dos filmes de SCC com diferentes número de bicamadas excitados nos máximos de absorção das bandas Soret e Q ......................... 61 Figura 37: Espectros de impedância e seus respectivos diagramas de Argand para os dispositivos ITO/(PAH/SCC)8/Al .................................................................................... 62 Figura 38: Espectros de impedância e seus respectivos diagramas de Argand para os dispositivos ITO/(PAH/SCC)16/Al .................................................................................. 63 Figura 39: Espectros de impedância e seus respectivos diagramas de Argand para os dispositivos ITO/(PAH/SCC)24/Al .................................................................................. 63 Figura 40: Espectros de impedância e seus respectivos diagramas de Argand para os dispositivos ITO/(PAH/SCC)32/Al .................................................................................. 63 Figura 41: Gráficos da condutividade em função da frequência de oscilação do campo aplicado para os dispositivos ITO/(PAH/SCC)n/Al, com n = 8, 16, 24 e 32 .................. 65 Figura 42: Componente real da capacitância (C') em função da frequência para os dispositivos (PAH/SCC)n ................................................................................................ 66 Figura 43: Curvas J-V para o dispositivo ITO/(PAH/SCC)80/Ca/Al no escuro e sob iluminação de 100 mW/cm² ............................................................................................ 67 Figura 44: Quarto quadrante das curvas J-V para o dispositivo ITO/(PAH/SCC)80/Ca/Al no escuro e sob iluminação de 100 mW/cm² .......................... 68 Figura 45: Curvas J-V para o dispositivo ITO/(PAH/SCC)80/Al no escuro e sob iluminação de 100 mW/cm² ............................................................................................ 69 Figura 46: Quarto quadrante das curvas J-V para o dispositivo ITO/(PAH/SCC)80/Al no escuro e sob iluminação de 100 mW/cm² .................................................................. 69 Figura 47: Circuito simplificado equivalente a um dispositivo fotovoltaico ideal. A resistência em paralelo RP representa todas as perdas na corrente enquanto a resistência em série RS representa a resistividade do circuito, resistividade nos contatos elétricos e a resistividade da camada ativa [52] ........................................................... 70 Figura 48: Quarto quadrante das curvas J-V características de um dispositivo fotovoltaico no caso ideal (RP = ∞ e RS = 0) e em casos onde existe variações em RS ou em RP [52] ....................................................................................................................... 70 Figura 49: Representação esquemática das bandas de energia da CuTsPc [83] comparadas às funções trabalho dos eletrodos utilizados neste trabalho ..................... 72 Figura 50: Curva J-t para o dispositivo ITO/(PAH/SCC)80/Ca/Al obtida alternando-se entre a incidência de luz branca (100 mW/cm²) e o escuro ........................................... 73 Figura 51: Curva J-t para o dispositivo ITO/(PAH/SCC)80/Al obtida alternando-se entre a incidência de luz branca (100 mW/cm²) e o escuro ........................................... 73 Figura 52: Ajustes das curvas (αhν)²-hν em energias próximas aos máximos de absorção, a fim de se calcular o gap energético envolvido em cada uma das transições

das bandas Soret e Q para a solução aquosa de SCC com concentração de 0,05 mg/mL ........................................................................................................................................ 75 Figura 53: Ajustes das curvas (αhν)²-hν em energias próximas aos máximos de absorção, a fim de se calcular o gap energético envolvido em cada uma das transições das bandas Soret e Q para o filme de (PAH/SCC)80 ...................................................... 76 Figura 54: Ajuste das curvas ln(α)-hν em energias próximas ao início da absorção óptica nas bandas Q e Soret para a solução aquosa de SCC com concentração de 0,05 mg/mL ............................................................................................................................. 77 Figura 55: Ajuste das curvas ln(α)-hν em energias próximas ao início da absorção óptica nas bandas Soret e Q para o filme (PAH/SCC)80 ................................................ 78 Figura 56: Ajustes das curvas J-t dos dispositivos ITO/(PAH/SCC)80/Ca/Al de acordo com a Equação 31, a fim de investigar aspectos da fotocondutividade persistente ...... 81 Figura 57: Ajustes das curvas J-t dos dispositivos ITO/(PAH/SCC)80/Ca/Al de acordo com a Equação31, a fim de investigar aspectos da fotocondutividade persistente ....... 82

LISTA DE TABELAS
Tabela 1: Relação entre o número de bicamadas e a espessura do filme de (PAH/SCC)n medida por perfilometria .......................................................................... 50 Tabela 2: Momento dipolar dos solventes utilizados nas soluções de SCC. ................. 52 Tabela 3: Máximos de absorção da banda Soret dos espectros de absorção da SCC em diferentes solventes e seus respectivos coeficientes de absorção molar obtidos através da Equação 4, sendo L = 1 cm o caminho óptico .......................................................... 53 Tabela 4: Cálculo da resistividade, da condutividade e da constante dielétrica dos filmes de (PAH/SCC)n a partir dos valores de impedância e capacitância obtidos através da espectroscopia de impedância. A constante dielétrica foi calculada a partir da Equação 16 ................................................................................................................ 66 Tabela 5: Valores das correntes de curto circuito, das tensões de circuito aberto, dos fatores de preenchimento e dos rendimentos para os dispositivos ITO/(PAH/SCC)80/Ca/Al e ITO/(PAH/SCC)80/Al ........................................................... 71 Tabela 6: Cálculo do Et das bandas Soret e Q para os filmes (PAH/SCC)n ................ 78 Tabela 7: Valores obtidos para os parâmetros τ e β (Equação 31) do ajuste das curvas J-t do dispositivo ITO/(PAH/SCC)80/Ca/Al .................................................................... 81 Tabela 8: Valores obtidos para os parâmetros τ e β (Equação 31) do ajuste das curvas J-t do dispositivo ITO/(PAH/SCC)80/Al .......................................................................... 83

ÍNDICE
1. Introdução................................................................................................................ 15
1.1. Moléculas orgânicas conjugadas ...................................................................... 16
1.2. Porfirinas .......................................................................................................... 17
1.2.1. Metaloporfirinas ............................................................................................... 21
1.2.2. Clorofila e Clorofilina cúprica de sódio (SCC) ................................................ 23
1.3. Processos Radiativos e não-radiativos em Moléculas Conjugadas .................. 24
1.4. Células solares orgânicas.................................................................................. 25
1.5. Dispositivo fotovoltaico ................................................................................... 26
1.5.1. Estrutura e funcionamento................................................................................ 26
2. Método experimental............................................................................................... 29
2.1. Fabricação dos dispositivos: ............................................................................. 29
2.1.1. Materiais: .......................................................................................................... 29
2.1.2. Processamento dos filmes: ............................................................................... 30
2.1.3. Limpeza e preparação do substrato: ................................................................. 33
2.1.4. Metalização dos eletrodos: ............................................................................... 34
3. Técnicas de caracterização ...................................................................................... 36
3.1. Espectroscopia de absorção: ............................................................................. 36
3.2. Espectroscopia de fluorescência:...................................................................... 37
3.3. Condutividade DC ............................................................................................ 38
3.4. Condutividade persistente ................................................................................ 42
3.5. Espectroscopia de impedância: ........................................................................ 42
4. Resultados e Discussões .......................................................................................... 48
4.1. Medidas Ópticas. .............................................................................................. 48
4.1.1. Espectroscopia de Absorção:............................................................................ 48
4.1.2. Espectroscopia de fluorescência:...................................................................... 54
4.2. Caracterizações elétricas (dc) ........................................................................... 62
4.2.1. Espectroscopia de impedância.......................................................................... 62
4.2.2. Medidas elétricas de tensão-corrente (J-V): ..................................................... 66
4.2.3. Condutividade persistente ................................................................................ 72
5. Análise dos resultados ............................................................................................. 74
5.1. Determinação do gap de energia da SCC ......................................................... 74
5.2. Grau de desordem do material .......................................................................... 76
5.3. Tempo de relaxação dos portadores de cargas ................................................. 79
6. Conclusão ................................................................................................................ 84

15
1. Introdução
A indústria eletrônica é um dos principais pilares da sociedade moderna, estando
presente de diversas formas na sociedade. Os principais elementos nos quais se baseiam
a indústria eletrônica são dispositivos eletrônicos à base de materiais semicondutores
inorgânicos (como o arseneto de gálio, o germano e o silício), tais como diodos,
transistores e dispositivos fotovoltaicos [1,2,3]. Entretanto, materiais orgânicos estão
sendo cada vez mais aplicados em dispositivos eletrônicos, sendo uma nova perspectiva
para o cenário tecnológico e dando origem a uma outra classe dentro da eletrônica: a
eletrônica orgânica. A eletrônica orgânica é um ramo da nanociência que permite que
materiais orgânicos semicondutores, principalmente as moléculas orgânicas conjugadas,
sejam utilizados em circuitos eletrônicos substituindo os tradicionais materiais
inorgânicos. Uma das grandes vantagens que os materiais orgânicos possuem sobre os
inorgânicos é a maior facilidade de processamento em substratos flexíveis, resultando
em inúmeras inovações tecnológicas. Alguns exemplos de aplicações, como em
displays de diodos orgânicos emissores de luz (OLEDs, do inglês organic light-emitting
diode) e dispositivos orgânicos fotovoltaicos (OPVs, do inglês organic photovoltaics),
são mostrados na Figura 1.
Figura 1: Exemplos de aplicação de materiais orgânicos semicondutores em OLEDs e em OPVs

16
1.1. Moléculas orgânicas conjugadas
Moléculas orgânicas conjugadas possuem em sua estrutura uma alternância
entre ligações simples e duplas na cadeia de carbono, o que as tornam materiais
semicondutores. Os elétrons de valência de um átomo de carbono são os elétrons
presentes nos orbitais 2s e 2p. Orbitais híbridos são obtidos pela sobreposição das
funções de onda dos orbitais 2s e 2p, sendo do tipo sp³, sp² e sp [4]. A estrutura
eletrônica de moléculas conjugadas é determinada basicamente pelas ligações entre
carbonos chamadas de ligações σ, formadas pela sobreposição de orbitais híbridos
sp² ao longo do plano da molécula. Em adição, existem as ligações π formadas pela
sobreposição lateral dos orbitais atômicos pz entre carbonos adjacentes,
perpendicular ao plano da molécula [ 5 ]. Os elétrons das ligações π são
deslocalizados, isto é, não estão presos a uma região específica da molécula,
podendo transitar livremente pela conjugação da molécula. A superposição dos
orbitais pz formam orbitais do tipo π (ligantes) e π* (anti-ligantes) e as interações
entre os orbitais π ligantes (ocupados) equivalem à banda de valência e as
interações entre os orbitais π* anti-ligantes (desocupados) equivalem à banda de
condução, análogo ao que ocorre em semicondutores inorgânicos [6,7]. A diferença
entre a energia que separa o orbital molecular preenchido mais alto energeticamente
(Highest Occupied Molecular Orbital – HOMO) e o orbital molecular vazio mais
baixo energeticamente (Lowest Unoccupied Molecular Orbital – LUMO) resulta na
energia de banda proibida do material (Eg). Nos semicondutores inorgânicos, a Eg é
definida como a diferença de energia entre o máximo da banda de valência e o
mínimo da banda de condução [8], enquanto em semicondutores orgânicos não são
observadas bandas definidas.
Em geral, moléculas orgânicas conjugadas apresentam propriedades de
absorção óptica e fluorescência na região visível do espectro eletromagnético. Esta
propriedade confere a estas moléculas características interessantes para aplicações
tecnológicas, como filtros ópticos e dispositivos optoeletrônicos como diodos
emissores de luz (OLEDs ) [9,10,11], transistores de efeito de campo (OFETs, do
inglês organic field-effect transistor) [ 12 , 13 ] e células solares (OPVs)
[14,15,16,17].

17
1.2. Porfirinas
Dentre as moléculas orgânicas conjugadas, as porfirinas (do grego
porphura, que significa cor púrpura) compreendem uma importante classe de
moléculas presentes na natureza, exercendo diversas funções biológicas e de grande
interesse em diversas áreas como oncologia, física de materiais, entre outros [18].
São compostos orgânicos e organometálicos formadas por quatro anéis pirrólicos
ligados por pontes de metil, formando uma estrutura planar e caráter aromático. A
porfirina de base livre é mostrada na Figura 2. O macrociclo pode sofrer
substituição dos seus doze hidrogênios por grupos funcionais, e, dependendo dos
grupos substituintes, o comportamento das porfirinas pode ser alterado, como por
exemplo, a solubilidade em determinados solventes. No centro desta estrutura, é
possível inserir prótons ou íons metálicos substituindo os dois hidrogênios centrais,
alterando de forma significativa suas propriedades estruturais e eletrônicas
[19,20,21].
Figura 2: Estrutura molecular da porfirina de base livre
O anel porfírico possui dezoito elétrons π deslocalizados em sua estrutura, e
esta extensa conjugação é responsável pela alta absorção de radiação na luz visível,
podendo ainda transmitir tal energia a moléculas vizinhas.
As porfirinas possuem elevado rendimento quântico de formação de estados
tripletos e geração de oxigênios singletos [22], além de uma alta biocompatibilidade
[23]. Tais propriedades são interessantes para o uso das porfirinas em sistemas
biológicos, sendo aplicadas, por exemplo, na terapia fotodinâmica (PDT, do inglês
photodynamic therapy). A PDT é utilizada em tratamento de cânceres e nela as
porfirinas atuam transferindo energia para outros compostos, auxiliando na necrose
de tecidos tumorais [24].

18
Em geral, as porfirinas e seus derivados possuem duas principais transições
eletrônicas quando excitadas com radiação visível, verificadas através de duas
principais bandas no espectro de absorção: banda Soret (B), na região de menores
comprimentos de onda e banda Q, em maiores comprimentos de onda (Figura 3).
Figura 3: Espectro de absorção na região visível típico de uma porfirina
Martin Gouterman [25] propôs pela primeira vez um modelo de orbitais que
explicasse o espectro de absorção das porfirinas. Neste modelo, as bandas de
absorção das porfirinas surgem de transições entre dois orbitais HOMO (a1u e a2u) e
dois orbitais LUMO (orbitais degenerados de eg) mostrados na Figura 4, sendo que
o íon central e os demais substituintes no anel afetam tais energias de transição.
Transições entre estes orbitais dão origem a dois estados excitados, ambos de
caráter 1Eu. A combinação dos orbitais divide 1Eu em dois estados de energia,
criando um estado 1Eu de maior energia com maior força de oscilador, dando
origem à banda Soret, e um estado 1Eu de menor energia, com menor força de
oscilador, dando origem à banda Q (ver Figura 5).

19
Figura 4: Orbitais HOMO (abaixo) e LUMO (acima) das porfirinas
Figura 5: a) Diagrama de orbitais mostrando as transições possíveis para as porfirinas; b) Diagrama de estados mostrando os possíveis estados excitados para as porfirinas.
Neste modelo, ambas as transições ocorrem entre os orbitais ligantes (π) e
os antiligantes (π*) da conjugação periférica do anel porfírico, sendo que a banda

20
Soret é observada no espectro de absorção UV-visível em torno de 350-500 e
bandas Q em torno de 500-750 nm [26].
Há uma outra teoria que explica as duas principais transições eletrônicas
observadas no espectro de absorção das porfirinas que considera, além da transição
do tipo π-π*, uma outra transição do tipo n-π*. A presença de um orbital n é devida
à presença de pares eletrônicos não ligantes dos heteroátomos (neste caso, o
nitrogênio), sendo n um orbital molecular não ligante [27,28,29]. A transição π-π* é
mais energética, sendo corresponde a banda Soret, enquanto a transição n-π*,
menos energética, caracteriza a banda Q. As transições estão ilustradas na Figura 6.
Figura 6: Espectro de absorção das porfirinas contendo os principais níveis de energia, sendo que a banda Soret corresponde a uma transição π-π* e a banda Q corresponde a
uma transição n-π*.
O número de estados e configurações utilizado para descrever estados
eletrônicos das porfirinas varia de acordo com a o método teórico utilizado, mas
algumas semelhanças entre eles ainda se mantêm. Trabalho realizado por Parusel e
Grimme [30] através da teoria do funcional da densidade e interação configuração
multireferencial (DFT/MRCI) atribui mais de um estado excitado eletrônico (S2, S3,
..., Sn) para moléculas da família das porfirinas, como observado
experimentalmente para o espectro de absorção linear de clorofila A (Chl-A) por
De Boni e co-autores [31].

21
1.2.1. Metaloporfirinas
Quando se substitui os hidrogênios centrais da porfirina por um íon
metálico, a porfirina passa a ser um composto denominado metaloporfirina. Tal
modificação no anel porfírico acarreta em alterações radicais do
comportamento da porfirina, alterações que dependerão fortemente do metal
substituinte [21]. Na maioria dos casos, a estrutura deixa de ser planar,
podendo então ser em forma de sela, cesta, onda ou cúpula. A presença do
metal no centro do anel porfírico faz com que haja uma reorientação dos
orbitais moleculares, alterando as energias dos orbitais HOMO e LUMO e,
consequentemente, os espectros de absorção e emissão [32]. Exemplos de
metaloporfirinas encontradas na natureza são: a clorofila, responsável pelo
processo de fotossíntese em plantas e bactérias, contendo Mg no centro do anel
porfírico e a heme, associada à hemoglobina com função de transporte de
oxigênio pelo sangue e ao citocromo C com função catalítica no processo de
respiração, tendo como íon metálico central o ferro [33,34,35]. As estruturas
moleculares da clorofila A e da heme podem ser verificadas nas Figura 7-a e
Figura 7-b, respectivamente. Importantes exemplos de metaloporfirinas são as
ftalocianinas, amplamente utilizadas como corantes e com crescente aplicação
em dispositivos e sensores eletrônicos [ 36 , 37 , 38 , 39]. Exemplos de
ftalocianinas são as ftalocianinas de cobre e manganês, mostradas
respectivamente na Figura 7-c e Figura 7-d.

22
Figura 7: Exemplos de metaloporfirinas: (a) clorofila, (b) heme, (c) ftalocianina de cobre e (d) ftalocianina de manganês.
As metaloporfirinas apresentam, além de propriedades semicondutoras,
alta estabilidade química e fácil processabilidade na forma de filmes finos,
podendo ser depositadas sobre grandes áreas e por diversas técnicas de
deposição. Tais propriedades fazem com que as metaloporfirinas sejam
consideradas alternativas de grande potencial de aplicação na eletrônica e na
optoeletrônica molecular [37, 38, 40 ]. Além disso, apresentam elevados
coeficientes de extinção para absorção próxima de 650 nm, onde ocorre o
máximo fluxo de fótons da radiação solar, resultando em uma eficiente
captação de fótons, tornando-as adequadas para a integração de sistemas de
conversão de luz em corrente elétrica [ 41 ] Outra grande vantagem das
metaloporfirinas é o fato de a maioria delas serem solúveis em água quando
modificadas, o que possibilita a confecção de filmes finos para aplicação em
dispositivos optoeletrônicos sem a utilização de solventes danosos ao meio
ambiente, [38,42]. Neste trabalho, o material de interesse dentro da classe das

23
metaloporfirinas é a clorofilina cúprica de sódio (SCC, do inglês Sodium
Copper Chlorophyllin).
1.2.2. Clorofila e Clorofilina cúprica de sódio (SCC)
A clorofila (Chl) é um pigmento verde de origem natural responsável
pelo processo de fotossíntese, processo em que a luz solar é convertida na
energia necessária às plantas e bactérias clorofiladas. Possui uma ampla
absorção de luz na região do visível e possui energia de ativação de
semicondutor entre 1,2 e 1,65 eV, característica que torna a clorofila e seus
derivados materiais de interesse para a aplicação em células solares [43,44]. As
clorofilinas são moléculas sintetizadas a partir da clorofila, obtidas pela
saponificação em meio alcoólico-alcalino (etanol contendo NaOH) [40,45,46].
Em particular, a clorofilina cúprica de sódio (SCC) é uma molécula derivada
da clorofila obtida através da substituição do átomo central de Mg por um
átomo de Cu, passando, posteriormente, pelo processo de saponificação. Tal
reação confere à estrutura da molécula de SCC a solubilidade em água,
propriedade de interesse neste trabalho. As moléculas de Chl-A e SCC são
mostradas na Figura 8.
Figura 8: Estrutura molecular da clorofila (a) e da clorofilina cúprica de sódio (SCC) (b)
A clorofilina cúpria de sódio (SCC) tem sido aplicada em várias áreas,
como por exemplo, na medicina [47], na indústria alimentícia [48] e em

24
sistemas eletrônicos como camada fotoativa de dispositivos fotovoltaicos
[40,49].
1.3. Processos Radiativos e não-radiativos em Moléculas Conjugadas
Em moléculas orgânicas conjugadas, a energia que separa os orbitais
HOMO e LUMO são de alguns elétron-volts (1,8 a 3,1 eV) [50], permitindo a
transição de eletrônica entre estes níveis através absorção do fóton. Após a
absorção, diversos mecanismos radiativos e não-radiativos ocorrem. O diagrama de
Jablonski (Figura 9) analisa de maneira simples os possíveis processos envolvidos
de transição energética na molécula: absorção de fótons, conversão interna,
fluorescência, cruzamento intersistema, fosforescência, entre outros.
Figura 9: Diagrama de Jablonski adaptado [29]
Como verifica-se no diagrama, S0 (estado fundamental), S1 e S2 são os
estados eletrônicos singletos, ou seja, estados com apenas um valor possível de
energia, e os estados T1 e T2 são os estados tripletos, com três valores possíveis
para energia. A cada um destes níveis energéticos, estão associados diversos
estados vibracionais.
Quando um elétron é excitado por radiação incidente, ocorre absorção da
energia desta radiação, o que promove o elétron de seu estado fundamental em S0
para um nível de maior energia Sn (n = 1, 2...), sendo que cada um dos níveis Sn

25
ainda possuem diversos níveis vibracionais. Como pode-se observar na Figura 9, o
elétron pode ser excitado de S0 para S1 ou S2. Do estado S2, o elétron pode decair
para S1 através da conversão interna, decaimento não radiativo que ocorre entre
dois níveis de mesma multiplicidade de spin. A partir de então, o elétron pode
decair para S0 de forma radiativa, ou seja, fluorescendo, ou pode se transferir para o
estado tripleto T1 através de um cruzamento intersistema e decair radiativamente
em forma de fosforescência ou ainda decair de forma não radiativa por meio de um
cruzamento intersistema de T1 a S0. Num cruzamento intersistema o elétron sofre
inversão de spin e a necessidade de inverter-se o spin torna o fenômeno de
fosforescência significativamente mais lento que o de fluorescência. Outro
fenômeno que ainda pode ocorrer após o elétron ser excitado é a relaxação
vibracional, já que um elétron pode ser excitado a qualquer um dos níveis
vibracionais de S1 ou S2, decaindo então de forma não radiativa entre estes níveis.
A excitação do estado S0 para T1 é uma transição proibida, com baixa probabilidade
de ocorrência.
Pela análise dos espectros de absorção e fluorescência da Figura 9, pode-se
observar que os comprimentos de onda da emissão são maiores que os da absorção,
ou seja, a fluorescência ocorre em energias menores que a absorção. Apesar de a
emissão de um fóton ocorrer tão rapidamente quando sua absorção (10-15 s), as
espécies excitadas permanecem em S1 por um determinado tempo antes de
emitirem um fóton ou decaírem de forma não radiativa. Este tempo é suficiente
para que o sistema assuma a configuração de equilíbrio do estado excitado e, como
este é menos energético, a emissão ocorre em comprimentos de onda maiores que o
absorvido [29]. Uma causa comum para que isso ocorra é a rápida relaxação das
espécies excitadas para níveis vibracionais abaixo de S1, para que então decaiam
radiativamente para S0. O deslocamento entre os máximos de absorção e
luminescência é chamado deslocamento de Stokes, estudado pela primeira vez pelo
fisico irlandês George G. Stokes em 1852. Em geral, a energia que separa os níveis
vibracionais são similares em S0 e S1 e, devido a isto, o espectro de fluorescência é
uma imagem especular do espectro de absorção.
1.4. Células solares orgânicas
A tecnologia tem se desenvolvido de forma acelerada, demandando cada
vez mais energia. Como as fontes convencionais de energia são, em geral,

26
esgotáveis (tais como o petróleo e o carvão mineral), existe a crescente necessidade
de buscar fontes renováveis de energia. Neste contexto, a energia solar se mostra
uma fonte abundante de energia alternativa, ecologicamente mais limpa e viável
tecnicamente [51]. Os dispositivos fotovoltaicos (capazes de converter energia
luminosa em elétrica) disponíveis no comércio atualmente são constituídos
principalmente por silício de alta pureza, apresentando um elevado custo de
produção. Como alternativa de baixo custo aos dispositivos de silício, materiais
semicondutores orgânicos são considerados candidatos potencialmente promissores
para aplicações no campo de dispositivos optoeletrônicos, em particular em células
solares. Apesar da atual baixa eficiência de conversão energética, as células
fotovoltaicas orgânicas representam uma solução de baixo custo para a necessidade
de geração de eletricidade através da radiação solar. Além disso, os dispositivos
fotovoltaicos orgânicos apresentam extenso intervalo de absorção luminosa, podem
ser fabricados tanto em substratos rígidos quanto em substratos flexíveis e a custo e
inferior aos tradicionais fotovoltaicos à base de materiais inorgânicos (Si)
[52,53,54].
As porfirinas, devido a suas propriedades semicondutoras, constituem uma
classe de moléculas orgânicas conjugadas com potencial de aplicação em células
solares à base de moléculas orgânicas pequenas (do inglês Small molecule organic
solar cells, SMOSC) [42, 55 , 56 ]. Recentemente, SMOSC alcançaram uma
eficiência recorde de conversão energética para este tipo de material de 6,7 % [16].
Entretanto, as propriedades importantes da maioria das porfirinas, tais como
características da estrutura molecular, interações com o meio, propriedades
fotofísicas, energia de ligação dos éxcitons, mecanismo de geração de portadores de
carga e dinâmica do estado excitado são muito recentes ou pouco estudados
[42,57,58]. O estudo destas propriedades é de extremo interesse na otimização dos
dispositivos eletrônicos à base de porfirinas.
1.5. Dispositivo fotovoltaico
1.5.1. Estrutura e funcionamento
Um típico dispositivo fotovoltaico orgânico possui como camada
fotoativa um material orgânico semicondutor depositado entre eletrodos
metálicos (cátodo e ânodo), sendo um desses eletrodos semitransparente (para

27
incidência de radiação), formando uma estrutura do tipo sanduíche
(eletrodo/camada fotoativa/eletrodo), como pode-se verificar na Figura 10. O
funcionamento e a eficiência de conversão energética do dispositivo dependem
das características dos materiais (incluindo todos os seus elementos – eletrodo
e camada fotoativa), da morfologia (incluído a forma de deposição das
camadas) e da arquitetura dos dispositivos (monocamada, multicamadas, com
heterojunções, etc) [52]. Portanto, os dispositivos fotovoltaicos são sistemas
complexos com múltiplos processos ocorrendo simultaneamente e com
diversas variáveis a serem ajustadas e otimizadas para garantir um bom
desempenho.
Figura 10: Dispositivo fotovoltaico orgânico com estrutura tipo sanduíche. Nessa arquitetura exemplificada, o dispositivo é composto por uma camada fotoativa (material orgânico) depositada entre o eletrodo Cátodo (Al) e o Ânodo (ITO)
semitransparente
O princípio de funcionamento dos dispositivos fotovoltaicos depende
do fenômeno físico conhecido como fotocondutividade, associado à variação
de condutividade do material semicondutor quando exposto à radiação
luminosa. A fotocondutividade se dá quando a densidade de elétrons
promovidos à banda de condução por meio da interação com os fótons
incidentes é significativamente superior à concentração dos elétrons excitados
termicamente. Sua magnitude depende do número de portadores produzidos ou
fotogerados (eficiência interna) e da mobilidade desses portadores durante o
transporte. O tempo de duração da fotocondutividade depende do tempo de
exposição do material à radiação e do tempo de vida das espécies excitadas.
No caso de um semicondutor cristalino, a energia dos fótons incidentes
é diretamente transferida para o sistema eletrônico do material, promovendo a
excitação dos elétrons da banda de valência para a banda de condução e dando

28
origem aos portadores de carga livres de sinais opostos (fotogeração intrínseca)
por meio da rápida dissociação dos pares elétron-buraco atraídos por força
eletrostática (éxcitons) fotogerados. Em semicondutores orgânicos, há a
formação de éxcitons com maior energia de ligação entre eles, sendo que neste
caso a dissociação ocorre por processos secundários (fotogeração extrínseca),
como por exemplo através da ação do campo elétrico externo, da interação dos
pares com as cargas na interface material/eletrodos com defeitos e impurezas
do próprio material e transferência de carga entre moléculas doadoras e
aceitadoras [59,60,61,62,63].
Em semicondutores orgânicos, entretanto, tais processos não são bem
definidos e há bastantes controvérsias no estudo dos mesmos, embora seja bem
aceita a ideia de que há formação de éxcitons em sua estrutura, apesar da falta
de informação acerca das suas características e sobre o valor de suas energias
de ligação [64,65]. Nestes materiais, após a dissociação dos éxcitons, as cargas
são conduzidas por meio de um mecanismo de transporte conhecido como
“saltos” (hopping) [66,67], sendo que a mobilidade dos portadores depende
principalmente da densidade de estados (sítios) eletrônicos do material. Num
dispositivo de monocamadas, as cargas livres são conduzidas ao circuito
externo pelo campo elétrico intrínseco do dispositivo gerado pela diferença de
função trabalho entre os eletrodos metálicos (conhecido como potencial de
built-in e responsável pelo efeito fotovoltaico), gerando corrente elétrica.
Durante este processo, cargas de sinais opostos (elétrons e buracos livres)
podem se recombinar, havendo aniquilação dos portadores [68]. A barreira
energética existente entre a interface eletrodo/material semicondutor (barreira
Schottky) deve ser minimizada, a fim de melhorar a eficiência de coleta de
cargas pelos eletrodos, garantindo que maior parte da energia luminosa seja
convertida em corrente ou tensão elétrica. A razão entre a corrente elétrica
gerada e o número fótons absorvido determina a eficiência de conversão
energética do dispositivo.

29
2. Método experimental
Neste capítulo estão descritos os materiais, as etapas e os procedimentos
experimentais utilizados na fabricação dos filmes e dos dispositivos fotovoltaicos. As
etapas foram realizadas em laboratórios do Departamento de Física da Universidade
Federal de Ouro Preto em colaboração com o Grupo de Polímeros "Prof. Bernhard
Gross", do Instituto de Física de São Carlos (IFSC).
2.1. Fabricação dos dispositivos:
Os dispositivos de monocamada confeccionados neste trabalho foram
preparados de acordo com as seguintes etapas de fabricação: preparação das
soluções para confecção da camada fotoativa e deposição da mesma, limpeza e
preparação do substrato e metalização dos eletrodos. Os detalhes de cada etapa
serão apresentados a seguir.
2.1.1. Materiais:
O material orgânico semicondutor da família das porfirinas e derivado da
clorofila utilizado neste trabalho foi a clorofilina cúprica de sódio (SCC)
adquirida da Sigma-Aldrich®, sem purificações posteriores. A estrutura química
da SCC está mostrada na Figura 11, bem como a estrutura do polieletrólito
hidrocloreto de polialilamina (PAH), ambos utilizados na confecção da camada
fotoativa.
SCC PAH
Figura 11: Estrutura química da clorofilina cúprica de sódio (SCC) e do hidrocloreto de polialilamina (PAH)

30
2.1.2. Processamento dos filmes:
A técnica escolhida para deposição da camada fotoativa do dispositivo
foi a automontagem ou camada-por-camada (LBL, do inglês layer-by-layer). É
uma técnica que permite controlar a espessura do filme, podendo então serem
produzidos filmes ultrafinos (nanométricos). Além disso, é uma técnica na qual
os filmes de SCC podem ser depositados a partir de soluções aquosas, já que a
água é um solvente pouco volátil, inviabilizando o emprego de outras técnicas de
deposição, como espalhamento casting ou spin coating [ 69 ]. A técnica de
automontagem é baseada em interações eletrostáticas entre grupos iônicos e
permite a adsorção sucessiva de camadas de material aniônico e catiônico sobre
um substrato. Um substrato carregado negativamente, quando imerso em uma
solução catiônica, adsorve uma certa quantidade de material, formando uma
camada de policátions. Em seguida, o sistema substrato/policátions é imerso na
solução aniônica, ocasionando adsorção de outra camada. A adsorção de duas
camadas de cargas opostas (policátion/poliânion) constitui uma bicamada e a
deposição do filme fino é obtida por sucessivos ciclos de imersão. Cada
bicamada possui uma espessura bastante definida, permitindo o controle da
espessura do filme depositado.
Apesar de a energia de ligação entre esses pares ser baixa, proporcional a
kT, sendo k a constante de Boltzmann e T a temperatura, a estabilidade das
camadas adsorvidas é grande [ 70 , 71 ] devido a diversos fatores como a
supercompensação de cargas, caracterizada pela inversão do sinal da carga
líquida inicial do substrato [ 72 ] e a interpenetração das cadeias dos
polieletrólitos adsorvidos [73].
No processo dos filmes por automontagem, utilizou-se como solução
catiônica a solução aquosa contendo o polieletrólito PAH e como solução
aniônica a solução aquosa de clorofilina cúprica de sódio (SCC). As soluções de
PAH (policátion) e de SCC (poliânion) foram preparadas com concentração de
0,5g/L em água destilada de baixa condutividade (Milli-Q). Para isso, pesou-se
15,0 mg do soluto em uma balança de precisão e posteriormente dissolvendo-o
em 30,0 ml de água Milli-Q. O pH das soluções preparadas foram ajustados para
pH neutros (próximos de 8,0). O polieletrólito PAH é um polímero utilizado

31
apenas como contra íon para o processo de deposição, é inerte opticamente e não
afeta as propriedades ópticas da SCC.
O substrato de vidro, carregado negativamente após o processo de
hidrofilização (ver item 2.1.3) foi primeiramente imerso na solução de PAH
durante três minutos. Em seguida, o substrato de vidro com o PAH adsorvido foi
imerso na solução de SCC durante três minutos, formando assim uma bicamada.
Este ciclo foi repetido a fim de conseguir o número de bicamadas desejado e,
entre cada uma das imersões, o substrato com as moléculas adsorvidas foi limpo
em água Milli-Q e seco com nitrogênio (N2) 99,9 %. A deposição foi realizada à
temperatura ambiente e sem controle da umidade na atmosfera local. Os filmes
depositados por esta técnica são referenciados como (PAH/SCC)n, sendo n o
número de bicamadas.
Figura 12: Ilustração do ciclo de deposição de bicamadas de clorofilina cúprica de sódio pela técnica de automontagem
Os parâmetros que determinam a qualidade dos filmes automontados e
influenciam nas propriedades fotofísicas dos filmes, como a concentração das
soluções de imersão, o pH das soluções e o tempo de imersão foram estudados
anteriormente [ 74 , 75 ]. Foi verificado que o filme cresce com maior
homogeneidade e ocorre maior adsorção das moléculas de SCC durante o
processo de automontagem quando o pH das soluções de imersão é básico, o que
pode ser observado nas curvas de crescimento mostrada na Figura 13 [75].

32
0 100 200 300 400 500
0,00
0,01
0,02
0,03
0,04
0,05
0,06
Abs
orbâ
ncia
/(u.
a.)
Tempo de imersao/s
PAH(pH 3,5)/SCC(pH 3,7) PAH(pH 7,7)/SCC(pH 7,5) PAH(pH 9,5)/SCC(pH10,0) PAH(pH 4,4)/SCC(pH 7,4)
Figura 13: Curva de crescimento dos filmes de (PAH/SCC) obtidos em soluções com diferentes pHs em função do tempo de imersão
De acordo com a Figura 13, verificou-se que após cerca de 180 segundos
(3 minutos), a intensidade da absorção óptica não sofreu variação, o que
significa que não há mais material sendo adsorvido, sendo este, então, o tempo
ideal de imersão do substrato em cada uma das soluções. Além disso, a
influência do pH na adsorção foi verificada nestas curvas de crescimento, já que,
quanto mais ácido o pH das soluções, menor a intensidade de absorção dos
filmes, o que significa menor adsorção de material ao substrato.
Na Figura 14 observam-se imagens dos filmes com 80 bicamadas
depositados em pH ácido e pH básico. É possível observar que o filme de
(PAH/SCC)80 fabricado com solução de PAH com pH básico (8,5) adsorveu
mais material que o filme fabricado com solução de PAH com pH ácido (4,4).

33
Figura 14: Filmes de (PAH/SCC)80 fabricados utilizando soluções de PAH com pH ácido (à esquerda) e pH básico (à direita)
Além disso, observou-se que, durante a automontagem dos filmes
(PAH/SCC) em soluções de pH ácido, pode ocorrer precipitação da clorofilina,
conforme pode ser observado na Figura 15 [74]. Isso ocorreu pois a solução de
PAH tende a ficar mais ácida devido à formação de ácido clorídrico na diluição
em água, na qual o íon Cl- interage com o hidrogênio presente no solvente. Da
mesma forma, a solução de SCC tende a ter maior basicidade devido à interação
do íon Na+ com o grupo OH- presente no solvente. Neste trabalho, os pHs foram
medidos pelo pHmetro PHtek PHS-3B utilizando soluções de NH3 e HCl em
água, ambas com concentração de 0,1 M para corrigir o pH das soluções.
Figura 15: Solução de SCC (a) a direita, em pH 7,05; (b) a esquerda, em pH 3,48 [74].
2.1.3. Limpeza e preparação do substrato:
Os substratos utilizados para a fabricação dos dispositivos fotovoltaicos
foram lâminas de vidro com uma camada de óxido de estanho índio (ITO)
(obtidas comercialmente), cortadas em tamanhos (2,0 x 1,0 x 0,1) cm, de
resistividade de 15-22 Ω/. Em seguida, parte do ITO foi removida através do
procedimento mostrado na Figura 16. Este procedimento é utilizado para definir

34
a geometria do eletrodo ITO e evitar que ocorram curtos circuitos durante o
funcionamento do dispositivo fotovoltaico. Neste procedimento, é colocada uma
proteção de fita adesiva sobre a parte da lâmina da qual o ITO não deve ser
removido (Figura 16-b). Em seguida, a solução aquosa de zinco é espalhada
sobre o ITO a ser removido e, em seguida, utilizou-se HCl 6 M para remover
completamente a camada de ITO (Figura 16-c). Feito isso, retira-se a proteção
ao ataque químico e obtém-se, então, o eletrodo de ITO na geometria desejada
(Figura 16-d). Na primeira etapa da limpeza, as lâminas passaram por uma
lavagem com sabão e água. Em sequência, as lâminas foram imersas em solução
de etanolamina 20%, que foi aquecida até a ebulição. Finalmente, as lâminas
foram imersas em acetona a 50°C e secas. Além da limpeza descrita
anteriormente, as lâminas passaram por um processo de hidrofilização, etapa
necessária para a deposição do filme pela técnica de automontagem, já que o
substrato deve conter carga para adsorver o material. Neste processo, as lâminas
são imersas em solução de NaOH com concentração de 0,1M em água e levadas
ao ultrassom por 15 minutos. Feito isso, as lâminas foram lavadas com água
Milli-Q para retirada do excesso de NaOH e estavam prontas para a deposição
da camada fotoativa.
Figura 16: Etapas do procedimento adotado para a remoção da camada de ITO da superfície do substrato: (a) lâmina de ITO,(b) proteção (fita adesiva) sobre a região da lâmina da qual não se deseja remover o ITO, (c) remoção do ITO por meio do ataque
químico e (d) obtenção do eletrodo de ITO na configuração desejada.
2.1.4. Metalização dos eletrodos:
Os eletrodos metálicos foram depositados por evaporação térmica em
alto vácuo (<10-6 mbar) sobre a camada fotoativa e a geometria do eletrodo foi
obtida utilizando máscara mecânica (ver Figura 17) sobre o dispositivo durante a
evaporação. Os metais utilizados nos eletrodos foram o cálcio (Ca) e o alumínio

35
(Al) e os eletrodos obtidos possuem espessura entre 100 e 110 nm. Para os casos
em que o cálcio foi utilizado, metalizou-se uma camada de Al sobre o eletrodo
de Ca (Ca_Al) para evitar a rápida oxidação desse, já que o Ca é muito reativo
ao oxigênio e à água. O Ca foi utilizado por possuir uma menor função trabalho
quando comparado ao Al. A geometria dos eletrodos no dispositivo está
mostrada na Figura 18. A etapa de metalização dos eletrodos foi realizada
utilizando a evaporadora Edwards, no Instituto de Física de São Carlos, da
Universidade de São Paulo, dentro de uma glove box.
Figura 17: Medidas (em mm) da máscara utilizada na metalização dos eletrodos dos dispositivos fotovoltaicos.
Figura 18: Geometria dos eletrodos de ITO sobre substrato de vidro e dos eletrodos metálicos utilizados na confecção do dispositivo.
Após a etapa de metalização, dispositivos com estruturas sanduíche do
tipo ITO/(PAH/SCC)n/Al e ITO/(PAH/SCC)n/Ca/Al foram obtidos e estavam
prontos para serem caracterizados óptica e eletricamente. A área efetiva dos
dispositivos é de cerca de 0,1 cm².

36
3. Técnicas de caracterização
A seguir, são descritas as técnicas e os instrumentos utilizados na caracterização
óptica das soluções e dos filmes e elétrica e dispositivos. Os filmes e as soluções foram
caracterizados através da espectroscopia de absorção e espectroscopia de fluorescência.
Os dispositivos foram caracterizados por meio de curvas J-V, medidas de condutividade
persistente e espectroscopia de impedância,
3.1. Espectroscopia de absorção:
A espectroscopia de absorção no ultravioleta e visível (UV-VIS) envolve a
espectroscopia de fótons na faixa de visível e de ultravioleta (UV), em que as
moléculas sofrem transições eletrônicas. Neste trabalho, utilizou-se o espectrômetro
Hitachi, modelo U-2001 para obter os espectros de absorção dos filmes de
PAH/SCC automontados com diferente número de bicamadas e de soluções de
SCC em diferentes solventes.
O princípio básico de absorção óptica é bastante simples. Considerando uma
molécula absorve luz visível e na região ultravioleta, quando a radiação incide
sobre esta molécula, ocorre uma transição eletrônica, na qual um elétron absorve
energia do fóton incidente e é promovido do orbital da molécula no seu estado
fundamental para um orbital desocupado de maior energia (ver item 1.2).
A luz ao passar através de um material (filme ou solução) sofre uma
redução na intensidade, proporcional à intensidade incidente (I0), à espessura (dx)
do material e o coeficiente de absorção (α), podendo ser escrita como:
dxIdI 0α−= (Equação 1)
Para obter a intensidade que emerge da amostra de espessura l, soma-se
sobre todas as intensidades do lado esquerdo da equação e sobre toda a espessura
do lado direito. Integrando a Equação 1, observa-se que a intensidade decai
exponencialmente com a espessura.
leII α−⋅= 0 (Equação 2)
A Equação 2 é a lei fundamental (Lei de Lambert-Beer) que governa a
absorção da luz por uma amostra, freqüentemente expressa como:

37
leII ε−⋅= 100 ou ( ) lII .log 0 ε−= (Equação 3)
A constante adimensional T = I / I0 é chamada transmitância e A = ε.l de
absorbância da amostra escrita como:
( ) lII .log 0 ε−= (Equação 4)
onde ε é chamado coeficiente de absorção molar que depende da molécula e da
freqüência da luz em questão e sua unidade é dada pelo inverso da concentração e
do comprimento (1/M.m).
No espectro de absorção de uma solução, tem-se que A = ε.l.c, onde c é a
concentração da solução, já que o número de partículas presentes no caminho ótico
irá influenciar na intensidade da absorção.
3.2. Espectroscopia de fluorescência:
A fluorescência é definida como a relaxação da molécula excitada através
da reemissão da energia absorvida. Na espectroscopia de fluorescência (ou
luminescência), obtém-se as intensidades de fluorescência em função do
comprimento de onda e o espectro é obtido fixando-se o comprimento de onda de
excitação. A espectroscopia de fluorescência é uma poderosa técnica de análise
química e estrutural, possuindo elevada sensibilidade às vizinhanças da molécula.
Sendo assim, efeitos como a polaridade das moléculas e o ordenamento do sistema
analisado são possíveis de ser detectados pela espectroscopia de luminescência
[29]. A fluorescência, entretanto, pode ser suprimida devido a diversos fatores,
como a interação entre o composto fluorescente e outro composto presente na
amostra ou mesmo a interação deste composto com impurezas.
Utilizando-se o espectrofluorímetro Shimadzu modelo RF-5301 PC
equipado com uma lâmpada de Xe foi possível medir o espectro de emissão (PL)
das soluções e dos filmes utilizados como camadas fotoativas nos dispositivos
fotovoltaicos. O feixe de excitação de comprimento de onda previamente
selecionado incide sobre a superfície da amostra e a intensidade da luz emitida pela
amostra (luminescência) em função do comprimento de onda é coletada por um
detector. O espectro de fotoluminescência dos filmes e soluções foram obtidos em

38
condições ambientes de pressão e temperatura e foram posicionados de modo que a
emissão fosse coletada na mesma face de incidência (front face).
3.3. Condutividade DC
A caracterização elétrica básica dos dispositivos fabricados foi realizada
através de medidas de corrente-tensão (J-V) em regime de corrente contínua (DC).
Para melhor explicar o comportamento da curva característica tensão-corrente de
um dispositivo fotovoltaico sob a ação de diferentes valores de tensão aplicada,
considere o esquema dos diagramas de bandas de um dispositivo de monocamada
ITO/camada fotoativa/Al sob iluminação, mostrado na Figura 19, sendo que a
camada fotoativa é formada por um material orgânico semicondutor.
Figura 19: Diagrama de bandas simplificado para um dispositivo onde os eletrodos são o ITO e o alumínio e a camada fotoativa composta por um material orgânico semicondutor.
(a) materiais antes do contato; (b) materiais após o contato; (c) em tensão reversa aplicada, diodo retificador; (d) tensão direta aplicada igual ao potencial de built-in; (e) com o aumento da polarização, surge a corrente elétrica em tensão direta. ΦITO / ΦAl: função trabalho dos eletrodos, Ip: potencial de ionização e χ: afinidade eletrônica da
camada ativa.

39
A Figura 19-a mostra a configuração energética de um material
semicondutor e dos eletrodos, separadamente, antes de haver um contato. Ao
estabelecer o contato entre os materiais (material orgânico e eletrodos), a tendência
é haver um balanceamento de cargas entre o material e os eletrodos até que se atinja
o equilíbrio. Isso gera uma deflexão das bandas, como se observa na Figura 19-b.
Quanto tal equilíbrio é atingido, os níveis de Fermi dos eletrodos se equiparam com
o nível de Fermi do material orgânico nas interfaces, formando o contato Schottky
[76]. O surgimento de um potencial de contato (VC) ocorre na interface entre o
material orgânico e o eletrodo e a diferença entre esses potenciais de contato é
denominada “potencial de built-in” VB, que é a diferença entre as funções trabalho
dos eletrodos em dispositivos de monocamada:
( ) qVV ÂNODOCÁTODOCB Φ−Φ=∆= (Equação 5)
onde Φ representa a função trabalho e q é a carga elementar.
O potencial de built-in gera um campo elétrico intrínseco (INTEr
)
perpendicular às superfícies dos eletrodos e é esse campo elétrico que conduz as
cargas fotogeradas em um dispositivo fotovoltaico, sendo responsável pelo efeito
fotovoltaico. Ao aplicar-se uma tensão externa, o dispositivo pode se polarizar no
modo direto ou reverso. No modo reverso, o ITO, polarizado negativamente, será o
injetor de elétrons e o Al, polarizado positivamente, será o injetor de buracos
(Figura 19-c). Entretanto, a energia necessária para os elétrons e buracos serem
injetados através das barreiras dadas pela diferença entre as funções-trabalho dos
eletrodos e das energias do HOMO/LUMO do material que constitui a camada
fotoativa, nesse caso, são muito altas, resultando em uma densidade de corrente
total reduzida devido à baixa densidade de portadores de carga disponíveis. A
densidade de corrente total é basicamente constituída pela corrente gerada pelas
cargas fotogeradas no interior do material orgânico, que foram conduzidas pelo
campo elétrico resultante (REr
), neste caso, devido ao campo aplicado (APLEr
)
somado ao campo elétrico intrínseco (INTEr
). Na curva J-V mostrada na Figura 20,
este regime de funcionamento é apresentado entre os pontos A e B. Quando a
tensão aplicada se aproxima do valor nulo, haverá uma corrente remanescente no
dispositivo (ponto B da curva J-V na Figura 20), que resulta da condução das cargas
fotogeradas pelo campo elétrico intrínseco criado pelos eletrodos. Esta corrente é

40
denominada corrente de curto circuito ou ISC. Ao aplicar uma tensão positiva, o
comportamento do dispositivo depende do valor do potencial aplicado. Para o caso
em que a tensão aplicada é menor que o potencial de built-in (V<VB), o
comportamento ainda é o mesmo do dispositivo polarizado no modo reverso.
Quando o potencial aplicado equivale ao potencial de built-in (V=VB), ocorrerá a
situação na qual as bandas ficam planas, como representado na Figura 19-d. Neste
momento (ponto C da curva da Figura 20), o campo resultante é nulo (
0=+ INTAPL EErr
), a corrente se torna nula, sendo então a tensão aplicada chamada de
tensão de circuito aberto (VOC). A tensão de circuito aberto em um dispositivo de
monocamada fotoativa é geralmente determinada pela diferença entre os valores
das funções trabalho dos eletrodos: ( ) BÂNODOCÁTODOOC VqV −=Φ−Φ= [66].
Entretanto, em dispositivos fotovoltaicos de heterojunção esta relação nem sempre
é verificada [77,78]
Para valores de tensão aplicada maiores que o VOC, o dispositivo se polariza
no modo direto, no qual o ITO, agora polarizado positivamente, é o injetor de
buracos e o Al, polarizado negativamente, é o injetor de elétrons (Figura 19-e.).
Neste caso, a barreira energética necessária para injetar elétrons e buracos é bem
menor quando comparada às energias no modo reverso, e as cargas injetadas pelos
eletrodos e fotogeradas pelo material orgânico se somam durante a condução pelo
campo elétrico aplicado, resultando em correntes consideravelmente maiores. Esta
etapa equivale ao ponto D da curva na Figura 20.
Figura 20: Curva característica de um dispositivo fotovoltaico, tanto no escuro quanto iluminado. A Figura mostra a tensão de circuito aberto (VOC) e a corrente de curto circuito
para o dispositivo iluminado. O ponto A representa a corrente do dispositivo durante a tensão reversa (negativa), o ponto B é conhecido como corrente de curto-circuito, o ponto
C é a tensão de circuito aberto e o ponto D a corrente no modo direto para V>VOC.

41
Um outro fator a ser analisado é o FF (fator de preenchimento), que dá a
potência máxima na qual o produto J.V atinge seu maior valor e representa a razão
entre a potência de entrada e a potência de saída. Graficamente, o FF é a razão entre
as áreas do retângulo verde e o retângulo cinza (de lados JSC e VOC), mostrados na
Figura 20. Na curva J-V ilustrada na Figura 20, pode-se observar dois retângulos;
no menor delimitado pela curva do dispositivo sob iluminação, a área representa o
máximo de energia por unidade de tempo (potência) fornecida pelo dispositivo; e
no maior retângulo, a área representa a potência nominal. O FF também é um
parâmetro de grande importância para análise do rendimento do dispositivo.
O rendimento (η) do dispositivo é definido pela razão da potência de saída
pela incidente [8]:
0
´
I
VJFF
P
P OCSC
incidente
saída ⋅⋅==η (Equação 6)
onde FF é o fator de preenchimento, JSC é a corrente de curto circuito, VOC é a
tensão de circuito aberto e I0 é a irradiância da lâmpada utilizada na caracterização.
Neste trabalho, a condutividade DC do material foi obtida registrando-se a
corrente que flui entre os eletrodos através do filme em função da tensão aplicada
pelo eletrômetro Keithley 617 ou fontes de tensão/corrente (modelos Keithley
2400). O estudo do comportamento fotocondutivo dos dispositivos foi realizado
medindo-se as curvas características J-V dos dispositivos no escuro e sob
iluminação, utilizando-se uma lâmpada de xenônio com irradiância de 100 mW/cm2
da Oriel como fonte de radiação luminosa.A lâmpada de xenônio foi utilizada
devido ao fato de possuir uma emissão semelhante à radiação solar que chega à
superfície terrestre, sendo, portanto, de uso conveniente na caracterização de
dispositivos fotovoltaicos. A incidência da iluminação se fez pelo lado do eletrodo
de ITO no dispositivo (Figura 21). As caracterizações óptica e elétrica dos
dispositivos foram realizadas no laboratório do Grupo de Polímero Bernhard Gross.

42
Figura 21: Representação de um dispositivo fotovoltaico de monocamada com estrutura tipo sanduíche durante a caracterização de condutividade DC, com
incidência de luz através do eletrodo de ITO
3.4. Condutividade persistente
As medidas de condutividade persistente (ou curvas densidade de corrente-
tempo J-t) são obtidas alternando entre a incidência de luz branca (100 mW/cm²)
proveniente da lâmpada de Xe e o escuro, verificando o comportamento da
fotocorrente em função do tempo de iluminação e após a lâmpada ser apagada. Esta
medida permite observar o comportamento e o tempo de vida dos portadores de
carga fotogerados.
A mesma instrumentação utilizada na caracterização de condutividade DC
foi utilizada para verificar a condutividade persistente nos dispositivos. Durante a
medida J-t, a tensão aplicada no dispositivo foi nula e o dispositivo estava em
curto-circuito.
3.5. Espectroscopia de impedância:
A espectroscopia de impedância permite analisar os processos de condução
que ocorrem dentro de um material sólido ou líquido que apresentam certa
resistividade elétrica. Ao aplicar uma diferença de potencial V(t) na amostra a ser
analisada, sendo que V(t) varia no tempo com frequência ω e amplitude V0:
(Equação 7)
A diferença de potencial aplicada gera uma corrente alternada no interior do
material, com uma diferença de fase θ (0 ≤ θ ≤ 2π) em relação ao potencial devida à

43
dificuldade que os portadores de carga encontram de se deslocarem dentro do
material. A corrente será, então, da seguinte forma:
I (Equação 8)
Dessa forma, tem-se a impedância complexa Z*:
∗
(Equação 9)
onde Z* = Z'(ω) - iZ"(ω), sendo que Z'(ω) representa a componente real da
impedância e Z"(ω) representa a componente imaginária.
Na espectroscopia de impedância, ao variar a frequência de oscilação do
potencial aplicado, o material também varia sua resposta, ou seja, a cada valor de
frequência de oscilação corresponde a um valor da impedância complexa na
amostra. O resultado é um espectro que fornece informações sobre os principais
processos envolvidos na relaxação dielétrica e sobre os tipos de portadores de
cargas no material.
Os resultados obtidos na espectroscopia de impedância também podem ser
expressos em termos da admitância complexa:
∗ !∗ (Equação 10)
onde Y'(ω) e Y''(ω) são:
!"!"#$!""# (Equação 11)
% !""!"#$!""# (Equação 12)
A condutividade complexa difere da admitância complexa apenas por
fatores geométricos, sendo L a distância percorrida pelos portadores de carga de um
eletrodo a outro (espessura da amostra) e A a área dos eletrodos. Desta forma, tem-
se que:
&∗ & &′ ∗ () (Equação 13)
onde &′ () e &′′ ().
Através da admitância complexa, ainda pode-se calcular a capacitância
complexa, sendo que:

44
*∗ * * +∗ (Equação 14)
sendo que * +" e * +""
.
Substituindo Y*(ω) na Equação 14:
*∗ -% ′′.′2′′201 % - 2
.′2′′201 (Equação 15)
Numa amostra que se comporta como um dielétrico ideal, pode-se
considerar que C'(ω) é constante em todo o intervalo de frequência utilizadas na
medida da espectroscopia de impedância. Portanto, extrapolando-se a curva de
C'(ω) obtida para o regime de altas frequências (ω→∞), é possível determinar a
constante dielétrica k do material pela seguinte relação:
* → ∞ 45)( (Equação 16)
na qual ε0 é a permissividade elétrica no vácuo, A é a área dos eletrodos da amostra
e L é a espessura da amostra.
Circuito equivalente
Para um sólido desordenado, a densidade de corrente possui a contribuição
da corrente de condução devida ao deslocamento das cargas livres e da corrente de
deslocamento, relacionada ao movimento de dipolos e a outros efeitos capacitivos.
A corrente total, pelas equações de Maxwell, é dada por:
678889 &:;89 < =>89= (Equação 17)
onde ;89 ;89 , σ0 está relacionado à condutividade do material e ϵ é a
permissividade do meio. Então:
678889 &: <;89 (Equação 18)
Chamando de σT = σ0 + iωϵ a condutividade total , tem-se que
678889 &7;89 (Equação 19)
Pode-se verificar que σT apresenta uma dependência da frequência de
oscilação do campo aplicado, o que é típico de materiais desordenados.

45
Considerando um material desordenado de comprimento l e área
longitudinal A, tem-se para este material que &: ? @A⁄ e < *? A⁄ , sendo R a
resistência do material e C sua capacitância. A impedância complexa ∗ ? &7A⁄ é, então:
∗ C $CD (Equação 20)
A Equação 20 é exatamente a que descreve a impedância de um circuito RC
em paralelo. A grandeza τ = RC é chamada de tempo de relaxação e é bastante
importante na análise do material, já que para materiais condutores τ→0 e para
isolantes τ→∞. Materiais com alto tempo de relaxação são chamados dielétricos. O
tempo de relaxação descreve o quão rápido as cargas espaciais se deslocam para
uma posição de equilíbrio, com menor energia potencial possível. O movimento de
dipolos gerados por moléculas polarizadas também contribuem ao tempo de
relaxação do material.
Seja ∗ |∗| , onde ∗ C $CD#F #⁄ e G tan KLM!∗N
CM!∗NO. Re[Z*] e
Im[Z*] são as partes real e imaginária de Z*, respectivamente, e suas expressões
são da seguinte forma:
@M∗N C $CD# (Equação 21)
PM∗N C#D $CD# (Equação 22)
onde Z* = Z' + Z" .
De maneira geral, a componente real da impedância complexa está
associada a resistência ao movimento das cargas que estão em fase com o campo
elétrico oscilante e a componente imaginária está relacionada a resistência à
corrente que está fora de fase. A corrente fora de fase com o campo elétrico
aplicado é devida a portadores de carga que não acompanham o campo devido a
dificuldades encontradas na estrutura do material e a moléculas polarizadas cujos
dipolos estejam sob influência do campo elétrico.
Ao analisar as equações 21 e 22 pode-se verificar que no limite em que
ω→0, Z'→R, já que o sistema aproxima-se do regime DC, sendo que R é
denominada resistência DC do material. No mesmo limite, tem-se que Z"→0, pois
não há corrente oscilando fora de fase quando não existe um campo oscilante. No

46
limite em que ω→∞, Z'→0 e Z"→0, pois neste caso o campo oscila tão rápido que
os portadores de carga se movem muito pouco em relação à sua posição original,
sendo desprezível a resistência do meio. Nesta mesma situação, os elementos
responsáveis pelos efeitos capacitivos entram em curto-circuito, sendo então a
impedância nula.
Desta forma, o comportamento universal dos sólidos desordenados é
verificado no espectro da Figura 22. Este comportamento é denominado
comportamento Debye e descreve o tempo de relaxação do material, que pode ser
de difícil descrição devido a heterogeneidade dos portadores de carga que
participam dos processos de condução elétrica. A frequência crítica, representada
por ω0, é um importante parâmetro para avaliar a condutividade do material, ou
seja, quanto maior a frequência crítica, maior a condutividade.
Figura 22: Espectro de impedância de um RC paralelo [79]
Na Figura 23, é representado o diagrama de Argand, que é útil na
identificação e comparação de processos de condução com diferentes tempos de
relaxação. Por meio das expressões de Re[Z'] e Im[Z"], tem-se que:
(Equação 23)
que corresponde à equação de um círculo de raio e centrado em e .
No diagrama de Argand só verifica-se a solução positiva para Z".

47
Figura 23: Espectro de impedância pelo diagrama de Argand [79]

48
4. Resultados e Discussões
Neste capítulo são apresentados os resultados experimentais obtidos a partir das
caracterizações óticas e elétricas dos filmes e dos dispositivos preparados neste trabalho.
4.1. Medidas Ópticas.
A seguir, são mostrados os resultados obtidos da caracterização das
propriedades ópticas dos filmes de (PAH/SCC)n, onde n é o número de bicamadas,
e de soluções de SCC em diferentes solventes e concentrações, através da
espectroscopia de absorção e de fluorescência. Os resultados da espectroscopia de
absorção e fluorescência para soluções de SCC para as concentrações de 0,01
mg/mL, 0,05 mg/mL e 0,1 mg/mL foram bastante similares. Optou-se por
apresentar os resultados da caracterização óptica apenas na concentração de 0,05
mg/mL.
4.1.1. Espectroscopia de Absorção:
Na Figura 24, é apresentado o espectro de absorção dos filmes de
PAH/SCC com diferentes números de bicamadas e da solução aquosa de SCC.
Para os filmes, pode-se verificar a presença das duas principais bandas de
absorção da SCC a banda Soret em torno de 408 nm e a banda Q, com máximos
próximos de 638 nm. Para a solução aquosa de SCC na concentração de 0,05
mg/mL também são observados os dois máximos de absorção característicos
(bandas Q e Soret), entretanto é observado um pequeno deslocamento
hipsocrômico (para a região do azul) do máximo das bandas Soret (∆λ = 6 nm) e
Q (∆λ = 12 nm) em relação à bandas Soret e Q do espectro de absorção do filme.
Tal deslocamento da banda Q também foi observado para filmes automontados e
solução aquosa de ftalocianina tetrassulfonada de ferro (FeTsPc) e duas
hipóteses foram consideradas por Zucolotto et al.: a primeira delas é que o
empilhamento colunar das moléculas de ftalocianina quando na forma de filme
aumenta a interseção entre os orbitais π das moléculas (π-stacking), diminuindo
a energia de gap e a segunda hipótese é a de que existe a interação do grupo não
protonado NH2 presente no PAH e o metal de coordenação da FeTsPc (neste
caso, o ferro), diminuindo a energia de transição da banda Q [80]. Porém, ainda
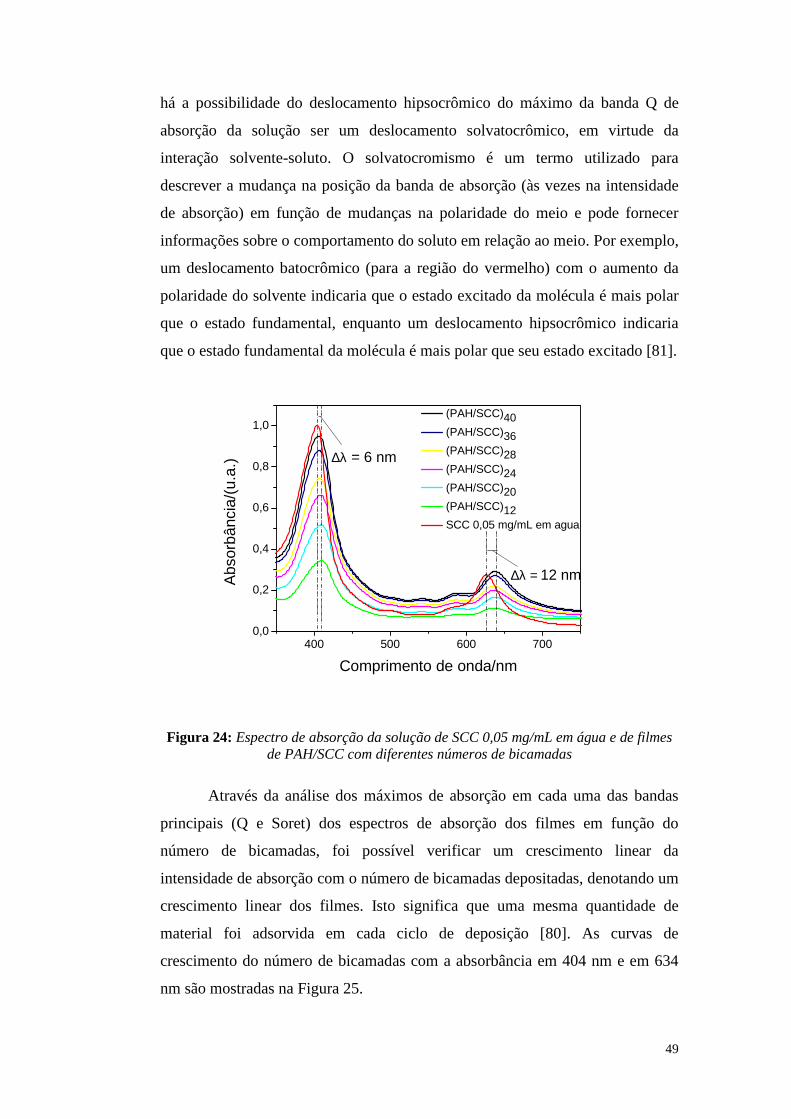
49
há a possibilidade do deslocamento hipsocrômico do máximo da banda Q de
absorção da solução ser um deslocamento solvatocrômico, em virtude da
interação solvente-soluto. O solvatocromismo é um termo utilizado para
descrever a mudança na posição da banda de absorção (às vezes na intensidade
de absorção) em função de mudanças na polaridade do meio e pode fornecer
informações sobre o comportamento do soluto em relação ao meio. Por exemplo,
um deslocamento batocrômico (para a região do vermelho) com o aumento da
polaridade do solvente indicaria que o estado excitado da molécula é mais polar
que o estado fundamental, enquanto um deslocamento hipsocrômico indicaria
que o estado fundamental da molécula é mais polar que seu estado excitado [81].
400 500 600 7000,0
0,2
0,4
0,6
0,8
1,0
Abs
orbâ
ncia
/(u.
a.)
Comprimento de onda/nm
(PAH/SCC)40 (PAH/SCC)36 (PAH/SCC)28 (PAH/SCC)24 (PAH/SCC)20 (PAH/SCC)12 SCC 0,05 mg/mL em agua
∆λ = 6 nm
∆λ = 12 nm
Figura 24: Espectro de absorção da solução de SCC 0,05 mg/mL em água e de filmes de PAH/SCC com diferentes números de bicamadas
Através da análise dos máximos de absorção em cada uma das bandas
principais (Q e Soret) dos espectros de absorção dos filmes em função do
número de bicamadas, foi possível verificar um crescimento linear da
intensidade de absorção com o número de bicamadas depositadas, denotando um
crescimento linear dos filmes. Isto significa que uma mesma quantidade de
material foi adsorvida em cada ciclo de deposição [80]. As curvas de
crescimento do número de bicamadas com a absorbância em 404 nm e em 634
nm são mostradas na Figura 25.

50
10 20 30 40
0,3
0,6
0,9
Abs
orbâ
ncia
/(u.
a.)
Numero de bicamadas
Banda Soret
10 20 30 40
0,1
0,2
0,3
Abs
orbâ
ncia
/(u.
a.)
Numero de bicamadas
Banda Q
Figura 25: Curvas de crescimento baseada nos máximos de absorção da banda Soret (406 nm) e da banda Q (638 nm) dos filmes de (PAH/SCC)n automontados em função
do número de bicamadas. A linha tracejada é guia para os olhos.
A relação entre o número de bicamadas n dos filmes (PAH/SCC)n e a
espessura dos filmes medidas pelo perfilômetro Veeco Dektak 150 está mostrada
na Tabela 1. Apesar de os filmes apresentarem um crescimento linear da
intensidade de absorção com o número de bicamadas, observou-se que as razões
entre as espessuras dos filmes e o número de bicamadas não permaneceram
constantes, embora não tenham se desviado de forma significativa. A causa disto
podem ser erros nas medidas de espessura devido à baixa precisão do
equipamentos para medidas de menores espessura.
Tabela 1: Relação entre o número de bicamadas e a espessura do filme de (PAH/SCC)n medida por perfilometria
Número de bicamadas Espessura (nm) Espessura por bicamada (nm)
8 10,0 1,25
12 17,0 1,42
16 20,0 1,25
20 23,0 1,15
24 40,0 1,67
28 45,0 1,61
36 54,0 1,50
40 62,0 1,55
Na Figura 26 estão os espectros de absorção de soluções de SCC em
diferentes solventes (água, etanol, isopropanol, clorofórmio e tolueno), na

51
concentração de 0,05 mg/mL. No preparo das soluções, observou-se que os
solventes capazes de diluir efetivamente a SCC foram a água e o etanol. No caso
do isopropanol, foi formada uma espécie de solução coloidal e para o tolueno e
clorofórmio não foi verificada solubilidade ou solubilidade muito baixa. Não
houve controle no pH das soluções de SCC nos diferentes solventes.
400 500 600 700
0,0
0,5
1,0
1,5
2,0
2,5
0,0 0,3 0,6 0,9 1,2 1,5 1,8 2,10
5
10
15
20
25
30
35
40
45
50
Co
ef.
Ab
s. M
ola
r (L
/g.c
m)
Momento Dipolar (D)
Banda Soret Banda Q
Comprimento de onda/nm
Abs
orbâ
ncia
/(u.
a.)
Agua Etanol Isopropanol Tolueno Cloroformio
Figura 26: Espectros de absorção das soluções de SCC em diferentes solventes com concentração de 0,05 mg/mL (em detalhe o gráfico do coeficiente de absorção molar
em função do momento dipolar do solvente)
A Tabela 2 mostra o momento dipolar dos solventes utilizados nas
soluções de SCC. O momento dipolar diz respeito à polaridade do solvente e,
quanto maior o momento dipolar, maior a sua polaridade. Uma molécula é dita
polar se o seu momento de dipolo Q9 é diferente de zero. Como a molécula de
SCC é polar (já que seu momento dipolar não é nulo), é previsível que ela seja
solúvel em solventes polares e que sua solubilidade decresca à medida que o
momento dipolar diminui.

52
Tabela 2: Momento dipolar dos solventes utilizados nas soluções de SCC [4].
Solvente Fórmula química Momento dipolar (D)
Tolueno C6H5-CH3 0,36
Clorofórmio CHCl3 1,04
Isopropanol CH3-CH(-OH)-CH3 1,66
Etanol CH3-CH2-OH 1,69
Água H2O 1,85
Inserido na Figura 26 está o gráfico dos coeficientes de absorção molar
das bandas Q e Soret em função do momento dipolar dos solventes (ver Tabela
2). De acordo com o gráfico, as bandas de absorção Q e Soret tem intensidades
similares e baixos valores de coeficiente de absorção molar ε (ver Tabela 3) para
solventes com baixa polaridade (0,36 ≤ D ≤ 1,66), enquanto para os solventes
mais polares (D ≥ 1,69), o coeficiente de absorção molar aumenta (ver Tabela 3)
e a intensidade da banda Soret é superior a banda Q, ou seja, a banda Soret
(transição S0-S2) é mais afetada pela polaridade do solvente. Entretanto, verifica-
se uma relação entre ε e a solubilidade da SCC nos solventes e visto que nenhum
dos solventes absorve na região do espectro eletromagnético analisado, quanto
menor a solubilidade, menor o número de moléculas de SCC no caminho óptico
e, consequentemente, menor a intensidade de absorção e o coeficiente de
absorção molar.

53
Tabela 3: Máximos de absorção da banda Soret dos espectros de absorção da SCC em diferentes solventes e seus respectivos coeficientes de absorção molar obtidos através
da Equação 4, sendo L = 1 cm o caminho óptico
Água Etanol Isopropanol Clorofórmio Tolueno
Ban
da
Sor
et Absorbância
(u.a) 2,57 1,22 0,26 0,15 0,11
ε (L/g.cm) 51,13 24,31 5,24 3,01 2,13
Ban
da Q
Absorbância
(u.a) 0,70 0,40 0,12 0,10 0,08
ε (L/g.cm) 14,08 8,08 2,48 1,94 1,67
Os espectros de absorção das soluções em diferentes solventes foram
normalizados em relação à banda Soret (Figura 27-a) e à banda Q (Figura 27-b),
a fim de se verificar eventuais deslocamentos na posição das bandas de absorção
com a mudança dos solventes.
350 400 450 5000,0
0,2
0,4
0,6
0,8
1,0
1,2 Agua Etanol Isopropanol Cloroformio Tolueno
Abs
orbâ
ncia
nor
mal
izad
aB
anda
Sor
et /(
u.a.
)
Comprimento de onda/nm
∆λ = 6 nm
(a)
550 600 650 700 7500,0
0,5
1,0
Agua Etanol Isopropanol Cloroformio Tolueno
Abs
orbâ
ncia
nor
mal
izad
aB
anda
Q /(
u.a.
)
Comprimento de onda/nm
∆λ = 14 nm
(b)
Figura 27: Espectros de absorção das soluções de SCC em diferentes solventes a 0,05 mg/mL, normalizados em relação às principais bandas (a) Soret e (b) Q.
Nos espectros de absorção das soluções, é possível observar um
deslocamento hipsocrômico da banda Q com o aumento da polaridade dos
solventes. O deslocamento solvatocrômico evidencia que há uma mudança da
energia de solvatação da SCC em solventes de diferentes polaridades, sendo que

54
esta energia está relacionada à distribuição eletrônica da molécula de SCC em
seus estados excitados quando interagem com o soluto. Supõe-se, então, que o
estado fundamental da SCC é cada vez mais polar à medida que o momento
dipolar do solvente aumenta, e que isso pode ser devido à mudança de
polaridade da molécula de SCC ocasionada pela ação das moléculas do solvente.
4.1.2. Espectroscopia de fluorescência:
A seguir, são mostrados os espectros de fluorescência das soluções de
SCC em diferentes solventes a uma mesma concentração (0,05 mg/mL). As
eventuais emissões por parte dos solventes utilizados foram subtraídas dos
espectros de fluorescência das soluções contendo SCC.
A Figura 28 mostra os espectros de fluorescência das soluções de SCC
em diferentes solventes quando excitadas nos comprimentos de onda de suas
respectivas bandas Soret. Para solventes menos polares, como o tolueno e o
clorofórmio, os picos de emissão estão localizados em regiões de menores
comprimentos de onda, quando comparados aos picos de emissão das soluções
em solventes mais polares (etanol e isopropanol), ou seja, há um deslocamento
batocrômico da emissão com o aumento da polaridade do solvente.
500 550 600 650 700 750
0
1
2
3
4
667 nm564 nm
572 nm agua etanol isopropanol cloroformio tolueno
Flu
ores
cênc
ia/(
u.a.
)
Comprimento de onda/nm
508 nm
Figura 28: Espectros de fluorescência de soluções de SCC em diferentes solventes à concentração de 0,05 mg/mL excitadas no comprimento de onda de suas respectivas
bandas Soret

55
De acordo com os resultados da Figura 28, observa-se que as
intensidades de emissão das soluções de SCC foram muito baixas. A energia da
emissão radiativa geralmente difere da energia de absorção e o efeito da
polaridade nesta diferença energética é explicado pelo efeito conhecido como
relaxação do solvente. Na maioria dos casos, o momento dipolo de uma
molécula aromática no estado excitado µe difere do momento dipolo quando no
estado fundamental µf, devido a uma redistribuição intramolecular de cargas
após a excitação. Após a excitação, as moléculas do solvente sofrem uma
reorganização, levando a um estado de relaxação cuja energia é minimizada
(Figura 29). Quanto maior a polaridade do solvente, menor a energia do estado
de relaxação e maior o deslocamento batocrômico (para o vermelho) no espectro
de emissão [29]. Na Figura 28, é possível verificar este efeito analisando-se os
comprimentos de onda dos máximos de emissão (valores mencionados na
figura) e relacionando-os com a polaridade dos solventes utilizados.
Figura 29: Diagrama de energias evidenciando o efeito da relaxação do solvente. As esferas centrais representam as moléculas do soluto e as periféricas representam as
moléculas do solvente, demonstrando a mudança do momento dipolar do estado excitado µe com relação ao do estado fundamental µf [29]
Não foi observada fluorescência em nenhum dos dois comprimentos de
onda de excitação para a solução aquosa de SCC. Supõe-se então que, após a
excitação, o decaimento da molécula de SCC em água para o estado fundamental
(S0) seja de forma não radiativa, por meio de conversões internas e cruzamentos
intersistemas. Há a hipótese de que a supressão de luminescência da solução
aquosa de SCC ocorre devido a presença dos íons de oxigênio dos grupos
periféricos da molécula solubilizada em água.

56
A Figura 30 mostra os espectros de fluorescência destas soluções quando
excitadas nos comprimentos de onda de suas respectivas bandas Q. De acordo
com os resultados, as soluções de SCC em tolueno, em clorofórmio e em
isopropanol apresentaram baixa intensidade de emissão e apenas uma emissão
de maior intensidade foi observada para o etanol em 667 nm.
650 675 700 725 750
0,0
0,5
1,0
1,5
2,0
2,5
3,0
Flu
ores
cênc
ia/(
u.a.
)
Comprimento de onda/nm
agua etanol isopropanol cloroformio tolueno
667 nm
Figura 30: Espectros de fluorescência de soluções de SCC em diferentes solventes à concentração de 0,05 mg/mL excitadas no comprimento de onda de suas respectivas
banda Q
A seguir são apresentados os espectros de absorção juntamente aos
espectros de fluorescência das soluções de SCC em diferentes solventes para
efeito de comparação.
Na Figura 31 estão os espectros de absorção e fluorescência da solução
de SCC em tolueno com concentração de 0,05 mg/mL. Para obtenção destes
espectros, os comprimentos de excitação utilizados foram de 410 nm (máximo
de absorção da banda Soret) e 645 nm (máximo de absorção da banda Q). Duas
bandas foram observadas no espectro de fluorescência quando a solução foi
excitada em 410 nm (azul escuro): uma com máximo em 508 nm e outra banda
mais larga com máximo em torno de 564 nm. De acordo com este resultado,
conclui-se que o decaimento radiativo da molécula de SCC em tolueno ocorre
apenas devido a transição do segundo estado excitado (S2) para o estado
fundamental (S0). Na excitação em 645 nm (azul claro), não houve

57
fluorescência, o que significa que o decaimento S1-S0 ocorre por meios não
radiativos.
400 500 600 700
0,00
0,01
0,02
0,03
0,04
Comprimento de onda/nm
Abs
orbâ
ncia
/(u.
a.)
0
1
2
3
4F
luorescência/(u.a.)
Figura 31: Espectro de absorção da solução de SCC em tolueno com concentração de 0,05 mg/mL (em preto) e seus espectros de fluorescência quando excitada em 410 nm
(azul escuro) e em 645 nm (azul claro)
Os espectros de absorção e fluorescência da solução de SCC em
clorofórmio com concentração de 0,05 mg/mL são mostrados na Figura 32 e
foram obtidos através da excitação nos máximos das bandas Soret (407 nm) e Q
(638 nm). Foi observada uma banda de fluorescência com máximo em 572 nm
quando a solução foi excitada no comprimento de onda do máximo da banda
Soret (azul escuro) e um ombro em torno de 665 nm. Na excitação no
comprimento de onda de absorção da banda Q não houve emissão. Observa-se
que o decaimento radiativo da molécula de SCC em clorofórmio ocorreu através
da transição S2-S0. Entretanto, o ombro em 665 nm sugere uma transição do tipo
S1-S0, após o elétron decair do estado excitado S2 para o S1 via conversão interna
(IC).

58
400 500 600 700
0,00
0,02
0,04
0,06
0,08
Comprimento de onda/nm
Abs
orbâ
ncia
/(u.
a.)
0
1
2
3
4
5
6
Fluorescência/(u.a.)
Figura 32: Espectro de absorção da solução de SCC em clorofórmio com concentração de 0,05 mg/mL (em preto) e seus espectros de fluorescência
quando excitada em 407 nm (azul escuro) e em 638 nm (azul claro)
Para a solução de SCC em isopropanol com concentração de 0,05 mg/mL
foram obtidos os espectros de absorção e fluorescência apresentados na Figura
33. As excitações ocorreram em 407 nm, máximo da banda Soret e em 630 nm,
máximo da banda Q. Quando excitada em 407 nm (azul escuro), a solução
apresentou uma banda larga de luminescência com máximo em 496 nm (relativa
ao decaimento S2-S0) e outra banda mais intensa e definida com máximo em
torno de 665 nm (relativa ao decaimento não radiativo S2-S1 seguido de um
decaimento radiativo S1-S0). Na excitação em 630 nm (azul claro), foram
verificadas bandas com máximos em 673 nm, 696 nm e 736 nm (transição S1-
S0).

59
400 500 600 700
0,00
0,05
0,10
0,15
0,20
Comprimento de onda/nm
Abs
orbâ
ncia
/(u.
a.)
0,0
0,3
0,6
0,9
1,2
Fluorescência/(u.a.)
Figura 33: Espectro de absorção da solução de SCC em isopropanol com concentração de 0,05 mg/mL (em preto) e seus espectros de fluorescência quando
excitada em 407 nm (azul escuro) e em 630 nm (azul claro)
Na Figura 34, estão apresentados os espectros de absorção e
fluorescência da solução de SCC em etanol, com concentração de 0,05 mg/mL,
excitada em seus máximos de absorção, sendo 405 nm para a banda Soret e 625
para a banda Q. Em ambos os comprimentos de onda de excitação foram
verificados uma banda de luminescência com máximo em torno de 667 nm.
Neste caso, as emissões ocorrem apenas devido as transições eletrônicas S1-S0.
Houve uma transição de S2-S1 por meio de conversão interna, ocorrendo, em
seguida, o decaimento radiativo via transição S1-S0.

60
400 500 600 700
0,0
0,3
0,6
0,9
1,2
Comprimento de onda/nm
Abs
orbâ
ncia
/(u.
a.)
0
1
2
3
Fluorescência/(u.a.)
Figura 34: Espectro de absorção da solução de SCC em etanol com concentração de 0,05 mg/mL (em preto) e seus espectros de fluorescência quando excitada em 405 nm
(azul escuro) e em 625 nm (azul claro)
Na Figura 35, são mostrados os espectros de fluorescência da solução de
SCC em água na concentração de 0,05 mg/mL excitada em seus máximos de
absorção: 404 nm (banda Soret) e 627 (banda Q). Nesta mesma figura é
apresentado o espectro de absorção da solução de aquosa de SCC a 0,05 mg/mL
para comparação. Nenhuma fluorescência foi verificada.
400 500 600 700
0,0
0,5
1,0
1,5
2,0
2,5
3,0
Comprimento de onda/nm
Abs
orbâ
ncia
/(u.
a.)
0,0
0,5
1,0
1,5
2,0
2,5
3,0
Fluorescência/(u.a.)
Figura 35: Espectro de absorção da solução de SCC em água com concentração de 0,05 mg/mL (em preto) e seus espectros de fluorescência quando excitada em 404 nm
(azul escuro) e em 627 nm (azul claro)

61
Observando-se o comportamento dos espectros de fluorescência, pode-se
deduzir que nas soluções de SCC em clorofórmio e tolueno ocorrem transições
do estado S2 para S0, já que o espectro destas soluções apresenta fluorescência
em menor comprimento de onda, ou seja, em maior energia. No caso das
soluções de SCC em etanol e isopropanol, as transições podem ser de S1 para S0,
resultando numa menor energia verificada no espectro de fluorescência.
Os espectros de fluorescência dos filmes de PAH/SCC com diferentes
números de bicamadas são mostrados na Figura 36 e foram obtidos através da
excitação nos máximos das bandas Soret (406 nm) e Q (635 nm). Não foi
observada luminescência em nenhum dos filmes quando excitados em ambos os
comprimentos de onda dos máximos de absorção. Da mesma forma que na
solução aquosa de SCC, os filmes podem ter sua emissão suprimida pela
presença de íons O- formados na solubilização da SCC em água para a confecção
dos filmes pela automontagem.
500 600 700 8000
100
200
Flu
ores
cênc
ia/(
u.a.
)
Comprimento de onda/nm
(PAH/SCC)40
(PAH/SCC)12
(PAH/SCC)8 λ
ex = 406 nm
700 725 750 775 8000
100
200
300
(PAH/SCC)40
(PAH/SCC)12
(PAH/SCC)8
Flu
ores
cênc
ia/(
u.a.
)
Comprimento de onda/nm
λex
= 635 nm
Figura 36: Espectros de fluorescência dos filmes de SCC com diferentes número de bicamadas excitados nos máximos de absorção das bandas Soret e Q
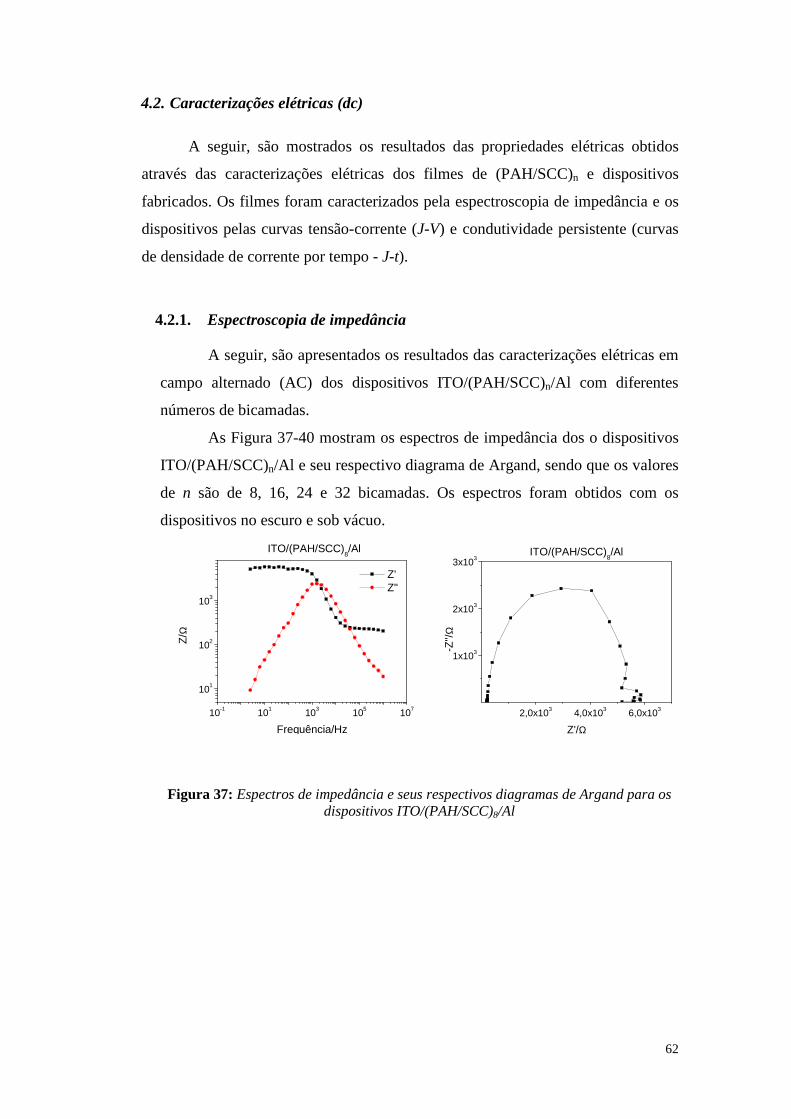
62
4.2. Caracterizações elétricas (dc)
A seguir, são mostrados os resultados das propriedades elétricas obtidos
através das caracterizações elétricas dos filmes de (PAH/SCC)n e dispositivos
fabricados. Os filmes foram caracterizados pela espectroscopia de impedância e os
dispositivos pelas curvas tensão-corrente (J-V) e condutividade persistente (curvas
de densidade de corrente por tempo - J-t).
4.2.1. Espectroscopia de impedância
A seguir, são apresentados os resultados das caracterizações elétricas em
campo alternado (AC) dos dispositivos ITO/(PAH/SCC)n/Al com diferentes
números de bicamadas.
As Figura 37-40 mostram os espectros de impedância dos o dispositivos
ITO/(PAH/SCC)n/Al e seu respectivo diagrama de Argand, sendo que os valores
de n são de 8, 16, 24 e 32 bicamadas. Os espectros foram obtidos com os
dispositivos no escuro e sob vácuo.
10-1 101 103 105 107
101
102
103
ITO/(PAH/SCC)8/Al
Z/Ω
Frequência/Hz
Z' Z"
2,0x103 4,0x103 6,0x103
1x103
2x103
3x103
-Z"/
Ω
Z'/Ω
ITO/(PAH/SCC)8/Al
Figura 37: Espectros de impedância e seus respectivos diagramas de Argand para os dispositivos ITO/(PAH/SCC)8/Al

63
10-2 10-1 100 101 102 103 104 105 106101
102
103
104
105
106
ITO/(PAH/SCC)16
/Al
Z/Ω
Frequência/Hz
Z' Z"
0 1x106 2x106 3x1060,0
5,0x105
1,0x106
-Z"/
Ω
Z'/Ω
ITO/(PAH/SCC)16
/Al
Figura 38: Espectros de impedância e seus respectivos diagramas de Argand para os dispositivos ITO/(PAH/SCC)16/Al
10-1 100 101 102 103 104 105 106
102
103
104
105
106
ITO/(PAH/SCC)24
/Al
Z/Ω
Frequência/Hz
Z' Z"
0,0 2,0x106 4,0x106 6,0x100,0
6,0x105
1,2x106
-Z"/
Ω
Z'/Ω
ITO/(PAH/SCC)24
/Al
Figura 39: Espectros de impedância e seus respectivos diagramas de Argand para os dispositivos ITO/(PAH/SCC)24/Al
10-2 100 102 104 106101
103
105
107
109ITO/(PAH/SCC)
32/Al
Z/Ω
Frequência/Hz
Z' Z"
0,0 2,0x107 4,0x107 6,0x100,0
2,0x107
4,0x107
-Z"/
Ω
Z'/Ω
ITO/(PAH/SCC)32
/Al
Figura 40: Espectros de impedância e seus respectivos diagramas de Argand para os dispositivos ITO/(PAH/SCC)32/Al
De acordo com os resultados das medidas AC, observa-se que os filmes
que constituem a camada fotoativa dos dispositivos ITO/(PAH/SCC)n/Al com

64
diferentes valores de n possuem um comportamento típico dos sólidos
desordenados, onde o mecanismo de condução é feito por saltos entre sítios
vizinhos (hopping), conforme discutido na seção 3.5 deste trabalho e verificado
por Farag et al [40]. Em baixas frequências, espera-se que as curvas de
impedância apresentem um patamar, entretanto este patamar só foi nitidamente
observado no espectro de impedância do dispositivo ITO/(PAH/SCC)8/Al
(Figura 37), sendo que o valor da impedância neste patamar é o valor da
resistência DC deste dispositivo. Nas curvas dos demais dispositivos, utilizou-se
o diagrama de Argand para obter-se o valor das resistências DC. Os valores das
resistências DC dos dispositivos ITO/(PAH/SCC)n/Al, bem como as
condutividades e resistividades calculadas, estão na Tabela 4. Os filmes se
mostraram bastante resistivos e verificou-se um aumento da resistividade com o
número de bicamadas, com uma diferença da ordem de 103 entre o filme mais
fino (8 bicamadas) e o mais espesso (32 bicamadas).
Na Figura 41 são mostrados os gráficos de condutividade em função da
frequência, sendo que a frequência crítica nos dispositivos com diferentes
números de bicamadas está evidenciada em cada um dos gráficos. Como a
frequência crítica está relacionada com a condutividade, sendo que o aumento de
condutividade acarreta em um aumento da frequência crítica, é possível analisar
a condutividade das camadas fotoativas por meio deste parâmetro. Comparando-
se os valores de frequência crítica para o dispositivo com filme (PAH/SCC) de 8
bicamadas (2109,9 Hz) e de 32 bicamadas (0,1 Hz), pode-se observar uma
expressiva diferença entre estes valores e desta forma concluir que o aumento do
número de bicamadas resulta em uma diminuição da condutividade do filme.

65
10-1 101 103 105 10710-10
10-8
10-6
ITO/(PAH/SCC)8/Al
Con
dutiv
idad
e co
mpl
exa/
(Ω m
)
Frequência/Hz
Real Imaginario
2109,9 Hz
10-2 100 102 104 10610-11
10-9
10-7
10-5
ITO/(PAH/SCC)16
/Al
Con
dutiv
idad
e co
mpl
exa/
(Ω m
)
Frequência/Hz
Real Imaginario
3,3 Hz
10-1 101 103 105
10-8
10-6
10-4ITO/(PAH/SCC)
24/Al
Con
dutiv
idad
e co
mpl
exa/
(Ω m
)
Frequência/Hz
Real Imaginario
5,04 Hz
10-2 100 102 104 10610-11
10-9
10-7
10-5
ITO/(PAH/SCC)32
/Al
Con
dutiv
idad
e co
mpl
exa/
(Ω m
)
Frequência/Hz
Real Imaginario
0,1 Hz
Figura 41: Gráficos da condutividade em função da frequência de oscilação do campo aplicado para os dispositivos ITO/(PAH/SCC)n/Al, com n = 8, 16, 24 e 32
Na Figura 42 é mostrado um gráfico de dependência da componente real
da capacitância C' com a frequência de oscilação do campo para todos os
dispositivos analisados na espectroscopia de impedância. A capacitância foi
obtida pela extrapolação do valor de C' a frequências médias (linha tracejada na
Figura 42), que tende a ser constante. A constante dielétrica k foi calculada para
cada um dos dispositivos pela Equação 16 e os valores obtidos estão na Tabela
4.
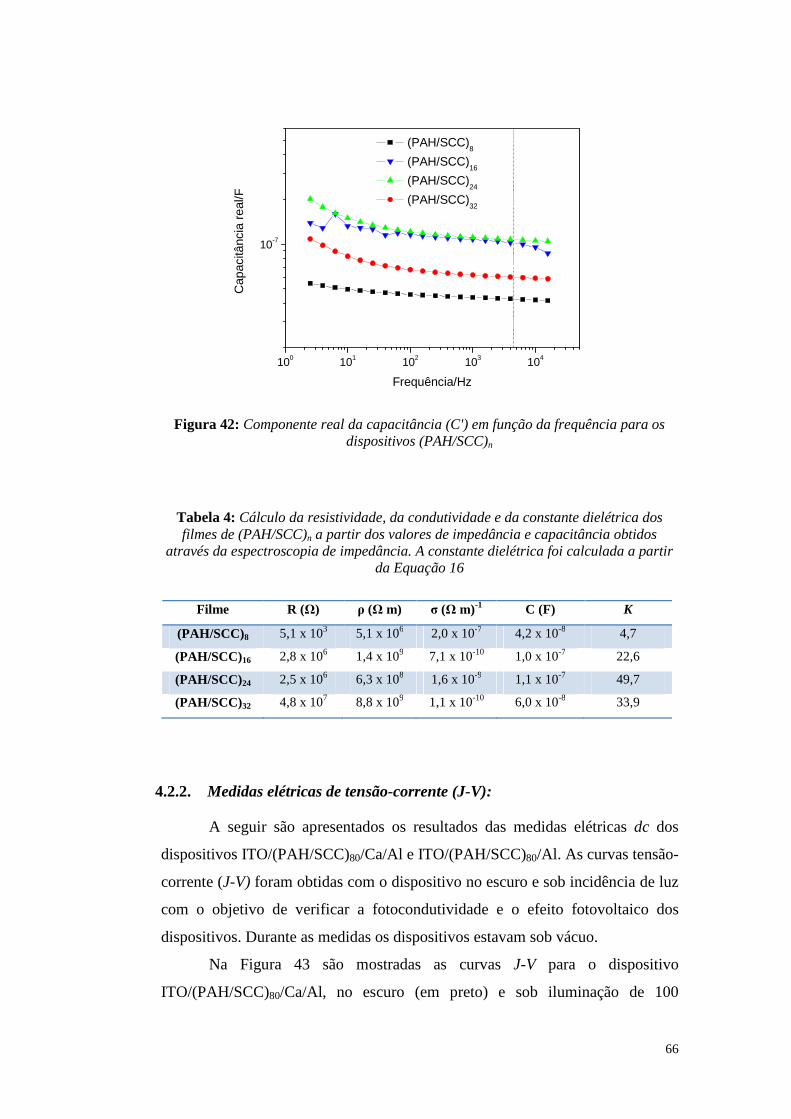
66
100 101 102 103 104
10-7
(PAH/SCC)8
(PAH/SCC)16
(PAH/SCC)24
(PAH/SCC)32
Cap
acitâ
ncia
rea
l/F
Frequência/Hz
Figura 42: Componente real da capacitância (C') em função da frequência para os dispositivos (PAH/SCC)n
Tabela 4: Cálculo da resistividade, da condutividade e da constante dielétrica dos filmes de (PAH/SCC)n a partir dos valores de impedância e capacitância obtidos
através da espectroscopia de impedância. A constante dielétrica foi calculada a partir da Equação 16
Filme R (Ω) ρ (Ω m) σ (Ω m)-1 C (F) K
(PAH/SCC)8 5,1 x 103 5,1 x 106 2,0 x 10-7 4,2 x 10-8 4,7
(PAH/SCC)16 2,8 x 106 1,4 x 109 7,1 x 10-10 1,0 x 10-7 22,6
(PAH/SCC)24 2,5 x 106 6,3 x 108 1,6 x 10-9 1,1 x 10-7 49,7
(PAH/SCC)32 4,8 x 107 8,8 x 109 1,1 x 10-10 6,0 x 10-8 33,9
4.2.2. Medidas elétricas de tensão-corrente (J-V):
A seguir são apresentados os resultados das medidas elétricas dc dos
dispositivos ITO/(PAH/SCC)80/Ca/Al e ITO/(PAH/SCC)80/Al. As curvas tensão-
corrente (J-V) foram obtidas com o dispositivo no escuro e sob incidência de luz
com o objetivo de verificar a fotocondutividade e o efeito fotovoltaico dos
dispositivos. Durante as medidas os dispositivos estavam sob vácuo.
Na Figura 43 são mostradas as curvas J-V para o dispositivo
ITO/(PAH/SCC)80/Ca/Al, no escuro (em preto) e sob iluminação de 100
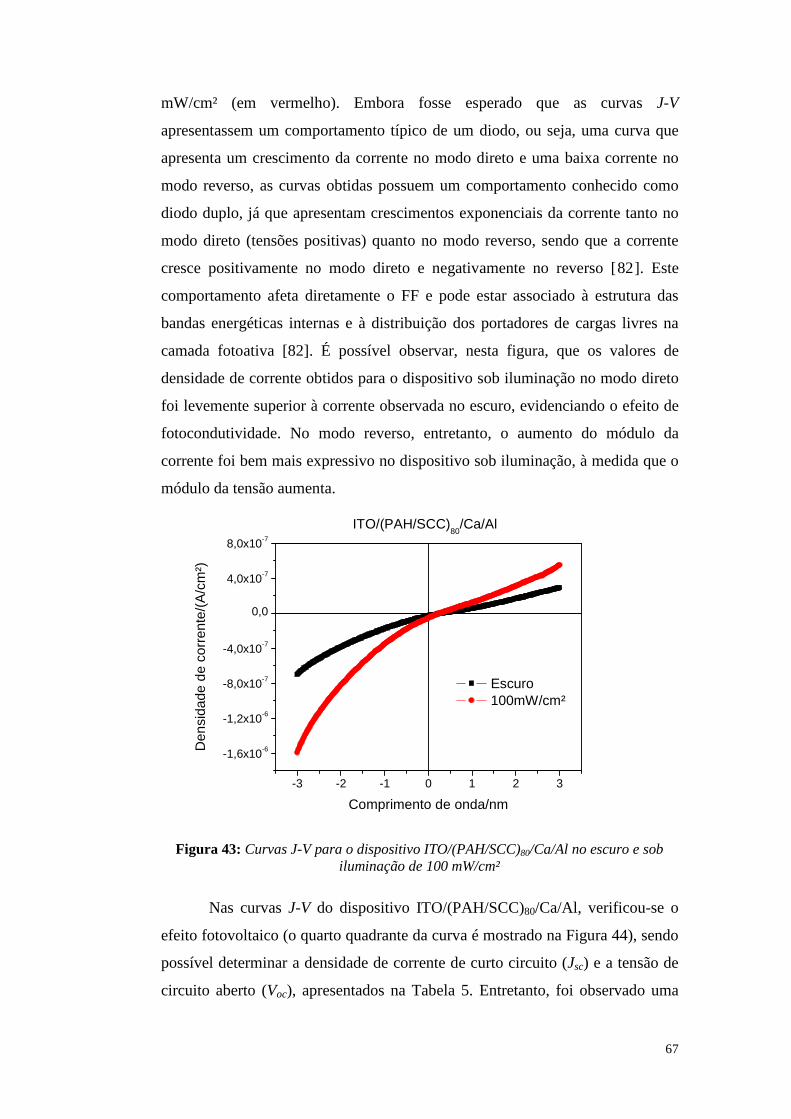
67
mW/cm² (em vermelho). Embora fosse esperado que as curvas J-V
apresentassem um comportamento típico de um diodo, ou seja, uma curva que
apresenta um crescimento da corrente no modo direto e uma baixa corrente no
modo reverso, as curvas obtidas possuem um comportamento conhecido como
diodo duplo, já que apresentam crescimentos exponenciais da corrente tanto no
modo direto (tensões positivas) quanto no modo reverso, sendo que a corrente
cresce positivamente no modo direto e negativamente no reverso [82]. Este
comportamento afeta diretamente o FF e pode estar associado à estrutura das
bandas energéticas internas e à distribuição dos portadores de cargas livres na
camada fotoativa [82]. É possível observar, nesta figura, que os valores de
densidade de corrente obtidos para o dispositivo sob iluminação no modo direto
foi levemente superior à corrente observada no escuro, evidenciando o efeito de
fotocondutividade. No modo reverso, entretanto, o aumento do módulo da
corrente foi bem mais expressivo no dispositivo sob iluminação, à medida que o
módulo da tensão aumenta.
-3 -2 -1 0 1 2 3
-1,6x10-6
-1,2x10-6
-8,0x10-7
-4,0x10-7
0,0
4,0x10-7
8,0x10-7
Den
sida
de d
e co
rren
te/(
A/c
m²)
Comprimento de onda/nm
Escuro 100mW/cm²
ITO/(PAH/SCC)80
/Ca/Al
Figura 43: Curvas J-V para o dispositivo ITO/(PAH/SCC)80/Ca/Al no escuro e sob iluminação de 100 mW/cm²
Nas curvas J-V do dispositivo ITO/(PAH/SCC)80/Ca/Al, verificou-se o
efeito fotovoltaico (o quarto quadrante da curva é mostrado na Figura 44), sendo
possível determinar a densidade de corrente de curto circuito (Jsc) e a tensão de
circuito aberto (Voc), apresentados na Tabela 5. Entretanto, foi observado uma

68
corrente de curto circuito na curva J-V do dispositivo no escuro, que pode ser
devida a um efeito capacitivo, no qual as cargas que não foram extraídas pelos
eletrodos são acumuladas na camada fotoativa.
0,0 0,2
-6,0x10-5
-3,0x10-5
0,0
Den
sida
de d
e co
rren
te/(
A/c
m²)
Comprimento de onda/nm
Escuro 100mW/cm²
ITO/(PAH/SCC)80
/Ca/Al
Figura 44: Quarto quadrante das curvas J-V para o dispositivo ITO/(PAH/SCC)80/Ca/Al no escuro e sob iluminação de 100 mW/cm²
A Figura 45 mostra as curvas J-V para o dispositivo
ITO/(PAH/SCC)80/Al no escuro (em preto) e sob iluminação de 100 mW/cm²
(em azul). Em ambas as curvas, o efeito retificador não foi observado, sendo
curvas J-V características de duplo diodo. Na curva J-V do dispositivo sob
iluminação, pôde-se verificar a ocorrência do efeito fotovoltaico e a presença de
uma JSC e de uma VOC (Tabela 5), melhor verificados no aumento do quarto
quadrante da curva J-V (Figura 46).

69
-2 -1 0 1 2 3-0,08
-0,04
0,00
0,04
0,08
Escuro 100 mW/cm2
Den
sida
de d
e co
rren
te/(
mA
/cm
2 )
Voltagem/V
ITO/(PAH/SCC)80
/Al
Figura 45: Curvas J-V para o dispositivo ITO/(PAH/SCC)80/Al no escuro e sob iluminação de 100 mW/cm²
0,0 0,1 0,2 0,3 0,4 0,5 0,6
-4,0x10-4
-2,0x10-4
0,0
2,0x10-4
Escuro 100 mW/cm2
Den
sida
de d
e co
rren
te/(
mA
/cm
2 )
Voltagem/V
ITO/(PAH/SCC)80
/Al
Figura 46: Quarto quadrante das curvas J-V para o dispositivo ITO/(PAH/SCC)80/Al no escuro e sob iluminação de 100 mW/cm²
Em dispositivos fotovoltaicos reais, o circuito simplificado equivalente é
mostrado na Figura 47. A resistência em série representa perdas ôhmicas nos
eletrodos e a resistência em paralelo representa a perda na corrente.

70
Figura 47: Circuito simplificado equivalente a um dispositivo fotovoltaico ideal. A resistência em paralelo RP representa todas as perdas na corrente enquanto a
resistência em série RS representa a resistividade do circuito, resistividade nos contatos elétricos e a resistividade da camada ativa [52]
A corrente do diodo para um dispositivo convencional é da seguinte
forma:
(Equação 24)
onde I0 é a corrente de saturação, RS é a resistência em série, RP é a resistência
em paralelo, n é o valor de idealidade do diodo e q/kT é o potencial da
temperatura (25 mV à temperatura ambiente) [52]. Quando varia-se os valores
de RS e RP, a forma da curva I-V se modifica, como mostrado na Figura 48.
Figura 48: Quarto quadrante das curvas J-V características de um dispositivo fotovoltaico no caso ideal (RP = ∞ e RS = 0) e em casos onde existe variações em RS ou
em RP [52]
A curva J-V dos dispositivos fabricados sugerem que houveram perdas
de corrente devido a RS e RP, evidenciando uma baixa extração de portadores de
cargas pelos eletrodos que, que pode ser devida a fatores que impedem o
transporte dos portadores aos eletrodos, como a recombinação dos pares
excitônicos, presença de armadilhas, ou mesmo a espessura do filme.

71
Comparando os rendimentos dos dispositivos calculados a partir da
Equação 6 (Tabela 5), foi possível verificar que o dispositivo
ITO/(PAH/SCC)80/Ca/Al apresentou menor rendimento comparado ao do
dispositivo ITO/(PAH/SCC)80/Al, evidenciando a influência do eletrodo de
cálcio na diminuição do rendimento do dispositivo. Na tabela ainda constam os
valores de JSC, VOC e FF para um dispositivo ITO/CuTsPc/C60/Al [38] para
comparação com os valores dos dispositivos fabricados neste trabalho.
Tabela 5: Valores das correntes de curto circuito, das tensões de circuito aberto, dos fatores de preenchimento e dos rendimentos para os dispositivos
ITO/(PAH/SCC)80/Ca/Al e ITO/(PAH/SCC)80/Al
Dispositivos JSC(mA/cm²) VOC(V) FF η(%)
ITO/(PAH/SCC)80/Ca/Al 5,43 x 10-5 0,26 0,24 3,39 x 10-6
ITO/(PAH/SCC)80/Al 2,00 x 10-4 0,50 0,32 3,20 x 10-5
ITO/CuTsPc/C60/Al [38] 1,56 0,29 0,50 0,30
A Figura 49 mostra uma representação esquemática das bandas de
energia da ftalocianina tetrassulfonada de cobre (CuTsPc) comparadas às
funções trabalho dos eletrodos utilizados na confecção dos dispositivos [83]. A
CuTsPc pode ser comparada energeticamente à SCC por serem porfirinas de
estruturas similares, com valores de energia dos orbitais HOMO e LUMO
parecidos. Ao analisar as energias dos eletrodos, era esperado que os
dispositivos fabricados com eletrodos de Ca apresentassem maior VOC (1,3 V) e
JSC, acarretando em maior rendimento quântico (η). Entretanto, isto não ocorre e
o VOC do dispositivo com eletrodo de Ca é bem menor que o esperado (0,26 V).
A falha destes dispositivos pode estar na baixa coleta de cargas pelo Ca,
ocasionando acúmulo de cargas na interface eletrodo/camada fotoativa e
favorecendo a recombinação de cargas nesta interface. Pela análise do diagrama
da Figura 49, uma sugestão de eletrodo metálico seria o magnésio, de função
trabalho entre 3,7 e 3,8 eV, que seria um melhor coletor de elétrons e
contribuiria para um maior valor de VOC.

72
Figura 49: Representação esquemática das bandas de energia da CuTsPc [83] comparadas às funções trabalho dos eletrodos utilizados neste trabalho
4.2.3. Condutividade persistente
A Figura 50 mostra a curva de fotocondutividade por tempo (J-t) para o
dispositivo ITO/(PAH/SCC)80/ Ca/Al e a Figura 51 mostra a curva J-t para o
dispositivo ITO/(PAH/SCC)80/Al. Ambas as curvas foram obtidas com intervalo
de alternância entre a incidência de luz branca (100 mW/cm²) e o escuro de
cerca de sessenta segundos.
Nas duas curvas, observou-se a densidade de corrente fotogerada diminui
ligeiramente com o aumento do número de ciclos de alternância entre a luz e o
escuro. Além disso, em ambos os gráficos verifica-se a presença de um a
corrente de fundo, já que quando a luz está apagada, a corrente fotogerada nunca
atinge o valor nulo.
Nas curvas de fotocondutividade por tempo, pode-se verificar que
quando a luz incidente na amostra é interrompida, a condutividade fotoinduzida
exibe uma relaxação lenta num regime temporal, o que é denominada
condutividade persistente.

73
0 100 200 300 400 500-1,4x10-5
-1,2x10-5
-1,0x10-5
-8,0x10-6
-6,0x10-6
-4,0x10-6
-2,0x10-6
0,0
Den
sida
de d
e co
rren
te/(
mA
/cm
²)
Tempo/s
liga desl.
Jfundo = 3,47 x 10-6 mA/cm2
Figura 50: Curva J-t para o dispositivo ITO/(PAH/SCC)80/Ca/Al obtida alternando-se entre a incidência de luz branca (100 mW/cm²) e o escuro
0 100 200 300 400 500 600 700 800
-4,0x10-4
-3,0x10-4
-2,0x10-4
-1,0x10-4
0,0
Den
sida
de d
e co
rren
te/(
mA
/cm
2 )
Tempo/s
liga desl.Jfundo = 2,87 x 10-5 mA/cm2
Figura 51: Curva J-t para o dispositivo ITO/(PAH/SCC)80/Al obtida alternando-se entre a incidência de luz branca (100 mW/cm²) e o escuro

74
5. Análise dos resultados
Neste capítulo é realizada a análise de alguns resultados experimentais a fim de
se obter os gaps energéticos da SCC, a eficiência dos dispositivos e o tempo de
decaimento dos portadores na fotocondutividade persistente.
5.1. Determinação do gap de energia da SCC
Para se obter informações acerca das transições entre bandas, pode-se
analisar o espectro de absorção nos intervalos onde ocorrem a maior absorção
através da teoria de um elétron [84], que possibilita a descrição de um sistema com
vários elétrons a partir da solução do problema de um único elétron. Esta teoria
pode ser usada para analisar a absorção de sólidos moleculares como derivados das
porfirinas [85,86].
A relação de Tauc [87] é utilizada para a determinação do gap óptico Eg
para materiais amorfos. Em seu trabalho inicial, Tauc obteve uma relação entre a
parte imaginária da constante dielétrica (ε2 = 2nk) e a energia dos fótons incidentes,
por meio da seguinte relação:
RSR AT % ;U (Equação 25)
O coeficiente de absorção α também pode ser escrito através da relação de
Tauc,
VWXY AWX % ;U (Equação 26)
onde Eg é a gap óptico correspondente à transição indicada, o fator A depende da
probabilidade de transição, podendo ser assumido como constante. O valor de r
depende do tipo de transição, sendo 2, 1/2 e 2/3 para transições direta, indireta e
proibida, respectivamente. Fazendo o gráfico de (αhν)r vs. hν em energias próximas
aos máximos de absorção das bandas Q e Soret e fazendo a extrapolação da reta até
o zero de absorção, é possível obter os valores dos gap energéticos.
Para o espectro de absorção da solução aquosa de SCC 0,05 mg/mL, a
dependência de (αhν)r da energia luminosa hν foram traçadas para diferentes
valores de r e o melhor ajuste foi obtido para r = 2, tendo, portanto,
comportamentos característicos de transições diretas. O caminho óptico foi de 1
cm, utilizado para calcular α. Os valores de energia correspondentes às transições

75
energéticas foram 2,93 eV para a banda Soret e 1,90 eV para a banda Q. Os ajustes
podem ser verificados na Figura 52.
2,75 2,80 2,85 2,90 2,95 3,000
10
20
30
40
(αhν
)2 /(eV
/cm
)2
hν/eV
Banda SoretSoluçao SCC 0,05 mg/mL
Eg=2,930 eV
1,75 1,80 1,85 1,90 1,95 2,000,0
0,5
1,0
1,5 Banda QSoluçao SCC 0,05 mg/mL
(αhν
)2 /(eV
/cm
)2
hν/eV
Eg=1,903 eV
Figura 52: Ajustes das curvas (αhν)²-hν em energias próximas aos máximos de absorção, a fim de se calcular o gap energético envolvido em cada uma das transições das bandas
Soret e Q para a solução aquosa de SCC com concentração de 0,05 mg/mL
Os ajustes utilizando a relação de Tauc também foram realizados para as
curvas (αhν)r-hν traçadas a partir dos espectro de absorção do filme de
(PAH/SCC)80 e foram melhor ajustados utilizando r = 2. O caminho óptico
considerado no cálculo de α foi de metade da espessura do filme, dispensando,
desta forma, a contribuição do PAH para a espessura do filme. As energias de gap
resultantes dos ajustes foram de 2,88 eV para a banda Soret e 1,815 eV para a
banda Q. Tais ajustes podem ser observados na Figura 53. Comparando-se estes
resultados aos obtidos nos ajustes da absorção da SCC em solução aquosa, verifica-
se que houve um pequeno decréscimo nas energias de gap das duas principais
bandas de absorção (Soret e Q), conforme verificado nos espectros de absorção (ver
Figura 24).
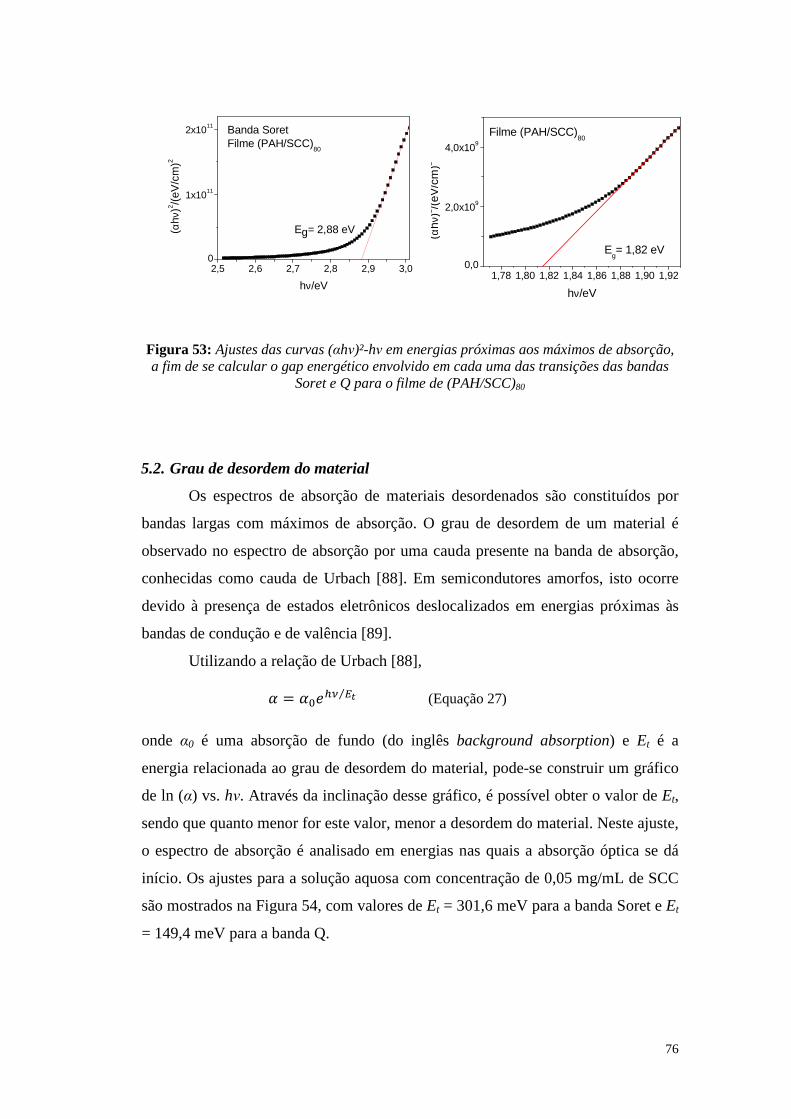
76
2,5 2,6 2,7 2,8 2,9 3,00
1x1011
2x1011 Banda SoretFilme (PAH/SCC)
80
(αhν
)2 /(eV
/cm
)2
hν/eV
Eg= 2,88 eV
1,78 1,80 1,82 1,84 1,86 1,88 1,90 1,920,0
2,0x109
4,0x109
Filme (PAH/SCC)80
(αhν
)2 /(eV
/cm
)2
hν/eV
Eg= 1,82 eV
Figura 53: Ajustes das curvas (αhν)²-hν em energias próximas aos máximos de absorção, a fim de se calcular o gap energético envolvido em cada uma das transições das bandas
Soret e Q para o filme de (PAH/SCC)80
5.2. Grau de desordem do material
Os espectros de absorção de materiais desordenados são constituídos por
bandas largas com máximos de absorção. O grau de desordem de um material é
observado no espectro de absorção por uma cauda presente na banda de absorção,
conhecidas como cauda de Urbach [88]. Em semicondutores amorfos, isto ocorre
devido à presença de estados eletrônicos deslocalizados em energias próximas às
bandas de condução e de valência [89].
Utilizando a relação de Urbach [88],
V VZ[ >⁄ (Equação 27)
onde α0 é uma absorção de fundo (do inglês background absorption) e Et é a
energia relacionada ao grau de desordem do material, pode-se construir um gráfico
de ln (α) vs. hν. Através da inclinação desse gráfico, é possível obter o valor de Et,
sendo que quanto menor for este valor, menor a desordem do material. Neste ajuste,
o espectro de absorção é analisado em energias nas quais a absorção óptica se dá
início. Os ajustes para a solução aquosa com concentração de 0,05 mg/mL de SCC
são mostrados na Figura 54, com valores de Et = 301,6 meV para a banda Soret e Et
= 149,4 meV para a banda Q.

77
2,76 2,78 2,80 2,82
-0,8
-0,6
-0,4
lnα/
cm-1
hν/eV
Banda Soret
Et= 301,6 meV
1,78 1,80 1,82 1,84
-2,2
-2,0
-1,8
-1,6
lnα/
cm-1
hν/eV
Et=149,4 meV
Banda Q
Figura 54: Ajuste das curvas ln(α)-hν em energias próximas ao início da absorção óptica nas bandas Q e Soret para a solução aquosa de SCC com concentração de 0,05 mg/mL
As curvas ln(α)-hν foram traçadas a partir dos espectros de absorção dos
filmes (PAH/SCC)20, (PAH/SCC)40 e (PAH/SCC)80 e foram ajustadas conforme a
relação de Urbach a fim de se obter os valores de Et em energias próximas aos
máximos das bandas Soret e Q. Os resultados obtidos no ajuste são mostrados na, e
os valores de Et são mostrados na Figura 55.

78
2,60 2,62 2,64 2,66 2,68 2,7010,8
10,9
11,0
11,1
11,2
Filme (PAH/SCC)20
Banda Soret
lnα/
cm-1
hν/eV
Et = 386,3 meV
1,77 1,80 1,8310,4
10,5
10,6
10,7 Filme (PAH/SCC)20
Banda Q
lnα/
cm-1
hν/eV
Et = 307,1 meV
2,60 2,62 2,64 2,66 2,68 2,70
10,4
10,5
10,6
10,7
Filme (PAH/SCC)40
Banda Soret
lnα/
cm-1
hν/eV
Et = 393,1 meV
1,77 1,80 1,839,9
10,0
10,1
10,2
10,3
Filme (PAH/SCC)40
Banda Q
lnα/
cm-1
hν/eV
Et = 248,6 meV
2,60 2,62 2,64 2,66 2,68 2,70
10,0
10,1
10,2
10,3
10,4
Filme (PAH/SCC)80
Banda Soret
lnα/
cm-1
hν/eV
Et= 365,6 meV
1,77 1,80 1,83
9,76
9,84
9,92
10,00
Filme (PAH/SCC)80
Banda Q
lnα/
cm-1
hν/eV
Et = 287,0 meV
Figura 55: Ajuste das curvas ln(α)-hν em energias próximas ao início da absorção óptica nas bandas Soret e Q para o filme (PAH/SCC)80
Tabela 6: Cálculo do Et das bandas Soret e Q para os filmes (PAH/SCC)n
Filmes Et Banda Soret (meV) Et Banda Q (meV)
(PAH/SCC)20 386,3 307,1
(PAH/SCC)40 393,1 248,6
(PAH/SCC)80 365,6 287,0

79
De acordo com os valores obtidos para Et com relação às bandas Soret e Q
dos filmes (Tabela 6) e da solução aquosa, observa-se que os filmes apresentaram
maior grau de desordem em relação à solução e que a energia de desordem da
banda Soret é superior a energia da banda Q.
5.3. Tempo de relaxação dos portadores de cargas
Nas curvas de densidade de corrente por tempo, foi verificado um
decaimento exponencial estendido no tempo, também observado para vários
materiais com estrutura desordenada, sendo, então, um comportamento geral de
materiais amorfos [90]. Nesta relaxação, incluem-se vários processos como difusão
dependente do tempo [91 ], presença de armadilhas [92 ] e recombinação dos
portadores de carga [93].
Para explicar este fenômeno da fotocondutividade persistente, foram
propostos dois modelos [93]. Um deles leva em consideração uma distribuição
estatística do tempo de vida dos portadores com graus de liberdade não
correlacionados, que reflete a complexidade da dinâmica em estruturas
desordenadas. Então, assumindo todas as contribuições de cada grau de liberdade, a
relaxação é expressa da seguinte forma:
\ ] ^_`% ^⁄ a^b (Equação 28)
onde τ é o tempo de decaimento característico, n(t) é o número de portadores de
carga livres que decai com o tempo e ω(t) é uma função de distribuição.
Entretanto, este modelo faz uma aproximação microscopicamente arbitrária
[94], onde a distribuição ω(τ) deve ter uma origem microscópica na correlação
entre diferentes graus de liberdade. O segundo modelo propõe uma relaxação em
série até o equilibrio, com uma taxa de relaxação que depende do tempo, no qual
processos envolvidos na relaxação não são independentes uns dos outros. O
decaimento dos portadores excitados pela luz pode ser descrito pela equação
cd c %e\, (Equação 29)
onde o decaimento exponencial deve estar relacionado com a função k(t).
Na maioria dos fotocondutores amorfos, a mobilidade dos portadores de
carga em excesso exibe um decaimento na forma k(t) ~ t-(1-β) [95]. Substituindo k(t)

80
~ t-(1-β) na Equação 29 e integrando, obtém-se a lei do decaimento exponencial
estendido no tempo:
\ \_`f% ^⁄ gh, 0<β<1 (Equação 30)
onde n0 é a numero de portadores de carga livres inicial e β é um parâmentro
associado à distribuição de energias de ativação para recombinação. Na equação
convencional de relaxação dielétrica de Debye considera-se β=1. Entretanto,
experimentalmente a relaxação é melhor descrita por uma equação cujo valor de β
está compreendido entre 0 e 1. Primeiramente, Kohlrausch [96] definiu esta
equação para descrever a descarga de capacitores. Mais tarde, Williams e Watts
[97] postularam a mesma equação para a descrição da relaxação dielétrica.
Em termos da densidade de corrente, a Equação 30 fica da seguinte forma:
6 6 expf%^ ⁄ gh (Equação 31)
Através da Equação 31, o decaimento da fotocorrente com o tempo foi
ajustado a fim de se obter os parâmetros β e τ.
Os ajustes das curvas de fotocondutividade persistente (J-t) para o
dispositivo ITO/(PAH/SCC)80/Ca/Al são mostrados na Figura 56 e os parâmetros
obtidos por meio destes ajustes estão na Tabela 7.

81
0 10 20 30 40 500,0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
0,8
0,9
1,0
Curva experimental Ajuste
J = J0exp(-t/τ)β
τ = 4,913sβ = 0,463
Den
sida
de d
e co
rren
te/(
A/c
m²)
Tempo/s
ITO/(PAH/SCC)80
/Ca/Al
0 10 20 30 40 50
0,0
0,2
0,4
0,6
0,8
1,0 Curva experimental Ajuste
J = J0exp(-t/τ)β
τ = 0,961sβ = 0,296
ITO/(PAH/SCC)80
/Ca/Al
Den
sida
de d
e co
rren
te/(
A/c
m²)
Tempo/s
0 10 20 30 40 50 60
0,0
2,0x10-1
4,0x10-1
6,0x10-1
8,0x10-1
1,0x100
Curva experimental Ajuste
ITO/(PAH/SCC)80
/Ca/Al
J = J0exp(-t/τ)β
τ = 5,160sβ = 0,562
Den
sida
de d
e co
rren
te/(
A/c
m²)
Tempo/s
0 20 40 60
0,0
0,2
0,4
0,6
0,8
1,0 Curva experimental Ajuste
J = J0exp(-t/τ)β
τ = 8,413sβ = 0,451
ITO/(PAH/SCC)80
/Ca/Al
Den
sida
de d
e co
rren
te/(
A/c
m²)
Tempo/s
Figura 56: Ajustes das curvas J-t dos dispositivos ITO/(PAH/SCC)80/Ca/Al de acordo com a Equação 31, a fim de investigar aspectos da fotocondutividade persistente
Tabela 7: Valores obtidos para os parâmetros τ e β (Equação 31) do ajuste das curvas J-t do dispositivo ITO/(PAH/SCC)80/Ca/Al
τ(s) β
4,913 0,463
0,961 0,296
5,160 0,562
8,413 0,451
Na Figura 57 são mostrados os ajustes dos decaimentos presentes na curva
J-t (Figura 51) para o dispositivo ITO/(PAH/SCC)80/Al. Os parâmetros obtidos
estão na Tabela 8.

82
0 10 20 30 40 50 600,0
0,2
0,4
0,6
0,8
1,0 Curva experimental Ajuste
ITO/(PAH/SCC)80
/Al
Den
sida
de d
e co
rren
te/(
mA
/cm
2 )
Tempo/s
J = J0exp(-t/τ)β
τ = 0,410sβ = 0,179
0 10 20 30 40 50 60 70
0,0
0,2
0,4
0,6
0,8
1,0 Curva experimental Ajuste
J = J0exp(-t/τ)β
τ = 0,490sβ = 0,318
ITO/(PAH/SCC)80
/Al
Den
sida
de d
e co
rren
te/(
mA
/cm
2 )
Tempo/s
0 10 20 30 40 50 60 70
0,0
0,2
0,4
0,6
0,8
1,0
Curva experimental Ajuste
J = J0exp(-t/τ)β
τ = 0,545sβ = 0,323
ITO/(PAH/SCC)80
/Al
Den
sida
de d
e co
rren
te/(
mA
/cm
2 )
Tempo/s
0 10 20 30 40 50 60
0,0
0,2
0,4
0,6
0,8
1,0
Curva experimental Ajuste
J = J0exp(-t/τ)β
τ = 0,455sβ = 0,307
ITO/(PAH/SCC)80
/Al
Den
sida
de d
e co
rren
te/(
mA
/cm
2 )
Tempo/s
0 10 20 30 40 50 60
0,0
0,2
0,4
0,6
0,8
1,0 Curva experimental Ajuste
J = J0exp(-t/τ)β
τ = 0,537sβ = 0,317
ITO/(PAH/SCC)80
/Al
Den
sida
de d
e co
rren
te/(
mA
/cm
2)
Tempo/s
Figura 57: Ajustes das curvas J-t dos dispositivos ITO/(PAH/SCC)80/Ca/Al de acordo com a Equação31, a fim de investigar aspectos da fotocondutividade persistente

83
Tabela 8: Valores obtidos para os parâmetros τ e β (Equação 31) do ajuste das curvas J-t do dispositivo ITO/(PAH/SCC)80/Al
τ(s) β
0,410 0,179
0,491 0,318
0,545 0,320
0,456 0,307
0,537 0,317

84
6. Conclusão
A técnica de automontagem mostrou-se bastante eficaz na confecção de filmes
ultrafinos homogêneos a partir de soluções aquosas. Além disso, a técnica permite um
controle da espessura dos filmes, conforme verificado, e adsorção de uma mesma
quantidade de material a cada ciclo de deposição. Observou-se que o pH das soluções
de imersão utilizadas na automontagem influenciam na quantidade de material
adsorvido e, neste trabalho, concluiu-se que o pH básico (em torno de 8,0) é ideal para a
obtenção de filmes de (PAH/SCC).
Pela espectroscopia de absorção, foi possível analisar a influência do solvente
nas soluções de SCC, evidenciando uma mudança no comportamento das moléculas
quando se varia a polaridade do solvente (deslocamento solvatocrômico). Foi também
possível verificar que, com o aumento do número de bicamadas, aumenta-se a
intensidade de absorção de forma linear. Através dos espectros de absorção foi possível
determinar o gap energético em cada uma das principais transições eletrônicas e
calcular o grau de desordem destas transições em cada uma das amostras analisadas.
Com a espectroscopia de fluorescência, constatou-se, além da influência dos
solventes, os tipos de transições envolvidas nos decaimentos radiativos. Verificou-se
também que a solução aquosa de SCC, bem como os filmes, não apresentaram
fluorescência.
A espectroscopia de impedância permitiu a análise da resistividade dos filmes
confeccionados como camada ativa dos dispositivos fabricados neste trabalho e, por
meio desta técnica, calculou-se a capacitância e a constante dielétrica de cada um dos
filmes. Por meio desta, foi possível verificar que os filmes automontados de SCC
possuem uma alta resistividade.
As medidas de condutividade dc foram úteis para verificar o comportamento
fotocondutivo e o caráter fotovoltaico dos dispositivos fabricados e determinar a
eficiência energética de cada um dos dispositivos.
Por meio das análises de condutividade persistente (curvas J-t), analisou-se o
decaimento exponencial da fotocorrente quando a luz incidente é interrompida e, a
partir do ajuste deste decaimento, foi possível calcular o tempo de vida dos portadores
de carga fotogerada.

85
REFERÊNCIAS BIBLIOGRÁFICAS
1 SZE, S. M.: Semiconductor devices: physics and technology. John Wiley & Sons,
2008. 2 WERNER, K.: Higher visibility for LEDs. Spectrum, IEEE, v. 31, n. 7, p. 30-34, 1994. 3 ANIL KUMAR, R.; SURESH, M. S.; NAGARAJU, J.: Measurement and comparison
of AC parameters of silicon (BSR and BSFR) and gallium arsenide (GaAs/Ge) solar cells used in space applications. Solar energy materials and solar cells, v. 60, n. 2, p. 155-166, 2000.
4 SOLOMONS, T. W.; FRYHLE, G. B.: Química Orgânica, vol. 1. LTC Editora: Rio de Janeiro, 1996. 5GREENHAM, N. C.; FRIEND, R. H.; PEIERLS, R. E.: Em Quantum Theory of Solids. Quantum Theory of Solids, v. 49, 1995. 6ATKINS, P. W.; JONES, L.: Princípios de Química: questionando a vida moderna e o
meio ambiente. Bookman, 2001. 7 HEEGER, A. J. et al.: Solitons in conducting polymers. Reviews of Modern Physics,
v. 60, n. 3, p. 781, 1988. 8 COUTINHO, D. J.: Estudo e caracterização de dispositivos fotovoltaicos orgânicos
(OPV) baseados em heterojunção de volume. Dissertação de Mestrado, Instituto de Física de São Carlos, Universidade de São Paulo, São Carlos, 2011.
9 BAEUERLE, P. et al.: Synthesis and characterization of mixed oligoheterocycles based on end-capped oligothiophenes suited for LED applications. In: SPIE's International Symposium on Optical Science, Engineering, and Instrumentation. International Society for Optics and Photonics, 1998. p. 32-39. 10 MONTES, V. A. et al.: Molecular-wire behavior of OLED materials: exciton dynamics in multichromophoric Alq3-oligofluorene-Pt (II) porphyrin triads. Journal of the American Chemical Society, v. 128, n. 38, p. 12436-12438, 2006. 11 BARKER, C. A. et al.: Porphyrin, Phthalocyanine and Porphyrazine Derivatives with Multifluorenyl Substituents as Efficient Deep‐Red Emitters. Chemistry-A European Journal, v. 13, n. 23, p. 6710-6717, 2007. 12 SONNTAG, M. et al.: Novel star-shaped triphenylamine-based molecular glasses and their use in OFETs. Chemistry of materials, v. 17, n. 11, p. 3031-3039, 2005. 13 MINARI, T. et al.: Molecular-packing-enhanced charge transport in organic field-effect transistors based on semiconducting porphyrin crystals. Applied Physics Letters, v. 91, n. 12, p. 123501, 2007. 14 GÜNES, S.; NEUGEBAUER, H.; SARICIFTCI, N. S.: Conjugated polymer-based
organic solar cells. Chemical reviews, v. 107, n. 4, p. 1324-1338, 2007.

86
15 SPANGGAARD, H.; KREBS, F. C.: A brief history of the development of organic
and polymeric photovoltaics. Solar Energy Materials and Solar Cells, v. 83, n. 2, p. 125-146, 2004.
16 SUN, Y. et al.: Solution-processed small-molecule solar cells with 6.7% efficiency. Nature materials, v. 11, n. 1, p. 44-48, 2012.
17 WALTER, M. G.; RUDINE, A. B.; WAMSER, C. C.: Porphyrins and phthalocyanines in solar photovoltaic cells. Journal of Porphyrins and Phthalocyanines, v. 14, n. 09, p. 759-792, 2010. 18 GAMEIRO Júnior, FERREIRA, A.: Filmes nanoestruturados de porfirinas com
aplicações em sensores de gases. Dissertação de Mestrado, Faculdade de Ciências e Tecnologia, Universidade Estadual Paulista Júlio Mesquita Filho, Presidente Prudente, 2006.
19 STILLMAN, M. J.; NYOKONG, T.: Phthalocyanines: Properties and Applications. VCH, New York, p. 133, 1989.
20 RAWLING, T.; MCDONAGH, A.: Ruthenium phthalocyanine and naphthalocyanine complexes: Synthesis, properties and applications. Coordination chemistry reviews, v. 251, n. 9, p. 1128-1157, 2007. 21 DRZEWIECKA-MATUSZEK, A. et al.: Effects of heavy central metal on the ground and excited states of chlorophyll. JBIC Journal of Biological Inorganic Chemistry, v. 10, n. 5, p. 453-462, 2005. 22 PINEIRO, M. et al. Photoacoustic measurements of porphyrin triplet-state quantum yields and singlet-oxygen efficiencies. Chemistry - A European Journal, v. 4, n. 11, p. 2299-2307, 1998. 23 CALLARI, F. L. et al.: Biocompatible nanoparticles of amphiphilic cyclodextrins
entangling porphyrins as suitable vessels for light-induced energy and electron transfer. Journal of Materials Chemistry, v. 18, n. 7, p. 802-805, 2008.
24 BONNETT, R.: Photosensitizers of the porphyrin and phthalocyanine series for photodynamic therapy. Chemical Society Reviews, v. 24, n. 1, p. 19-33, 1995.
25 GOUTERMAN, M.: Spectra of porphyrins. Journal of Molecular Spectroscopy, v. 6, p. 138-163, 1961.
26 VALICSEK, Z.; HORVÁTH, O.: Application of the electronic spectra of porphyrins for analytical purposes: The effects of metal ions and structural distortions. Microchemical Journal, v. 107, p. 47-62, 2013. 27 LIM, E. C.; LI, Y. H.: Non-planar distortions of, and polarization of luminescence
from, ππ* states of some nitrogen heterocyclic compounds. Chemical Physics Letters, v. 4, n. 2, p. 68-70, 1969.
28 LI, R.; LIM, E. C.: Quantitative Study of Luminescence in Aromatic and Heteroaromatic Molecules. The Journal of Chemical Physics, v. 57, p. 605, 1972.

87
29 VALEUR, B: Molecular fluorescence - Principles and Applications. Wiley-VCH
Germany (2002). 30 PARUSEL, A. B. J.; GRIMME, S.: A theoretical study of the excited states of
chlorophyll a and pheophytin a. The Journal of Physical Chemistry B, v. 104, n. 22, p. 5395-5398, 2000.
31 DE BONI, L. et al.: Excited state absorption spectrum of chlorophyll a obtained with white-light continuum. Journal of Chemical Physics, v. 126, n. 16, p. 165102-165102, 2007.
32 PEREIRA, Mário R.: Fotofísica de porfirinas e ftalocianinas em matrizes de TiO2 preparadas pelo método de Sol-Gel. Tese de Doutorado, Universidade do Minho, Braga, 2004. 33 KRAUSE, G. H.; WEIS, E.: Chlorophyll fluorescence and photosynthesis: the
basics. Annual review of plant biology, v. 42, n. 1, p. 313-349, 1991. 34 MCCARTHY, M. R.; VANDEGRIFF, K. D.; WINSLOW, R. M.: The role of
facilitated diffusion in oxygen transport by cell-free hemoglobins: implications for the design of hemoglobin-based oxygen carriers. Biophysical chemistry, v. 92, n. 1, p. 103-117, 2001.
35 POULOS, T. L.; KRAUT, J.: The stereochemistry of peroxidase catalysis. Journal of Biological Chemistry, v. 255, n. 17, p. 8199-8205, 1980.
36 NABOK, A. V. et al.: Nitrogen oxide gas sensor based on tetra-tertbutyl copper phthalocyanine Langmuir-Blodgett films. International journal of electronics, v. 78, n. 1, p. 129-133, 1995.
37 ZHOU, R. et al.: Phthalocyanines as sensitive materials for chemical sensors. Applied Organometallic Chemistry, v. 10, n. 8, p. 557-577, 1996.
38 SCHUMANN, S.; HATTON, R. A.; JONES, T. S.: Organic photovoltaic devices based on water-soluble copper phthalocyanine. The Journal of Physical Chemistry C, v. 115, n. 11, p. 4916-4921, 2011. 39 BOTTARI, G. et al.: Covalent and noncovalent phthalocyanine− carbon nanostructure systems: synthesis, photoinduced electron transfer, and application to molecular photovoltaics. Chemical reviews, v. 110, n. 11, p. 6768-6816, 2010. 40 FARAG, A. A. M. et al.: Electrical conductivity, dielectric properties and optical
absorption of organic based nanocrystalline sodium copper chlorophyllin for photodiode application. Journal of Alloys and Compounds, v. 513, p. 404-413, 2012.
41 CLAESSENS, C. G.; HAHN, U.; TORRES, T.. Phthalocyanines: from outstanding electronic properties to emerging applications. The Chemical Record, v. 8, n. 2, p. 75-97, 2008.

88
42 RYAN, J. W. et al.: Small molecule solar cells based on a series of water-soluble zinc phthalocyanine donors. Chemical Communications, v. 48, n. 49, p. 6094-6096, 2012. 43 TANG, C. W.; ALBRECHT, A. C.: Transient photovoltaic effects in metal–
chlorophyll‐a–metal sandwich cells. The Journal of Chemical Physics, v. 63, n. 2, p. 953-961, 1975.
44 TANG, C. W.; ALBRECHT, A. C.: Photovoltaic effects of metal–chlorophyll‐a–metal sandwich cells. The Journal of Chemical Physics, v. 62, n. 6, p. 2139-2149, 1975.
45 ISMAIL, S. I. et al.: Charge transfer complex formation constants of edta with methylviologen And its zwitterionic derivatives. Qatar University Science Journal, v. 14, p. 161,1994.
46 IRIYAMA, K.; OGURA, N.; TAKAMIYA, A.: A simple method for extraction and
partial purification of chlorophyll from plant material, using dioxane. Journal of biochemistry, v. 76, n. 4, p. 901-904, 1974.
47 EGNER, P. A. et al.: Chlorophyllin intervention reduces aflatoxin–DNA adducts in individuals at high risk for liver cancer. Proceedings of the National Academy of Sciences, v. 98, n. 25, p. 14601-14606, 2001. 48 GOMES, B. B. et al.: Bioavailability of dietary sodium copper chlorophyllin and its effect on antioxidant defence parameters of Wistar rats. Journal of the Science of Food and Agriculture, v. 89, n. 12, p. 2003-2010, 2009. 49 AYDIN, M. E. et al.: Device characterization of organic nanostructure based on
sodium copper chlorophyllin (SCC). Synthetic Metals, v. 161, n. 23, p. 2700-2707, 2012.
50 NALWA, H. S.; ROHWER, L. S.: Handbook of luminescence, display materials, and devices. Vol. 1, Organic light-emitting diodes. American scientific publishers, 2003.
51 ARUNACHALAM, V. S.; FLEISCHER, E. L.: Harnessing materials for energy. MRS Bulletin, v. 33, n. 04, p. 355-371, 2008.
52 BRABEC, C. J.: Organic photovoltaics: concepts and realization. Springer, 2003. 53 BRABEC, C. J. Organic photovoltaics: technology and market. Solar energy
materials and solar cells, v. 83, n. 2, p. 273-292, 2004. 54 GREGG, B. A.; HANNA, M. C.: Comparing organic to inorganic photovoltaic cells:
Theory, experiment, and simulation. Journal of Applied Physics, v. 93, n. 6, p. 3605-3614, 2003.

89
55 SÁNCHEZ-DÍAZ, A. et al.: Charge transfer reactions in near IR absorbing small
molecule solution processed organic bulk-heterojunction solar. Organic Electronics, v. 12, n. 2, p. 329-335, 2011.
56 UCHIDA, S. et al.: Organic small molecule solar cells with a homogeneously mixed copper phthalocyanine: C60 active layer. Applied physics letters, v. 84, n. 21, p. 4218-4220, 2004.
57 GONÇALVES, P. J. et al.: Effects of environment on the photophysical characteristics of mesotetrakis methylpyridiniumyl porphyrin (TMPyP). Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy, v. 79, n. 5, p. 1532-1539, 2011.
58 HOWE, L.; ZHANG, J. Z.: Ultrafast studies of excited-state dynamics of phthalocyanine and zinc phthalocyanine tetrasulfonate in solution. The Journal of Physical Chemistry A, v. 101, n. 18, p. 3207-3213, 1997.
59 KHAN, M. I.; BAZAN, G. C.; POPOVIC, Z. D.: Evidence for electric field-assisted dissociation of the excited singlet state into charge carriers in MEH–PPV. Chemical physics letters, v. 298, n. 4, p. 309-314, 1998. 60 PERSSON, B. N. J.; LANG, N. D.: Electron-hole-pair quenching of excited states
near a metal. Physical Review B, v. 26, p. 5409-5415, 1982. 61 ANTONIADIS, H. et al.: Enhanced carrier photogeneration by defects in conjugated
polymers and its mechanism. Physical Review B, v. 50, n. 20, p. 14911, 1994. 62 CHAWDHURY, N. et al.: The effects of H2O and O2 on the photocurrent spectra of
MEH-PPV. Synthetic metals, v. 102, n. 1, p. 871-872, 1999. 63 TANG, C. W. Two‐layer organic photovoltaic cell. Applied Physics Letters, v. 48, p.
183, 1986. 64 FARCHIONI, R.; GROSSO, G.: Organic Electronic Materials: Conjugted Polymers
and Low Molecular Weight Electronic Solids. Springer, 2001. 65 HENDRY, E. et al. Efficiency of exciton and charge carrier photogeneration in a
semiconducting polymer. Physical review letters, v. 92, n. 19, p. 196601, 2004. 66 NAGASHIMA, H. N.: Simulação de condutividade alternada em sistemas poliméricos e aplicações em polímeros condutivos. Tese de Doutorado, Instituto de Física de São Carlos, Universidade de São Paulo, São Carlos, 2000. 67 BIANCHI, C.; CECCHETTO, E.; FRANÇOIS, B.: Synthesis and characterisation of
poly (ethylene oxide)-block-poly(para-phenylene vinylene). Synthetic metals, v. 102, n. 1, p. 916-917, 1999.
68 CAZATI, T.: Efeito da fotocondução em diodos com camada ativa de derivados de poli (p-fenileno vinileno)(PPV). Tese de Doutorado. Instituto de Física de São Carlos, Universidade de São Paulo, São Carlos, 2008.

90
69 LOWACK, K.; HELM, C. A.: Molecular mechanisms controlling the self-assembly
process of polyelectrolyte multilayers. Macromolecules, v. 31, n. 3, p. 823-833, 1998. 70 PATERNO, L. G.; MATTOSO, L. H. C.; OLIVEIRA Júnior, O. N.: Filmes
poliméricos ultrafinos produzidos pela técnica de automontagem: preparação, propriedades e aplicações. Química Nova, v. 24, n. 2, p. 228-235, 2001.
71 LVOV, Y.; DECHER, G.; MOEHWALD, H.: Assembly, structural characterization, and thermal behavior of layer-by-layer deposited ultrathin films of poly (vinyl sulfate) and poly (allylamine). Langmuir, v. 9, n. 2, p. 481-486, 1993.
72 HOOGEVEEN, N. G. et al.: Formation and stability of multilayers of polyelectrolytes. Langmuir, v. 12, n. 15, p. 3675-3681, 1996.
73 SCHMITT, J. et al.: Internal structure of layer-by-layer adsorbed polyelectrolyte films: a neutron and X-ray reflectivity study. Macromolecules, v. 26, n. 25, p. 7058-7063, 1993.
74 ARAÚJO, M. H.: Filmes automontados contendo derivados de clorofila. Relatório de iniciação científica, Universidade Federal de Ouro Preto, 2012.
75 FERREIRA, L. L.: Filmes automontados contendo derivados de clorofila. Relatório de iniciação científica, Universidade Federal de Ouro Preto, 2013.
76 RHODERICK, Emlyn Huw; WILLIAMS, R. H.: Metal-semiconductor contacts. Oxford: Clarendon Press, 1988.
77 YAMANARI, Toshihiro et al.: Origin of the open-circuit voltage of organic thin-film solar cells based on conjugated polymers. Solar Energy Materials and Solar Cells, v. 93, n. 6, p. 759-761, 2009.
78 BRABEC, C. J. et al.: Origin of the open circuit voltage of plastic solar cells. Advanced Functional Materials, v. 11, n. 5, p. 374-380, 2001
79 MACIEL, A. C.: Propriedades elétricas de sólidos desordenados: aplicação em estruturas metal/polímero/metal. Trabalho de conclusão de curso, Universidade Federal do Piauí, 2006.
80 ZUCOLOTTO, V. et al.: Unusual interactions binding iron tetrasulfonated phthalocyanine and poly (allylamine hydrochloride) in layer-by-layer films. The Journal of Physical Chemistry B, v. 107, n. 16, p. 3733-3737, 2003.
81 DE MIRANDA, J. A.: Caracterização fotofísica de derivados de cumarinas. Tese de Doutorado, Universidade Federal de Uberlândia, 2001
82 WAGENPFAHL, A. et al. S-shaped current-voltage characteristics of organic solar devices. Physical Review B, v. 82, n. 11, p. 115306, 2010.
83 BENTEN, H. et al.: Layer-by-layer deposition films of copper phthalocyanine derivative; their photoelectrochemical properties and application to solution-processed thin-film organic solar cells. Thin Solid Films, v. 517, n. 6, p. 2016-2022, 2009.

91
84 DYRE, J. C., SCHRODER, T. B.: Hopping Models and ac Universality. Physica
Status Solidi, v. 230, p. 5-13, 2012. 85 FAKIR, M. S.; AHMAD, Z.; SULAIMAN, K.: Modification of Optical Band Gap
and Surface Morphology of NiTsPc Thin Films. Chinese Physics Letters, v. 29, n. 12, p. 126802, 2012.
86 EL-NAHASS, M. M. et al.: Dispersion studies and electronic transitions in nickel phthalocyanine thin films. Optics & Laser Technology, v. 37, n. 7, p. 513-523, 2005.
87 TAUC, J.; GRIGOROVICI, R.; VANCU, A.: Optical properties and electronic structure of amorphous germanium. Physica status solidi (b), v. 15, n. 2, p. 627-637, 1966.
88 URBACH, F.: The long-wavelength edge of photographic sensitivity and of the electronic absorption of solids. Physical Review, v. 92, p. 1324-1324, 1953.
89 CAPIZZI, M.; FROVA, A.: Optical gap of strontium titanate (deviation from Urbach tail behavior). Physical Review Letters, v. 25, n. 18, p. 1298, 1970.
90 SAITO, R.; MURAYAMA, K.: A universal distribution function of relaxation in amorphous materials. Solid state communications, v. 63, n. 7, p. 625-627, 1987.
91 KAKALIOS, J.; STREET, R. A.; JACKSON, W. B.: Stretched-exponential relaxation arising from dispersive diffusion of hydrogen in amorphous silicon. Physical review letters, v. 59, n. 9, p. 1037, 1987.
92 SANTOS, L. F. et al.: Observation of persistent photoconductivity in vanadyl phthalocyanine. Journal of Physics D: Applied Physics, v. 41, n. 12, p. 125107, 2008.
93 LEE, C. H.; YU, G.; HEEGER, A. J.: Persistent photoconductivity in poly (p-phenylenevinylene): Spectral response and slow relaxation. Physical Review B, v. 47, n. 23, p. 15543, 1993.
94 PALMER, R. G. et al.: Models of hierarchically constrained dynamics for glassy relaxation. Physical Review Letters, v. 53, n. 10, p. 958, 1984.
95 SCHER, H.; MONTROLL, E. W.: Anomalous transit-time dispersion in amorphous solids. Physical Review B, v. 12, n. 6, p. 2455, 1975.
96 KOHLRAUSCH, R.: Theorie des elektrischen Rückstandes in der Leidener Flasche. Annalen der Physik, v. 167, n. 2, p. 179-214, 1854.
97 WILLIAMS, G.; WATTS, D. C.: Non-symmetrical dielectric relaxation behaviour arising from a simple empirical decay function. Transactions of the Faraday Society, v. 66, p. 80-85, 1970.